

Abstract
Living systems achieve robust self-assembly across a wide range of length scales. In the synthetic realm, nanofabrication strategies such as DNA origami have enabled robust self-assembly of submicron-scale shapes from a multitude of single-stranded components. To achieve greater complexity, subsequent hierarchical joining of origami can be pursued. However, erroneous and missing linkages restrict the number of unique origami that can be practically combined into a single design. Here we extend crisscross polymerization, a strategy previously demonstrated with single-stranded components, to DNA-origami “slats” for fabrication of custom multi-micron shapes with user-defined nanoscale surface patterning. Using a library of ~2000 strands that are combinatorially arranged to create unique DNA-origami slats, we realize finite structures composed of >1000 uniquely addressable slats, with a mass exceeding 5 GDa and with lateral dimensions of roughly 2 μm, as well as a multitude of periodic structures. Robust production of target crisscross structures is enabled through strict control over initiation, rapid growth and minimal premature termination, and highly orthogonal binding specificities. Thus crisscross growth provides a route for prototyping and scalable production of structures integrating thousands of unique components (i.e. origami slats) that each are sophisticated and molecularly precise.
In structural DNA nanotechnology1–27, the scaffolded DNA origami method affords robust self-assembly of arbitrary two- and three-dimensional nanoscale objects1–6. The oligonucleotide “staple” strands are designed to lack complementarity to each other, and folding is exactly controlled by a long single-stranded DNA (ssDNA) scaffold that is substoichiometric with respect to the staple strands, resulting in one-to-one conversion of scaffold particles into origami. This absolute scaffold dependence enables assembly over a broad range of temperatures and salt concentrations, while circumventing accumulation of incomplete assemblies or byproducts. Due to their robust folding performance, origami offer a user-friendly approach — suitable for specialists and non-specialists alike — for creating structures with addressable features. Applications have included plasmonic devices relying on placement of nanoparticles28,29, therapeutic devices with spatial control over cargos that sense the in vivo environment30,31, and research tools that place biomolecules in specified arrangements to deduce their biophysical properties32,33.
One limitation of DNA origami is that their maximal size is limited to about five megadaltons because the shape is bounded by the length of the scaffold DNA. While it is possible to use longer scaffold sequences, they are difficult to obtain in ssDNA form, and are delicate to handle because they are prone to shearing7–9. DNA bricks10–13 may instead be used to create structures significantly larger than a single origami, with as many as 30,000 unique monomers and a total mass of ~0.5 GDa per assembled particle. In contrast to origami “staple” strands, DNA tiles and bricks are complementary to each other thus eliminating the scaffold dependence of the assembly. However, spontaneous association of building blocks effectively limits the yield for such single-pot growth processes and increases the burden for post-assembly purification26. Additionally, for the largest of such structures, current pricing on nanomole-scale oligonucleotide synthesis can be cost-prohibitive. For example, assembly of a 0.5 gigadalton megastructure required ~30,000 distinct 52mers, at a cost of roughly USD$150,000, with a final yield of ~1%.
Hierarchical approaches can be used, however achieving assemblies containing more than a few distinct DNA nanostructures has been challenging14–24 (see Supplementary Text 1 for discussion of hierarchical and periodic DNA-origami assembly methods). In the most complex demonstration in terms of the number of unique DNA origamis to date, structures consisting of 64 unique DNA-origami components were constructed using a method termed fractal assembly. To suppress off-target joining of DNA-origami monomers, three sequential steps were employed to build 4-component, then 16-component, and finally 64-component supershapes25. Fine-tuning of monomer stoichiometries and reaction temperatures were required, nevertheless reported yields dropped from ~93% to ~48% to ~2% for the three steps, respectively. Despite only four sub-assemblies coming together per stage and precise care in preparation, unfinished, erroneous, and aggregated by-products led to rapidly diminishing yields as more unique monomers and assembly stages were added. Thus fractal assembly in this way appears effective for hierarchical constructions with dozens of components, but may face severe yield issues when larger numbers of parts are desired.
Previously we introduced crisscross polymerization of ssDNA slats for robust control over nucleation26; here we generalize this method to operate with DNA-origami slats — which are over two orders of magnitude larger than their ssDNA counterparts — for synchronous initiation of growth of target supershapes from relatively small seeds. Using six-helix bundle (6HB)22,34,35 and twelve-helix bundle (12HB) nanorods extending weak binding handles along their lengths, we created a diversity of finite and periodic assemblies. The number of fully formed assemblies is controlled exactly by the amount of seed added, with the robustness of growth from hundreds of origami-based parts comparable to that of origami folding itself from similar numbers of much smaller components.
Design of the DNA-origami slats
In crisscross polymerization, an incoming slat must engage with a large number of other slats (up to either eight or sixteen in this study) for stable attachment to the edge of a growing structure; this requirement for a high level of coordination is the basis for the robustness of crisscross against unwanted spurious nucleation26 (Fig. S1). To meet this design criterion, each individual pairwise interaction must be quite weak at the desired temperature of growth, and ideally far below this temperature as well. For ssDNA slats, this was achieved with interactions that span just a half turn of DNA (i.e. 5 or 6 bp)26. We hypothesized that crisscross assembly could also be implemented for DNA-origami slats by engineering sufficiently weak binding handles. Then to bypass the nucleation barrier in a controlled fashion, pre-formed seeds could be employed that use much stronger binding interactions to capture an initial set of “nucleating” slats in an arrangement that resembles a critical nucleus (Fig. S1Bi–ii). Consequently, a cascade of energetically favorable downstream assembly steps could propagate growth (Fig. 1A, Fig. S1Biii–v).
Figure 1.
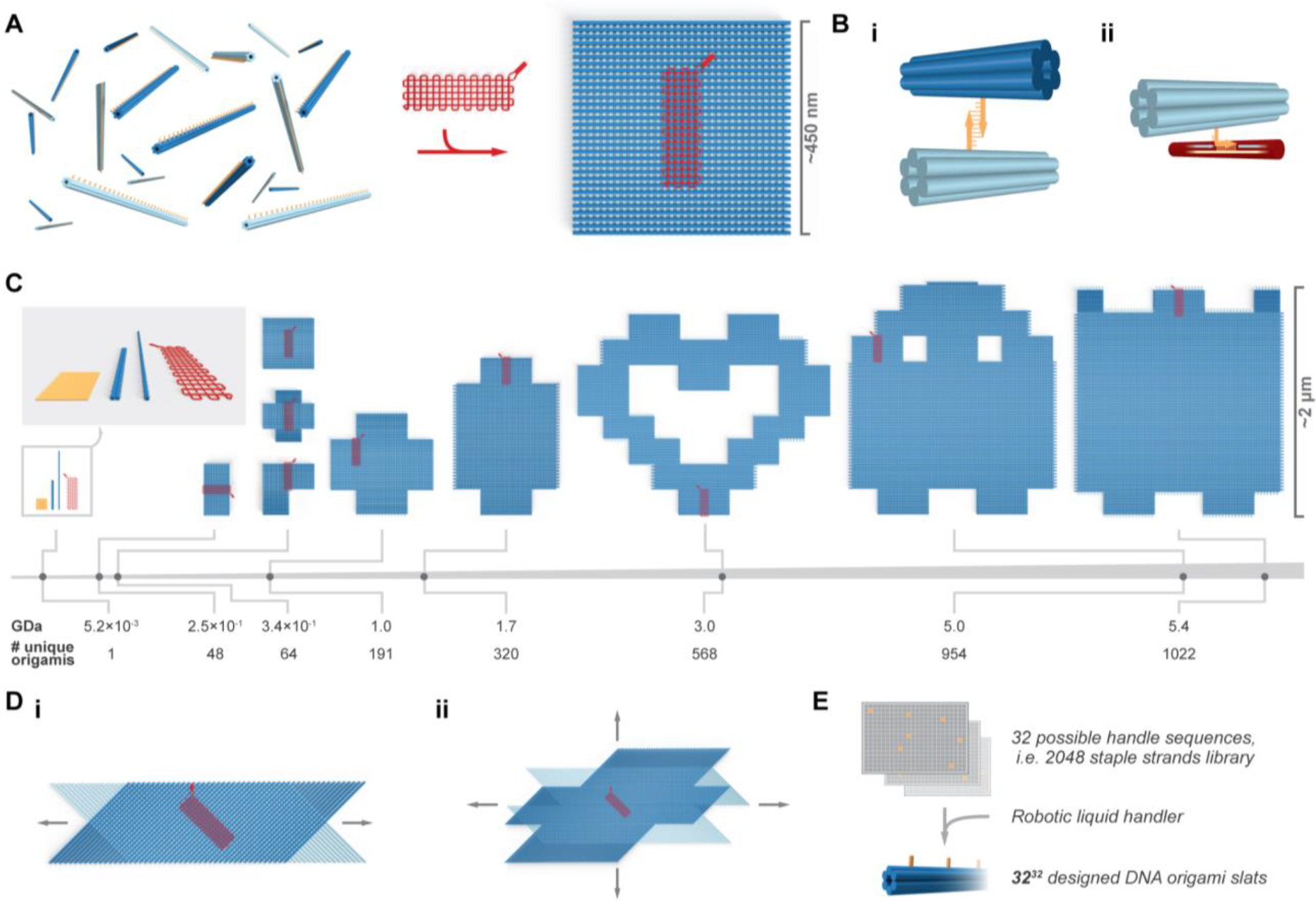
Overview of crisscross assembly of DNA-origami slats. A, left, a pool of 64 unique free 6HB slats. A 6HB slat functionally comprises a linear arrangement of 32 binding-site sequences, each selected from the same set of 32 distinct sequences; a square megastructure with 64 unique slats is triggered to form only when the gridiron-origami seed is added. Bi, binding of a pair of complementary weak 7-nt handles on two perpendicular 6HB slats. Bii, a strong 10-nt “plug” handle on a 6HB slat engaged with an exposed region of scaffold (i.e. a “socket”) on the gridiron seed. C, the breadth and relative scale of the megastructures tested, versus the leftward single DNA-origami slats, seed, and origami reference square. Di–ii, renderings of periodic one- and two-dimensionally growing ribbons and sheets. E, schematic for how subsets of the strand library are combinatorially collected to yield unique slats.
As a convenient shorthand, we refer to any designed DNA-based assembly that consists of roughly a million or more nucleotides as “megastructures”. To achieve crisscross growth of megastructures from DNA-origami building blocks, we designed 6HB and 12HB slats that assemble by nucleating upon a gridiron-origami6 seed (Fig. 1A, Fig. S2A). The 3′ ends of staple strands on the top and bottom helices of each slat were encoded with ssDNA binding handles to link the slats to one another, though each could alternatively be programmed as an addressable “node” that engages a desired cargo (Fig. 1Bi, Fig. S2Bi–ii). The 6HB is ~450 nm long and features 32 handle positions spaced ~14 nm apart along its length; the 12HB is ~225 nm long with 16 positions along its length (see Supplementary Text 2 and Fig. S3 for more discussion of the 12HB design). As depicted in red in Fig. 1A and in Fig. S2Aiii, the seed has 16 columns of five “sockets”, where each column captures an individual “nucleating” slat with five 10-nucleotide (nt) handles that each “plug” into its complementary socket (Fig. S2Biii, Fig. 1Bii). Spacings of the plug handles on a nucleating slat, at 42 bp intervals, are commensurate with spacing of seed sockets. We validated folding of 6HB slats, 12HB slats, and seeds by imaging with negative-stain transmission electron microscopy (TEM, Fig. S4–Fig. S5).
We tested unseeded formation of sample crisscross structures with 10-, 9-, 8-, 7-, or 6-nt handles to explore how handle length affects spurious nucleation as a function of temperature. The 10-, 9-, and 8-nt handles were found to yield significant unseeded assembly at relevant temperatures (Fig. S6A–Ci). However, this was not observed with either the 7- or 6-nt handles (Fig. S6Cii–iii). Hence, we narrowed our focus to 7-nt handles, affording greater thermal stability versus 6-nt handles, for creating origami-slat megastructures (Fig. 1C–D). The algorithm for designing the sequence handles, computed energies versus handle length, and need for poly-T linkers are described in Supplementary Text 3, Fig. S7, and Fig. S8, respectively.
Finite megastructures
We conceived of many finite and periodic megastructures that could be made from two perpendicular layers of the DNA slats (Fig. 1C–D). Initially, we considered creating these designs using unique handles at the slat intersections, but such an approach would have been cost prohibitive since a distinct set of staple strands would need to be purchased for each design. We hypothesized that with the same handle sequence appearing on distinct slats, or even on the same slat, any transient non-target interaction would readily dissociate except in cases where several matching handles could be engaged simultaneously. We selected 32 isoenergetic 7-nt handle sequences and purchased a library of 2048 staple strands (i.e. 32 positions × 32 handle sequences × 2 for complementary handles) that would allow any one of these handles to be encoded at any possible perpendicular slat intersection. In principle, subsets of staple strands could be selected from this library to create up to 3232 (~1048) distinct 6HB (or 3216 ≈ 1024 half-length 12HB using an analogous library strategy) slats (Fig. 1E).
We tested this library for growth of finite megastructures to determine the extent to which the strands could be rearranged to make novel shapes. The relative area, molecular mass, and number of unique DNA origamis in each are shown in Fig. 1C. The smaller shapes with 48 and 64 slats have maximum lateral dimensions limited to 450 nm, the length of a 6HB slat (Fig. S9A). In order to achieve larger dimensions, we also designed assembly trajectories where the 6HB slats join in a zigzagging raster-fill pattern where for a typical step, sixteen parallel slats bind to each of two growth fronts that rotates 90 degrees clockwise or counterclockwise (Fig. S9B–D, Fig. S10). Using this raster-fill growth paradigm, we created larger megastructures including a 191-slat plus symbol, a 320-slat elongated plus symbol, a 568-slat heart, a ghost caricature with 954 slats, and a sheet with 1022 slats. The largest 1022-slat sheet has lateral dimensions ~2 μm and a molecular mass of ~5.4 gigadaltons, which is over an order of magnitude greater number of unique DNA origamis compared to fractal tiles as previously published25 (Fig. 1C). We juxtaposed a single DNA-origami square in Fig. 1C (i.e. the 85 nm × 85 nm shape as shown with leftmost orange render) to illustrate how origami crisscross overcomes the size limits attainable with single origamis.
We selected strands from the handle library for independent folding of each slat. We then combined the folded slats into pools with maximally 100 unique members, and concentrated each pool into a smaller volume (Fig. S11). Crisscross growth was initiated by mixing a seed with top- and bottom-layer slat pools. The larger finite shapes with rastering growth (Fig. S9C–D, Fig. S10, Supplementary Video 1) were assembled in several stages, where ~200 of the slats were added and incubated for ~60 hours before 2.5-fold dilution into a pool of the subsequent series of slats. We successfully assembled the panel of finite megastructures, as shown by TEM in Fig. 2. All the megastructures formed dispersed single particles in a seed-dependent fashion (Fig. S12–Fig. S15). The structures in Fig. 2Ai–v were formed exclusively with 6HB slats, with the exception of the 1022-slat sheet in Fig. 2Avi that also had 28 12HB slats. Half or more of the total slats in the Fig. 2B shapes were the shorter 12HB slats, allowing for the megastructures to have features finer than the length of a 6HB slat (Supplementary Text 2, Fig. S3).
Figure 2.
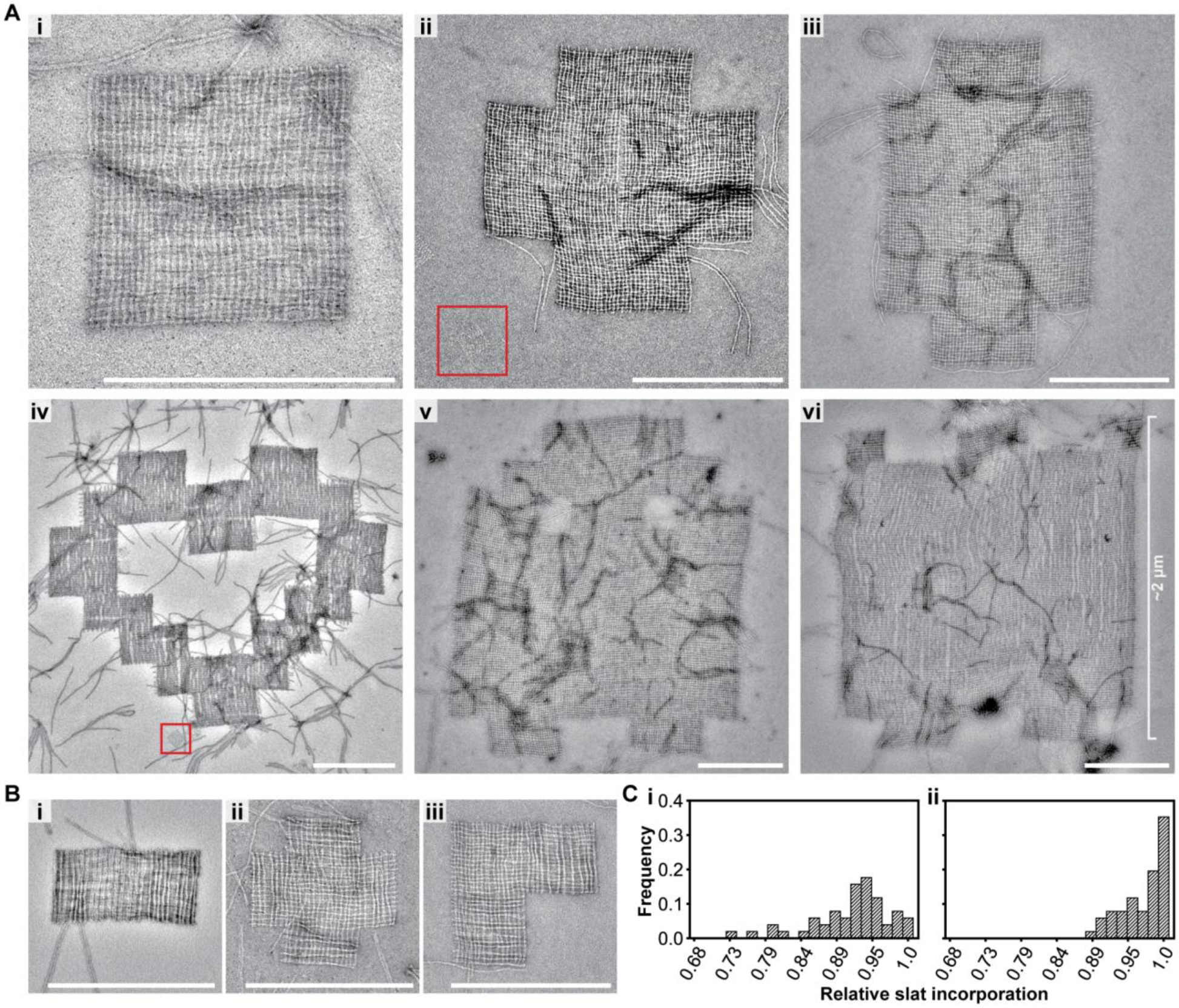
Assembly of finite megastructures from DNA-origami slats, where every slat is unique and addressable (see the designs in Fig. 1C). A, TEM images of megastructures composed entirely of 6HB slats, except for the 1022-slat rectangle that has 28 horizontal 12HB slats in Avi. The red boxed regions are a single origami reference square for size comparison, which is the largest area structure attainable with the same scaffold used for each slat. B, TEM images of finite megastructures where half or more of the slats are 12HBs. C, histogram for the number of slats counted in close-up TEM images of 50 randomly selected finite squares. The squares were assembled at 34°C overnight (Ci) versus assembled for an additional two days at room temperature (Cii). Scale bars are 500 nm.
To assess the relative incorporation of the 6HB slats into the megastructures, we counted the number of slats in higher-magnification TEM images in 64-slat squares. We determined an average incorporation of ~90% of the slats after overnight isothermal assembly, increasing to ~97% after an additional two days at room temperature (Fig. 2Ci–ii), which is comparable to the ~80–90% full incorporation of a given staple strand previously reported for some DNA origami structures36. We also assessed the relative completion of the largest finite megastructures by concentrating the final samples and looking to see if the features of each shape (i.e. corners and middle sections of the shape were appropriately filled with slats) could be observed in lower-magnification TEM images. Greater than 22% of the megastructures for the 954-slat ghost and 1022-slat sheet were fully grown as opposed to prematurely terminated (i.e. all the major morphological features could be observed, as explained in Method 15) by the last assembly stage (N954_ghost = 225, N1022_sheet = 267, see Fig. S15). This suggests that over 75% of the assemblies at each stage were competent for continuing growth (i.e. 0.78n = 0.225, where n = 6 growth stages, see caption of Fig. S15).
Periodic ribbon and sheet megastructures
We used the strand library to create periodic 6HB-based crisscross ribbons and sheets (Fig. 3). We first explored the ribbons depicted in Fig. 3A, which grew bidirectionally from the first series of slats bound to the seed. In Fig. S16A–B, each slat added is staggered one binding site unit compared to the parallel slat that preceded it, as similar to previously established for ssDNA slats26. The ribbons in Fig. 3Ai versus Fig. S17 are termed version 16 (v16) and version 8 (v8), respectively. In v16, a given slat has 32 perpendicular slats bound to all of its 32 possible binding handles, versus v8 which only has 16 slats bound to every other of its 32 possible binding sites. As observed by TEM, v16 ribbons appeared as relatively uniform flat ribbons as expected. In contrast, v8 ribbons were much more flexible, and exhibited pronounced fluctuations in width along their lengths due to an accordion-style stretch. Furthermore, many v8 ribbons underwent full conversion to elongated spindles, although it is unclear from the images what the structure is (e.g. whether this a simple accordion-style stretch taken to an extreme, or instead these are twisted as well) (Fig. S18A–B). We also created ribbons with zigzag raster growth, where alternating clockwise then counterclockwise sets of 16 slat additions creates jagged edges, while alternating two clockwise sets with two counter-clockwise sets creates flush edges (Fig. 3Aii versus Fig. 3Aiii, also see Fig. S16C–D and Fig. S18C–D). Ribbons of all three design types attained comparable mean lengths after 16 hours of isothermal incubation, despite the differences in programmed stagger (Fig. S19).
Figure 3.
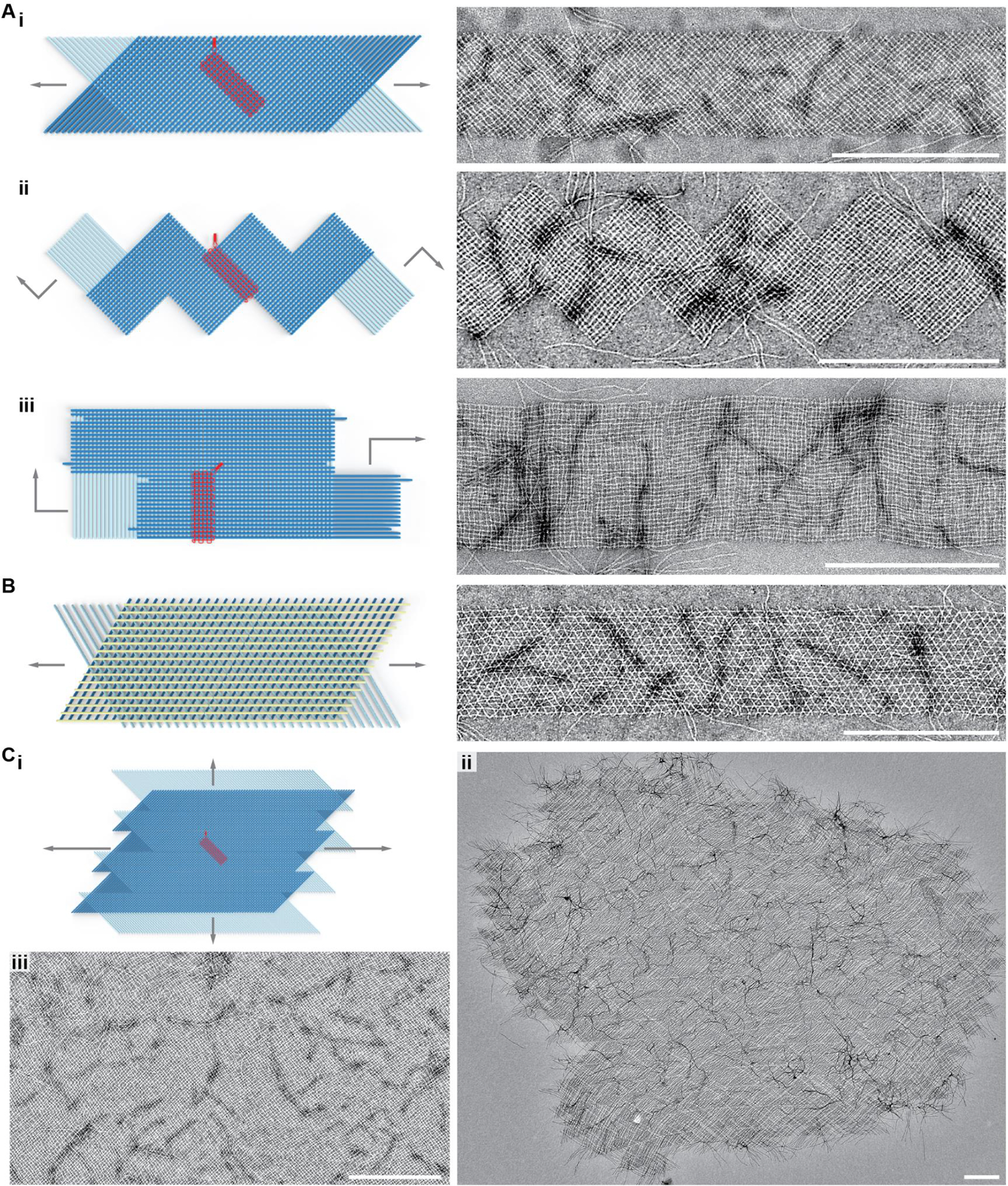
Assembly of periodic ribbons and sheets that grow with 6HB slats in one- and two-dimensions. Ai, v16 ribbon where top and bottom-layer slats are staggered such that addition occurs in alternating order. Aii, v16 ribbon where the slats are added in a zigzag raster-fill pattern that creates jagged edges. Aiii, v16 ribbon where the slats are added in a zigzag raster-fill pattern that creates flush edges. B, tri-layer arrangement of slats, where a top-layer of yellow slats rigidifies the otherwise flexible v8 ribbon. Ci, rendering of v16 growth from Ai where slats are shifted to enable formation of a sheet that grows in two dimensions. Cii–iii, TEM images showing an entire sheet positively stained with uranyl formate in ii, and a subset of another sheet with negative staining in iii. Scale bars are 500 nm.
For all periodic designs, the size of the repeating set of slats was explored from 8 to 64 unique slats in top and bottom layers each (Fig. S20). However, most designs in this study were composed of 8 or 16 unique slats in the top and bottom layers each. We found that the apparent second-order rate constant for slat addition became progressively smaller as the overall slat concentration was increased to over 1 – 2 μM (Supplementary Text 4.1, Fig. S21, Fig. S22). Consideration of this limiting behavior motivated our strategy to grow our larger megastructures in multiple stages, sequentially adding subpools with only ~200 slats at a time to avoid lower than ~4 nM concentration of any one slat while maintaining total slat concentration close to 1 μM (Supplementary Text 4.2; rightward panels of Fig. S9C–D and Fig. S10; the megastructure in Fig. 2Aiii grew faster using the multi-stage protocol as shown in Fig. S14).
We also created periodic 2D structures with the 6HB slats. One approach was to incorporate an additional layer of slats to a v8 ribbon to show that megastructures with more than two layers are possible (Fig. 3B, Fig. S23). In this particular design, the bottom two layers of the v8 ribbon are locked into a rigid conformation where slats between layers are positioned 60° to one another. Next, we tested 2D sheets with two layers of slats (Fig. 3C). The design of the sheets is equivalent to the v16 staggered 1D ribbons shown in Fig. 3Ai, but with the bottom layer of slats shifted half of their lengths with respect to the top layer (Fig. S24). These sheets, in this case defined by 16 + 16 slat unit cells with lateral dimensions of ~320 nm and containing 512 addressable nodes, typically grew to significant dimensions after three days of isothermal growth; the rightward example in Fig. 3Cii is composed of ~10,000 slats, with a molecular mass exceeding 50 GDa and lateral dimensions of ~10 μm (also see additional sheets in Fig. S25). The higher-magnification TEM image Fig. 3Ciii shows a typical middle region of a sheet, with a fabric-like character where defects such as missing slats were infrequent. Both the 1D ribbons and 2D sheets were of sufficiently large molecular mass that they could be enriched over the excess unpolymerized slats by sedimentation into a pellet via centrifugation at 2500 RCF (Fig. S26 includes a discussion of the limitations of this centrifugation method, as well as a potential strategy for further enrichment).
We also note that the ribbons were stable at room temperature for two days in MgCl2 as low as 4 mM (versus the 15 mM condition during growth, see Fig. S27).
Addressability of megastructures
To illustrate that origami-crisscross megastructures can be functionalized as large addressable canvases, we designed the 1022-slat sheet and periodic sheets to display custom patterns of handles on their top faces (Fig. 4A–B). We assembled a 10 nm DNA nanocube37 contrast agent bearing a single complementary handle, incubated the patterned sheets with the purified nanocube, and visualized the resulting patterns using negative-stain TEM (Fig. S28). We note that any ssDNA-tagged guest molecule could in principle be incorporated into the assembly either via a single handle (as with the DNA nanocube), or multiple weaker handles (more akin to the addition of the slats themselves). This incorporation could occur both during megastructure assembly or after via the incorporation of the complementary handles on the slats. The 1022-slat sheets in Fig. 4A were observed to display the programmed patterning of intricate designs including a jigsaw puzzle piece, a happy face, and institutional logos for some of our affiliations. Each ~2.0 μm × ~1.8 μm DNA canvas contains 16,128 addressable nodes, spaced ~14 nm apart. The nanocube was further used to decorate patterned 2D sheets, with the left panel of Fig. 4B showing the smaller 512-node canvas and the right panel showing a TEM overview of a checkerboard, a polkadot sheet, and a continuous jagged line. We additionally characterized the periodic 2D sheets and 1D ribbons using DNA-PAINT. We resolved single handles when spaced ~57 and ~43 nm (i.e. 168 and 126 bp, respectively) between adjacent handles (Fig. 4C, Fig. S29, Fig. S30, Fig. S31). Some lines of eight consecutive missing sites likely correspond to missing slats. In cases where washing steps are stringent enough to strip off slats to an appreciable extent, a future solution may be to crosslink handle interactions post-assembly. Other contiguous missing sites may be due to material lying on the megastructure, such as additional slats, that block access to probes.
Figure 4.
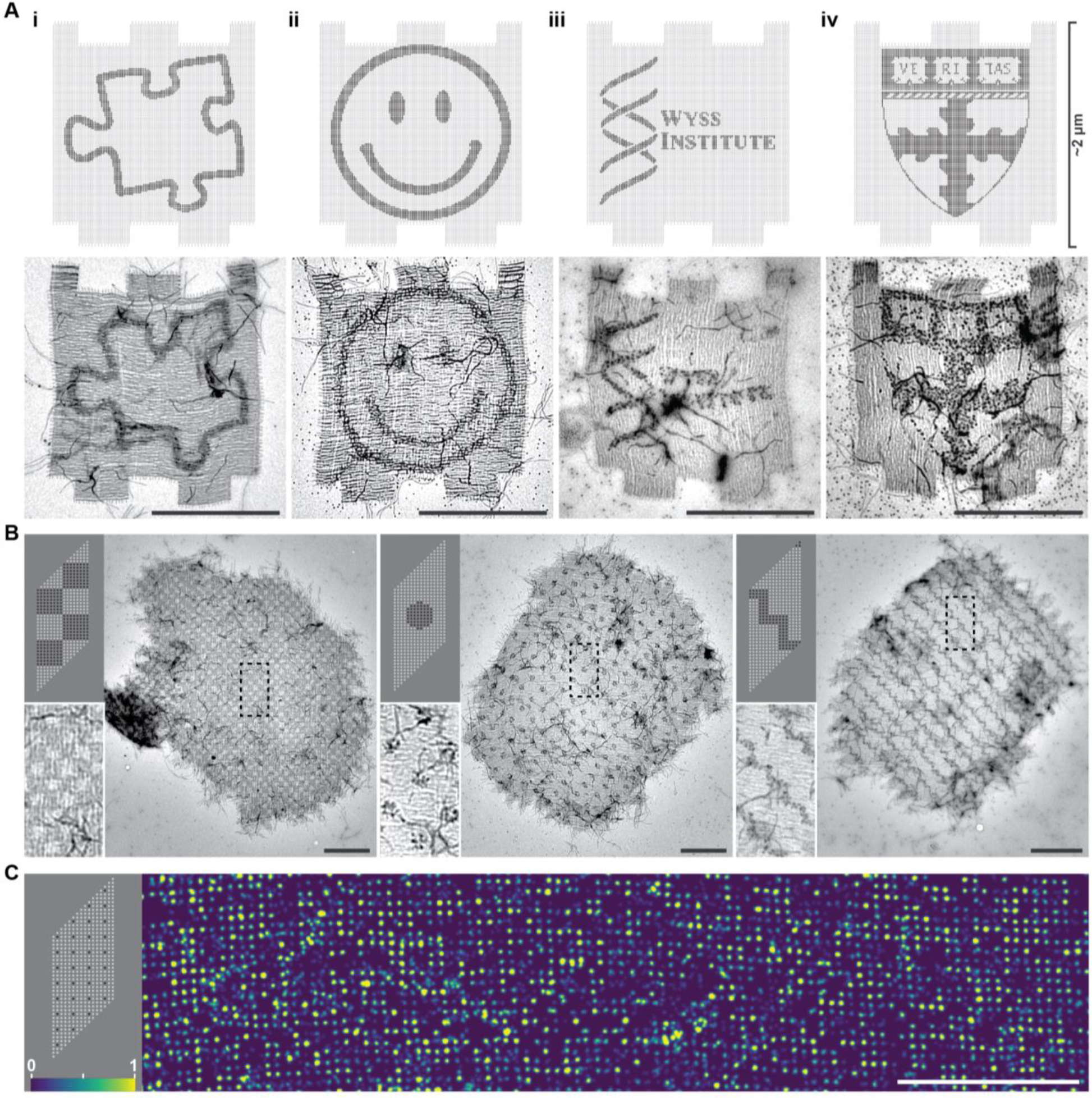
Finite and periodic crisscross megastructures as addressable DNA canvases to pattern arbitrary cargo. A, top row, designs of the finite 1022-slat sheet, with darker dots indicating sites that were programmed with a handle sequence to bind a DNA-nanocube contrast agent, with patterns including the outline of a jigsaw puzzle piece (i), a happy face (ii), and the crests for the Wyss Institute of Harvard University (iii) and the Harvard John A. Paulson School of Engineering and Applied Sciences (iv). Bottom row, TEM images of the patterned finite sheets. B, TEM images of 512-node periodic-sheet canvases patterned with DNA nanocubes, with the upper-left panels showing the designs. Boxed regions are shown more closely in the bottom-left panels. C, DNA-PAINT image of single handles on the sheets, as indicated in the top left design panel. Relative imager strand on-time is denoted by color as shown in bottom-left. Scale bars are 1 μm.
Nucleation, kinetics, and design rules
There was no observable formation of either finite or periodic megastructures in the absence of an added seed (Fig. 5A–B, Fig. S32). As a benchmark, we folded a single DNA-origami square with a constant concentration of staple strands (400 nM each) and variable concentrations of scaffold (0.5–50 nM), and observed a linear one-for-one dependence in the concentration of squares formed (Fig. 5Ci, Fig. S33). We proceeded to mix constant concentrations of slats with variable concentrations of seed and indeed observed a similar one-for-one stoichiometric dependence in the megastructures formed (Fig. 5ii–iii, Fig. S34).
Figure 5.
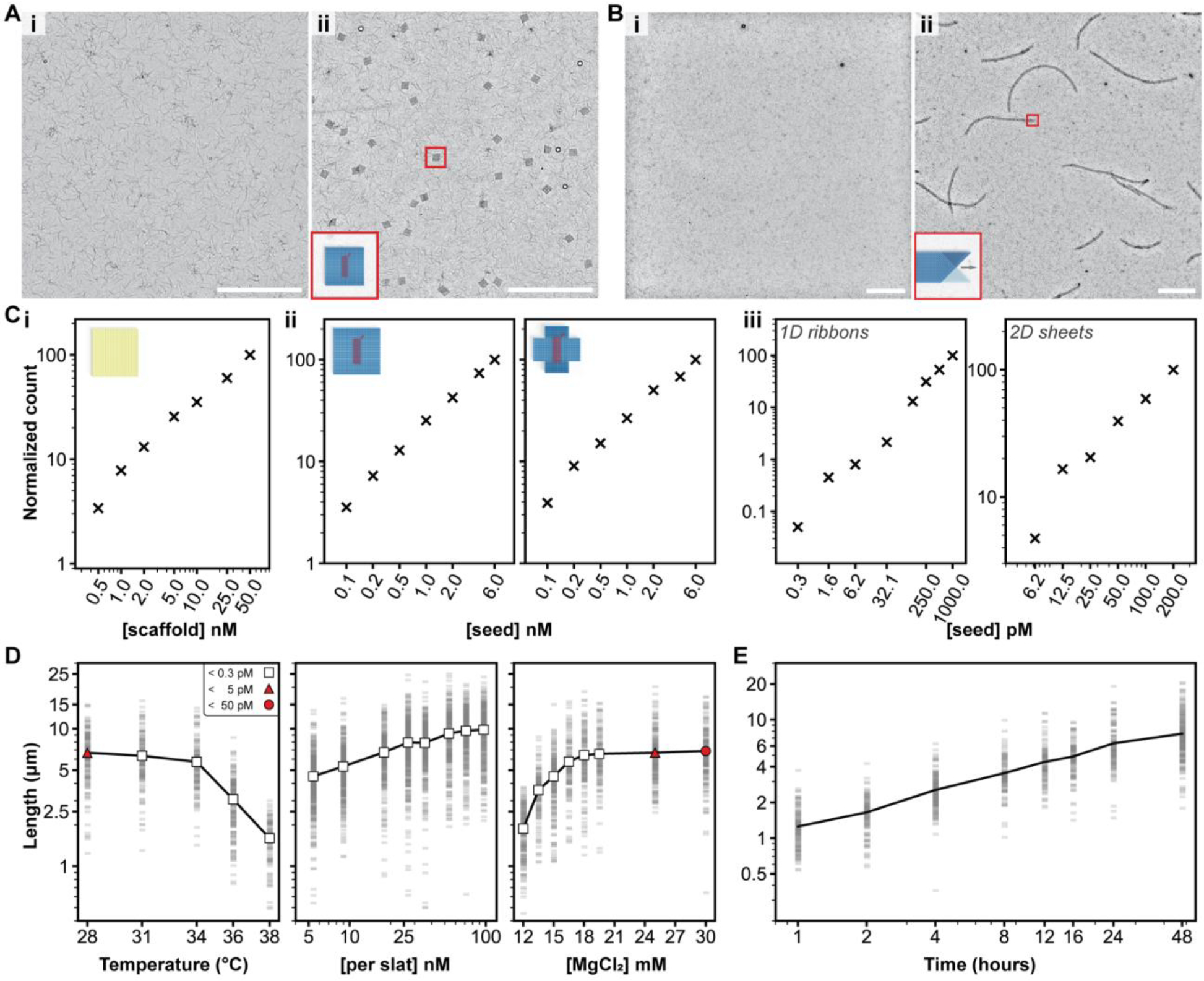
Characterization of growth versus reaction parameters. A and B, seed-controlled assembly of the finite 64-slat square and the periodic 1D ribbon in low magnification TEM images, with no seed versus seed in i and ii, respectively. Ci, the number of DNA-origami squares formed versus the amount of scaffold added, Cii, the number of finite squares (left) or plus symbols (right) versus amount of seed added, and Ciii, the number of periodic megastructures versus the amount of seed added. The relative number of particles per condition is shown with the ‘×’ marker. D, how the length of v16 1D ribbons with 7-nt binding sites varies as a function of temperature, concentration of slats, and concentration of MgCl2. Each faint gray bar is a single ribbon measurement. The white markers indicate no spontaneous assembly above the detection limit, versus red markers where spontaneous assembly was observed to the degree shown in the legend in the leftmost plot. E, the length of the v16 1D ribbons versus time, grown at 20 nM of each slat. Axes in all plots are on a log10 scale, with the exception of the temperature and MgCl2 scale in D. All scale bars are 5 μm, particle counts in C were determined by counting structures in ten low-magnification TEM images, and ~150 ribbons were measured per condition in D and E.
To quantitatively assess spontaneous nucleation under various reaction conditions, we compared seeded v16 7-nt ribbons to unseeded control reactions with variations to temperature, concentration of slats, or concentration of Mg2+ (Fig. 5D). Any unseeded control reactions where no ribbons were observed were presumed to have fewer than 0.3 pM ribbons, as per our limit of detection (Fig. S35, Fig. S36). No spontaneously formed ribbons were observed above 28°C (leftmost Fig. 5D). Growth could not be observed above 38°C, and fully formed ribbons were observed to shrink at temperatures above ~42°C (Fig. S37). Thus we selected 34°C as a reliable temperature for robust and fast seed-controlled growth. Spontaneous nucleation was also not evident at this temperature across a broad window of concentrations of the slats and Mg2+ (middle and rightmost Fig. 5D). Hence, growth of the v16 7-nt ribbons is strictly seed-dependent at temperatures well below where ribbons begin to shrink, using high concentrations of each slat and Mg2+ of up to 100 nM and ~20 mM, respectively. We further characterized relative spontaneous nucleation of v8 and v16 ribbons using weaker 6-nt and stronger 8-nt handles (Supplementary Text 5, Fig. S37–Fig. S39).
The apparent second-order rate constant for slat addition, which we estimated at ~106 M−1s−1, was remarkably high (Supplementary Text 6, Fig. 5E, Fig. S40). It is notable that these observed kinetics are comparable to those for hybridization of DNA strands38, or up to two-orders of magnitude faster than some other approaches to assemble DNA origami, such as blunt-end stacking of shape-complementary features (although these carry the advantage of stronger penalization of non-cognate interactions)39. Finally, we explored principles for combinatorial design of slats — using our 2048 7-nt handle library allowing for 32 possible handles at each of 32 positions — that are sufficiently orthogonal to support robust growth of complex megastructures (Supplementary Text 7, Fig. S41–Fig. S45).
Conclusion
We generalized crisscross polymerization to DNA-origami slats for growth of diverse finite shapes including an addressable canvas from 1022 unique slats that spans about two micrometers per side, periodic ribbons with several different extension patterns, and periodic sheets that attained lateral dimensions exceeding ten micrometers. Hierarchical self-assembly with these building blocks exhibited several features that are advantageous for rapid and robust nanoconstruction: (i) strict seed dependence of initiation, compatible with addition of slats at relatively high concentrations and in stages; (ii) relatively low defect rate in terms of missing slats and prematurely terminated megastructures, implying robustness in spite of inherent defects in the origami building blocks (e.g. unavailable handles)36, (iii) highly orthogonal behavior that enables hundreds of distinct slats to assemble correctly in a single pot; (iv) relatively large second-order rate constant for growth (106 M−1s−1) despite the 5 MDa size of the building blocks. Moreover, structural diversity was created by mixing and matching strands from a library of only 2048 staple strands, where each binding handle of the slats was encoded with one of 32 possible sequences. Therefore prototyping diverse megastructures in this way is cost-effective.
In future studies, the design of the DNA-origami slats could be tailored to create a larger diversity of megastructure architectures22, and various routes to more three-dimensional structures — that would be more rigid than the two-layer structures predominately investigated in this study — should be accessible3,40–42. As with other tiling approaches, it may be possible to program growth with sophisticated algorithmic behaviors43–45. The resulting megastructures could provide access to templates for patterning of diverse guests, such as functional proteins and optically active nanoparticles, on length scales comparable to those of biological cells (Supplementary Text 8).
Methods
M1 Design and purchasing of handle staple strands:
Binding sequences for the various handles were selected as explained in Supplementary Text 3. The handle sequences (and the complementary handle sequences) were appended to the 3′ end of either the top or bottom helix staple strands of the 6HB slat, where they were separated with a thymine (2T) linker. The binding handle sequences are reported in Table 6 and Table 7 (and also in Supplementary Data S1 as a spreadsheet), and the importance of the linker sequence is shown in Fig. S8. All strands were purchased dry at full yield at the 10 nmol scale from Integrated DNA Technologies (IDT). We preferred to purchase the strands on Echo 525 compatible source plates (Labcyte #PP-0200) directly from IDT, though sometimes ordered the strands on other plates and later transferred them to Echo 525 compatible source plates using a manual multichannel pipette. All strands were rehydrated in 50 μL of water, with their concentration assumed to be ~200 μM.
M2 Design of sequence assignments (i.e. permutations) of the handles from the library:
See Supplementary Text 7 for an explanation of why care had to be taken in choosing permutations as described below, and Supplementary Data S2–S3 for the final permutations. Blank layouts of top- and bottom-layer slats were drawn out in Microsoft Excel sheets after the megastructure design was initially conceived. Python scripts were then used to populate each cell of the blank Excel sheet randomly with a number ranging from 1 to 32, corresponding to a specific handle sequence from the 2048 strand library. Next, the script converted the assigned sequences into a set of top-layer slats and bottom-layer slats, where each slat is defined as a one-dimensional list that is 32 numbers long. The Hamming distances between the slats were measured to determine the number of handles on a given slat from one layer that matched together with complementary handles with each slat in the other layer. The process of random assignment and measurement of the Hamming distances in the resulting slats was repeated until some arbitrary maximum threshold of allowable kinetic-trap strength (i.e. the minimum Hamming distance) was attained. We note that megastructure designs that were composed of larger numbers of unique slats tended to have more undesired matched complementarity compared to designs composed of smaller numbers of unique slats. One possible solution to further maximize the Hamming distances for a design would be to increase the size of the handle library (i.e. using > 32 different possible 7-nt handles at each slat intersection with a strand library that has > 2048 unique handle strands versus what was tested here). Nonetheless, we were able to attain satisfactory growth of megastructures that were composed with up to 1022 unique DNA slats that were generated from the 2048 7-nt handle library.
M3 Pooling handle strands for each 6HB and 12HB slat and other folding details for preparation of slats on 96-well plates:
Liquid handling protocols for each design were written to a csv file using Python scripts referencing the Excel sheet of the megastructure design (see Method 2). Staple strands with or without handles were added into 96-well PCR plates (Eppendorf E0030129512) from the 384-well strand library using a Labcyte Echo 525 acoustic liquid handler, which read the csv file using Echo Cherry Pick v1.7.2. Each particular 6HB or 12HB slat respectively required transfer of 64 or 32 strands for the customizable top and bottom helices of the slat. We note that handle staple strands could also be pooled together manually using single or multichannel pipettes, though such preparations could require an untenable amount of time depending on the number of slats in the megastructure. See Fig. S11 for a time comparison of such approaches. After strand transfer into the 96-well plates, any droplets along the rim of the wells were spun down, at which point a slat-folding mixture containing other core strands, scaffold, and buffer as specified in Method 4 was added to each well and mixed using a manual multichannel pipette. The 96-well plates were then thermally sealed with plastic films using an ABgene ALPS-300 microplate sealer and spun down one final time on a centrifuge, at which point plates were placed on a thermocycler and the origami folded using the temperature gradient in Method 4.
M4 Design and folding of DNA origami:
All DNA origamis were designed using legacy caDNAno46 v0. See Supplementary Data S5–S8 for caDNAno json files of the 6HB, 12HB, gridiron seed, and single origami reference square. Staple and scaffold DNA sequences are reported in Table 1–Table 5 (and also in Supplementary Data S1 as a spreadsheet). Unpurified dehydrated staple oligonucleotides were purchased from Integrated DNA Technologies (IDT) at 100 or 10 nmol scale. For the slats, each unpurified core staple strand (i.e. strands other than the 64 or 32 customizable handle strands for the top and bottom helices of the 6HB and 12HB slats, respectively) were rehydrated water at ~1 mM and pooled together with equal volumes of each strand. For the gridiron seed and origami reference square, each dehydrated strand was resuspended in water at ~100 μM and pooled together with equal volumes of each strand. The p8064 and p8634 scaffold strands were produced from M13 phage replication in Escherichia coli. All folding was done in 1x TE buffer (5 mM Tris pH 8.0, 1 mM EDTA) with the MgCl2 as specified below. The gridiron seed was mixed with 40 nM p8634 scaffold, ~250 nM of each staple strand, 10 mM MgCl2, and folded with a 12-hour temperature gradient: 94–86°C in 5 min steps less 4°C/step; 85–70°C in 5 min steps less 1°C/step; 70–40°C in 15 min steps less 1°C/step; 40–25°C in 10 min steps less 1°C/step; and 16°C thereafter until the sample was collected. The 6HB slats were mixed with 50 nM p8064 scaffold, ~500 nM of each staple strand, 6 mM MgCl2, and folded with an 18 hour temperature gradient: 80°C/10 minutes in a single step; 60–45°C in 160 6.75 min steps less 0.1°C/step; 16°C thereafter until collection of the sample. The 12HB slats were mixed with 50 nM p8064 scaffold, ~500 nM of each staple strand, 8 mM MgCl2, and folded with an 18 hour temperature gradient: 80°C/10 minutes in a single step; 75–45°C in 310 3.48 min steps less 0.1°C/step; 16°C thereafter until collection of the sample. The origami reference square was mixed with 40 nM p8064 scaffold, ~400 nM of each staple strand, 6 mM MgCl2, and folded with an 18 hour temperature gradient: 80°C/10 minutes in a single step; 60–45°C in 160 6.75 min steps less 0.1°C/step; 16°C thereafter until collection of the sample. We note that there were special considerations for preparation of the slats in 96-well plates, as explained in Method 3.
M5 Preparation of the DNA nanocube contrast agent:
Unpurified dehydrated nanocube oligonucleotides were purchased from Integrated DNA Technologies (IDT) at 10-nmol scale. Unpurified nanocube strands were rehydrated in water at ~100 μM each and pooled together with equal volumes per strand. Out of the 28 total nanocube strands as published previously37, a single strand was selected and appended with a 4T linker and 16-nt handle to its 3′ end (see sequences in Table 8 or Supplementary Data S1). The nanocube was prepared with ~1 μM of each strand (with the handle-tagged strand at ~2 μM), 40 mM MgCl2, and folded with a 42-hour temperature gradient: 80°C/10 min in a single step; 65–37°C in 290 8.69 min steps less 0.1°C/step; 16°C thereafter until collection of the sample. The folded nanocube was separated on an agarose gel and purified as described in Method 8, and bound to the megastructures without any further downstream assembly.
M6 Agarose-gel electrophoresis:
Gel characterization of the DNA-origami gridiron seed, 6HB slats, or 12HB slats was performed using the Thermo Scientific™ Owl™ EasyCast™ B2 electrophoresis system. UltraPure agarose (Life Technologies, 16500500) was melted in 0.5x TBE (45 mM Tris, 45 mM boric acid, 0.78 mM EDTA, 11 mM MgCl2) to a concentration of 1.0 % (w/v). The molten agarose was cooled to 65°C and 6.25*10−5 % (w/v) ethidium bromide was added. To assess folding, ~50 fmol of DNA-origami sample was mixed in an excess of agarose-gel loading buffer (5 mM Tris, 1 mM EDTA, 30% w/v glycerol, 0.025% w/v xylene cyanol, 10 mM MgCl2; with typically 4 μL loading buffer added to 1 μL of each sample that was folded with 50 nM scaffold). The mixed samples were loaded onto the gel and separated for 3–4 hours at 60 V at room temperature. Control samples for size and densitometry included one or both of the following: first, ~50 fmol of the same scaffold from which the DNA origami was folded; or second, ~0.5 μg of Gene Ruler 1kb Plus DNA Ladder (Thermo Scientific™ SM1331). Gel images were captured on a GE Typhoon FLA 9500 fluorescent imager using the ethidium-bromide parameters as given in the Typhoon control software. The photo-multiplier tube (PMT) was set to 500V. Densitometry to quantify relative assembly of DNA bands was performed with FIJI ImageJ (v1.53c)47. Background subtraction with a rolling ball radius of 30–60 pixels was performed on linear TIFF images. The GelAnalyzer plugin in ImageJ and wand tool was used to integrate total pixel intensities from lanes of interest. DNA-origami yields were determined by taking the ratio of the intensity of the band of interest with respect to all the species with a molecular weight larger than the excess staple strands.
M7 PEG precipitation to concentrate pools of 6HB slats:
The slats as folded on 96-well PCR plates were collected and combined into pools using a manual multichannel pipette. Each pool maximally had ~100 slats, though the number of slats in a given pool was variable depending on the design of the megastructure. We generally kept the slats in one layer of the megastructure in a separate pool from the perpendicular slats in the other layer of the megastructure, so that slats with complementary handles would not be concentrated in the same mixture together. Our rationale was that spontaneous interactions between the slats during concentration could be deleterious to the yield, though we did not study this carefully to determine whether such care was necessary. The pooled slats were subsequently concentrated using two rounds of PEG precipitation, as adapted from as published previously48. The Mg2+ in the slat pool was increased from 6 mM to 20 mM by adding the appropriate volume of 1 M MgCl2. One volume of 2x PEG-purification buffer (5 mM Tris, 1 mM EDTA, 15% w/v PEG-8000, 510 mM NaCl) was added and mixed with the equal volume of pooled slats in a 2 mL round-bottom tube. The mixture was spun at 16k × g for 25 min, the supernatant was gently extracted using a pipette, and the pellet was resuspended in 50 μL 1x TE buffer with 20 mM MgCl2. The second round of PEG precipitation was performed using an equal volume of 2x PEG-purification buffer and the final slat pellet resuspended in a small volume of 1x TE buffer with 10 mM MgCl2 so that the total concentration of slats was ~2 μM. However, the volume was adjusted as needed so that the pool was sufficiently concentrated to achieve the desired final per slat concentration in Method 9 or Method 10. The final slat pool was placed on a shaking incubator set to 1000 rpm at 33°C for about one hour. Finally, the concentration of DNA was measured on a Nanodrop 2000c Spectrophotometer (Thermo Scientific™) so as to estimate the final nM concentration of slats.
M8 Agarose-gel extraction to purify pools of 12HB slats:
The 12HB slats in raw folded samples were low yielding and insufficient to prepare as described exactly in Method 7. See Supplementary Text 2, Fig. S3 and Fig. S4 where the yield and resulting challenges are discussed in detail. The 12HB slats were pooled as described in Method 7 and then separated on an agarose gel as described in Method 6. The gel was examined on a UV transilluminator to identify the monomer band which was then excised from the gel using a razor blade. The gel-band pieces were transferred to a 1.5 mL eppendorf tube, crushed using a plastic pestle, and purified using Freeze N’ Squeeze spin columns (Bio-Rad, 732–6166) as published previously48. The gel-purified samples was typically too dilute, such that it was necessary to concentrate them into a smaller volume of 1x TE buffer with 10 mM MgCl2 using one round of PEG precipitation as explained in Method 7.
M9 Preparation of megastructure assembly reactions with fewer than 200 unique slats:
The raw gridiron seed (folded with 40 nM scaffold), purified slats (generally ~1 μM total slats per pool depending on the extent to which it was concentrated during purification), and 4x Megastructure buffer (5 mM Tris pH 8.0, 1 mM EDTA, 30 mM MgCl2, 0.04% Tween-20) were mixed together in 5–20 μL at room temperature. We generally prepared the reactions in 0.1 mL Eppendorf PCR tubes (E0030124812) because of their tightly fitted lids which guarded against evaporative loss during extended growth periods. The final reaction generally contained ~0.5–2 nM seed, ~5–20 nM of each unique slat, 1x Megastructure buffer, with any excess volume filled with 1x TE buffer with 10 mM MgCl2. We assumed that the seed and slats would be in 1x TE buffer with 10 mM MgCl2, such that the final megastructure assembly reaction would contain 15 mM MgCl2. Next, the reactions were placed on a thermocycler and incubated for 4 hours at a high temperature where slats could only bind the seed (i.e. 45°C or 55°C for the 6/7-nt or 8-nt designs, respectively), before the temperature was lowered for slat growth for 1–72 hours. We used 34°C for typical growth of the v16 periodic and finite megastructures using the 7-nt handle library, versus other designs which used growth temperatures as summarized with cyan markers in the Fig. S39 plot.
M10 Preparation of megastructure assembly reactions with more than 200 unique slats:
As explained in Supplementary Text 4, megastructures composed of a large number of unique slats (i.e. > 100 slats) generally grew slowly if all the slats were mixed simultaneously. Hence, designs with more than 100 unique slats were either incubated for a longer growth time, or grown in stages with several additions of slats. The first growth stage with ~200 of the slats most proximal to the seed were prepared as in Method 9. We prepared a 4x MultiMegastructure buffer (5 mM Tris pH 8.0, 1 mM EDTA, 22 mM MgCl2, 0.024% Tween-20). In each stage thereafter, 5 parts of 4x MultiMegastructure buffer were added to 8 parts of an aliquot of the reaction from the prior stage with ~200 of the next slats in 20 parts total. The purpose of the 4x MultiMegastructure buffer was to maintain the buffer with 5 mM Tris pH 8.0, 1 mM EDTA, 15 mM MgCl2, and 0.01% Tween-20 as more slats were added.
M11 Binding of the DNA nanocube to 6HB-slat canvases:
An aliquot of the sheet or 1022-slat sheet was diluted 10–50 fold with agarose-gel purified nanocube solution from Method 5. The sample was incubated at room temperature overnight to bind the nanocube to the megastructures. The subsequent day, the sample was purified from excess slats and nanocubes using two rounds of centrifugation, as outlined in Method 12. The sample was resuspended in 1x TE buffer with 10 mM MgCl2 and 10% trehalose (w/v) and negatively stained with 1% uranyl formate, as described in Method 13. We qualitatively noted that trehalose and the lower percentage (i.e. versus 2%) uranyl formate provided better contrast for the nanocubes on the megastructure canvas. The locations of nanocube handles on slats to create the desired patterns are shown in Supplementary Data S4.
M12 Purification of periodic-ribbon and sheet megastructures by centrifugation:
The largest periodic ribbons and sheets were purified from excess free monomers by centrifuging the samples at low speed. See Fig. S26 for results using this method. Ribbons or sheets were prepared as in Method 9, diluted ~10 fold in 1x TE buffer with 10 mM MgCl2, and then spun in a 1.5 mL eppendorf tube at 2.5k RCF for 15 minutes. The supernatant was gently extracted with a pipette and the pellet resuspended in 1x TE buffer with 10 mM MgCl2 with the final volume as desired for the particular application. This procedure of centrifugation and resuspension could be repeated multiple times to further deplete excess free slats.
M13 Transmission electron microscopy (TEM):
Raw assembled megastructure reactions were diluted 1:250–1:1000 in 1x TE buffer with either 10 mM or 15 mM MgCl2. TEM grids (Electron Microscopy Sciences; FCF400-CU, FCF200-CU-TA, or FCF100-CU-TA) were negatively glow discharged at 15 mA for 25 s in a PELCO easiGlow. The diluted sample (4 μL) was applied to the glow-discharged grid, incubated for 2 min, and wicked off gently into Whatman paper (Fisher Scientific, 09–874-16B). We used one of two possible approaches to stain the sample with 2% uranyl formate: (1), When closer-up structural detail of individual slats was desired, we used a negative-stain approach where 4 μL of 2% aqueous filtered uranyl formate was applied, incubated for 1–2 seconds, and gently wicked from its side into Whatman paper so as to leave a thicker deposit of uranyl formate around the sample. Or (2), when clarity of the overall forms and length of the megastructure was desired in low-magnification images, we used a positive stain approach where 4 μL of 2% aqueous filtered uranyl formate was applied, incubated for 1–2 seconds, and wicked off completely into Whatman paper so as to leave a thinner layer of uranyl formate, which darkened each particle with respect to the grid substrate. All imaging was performed at 80 kV on a JEOL JEM 1400 Plus microscope. All images presented in this work were imported into FIJI ImageJ (v1.53c)47, corrected for background noise using a pseudo flat-field image, and then contrast and brightness adjusted for clarity in publication.
M14 Quantification of slat incorporation in TEM images:
We used the finite 64-slat square as a model system to assess the relative incorporation of a given slat into a megastructure (see Fig. 2). Each sample was assembled for a given period of time, diluted 750-fold so that single squares could be differentiated from excess free slats on the TEM substrate, negatively stained with uranyl formate, and then imaged by TEM. Images of ~50 well stained squares were collected at a magnification where the single slats could be differentiated. The slats in each layer of the square were quantified using the Cell Counter plugin in FIJI ImageJ (v1.53c)47.
M15 Quantification of full growth of large finite megastructures in TEM images:
Aliquots of megastructures with ~1000 unique were concentrated ~10-fold and applied to TEM grids using a positive stain approach (see Method 12 and Method 13, respectively). Sufficient numbers of low magnification TEM images were collected so that ~200–300 individual megastructures could be assessed for their relative completion (i.e. determining if the larger morphological features of the design—including edges and corners of the overall two-layer structure—were present with no observable missing segments). We alternatively considered doing Method 14 for the largest finite structures and periodic ribbons and sheets; however, the large number of slats in the megastructures, variable sizes of the periodic designs, and manual approach to identifying the slats in images made this approach untenable. Thus sporadic missing slats were not considered in classifying megastructures as “fully grown”. We view this analogous to conventional analysis of DNA origami, where missing staple strands are hard to quantify and usually not considered as long as the overall structural integrity is intact.
M16 Quantification of relative ribbon growth in TEM images:
We used periodic ribbons to determine how parameters including temperature, concentration of Mg2+, concentration of the slats, time, strength of each binding site, the number of crisscross binding sites, growth pattern, and sequence assignment influences growth (see Fig. 5D–E, Fig. S37–Fig. S38, Fig. S19, Fig. S42–Fig. S43). We assembled ribbons using 0.5 nM seed as explained in Method 9, diluted the initial sample 250-fold, and applied it to a grid where the structures were positively stained with uranyl formate. Each sample was then imaged at low magnification with TEM. We collected enough images so that at least 150 ribbons could be measured. The lengths of the ribbons were calculated using the NeuronJ plugin in FIJI ImageJ (v1.53c)47,49.
M17 Quantification of stoichiometric control of megastructure formation from the seed in TEM images:
We counted the number of megastructures in low-magnification TEM images of reactions where different concentrations of seed were added (see results in Fig. 5C). The raw samples were diluted so as to be able to see single particles in the condition where the highest amount of seed was added. We positively stained the sample with uranyl formate, and collected ten TEM images at a suitable magnification where the structure in question could be identified, but still allowed us to observe a reasonably large number of particles. The number of megastructures in each image was counted using the Cell Counter plugin in FIJI ImageJ (v1.53c)47. We normalized the number of structures counted for a given seed concentration with respect to one of the concentrations tested and plotted the normalized count versus the concentration of seed (or scaffold) added.
M18 Quantification of spontaneous nucleation of ribbons in TEM images:
We counted the formation of periodic ribbons in control reactions with no seed to quantify the amount of spontaneous nucleation. We considered the ribbons useful as a model for spontaneous nucleation of megastructures because they were easy to identify as micrometer-sized objects that could be readily counted in low-magnification TEM images (see results in Fig. 5D and Fig. S38). Samples were incubated at the reaction conditions that we desired to test, diluted ~250 fold, and added to the TEM substrate, positively stained with uranyl formate, and then ten low-magnification TEM images were collected. The number of ribbons in each image was counted using the Cell Counter plugin in FIJI ImageJ (v1.53c)47. The ribbons counted in each image area were normalized to the dilution and magnification, and then converted into a molar amount by comparing it to a standard curve of ribbons (see Fig. S35). The recorded ribbon detection limit was ~0.3 pM, which corresponded to observing a single ribbon in the ten images.
M19 Preparation of megastructures for DNA-PAINT imaging:
The outside faces of slats on ribbons and sheets were decorated on one side with 3′ complementary handle sequences for PAINT imager strands and on the other side with 5′ biotin sites. Slats were folded, purified, assembled into ribbons and sheets, and purified from excess slat monomers and resuspended in buffer B (5 mM Tris-HCl pH 8.0, 10 mM MgCl2, 1 mM EDTA, 0.05% v/v Tween-20) at a final concentration of ~0.1 nM per megastructure as described in Method 4, Method 7, Method 9, and Method 12, respectively. The megastructure sample was further prepared as described previously50, where an imaging chamber with an inner volume of ∼20 μl was created by adhering one coverslip (#1.5, 18 × 18 mm2, ∼0.17 mm thick) to a glass slide (75 × 26 mm2, 1 mm thick) with double-sided tape. Surfaces of the chamber were prepared by the following steps: (1) flowing 20 μl of 1 mg/ml biotin-labeled bovine albumin (Sigma-Aldrich, catalog number A8549) dissolved in buffer A (10 mM Tris-HCl (pH 8.0), 100 mM NaCl, 0.05% (v/v) Tween-20) and incubating it for 2 minutes; (2) washing with 40 μl of buffer A; (3) flowing 20 μl of 0.5 mg/ml streptavidin (Invitrogen, catalog number S-888) dissolved in buffer A and incubating it for 2 minutes; (4) washing with 40 μl of buffer A; (5) equilibrating with 40 μl of buffer B. Twenty microliter of the purified biotin-labeled megastructure sample was then flown into the chamber and incubated for 2 minutes followed by washing with 40 μl of buffer B. For the purpose of drift correction, fiducial markers (40 nm gold nanoparticles; Sigma-Aldrich, catalog number 753637) were diluted to 1:10 in buffer B, flown into the chamber, and incubated for 10 minutes followed by washing with 40 μl buffer B. Finally, imaging buffer (buffer B containing 2 U/ml PCD (OYC Americas, sold as rPCO), 2.5 mM PCA (Sigma-Aldrich, catalog number 37580), 1 mM Trolox (Sigma-Aldrich, catalog number 238813)) was flown into the chamber. The imaging chamber was sealed with nail polish before imaging.
M20 DNA-PAINT super-resolution imaging:
DNA-PAINT imaging was performed on a Nikon Ti Eclipse inverted microscope with a Perfect Focus System and a custom-build TIRF illuminator. Laser excitation with a 532-nm laser (MPB Communications Inc., 1 W, DPSS-system) was used for excitation with a 100 mW input at an effective power density of ~2 kW/cm2. The excitation laser was passed through a quarter-wave plate (Thorlabs, WPQ05M-532), placed at 45° to the polarization axis, and directed to the objective through an excitation filter (Chroma ZET532/10x) via a long-pass dichroic mirror (Chroma ZT532RDC_UF2). The laser beam was then expanded using a commercial variable beam expander (Edmund Optics, Broadband VIS 2X-8X) and a custom-built Galilean telescope followed by coupling into the microscope objective using a motorized mirror to generate a total internal reflection illumination. Emission light was spectrally filtered (Chroma ET542LP and Chroma ET550 LP), directed into a 4f adaptive optics system containing a deformable mirror (Imagine Optic, MicAO 3DSR) to correct optical aberration and optimize point spread functions (PSFs), and imaged on scientific complementary metal-oxide–semiconductor sCMOS camera (Andor Technologies, Zyla 4.2+) using rolling shutter readout at a bandwidth of 200 MHz at 16 bit and a 150 ms exposure time with 6.5-μm pixels, resulting in an effective pixel size of 65 nm. At each imaging session, 10,000 frames with an exposure time of 150 ms per frame were captured. DNA-PAINT image data were processed and rendered using Picasso51. The lateral positions of single-molecule localization events were determined using Picasso Localize followed by drift correction using imaged gold nanoparticles as fiducial markers.
Supplementary Material
S1–S4 Spreadsheets of sequences (S1), finite sequence assignments (S2), periodic sequences assignments (S3), and nanocube/PAINT patterns (S4), respectively.
S5–S8 DNA origami caDNAno v0 designs of the 6HB slat (S5), 12HB slat (S6), gridiron seed (S7), and single reference square (S8), respectively.
Acknowledgments:
The authors would like to thank the following individuals: Jocelyn (Josie) Kishi for suggesting and helping write a grant for the Echo Acoustic Liquid Handler that made this work possible; Serkan Cabi and Tao Zhang for helping test early designs of crisscross origamis; Prof. Vinothan Manoharan, Prof. Michael Brenner, Rasmus Sørensen, and Jaeseung Hahn for fruitful discussions.
Funding:
Wyss Core Faculty Award (WS, PY)
Wyss Molecular Robotics Initiative Award (WS, PY)
National Science Foundation DMREF Award 1435964 (WS)
National Science Foundation Award CCF-1317291 (WS)
Office of Naval Research Award N00014-15-1-0073 (WS)
Office of Naval Research Award N00014-18-1-2566 (WS)
Office of Naval Research DURIP Award N00014-19-1-2345 (WS)
NIH NIGMS Award 5R01GM131401 (WS)
NSERC PGSD3-502356-2017 (CMW)
Alexander S. Onassis Scholarship for Hellenes (AE)
Footnotes
Competing interests: A patent (PCT/US2017/045013) entitled “Crisscross Cooperative Self-Assembly” has been filed based on this work.
Code availability: Scripts that were used to make various assignments of handle staple oligonucleotides, and scripts that were used to measure Hamming distances of sequence assignments, are available at https://github.com/aersh/origamicrisscross.
Data availability:
All raw TEM image data that was measured to determine growth and nucleation of origami crisscross megastructures are available upon request from WMS.
References
- 1.Rothemund PWK Folding DNA to create nanoscale shapes and patterns. Nature 440, 297–302 (2006). [DOI] [PubMed] [Google Scholar]
- 2.Douglas SM et al. Self-assembly of DNA into nanoscale three-dimensional shapes. Nature 459, 414–418 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andersen ES et al. Self-assembly of a nanoscale DNA box with a controllable lid. Nature 459, 73–76 (2009). [DOI] [PubMed] [Google Scholar]
- 4.Benson E et al. DNA rendering of polyhedral meshes at the nanoscale. Nature 523, 441–444 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Han D et al. DNA Origami with Complex Curvatures in Three-Dimensional Space. Science 332, 342–346 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Han D et al. DNA Gridiron Nanostructures Based on Four-Arm Junctions. Science 339, 1412–1415 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Marchi AN, Saaem I, Vogen BN, Brown S & LaBean TH Toward Larger DNA Origami. Nano Lett. 14, 5740–5747 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Nickels PC et al. DNA origami structures directly assembled from intact bacteriophages. Small 10, 1765–1769 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Zhang H et al. Folding super-sized DNA origami with scaffold strands from long-range PCR. Chem. Commun. 48, 6405–6407 (2012). [DOI] [PubMed] [Google Scholar]
- 10.Wei B, Dai M & Yin P Complex shapes self-assembled from single-stranded DNA tiles. Nature 485, 623–626 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ke Y et al. DNA brick crystals with prescribed depths. Nat. Chem. 6, 994–1002 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ong LL et al. Programmable self-assembly of three-dimensional nanostructures from 10,000 unique components. Nature 552, 72–77 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ke Y, Ong LL, Shih WM & Yin P Three-Dimensional Structures Self-Assembled from DNA Bricks. Science 338, 10.1126/science.1227268 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pfeifer W & Saccà B From Nano to Macro through Hierarchical Self-Assembly: The DNA Paradigm. ChemBioChem 17, 1063–1080 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Zhao Z, Liu Y & Yan H Organizing DNA Origami Tiles into Larger Structures Using Preformed Scaffold Frames. ACS Publ. (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wagenbauer KF, Sigl C & Dietz H Gigadalton-scale shape-programmable DNA assemblies. Nature 552, 78–83 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Gerling T, Wagenbauer KF, Neuner AM & Dietz H Dynamic DNA devices and assemblies formed by shape-complementary, non–base pairing 3D components. Science 347, 1446–1452 (2015). [DOI] [PubMed] [Google Scholar]
- 18.Rajendran A, Endo M, Katsuda Y, Hidaka K & Sugiyama H Programmed Two-Dimensional Self-Assembly of Multiple DNA Origami Jigsaw Pieces. ACS Nano 5, 665–671 (2011). [DOI] [PubMed] [Google Scholar]
- 19.Liu W, Zhong H, Wang R & Seeman NC Crystalline two-dimensional DNA-origami arrays. Angew. Chem. Int. Ed. 50, 264–267 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Woo S & Rothemund PWK Programmable molecular recognition based on the geometry of DNA nanostructures. Nat. Chem. 3, 620–627 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Sigl C et al. Programmable icosahedral shell system for virus trapping. Nat. Mater. 20, 1281–1289 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yao G et al. Meta-DNA structures. Nat. Chem. 12, 1067–1075 (2020). [DOI] [PubMed] [Google Scholar]
- 23.Berengut JF et al. Self-Limiting Polymerization of DNA Origami Subunits with Strain Accumulation. ACS Nano (2020) doi: 10.1021/acsnano.0c07696. [DOI] [PubMed] [Google Scholar]
- 24.Wickham SF et al. Complex multicomponent patterns rendered on a 3D DNA-barrel pegboard. Nat. Commun. 11, 1–10 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tikhomirov G, Petersen P & Qian L Fractal assembly of micrometre-scale DNA origami arrays with arbitrary patterns. Nature 552, 67–71 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Minev D, Wintersinger CM, Ershova A & Shih WM Robust nucleation control via crisscross polymerization of highly coordinated DNA slats. Nat. Commun. 12, 1741 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seeman NC Nanomaterials based on DNA. Annu. Rev. Biochem. 79, 65 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuzyk A et al. DNA-based self-assembly of chiral plasmonic nanostructures with tailored optical response. Nature 483, 311–314 (2012). [DOI] [PubMed] [Google Scholar]
- 29.Acuna GP et al. Fluorescence Enhancement at Docking Sites of DNA-Directed Self-Assembled Nanoantennas. Science 338, 506–510 (2012). [DOI] [PubMed] [Google Scholar]
- 30.Douglas SM, Bachelet I & Church GM A Logic-Gated Nanorobot for Targeted Transport of Molecular Payloads. Science 335, 831–834 (2012). [DOI] [PubMed] [Google Scholar]
- 31.Li S et al. A DNA nanorobot functions as a cancer therapeutic in response to a molecular trigger in vivo. Nat. Biotechnol. 36, 258–264 (2018). [DOI] [PubMed] [Google Scholar]
- 32.Shaw A et al. Binding to nanopatterned antigens is dominated by the spatial tolerance of antibodies. Nat. Nanotechnol. 14, 184–190 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Derr ND et al. Tug-of-War in Motor Protein Ensembles Revealed with a Programmable DNA Origami Scaffold. Science 338, 662–665 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mathieu F et al. Six-helix bundles designed from DNA. Nano Lett. 5, 661–665 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Douglas SM, Chou JJ & Shih WM DNA-nanotube-induced alignment of membrane proteins for NMR structure determination. Proc. Natl. Acad. Sci. 104, 6644–6648 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Strauss MT, Schueder F, Haas D, Nickels PC & Jungmann R Quantifying absolute addressability in DNA origami with molecular resolution. Nat. Commun. 9, 1600 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scheible MB et al. A Compact DNA Cube with Side Length 10 nm. Small 11, 5200–5205 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang DY & Winfree E Control of DNA strand displacement kinetics using toehold exchange. J. Am. Chem. Soc. 131, 17303–17314 (2009). [DOI] [PubMed] [Google Scholar]
- 39.Bruetzel LK, Walker PU, Gerling T, Dietz H & Lipfert J Time-Resolved Small-Angle X-ray Scattering Reveals Millisecond Transitions of a DNA Origami Switch. Nano Lett. 18, 2672–2676 (2018). [DOI] [PubMed] [Google Scholar]
- 40.Zhang T et al. 3D DNA Origami Crystals. Adv. Mater. 30, 1800273 (2018). [DOI] [PubMed] [Google Scholar]
- 41.Zheng J et al. From molecular to macroscopic via the rational design of a self-assembled 3D DNA crystal. Nature 461, 74–7 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tikhomirov G, Petersen P & Qian L Triangular DNA origami tilings. J. Am. Chem. Soc. 140, 17361–17364 (2018). [DOI] [PubMed] [Google Scholar]
- 43.Rothemund PWK, Papadakis N & Winfree E Algorithmic Self-Assembly of DNA Sierpinski Triangles. PLOS Biol. 2, e424 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barish RD, Schulman R, Rothemund PWK & Winfree E An information-bearing seed for nucleating algorithmic self-assembly. Proc. Natl. Acad. Sci. 106, 6054–6059 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Woods D et al. Diverse and robust molecular algorithms using reprogrammable DNA self-assembly. Nature 567, 366–372 (2019). [DOI] [PubMed] [Google Scholar]
- 46.Douglas SM et al. Rapid prototyping of 3D DNA-origami shapes with caDNAno. Nucleic Acids Res. gkp436 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schindelin J et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wagenbauer KF et al. How We Make DNA Origami. ChemBioChem 18, 1873–1885 (2017). [DOI] [PubMed] [Google Scholar]
- 49.Meijering E et al. Design and validation of a tool for neurite tracing and analysis in fluorescence microscopy images. Cytom. Part J. Int. Soc. Anal. Cytol. 58, 167–176 (2004). [DOI] [PubMed] [Google Scholar]
- 50.Dai M, Jungmann R & Yin P Optical imaging of individual biomolecules in densely packed clusters. Nat. Nanotechnol. 11, 798–807 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schnitzbauer J, Strauss MT, Schlichthaerle T, Schueder F & Jungmann R Super-resolution microscopy with DNA-PAINT. Nat. Protoc. 12, 1198–1228 (2017). [DOI] [PubMed] [Google Scholar]
- 52.Zadeh JN, Wolfe BR & Pierce NA Nucleic acid sequence design via efficient ensemble defect optimization. J. Comput. Chem. 32, 439–452 (2011). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
S1–S4 Spreadsheets of sequences (S1), finite sequence assignments (S2), periodic sequences assignments (S3), and nanocube/PAINT patterns (S4), respectively.
S5–S8 DNA origami caDNAno v0 designs of the 6HB slat (S5), 12HB slat (S6), gridiron seed (S7), and single reference square (S8), respectively.
Data Availability Statement
All raw TEM image data that was measured to determine growth and nucleation of origami crisscross megastructures are available upon request from WMS.