

Abstract
Background
Neuroligin 4 X-linked (NLGN4X) harbors a human leukocyte antigen (HLA)-A*02-restricted tumor-associated antigen, overexpressed in human gliomas, that was found to induce specific cytotoxic T cell responses following multi-peptide vaccination in patients with newly diagnosed glioblastoma.
Methods
T cell receptor (TCR) discovery was performed using droplet-based single-cell TCR sequencing of NLGN4X-tetramer-sorted T cells postvaccination. The identified TCR was delivered to Jurkat T cells and primary human T cells (NLGN4X-TCR-T). Functional profiling of NLGN4X-TCR-T was performed by flow cytometry and cytotoxicity assays. Therapeutic efficacy of intracerebroventricular NLGN4X-TCR-T was assessed in NOD scid gamma (NSG) major histocompatibility complex (MHC) I/II knockout (KO) (NSG MHC I/II KO) mice bearing NLGN4X-expressing experimental gliomas.
Results
An HLA-A*02-restricted vaccine-induced T cell receptor specifically binding NLGN4X131–139 was applied for preclinical therapeutic use. Reactivity, cytotoxicity, and polyfunctionality of this NLGN4X-specific TCR are demonstrated in various cellular models. Intracerebroventricular administration of NLGN4X-TCR-T prolongs survival and leads to an objective response rate of 44.4% in experimental glioma-bearing NSG MHC I/II KO mice compared to 0.0% in control groups.
Conclusion
NLGN4X-TCR-T demonstrate efficacy in a preclinical glioblastoma model. On a global scale, we provide the first evidence for the therapeutic retrieval of vaccine-induced human TCRs for the off-the-shelf treatment of glioblastoma patients.Keywords cell therapy | glioblastoma | T cell receptor | tumor antigen
Keywords: Keywords cell therapy, glioblastoma, T cell receptor, tumor antigen
Key Points.
NLGN4X-specific TCR T cells specifically lyse glioma cells.
Intracerebroventricular NLGN4X-TCR-T are efficacious in a preclinical glioblastoma model.
Vaccine-induced TCRs targeting glioblastoma-associated antigens can be leveraged for the off-the-shelf treatment.
Importance of the Study.
The glioblastoma-associated antigen NLGN4X was targeted in multi-peptide vaccine trials in HLA-A*02+ patients with newly diagnosed and recurrent glioblastoma with robust antigen-specific T-cell responses. We apply a vaccine-induced patient-derived T cell receptor (TCR) targeting the NLGN4X131–139 epitope from the Glioma Actively Personalized Vaccine Consortium (GAPVAC) trial for adoptive T cell therapy.
Human T cells engineered to express the NLGN4X TCR efficiently recognized and lysed NLGN4X-expressing tumor cells and induced temporary rejection of experimental gliomas in an immunodeficient mouse model after intracerebroventricular adoptive transfer.
In summary, we demonstrate that adoptive transfer of T cells transgenic for a patient-derived vaccine-induced TCR is an effective therapeutic strategy to target glioblastoma-associated antigens.
The median survival of patients with MGMT-methylated glioblastomas following standard-of-care (SOC) therapy is still limited to approximately 32 months with no improvements when applying established immunotherapeutic concepts, thus highlighting the urgent need for novel efficacious treatments.1,2 Conversely, recent early clinical trials have demonstrated the ability of cellular products to induce clinical responses in human brain malignancies.3,4 However, durable clinical responses are often not achieved despite signs of local immune cell activation.5–7
Current immunotherapeutic approaches mostly use chimeric antigen receptor (CAR) engineered T cells to target surface antigens expressed on human brain tumors.8 Preclinical studies targeting EPHA2, HER2, EGFR, B7-H3, and IL13Rα2 with CAR T cells showed efficient and persistent tumor eradication in different experimental glioblastoma models.9–12 The tumor-associated antigen disialoganglioside GD2 is frequently overexpressed in neuroblastoma13 and highly expressed on histone H3K27M-mutated glioma cells. In a first-in-human phase I dose-escalation trial, GD2-directed CAR T cells were manufactured and administered to patients with H3K27M-mutant midline gliomas such as diffuse intrinsic pontine gliomas, after cyclophosphamide/fludarabine-based lymphodepletion. A remarkable fraction of patients experienced a reduction in clinical symptoms and radiographic response.3 Similarly, a recent report on intracerebroventricular transfer of B7-H3 CAR T cells in 3 patients with diffuse intrinsic pontine glioma resulted in clinical and radiographic improvement in one patient,14 further providing evidence of efficacy of immune-receptor engineered T cells in primary central nervous system tumors that typically only harbor few targetable surface molecules.
A main advantage of T cell receptor (TCR) engineered T cells is their ability to target not only extracellular but also intracellular antigens that are naturally processed and presented on MHC classes I and II molecules. T cells modified to express TCRs targeting tumor-associated antigens led to tumor regressions and T cell persistence in patients with different solid tumors.15,16
The GAPVAC study conducted by Hilf et al. provided first evidence that the administration of personalized multi-peptide vaccines targeting glioblastoma-associated antigens in patients with newly diagnosed glioblastoma was feasible and safe. APVAC1 peptides contained wildtype antigens that were previously identified in the peptidome of glioblastoma specimens17, selected for the off-the-shelf use, and ranked by HLA-ligandome analysis for each patient, resulting in an individualized multi-peptide composition.1 Sixteen patients were enrolled in the study and 12/13 (92.3%) developed MHCI-restricted T cell responses to at least one of the APVAC1 peptides after administration of APVAC1.1 Nine of 15 (60.0%) vaccinated patients received a multi-peptide vaccine containing the tumor-associated epitope neuroligin 4, X-linked NLGN4X131-139.1 Immunogenicity for this particular peptide was 86.0%. One patient with a favorable clinical course (PFS > 27.4 months, censored) showed an expansion of NLGN4X131–139-specific CD8+ T cells after vaccination and specific lysis of NLGN4X-expressing glioma cells by these cells.1
NLGN4X is overexpressed in human gliomas with limited expression in healthy tissue.18 Publicly available single-cell RNA expression data show low expression in healthy tissue with predominant expression on neurons and precursor oligodendrocytes.18 Neuroligins (NLGNs) are postsynaptic adhesion molecules binding to neurexins on the presynaptic membrane19 and are essential for synaptic formation.20 NLGN4X is predominantly expressed on excitatory neurons regulating excitatory synaptic transmission.21
Here, we investigated the therapeutic potential of a patient-derived vaccine-induced HLA-A*02+-restricted TCR directed against NLGN4X131–139, providing the first evidence that vaccine-induced TCRs targeting glioblastoma-associated antigens can be exploited to develop off-the-shelf TCR-engineered T cell therapies.
Methods
Mice
NOD.Cg-PrkdcscidH2-K1tm1BpeH2-Ab1em1MvwH2-D1tm1BpeIl2rgtm1Wjl/SzJ (NSG MHCI/II KO) mice were bred at the DKFZ animal facility. All animal procedures were performed following the institutional laboratory animal research guidelines and were approved by the governmental institutions (Regional Administrative Authority Karlsruhe, Germany, file number: G-37/18). For experimental groups, 6- to 28-weeks old mice were matched by age and sex.
Electroporation of Jurkat Cells
Jurkat76 TCR-deficient T cells with permanent expression of CD8 (Jurkat76 CD8+) with 2 × 106 per electroporation were used and 5 μg each of TCR- and NFAT reporter-encoding DNA vectors were delivered (neon electroporation system: settings: 1325 V, 3 pulses, 10 ms). After 24 h incubation, TCR expression levels were determined by flow cytometric analysis of murine constant TCR beta chain (mTCRb) positive cells compared to untransfected Jurkat76 T cells.
Viral Transduction of Human T Cells
Human T cells obtained from healthy donors were activated and transduced with either a pLEX307 lentiviral vector or a TCR-encoding SFG-IRES-GFP retroviral vector22 and expanded in vitro (comp. Supplementary Material and Methods). Both vectors carry a murine constant TCR beta chain (mTCRb) that prevents endogenous mispairing of the TCR chains allowing TCR delivery without a genetic deletion of the endogenous TCR. As a transduction control, T cells were transduced with the helper plasmids without the encoding vector (= Mock control).
Intracranial Tumor Experiments and Intracerebroventricular Injection of T Cells
For orthotopic tumor cell injection, 1 × 105 U87 tandem-minigene (TMG) or U87 NLGN4X glioma cells were injected at a concentration of 5 × 107 cells per ml in 2 μl PBS into the right hemisphere of NSG MHCI/II KO mice 1 mm anterior to the coronal suture, 2 mm lateral to the bregma and in 3 mm depth. T cells were transduced and expanded as described above. After confirmation of tumor growth using magnetresonance imaging (MRI), up to 5 × 106 T cells were injected into the left lateral ventricle at 0.5 mm lateral to the bregma and 2.2 mm depth of tumor bearing mice in a total volume of 4 μl PBS. For survival experiments, T cells were injected on days 15 and 22, and for assessment of intratumoral phenotype one injection on day 48 was performed. Tumor growth was monitored with repetitive MRI (comp. Supplementary Material and Methods) and mice were checked daily for tumor-related symptoms and sacrificed when the humane or experimental stop criteria (Regional commission Karlsruhe, file number: G-37/18) were met. Obtained tissue was either directly processed for flow cytometry or cryo-fixed and assessed by immunofluorescence (comp. Supplementary Material and Methods).
NFAT Reporter-Based Assays and Jurkat-BOLETH Co-cultures
Jurkat76 T cells were transfected with a TCR-encoding vector and activation was assessed by an NFAT reporter assay (comp. Supplementary Material and Methods) or a flow cytometric assessment of cluster of differentiation (CD)69 expression for Jurkat-BOLETH cells co-cultures (comp. Supplementary Material and Methods).
Human T Cell Activation Assays
Human T cells were isolated and activated as described above and the respective TCRs were delivered by retroviral transduction. After confirmation of TCR surface expression, T cells were used for overnight in vitro T cell activation and cytotoxicity assays using NLGN4X131–139-peptide-loaded U87 glioma cells and NLGN4X-overexpressing U87 glioma cells as target cells. Subsequently, cytotoxicity was measured using the CytoTox96® Non-Radioactive Cytotoxicity Assay (Promega) according to manufacturer's instrucitons and T cells were stained extra- and intracellularly for flow cytometric analysis as described above.
Vital-FR Cytotoxicity Assay and Flow Cytometry-Based Cytotoxicity Assays
TCR- transduced human T cells were used on day 4 after transduction and co-cultured overnight at different effector-to-target (E:T) cell ratios with different target cell lines that had been previously labeled with CellTraceTM Far Red or Violet reagents at 0.2 µM according to manufacturer’s instructions. If peptide-pulsed target cells were used as antigen-presenting cells (APC), APC were loaded at the indicated peptide dilution for 1 hour at 37°C, 5% CO2 prior to the assay. Following overnight co-culture, plates were centrifuged and resuspended in PBS. T cells were removed, and 30 µl trypsin per well was added. After 10 minutes, target cells were resuspended and subjected to flow cytometry using eFluor780-conjugated fixable viability dye and PE-conjugated anti-human CD3 for negative selection of tumor cells. One hundred and twenty-three counting beads were added in 50 µl at a 1:5 dilution. Cytotoxicity was calculated using the normalized count of tumor cells. Cytotoxicity (in %) was calculated as the quotient of live tumor cells after T cell co-culture to either a tumor cell only or unloaded tumor cell control.
Statistical Analysis
All results were analyzed with Prism version 9.4.0. Statistical tests are indicated in the respective figure legends. ANOVA tests were corrected for multiple testing using Sidak’s multiple comparisons correction. Results were considered as significant if the p-value was below 0.05.
Study Approval
All animal procedures followed the institutional laboratory animal research guidelines and were approved by the governmental authorities (Regional Administrative Authority Karlsruhe, Germany, file number: G-37/18).
Data Availability
Cell lines will be provided by the corresponding author upon reasonable request. All additional datasets generated or analyzed during this study are included in this published article (and its Supplementary Data files).
Results
NLGN4X131–139-Reactive TCR Identified by Single-Cell TCR-Seq From a Vaccinated Glioblastoma Patient
NLGN4X131–139-HLA-A*02 tetramer-enriched CD45+ CD3+ T cells from a patient with expansion of NLGN4X131–139-reactive T cells following administration of a multi-peptide vaccine including NLGN4X131–139 were subjected to single-cell VDJ sequencing (scVDJ-seq). The tetramer-enriched T cell pool was highly clonal and one single clonotype made up 90.82% of the TCR repertoire (Figure 1A). Next, we generated episomal nano scaffold matrix attachment region (nano-S/MAR) DNA vectors with the respective variable alpha and beta chains of the top 4 TCR clones (frequotype 1-4 = ft1-4) and transfected TCR-deficient Jurkat T cells (Jurkat76). TCR surface expression was confirmed by flow cytometric analysis of the murine TCR beta chain (mTCRb) allowing specific detection of the transgenic TCR and excluding mispairing with the endogenous TCR in primary T cells (Figure 1B). TCR-transfected Jurkat76 were then co-cultured with peptide-loaded BOLETH HLA-A*02+ presenter cells and CD69 expression was assessed by flow cytometry. The T cell activation marker CD69 was upregulated only in TCRft1-expressing Jurkat76 when exposed to the NLGN4X131–139-epitope (Figure 1C). Thus, TCRft1 was consequently used for downstream reactivity assessment. As a mode of action for antigen-dependent T-cell stimulation, nuclear translocation of nuclear factor of activated T cells (NFAT) represents a hallmark of TCR signaling. Therefore, TCRft1-expressing Jurkat76 were co-transfected with a luciferase NFAT-reporter and subsequently co-cultured with peptide-loaded HLA-A*02+ peripheral blood mononuclear cells (PBMC) and HLA-A*02+ U87 glioma cells (Figure 1D and E) that do not endogenously express the NLGN4X protein (Supplementary Figure S1 A–C). Using PBMCs as antigen-presenting cells, the NLGN4X131–139-induced a luminescence signal intensity was similar to TCR stimulation by anti-CD3/CD28 beads (Figure 1D).
Figure 1.
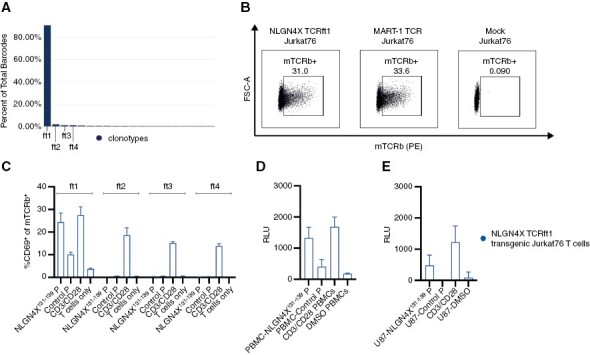
NLGN4X ft1 TCR is specifically reactive to the Neuroligin-4, X-linked target epitope (A) VDJ analysis from single-cell TCR sequencing of multimer-sorted patient T cells after vaccination. Frequencies: ft1—90.82%; ft2—1.78%; ft3—0.96%; ft4—0.96%. (B) Exemplary flow cytometric analysis of the transfection efficiency of TCR-transfected Jurkat T cells. (C) NLGN4X ft1-4 TCR transfected Jurkat76 co-cultured with peptide-loaded BOLETH APC. Myelin oligodendrocyte glycoprotein (MOG35-55) was used as the control peptide. Mean with SEM of 3 technical replicates. Transfection efficiencies assessed by mTCRb expression of the respective TCRs ft1-4: ft1: 15.56% ft2: 12.13%, ft3: 14.63%, ft4: 17.93% (data not shown). (D) NFAT-reporter assay of NLGN4X TCR transfected Jurkat76 T cells co-cultured with peptide-loaded HLA-A*02+ PBMCs. RLU = relative luminescence units. Mean with SEM of 3 technical replicates. (E) NFAT-reporter assay of NLGN4X TCR transfected Jurkat76 T cells co-cultured with peptide-loaded HLA-A*02+ U87 glioma cells. RLU = relative luminescence units. Mean with SEM of 3 technical replicates.
Generation and Phenotypic Characterization of NLGN4X131–139-Reactive TCR-Transduced Human T Cells
For efficient delivery of the transgene TCRft1, we first assessed lentiviral delivery and therefore primarily evaluated the expression of a transgenic green fluorescent protein (GFP) under cytomegalovirus (CMV), spleen focus-forming virus (SFFV), human phosphoglycerate kinase (hPGK), and the human polyubiquitin (hUBC) promoters (Supplementary Figure S2A). In comparison to CMV, SFFV, hUBC, and the hPGK promotors, target expression under the elongation factor 1 (EF1)-alpha promoter resulted in higher GFP median fluorescence intensity (MFI; Supplementary Figure S2A) and 40.9% of T cells were positive for GFP (Supplementary Figure S2B). However, by assessing the fraction of TCRft1-expressing primary human T cells, we only achieved up to 7.75% TCR-engineered T cells using the pLEX307 EF1-alpha lentiviral vector (Figure 2A and B; Supplementary Figure S2C). Thus, we made use of a retroviral SFG-IRES-GFP vector (Figure 2C), that is currently being investigated in a clinical trial evaluating anti-CD19 CAR T cells in patients with relapsed or refractory CD19+ lymphoid disease,23 and used with RD-114 and PeqPam, a different packaging plasmid system.24 Importantly, the transduction process of primary human T cells using the retroviral TCRft1-SFG-IRES-GFP vector (Figure 2C) resulted in up to 93.2% of GFP+ CD3+ T cells and led to a transduction efficiency of up to 90.4% (Figure 2D–F). Consequently, the SFG-IRES-GFP vector was used for all subsequent in vitro and in vivo experiments. Prior to re-infusion, engineered autologous T cells are routinely expanded after transduction.25 To assess the phenotype of TCRft1-SFG-IRES-GFP-expressing primary human T cells during in vitro expansion, we subjected the TCR-transduced T cells to longitudinal flow cytometric analysis by using phenotypic markers for T cell differentiation. TCRft1-SFG-IRES-GFP-expressing T cells acquired a CD45RA+ CD45RO− CCR7+ T cell phenotype (Figure 2G; Supplementary Figure S3B) that resembles naïve T cell states26 and suggests that transduction with TCRft1-SFG-IRES-GFP does not lead to differentiation of T cells into terminal effector cells. Moreover, we did not observe a relevant increase in the expression of the exhaustion markers LAG3, PD-1, and TIM-3. Collectively, our flow cytometric profiling suggests that transduced TCRft1-SFG-IRES-GFP primary human T cells display a naïve T-cell phenotype that has recently been described to provide superior anti-tumor capacities.5
Figure 2.
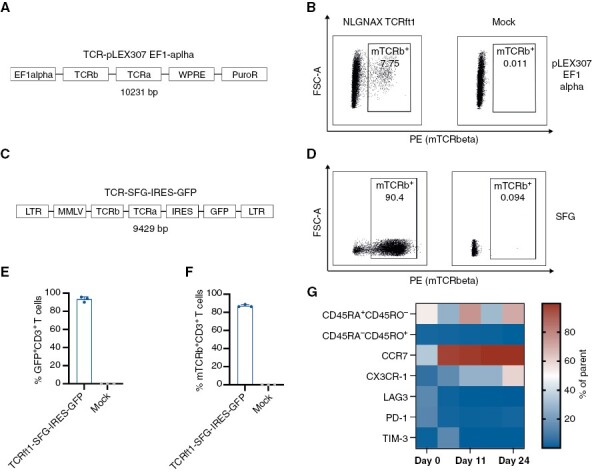
Development of the manufacturing process of the T cell product. (A) Schematic overview of the TCRft1-pLEX307 EF1-alpha: EF1-alpha promoter—TCR beta chain (including the murine TCR beta constant region)—TCR alpha chain—Woodchuck hepatitis virus posttranscriptional regulatory element—puromycin resistance. (B) Exemplary transduction efficiency of TCRft1-pLEX307 EF1-alpha transduced human T cells by flow cytometric analysis of mTCRb, compared to Mock-transduced T cells. (C) Schematic overview of the TCR-SFG-IRES-GFP vector: long terminal repeat sequence—Moloney murine leukemia virus—TCR beta chain (including the murine TCR beta constant region)—TCR alpha chain—internal ribosomal entry site—green fluorescent protein—long terminal repeat sequence. (D) Exemplary transduction efficiency of TCRft1-SFG-IRES-GFP transduced human T cells by flow cytometric analysis of mTCRb, compared to Mock-transduced T cells. (E) GFP+ human T cells after transduction with the TCRft1-SFG-IRES-GFP retroviral vector. Mean with SEM of 3 technical replicates. (F) mTCRb expression of primary T cells 4 days after transduction with the TCRft1-SFG-IRES-GFP retroviral vector. Mean with SEM of 3 technical replicates. (G) Multi-color flow cytometry assessment of different phenotypic markers in human T cells after transduction with a TCRft1-SFG-IRES-GFP vector. n = 2 biological replicates.
NLGN4X-TCR-T Lyse Glioma Cells In Vitro
We then aimed to assess the functionality of NLGN4X131–139-specific TCR-expressing primary human T cells in vitro using flow cytometric profiling following co-culture with exogenously peptide-loaded nonadherent HLA-A*02+ K562 leukemia cells. As the most relevant effector proteins, Granzyme-B (GrzB), interferon-γ (IFN-γ), and tumor necrosis factor-α (TNF-α) production was assessed. Independent of healthy T cell donors, we found robust expression of the highly cytotoxic cytokines TNF-α and GrzB by NLGN4X131–139-specific TCR-expressing T cells (Figure 3A–D). Strikingly, when benchmarking NLGN4X131–139-specific TCR-expressing primary human T cells with the well-established high-affinity melanoma antigen recognized by T cells 1 (MART-1)-specific TCR that has been used for phase I and II clinical trials,15,28 we observed similar expression levels of GrzB, IFN-γ, and TNF-α in our co-culture systems (Figure 3B–D). To confirm that the robust expression of cytotoxic proteins by NLGN4X131–139-specific TCR-expressing primary human T cells leads to target cell killing, we utilized a modified version of the Vital-FR assay to detect specific lysis of peptide-loaded K562 target cells (Figure 3E).27 Importantly, NLGN4X131–139-specific and MART1-specific T cells (MART-1-TCR-T) showed comparable cytotoxic activity against peptide-loaded K562 target cells (Figure 3F).
Figure 3.

NLGN4X-TCR-T showed comparable effective in vitro recognition and lysis of target cells as a clinically used control TCR. (A) Heatmap of the functional response (IFN-γ, TNF-α, GrzB) of primary human T cells from 3 different donors (D1-3) transduced with the NLGN4X131–139 TCR and co-cultured with peptide-loaded HLA-A*02+ K562 leukemia cells. For statistical analysis compare Figure 3B–D. n = 3 biological replicates. (B) TNF-α production of NLGN4X-TCR-T and Mart-1-TCR-T cultured with peptide-loaded HLA-A*02+ K562 leukemia cells. Target peptide: NLGN4X-TCR-T versus MART-1 TCR T cells, p = 0.0863. Mean with SEM of n = 3 biological replicates, 2-way ANOVA. (C) IFN-γ production of NLGN4X-TCR-T and MART-1-TCR-T cultured with peptide-loaded HLA-A*02+ K562 leukemia cells. Target peptide: NLGN4X-TCR-T versus MART-1-TCR-T, p = 0.2926. Mean with SEM of n = 3 biological replicates, 2-way ANOVA. (D) Granzyme B expression of NLGN4X-TCR-T versus MART-1-TCR-T cultured with peptide-loaded HLA-A*02+ K562 leukemia cells. Target peptide: NLGN4X-TCR-T versus MART-1-TCR-T, p = 0.3461. Mean with SEM of n = 3 biological replicates, 2-way ANOVA. (E) Exemplary overview of the modified Vital-FR assay28 used in this study: target cells that either endogenously expressed the target epitope or were exogenously loaded with the respective peptide were labeled with CellTraceTM FarRed and nontarget cells (irrelevant peptide or no target) were labeled with CellTraceTM Violet and cultured with target-specific TCR transduced T cells in the same well. Created with Biorender.com. (F) Live cells of either target peptide-loaded or unloaded K562 cells after overnight co-culture with NLGN4X-TCR-T or MART-1-TCR-T at an effector—target (E:T) cell ratio of 10:1 assessed by flow cytometric analysis. Mean with SEM of n = 3 biological replicates, 2-way ANOVA. Target peptide for NLGN4X-TCR-T is the NLGN4X131–139 peptide, and for MART-1-TCR-T the MART-127-35 peptide. Control peptide is the MOG35–55(myelin oligodendrocyte glycoprotein) peptide.
Next, we aimed to evaluate the cytotoxic capacity of NLGN4X-TCR-T against the HLA-A*02+ adherent glioma cell line U87. U87 wildtype cells loaded with the NLGN4X131–139 peptide induced specific upregulation of GrzB, CD69, and 4-1BB (CD137) in NLGN4X-TCR-T (Figure 4A; Supplementary Figures S4, S5A–C). In line with these findings, NLGN4X131–139-specific T cells were able to specifically lyse U87 cells loaded with the target peptide in lactate dehydrogenase (LDH)-based and flow cytometry-based killing assays (Figure 4B and C). In co-culture assays applying synthetic MHC class I-restricted peptides for reactivity testing, MHC molecules are usually saturated, and external MHC loading might occur. Although the NLGN4X131–139 was previously identified by HLA-ligandome analysis in some glioblastoma patients,1 we aimed at demonstrating endogenous presentation of NLGN4X131–139 in our tumor model system. Thus, we expressed the NLGN4X131–139 antigen by using either a tandem-minigene (TMG) with different MHC class I epitopes including NLGN4X (U87 TMG) or a retroviral vector containing full-length NLGN4X (U87 NLGN4X), confirming endogenous processing. Overexpression of the NLGN4X antigen was confirmed by qPCR and Western blot (Supplementary Figure S1A–C). Similar to our findings with K562 cells as target cells, we found specific upregulation of GrzB, CD69, and 4-1BB when we co-cultured NLGN4X-TCR-T with U87 NLGN4X or U87 TMG target cells (Figure 4D, Supplementary Figure S5D–F). Moreover, U87 TMG and U87 NLGN4X were specifically lysed by NLGN4X-TCR-T (Figure 4E). Next, we aimed to assess the reactivity of NLGN4X-TCR-T against a patient-derived glioblastoma cell line that endogenously expresses NLGN4X (Figure 4F). When we co-cultured these PB-1 cells with NLGN4X-TCR-T, we observed specific upregulation of the activation marker 4-1BB and the effector protein perforin (Figure 4G; Supplementary Figure S5G) and ratio-dependent killing of the target cell line (Figure 4H). In summary, we demonstrate that TCRft1-SFG-IRES-GFP-expressing primary human T cells specifically recognize and lyse tumor cells expressing the NLGN4X131–139 epitope including a patient-derived glioblastoma cell line with natural processing of NLGN4X protein in vitro.
Figure 4.

The NLGN4X TCR specifically recognizes and lyses glioma cells expressing the NLGN4X target epitope. (A) Heatmap of the functional response (CD69, 4-1BB, GrzB) of 3 different donors transduced with the NLGN4X TCRft1 and co-cultured with peptide-loaded HLA-A*02+ U87 glioma cells. For statistical analysis compare Supplementary Figure S5A–C. (B) Optical density (OD) measuring LDH release after overnight co-culture of NLGN4X-TCR-T and Flu (MHCI epitope) TCR transgenic T cells (Flu-TCR-T) with NLGN4X131-139 or Flu58-66 peptide-loaded U87 glioma cells. The E:T ratio was 2:1. Mean with SEM of n = 3 biological replicates, 2-way ANOVA. (C) NLGN4X-TCR-T were co-cultured with either peptide-loaded U87 glioma cells or unloaded cells and specific cytotoxicity was calculated using FACS-based counting of tumor cells. The E:T ratio was 2:1. Mean with SEM of n = 3 biological replicates, unpaired t-test. (D) Heatmap of the functional response (CD69, 4-1BB, GrzB) of 3 different donors transduced with an NLGN4X TCR or Flu TCR (here: negative control TCR) and co-cultured with U87 glioma endogenously expressing the NLGN4X protein sequence including the relevant epitope (U87 NLGN4X) or a tandem-minigene (U87 TMG) containing the antigenic sequence of NLGN4X. For statistical analysis compare Supplementary Figure S5D–F. (E) LDH release assay of NLGN4X-TCR-T versus Flu-TCR-T (TCR negative control) targeting either U87 NLGN4X, U87 TMG, or U87 (target negative control) glioma cells. The E:T ratio was 2:1. Mean with SEM of n = 3 biological replicates, 2-way RM-ANOVA. (F) Different patient-derived glioblastoma cell lines were evaluated for NLGN4X expression by Taqman quantitative PCR. PB1 was used for further in vitro testing. Normalized to Bestkeeper. Mean with SEM of 2-5 technical replicates. (G) Heatmap of the functional response (4-1BB, TNF-α, Perf, GrzB) of TCR-transgenic priamry human T cells from 5 different donors expressing either the NLGN4X TCR or Flu TCR and co-cultured with PB-1 patient-derived glioblastoma cells naturally expressing the NLGN4X131–139 epitope. Perf, perforin. For statistical analysis see Supplementary Figure S5G. (H) OD measuring LDH release of an overnight co-culture of NLGN4X-TCR-T versus Flu-TCR-T (TCR negative control) with PB-1 glioma cells. E:T ratio as indicated below. Mean with SEM of n = 3 biological replicates, 2-way RM-ANOVA. Target peptide for NLGN4X-TCR-T is the NLGN4X131–139 peptide. Control peptide is the Flu (influenza)58–66 MHC class I peptide, to which Flu-TCR-T are reactive.
NLGN4X-TCR-T Promote Tumor Regression and Improve Survival of HLA-A*02+ Glioma-Bearing Mice
Having demonstrated specific recognition of the NLGN4X131–139 epitope and tumor cell lysis of the NLGN4X-expressing U87 human glioma cell line by NLGN4X131–139-specific TCR-engineered human T cells, we next aimed to assess their therapeutic potential in vivo. Therefore, we challenged NOD scid gamma (NSG) MHC class I and II knockout (NSG MHCI/II KO) mice, that do not develop graft versus host disease after T cell transfer, with intracranial U87 TMG experimental gliomas. Mice received either NLGN4X131–139-specific TCR- (NLGN4X-TCR-T) or negative control (influenza [Flu]) TCR-engineered human T cells (Flu-TCR-T) via intracerebroventricular transfer or did not receive any T cell treatment ((NTC) = No T cell control; Figure 5A). Two injections of 5 × 106 TCR-engineered primary human HLA-A*02+ T cells with 86.6–87.4% (Supplementary Figure S6) mTCRb surface expression after transduction were performed at days 15 and 22 (Figure 5A) into the lateral ventricle of the nontumor-bearing hemisphere. Treatment with NLGN4X-TCR-T resulted in prolonged survival of glioma-bearing animals compared to Flu-TCR-T or NTC mice (Figure 5B). By using longitudinal MRI, we aimed to investigate if treatment with NLGN4X-TCR-T leads to objective radiographic responses, therefore we assessed tumor volumes between days 11 and 67 according to the modified RANO criteria.29 At day 67 as the timepoint of best response, we observed stable disease in 11.1%, partial responses (PR) in 22.2%, and complete response (CR) in 22.2% of NLGN4X-TCR-T treated mice resulting in an objective response rate (ORR: CR + PR) of 44.4% in comparison to 0.0% in both Flu-TCR-T and NTC mice (Figure 5C; Supplementary Figure S7A and C). In addition to the assessment of radiographic responses upon NLGN4X131–139-specific TCR-engineered T-cell therapy, longitudinal MRI enabled the local assessment of tumor growth (Figure 5D and E). Interestingly, 2 mice with late recurrence of U87 TMG tumors (Figure 5E; Supplementary Figure S7B) had shown radiographic responses (one PR, one CR) in previous MRI (Figure 5E). Thus, we performed a flow cytometric analysis of tumor-infiltrating leukocytes from recurrent late-stage tumors (Figure 5F). In these tumors, 78 days after the second administration of NLGN4X-TCR-T, we found predominantly CD4+ T cells with low GFP expression within the experimental tumors. Moreover, T cells were CD45RA− CCR7− and had low expression of the proliferation marker Ki67 and high PD-1 expression (Figure 5F).
Figure 5.

Intraventricular delivery of NLGN4X-TCR-T mediates temporary tumor regression and increased survival in an intracranial tumor model. (A) Schematic experimental overview: NSG MHC I/II KO mice were challenged with intracranial U87 NLGN4X antigen overexpressing gliomas and after confirmation of tumor growth NLGN4X-TCR-T or Flu-TCR-T were injected at days 15 and 22 after tumor inoculation. Created with Biorender.com. (B) Preclinical survival of U87 TMG-bearing mice treated either with NLGN4X-TCR-T or Flu-TCR-T. NTC = No T cell control. n = 9 mice for NLGN4X-TCR-T, n = 8 mice for Flu-TCR-T, n = 7 for NTC, log-rank-test. (C) Radiographic response assessment according to the mRANO criteria29: between days 11 and 67 CR was defined as a change in tumor volume of −100%, PR as < -65%, SD between −65% and +40% and PD as >+40%. (D) MRI image of one long-term surviving NLGN4X-TCR-T treated animal showing tumor regression at the initial tumor site until day 67 and tumor progression at day 98. (E) Individual growth curves of U87 TMG glioma cells of NLGN4X-TCR-T (I) and Flu-TCR-T (II) treated animals. Circled mice were analyzed by FACS as shown in Figure 5F. Log10-scaled growth. Thus, tumor volumes with V = 0 μl are not displayed in the graph. For visualization of tumor growth, the detection limit for tumor volumes was set to 0.1 μl. CR = complete response, PR = partial response, SD = stable disease, PD = progressive disease, D = death. (F) Representative flow cytometric analysis from 2 animals (M1 = mouse 1, M2 = mouse 2) with late-stage recurrence of the tumor showing persistence of primarily CD4+ T cells at the tumor site with a predominantly CCR7−CD45RA− effector memory phenotype and impaired proliferation with high PD-1 expression. Gated on live hCD3+ T cells. (G) Realtime quantitative PCR of the U87 TMG plasmid sequence in tumors of NLGN4X-TCR-T, Flu-TCR-T treated or NTC animals at late-stage time point compared to in vitro cultured U87 TMG and U87 cells. Relative expression to hGAPDH or hβ-actin, log10-scaled. n = 3 biological replicates. (H) Exemplary immunofluorescence staining of HLA-A expression: One NLGN4X-TCR-T-treated animal and one untreated animal at late stage timepoint shown. Immunofluorescence images of additional animals are shown in Supplementary Figure S8C.
Notably, recurrent U87 TMG tumors maintained expression of the NLGN4X131–139 antigen as demonstrated by qPCR, and in addition, MHC class I expression was still detectable by immunofluorescent staining (Figure 5G and H; Supplementary Figure S8A–C). Overall, our findings suggest that late recurrence results from the absence of intratumoral cytotoxic NLGN4X131–139-specific CD8+ T cells (Figure 5F–H; Supplementary Figure S8).
Furthermore, we evaluated if NLGN4X-TCR-T were also able to target the U87 NLGN4X glioma cell line carrying the full-length NLGN4X protein in vivo (Supplementary Figure S9A). Mice treated with NLGN4X-TCR-T showed reduced tumor growth at day 42 (Supplementary Figure S9B), leading to improved survival of NLGN4X-TCR-T treated mice compared to NTC animals (Supplementary Figure S9C). Thus, we demonstrate that NLGN4X-TCR-T also recognize the NLGN4X antigen when processed from the full-length NLGN4X in vivo.
NLGN4X-TCR-T Phenotypically Adapt Within the Tumor Microenvironment
At late recurrence, we found exhausted CD4+ T cells with low TCR transgene expression. Hence, this observation prompted us to assess the intratumoral phenotype of NLGN4X-TCR-T at an early timepoint after intracerebroventricular delivery in U87 TMG glioma-bearing animals (Figure 6A). Six days after intracerebroventricular transfer, T cells were present in the contralateral experimental glioma and NLGN4X131–139-specific TCR positivity was confirmed on the CD8+ T-cell subset (Figure 6B). Quantitatively, U87 TMG gliomas treated with NLGN4X-TCR-T and Flu-TCR-T were not differentially infiltrated by human CD3+ T cells in immunocompromised mice (Figure 6C). CD8+ T cells mainly showed a T-effector memory (TEM) phenotype with expression of CD45RA and high expression of Ki67 compared to low expression of the exhaustion markers PD-1 and TIM-3, regardless of which TCR was expressed (Figure 6D and E). However, specific upregulation of various activation and effector cell markers in NLGN4X-TCR-T compared to Flu-TCR-T (Figure 6F) suggests antigen recognition and specific intratumoral T cell activation. However, the fundamental difference between the early phenotypes of intratumoral NLGN4X-TCR-T (Figure 6D) and those found at late recurrence (Figure 5E) supports the hypothesis that loss of cytotoxic CD8+ NLGN4X-TCR-T results in late tumor recurrence. Altogether, we demonstrate that human T cells engineered with a patient-retrieved off-the-shelf TCR targeting the NLGN4X131–139 antigen are capable of lysing tumor cells in vitro and mediating temporary tumor control in experimental gliomas.
Figure 6.

NLGN4X-TCR-T exhibits an effector phenotype in the tumor microenvironment after intracerebroventricular delivery. (A) Experimental overview: U87 TMG gliomas were injected intracranially and NLGN4X-TCR-T or Flu-TCR-T were injected into the contralateral ventricle. After 6 days T cells were analyzed by flow cytometry and ex vivo activation was assessed. Created with Biorender.com. (B) Exemplary flow cytometry plots showing intratumoral CD3+ T cells, CD4–CD8 distribution, and mTCRb and GFP expression. Middle and right plots gated on CD3+ and CD8+ cells, respectively. (C) Normalized (to tumor volume) count of CD3+CD8+ T cells in the TME. n = 8 (NLGN4X-TCR-T), n = 7 (Flu-TCR-T). Unpaired t-test. (D) Exemplary FACS plots showing the CD45RA and CCR7 as well as Ki67 and PD-1 expression on intratumoral CD3+CD8+ T cells (TCM = T central memory cells, TN = naïve T cells, TEM = T effector memory cells, TEM-CD45RA+ = TEM re-expressing CD45RA). (E) Heatmap of phenotypic markers of intratumoral CD3+CD8+ T cells. n = 8 (NLGN4X-TCR-T), n = 7 (Flu-TCR-T). (F) Assessment of activation and effector cell markers of intratumoral CD8+ T cells. n = 8 (NLGN4X-TCR-T), n = 7 (Flu-TCR-T), 2-way ANOVA.
Discussion
The overall concept that TCR-expressing T cells are of great therapeutic potential has been underpinned by the recently published phase 3 study on patients with advanced melanoma who received TIL therapy (TIL-T). PFS was significantly longer among TIL-T-treated patients than among those who received the checkpoint inhibitor ipilimumab.30 Here, we show that an HLA-A*02-restricted TCR that was identified and retrieved from the blood of a glioblastoma patient following multi-peptide vaccination targeting—among other tumor-associated antigens—NLGN4X, and exogenously expressed in primary human T cells, can specifically recognize and lyse tumor cells and mediate objective responses in experimental glioblastoma.
Although still in its infancy, adoptive T-cell therapies using TCR-engineered T cells have already proven to be clinically beneficial in metastatic melanoma.15,31 However, until now, most experimental T-cell therapies in brain malignancies utilize CAR T cells.3–5 A major limitation of CAR T cells is that the extracellular antibody domain of CARs is only able to bind extracellular surface antigens, thereby excluding all intracellularly processed epitopes that are presented on MHC class I molecules. TCR-transgenic T cells, in contrast, are able to specifically target intracellularly expressed antigens, but rely on antigen presentation on MHC molecules. While neoantigens, mostly highly patient-individual, are of great interest for the future development of TCR-engineered T-cell therapy, we choose here to develop an off-the-shelf HLA-A*02-restricted TCR-engineered T-cell therapy for the potential use in glioblastoma patients. Antigen-specific vaccines1 can induce such high-affinity TCRs as a potential source for transgenic T cell modification. We exploited this concept to retrieve a TCR robustly recognizing NLGN4X131–139, a glioblastoma-associated antigen with a limited off-tumor expression that is presented on HLA-A*02. We expressed the NLGN4X131-139-specific TCR in human T cells using a retroviral vector that is already in clinical use. The transduction process applied here resulted in a TCR-T cell product that resembles a naïve T-cell phenotype associated with favorable anti-tumor properties.5 To date, the only comparable study assessing the preclinical potential of TCR-engineered T cells in glioma targeting an MHC class I-restricted antigen is the study by Chheda et al., proposing the use of an anti-H3.3K27M TCR for glioma therapy.32 Chheda et al. report a cytotoxicity against peptide-loaded U87 glioma cells of about 80.0% at an effector ratio of 5:1. While this is a highly innovative neoantigen-targeting concept, endogenous presentation of the H3.3K27M antigen in patients will determine the efficacy of this cellular concept in future clinical trials.33 A limitation of our study is the use of the model cell line U87 genetically engineered to express the NLGN4X antigen. Using a patient-derived glioblastoma cell line, we provide evidence that NLGN4X-TCR-T cells are able to recognize their target when naturally expressed and processed. In addition to MHC class I-restricted TCR-engineered T cell therapy, we have previously described the first MHC class II neoantigen-specific TCR-engineered preclinical T cell therapy targeting mutant Capicua transcriptional repressor (CICR215W) in syngeneic and MHC-humanized experimental gliomas.34 While this represents another promising neoantigenic concept for patients with CICR215W-mutated oligodendrogliomas, direct cytotoxicity of CICR215W-specific TCR-engineered MHC-humanized murine T cells was not observed; hence, in vivo efficacy was strongly dependent on auxiliary signals from, for example, the irradiated tumor microenvironment.34 Irradiation leads to upregulation of intratumoral NF-κB and the type I interferon responses and enhances myeloid recruitment. While irradiation can potentially augment or suppress TCR-engineered T cells depending on treatment schedules and T cell phenotypes,35,36 we observed an improved efficacy when combining preclinical irradiation with CICR215W-TCR-T; hence, it is tempting to speculate that the efficacy of NLGN4X-TCR-T can also be improved further.
NLGN4X shows limited mRNA expression in healthy neurons and oligodendrocytes; however, in humans, NLGN4X is detectable on protein level only in the tumor and not in healthy tissue.17 Up to now, it remains unclear if this results from limited sensitivity or if there is indeed no biologically meaningful target antigen processing and presentation in noncancerous cells. Of note, the NLGN4X antigen is not conserved between mouse and human37, limiting meaningful preclinical off-tumor cytotoxicity assessment. A solution to this would be to conduct safety studies in NLGN4X-humanized mouse model systems. However, such mice would only allow for the assessment of on-target off-tumor side effects. Off-target off-tumor effects would not be reliable in such testing systems. Ultimately, a clinical dose-escalation trial will be needed to assess the safety of NLGN4X131–139 TCR-engineered T cells. Similar to the multifaceted strategies in the CAR-T field, safety concepts such as the SynNotch system,38 or safety switches using reversible control of cell products either by supply or removal of small molecules, by supply of protein-based regulators, or by physical stimuli such as light, ultrasound, or heat39 are in principle adaptable for TCR-engineered T cells as well. In addition, local administration of such T cell products could increase efficacy and minimize off-target side effects. Lastly, in our model, late-stage tumor recurrence limits the long-term clinical response and is most probably driven by the loss of cytotoxic CD8+ T cells with persistent expression of the transgenic TCR. Here, repetitive infusions over a longer period of time or an innovative approach using co-expression of a co-stimulatory receptor offer promising solutions.40 Collectively, with the constantly increasing technical solutions in the field of synthetic immunology, TCR-engineered cellular therapies that target patient-individual or patient-associated antigens could pave a future path in the treatment of malignant brain tumors such as glioblastoma.
Supplementary Material
Contributor Information
Christoper Krämer, German Cancer Consortium (DKTK) Clinical Cooperation Unit (CCU) Neuroimmunology and Brain Tumor Immunology, German Cancer Research Center (DKFZ), Heidelberg, Germany; Department of Neurology, MCTN, Medical Faculty Mannheim, Heidelberg University, Mannheim, Germany.
Michael Kilian, German Cancer Consortium (DKTK) Clinical Cooperation Unit (CCU) Neuroimmunology and Brain Tumor Immunology, German Cancer Research Center (DKFZ), Heidelberg, Germany; Department of Neurology, MCTN, Medical Faculty Mannheim, Heidelberg University, Mannheim, Germany.
Yu-Chan Chih, German Cancer Consortium (DKTK) Clinical Cooperation Unit (CCU) Neuroimmunology and Brain Tumor Immunology, German Cancer Research Center (DKFZ), Heidelberg, Germany; Faculty of Bioscience, Heidelberg University, Heidelberg, Germany.
Alexandros Kourtesakis, Faculty of Bioscience, Heidelberg University, Heidelberg, Germany; Neurology Clinic, Heidelberg University Hospital, University of Heidelberg, Heidelberg, Germany; DKTK CCU Neurooncology, DKFZ, Heidelberg, Germany.
Dirk C Hoffmann, Faculty of Bioscience, Heidelberg University, Heidelberg, Germany; Neurology Clinic, Heidelberg University Hospital, University of Heidelberg, Heidelberg, Germany; DKTK CCU Neurooncology, DKFZ, Heidelberg, Germany.
Tamara Boschert, German Cancer Consortium (DKTK) Clinical Cooperation Unit (CCU) Neuroimmunology and Brain Tumor Immunology, German Cancer Research Center (DKFZ), Heidelberg, Germany; Faculty of Bioscience, Heidelberg University, Heidelberg, Germany; Helmholtz Institute of Translational Oncology (HI-TRON), Mainz, Germany.
Philipp Koopmann, German Cancer Consortium (DKTK) Clinical Cooperation Unit (CCU) Neuroimmunology and Brain Tumor Immunology, German Cancer Research Center (DKFZ), Heidelberg, Germany; Department of Neurology, MCTN, Medical Faculty Mannheim, Heidelberg University, Mannheim, Germany.
Khwab Sanghvi, German Cancer Consortium (DKTK) Clinical Cooperation Unit (CCU) Neuroimmunology and Brain Tumor Immunology, German Cancer Research Center (DKFZ), Heidelberg, Germany; Faculty of Bioscience, Heidelberg University, Heidelberg, Germany.
Alice De Roia, German Cancer Consortium (DKTK) Clinical Cooperation Unit (CCU) Neuroimmunology and Brain Tumor Immunology, German Cancer Research Center (DKFZ), Heidelberg, Germany; Department of Neurology, MCTN, Medical Faculty Mannheim, Heidelberg University, Mannheim, Germany; Faculty of Bioscience, Heidelberg University, Heidelberg, Germany; DNA Vector Laboratory, DKFZ, Heidelberg, Germany.
Stefanie Jung, German Cancer Consortium (DKTK) Clinical Cooperation Unit (CCU) Neuroimmunology and Brain Tumor Immunology, German Cancer Research Center (DKFZ), Heidelberg, Germany.
Kristine Jähne, German Cancer Consortium (DKTK) Clinical Cooperation Unit (CCU) Neuroimmunology and Brain Tumor Immunology, German Cancer Research Center (DKFZ), Heidelberg, Germany.
Bryan Day, Faculty of Medicine, University of Queensland, Herston, Australia; Cell and Molecular Biology Department, Sid Faithfull Brain Cancer Laboratory, QIMR Berghofer MRI, Brisbane, Australia; School of Biomedical Sciences, Faculty of Health, Queensland University of Technology, Gardens Point, Australia.
Lenny D Shultz, Department of Immunology, The Jackson Laboratory, Bar Harbor, Maine, USA.
Miriam Ratliff, Department of Neurosurgery, University Hospital Mannheim, Mannheim, Germany.
Richard Harbottle, DNA Vector Laboratory, DKFZ, Heidelberg, Germany.
Edward W Green, German Cancer Consortium (DKTK) Clinical Cooperation Unit (CCU) Neuroimmunology and Brain Tumor Immunology, German Cancer Research Center (DKFZ), Heidelberg, Germany.
Rainer Will, Neurology Clinic, Heidelberg University Hospital, University of Heidelberg, Heidelberg, Germany; DKTK CCU Neurooncology, DKFZ, Heidelberg, Germany; Core Facility Cellular tools, DKFZ, Heidelberg, Germany.
Michael Platten, German Cancer Consortium (DKTK) Clinical Cooperation Unit (CCU) Neuroimmunology and Brain Tumor Immunology, German Cancer Research Center (DKFZ), Heidelberg, Germany; Department of Neurology, MCTN, Medical Faculty Mannheim, Heidelberg University, Mannheim, Germany; Helmholtz Institute of Translational Oncology (HI-TRON), Mainz, Germany; Immune Monitoring Unit, National Center for Tumor Diseases (NCT), Heidelberg, Germany; DKFZ Hector Cancer Institute at the University Medical Center Mannheim, Mannheim, Germany.
Lukas Bunse, German Cancer Consortium (DKTK) Clinical Cooperation Unit (CCU) Neuroimmunology and Brain Tumor Immunology, German Cancer Research Center (DKFZ), Heidelberg, Germany; Department of Neurology, MCTN, Medical Faculty Mannheim, Heidelberg University, Mannheim, Germany; DKFZ Hector Cancer Institute at the University Medical Center Mannheim, Mannheim, Germany.
Funding
This study was supported by grants from the Swiss Cancer Foundation (Swiss Bridge Award), the Else Kröner Fresenius Foundation (2019_EKMS.49), the University Heidelberg Foundation (Hella Bühler Award), the DFG (German Research Foundation), project 404521405(CRC1389 UNITE Glioblastoma B03), the DKFZ-Hector institute (T-SIRE), the Hertie Foundation, and the Sybille Assmus Foundation to L.B.; the German Cancer Aid (Deutsche Krebshilfe, reference number 70114417), the DFG, project 404521405 (CRC1389 UNITE Glioblastoma B01), DFG, project PL-315/9-1 (RTG2727 Innate Immune Checkpoints in Cancer and Tissue Damage), the Helmholtz Institute for Translational Oncology (HI-TRON) Mainz, the project BWST_ISF2018-046 from the Baden-Württemberg-Stiftung and the DKTK Joint Funding AMI2GO to M.P.; the Rolf Schwiete Foundation (2021-009) to L.B and M.P.;the University of Heidelberg, ExploreTech! Grant to E.W.G . National Institutes of Health grant CA034196 and AI32963, DFG, SFB 1309, and University of Heidelberg, Olympia-Morata Scholarship to M.R.; and MD Fellowship RTG 2099 “Hallmarks of Skin Cancer” to P.K. M.K. received funding from the University Medical Centre Mannheim (UMM).
Conflict of interest statement
T-cell receptor-derived binding polypeptides PCT/EP2023/061716 to L.B., E.G., M.P, Y.C.C, W.W., M.K., and C.K. Non-integrating DNA vectors for the genetic modification of cells WO2019060253A1 to R.H. Honoraria from Adaptive Biotechnologies to L.B. Consulting fees from Servier, Bayer, GSK, MSD, and Roche to W.W. Royalties are received from The Jackson Laboratory from their licensing NSG MHC class I/II knockout mice to For Profit Companies by L.D.S.
Authorship statement
Performed experiments and wrote the manuscript: C.K. Performed experiments: M.K., Y.C.C., A.K., T.B., P.K., D.C.H., K.S., A.d.R., S.J, K.J., R.W., E.G. Analyzed and interpreted data: C.K., E.W.G., R.W., W.W. Provided resources: Y.C.C., A.K., T.B., A.d.R., R.H., B.D., L.D.S., M.R., R.W., W.W. Edited manuscript, analyzed and interpreted data: M.K., Y.C.C., A.K., T.B., K.S., A.d.R., S.J, K.J., L.D.S., R.H., E.W.G., R.W., W.W., M.P., L.B. Methodology: R.W., R.H., E.W.G, L.B. Conceptualized the study, wrote the manuscript, analyzed and interpreted data: M.P. and L.B.
References
- 1. Hilf N, Kuttruff-Coqui S, Frenzel K, et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature. Jan 2019;565(7738):240–245. [DOI] [PubMed] [Google Scholar]
- 2. Lim M, Weller M, Idbaih A, et al. Phase 3 trial of chemoradiotherapy with temozolomide plus nivolumab or placebo for newly diagnosed glioblastoma with methylated MGMT promoter. Neuro Oncol. 2022;24(11):1935–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Majzner RG, Ramakrishna S, Yeom KW, et al. GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature. 2022;603(7903):934–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brown CE, Alizadeh D, Starr R, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med. 2016;375(26):2561–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vitanza NA, Johnson AJ, Wilson AL, et al. Locoregional infusion of HER2-specific CAR T cells in children and young adults with recurrent or refractory CNS tumors: an interim analysis. Nat Med. 2021;27(9):1544–1552. [DOI] [PubMed] [Google Scholar]
- 6. O’Rourke DM, Nasrallah MP, Desai A, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. 2017;9(399):eaaa0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ahmed N, Brawley V, Hegde M, et al. HER2-specific chimeric antigen receptor-modified virus-specific T cells for progressive glioblastoma: a phase 1 dose-escalation trial. JAMA Oncol. 2017;3(8):1094–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kilian M, Bunse T, Wick W, Platten M, Bunse L.. Genetically modified cellular therapies for malignant gliomas. Int J Mol Sci . 2021;22(23):12810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Theruvath J, Sotillo E, Mount CW, et al. Locoregionally administered B7-H3-targeted CAR T cells for treatment of atypical teratoid/rhabdoid tumors. Nat Med. 2020;26(5):712–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Donovan LK, Delaidelli A, Joseph SK, et al. Locoregional delivery of CAR T cells to the cerebrospinal fluid for treatment of metastatic medulloblastoma and ependymoma. Nat Med. 2020;26(5):720–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Johnson LA, Scholler J, Ohkuri T, et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci Transl Med. 2015;7(275):275ra22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brown CE, Starr R, Aguilar B, et al. Stem-like tumor-initiating cells isolated from IL13Ralpha2 expressing gliomas are targeted and killed by IL13-zetakine-redirected T Cells. Clin Cancer Res. 2012;18(8):2199–2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Heczey A, Courtney AN, Montalbano A, et al. Anti-GD2 CAR-NKT cells in patients with relapsed or refractory neuroblastoma: an interim analysis. Nat Med. 2020;26(11):1686–1690. [DOI] [PubMed] [Google Scholar]
- 14. Vitanza NA, Wilson AL, Huang W, et al. Intraventricular B7-H3 CAR T cells for diffuse intrinsic pontine glioma: preliminary first-in-human bioactivity and safety. Cancer Discov. 2022;13(1):114–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Morgan RA, Dudley ME, Wunderlich JR, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314(5796):126–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Parkhurst MR, Yang JC, Langan RC, et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther. 2011;19(3):620–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dutoit V, Herold-Mende C, Hilf N, et al. Exploiting the glioblastoma peptidome to discover novel tumour-associated antigens for immunotherapy. Brain. 2012;135(Pt 4):1042–1054. [DOI] [PubMed] [Google Scholar]
- 18. The Human Protein Atlas. NLGN4X protein expression summary.https://www.proteinatlas.org/ENSG00000146938-NLGN4X. Date accessed February 28, 2022.
- 19. Ichtchenko K, Hata Y, Nguyen T, et al. Neuroligin 1: a splice site-specific ligand for beta-neurexins. Cell. 1995;81(3):435–443. [DOI] [PubMed] [Google Scholar]
- 20. Varoqueaux F, Aramuni G, Rawson RL, et al. Neuroligins determine synapse maturation and function. Neuron. 2006;51(6):741–754. [DOI] [PubMed] [Google Scholar]
- 21. Marro SG, Chanda S, Yang N, et al. Neuroligin-4 regulates excitatory synaptic transmission in human neurons. Neuron. 2019;103(4):617–626.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sauer T, Parikh K, Sharma S, et al. CD70-specific CAR T cells have potent activity against acute myeloid leukemia without HSC toxicity. Blood. 2021;138(4):318–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schmitt PDM. Treatment of patients with relapsed or refractory CD19+ lymphoid disease with T cells expressing a third-generation CAR. 2022. https://clinicaltrials.gov/ct2/show/study/NCT03676504. Date accessed August 12, 2022,
- 24. Brewin J, Mancao C, Straathof K, et al. Generation of EBV-specific cytotoxic T cells that are resistant to calcineurin inhibitors for the treatment of posttransplantation lymphoproliferative disease. Blood. 2009;114(23):4792–4803. [DOI] [PubMed] [Google Scholar]
- 25. Blache U, Popp G, Dunkel A, Koehl U, Fricke S.. Potential solutions for manufacture of CAR T cells in cancer immunotherapy. Nat Commun. 2022;13(1):5225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Alspach E, Lussier DM, Miceli AP, et al. MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature. 2019;574(7780):696–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stanke J, Hoffmann C, Erben U, et al. A flow cytometry-based assay to assess minute frequencies of CD8+ T cells by their cytolytic function. J Immunol Methods. 2010;360(1-2):56–65. [DOI] [PubMed] [Google Scholar]
- 28. Rosenberg SA. Phase II study of metastatic melanoma with lymphodepleting conditioning and infusion of anti-MART-1 F5 TCR-gene-engineered lymphocytes. 2022. https://clinicaltrials.gov/ct2/show/results/NCT00509288. Date accessed September 5, 2022. [Google Scholar]
- 29. Ellingson BM, Wen PY, Cloughesy TF.. Modified criteria for radiographic response assessment in glioblastoma clinical trials. Neurotherapeutics. 2017;14(2):307–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rohaan MW, Borch TH, van den Berg JH, et al. Tumor-infiltrating lymphocyte therapy or ipilimumab in advanced melanoma. N Engl J Med. 2022;387(23):2113–2125. [DOI] [PubMed] [Google Scholar]
- 31. Morgan RA, Chinnasamy N, Abate-Daga D, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother. 2013;36(2):133–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chheda ZS, Kohanbash G, Okada K, et al. Novel and shared neoantigen derived from histone 3 variant H3.3K27M mutation for glioma T cell therapy. J Exp Med. 2018;215(1):141–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Immisch L, Papafotiou G, Popp O, et al. H3.3K27M mutation is not a suitable target for immunotherapy in HLA-A2+ patients with diffuse midline glioma. J ImmunoTher Cancer. 2022;10(10):e005535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kilian M, Friedrich M, Sanghvi K, et al. T-cell receptor therapy targeting mutant capicua transcriptional repressor in experimental gliomas. Clin Cancer Res. 2022;28(2):378–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Spiotto M, Fu YX, Weichselbaum RR.. The intersection of radiotherapy and immunotherapy: mechanisms and clinical implications. Sci Immunol. 2016;1(3):EAAG1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Grassberger C, Ellsworth SG, Wilks MQ, Keane FK, Loeffler JS.. Assessing the interactions between radiotherapy and antitumour immunity. Nat Rev Clin Oncol. 2019;16(12):729–745. [DOI] [PubMed] [Google Scholar]
- 37. Bolliger MF, Pei J, Maxeiner S, et al. Unusually rapid evolution of Neuroligin-4 in mice. Proc Natl Acad Sci U S A. 2008;105(17):6421–6426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Choe JH, Watchmaker PB, Simic MS, et al. SynNotch-CAR T cells overcome challenges of specificity, heterogeneity, and persistence in treating glioblastoma. Sci Transl Med. 2021;13(591):eabe7378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sahillioglu AC, Schumacher TN.. Safety switches for adoptive cell therapy. Curr Opin Immunol. 2022;74:190–198. [DOI] [PubMed] [Google Scholar]
- 40. Alizadeh D, Wong RA, Yang X, et al. IL15 Enhances CAR-T cell antitumor activity by reducing mTORC1 activity and preserving their stem cell memory phenotype. Cancer Immunol Res. 2019;7(5):759–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Cell lines will be provided by the corresponding author upon reasonable request. All additional datasets generated or analyzed during this study are included in this published article (and its Supplementary Data files).