

Summary
Leveraging protein structural information to evaluate pathogenicity has been hindered by the scarcity of experimentally determined 3D protein. With the aid of AlphaFold2 predictions, we developed the structure-informed genetic missense mutation assessor (SIGMA) to predict missense variant pathogenicity. In comparison with existing predictors across labeled variant datasets and experimental datasets, SIGMA demonstrates superior performance in predicting missense variant pathogenicity (AUC = 0.933). We found that the relative solvent accessibility of the mutated residue contributed greatly to the predictive ability of SIGMA. We further explored combining SIGMA with other top-tier predictors to create SIGMA+, proving highly effective for variant pathogenicity prediction (AUC = 0.966). To facilitate the application of SIGMA, we pre-computed SIGMA scores for over 48 million possible missense variants across 3,454 disease-associated genes and developed an interactive online platform (https://www.sigma-pred.org/). Overall, by leveraging protein structure information, SIGMA offers an accurate structure-based approach to evaluating the pathogenicity of missense variants.
Keywords: variant effect predictor, protein structure, machine learning, missense variant
Graphical abstract
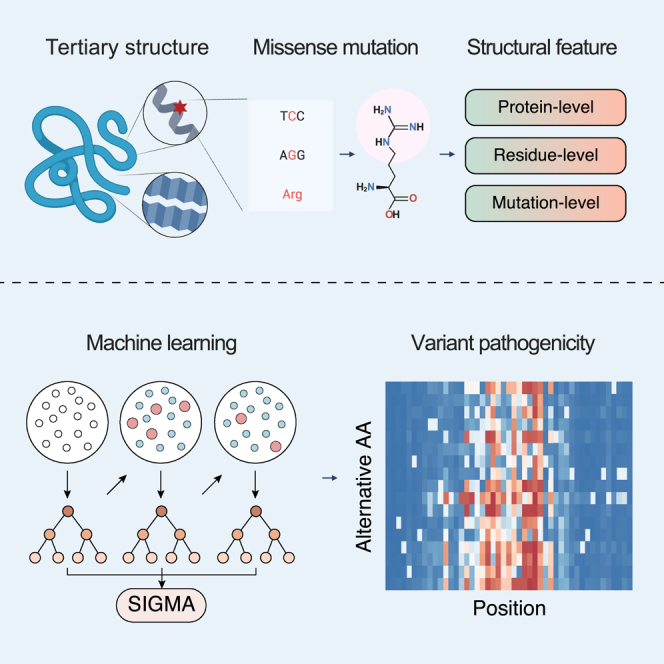
Highlights
-
•
Protein structural features are significantly associated with variant pathogenicity
-
•
SIGMA leverages structural features to improve pathogenic variant prediction
-
•
An interactive website provides access to pre-computed scores for millions of variants
Motivation
The task of assessing the pathogenicity of missense variants is key to interpreting genetic data. A promising strategy involves evaluating variant effects within the context of protein structure. However, the scarcity of known 3D protein structures has hindered the exploitation of structural information, as protein structure is critically important for ensuring proper molecular function. The advent of AlphaFold2 has begun to change this situation by enabling extensive, accurate predictions of 3D structures. Here, we introduce SIGMA, which utilizes AlphaFold2 predictions to evaluate the effects of missense variants in the context of predicted protein structures.
Zhao et al. present a machine learning method—SIGMA—to predict pathogenic missense mutations using features extracted from protein structures predicted by AlphaFold2. Through benchmarking, they demonstrate performance improvements over existing predictors, and they also provide a website to query pre-computed pathogenicity predictions and to download the entire dataset.
Introduction
Interpreting human genetic variants relies heavily on predicting their impacts on the protein products. For example, protein-truncating variants (e.g., nonsense variants and frame-shifting variants) often lead to loss of function of the protein and are likely to be classified as pathogenic variants. Variants that affect mRNA splicing could lead to the loss of function or altered function of the resulting protein. In contrast, missense variants, which are the majority of clinically relevant variants, produce highly variable effects on protein structure and function, and only a small proportion of them are pathogenic.1,2 There are a huge number of potential missense variants (∼76 million) in the human exome. Recently, many community efforts have been made to characterize these missense mutations experimentally in a high-throughput manner, commonly known as deep mutational scanning.3,4,5 However, this approach is extremely time consuming and remains constrained in its scope. Notably, most proteins do not have such measurements available, leaving a significant portion of variants whose significance remains uncertain.
In order to assess the pathogenicity of missense variants in silico, a number of state-of-the-art variant effect predictors (VEPs) have been developed, such as SIFT, REVEL, and EVE.6,7,8 Features commonly used to train the VEPs include variant frequency, evolutionary conservation, and the physiochemical properties of the amino acids.8,9,10 In addition to using a single source of information, integrating the results of multiple VEPs can significantly enhance the performance.11 Given the close relation between the structure and function of proteins, the structural context of a variant represents promising information independent of variant frequency and evolutionary conservation.12,13 However, the scarcity of high-resolution 3D protein structures has hindered the development of VEPs that exploit protein structural information.14
Recent advances in computational technology have provided breakthroughs in protein structure modeling.15,16,17 Among the most successful achievements, AlphaFold2 predicts 3D protein structures with near-experimental accuracy and has expanded the structural coverage of the human proteome from 17% to 98.5%.18,19 The extensive and accurate structural information has an unprecedented potential to aid variant effect prediction.
This study aimed to assess the pathogenicity of missense variants using structural information derived from predicted protein 3D structures. A machine learning model, structure-informed genetic missense mutation assessor (SIGMA), was trained on high-quality variant data labeled with clinical significance. We assembled the outputs of deep mutational scanning (DMS) experiments as an independent dataset to benchmark the performance of VEPs. We also explored the potential application of combining SIGMA with other VEPs. For easy access, we pre-computed SIGMA scores for all possible missense variants in the disease-associated genes and accommodated them in an interactive online platform (https://www.sigma-pred.org/).
Results
Structure-informed features are associated with variant pathogenicity
The design and workflow of our study are shown in Figure 1. A total of 27,165 benign and 22,957 pathogenic missense variants were retrieved from the gnomAD and the ClinVar databases (Table S1). In addition, 27,928 variants in six proteins systematically characterized by DMS experiments were included to independently assess the VEPs. For each variant, we extracted 57 features derived from the 3D protein structures predicted by AlphaFold219 (Table S2). Of the 57 features included, the variant pathogenicity was most significantly associated with two of them: the relative solvent accessibility (RSA) and the free energy difference (ΔΔG) between the wild-type and mutant proteins (Figure S1; Table S2). The RSA of pathogenic variants was significantly lower compared to that of benign variants (p = 2.89e–2,276, Wald test; Figure 2A), consistent with the fact that most proteins are less tolerant of buried mutations than exposed mutations.20 ΔΔG measures the effect of a single amino acid substitution on protein stability. Our findings, showing that pathogenic variants are often associated with more substantial changes in protein stability than benign variants (p = 6.99e–1,121, Wald test; Figure 2B), are in line with previous research using FoldX and Rosetta. This observation further emphasizes the crucial role of protein stability in the pathogenicity of missense variants. By affecting the protein’s stability, these variants can alter the protein’s function, leading to potential pathogenic consequences. It is, therefore, essential to consider the protein stability changes when assessing the potential pathogenicity of missense variants.
Figure 1.

Schematic illustration of the study design
Step 1: data collection of variant datasets. A total of 27,165 benign and 22,957 pathogenic missense variants were retrieved from gnomAD and ClinVar databases as the “gold standard.” In addition, 27,928 variants in six proteins systematically characterized by DMS experiments were included to provide an independent measurement for assessing predictors. Step 2: extraction of structure-informed features. For each variant, 57 features were extracted based on protein 3D structure predictions. Step 3: development and evaluation of SIGMA. The gold standard dataset was sorted in chronological order and divided with 80% used for training and 20% for testing (see STAR Methods). Using the GBM algorithm, we developed SIGMA to predict the variant pathogenicity. ROC analysis and correlation analysis were used to evaluate the performance of SIGMA on the test dataset and the DMS dataset, respectively. DMS, deep mutational scanning; 3D, three-dimensional; SIGMA, structure-informed genetic missense mutation assessor; GBM, gradient boosting machine; ROC, receiver operating characteristic; RSA, relative solvent accessibility; ΔΔG, the unfolding free energy difference between the wild-type and mutant protein; SS-bond: disulfide bond.
Figure 2.

The associations of structure-informed features with variant pathogenicity
(A and B) The differences of RSA (A) and ΔΔG (B) between 27,165 benign and 22,957 pathogenic variants (Mann-Whitney U test). The violin plot depicts the distribution of specific features using density curves. The boxes show the median ± one quartile. Whiskers extend 1.5 times the interquartile range.
(C) Mutations that broke the disulfide bond (blue = benign, red = pathogenic).
(D) Forest plot for effect sizes of mutations with eight secondary structure types assigned by the DSSP program. Center values represent OR, and error bars represent the 95% CI for the OR. p values were calculated by using Pearson’s chi-squared test. RSA, relative solvent accessibility; ΔΔG, the unfolding free energy difference between the wild-type and mutant protein; OR, odds ratio; SS-bond, disulfide bond; DSSP codes: C, loop; H, alpha helix; E, beta sheet; S, bend; G, 3-helix; I, 5-helix; T, hydrogen-bonded turn; B, residue in isolated beta bridge.
As expected, the breakage of disulfide bonds had the highest positive predictive value among all structural features (odds ratio [OR] = 93.8, 95% confidence interval [CI] = 44.5–198, p = 3.23e–138, Pearson’s chi-squared test; Figure 2C). Almost all (98.72%) missense variants that disrupted a disulfide bond were pathogenic, supporting the essential role of disulfide bonds in protein function.
The association between the type of secondary structure where a variant was located and its pathogenicity is also consistent with prior knowledge. Mutations in loops or irregular stretches (Dictionary of Secondary Structure of Proteins [DSSP]-C) of a protein tended to be benign (OR = 0.32, 95% CI = 0.31–0.34, p = 2.17e-714, Pearson’s chi-squared test). In contrast, mutations in regular secondary structures tended to be pathogenic, especially those in alpha helices (DSSP-H; OR = 1.73, 95% CI = 1.66–1.79, p = 1.48e–173, Pearson’s chi-squared test) or beta-sheets (DSSP-E; OR = 1.97, 95% CI = 1.87–2.08, p = 1.55e–141, Pearson’s chi-squared test; Figure 2D; Table S2).
SIGMA outperformed existing VEPs in predictive ability
Using the training dataset of 40,195 variants with structural features, we developed SIGMA, a gradient boosting machine (GBM)-based model to predict the pathogenicity of missense variants. The quantitative SIGMA scores for potential pathogenicity of the variants (from 0 to 1) showed a bimodal distribution (Figures 3A and S2A), suggesting a strong discriminative ability of SIGMA. For the training set, an area under the receiver operating characteristic (ROC) curve (AUC) of 0.944 (95% CI = 0.942–0.946; Figures 3B and S3A) was yielded with out-of-fold predictions. Consistently, high prediction accuracy was obtained on the test set (AUC = 0.933, 95% CI = 0.928–0.938; Figure 3C). When compared to 16 individual predictors, SIGMA performed better than all the individual predictors, and its performance was significantly better than 15 out of the 16 predictors, whose AUCs ranged from 0.779 (FATHMM) to 0.929 (MutPred) (Figure 3D).
Figure 3.

The performance of SIGMA
(A) The distribution of SIGMA scores for 20,047 pathogenic (red) and 20,148 benign (blue) variants in the training set. SIGMA scores were calculated using out-of-fold predictions.
(B and C) Receiver operating characteristic (ROC) curves of SIGMA scores in the training set (B) and test set (C).
(D) Comparison of area under the ROC curves (AUCs) of SIGMA and 16 individual variant effect predictors (VEPs) using the test set.
(E) Rank of correlation between individual VEPs and deep mutational scanning (DMS) measurements. Spearman’s correlation was calculated between functional scores from DMS experiments and prediction scores from SIGMA as well as 17 individual VEPs. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001.
To limit the data circularity that may exaggerate the performance of predictors, the DMS dataset for six proteins was assembled as an independent test dataset. As a result, SIGMA achieved the highest correlation with DMS dataset among all individual predictors (overall rho = 0.419, Spearman’s correlation analysis), especially for BRCA1 (rho = 0.286), PTEN (rho = 0.519), and HRAS (rho = 0.404; Figures 3E and S4A; Table S3). The closest competitor was DEOGEN2 (overall rho = 0.387, Spearman’s correlation analysis; Figure 3E), which incorporated extensive heterogeneous information. Remarkably, the performance ranking of the recently developed predictor, EVE, improved from number six on the labeled dataset to number three on the DMS dataset (Figure 3E). The robustness of EVE, an unsupervised deep learning tool, could likely account for its enhanced performance when assessed using independent DMS datasets. To conduct a more in-depth investigation during the benchmarking against other methods, we specifically focused on the BRCA1 DMS dataset, which has predefined threshold (considering variants with scores greater than one as pathogenic) calibrated in the original publication.21 Assessing the performance of predictors on the BRCA1 DMS dataset through ROC analysis, SIGMA demonstrated superior performance with an impressive AUC of 0.911 (Figure S4C). These results underscore the proficiency and robustness of SIGMA in predicting pathogenicity, particularly when compared to existing methods.
Systematic integration of structural features optimized the performance of SIGMA
Through a feature importance analysis of the SIGMA model, we found that the residue-level feature, RSA, contributed most to the discriminative ability of SIGMA (Figure 4A; Table S2), followed by two mutation-level features (ΔΔG and Δvan der Waals clashes). These findings were consistent with our initial feature-level analysis. Intriguingly, seven of the ten most important features were protein-level features, demonstrating the essential role of wild-type protein stability in predicting variant pathogenicity.
Figure 4.

The importance of RSA in predicting variant pathogenicity
(A) The top ten most important features that contributed to the discriminative ability of SIGMA.
(B and C) Gene set enrichment analysis plot for the ion channel genes (B) and the collagen family genes (C). The predictive ability of the RSA for the genes with at least ten pathogenic and ten benign variants. RSA, relative solvent accessibility; ΔΔG, the unfolding free energy difference between the wild-type and mutant protein; ΔG, the unfolding free energy of the wild-type protein; VdW, van der Waals.
Notably, in 200 genes with at least ten pathogenic and ten benign variants for evaluation, it was found that RSA alone achieved an accuracy of greater than 0.8 in 72 genes, whereas SIGMA achieved the same level of accuracy in 147 genes (Table S4). RSA had the highest classification accuracy for the ion channel genes (p = 8.67e−04; Figure 4B; Table S5). Thus, while SIGMA is more effective overall, RSA alone still has some utility in predicting variant pathogenicity in specific genes.
However, using RSA alone resulted in unsatisfactory performance for 21 out of 200 genes, with an accuracy of less than 0.5, which was enriched in the collagen gene family (p = 1.99e−06; Figure 4C; Table S5). Misclassification of mutations in the triple-helix domains of collagen genes by RSA could be attributed to the use of structures of collagen monomers rather than triple helices for RSA value calculations. Despite the limitations of RSA, SIGMA was able to accurately predict the pathogenicity of variants in collagen genes by incorporating additional structural features such as glycine-associated features. An illustrative instance is the poor performance of RSA for mutations in the COL6A2 protein (accuracy = 0.3, specificity = 0.45, sensitivity = 0.18), which were accurately classified (accuracy = 0.91, specificity = 0.79, sensitivity = 1) by SIGMA (Table S5). In summary, by integrating multiple structural features, particularly RSA, SIGMA achieved superior performance.
Application of SIGMA in variant interpretation
Based on the predictions of SIGMA in the training set, we examined its performance using different thresholds and found an optimal threshold of 0.498 for determining the pathogenicity of variants, yielding an accuracy of 85%, a sensitivity of 88%, and a specificity of 83% in the test dataset (Table S6).
We further compared the gene-level performance of SIGMA and two state-of-art VEPs (EVE and REVEL) for every gene with at least ten pathogenic variants and ten benign variants. SIGMA performed exceptionally well in predicting variant pathogenicity in certain genes that are difficult to assess using other methods (i.e., EVE and REVEL) (Table S5). This may be because SIGMA relies mainly on protein 3D structure to obtain structural information, which provides a different perspective compared to other methods. One example is the TSC2 protein, a well-studied protein associated with tuberous sclerosis (MIM: 613254). Our findings indicated that SIGMA demonstrated a superior bimodal distribution of SIGMA scores for variants in the TSC2 protein compared to REVEL and EVE. The consistency between the distribution of variant pathogenicity and the RSA level emphasizes the significance of incorporating structural features to evaluate variant pathogenicity. Furthermore, we observed that the recommended cutoff values of REVEL and EVE were not suitable for the TSC2 gene, highlighting the necessity of gene-level cutoffs for these methods (Figure 5A). We performed SIGMA score calculations for all potential single amino acid substitutions in the TSC2 protein. The data also showed that mutations of buried amino acids tended to be more pathogenic, but those replaced by hydrophobic amino acids were predicted to be less severe (Figure 5B). The results revealed that SIGMA scores were capable of distinguishing between different amino acid substitutions of buried residues by incorporating additional features.
Figure 5.

Predictions for variants in TSC2 protein
(A) The SIGMA, EVE, and REVEL scores for all labeled variants in TSC2 protein.
(B) The heatmap for SIGMA scores of all possible amino acid substitutions in TSC2 protein.
Sequence conservation, population frequency, and regional missense constraint have been demonstrated to be predictive of variant pathogenicity.8,22,23 To investigate the relationship of SIGMA with these well-established predictors, and to analyze the association between structural information and other aspects of information sources, we examined the distribution of SIGMA scores in various subgroups of variants. These subgroups were classified according to the degree of sequence conservation, population frequency, and regional missense mutational constraint. We found that variants with high SIGMA scores were prone to occur in highly conserved regions (Figure 6A), to have low population frequencies (Figure 6B), and to be present in regions intolerant to missense variations (Figure 6C). Moreover, SIGMA distinguished well between pathogenic and benign variants in each subgroup (Figures 6A–6C). These results implied that SIGMA might serve as a valuable addition to the current predictors by providing a separate, independent source of information, thus enhancing the ability to predict the variant pathogenicity.
Figure 6.

The application of SIGMA in variant interpretation
(A–C) The distribution of SIGMA scores in various subgroups of variants classified by the degree of sequence conservation (A), population frequency (B), and missense mutational constraint (C). The boxes show the median ± one quartile. Whiskers extend 1.5 times the interquartile range. The Mann-Whitney U test was performed between the SIGMA scores of benign variants and pathogenic variants. A two-sided p < 0.05 is considered a statistically significant difference.
(D) The heatmap shows a pathogenicity matrix for each type of amino acid substitution. The pathogenicity for each type of amino acid substitution was measured using mean SIGMA scores based on results from computational saturation mutagenesis in all disease-associated genes. The colors range from yellow (low pathogenicity) to black (high pathogenicity). ∗∗∗∗p < 0.0001. AA, amino acid.
We also determined the predictive ability of SIGMA on gain-of-function (GoF) and loss-of-function (LoF) variants since these variants may have different structural properties. SIGMA performed better in the LoF group (recall rate = 86.43%) than in the GoF group (recall rate = 74.09%; p = 3.01e−05, Pearson’s chi-squared test; Figure S5), probably because GoF variants often affect protein-protein interaction sites, which is not captured by our features.
In silico saturation mutagenesis by SIGMA
To facilitate the use of SIGMA by clinicians and researchers, we have pre-computed SIGMA scores for all possible missense variants (more than 48 million variants) across disease-associated genes (n = 3,454) that have at least one qualified pathogenic variant in ClinVar database, and we developed an interactive online platform to disseminate them (https://sigma-pred.org/). Based on the results from computational saturation mutagenesis in all disease-associated genes, we determined a pathogenicity matrix by calculating mean SIGMA scores for each type of substitution. On average, variants were more likely to be pathogenic when the wild-type residues were replaced by proline or when hydrophobic residues were changed to hydrophilic residues (Figures 6D and S6).
Combining SIGMA with other predictors further enhanced its performance
When we combined SIGMA with the top individual predictors (i.e., DEOGEN2, EVE, PROVEAN, and MutPred) ranked by performance on the DMS dataset, various combinations of these five predictors enhanced the predictive performance (AUC ranged from 0.942–0.954; Figure 7A). SIGMA+, the combination of all five predictors, significantly discriminated between benign and pathogenic variants (Figure S2B and S2C), with the highest accuracy on the training set (AUC = 0.96, 95% CI = 0.957–0.964, p < 2.2e−16; Figures 7A, 7B, and S3B) and the test set (AUC = 0.966, 95% CI = 0.957–0.974, p < 2.2e−16; Figure 7C). Importantly, SIGMA made the most contribution to the combination, whereas DEOGEN2 contributed the least (Figure S7).
Figure 7.

The performance of SIGMA+
(A) Combination of SIGMA with four other predictors (i.e., DEOGEN2, EVE, PROVEAN, and MutPred).
(B and C) Receiver operating characteristic (ROC) curves of SIGMA+ scores in the training set (B) and the test set (C).
(D) Comparison of AUCs of SIGMA+ with 11 meta-predictors using the test set.
(E) Rank of correlation between VEPs and deep mutational scanning (DMS) measurements. Spearman’s correlation was calculated between functional scores from DMS experiments and prediction scores from SIGMA+ as well as 11 meta-predictors. AUC, the area under the ROC curve; SPE, specificity; SEN, sensitivity. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001.
We also compared SIGMA+ to 11 meta-predictors. Overall, SIGMA+ outperformed all other meta-predictors, whose AUC values ranged from 0.81 (DANN) to 0.95 (VEST4) (Figure 7D). An assessment using the independent DMS dataset also supported the superior performance of SIGMA+ (overall rho = 0.421, Spearman’s correlation analysis). Notably, SIGMA+ achieved the highest correlation with DMS data for all proteins except P53, for which MetaLR performed best (Figures 7E and S4B; Table S3). When assessing the performance of predictors on the BRCA1 DMS dataset through ROC analysis, SIGMA+ demonstrated superior performance with an AUC of 0.87 (Figure S4D). Therefore, digging into additional sources of information may be more advantageous than iteratively constructing meta-predictors using an increasing number of existing predictors.
Discussion
In this study, we developed SIGMA, a machine learning model that predicts the pathogenicity of missense variants based on structural information. We demonstrate the superior performance of SIGMA and SIGMA+ compared to existing VEPs when evaluated on a labeled variant dataset and an experimental dataset.
The protein structural features used to train SIGMA are mainly based on empirical knowledge that has long been used to evaluate the pathogenicity of amino acid changes. For example, a missense variant in a cysteine residue could destroy a disulfide bond and disrupt protein stability,24 or a missense variant affecting a residue in defined secondary structures (such as alpha helices and beta sheets) is more likely to be pathogenic.13 However, in the absence of a high-resolution protein structure, it is difficult to assess such protein-level features for clinical variant interpretation accurately.13,14 The many accurate protein structural models provided by AlphaFold2 enabled us to extract a wide range of features for most proteins,25 which would not be possible in years if relying on crystallographic protein structures.
The prominent contributory feature in SIGMA is RSA, which measures the extent of burial or exposure of the residue in the 3D structure that is essential for the folding and stability of the protein. Thus, the variant’s position, regardless of the specific amino acid change, is a strong predictor of variant pathogenicity. This is in line with a recent study that identified significant positional clustering of pathogenic variants.26 By identifying the key regions that are vulnerable to genetic variants (i.e., regions with a low RSA), SIGMA provides additional insights into the mechanisms that underlie this clustering. In addition, structural features calculated from the 3D structure could better reflect the spatial clustering of pathogenic variants compared with the reported positional clustering, which was based on a linear protein model.26 However, a single structural feature is insufficient to predict variant pathogenicity. Although RSA contributed most to the discriminative ability of SIGMA, its predictive accuracy for collagen family genes was limited possibly due to the distinct structural features of collagen triple helices. However, the implementation of SIGMA was successful in correcting the prediction errors of RSA. Therefore, the systematic integration of all structural features by the machine learning algorithm optimized the performance of SIGMA.
The presence of data circularity may exaggerate the performance of predictors and thus hinder the performance evaluation of VEPs.27 Type I circularity occurs when variants in the training phase are reused in the testing phase, which is especially common for meta-predictors. To avoid type I circularity, we sorted the variation data in chronological order, and more recent variants, which were entirely naive for existing predictors, were used as the test set. Besides, we used an experimental dataset assembled from the outputs of DMS studies to provide an independent measurement for benchmarking the performances of various predictors, which also limits the data circularity.
While AlphaFold2 has revolutionized the field of protein structure prediction by increasing the coverage of the human proteome to an impressive 98.5%, it is important to note that a non-negligible portion of these predicted structures are still of low confidence. AlphaFold2 is known to produce low-confidence predictions for regions that are intrinsically disordered or unstructured in isolation. Despite this limitation, our study found that variants located in these regions without secondary structure are more likely to be benign. Therefore, the impact of low-confidence regions on variant prediction is limited. Additionally, for areas in which AlphaFold may not provide reliable predictions, including regions with secondary structures, we recommend that users of our SIGMA tool should consider not just the predicted pathogenicity of variants but also the pLDDT confidence level provided by AlphaFold2. This more integrative approach is expected to help users make more informed decisions, especially in cases where the AlphaFold2 predictions might be of lower confidence. Overall, while AlphaFold2 represents a significant advancement in the field of protein structure prediction, further improvements are needed to accurately predict the structure of all proteins, including those that are intrinsically disordered or unstructured.
In conclusion, we provide an independent source of information for predicting the pathogenicity of missense variants by leveraging protein structure information. We expect SIGMA and SIGMA+ to aid in interpreting genetic and genomic data and to contribute to the future development of more powerful meta-predictors.
Limitations of the study
Our study has several limitations. We were unable to model the mutant protein structure based on the current AlphaFold2 algorithm, which precluded an accurate characterization of the structural alterations caused by the genetic mutations.28,29 In addition, SIGMA did not incorporate protein-protein interactions, which are often involved in the mechanism of GoF variants.30 Currently, we focus on the missense variants, which account for most of the overall genetic variations seen in humans. Other variation types are more complicated and require more accurate protein structure predictions. Furthermore, state-of-the-art algorithms, typically supervised and reliant on clinical labels, often exhibit inflated accuracy in real-world prediction scenarios. Recent developments in this field have shown that unsupervised models utilizing sequence data alone, such as ESM-1v31 and EVE,8 have achieved notable success in predicting the effects of variants. These models have been proved to be fundamentally generalizable as they avoid learning from clinical labels. This highlights the potential of unsupervised learning on protein 3D structure data to provide more accurate and robust predictions. Thus, building a comprehensive and unsupervised predictor based on more accurate protein structure prediction has become one of the priorities in our future studies.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| 3D protein structures | AlphaFold2 | https://alphafold.ebi.ac.uk/ |
| Variant data | ClinVar | https://www.ncbi.nlm.nih.gov/clinvar/ |
| Variant data | gnomAD | https://gnomad.broadinstitute.org/ |
| Variant annotation | dbNSFP | http://database.liulab.science/dbNSFP |
| Deep mutational scanning data | MaveDB | https://www.mavedb.org/#/ |
| Gain-of-function and loss-of-function pathogenic missense variants | GOF/LOF | https://itanlab.shinyapps.io/goflof/ |
| Pre-computed SIGMA scores | This paper | https://sigma-pred.org/ |
| Software and algorithms | ||
| Ensembl Variant Effect Predictor program | McLaren et al.32 | https://useast.ensembl.org/info/docs/tools/vep/index.html |
| SpliceAI | Jaganathan et al.33 | https://github.com/Illumina/SpliceAI |
| FoldX | Schymkowitz et al.34 | https://foldxsuite.crg.eu/ |
| DSSP (Dictionary of Secondary Structure of Proteins) program | Kabsch et al.35 | http://www.csb.yale.edu/userguides/databases/dssp/dssp_man.html |
| ProteinVolume | Chen et al.36 | https://gmlab.bio.rpi.edu/PVolume.php |
| R | The R Project for Statistical Computing | https://www.r-project.org/ |
| R scripts for data analysis | This paper |
https://github.com/zhq921/SIGMA https://doi.org/10.5281/zenodo.10373120 |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Nan Wu (dr.wunan@pumch.cn).
Materials availability
This study did not generate any new reagents and is based on in silico computations relying on publicly available data.
Data and code availability
-
•
Data curated or generated here can be found in the supplementary tables. Publicly available datasets used are listed in the key resources table. The SIGMA scores for over 48 million possible missense variants across 3,454 disease-associated genes have been deposited at https://sigma-pred.org/.
-
•
The original code has been deposited at github and is publicly available as of the date of publication. DOIs are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Method details
Study design
Benign/likely benign and pathogenic/likely pathogenic missense variants from the gnomAD and the ClinVar databases were obtained and split into the training and test datasets. In addition, variants in six proteins systematically characterized by DMS experiments were used as an independent test dataset. For each variant, we extracted 57 features derived from the 3D protein structures predicted by AlphaFold2. Using the gradient boosting machine algorithm, we developed the Structure-Informed Genetic Missense mutation Assessor (SIGMA) that predicts the pathogenicity of missense variants.
Training and testing datasets
The variant data were obtained from the ClinVar database (version 2021/10/30, hg38)37 and the gnomAD database (version 3.1.2, hg38).1 For ClinVar variants, we retained the single-nucleotide missense variants with a review status of at least one star (practice guideline; reviewed by expert panel; criteria provided, multiple submitters, no conflicts; criteria provided, single submitter). Variants with a clinical significance of “conflicting interpretations of pathogenicity” were excluded. The pathogenic/likely pathogenic variants were labeled as positive, while the benign/likely benign variants were labeled as negative. In addition, 193 gain-of-function (GoF) and 921 loss-of-function (LoF) pathogenic missense variants were labeled by the GOF/LOF database (https://itanlab.shinyapps.io/goflof). For the gnomAD variants, we labeled the common missense variants (with a maximum allele frequency >0.05 across all populations in gnomAD) as negative. Any variants that overlapped with the clinvar dataset were removed from the gnomAD. Variants that introduce cryptic splice sites primarily affect mRNA splicing rather than protein structure. Hence, we predicted the effect on splicing for all variants using the SpliceAI algorithm33 and excluded variants with SpliceAI scores >0.5. The final labeled dataset that included 27,165 benign (negative) and 22,957 pathogenic (positive) missense variants was considered the “gold standard” dataset.
The dataset was divided with 80% used for training and 20% for testing. The training set included ClinVar pathogenic variants last evaluated before 11/26/2020 and benign variants last evaluated before 01/14/2021, while variants evaluated after those dates were used for testing. All the gnomAD variants were added to the training dataset. The allele frequency for each variant was retrieved from the gnomAD project by the Ensembl Variant Effect Predictor program (version 104).32 The regional missense constraint for each gene was obtained from the ExAC database.
To avoid the issue of data circularity in model evaluation, we assembled an additional test dataset using the outputs of deep mutational scanning (DMS) studies, which are independent of the labeled dataset. DMS studies use high-throughput functional assays to quantify with continuous scores the functional impacts of all possible variants in proteins/protein domains.3 We obtained DMS studies archived in the MaveDB database (accession date: 12/05/2021),38 excluding data from studies that used non-human cells or imputations to generate functional scores. To evaluate the robustness of predictors’ performance, we incorporated two high-quality PTEN DMS data measuring different aspects of variant effects (One is about functional readout based on enzyme activity; the other is about protein abundance). The correlation of these two DMS studies with the predictors are consistent (Figure S8), suggesting that the performance is robust to different DMS data of the same protein. Thus, we selected the study with the broadest coverage for different DMS studies investigating the same protein. High-quality DMS studies with 28,293 variants for six proteins (BRCA1, P53, MSH2, PTEN, VKORC1, and HRAS) were retained. To avoid the presence of data circularity that may exaggerate the performance of predictors when variants in the training phase are reused in the testing phase, we have excluded DMS variants that were present in the labeled dataset or had SpliceAI scores >0.5, keeping 27,928 variants for independent assessment.
Protein structure dataset
The wild-type protein structure predictions in PDB format were retrieved from the AlphaFold Protein Structure Database (AlphaFold DB, https://alphafold.ebi.ac.uk/, accession date: 11/05/2021). All variants were mapped to the predicted protein 3D structures. Variants that could not be mapped to the structures due to inconsistent isoforms were also excluded.
Structural feature extraction
For the assessment of each variant, our methodology stands distinct from conventional methods, as it capitalizes on 3D protein structures predicted by AlphaFold2. This approach enables us to gain structural insights into the potential impact of the variant, adding a new dimension to the assessment of pathogenicity. From these 3D protein structures, we have identified and extracted 57 unique features. Among these, several features such as the relative solvent accessibility (RSA) and the free energy difference (ΔΔG) between the wild-type and mutant proteins have been previously demonstrated to hold predictive value for variant pathogenicity.39,40 In combination, these 57 features offer a comprehensive toolset for a more accurate pathogenicity prediction. These structural features from the predicted protein 3D structures can be classified into three categories.
-
(1)
Protein-level features that characterize the general properties of the wild-type protein structures included 16 thermodynamic features and three protein volume features. We used the “Stability” command of the FoldX program (version 5.0) to estimate the thermodynamic features,34 including the total unfolding energy and its 15 components such as van der Waals clash, hydrogen bond energy, sidechain entropy, etc. The void volume of proteins, i.e., the volume of the internal cavities of proteins, is also related to protein stability.41 We used the ProteinVolume software (version 1.3) to calculate the overall volume, the void volume, and the van der Waals volume of wild-type proteins.36.
-
(2)
Residue-level features that characterize the structural context of the mutated residues included eight secondary structure-related features and the relative solvent accessibility (RSA) of the mutated residues. For each mutated residue, its secondary structure was assigned using the DSSP (Dictionary of Secondary Structure of Proteins) program,35 yielding eight features corresponding to the eight secondary structure types. The RSA quantifies a residue’s exposure status, and mutations of buried residues (i.e., residues with low RSA) are more likely to be pathogenic than mutations of exposed residues (i.e., residues with high RSA).39 We calculated the RSA of the mutated residue by dividing the solvent accessible area from DSSP by the maximal possible solvent accessible surface area of the amino acid.35,42.
-
(3)
Mutation-level features that characterize the effect of variants on the protein included 16 thermodynamic features that describe the change in the protein stability after mutation and 13 features derived from empirical rules that determine whether a variant has a significant impact on the protein structure. We used the “PositionScan” command of the FoldX program to estimate the 16 thermodynamic features, including the free energy difference (ΔΔG) between the wild-type and mutant protein and its 15 components.34 The other 13 features are combinations of the structural context and the physiochemical properties of the mutated/mutant residues.
A detailed description of these features, including the rationale behind their selection, is provided in Table S2. For instance, the feature “Buried proline introduced” was incorporated as we hypothesized that substitutions within the core of a protein are generally damaging. The introduction of a proline with its uniquely restricted backbone conformation could potentially disrupt the maintenance of the wild-type protein structure, leading to harmful effects. Another feature, “Proline in alpha helix” was also included. Even though proline residues are typically unfavorable in an alpha helix, they can indeed exist in such helices. In these scenarios, the rigidity of the proline residue may be essential for protein function. Consequently, a substitution that replaces a proline residue in an alpha helix with a non-proline residue is deemed potentially damaging.
Model development and evaluation
We first normalized each feature for the variants in the training dataset using the one-hot encoding technique for dichotomous variables (e.g., disulfide bond cleavage and secondary structures) and Z score normalization for continuous variables (e.g., RSA and ΔΔG). The normalization parameters derived from the training set were used for the test datasets. After data preprocessing, a gradient boosting machine (GBM) model was used to train a classifier that distinguishes pathogenic versus benign variants. The cartesian grid search procedure was used for tuning hyperparameters (e.g., learning rate, maximum tree depth, and sample rate per tree) with a 5-fold cross-validation strategy. Various models were built using different hyperparameters. Out-of-fold predictions (i.e., predictions made on data not used to train the model) were used to estimate the generalization performance of the models. The area under the receiver operating characteristic (ROC) curve (AUC) was used to evaluate the ability of the models to discriminate between benign and pathogenic variants. All models in the grid space were sorted by the AUC metric, and the GBM model with the highest AUC in the validation set was kept as the final predictor.
To determine each feature’s contribution to the predictor’s discriminative ability, we retrieved the importance scores from boosted trees in the GBM model. The importance scores for the features were calculated by the average amount that the selected feature improved the performance measure (squared error) for all trees, and the importance scores were rescaled into a fixed range between 0 and 1.
The optimal threshold for binary classification was determined by achieving the maximum Youden index. Evaluation metrics including AUC, accuracy, sensitivity, specificity, positive predictive value, and negative predictive value were used to measure the model’s performance.
Combining SIGMA with other VEPs
To obtain a more comprehensive predictor for variant pathogenicity, we combined SIGMA with four individual VEPs with high performances, DEOGEN2, EVE, PROVEAN, and MutPred. The combined predictor (SIGMA+) was constructed using elastic-net-penalized logistic regression with a 5-fold cross-validation strategy based on the same training set.
Comparing SIGMA and SIGMA+ with in silico VEPs
To evaluate SIGMA’s performance, we collected 28 in silico VEPs (Table S7), such as EVE,8 SIFT,6 and DEOGEN2.10 The EVE score for each variant was obtained from https://evemodel.org/. The predicted results from other VEPs for each variant were retrieved from the dbNSFP database (version 4.1a).43 These VEPs fell into two categories, individual predictors (n = 17) that do not rely on other VEPs and meta-predictors (n = 11) that integrate the results of other VEPs as input features. We compared the performance of SIGMA with that of the individual predictors and the performance of SIGMA+ with that of the meta-predictors. gMVP was excluded from the performance comparison with SIGMA on the ClinVar test dataset, owing to the significant overlap of variants between the test dataset used in our study and the dataset on which gMVP was trained.
We also compared the performance of these VEPs on the DMS dataset that was independent of the labeled dataset. We calculated Spearman’s correlation coefficient between functional scores from the DMS dataset and prediction scores from VEPs for each of the six proteins with DMS data. The overall performance of a VEP was defined as the mean of the correlation coefficients of all six proteins.
Quantification and statistical analysis
The associations of the structural features described above with variant pathogenicity were assessed based on the data type. For dichotomous features, the contingency table was built, and the chi-squared test was used to determine whether there is an association between two dichotomous variables. For continuous features, the associations of features with the variant pathogenicity were examined by logistic regression analysis. The strength of the association between a feature and variant pathogenicity was quantified using the odds ratio (OR) and a 95% confidence interval (CI). All statistical analyses and data visualizations were conducted using R software (version 3.6.3) packages: h2o, caret, pROC, forestplot, ggpubr, ggsci, viridis, and cutpointr. A p value <0.05 was considered statistically significant.
Acknowledgments
We thank Beijing Ekitech Co., Ltd., for the technical support in database and data management. We thank Ms. Kexin Xu for the support in preparing the manuscript. We thank Dr. Jianzhong Su for the constructive suggestions. This research was funded by the National Key Research and Development Program of China (2022YFC2703102 to N.W.), CAMS Innovation Fund for Medical Sciences (CIFMS, 2021-I2M-1-051 to T.J.Z. and N.W. and 2021-I2M-1-052 to Z.W.), the National Natural Science Foundation of China (82072391 to N.W., 81930068 and 81772299 to Z.W., 81972037 and 82172382 to T.J.Z., and 81972132 to G.Q.), Beijing Natural Science Foundation (JQ20032 to N.W. and 7191007 to Z.W.), and the Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences (no. 2019PT320025 to N.W.).
Author contributions
Conceptualization, N.W., S.Z., P.L., H.Z., and H.D.; methodology, H.Z., H.D., Z.C., Y.L., X.C., W.W., G.L., and G.C.; investigation, H.Z., H.D., Z.C., Y.L., B.L., X.C., W.W., G.L., G.C., and Z.Z.; visualization, H.Z., H.D., S.Z., Z.C., and Y.L.; project administration, G.Q., T.J.Z., Z.W., and N.W.; writing – original draft, H.Z., N.W., S.Z., H.D., and K.X.
Declaration of interests
The authors declare no competing interests.
Published: January 10, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.crmeth.2023.100687.
Contributor Information
Terry Jianguo Zhang, Email: jgzhang_pumch@yahoo.com.
Zhihong Wu, Email: orthoscience@126.com.
Nan Wu, Email: dr.wunan@pumch.cn.
Supplemental information
References
- 1.Karczewski K.J., Francioli L.C., Tiao G., Cummings B.B., Alföldi J., Wang Q., Collins R.L., Laricchia K.M., Ganna A., Birnbaum D.P., et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–443. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Landrum M.J., Kattman B.L. ClinVar at five years: Delivering on the promise. Hum. Mutat. 2018;39:1623–1630. doi: 10.1002/humu.23641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fowler D.M., Fields S. Deep mutational scanning: A new style of protein science. Nat. Methods. 2014;11:801–807. doi: 10.1038/nmeth.3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Majithia A.R., Tsuda B., Agostini M., Gnanapradeepan K., Rice R., Peloso G., Patel K.A., Zhang X., Broekema M.F., Patterson N., et al. Prospective functional classification of all possible missense variants in PPARG. Nat. Genet. 2016;48:1570–1575. doi: 10.1038/ng.3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matreyek K.A., Starita L.M., Stephany J.J., Martin B., Chiasson M.A., Gray V.E., Kircher M., Khechaduri A., Dines J.N., Hause R.J., et al. Multiplex assessment of protein variant abundance by massively parallel sequencing. Nat. Genet. 2018;50:874–882. doi: 10.1038/s41588-018-0122-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vaser R., Adusumalli S., Leng S.N., Sikic M., Ng P.C. SIFT missense predictions for genomes. Nat. Protoc. 2016;11:1–9. doi: 10.1038/nprot.2015.123. [DOI] [PubMed] [Google Scholar]
- 7.Ioannidis N.M., Rothstein J.H., Pejaver V., Middha S., McDonnell S.K., Baheti S., Musolf A., Li Q., Holzinger E., Karyadi D., et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016;99:877–885. doi: 10.1016/j.ajhg.2016.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frazer J., Notin P., Dias M., Gomez A., Min J.K., Brock K., Gal Y., Marks D.S. Disease variant prediction with deep generative models of evolutionary data. Nature. 2021;599:91–95. doi: 10.1038/s41586-021-04043-8. [DOI] [PubMed] [Google Scholar]
- 9.Alirezaie N., Kernohan K.D., Hartley T., Majewski J., Hocking T.D. ClinPred: Prediction Tool to Identify Disease-Relevant Nonsynonymous Single-Nucleotide Variants. Am. J. Hum. Genet. 2018;103:474–483. doi: 10.1016/j.ajhg.2018.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Raimondi D., Tanyalcin I., Ferté J., Gazzo A., Orlando G., Lenaerts T., Rooman M., Vranken W. DEOGEN2: Prediction and interactive visualization of single amino acid variant deleteriousness in human proteins. Nucleic Acids Res. 2017;45:W201–W206. doi: 10.1093/nar/gkx390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kircher M., Witten D.M., Jain P., O’roak B.J., Cooper G.M., Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014;46:310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kucukkal T.G., Petukh M., Li L., Alexov E. Structural and physico-chemical effects of disease and non-disease nsSNPs on proteins. Curr. Opin. Struct. Biol. 2015;32:18–24. doi: 10.1016/j.sbi.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iqbal S., Pérez-Palma E., Jespersen J.B., May P., Hoksza D., Heyne H.O., Ahmed S.S., Rifat Z.T., Rahman M.S., Lage K., et al. Comprehensive characterization of amino acid positions in protein structures reveals molecular effect of missense variants. Proc. Natl. Acad. Sci. USA. 2020;117:28201–28211. doi: 10.1073/pnas.2002660117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ittisoponpisan S., Islam S.A., Khanna T., Alhuzimi E., David A., Sternberg M.J.E. Can Predicted Protein 3D Structures Provide Reliable Insights into whether Missense Variants Are Disease Associated? J. Mol. Biol. 2019;431:2197–2212. doi: 10.1016/j.jmb.2019.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baek M., DiMaio F., Anishchenko I., Dauparas J., Ovchinnikov S., Lee G.R., Wang J., Cong Q., Kinch L.N., Schaeffer R.D., et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science (80-. ) 2021;373:871–876. doi: 10.1126/science.abj8754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Senior A.W., Evans R., Jumper J., Kirkpatrick J., Sifre L., Green T., Qin C., Žídek A., Nelson A.W.R., Bridgland A., et al. Improved protein structure prediction using potentials from deep learning. Nature. 2020;577:706–710. doi: 10.1038/s41586-019-1923-7. [DOI] [PubMed] [Google Scholar]
- 17.Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., Tunyasuvunakool K., Bates R., Žídek A., Potapenko A., et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–589. doi: 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tunyasuvunakool K., Adler J., Wu Z., Green T., Zielinski M., Žídek A., Bridgland A., Cowie A., Meyer C., Laydon A., et al. Highly accurate protein structure prediction for the human proteome. Nature. 2021;596:590–596. doi: 10.1038/s41586-021-03828-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Varadi M., Anyango S., Deshpande M., Nair S., Natassia C., Yordanova G., Yuan D., Stroe O., Wood G., Laydon A., et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022;50:D439–D444. doi: 10.1093/nar/gkab1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chothia C., Lesk A.M. The relation between the divergence of sequence and structure in proteins. EMBO J. 1986;5:823–826. doi: 10.1002/j.1460-2075.1986.tb04288.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Starita L.M., Islam M.M., Banerjee T., Adamovich A.I., Gullingsrud J., Fields S., Shendure J., Parvin J.D. A Multiplex Homology-Directed DNA Repair Assay Reveals the Impact of More Than 1,000 BRCA1 Missense Substitution Variants on Protein Function. Am. J. Hum. Genet. 2018;103:498–508. doi: 10.1016/j.ajhg.2018.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Whiffin N., Minikel E., Walsh R., O’Donnell-Luria A.H., Karczewski K., Ing A.Y., Barton P.J.R., Funke B., Cook S.A., MacArthur D., Ware J.S. Using high-resolution variant frequencies to empower clinical genome interpretation. Genet. Med. 2017;19:1151–1158. doi: 10.1038/gim.2017.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Samocha K.E., Kosmicki J.A., Karczewski K.J., O’Donnell-Luria A.H., Pierce-Hoffman E., MacArthur D.G., Neale B.M., Daly M.J. Regional Missense Constraint Improves Variant Deleteriousness Prediction. bioRxiv. 2017 Preprint at. [Google Scholar]
- 24.Betz S.F. Disulfide bonds and the stability of globular proteins. Protein Sci. 1993;2:1551–1558. doi: 10.1002/pro.5560021002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burley S.K., Arap W., Pasqualini R. Predicting Proteome-Scale Protein Structure with Artificial Intelligence. N. Engl. J. Med. 2021;385:2191–2194. doi: 10.1056/NEJMcibr2113027. [DOI] [PubMed] [Google Scholar]
- 26.Quinodoz M., Peter V.G., Cisarova K., Royer-Bertrand B., Stenson P.D., Cooper D.N., Unger S., Superti-Furga A., Rivolta C. Analysis of missense variants in the human genome reveals widespread gene-specific clustering and improves prediction of pathogenicity. Am. J. Hum. Genet. 2022;109:457–470. doi: 10.1016/j.ajhg.2022.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grimm D.G., Azencott C.A., Aicheler F., Gieraths U., Macarthur D.G., Samocha K.E., Cooper D.N., Stenson P.D., Daly M.J., Smoller J.W., et al. The evaluation of tools used to predict the impact of missense variants is hindered by two types of circularity. Hum. Mutat. 2015;36:513–523. doi: 10.1002/humu.22768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Buel G.R., Walters K.J. Can AlphaFold2 predict the impact of missense mutations on structure? Nat. Struct. Mol. Biol. 2022;29:1–2. doi: 10.1038/s41594-021-00714-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thornton J.M., Laskowski R.A., Borkakoti N. AlphaFold heralds a data-driven revolution in biology and medicine. Nat. Med. 2021;27:1666–1669. doi: 10.1038/s41591-021-01533-0. [DOI] [PubMed] [Google Scholar]
- 30.Sevim Bayrak C., Stein D., Jain A., Chaudhary K., Nadkarni G.N., Van Vleck T.T., Puel A., Boisson-Dupuis S., Okada S., Stenson P.D., et al. Identification of discriminative gene-level and protein-level features associated with pathogenic gain-of-function and loss-of-function variants. Am. J. Hum. Genet. 2021;108:2301–2318. doi: 10.1016/j.ajhg.2021.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meier J., Rao R., Verkuil R., Liu J., Sercu T., Rives A. In: Advances in Neural Information Processing Systems. Ranzato M., Beygelzimer A., Dauphin Y., Liang P.S., Vaughan J.W., editors. Curran Associates, Inc.); 2021. Language models enable zero-shot prediction of the effects of mutations on protein function; pp. 29287–29303. [Google Scholar]
- 32.McLaren W., Gil L., Hunt S.E., Riat H.S., Ritchie G.R.S., Thormann A., Flicek P., Cunningham F. The Ensembl Variant Effect Predictor. Genome Biol. 2016;17:122. doi: 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jaganathan K., Kyriazopoulou Panagiotopoulou S., McRae J.F., Darbandi S.F., Knowles D., Li Y.I., Kosmicki J.A., Arbelaez J., Cui W., Schwartz G.B., et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell. 2019;176:535–548.e24. doi: 10.1016/j.cell.2018.12.015. [DOI] [PubMed] [Google Scholar]
- 34.Schymkowitz J., Borg J., Stricher F., Nys R., Rousseau F., Serrano L. The FoldX web server: an online force field. Nucleic Acids Res. 2005;33:W382–W388. doi: 10.1093/nar/gki387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kabsch W., Sander C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 36.Chen C.R., Makhatadze G.I. ProteinVolume: calculating molecular van der Waals and void volumes in proteins. BMC Bioinf. 2015;16:101. doi: 10.1186/s12859-015-0531-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Landrum M.J., Lee J.M., Benson M., Brown G.R., Chao C., Chitipiralla S., Gu B., Hart J., Hoffman D., Jang W., et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46:D1062–D1067. doi: 10.1093/nar/gkx1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Esposito D., Weile J., Shendure J., Starita L.M., Papenfuss A.T., Roth F.P., Fowler D.M., Rubin A.F. MaveDB: An open-source platform to distribute and interpret data from multiplexed assays of variant effect. Genome Biol. 2019;20:223. doi: 10.1186/s13059-019-1845-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Savojardo C., Manfredi M., Martelli P.L., Casadio R. Solvent Accessibility of Residues Undergoing Pathogenic Variations in Humans: From Protein Structures to Protein Sequences. Front. Mol. Biosci. 2020;7:626363–626369. doi: 10.3389/fmolb.2020.626363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pancotti C., Benevenuta S., Birolo G., Alberini V., Repetto V., Sanavia T., Capriotti E., Fariselli P. Predicting protein stability changes upon single-point mutation: a thorough comparison of the available tools on a new dataset. Brief. Bioinform. 2022;23 doi: 10.1093/bib/bbab555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xue M., Wakamoto T., Kejlberg C., Yoshimura Y., Nielsen T.A., Risør M.W., Sanggaard K.W., Kitahara R., Mulder F.A.A. How internal cavities destabilize a protein. Proc. Natl. Acad. Sci. USA. 2019;116:21031–21036. doi: 10.1073/pnas.1911181116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tien M.Z., Meyer A.G., Sydykova D.K., Spielman S.J., Wilke C.O. Maximum allowed solvent accessibilites of residues in proteins. PLoS One. 2013;8:e80635. doi: 10.1371/journal.pone.0080635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu X., Jian X., Boerwinkle E. dbNSFP: A lightweight database of human nonsynonymous SNPs and their functional predictions. Hum. Mutat. 2011;32:894–899. doi: 10.1002/humu.21517. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Data curated or generated here can be found in the supplementary tables. Publicly available datasets used are listed in the key resources table. The SIGMA scores for over 48 million possible missense variants across 3,454 disease-associated genes have been deposited at https://sigma-pred.org/.
-
•
The original code has been deposited at github and is publicly available as of the date of publication. DOIs are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.