

Abstract
Despite progress in the development of antiseizure medications (ASMs), a third of people with epilepsy have drug resistant epilepsy (DRE). The working definition of DRE, proposed by the International League Against Epilepsy (ILAE) in 2010, helped identify individuals who might benefit from presurgical evaluation early on. As the incidence of DRE remains high, the TASK1 workgroup on DRE of the ILAE/AES Joint Translational Task Force discussed the heterogeneity and complexity of its presentation and mechanisms, the confounders in drawing mechanistic insights when testing treatment responses, barriers in modeling DRE across the lifespan and translating across species. We propose that it is necessary to revisit the current definition of DRE, in order to transform the preclinical and clinical research of mechanisms and biomarkers, to identify novel, effective, precise, pharmacologic treatments, allowing for earlier recognition of drug resistance and individualized therapies.
Introduction
The number of people for whom treatment fails to control their seizures remains at about 30% 1–3. This has not shifted appreciably even though many new antiseizure medications (ASMs) have been introduced. In 2010, the International League Against Epilepsy (ILAE) proposed a working definition of drug resistant epilepsy (DRE) as the persistence of seizures after “adequate trials of two tolerated and appropriately chosen and used ASM schedules”.4 The definition of drug resistance extends to the persistence of auras that may continue even after more disruptive seizure-types are controlled.4 A variety of risk factors for DRE have been proposed (Table 1). A subsequent observational study also reported that the availability of newer ASMs did not improve the likelihood of one year seizure freedom.3 The limitation that the DRE “definition must be based on the probability of subsequent remission after each drug failure” ideally ascertained by “large scale prospective, long-term, population-based studies including both adults and children”, when “few, if any, such studies met such requirement” at the time was already recognized.4
Table 1.
Factors proposed in the literature to increase risk for DRE in certain human epilepsies
| DRE predictors | References |
|---|---|
| Clinical | |
| Younger age at epilepsy onset | 1, 147–153 |
| Short latency to epilepsy development (i.e., after stroke) | 154 |
| Neurodevelopmental abnormalities | 1, 147, 149 |
| Neuropsychiatric comorbidities | 1, 26, 148, 149, 155, 156 |
| Recreational drug use | 26 |
| Focal seizure related comorbidities, e.g., migraine | 1 |
| History of febrile seizures or complex febrile seizures | 1, 26, 147–149 |
| Seizure types | |
| e.g., focal, infantile and epileptic spasms, initial myoclonic seizures | 147, 150, 156 |
| Focal or mixed (vs generalized) | 1, 153 |
| Multiple seizure types | 1, 147, 148, 157, 158 |
| Status epilepticus at epilepsy onset | 159 |
| Status epilepticus | 147–149, 152, 153 |
| Photoparoxysmal response, seizure triggers | 1 |
| Seizure clusters | 1 |
| History of CAE progressing to JME | 148 |
| High seizure frequencies | 147, 150 |
| High baseline seizure frequency | 1 |
| Poor response to first ASM | 1, 147, 150 |
| Number of previous ASM | 1 |
| Number of seizures prior to starting ASM | 26, 158 |
| Ethnicity, socioeconomic factors | 1, 148, 160 |
| History of catamenial epilepsy (JME, GGE) | 1, 148, 156 |
| Family history of epilepsy | 26, 148 |
| Epilepsy etiology | |
| Structural, metabolic, infectious | 1, 147, 149, 150, 158 |
| Traumatic brain injury | 26 |
| Intracerebral hemorrhage | 159 |
| Severe stroke | 159 |
| Neuroimaging (brain) | |
| MRI brain abnormalities | 1, 147, 149 |
| Electrophysiological | |
| Slow background, Multifocal epileptiform EEG | 147, 152 |
| Epileptiform EEG | 1, 153 |
| Epileptiform focality (JME) | 148 |
| Abnormal EEG | 1, 149, 150, 152 |
| Increased generalized spike wave discharges in sleep, generalized polyspikes (IGEs) | 157 |
| Genetic | |
| Gene variants etiologically associated with DREs (multiple, e.g. SCN1A variants) | 161–164 |
| Gene variants associated with drug resistance | |
| ABCB1, ABCC2, CCL2 variants | 150 |
| GABAA receptor subunit variants conferring resistance to benzodiazepines | 62 |
| Biomarkers (protein, miRNAs) | |
| Plasma, serum or CSF biomarkers: multiple, validation needed | Reviewed in 150 |
The list of risk factors for DRE is based on published studies (research studies or reviews) on predictors or risk factors of DRE, focal or generalized onset.
Abbreviations: ABCB1: ATP binding cassettes subfamily B member 1, p-glycoprotein; ABCC2: ATP binding cassettes subfamily C member 2, multidrug resistance protein 2; ASM: antiseizure medication; CAE: childhood absence epilepsy; CCL2: C-C motif chemokine ligand 2; GABA: gamma aminobutyric acid; GGE: genetic generalized epilepsy; JME: juvenile myoclonic epilepsy; IGE: idiopathic generalized epilepsy; SCN1A: sodium channel 1A.
The existing definition of DRE has been valuable for the purposes it was created, namely to improve patient care by recognizing individuals who may need prompt referral to specialized centers for presurgical evaluation and to facilitate clinical research. However, the 2010 definition does not consider the potential causes or mechanisms of DRE; drug-related or individualized factors that may manifest as DRE; or epilepsies that do not have two appropriate therapeutic treatments by which we mean treatments that have a realistic chance of achieving seizure freedom. Further, the binary definition of drug efficacy in epilepsy (persistence of any ongoing seizure vs seizure freedom), while important in urging for earlier consideration of epilepsy surgery, may potentially ignore mechanistic insights from treatments that are only partially effective in controlling seizures. The latter could inform the design of more effective future treatments used either as monotherapies or as combination treatment strategies to further improve seizure control. Also, waiting for two ASM schedules to fail can have severe consequences in developmental and epileptic encephalopathies (DEEs), where early control of seizures may be important for better developmental outcomes.5–7 Preclinical studies have offered invaluable insights into mechanisms, treatments and biomarkers aimed at identifying better medical interventions. Nevertheless, the absence of a definition of drug resistance that is easily applicable to both preclinical and clinical research has hampered the translation of preclinical discoveries to the clinic. It is thus important to revisit the definition of DRE, to address the mechanistic complexity across the lifespan and guide translational research to identify novel, mechanism-targeted effective therapies.
The TASK1 group of the Joint Translational Task Force of the ILAE and the American Epilepsy Society (AES), tasked to re-evaluate preclinical models of epilepsies used for therapy development, initiated a discussion among its members to outline some of the issues to propose a road forward by adopting a new DRE definition that can better serve clinical and preclinical needs leading to better therapies for epilepsies. In this article, we outline the complexity of clinical resistance, discuss the potential underlying mechanisms, and the preclinical models and strategies used to study DRE, emphasize the challenges associated with DEEs and outline the reasons why revisiting the DRE definition would be important to align preclinical and clinical studies so as to provide new directions for mechanistic studies and develop more effective treatments. The terms used in this report and some of their current limitations are outlined in Box 1. Ultimately, this paper should be viewed as an opening conversation in finding solutions to this unresolved problem.
Box 1.
| Definitions | |
|---|---|
| Treatment failure | Treatments have no effect on seizures. This can be due to drug resistance, toxicity, pharmacokinetics / pharmacodynamics, noncompliance. Treatment failure is not necessarily drug resistance. |
| Resistance to a treatment | Lack or reduction in efficacy of a treatment to control seizures, at treatment schedules that would be expected to have the desired biologic effect. Limitations: Effective treatment schedules are usually deduced by population responses and corresponding peripheral blood levels, as target exposure and modification cannot easily be documented in vivo, particularly in humans. Peripheral blood levels do not however reflect accurately the presence or effects of a treatment in the targeted brain regions of an individual. |
| Drug resistant epilepsy (DRE) | “Failure of adequate trials of two tolerated and appropriately chosen and used antiseizure medication (ASM) schedules (whether as monotherapies or in combination) to achieve sustained seizure freedom”. 4 It is assumed that DRE mechanisms may be independent of a specific treatment’s mechanism of action and extend across various medical treatments. Limitations: An individual may still respond to a different treatment, albeit the probability is significantly lower. Partial seizure response may still be a welcome effect for certain individuals or guide the design of more effective treatments. |
| Tolerance | A subject’s diminished response to a treatment after repeated exposure to the treatment, which occurs when the body adapts to the treatment. |
| Therapeutic levels | Levels of a treatment that can affect the desired biologic effect at the target organ. Limitations: Brain levels cannot be usually measured in live subjects. Therapeutic blood levels may not always reflect the levels of a treatment at the target brain region that generates seizures; lack of effect may be also due to inability to reach and modify the function of the target organ or brain region. |
The Clinical Complexity of Drug Resistant Epilepsy
The ILAE report on the definition of drug resistance in epilepsy, acknowledges the complexity of this area and arrived at a pragmatic definition.4 From the mechanistic perspective, it may be wrong to consider DRE as a binary concept, because some people who are resistant to two drugs may fully respond to the next drug tried. Evidence indicates that even after failing 2–5 different ASMs, there can still be a ~17% chance of responding to the next.8 Moreover, someone may apparently respond well to medication initially with drug resistance occurring many years after the first seizure; the delay before drug failure can be over 30 years.9, 10 This suggests that the response of an epilepsy to an ASM can change over time, even in people with non-progressive etiologies.11 Conversely, 20–38% 9, 12–14 people with DRE may go into remission, with 4–5% entering seizure remission per year, 12, 13 but 16–58% 12–14 of these patients later relapse.9, 13–15 These observational studies demonstrate the complexity of drug resistance and indicate that it is a temporally dynamic process. Treatment response may be partial, manifesting as a reduction of seizures or cessation of some but not all of the seizure types.16, 17 Although seizure freedom is the ultimate treatment goal in patient care, when this is not feasible, control of selected seizure types, prevention of prolonged seizure activity (status epilepticus) or the reduction in seizure frequency can have a relevant impact on quality of life and seizure-associated risks. 18–20 The partial treatment response may also help select more effective combination treatments or develop optimized therapies.
DRE may not result from one mechanism only as multiple mechanisms can contribute to drug resistance. Etiology may play a key role. The cause of the epilepsy regardless of the epilepsy syndrome could be responsible for a DRE in all patients (eg CDKL5-DEE).21, 22 But, genetic causes are not synonymous with DRE for all patients. For example, pathogenic variants in KCNQ2 or SCN2A may be associated with either self-limited epilepsies or with a DEE.23, 24 Some syndromes respond better to medication, e.g. the idiopathic generalized epilepsies or the childhood onset self-limited epilepsies which are less associated with resistance than the focal epilepsies or most of the DEEs.10, 11 Also, certain pathologies (such as malformations of cortical development) are more likely to be drug resistant, while post-stroke epilepsies are less likely.21 The two most robust epidemiological predictors of resistance are the number of previous ASMs tried (irrespective of the class of medication) and the number of seizures prior to starting medication, which is more likely a measure of disease severity rather than seizures influencing drug response.8, 25, 26
The diversity of the underlying etiology of early onset DRE is an opportunity for the development of personalized medicine (i.e. targeting the mechanism of the epilepsy, when known, for each patient). Specific non-surgical treatment of the underlying cause has been successful for such diseases as glucose transporter deficiency (GLUT1-DS) and some of the autoimmune disorders.27, 28 Other examples of personalized medicine approaches directed at the underlying cause include everolimus for tuberous sclerosis complex (TSC) epilepsy.29–32 However, patients with similar genetic etiology epilepsies, e.g., with the same TSC genotype, may not always have a positive response to everolimus.29, 31 Indeed, in humans, it is not usually possible to confirm in vivo target modification.
Differences in etiology and resultant syndromes cannot be the whole explanation because epilepsies with essentially identical etiologies in identical brain areas may respond very differently to medications with some becoming seizure free after a single drug and others proving resistant to trials of multiple medications. It may be that genes (perhaps independent of the epilepsy) are contributing to drug responsiveness.
Length of time with epilepsy may also be a determining factor. In children with focal epilepsy, the time to DRE might take years (up to 12 years in one epidemiological study) after initial good seizure control.9 In this clinical pattern, the mechanism of resistance to ASM seems to develop progressively over time or appear after initial efficacy of ASMs. 9
Epilepsies occurring early in life pose another problem as maturational changes may present a pharmacological “moving target” as the networks, channel and receptor expression/function are changing over time.33. A further important consideration is that early life epilepsies may exhibit different seizure types (spasms, tonic or myoclonic), as often observed in DEEs. Achieving seizure freedom will depend upon the efficacy of the drug on all seizure types as well as on the underlying pathogenic epileptogenic mechanisms. Further, there is an urgency to achieve early seizure control to improve the chances for better developmental outcomes in DEEs like infantile epileptic spasms syndrome (IESS).5, 34 To achieve this, we will need better strategies to predict treatment response, even before two treatments fail.
Potential Mechanisms of DRE
For an ASM to be effective in treating epilepsy, it must reach its targets in the brain, affect only critical components of the seizure-related network, maintain its efficacy in the short- or long-term, and not cause intolerable adverse effects. In the following we provide an overview of the hypotheses on the possible mechanisms of DRE. Moreover, we provide an overview of modulatory factors that can influence these mechanisms. Please note that this Task Force report does not aim to provide a comprehensive overview concerning the current state-of-knowledge for all possible mechanisms and all regulatory and modulatory factors. For more details, readers are referred to earlier reviews.35–43
Intrinsic severity hypothesis:
The ‘intrinsic severity hypothesis’ states that epilepsy with a high intrinsic severity is more difficult to treat.44, 45 This hypothesis is based on the clinical observation that a high seizure frequency before epilepsy onset is one of the most important predictors of a poor ASM response. A high intrinsic severity is reflected not only by seizure frequencies but also by other clinical factors including psychiatric comorbidities and neuropathological alterations such as hippocampal sclerosis.44 In the context of the ‘intrinsic severity hypothesis’, it has been pointed out that DRE is not necessarily caused by pharmacokinetic and pharmacodynamic changes, but that it may simply be related to an epileptic network that generates seizures that are difficult to control.
Network hypothesis:
Epilepsy is a disease of altered neuronal networks and their propensity to generate seizures.46, 47 If not all, many types of epilepsy are associated with changes in connectivity patterns between the components of the primary seizure circuit and surrounding or inter-connected cortical and/or subcortical regions.36, 48 Alterations in connectivity can change the physiology of the network and its response to an ASM. Even in well-defined syndromes, such as mesial temporal lobe epilepsy, there is variability in the pathology and connectivity. 49 Thus, interindividual differences in connectivity could affect individual drug-responsiveness.49 Evidence for the network hypothesis comes from the possible clinical success of surgical interventions that can result in seizure control or convert a DRE into a drug-sensitive epilepsy.50–52 Moreover, a clinical association between drug-responsiveness and the connectome has been suggested by neuropathological studies, structural imaging studies, and electroencephalographic functional network topology analysis.49, 53–56 Despite this evidence, it seems crucial to expand our knowledge and further explore the influence of aberrant connectivity on areas and brain circuits involved in ictogenesis and how ASMs may or may not work to prevent ictal activity in critical brain regions.
Target hypothesis:
Epilepsy can be associated with comprehensive changes in the expression of targets, including ion channel isoforms and receptor subunits, and of associated downstream signaling factors. These changes can alter the function of specific targets, which in turn may lead to an altered pharmacological response to ASMs and may make these drugs less effective.41, 57, 58 This situation could explain the lack of efficacy of many ASMs with specific mechanisms of action. While numerous epilepsy- or epileptogenesis-associated alterations of various ASM targets have been suggested by preclinical and clinical studies, a functional link with drug resistance in chronic epilepsy management has been intensely studied for modulators of voltage-gated sodium channels. Electrophysiological studies revealed a reduced sensitivity to carbamazepine in brain tissue from patients with drug-resistant temporal lobe epilepsy.58, 59 Preclinical epilepsy models confirmed this alteration in the responsiveness of voltage-gated sodium channels.60
DRE in patients has also been associated with changes in GABAergic neurotransmission, including subunit configuration,61 binding dynamics62 and signaling of the GABAA receptors. 63
It has been stated that the target hypothesis does not explain why patients are resistant to multiple ASMs with different mechanisms.41 However, in view of the multitude of molecular alterations in the epileptic brain, it is likely that functionally relevant alterations can affect different ASM targets in parallel. Of course, this assumption requires further evidence including more preclinical and clinical research efforts to explore the functional consequences of alterations in various ASM targets.
Aberrant expression of homo- or heteroreceptors mosaics has been reported for different neurological and neuropsychiatric disorders such as Parkinson’s disease, schizophrenia, addiction, and depression.64 Mosaics result from the physical interactions at <40 nm between receptors and ion channels. They can modify transduction signals induced by receptor or channel activation, i.e., a drug that typically induces inhibition can produce excitatory effects when acting on a mosaic. While a possible role of homo- or heteroreceptors mosaic formation has been suggested for DRE,41 the hypothesis remains speculative up to now requiring further studies to explore the hypothesis in the context of DRE.
Blood-brain barrier transporter hypothesis:
Increased expression of efflux transporters (such as P-glycoprotein) at the blood-brain barrier can be associated with a reduced brain penetration of selected ASMs thereby decreasing their efficacy. While several modulatory factors may be involved in the regulation of blood-brain barrier transporters, a series of studies suggested that the seizure-associated increase in P-glycoprotein is related to a glutamate-mediated activation of arachidonic acid signaling involving NMDA receptors, cyclooxygenase-2, and EP1 receptors.65, 66 Studies support that overexpression of drug transporter proteins controlling ASMs penetration into the brain parenchyma is rather restricted to the epileptogenic focus.57, 67 However, the evidence does not exclude an increased expression of drug transporter proteins in non-epileptogenic brain regions.68
Several studies have reported associations of polymorphisms of ABCB1 (p-glycoprotein; ATP-binding cassette subfamily B member 1) or, less frequently, ABCC2 (ATP-binding cassette subfamily C member 2; multidrug resistance protein 2) genes with DRE, although others have not confirmed such associations.69–72 Methodological and population differences may contribute to these different results.
A general limitation of the transporter hypothesis is that it only explains resistance to ASMs that are substrates of human drug efflux transporters. While preclinical and clinical data confirm an overexpression of blood-brain barrier efflux transporters, proof that a prevention of transporter up-regulation can help to overcome DRE is limited to preclinical studies in laboratory rodents. Ex vivo and in vitro studies in human brain capillaries as well as human endothelial cells and astrocytes suggest that translation of different strategies preventing transporter and enzyme induction at the blood-brain barrier may be possible, but further evidence is needed.73
Pharmacokinetic hypothesis:
This hypothesis suggests that DRE can be associated not only with pharmacokinetic alterations in ASM distribution to the brain, but also with increased metabolism and/or elimination of ASMs, leading to lower brain drug concentrations.74 At the systemic level, increased metabolism and elimination of ASMs in the liver, intestine, and kidney can reduce the availability of the drug to penetrate the brain parenchyma and reach epileptic tissue.57 Among others, clinical evidence comes from case reports with persistent low levels of AMSs in plasma and from a molecular imaging study describing increased liver clearance.74
In addition, lower ASM brain concentrations may result from an increased expression of metabolizing enzymes at the blood-brain barrier, which can further increase drug metabolism and reduce drug efficacy.75, 76
The pharmacokinetic hypothesis is likely not a universal explanation for drug resistance, as not all ASMs share the same pharmacokinetic mechanisms. Moreover, peripheral changes in pharmacokinetics would be reflected by reduced plasma concentrations, and it is well known that DRE occurs despite ASM concentrations in the therapeutic range in the majority of patients.
Modulatory factors:
The possible mechanisms of DRE discussed above can be influenced by a variety of factors and can be regulated at different levels. These mechanisms include genetic, epigenetic, and other endogenous modulators, such as inflammatory mediators or metabolic factors. Different pharmacogenetic studies reported an association between ASM response and genetic variants (mutations or polymorphisms) that modify the pharmacological response to ASMs in different types of epilepsy.69, 77 These genetic variants can affect the pharmacokinetics or pharmacodynamics of ASMs.
Gene expression can be modified by different epigenetic mechanisms including histone modification, DNA methylation, and regulatory RNAs including microRNAs (short non-coding RNAs that regulate gene expression).78 An association between changes in different epigenetic mechanisms has been associated with DRE. For example, several studies explored differences in the expression pattern of circulating miRNAs between patients with DRE and drug-sensitive epilepsy.79–81 These differences may for example influence inflammatory mediators as regulatory factors or may more directly contribute to alterations in expression of ASM targets.82
Evidence exists that pro-inflammatory mediators can not only contribute to intrinsic severity but also directly affect pharmacokinetics and pharmacodynamics e.g. by effects on transporter expression or target subunit composition. However, so far evidence for a functional relevance for drug resistance is limited to preclinical studies in a rodent epilepsy model reporting prevention of seizure-associated transporter induction along with an improvement of ASM efficacy.83, 84
Exploratory and descriptive clinical studies also revealed an association between the metabolome as well as the intestinal microbiome and ASM responsiveness.43, 85–87 However, to our knowledge there is a lack of studies exploring the functional relevance of these differences.
In this context, it is important to keep in mind that epilepsy and recurrent seizures can cause a multitude of alterations at the molecular, cellular, and network level. Thus, differences between patients with DRE and drug sensitive epilepsy can always reflect the different level of intrinsic severity and can be driven by uncontrolled seizure activity. Keeping in mind that correlation does not necessarily imply causation, one should be extremely cautious when it comes to conclusions from studies that did not assess the functional consequences of a possible mechanism of DRE and did not test whether it is possible to overcome DRE by targeting this mechanism.
Thus, future preclinical and clinical studies are urgently needed to further study the functional relevance of possible mechanisms and regulatory factors. Thereby, mechanisms and influencing factors need to be analyzed across different patient populations (including pediatric, elderly) and across different etiologies.
DRE as a multifactorial phenomenon
No single theory can explain all cases of DRE. The different hypotheses involve mechanisms that are initially seen as independent, but actually can be linked in many ways.40, 88 For instance, excessive release of glutamate in the brain may contribute to development of DRE because it contributes to neuronal cell loss and network alterations,89 to neuroinflammation,90 and induction of the efflux transporter P-glycoprotein.66, 91 As another example vascular changes, gliosis, and changes in cerebrospinal fluid dynamics in the seizure onset zone may all result in reduced (subtherapeutic) ASM concentrations. Along this line, hippocampal sclerosis is associated with gliosis and abnormal vascular formations with a decreased or absent lumen. This situation reduces blood perfusion in the epileptic zone of patients with temporal lobe epilepsy.92 Then, seizure-associated induction of the efflux transporter P-glycoprotein has not only been reported at the blood-brain barrier but also in neurons with a functional link to increased cell membrane depolarization in the hippocampus and cortex. Thus, P-glycoprotein regulation not only may affect brain pharmacokinetics but also contributes to intrinsic severity.93
Treatments are aimed at controlling seizures and/or modifying disease processes (disease modification/antiepileptogenesis) but, conversely, seizures and disease processes can modify treatment targets and so treatment efficacy.94 DRE can refer to the failure not only of the treatment to control the symptom, the seizures, but also to modify the disease. Resistance to disease modifying effects, other than those directly assessed by seizure outcomes, are however more difficult to evaluate in clinical, and, sometimes preclinical, trials.
As indicated by these examples, research exploring mechanisms of DRE and factors regulating these mechanisms needs to consider the complexity and interconnections. The multifactorial nature implies that it is unlikely that drug resistance can be overcome by targeting one selected mechanism or factor. Moreover, there will not be one single biomarker predicting drug responsiveness. Along this line a recent artificial intelligence case study reported machine-learning based integration of multivariate clinical and genetic data into a multimodal model for prediction of brivaracetam responsiveness.95 This study underlines the need to better consider the highly complex nature of the drug response.
Moreover, DRE needs to be carefully distinguished from other causes of therapeutic failure including tolerance development. Repeated administration of ASMs may cause pharmacodynamic or pharmacokinetic tolerance due to alterations in receptors, such as desensitization, downregulation, internalization, uncoupling from their signal transducers, or metabolic enzyme induction with consequent lower drug efficacy.96, 97 Pharmacokinetic studies support that phenytoin, carbamazepine, or phenobarbital upregulate efflux transporters in the brain and peripheral organs, affecting the bioavailability and disposition of ASMs.98 In this context, it is of interest that administration of ASMs can also result in epigenetic changes that negatively modify the course of the disease or may facilitate excitatory neurotransmission.99,100 Respective alterations can contribute to intrinsic severity of the disease and therapeutic failure.
Using animal models to understand drug resistance
Animal models of seizures or epilepsies have been used to screen for therapies that may be more effective in controlling seizures than existing treatments or to identify the mechanisms that prevent or control seizures or, in contrast, that underlie treatment resistance.101–104 As drug resistance in epilepsy is a multifactorial phenomenon, models of DRE should ideally reflect one or more of the different mechanisms of drug resistance found in humans.35, 104
Several of the animal models that have been used so far are based on the use of young adult or adult animals (Supplementary Table 1).40, 105 These models are therefore more relevant for DRE in young adult patients and extrapolation to both pediatric and elderly DRE populations may be inappropriate. Another limitation of many models and studies is the frequent focus on one sex, further limiting generalizability.40
Among available models, acute rodent seizure models are used for the early phase of efficacy testing to allow high throughput in the selection of promising drug candidates.35, 104 Considering the complexity of probable drug resistance mechanisms affecting the pharmacokinetics and pharmacodynamics of ASMs and the variety of factors that can modulate these mechanisms over time (e.g. genetic, epigenetic etc.),35, 40, 41 these models can be characterized by a poor ASM responsiveness, but are unlikely to reflect the complexity of possible mechanisms for DRE. However, acute seizure models may reflect the intrinsic disease severity and the nature of the seizure networks that have a major impact on initial (screening) therapeutic responses.44, 45, 53, 106. This aspect has also been taken into account for the reorganization of the National Institute of Neurological Disorders and Stroke (NINDS) Epilepsy Therapy Screening Project (ETSP), resulting in the decision to avoid the use of acute seizure models as the only filter and to give drug candidates another chance in a chronic paradigm even in case of failure to exert antiseizure effects in an acute model.104 Among acute seizure models, the 6-Hz model, commonly used in young adult mice, stands out for its particularly poor responsiveness to various ASMs.107 It has been proposed as a model for drug-resistant seizures and is an early test in the ETSP.104
The chronic rodent models, models with repeated seizure induction, i.e. electrical or chemical kindling models, are characterized by multiple molecular, cellular and network alterations.101 Thus, these models can reflect selected mechanisms of drug resistance in epilepsy. However, as they do not have spontaneous seizures, they cannot be considered true models of DRE. In this context, it is also emphasized that models, in which exposure to an ASM during the phase with repeated seizure induction results in a poor ASM responsiveness (e.g., lamotrigine-resistant kindled rats or mice),108–110 may model contingent tolerance rather than drug resistance.
There are rodent models with spontaneous seizure development that have been partly characterized concerning their responsiveness to ASMs.35 While selected probable mechanisms of drug resistance and influencing factors have been studied, major gaps-in-knowledge are evident within the long list of potential mechanisms and modulatory factors.101 Considering the time-consuming and elaborate nature of seizure recording in chronic models, the duration of video-EEG based seizure monitoring is often limited without long-term follow up. Thus, the majority of studies do not provide information about the course and pattern of responsiveness or resistance.111–113 This is particularly relevant to models that do not have frequent spontaneous seizures. Another potential limitation may arise from an emphasis on the detection of generalized motor seizures, which may result in the failure to assess the responsiveness of all seizure types. The list of chronic rodent models with a relevant pharmacological characterization is still quite short,35 and by no means reflects the full range of possible etiologies and epilepsy types.
A DRE model may have been proposed because of its origin, e.g., resected human epileptogenic tissue, or its relevance to pathologies associated with DRE, or because its seizures fail to respond to treatments appropriate for the syndrome it models (Supplementary Table 1, Supplementary Tables 2). Typically, testing of such drugs in animal models has been done independently for each drug, rather than consecutively exposing animals to different appropriate drugs. While some of the tested compounds that showed promise in such models of DRE have eventually entered clinical practice or clinical testing,32, 114–121 there are lessons that need to be considered to improve our future approaches to therapy development.
Issues related to pediatric models of DRE:
Aligning the goals of preclinical and clinical research will facilitate translation and repurposing of preclinical findings into the clinics. The current definition of DRE poses however several challenges. There are unique characteristics, needs, and challenges regarding pediatric DRE. Age-specific models are necessary to address age- and sex-specific influences on the expression or function of the therapy targets, effects of treatments, or the pharmacosensitivity of age-specific seizures or epilepsies. Models of age-specific seizures are also constrained by the short developmental period when such seizures are expressed, i.e. days, and the rapid trajectories of developmental changes that occur in the brain, which also influence the efficacy of the drugs. Therefore, the developmental age-, region-, and sex-specific structural and functional changes in innate networks and mechanisms involved in epilepsy may manifest as apparent transient periods of remission or resistance, irrespective of the drug’s therapeutic potential (Figure 1).33, 122–126 Maturation may also alter systems affecting drug bioavailability, metabolism and clearance, requiring adaptation of the treatment protocols for specific ages.127, 128 A drug’s ability to modify a critical pathway in epileptogenesis may manifest during tight developmental periods only.129 Conversely its inability to control seizures during a period when the drug’s target is not operative130 or relevant,129 or leads to paradoxical responses,131, 132 may not preclude its therapeutic potential if used in more appropriate developmental periods. Drug resistance in epilepsy may therefore not be a static, persistent feature, and knowing the complex factors, developmental or other, that govern it may help optimize epilepsy treatments. Correlation of the findings between animal models and humans is complicated by the species differences in the temporal trajectories of developmental processes, as well as in drug effects and targets. To deliver effective specific screening platforms and treatments for early life drug-resistant seizures and epilepsies with specific purposes, it is important to develop biomarkers to guide the selection and implementation of treatments across species.
Figure 1. Challenges in interpreting drug responsiveness and resistance in models of early life epilepsies.
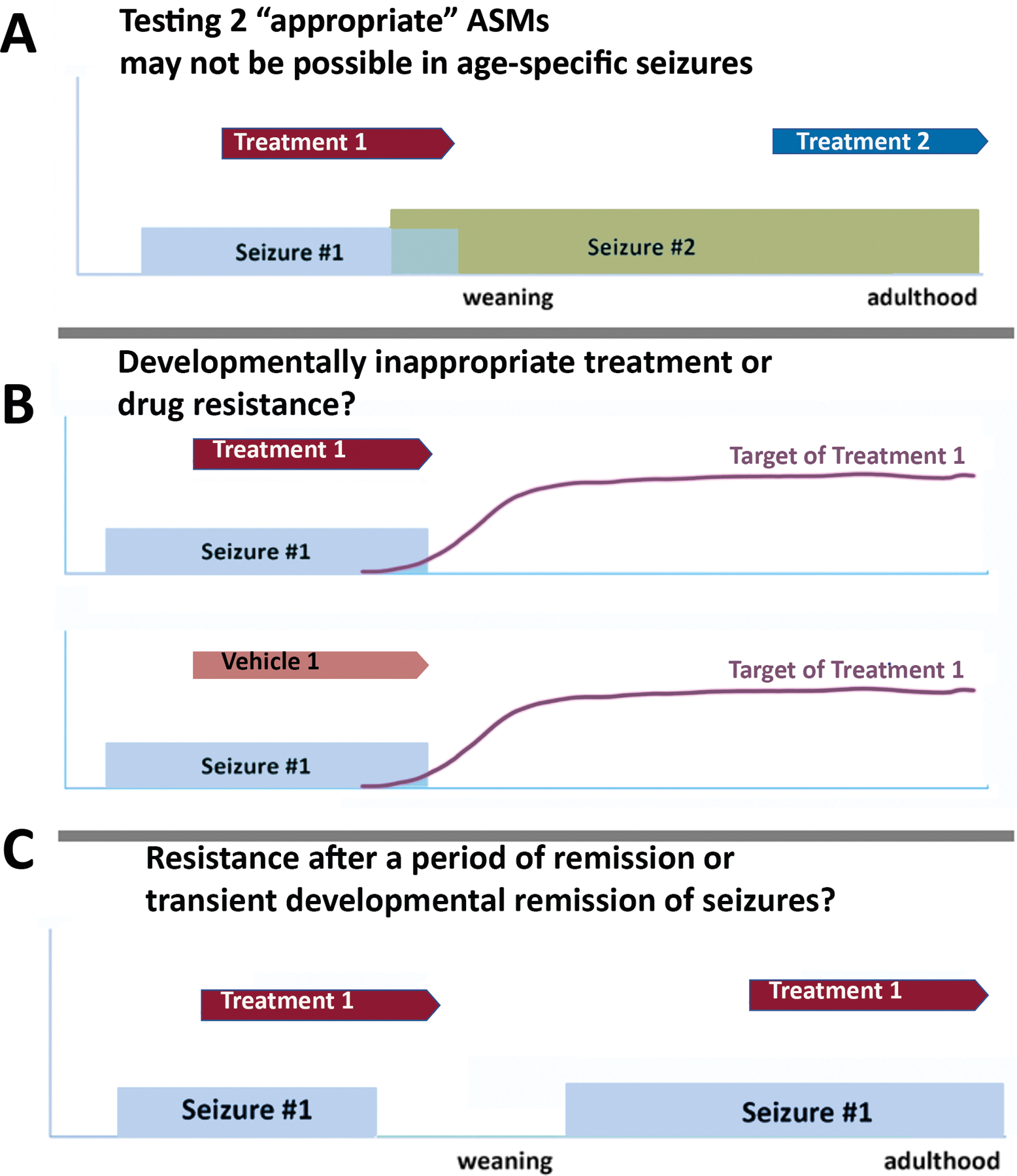
Development, i.e., the period till the time adulthood is reached, can be relatively short in rodents where puberty is reached around postnatal days 32–36 (PN32–36) and age-specific seizures (seizure #1 in the graphs) may occur during shorter developmental periods. Further, the ongoing brain development leads to continuous developmental changes, including of potential treatment targets. Assessing drug responsiveness in this setting, agnostic of how target expression/function or seizure natural history is, can be challenging, as presented by the following scenarios.
A. In a model with an evolving phenotype with early age-specific seizure #1 and late onset of seizure #2, testing two “appropriate” treatments can be challenging as these are likely to be tested upon different types of seizures, and at different stages of brain development. Seizures #1 and #2 may have known different pharmacosensitivities (i.e., spasms vs focal seizures) and therefore effects of drug on seizure #2 may not necessarily predict its effects on seizure #1. Furthermore, testing the treatment #2 in adulthood may not predict the treatment effects in early development if the drug’s target expression or relevance change with age.
B. Testing of treatment #1 on age-specific seizure #1 has no effect compared to vehicle, yet the target of treatment #1 is not yet expressed or functional during the period when seizure #1 is present. Is this seizure type resistant to treatment #1 or is the treatment developmentally inappropriate? In the clinical setting we cannot always test this possibility.
C. A model has an early appearance of seizure #1, a period without seizures and late reappearance of seizure #1. Treatment #1 appears to stop seizure #1 when given early in life but not when given at the late recurrence. Is this late resistance after a period with drug-sensitive seizures, or a transient developmental remission of seizures treated with an ineffective treatment, or apparent late resistance of seizures because its target (and hence efficacy) is only present during the early developmental period?
There are several in vivo and in vitro models used to study early life seizures or epilepsy; a variety of assessments of drug efficacy have been used, including outcomes relevant to age-specific seizure types (e.g., epileptic spasms, thermally-induced seizures) (Supplementary Table 2). 133–140 Methods of induction resemble etiologies of DREs and render seizures or epilepsies refractory to seizures from the start.15, 141 Testing two consecutive treatments may result in testing them against different types of seizures, with distinct pharmacosensitivity, or during different developmental periods with distinct target relevance (Figure 1). Tools to track in live mode epileptogenic or seizure-generating mechanisms and target modification by the drugs would be needed. Validation of a model as a model of DRE, based on its pharmacosensitivity to at least two ASMs that achieve seizure freedom in the respective human epilepsy syndrome is difficult to translate. Waiting for two “appropriate” medications, when they exist, to declare their effects may result in valuable loss of time and potentially diminish the likelihood of clinical success, as suggested by the experience in treating IESS.5 Testing seizure freedom in animal models with the expectations of “at least 3x the longest pre-intervention interseizure interval or 12 months, whichever is the longest” may be inappropriate in the setting of DEEs.
Tolerability:
Conclusions about drug responsiveness in animal models also need to consider tolerability. In the context of the elaborate nature of studies in chronic models with spontaneous recurrent seizures, titration of the dosage to the maximum tolerated dose is often omitted in preclinical studies. Thus, it is frequently not possible to conclude about an actual drug resistance. Further limitations of the assessment of protective indices (toxic dose of a drug for 50% of the population divided by the effective dose for 50% of the population) of ASMs in rodents are based on the fact that an assessment of the adverse effect potential is often focused on motor function, well-being or mortality, and neglects other relevant tolerability issues.
Pharmacokinetics and pharmacodynamics:
In chronic models, long-term, consistent dosing must be ensured to maintain drug levels for weeks or months. As animals tend to metabolize drugs more rapidly, careful consideration of species-, age- and sex-differences in pharmacokinetics, and special delivery methods to achieve steady-state drug levels may be needed. Studies in young rodents face particular challenges related to limitations in the miniaturization of the delivery devices. In addition, one needs to consider species differences as well as age- and model-associated alterations in pharmacodynamics. For instance, target expression, function or downstream signaling changing over time can significantly affect pharmacodynamics despite stable drug levels at the target sites.
In vitro models of DRE:
In vitro and ex vivo testing approaches have been developed using slices, cells or capillaries from surgical specimen or from experimental animals, cell lines or patient-derived iPSCs and organoids.142–144 These approaches can serve as valuable additional tools to screen ASMs and test whether the drug candidate is affected by a selected specific drug resistance mechanism. However, these approaches have limitations as they cannot consider the variety and complexity of potential drug resistance mechanisms and influencing factors. A major limitation of studies in surgical specimens is that they are only available from patients with DRE not allowing a comparison between patient subgroups with differences in responsiveness. Interestingly, different drug responses across different slices from the same patient have been documented, highlighting the complexity of seizure-controlling networks and drug resistance mechanisms.
Conclusions
We propose that it is important to revisit DRE as a concept to provide a roadmap to address future clinical needs as well as harmonize clinical and preclinical research towards prompter, more effective, mechanism-informed, diagnostic, monitoring and therapeutic approaches for DRE. The DRE concept should ideally consider the heterogeneity, complexity of epilepsies and drug resistance mechanisms across the lifespan, as well as allow prompt recognition of drug resistance at earlier timepoints to facilitate earlier implementation of effective treatment strategies as they become available (Box 2 and Box 3). Agreeing on priorities for future clinical and preclinical research on DRE and aligning the goals and endpoints would potentially be important in improving transfer of knowledge and tools from the bench to the clinics. It would be important not only to define the causes of drug resistance in specific animal models but also to have biomarkers of those causes that can be used to predict drug resistance in humans and, importantly, to indicate the potential underlying mechanisms of drug resistance in an individual that can be used to develop better, more effective treatment approaches. In addition, it is important to consider the patients’ seizure susceptibility patterns.145, 146 This knowledge can facilitate the design of chronotherapy strategies focused to improve the efficacy of ASMs and reduce their side effects in patients with DRE.
Box 2.
| A. Key points from the clinical management of DRE |
|---|
| 1. Failure of two appropriate and tolerated treatments reduces but does not preclude seizure freedom in response to a different antiseizure medication. |
| 2. Some epilepsies or etiologies are drug resistant from the start or do not have treatments leading to seizure freedom and are therefore not addressed by the current DRE definition. |
| 3. Earlier diagnosis of resistance to a drug or of DRE, before the failure of two appropriate and tolerated treatments, could accelerate decisions to direct care to more effective treatment choices, when these are available. |
| 4. Clinical assessments of treatment response are not absolute and may be confounded by the natural history of epilepsy or comorbidities, or other pharmacological or contextual factors influencing target relevance or modification by the drug. |
| 5. “Appropriate treatment” is usually extrapolated by data from responses of populations with similar seizure types and may not necessarily be effective for a given individual with such seizures or across etiologies. |
| 6. The efficacy of a treatment in a patient may change over time, as diverse factors that control the biological effects of a drug may change. |
| 7. The current DRE definition relies on seizure freedom as readout of success which is a delayed endpoint; search for tools to monitor earlier effects on the epileptogenic substrate that predict treatment response might accelerate the diagnosis of DRE. |
| 8. Treatments with incomplete efficacy on seizures may still be useful in improving quality of life or informing on future successful combination treatments. |
| B. Framework for future research on DRE |
| 1. Further research on the multifactorial mechanisms of DRE is needed to expand current knowledge, on: (a) drug pharmacokinetics-pharmacodynamics, (b) transport and access to brain targets, (c) effects on drug targets, networks and off target sites, (d) genetic and epigenetic mechanisms, (e) other co-occurring biological processes or medical conditions, (f) exogenous or environmental factors. |
| 2. Better define appropriate and effective treatments for a person with epilepsy at early stages after diagnosis, so as to inform and accelerate treatment decisions. |
| 3. Define and monitor the biological substrates and biomarkers of drug response and drug resistance for a given individual with epilepsy, to enable early prediction of treatment response. |
| 4. Determine patterns of DRE and critical windows for interventions to maximize therapeutic effects and prevent adverse consequences (cognitive, SUDEP, etc) for DRE across the lifespan. |
| 5. Develop treatments targeting drug resistance substrates that are also effective in the context of specific epileptogenic processes. |
Box 3.
| Reasons for redefining DRE |
|---|
| The current definition of DRE has increased awareness of the value of prompt referrals for presurgical evaluation of individuals whose epilepsy did not respond to antiseizure medications. However, as the incidence of DRE remains high, we may need to revisit DRE to steer new research and clinical efforts towards more effective, rational, and precise treatments. We propose to revisit the DRE concept to incorporate the following elements. |
|
|
|
|
|
Supplementary Material
Key Points.
The ILAE definition of drug resistant epilepsy (DRE) aimed to improve patient care but does not address the clinical complexity of DRE.
The DRE concept should ideally consider the heterogeneity, complexity of epilepsies and drug resistance mechanisms across the lifespan.
Earlier recognition of drug resistance could facilitate earlier implementation of effective and individualized treatment strategies.
A DRE concept that aligns the goals of preclinical and clinical research could facilitate translation of preclinical findings into the clinics
Acknowledgements
This report was written by experts selected by the International League Against Epilepsy (ILAE) and American Epilepsy Society (AES) and was approved for publication by the ILAE and AES. Opinions expressed by the authors, however, do not necessarily represent the policy or position of the ILAE or AES. The authors acknowledge the support of AES and ILAE for sponsoring the activities of the task force.
S. Auvin acknowledges research support by the Institut Universitaire de France, French Dravet Association and Fondation Française de Recherche sur l’épilepsie.
A.S. Galanopoulou acknowledges grant support by NINDS U54 NS100064, NINDS R01 NS127524, a pilot grant from NICHD center grant (P50 HD105352) for the Rose F. Kennedy Intellectual and Developmental Disabilities Research Center (RFK-IDDRC), U.S. Department of Defense (W81XWH-22-1-0510, W81XWH-22-1-0210), the Heffer Family and the Segal Family Foundations, and the Abbe Goldstein/Joshua Lurie and Laurie Marsh/Dan Levitz families.
S.L. Moshé is the Charles Frost Chair in Neurosurgery and Neurology and acknowledges grant support by NIH U54 NS100064, NINDS R01 NS127524, and NS43209, a pilot grant from NICHD center grant (P50 HD105352) for the Rose F. Kennedy Intellectual and Developmental Disabilities Research Center (RFK-IDDRC), U.S. Department of Defense (W81XWH-22-1-0510, W81XWH-22-1-0210), the Heffer Family and the Segal Family Foundations, and the Abbe Goldstein/Joshua Lurie and Laurie Marsh/Dan Levitz families.
H. Potschka acknowledges research support by Deutsche Forschungsgemeinschaft (e.g. PO 681/12-1) and EU-IMI (grant agreement no. 777364).
L. Rocha acknowledges research support by the National Council of Science and Technology (CONACyT grant A3-S-26782).
M. Walker receives support from the Department of Health’s National Institute for Health Research University College London/University College London Biomedical Research Centre.
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Disclosure of conflicts of interest
S. Auvin is Deputy Editor for Epilepsia. He has served as consultant or gave lectures for Angelini, Biocodex, Eisai, Encoded, Grintherapeutics, Jazz Pharmaceuticals, Neuraxpharm, Orion, Nutricia, Proveca, UCB Pharma, Vitaflo, Xenon, Zogenix. He has been investigator for clinical trials for Eisai, Jazz Pharmaceuticals, Marinus, Proveca, Takeda, UCB Pharma and Zogenix.
A.S. Galanopoulou is the Editor-in-Chief of Epilepsia Open, associate editor of Neurobiology of Disease, and receives royalties from Elsevier (publications, journal editorial board participation) and Medlink (publications).
S.L. Moshé is on the editorial board of Brain and Development, Pediatric Neurology, Annals of Neurology, MedLink and Physiological Research. He received royalties from Elsevier for his work as Associate Editor in Neurobiology of Disease; annual compensation from MedLink; and royalties from 2 books he co-edited.
H. Potschka has received funding for consulting, talks and research collaborations from Eisai, Zogenix, Bayer/Elanco, Roche, Lario/Exeed Epidarex, Angelini, Galapagos, MSD, and Jazz (GW) Pharmaceuticals.
L. Rocha has no disclosures to declare.
M. Walker has received funding for consulting and for talks from Eisai, UCB pharma, Angelini, Seer, Marinus and Jazz. He has shares in EpilepsyGtx.
Footnotes
Disclaimer: The above examples are given to highlight the ambivalence in concluding whether a treatment is effective or appropriate under different scenarios, given some features peculiar to pediatric epilepsies. The discussion herein does not exclude that additional factors or explanations may also contribute to these patterns.
References
- 1.Sultana B, Panzini MA, Veilleux Carpentier A, et al. Incidence and Prevalence of Drug-Resistant Epilepsy: A Systematic Review and Meta-analysis. Neurology. 2021;96:805–17. [DOI] [PubMed] [Google Scholar]
- 2.WHO. Epilepsy: a public health imperative. Geneva: World Health Organization; 2019. [Google Scholar]
- 3.Chen Z, Brodie MJ, Liew D, et al. Treatment Outcomes in Patients With Newly Diagnosed Epilepsy Treated With Established and New Antiepileptic Drugs: A 30-Year Longitudinal Cohort Study. JAMA Neurol. 2018;75:279–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kwan P, Arzimanoglou A, Berg AT, et al. Definition of drug resistant epilepsy: consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia. 2010;51:1069–77. [DOI] [PubMed] [Google Scholar]
- 5.O'Callaghan FJ, Lux AL, Darke K, et al. The effect of lead time to treatment and of age of onset on developmental outcome at 4 years in infantile spasms: evidence from the United Kingdom Infantile Spasms Study. Epilepsia. 2011;52:1359–64. [DOI] [PubMed] [Google Scholar]
- 6.Kivity S, Lerman P, Ariel R, et al. Long-term cognitive outcomes of a cohort of children with cryptogenic infantile spasms treated with high-dose adrenocorticotropic hormone. Epilepsia. 2004;45:255–62. [DOI] [PubMed] [Google Scholar]
- 7.Riikonen R A long-term follow-up study of 214 children with the syndrome of infantile spasms. Neuropediatrics. 1982;13:14–23. [DOI] [PubMed] [Google Scholar]
- 8.Schiller Y, Najjar Y. Quantifying the response to antiepileptic drugs: effect of past treatment history. Neurology. 2008;70:54–65. [DOI] [PubMed] [Google Scholar]
- 9.Berg AT, Vickrey BG, Testa FM, et al. How long does it take for epilepsy to become intractable? A prospective investigation. Ann Neurol. 2006;60:73–9. [DOI] [PubMed] [Google Scholar]
- 10.Ramos-Lizana J, Rodriguez-Lucenilla MI, Aguilera-Lopez P, et al. A study of drug-resistant childhood epilepsy testing the new ILAE criteria. Seizure. 2012;21:266–72. [DOI] [PubMed] [Google Scholar]
- 11.Geerts A, Brouwer O, Stroink H, et al. Onset of intractability and its course over time: the Dutch study of epilepsy in childhood. Epilepsia. 2012;53:741–51. [DOI] [PubMed] [Google Scholar]
- 12.Choi H, Heiman G, Pandis D, et al. Seizure remission and relapse in adults with intractable epilepsy: a cohort study. Epilepsia. 2008;49:1440–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Callaghan B, Schlesinger M, Rodemer W, et al. Remission and relapse in a drug-resistant epilepsy population followed prospectively. Epilepsia. 2011;52:619–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brodie MJ, Barry SJ, Bamagous GA, et al. Patterns of treatment response in newly diagnosed epilepsy. Neurology. 2012;78:1548–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schmidt D, Loscher W. Drug resistance in epilepsy: putative neurobiologic and clinical mechanisms. Epilepsia. 2005;46:858–77. [DOI] [PubMed] [Google Scholar]
- 16.Gilioli I, Vignoli A, Visani E, et al. Focal epilepsies in adult patients attending two epilepsy centers: classification of drug-resistance, assessment of risk factors, and usefulness of “new” antiepileptic drugs. Epilepsia. 2012;53:733–40. [DOI] [PubMed] [Google Scholar]
- 17.French JA. Refractory epilepsy: one size does not fit all. Epilepsy Curr. 2006;6:177–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alnaamani A, Ahmad F, Al-Saadoon M, et al. Assessment of quality of life in children with epilepsy in Oman. J Patient Rep Outcomes. 2023;7:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tayeb HO, Alsawwaf Y, Khoja AA, et al. Determinants of Health-Related Quality of Life of Epilepsy Patients in Jeddah, Saudi Arabia. Cureus. 2022;14:e24118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guekht AB, Mitrokhina TV, Lebedeva AV, et al. Factors influencing on quality of life in people with epilepsy. Seizure. 2007;16:128–33. [DOI] [PubMed] [Google Scholar]
- 21.Muller A, Helbig I, Jansen C, et al. Retrospective evaluation of low long-term efficacy of antiepileptic drugs and ketogenic diet in 39 patients with CDKL5-related epilepsy. Eur J Paediatr Neurol. 2016;20:147–51. [DOI] [PubMed] [Google Scholar]
- 22.Lim Z, Wong K, Olson HE, et al. Use of the ketogenic diet to manage refractory epilepsy in CDKL5 disorder: Experience of >100 patients. Epilepsia. 2017;58:1415–22. [DOI] [PubMed] [Google Scholar]
- 23.Wolff M, Johannesen KM, Hedrich UBS, et al. Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders. Brain. 2017;140:1316–36. [DOI] [PubMed] [Google Scholar]
- 24.Kato M, Yamagata T, Kubota M, et al. Clinical spectrum of early onset epileptic encephalopathies caused by KCNQ2 mutation. Epilepsia. 2013;54:1282–7. [DOI] [PubMed] [Google Scholar]
- 25.Regesta G, Tanganelli P. Clinical aspects and biological bases of drug-resistant epilepsies. Epilepsy Res. 1999;34:109–22. [DOI] [PubMed] [Google Scholar]
- 26.Hitiris N, Mohanraj R, Norrie J, et al. Predictors of pharmacoresistant epilepsy. Epilepsy Res. 2007;75:192–6. [DOI] [PubMed] [Google Scholar]
- 27.Helbig I, Ellis CA. Personalized medicine in genetic epilepsies - possibilities, challenges, and new frontiers. Neuropharmacology. 2020;172:107970. [DOI] [PubMed] [Google Scholar]
- 28.Meador KJ, Shin C. Pitfalls in developing precision medicine for genetic epilepsy. Neurology. 2018;90:16–7. [DOI] [PubMed] [Google Scholar]
- 29.Franz DN, Lawson JA, Yapici Z, et al. Adjunctive everolimus therapy for tuberous sclerosis complex-associated refractory seizures: Results from the postextension phase of EXIST-3. Epilepsia. 2021;62:3029–41. [DOI] [PubMed] [Google Scholar]
- 30.Franz DN, Lawson JA, Yapici Z, et al. Everolimus for treatment-refractory seizures in TSC: Extension of a randomized controlled trial. Neurol Clin Pract. 2018;8:412–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.French JA, Lawson JA, Yapici Z, et al. Adjunctive everolimus therapy for treatment-resistant focal-onset seizures associated with tuberous sclerosis (EXIST-3): a phase 3, randomised, double-blind, placebo-controlled study. Lancet. 2016;388:2153–63. [DOI] [PubMed] [Google Scholar]
- 32.Samueli S, Dressler A, Groppel G, et al. Everolimus in infants with tuberous sclerosis complex-related West syndrome: First results from a single-center prospective observational study. Epilepsia. 2018;59:e142–e6. [DOI] [PubMed] [Google Scholar]
- 33.Galanopoulou AS, Moshe SL. In search of epilepsy biomarkers in the immature brain: goals, challenges and strategies. Biomark Med. 2011;5:615–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Auvin S, Hartman AL, Desnous B, et al. Diagnosis delay in West syndrome: misdiagnosis and consequences. Eur J Pediatr. 2012;171:1695–701. [DOI] [PubMed] [Google Scholar]
- 35.Loscher W, Potschka H, Sisodiya SM, et al. Drug Resistance in Epilepsy: Clinical Impact, Potential Mechanisms, and New Innovative Treatment Options. Pharmacol Rev. 2020;72:606–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gesche J, Beier CP. Drug resistance in idiopathic generalized epilepsies: Evidence and concepts. Epilepsia. 2022;63:3007–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Servilha-Menezes G, Garcia-Cairasco N. A complex systems view on the current hypotheses of epilepsy pharmacoresistance. Epilepsia Open. 2022;7 Suppl 1:S8–S22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Perucca E, Perucca P, White HS, et al. Drug resistance in epilepsy. Lancet Neurol. 2023. [DOI] [PubMed] [Google Scholar]
- 39.Santana-Gomez CE, Engel J Jr., Staba R Drug-resistant epilepsy and the hypothesis of intrinsic severity: What about the high-frequency oscillations? Epilepsia Open. 2022;7 Suppl 1:S59–S67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Perez-Perez D, Frias-Soria CL, Rocha L. Drug-resistant epilepsy: From multiple hypotheses to an integral explanation using preclinical resources. Epilepsy Behav. 2021;121:106430. [DOI] [PubMed] [Google Scholar]
- 41.Fonseca-Barriendos D, Frias-Soria CL, Perez-Perez D, et al. Drug-resistant epilepsy: Drug target hypothesis and beyond the receptors. Epilepsia Open. 2022;7 Suppl 1:S23–S33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Potschka H, Fischer A, Loscher W, et al. Pathophysiology of drug-resistant canine epilepsy. Vet J. 2023;296–297:105990. [DOI] [PubMed] [Google Scholar]
- 43.Chatzikonstantinou S, Gioula G, Kimiskidis VK, et al. The gut microbiome in drug-resistant epilepsy. Epilepsia Open. 2021;6:28–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rogawski MA. The intrinsic severity hypothesis of pharmacoresistance to antiepileptic drugs. Epilepsia. 2013;54 Suppl 2:33–40. [DOI] [PubMed] [Google Scholar]
- 45.Rogawski MA, Johnson MR. Intrinsic severity as a determinant of antiepileptic drug refractoriness. Epilepsy Curr. 2008;8:127–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Capovilla G, Moshe SL, Wolf P, et al. Epileptic encephalopathy as models of system epilepsy. Epilepsia. 2013;54 Suppl 8:34–7. [DOI] [PubMed] [Google Scholar]
- 47.Wolf P, Yacubian EM, Avanzini G, et al. Juvenile myoclonic epilepsy: A system disorder of the brain. Epilepsy Res. 2015;114:2–12. [DOI] [PubMed] [Google Scholar]
- 48.Johnson GW, Doss DJ, Morgan VL, et al. The interictal suppression hypothesis in focal epilepsy: network-level supporting evidence. Brain. 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Johnson GW, Doss DJ, Englot DJ. Network dysfunction in pre and postsurgical epilepsy: connectomics as a tool and not a destination. Curr Opin Neurol. 2022;35:196–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wiebe S, Blume WT, Girvin JP, et al. A randomized, controlled trial of surgery for temporal-lobe epilepsy. N Engl J Med. 2001;345:311–8. [DOI] [PubMed] [Google Scholar]
- 51.Dwivedi R, Ramanujam B, Chandra PS, et al. Surgery for Drug-Resistant Epilepsy in Children. N Engl J Med. 2017;377:1639–47. [DOI] [PubMed] [Google Scholar]
- 52.Piper RJ, Richardson RM, Worrell G, et al. Towards network-guided neuromodulation for epilepsy. Brain. 2022;145:3347–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fang M, Xi ZQ, Wu Y, et al. A new hypothesis of drug refractory epilepsy: neural network hypothesis. Med Hypotheses. 2011;76:871–6. [DOI] [PubMed] [Google Scholar]
- 54.Gleichgerrcht E, Kocher M, Bonilha L. Connectomics and graph theory analyses: Novel insights into network abnormalities in epilepsy. Epilepsia. 2015;56:1660–8. [DOI] [PubMed] [Google Scholar]
- 55.Kreilkamp BAK, McKavanagh A, Alonazi B, et al. Altered structural connectome in non-lesional newly diagnosed focal epilepsy: Relation to pharmacoresistance. Neuroimage Clin. 2021;29:102564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pegg EJ, Taylor JR, Laiou P, et al. Interictal electroencephalographic functional network topology in drug-resistant and well-controlled idiopathic generalized epilepsy. Epilepsia. 2021;62:492–503. [DOI] [PubMed] [Google Scholar]
- 57.Tang F, Hartz AMS, Bauer B. Drug-Resistant Epilepsy: Multiple Hypotheses, Few Answers. Front Neurol. 2017;8:301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Remy S, Beck H. Molecular and cellular mechanisms of pharmacoresistance in epilepsy. Brain. 2006;129:18–35. [DOI] [PubMed] [Google Scholar]
- 59.Jandova K, Pasler D, Antonio LL, et al. Carbamazepine-resistance in the epileptic dentate gyrus of human hippocampal slices. Brain. 2006;129:3290–306. [DOI] [PubMed] [Google Scholar]
- 60.Remy S, Gabriel S, Urban BW, et al. A novel mechanism underlying drug resistance in chronic epilepsy. Ann Neurol. 2003;53:469–79. [DOI] [PubMed] [Google Scholar]
- 61.Sharma D, Dixit AB, Dey S, et al. Increased levels of alpha4-containing GABA(A) receptors in focal cortical dysplasia: A possible cause of benzodiazepine resistance. Neurochem Int. 2021;148:105084. [DOI] [PubMed] [Google Scholar]
- 62.Chakraborty A, Dey S, Kumar K, et al. Novel variants in GABA(A) receptor subunits: A possible association with benzodiazepine resistance in patients with drug-resistant epilepsy. Epilepsy Res. 2023;189:107056. [DOI] [PubMed] [Google Scholar]
- 63.Cohen I, Navarro V, Clemenceau S, et al. On the origin of interictal activity in human temporal lobe epilepsy in vitro. Science. 2002;298:1418–21. [DOI] [PubMed] [Google Scholar]
- 64.Fuxe K, Borroto-Escuela D, Fisone G, et al. Understanding the role of heteroreceptor complexes in the central nervous system. Curr Protein Pept Sci. 2014;15:647. [DOI] [PubMed] [Google Scholar]
- 65.Salvamoser JD, Avemary J, Luna-Munguia H, et al. Glutamate-Mediated Down-Regulation of the Multidrug-Resistance Protein BCRP/ABCG2 in Porcine and Human Brain Capillaries. Mol Pharm. 2015;12:2049–60. [DOI] [PubMed] [Google Scholar]
- 66.Bauer B, Hartz AM, Pekcec A, et al. Seizure-induced up-regulation of P-glycoprotein at the blood-brain barrier through glutamate and cyclooxygenase-2 signaling. Mol Pharmacol. 2008;73:1444–53. [DOI] [PubMed] [Google Scholar]
- 67.Tishler DM, Weinberg KI, Hinton DR, et al. MDR1 gene expression in brain of patients with medically intractable epilepsy. Epilepsia. 1995;36:1–6. [DOI] [PubMed] [Google Scholar]
- 68.Langer O, Bauer M, Hammers A, et al. Pharmacoresistance in epilepsy: a pilot PET study with the P-glycoprotein substrate R-[(11)C]verapamil. Epilepsia. 2007;48:1774–84. [DOI] [PubMed] [Google Scholar]
- 69.Urzi Brancati V, Pinto Vraca T, Minutoli L, et al. Polymorphisms Affecting the Response to Novel Antiepileptic Drugs. Int J Mol Sci. 2023;24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Smolarz B, Makowska M, Romanowicz H. Pharmacogenetics of Drug-Resistant Epilepsy (Review of Literature). Int J Mol Sci. 2021;22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Leschziner GD, Andrew T, Pirmohamed M, et al. ABCB1 genotype and PGP expression, function and therapeutic drug response: a critical review and recommendations for future research. Pharmacogenomics J. 2007;7:154–79. [DOI] [PubMed] [Google Scholar]
- 72.Boughrara W, Chentouf A. The ABCB1, ABCC2 and RALBP1 polymorphisms are associated with carbamazepine response in epileptic patient: a systematic review. Acta Neurol Belg. 2022;122:871–80. [DOI] [PubMed] [Google Scholar]
- 73.Avemary J, Salvamoser JD, Peraud A, et al. Dynamic regulation of P-glycoprotein in human brain capillaries. Mol Pharm. 2013;10:3333–41. [DOI] [PubMed] [Google Scholar]
- 74.Lazarowski A, Czornyj L, Lubienieki F, et al. ABC transporters during epilepsy and mechanisms underlying multidrug resistance in refractory epilepsy. Epilepsia. 2007;48 Suppl 5:140–9. [DOI] [PubMed] [Google Scholar]
- 75.Williams S, Hossain M, Ferguson L, et al. Neurovascular Drug Biotransformation Machinery in Focal Human Epilepsies: Brain CYP3A4 Correlates with Seizure Frequency and Antiepileptic Drug Therapy. Mol Neurobiol. 2019;56:8392–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ghosh C, Marchi N, Desai NK, et al. Cellular localization and functional significance of CYP3A4 in the human epileptic brain. Epilepsia. 2011;52:562–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wolking S, Moreau C, Nies AT, et al. Testing association of rare genetic variants with resistance to three common antiseizure medications. Epilepsia. 2020;61:657–66. [DOI] [PubMed] [Google Scholar]
- 78.Van Loo KMJ, Carvill GL, Becker AJ, et al. Epigenetic genes and epilepsy - emerging mechanisms and clinical applications. Nat Rev Neurol. 2022;18:530–43. [DOI] [PubMed] [Google Scholar]
- 79.Zahra MA, Kamha ES, Abdelaziz HK, et al. Aberrant Expression of Serum MicroRNA-153 and -199a in Generalized Epilepsy and its Correlation with Drug Resistance. Ann Neurosci. 2022;29:203–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang Y, Wang Y, Chen Y, et al. Circulating MicroRNAs From Plasma Small Extracellular Vesicles as Potential Diagnostic Biomarkers in Pediatric Epilepsy and Drug-Resistant Epilepsy. Front Mol Neurosci. 2022;15:823802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.De Benedittis S, Fortunato F, Cava C, et al. Circulating microRNA: The Potential Novel Diagnostic Biomarkers to Predict Drug Resistance in Temporal Lobe Epilepsy, a Pilot Study. Int J Mol Sci. 2021;22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kobow K, Baulac S, von Deimling A, et al. Molecular diagnostics in drug-resistant focal epilepsy define new disease entities. Brain Pathol. 2021;31:e12963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Enrique AV, Di Ianni ME, Goicoechea S, et al. New anticonvulsant candidates prevent P-glycoprotein (P-gp) overexpression in a pharmacoresistant seizure model in mice. Epilepsy Behav. 2021;121:106451. [DOI] [PubMed] [Google Scholar]
- 84.Schlichtiger J, Pekcec A, Bartmann H, et al. Celecoxib treatment restores pharmacosensitivity in a rat model of pharmacoresistant epilepsy. Br J Pharmacol. 2010;160:1062–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ozcan E, Lum GR, Hsiao EY. Interactions between the gut microbiome and ketogenic diet in refractory epilepsy. Int Rev Neurobiol. 2022;167:217–49. [DOI] [PubMed] [Google Scholar]
- 86.Olson CA, Vuong HE, Yano JM, et al. The Gut Microbiota Mediates the Anti-Seizure Effects of the Ketogenic Diet. Cell. 2018;173:1728–41 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Murgia F, Muroni A, Puligheddu M, et al. Metabolomics As a Tool for the Characterization of Drug-Resistant Epilepsy. Front Neurol. 2017;8:459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Juvale IIA, Che Has AT. Possible interplay between the theories of pharmacoresistant epilepsy. Eur J Neurosci. 2021;53:1998–2026. [DOI] [PubMed] [Google Scholar]
- 89.Yi JH, Hazell AS. Excitotoxic mechanisms and the role of astrocytic glutamate transporters in traumatic brain injury. Neurochem Int. 2006;48:394–403. [DOI] [PubMed] [Google Scholar]
- 90.Chaparro-Huerta V, Rivera-Cervantes MC, Flores-Soto ME, et al. Proinflammatory cytokines and apoptosis following glutamate-induced excitotoxicity mediated by p38 MAPK in the hippocampus of neonatal rats. J Neuroimmunol. 2005;165:53–62. [DOI] [PubMed] [Google Scholar]
- 91.Bankstahl JP, Hoffmann K, Bethmann K, et al. Glutamate is critically involved in seizure-induced overexpression of P-glycoprotein in the brain. Neuropharmacology. 2008;54:1006–16. [DOI] [PubMed] [Google Scholar]
- 92.Alonso-Nanclares L, DeFelipe J. Alterations of the microvascular network in the sclerotic hippocampus of patients with temporal lobe epilepsy. Epilepsy Behav. 2014;38:48–52. [DOI] [PubMed] [Google Scholar]
- 93.Auzmendi J, Buchholz B, Salguero J, et al. Pilocarpine-Induced Status Epilepticus Is Associated with P-Glycoprotein Induction in Cardiomyocytes, Electrocardiographic Changes, and Sudden Death. Pharmaceuticals (Basel). 2018;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Galanopoulou AS, Loscher W, Lubbers L, et al. Antiepileptogenesis and disease modification: Progress, challenges, and the path forward-Report of the Preclinical Working Group of the 2018 NINDS-sponsored antiepileptogenesis and disease modification workshop. Epilepsia Open. 2021;6:276–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.de Jong J, Cutcutache I, Page M, et al. Towards realizing the vision of precision medicine: AI based prediction of clinical drug response. Brain. 2021;144:1738–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rocha L Subchronic treatment with antiepileptic drugs modifies pentylenetetrazol-induced seizures in mice: Its correlation with benzodiazepine receptor binding. Neuropsychiatr Dis Treat. 2008;4:619–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Loscher W, Schmidt D. Experimental and clinical evidence for loss of effect (tolerance) during prolonged treatment with antiepileptic drugs. Epilepsia. 2006;47:1253–84. [DOI] [PubMed] [Google Scholar]
- 98.Kim RB. Drugs as P-glycoprotein substrates, inhibitors, and inducers. Drug Metab Rev. 2002;34:47–54. [DOI] [PubMed] [Google Scholar]
- 99.Navarrete-Modesto V, Orozco-Suarez S, Feria-Romero IA, et al. The molecular hallmarks of epigenetic effects mediated by antiepileptic drugs. Epilepsy Res. 2019;149:53–65. [DOI] [PubMed] [Google Scholar]
- 100.Bohosova J, Vajcner J, Jabandziev P, et al. MicroRNAs in the development of resistance to antiseizure drugs and their potential as biomarkers in pharmacoresistant epilepsy. Epilepsia. 2021;62:2573–88. [DOI] [PubMed] [Google Scholar]
- 101.Loscher W Animal Models of Seizures and Epilepsy: Past, Present, and Future Role for the Discovery of Antiseizure Drugs. Neurochem Res. 2017;42:1873–88. [DOI] [PubMed] [Google Scholar]
- 102.Galanopoulou AS, Moshe SL. Neonatal and Infantile Epilepsy: Acquired and Genetic Models. Cold Spring Harb Perspect Med. 2015;6:a022707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Simonato M, Brooks-Kayal AR, Engel J Jr., et al. The challenge and promise of antiepileptic therapy development in animal models. Lancet Neurol. 2014;13:949–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kehne JH, Klein BD, Raeissi S, et al. The National Institute of Neurological Disorders and Stroke (NINDS) Epilepsy Therapy Screening Program (ETSP). Neurochem Res. 2017;42:1894–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Loscher W, White HS. Animal Models of Drug-Resistant Epilepsy as Tools for Deciphering the Cellular and Molecular Mechanisms of Pharmacoresistance and Discovering More Effective Treatments. Cells. 2023;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Vega-Garcia A, Guevara-Guzman R, Garcia-Gomez O, et al. Aberrant Connection Formation and Glia Involvement in the Progression of Pharmacoresistant Mesial Temporal Lobe Epilepsy. Curr Pharm Des. 2022;28:2283–97. [DOI] [PubMed] [Google Scholar]
- 107.Barton ME, Klein BD, Wolf HH, et al. Pharmacological characterization of the 6 Hz psychomotor seizure model of partial epilepsy. Epilepsy Res. 2001;47:217–27. [DOI] [PubMed] [Google Scholar]
- 108.Postma T, Krupp E, Li XL, et al. Lamotrigine treatment during amygdala-kindled seizure development fails to inhibit seizures and diminishes subsequent anticonvulsant efficacy. Epilepsia. 2000;41:1514–21. [DOI] [PubMed] [Google Scholar]
- 109.Koneval Z, Knox KM, White HS, et al. Lamotrigine-resistant corneal-kindled mice: A model of pharmacoresistant partial epilepsy for moderate-throughput drug discovery. Epilepsia. 2018;59:1245–56. [DOI] [PubMed] [Google Scholar]
- 110.Metcalf CS, Huff J, Thomson KE, et al. Evaluation of antiseizure drug efficacy and tolerability in the rat lamotrigine-resistant amygdala kindling model. Epilepsia Open. 2019;4:452–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bethmann K, Brandt C, Loscher W. Resistance to phenobarbital extends to phenytoin in a rat model of temporal lobe epilepsy. Epilepsia. 2007;48:816–26. [DOI] [PubMed] [Google Scholar]
- 112.Glien M, Brandt C, Potschka H, et al. Effects of the novel antiepileptic drug levetiracetam on spontaneous recurrent seizures in the rat pilocarpine model of temporal lobe epilepsy. Epilepsia. 2002;43:350–7. [DOI] [PubMed] [Google Scholar]
- 113.West PJ, Thomson K, Billingsley P, et al. Spontaneous recurrent seizures in an intra-amygdala kainate microinjection model of temporal lobe epilepsy are differentially sensitive to antiseizure drugs. Exp Neurol. 2022;349:113954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Briggs SW, Mowrey W, Hall CB, et al. CPP-115, a vigabatrin analogue, decreases spasms in the multiple-hit rat model of infantile spasms. Epilepsia. 2014;55:94–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Doumlele K, Conway E, Hedlund J, et al. A case report on the efficacy of vigabatrin analogue (1S, 3S)-3-amino-4-difluoromethylenyl-1-cyclopentanoic acid (CPP-115) in a patient with infantile spasms. Epilepsy Behav Case Rep. 2016;6:67–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ceulemans B, Boel M, Leyssens K, et al. Successful use of fenfluramine as an add-on treatment for Dravet syndrome. Epilepsia. 2012;53:1131–9. [DOI] [PubMed] [Google Scholar]
- 117.Zhang Y, Kecskes A, Copmans D, et al. Pharmacological characterization of an antisense knockdown zebrafish model of Dravet syndrome: inhibition of epileptic seizures by the serotonin agonist fenfluramine. PLoS One. 2015;10:e0125898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Raffo E, Coppola A, Ono T, et al. A pulse rapamycin therapy for infantile spasms and associated cognitive decline. Neurobiol Dis. 2011;43:322–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Li J, Nelis M, Sourbron J, et al. Efficacy of Fenfluramine and Norfenfluramine Enantiomers and Various Antiepileptic Drugs in a Zebrafish Model of Dravet Syndrome. Neurochem Res. 2021;46:2249–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gogou M, Cross JH. Fenfluramine as antiseizure medication for epilepsy. Dev Med Child Neurol. 2021;63:899–907. [DOI] [PubMed] [Google Scholar]
- 121.Dinday MT, Baraban SC. Large-Scale Phenotype-Based Antiepileptic Drug Screening in a Zebrafish Model of Dravet Syndrome. eNeuro. 2015;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Giorgi FS, Galanopoulou AS, Moshe SL. Sex dimorphism in seizure-controlling networks. Neurobiol Dis. 2014;72 Pt B:144–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lam J, Baello S, Iqbal M, et al. The ontogeny of P-glycoprotein in the developing human blood-brain barrier: implication for opioid toxicity in neonates. Pediatr Res. 2015;78:417–21. [DOI] [PubMed] [Google Scholar]
- 124.Soares RV, Do TM, Mabondzo A, et al. Ontogeny of ABC and SLC transporters in the microvessels of developing rat brain. Fundam Clin Pharmacol. 2016;30:107–16. [DOI] [PubMed] [Google Scholar]
- 125.Veliskova J, Moshe SL. Update on the role of substantia nigra pars reticulata in the regulation of seizures. Epilepsy Curr. 2006;6:83–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Verscheijden LFM, van Hattem AC, Pertijs J, et al. Developmental patterns in human blood-brain barrier and blood-cerebrospinal fluid barrier ABC drug transporter expression. Histochem Cell Biol. 2020;154:265–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Fernandez E, Perez R, Hernandez A, et al. Factors and Mechanisms for Pharmacokinetic Differences between Pediatric Population and Adults. Pharmaceutics. 2011;3:53–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sinha J, Karatza E, Gonzalez D. Physiologically-based pharmacokinetic modeling of oxcarbazepine and levetiracetam during adjunctive antiepileptic therapy in children and adolescents. CPT Pharmacometrics Syst Pharmacol. 2022;11:225–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Olivetti PR, Maheshwari A, Noebels JL. Neonatal estradiol stimulation prevents epilepsy in Arx model of X-linked infantile spasms syndrome. Sci Transl Med. 2014;6:220ra12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Jequier Gygax M, Klein BD, White HS, et al. Efficacy and tolerability of the galanin analog NAX 5055 in the multiple-hit rat model of symptomatic infantile spasms. Epilepsy Res. 2014;108:98–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Galanopoulou AS. GABA(A) receptors in normal development and seizures: friends or foes? Curr Neuropharmacol. 2008;6:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ben-Ari Y, Khalilov I, Kahle KT, et al. The GABA excitatory/inhibitory shift in brain maturation and neurological disorders. Neuroscientist. 2012;18:467–86. [DOI] [PubMed] [Google Scholar]
- 133.Wahab A, Albus K, Gabriel S, et al. In search of models of pharmacoresistant epilepsy. Epilepsia. 2010;51 Suppl 3:154–9. [DOI] [PubMed] [Google Scholar]
- 134.Scantlebury MH, Galanopoulou AS, Chudomelova L, et al. A model of symptomatic infantile spasms syndrome. Neurobiol Dis. 2010;37:604–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Griffin A, Hamling KR, Hong S, et al. Preclinical Animal Models for Dravet Syndrome: Seizure Phenotypes, Comorbidities and Drug Screening. Front Pharmacol. 2018;9:573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Han Z, Chen C, Christiansen A, et al. Antisense oligonucleotides increase Scn1a expression and reduce seizures and SUDEP incidence in a mouse model of Dravet syndrome. Sci Transl Med. 2020;12. [DOI] [PubMed] [Google Scholar]
- 137.Serbanescu I, Cortez MA, McKerlie C, et al. Refractory atypical absence seizures in rat: a two hit model. Epilepsy Res. 2004;62:53–63. [DOI] [PubMed] [Google Scholar]
- 138.Siehr MS, Massey CA, Noebels JL. Arx expansion mutation perturbs cortical development by augmenting apoptosis without activating innate immunity in a mouse model of X-linked infantile spasms syndrome. Dis Model Mech. 2020;13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Cepeda C, Levinson S, Nariai H, et al. Pathological high frequency oscillations associate with increased GABA synaptic activity in pediatric epilepsy surgery patients. Neurobiol Dis. 2020;134:104618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Cepeda C, Levinson S, Yazon VW, et al. Cellular antiseizure mechanisms of everolimus in pediatric tuberous sclerosis complex, cortical dysplasia, and non-mTOR-mediated etiologies. Epilepsia Open. 2018;3:180–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.A clinical trial of induction of labor versus expectant management in postterm pregnancy. The National Institute of Child Health and Human Development Network of Maternal-Fetal Medicine Units. Am J Obstet Gynecol. 1994;170:716–23. [PubMed] [Google Scholar]
- 142.Sandow N, Kim S, Raue C, et al. Drug resistance in cortical and hippocampal slices from resected tissue of epilepsy patients: no significant impact of p-glycoprotein and multidrug resistance-associated proteins. Front Neurol. 2015;6:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Que Z, Olivero-Acosta MI, Zhang J, et al. Hyperexcitability and Pharmacological Responsiveness of Cortical Neurons Derived from Human iPSCs Carrying Epilepsy-Associated Sodium Channel Nav1.2-L1342P Genetic Variant. J Neurosci. 2021;41:10194–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hirose S, Tanaka Y, Shibata M, et al. Application of induced pluripotent stem cells in epilepsy. Mol Cell Neurosci. 2020;108:103535. [DOI] [PubMed] [Google Scholar]
- 145.Baud MO, Rao VR. Gauging seizure risk. Neurology. 2018;91:967–73. [DOI] [PubMed] [Google Scholar]
- 146.Thome-Souza S, Klehm J, Jackson M, et al. Clobazam higher-evening differential dosing as an add-on therapy in refractory epilepsy. Seizure. 2016;40:1–6. [DOI] [PubMed] [Google Scholar]
- 147.Abokrysha NT, Taha N, Shamloul R, et al. Clinical, radiological and electrophysiological predictors for drug-resistant epilepsy. Egypt J Neurol Psychiatr Neurosurg. 2023;59:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Stevelink R, Al-Toma D, Jansen FE, et al. Individualised prediction of drug resistance and seizure recurrence after medication withdrawal in people with juvenile myoclonic epilepsy: A systematic review and individual participant data meta-analysis. EClinicalMedicine. 2022;53:101732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kalilani L, Sun X, Pelgrims B, et al. The epidemiology of drug-resistant epilepsy: A systematic review and meta-analysis. Epilepsia. 2018;59:2179–93. [DOI] [PubMed] [Google Scholar]
- 150.Li Z, Cao W, Sun H, et al. Potential clinical and biochemical markers for the prediction of drug-resistant epilepsy: A literature review. Neurobiol Dis. 2022;174:105872. [DOI] [PubMed] [Google Scholar]
- 151.Weil AG, Dimentberg E, Lewis E, et al. Development of an online calculator for the prediction of seizure freedom following pediatric hemispherectomy using HOPS. Epilepsia. 2023. [DOI] [PubMed] [Google Scholar]
- 152.Lob K, Hou T, Chu TC, et al. Clinical features and drug-resistance in pediatric epilepsy with co-occurring autism: A retrospective comparative cohort study. Epilepsy Behav. 2023;143:109228. [DOI] [PubMed] [Google Scholar]
- 153.Yu T, Liu X, Sun L, et al. Risk factors for Drug-resistant Epilepsy (DRE) and a nomogram model to predict DRE development in post-traumatic epilepsy patients. CNS Neurosci Ther. 2022;28:1557–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Lattanzi S, Trinka E, Turcato G, et al. Latency of poststroke epilepsy can predict drug resistance. Eur J Neurol. 2022;29:2481–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Zhong R, Chen Q, Li N, et al. Psychiatric symptoms predict drug-resistant epilepsy in newly treated patients. Seizure. 2022;103:86–91. [DOI] [PubMed] [Google Scholar]
- 156.Choi H, Detyniecki K, Bazil C, et al. Development and validation of a predictive model of drug-resistant genetic generalized epilepsy. Neurology. 2020;95:e2150–e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kamitaki BK, Janmohamed M, Kandula P, et al. Clinical and EEG factors associated with antiseizure medication resistance in idiopathic generalized epilepsy. Epilepsia. 2022;63:150–61. [DOI] [PubMed] [Google Scholar]
- 158.Janmohamed M, Hakeem H, Ooi S, et al. Treatment Outcomes of Newly Diagnosed Epilepsy: A Systematic Review and Meta-analysis. CNS Drugs. 2023;37:13–30. [DOI] [PubMed] [Google Scholar]
- 159.Lattanzi S, Meletti S, Trinka E, et al. Individualized Prediction of Drug Resistance in People with Post-Stroke Epilepsy: A Retrospective Study. J Clin Med. 2023;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Gregerson CHY, Bakian AV, Wilkes J, et al. Disparities in Pediatric Epilepsy Remission Are Associated With Race and Ethnicity. J Child Neurol. 2019;34:928–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Guerrini R, Conti V, Mantegazza M, et al. Developmental and epileptic encephalopathies: from genetic heterogeneity to phenotypic continuum. Physiol Rev. 2023;103:433–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Specchio N, Wirrell EC, Scheffer IE, et al. International League Against Epilepsy classification and definition of epilepsy syndromes with onset in childhood: Position paper by the ILAE Task Force on Nosology and Definitions. Epilepsia. 2022;63:1398–442. [DOI] [PubMed] [Google Scholar]
- 163.Riney K, Bogacz A, Somerville E, et al. International League Against Epilepsy classification and definition of epilepsy syndromes with onset at a variable age: position statement by the ILAE Task Force on Nosology and Definitions. Epilepsia. 2022;63:1443–74. [DOI] [PubMed] [Google Scholar]
- 164.Zuberi SM, Wirrell E, Yozawitz E, et al. ILAE classification and definition of epilepsy syndromes with onset in neonates and infants: Position statement by the ILAE Task Force on Nosology and Definitions. Epilepsia. 2022;63:1349–97. [DOI] [PubMed] [Google Scholar]
- 165.Enrique A, Goicoechea S, Castano R, et al. New model of pharmacoresistant seizures induced by 3-mercaptopropionic acid in mice. Epilepsy Res. 2017;129:8–16. [DOI] [PubMed] [Google Scholar]
- 166.Leclercq K, Matagne A, Kaminski RM. Low potency and limited efficacy of antiepileptic drugs in the mouse 6 Hz corneal kindling model. Epilepsy Res. 2014;108:675–83. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.