

Abstract
By deciphering information encoded in degraded ancient DNA extracted from up to million‐years‐old samples, molecular paleomicrobiology enables to objectively retrace the temporal evolution of microbial species and communities. Assembly of full‐length genomes of ancient pathogen lineages allows not only to follow historical epidemics in space and time but also to identify the acquisition of genetic features that represent landmarks in the evolution of the host–microbe interaction. Analysis of microbial community DNA extracted from essentially human paleo‐artefacts (paleofeces, dental calculi) evaluates the relative contribution of diet, lifestyle and geography on the taxonomic and functional diversity of these guilds in which have been identified species that may have gone extinct in today's human microbiome. As for non‐host‐associated environmental samples, such as stratified sediment cores, analysis of their DNA illustrates how and at which pace microbial communities are affected by local or widespread environmental disturbance. Description of pre‐disturbance microbial diversity patterns can aid in evaluating the relevance and effectiveness of remediation policies. We finally discuss how recent achievements in paleomicrobiology could contribute to microbial biotechnology in the fields of medical microbiology and food science to trace the domestication of microorganisms used in food processing or to illustrate the historic evolution of food processing microbial consortia.
By deciphering information encoded in degraded ancient DNA extracted from up to million years‐old samples, molecular paleomicrobiology objectively retrace the temporal evolution of individual microbial species and also of complex communities (either host‐associated or free living). Paleomicrobiology is likely to make major contributions to various fields of microbiology, including medical and food microbiologyby providing unique insights into the taxonomic and functional diversity of past microbial species and communities.

INTRODUCTION
Archaea, bacteria and unicellular eukaryotes are certainly the oldest forms of life on Earth. They originated about 3.5 Gyr ago and participated in the formation of the original biosphere. They first inhabited the earth's anoxic environment and then enabled its oxygenation (Falkowski et al., 2008; Fischer et al., 2016).
Although they played essential roles in ancient and modern environments, temporal patterns of evolution and diversification of microorganisms are poorly known because of the scarcity of fossil records whose taxonomic assignments are often problematic. With few notable exceptions (e.g. foraminifera, diatoms), most microbial cells do not produce mineralized structures and their often indistinctive shapes and structures preclude precise identification of fossilized microbial taxa based on morphological examination (Xie & Kershaw, 2012). Furthermore, even for taxa well represented in the fossil records, such as the Foraminifera, the existence of clades comprising naked unfossilized species may prevent a correct interpretation of their patterns of evolution over time when exclusively based on fossil data (Pawlowski et al., 2003). Thus, as opposed to ancient fauna and flora (McElwain & Punyasena, 2007; Raup & Sepkoski, 1982; Signor, 1994), the rarity of taxonomically assignable paleontological specimens illuminates only a small sliver of the real past microbial diversity and makes it difficult to investigate microbial evolution, diversification and functional significance across the different geological eras.
For these reasons, the evolution and diversification of microorganisms have been largely inferred from molecular phylogenetic reconstructions that make use of DNA/protein sequences of extant species (Louca et al., 2018; Sanderson & Donoghue, 1996; Morlon, 2014; Nee, Holmes, et al., 1994; Nee, May, et al., 1994). However, these phylogenies are difficult to time calibrate, precisely because of the absence, gaps or imprecision of microbial fossil records that could be used as landmarks. Molecular phylogenies also hardly predict the potential existence and functions (ecological roles) of extinct clades that are thought to exceed extant ones in number (Stilianos Louca et al., 2018; Tricou et al., 2022). To date, a global microbial life pattern of evolution remains largely unresolved and only few studies focus on their past diversification patterns (Gubry‐Rangin et al., 2015; Lebreton et al., 2017; Lorén et al., 2014; Louca et al., 2018; Marin et al., 2017; Morlon et al., 2012).
Furthermore, until a very recent past, most of diversity and global phylogenetic studies also suffered from the lack of a global view regarding the magnitude of extant microbial diversity not only in terms of absolute number of taxa whose estimates vary by several orders of magnitude in the case of Bacteria and Archaea (Lennon & Locey, 2020; Loucaid et al., 2019), but also in terms of phylogenetic diversity. Regarding this last aspect, the gap is rapidly closing thanks to the multiplication of metagenomics studies that give access to the genome sequences (so‐called Metagenome assembled genomes, MAGs) of species belonging to, thus far, overlooked microbial clades (Hug et al., 2016; Nayfach et al., 2021).
While the previous paragraphs exposed the obstacles to which one is confronted when addressing microbial evolution, past diversity and contribution to ecosystem processes in a ‘distant past’, these obstacles partially vanish when referring to a ‘recent past’. This is made possible thanks to the emergence of paleogenetics/paleogenomics that analyses microbial DNA preserved in ancient environmental samples as diverse as sediment cores, archaeological artefacts, long‐buried animal/human skeletons, or items preserved in natural history collections (Figure 1). Indeed, ancient DNA (aDNA, but also RNA or proteins) that survives through time to the death of any organism can be regarded as a fossil trace of the corresponding organism and can be interrogated to retrace the ‘recent’ evolution of the corresponding species or group of species (Arning & Wilson, 2020; Kistler et al., 2020; Orlando et al., 2021; Raxworthy & Smith, 2021; Siano et al., 2021). Besides specific taxa, aDNA studies also make the exploration of entire communities of (micro)organisms from the past possible, providing a comprehensive vision of their diversity and functional roles in their original ecosystems.
FIGURE 1.

Main types of historical samples used as sources of ancient microbial DNA. These samples can be extremely ancient as Bronze Age teeth or dental calculi or much more recent such as often less than 100‐year‐old herbarium plants. These samples have been used to study either specific (often pathogenic) microbial species or entire communities.
The aim of this review is to highlight, through the description of selected examples, the different facets and main achievements of molecular paleomicrobiology. In this article, which does not claim to be exhaustive, technical and theoretical issues of paleogenetics/paleogenomics that have been reviewed many times and that are not at the heart of this article, will only be briefly mentioned (Brunson & Reich, 2019; Warinner et al., 2017). We separately illustrate and discuss studies that focus (i) on individual microbial taxa and those that probe (ii) entire microbial communities whatever the environment they originate from. Finally, we will conclude by suggesting what could be the potential contribution of molecular paleomicrobiology to the field of microbial biotechnology.
MOLECULAR PALEOMICROBIOLOGY AS A DISCIPLINE
Time frame
Molecular paleomicrobiology, based on the analysis of degraded ancient DNA (or eventually RNA) is a discipline that presents several specificities. It is based on a diachronic approach that documents the occurrence of microorganisms and their original genetic make‐up directly in ancient samples as diverse as environmental (e.g. soils or sediments) or archaeological ones to compare the ancient samples to those of today. It is thus different from phylogenetic‐based approaches that reconstruct past evolutionary events in silico, using information obtained on extant organisms. Although the prefix ‘paleo’ commonly refers to a ‘distant’ past, it seems preferable not to impose a minimum temporal threshold to molecular paleomicrobiology that studies organisms with often very short generation times that can acquire adaptive mutations in far shorter time spans compared to most ‘macroorganisms’. Thus, a study published in 2016 (Worobey et al., 2016) that investigated the initial events of the AIDS epidemics in North America in the 1970s based on the extraction and sequencing of degraded RNA molecules from circa 40‐year‐old archived blood samples to reconstruct HIV genomes clearly belongs to the research field from both a technical and scientific point of view.
Ancient DNA (aDNA)
Indeed, a second specificity of the discipline, detailed in several technical reviews (Afouda et al., 2020; Orlando et al., 2021; Pedersen et al., 2015; Rivera‐Perez et al., 2016; Warinner et al., 2017), is to deal with aDNA, a material often difficult to access, limiting in quantity and degraded. Its extraction and manipulation require the implementation, by specifically trained scientists, of particular protocols in dedicated laboratories (clean rooms, Cooper & Poinar, 2000; Orlando et al., 2021). As in 2023, the oldest DNA sample ever analysed was extracted from two‐million‐year‐old frozen sediments in Greenland and allowed reconstructing the entire ecosystem that shaped this region at that time (Kjær et al., 2022).
Ancient DNA is more complex to analyse compared to modern one due to the presence of postmortem damages (PMDs) resulting in DNA fragmentation and base modifications (Dabney et al., 2013, Figure 2). These PMDs are essentially the result of depurination and deamination. Depurination, that is the loss of adenine and guanine bases, is the consequence of cleavage of β‐N‐glycosidic bonds and is at the origin of DNA fragmentation producing very short fragments of mostly less than 100 bp in length. The rate of DNA fragmentation is however environment‐dependent. While aDNA extracted from 100‐year‐old plant herbarium specimens is mostly made of fragments in the range 40–150 bp (Staats et al., 2011; Yoshida et al., 2013), aDNA extracted from more than 10,000 years‐old lake sediments can still be mostly made of fragments larger than 100 bp (Talas et al., 2021). Consequently, studies carried out on sedimentary DNA can implement metabarcoding approaches based on the amplification of barcode DNA sequences sometimes larger than 200 bp. This approach would be inoperative for highly degraded DNA samples.
FIGURE 2.
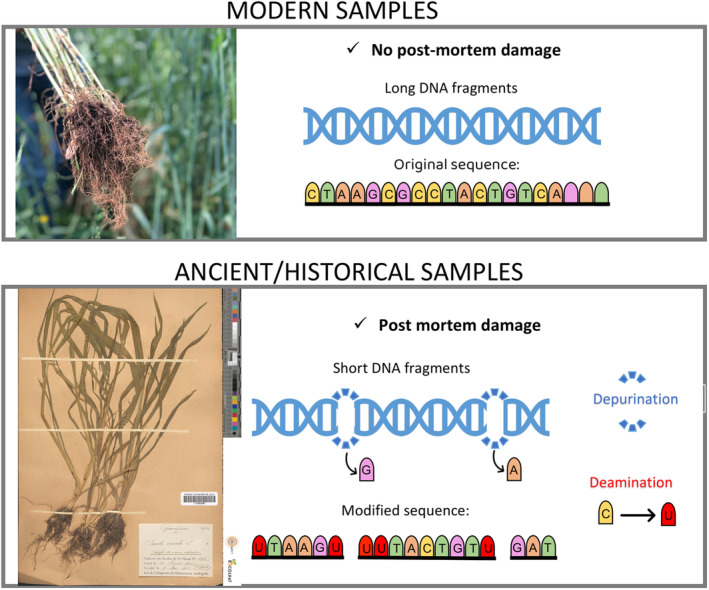
Main characteristics of ancient DNA. While DNA extracted from living organisms is made of long molecules, often exceeding 1 kbp in length and perfectly representative of the original organism's genomic sequence, ancient DNA extracted from historical samples has experienced post‐mortem damages. They lead to DNA fragmentation (fragments often shorter than 100 bp) and base modifications altering the original sequence. Thus, cytosine deamination produces uracil, preferentially at the ends of the molecules. Photo credits: modern plant samples: ©Herbier LY, FR‐CERESE, UCBLyon1 Recolnat portal (https://explore.recolnat.org/) under accessions LY0662689.
As for ‘spontaneous’ cytosine deamination, it leads to the conversion of cytosine to uracil and results in the incorporation of adenine on the complementary strand instead of guanine during in vitro DNA synthesis. Cytosine deamination occurs at a higher frequency in single‐stranded ends of degraded DNA molecules. While the age of the specimen represents one of the main factors that control the magnitude of PMDs, other factors like local temperature, depositional conditions, post‐excavation handling, specimen treatment and source tissue also influence aDNA conservation (Arning & Wilson, 2020).
Thus, besides the ‘wet lab’ manipulation of degraded DNA, paleogenetics, including molecular paleomicrobiology, relies on a suite of adapted bioinformatics tools to extract relevant information from very short DNA sequences and to quantify chemical alterations that represent signatures of the ancient origin of the studied nucleic acids. Detection of these chemical modifications is performed by specific software like PMDTools (Skoglund et al., 2014) or mapDamage (Jónsson et al., 2013) which quantify C‐T and G‐A transitions at the ends of the aDNA fragments.
A dialogue between disciplines
Finally, a third specificity of paleomicrobiology frequently requires close collaborations between microbiologists and specialists of other disciplines sometimes from humanities, such as historians, archaeologists, paleoanthropologists, paleopathologists, paleoclimatologists, geologists or curators of collections of natural history or anthropology. These collaborations are necessary to localize, identify, date and access relevant historical samples with the highest probability to contain usable DNA traces of the studied microbial species or microbial communities and also to ‘contextualize’ these samples in their original, historical environment (van der Kuyl, 2022).
SINGLE SPECIES APPROACH
Numerous molecular paleomicrobiology studies focus on a specific microbial taxon. Most of them are pathogens, primarily human ones, bacteria or viruses. The field of investigation is likely to widen rapidly, with the emergence of studies in the field of plant pathology as well as the availability of genomes of non‐pathogenic, often uncultivable species reconstructed in silico (so‐called MAGs, for metagenome‐assembled genomes) from massive sequencing data of ‘microbial paleo‐communities’ (Bos et al., 2019; Granehäll et al., 2021; Marx, 2016; Wibowo et al., 2021). This section of the manuscript takes as a main example the studies on the human plague that illustrate a posteriori the specificity of the discipline presented in the introductory section.
The history of the plague bacterium
Before paleogenomic studies of plague, there were a number of certainties about the nature of this disease, but also a number of uncertainties that could be not addressed using currently circulating bacterial strains. First of all, it is caused by Yersinia pestis, which was first isolated in China in 1894 during the last great plague epidemic (the so‐called 3rd pandemic). The main reservoir of this disease is wild rodents, and it is essentially transmitted from animals to humans through the bites of infected fleas. This form of transmission leads to the bubonic form of the disease, which takes its name from the swelling of lymph nodes. The disease, in rarer cases, can be transmitted directly from humans to humans through the respiratory tract where it leads to a pulmonary infection that is also usually fatal. In the absence of appropriate treatment, the infection leads to very high mortality rates during epidemic episodes. Y. pestis, whose genome has been sequenced many times (more than 600 genomes available in GenBank in 2023), has been the subject of numerous experimental studies which have identified many genes involved in the virulence and aggressiveness of this bacterium in humans as well as in the insect vector (Demeure et al., 2019; Hinnebusch et al., 2021). Some of these genes are carried by plasmids.
None of the studies conducted on modern strains of Y. pestis could however confirm that the so‐called ‘plague’ epidemics, prior to the late 19th century one, were caused by the same pathogen despite the similarity of symptoms reported in historical texts and illustrations. The pandemic that broke out in China in the second half of the 19th century is referred to as the 3rd pandemic. It followed a first one called the Justinian pandemic (6th‐8th century AD) and a second one (14th‐18th century) that peaked in Europe as the Black Death in 1347–1351, decimating up to 60% of the population locally. After a phase of decline, each of these pandemics gave rise to more localized epidemic episodes.
In this context, the paleomicrobiological approach is a priori the only one that could answer a set of questions, common to many other infectious diseases. (i) Are the historical cases of the disease (here the 1st and 2nd pandemics) attributable to the same infectious agent? (ii) Did several distinct strains/evolutionary lineages of the pathogen circulate during a single pandemic? (iii) Are successive pandemics/epidemics due to the re‐emergence of the evolutionary lineage that predominated during the previous pandemic? (iv) Can infections be documented in humans at earlier dates for which documentary sources do not exist? And (v), what can the analysis of ancient strains teach us about the temporal evolution of the pathogen's virulence?
If certain diseases lead to bone alterations such as tuberculosis (bone tuberculosis) or leprosy, thus allowing human remains to be targeted for paleomicrobiological studies, this is not the case for plague. Nevertheless, the high mortality induced by the disease is known to lead to a modification of funerary practices characterized by the burial of bodies not in individual, but in collective graves. aDNA of Y. pestis has thus been successfully extracted essentially from the dental pulp of teeth, a densely vascularized tissue, taken from skeletons found in collective graves of the different pandemics (Bos et al., 2016; Harbeck et al., 2013; Spyrou et al., 2016; Susat et al., 2020). Several research groups have independently been able to reconstitute entire genomes of Y. pestis either directly from systematic sequencing of the extracted aDNA, or after enrichment in Y. pestis DNA by sequence capture (Bos et al., 2016; Spyrou et al., 2016). This first set of results validated the hypothesis that the first two plague pandemics (6th‐8th century and 14th‐18th century) were indeed due to Y. pestis found in numerous sites covering a vast territory in Eurasia and several decades (Bos et al., 2016; Bramanti et al., 2021; Harbeck et al., 2013; Spyrou et al., 2022; Susat et al., 2020).
Integration of ancient and modern genomic sequences into a global phylogeny of the Y. pestis species showed that strains from the same pandemic tend to cluster together to form distinct lineages, suggesting that major pandemics do not result from the re‐emergence of strains that produced the previous ones (Bramanti et al., 2021; Hinnebusch et al., 2021). The earliest evidence of the evolutionary lineage of the second pandemic dates from 1338 to 1339 and was found in Kyrgyzstan, suggesting that it originated in Central Asia (Spyrou et al., 2022). It was always members of this lineage that caused epidemic rebounds more than 3–4 centuries later in Europe (Bos et al., 2016; Bramanti et al., 2021; Spyrou et al., 2022).
To address the diffusion of Y. pestis before the first documented pandemics, a systematic screening for the presence of the bacterium sequences in DNA extracted from human remains buried individually or collectively was necessary. This more tedious approach led to the reconstruction of 17 ancient Y. pestis genomes among a set of 252 human skeletons dating from 5000 to 2500 years BP and over a territory ranging from the Iberian Peninsula in the West to Mongolia in the East (Valtueña et al., 2022). The ability to infect humans thus predates the recorded three pandemics (Rascovan et al., 2019; Spyrou et al., 2018; Valtueña et al., 2017, 2022). The fact that many infected bodies were buried individually suggested that these cases of historical infections may not have led to mass mortality.
We thus now have a substantial number of genomes of historical Y. pestis strains covering a period of time of more than 5000 years. As we have just summarized, the comparison of these genomes allows the construction of time‐calibrated molecular phylogenies and traces ‘objectively’ the course of epidemics and movements of these pathogens on different time and spatial scales (Rascovan et al., 2019; Spyrou et al., 2018; Valtueña et al., 2022). Besides phylogenetic reconstructions, confrontation of the gene content of ancient genomes with our current knowledge of the molecular basis of the modes of infection and virulence of the pathogen offers a unique opportunity to address the evolution of the modes of transmission and of the virulence of the pathogen over time.
In the case of Y. pestis, several of the key genes necessary for this bacterium to infect fleas and make them effective in transmitting the pathogen to humans and causing the bubonic form of the disease are known (Hinnebusch et al., 2021). The ability to infect these insects requires for instance the presence of the ymt gene and a mutation in the ure2 gene that inhibits the production of a functional urease. The key ymt gene was absent in most strains found in the Late Neolithic‐Early Bronze age (5000–3500 BP) which also seems to be able to produce a functional urease (Spyrou et al., 2018; Vågene et al., 2022; Valtueña et al., 2017). During this period the plague could have thus been predominantly transmitted from human to human without an intermediary insect host. However, the mode of transmission via flea bites is by far the most efficient and determinant for a massive diffusion of the pathogen. While early studies on Bronze Age strains suggested that acquisition of the ymt gene occurred later, two recent studies independently found two Y. pestis strains carrying this gene as well as mutations in the ure2 gene and other genes promoting flea infection. These two observations were made on skeletons excavated, one in Spain (3200 years BP) (Valtueña et al., 2022) and the other in Russia (3800 years BP) (Spyrou et al., 2018). These strains potentially capable of being transmitted by fleas may therefore have spread unnoticed over a large geographical area at a time when other forms of pathogen transmission were predominant.
A second piece of information that may shed light on the aggressiveness on mammals of historical plague strains concerns the pla gene, a major virulence factor carried on the Y. pestis pPCP1 plasmid. Independent studies on different isolates from different geographical origins reported a specific ‘erosion’ of this gene in several strains of the second pandemics, after the end of the Black Death episode (after the 15th century, Bramanti et al., 2021; Susat et al., 2020). This ‘erosion’ corresponds to a lower sequence coverage of this gene compared to the rest of the pPCP1 plasmid sequence. Thus, only a small percentage of plasmids may have carried the pla gene. This could indicate that the strains at the end of the epidemic peak were affected in their virulence, although this remains to be experimentally demonstrated.
Beyond plague, other pathosystems
While plague has, thus far, certainly been the focus of the largest number of studies in the field of paleomicrobiology; similar scientific questioning has been elaborated on other pathogens. They belong to the bacteria (e.g. Mycobacterium sp.) but also to the viruses (e.g. the Hepatitis B virus) or the Eukarya (e.g. the Oomycete Phytophthora infestans) and infect either animal/humans or plant species. In different instances, the results reported contrast with those obtained for plague.
In the case of bone tuberculosis, essentially resulting nowadays from infections by Mycobacterium tuberculosis, it has been demonstrated that the symptoms observed on human skeletons from 950 to 1550 CE along the coasts of South America, prior to the European invasions that may have brought M. tuberculosis to this region, were the result of infections by the species M. pinnipedii (Vågene et al., 2022). This species preferentially infects Pinnipeds, such as fur seals abundant in this region. The presence of this infection in human communities along the coast can be explained by recurrent transmissions from animals to humans resulting from the proximity between these two species, the former of which being hunted for its meat. However, the presence of such infections on skeletons found further inland raises questions about possible human‐to‐human transmission by one or more lines of M. pinnipedii that would have adapted to this new host and that may have gone extinct (Vågene et al., 2022).
Regarding the progression of ‘successive waves’ of different genotypes of a pathogen in the same geographic area, it has been reported for other microbial species belonging to different taxonomic groups in different time periods. Thus, the systematic screening of more than 130 human remains from Eurasia and America covering a period from 10,500 BP to 400 BP has allowed to trace the evolutionary history of the hepatitis B virus whose infection does not leave any visible trace on skeletons and does not lead to mass mortality (Kocher et al., 2021). In Western Europe, at least 5 evolutionary lineages of the virus followed one another over this period. One of them in particular, called WENBA, prevailed for nearly 4000 years from about 7500 to 3500 years ago, that is a period straddling the Neolithic and the Bronze Age, before being apparently eliminated and replaced by a new genotype that is still found today in Europe. The dissemination of the WENBA lineage found on several Early European Farmer's skeletons coincides with the Hunter‐Gatherers/Farmers transition in Europe. It is interesting to note that a descendant of the WENBA lineage, not found for nearly 3500 years, has recently reappeared in today's human populations, often in association with HIV carriers. The source of this re‐emergence of a lineage thought to be extinct remains to be identified (Kocher et al., 2021).
Another example of historical epidemiological monitoring concerns the Oomycete Phytophthora infestans, agent of the Potato blight. Its introduction from America to Europe in the first half of the 19th century led to the destruction of this crop and triggered the great famine in Ireland, which caused more than a million deaths and resulted in a wave of emigration from Europe to North America. Genome sequencing of this species was performed using (degraded) aDNA extracted from potato leaves, stored in herbaria, which displayed typical leaf lesions (Martin et al., 2013; Yoshida et al., 2013). These specimens were collected mainly in Western Europe but also in North America between 1845, the probable date of the first introduction of the pathogen into Europe, and the end of the 19th century. All isolates collected along the 19th century had genomes very similar to each other but distinct from the genomes of strains currently circulating in Europe. Thus, it appears that the initial introduction into Europe was from a single strain or from closely related strains that circulated throughout the 19th century only to be replaced by new, genetically distinct isolates (Yoshida et al., 2013).
As in the case of Y. pestis, functional information was also deduced from the analysis of P. infestans genomes that circulated in Europe in the 19th century. They possessed a functional AVR3KI avirulence gene, whereas modern strains possess the AVR3EM allele of this gene (Martin et al., 2013; Yoshida et al., 2013). When a P. infestans strain carries a AVR3KI allele it cannot infect a potato cultivar carrying the cognate R3a resistance gene (Bieker et al., 2020; Yoshida et al., 2013). This R3a resistance gene was however absent in potato lines grown in Europe in the 19th century. Its introgression into modern potato cultivars to fight P. infestans led however to the emergence of strains of the pathogen carrying the new AVR3EM allele that bypassed the resistance conferred by the R3a gene.
ANCIENT MICROBIAL COMMUNITIES—SPECIFIC ISSUES
Unlike studies targeting a single species, the analyses of ancient microbial communities must take several factors into account that may have biased the relative abundance of the different species within the pool of ancient DNA extracted from the samples under study.
The first factor is the contamination of ancient material by an external source, which may itself be ancient and therefore characterized by the presence of DNA with post‐mortem damages. For example, in the case of buried animal/human remains, the surrounding substrate (soil) may have contaminated these remains. These contaminations can be evaluated and removed a posteriori from the sequence datasets using bioinformatics approaches. Thus, to validate microbiome preservation in different ancient dental biofilm samples, Fellows Yates, Velsko, et al. (2021), developed a multistep procedure that included (i), metagenomic binning of the data to the NCBI nucleotide database for a taxonomic assignation and, (ii) subsequent validation of the identified microbial taxa by comparison with oral and non‐oral reference microbiomes. Another popular approach is the use of the SourceTracker software (Knights et al., 2011), a tool based on Bayesian methods that compare the microbiome dataset under study with datasets of published microbiomes from the same and different environments. Similarly, the R decontam package allows for the removal of laboratory and environmental contaminants prior to subsequent analysis of microbiomes (Davis et al., 2018).
In certain environments, such as lake or marine sediments, active or dormant microorganisms may also naturally cohabit with ancient nucleic acids from microorganisms that have disappeared. In paleomicrobiology, it is not easy to distinguish between these different sources and this can lead to erroneous conclusions about the temporal evolution of microbial communities. In environmental microbiology, it is traditionally considered that DNA persists longer in the environment after the death of cells than RNA, which is a more labile molecule whose presence would indicate the presence of active living cells. Thus the detection within an environmental archive of RNA associated with a specific taxon should prompt its exclusion from downstream analyses. However, recently published results suggest that RNAs may themselves persist in the environment for several decades (Pearman et al., 2022). Therefore, as a precautionary measure, paleoenvironmental analyses, particularly on sediment cores, should primarily focus on microbial taxonomic groups whose short‐ to medium‐term survival in sediments is unlikely. This is the case, for example, for many strictly photoautotrophic organisms such as cyanobacteria or unicellular eukaryotic algae.
Contamination of ancient material may also be endogenous, due to the secondary development of microorganisms during the conservation or fossilization process. This was suggested in the study of herbarium samples of Ambrosia and Arabidopsis plants (Bieker et al., 2020). Most of the plants preserved in herbaria contained sequences attributed to the ascomycete fungus Alternaria alternata that were never found in modern samples of plants of these species. This observation led the authors to suggest that this fungus colonized the plants in the herbaria after they had been collected.
Another factor that is difficult to assess is the differential conservation (of DNA) of the different species in the microbial communities, leading to artifactual changes in their relative abundance. These changes could occur at different stages in the conservation process. In aquatic ecosystems, in the case of lacustrine cyanobacterial communities, differential sedimentation of planktonic cells has been reported. Nwosu et al. (2021) observed an over‐representation in sediment traps, placed at the bottom of a lake, of species producing aggregates or colonies of cells compared with other species, which, although abundant in the water column, are characterized by small individual cells producing gas vesicles. These latter species are not only less likely to sediment rapidly, but are also more vulnerable to predation and the rapid degradation of their DNA (Nwosu et al., 2021). In addition to this direct evidence made on extant samples, observations made on ancient material also suggest differential conservation of the genetic material of certain taxa after their death. This was reported for a desiccated microbial mat dated around 1000 yr BP collected in Antarctica. Analysis of the DNA and proteins extracted from this mat revealed very different taxonomic profiles of the microbial communities (Lezcano et al., 2022). Among the DNA sequences, the authors observed a high prevalence of Clostridiales and Actinomycetales and a virtual absence of cyanobacteria. The taxonomic affiliation of the extracted proteins, on the other hand, reveals a high abundance of cyanobacteria and a quasi‐absence of Clostridiales and Actinomycetales. This latter observation reflected better the presumed nature of the studied material. It was hypothesized that certain microbial taxa, particularly those producing spores such as the Clostridiales, are characterized by a slower postmortem degradation of their DNA and are therefore over‐represented in the archives. It should however be noted that this study was conducted using a metabarcoding approach, which only allows analysis of the fraction of extracted fragments whose size exceeds the size of the amplified marker. In that way, DNA less degraded is favoured over the very short fragments that may be more representative of ancient communities.
To minimize the complex problem of artefactual modification of the relative abundance of taxa over time, it is possible to discuss the results only in terms of the presence or absence of taxa within the communities. However, this only makes sense in cases where these taxa play a very specific role, as is the case for pathogenic species. Thus, Bonczarowska et al. (2022) systematically searched for human pathogen sequences (bacteria and viruses) in DNA extracted from the teeth of 70 individuals buried in the same German village during the Merovingian period (fifth‐eighth century CE). On twenty‐two individuals (31%) were found at least one of the following 4 pathogens: Hepatitis B virus, Smallpox virus or Parvovirus B and the leprosy agent, Mycobacterium leprae. Seven cases of double infection and one case of triple infection suggested that several of the pathogens were endemic within the population and that overall this village community had a poor health status.
Despite these warnings, molecular paleomicrobiology remains the only approach for probing the diversity of past microbial communities, whose members, sometimes all of them, have left no fossil record. Nevertheless, this assertion is also true in the case of the very few microbial groups that are widely studied in micropaleontology, such as the Foraminifera. Thus, a metabarcoding study targeting this taxon in a marine sediment core covering more than 1000 years of sedimentary deposits identified nine times more molecular taxa than morphological ones producing hard shells preserved in sediment (Pawłowska et al., 2014). This ‘excess’ of molecular taxa can be explained by the existence of cryptic species producing morphologically similar hard shells, as well as by the existence of numerous taxa that do not produce these fossilized structures.
In this section dedicated to communities, we will distinguish studies exploring free‐living microbial communities from studies of ‘host‐associated’ microbial communities. This distinction is justified by the fact that to date, studies of free‐living communities have mainly involved the analysis of DNA extracted from sediment cores using metabarcoding approaches. As for studies on ‘host‐associated’ communities, besides exploring more diverse sample types (Figure 1), they mainly follow a systematic high‐throughput DNA sequencing approach. This approach allows de novo reconstruction of microbial genomes and detailed functional analysis of the role of microorganisms in ancient ecosystems.
FREE‐LIVING ANCIENT MICROBIAL COMMUNITIES
Free‐living ancient microbiomes, or ‘non‐host associated microbiomes’, encompass microbial communities entrapped in environmental matrices as diverse as freshwater or marine sediments, ancient soils, permafrost, or ice cores (Figure 1). The study of aDNA from these samples, often referred to as sedimentary DNA (sedaDNA), makes it possible to reconstruct the history of ecosystems over geologic times and to reveal shifts and changes in microbial communities that have occurred in response to natural or anthropogenic constraints. At present, most studies have been performed on sediments and few data are available on ancient soils (Clark & Hirsch, 2008) and permafrost (see below ‘Future perspectives’).
Freshwater and marine sediments
Freshwater (e.g. lake), and marine sediments archive DNA not only from aquatic benthic and pelagic (micro)organisms, but also from terrestrial ones encapsulated in wind‐dispersed propagules (e.g. fungal spores) or that are transported horizontally by rivers to their estuaries where they sediment. Therefore, the study of sedimentary DNA (sedaDNA) composition does not only illustrate past aquatic biodiversity but also the global diversity of surrounding terrestrial ecosystems, including their fauna and flora (Kjær et al., 2022; Wang et al., 2021). Thus, the analysis of sedaDNA extracted from a Latvian lake sediment core, allowed Talas et al. (2021) to retrace the evolution of both aquatic (23% of the molecular taxa) and terrestrial (40%) fungal communities over a period of more than 10,000 years encompassing the Holocene. Besides taxonomic assignation, functional assignation to different trophic modes (e.g. saprotrophs, animal of plant pathogens or symbionts) provided indirect information regarding the occurrence of plant genera (Alnus, Salix,…) known to be specifically associated with particular fungal pathogenic or mutualistic species.
Studies focusing on restricted geographic areas allowed monitoring on a fine temporal scale how human activities strongly, and maybe irreversibly, impacted coastal microbial, and more specifically microeukaryotic communities. In the bay of Brest, on the French Atlantic coast, after a long period of global stability since the Middle Ages, communities of eukaryotic microorganisms ‘suddenly’ changed from the Second World War onwards and since then never turned back to their initial composition (Siano et al., 2021). Dinoflagellates and Stamenopiles were the most affected groups with the almost complete disappearance of taxa that durably dominated the communities since the middle age and their replacement by other taxa such as the potentially harmful, toxin‐producing Alexandrium algae. Such changes parallel and can certainly be attributed to simultaneous abrupt changes in local human activities that also left identifiable traces in sediments in the form of accumulation of inorganic and organic pollutants or changes in sediment accumulation patterns attributable to agricultural practices. Similar dramatic changes in the composition of coastal microbial communities have also been reported by Barrenechea Angeles et al. (2023) who studied the temporal dynamics of bacterial, eukaryotic and Foraminiferal communities in a sediment core sampled in the bay of Pozzoli (Mediterranean Sea, SW Italy). Changes in microbial communities recapitulated the different phases of the heavy industrial development of the surrounding area in the period 1851–1992 that left geochemical signatures in the core layers. Both these studies (Barrenechea Angeles et al., 2023; Siano et al., 2021) highlight the initial status of microbial communities before human impact. This status could be considered as a baseline value that represents the target to reach in a restoration operation of the polluted sites.
Besides studies targeting a single geographic site that recapitulates its local history, multisite studies allow evaluating the impact of more widespread environmental changes. By studying sediment cores sampled in ca 50 lakes in France along a ca 2000 m elevation gradient (Barouillet et al., 2022; Keck et al., 2020) aimed at assessing the impact of the so‐called Anthropocene ‘great acceleration’ on freshwater ecosystems. For each core, DNA was extracted from one layer that was deposited in the 19th century and from a second more modern one representative of extant microbial communities. Metabarcoding analyses targeting either the whole eukaryotic microbial communities (Keck et al., 2020) or more specifically the ciliates (Barouillet et al., 2022), gave convergent results. In a global comparative analysis, it emerged that modern eukaryotic communities of the 50 lakes displayed greater similarity between them than their 19th‐century counterparts. This spatial homogenization was stronger for lakes located below 1400 m above sea level than for those located above. In France, this altitude globally marks the upper limit of permanent human settlements and territories above this line are unlikely to be directly impacted by intensive farming practices that may represent the main causes of diversity changes. Functional assignation of molecular taxa also highlighted pervasive shifts in the functional profile of lake eukaryotic microbial communities. Modern communities were significantly enriched in mixotrophic and photosynthetic taxa at the expense of primary consumers, parasitic and saprotrophic species that were possibly counter‐selected by eutrophication of the lacustrine ecosystems indicated by the higher organic carbon concentrations found in the most recent sediment layers.
HOST‐ASSOCIATED ANCIENT MICROBIAL COMMUNITIES
Host‐associated microbial communities can be affected by the characteristics of their host (species, genotype), its lifestyle (e.g. its diet, its health status) and the environment in which it evolves and where part or all of its microbiome is recruited horizontally. Several studies have examined the relative importance of the host, geography, ecology and environment in the evolution of these communities, which contribute to the fitness and health of their host and partly determine its selective value.
Diversity of source materials
Dental calculus
As in the case of studies targeting a specific microbial species, many paleomicrobiology studies of host‐associated communities regard humans and related species (hominids, monkeys). The most numerous studies focus on the oral ecosystem where mineralization of dental plaque leads to the formation of dental calculus (Figures 1 and 2). It protects the microorganisms that are present in this environment, whose DNA remains preserved over very long periods of time (Ozga & Ottoni, 2023). Interestingly, the ancient DNA of dental calculi from skulls preserved in natural history collections is now being studied not in a historical context, but simply because it allows studying oral microbial communities associated with rare animal species in danger of extinction, whose extant populations have become difficult to sample. An illustration of this approach is the study carried out on three extant species/sub‐species of Gorillas, whose skulls were preserved in various natural history collections (Moraitou et al., 2022). Sequencing of degraded DNA from the corresponding dental calculi indicated that the nature of the Gorilla's diet (ecology), more than the phylogenetic proximity of Gorilla species, determined the composition of the oral microbiome.
Paleofeces and coprolites
These remains represent dried and mineralized fossilized faeces respectively. A paleomicrobiological analysis of coprolites has already provided information about a variety of organisms, including micro‐eukaryotes, bacteria, and archaea, present in this material, thus enhancing our understanding of ancient human diet, gut microbiota, and intestinal and systemic diseases (Appelt et al., 2016). Faeces are more rarely preserved with their original intestinal microbiome, but exceptional cases of preservation do exist, such as in salt‐rich environments (salt mines) (Maixner et al., 2021) or dry desert ones (Wibowo et al., 2021) that allow for a rapid desiccation of the samples.
Zoological collections
In the animal kingdom, studies are however not limited to mammals, several papers have for instance reported the composition of the gut microbiome of specimens of other animal taxa preserved in alcohol in natural history collections. This is the case of related Mexican Herichthys endemic fish species that; for several specimens; were stored for about 50 years in alcohol (Mejía et al., 2022), or for terrestrial snails, several of which collected 98 years before analysis (Chalifour et al., 2022). As in the case of dental calculus, mineralized structures are however more likely to preserve DNA for longer periods. Thus, Scott et al. (2022) successfully extracted and sequenced DNA from millennia‐old corals that revealed their original microbiomes which showed similarities to those from today. However, they identified very few sequences that could be affiliated to Symbiodiniaceae, which are essential eukaryotic phototrophic symbionts of corals.
Herbarium collections
In the case of plants, both aerial and underground (roots) organs of dried herbarium plants have been examined for their microbiome (Bieker et al., 2020; Heberling & Burke, 2019). As most herbaria have been constituted in the 19th and 20th centuries with very few specimens from the 17th and 18th centuries, other sources of plant material have to be looked for to address plant microbiome evolution across longer periods. Ancient plant DNA has been extracted from up to ca 10,000‐year‐old woods preserved in waterlogged environments; however, their associated microbiomes probably correspond to ‘post‐mortem’ communities recruited from the surrounding sediments, and not to the original endophytic ones (Wagner et al., 2018). DNA has also been extracted from seeds or inflorescence of cultivated plants collected in archaeological sites (Kistler et al., 2020; Trucchi et al., 2021), but their microbiome has, thus far, not been specifically reported.
Monitoring host‐associated‐microbiome taxonomic and functional shifts
In this section, using selected examples, we describe studies on past host‐associated microbial communities that investigate either their taxonomic or functional diversity. Both approaches illustrate how microbial communities evolve over time and how environmental factors can influence the microbiome composition, its metabolism and ultimately its activities and roles in its original ecosystem.
Evolution of animal/human microbiomes and taxonomic diversity
Focusing on human dental calculus, evolution of its microbiome has been addressed at different time and geographic scales. Deep in time, Fellows Yates, Velsko, et al. (2021) compared calculus microbiomes of different extant (Gorillas, Chimpanzees, Homo sapiens) and extinct (H. neanderthalensis) Hominids to delineate the set of microbial species shared between the different species (i.e. the Hominid calculus core microbiome) and the set of taxa specific, or more frequent, in one or more species.
As opposed to metabarcoding, thus far commonly used to explore non‐host associated paleo communities, systematic sequencing allows either the de novo assembly of entire microbial genomes (MAGs for Metagenome Assembled Genomes) or the mapping of sequencing reads against already known microbial genomes. Identification of different genomes of the same microbial taxon in different samples allows delineating intraspecific lineages whose distribution in time and space can be studied. This latter approach was presented by Fellows Yates, Velsko, et al. (2021) for three bacterial species found in all studied Hominid species. Phylogenetic analyses based on Single Nucleotide Polymorphisms (SNPs) identified in whole genome sequence data highlighted a greater proximity between human‐associated lineages (from either modern or ancient individuals) that were distinct from Gorilla and Chimpanzee ones that grouped together. Thus, although these three bacterial species belonged to the core Hominid oral microbiome, genome‐level analyses split each of these taxa into different host‐specific groups. A number of potential confounding factors can however contribute to these observed associations between hosts. One of them is geography as almost all human samples were of European origin while Gorillas and Chimpanzees co‐occur in Central Africa.
Data interpretation in paleomicrobiology, especially when referring to microbial communities, is indeed often subject to caution since available archaeological samples (e.g. human remains) are often rare and not evenly distributed (and therefore available for analysis) across time, space and ecological gradients. It is thus often difficult to disentangle the relative contribution of each of these factors and others on the genetic makeup, diversity and evolution of past host‐associated microbial communities. This may explain some conflicting results in studies addressing for example the consequences of the Neolithic transition to agriculture in Europe on the human oral (dental calculus as a proxy) microbiome. Because this progressive transition lasted thousands of years, involved human migrations, episodes of cohabitation between different human groups and did not probably proceed at the same pace in different geographic areas, differences in sample selection may lead to discrepancies across studies. Thus, while Ottoni et al. (2021) concluded that the introduction of farming did not significantly alter the oral microbiome present in ancient foragers, Quagliariello et al. (2022) concluded the opposite. Both studies followed the same approach based on the systematic sequencing of aDNA and the assembly of ancient bacterial genomes. They shared nevertheless a number of common observations, such as the higher frequency of specific taxa (e.g. Olsenella sp. Oral taxon 807) in Neolithic farmers compared to earlier hunter‐gatherers.
At the intraspecific level, as exposed above for different Hominid species (Fellows Yates, Velsko, et al., 2021), association between specific bacterial lineages and specific human groups has also been reported. In the case of commensal Anaerolineaceae oral taxon 438 specific lineages each associated with a specific geographical and chronological group (Mesolithic‐Neolithic) of Humans in Europe were identified (Ottoni et al., 2021) as well as others, specific to Japanese Jomon hunter‐gatherers (−3000 years BP), Japanese Edo agriculturalists (400–140 BP) (Eisenhofer et al., 2020) or native Wichita north Americans (1250–1450 CE) (Honap et al., 2023).
This last observation was part of a study that aimed at understanding the impact of colonization by Europeans of North America on the native North American populations' microbiomes (Honap et al., 2023). Since this historical event is far better documented and took place over a shorter time span (a few centuries) compared to the Mesolithic‐Neolithic transition, it could represent a more appropriate framework to disentangle the relative contribution of geography, nutrition and host (Human) genetics on the Human microbiome.
Besides dental calculi, reconstruction of nearly 500 MAGs has been reported for aDNA extracted from exceptionally well‐preserved 1000–2000 years‐old Native American human faeces found in desert areas of S‐W USA and nearby Mexico (Wibowo et al., 2021). In multivariate meta‐analysis these ancient microbiomes grouped with modern ones from individuals living in ‘non‐industrialized societies’, while microbiomes from individuals from ‘industrialized societies’ grouped together in a distinct cluster. This observation lends support to the hypothesis that the microbiome of extant humans of ‘non‐industrialized societies’ represents an ancestral state that may be explained by similarities in diets between these extant human populations and ancestral ones. Although ancient microbiomes shared similarities with extant ones, the authors reported that 39% of the reconstructed genome sequences corresponded to microbial taxa that had not been reported previously although several thousands of reference genomes are available for the human microbiome (Wibowo et al., 2021). While these microbial taxa may no longer be associated with extant humans, they may still be present, but associated with underexplored human populations (Almeida et al., 2021).
Beyond taxonomic diversity, functional diversity
As opposed to metabarcoding, commonly used to explore non‐host associated paleo communities, systematic sequencing of aDNA offers the opportunity of exploring not only the taxonomic, but also the functional and metabolic diversity of ancient microbial communities. Thus, in their study of the oral microbiome of Hominids, Fellows Yates, Velsko, et al. (2021) identified the putative acquisition of a ‘salivary amylase‐binding capability’ by oral streptococci as a potential functional marker that distinguishes oral Homo sp. Microbiomes from other Hominid ones. Acquisition of this marker could be explained by the adoption of a starch‐rich alimentation by Homo species. Similarly, regarding the gut microbiome, paleofeces and faeces from non‐industrialized extant humans are enriched in genes encoding enzymes degrading starch and glycogen (Wibowo et al., 2021). This difference with ‘industrialized humans’ could result from a larger intake of food products enriched in complex carbohydrates by ancestral and non‐industrialized populations.
Among gene categories that have been scrutinized in both oral (dental calculi) and intestinal (faeces) microbiomes, a special attention has been paid to those coding for antibiotic resistance. In accordance with the hypothesis that the prevalence and diversity of these categories increased as a result of post‐World War II antibiotic massive diffusion, it was observed that tetracycline resistance gene categories were the most enriched ones in comparisons between extant human (from both industrialized and non‐industrialized societies) and ancestral one paleofeces (Wibowo et al., 2021). Similarly, Ottoni et al. (2021) reported the exclusive presence of three antibiotic resistance gene categories in modern dental calculus, the increase in frequency of a fourth one between ancient and modern calculus, but also the disappearance of a fifth one (coding for vancomycin resistance) in the modern calculus.
All studies cited thus far used high‐throughput sequence data to probe both the taxonomic and the functional diversity of ancient microbiomes, it is however conceivable to focus exclusively on functional analyses to address a specific scientific issue. Thus, Brealey et al. (2021) addressed the historical impact of antibiotic use by humans and domestic animals on the distribution and prevalence of antibiotic resistance genes (ARGs) among bacterial communities associated with non‐target wild animal species. This was achieved through the specific identification and annotation of these genes in DNA samples extracted from dental calculi of Brown Bears whose skulls were deposited in natural history collections. These samples, all from Sweden, covered a period of time ranging from 1842 to 2016. It encompassed several phases of antibiotic use in this country. While prior to 1951 antibiotics were not yet available, the 1951–1985 period corresponded to their diffusion and massive use in humans and livestock. After that date, measures were implemented to reduce their use, including their ban as growth promoters. The authors observed that the prevalence, but also diversity of ARGs significantly changed over time, seemingly reflecting both the different phases of antibiotic use in Sweden and their widespread impact even on species remotely associated with humans. Schematically, ARGs increased in abundance during the 1951–1985 period when compared to the pre‐1951 one, to then regress, especially in bear specimens who died after the year 2000 and spent their entire life after antibiotic restrictive measures entered into force. This study represents an additional example of the strength of molecular paleomicrobiology that provides a dynamic view of past events and can be used to evaluate the impact and validity of environmental policies.
FUTURE PROSPECTS
As illustrated in the manuscript, molecular paleomicrobiology is revolutionizing several aspects of microbiology by inserting microorganisms in a historical framework that predates the development of microbiology as a scientific discipline (Figure 3). The large number of studies on human‐associated microbial species and communities identified both long‐term (e.g. Y. pestis or Hepatitis B virus) and occasional (e.g. M. pinnipedii) human ‘companion’ species. Besides revealing the long history of several human/microbe associations, by giving access to historical microbial genomes, paleomicrobiology highlights the temporal population dynamics of major pathogens and the successive acquisition of key functional attributes that are susceptible to affect the outcome of these associations. However, future research should clarify whether these temporal and functional changes are due to co‐evolutionary processes between host and microbe or to competition between microbial lineages that have inadvertently entered into contact as a result of host migration.
FIGURE 3.

Main achievements and research fields explored by molecular paleomicrobiology.
Several studies also identified microbial species or species lineages (in the case of pathogens) that have never been reported in studies on modern samples (Granehäll et al., 2021; Wibowo et al., 2021). These observations are likely to fuel the debate on species extinction in the microbial world and to stimulate systematic surveys to identify potential refuges where microbial species could ‘hide’ for long periods of time. This is particularly relevant in the case of pathogens as illustrated by the apparent reemergence of the WENBA genotype of the hepatitis B virus that prevailed in Bronze Age human populations to then seemingly vanished (Kocher et al., 2021).
Several studies suggest that permafrost, which covers 11% of Earth's land surface (Obu, 2021), could represent a reservoir for ‘ancient’, but still alive microorganisms, including pathogens (Miner et al., 2021). For example, Liang et al. (2021) assembled similar MAGs from both intracellular iDNA and extracellular eDNA extracted from Siberian permafrost samples up to 43,000 years‐old (Pleistocene–Holocene). This observation lends support to the persistence of intact bacterial cells for very long periods of time in this environment from which a variety of viruses and bacteria have been identified and put into culture (Liang et al., 2019, 2021). Thus, a recent outbreak of anthrax caused by the spore‐forming Bacillus anthracis, which severely affected reindeer herds in Siberia, is probably connected with the thawing of the permafrost that is intensifying in the Arctic (Stella et al., 2020).
With the rapid accumulation of data, molecular paleomicrobiology could certainly benefit from the set‐up of specific data repositories that would facilitate comparisons across studies and meta‐analyses. Such a scientific endeavour has been undertaken in the case of ancient human genomes as illustrated by the ancient mtDNA database (amtdb, https://amtdb.org/, (Ehler et al., 2019), the Poseidon framework (https://www.poseidon‐adna.org/#/) or the Allen Ancient DNA Resource (AADR), a curated version of the world's published ancient and modern human DNA data (Mallick et al., 2023). In the case of paleomicrobiology, the AncientMetagenomeDir of SPAAM community represents the first initiative to collect published ancient metagenomics data (Fellows Yates, Andrades Valtueña, et al., 2021).
Another perspective in molecular paleomicrobiology is to go beyond the analysis of ancient DNA extracted from ancient materials and to integrate additional genetic information deduced from the analysis of other biomolecules such as proteins or RNA. Thus, mass spectrometry analysis of proteins extracted from ancient human bones and dental calculi has identified signature peptide sequences of viruses as well as of pathogenic bacteria, such as Hepatitis virus B (Krause‐Kyora et al., 2018), Y. pestis (Barbieri et al., 2017) or Mycobacterium leprae (Fotakis et al., 2020). Regarding RNA, usually considered as a highly labile molecule rapidly degraded upon cell death, it has been shown that plant virus small RNAs are surprisingly more stable than long RNAs and DNA molecules (Hartung et al., 2015; Rieux et al., 2021; Smith et al., 2014). This property was used by Rieux et al. (2021) to assemble a genome of Cassava mosaic virus (ACMV) using small RNAs extracted from a 90 years‐old Cassava herbarium plant herbarium samples. Analysis of small RNAs from ancient cellular microorganisms has not yet been reported.
A FUTURE CONTRIBUTION TO MICROBIAL BIOTECHNOLOGIES?
In addition, the study of ancient metabolites and natural compounds could provide new insights into the evolution and functions of ancient microbiomes through approaches that bring together paleomicrobiology and biochemistry (Klapper et al., 2023; Velsko et al., 2017). Paleobiotechnology is a recently developed discipline that enables the study of the evolution of natural products extracted directly from ancient materials, such as dental calculus (Velsko et al., 2017). By exploiting their potential biological activities, these molecules could represent new drugs to be used in medicine and pharmacology. Besides the direct extraction and characterization of metabolites from ancient samples, ‘paleometabolites’ can also be produced by genetic engineering. This approach was followed by Klapper et al. (2023) who assembled high‐quality MAGs from aDNA extracted from dental calculi of seven hominids (H. neanderthalensis and H. sapiens) who lived between the Middle and Upper Palaeolithic. In MAGs affiliated to the genus Chlorobium the authors identified novel putative biosynthetic gene clusters (BCGs). Heterologous co‐expression in Pseudomonas protegens of two enzyme‐coding genes (plfA and plfB) from one of these clusters resulted in the production of two novel furan‐like molecules called ‘paleofurans’. This proof‐of‐concept study, which makes use of ancient microbial DNA information to characterize experimentally the metabolites produced by the corresponding microbes represents a novel way for discovering new metabolites and to address the biology of past microbial communities.
For millennia, humans have used microorganisms (bacteria, fungi) to transform raw agricultural products (e.g. grains, flour, milk, meat or fruit juice) into elaborated, often more digestible, and tasty food products that can be kept for longer periods of time (e.g. bread, cheese, fermented beverages, processed meat). Extensive studies on several of these microorganisms, as in the case of Saccharomyces cerevisiae or Penicillium spp., clearly show that strains participating in food transformation differ from wild ones (De Chiara et al., 2022; Legras et al., 2018; Peter et al., 2018; Ropars & Giraud, 2022). Furthermore, whole genome phylogenetic analyses also tend to cluster strains according to the food product they have been isolated from (Legras et al., 2018; Peter et al., 2018; Ropars & Giraud, 2022), thus defining in the case of S. cerevisiae so‐called ‘wine’, ‘sake’, ‘cheese’, or ‘beer and bread’ lineages (Legras et al., 2018; Peter et al., 2018). Besides phylogeny, genome comparisons coupled to reverse genetics and large‐scale phenotyping have identified several key genetic determinants and phenotypes that distinguish on the one hand food‐adapted strains from wild ones, and, on the other hand, strains adapted to different foodstuffs (De Chiara et al., 2022; Legras et al., 2018). These observations support the idea that some sort of inadvertent domestication process has led to the selection of these very specific microbial lineages.
We suggest that a molecular paleomicrobiological approach could well be implemented to explore the history and main stages of these domestication processes. Among the main questions that could be addressed, we can cite: (i) Where and to which date can we trace the first signs of domestication? (ii) For a given microbial species and a specific transformation process, has domestication occurred several times independently? (iii) Can we establish a time‐calibrated chronology of the genome modifications that lead to extant domesticated lineages? (iv) Can we identify extinct domesticated lineages characterized by specific genome features? (v) Had other, unsuspected, microbial species been domesticated in the past for the transformation of a specific foodstuff?
Although these questions regard individual species, paleomicrobiological investigations could be extended to microbial communities that participate in the maturation of food products and sometimes define their specificity. In this framework, paleomicrobiological investigations could be carried out on a few century/decade‐old food residues to evaluate recent evolutions in the preparation of so‐called ‘traditional’ local food products.
Archaeological food remains, storage vessels or tools for their preparation could be obvious sources of aDNA from foodborne microorganisms. Human paleofeces have recently been shown to, sometimes, contain significant amounts of DNA from two of these microorganisms, Penicillium spp. and S. cerevisiae, that displayed typical post‐mortem damage features of aDNA (Maixner et al., 2021). Mapping of aDNA reads to modern genomes affiliated the ancient yeast sequences to a ‘beer clade’ and the Penicillium ones to ‘non‐Roquefort’ P. roqueforti strain used nowadays for blue cheese ripening. From a historical point of view, these results suggest that Iron‐age European populations may have already consumed beer and blue cheese and therefore mastered to some extent their preparation. From a technical point of view, this study demonstrates how molecular paleomicrobiology can indeed contribute to microbial biotechnology.
AUTHOR CONTRIBUTIONS
Gianluca Grasso: Visualization (equal); writing ‐ original draft (equal); writing ‐ review and editing (equal). Valeria Bianciotto: Visualization (equal); writing – original draft (equal); writing – review and editing (equal). Roland Marmeisse: Visualization (equal); writing – original draft (equal); writing – review and editing (equal).
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of Interest.
ACKNOWLEDGEMENTS
Work on ancient plant‐associated microbiota at the Muséum National d'Histoire Naturelle (MNHN) was supported by grant ATM 2021 (HoloHerbier) and the Emergence program of Sorbonne Université. GG was supported by a PhD grant from the University of Turin and a mobility grant from the French‐Italian University (program da Vinci 2022). VB was supported by the Short Term Mobility (STM) 2023 program of the National Council of Research (CNR ‐ Italy). We would like to thank our colleague William Conrad Ledford for improving the quality of the manuscript.
Grasso, G. , Bianciotto, V. & Marmeisse, R. (2024) Paleomicrobiology: Tracking the past microbial life from single species to entire microbial communities. Microbial Biotechnology, 17, e14390. Available from: 10.1111/1751-7915.14390
Contributor Information
Gianluca Grasso, Email: gianluca.grasso@unito.it.
Roland Marmeisse, Email: roland.marmeisse@mnhn.fr.
REFERENCES
- Afouda, P. , Dubourg, G. & Raoult, D. (2020) Archeomicrobiology applied to environmental samples. Microbial Pathogenesis, 143, 104140. Available from: 10.1016/j.micpath.2020.104140 [DOI] [PubMed] [Google Scholar]
- Almeida, A. , Nayfach, S. , Boland, M. , Strozzi, F. , Beracochea, M. , Shi, Z.J. et al. (2021) A unified catalog of 204,938 reference genomes from the human gut microbiome. Nature Biotechnology, 39, 105–114. Available from: 10.1038/s41587-020-0603-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appelt, S. , Drancourt, M. & Le Bailly, M. (2016) Human coprolites as a source for Paleomicrobiology. Microbiology Spectrum, 4(4), 1–11. Available from: 10.1128/microbiolspec.poh-0002-2014 [DOI] [PubMed] [Google Scholar]
- Arning, N. & Wilson, D.J. (2020) The past, present and future of ancient bacterial DNA. Microbial Genomics, 6(7), 1–19. Available from: 10.1099/mgen.0.000384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbieri, R. , Mekni, R. , Levasseur, A. , Chabrière, E. , Signoli, M. , Tzortzis, S. et al. (2017) Paleoproteomics of the dental pulp: the plague paradigm. PLoS ONE, 12(7), e0180552. Available from: 10.1371/journal.pone.0180552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barouillet, C. , Vasselon, V. , Keck, F. , Millet, L. , Etienne, D. , Galop, D. et al. (2022) Paleoreconstructions of ciliate communities reveal long‐term ecological changes in temperate lakes. Scientific Reports, 12(1), 1–12. Available from: 10.1038/s41598-022-12041-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrenechea Angeles, I. , Romero‐Martínez, M.L. , Cavaliere, M. , Varrella, S. , Francescangeli, F. , Piredda, R. et al. (2023) Encapsulated in sediments: eDNA deciphers the ecosystem history of one of the most polluted European marine sites. Environment International, 172, 107738. Available from: 10.1016/J.ENVINT.2023.107738 [DOI] [PubMed] [Google Scholar]
- Bieker, V.C. , Sánchez Barreiro, F. , Rasmussen, J.A. , Brunier, M. , Wales, N. & Martin, M.D. (2020) Metagenomic analysis of historical herbarium specimens reveals a postmortem microbial community. Molecular Ecology Resources, 20(5), 1206–1219. Available from: 10.1111/1755-0998.13174 [DOI] [PubMed] [Google Scholar]
- Bonczarowska, J.H. , Susat, J. , Mühlemann, B. , Jasch‐Boley, I. , Brather, S. , Höke, B. et al. (2022) Pathogen genomics study of an early medieval community in Germany reveals extensive co‐infections. Genome Biology, 23, 250. Available from: 10.1186/S13059-022-02806-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos, K.I. , Herbig, A. , Sahl, J. , Waglechner, N. , Fourment, M. , Forrest, S.A. et al. (2016) Eighteenth century Yersinia pestis genomes reveal the long‐term persistence of an historical plague focus. eLife, 5, e12994. Available from: 10.7554/ELIFE.12994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos, K.I. , Kühnert, D. , Herbig, A. , Esquivel‐Gomez, L.R. , Andrades Valtueña, A. , Barquera, R. et al. (2019) Paleomicrobiology: diagnosis and evolution of ancient pathogens. Annual Review of Microbiology, 73, 639–666. Available from: 10.1146/annurev-micro-090817 [DOI] [PubMed] [Google Scholar]
- Bramanti, B. , Wu, Y. , Yang, R. , Cui, Y. & Stenseth, N.C. (2021) Assessing the origins of the european plagues following the black death: a synthesis of genomic, historical, and ecological information. Proceedings of the National Academy of Sciences of the United States of America, 118(36), e2101940118. Available from: 10.1073/pnas.2101940118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brealey, J.C. , Leitão, H.G. , Hofstede, T. , Kalthoff, D.C. & Guschanski, K. (2021) The oral microbiota of wild bears in Sweden reflects the history of antibiotic use by humans. Current Biology, 31(20), 4650–4658.e6. Available from: 10.1016/J.CUB.2021.08.010 [DOI] [PubMed] [Google Scholar]
- Brunson, K. & Reich, D. (2019) The promise of paleogenomics beyond our own species. Trends in Genetics, 35(5), 319–329. Available from: 10.1016/J.TIG.2019.02.006 [DOI] [PubMed] [Google Scholar]
- Chalifour, B.N. , Elder, L.E. & Li, J. (2022) Gut microbiome of century‐old snail specimens stable across time in preservation. Microbiome, 10(1), 1–16. Available from: 10.1186/S40168-022-01286-Z/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark, I. & Hirsch, P. (2008) Survival of bacterial DNA and culturable bacteria in archived soils from the Rothamsted broadbalk experiment. Soil Biology and Biochemistry, 40(5), 1090–1102. Available from: 10.1016/j.soilbio.2007.11.021 [DOI] [Google Scholar]
- Cooper, A. & Poinar, H.N. (2000) Ancient DNA: do it right or not at all. Science, 289(5482), 1139. Available from: 10.1126/SCIENCE.289.5482.1139B [DOI] [PubMed] [Google Scholar]
- Dabney, J. , Meyer, M. & Pääbo, S. (2013) Ancient DNA damage. Cold Spring Harbor Perspectives in Biology, 5(7), 1–7. Available from: 10.1101/CSHPERSPECT.A012567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, N.M. , Proctor, D.M. , Holmes, S.P. , Relman, D.A. & Callahan, B.J. (2018) Simple statistical identification and removal of contaminant sequences in marker‐gene and metagenomics data. Microbiome, 6, 226. Available from: 10.1186/s40168-018-0605-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Chiara, M. , Barré, B.P. , Persson, K. , Irizar, A. , Vischioni, C. , Khaiwal, S. et al. (2022) Domestication reprogrammed the budding yeast life cycle. Nature Ecology & Evolution, 6(4), 448–460. Available from: 10.1038/s41559-022-01671-9 [DOI] [PubMed] [Google Scholar]
- Demeure, C.E. , Dussurget, O. , Guillem, F.M. , Le Guern, A.‐S. , Savin, C. & Pizarro‐Cerdá, J. (2019) Yersinia pestis and plague: an updated view on evolution, virulence determinants, immune subversion, vaccination, and diagnostics. Genes & Immunity, 20, 357–370. Available from: 10.1038/s41435-019-0065-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehler, E. , Novotńy, J. , Juras, A. , Chylénski, M. , Moravčik, O. & Pačes, J. (2019) AmtDB: a database of ancient human mitochondrial genomes. Nucleic Acids Research, 47(D1), D29–D432. Available from: 10.1093/nar/gky843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenhofer, R. , Kanzawa‐Kiriyama, H. , Shinoda, K.I. & Weyrich, L.S. (2020) Investigating the demographic history of Japan using ancient oral microbiota. Philosophical Transactions of the Royal Society B: Biological Sciences, 375(1812), 20190578. Available from: 10.1098/RSTB.2019.0578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falkowski, P.G. , Fenchel, T. & Delong, E.F. (2008) The microbial engines that drive earth's biogeochemical cycles. Science, 320(5879), 1034–1039. Available from: 10.1126/science.1153213 [DOI] [PubMed] [Google Scholar]
- Fellows Yates, J.A. , Andrades Valtueña, A. , Vågene, Å.J. , Cribdon, B. , Velsko, I.M. , Borry, M. et al. (2021) Community‐curated and standardised metadata of published ancient metagenomic samples with AncientMetagenomeDir. Scientific Data, 8, 31. Available from: 10.1038/s41597-021-00816-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellows Yates, J.A. , Velsko, I.M. , Aron, F. , Posth, C. , Hofman, C.A. , Austin, R.M. et al. (2021) The evolution and changing ecology of the African hominid oral microbiome. Proceedings of the National Academy of Sciences of the United States of America, 118(20), e2021655118. Available from: 10.1073/pnas.2021655118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer, W.W. , Hemp, J. & Johnson, J.E. (2016) Evolution of oxygenic photosynthesis. Annual Review of Earth and Planetary Sciences, 44, 647–683. Available from: 10.1146/annurev-earth-060313-054810 [DOI] [Google Scholar]
- Fotakis, A.K. , Denham, S.D. , MacKie, M. , Orbegozo, M.I. , Mylopotamitaki, D. , Gopalakrishnan, S. et al. (2020) Multi‐omic detection of Mycobacterium leprae in archaeological human dental calculus: M. leprae from dental calculus. Philosophical Transactions of the Royal Society B: Biological Sciences, 375(1812), 20190584. Available from: 10.1098/rstb.2019.0584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granehäll, L. , Huang, K.D. , Tett, A. , Manghi, P. , Paladin, A. , O'Sullivan, N. et al. (2021) Metagenomic analysis of ancient dental calculus reveals unexplored diversity of oral archaeal Methanobrevibacter . Microbiome, 9(1), 1–18. Available from: 10.1186/S40168-021-01132-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubry‐Rangin, C. , Kratsch, C. , Williams, T.A. , Mchardy, A.C. , Martin Embley, T. , Prosser, J.I. et al. (2015) Coupling of diversification and pH adaptation during the evolution of terrestrial Thaumarchaeota. Proceedings of the National Academy of Sciences of the United States of America, 112(30), 9370–9375. Available from: 10.5061/dryad.0nv00 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbeck, M. , Seifert, L. , Hänsch, S. , Wagner, D.M. , Birdsell, D. , Parise, K.L. et al. (2013) Yersinia pestis DNA from skeletal remains from the 6th century AD reveals insights into Justinianic plague. PLoS Pathogens, 9(5), e1003349. Available from: 10.1371/JOURNAL.PPAT.1003349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartung, J.S. , Roy, A. , Fu, S. , Shao, J. , Schneider, W.L. & Brlansky, R.H. (2015) History and diversity of citrus Leprosis virus recorded in herbarium specimens. Phytopathology, 105(9), 1277–1284. Available from: 10.1094/PHYTO-03-15-0064-R [DOI] [PubMed] [Google Scholar]
- Heberling, J.M. & Burke, D.J. (2019) Utilizing herbarium specimens to quantify historical mycorrhizal communities. Applications in Plant Sciences, 7(4), e01223. Available from: 10.1002/APS3.1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinnebusch, B.J. , Jarrett, C.O. & Bland, D.M. (2021) Molecular and genetic mechanisms that mediate transmission of yersinia pestis by fleas. Biomolecules, 11(2), 1–13. Available from: 10.3390/BIOM11020210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honap, T.P. , Monroe, C.R. , Johnson, S.J. , Jacobson, D.K. , Abin, C.A. , Austin, R.M. et al. (2023) Oral metagenomes from native American ancestors reveal distinct microbial lineages in the pre‐contact era. American Journal of Biological Anthropology, 182, 542–556. Available from: 10.1002/AJPA.24735 [DOI] [PubMed] [Google Scholar]
- Hug, L.A. , Baker, B.J. , Anantharaman, K. , Brown, C.T. , Probst, A.J. , Castelle, C.J. et al. (2016) A new view of the tree of life. Nature Microbiology, 1, 16048. Available from: 10.1038/NMICROBIOL.2016.48 [DOI] [PubMed] [Google Scholar]
- Jónsson, H. , Ginolhac, A. , Schubert, M. , Johnson, P.L.F. & Orlando, L. (2013) MapDamage2.0: fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics, 29(13), 1682–1684. Available from: 10.1093/bioinformatics/btt193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck, F. , Millet, L. , Debroas, D. , Etienne, D. , Galop, D. , Rius, D. et al. (2020) Assessing the response of micro‐eukaryotic diversity to the great acceleration using lake sedimentary DNA. Nature Communications, 11(1), 1–8. Available from: 10.1038/s41467-020-17682-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kistler, L. , Thakar, H.B. , VanDerwarker, A.M. , Domic, A. , Bergström, A. , George, R.J. et al. (2020) Archaeological central American maize genomes suggest ancient gene flow from South America. Proceedings of the National Academy of Sciences of the United States of America, 117(52), 33124–33129. Available from: 10.1073/PNAS.2015560117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjær, K.H. , Winther Pedersen, M. , De Sanctis, B. , De Cahsan, B. , Korneliussen, T.S. , Michelsen, C.S. et al. (2022) A 2‐million‐year‐old ecosystem in Greenland uncovered by environmental DNA. Nature, 612(7939), 283–291. Available from: 10.1038/s41586-022-05453-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klapper, M. , Hübner, A. , Ibrahim, A. , Wasmuth, I. , Borry, M. , Haensch, V.G. et al. (2023) Natural products from reconstructed bacterial genomes of the middle and upper Paleolithic. Science, 380(6645), 619–624. Available from: 10.1126/science.adf5300 [DOI] [PubMed] [Google Scholar]
- Knights, D. , Kuczynski, J. , Charlson, E.S. , Zaneveld, J. , Mozer, M.C. , Collman, R.G. et al. (2011) Bayesian community‐wide culture‐independent microbial source tracking. Nature Methods, 8(9), 761–763. Available from: 10.1038/nmeth.1650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocher, A. , Papac, L. , Barquera, R. , Key, F.M. , Spyrou, M.A. , Hübler, R. et al. (2021) Ten millennia of hepatitis B virus evolution. Science, 374(6564), 132–188. Available from: 10.1126/science.abi5658 [DOI] [PubMed] [Google Scholar]
- Krause‐Kyora, B. , Susat, J. , Key, F.M. , Kühnert, D. , Bosse, E. , Immel, A. et al. (2018) Neolithic and medieval virus genomes reveal complex evolution of hepatitis B. eLife, 7, e36666. Available from: 10.7554/ELIFE.36666.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebreton, F. , Manson, A.L. , Saavedra, J.T. , Straub, T.J. , Earl, A.M. & Gilmore, M.S. (2017) Tracing the enterococci from Paleozoic origins to the hospital. Cell, 169(5), 849–861.e13. Available from: 10.1016/j.cell.2017.04.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legras, J.L. , Galeote, V. , Bigey, F. , Camarasa, C. , Marsit, S. , Nidelet, T. et al. (2018) Adaptation of S. cerevisiae to fermented food environments reveals remarkable genome plasticity and the footprints of domestication. Molecular Biology and Evolution, 35(7), 1712–1727. Available from: 10.1093/molbev/msy066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennon, J.T. & Locey, K.J. (2020) More support for Earth's massive microbiome. Biology Direct, 15(1), 1–6. Available from: 10.1186/S13062-020-00261-8/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lezcano, M.Á. , Sánchez‐García, L. , Quesada, A. , Carrizo, D. , Fernández‐Martínez, M.Á. , Cavalcante‐Silva, E. et al. (2022) Comprehensive metabolic and taxonomic reconstruction of an ancient microbial mat from the McMurdo ice shelf (Antarctica) by integrating genetic, Metaproteomic and lipid biomarker analyses. Frontiers in Microbiology, 13, 799360. Available from: 10.3389/FMICB.2022.799360/BIBTEX [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang, R. , Lau, M. , Vishnivetskaya, T. , Lloyd, K.G. , Wang, W. , Wiggins, J. et al. (2019) Predominance of anaerobic, spore‐forming bacteria in metabolically active microbial communities from ancient Siberian permafrost. Applied Environmental Microbiology, 85(15), 00560‐19 https://journals.asm.org/journal/aem [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang, R. , Li, Z. , Lau Vetter, M.C.Y. , Vishnivetskaya, T.A. , Zanina, O.G. , Lloyd, K.G. et al. (2021) Genomic reconstruction of fossil and living microorganisms in ancient Siberian permafrost. Microbiome, 9, 110. Available from: 10.1186/s40168-021-01057-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorén, J.G. , Farfán, M. & Fusté, M.C. (2014) Molecular phylogenetics and temporal diversification in the genus Aeromonas based on the sequences of five housekeeping genes. PLoS ONE, 9(2), e88805. Available from: 10.1371/journal.pone.0088805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louca, S. , Shi, P.M. , Pennell, M.W. , Fischer, W.W. , Parfrey, L.W. & Doebeli, M. (2018) Bacterial diversification through geological time. Nature Ecology and Evolution, 2, 1458–1467. [DOI] [PubMed] [Google Scholar]
- Loucaid, S. , Mazelid, F. , Doebeli, M. & Parfrey, L.W. (2019) A census‐based estimate of Earth's bacterial and archaeal diversity. PLoS Biology, 17(2), e3000106. Available from: 10.1371/journal.pbio.3000106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maixner, F. , Sarhan, M.S. , Huang, K.D. , Tett, A. , Schoenafinger, A. , Zingale, S. et al. (2021) Hallstatt miners consumed blue cheese and beer during the iron age and retained a non‐westernized gut microbiome until the baroque period. Current Biology, 31(23), 5149–5162.e6. Available from: 10.1016/j.cub.2021.09.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallick, S. , Micco, A. , Mah, M. , Ringbauer, H. , Lazaridis, I. , Olalde, I. et al. (2023) The Allen ancient DNA resource (AADR): a curated compendium of ancient human genomes. BioRxiv, preprint 10.1101/2023.04.06.535797 [DOI] [PMC free article] [PubMed]
- Marin, J. , Battistuzzi, F.U. , Brown, A.C. & Hedges, S.B. (2017) The Timetree of prokaryotes: new insights into their evolution and speciation. Molecular Biology and Evolution, 34(2), 437–446. Available from: 10.1093/molbev/msw245 [DOI] [PubMed] [Google Scholar]
- Martin, M.D. , Cappellini, E. , Samaniego, J.A. , Zepeda, M.L. , Campos, P.F. , Seguin‐Orlando, A. et al. (2013) Reconstructing genome evolution in historic samples of the Irish potato famine pathogen. Nature Communications, 4(1), 1–7. Available from: 10.1038/ncomms3172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx, V. (2016) Microbiology: the road to strain‐level identification. Nature Methods, 13(5), 401–404. Available from: 10.1038/nmeth.3837 [DOI] [PubMed] [Google Scholar]
- McElwain, J.C. & Punyasena, S.W. (2007) Mass extinction events and the plant fossil record. Trends in Ecology & Evolution, 22(10), 548–557. Available from: 10.1016/j.tree.2007.09.003 [DOI] [PubMed] [Google Scholar]
- Mejía, O. , Sánchez‐Quinto, A. , Gómez‐Acata, E.S. , Pérez‐Miranda, F. & Falcón, L.I. (2022) Unraveling the gut microbiome of the genus Herichthys (Pisces: Cichlidae): what can we learn from museum specimens? Current Microbiology, 79(11), 346. Available from: 10.1007/S00284-022-03047-5 [DOI] [PubMed] [Google Scholar]
- Miner, K.R. , D'Andrilli, J. , Mackelprang, R. , Edwards, A. , Malaska, M.J. , Waldrop, M.P. et al. (2021) Emergent biogeochemical risks from Arctic permafrost degradation. Nature Climate Change, 11(10), 809–819. Available from: 10.1038/s41558-021-01162-y [DOI] [Google Scholar]
- Moraitou, M. , Forsythe, A. , Fellows Yates, J.A. , Brealey, J.C. , Warinner, C. & Guschanski, K. (2022) Ecology, not host phylogeny, shapes the Oral microbiome in closely related species. Molecular Biology and Evolution, 39(12), 1–20. Available from: 10.1093/MOLBEV/MSAC263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morlon, H. (2014) Phylogenetic approaches for studying diversification. Ecology Letters, 17(4), 508–525. Available from: 10.1111/ele.12251 [DOI] [PubMed] [Google Scholar]
- Morlon, H. , Kemps, B.D. , Plotkin, J.B. & Brisson, D. (2012) Explosive radiation of a bacterial species group. Evolution, 66(8), 2577–2586. Available from: 10.1111/j.1558-5646.2012.01598.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayfach, S. , Roux, S. , Seshadri, R. , Udwary, D. , Varghese, N. , Schulz, F. et al. (2021) A genomic catalog of Earth's microbiomes. Nature Biotechnology, 39, 499–509. Available from: 10.1038/s41587-020-0718-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nee, S. , Holmes, E.C. , May, R.M. & Harvey, P.H. (1994) Extinction rates can be estimated from molecular phylogenies. Philosophical Transactions of the Royal Society B: Biological Sciences, 344, 77–82 https://royalsocietypublishing.org/ [DOI] [PubMed] [Google Scholar]
- Nee, S. , May, R.M. & Harvey, P.H. (1994) The reconstructed evolutionary process. Philosophical Transactions of the Royal Society B: Biological Sciences, 344, 305–311 https://royalsocietypublishing.org/ [DOI] [PubMed] [Google Scholar]
- Nwosu, E.C. , Roeser, P. , Yang, S. , Ganzert, L. , Dellwig, O. , Pinkerneil, S. et al. (2021) From water into sediment—tracing freshwater cyanobacteria via DNA analyses. Microorganisms, 9(8), 1778. Available from: 10.3390/MICROORGANISMS9081778/S1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obu, J. (2021) How much of the Earth's surface is underlain by permafrost? Journal of Geophysical Research: Earth Surface, 126(5), 1–5. Available from: 10.1029/2021JF006123 [DOI] [Google Scholar]
- Orlando, L. , Allaby, R. , Skoglund, P. , Der Sarkissian, C. , Stockhammer, P. W. , Ávila‐Arcos, M. C. , Fu, Q. , Krause, J. , Willerslev, E. , Stone, A. C. , & Warinner, C. (2021) Ancient DNA analysis. Nature reviews methods primers, 1(1), 14. Available from: 10.1038/s43586-020-00011-0 [DOI] [Google Scholar]
- Ottoni, C. , Boric, D. , Cheronet, O. , Sparacello, V. , Dori, I. , Coppa, A. et al. (2021) Tracking the transition to agriculture in southern Europe through ancient DNA analysis of dental calculus. Proceedings of the National Academy of Sciences of the United States of America, 118(32), e2102116118. Available from: 10.1073/pnas.2102116118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozga, A.T. & Ottoni, C. (2023) Dental calculus as a proxy for animal microbiomes. Quaternary International, 653–654, 47–52. Available from: 10.1016/J.QUAINT.2021.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawłowska, J. , Lejzerowicz, F. , Esling, P. , Szczuciński, W. , Zajaogonekczkowski, M. & Pawlowski, J. (2014) Ancient DNA sheds new light on the Svalbard foraminiferal fossil record of the last millennium. Geobiology, 12(4), 277–288. Available from: 10.1111/GBI.12087 [DOI] [PubMed] [Google Scholar]
- Pawlowski, J. , Holzmann, M. , Berney, C. , Fahrni, J. , Gooday, A.J. , Cedhagen, T. et al. (2003) The evolution of early foraminifera. Proceedings of the National Academy of Sciences of the United States of America, 100(20), 11494–11498. Available from: 10.1073/pnas.2035132100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearman, J.K. , Biessy, L. , Howarth, J.D. , Vandergoes, M.J. , Rees, A. & Wood, S.A. (2022) Deciphering the molecular signal from past and alive bacterial communities in aquatic sedimentary archives. Molecular Ecology Resources, 22(3), 877–890. Available from: 10.1111/1755-0998.13515 [DOI] [PubMed] [Google Scholar]
- Pedersen, M.W. , Overballe‐Petersen, S. , Ermini, L. , der Sarkissian, C. , Haile, J. , Hellstrom, M. et al. (2015) Ancient and modern environmental DNA. Philosophical Transactions of the Royal Society B: Biological Sciences, 370(1660), 20130383. Available from: 10.1098/rstb.2013.0383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter, J. , De Chiara, M. , Friedrich, A. , Yue, J.X. , Pflieger, D. , Bergström, A. et al. (2018) Genome evolution across 1,011 Saccharomyces cerevisiae isolates. Nature, 556(7701), 339–344. Available from: 10.1038/s41586-018-0030-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quagliariello, A. , Modi, A. , Innocenti, G. , Zaro, V. , Conati Barbaro, C. , Ronchitelli, A. et al. (2022) Ancient oral microbiomes support gradual Neolithic dietary shifts towards agriculture. Nature Communications, 13(1), 1–14. Available from: 10.1038/s41467-022-34416-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rascovan, N. , Sjögren, K.G. , Kristiansen, K. , Nielsen, R. , Willerslev, E. , Desnues, C. et al. (2019) Emergence and spread of basal lineages of Yersinia pestis during the Neolithic decline. Cell, 176(1–2), 295–305.e10. Available from: 10.1016/J.CELL.2018.11.005 [DOI] [PubMed] [Google Scholar]
- Raup, D.M. & Sepkoski, J.J. (1982) Mass extinctions in the marine fossil record. Science, 215(19), 1501–1503. [DOI] [PubMed] [Google Scholar]
- Raxworthy, C.J. & Smith, B.T. (2021) Mining museums for historical DNA: advances and challenges in museomics. Trends in Ecology & Evolution, 36(11), 1049–1060. Available from: 10.1016/j.tree.2021.07.009 [DOI] [PubMed] [Google Scholar]
- Rieux, A. , Campos, P. , Duvermy, A. , Scussel, S. , Martin, D. , Gaudeul, M. et al. (2021) Contribution of historical herbarium small RNAs to the reconstruction of a cassava mosaic geminivirus evolutionary history. Scientific Reports, 11, 21280. Available from: 10.1038/s41598-021-00518-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera‐Perez, J.I. , Santiago‐Rodriguez, T.M. & Toranzos, G.A. (2016) Paleomicrobiology: a snapshot of ancient microbes and approaches to forensic microbiology. Microbiology Spectrum, 4(4), 2165‐0497. Available from: 10.1128/microbiolspec.emf-0006-2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ropars, J. & Giraud, T. (2022) Convergence in domesticated fungi used for cheese and dry‐cured meat maturation: beneficial traits, genomic mechanisms, and degeneration. Current Opinion in Microbiology, 70, 102236. Available from: 10.1016/J.MIB.2022.102236 [DOI] [PubMed] [Google Scholar]
- Sanderson, M.J. & Donoghue, M.J. (1996) Reconstructing shifts in diversification rates on phylogenetic trees. Trends in ecology & evolution, 11(1), 15–20. Available from: 10.1016/0169-5347(96)81059-7 [DOI] [PubMed] [Google Scholar]
- Scott, C.B. , Cárdenas, A. , Mah, M. , Narasimhan, V.M. , Rohland, N. , Toth, L.T. et al. (2022) Millennia‐old coral holobiont DNA provides insight into future adaptive trajectories. Molecular Ecology, 31(19), 4979–4990. Available from: 10.1111/MEC.16642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siano, R. , Lassudrie, M. , Cuzin, P. , Briant, N. , Loizeau, V. , Schmidt, S. et al. (2021) Sediment archives reveal irreversible shifts in plankton communities after World War II and agricultural pollution. Current Biology, 31(12), 2682–2689.e7. Available from: 10.1016/J.CUB.2021.03.079 [DOI] [PubMed] [Google Scholar]
- Signor, P.W. (1994) Biodiversity in geological time. American Zoology, 34, 23–32 https://academic.oup.com/icb/article/34/1/23/111698 [Google Scholar]
- Skoglund, P. , Northoff, B.H. , Shunkov, M.V. , Derevianko, A.P. , Pääbo, S. , Krause, J. et al. (2014) Separating endogenous ancient DNA from modern day contamination in a Siberian Neandertal. Proceedings of the National Academy of Sciences of the United States of America, 111(6), 2229–2234. Available from: 10.1073/pnas.1318934111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, O. , Clapham, A. , Rose, P. , Liu, Y. , Wang, J. & Allaby, R.G. (2014) A complete ancient RNA genome: identification, reconstruction and evolutionary history of archaeological barley stripe mosaic virus. Scientific Reports, 4, 4003. Available from: 10.1038/srep04003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spyrou, M.A. , Musralina, L. , Gnecchi Ruscone, G.A. , Kocher, A. , Borbone, P.G. , Khartanovich, V.I. et al. (2022) The source of the black death in fourteenth‐century Central Eurasia. Nature, 606(7915), 718–724. Available from: 10.1038/s41586-022-04800-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spyrou, M.A. , Tukhbatova, R.I. , Feldman, M. , Herbig, A. , Bos, K.I. , Krause, J.M. et al. (2016) Historical Y. Pestis genomes reveal the European black death as the source of ancient and modern plague pandemics. Cell Host & Microbe, 19, 874–881. Available from: 10.1016/j.chom.2016.05.012 [DOI] [PubMed] [Google Scholar]
- Spyrou, M.A. , Tukhbatova, R.I. , Wang, C.C. , Valtueña, A.A. , Lankapalli, A.K. , Kondrashin, V.V. et al. (2018) Analysis of 3800‐year‐old Yersinia pestis genomes suggests bronze age origin for bubonic plague. Nature Communications, 9(1), 1–10. Available from: 10.1038/s41467-018-04550-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staats, M. , Cuenca, A. , Richardson, J.E. , Vrielink‐Van Ginkel, R. & Petersen, G. (2011) DNA damage in plant herbarium tissue. PLoS ONE, 6(12), 28448. Available from: 10.1371/journal.pone.0028448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stella, E. , Mari, L. , Gabrieli, J. , Barbante, C. & Bertuzzo, E. (2020) Permafrost dynamics and the risk of anthrax transmission: a modelling study. Scientific Reports, 10, 16460. Available from: 10.1038/s41598-020-72440-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susat, J. , Bonczarowska, J.H. , Pētersone‐Gordina, E. , Immel, A. , Nebel, A. , Gerhards, G. et al. (2020) Yersinia pestis strains from Latvia show depletion of the pla virulence gene at the end of the second plague pandemic. Scientific Reports, 10(1), 1–10. Available from: 10.1038/s41598-020-71530-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talas, L. , Stivrins, N. , Veski, S. , Tedersoo, L. & Kisand, V. (2021) Sedimentary ancient DNA (Sedadna) reveals fungal diversity and environmental drivers of community changes throughout the holocene in the present boreal lake Lielais Svētin, U (eastern Latvia). Microorganisms, 9(4), 719. Available from: 10.3390/MICROORGANISMS9040719/S1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tricou, T. , Tannier, E. & de Vienne, D.M. (2022) Ghost lineages can invalidate or even reverse findings regarding gene flow. PLoS Biology, 20(9), e3001776. Available from: 10.1371/JOURNAL.PBIO.3001776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trucchi, E. , Benazzo, A. , Lari, M. , Iob, A. , Vai, S. , Nanni, L. et al. (2021) Ancient genomes reveal early Andean farmers selected common beans while preserving diversity. Nature Plants, 7(2), 123–128. Available from: 10.1038/s41477-021-00848-7 [DOI] [PubMed] [Google Scholar]
- Vågene, Å.J. , Honap, T.P. , Harkins, K.M. , Rosenberg, M.S. , Giffin, K. , Cárdenas‐Arroyo, F. et al. (2022) Geographically dispersed zoonotic tuberculosis in pre‐contact south American human populations. Nature Communications, 13(1), 1–12. Available from: 10.1038/S41467-022-28562-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valtueña, A.A. , Mittnik, A. , Key, F.M. , Haak, W. , Allmäe, R. , Belinskij, A. et al. (2017) The Stone age plague and its persistence in Eurasia. Current Biology, 27(23), 3683–3691.e8. Available from: 10.1016/J.CUB.2017.10.025 [DOI] [PubMed] [Google Scholar]
- Valtueña, A.A. , Neumann, G.U. , Spyrou, M.A. , Musralina, L. , Aron, F. , Beisenov, A. et al. (2022) Stone age Yersinia pestis genomes shed light on the early evolution, diversity, and ecology of plague. Proceedings of the National Academy of Sciences of the United States of America, 119(17), e2116722119. Available from: 10.1073/pnas.2116722119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Kuyl, A.C. (2022) Historic and prehistoric epidemics: an overview of sources available for the study of ancient pathogens. Epidemiologia, 3(4), 443–464. Available from: 10.3390/EPIDEMIOLOGIA3040034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velsko, I.M. , Overmyer, K.A. , Speller, C. , Klaus, L. , Collins, M.J. , Loe, L. et al. (2017) The dental calculus metabolome in modern and historic samples. Metabolomics, 13(11), 134. Available from: 10.1007/s11306-017-1270-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner, S. , Lagane, F. , Seguin‐Orlando, A. , Schubert, M. , Leroy, T. , Guichoux, E. et al. (2018) High‐throughput DNA sequencing of ancient wood. Molecular Ecology, 27(5), 1138–1154. Available from: 10.1111/MEC.14514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Pedersen, M.W. , Alsos, I.G. , De Sanctis, B. , Racimo, F. , Prohaska, A. et al. (2021) Late quaternary dynamics of Arctic biota from ancient environmental genomics. Nature, 600(7887), 86–92. Available from: 10.1038/s41586-021-04016-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warinner, C. , Herbig, A. , Mann, A. , Fellows Yates, J.A. , Weiß, C.L. , Burbano, H.A. et al. (2017) A robust framework for microbial archaeology. Annual Review of Genomics and Human Genetics, 18, 321–356. Available from: 10.1146/annurev-genom-091416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wibowo, M.C. , Yang, Z. , Borry, M. , Hübner, A. , Huang, K.D. , Tierney, B.T. et al. (2021) Reconstruction of ancient microbial genomes from the human gut. Nature, 594(7862), 234–239. Available from: 10.1038/s41586-021-03532-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worobey, M. , Watts, T.D. , McKay, R.A. , Suchard, M.A. , Granade, T. , Teuwen, D.E. et al. (2016) 1970s and ‘patient 0’ HIV‐1 genomes illuminate early HIV/AIDS history in North America. Nature, 539(7627), 98–101. Available from: 10.1038/nature19827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, S. & Kershaw, S. (2012) Microbes and paleoenvironments. Geobiology, 10(1), 1–2. Available from: 10.1111/j.1472-4669.2011.00311.x [DOI] [PubMed] [Google Scholar]
- Yoshida, K. , Schuenemann, V.J. , Cano, L.M. , Pais, M. , Mishra, B. , Sharma, R. et al. (2013) The rise and fall of the Phytophthora infestans lineage that triggered the Irish potato famine. eLife, 2, e00731. Available from: 10.7554/ELIFE.00731 [DOI] [PMC free article] [PubMed] [Google Scholar]