

Abstract
Cytotoxic CD8+ T cells are potent killers of diseased cells, but their functional capacity is often compromised in cancer. The quality of anti-tumor T cell immunity is determined during T cell priming in the lymph node and further influenced by the local microenvironment of the tumor. Increasing evidence indicates that dendritic cells (DCs) have the capacity to precisely regulate the functional quality of anti-tumor T cell responses in both locations. In this review, we discuss recent advances in our understanding of how distinct DC-derived signals influence CD8+ T cell differentiation and anti-tumor functions. Insight into the mechanisms of DC-mediated regulation of anti-tumor immunity could inspire development of improved approaches to prevent and reverse T cell dysfunction in cancer.
Keywords: Dendritic cells, cytotoxic T cells, anti-tumor immunity, stimulatory signals, T cell priming, cancer immunotherapy
Anti-tumor T cell responses require dendritic cells
Cytotoxic CD8+ T cells can eliminate cancer cells in a highly specific, antigen-dependent manner. This unique function has made them a central focus of cancer immunotherapy approaches such as immune checkpoint blockade therapy (ICB) and cellular therapies, which act directly on T cells to reinvigorate pre-existing anti-tumor responses or engineer improved specificity and functionality into patient-derived cells, respectively [1, 2]. These T cell-focused therapies have revolutionized the treatment of cancer, resulting in unprecedented long-term survival benefits in patients with late-stage disease across a wide range of cancer types [1]. However, only a fraction of patients respond [1]. Responders are often characterized by the presence of a T cell infiltrate in tumors [3, 4]. CD8+ T cell infiltration can serve as a predictive biomarker [5], underscoring the critical role of cytotoxic T cells in tumor control. However, tumors often progress despite the presence of CD8+ T cells [6]. It has become apparent that tumor-reactive T cells can become dysfunctional [7] or fail to acquire cytotoxic function altogether [8, 9], and both scenarios significantly limit tumor control. Thus, a more nuanced understanding of factors that dictate the quality of CD8+ T cell responses in cancer is needed to design more effective therapeutic solutions.
Anti-tumor T cell responses do not occur in isolation but are directly regulated by dendritic cells (DCs; see Box 1 for a summary of critical DC subsets). The interplay of T cells and DCs in anti-tumor immunity can be illustrated in the Cancer Immunity Cycle (Figure 1) [10]. Tumor-reactive T cell responses begin in the tumor-draining lymph node (tdLN), where T cells encounter tumor-derived antigens presented on DCs [10, 11]. If the peptide presented on type I major histocompatibility complex (MHC-I) on the DC is recognized by the CD8+ T cell receptor (TCR) on the T cell, the two cells engage in an immunological synapse, an interaction during which DCs instruct T cells to proliferate and differentiate [12]. This process of T cell activation is referred to as T cell priming. In the case of a productive anti-tumor immune response, this interaction prompts T cell activation, characterized by clonal expansion and acquisition of effector functions [13]. The activated T cells then leave the tdLN, enter circulation and can become recruited to the tumor site by a chemokine gradient produced by tumor-resident DCs [14]. Once T cells reach the tumor, tumor-resident DCs further support their effector functions [15–21], thereby promoting T cell-driven clearance of cancer cells. The Cancer Immunity Cycle restarts when tumor-resident DCs bring newly acquired tumor-derived antigens into the LN. Thus, DCs are indispensable for T cell progression through the Cancer Immunity Cycle. In this review, we will examine the recent advances in our understanding of DC-mediated control of anti-tumor CD8+ T cell responses and the implications of these findings for the development of improved immunotherapies. It should be noted that DCs can likewise regulate the quality of both helper and regulatory CD4+ T cell responses, which in turn can affect the stimulatory potential of DCs. Due to the complexity of these interactions (previously reviewed in [22–26]), we will focus this review on the mechanisms of DC-mediated regulation of CD8+ T cell immunity in cancer.
Box 1. Dendritic cell subsets in cancer.
DCs are heterogeneous and comprise several subsets with distinct functional specializations. Major DC subsets are defined based on a combination of ontogenic and phenotypic criteria and include conventional type-1 (cDC1) and type-2 (cDC2) DC, plasmacytoid DC (pDC) and monocyte-derived DC (moDC) [28, 124]. Beyond these subset definitions, recent studies indicate that DC can exist in distinct activation states [21, 64, 65]. Herein, we specifically focus on the role of cDC in guiding T cell responses against cancer.
cDC1
cDC1 are critical drivers of T cell responses against cancer. cDC1 are required for tumor control and immunotherapy response in most preclinical models [14, 15, 32, 42, 61, 125] and their presence in tumors has been linked to improved survival in patients [14, 15, 125, 126]. Due to their specialized ability to cross-present antigens and superior interleukin 12 (IL-12) production, cDC1 are regarded as the primary inducers of CD8+ T cell responses [15, 32, 127]. Additionally, recent work suggests the importance of cDC1-mediated priming of CD4+ T cells in cancer [66].
cDC2
Unlike cDC1, the role of cDC2 in anti-tumor immunity has been less clear. Limited data suggests that the presence of cDC2 signatures in patient tumors can also correlate with improved survival outcomes [126, 128, 129], indicating that cDC2 can contribute to protective anti-tumor immunity. cDC2 are preferentially specialized to present MHC-II antigens and prime CD4+ T cells [72]. Additionally, recent studies demonstrate that in inflammatory conditions cDC2 can acquire the functional ability to drive productive CD8+ T cell responses and even mediate tumor regression [21, 65].
Migratory DC
Activated tissue-resident cDC1 and cDC2 can upregulate the chemokine receptor CCR7, which allows them to traffic to the draining LN [11]. This migratory activation state of DC has been observed in preclinical models and human tumors [21, 64, 129–131].
Figure 1. DCs guide T cell progression through the Cancer Immunity Cycle.
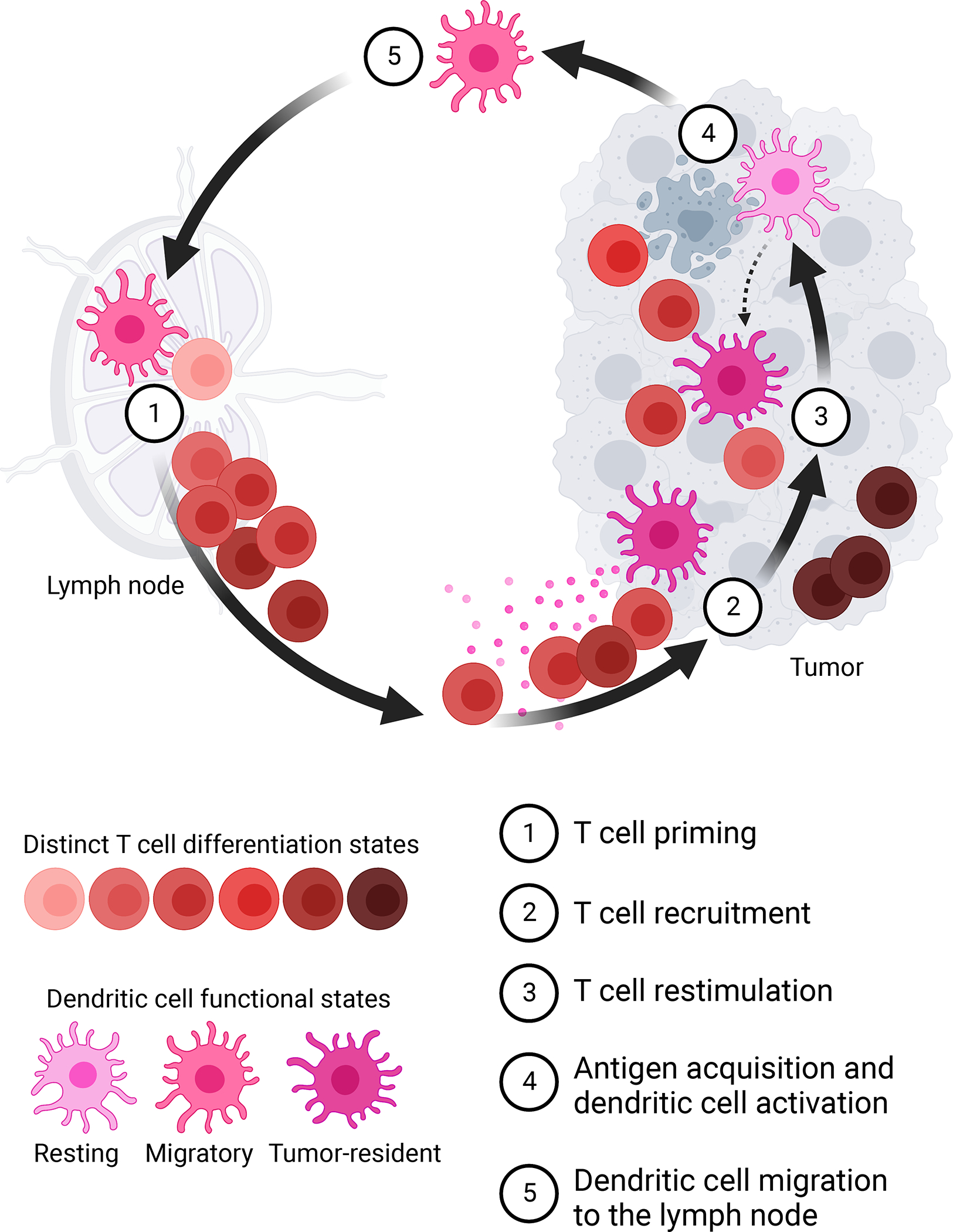
DCs initiate and support anti-tumor CD8+ T cell responses by (1) priming naïve tumor-reactive T cells in the tdLN, (2) recruiting activated T cells from circulation to the tumor site, (3) restimulating effector T cells within the tumor microenvironment to promote cancer cell elimination, (4) acquiring tumor-derived antigens from dying cancer cells and becoming activated, (5) migrating to the tdLN to prime new T cells and thereby restart the Cancer Immunity cycle. Created with Biorender.com.
Dendritic cells provide key stimulatory signals to activate T cells
DCs have the unique ability to sense danger signals and convert this information into specific instructions that guide appropriate T cell responses [27, 28]. Essential DC-derived signals for effective T cell engagement include the peptide antigen presented on the MHC (Signal 1), costimulatory ligands (Signal 2), and cytokines and chemokines (Signal 3) (Figure 2). In this review, we discuss how distinct DC signals regulate the quality of anti-tumor T cell responses. Understanding how DC-derived signals shape T cell responses in cancer is instrumental for the design of rational therapeutic strategies to prevent and reverse T cell dysfunction.
Figure 2. DCs provide distinct stimulatory signals to activate CD8+ T cells in the LN and tumor.
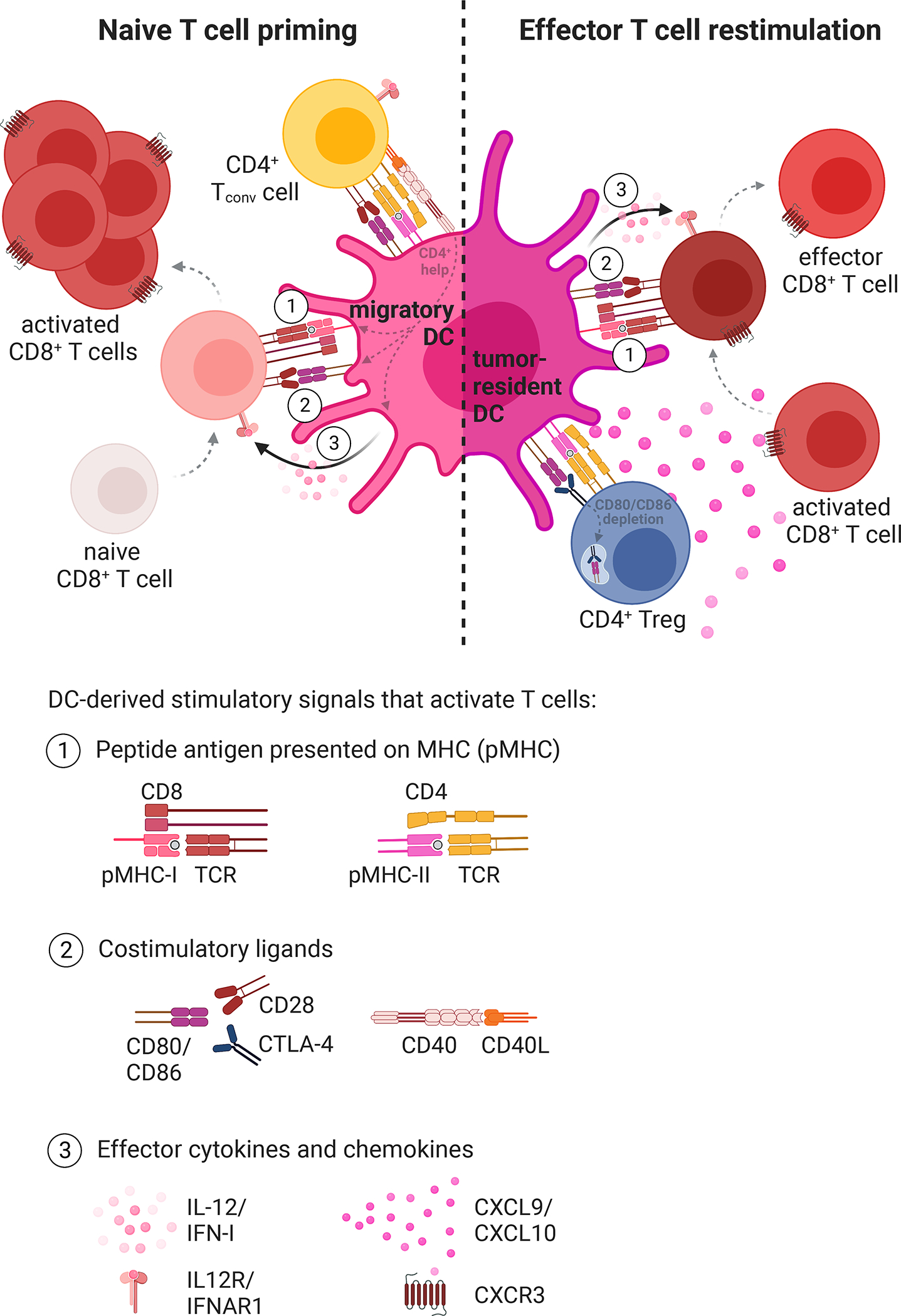
(Left) Migratory DCs in the tdLN prime naïve tumor-reactive CD8+ T cells and (right) tumor-resident DCs in the tumor microenvironment restimulate effector CD8+ T cell responses to promote anti-tumor immunity. In both cases, DCs can provide three distinct stimulatory signals to enable optimal T cell engagement: (1) the tumor-derived antigen presented on MHC-I, (2) costimulatory ligands, and (3) cytokines and chemokines. DCs also interact with CD4+ Tconv and Treg cells, which can influence DC stimulatory capacity and the resulting CD8+ T cell responses. Created with Biorender.com.
Dendritic cells acquire and present tumor cell-derived antigens
As professional antigen-presenting cells, DCs can acquire peptide antigens from their surroundings for presentation on surface MHC molecules [29, 30]. Antigen-specific T cells can recognize cognate peptide-MHC (pMHC) complexes on DCs, and the resulting pMHC:TCR interaction is the first signal necessary for naïve T cell activation or effector T cell re-stimulation (Figure 2) [12, 13]. CD4+ T cell responses rely on conventional MHC-II presentation, a process that has been extensively studied (reviewed in detail in [29]). However, CD8+ T cell responses require that DCs present exogenous antigens on MHC-I by cross-presentation or MHC class I dressing [30]. These two mechanisms are less well defined and represent areas of active investigation and are discussed here.
Cross-presentation
Cross-presentation refers to the process by which DCs engulf exogenous debris, process it into peptides and present the resulting peptides on their surface [30, 31]. Among the distinct DC subsets, cDC1 excel at cross-presentation [15, 32–34], a function that has been linked to their high expression of the C-type lectin receptor DNGR-1 (encoded by CLEC9A) [35, 36]. Specifically, DNGR-1 binds actin filaments exposed on dead cell corpses encountered or ingested by cDC1 [36–38]. Two mechanisms of cross-presentation have been proposed: the ‘vacuolar’ model, wherein exogenous antigens are processed and loaded on MHC-I within endosomes, and the ‘cytosolic’ model, which implies that exogenous antigens from the endosomes gain access to the cytosol for entry into the endogenous MHC-I processing pathway [30, 31]. Previous studies have suggested that the cytosolic pathway of cross-presentation is favored in vivo [39], and recent work further clarified the mechanistic details of this process. DNGR-1 was shown to promote cross-presentation upon ligand binding, specifically by inducing damage to endocytic vesicles [38]. The resulting ruptured phagosomes could release their cargo into the cytosol, enabling the exogenous antigens to undergo proteasomal cleavage and eventual MHC-I loading [38]. Intriguingly, some tumors evolve to escape DNGR-1-dependent immune effects by producing secreted gelsolin, an extracellular protein which inhibits DNGR-1 binding to F-actin and thereby dampens cDC1 cross-presentation [40]. However, while cDC1 ablation can lead to a profound decrease in cross-presentation and anti-tumor immunity [21, 32, 34, 41, 42], DNGR-1 deficiency results in only a partial defect [35, 38], indicating DNGR-1-independent mechanisms of cross-presentation likely also exist and could be uncovered in future studies. Beyond DNGR-1, recent studies have identified additional regulators of cross-presentation in cDC1. For instance, mRNA modifications were found to directly control expression of lysosomal proteases via the protein YTHDF1, which can bind protease transcripts and accelerate their translation [43]. Loss of YTHDF1 in cDC1 caused delayed antigen degradation, resulting in enhanced cross-presentation and CD8+ T cell priming [43]. Similarly, palmitoyl-protein thioesterase 1 was found promote antigen degradation in cDC1 and its downregulation enabled efficient cross-priming [44]. Additional cDC1 adaptations for cross-presentation are thought to include specialized vesicular trafficking mechanisms, which could be regulated by the SNARE family member Sec22b [45] and the GTP-binding protein RAB43 [46]. Recent work further identified the BEACH domain-containing WDFY4 protein as essential for cross-presentation, since WDFY4 loss inhibited CD8+ T cell cross-priming and abrogated tumor control despite normal development of cDC1 [41]. Based on its endosomal localization and interactions with endocytic and cytoskeletal proteins, WDFY4 was proposed to play a role in vesicular trafficking, however, its cellular function remains to be defined [41]. Due to the key requirement of cross-presenting cDC1 for induction of anti-tumor T cell responses, there is significant interest in understanding the precise mechanisms regulating cross-presentation and whether this function could be engineered into other cells or therapeutically induced to promote anti-tumor immunity.
MHC-dressing
MHC-dressing (or cross-dressing) is the phenomenon whereby a DC (or another cell) acquires a preformed pMHC-I molecule from a nearby donor cell [30, 47]. In cancer, DC acquisition of tumor antigens via this mechanism would imply the transfer of an intact pMHC-I from a cancer cell directly to a DC. Mechanistically, cross-dressing is thought to occur in both contact-dependent and contact-independent ways. pMHC transfer between immune cells can be mediated by trogocytosis, a process in which one cell directly contacts and nibbles the membrane of another cell [48]. Alternatively, pMHC-I transfer has also been observed to occur between two DCs via dynamic contact-mediated vesicle exchange [49]. Contact-independent cross-dressing has been proposed to occur upon acquisition of pMHC-containing extracellular vesicles derived from tumor cells [50]. Although the mechanism of MHC-dressing is still under investigation, it is becoming increasingly apparent that this mode of antigen acquisition can have important implications for anti-tumor immunity. Recent studies in preclinical cancer models have demonstrated that in the absence of cross-presentation both cDC1 and cDC2 can acquire tumor-derived pMHC complexes by cross-dressing and consequently mediate productive tumor-reactive CD8+ T cell responses [21, 49, 51, 52]. Cross-dressing in cDC1 was detected in both tumors and tdLN [51], while cross-dressing in cDC2 was specifically associated with a subset enriched in IFN-stimulated genes, termed ISG+ DC, which was restricted to the tumor [21]. The contribution of DC cross-dressing to CD8+ T cell activation was found to vary across different tumor models and could be further enhanced by IFN-I, indicating that context-specific factors may regulate the ability of cross-dressing DCs to elicit anti-tumor immunity [21, 51]. Consequently, strategies to therapeutically exploit the anti-tumor potential of cross-dressing could include IFN-I administration or vaccination approaches using DCs engineered to express chimeric receptors that promote selective uptake of cancer-cell-derived extracellular vesicles [21, 53]. Further research is needed to uncover the mechanistic details of cross-dressing and determine additional factors that control the ability of this process to drive protective anti-tumor immunity.
Mode of tumor-derived antigen acquisition by DCs can modulate T cell responses
The mechanism of tumor antigen acquisition by DCs could influence features of downstream T cell responses, including the clonal repertoire and functional quality. Direct transfer of intact pMHC complexes to DCs could enable faithful representation of the tumor antigen repertoire, resulting in activation of T cell clones specific for antigens presented on cancer cells. However, indirect acquisition of tumor antigens by the complex process of cross-presentation could result in a skewed tumor-derived antigen repertoire on DCs. In fact, recent work revealed that DC-mediated cross-presentation favors antigens derived from cytosolic proteins and is specifically biased against antigens derived from plasma membrane proteins [54]. Moreover, cancer cells can change their antigen presentation pathways, for instance, by suppressing expression of critical elements of antigen presentation machinery such as the transporter associated with antigen processing (TAP) complexes, to globally alter their immunopeptidome and evade immune responses [55]. Even though Barbet et al. recently demonstrated that TAP-deficient DCs cross-present peptides that mirror antigens presented on TAP-deficient diseased cells [56], DCs in tumors would be unlikely to lose TAP and thus would fail to activate T cell clones needed to effectively target TAP-deficient tumor cells. Thus, both DC-intrinsic biases in cross-presentation and systemic alterations in the antigen presentation in tumor cells could cause discrepancies between cross-presented and tumor antigen repertoires, which would limit the number of activated T cell clones capable of recognizing antigens presented on the tumor cells.
In addition to influencing the T cell repertoire, mode of tumor antigen presentation by DCs could also impact the magnitude and functional quality of downstream T cell responses. In support of this notion, recent studies indicate that MHC-dressing and cross-presenting DC subsets induce qualitatively distinct T cell responses [21, 49, 51]. Compared to cross-presenting cDC1, ISG+ DC and cDC1 that cross-dressed with tumor-derived MHC-I complexes appeared to induce slower T cell proliferation and were correlated with sustained T cell immunity [21, 51], although additional studies will be needed to decouple the mode of antigen presentation from DC subset identity. The precise mechanisms leading to these differences in T cell activation warrant further investigation. It is plausible that the two routes of antigen acquisition influence the density of tumor antigens presented on MHC of DCs, resulting in differential T cell stimulation. In fact, cross-presented antigen density was shown to directly correlate with the size of the resulting T cell response and can influence memory responses [57, 58]. Future studies quantifying the relative efficiency of tumor antigen acquisition via cross-presentation and cross-dressing will determine how these distinct processes influence DC antigen density. Interestingly, cross-dressing has been proposed to be more efficient than cross-presentation for priming T cells when antigen levels are low [59], suggesting that the distinct modes of antigen acquisition may be selectively advantageous in different contexts. Our current knowledge of how cross-dressing and cross-presentation differentially influence CD8+ T cell function is limited, and further research is needed to delineate the individual contributions of the two processes to anti-tumor immunity. Since cross-presentation and cross-dressing have intrinsic differences and can complement each other, optimal immune responses may occur when both modes of antigen presentation are happening simultaneously.
Dendritic cell activation state is key
Beyond presentation of tumor-derived antigens, DCs can also provide the necessary stimulatory signals to drive effector T cell responses. These stimulatory signals comprise costimulatory ligands, such as CD80, CD86 and CD40, as well as secreted cytokines, including IL-12 and IFN-I (Figure 2) [28]. DCs that robustly express stimulatory signals are termed mature, due to their superior immunostimulatory capacity [28]. The role of DC-derived stimulatory signals in T cell activation is well accepted, and numerous studies have demonstrated that treatment with DC expansion and maturation agents can enhance anti-tumor T cell responses and potentiate tumor control [17, 18, 42, 60–63]. Recent studies further underscore the importance of DC activation states for determining the functional quality of anti-tumor T cell responses.
DCs are plastic and can exist in distinct context-dependent activation states, which are characterized by specific expression patterns of immunomodulatory signals. For instance, a recent study identified that mature DCs in lung tumors could acquire an immunoregulatory program termed mregDC, which dampened their ability to activate effector T cells and promote anti-tumor immunity [64]. The mregDC program was distinguished by the concurrent upregulation of maturation and immunoregulatory transcripts and could be acquired by both cDC1 and cDC2 upon tumor antigen uptake [64]. Maier et al. could reverse the inhibitory effects of mregDC by administering in vivo IL-4 blockade, which led to increased IL-12 production by tumor DCs, augmented effector T cell responses and enhanced tumor control [64]. Additionally, two recent studies have shown that in the context of inflammation, cDC2 can acquire a novel activation state that enables them to effectively prime CD8+ T cells [21, 65]. Bosteels et al. termed this activation state inflammatory cDC2 (inf-cDC2) in the context of a viral infection [65], while Duong et al. identified a similar cDC2 state enriched in IFN-stimulated genes (ISG+ DC) in regressing tumors [21]. Specifically, IFN-I was found to drive ISG+ DC activation, resulting in the upregulation of the costimulatory ligand CD86 and thereby enabling these cDC2 to cross-prime CD8+ T cells [21]. Notably, ISG+ DC-mediated priming of CD8+ T cell responses could drive spontaneous tumor regression even in the absence of cDC1 [21]. Moreover, IFN-I administration could induce cDC1-independent CD8+ T cells responses against progressing tumors as well [21]. Thus, the detailed understanding of DC activation states and their implications for T cell immunity can inform approaches for therapeutic intervention.
DC activation states can be directly influenced by other immune cells, particularly, CD4+ T cells. Using a tumor model that requires both CD4+ and CD8+ T cell responses for mediating tumor control, recent work clarified the importance of CD4+ help for enabling cDC1 functional ability to drive protective CD8+ T cell immunity (a phenomenon termed licensing) [66]. Ferris et al. demonstrate that optimal anti-tumor CD8+ T cell responses require that cDC1 receive CD4+ help in an MHC-II-specific interaction that involves CD40 signaling [66]. In fact, cDC1-specific ablation of MHC-II or CD40 blunted anti-tumor immune responses and abrogated tumor control [66]. These findings build on prior work showing that CD40 signaling can induce DC maturation and enhance their immunostimulatory capacity [67]. In preclinical tumor models, CD40 agonist treatment can boost anti-tumor immunity by mimicking CD4+ help and enhancing DC ability to activate T cells [68–71]. However, in contrast to the ability of CD4+ conventional T (Tconv) cells to activate DCs, CD4+ regulatory T cells (Tregs) can exert the converse effect of suppressing DC stimulatory capacity. Treg-mediated suppression was shown to restrain DC ability to drive CD4+ Tconv and CD8+ T cell responses in cancer, an effect that could be rescued by Treg depletion [72–75]. Specifically, Tregs can induce downregulation of CD80 and CD86 costimulatory ligand expression on cDC1 and cDC2 within tumors [76]. The best-studied mechanism of Treg-mediated DC suppression involves the CTLA-4 molecule, which can capture and degrade CD80/CD86 ligands from DCs in a process of trans-endocytosis [77, 78]. Although CTLA-4 blockade (using a non-depleting antibody) can reverse the observed effects of Treg-mediated DC suppression in tumors [76], it does not always effectively control tumors [9, 79]. Since Tregs can suppress in a myriad of ways [80], a more refined understanding of how Tregs impact DC-mediated T cell responses could inspire development of novel therapeutic perturbations.
Although it is known that the DC activation state can directly influence the quality of downstream anti-tumor T cell responses, a precise understanding of how DC instruct distinct T cell programs is lacking. Both effector and memory CD8+ T cells are required for effective tumor control, yet it remains unclear which DC-derived activation signals are needed to induce and maintain these distinct functional states of T cells in cancer. It is well accepted that CD4+ help plays an important role in promoting both memory and effector T cell responses [66, 67, 81], but therapeutic delivery of CD4+ help via CD40 agonists is not always sufficient for inducing protective anti-tumor immunity [71]. Thus, additional factors and pathways beyond CD40 signaling must regulate T cell effector and memory responses in cancer. Effector T cell differentiation was shown to specifically require DC-derived cytokines such as IFN-I and IL-12 [13], the latter of which can be induced upon CD40 engagement [28]. However, the timing and context of cytokine signals can influence T cell function in complex ways [9, 82–85] and our incomplete understanding of this biological complexity has thus far hindered the success of cytokine therapies [86]. Recent studies of tumor-reactive CD8+ T cell activation states in the context of ICB therapy have suggested that ICB response is specifically linked to the expansion of the TCF1+ stem-like memory CD8+ T cell subset [87–89]. Importantly, work by Schenkel et al. demonstrated that cDC1 in the tdLN are critical for the maintenance of proliferative tumor-reactive TCF1+ CD8+ T cells [90]. Treatment-induced expansion and activation of cDC1 could increase the abundance of this critical T cell subset and decreased tumor burden [90]. Although the specific DC-derived signals necessary for this effect warrant further investigation, insights from a model of chronic viral infection suggest that reduced expression of costimulatory molecules on DCs along with diminished TCR signaling on T cells can drive differentiation of TCF1+ CD8+ T cells [91]. Conversely, enhanced TCR signaling downstream of high affinity pMHC-I:TCR engagement was associated with the differentiation of effector CD8+ T cells that exhibited increased cytotoxic capacity and could mediate improved tumor control [92, 93]. This observation is particularly important in the context of therapeutic vaccination as neoantigen-based cancer vaccines are frequently designed to engage CD8+ T cells with high affinity TCR [94]. In addition to the ability of DCs to tune effector and stem-like memory T cell differentiation, several studies have highlighted the importance of specific T cell and DC interactions for the establishment of the tissue-resident memory CD8+ T cell fate and functional reactivation of these T cells [95–98]. The diverse outcomes of DC-mediated regulation of T cell function are further highlighted by recent reports of inhibitory checkpoint molecule expression on DCs. Several studies have pointed out that IFN-II-driven expression of PD-L1 on DCs restrains effector CD8+ T cell responses, and specific blockade of PD-L1 on DCs, but not macrophages, is required to reinvigorate tumor-reactive T cells [99–101]. Similarly, DC-specific expression of TIM-3 was shown to limit anti-tumor immunity, and TIM-3 deletion or blockade could promote DC stimulatory capacity, resulting in enhanced effector CD8+ T cell responses and improved tumor control [19, 20, 102]. Thus, further research clarifying how DC-derived signals regulate induction of distinct CD8+ T cell activation states will enable approaches to rewire anti-tumor immunity with increased precision.
Dendritic cells spatially regulate T cell responses
Increasing evidence suggests that the spatial regulation of T cell responses can have important implications for anti-tumor immunity and immunotherapy response [103]. DCs can produce chemokines to attract T cells and thereby directly control T cell recruitment and localization within distinct tissues during an immune response (Figure 1 and Figure 2). The anatomic site of tumor growth can also influence DC-mediated instruction of T cell responses. Recent studies have begun to shed light on how CD8+ T cell responses are spatially coordinated by local DCs.
T cell responses are coordinated within microanatomical niches of LN and tumors and the resulting T cell activation states can be regulated by the physical proximity of DC-derived signals. Duckworth et al. recently demonstrated that effector and stem-like memory CD8+ T cells are generated in different microanatomical niches of the LN and associated with spatially distinct DC subsets [104]. Specifically, T-bet+ short-lived effector-like T cells were induced in the LN periphery in a CXCR3-dependent manner and were associated with the CXCL10-producing inflammatory cDC2 [104]. In contrast, TCF1+ stem-like memory precursors were preferentially generated in the LN paracortex and spatially colocalized with CXCL9-expressing cDC1 [104]. This spatial distribution of DCs was further shown to be dynamic and tunable, as inflammation-induced DC repositioning within the LN or specific perturbation of DC-derived chemokine signals could directly influence the phenotype of primed CD8+ T cells [104, 105]. Deficiencies in both CXCL10 and IFN-I signaling were found to skew differentiation towards more TCF1+ stem-like T cell memory precursors, which expressed higher levels of CCR7 and accumulated within the LN center [104]. Additionally, Leal et al. found that vaccination with type I inflammatory toll-like receptor agonists induced cDC to upregulate CCR7 and relocate into the LN T cell zone, which was enriched for TCF1+ CD8+ T cells [105]. Outside of the orderly structure of the LN, DCs within the tumor microenvironment can also influence the spatial organization of T cell responses. The critical role of tumor-resident cDC1 is underscored by studies showing that tumor-mediated suppression of cDC1 recruitment, accumulation or activation can enable tumor evasion of T cell responses [106–108]. For instance, tumor cDC1 can produce chemokines CXCL9 and CXCL10 and thereby recruit activated T cells to the tumor site [14]. Consequently, tumors devoid of cDC1 were found to have low levels of T cell infiltration [14, 106]. CXCL9 production by cDC1 was also shown to induce colocalization of cDC1 and CD8+ T cells within tumors [20]. As a result of this increased proximity, CD8+ T cells were exposed to higher levels of cDC1-derived IL-12, which induced T cells to produce the effector cytokine IFN-II [20, 109]. This cDC1 function could be therapeutically induced with TIM-3 blockade, which was found to increase cDC1 expression of CXCL9 and thus enhance CD8+ T cell production of IFN-II [19, 20]. DCs were further found to provide critical survival signals to CD8+ T cells within the perivascular niche of the tumor stroma [110]. This DC-mediated effect was spatially coordinated via the CXCL16-CXCR6 axis, whereby CXCL16+ DC trans-presented the survival cytokine IL-15 to CXCR6+ T cells to promote local expansion of effector CD8+ T cell responses within the tumor microenvironment [110]. Additionally, tumor-residing DCs and T cells can interact within tertiary lymphoid structures (TLS), which are organized aggregates of immune cells that often form within tumors [111]. The density of mature DCs within TLS was found to correlate with CD8+ T cell infiltration and long-term survival in patients with lung cancer [112]. Since TLS are generally correlated with better survival across a range of cancer types [111], there is significant interest in advancing our mechanistic understanding of their role in anti-tumor immunity. Further research will uncover additional spatiotemporal requirements of optimal anti-tumor T cell responses and determine how these are influenced by local DCs.
DCs can also provide anatomic site-specific instructions to guide T cell function [113]. Studies of lung and gut immunity have previously indicated that DCs can induce T cells to upregulate tissue-specific homing receptors, which enable T cells to traffic into the appropriate organ sites [114, 115]. Recent studies have further found that DCs can polarize T cell responses towards effector, tolerance or residence memory phenotypes in a tissue-specific manner [95, 116]. Thus, the local tissue microenvironment of the tumor can play an important role in shaping the quality of DC-mediated T cell responses [117–119]. Although further research is needed to uncover how tissue-specific factors regulate DC functions in cancer, several studies provide evidence that the tissue site of tumor growth can influence the quality of anti-tumor T cell immunity [9, 120–123]. Recent work focused on colorectal cancer metastases revealed that liver tumors contained fewer DCs and T cells and had worse ICB responses compared to subcutaneous colorectal tumors [120]. Induction of local cDC development and expansion was found to sensitize liver tumors to ICB [120], suggesting that liver-specific suppression of DC recruitment and expansion within tumors can blunt anti-tumor immunity. In addition to impacting local immune function, tissue site of tumor growth could also influence immune responses in the draining LN. In fact, Horton et al. recently determined that lung cancer cells growing orthotopically in the lungs induced priming of dysfunctional CD8+ T cell responses in the lung-draining LN [9]. The resulting dysfunctional T cells lacked cytotoxic effector function and were resistant to ICB reinvigoration [9]. In contrast, the same cancer cells growing subcutaneously induced effector CD8+ T cell responses in the iLN, and these T cells were responsive to ICB [9]. Since qualitatively different T cell fates were encoded during the T cell priming event in the tdLN, prior to T cell exposure to the local tumor microenvironment [9], it is plausible that the distinct priming outcomes resulted from tissue-specific differences in function of LN DCs. Thus, tissue-specific factors in the tumor microenvironment as well as the draining LN may impact DC ability to activate, support and maintain anti-tumor T cell responses. Such tissue site-specific immune regulation represents an additional layer of DC-mediated control of T cell responses.
Concluding remarks
Cancer-associated T cell dysfunction has emerged as a key barrier to anti-tumor immunity. As T cells can become dysfunctional within tumors [7] as well as early on during T cell priming in the tdLN [8, 9], it is plausible that exposure to DCs equipped with appropriate stimulatory signals could reverse these effects and/or promote differentiation of alternative T cell states needed for tumor control and immunotherapy response. Although prior studies have correlated anti-tumor immunity with the presence of DCs in tumors, it has become apparent that DC effects on T cell function are complex and multifactorial. DCs instruct T cell differentiation by providing different signals at different times and across different anatomic locations. As a result, DC-derived signals can have diverse, time-dependent, and context-specific effects on the functional capacity of tumor-reactive T cell responses. Moreover, different DC subsets and activation states can have distinct functional specializations with respect to T cell stimulation. In cancer, DC-mediated regulation of T cell function is further complicated by the fact that DCs themselves often become suppressed. Tumor development can result in both decreased abundance and diminished stimulatory capacity of DCs [19, 20, 64, 72–76, 99–102, 106–108], and these suppression effects can be local or systemic and likely change over time. Thus, careful studies are needed to dissect the complexity of DC functions in cancer and their impact on the quality of resulting anti-tumor T cell immunity (see Outstanding questions). A refined understanding of how DC-derived signals shape the functional quality of anti-tumor T cell responses could enable the development of improved approaches to therapeutically counteract and reinvigorate dysfunctional T cell responses in cancer.
Acknowledgements
M.Z. is supported by the Virginia and D.K. Ludwig Fund for Cancer Research and S.S. is a Pew-Steward Scholar of the Pew Charitable Trust and holds the Howard S. (1953) and Linda B. Stern Career Development Professorship.
Footnotes
Declaration of interests
The authors declare no competing interests. S.S. is a SAB member for Related Sciences, Arcus Biosciences and Venn Therapeutics. S.S. is a co-founder of Danger Bio. S.S. is a consultant for TAKEDA, Merck, Tango Therapeutics, and Ribon Therapeutics and receives funding for unrelated projects from Leap Therapeutics.
References
- 1.Ribas A and Wolchok JD (2018) Cancer immunotherapy using checkpoint blockade. Science 359 (6382), 1350–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Finck AV et al. (2022) Engineered cellular immunotherapies in cancer and beyond. Nat Med 28 (4), 678–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tumeh PC et al. (2014) PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515 (7528), 568–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li F et al. (2021) The association between CD8+ tumor-infiltrating lymphocytes and the clinical outcome of cancer immunotherapy: A systematic review and meta-analysis. EClinicalMedicine 41, 101134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen DS and Mellman I (2017) Elements of cancer immunity and the cancer-immune set point. Nature 541 (7637), 321–330. [DOI] [PubMed] [Google Scholar]
- 6.Herbst RS et al. (2014) Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 515 (7528), 563–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Philip M and Schietinger A (2022) CD8(+) T cell differentiation and dysfunction in cancer. Nat Rev Immunol 22 (4), 209–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nussing S et al. (2020) Revisiting T Cell Tolerance as a Checkpoint Target for Cancer Immunotherapy. Front Immunol 11, 589641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Horton BL et al. (2021) Lack of CD8(+) T cell effector differentiation during priming mediates checkpoint blockade resistance in non-small cell lung cancer. Sci Immunol 6 (64), eabi8800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen DS and Mellman I (2013) Oncology meets immunology: the cancer-immunity cycle. Immunity 39 (1), 1–10. [DOI] [PubMed] [Google Scholar]
- 11.Roberts EW et al. (2016) Critical Role for CD103(+)/CD141(+) Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer Cell 30 (2), 324–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dustin ML (2014) The immunological synapse. Cancer Immunol Res 2 (11), 1023–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mescher MF et al. (2007) Molecular basis for checkpoints in the CD8 T cell response: tolerance versus activation. Semin Immunol 19 (3), 153–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Spranger S et al. (2017) Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell 31 (5), 711–723 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Broz ML et al. (2014) Dissecting the Tumor Myeloid Compartment Reveals Rare Activating Antigen-Presenting Cells Critical for T Cell Immunity. Cancer Cell 26 (6), 938. [DOI] [PubMed] [Google Scholar]
- 16.Ruffell B et al. (2014) Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell 26 (5), 623–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Diamond MS et al. (2011) Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med 208 (10), 1989–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fuertes MB et al. (2011) Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med 208 (10), 2005–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Mingo Pulido A et al. (2018) TIM-3 Regulates CD103(+) Dendritic Cell Function and Response to Chemotherapy in Breast Cancer. Cancer Cell 33 (1), 60–74 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gardner A et al. (2022) TIM-3 blockade enhances IL-12-dependent antitumor immunity by promoting CD8(+) T cell and XCR1(+) dendritic cell spatial co-localization. J Immunother Cancer 10 (1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duong E et al. (2022) Type I interferon activates MHC class I-dressed CD11b(+) conventional dendritic cells to promote protective anti-tumor CD8(+) T cell immunity. Immunity 55 (2), 308–323 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Borst J et al. (2018) CD4(+) T cell help in cancer immunology and immunotherapy. Nat Rev Immunol 18 (10), 635–647. [DOI] [PubMed] [Google Scholar]
- 23.Ahrends T and Borst J (2018) The opposing roles of CD4(+) T cells in anti-tumour immunity. Immunology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yin X et al. (2021) Dendritic Cell Regulation of T Helper Cells. Annu Rev Immunol 39, 759–790. [DOI] [PubMed] [Google Scholar]
- 25.Waisman A et al. (2017) Dendritic cells as gatekeepers of tolerance. Semin Immunopathol 39 (2), 153–163. [DOI] [PubMed] [Google Scholar]
- 26.Basu A et al. (2021) Differentiation and Regulation of TH Cells: A Balancing Act for Cancer Immunotherapy. Front Immunol 12, 669474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lanzavecchia A and Sallusto F (2001) Regulation of T cell immunity by dendritic cells. Cell 106 (3), 263–6. [DOI] [PubMed] [Google Scholar]
- 28.Cabeza-Cabrerizo M et al. (2021) Dendritic Cells Revisited. Annu Rev Immunol 39, 131–166. [DOI] [PubMed] [Google Scholar]
- 29.Pishesha N et al. (2022) A guide to antigen processing and presentation. Nat Rev Immunol. [DOI] [PubMed] [Google Scholar]
- 30.Embgenbroich M and Burgdorf S (2018) Current Concepts of Antigen Cross-Presentation. Front Immunol 9, 1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Childs E et al. (2021) Maintenance and loss of endocytic organelle integrity: mechanisms and implications for antigen cross-presentation. Open Biol 11 (11), 210194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hildner K et al. (2008) Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science 322 (5904), 1097–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Iyoda T et al. (2002) The CD8+ dendritic cell subset selectively endocytoses dying cells in culture and in vivo. J Exp Med 195 (10), 1289–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Edelson BT et al. (2010) Peripheral CD103+ dendritic cells form a unified subset developmentally related to CD8alpha+ conventional dendritic cells. J Exp Med 207 (4), 823–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sancho D et al. (2009) Identification of a dendritic cell receptor that couples sensing of necrosis to immunity. Nature 458 (7240), 899–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cueto FJ et al. (2019) DNGR-1, a Dendritic Cell-Specific Sensor of Tissue Damage That Dually Modulates Immunity and Inflammation. Front Immunol 10, 3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schulz O et al. (2018) Myosin II Synergizes with F-Actin to Promote DNGR-1-Dependent Cross-Presentation of Dead Cell-Associated Antigens. Cell Rep 24 (2), 419–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Canton J et al. (2021) The receptor DNGR-1 signals for phagosomal rupture to promote cross-presentation of dead-cell-associated antigens. Nat Immunol 22 (2), 140–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Palmowski MJ et al. (2006) Role of immunoproteasomes in cross-presentation. J Immunol 177 (2), 983–90. [DOI] [PubMed] [Google Scholar]
- 40.Giampazolias E et al. (2021) Secreted gelsolin inhibits DNGR-1-dependent cross-presentation and cancer immunity. Cell 184 (15), 4016–4031 e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Theisen DJ et al. (2018) WDFY4 is required for cross-presentation in response to viral and tumor antigens. Science 362 (6415), 694–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sanchez-Paulete AR et al. (2016) Cancer Immunotherapy with Immunomodulatory Anti-CD137 and Anti-PD-1 Monoclonal Antibodies Requires BATF3-Dependent Dendritic Cells. Cancer Discov 6 (1), 71–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Han D et al. (2019) Anti-tumour immunity controlled through mRNA m(6)A methylation and YTHDF1 in dendritic cells. Nature 566 (7743), 270–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ou P et al. (2019) Thioesterase PPT1 balances viral resistance and efficient T cell crosspriming in dendritic cells. J Exp Med 216 (9), 2091–2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alloatti A et al. (2017) Critical role for Sec22b-dependent antigen cross-presentation in antitumor immunity. J Exp Med 214 (8), 2231–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kretzer NM et al. (2016) RAB43 facilitates cross-presentation of cell-associated antigens by CD8alpha+ dendritic cells. J Exp Med 213 (13), 2871–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Martinez-Usatorre A and De Palma M (2022) Dendritic cell cross-dressing and tumor immunity. EMBO Mol Med, e16523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miyake K et al. (2017) Trogocytosis of peptide-MHC class II complexes from dendritic cells confers antigen-presenting ability on basophils. Proc Natl Acad Sci U S A 114 (5), 1111–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ruhland MK et al. (2020) Visualizing Synaptic Transfer of Tumor Antigens among Dendritic Cells. Cancer Cell 37 (6), 786–799 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zeng F and Morelli AE (2018) Extracellular vesicle-mediated MHC cross-dressing in immune homeostasis, transplantation, infectious diseases, and cancer. Semin Immunopathol 40 (5), 477–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.MacNabb BW et al. (2022) Dendritic cells can prime anti-tumor CD8(+) T cell responses through major histocompatibility complex cross-dressing. Immunity 55 (6), 982–997 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Das Mohapatra A et al. (2020) Cross-dressing of CD8alpha(+) Dendritic Cells with Antigens from Live Mouse Tumor Cells Is a Major Mechanism of Cross-priming. Cancer Immunol Res 8 (10), 1287–1299. [DOI] [PubMed] [Google Scholar]
- 53.Squadrito ML et al. (2018) EVIR: chimeric receptors that enhance dendritic cell cross-dressing with tumor antigens. Nat Methods 15 (3), 183–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fessenden TB et al. (2022) Dendritic cell-mediated cross presentation of tumor-derived peptides is biased against plasma membrane proteins. J Immunother Cancer 10 (7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Marijt KA et al. (2019) TEIPP antigens for T-cell based immunotherapy of immune-edited HLA class I(low) cancers. Mol Immunol 113, 43–49. [DOI] [PubMed] [Google Scholar]
- 56.Barbet G et al. (2021) TAP dysfunction in dendritic cells enables noncanonical cross-presentation for T cell priming. Nat Immunol 22 (4), 497–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bullock TN et al. (2003) Antigen density presented by dendritic cells in vivo differentially affects the number and avidity of primary, memory, and recall CD8+ T cells. J Immunol 170 (4), 1822–9. [DOI] [PubMed] [Google Scholar]
- 58.Anikeeva N et al. (2012) Evidence that the density of self peptide-MHC ligands regulates T-cell receptor signaling. PLoS One 7 (8), e41466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dolan BP et al. (2006) Dendritic cells cross-dressed with peptide MHC class I complexes prime CD8+ T cells. J Immunol 177 (9), 6018–24. [DOI] [PubMed] [Google Scholar]
- 60.Corrales L et al. (2015) Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep 11 (7), 1018–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Salmon H et al. (2016) Expansion and Activation of CD103(+) Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity 44 (4), 924–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Saxena M and Bhardwaj N (2017) Turbocharging vaccines: emerging adjuvants for dendritic cell based therapeutic cancer vaccines. Curr Opin Immunol 47, 35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wculek SK et al. (2020) Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol 20 (1), 7–24. [DOI] [PubMed] [Google Scholar]
- 64.Maier B et al. (2020) A conserved dendritic-cell regulatory program limits antitumour immunity. Nature 580 (7802), 257–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bosteels C et al. (2020) Inflammatory Type 2 cDCs Acquire Features of cDC1s and Macrophages to Orchestrate Immunity to Respiratory Virus Infection. Immunity 52 (6), 1039–1056 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ferris ST et al. (2020) cDC1 prime and are licensed by CD4(+) T cells to induce anti-tumour immunity. Nature 584 (7822), 624–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wu R and Murphy KM (2022) DCs at the center of help: Origins and evolution of the three-cell-type hypothesis. J Exp Med 219 (7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Diamond MS et al. (2021) Site-Dependent Immune Escape Due to Impaired Dendritic Cell Cross-Priming. Cancer Immunol Res 9 (8), 877–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stevens AD and Bullock TNJ (2021) Therapeutic vaccination targeting CD40 and TLR3 controls melanoma growth through existing intratumoral CD8 T cells without new T cell infiltration. Cancer Immunol Immunother 70 (8), 2139–2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bullock TNJ (2022) CD40 stimulation as a molecular adjuvant for cancer vaccines and other immunotherapies. Cell Mol Immunol 19 (1), 14–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vonderheide RH (2020) CD40 Agonist Antibodies in Cancer Immunotherapy. Annu Rev Med 71, 47–58. [DOI] [PubMed] [Google Scholar]
- 72.Binnewies M et al. (2019) Unleashing Type-2 Dendritic Cells to Drive Protective Antitumor CD4(+) T Cell Immunity. Cell 177 (3), 556–571 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bauer CA et al. (2014) Dynamic Treg interactions with intratumoral APCs promote local CTL dysfunction. J Clin Invest 124 (6), 2425–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Joshi NS et al. (2015) Regulatory T Cells in Tumor-Associated Tertiary Lymphoid Structures Suppress Anti-tumor T Cell Responses. Immunity 43 (3), 579–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jang JE et al. (2017) Crosstalk between Regulatory T Cells and Tumor-Associated Dendritic Cells Negates Anti-tumor Immunity in Pancreatic Cancer. Cell Rep 20 (3), 558–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Marangoni F et al. (2021) Expansion of tumor-associated Treg cells upon disruption of a CTLA-4-dependent feedback loop. Cell 184 (15), 3998–4015 e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Qureshi OS et al. (2011) Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science 332 (6029), 600–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kennedy A et al. (2022) Differences in CD80 and CD86 transendocytosis reveal CD86 as a key target for CTLA-4 immune regulation. Nat Immunol 23 (9), 1365–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Grosso JF and Jure-Kunkel MN (2013) CTLA-4 blockade in tumor models: an overview of preclinical and translational research. Cancer Immun 13, 5. [PMC free article] [PubMed] [Google Scholar]
- 80.Sakaguchi S et al. (2009) Regulatory T cells: how do they suppress immune responses? Int Immunol 21 (10), 1105–11. [DOI] [PubMed] [Google Scholar]
- 81.Ahrends T et al. (2019) CD4(+) T cell help creates memory CD8(+) T cells with innate and help-independent recall capacities. Nat Commun 10 (1), 5531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Urban SL and Welsh RM (2014) Out-of-sequence signal 3 as a mechanism for virus-induced immune suppression of CD8 T cell responses. PLoS Pathog 10 (9), e1004357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zemek RM et al. (2022) Temporally restricted activation of IFNβ signaling underlies response to immune checkpoint therapy in mice. Nature Communications 13 (1), 4895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sckisel GD et al. (2015) Out-of-Sequence Signal 3 Paralyzes Primary CD4(+) T-Cell-Dependent Immunity. Immunity 43 (2), 240–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Whyte CE et al. (2022) Context-dependent effects of IL-2 rewire immunity into distinct cellular circuits. J Exp Med 219 (7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Propper DJ and Balkwill FR (2022) Harnessing cytokines and chemokines for cancer therapy. Nat Rev Clin Oncol 19 (4), 237–253. [DOI] [PubMed] [Google Scholar]
- 87.Kurtulus S et al. (2019) Checkpoint Blockade Immunotherapy Induces Dynamic Changes in PD-1(−)CD8(+) Tumor-Infiltrating T Cells. Immunity 50 (1), 181–194 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Siddiqui I et al. (2019) Intratumoral Tcf1(+)PD-1(+)CD8(+) T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity 50 (1), 195–211 e10. [DOI] [PubMed] [Google Scholar]
- 89.Connolly KA et al. (2021) A reservoir of stem-like CD8(+) T cells in the tumor-draining lymph node preserves the ongoing antitumor immune response. Sci Immunol 6 (64), eabg7836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Schenkel JM et al. (2021) Conventional type I dendritic cells maintain a reservoir of proliferative tumor-antigen specific TCF-1(+) CD8(+) T cells in tumor-draining lymph nodes. Immunity 54 (10), 2338–2353 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Snell LM et al. (2018) CD8(+) T Cell Priming in Established Chronic Viral Infection Preferentially Directs Differentiation of Memory-like Cells for Sustained Immunity. Immunity 49 (4), 678–694 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Schmid DA et al. (2010) Evidence for a TCR affinity threshold delimiting maximal CD8 T cell function. J Immunol 184 (9), 4936–46. [DOI] [PubMed] [Google Scholar]
- 93.Martinez-Usatorre A et al. (2018) PD-1 Blockade Unleashes Effector Potential of Both High- and Low-Affinity Tumor-Infiltrating T Cells. J Immunol 201 (2), 792–803. [DOI] [PubMed] [Google Scholar]
- 94.Blass E and Ott PA (2021) Advances in the development of personalized neoantigen-based therapeutic cancer vaccines. Nat Rev Clin Oncol 18 (4), 215–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mani V et al. (2019) Migratory DCs activate TGF-beta to precondition naive CD8(+) T cells for tissue-resident memory fate. Science 366 (6462). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Low JS et al. (2020) Tissue-resident memory T cell reactivation by diverse antigen-presenting cells imparts distinct functional responses. J Exp Med 217 (8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Iborra S et al. (2016) Optimal Generation of Tissue-Resident but Not Circulating Memory T Cells during Viral Infection Requires Crosspriming by DNGR-1(+) Dendritic Cells. Immunity 45 (4), 847–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Enamorado M et al. (2017) Enhanced anti-tumour immunity requires the interplay between resident and circulating memory CD8(+) T cells. Nat Commun 8, 16073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mayoux M et al. (2020) Dendritic cells dictate responses to PD-L1 blockade cancer immunotherapy. Sci Transl Med 12 (534). [DOI] [PubMed] [Google Scholar]
- 100.Peng Q et al. (2020) PD-L1 on dendritic cells attenuates T cell activation and regulates response to immune checkpoint blockade. Nat Commun 11 (1), 4835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Oh SA et al. (2020) PD-L1 expression by dendritic cells is a key regulator of T-cell immunity in cancer. Nat Cancer 1 (7), 681–691. [DOI] [PubMed] [Google Scholar]
- 102.Dixon KO et al. (2021) TIM-3 restrains anti-tumour immunity by regulating inflammasome activation. Nature 595 (7865), 101–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tooley KA et al. (2022) Spatial determinants of CD8(+) T cell differentiation in cancer. Trends Cancer 8 (8), 642–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Duckworth BC et al. (2021) Effector and stem-like memory cell fates are imprinted in distinct lymph node niches directed by CXCR3 ligands. Nat Immunol 22 (4), 434–448. [DOI] [PubMed] [Google Scholar]
- 105.Leal JM et al. (2021) Innate cell microenvironments in lymph nodes shape the generation of T cell responses during type I inflammation. Sci Immunol 6 (56). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Spranger S et al. (2015) Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 523 (7559), 231–5. [DOI] [PubMed] [Google Scholar]
- 107.Zelenay S et al. (2015) Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell 162 (6), 1257–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hegde S et al. (2020) Dendritic Cell Paucity Leads to Dysfunctional Immune Surveillance in Pancreatic Cancer. Cancer Cell 37 (3), 289–307 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Garris CS et al. (2018) Successful Anti-PD-1 Cancer Immunotherapy Requires T Cell-Dendritic Cell Crosstalk Involving the Cytokines IFN-gamma and IL-12. Immunity 49 (6), 1148–1161 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Di Pilato M et al. (2021) CXCR6 positions cytotoxic T cells to receive critical survival signals in the tumor microenvironment. Cell 184 (17), 4512–4530 e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Schumacher TN and Thommen DS (2022) Tertiary lymphoid structures in cancer. Science 375 (6576), eabf9419. [DOI] [PubMed] [Google Scholar]
- 112.Goc J et al. (2014) Dendritic cells in tumor-associated tertiary lymphoid structures signal a Th1 cytotoxic immune contexture and license the positive prognostic value of infiltrating CD8+ T cells. Cancer Res 74 (3), 705–15. [DOI] [PubMed] [Google Scholar]
- 113.Poholek AC (2021) Tissue-Specific Contributions to Control of T Cell Immunity. Immunohorizons 5 (6), 410–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mikhak Z et al. (2013) Lung dendritic cells imprint T cell lung homing and promote lung immunity through the chemokine receptor CCR4. J Exp Med 210 (9), 1855–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Svensson M et al. (2008) Retinoic acid receptor signaling levels and antigen dose regulate gut homing receptor expression on CD8+ T cells. Mucosal Immunol 1 (1), 38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Esterhazy D et al. (2019) Compartmentalized gut lymph node drainage dictates adaptive immune responses. Nature 569 (7754), 126–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Horton BL et al. (2019) Tissue Site and the Cancer Immunity Cycle. Trends Cancer 5 (10), 593–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zagorulya M et al. (2020) Impact of anatomic site on antigen-presenting cells in cancer. J Immunother Cancer 8 (2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ho WW et al. (2022) The local microenvironment matters in preclinical basic and translational studies of cancer immunology and immunotherapy. Cancer Cell 40 (7), 701–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ho WW et al. (2021) Dendritic cell paucity in mismatch repair-proficient colorectal cancer liver metastases limits immune checkpoint blockade efficacy. Proc Natl Acad Sci U S A 118 (45). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lee JC et al. (2020) Regulatory T cell control of systemic immunity and immunotherapy response in liver metastasis. Sci Immunol 5 (52). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Jiao S et al. (2019) Differences in Tumor Microenvironment Dictate T Helper Lineage Polarization and Response to Immune Checkpoint Therapy. Cell 179 (5), 1177–1190 e13. [DOI] [PubMed] [Google Scholar]
- 123.Yu J et al. (2021) Liver metastasis restrains immunotherapy efficacy via macrophage-mediated T cell elimination. Nat Med 27 (1), 152–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Merad M et al. (2013) The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol 31, 563–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bottcher JP et al. (2018) NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell 172 (5), 1022–1037 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Michea P et al. (2018) Adjustment of dendritic cells to the breast-cancer microenvironment is subset specific. Nat Immunol 19 (8), 885–897. [DOI] [PubMed] [Google Scholar]
- 127.Mashayekhi M et al. (2011) CD8alpha(+) dendritic cells are the critical source of interleukin-12 that controls acute infection by Toxoplasma gondii tachyzoites. Immunity 35 (2), 249–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Iwanowycz S et al. (2021) Type 2 dendritic cells mediate control of cytotoxic T cell resistant tumors. JCI Insight 6 (17). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zilionis R et al. (2019) Single-Cell Transcriptomics of Human and Mouse Lung Cancers Reveals Conserved Myeloid Populations across Individuals and Species. Immunity 50 (5), 1317–1334 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Gerhard GM et al. (2021) Tumor-infiltrating dendritic cell states are conserved across solid human cancers. J Exp Med 218 (1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Cheng S et al. (2021) A pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells. Cell 184 (3), 792–809 e23. [DOI] [PubMed] [Google Scholar]