

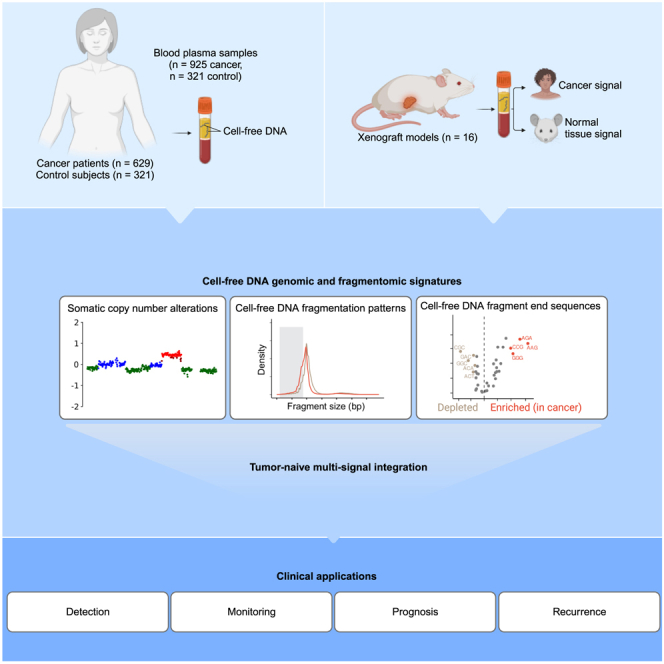
Summary
The structure of cell-free DNA (cfDNA) is altered in the blood of patients with cancer. From whole-genome sequencing, we retrieve the cfDNA fragment-end composition using a new software (FrEIA [fragment end integrated analysis]), as well as the cfDNA size and tumor fraction in three independent cohorts (n = 925 cancer from >10 types and 321 control samples). At 95% specificity, we detect 72% cancer samples using at least one cfDNA measure, including 64% early-stage cancer (n = 220). cfDNA detection correlates with a shorter overall (p = 0.0086) and recurrence-free (p = 0.017) survival in patients with resectable esophageal adenocarcinoma. Integrating cfDNA measures with machine learning in an independent test set (n = 396 cancer, 90 controls) achieve a detection accuracy of 82% and area under the receiver operating characteristic curve of 0.96. In conclusion, harnessing the biological features of cfDNA can improve, at no extra cost, the diagnostic performance of liquid biopsies.
Keywords: liquid biopsy, cell-free DNA, cancer, fragmentomics, multi-modal, sequencing
Graphical abstract
Highlights
-
•
The structural patterns of cfDNA are altered in the blood of patients with cancer
-
•
Xenograft models (n = 16) confirm these patterns are cancer derived
-
•
Integrating cfDNA patterns yields 72% detection at 95% specificity (n = 628)
-
•
Combining cfDNA patterns can predict shorter survival
Moldovan et al. demonstrate that structural patterns of cfDNA are altered in the blood of patients with cancer. In a cell-line-derived xenograft model, this study ties these alterations to cancer. Integrating cfDNA patterns achieves 72% cancer detection at 95% specificity (n = 628, including 220 early stage) and can predict shorter survival.
Introduction
Liquid biopsies, and cell-free DNA (cfDNA) in particular, are actively investigated in clinical oncology. Genetic approaches, including screening for mutations and somatic copy-number aberrations (SCNAs), are promising biomarker candidates for precision oncology.1,2,3,4 Mutation-based detection of tumor-derived cfDNA is often hampered by technical and biological noise (the latter linked to the accumulation of mutations in normal cells).5,6 For example, in elderly patients with TP53 mutant tumors, such as in esophageal adenocarcinoma (EAC), clonal hematopoiesis of indeterminate potential (CHIP) hinder the determination of the origin of cfDNA variants.6,7 In stage I–III patients with a low tumor fraction of cfDNA, this requires a high sequencing depth for cfDNA, availability of buffy coat samples, and tumor-informed sequencing or computational strategies to filter CHIP-derived variants.8,9 However, the complexity and costs of these methods are still high, and their clinical applicability remains limited.10 Methylation and fragmentomic sequencing have recently emerged as potentially sensitive and cost-effective alternatives.11,12,13,14
During cell death and mitosis, DNA can be cleaved at non-random locations and is subsequently released into the bloodstream.15,16,17,18,19 This pool of cfDNA bears information about their cells of origin and mechanism of release.19,20,21 The type of DNase cleaving the DNA is dependent on the nucleosome organization and the presence or absence of cofactors, resulting in distinct fragment sizes and fragment end sequences.15,16,19 The size distribution of cfDNA, with a mode of ∼167 bp and multiples thereof, is related to the wrapping of DNA around the nucleosomes.11,22 An increase in the proportion of shorter fragment sizes (<150 bp) can be observed in the presence of tumor, which correlates with tumor fraction measured by mutation analysis, and may help monitor or forecast disease outcome.23,24,25 Furthermore, a genome-wide analysis of the cfDNA size profile can identify cancer from different types and stages,26,27 which could complement methylation or nucleosome footprinting analysis.14,28 Studies of fragment end sequence profiles revealed the predominance of C-rich 5′ end motifs, linked to the activity of DNASE1L3 in apoptotic cells and in plasma.15,29 The proportion of fragments ending with a C-rich motif is decreased in patients with cancer, and they show a higher sequence diversity in their fragment ends.30 Information on the diversity of fragment end sequences and the clinical utility of biological features retrievable from cfDNA remains limited in oncology.30 The cancer signal carried by these cfDNA biological features is diluted by fragments originating from other tissues. No direct evidence is available for the cancer-specific nature of these signals.
We aim to improve the sensitivity of cfDNA-based non-invasive cancer analysis by mining and combining genetic and fragmentomic patterns. We hypothesized that changes in the cfDNA fragment-end patterns, the proportion of short fragments, and the SCNA tumor fraction (ichorCNA TF), all obtainable from the same low-coverage whole-genome sequencing (WGS) sample, can be utilized to improve the detection and management of patients with cancer. To test this, we established a genome-wide catalog of cfDNA biological signatures of 3 large independent cohorts of patients with cancer. For the extraction of fragment end sequences, we developed the fragment end integrated analysis (FrEIA) score to quantitatively evaluate liquid biopsy samples using low-coverage WGS. In a xenograft mouse model grafted with human colorectal cancer cells, we show that fragments originating from the graft exhibit an increase of these signatures. We determined that the combination of cfDNA biological features can enhance the detection and monitoring of cancer in patients. Combined with a mutation-based tumor fraction detection, these metrics improved sensitivity for cancer detection. Furthermore, we demonstrated the prognostic and predictive value of these integrated cfDNA metrics in patients with lung cancer and EAC, respectively.
Results
The biological signatures of cfDNA are altered in cancer
We generated a catalog of cfDNA biological signatures (Figure 1A) using 925 plasma samples from 629 patients with 21 different cancer types, 306 control samples, and 15 samples from patients with lung nodules (or other lung lesions) not otherwise classified (Table S1). In total, 628 cancer samples were acquired at baseline prior to any treatment, while 297 were collected after various lines of treatment. These samples originate from 3 datasets: sequencing data for 243 of the samples were retrieved from a previous study (cohort #1),26 500 are newly collected (cohort #2), and 503 were retrieved from a public dataset27 (see STAR Methods) (cohort #3).
Figure 1.

Measures of cfDNA biological features are altered in cancer
(A) The number of cancer, nodule, and control samples, the biological signatures of cfDNA, and the extracted measures used in this study.
(B) Log10 cancer/control fold changes (FCs) of the 5′ trinucleotide fragment end sequence proportions. Trinucleotides with a p < 0.01 and a log10FC below the 25th percentile (red) or above the 75th percentile (blue) are shown.
(C) The log10FC of trinucleotides significantly altered (∗p < 0.01) in various cancer types pre-treatment.
(D and E) The increase in (D) the FrEIA score and (E) the Gini diversity index by cancer type in pre-treatment samples.
(F) Aberrant normalized size distribution of cfDNA fragments in pre-treatment cancer samples compared to control samples. The vertical dashed lines outline the size interval used to calculate the P20-150 measure.
(G and H) The (G) P20-150 and (H) the ichorCNA TF increased by cancer type in pre-treatment samples. Bd, bile duct cancer; Br, breast cancer; Cr, colorectal cancer; Es, esophageal cancer; Ga, gastric cancer; Gl, glioblastoma; Lu, lung cancer; Ov, ovarian cancer; Pa, pancreatic cancer. Numbers below the cancer type abbreviation represent the sample count. Cancer types with less than 10 samples are in the “other” category. p values were calculated using two-sided Mann-Whitney U test: ns, not significant, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.005, ∗∗∗∗p < 0.001. When multiple hypotheses were tested, alpha values were adjusted using the Bonferroni method. #, mean passing the threshold of 3% tumor fraction. No biological or technical replicates were used.
To characterize and assess the cfDNA fragment-end trinucleotide patterns from genome-wide sequencing, we developed the FrEIA toolkit (see STAR Methods). cfDNA fragment ends were categorized as pan-cancer based on the frequency of the first three bases on the 5′ end (64 features). The relative proportion of 14 out of the 64 possible 5′ trinucleotide fragment end sequences decreased significantly in cancer samples compared to control samples, while 26 increased (alpha = 0.01, two-sided Mann-Whitney U test; Figure 1B; Table S2). We detected deviations between the mean cancer/control fold changes of the 40 significantly altered fragment-end trinucleotides of 9 cancer types with more than 10 pre-treatment samples (Figure 1C). All cancer types showed a similar trend in fragment end sequence fold changes, with fragments starting with CCA, CCC, or CCT decreasing the most compared to healthy control fragments, while fragments starting with TTC, TTA, or ATG increased the most, suggesting common mechanisms of cfDNA cleavage in cancer irrespective of the cancer type.
Accurate detection of fragment end sequences primarily depends on the base-calling accuracy of the sequencing. Our sequencing batches in cohorts #1 and #2 show high per-base sequence quality with a mean accuracy greater than 30 (1 incorrect base call/1,000 bases) on their 5′ end nucleotides (Figure S1). As reads in cohort 3 were inferred from genomic locations retrieved from finaleDB (see STAR Methods), sequencing accuracy is not applicable for these. These results suggest that fragment end sequences are high fidelity and can be used for the analysis.
As plasma cfDNA from patients with cancer exhibited alterations in the proportion of 5′ end sequences compared to healthy individuals, we integrated these proportions into a single quantitative measurement called the FrEIA score (see STAR Methods). The FrEIA score measures a sample’s relative distance in 5′ end trinucleotide composition from a panel of case and control samples. The FrEIA score is increased in every cancer type for each cohort compared to control individuals (p < 0.001; two-sided Mann-Whitney U tests; Figures 1D and S2A). The diversity in the 5′ end trinucleotide sequences, evaluated using the Gini index (see STAR Methods), is increased for seven out of nine cancer types in comparison to control samples (p < 0.01; two-sided Mann-Whitney U tests; Figures 1E and S2B).
We detected a mode of ∼167 bp and an enrichment of short fragments for patients with cancer compared to healthy control individuals in all three cohorts (Figures 1F and S2C). Based on this, we selected a range between 20 and 150 bp and calculated its proportion (P20-150), resulting in a single metric per sample. P20-150 increased in 7 cancer types (p < 0.05; two-sided Mann-Whitney U tests; Figures 1G and S2D).
We used the copy-number alterations detectable using low-coverage samples to estimate tumor fraction (ichorCNA TF) (Figure S2E). All cancer types passing the threshold of 3% mean tumor fraction showed an increased ichorCNA TF (p < 0.05; two-sided Mann-Whitney U tests) compared to control samples (Figures 1H and S2F).
To demonstrate the strong association between cfDNA biological features and cancer, we conducted an analysis on the plasma samples obtained from 16 xenograft mice, engrafted with a human colorectal cancer cell line (Figure 2A). We observed a robust correlation (Spearman’s R = 0.96, p < 0.01) between the trinucleotide end proportions in patients with cancer and the colorectal graft. Specifically, the graft exhibited a higher frequency of A- and T-rich trinucleotides, whereas C-rich trinucleotides were more prevalent in cancer samples (Figure 2B). Furthermore, the FrEIA score, Gini diversity index, and P20-150 in the grafts showed a significant increase when compared to control patient samples (Figures 2C–2E). These findings are consistent with previous observations in both control individuals and patients with cancer. It is important to note that human patient samples comprise a combination of signals originating from both cancerous and healthy tissues. In contrast, the graft represents the purest possible form of the signal, providing valuable insights into the underlying molecular characteristics. Overall, our results strongly support the connection between the cfDNA biological features and cancer.
Figure 2.

cfDNA biological signatures from xenograft mouse models (n = 16) grafted with a human colorectal cancer cell line
(A) The schematic of the workflow. Signatures were computed from the reads aligning to the human reference genome (GRCh38; graft).
(B) Spearman correlation of trinucleotide fragment-end proportions from the patient-derived samples (cancer) and the xenograft-derived samples (graft). Trinucleotides with a p < 0.01 and a log10FC below the 25th percentile (red) or above the 75th percentile (blue) are shown, as computed from the patient-derived samples with a tumor fraction >10% (see Figure 1B).
(C–E) The increase in (C) the FrEIA score, (D) the Gini diversity index, and (E) the proportion of short fragments (P20-150) of the graft (in magenta) compared to patient-derived control and cancer samples. No biological or technical replicates were used.
To evaluate if the cfDNA features carry the tumor signal independently, we computed their correlation in pre-treatment cancer samples (n = 628). Measures show that they have a moderate positive correlation with each other (Spearman R > 0.25, p < 0.01) (Figure 3A) and—where the mutant allele fraction (MAF) is available—with the MAF (Spearman R > 0.31, p < 0.01, n = 196 pre-treatment cancer samples) (Figure 3B). No correlation was found between the measures for healthy individuals (n = 306) (Figure 3C).
Figure 3.

Correlation between cfDNA biological features and with physiological variables
(A) Spearman correlation between cfDNA measures in pre-treatment cancer samples.
(B) Spearman correlation of cfDNA biological variables with the mutant allele fraction, where available (n = 196 samples).
(C) Spearman correlation between cfDNA measures in control samples. ns, not significant, other values p < 0.01.
(D–F) Spearman correlation of age and (D) the FrEIA score, (E) the Gini diversity index, and (F) the P20-150 of controls.
(G–I) The FrEIA score (G), the Gini diversity index (H), and the P20-150 (I) by gender of control individuals. p values were calculated using two-sided Mann-Whitney U test: ns, not significant, ∗∗∗∗p < 0.001. No biological or technical replicates were used.
Evaluated against physiological variables in healthy control individuals, the cfDNA measures have weak or no correlation with the age of healthy individuals (FrEIA score: Spearman R = 0.058, p = 0.38; Gini diversity index: Spearman R = −0.24, p > 0.001; P20-150: Spearman R = 0.065, p = 0.32; ichorCNA TF below detection threshold thus not evaluated; Figures 3D–3F). The Gini diversity index is higher for female patients (p < 0.001; two-sided Mann-Whitney U tests), while the other measures show no differences between sexes (Figures 3G–3I).
These results suggest that the FrEIA score, the fragment-end trinucleotide diversity, the proportion of short cfDNA fragments, and the tumor fraction derived from SCNAs can be altered in multiple cancer types. A moderate correlation with the MAF (where available) and the fact that they correlate with each other only in samples from patients with cancer show their link to cancer but also indicate that these measures may be under the influence of other, yet unknown physiological factors.
Integration of measures from cfDNA biological signatures improves cancer detection
The primary use of cfDNA fragmentation features in oncology was improving the detection of cancer26,27,31 or genetic alterations.26 Fragment end sequences were used to distinguish cfDNA from patients with cancer and healthy control individuals in a cohort of patients with hepatocellular carcinoma.30 As cancer signal is retrievable from cfDNA biological signatures in various forms, we tested if their combined use would improve cancer detection. At 95% specificity, 269/628 (43%) pre-treatment cancer samples were detected by the FrEIA score (detection threshold of 1.9), 324/628 (52%) by the Gini diversity index (detection threshold of 0.976), and 180/628 (29%) by the P20-150 (detection threshold of 0.216). For the ichorCNA TF, assuming a 3% TF detection threshold,32 we detected 199/628 (32%) pre-treatment cancer samples (Figures 4A and S3A). Altogether, 454/628 (72%) pre-treatment cancer samples were detected by at least one measure with a specificity of 95%. Also, 16/26 (61%) stage I, 11/14 (79%) stage II, 27/40 (68%) stage III, and 111/146 (76%) stage IV lung cancer samples were detected by at least one of the cfDNA measures. Similar detection rates were shown for stage II (27/44; 61%) and III (48/62; 77%) resectable EAC (rEAC) samples, a challenging cancer type for mutation-based detection methods,33,34 and other cancer types (Figures 4B and S3B–S3E; Table S3). We also detected 6/15 samples from patients with nodule/lung lesions. Among them, one patient was suffering from pancreatic cancer when the lung lesions were detected. None of the 9/15 non-detected patients were diagnosed with cancer at follow-up.
Figure 4.

Cancer detection and classification using cfDNA biological features
(A) Receiver operating characteristic (ROC) curve of the detection performance of pre-treatment samples using distinct cfDNA measures individually or in combination (all metrics). The vertical dashed line marks 95% specificity.
(B) The proportion of detected pre-treatment lung and esophageal adenocarcinoma samples by stage. The numbers below the stages represent the detection rate.
(C) Detection rates by at least one of the cfDNA measure or by the MAF, where available, of pre-treatment samples (n = 196 samples).
(D) Schematic representation of the machine learning approach.
(E) ROC curve from predictions on an independent dataset of a logistic regression classifier based on individual or the combination of cfDNA measures. The vertical dashed line marks 95% specificity.
(F) Prediction probabilities of the logistic regression classifier of pre-treatment lung and esophageal adenocarcinoma samples by stage. Samples above the detection threshold (the horizontal dashed line) are considered detected. C, controls; NC, nodules; I, stage I; II, stage II; III, stage III; IV, stage IV. Numbers below the stages represent detection rates. No biological or technical replicates were used.
The integration of cfDNA biological signatures performed slightly better at detecting cancer samples than the mutation-based technique. Out of 196 baseline cancer samples with a MAF available from previous studies,26,27 143 (73%) were detected by at least one and 97 (50%) by multiple measures, while 140 (71%) were detected by MAF (specificity 95%, detection thresholds: FrEIA score: 1.9, Gini diversity score: 0.98, P20-150: 0.22, ichorCNA TF: 3%) (Figure 4C). Out of 57 samples with a MAF <0.1%, 33 (58%) were detected by at least one and 19 (33%) by multiple measures.
Next, we tested if cfDNA feature integration via machine learning approaches would improve cancer classification. To select the best estimator and hyper parameters, we used the pre-treatment samples from cohort #3 (n = 232 cancer and n = 231 control samples), iteratively randomly split into 9 training sets and 1 validation set, with 80% data in a training set and the remaining 20% in the corresponding validation set, using 10-fold cross-validation and 100-fold re-training (Figure 4D; see STAR Methods). Benchmarking of 4 supervised machine learning approaches (k-neighbors, logistic regression, random forest, support vector classifier) using the four cfDNA metrics indicated the highest estimation of classification performance (accuracy: 0.82) was with logistic regression. To test the best model, we used pre-treatment cancer samples (n = 396) and samples from healthy control individuals (n = 75) and patients with nodules (n = 15) from cohorts #1 and #2, all collected independently from the training/validation sets. With a limit of detection probability set to 50%, the model using the combination of metrics performed the best (AUC = 0.96, positive predictive value [PPV] = 0.99, and negative predictive value [NPV] = 0.49), followed by the Gini diversity index (AUC = 0.89, PPV = 0.96, and NPV = 0.39) and the ichorCNA TF (AUC = 0.85, PPV = 0.96, and NPV = 0.39). The P20-150 and the FrEIA score showed lower performance (AUC = 0.82, PPV = 0.96, and NPV = 0.26 and AUC = 0.76, PPV = 0.92, and NPV = 0.32, respectively) (Figures 3E and S3B). At a specificity of 95%, our classifier based on the combination of metrics detected 12/19 (63%) stage I, 2/3 (66%) stage II, 28/38 (74%) stage III, and 105/127 (83%) stage IV lung cancer and 34/44 (77%) stage II and 50/62 (81%) stage III rEAC samples (Figure 3F). These results suggest that the integration of metrics from cfDNA biological signatures can improve the detection of cancer from shallow WGS data even at early stages of the disease.
Combining cfDNA biological signatures for improved clinical management of patients with cancer
To evaluate the significance of the integrated measures of cfDNA biological signatures in a “real-world” clinical setting, we tested cfDNA from patients with rEAC, where serial circulating tumor DNA (ctDNA) detection has been shown to predict adverse outcome.7 Here, we assessed the potential of the combined measures in 293 rEAC samples from 2 clinical cohorts: a neoadjuvant chemoradiotherapy (CRT) cohort (BIOES cohort, n = 70 patients, n = 149 plasma samples) receiving standard-of-care carboplatin combined with paclitaxel-based CRT, and a cohort of patients who participated in the phase II PERFECT trial (n = 40 patients, n = 144 samples; see STAR Methods) received CRT in combination with a PD-L1 inhibitor.35 Both cohorts included EAC stage II (n = 125 samples) and stage III (n = 168 samples). Plasma samples were collected longitudinally before and after chemoradiation, and also postoperatively, in the PERFECT cohort (Figure 4A).
The detection of treatment response can help stratify patients for surgery or further adjuvant treatment. However, detection of treatment response using liquid biopsies is challenging and most commonly requires more complex and elaborate approaches such as tumor-guided and personalized sequencing36,37 (Figures S4 and S5). The FrEIA score post-CRT and pre-surgery was significantly increased compared to pre-treatment for patients with an incomplete response (pT+N+/−, or pT0N+) as determined by a pathologist from the resection specimen (p = 0.0015 and 0.04, two-sided Mann-Whitney U test), while patients with a pathological complete response (pCR; pT0N0) showed no difference (p = 0.77 and 0.62, two-sided Mann-Whitney U test) (Figure 5B). Moreover, the FrEIA score post-CRT and pre-surgery was significantly increased compared to pre-treatment for patients with a tumor regression (TR) grade (Mandard) score of 3–5 (partial/no response) (p < 0.0023 and p = 0.011, two-sided Mann-Whitney U test), while patients with a low TR score of 1–2 (complete/suboptimal response) showed no significant difference (p = 0.38 and 0.56, two-sided Mann-Whitney U test) (Figure 5B). In line with the FrEIA score, mean ichorCNA TF and the Gini diversity index also increased between the pre-CRT and the post-CRT samples for incomplete responders, while for patients with a TR score of 3–5, the Gini diversity index increased significantly (Table S4). These findings are surprising, as there was no significant difference between the measures of patients with or without a pCR or a high or low Mandard score at any of the time points. These results suggest that dynamic changes compared to the pre-treatment quantification of multiple cfDNA metrics were related to the prediction of treatment response prior to resection and histological assessment.
Figure 5.

cfDNA biological patterns enable monitoring and prediction of recurrence for esophageal carcinoma
(A) Schematic representation of the clinical timeline and sampling of patients with EAC.
(B) The change in FrEIA score between pre-CRT and post-CRT samples based on the pathological complete response (pCR). p values were calculated using two-sided Mann-Whitney U test: ns, not significant, ∗∗p < 0.01, ∗∗∗p < 0.005.
(C) Clinical timeline of patients with EAC undergoing resection from the PERFECT subcohort (n = 33) centered around the time point of resection. EOT, end of treatment; CSD, cancer-specific death.
(D) Kaplan-Meier curves of the recurrence-free survival probabilities for patients with EAC from the PERFECT subcohort from the postresection time point. Samples with one of the measures higher than the threshold were considered “detected” (FrEIA score: 2.54, Gini diversity index: 0.98, P20-150: 0.26, ichorCNA: 3% sensitivity threshold). p values were calculated using log-rank test statistics. Dashed lines show the median survival time. No biological or technical replicates were used.
The prediction of recurrence after surgery is challenging for tumor-naive liquid biopsy assay due to the minute amount of tumor signal in circulation following surgery. Patients from the PERFECT trial had one plasma samples collected ∼3 months after surgery (n = 31), and 17 showed recurrence within 2 years of sampling (Figure 5C). Using low-coverage WGS, we detected one of the 4 cfDNA features in 6/11 (55%) early recurrent patients (<365 days postsurgery) in follow-up samples and in 1/12 (8%) patients that are not experiencing clinical recurrence. A total of 53% patients with recurring disease were detected postsurgery, which was associated with a shorter recurrence-free survival (RFS) from the time of surgery (hazard ratio = 4.08; log-rank p = 0.017), with stage III patients having a higher chance of recurrence (RFS; hazard ratio = 2.25; log-rank p = 0.017) (Figure 5D).
We further assessed the prognostic potential of cfDNA features from the postresection samples of 31 patients with rEAC and 101 pre-treatment patients with lung cancer with available survival data (stage I = 11, stage II = 2, stage III = 24, stage IV = 64). Patients with rEAC with at least one or more cfDNA measures above the detection threshold have a shorter survival from the time of surgery than patients who had undetected levels of cfDNA (hazard ratio [HR] = 4, log-rank p = 0.0086) and stage III patients showing a slightly increased risk of death (HR = 0.7, log-rank p = 0.0086) (Figure 6A). Similarly, patients with lung cancer with multiple cfDNA features above the detection threshold pre-treatment displayed significantly lower overall survival (OS) from the time of first sampling (HR = 1.56, log-rank p = 0.03), with stage IV patients having a higher risk of death (HR = 1.66, log-rank p = 0.03) (Figure 6B). This demonstrates the potential clinical utility of multi-signal profiling of different malignancies.
Figure 6.

Integrating cfDNA biological patterns improve survival prognostication
(A) Kaplan-Meier curves of the survival probability for patients with EAC from the postsurgery time point. Dashed lines represent the median survival.
(B) Kaplan-Meier curves of the survival probability for patients with lung cancer from the time of initial sampling. Samples with one of the measures higher than the threshold were considered “detected” (FrEIA score: 2.54, Gini diversity index: 0.98, P20-150: 0.26, ichorCNA: 3% sensitivity threshold). p values were calculated using log-rank test statistics. Dashed lines show the median survival time. No biological or technical replicates were used.
Discussion
The combination of different analytes from the blood plasma can improve the sensitivity of liquid biopsy for low-tumor-burden patients in a tumor-naive context but requires an accumulation of expensive tests and skills for their analysis.38,39 Here, we evaluated if multiple biological signatures obtained from the same sample and sequencing data can be harnessed to enhance the sensitivity of detecting cancer signals in a range of clinical applications. Using a pan-cancer dataset of 925 plasma samples from three independent cohorts of cost-effective, low-coverage WGS, we demonstrated that integrating genomic and fragmentomic features can enhance the detection of early-stage cancer, providing value as a prognostic biomarker as well as for monitoring recurrence in serial samples.
Despite biological unknowns, the cfDNA fragmentation patterns are being evaluated for cancer detection.26,27,31,40 Other cfDNA biological features, notably their fragment end sequences and positions, remain to be extensively characterized and their potential for cancer diagnostics to be determined.41,42 Our new open pipeline, called FrEIA, allows the recovery of cfDNA fragment end sequences from genome-wide sequencing data in a reproducible way. FrEIA can be used on low-coverage WGS,26,32,43 as well as higher-depth WGS27,28,44 or other forms of paired-end sequencing.37 Our work provides strong evidence that the composition in bases at the end of cfDNA fragments in the plasma of multiple cancer types is altered in comparison to healthy control individuals, resulting in increased diversity of cfDNA fragment ends in cancer. A previous report observed such bias in hepatocellular carcinoma but did not verify its tumor-specific nature.30 We confirmed specifically that such modifications can be cancer derived using xenograft models, allowing a separation of tumor (human) and non-tumor (mouse) DNA.23,26,45,46,47 Based on these observations, we developed the FrEIA score, which is increased for cancer samples irrespective of cancer type and stage. Plasma cfDNA from patients with cancer are enriched in shorter fragments.23,48,49 We observed an increase of the proportion of short cfDNA fragments in the different datasets. These fragmentation-related biological features correlate moderately with ctDNA proportion estimates (MAF or ichorCNA TF) in plasma samples of patients with cancer but not healthy control individuals. Solid tumors exhibit heterogeneity at the genetic and non-genetic levels. Focusing on a single biological characteristic, such as point mutations or copy-number alterations, may not fully capture this heterogeneity. Thus, the limited correlation observed between cfDNA biological features and the disease underscores the necessity of integrating multiple cancer-related signals from diverse sources to enhance detection accuracy.50
Pre-analytical conditions pose potential limitations to the usage of cfDNA fragmentation features and could bias conclusions if not carefully examined.51 Similarly, the choice of library preparation with either single-stranded and double-stranded DNA,52,53 or PCR and PCR-free,54 could impact the size distribution of cfDNA, with an anticipated method-dependent bias in the fragment-end composition. Sequencing quality and read filtering can affect the fragment end sequence composition. Furthermore, cfDNA fragment-end analysis could be potentially obscured by other clinical conditions that affect cfDNA release in the bloodstream.55,56,57 Another limitation is the number of nucleotides chosen for analysis, as the stretch of DNA on the fragment end carrying the tumor-specific signal is currently not clearly defined. Previous studies observed cancer-type-specific cfDNA signatures, which may be the case for fragment end sequence patterns.58 However, the cohorts used in our study have an unbalanced distribution of samples across different cancer types, which limits the statistical power of our results for cancer types with lower sample counts.
The combination of cfDNA features improved the detection of cancer with machine learning. A classifier tested on a cohort of 528 samples (396 cancer, 75 control, and 15 nodule samples) leads to an area under the receiver operating characteristic curve (AUROC) = 0.96 when classifying cancer from control samples. In contrast, separate use of the FrEIA score, the fragment-end diversity, the proportion of short fragments, or the ichorCNA TF with a logistic-regression-based classifier for the classification of cancer from control decreases the classification power (AUROCs of 0.76, 0.89, 0.82, and 0.85, respectively). Using a classifier based on the combination of cfDNA features in a cohort of 187 patients with lung cancer, we could detect 14/22 early-stage patients, while in a subcohort of 106 pre-CRT rEAC samples from two different studies, 34/44 early-stage patients were detected. The availability of early-stage cancer samples is limited in our study; thus, additional confirmation will be needed to determine the clinical utility for early detection. The analysis of fragment sizes and fragment end sequences in genomic bins could have potential for cancer classification.59 A combined use of cfDNA features extracted from subgenomic bins or from genomic regions of interest could improve detection rates and thus needs further evaluation.
Beyond the classification of cancer beyond a cancer diagnostic, cfDNA biological features can be used in realistic clinical scenarios. Here, we show that a multi-signal cfDNA approach can be a sensitive, cost-effective, and flexible tool for a range of clinical applications in the tumor-naive context. The recovery of cfDNA fragment end sequences has a prognostic value for treatment response. In a cohort of 46 patients with rEAC, 75% patients with suboptimal TR (TR score 3–5) have an increase in their FrEIA score, 64% in their Gini diversity index, 47% in their P20-150, and 44% in their irchorCNA TF post-CRT compared to their baseline values. Patients with a complete pathological response or with an optimal TR (TR score 1–2) also show a non-significant increase, explained by the transient release of ctDNA after radiotherapy or by other physiological and clinical conditions.57 However, these results are limited by the low number of patients with a pCR in this cohort. When analyzing the 30 samples collected post-surgery, we demonstrated that 55% patients detected by one of the cfDNA features (6 out of 11 patients) showed recurrence in a year. In a recent publication, a ctDNA panel consisting of 77 genes was tested in 97 patients with EAC. After filtering of CHIP variants, the panel showed high prognostic potential for disease-free survival (HR = 5.35, 95% confidence interval [CI] 2.10–13.63; p ≤ 0.0001) based on the post-surgery samples.7 Another study used an EAC tumor-guided sequencing approach and found ctDNA status (positive vs. negative) to be prognostic at baseline for disease-free survival (p = 0.042).8 In contrast to these two approaches, the metrics derived from cfDNA features do not require a buffy coat or a tumor biopsy and have the potential to be easily implementable in the clinic, costing a fraction of tumor-informed sequencing. However, the specificity of metrics derived from cfDNA features is below that of tumor-informed sequencing methods (and bespoke sequencing panels that can reach parts per million fragments).36,37 Furthermore, due to the nature of hybrid-capture sequencing, a combination of mutation analysis with cfDNA biological signatures is possible and could result in improved tumor signal detection.60,61 The armamentarium of cfDNA fragmentomic signals is increasing quickly, and we can foresee that some of these features could have a diagnostic potential in combination with other cfDNA signals.11,12,62
Our results highlight that a multi-signal combination of cfDNA genomic and fragmentomic features has the potential to deliver sensitive detection of tumor-derived cfDNA using genome-wide sequencing. Although further validation in larger cohorts is needed, cfDNA multi-signal integration can inform on the early detection of cancer and could contribute to addressing, at a competitive cost, the unmet need of residual disease therapy decision-making in oncology.
Limitations of the study
The following limitations should be considered when interpreting the results. cfDNA fragmentomic signals may be biased by the pre-analytical variables, computational pre-processing, and individual genetic diversity or comorbidities. The current study enrolls a wide range of cancer types (n = 21), which may have variable ctDNA release, potentially different fragmentomic signatures, and a limited number of samples per condition. Treatment response monitoring may be affected by the sampling time because of transient release of ctDNA after treatment. Additionally, the effects of treatment and drug toxicity on cfDNA fragmentomic signatures are unknown. The necessity of data harmonization when comparing multiple cohorts may blur small differences between cancer types or sampling time points. Finally, there are no clear guidelines on how multi-modal liquid biopsy analysis could be integrated in clinical settings.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Human plasma samples | Amsterdam Liquid Biopsy Center | http://www.liquidbiopsycenter.nl/ |
| Critical commercial assays | ||
| ThruPLEX Plasma-Seq | Takara | Cat #: R400492 |
| Deposited data | ||
| Esophageal adenocarcinoma dataset | This paper | EGA: EGAD00001008316 |
| Lung cancer and healthy control dataset #1 | This paper | EGA: EGAD00001008321 |
| Lung cancer dataset #2 | This paper | EGA: EGAD00001008666 |
| Healthy control dataset | This paper | EGA: EGAD00001008322 |
| Retrieved dataset | Mouliere et al.26 | EGA: EGAS00001003258 |
| Xenograft mouse dataset | This paper | EGA: EGAD00001011128 |
| Experimental models: Cell lines | ||
| MDST8 | Sanger Institute (Cambridge, UK) | N/A |
| Experimental models: Organisms/strains | ||
| Mouse model (Hsd:Athymic Nude-Fox1nu) | Envigo | N/A |
| Software and algorithms | ||
| FrEIA tool | This paper | Github: https://github.com/mouliere-lab/FrEIA.git |
| Machine learning classifier pipeline | This paper | Github: https://github.com/mouliere-lab/FrEIA.git |
| ichorCNA | Adalsteinsson et al.32 | Github: https://github.com/broadinstitute/ichorCNA |
| bwa-mem | Li and Durbin63 | Github: https://github.com/lh3/bwa |
| Samtools | Li et al64 | https://github.com/samtools/samtools |
| ComBat | Johnson et al.65 | http://www.bioconductor.org/packages/release/bioc/html/sva.html |
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Florent Mouliere (f.mouliere@amsterdamumc.nl).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The cfDNA sequencing data have been deposited in the European Genome-Phenome Archive (EGA) and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
The code for the FrEIA tool and of the machine learning pipeline has been deposited on Github and is publicly available as of the date of publication. Links to the code are listed in the key resources table.
Experimental model and study participant details
Human participants
A total of 925 plasma samples from 629 patients were analyzed across 21 cancer types, together with samples of 306 healthy controls and 15 plasma samples from patients with lung nodules or other lesions in three independent cohorts (Table S1). Data for cohort #1 (n = 243) was retrieved from a previous study from a public database (EGA accession number: EGAS00001003258).26 Cohort #2 (n = 500) was recruited following informed consent via the Liquid Biopsy Center at the Amsterdam UMC, location VUmc and location AMC (study approved by the Amsterdam UMC ethics board, METC U2019_035). Esophageal adenocarcinoma patients were recruited as part of the PERFECT trial or the BIOES esophageal and gastric cancer biobank (nCRT cohort).35 The PERFECT trial (study approved by the Amsterdam UMC ethics board, METC 2016_325) and the BIOES biobank (study approved by the Amsterdam UMC ethics board, METC 2013_241) have both received local approval from the medical ethical committee, resp. biobanking committee of the Academic Medical Center. Data from cohort #3 (n = 503) was retrieved from the public finaleDB database as described in the methods section.
Cell lines
Colorectal cancer cell line MDST8 was obtained from the Sanger Institute (Cambridge, UK) and cultured in Dulbecco’s modified Eagle’s medium/F-12 medium with L-glutamine, 15 mM HEPES (Thermo-Fisher Scientific, Bleiswijk, The Netherlands) supplemented with 10% v/v fetal bovine serum (Life Technologies), penicillin and streptomycin. The cell line was authenticated by STR Genotyping and regularly tested for mycoplasma infection.
Animals
Animal experiments were approved by the Animal Experimentation Committee at the Amsterdam UMC (location AMC) and conducted in accordance with the national guidelines. 16 female nude (Hsd:Athymic Nude-Fox1nu) mice (6–12 weeks old) were purchased from Envigo. Human MDST8 CRC cells (10,000 cells/mice) in medium containing 50% matrigel (Corning) were injected intraperitoneally. Five weeks after tumor cell injection, blood collection via cardiac puncture under anesthesia was performed, immediately followed by euthanasia.
Method details
Blood processing and DNA extraction
Blood samples for cohort #2 were collected into EDTA-containing tubes and processed by a double-centrifugation protocol (1600 g for 10 min; 16000 g for 10 min) before storage at −80°C. Blood samples collected locally in Amsterdam in EDTA coated tubes were processed using a double-centrifugation protocol (900 g for 15 min; 2500 g for 10 min). Supernatant plasma was carefully aliquoted in 0.5mL Nunc tubes before being stored at −80°C.Plasma cfDNA was extracted using either the QIAamp Circulating Nucleic Acid Kit (QIAGEN; silica column-based) in the EAC cohort or QIAsymphony DSP Circulating Nucleic Acid Kit (QIAGEN) for the lung cohort.
Library preparation and sequencing
Plasma cfDNA was quantified using the cell-free DNA screentape kit and a Tapestation 4200 system (Agilent) or a BioAnalyzer HS chip and system (Agilent). Indexed sequencing libraries were prepared using 1–10 ng of DNA and the ThruPLEX-Plasma Seq kit or ThruPLEX-Tag Seq kit (Takara). Libraries were pooled in equimolar amounts and sequenced to <1x depth of coverage on a NovaSeq 6000 (Illumina) generating 150-bp paired-end reads from an S4 flowcell.
Fragment inference from genomic locations
For cohort #3 we inferred the fragments based on the start and end positions from the fragment.tsv files retrieved from finalDB [http://finaledb.research.cchmc.org]. In brief, we queried “Cristiano et al., 2019, “blood plasma” and “WGS” on the finalDB database and retrieved the fragment.tsv files containing the genomic locations of fragments for the GRCh38 human genome assambly. We first converted the fragment.tsv to a Browser Extensible Data (bed) file format using AWK “awk -v OFS = '∖t' '$4≥5 {{print $1, $2, $3, ".", $4, $5}}' {input_fragment.tsv} > {output_fragment.bed}”, selecting fragments with a mapping quality ≥5. Next we converted the bed files to fasta using “bedtools getfasta -fi {GRCh38.fna} -bed {output_fragment.bed} -s | gzip > {output_fragment.fa.gz}” (bedtools v.2.30.0 [https://bedtools.readthedocs.io/en/latest/]) and the GRCh38 human genome assembly. The resulting fasta files were used in further analysis.
Fragment end analysis
Sequencing data were processed using a pipeline controlled by Snakemake (v. 5.14.0), and fragment ends were analyzed using the FrEIA toolkit developed in our group [https://github.com/mouliere-lab/FrEIA.git]. In brief, adapters and indexes were trimmed using the bbduk.sh (v. 38.79) [https://sourceforge.net/projects/bbmap/] in paired mode with the ‘ktrim = r k = 23 mink = 11 hdist = 1’ parameters and the adapter reference dataset provided with the software. For the xenograft model samples, trimmed human derived reads were split from trimmed mouse derived reads by using bbsplit (v 38.79) aligned to the human reference genome GRCh38 (GeneBank accession: GCA_000001405.28) and the mouse reference genome GRCmm10 (GeneBank accession: GCA_000001635.9). The trimmed reads from the three clinical cohorts were mapped to the GRCh38 human genome assembly (GeneBank accession: GCA_000001405.28) using the bwa-mem (v. 0.7.17) [https://github.com/lh3/bwa]. Reads with a mapping quality lower than 5, unmapped reads, secondary mappings, chimeric and PCR duplicates were filtered with samtools (v. 1.12) [https://github.com/samtools/samtools]. Reads passing the filtration step were submitted for our custom pysam (v. 0.16.0.1) implementation, extracting the first 3 mapped bases from the 5′ end of the remaining paired reads. Fragments were categorized based on their first mapped 5′ trinucleotide sequence. Fractions of these fragment categories were calculated for every sample.
Data harmonization
We observed batch-effect in the 64 trinucleotide counts of both healthy and cancer samples, supposedly caused by pre-analytical conditions51 (Figure S5A). To eliminate this, data harmonization was performed using the ComBat-Seq module65 from the R package SVA (v.3.42.0) with 6 batches as covariates (B1 n = 293, B2 n = 42, B3 n = 77, B4 n = 88, B5 n = 243 and B6 n = 503) (Figure S5B). ComBat-Seq works by modeling and adjusting for batch effects using an empirical Bayes framework, enabling the harmonization of data across different sequencing experiments while preserving biological variability. The 6 batches represent 4 rounds of sequencing belonging in Cohort #2 (B1, B2, B3 and B4), and Cohort #1 and #3 considered as two separate batch (B5 and B6 respectively). The resulting fragment end trinucleotide counts were used in further analysis.
The FrEIA score calculation
Based on the observation that cfDNA fragment endings are non-random, and that cancer patients show a shift in fragment end sequences, we developed a single quantitative metric, designated the FrEIA score (F), with the following formula:
where dn is the Euclidean distance in fragment end trinucleotide pattern of a given sample from the median vector of a panel of control samples, while dc is that from a panel of cancer samples. The fragment end trinucleotide pattern is represented by vectors that are composed of selected trinucleotide proportions with a significant increase or decrease in cancer. The distances were computed using the dist function from the R package stats v.4.1.2. To select these trinucleotides, we first picked samples with a ichorCNA TF higher than 10% to ensure the tumor signal to noise ratio is high, and used these samples to calculate the log10-fold change of each trinucleotide proportion with the following formula:
where Px is the proportion of a given trinucleotide. Following this, we compared the mean proportion of each trinucleotide of the cancer cohort to the mean proportion of the same trinucleotide of the healthy cohort using the Wilcoxon Rank-Sum Test and selected those that passed the alpha = 0.01 significance threshold. Those that had an FC lower than −0.018, the 25% percentile were considered “significantly decreased in cancer”, while those that had an FC higher than 0.056, the 75% percentile were considered “significantly increased in cancer”. As panel of controls, we used the 117 control samples while the panel of cancer samples was composed of 396 baseline cancer samples.
Fragment end trinucleotide diversity analysis
The 5′ trinucleotide fragment end sequence diversity was calculated for every sample as the Gini index using the formula:
where Pi is the frequency of a specific i trinucleotide ending.
Somatic copy number analysis
The ichorCNA software (commit 5bfc03e) was used to perform the copy number analysis and estimate the ctDNA tumor fraction.32 Exceptions to the software’s default settings are as follows: (1) An in-house panel-of-normals from shallow Whole Genome Sequencing (sWGS) was created; (2) non-tumor fraction parameter restart values were increased to c(0.95,0.99,0.995,0.999); (3) ichorCNA ploidy parameter restart value was set to 2; (4) no states were used for subclonal copy number and (5) the maximum copy number to use was lowered to 3. The tumor fraction with the highest log likelihood was retrieved and reported.
Classification and predictive model
For the classification of baseline cancer samples from control samples we trained, validated and tested a machine learning model, using the combination of the FrEIA score, the Gini diversity index, the P20-150 and the ichorCNA TF, and the scores separately. To test the robustness of our model we split our dataset into two: one training/validation set encompassing pre-treatment cancer samples (n = 232) and controls (n = 231) of cohort #3 and one independent test set including pre-treatment cancer samples (n = 396), controls (n = 75) and nodules (n = 15) of cohorts #1 and #2. To select the best model, we performed hyper-parameter tuning coupled with estimator selection using Optuna (v. 3.0.5). In brief, we performed 10-fold cross-validation with random sample selection on the training/validation set scaled with StandardScaler, splitting the data into 80% training and 20% validation sets - stratified by the ‘cancer’ and ‘control’ categories. We surveyed the parameter landscape of the KNeighborsClassifier, LogisticRegression, SupportVectorClassifier and RandomForestClassifier estimators throughout 1000 trials, encompassing 100-fold re-training. We pruned trials with mean intermediate accuracy smaller than the best accuracy. The model with the highest accuracy was selected and used to classify the independent testing set, namely the LogisticRegression with the parameters: solver: lbfgs, c: 24.986504780795247, maxitter: 8332, classweight: balanced. Samples with a score above 0.5 were classified as ‘cancer’, why those below were classified as ‘control’. For a graphical representation of the classification sequence see Figure 3D.
Statistical analysis and plotting
For hypothesis testing we used the two-sided Mann-Whitney U test with a significance level of 0.05, where not stated otherwise. When multiple hypotheses were tested, alpha values were adjusted using the Bonferroni method. Figures were plotted in RStudio (v. 1.3.1093) running R (v. 3.6.3) using ‘ggplot2’ (v. 3.3.3), ‘ggpubr’ (v. 0.4.0), ‘ggsci’ (v. 2.9) and ‘ggfortify’ (v. 0.4.11). The Kaplan-Meier analysis was performed using the R packages ‘survival’ (v 3.1–8) and visualized using ‘survminer’ (v. 0.4.9). We used a detection threshold of 95% specificity, except for overall and recurrence-free survival, which were calculated using a detection threshold of 99% specificity. Survival curves were calculated using the overall survival of the patients with a detection threshold of 99% specificity. The survival of patients who survived beyond the end of the study or the recurrence free survival of patients without recurrence before the end of the study was censored.
Additional resources
No additional resources.
Acknowledgments
The authors are thankful to Mai Tran, Dr. Wendy Onstenk, and the Amsterdam UMC Liquid Biopsy Center for the logistical support and advice. The authors are also thankful to Ilias Houda, Rimsha Shaikh, and Ezgi Ulas for their help in annotating clinical information. Y.v.d.P. and F.M. are funded by the Amsterdam UMC Liquid Biopsy Center, an initiative made possible through the Stichting Cancer Center Amsterdam. The authors would like to thank Dr. Dineika Chandrananda for comments and discussions to improve the analysis of the ichorCNA algorithm. The authors would like to thank Dr. Caitrin Crudden, Dr. Steven Wang, Dr. Yongsoo Kim, Ignas Krikstaponis, and Francesco Orlando for comments and discussions. This work was carried out on the Dutch national e-infrastructure with the support of SURF Cooperative. N.M. and F.M. are supported by a Dutch Cancer Fund (KWF-12822). The PERFECT study was financially supported by Hoffmann-La Roche, Ltd., Basel, Switzerland. Analysis of cfDNA of the neoadjuvant CRT (nCRT) cohort was made possible through a grant of the Maag Lever Darm Stichting (SK18-32). Funders have no role in the design of the study.
Author contributions
Conception and design, N.M. and F.M.; experiments and data collection, Y.v.d.P., J.R., S.V., and T.V.; data processing, N.M., Y.v.d.P., D.B., and F.M.; software development, N.M.; data analysis, N.M. and F.M.; sample acquisition, T.v.d.E., A.C., M.F.F., H.v.L., and I.B.; funding acquisition, M.P., H.v.L., I.B., and F.M.; manuscript draft, N.M., Y.v.d.P., and F.M.; manuscript revisions and comments, N.M., Y.v.d.P, T.v.d.E., D.B., S.V., J.R., A.C., T.V., M.F.F., M.P., H.v.L., I.B., and F.M.; supervision, F.M.
Declaration of interests
F.M. is co-inventor on multiple patents related to cfDNA analysis. Other co-authors have no relevant conflict of interests.
Published: January 16, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.101349.
Supplemental information
P-values were calculated using the two-sided Mann-Whitney U test.
References
- 1.Bettegowda C., Sausen M., Leary R.J., Kinde I., Wang Y., Agrawal N., Bartlett B.R., Wang H., Luber B., Alani R.M., et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014;6:224ra24. doi: 10.1126/scitranslmed.3007094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Garcia-Murillas I., Schiavon G., Weigelt B., Ng C., Hrebien S., Cutts R.J., Cheang M., Osin P., Nerurkar A., Kozarewa I., et al. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci. Transl. Med. 2015;7:302ra133. doi: 10.1126/scitranslmed.aab0021. [DOI] [PubMed] [Google Scholar]
- 3.Heitzer E., Auer M., Hoffmann E.M., Pichler M., Gasch C., Ulz P., Lax S., Waldispuehl-Geigl J., Mauermann O., Mohan S., et al. Establishment of tumor-specific copy number alterations from plasma DNA of patients with cancer. Int. J. Cancer. 2013;133:346–356. doi: 10.1002/ijc.28030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Creemers A., Krausz S., Strijker M., van der Wel M.J., Soer E.C., Reinten R.J., Besselink M.G., Wilmink J.W., van de Vijver M.J., van Noesel C.J.M., et al. Clinical value of ctDNA in upper-GI cancers: A systematic review and meta-analysis. Biochim. Biophys. Acta Rev. Canc. 2017;1868:394–403. doi: 10.1016/j.bbcan.2017.08.002. [DOI] [PubMed] [Google Scholar]
- 5.Razavi P., Li B.T., Brown D.N., Jung B., Hubbell E., Shen R., Abida W., Juluru K., De Bruijn I., Hou C., et al. High-intensity sequencing reveals the sources of plasma circulating cell-free DNA variants. Nat. Med. 2019;25:1928–1937. doi: 10.1038/s41591-019-0652-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abbosh C., Swanton C., Birkbak N.J. Clonal haematopoiesis: a source of biological noise in cell-free DNA analyses. Ann. Oncol. 2019;30:358–359. doi: 10.1093/annonc/mdy552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ococks E., Frankell A.M., Masque Soler N., Grehan N., Northrop A., Coles H., Redmond A.M., Devonshire G., Weaver J.M.J., Hughes C., et al. Longitudinal tracking of 97 esophageal adenocarcinomas using liquid biopsy sampling. Ann. Oncol. 2021;32:522–532. doi: 10.1016/j.annonc.2020.12.010. [DOI] [PubMed] [Google Scholar]
- 8.Ococks E., Sharma S., Ng A.W.T., Aleshin A., Fitzgerald R.C., Smyth E. Preprint at Elsevier; 2021. Serial Circulating Tumor DNA Detection Using a Personalized, Tumor-Informed Assay in Esophageal Adenocarcinoma Patients Following Resection. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Azad T.D., Chaudhuri A.A., Fang P., Qiao Y., Esfahani M.S., Chabon J.J., Hamilton E.G., Yang Y.D., Lovejoy A., Newman A.M., et al. Circulating Tumor DNA Analysis for Detection of Minimal Residual Disease After Chemoradiotherapy for Localized Esophageal Cancer. Gastroenterology. 2020;158:494–505.e6. doi: 10.1053/j.gastro.2019.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ignatiadis M., Sledge G.W., Jeffrey S.S. Liquid biopsy enters the clinic — implementation issues and future challenges. Preprint. 2021;18:297–312. doi: 10.1038/s41571-020-00457-x. [DOI] [PubMed] [Google Scholar]
- 11.van der Pol Y., Mouliere F. Toward the Early Detection of Cancer by Decoding the Epigenetic and Environmental Fingerprints of Cell-Free DNA. Cancer Cell. 2019;36:350–368. doi: 10.1016/j.ccell.2019.09.003. [DOI] [PubMed] [Google Scholar]
- 12.Lo Y.M.D., Han D.S.C., Jiang P., Chiu R.W.K. Epigenetics, fragmentomics, and topology of cell-free DNA in liquid biopsies. Science (1979) 2021;372:eaaw3616. doi: 10.1126/science.aaw3616. [DOI] [PubMed] [Google Scholar]
- 13.Burgener J.M., Zou J., Zhao Z., Zheng Y., Shen S.Y., Huang S.H., Keshavarzi S., Xu W., Liu F.F., Liu G., et al. Tumor-Naïve multimodal profiling of circulating tumor DNA in head and neck squamous cell carcinoma. Clin. Cancer Res. 2021;27:4230–4244. doi: 10.1158/1078-0432.CCR-21-0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shen S.Y., Singhania R., Fehringer G., Chakravarthy A., Roehrl M.H.A., Chadwick D., Zuzarte P.C., Borgida A., Wang T.T., Li T., et al. Preprint at Nature Publishing Group; 2018. Sensitive Tumour Detection and Classification Using Plasma Cell-free DNA Methylomes. [DOI] [PubMed] [Google Scholar]
- 15.Chandrananda D., Thorne N.P., Bahlo M. High-resolution characterization of sequence signatures due to non-random cleavage of cell-free DNA. BMC Med. Genomics. 2015;8:29. doi: 10.1186/s12920-015-0107-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Han D.S.C., Ni M., Chan R.W.Y., Chan V.W.H., Lui K.O., Chiu R.W.K., Lo Y.M.D. The Biology of Cell-free DNA Fragmentation and the Roles of DNASE1, DNASE1L3, and DFFB. Am. J. Hum. Genet. 2020;106:202–214. doi: 10.1016/j.ajhg.2020.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jahr S., Hentze H., Englisch S., Hardt D., Fackelmayer F.O., Hesch R.D., Knippers R. DNA fragments in the blood plasma of cancer patients: Quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001;61:1659–1665. [PubMed] [Google Scholar]
- 18.Jiang P., Sun K., Tong Y.K., Cheng S.H., Cheng T.H.T., Heung M.M.S., Wong J., Wong V.W.S., Chan H.L.Y., Chan K.C.A., et al. Preferred end coordinates and somatic variants as signatures of circulating tumor DNA associated with hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA. 2018;115:E10925–E10933. doi: 10.1073/pnas.1814616115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mouliere F. A hitchhiker’s guide to cell-free DNA biology. Neurooncol. Adv. 2022;4:ii6–ii14. doi: 10.1093/noajnl/vdac066. ii6–ii14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thierry A.R., El Messaoudi S., Gahan P.B., Anker P., Stroun M. Origins, structures, and functions of circulating DNA in oncology. Cancer Metastasis Rev. 2016;35:347–376. doi: 10.1007/s10555-016-9629-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lehmann-Werman R., Neiman D., Zemmour H., Moss J., Magenheim J., Vaknin-Dembinsky A., Rubertsson S., Nellgård B., Blennow K., Zetterberg H., et al. Identification of tissue-specific cell death using methylation patterns of circulating DNA. Proc. Natl. Acad. Sci. USA. 2016;113:E1826–E1834. doi: 10.1073/pnas.1519286113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lo Y.M.D., Chan K.C.A., Sun H., Chen E.Z., Jiang P., Lun F.M.F., Zheng Y.W., Leung T.Y., Lau T.K., Cantor C.R., Chiu R.W.K. Maternal plasma DNA sequencing reveals the genome-wide genetic and mutational profile of the fetus. Sci. Transl. Med. 2010;2:61ra91. doi: 10.1126/scitranslmed.3001720. [DOI] [PubMed] [Google Scholar]
- 23.Mouliere F., Robert B., Arnau Peyrotte E., Del Rio M., Ychou M., Molina F., Gongora C., Thierry A.R. High fragmentation characterizes tumour-derived circulating DNA. PLoS One. 2011;6 doi: 10.1371/journal.pone.0023418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.El Messaoudi S., Mouliere F., Du Manoir S., Bascoul-Mollevi C., Gillet B., Nouaille M., Fiess C., Crapez E., Bibeau F., Theillet C., et al. Circulating DNA as a strong multimarker prognostic tool for metastatic colorectal cancer patient management care. Clin. Cancer Res. 2016;22:3067–3077. doi: 10.1158/1078-0432.CCR-15-0297. [DOI] [PubMed] [Google Scholar]
- 25.Lapin M., Oltedal S., Tjensvoll K., Buhl T., Smaaland R., Garresori H., Javle M., Glenjen N.I., Abelseth B.K., Gilje B., Nordgård O. Fragment size and level of cell-free DNA provide prognostic information in patients with advanced pancreatic cancer. J. Transl. Med. 2018;16:300. doi: 10.1186/s12967-018-1677-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mouliere F., Chandrananda D., Piskorz A.M., Moore E.K., Morris J., Ahlborn L.B., Mair R., Goranova T., Marass F., Heider K., et al. Enhanced detection of circulating tumor DNA by fragment size analysis. Sci. Transl. Med. 2018;10 doi: 10.1126/scitranslmed.aat4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cristiano S., Leal A., Phallen J., Fiksel J., Adleff V., Bruhm D.C., Jensen S.Ø., Medina J.E., Hruban C., White J.R., et al. Genome-wide cell-free DNA fragmentation in patients with cancer. Nature. 2019;570:385–389. doi: 10.1038/s41586-019-1272-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Snyder M.W., Kircher M., Hill A.J., Daza R.M., Shendure J. Cell-free DNA Comprises an in Vivo Nucleosome Footprint that Informs Its Tissues-Of-Origin. Cell. 2016;164:57–68. doi: 10.1016/j.cell.2015.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Han D.S.C., Lo Y.M.D. The Nexus of cfDNA and Nuclease Biology. Trends Genet. 2021;37:758–770. doi: 10.1016/j.tig.2021.04.005. [DOI] [PubMed] [Google Scholar]
- 30.Jiang P., Sun K., Peng W., Cheng S.H., Ni M., Yeung P.C., Heung M.M.S., Xie T., Shang H., Zhou Z., et al. Plasma DNA end-motif profiling as a fragmentomic marker in cancer, pregnancy, and transplantation. Cancer Discov. 2020;10:664–673. doi: 10.1158/2159-8290.CD-19-0622. [DOI] [PubMed] [Google Scholar]
- 31.Peneder P., Stütz A.M., Surdez D., Krumbholz M., Semper S., Chicard M., Sheffield N.C., Pierron G., Lapouble E., Tötzl M., et al. Multimodal analysis of cell-free DNA whole-genome sequencing for pediatric cancers with low mutational burden. Nat. Commun. 2021;12:1–16. doi: 10.1038/s41467-021-23445-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Adalsteinsson V.A., Ha G., Freeman S.S., Choudhury A.D., Stover D.G., Parsons H.A., Gydush G., Reed S.C., Rotem D., Rhoades J., et al. Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat. Commun. 2017;8:1324. doi: 10.1038/s41467-017-00965-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maron S.B., Chase L.M., Lomnicki S., Kochanny S., Moore K.L., Joshi S.S., Landron S., Johnson J., Kiedrowski L.A., Nagy R.J., et al. Circulating tumor DNA sequencing analysis of gastroesophageal adenocarcinoma. Clin. Cancer Res. 2019;25:7098–7112. doi: 10.1158/1078-0432.CCR-19-1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Spoor J., Eyck B.M., Atmodimedjo P.N., Jansen M.P.H.M., Helmijr J.C.A., Martens J.W.M., Van Der Wilk B.J., van Lanschot J.J.B., Dinjens W.N.M., Dinjens W.N.M. Liquid biopsy in esophageal cancer: a case report of false-positive circulating tumor DNA detection due to clonal hematopoiesis. Ann. Transl. Med. 2021;9:1264. doi: 10.21037/atm-21-525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van den Ende T., de Clercq N.C., van Berge Henegouwen M.I., Gisbertz S.S., Geijsen E.D., Verhoeven R.H.A., Meijer S.L., Schokker S., Dings M.P.G., Bergman J.J.G.H.M., et al. Neoadjuvant Chemoradiotherapy Combined with Atezolizumab for Resectable Esophageal Adenocarcinoma: A Single-arm Phase II Feasibility Trial (PERFECT) Clin. Cancer Res. 2021;27:3351–3359. doi: 10.1158/1078-0432.CCR-20-4443. [DOI] [PubMed] [Google Scholar]
- 36.Zviran A., Schulman R.C., Shah M., Hill S.T.K., Deochand S., Khamnei C.C., Maloney D., Patel K., Liao W., Widman A.J., et al. Genome-wide cell-free DNA mutational integration enables ultra-sensitive cancer monitoring. Nat. Med. 2020;26:1114–1124. doi: 10.1038/s41591-020-0915-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wan J.C.M., Heider K., Gale D., Murphy S., Fisher E., Mouliere F., Ruiz-Valdepenas A., Santonja A., Morris J., Chandrananda D., et al. ctDNA monitoring using patient-specific sequencing and integration of variant reads. Sci. Transl. Med. 2020;12 doi: 10.1126/scitranslmed.aaz8084. [DOI] [PubMed] [Google Scholar]
- 38.Cohen J.D., Li L., Wang Y., Thoburn C., Afsari B., Danilova L., Douville C., Javed A.A., Wong F., Mattox A., et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science. 2018;359:926–930. doi: 10.1126/science.aar3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wan N., Weinberg D., Liu T.-Y., Niehaus K., Ariazi E.A., Delubac D., Kannan A., White B., Bailey M., Bertin M., et al. Machine learning enables detection of early-stage colorectal cancer by whole-genome sequencing of plasma cell-free DNA. BMC Cancer. 2019;19:832. doi: 10.1186/s12885-019-6003-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mouliere F., Smith C.G., Heider K., Su J., van der Pol Y., Thompson M., Morris J., Wan J.C.M., Chandrananda D., Hadfield J., et al. Fragmentation patterns and personalized sequencing of cell-free DNA in urine and plasma of glioma patients. EMBO Mol. Med. 2021;13 doi: 10.15252/emmm.202012881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jiang P., Xie T., Ding S.C., Zhou Z., Cheng S.H., Chan R.W.Y., Lee W.S., Peng W., Wong J., Wong V.W.S., et al. Detection and characterization of jagged ends of double-stranded DNA in plasma. Genome Res. 2020;30:1144–1153. doi: 10.1101/gr.261396.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Markus H., Zhao J., Contente-Cuomo T., Stephens M.D., Raupach E., Odenheimer-Bergman A., Connor S., McDonald B.R., Moore B., Hutchins E., et al. Analysis of recurrently protected genomic regions in cell-free DNA found in urine. Sci. Transl. Med. 2021;13 doi: 10.1126/scitranslmed.aaz3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stover D.G., Parsons H.A., Ha G., Freeman S.S., Barry W.T., Guo H., Choudhury A.D., Gydush G., Reed S.C., Rhoades J., et al. Association of Cell-Free DNA Tumor Fraction and Somatic Copy Number Alterations With Survival in Metastatic Triple-Negative Breast Cancer. J. Clin. Oncol. 2018;36:543–553. doi: 10.1200/JCO.2017.76.0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ulz P., Belic J., Graf R., Auer M., Lafer I., Fischereder K., Webersinke G., Pummer K., Augustin H., Pichler M., et al. Whole-genome plasma sequencing reveals focal amplifications as a driving force in metastatic prostate cancer. Nat. Commun. 2016;7 doi: 10.1038/ncomms12008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.De Sarkar N., Patton R.D., Doebley A.L., Hanratty B., Adil M., Kreitzman A.J., Sarthy J.F., Ko M., Brahma S., Meers M.P., et al. Nucleosome Patterns in Circulating Tumor DNA Reveal Transcriptional Regulation of Advanced Prostate Cancer Phenotypes. Cancer Discov. 2023;13:632–653. doi: 10.1158/2159-8290.CD-22-0692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rao S., Han A.L., Zukowski A., Kopin E., Sartorius C.A., Kabos P., Ramachandran S. Transcription factor-nucleosome dynamics from plasma cfDNA identifies ER-driven states in breast cancer. Sci. Adv. 2022;8:4358. doi: 10.1126/sciadv.abm4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mair R., Mouliere F., Smith C.G., Chandrananda D., Gale D., Marass F., Tsui D.W.Y., Massie C.E., Wright A.J., Watts C., et al. Measurement of plasma cell-free mitochondrial tumor DNA improves detection of glioblastoma in patient-derived orthotopic xenograft models. Cancer Res. 2019;79:220–230. doi: 10.1158/0008-5472.CAN-18-0074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thierry A.R., Mouliere F., Gongora C., Ollier J., Robert B., Ychou M., del Rio M., Molina F. Origin and quantification of circulating DNA in mice with human colorectal cancer xenografts. Nucleic Acids Res. 2010;38:6159–6175. doi: 10.1093/nar/gkq421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Underhill H.R., Kitzman J.O., Hellwig S., Welker N.C., Daza R., Baker D.N., Gligorich K.M., Rostomily R.C., Bronner M.P., Shendure J. Fragment Length of Circulating Tumor DNA. PLoS Genet. 2016;12 doi: 10.1371/journal.pgen.1006162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Boehm K.M., Khosravi P., Vanguri R., Gao J., Shah S.P. Harnessing multimodal data integration to advance precision oncology. Nat. Rev. Cancer. 2021;22:114–126. doi: 10.1038/s41568-021-00408-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van der Pol Y., Moldovan N., Verkuijlen S., Ramaker J., Boers D., Onstenk W., de Rooij J., Bahce I., Pegtel D.M., Mouliere F. The Effect of Preanalytical and Physiological Variables on Cell-Free DNA Fragmentation. Clin. Chem. 2022;68:803–813. doi: 10.1093/clinchem/hvac029. [DOI] [PubMed] [Google Scholar]
- 52.Burnham P., Kim M.S., Agbor-Enoh S., Luikart H., Valantine H.A., Khush K.K., De Vlaminck I. Single-stranded DNA library preparation uncovers the origin and diversity of ultrashort cell-free DNA in plasma. Sci. Rep. 2016;6 doi: 10.1038/srep27859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hudecova I., Smith C.G., Hänsel-Hertsch R., Chilamakuri C.S., Morris J.A., Vijayaraghavan A., Heider K., Chandrananda D., Cooper W.N., Gale D., et al. Characteristics, origin, and potential for cancer diagnostics of ultrashort plasma cell-free DNA. Genome Res. 2022;32:215–227. doi: 10.1101/gr.275691.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Beagan J.J., Drees E.E.E., Stathi P., Eijk P.P., Meulenbroeks L., Kessler F., Middeldorp J.M., Pegtel D.M., Zijlstra J.M., Sie D., et al. PCR-free shallow whole genome sequencing for chromosomal copy number detection from plasma of cancer patients is an efficient alternative to the conventional PCR-based approach. J. Mol. Diagn. 2021;23:1553–1563. doi: 10.1016/j.jmoldx.2021.08.008. [DOI] [PubMed] [Google Scholar]
- 55.Im Y.R., Tsui D.W.Y., Diaz L.A., Wan J.C.M. Next-Generation Liquid Biopsies: Embracing Data Science in Oncology. Trends Cancer. 2021;7:283–292. doi: 10.1016/j.trecan.2020.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Heitzer E., Auinger L., Speicher M.R. How Dead Cells Inform about the Living. 2020. Cell-Free DNA and Apoptosis. Preprint, [DOI] [PubMed] [Google Scholar]
- 57.Rostami A., Lambie M., Yu C.W., Stambolic V., Waldron J.N., Bratman S.V. Senescence, Necrosis, and Apoptosis Govern Circulating Cell-free DNA Release Kinetics. Cell Rep. 2020;31:107830. doi: 10.1016/j.celrep.2020.107830. [DOI] [PubMed] [Google Scholar]
- 58.Qi T., Pan M., Shi H., Wang L., Bai Y., Ge Q. Cell-Free DNA Fragmentomics: The Novel Promising Biomarker. Int. J. Mol. Sci. 2023;24 doi: 10.3390/ijms24021503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Budhraja K.K., McDonald B.R., Stephens M.D., Contente-Cuomo T., Markus H., Farooq M., Favaro P.F., Connor S., Byron S.A., Egan J.B., et al. Genome-wide analysis of aberrant position and sequence of plasma DNA fragment ends in patients with cancer. Sci. Transl. Med. 2023;15:eabm6863. doi: 10.1126/scitranslmed.abm6863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chabon J.J., Hamilton E.G., Kurtz D.M., Esfahani M.S., Moding E.J., Stehr H., Schroers-Martin J., Nabet B.Y., Chen B., Chaudhuri A.A., et al. Integrating genomic features for non-invasive early lung cancer detection. Nature. 2020;580:245–251. doi: 10.1038/s41586-020-2140-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wan J.C.M., Heider K., Gale D., Murphy S., Fisher E., Mouliere F., Ruiz-Valdepenas A., Santonja A., Morris J., Chandrananda D., et al. ctDNA Monitoring to Parts Per Million Using Patient-specific Sequencing and Integration of Variant Reads. bioRxiv. 2019 doi: 10.1126/scitranslmed.aaz8084. Preprint at. [DOI] [PubMed] [Google Scholar]
- 62.van der Pol Y., Moldovan N., Ramaker J., Bootsma S., Lenos K.J., Vermeulen L., Sandhu S., Bahce I., Pegtel D.M., Wong S.Q., et al. The landscape of cell-free mitochondrial DNA in liquid biopsy for cancer detection. Genome Biol. 2023;24:229. doi: 10.1186/s13059-023-03074-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Johnson W.E., Li C., Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
P-values were calculated using the two-sided Mann-Whitney U test.
Data Availability Statement
The cfDNA sequencing data have been deposited in the European Genome-Phenome Archive (EGA) and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
The code for the FrEIA tool and of the machine learning pipeline has been deposited on Github and is publicly available as of the date of publication. Links to the code are listed in the key resources table.