

Abstract
Chimeric antigen receptor T cells (CART) have demonstrated curative potential for hematological malignancies, but the optimal manufacturing has not yet been determined and may differ across products. The first step, T cell selection, removes contaminating cell types that can potentially suppress T cell expansion and transduction. While positive selection of CD4/CD8 T cells after leukapheresis is often used in clinical trials, it may modulate signaling cascades downstream of these co-receptors; indeed, the addition of a CD4/CD8-positive selection step altered CD22 CART potency and toxicity in patients. While negative selection may avoid this drawback, it is virtually absent from good manufacturing practices. Here, we performed both CD4/CD8-positive and -negative clinical scale selections of mononuclear cell apheresis products and generated CD22 CARTs per our ongoing clinical trial (NCT02315612NCT02315612). While the selection process did not yield differences in CART expansion or transduction, positively selected CART exhibited a significantly higher in vitro interferon-γ and IL-2 secretion but a lower in vitro tumor killing rate. Notably, though, CD22 CART generated from both selection protocols efficiently eradicated leukemia in NSG mice, with negatively selected cells exhibiting a significant enrichment in γδ CD22 CART. Thus, our study demonstrates the importance of the initial T cell selection process in clinical CART manufacturing.
Keywords: chimeric antigen receptor, T cell selection, cytokines, γδ T cells, positive selection, negative selection, CD22 CAR, B-ALL, CAR T cell manufacturing
Graphical abstract
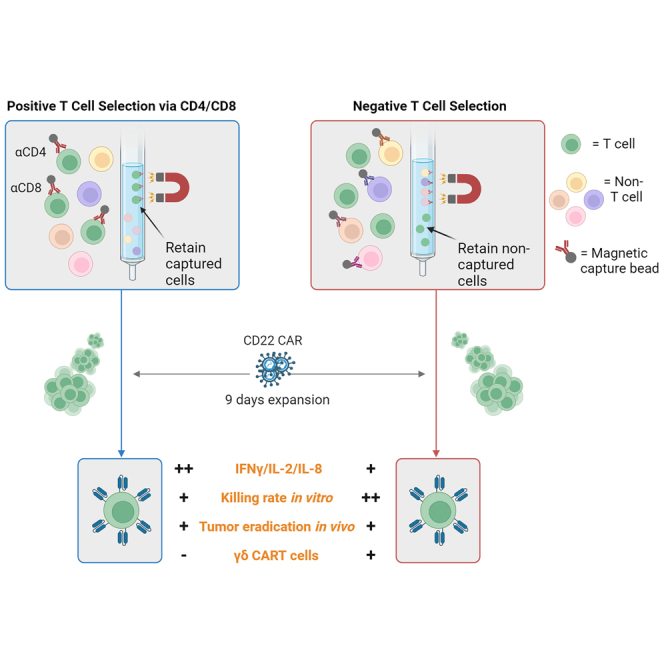
Song, Benzaoui, and colleagues describe a detailed comparison of CD22 CART cells manufactured from either positively or negatively selected T cells, outlining a number of phenotypic and functional differences relevant to CART function.
Introduction
Chimeric antigen receptor T cell (CART) therapy has shown much promise in the treatment of B cell hematological malignancies and is being further applied to solid tumors,1,2,3 autoimmune diseases,4,5,6 and regenerative therapies.7 Though initial studies demonstrated the important potential of CART,8,9,10 the parameters affecting the overall effectiveness of this therapy are still being investigated. Multiple clinical studies have highlighted the role of CART manufacturing on patient outcomes. For example, process variations performed at the beginning of the manufacturing process, including T cell selection and activation, can alter the functional properties of the resulting CART.11,12 To manufacture therapeutically relevant doses of CART, large numbers of T cells with high purity and functionality are generally required. To obtain these numbers of cells, patients undergo mononuclear cell (MNC) apheresis, which then requires additional processing to remove red blood cells and platelets. One of the simplest and most widely used methods to obtain MNCs is density gradient separation (i.e., Ficoll), which has been extensively used as a starting product for CART manufacturing.13,14,15,16 However, residual tumor cells and monocytes can persist in the cultures, potentially leading to manufacturing failures because of attenuated T cell expansion and transduction.17,18 In addition, while anti-CD3/CD28 paramagnetic beads can be used to simultaneously enrich and activate T cells for streamlined manufacturing,19,20 this approach can yield variable purities.11
Immunomagnetic T cell selection results in the isolation of purified T cells or T cell subsets in both preclinical and clinical settings.21,22,23 Indeed, many CART trials incorporate positive selection as a means of obtaining starting material with high T cell purity. Positive selection of T cells for CART manufacturing can be performed with as few as one (CD3) or two antibodies (CD4 and CD8),3,11,24,25,26 but binding of these receptors can potentially stimulate the T cells prematurely or prevent co-receptor activity.27 Conversely, negative T cell selection uses multiple antibodies to eliminate non-T cells. While negative selection is known to yield lower purities than positive selection,28 it has the benefit of leaving the T cells untouched by antibody-labeled beads. As such, negative selection is preferred in most functional preclinical studies, but importantly, it is less amenable for clinical manufacturing because of the quantity and expense of the multiple good manufacturing process (GMP)-grade antibodies required to remove contaminating cell types.
In the context of a CD22 CART trial carried out at the National Institutes of Health (NCT02315612), the addition of a positive CD4/CD8 selection step, which replaced a CD3/CD28 Dynabead enrichment step—with no other changes in manufacturing—was associated with increased potency in patients with B cell acute lymphoblastic leukemia (B-ALL), but also with an augmented toxicity profile.11 As such, it was of particular interest to evaluate the feasibility of a clinical scale CD22 CART manufacturing protocol based on the engineering of untouched, negatively selected T cells. To this end, we directly compared apheresis products manufactured via positive CD4/CD8 selection with negatively selected T cells selected using clinical scale (i.e., >2 × 109 starting cells) manufacturing methods. Selected T cells were activated and expanded in closed-system culture bags to generate CD22 CART, using the 9-day expansion protocol employed in our ongoing clinical trial.11 Although negative T cell selection resulted in a lower purity of the starting product, this method significantly increased CD3 T cell recovery compared with positive T cell selection. Furthermore, CD22 CART generated from both positive and negative selection (CARTpos and CARTneg) exhibited similar expansion, T cell subsets, activation markers, and CAR expression in the final product. Functionally, CARTneg cells exhibited lower in vitro cytokine production, but were characterized by a more rapid killing rate. Notably, both CARTpos and CARTneg displayed high in vivo cytotoxicity in a humanized leukemia mouse model, with the latter showing enrichment in γδ CART, a population of interest in anti-tumor immunotherapy strategies.
Results
Negative selection of apheresis products results in a lower purity and higher recovery of CD3+ T cells
Negative and positive immunomagnetic-based T cell selections (Figure 1A) were performed on thawed MNC apheresis products from five healthy adult donors and one pediatric donor with B-ALL, with the initial percentages of CD3+ T cells ranging from 20.8% to 57.9% (Figure 1B). T cell purity was higher after positive selection as compared with negative selection (84.0% ± 7.8% vs. 77.5% ± 10.0% CD3+, respectively; p < 0.05). In contrast, the recovery of CD3+ T cells was improved by an average of 20% after negative selection (72.7% ± 9.0% vs. 52.6% ± 6.0%, respectively; p < 0.05) (Figure 1B), with similar viabilities (87%–96%) (Figure 1B). The most abundant non-CD3+ cell type present after both positive and negative selection was CD14+/CD16+ monocytes, comprising approximately 10% of total cells in both selected products (Figures 1C and 1D). Interestingly, a 2-fold increase in the antibody cocktail used for negative selection resulted in a higher elimination of the CD14+/CD16+ monocyte population (Figure 1C, HD4, HD5, Pt), suggesting a non-saturating level of negative-selecting antibodies in the initial selections (Figures 1C and 1D). CD56+ natural killer cells were present at only minimal levels in both selections (0.6% ± 0.4% vs. 1.9% ± 1.3%) while CD19+ B cell contamination trended to higher levels after negative selection (6.2% ± 5.6% vs. 0.3% ± 0.5%) (Figure 1D). Overall, both positive and negative selection methods resulted in the successful isolation of T cells, albeit with differences in both purity and recovery.
Figure 1.

T cell selection using positive vs. negative selection
(A) Schematic of selection methods compared in this study; positive selection (anti-CD4 and anti-CD8 immunomagnetic beads) and negative selection (immunomagnetic beads targeting non-T cell subsets). Image created with biorender.com. (B) Different parameters were used to evaluate T cells obtained by positive and negative selection: (i) Purity of CD3+ cells; (ii) recovery of CD3+ cells calculated as the number of CD3+ T cells obtained after selection normalized to input CD3+ cells; and (iii) viability, measured via propidium iodide and acridine orange (AO/PI) staining immediately after selection. The B-ALL patient sample is indicated by an x. (C and D) Flow cytometry was performed on individual samples before and after selection using anti-CD19, anti-CD14/CD16, anti-CD3, and anti-CD56 monoclonal antibodies (mAbs), to distinguish B, monocyte, T, and natural killer cells, respectively. Quantifications are presented for each donor (C) as well as for each subset (D). The antibody concentrations used for negative selection were increased by 2-fold for healthy donor (HD) 4, HD5, and the patient (Pt) as compared with HD1, HD2, and HD3. ∗p < 0.05 using paired t test.
CD22 CART cells generated after positive and negative T cell selection exhibit similar phenotypes and gene expression profiles
After selection, T cells were activated with anti-CD3/CD28 Dynabeads in the presence of interleukin 2 (IL-2). T cells were transduced with clinical-grade lentivirus encoding CD22 CAR29 and expanded for 9 days as per the clinical manufacturing protocol (Figure 2A).11 T cells (30 × 106) were transduced on day 2, yielding more than 600 × 106 cells on day 9 (Figure 2B), corresponding with an average 38-fold expansion following both positive and negative selection (Figure 2C). Despite differences in T cell purity immediately after selection, cultures from both positive and negative selection reached 99.4% or greater CD3+ purity by day 9 (Figure 2D). The CD4:CD8 T cell ratio differed substantially between donors, but was not statistically different between CARTpos and CARTneg at any of the time points, whether gating on CD3+ T cells or transduced only (Figures 2Ei and S1). Of note, CD22CAR transduction, measured as a function of protein L staining, was similar between groups on days 4, 7, and 9, increasing to more than 35% by day 9 (Figure 2Eii). Thus, the T cell selection method did not affect the potential for efficient lentiviral transduction.
Figure 2.

Generation of CD22 CART from positively and negatively selected T cells
(A) Schema of the manufacturing protocol used to generate CD22 CART. TR, transduction. (B) Viable total nucleated cells (TNC) are shown between days 2 and 9 of culture. The B-ALL patient sample is indicated with an x. (C) Fold expansion was calculated as TNC on day 9 normalized to TNC on day 2. Lines are used to connect data points from the same donor. (D) CD3+ T cell purity was assessed on day 9 after gating on viable cells. (E) (i) The ratio of CD4:CD8 cells was assessed after gating on viable cells. (ii) Transduction was measured as a function of protein L staining on viable CD3+ cells, plotted as the mean ± SD of six donors. (iii) %CD25+ T cells was assessed on viable CD3+ cells, plotted as the mean ± SD of six donors. (iv) PD1 expression was assessed on viable CD3+ cells and means ± SDs of PD1+ T cells from the six donors at days 2–9 are presented. (F) Naive/stem/central memory-like (Tn/scm-like), central memory (Tcm), effector memory (Tem), and terminal effector (Teff) T cells were monitored by flow cytometry as CCR7+CD45RA+, CCR7+CD45RA−, CCR7−CD45RA−, and CCR7−CD45RA+, respectively and gated from the CD3+CAR+ population. Day 9 means ± SEM (n = 6) are shown. (G) PCA and (H) pathway analysis of gene expression data assessed at days 2, 4, 7, and 9 of culture. Means ± SEM for the six donors are shown. Statistical significance was calculated using a multiple paired t test; ns > 0.05.
To determine whether the T cells were differentially activated during the expansion phase, we monitored the cell surface expression of CD25 and PD1 activation markers via flow cytometry. Nearly identical expression patterns of CD25 were detected on CARTpos and CARTneg; CD25 levels increased from 32% on day 0 to 99% or greater on day 4 before decreasing to approximately 80% positive on day 9 (Figures 2Eiii and S2A). Similarly, PD1 expression increased from approximately 40% on day 0 to 100% positive on day 4, before decreasing to 60% on days 7–9, again with no difference between CARTpos and CARTneg (Figures 2Eiv and S2A). T cell subsets, including naive/stem cell memory-like (Tn/scm), central memory (Tcm), effector memory (Tem), and terminal effector (Teff), were monitored as CD45RA+CCR7+, CD45RA−CCR7+, CD45RA−CCR7–, and CD45RA+CCR7–, respectively. The distribution of these subsets was similar in both groups, with Tn/scm and Teff population comprising the largest subsets, irrespective of whether total T cell or CAR+ populations were evaluated (Figures 2F, S2B, and S2C). Thus, the phenotype of CART cells generated via positive and negative selection seemed to be largely similar.
To further probe potential differences in T cells during the expansion period, transcriptional profiling of a panel of 780 CART-related genes was performed. Principal component analysis (PCA) of gene expression levels at days 0, 2, 4, 7, and 9 of culture revealed distinct clustering, but these were based on the kinetics of activation rather than positive vs. negative selection (Figure 2G). Analyses of these signatures revealed a decrease in cytotoxicity and chemokine signaling signatures at later phases of the expansion period (Figure 2H). Interestingly, chemokine signaling was more highly induced in CARTneg than CARTpos at day 4 (Figure 2H). Activation and exhaustion gene signatures increased during the 9-day expansion phase, but there were no significant differences as a function of the selection method (Figure S3).
CARTneg produce lower levels of cytokines in response to CD22+ leukemia, but exhibit a more rapid ex vivo killing rate compared with CARTpos
We further characterized the differentially selected CD22 CART products by evaluating their ability to produce interferon γ (IFNγ), IL-17A, tumor necrosis factor α, IL-2, IL-4, IL-10, IL-6, IL-8, RANTES, MIG, IP-10, and MCP-1 after ex vivo stimulation by CD22+ NALM6 leukemia at an effector-to-target (E:T) ratio of 1:1. Both CARTpos and CARTneg produced high levels of cytokines after co-culture with CD22+ NALM6 leukemia, at levels significantly elevated as compared with co-culture with CD22– leukemia (Figure 3A). Interestingly, though, IFNγ, IL-2, IL-8, and RANTES were secreted at significantly higher levels by CARTpos than CARTneg (Figure 3A). To better understand the influence of positive and negative selection on CD22 CART antileukemic activity, a PCA was performed based on the cytokine secretion profiles obtained following leukemia co-culture (Figures S4A–S4C). By determining PCA distances between CARTpos and CARTneg manufactured from each donor, considerable heterogeneity between donors was detected (Figures S4E and S4F), showing a differential impact of positive and negative selection on the cytokine secretion of CART generated from different donors.
Figure 3.

CD22 CARTpos and CARTneg eradicate NALM6 leukemia in vitro
(A) Production of IFNγ, IL-17A, tumor necrosis factor α (TNFα), IL-2, IL-4, IL-10, and IL-6 cytokines and IL-8, RANTES, MIG, IP-10, and MCP-1 chemokines by CD22 CARTpos and CARTneg were monitored over 72 h following co-culture with CD22+ or CD22– (CD22KO) NALM6. Average cytokine concentrations were min/max scaled by the lower and upper detection limits of each cytokine across all samples and divided by the number of cytokines to yield a polyfunctionality index, presented as an average of six individual donors (top). Cumulative concentrations of each cytokine and chemokine are presented (bottom). The ALL patient sample is indicated with an x. (B) In vitro anti-leukemic activity of CD22 CARTpos and CARTneg were monitored against NALM6WT and NALM6CD22KO at a 1:1 effector/target ratio, evaluated as a function of GFP expression. Killing indices (left) and killing rates (right) from six individual donors are presented. Statistical significance was calculated using ratio paired t tests; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p<0.0001.
We next assessed the ex vivo cytotoxicity of CARTpos and CARTneg against CD22+ leukemia (Figure 3B). CART generated by either method did not kill CD22-negative NALM6, but exhibited potent cytotoxicity against CD22+ leukemia. Unexpectedly, though, CARTneg exhibited a significantly higher rate of killing than CARTpos (Figure 3B), pointing to differences in the regulation of cytokine secretion and anti-tumor cytotoxicity by CD22 CART.
CARTneg and CARTpos exhibit similar in vivo efficacy against B-ALL
As CARTneg and CARTpos exhibited high in vitro cytotoxicity, we tested the ability of these CART products to eradicate NALM6 in vivo. CARTpos and CARTneg manufactured from two healthy donors and one patient with B-ALL were adoptively transferred into NSG mice 4 days after inoculation of luciferase-expressing NALM6 (1 × 106). A limiting dose of CART (3 × 106), where relapse is expected, was used to potentially discriminate between potential functional differences between CARTpos and CARTneg. Interestingly, the in vivo efficacy of CART products varied significantly between donors, with mice adoptively transferred with CD22 CART from HD3 relapsing more rapidly than mice receiving CD22 CART products from healthy donor 4 or a patient, irrespective of the selection method (Figures 4A and 4B). There was no significant difference in leukemia growth or overall survival of NSG mice adoptively transferred with CARTpos or CARTneg, although intriguingly, CARTneg generated from the patient had lower overall disease burden at day 48 as compared with CARTpos. Moreover, consistent with the high donor-to-donor variability, the phenotype of T cells persisting in the spleens and bone marrow of these mice exhibited high variability; the percentages of PD1+CD39+ T cells, a phenotype of T cell exhaustion, as well as CD4:CD8 ratios varied markedly in mice treated with CD22 CART from the different donors (Figure S5). Thus, variability, even between healthy donors, resulted in significant differences in the ability of CD22 CART to control in vivo leukemic growth.
Figure 4.

Both CD22 CARTpos and CARTneg exhibit in vivo anti-leukemic activity
(A) On day 0, luciferase+ NALM6 cells (1 × 106) were injected via tail vein into NSG mice, followed by the injection of CD22 CARTpos and CARTneg (3 × 106) at day 4. Leukemia growth was evaluated over 50 days by bioluminescent imaging and images at the indicated time points are presented. (B) Bioluminescence ±SEM at each time point was quantified and tumor progression in each mouse is shown.
CD22 CART generated from negatively selected T cells include CD22 CAR+ γδ T cells
As in vivo leukemia eradication experiments demonstrated high donor-to-donor variability, transcriptional profiling analysis was used to assess potential donor-independent differences between CARTneg and CARTpos. Interestingly, only 9 of 780 genes were differentially expressed in CARTneg and CARTpos at the end of the expansion phase (day 9, p < 0.05) (Figure 5A). Of these nine genes, only two, TRDC and TRDV2, were differentially expressed on day 7 as well as day 9, both upregulated in CARTneg as compared with CARTpos (Figures 5A and S6). TRDC and TRDV2 are specifically expressed on γδ T cells, encoding the T cell receptor delta chain. Therefore, we investigated whether γδ T cells were present at different levels in CARTneg and CARTpos.
Figure 5.

Negative selection results in a final CART product enriched in γδ T cells
(A) Volcano plot representation of differential gene expression between CD22 CARTpos and CARTneg (day 9). Fold changes of >2 and p < 0.05 were considered significant (dotted lines). The identities of several differentially expressed transcripts are indicated. (B) A representative dot plot showing the percentages of TCRαβneg cells within the viable cell gate is shown for one donor (left). Quantification of TCRαβneg cells in all donors is shown (right). The B-ALL patient sample is indicated with an “x.” (C) T cells expressing TCRVδ1 or TCRVδ2 were monitored by flow cytometry. Representative plots (left) and quantifications (right) are shown. (D) Expression of TCRVδ1 and TCRVδ2 were assessed in CD4+, CD8+, and CD4−CD8– compartments. A representative TCRVδ1/TCRVδ2 profile in the CD4−CD8- subset (left) and quantifications (right) are shown. (E) Representative dot plots showing the percentages of CD22 CAR-transduced cells within the TCRVδ1 and TCRVδ2 subsets are shown for one donor (left). Quantifications of the percentages of CD22 CAR-transduced cells within the αβ and γδ populations in the 6 donors, following positive and negative selection, are shown (right). (F) The percentages of γδ (TCRVδ1/TCRVδ2) T cells within the total CAR+ population (left) as well as the percentages of γδ T cells within the CD4−CD8−CAR+ population (right) are presented. (G) T cells were isolated from elutriated lymphocytes using either CD3/CD28 Dynabeads (positive enrichment) or a non-GMP negative T cell selection (EasySep) as compared with CD4/CD8-selected cells. Quantifications of the percentages of γδ T cells within the CD3 populations are indicated (n = 3). (H) Schematic showing that only αβ T cells are retained by CD4/CD8 positive selection whereas γδ T cells are also retained during negative selection (image created with biorender.com). ∗p < 0.05. ∗∗p < 0.01, ∗∗∗p < 0.001, ratio paired t test (B, C, and F); ns > 0.05, ∗p < 0.05, ∗∗p < 0.01, two-way ANOVA with multiple comparisons (E); ∗∗∗∗p < 0.0001, one-way ANOVA with multiple comparison (G).
As expected, αβ T cells made up the vast majority of T cells in the final products,30 accounting for at least 97% of total cells on day 9 of culture (Figure 5B). Notably, though, there was a 4.4-fold higher level of αβneg cells in negatively selected as compared with positively selected T cells (0.9% ± 0.8% vs. 0.2% ± 0.2%; p < 0.001) (Figure 5B). Consistent with the gene expression data, a 6-fold higher level of αβneg cells were γδ T cells, identified as Vδ1+ or Vδ2+ (p < 0.001) (Figure 5C), with Vδ2 encoded by the TRDV2 gene (Figure 5A). Importantly, the Vδ1+ and Vδ2+ subsets corresponded with cells in the CD4−CD8− compartment (Figure 5D). It was interesting to note that αβ T cells were overall transduced at higher levels (37.4% ± 3.3% vs. 35.9% ± 4.0% for positive and negative selection, respectively) than γδ T cells (p = 0.02 and 0.006), but the percentage transduction for γδ T cells isolated by the two separation methods were similar (17.1% ± 5.1% vs. 19.5% ± 5.8%) (Figure 5E). Nonetheless, because the initial frequencies of γδ T cells were significantly higher in the negatively selected samples (Figure 5B), this resulted in 4.6-fold higher percentage of γδ T cells in the transduced cells within the negatively selected subset (1.0% ± 0.6% vs. 0.2% ± 0.2%; p = 0.002) (Figure 5F). Furthermore, this translated to a significantly higher frequency of γδ T cells within the CAR-transduced CD4−CD8– compartment, increasing from 11.3% ± 4.8% to 30.5% ± 5.0% in the negatively selected population (p < 0.05) (Figure 5F). The higher percentage of γδ T cells was not specific to the negative T cell selection performed here, as γδ T cells were also recovered at higher frequencies when lymphocytes were selected by both positive CD3/CD28 enrichment (3.2% ± 0.03%) and a non-GMP negative enrichment (1.8% ± 0.06%, EasySep). These data demonstrate the potential for diverse T cell isolation processes to recover γδ T cells in the selected product (Figure 5G). Thus, T cell selection––either via a clinical scale negative selection or a CD3/CD28 enrichment––but not via a CD4/CD8 T cell selection, allows for an enrichment of γδ T cells in the final expanded product, and these cells are successfully transduced by the CD22 CAR vector (Figure 5H).
Discussion
Given the impressive success of CARTs in the treatment of hematological malignancies, there are an expanding number of tools and reagents available for clinical-grade CART manufacturing. As such, investigators can better tailor their manufacturing methods to suit their desired product characteristics.15 Our group, as well as others, have found that T cell selection is a key step in the success of CART manufacturing11,17,31,32; it defines the subsets of cells that are present, or absent, both in the initial culture as well as in the final infusion product. The positive selection of T cells, based on their binding to anti-CD4 and anti-CD8 monoclonal antibodies, has become a commonly used method in both hematological and solid tumor CART applications, leading to robust manufacturing.3,11,24,25
Our ongoing CD22 CART clinical trial demonstrated that switching from a CD3/CD28 enrichment to a CD4/CD8-positive selection improved the reproducibility of CART manufacturing. Interestingly, this change led to a greater efficacy of the CD22 CART but also to an increased incidence of hemophagocytic lymphohistiocytosis (HLH)-like toxicity. The toxicity resulted in a dose de-escalation from 1 × 106/kg to 3 × 105/kg; while this dose had been ineffective in the initial manufacturing, it was effective after implementation of the positive selection step.11 These data led us to hypothesize that CD4/CD8-positive selection may impact the functional characteristics of CART cells, either by modulating signaling through CD4/CD8, by enriching for T cell subsets with different functional properties, and/or by the impact of non-T cell subsets in the initial culture. Indeed, previous studies have shown that CD4/CD8 binding alters cytokines including IL-4,33 and binding of CD4 selection beads increases mitogen-activated protein kinase signaling.27
Here we tested whether purification of unmanipulated T cells after a negative selection protocol using the RoboSep-C platform would alter the functional properties of CD22 CART and compared these cells to CD4/CD8 selection via CliniMACS Plus. Negative T cell selection resulted in a CD22 CART final product with similar expansion, transduction efficiency, and phenotype compared with CD4/CD8 positive selection. Notably, though, differences in cytokine secretion and in vivo anti-tumor responses revealed important donor-to-donor variability, outweighing changes in the selection protocol. Indeed, very recent work has shown that the level of stimulation used during the generation of CART should be tailored to individual differences in the donor cells.34 That being said, the selection protocol did result in some significant differences; positive selection was associated with higher levels of several proinflammatory cytokines such as IFNγ and IL-8. High levels of IFNγ have previously been associated with multiple systemic toxicities, most notably as a marker of cytokine release syndrome,35,36,37 as well as HLH-like toxicities in patients receiving CD22 CART11 and CD19 CART.38 Thus, negative T cell selection might translate into decreased HLH incidence or severity of inflammatory toxicities.
Because the majority of γδ T cells lack both CD4 and CD8 receptors, CD4/CD8-positive selection significantly reduced the numbers of these immune effectors in the final product, compared with both our negative selection protocol using the RoboSep-C platform as well as to T cells positively enriched using CD3/CD28 Dynabeads. While comprising only 1%–5% of the T cells in the circulation,30 γδ T cells are primed for rapid toxicity against both foreign antigens and tumor cells. Strikingly, a pan-cancer gene expression analysis revealed that intratumoral γδ T cells are strongly associated with a favorable prognosis.39 Although non-transduced γδ T cells naturally exert anti-tumor function, transduction of γδ T cells with a CAR may direct their cytotoxicity toward tumor targets while retaining their ability to present antigen, resist exhaustion, and home to the tumor microenvironment.40 It will be important to elucidate the ex vivo expansion conditions that allow for an optimal transduction of γδ T cells, as our data show that the expansion conditions used here resulted in a significantly higher transduction efficiency of αβ as compared with γδ T cells. This is especially pertinent as a longitudinal analysis of CD19 CART in a CLL patient who achieved a durable complete response revealed the expansion of a γδ T cell population, accounting for up to 33% of all CAR+ cells by 3 months after infusion. This context was associated with the emergence of a proliferative CD4 CART population that persisted over a decade,41 suggesting that γδ CART can contribute to both the initial and later phases of tumor elimination. Long-term studies in appropriate animal models with cross-reactive immune cells and cytokines, supporting human CART survival and persistence, are therefore needed to specifically study the role of γδ CART and its interplay with αβ CART. Given the high level of in vivo expansion capability, even limited numbers of γδ CART cells may favorably impact patient outcomes.
In conclusion, our study demonstrates that negative T cell selection is a viable manufacturing strategy that may improve therapeutic outcomes, particularly in certain donors, and should be tested clinically. We find that negative selection reduces cytokine production upon tumor re-stimulation while also increasing γδ CART output, potentially reducing the severity of inflammatory toxicities and improving tumor eradication.
Materials and methods
Cell selection
Cryopreserved MNC apheresis products from five healthy donors and one pediatric B-ALL patient were thawed and washed once in Plasmalyte A (Baxter) + 0.5% human serum albumin (HSA) (Baxalta) and split to perform positive and negative T cell selections in parallel; between 2.5 and 3.3 × 109 MNCs were selected in each process. Of note, cryopreserved MNC products have been found to be equivalent to their freshly isolated counterparts as regards expansion, CAR transduction, and patient responses.42,43 Positive T cell selection was performed using CliniMACS Plus (Miltenyi Biotec) with anti-CD4 and anti-CD8 GMP magnetic selection beads (Miltenyi Biotec) for a combined CD4/CD8 selection. Briefly, thawed cells were washed twice with PBS/EDTA + 0.5% HSA to remove platelets, blocked with 1.5 mg/mL intravenous immunoglobulin (Baxter Healthcare) for 5 min and then labeled with magnetic capture beads for 30 min at room temperature. Labeled cells were then washed twice with PBS/EDTA + 0.5% HSA to remove unbound beads before proceeding to automatic selection on the CliniMACS Plus instrument. For negative selection, MNC apheresis products were loaded onto the RoboSep-C instrument (Stem Cell Technologies), for automated antibody/bead labeling and magnetic T cell selection. The instrument performed incubation with a cocktail of seven proprietary antibodies against non-T cell targets followed by incubation with magnetic beads to capture labeled cells. Healthy donors 1, 2, and 3 were selected with one-half the antibody concentration used for healthy donors 4 and 5 and the B-ALL patient sample. All selections were performed in the absence of human serum. For comparisons with CD3/CD28-enriched cells and non-GMP-negative selection, elutriated lymphocytes were enriched using the Dynabead Human T-expander (Gibco) and EasySep T cell isolation kits (Stem Cell Technologies), respectively, according to the manufacturers’ recommendations. Cell counts prior to positive and/or negative selection were performed on the Advia 120 (Siemens), while cell counts and viability after T cell selection were performed on the Cellometer instrument using propidium iodide and acridine orange staining (Nexcelom Biosciences).
CD22 CART generation
T cells enriched via positive and negative selection were placed into culture immediately after selection. Approximately 100 million T cells were activated using Dynabeads (Thermo Fisher Scientific) at a bead to T cell ratio of 3:1 and cultured in bags (Origen Biomedical) at 37°C and 5% CO2. Media on day 0 was composed of AIM-V (Thermo Fisher Scientific) supplemented with 5% human AB serum (Valley Biomedical), 2 mM GlutaMAX (Thermo Fisher Scientific) and 40 IU/mL IL-2 (Clinigen). On day 2, 30 million T cells were transduced for 24 h with 10 μg/mL protamine sulfate (Fresenius Kabi) at a multiplicity of infection of 10 using clinical-grade lentiviral vector encoding CD22 CAR with 41BB and CD3ξ co-stimulatory domains (Lentigen Inc.). Cells were de-beaded on day 4 and diluted to a density of 0.4 × 106/mL with fresh media containing a higher IL-2 concentration (100 IU/mL). On day 7, cells were diluted again with fresh media to a density of 0.4 × 106/mL and harvested on day 9. At harvest, cells were washed twice with Plasmalyte A + 0.5% HSA and cryopreserved in Cryostor CS10 (BioLife Solutions) for subsequent downstream analyses.
Flow cytometry
Cells were resuspended in PBS + 0.5% HSA and stained for 30 min at 4°C. Cells were washed once and analyzed on either a FACSCanto (BD Biosciences) or Cytoflex flow cytometer (Beckman Coulter). Antibodies used for flow cytometry are listed in Table S1.
Gene expression analysis
Gene expression was measured using the nCounter Analysis Pipeline (NanoString Technologies) with the CART characterization gene panel (n = 780 genes), and data were analyzed using nSolver and R environment. Pathway enrichment was performed using a GSVA package in R with a custom script.
High-throughput cytokine secretion analysis
CART (2.5 × 104) were co-cultured with NALM6 (harboring the GFP and luciferase reporter genes) in 200 μL RPMI media in v-bottom 96-well plates at a 1:1 E:T ratio. The Immunotron robotic platform44 was programmed to perform automatic collection and replenishment of supernatants at given time points (typically, 1, 3, 6, 12, 18, 24, 30, 36, 42, 48, 60, and 72 h after initiation). Briefly, cell culture plates and medium plates were retrieved from the incubator, 10 μL culture supernatants were collected with minimal disruption of cell pellet and replenished with 10 μL fresh medium. Collected supernatants were stored at −20°C until analysis by cytokine bead array.
Multiplexed cytokine bead array
Human Th1/Th2/Th17 cytokine and human chemokine bead array kits (BD Biosciences) were used to quantify 7 cytokines and 5 chemokines in each collected supernatant. Collected supernatants were thawed at room temperature, loaded with cytokine bead array (CBA) reagents incubated at room temperature for 15 min. To multiplex acquisition by flow cytometry, plates were then barcoded using fluorescently labeled Fab fragments. Typically, Dy405 or fluorescein isothiocyanate-labeled anti-Rat Fab fragment (Jackson Immunoresearch) were added to a final concentration of 2 μg/mL and incubated for 15 min at room temperature. This reaction was then quenched by adding an excess of Rat IgG (Jackson Immunoresearch) to a final concentration of 30 μg/mL and incubated for 15 min at room temperature. Fab-labeled CBA plates were then consolidated into one plate, washed, and resuspended in fluorescence-activated cell sorting buffer for acquisition on a Fortessa flow cytometer (BD Biosciences).
Cytotoxicity assay
CARTs (2.5 × 104) were co-cultured with NALM6 or NALM6CD22KO at a 1:1 E:T ratio in 200 μL RMPI media in poly-D-lysine-coated (0.05 mg/mL) flat-bottom 96-well plates. Plates were then imaged every 2 h in an S3 IncuCyte live-cell analysis instrument (Sartorius). The killing index was determined as a ratio of the green area at every time point normalized to the first measurement. The killing rate (k) was calculated based on the slope of the natural logarithmic transformation of the killing index curve (y) as a function of time (x) wherein k = –ln(y)/x. After plotting the function ln(y) = –k∗x, the ordinate and corresponding abscissa (wherein x > 0) needed to calculate the killing rate were determined using Python.
In vivo analysis of CD22 CART function
The potential for positively and negatively selected CD22 CART to inhibit in vivo leukemia growth was assessed in NSG mice as previously described.24 All animal experiments were conducted under protocols approved by the National Cancer Institute Institutional Animal Care and Use Committees and in accordance with National Institutes of Health laboratory animal guidelines. Bioluminescent NALM6 (1 × 106) were adoptively transferred by tail vein injection at day 0 and CD22 CART (3 × 106) were then injected at day 4. Tumor engraftment and growth were assessed by bioluminescent imaging after intraperitoneal injection of D-luciferin (3 mg; Caliper Life Sciences) using the IVIS Lumina Imaging (PerkinElmer). Luminescence was quantified using the RadianceQuantifier Graphical User Interface (available at https://github.com/soorajachar/radianceQuantifier). At study termination, murine bone marrow and spleen were harvested for analysis of leukemia and CD22 CART cells by flow cytometry as described.24
Data and statistical analysis
CBA robotic data were processed as previously described.44 Briefly, a custom-designed Python pipeline (named Plateypus) was used to simultaneously capture plate-formatted settings, compile high-throughput bulk cytokine/surface marker/single cell data, and perform initial data visualization for quality control (https://doi.org/10.5281/zenodo.5759171). Plateypus is available at https://pypi.org/project/plateypus/. A p value of less than 0.05 was considered significant. Statistical analyses were performed using GraphPad Prism. Images were created using BioRender.
Data and code availability
The datasets analyzed in the current study are available from the corresponding authors upon reasonable request.
Acknowledgments
This study was supported by the Center for Cancer Research, Intramural Program, National Cancer Institute (Z99CL999999, ZIABC 011923, and ZIABC011807) and an NCI FLEX award (G.A-B., N.T., and N.N.S.). The views expressed by the authors do not necessarily represent the views of the NIH, Department of Health and Human Services, or the US federal government.
Author contributions
H.W.S., M.B., C.D.C., N.T., and S.L.H. conceived the study; H.W.S., M.B., A.D., S.U., L.S., S.A., P.J., R.P.S., D.F.S., G. A.-B., N.N.S., C.D.C., N.T., and S.L.H. designed the experiments; H.W.S., M.B., A.D., S.U., L.S., S.A., V.A.R., M.P., and Y.C. performed experiments and V.P. designed and formulated the negative selection antibody cocktail; all authors analyzed and interpreted data; H.W.S., M.B., C.D.C., N.T., and S.L.H. wrote the first version of the manuscript and all authors edited the manuscript; N.T. and S.L.H. supervised the project. All authors approved the final manuscript.
Declaration of interests
V.P. is an employee of STEMCELL Technologies, Inc.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2023.101171.
Contributor Information
Naomi Taylor, Email: taylorn4@mail.nih.gov.
Steven L. Highfill, Email: steven.highfill@nih.gov.
Supplemental information
References
- 1.Srour S., Kotecha R., Curti B., Chahoud J., Drakaki A., Tang L., Goyal L., Prashad S., Szenes V., Norwood K., Pal S. Abstract CT011: A phase 1 multicenter study (TRAVERSE) evaluating the safety and efficacy of ALLO-316 following conditioning regimen in pts with advanced or metastatic clear cell renal cell carcinoma (ccRCC) Cancer Res. 2023;83(Supplement):CT011. [Google Scholar]
- 2.Del Bufalo F., De Angelis B., Caruana I., Del Baldo G., De Ioris M.A., Serra A., Mastronuzzi A., Cefalo M.G., Pagliara D., Amicucci M., et al. GD2-CART01 for Relapsed or Refractory High-Risk Neuroblastoma. N. Engl. J. Med. 2023;388:1284–1295. doi: 10.1056/NEJMoa2210859. [DOI] [PubMed] [Google Scholar]
- 3.Majzner R.G., Ramakrishna S., Yeom K.W., Patel S., Chinnasamy H., Schultz L.M., Richards R.M., Jiang L., Barsan V., Mancusi R., et al. GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature. 2022;603:934–941. doi: 10.1038/s41586-022-04489-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mackensen A., Müller F., Mougiakakos D., Böltz S., Wilhelm A., Aigner M., Völkl S., Simon D., Kleyer A., Munoz L., et al. Anti-CD19 CAR T cell therapy for refractory systemic lupus erythematosus. Nat. Med. 2022;28:2124–2132. doi: 10.1038/s41591-022-02017-5. [DOI] [PubMed] [Google Scholar]
- 5.Oh S., Mao X., Manfredo-Vieira S., Lee J., Patel D., Choi E.J., Alvarado A., Cottman-Thomas E., Maseda D., Tsao P.Y., et al. Precision targeting of autoantigen-specific B cells in muscle-specific tyrosine kinase myasthenia gravis with chimeric autoantibody receptor T cells. Nat. Biotechnol. 2023;41:1229–1238. doi: 10.1038/s41587-022-01637-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee J., Lundgren D.K., Mao X., Manfredo-Vieira S., Nunez-Cruz S., Williams E.F., Assenmacher C.A., Radaelli E., Oh S., Wang B., et al. Antigen-specific B cell depletion for precision therapy of mucosal pemphigus vulgaris. J. Clin. Invest. 2020;130:6317–6324. doi: 10.1172/JCI138416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rurik J.G., Tombácz I., Yadegari A., Méndez Fernández P.O., Shewale S.V., Li L., Kimura T., Soliman O.Y., Papp T.E., Tam Y.K., et al. CAR T cells produced in vivo to treat cardiac injury. Science. 2022;375:91–96. doi: 10.1126/science.abm0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kochenderfer J.N., Dudley M.E., Kassim S.H., Somerville R.P.T., Carpenter R.O., Stetler-Stevenson M., Yang J.C., Phan G.Q., Hughes M.S., Sherry R.M., et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J. Clin. Oncol. 2015;33:540–549. doi: 10.1200/JCO.2014.56.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Porter D.L., Levine B.L., Kalos M., Bagg A., June C.H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011;365:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brentjens R.J., Davila M.L., Riviere I., Park J., Wang X., Cowell L.G., Bartido S., Stefanski J., Taylor C., Olszewska M., et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci. Transl. Med. 2013;5:177ra38. doi: 10.1126/scitranslmed.3005930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shah N.N., Highfill S.L., Shalabi H., Yates B., Jin J., Wolters P.L., Ombrello A., Steinberg S.M., Martin S., Delbrook C., et al. CD4/CD8 T-Cell Selection Affects Chimeric Antigen Receptor (CAR) T-Cell Potency and Toxicity: Updated Results From a Phase I Anti-CD22 CAR T-Cell Trial. J. Clin. Oncol. 2020;38:1938–1950. doi: 10.1200/JCO.19.03279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ceppi F., Wilson A.L., Annesley C., Kimmerly G.R., Summers C., Brand A., Seidel K., Wu Q.V., Beebe A., Brown C., et al. Modified Manufacturing Process Modulates CD19CAR T-cell Engraftment Fitness and Leukemia-Free Survival in Pediatric and Young Adult Subjects. Cancer Immunol. Res. 2022;10:856–870. doi: 10.1158/2326-6066.CIR-21-0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kochenderfer J.N., Dudley M.E., Feldman S.A., Wilson W.H., Spaner D.E., Maric I., Stetler-Stevenson M., Phan G.Q., Hughes M.S., Sherry R.M., et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119:2709–2720. doi: 10.1182/blood-2011-10-384388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brudno J.N., Lam N., Vanasse D., Shen Y.W., Rose J.J., Rossi J., Xue A., Bot A., Scholler N., Mikkilineni L., et al. Safety and feasibility of anti-CD19 CAR T cells with fully human binding domains in patients with B-cell lymphoma. Nat. Med. 2020;26:270–280. doi: 10.1038/s41591-019-0737-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Song H.W., Somerville R.P., Stroncek D.F., Highfill S.L. Scaling up and scaling out: Advances and challenges in manufacturing engineered T cell therapies. Int. Rev. Immunol. 2022;41:638–648. doi: 10.1080/08830185.2022.2067154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Till B.G., Jensen M.C., Wang J., Chen E.Y., Wood B.L., Greisman H.A., Qian X., James S.E., Raubitschek A., Forman S.J., et al. Adoptive immunotherapy for indolent non-Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood. 2008;112:2261–2271. doi: 10.1182/blood-2007-12-128843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stroncek D.F., Ren J., Lee D.W., Tran M., Frodigh S.E., Sabatino M., Khuu H., Merchant M.S., Mackall C.L. Myeloid cells in peripheral blood mononuclear cell concentrates inhibit the expansion of chimeric antigen receptor T cells. Cytotherapy. 2016;18:893–901. doi: 10.1016/j.jcyt.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deng B., Pan J., Liu Z., Liu S., Chen Y., Qu X., Zhang Y., Lin Y., Zhang Y., Yu X., et al. Peripheral leukemia burden at time of apheresis negatively affects the clinical efficacy of CART19 in refractory or relapsed B-ALL. Mol. Ther. Methods Clin. Dev. 2021;23:633–643. doi: 10.1016/j.omtm.2021.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Neurauter A.A., Bonyhadi M., Lien E., Nøkleby L., Ruud E., Camacho S., Aarvak T. Cell isolation and expansion using Dynabeads. Adv. Biochem. Eng. Biotechnol. 2007;106:41–73. doi: 10.1007/10_2007_072. [DOI] [PubMed] [Google Scholar]
- 20.Hollyman D., Stefanski J., Przybylowski M., Bartido S., Borquez-Ojeda O., Taylor C., Yeh R., Capacio V., Olszewska M., Hosey J., et al. Manufacturing validation of biologically functional T cells targeted to CD19 antigen for autologous adoptive cell therapy. J. Immunother. 2009;32:169–180. doi: 10.1097/CJI.0b013e318194a6e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Priesner C., Aleksandrova K., Esser R., Mockel-Tenbrinck N., Leise J., Drechsel K., Marburger M., Quaiser A., Goudeva L., Arseniev L., et al. Automated Enrichment, Transduction, and Expansion of Clinical-Scale CD62L(+) T Cells for Manufacturing of Gene Therapy Medicinal Products. Hum. Gene Ther. 2016;27:860–869. doi: 10.1089/hum.2016.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sommermeyer D., Hudecek M., Kosasih P.L., Gogishvili T., Maloney D.G., Turtle C.J., Riddell S.R. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia. 2016;30:492–500. doi: 10.1038/leu.2015.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Terakura S., Yamamoto T.N., Gardner R.A., Turtle C.J., Jensen M.C., Riddell S.R. Generation of CD19-chimeric antigen receptor modified CD8+ T cells derived from virus-specific central memory T cells. Blood. 2012;119:72–82. doi: 10.1182/blood-2011-07-366419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shalabi H., Qin H., Su A., Yates B., Wolters P.L., Steinberg S.M., Ligon J.A., Silbert S., DéDé K., Benzaoui M., et al. CD19/22 CAR T cells in children and young adults with B-ALL: phase 1 results and development of a novel bicistronic CAR. Blood. 2022;140:451–463. doi: 10.1182/blood.2022015795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhu F., Shah N., Xu H., Schneider D., Orentas R., Dropulic B., Hari P., Keever-Taylor C.A. Closed-system manufacturing of CD19 and dual-targeted CD20/19 chimeric antigen receptor T cells using the CliniMACS Prodigy device at an academic medical center. Cytotherapy. 2018;20:394–406. doi: 10.1016/j.jcyt.2017.09.005. [DOI] [PubMed] [Google Scholar]
- 26.Turtle C.J., Hanafi L.A., Berger C., Gooley T.A., Cherian S., Hudecek M., Sommermeyer D., Melville K., Pender B., Budiarto T.M., et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J. Clin. Invest. 2016;126:2123–2138. doi: 10.1172/JCI85309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bernard F., Jaleco S., Dardalhon V., Steinberg M., Yssel H., Noraz N., Taylor N., Kinet S. Ex vivo isolation protocols differentially affect the phenotype of human CD4+ T cells. J. Immunol. Methods. 2002;271:99–106. doi: 10.1016/s0022-1759(02)00412-x. [DOI] [PubMed] [Google Scholar]
- 28.Lyons P.A., Koukoulaki M., Hatton A., Doggett K., Woffendin H.B., Chaudhry A.N., Smith K.G.C. Microarray analysis of human leucocyte subsets: the advantages of positive selection and rapid purification. BMC Genom. 2007;8:64. doi: 10.1186/1471-2164-8-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fry T.J., Shah N.N., Orentas R.J., Stetler-Stevenson M., Yuan C.M., Ramakrishna S., Wolters P., Martin S., Delbrook C., Yates B., et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat. Med. 2018;24:20–28. doi: 10.1038/nm.4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vantourout P., Hayday A. Six-of-the-best: unique contributions of γδ T cells to immunology. Nat. Rev. Immunol. 2013;13:88–100. doi: 10.1038/nri3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang X., Borquez-Ojeda O., Stefanski J., Du F., Qu J., Chaudhari J., Thummar K., Zhu M., Shen L.B., Hall M., et al. Depletion of high-content CD14(+) cells from apheresis products is critical for successful transduction and expansion of CAR T cells during large-scale cGMP manufacturing. Mol Ther Methods Clin Dev. 2021;22:377–387. doi: 10.1016/j.omtm.2021.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arcangeli S., Falcone L., Camisa B., De Girardi F., Biondi M., Giglio F., Ciceri F., Bonini C., Bondanza A., Casucci M. Next-Generation Manufacturing Protocols Enriching T(SCM) CAR T Cells Can Overcome Disease-Specific T Cell Defects in Cancer Patients. Front. Immunol. 2020;11:1217. doi: 10.3389/fimmu.2020.01217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stanciu L.A., Shute J., Holgate S.T., Djukanović R. Production of IL-8 and IL-4 by positively and negatively selected CD4+ and CD8+ human T cells following a four-step cell separation method including magnetic cell sorting (MACS) J. Immunol. Methods. 1996;189:107–115. doi: 10.1016/0022-1759(95)00240-5. [DOI] [PubMed] [Google Scholar]
- 34.Zhang D.K.Y., Adu-Berchie K., Iyer S., Liu Y., Cieri N., Brockman J.M., Neuberg D., Wu C.J., Mooney D.J. Enhancing CAR-T cell functionality in a patient-specific manner. Nat. Commun. 2023;14:506. doi: 10.1038/s41467-023-36126-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Teachey D.T., Lacey S.F., Shaw P.A., Melenhorst J.J., Maude S.L., Frey N., Pequignot E., Gonzalez V.E., Chen F., Finklestein J., et al. Identification of Predictive Biomarkers for Cytokine Release Syndrome after Chimeric Antigen Receptor T-cell Therapy for Acute Lymphoblastic Leukemia. Cancer Discov. 2016;6:664–679. doi: 10.1158/2159-8290.CD-16-0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Giavridis T., van der Stegen S.J.C., Eyquem J., Hamieh M., Piersigilli A., Sadelain M. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat. Med. 2018;24:731–738. doi: 10.1038/s41591-018-0041-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McNerney K.O., DiNofia A.M., Teachey D.T., Grupp S.A., Maude S.L. Potential Role of IFNγ Inhibition in Refractory Cytokine Release Syndrome Associated with CAR T-cell Therapy. Blood Cancer Discov. 2022;3:90–94. doi: 10.1158/2643-3230.BCD-21-0203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hines M.R., Keenan C., Maron Alfaro G., Cheng C., Zhou Y., Sharma A., Hurley C., Nichols K.E., Gottschalk S., Triplett B.M., Talleur A.C. Hemophagocytic lymphohistiocytosis-like toxicity (carHLH) after CD19-specific CAR T-cell therapy. Br. J. Haematol. 2021;194:701–707. doi: 10.1111/bjh.17662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gentles A.J., Newman A.M., Liu C.L., Bratman S.V., Feng W., Kim D., Nair V.S., Xu Y., Khuong A., Hoang C.D., et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 2015;21:938–945. doi: 10.1038/nm.3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Capsomidis A., Benthall G., Van Acker H.H., Fisher J., Kramer A.M., Abeln Z., Majani Y., Gileadi T., Wallace R., Gustafsson K., et al. Chimeric Antigen Receptor-Engineered Human Gamma Delta T Cells: Enhanced Cytotoxicity with Retention of Cross Presentation. Mol. Ther. 2018;26:354–365. doi: 10.1016/j.ymthe.2017.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Melenhorst J.J., Chen G.M., Wang M., Porter D.L., Chen C., Collins M.A., Gao P., Bandyopadhyay S., Sun H., Zhao Z., et al. Decade-long leukaemia remissions with persistence of CD4(+) CAR T cells. Nature. 2022;602:503–509. doi: 10.1038/s41586-021-04390-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dreyzin A., Panch S.R., Shalabi H., Yates B., Highfill S.L., Jin P., Stroncek D., Shah N.N. Cryopreserved anti-CD22 and bispecific anti-CD19/22 CAR T cells are as effective as freshly infused cells. Mol. Ther. Methods Clin. Dev. 2023;28:51–61. doi: 10.1016/j.omtm.2022.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Panch S.R., Srivastava S.K., Elavia N., McManus A., Liu S., Jin P., Highfill S.L., Li X., Dagur P., Kochenderfer J.N., et al. Effect of Cryopreservation on Autologous Chimeric Antigen Receptor T Cell Characteristics. Mol. Ther. 2019;27:1275–1285. doi: 10.1016/j.ymthe.2019.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Achar S.R., Bourassa F.X.P., Rademaker T.J., Lee A., Kondo T., Salazar-Cavazos E., Davies J.S., Taylor N., François P., Altan-Bonnet G. Universal antigen encoding of T cell activation from high-dimensional cytokine dynamics. Science. 2022;376:880–884. doi: 10.1126/science.abl5311. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets analyzed in the current study are available from the corresponding authors upon reasonable request.