

Abstract
Transforming growth factor-βs (TGF-βs) and bone morphometric proteins (BMPs) belong to the TGF-β superfamily and perform essential functions during osteoblast and chondrocyte lineage commitment and differentiation, skeletal development, and homeostasis. TGF-βs and BMPs transduce signals through SMAD-dependent and -independent pathways; specifically, they recruit different receptor heterotetramers and R-Smad complexes, resulting in unique biological readouts. BMPs promote osteogenesis, osteoclastogenesis, and chondrogenesis at all differentiation stages, while TGF-βs play different roles in a stage-dependent manner. BMPs and TGF-β have opposite functions in articular cartilage homeostasis. Moreover, TGF-β has a specific role in maintaining the osteocyte network. The precise activation of BMP and TGF-β signaling requires regulatory machinery at multiple levels, including latency control in the matrix, extracellular antagonists, ubiquitination and phosphorylation in the cytoplasm, nucleus-cytoplasm transportation, and transcriptional co-regulation in the nuclei. This review weaves the background information with the latest advances in the signaling facilitated by TGF-βs and BMPs, and the advanced understanding of their diverse physiological functions and regulations. This review also summarizes the human diseases and mouse models associated with disordered TGF-β and BMP signaling. A more precise understanding of the BMP and TGF-β signaling could facilitate the development of bona fide clinical applications in treating bone and cartilage disorders.
Subject terms: Cell biology, Developmental biology
Introduction
Transforming growth factor-βs (TGF-βs) and bone morphometric proteins (BMPs) are cytokines belonging to the TGF-β superfamily. Around the 1970s, TGF-β was discovered as a growth factor (GF) that can transform mammalian fibroblasts.1 At the same time, BMP was found to be capable of inducing ectopic bone formation.2 TGF-β and BMP signaling regulates a variety of physiological and pathological processes. TGF-β and BMP signaling is also critical for skeletal system development and homeostasis, which has been comprehensively investigated by using cell and animal models and clinical studies. Numerous mutations of the genes in TGF-β and BMP signaling are associated with human skeletal disorders. Many mouse models with dysregulated TGF-β and BMP signaling displayed certain skeleton defects. In this review paper, we summarize the genetic mouse models (Table 1) and human diseases (Table 2) related to TGF-β and BMP signaling in the skeleton. We also comprehensively review the essential roles and dynamic regulatory functionality of TGF-β and BMP signaling in the skeletal system during embryonic development and postnatal homeostasis, mostly focusing on chondrocytes, osteoblasts, osteocytes, and osteoclasts.
Table 1.
Mouse models of TGF-β and BMP signaling in bone.
| Classification | Gene | KO/CKO/Tg/knock-in | Phenotype | References |
|---|---|---|---|---|
| TGF-β ligands | Tgfb1 | KO | Early death (1 month) | 88 |
| Tgfb1−/−Rag2−/− | Reduced bone density; OB↓ | 152 | ||
| Col1α1 Prom-Tgfb1 H222D Tg (1) | Diaphyseal thickening, fluctuating bone volume, increased bone remodeling, prone to fracture; OB↑; OC↑ | 152 | ||
| Col1α1 Prom-Tgfb1 H222D Tg | Knee and temporomandibular joint osteoarthritis↑ | 221,222 | ||
| Tgfb2 | KO | Perinatal mortality; neural arch defect; bifurcated sternum; shortened radius and ulna | 85 | |
| Tgfb3 | KO | Die within 20 h of birth; failure of the palatal shelves to fuse leading to cleft palate | 86,87 | |
| BMP ligands | Bmp2 | Col2α1-CreER | Severe chondrodysplasia; shortened stature and limbs; chondrocyte proliferation↓ & hypertrophy↓ | 50 |
| Prx1-Cre | Normal limb patterning | 51 | ||
| Bmp7 | KO | Die at birth; skull base defects; rib & sternum malformation; hindlimb polydactyly | 326 | |
| Bmp7, Alk6 | DKO | Malformed and shortened appendicular bones compared to Bmp7−/− | 25 | |
| Bmp2, 7 | Prx1-Cre;Bmp2f/f;Bmp7−/− | Slightly diminished appendicular skeleton; missing the last phalanx in digit III; malformed fibulae | 51 | |
| Bmp4 | Col2α1-CreER | Mild chondrodysplasia | 50 | |
| Prx1-Cre | Polydactyly | 51,52 | ||
| Bmp2, 4 | Col2α1-CreER | Severe chondrodysplasia; severely shortened and malformed or missing long bone skeleton element & fused joints; chondrocyte proliferation↓ & hypertrophy↓ | 50 | |
| Prx1-Cre | Polydactyly; complete syndactyly; delayed mineralization; chondrogenesis ↓ ; osteogenesis↓ | 51 | ||
| BMP3/GDF10 | Col1 Prom-Bmp3 Tg | Late hypertrophic chondrocyte differentiation↓; thinner cortical bone; mineralization ↓ ; rib fracture | 115 | |
| KO | Increased bone density | 28 | ||
| Bmp14/GDF5 | bpJ (2) | Normal axial bones; shortened limbs and digits; missing joints of autopods; missing phalange elements | 25,327 | |
| No delay in fracture healing | 328 | |||
| Increased joint damage in collagen-induced arthritis; reduced bone density | 329 | |||
| GDF5, Alk6 | bpJ/bpJ;Alk6−/− | Same as bpJ | 25 | |
| Type I receptors | Alk2/Acvr1 | Col2α1-Cre | Shortened cranial base; hypoplastic cervical vertebrae | 49 |
| Osx-Cre | Mandibular bone density ↓ ; OB ↓ ; sRANKL ↑ ; OC↑ | 113 | ||
| Q207D Tg | Fibrodysplasia ossificans progressive | 127,130,132 | ||
| Acvr1tnR206H/+;Tie2-Cre | Fibrodysplasia ossificans progressive | 128 | ||
| R206H knock-in | Fibrodysplasia ossificans progressive | 129 | ||
| Alk3/Bmpr1A | Col2α1-Cre | Split dorsal arches; shortened limbs; hypoplastic scapula; chondrodysplasia, chondrocyte proliferation ↓ , hypertrophy ↓ , terminal differentiation ↓ , and apoptosis↑ | 48,49,65 | |
| Gdf5-Cre | Automatically develop osteoarthritis | 330 | ||
| Col1α1-CreER | bone mass in long bones and ribs ↑ ; strength ↑ ; OC↓ | 148,149 | ||
| Sp7-Cre | Trabecular bone mass↑ | 147 | ||
| Dmp-Cre | Trabecular bone mass↑ (13-fold); OB proliferation↑ and activity ↓ ; RANKL & SOST ↓ ; OC↓ | 147,176 | ||
| Alk6/Bmpr1B | KO | Brachypodism; reduced phalangeal elements; the fusion of appendicular joints, similar to GDF5 mutant (bpJ) mice | 25,48,49,65 | |
| KO | Transient and gender-specific osteopenia caused by reduced osteogenesis from MSCs | 331 | ||
| Col1α1 Prom-truncated Alk6 Tg | Reduced BMD and bone volume; reduced osteoblast and osteoclast number | 332 | ||
| Alk3, Alk6 | Col2α1-Cre;Alk3f/f;Alk6+/− | Phenotype resembling and more severe than Alk3 CKO mice | 48,65 | |
| Col2-Cre;Alk3f/f;Alk6−/− | Severe defects in cartilage formation and skeletogenesis | 48 | ||
| Alk2, Alk3 | Col2-Cre | Malformed axis skeleton (vertebra, cervical and thoracic regions); more severe appendicular defects than Alk3 CKO mice | 49 | |
| Alk2, Alk6 | Col2-Cre;Alk2f/f;Alk6−/− | More severe axis and appendicular defects than each single KO | 49 | |
| Alk5/TGFBR1 | Dermo-Cre | Short and wide long bones, ectopic cartilaginous protrusions, reduced bone volumes | 83 | |
| Col2α1 Prom-Alk5 DN Tg | Elongated limbs; chondrocyte proliferation↑ | 68 | ||
| Col2α1-CreERT | Automatic osteoarthritis | 208,333 | ||
| Type II receptors | Tgfbr2 | Nestin-CreER | Knee osteoarthritis↓ | 221 |
| ColX-Cre | Delayed chondrocyte terminal differentiation; impeded mineralization | 84 | ||
| Prx-Cre | Die at birth; reduced periodontal and frontal bone; shortened limbs; split sternum; autopod joint fusion; reduced mineralization; chondrocyte proliferation↓ & hypertrophy↑ | 78,81 | ||
| Col2α1-Cre | Survive; neural arch defect; missing/incomplete intervetebral discs; progressive reduction in long bone length | 79,80 | ||
| Sp7-Cre (Dox) | Early death (1 M); reduced body size; reduced bone volume; increased bone marrow adipose tissue; disrupted molar tooth formation; OB↓ | 157,334 | ||
| Ocn-Cre | bone density ↑ ; OB ↑ ; OC↑ | 172 | ||
| Bmpr2 | Prx1-Cre | Trabecular bone volume ↑ ; BFR ↑ ; selectively abolish Activin-Smad2/3 but not Bmp-Smad1/5/8 signaling | 335 | |
| Col1α1 Prom-Bmpr2 DN Tg | Dwarfism; delayed mineralization; bone volume ↓ ; no change in cortical bone | 114 | ||
| ActRIIA/Acvr2A | Ocn-Cre | Trabecular bone volume↑ | 336 | |
| ActRIIB/Acvr2B | KO | Late hypertrophic chondrocyte differentiation ↓ ; thinner cortical bone; mineralization ↓ ; rib fracture | 115 | |
| Ocn-Cre | Normal | 336 | ||
| ActRIIA, ActRIIB | Ocn-Cre | Trabecular bone volume↑ like ActRIIA−/− | 336 | |
| Canonical pathway | Smad1 | Col1α1-Cre | Osteopenia; OB proliferation and differentiation↓ | 337 |
| Col2α1-Cre | Shortened growth plate; chondrocyte hypertrophy↓ and proliferation↓ | 68 | ||
| Smad1/5 | Col2α1-Cre;Smad1f/f;Smad5+/− | Similar and more severe phenotype than Smad1 CKO | 68 | |
| Smad1/5 | Col2α1-Cre | Chondrodysplasia; shortened limbs; thicker perichondrium; matrix production ↓ ; hypertrophy↓ | 69 | |
| Smad8 | KO | Normal | 69 | |
| Smad1/5/8 | Col2α1-Cre;Smad1f/f;Smad5f/f;Smad8−/− | Absence of an axial skeleton; severely disorganized appendicular bones | 69 | |
| Smad2 | Col2α1-Cre | Similar and more severe phenotype than Smad3 KO mice | 89 | |
| Smad2/3 | Col2α1-Cre;Smad2f/f;Smad3−/− | Similar and more severe phenotype than Smad3 KO mice | 89 | |
| Smad3 | KO | Postnatal dwarfism; expanded columnar and hypertrophic zone; chondrocyte proliferation↑ and hypertrophy↑ | 89 | |
| KO | Knee and temporomandibular joint osteoarthritis | 214,215 | ||
| KO | Osteopenia; OB and OCY apoptosis↑ | 156 | ||
| Smad4 | Tbx18-Cre | Short limbs, chondrogenesis↓ & hypertrophy ↓ , missing stylopod | 40 | |
| Sp7-Cre (Dox) | Increased trabecular bone mass | 147 | ||
| Sp7-Cre | Stunted growth; spontaneous fractures; increased trabecular bone volume; decreased BMD; a combination of features seen in osteogenesis imperfecta, cleidocranial dysplasia, and Wnt-deficiency syndromes | 118 | ||
| Dmp-Cre | Increased trabecular bone mass (~2-fold) | 147 | ||
| Col1α1-Cre | Increased trabecular bone mass; protection from tail suspension-induced bone loss; OB & OCY number ↑ ; OB & OCY apoptosis↓ | 178 | ||
| Ocn-Cre | Lower bone mass < 6-month, more bone mass > 7-month | 177 | ||
| Ctsk-Cre | Reduced bone mass; OC↑ | 186 | ||
| Non-canonical pathway | TAK1 | Col2α1-CreER | Growth retardation; osteoarthritis markers ↑ ; no osteoarthritis histological signs | 71 |
| Osx-Cre | Cleidocranial dysplasia (CCD)-like phenotype (clavicular hypoplasia and delayed fontanelle fusion); OB ↓ ; reduced cancellous and trabecular bone volume | 116 | ||
| Col2α1-Cre | Shorter limbs; chondrocyte proliferation ↓ , survival↓ & hypertrophy ↓ ; failure to maintain interzone cells of the elbow joint | 338 | ||
| Prx1-Cre | Widespread joint fusions; chondrocyte hypertrophy and proliferation | 338 | ||
| p38 | Ocn-Cre | OB activity and BFR ↓ ; reduced cancellous and trabecular bone volume | 117 | |
| Col2α1 Prom-p38DN Tg | Shortened limbs; knee joint osteoarthritis↑ | 210 | ||
| p38b−/− | A substantial decrease in long bone mineralization and a more modest effect on the calvarium | 116 | ||
| MKKs | Mkk3−/−Mkk6+/− and Mkk3−/− | Similar phenotype to Tak1f/f;Osx-Cre mice | 116 | |
| I-SMAD and ubiquitin-related regulation | Smad6 | Col11α1 Prom-Smad6 Tg | Dwarfism and osteopenia; chondrocyte hypertrophy↓ | 279 |
| KO | Dwarfism; defects in axial and appendicular bones; delayed onset of hypertrophy | 280 | ||
| Smad6;Smurf1 | Col11α1::Tg | More severely delayed endochondral ossification than Smad6 Tg | 279 | |
| Smad7 | ΔExon1 | Osteopenia; BFR ↓ ; OC↑ | 282 | |
| KO | Chondrocyte proliferation and hypertrophy ↓ ; shortened growth plate | 339 | ||
| Prx1 Prom-Tg; Col11 Enh-Tg; Col11 Prom-Tg | Chondrodysplasia; mesenchymal condensation ↓ ; chondrocyte proliferation and hypertrophy↓ | 283 | ||
| Smurf1 | KO | Bone mass ↑ ; BFR↑ | 286 | |
| Col1α1 Prom-Tg | Osteopenia; BFR↓ | 285 | ||
| Smurf2 | Col2α1 Prom-Tg | Osteoarthritis; intervertebral disc degeneration | 291,292 | |
| KO | Protection from age-related and DMM-induced osteoarthritis | 293 | ||
| KO | Osteopenia ↓ ; OC↑ | 296 | ||
| KO | Enhanced BMP-induced ectopic bone formation | 294 | ||
| PLEKHO1 | Osterix-Cre | Protection from age-related bone loss | 289 | |
| Osterix Prom-Tg | Age-related bone loss | 289 | ||
| NEDD4 | Col1α1-Cre | Bone mass ↓ ; OB↓ | 298 | |
| Col1α1 Prom-Tg | Bone mass ↑ ; OB↑ | 298 | ||
| Jab1 | Osx-Cre | Dwarfism; trabecular bone mass↓ | 303 | |
| Antagonists | Noggin | Ocn Prom-Tg | Osteopenia; BFR↓ | 250,253 |
| Ocn-Cre | Osteopenia | 254 | ||
| KO | Hyperplasia of cartilage; joint development failure; multiple skeletal defects related to neural tube and somite patterning (failure of neural tube closure, broad club-shaped limbs, loss of caudal vertebrae, a shortened body axis, and retention of a small vestigial tail) | 66,67 | ||
| Grem1 | Ocn-Cre | Bone mass ↑ ; BFR↑ | 258 | |
| Ocn Prom-Tg | Bone fractures; bone mass ↓ ; BFR↓ | 259 | ||
| FS | Tg | Bone mass ↓ ; bone fractures | 262 | |
| Co-receptors | β-glycan | KO | Defective palate development with OB↓ | 267 |
| Nrps | KO | Bone mass ↓ ; OB ↓ ; OC↓ | 268 | |
| Neogenin | KO | Elongated growth plate; chondrocyte proliferation & apoptosis ↓ ; endochondral ossification↓ | 276 | |
| Other regulators | Tmem53 | KO | Sclerosing bone | 308 |
| Endofin | Col1 Prom-Endofin F872A Tg | Bone mass ↑ ; BFR↑ | 304 |
Prom promoter, Enh Enhancer, Tg transgenic, KO knockout, CKO conditional knockout, BFR bone formation rate, OB osteoblast, OC osteoclast, OCY osteocyte, BMD bone mineral density, DN dominant negative
Annotations.
(1) Col1α1 Prom-Tgfb1 H222D Tg, transgenic mice carrying a Tgfb1 H222D mutant under the control of Col1α1 promoter; Tgfb1 H222D mutant is found in human Camurati-Engelmann disease (CED). This mouse model is also named as Tgfb1-CED or CED mice.
(2) BpJ is the mouse model carrying Gdf5 mutation (bp-J allele) which occurs spontaneously in the A/J strain. Bp is short for brachypodism.
Table 2.
Human diseases related to TGF-β and BMP signaling in bone.
| Gene | Disease | MIM# | Bone disorders | References |
|---|---|---|---|---|
| NOGGIN, GDF5 | Symphalangism | 185800, 186500, 184460, 615298 | Ankylosis or synostosis of the interphalangeal joints | 59,60 |
| NOGGIN | Tarsal–carpal coalition syndrome | 186570 | Fusion of the carpals, tarsals, and phalanges; short first metacarpals causing brachydactyly; humeroradial fusion | 61 |
| NOGGIN, BMP2, BMPR1B, GDF5 | Brachydactyly | 611377, 112600, 113100 | Brachydactyly | 60,62–64 |
| TGFΒR1, TGFΒR2, TGFΒ2, TGFΒ3, SMAD2, SMAD3 | Loeys-Dietz syndrome | 609192, 610168, 613795, 614816, 615582, 619656 | Variable skeletal anomalies (including skeletal overgrowth, pectus deformity, osteoarthritis, hernias, etc.) | 72–76 |
| ACVR1 | Fibrodysplasia ossificans progressiva | 135100 | Progressive heterotopic bone formation in muscles, tendons, ligaments, and joints | 122,123 |
| TGFB1 | Camurati-Engelmann disease | 131300 | Osteosclerotic lesions in the long bones and skull with increased remodeling; osteoarthritis | 158 |
| SMAD3, MAP2K1, LEMD3 | Melorheostosis | 155950 | Melorheostosis (special sclerosing bone disease) | 159–161,306,307 |
| LEMD3 | Osteopoikilosis; Buschke-Ollendorff syndrome | 166700 | Sclerosing bone | 306,307 |
| TMEM53 | Craniotubular dysplasia, Ikegawa type | 619727 | Hyperostosis; short stature in association with macrocephaly, dolichocephaly, or a prominent forehead | 308 |
| FBN-1 | Marfan syndrome | 154700 | Variable skeletal anomalies including long bone overgrowth | 77 |
| FBN-2 | Congenital contractural arachnodactyly | 121050 | Long limbs (dolichostenomelia) and long, slender fingers and toes (arachnodactyly), permanently bent joints (contractures) | 230 |
| ADAMTSL2 | Geleophysic dysplasia | 231050 | Short stature, short extremities, and skeletal abnormalities | 243 |
| ADAMTS10, ADAMTS17 | Weill-Marchesani syndrome | 277600, 608328 | Short stature brachydactyly, and ectopia lentis | 245 |
| COL1A1, COL1A2 | Osteogenesis imperfecta | 259420 | A bone dysplasia characterized by bone deformities, fractures, and a high un-union rate caused by low bone mass and impaired bone quality | 235 |
| EXT1, EXT2 | hereditary multiple exostoses | 33700, 133701 | Formation of cartilage-capped bony growths (osteochondroma) at the ends of the bones | 238 |
| SKI | Shprintzen-Goldberg syndrome | 182212 | A wide range of skeletal abnormalities including craniosynostosis, distinctive facial features, arachnodactyly, long limbs, pectus excavatum or carinatum, and scoliosis | 313 |
Overview of TGF-β and BMP Signaling Pathways
In the TGF-β and BMP signaling pathways, the dimeric ligands bind to heterotetrameric receptors comprising two type I and two type II receptors.1–3 This binding ultimately results in the phosphorylation and activation of a glycine-serine-rich domain within the type I receptor by the constitutively active type II receptor, transducing signals downstream through both suppressor of mothers against decapentaplegic homolog (SMAD)-dependent and -independent pathways (Figs. 1 and 2).1–3 The heterogenous ligand–receptor combinations and the dynamic regulations of TGF-β and BMP signaling result in versatile outcomes.
Fig. 1. BMP signaling in bone remodeling.

Pro-BMP proteins are bound with matrix proteins and are processed into active GF dimers through the proteolytic degradation of the PD by ADAMTSs and MMPs. BMP activity is regulated by bone matrix proteins in the extracellular region (FBN-1, COL1, HS) and by extracellular antagonists (Noggin, Grem1, Grem2, Chordin). Active BMPs bind to a receptor heterotetramer comprising of Type I and II receptors. Co-receptors such as Neogenin and endoglin might cooperatively bind to BMP receptors or ligands. The bindings ultimately result in the phosphorylation of type I receptors to transduce downstream signals through canonical and non-canonical pathways. In the canonical pathway, BMP-specific R-SMADs (SMAD-1/5/8) are activated by phosphorylation at C-terminal SSXS domains and form a complex with the Co-SMAD SMAD4 through the C-terminal MH2 domains. The activated SMAD complex then translocates into the nucleus to regulate the transcription of target genes. In the cytoplasm, I-SMAD SMAD6 inhibits the signaling by interfering with receptor–R-SMAD or SMAD complex formation. SMAD6 also cooperates with ubiquitin ligases (Smurf1 and Nedd4) to induce the ubiquitination and degradation of R-SMADs. Deubiquitinases such as Usp15 and LMP-1 positively regulate BMP signaling by antagonizing R-SMAD degradation. The nuclear translocation of the SMAD complex is regulated by nuclear envelope proteins such as TMEM53 and LEMD. In the osteoblast, the transcription function of the SMAD complex is regulated by co-transcription factors (p300, β-catenin, CBP, TCF4, Runx2) or repressors (HDAC4/5–SnoN; Ski complex, HDAC1–Nkx3.2 complex, Tob, Foxc1). In the non-canonical pathway, TRAF6 is recruited to the receptor to activate downstream factors, including MAPKs, PI3K, and small GTPases (Rho, Rac, Cdc42). MAPK signaling positively regulates activity of transcriptional factors, including Runx2 in osteoblasts and NF-кB in osteoclasts. Ultimately, BMP signaling promotes both osteoblast and osteoclast differentiation at all stages. OB osteoblast, pre-OB pre-osteoblast, BMM bone marrow monocyte, OC osteoclast.
Fig. 2. TGF-β signaling in bone remodeling.

Besides bone matrix proteins, the latency of TGF-βs is also maintained by LTBPs, which bind TGF-β precursors to form the LLC. Active TGF-β peptides are released by osteoclastic bone resorption and proteolytic degradation by ADAMTSs and MMPs. Active TGF-β binds with a receptor heterotetramer, which transduces signals through canonical and non-canonical pathways like BMPs. Co-receptors β-glycan and Nrps facilitate ligand–receptor binding. In the canonical pathway, TGF-β-specific R-SMADs (SMAD-2/3) are activated by phosphorylation at C-terminal SSXS domains and form a complex with the Co-SMAD SMAD4 through the C-terminal MH2 domains. The activated SMAD complex would then translocate into the nucleus to regulate the transcription of target genes. I-SMAD SMAD7 and Smurf2 antagonize signaling activation in the cytoplasm. The nuclear translocation of the SMAD complex is regulated by nuclear envelope protein LEMD. In the osteoblast, the SMAD complex drives osteogenic gene expression (Dlx5, Runx2); however, it recruits HDAC4/5 to antagonize Runx2 activity and drives the expression of genes that inhibit osteoblast formation (HDAC6, Smurf1). The SMAD complex also plays dual roles in osteoclastogenic gene expression in the osteoclast. In the non-canonical pathway, TRAF6 is recruited to the receptor to activate downstream factors, including MAPKs, PI3K, and small GTPases (Rho, Rac, Cdc42). MAPK signaling positively regulates activity of transcriptional factors, including Runx2 in osteoblasts and NF-кB in osteoclasts. Ultimately, TGF-β promotes osteoblast and osteoclast early differentiation, limiting their later maturation. TGF-β also maintains the formation and property of osteocytes, while its mechanism remains unclear. OB osteoblast, pre-OB pre-osteoblast, OCY osteocyte, BMM bone marrow monocyte, OC osteoclast, pre-OC pre-osteoclast.
Ligands and receptors: structure, diversity, and selectivity
More than 30 TGF-β superfamily members have been identified in mammals, including TGF-βs, BMPs/growth differentiation factors (GDFs), Nodals, and Activins. There are only a few studies addressing the roles of Nodals and Activins in the skeleton,4–6 which indicate that they play a negative role in osteogenesis. Activin A signaling was reported to increase in the skeleton of patients with chronic kidney disease-mineral bone disorder and might contribute to deranged bone turnover.5 In contrast, the functions of BMPs and TGF-βs in the skeleton have been more extensively investigated, and this review will mostly focus on them.
So far, more than 15 BMPs have been discovered in both humans and rodents. The skeletal system synthesizes many different BMPs, including BMP2, BMP3b/GDF10, BMP4, BMP5, BMP6, BMP7, BMP9/GDF2, BMP13/GDF6, and BMP14/GDF5.7 All three TGF-β ligands (TGF-β1, TGF-β2, and TGF-β3) are expressed in the skeleton.8 TGF-βs and BMPs are synthesized and secreted as pro-protein complexes which contain two N-terminal prodomains (PDs) non-covalently interacting with the C-terminal mature GF dimer (Fig. 3a, b).7,8 The PDs control the activity of GFs in different ways, including latency, localization, stability, and proper dimer formation.9 PDs of TGF-βs keep GFs latent in extracellular matrix (ECM) and control their bioavailability.8,10 Pro-TGF-β is also known as the small latent complex (SLC), with its PD known as latency-associated peptide (LAP). LAP interacts with a latent binding protein (LTBP) to form the large latent complex (LLC), which binds to ECM proteins such as fibrillin (FBN).8,10 The release and activation of TGF-βs from ECM involves dissociation at acidic pH or proteolysis by matrix metalloproteinases (MMPs) of osteoclasts.11–13 Our work showed that activation of TGF-β is abolished in ATP6i-deficient mice, whose osteoclasts were dysfunctional.14 In contrast, PDs of some BMPs do not convey their latency, including BMP4, BMP5, BMP7, and BMP9.15–17 Among them, BMP7 pro-protein is bound with FBNs in ECM to form proper signaling gradients,18 while BMP9 pro-protein is circulating,15 and the PD of BMP4 is also essential for the generation of active BMP4/7 heterodimer.16 Therefore, the activity of BMPs and TGF-βs are controlled by endopeptidases, and might also be controlled by matrix composites or matrix degradation enzymes if they are ECM-bound (discussed in more detail below).
Fig. 3. Structure and selectivity of TGF-β and BMP ligands and receptors.

a–c Structures of pro-TGF-β1, pro-BMP9, and TGF-β1–TGFBR1–ALK5 complex were re-created from PDB files with accession codes 3RJR, 4YCI and 3KFD, respectively. Pro-TGF-β1 and pro-BMP9 both contain PD and GF dimer which non-covalently interact with each other. PD of pro-TGF-β1 interacts with LTBPs and conveys the latency of its GF. Unlike TGF-β1, PD of pro-BMP9 does not convey latency of its GF, and leave GF’s receptor-interacting domain ‘open’ (a, b). The active TGF-β1 is a GF dimer. Each monomer is like a “hand” with two β-strand “fingers” protruding from an α-helix “wrist”. The dimer binds the receptor complex at an interface composed of the “wrist” of one monomer and ‘fingers’ of the other monomer (c). d, e Ligands and receptors of TGF-β and BMP signaling in bone.
Most TGF-β and BMP GF dimers are connected by a disulfide bond, although this is absent in a few BMPs (GDF3, GDF9, and BMP15).19 The disulfide-bonded dimeric structure is classically portrayed as a “hand”, in which two sets of anti-parallel β-strands form “finger extensions” that protrude from a central stabilizing “wrist” α-helix.9 They bind their receptors at a composite binding interface, which is formed by the “wrist” epitope of one monomer and the convex “knuckle” epitope of the “finger extensions” of the other monomer (Fig. 3c).9 Despite their structural similarity, TGF-β and BMP ligands possess different interfaces, also called hotspot regions or sites, to recognize diverse pairings of type I and type II receptor complexes (Fig. 3d, e). In the skeleton, TGF-βs usually bind to heterotetrameric receptors comprising of TGF-β type I receptor (TGFBR1)/Anaplastic lymphoma kinase 5 (ALK5) and TGF-β type II receptor (TGFBR2).8 Some studies also identified ALK1 as a second type I TGF-β receptor.20,21 The receptor-binding nature of BMPs is more heterogeneous than that of TGF-βs. In the skeleton exist three type II receptors for BMPs, including BMP type II receptor (BMPR2), Activin type IIA receptor (ActRIIA, ACVR2A), and Activin type IIB receptor (ActRIIB, ACVR2B). Moreover, there exist four type I receptors, including BMP type IA receptor (BMPRIA)/ALK3, BMP type IB receptor (BMPRIB)/ALK6, Activin type I receptor (ACVR1)/ALK2, and ALK1.7,22–24 Combinations of those type I and type II receptors form various heterotetrameric complexes, which possess different binding affinities for certain BMP ligands. For example, while both BMP7 and BMP14 bind to ALK6, only BMP7 binds to ALK2 and ALK3. Therefore, BMP7 and BMP14 play distinct but overlapping roles in skeletal development.25 Furthermore, BMP-2/4/9 stimulates bone formation preferably through ALK-1/3/6,22,26,27 while BMP3 antagonizes osteogenesis through binding to ActRIIB.24,28
Canonical and non-canonical signaling
Upon binding to their receptors, the TGF-β superfamily transduces signals through canonical (Smad-dependent) and non-canonical (Smad-independent) signaling pathways (Figs. 1 and 2). In the canonical signaling pathways, eight SMAD proteins have been characterized in mammals (SMAD1–8), which could be classified into three subtypes: common partner SMAD (Co-SMAD, SMAD4), receptor-regulated SMADs (R-SMADs, SMAD-1, -2, -3, -5, and -8), and inhibitory SMADs (I-SMADs, SMAD-6 and -7). Binding of TGF-βs or BMPs with their receptors results in the phosphorylation and activation of R-SMADs via interaction with the C-terminal SSXS motif.3,29 The phosphorylated R-SMADs then form a complex with the Co-SMAD SMAD4 through their C-terminal MH2 domains, and translocate to the nucleus to regulate the transcription of target genes via binding to DNA through their N-terminal MH1 domains.3,29 In most cases, BMPs elicit the activation of R-SMADs SMAD-1, -5, and -8. In contrast, TGF-βs elicit the activation of R-SMADs SMAD-2 and -3 (Fig. 3d, e). Alternatively, TGF-βs also bind to ALK1 to transduce signals to SMAD-1, -5 and -8.20,21 Unlike R-SMADs and Co-SMAD, I-SMADs lack the DNA-binding MH1 domain and coordinate the negative regulation of canonical signaling, which is discussed in more detail in this review.
Alternatively, TGF-β or BMP receptors can transmit signals independent of SMAD proteins (Figs. 1 and 2).3 Upon ligand binding, TGF-β or BMP receptors associate with TNF receptor-associated factors (TRAFs) to promote their polyubiquitylation, which activates TGF-β activated kinase 1 (TAK1). TAK1 subsequently phosphorylates mitogen-activated protein kinases (MAPKs) or phosphoinositide 3-kinase (PI3K), which in turn phosphorylates and activates target transcription factors (i.e., nuclear factor kappa-B (NF-κB), runt-related transcription factor 2 (RUNX2)). TAK1 might also activate small G proteins, including Rac1 and Cdc42. Canonical and non-canonical signaling activation reciprocally regulates each other. On the one hand, the activation of non-canonical signaling could potentiate canonical signaling. For example, PI3K was shown to stabilize SMAD1 protein through GSK3 activation in vivo and in vitro, enhancing osteogenesis;30 furthermore, knockdown of extracellular signal-regulated kinase 1 (ERK1) was shown to inhibit TGF-β1-induced Smad3 phosphorylation in rat chondrocytes.31 On the other hand, non-canonical signaling could also antagonize SMAD activity. For example, MAPK might phosphorylate Smad1 to recruit Smurf1 for its cytoplasm retention and degradation.32 NF-κB could interact with Smad4 and antagonize its transcriptional activity to suppress BMP2-induced bone formation.33 ERK signaling is reported to increase the expression of Smurf1 to inhibit BMP’s function in osteoblasts.34 An uneven activation of TAK1 over SMADs by c-Abl directs the expression of p16(INK4a) to control mesenchymal stem cell (MSC) maintenance and inhibit osteoblast differentiation.35
Target transcriptome
The Smad complex recognizes consensus DNA sequences, namely Smad-binding element (SBE) or BMP-responsive element (BRE), to regulate gene expression. The SBE element, also known as the GTCT motif or its complementary extended CAGAC sequence, has been previously identified.36 Smad1 and Smad5 were shown to also recognize GC-rich motifs (GGCGC), termed BRE, in certain BMP-responsive genes.36 As such, some target genes have been identified for TGF-β and BMP signaling, including Id-1, Gremlin, noggin, follistatin (FS), Smad6, and BambI.37 However, the target transcriptome of TGF-β and BMP signaling also varies greatly among cell types and pathological conditions, due to variable cofactors and chromatin structure and accessibility. Therefore, the development of chromatin immunoprecipitation followed by sequencing (ChIP-seq), formaldehyde-assisted isolation of regulatory elements followed by sequencing (FAIRE-seq) or CUT&Tag-seq, as well as ATAC-seq and RNA-seq techniques enables the genome-wide analysis of SMAD-binding and SMAD-responsive sites in a cell type-specific manner. Omata et al. 38 performed ChIP-seq combined with FAIRE-seq in osteoclasts to analyze the TGF-β-responsive and receptor activator of nuclear factor-κB ligand (RANKL)-regulated genes. Their results indicated the cooperation of Smad2/3 with c-Fos during osteoclastogenesis.38 Yu et al. 39 used RNA-seq, ATAC-seq combined with H3K27Ac CUT&Tag-seq, to analyze deregulated transcription factor networks in Bmp2-deficient osteoblasts, revealing that over 80% of deregulated elements are directly targeted by transcription factors such as RUNX2, DLX5 (Distal-Less Homeobox 5), MEF2C (MADS box transcription enhancer factor 2), OASIS (old astrocyte specifically induced substance), and KLF4 (Krüppel-like factor 4). These transcriptional factors may function together with or downstream of SMAD proteins to regulate the biological outcomes induced by BMP2. With RNA-seq and ChIP-seq techniques, Yan et al. 40 identified that Smad4 directly binds to the regulatory region of the Runx2 promoter, which contributes to osteoblast differentiation and chondrocyte hypertrophy. The diverse target transcriptome could be explained by the notion that SMAD proteins recruit different transcription co-regulators on the chromosomes, which is discussed in more detail in this review.
TGF-β and BMP signaling in skeleton development
The mammalian skeleton is formed through intramembranous ossification (i.e., calvarial bones) or endochondral ossification (i.e., appendicular bones and axis bones).41,42 During intramembranous ossification, condensed mesenchymal cells are directly differentiated into osteoblasts and osteocytes.41,42 During endochondral ossification, condensed mesenchyme undergoes chondrogenesis to form cartilage primordium, which develops into a cartilage anlage of embryonic bone shape, surrounded by the perichondrium.41,42 The cartilage anlage further develops into growth plates at the two epiphyseal ends, which are layered with chondrocytes in continuous differentiation stages (resting, proliferative, pre-hypertrophic, and hypertrophic).41,42 The hypertrophic chondrocytes undergo terminal differentiation and apoptosis and are gradually replaced by bone structures in the metaphyseal part. Multiple signaling pathways (i.e., Hedgehog, fibroblast growth factor (FGF), parathyroid hormone-related protein (PTHrP), BMP, and TGF-β) cooperate to determine the morphology of the skeleton in the cartilage primordium stage and modulate bone growth and maturation in the growth plates.41,42 Our work showed that transcriptional factor complexes Runx1/Cbfβ and Runx2/Cbfβ control chondrocyte proliferation and hypertrophy during growth plate development.43–47 Here, we will discuss the specific roles of BMP and TGF-β signaling in skeleton development, especially endochondral ossification.
BMP signaling in skeleton development
BMP signaling plays critical roles in multiple stages of skeletogenesis, including MSC condensation, cartilage primordium formation, skeleton patterning, and growth plate development (Fig. 4). As mentioned earlier, BMP signaling consists of a variety of ligands and receptors with heterogeneous binding affinities and patterns, which produce variable physiological outcomes. BMP ligands have different expression patterns during skeleton development, delineating their diverse physiological functions. For example, Bmp14 and its receptor Alk6 have a restricted expression pattern in appendicular bones.25 Consistently, mice carrying the Bmp14 mutation, Alk6 null mutation, or both display malformation of appendicular bones but not axis bones.25,48,49 Bmp-2, -4, -7, and -14 (GDF5) are expressed in the early stage of skeletal development, indicating their roles in the initiation of skeletogenesis.25,50–52 Consistently, embryonic deletions of Bmp-2, -4, -7, or -14 genes result in malformed skeletons.25,50–52 Among them, MSC-specific Bmp-2 and -4 double knockout (DKO) mice displayed the most severe malformation, highlighting their critical functions during embryonic skeletal development.51 At the molecular level, loss of BMP impairs prechondrogenic differentiation at mesenchyme condensations due to expressional loss of key chondrogenic transcription factors, including Sox-5, -6, and -9.48
Fig. 4. TGF-β and BMP signaling in endochondral bone development.

Endochondral bone development begins with the condensation of mesenchyme, which develops into limb bud, cartilage analogue, and embryonic bone with a well-organized growth plate in a step-wise fashion. In the early stage, BMPs are expressed in the anterior and posterior margins of the limb bud. IHH induces the expression of BMP antagonist Gremlin in the posterior margins. Gremlin prevents BMPs from downregulating FGF production which feeds back to maintain IHH production. The BMP-IHH-FGF regulatory loop establishes the dorsal-ventral and anterior-posterior axes of the limb bud and determines limb patterning. In the growth plate, BMP signaling promotes chondrocyte proliferation and differentiation at all stages, while TGF-β promotes the terminal differentiation of chondrocytes while inhibiting hypertrophic differentiation. BMP positively regulates IHH signaling to promote chondrocyte proliferation through the IHH-PTHrP loop, negatively regulates FGF signaling, a negative regulator of chondrocyte proliferation and hypertrophy, and promotes Runx2 activity to enhance hypertrophic and terminal differentiation. In contrast, TGF-β decreases IHH expression. BMP and TGF-β promote Sox9 expression or activity, favoring cartilage matrix production.
Moreover, BMP signaling is critical in early limb bud development (Fig. 4). BMP-2, -4, and -7 are expressed in both the anterior and posterior margins of limb bud mesenchyme.51 BMP antagonist Gremlin is also expressed in the posterior margins of the limb bud.53 Msx2-Cre-mediated Bmp4, Bmp2, and Bmp7 deletion in apical ectodermal ridge (AER) cells resulted in the disruption of dorsal-ventral polarization of mesenchyme and AER disorganization.54–57 During distal progression of limb bud development, Sonic Hedgehog (SHH) activity leads to the upregulation of the BMP antagonist Gremlin in the posterior mesenchyme (or zone of polarizing activity) to prevent BMPs from downregulating FGF production in the AER, which feeds back to maintain SHH production.58 Mutations of genes in BMP signaling, including NOGGIN, GDF5, BMP2, and BMPR1b, are associated with human diseases characterized by symphalangism or brachydactyly59–64 (Table 2). Manipulating the aforementioned genes in mice emulates these human disease phenotypes while also having additional autopod patterning defects, such as polydactyly and missing phalange elements.25,48–52,65–67
BMP signaling promotes chondrocyte proliferation and differentiation at all stages of growth plate development (Fig. 4). In the growth plate, expression is found for Bmp2, Bmp4, and Bmp5 in the perichondrium, Bmp2 in hypertrophic chondrocytes, and Bmp7 in proliferating chondrocytes.7 Severe chondrodysplasia and shortened long bones are observed in chondrocyte-specific Bmp2 conditional knockout (CKO) and Bmp-2 and -4 DKO mice but not in Bmp4 CKO mice, indicating that Bmp2 outweighs Bmp4 in regulating chondrocyte differentiation during growth plate formation.50 As for their receptors, ALK-2, -3, and -6 play redundant roles during skeleton development since all the chondrocyte-specific DKO mice display a more severe, perinatal, lethal chondrodysplasia phenotype than the single gene CKO mice, exhibited by delayed chondrocyte proliferation, matrix production, hypertrophic differentiation, and terminal differentiation.25,48,49,65 Similar phenotypes could also be observed in mice with R-Smad or Co-Smad proteins deleted specifically in chondrocytes.40,50,68,69 Conversely, augmentation of BMP signaling accelerates chondrocyte maturation and cartilage expansion, as observed in chick limbs loaded with constitutive active (CA) forms of BMP receptors and mouse models with activated BMP signaling.66,67,70 BMP regulates chondrocyte proliferation and hypertrophic differentiation through several different mechanisms. First, BMPs maintain the expression of Sox9, a master chondrogenic transcription factor.48,71 Second, BMP signaling induces the expression of Indian Hedgehog (Ihh),27,65,69 a cytokine critical for maintaining proliferating chondrocyte pool. Third, BMPs negatively regulate FGF signaling by inhibiting the expression of FGFR1.65 FGF signaling was shown to inhibit chondrocyte proliferation and hypertrophy through STAT and MAPK signaling.65 Furthermore, BMP/Smad4 promotes the expression and activity of Runx2, which positively regulates chondrocyte hypertrophy and ossification.40
TGF-β signaling in skeleton development
Like BMPs, TGF-β signaling is also indispensable for skeleton development. In humans, deregulated TGF-β signaling caused by the mutations of TGFΒR2, TGFΒ2, TGFΒ3, SMAD2, SMAD3, and FBN-1 is associated with Loeys-Dietz syndrome or Marfan syndrome, both of which are characterized by various skeletal anomalies such as long bone overgrowth72–77 (Table 2). In mice, deletion of Tgfbr2 abolished TGF-β signaling and resulted in severe defects in calvarial, appendicular, and axis bones.78 TGF-β also plays a critical role in joint morphogenesis. Tgfbr2 deficiency results in ankylosis of the interphalangeal joints and missing or incomplete intervertebral discs (IVDs).78–81 TGF-βs regulate the expression of several joint morphogenic genes, including Noggin, Wnt9a, GDF5, and MCP-5 (monocyte chemotactic protein-5).81,82
Unlike BMPs, the roles of TGF-β in chondrogenesis are differentiation stage-dependent. At an early stage of differentiation, TGF-β signaling is not required to initiate chondrogenesis but limits chondrogenesis for osteoblast lineage commitment. Neither MSC-specific nor chondrocyte-specific Tgfbr2 CKO mice experience difficulty in forming the primordium.78–80 Deletion of Alk5 in mice also led to a thinner perichondrium accompanied by ectopic cartilaginous tissues protruding into the perichondrium.83 At a later stage of differentiation, TGF-β signaling prevents chondrocyte hypertrophy while promoting terminal differentiation.78,81,84 TGFBR2 is the only type II receptor for TGF-βs, and deletion of Tgfbr2 effectively abolished TGF-β signaling and resulted in severe defects in calvarial, appendicular, and axis bones.78 However, severe skeleton defects were not observed in TGF-β1, 2, and 3 single gene KO mice,85–88 indicating that they play redundant roles. During terminal differentiation, Tgfbr2 deficiency accelerated the transition from pre-hypertrophic to hypertrophic chondrocyte while delaying ossification.78,81,84 Similar defects were observed in the chondrocyte-specific Smad-2 and -3 CKO and DKO mice, while Smad2-deficient mice displayed a more severe phenotype, indicating that Smad2 plays a more critical role than Smad3 in endochondral bone development.89 Smad2 is shown to inhibit the expression of Ihh at the transcriptional level to a greater extent than Smad3.89
TGF-β and BMP signaling in bone formation and remodeling
Throughout the life of humans, bone tissues undergo continuous remodeling, with bone resorption carried out by osteoclasts and bone formation by osteoblasts.90,91 The differentiation program from skeletal MSCs to osteoblasts is regulated by multiple signaling pathways (i.e., IGF, WNT, Hedgehog, parathyroid hormone (PTH), TGF-β, and BMP) and transcription factors (i.e., Runx2, Dlx5, Osterix, β-catenin).91 Our works showed that Runx1/Cbfβ and Runx2/Cbfβ control osteoblast differentiation and lineage commitment.44,92–94 Osteoclasts differentiate from bone marrow monocytes/macrophages, a process driven by two cytokines: M-CSF and RANKL.90 Osteoclast differentiation is also controlled by key transcription factors like c-Fos, NF-кB, and nuclear factor-activated T-cells 1 (NFATc1).90 Osteocytes are terminally differentiated osteoblasts embedded in the mineralized matrix.95 Osteocytes localized in the lacuna of bones have multiple dendritic extensions to connect with nearby osteocytes and cells on the bone surface, forming a specialized structure called the lacuna-canalicular network.95 Osteocytes directly participate in perilacuna bone remodeling and modulate osteoclast and osteoblast functions through paracrine pathways.95 An imbalance between osteoclast and osteoblast activity and dysregulated osteocyte function will disturb bone homeostasis, resulting in bone metabolic diseases like osteopenia and osteosclerosis. Here, we summarize the role of TGF-β and BMP signaling in regulating osteoclast, osteoblast, and osteocyte formation and function. Multiple genetic mutations in TGF-β and BMP signaling are associated with various human sclerosing symptoms (Table 2). Genome-wide studies and single-gene analysis also identified genetic polymorphisms of several genes in both pathways associated with bone mass, including TGF-β1, BMP2, BMP4, SMAD9, SMAD2, Noggin, SOSTDC1, GREM2, NAB1, and SPON1.96–106 The involvement of TGF-β and BMP signaling in postnatal bone homeostasis is also substantiated by extensive in vivo, in vitro and ex vivo studies.
BMP signaling in bone formation and osteoblast differentiation
BMPs were first discovered and mainly referred to as osteogenic proteins (Fig. 1). BMP2 is considered the gold standard for bone regeneration and has been clinically applied to promote fracture healing and spinal fusion.107,108 Additionally, BMP-2, -4, -6, -7, and -9 are also osteogenic in vitro and in vivo.107–110 However, endogenous BMP2 might have a unique and indispensable function in fracture healing since BMP2 CKO mice also have frequent fractures that fail to heal, which is not observed in BMP4 CKO mice.111,112 BMP9 has been recently found to be resistant to endogenous antagonists such as Noggin and BMP3b, providing a candidate alternative to BMP2 for treating fracture healing.109,110 In addition, mouse models were generated with BMP canonical and non-canonical signaling suppressed in osteoblasts, including Alk2 CKO mice,113 BmprII dominant-negative transgenic mice,114 ActRIIB-null mice,115 Smad1 CKO mice,50 Tak1 CKO mice,116 p38 CKO mice,117 and Smad4-deficient mice.118,119 All aforementioned mice exhibited osteopenia phenotypes, further substantiating the osteogenic role of BMP signaling in promoting osteoblast differentiation and matrix production.
Moreover, hyperactivated BMP signaling leads to heterotopic ossifications (HO). One of the major side effects of BMP implementation in bone healing is inducing HO in muscle tissues.120 Musculoskeletal trauma-induced HO in muscles and tendons at a high ratio is associated with hyperactivated BMP signaling.121 Antagonizing BMP signaling activation is proposed to be a potential treatment preventing trauma-induced HO.121 Fibrodysplasia ossificans progressiva (FOP; MIM #135100), a genetic disorder manifesting progressive HO, is caused by gain-of-function mutations (R260H and G356D) of ALK2/ACVR1, the type I receptor of BMPs (Table 2).122,123 In animal and cell models of FOP, the ACVR1 mutants transduce hyperactivated Smad1/5/8-dependent signals downstream of either BMP or ALK2, explaining the pathomechanism of FOP (Table 1).122,124–130 In contrast, under normal circumstances, BMP-ACVR1 activates Smad1/5/8 and Activin A-ACVR1 activates Smad2/3 signaling.131 Retinoic acid receptor γ (RARγ) agonist Palovarotene, which suppresses BMP signaling,132 has been recently approved by the U.S. Food and Drug Administration for FOP treatment based on its Phase III trial.133 Besides Palovarotene, selective ALK2 inhibitors (BLU-782, Phase I; INCB00928, Phase II; Saracatinib) and the Activin A neutralizing antibody also showed potential to alleviate FOP symptons.128,134,135
At the molecular level, BMPs promote osteogenesis through several different mechanisms. Firstly, BMP signaling positively regulates the activity of Runx2, an osteoblast master transcription factor. Smad1 physically interacts with Runx2 to bind to OSE2 sites on its target gene.136 Runx2 is also phosphorylated by BMP non-canonical signaling (TAK1-MEK-p38 or ERK), promoting its association with the coactivator CREB-binding protein (CBP).116 BMP also stabilizes Runx2 through promoting its acetylation by p300.137,138 Secondly, frequent crosstalk between BMP and WNT signalings promotes the osteogenic program. For example, transcription factor 4 (TCF4)/β-catenin complex physically interacts with the SMAD complex on the corresponding DNA-binding sites139; ablation of Smad4 causes cleavage of β-catenin and depletion of the WNT receptor, a low-density lipoprotein receptor (Lrp5)118; expression of LGR4, an orphan receptor and WNT regulator, is also induced by BMP2.140 Thirdly, BMP signaling induces the expression of several osteogenesis-related transcription factors, including Msx2, Runx2, Dlx5, KLF10, Forkhead box C1 (Foxc1), Foxc2, and Dlx3.139,141–143 Fourthly, BMP2 also induces the expression of PLCβ1 (phospholipase C β1) and IHH, both of which promote osteoblast differentiation.144,145 Additionally, SMAD1 dislodges Hoxc-8 from its DNA-binding sites to induce osteoblastic gene expression.146 Moreover, BMP signaling positively regulates mTORC1 activity to promote osteoblast activity.147 Our work showed that Runx1 regulates osteoblast differentiation through promoting BMP signaling, by controlling Bmp7 and Alk3 expression at transcriptional level.94
However, BMP signaling also has adverse effects on bone formation. BMP limits the proliferation of preosteoblasts and antagonizes osteogenesis in osteoblast progenitors.147 BMP signaling might also negatively regulate mineralization and collagen maturation.148,149 At the molecular level, Alk3 induces the expression of WNT antagonists, DKK1 (Dickkopf-related protein 1), and sclerostin (SOST).150 BMP2 promotes an interaction between Smad1 and Dvl-1 (Drosophila dishevelled gene) that restricts β-catenin activation.151 Smad4 also competitively interacts with Tcf and Lef (lymphoid enhancer binding factor) proteins to inhibit the transcriptional activity of β-catenin.119 Collectively, BMP antagonizes bone formation through perhaps inhibiting WNT/β-catenin signaling.
TGF-β signaling in bone formation and osteoblast differentiation
As discussed above, BMP signaling limits osteoprogenitor proliferation while promoting osteogenesis afterward. In contrast, TGF-β signaling promotes osteoprogenitor proliferation and osteogenesis at the early stage of differentiation while inhibiting bone formation at the later stage (Fig. 2). Many mouse models with impaired TGF-β signaling have been generated, including Tgfb1-null mice, MSC-specific and osteoprogenitor-specific Tgfbr2 CKO mice, Alk5-null mice, and Smad3-null mice.83,152–157 Those TGF-β signaling-dificient mice displayed significant bone loss with reduced osteoblast number, suggesting that TGF-β is anabolic for bone formation.
Conversely, hyperactivated TGF-β signaling increased bone mass. In humans, gain-of-function mutations in TGFB1 are associated with Camurati-Engelmann disease (CED; MIM #131300), characterized by osteosclerotic lesions within the long bones and the skull.158 Mice carrying the same tgfb1 mutation mirror the phenotype seen in humans.152 Somatic SMAD3-activating mutations in humans are associated with endosteal pattern melorheostosis (Leri’s disease; MIM #155950), characterized by asymmetric exuberant bone formation.159,160 Interestingly, osteogenesis of SMAD3-activating mutant cells is stimulated by TGF-β while inhibited by BMP2,159,160 indicating that SMAD3 links the reciprocal regulation between BMP and TGF-β. Furthermore, activating mutations of mitogen-activated protein kinase kinase 1 (MAP2K1) in non-canonical TGF-β signaling also caused sporadic melorheostosis.161 At the molecular level, TGF-β positively regulates the expression of Runx2, Osterix, Dlx5, and Msx2 to initiate the osteogenic program.153 TGF-β1 induces the expression of integrin Vα5 to promote osteoblast adhesion.162 TGF-β1-SMAD signaling also regulates the expression of connective tissue growth factor (CTGF), a matrix protein that positively regulates osteoblast differentiation and function.163
During the late stage of osteoblast differentiation, TGF-β signaling inhibits bone formation. TGF-β, SMAD3, and SMAD2 are shown to inhibit osteogenesis in vitro.164–167 Smad3 interacts with Runx2 and recruits histone deacetylase 4 (HDAC4) and 5 (HDAC5).166 HDAC4 and HDAC5 deacetylate Runx2 to facilitate its degradation.137 TGF-β regulates the expression of various signaling proteins involved in osteoblast formation. TGF-β induces the expression of vimentin, which negatively regulates the activity of ATF4, an osteogenesis-related transcription factor.168 TGF-β induces the expression of HDAC6, which distorts primary cilia to impair mechanical-stimulated osteogenesis.169 TGF-β induces the expression of Smurf1, which antagonizes osteogenic signaling such as BMP.34,170 TGF-β also inhibits the expression of IGF-1, a bone anabolic cytokine.171 In vivo, CKO of Tgfbr2 in mature osteoblasts results in high bone mass in mice.172 Qiu et al.172 revealed that Tgfbr2 forms a complex with PTHrP for endocytosis. With the deletion of Tgfbr2, PTH signaling is hyperactivated to produce excessive bone mass.172 PTH signaling also reciprocally regulates TGF-β signaling by inducing LTBP-1, TGF-β1, and Smad3 expression.173,174
BMP and TGF-β signaling in osteoclast differentiation
BMP signaling promotes osteoclast differentiation both directly and indirectly. BMP promotes osteoblast-induced osteoclast formation through upregulating the RANKL/osteoprotegerin (OPG) ratio (Fig. 1). Disruption of Alk3, Alk2, or Smad4 in osteoblasts or osteocytes results in an unexpected increase of bone mass in mice due to the decreased RANKL/OPG ratio causing less osteoclast formation.150,175–178 Alk2 and Alk3 signaling upregulate WNT antagonists (i.e., Sost) to inhibit WNT activation, and the latter regulates osteoclast formation by inhibiting the RANKL/OPG ratio.150,175,176 Therefore, BMP might be essential to promote osteoblast-osteoclast coupling in bones requiring extremely active remodeling, such as during regeneration.145
BMP signaling also stimulates osteoclast formation directly (Fig. 1). BMPs (i.e., BMP2, BMP7) stimulate and BMP inhibitor dorsomorphin blocks osteoclast formation and bone resorption.179–182 Consistently, deletion of ALK2, ALK3, SMAD1/5, or SMAD4 also impairs osteoclastogenesis.180,182,183 At the molecular level, BMP signaling promotes the expression or activity of osteoclastic transcription factors. BMPRII couples with RANK to activate p-Smad1/5/8 and NF-κB signaling simultaneously.181 Smad1/5/8 promotes the nuclear translocation of NFATc1.182 Moreover, Smad1/5 induces the expression of c-Fos and Nfatc1.180
Unlike BMPs, TGF-β regulates osteoclast formation in a dose- and stage-dependent manner (Fig. 2). Low-dose TGF-β induces, whereas high-dose TGF-β inhibits, migration of osteoclast precursors to the bone resorption pits.184 TGF-β at the monocyte stage promotes, while at the later differentiation stage antagonizes, osteoclast formation. TGF-β regulates multiple signalings in regulating osteoclast differentiation. TGF-β-induced p38 activation and Smad2/3 cooperation with c-Fos as a co-transcription factor favor osteoclast differentiation.38,185 TGF-β inhibits RANK expression, blocks Prdm1 activity to induce Irf8 and Bcl6 expression, and upregulates ROS production to block MAPK signaling to antagonize osteoclast differentiation.185–187 TGF-β also upregulates Bim expression to induce osteoclast apoptosis.188,189
The role of TGF-β signaling in osteocytes
While the well-known osteocyte marker SOST is an antagonist of WNT and BMP signaling and a critical regulator of skeletal homeostasis, knowledge about the role of BMP signaling in osteocytes is very limited. In contrast, recent studies brought up the physiological role of TGF-β in regulating osteocyte formation and function. TGF-β-Smad3 signaling has been previously shown to inhibit the transition of osteoblasts into osteocytes.156 In mature osteocytes, TGF-β signaling was recently demonstrated to play a critical role in maintaining its perilacunar-canalicular network and function (Fig. 2). In mice, intrinsic osteocytic TGF-β signaling promotes the perilacunar-canalicular remodeling of the osteocyte to control bone quality.190,191 Specific loss of TGF-β signaling in the osteocyte reduces osteocyte connectivity, impairing fluid dynamics and osteocyte exposure to mechanical stimulation.192 Conversely, administration of TGF-β1 increases osteocyte connectivity in bone tissue and an MLO-Y4 cell line by inducing connexin43 and pannexin1 expression.193 TGF-β3 was also shown to maintain the osteocyte differentiation of MLO-Y4 cells in an osteoblast-osteocyte co-culture 3D system as determined by stable E11 and osteocalcin mRNA expression.194 Furthermore, intrinsic osteocytic TGF-β signaling is also essential for the mechanosensing property of articular cartilage. Mice with impaired TGF-β signaling in osteocytes have thicker subchondral bone plates, high SOST levels, and more severe cartilage degeneration in an injury-induced osteoarthritis (OA) model.195
TGF-β and BMP signaling in articular cartilage homeostasis
Joints are organized structures allowing constrained motion. They are formed by adjacent bones with articular cartilage covering the bone surface and contain the synovial lining of the joint cavity. Articular chondrocytes govern articular cartilage homeostasis via their ability to modulate ECM production and degradation, whose imbalance causes degenerative joint diseases such as OA. In the diseased joint, chondrocytes undergo abnormal hypertrophic and terminal differentiation, followed by tearing of the cartilage matrix, focal calcification, and ectopic bone (osteophyte) formation.
On the one hand, TGF-β signaling plays a critical role in maintaining articular homeostasis (Fig. 5). TGF-β1-coupled biomaterials have been proposed as a therapeutic method for cartilage repair.196,197 TGF-β signalings protect articular cartilage by inhibiting chondrocyte hypertrophy and apoptosis,198,199 promoting cartilage matrix synthesis,200–202 and antagonizing inflammatory cytokine production.203,204 In humans with grade 3 OA, genetically modified allogeneic human chondrocytes that express TGF-β1 show significant improvement in knee joint function and reduce pain severity.205 Animal models with inhibited canonical and non-canonical TGF-β signaling are prone to developing OA, including dominant-negative Tgbr2 transgenic mice,206 mice with postnatal cartilage-specific deletion of Alk5, Tgfbr2, or Tak1,71,207–209 Smad3-null mice,199 and dominant-negative p38 transgenic mice.210 Pharmacological inhibition of TGF-β signaling also leads to an OA-like phenotype in rodents.71,211,212 At the molecular level, the reduction of TGF-β canonical signaling induces the death of articular chondrocytes.198 TGF-β non-canonical signaling induces the phosphorylation and activation of ATF2 and FoxO, which inhibits OA by upregulating the expression of Sox9 and autophagy proteins.71,213 Inhibition of TGF-β activity enhances BMP and S1P (sphingosine 1-phosphate) signaling, which accelerates chondrocyte maturation and matrix degradation.199,214 Abolished TGF-β activity also alters IGF and FGF signaling and upregulates the expression of biosynthesis-related genes and electron transport chain-related genes, contributing to chondrocyte hypertrophy.215 Our work showed that Runx1 protects cartilage homeostasis through promoting TGF-β signaling.216
Fig. 5. TGF-β and BMP signaling in cartilage homeostasis.
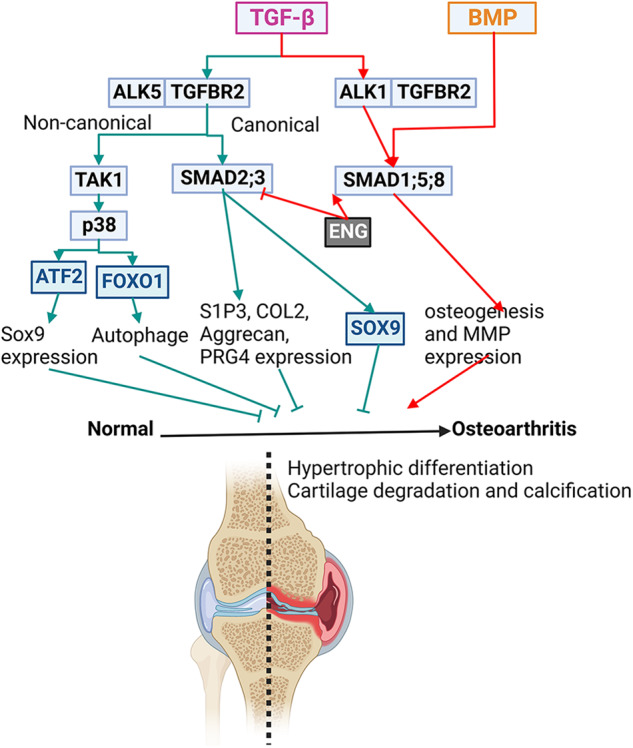
Postnatal cartilage homeostasis is maintained by matrix production and degradation balance, and the imbalance results in cartilage tearing and joint diseases like osteoarthritis. TGF-β plays dual roles in cartilage homeostasis. To protect cartilage health, TGF-β, through binding ALK5, activates SMAD-2 and -3 and TAK1-p38 signaling, which enhances the Sox9 expression and activity and promotes autophagy activity and matrix protein production. Conversely, TGF-β, through binding ALK1, activates SMAD-1, -5, and -8, like BMPs, which promotes MMP production and osteogenesis to aggravate cartilage degeneration.
On the other hand, TGF-βs also promote the progression of OA. TGF-β expression is increased in osteoarthritic cartilage and joints with ankylosing spondylitis.217–220 Furthermore, mechanical loading during OA could induce TGF-β1 secretion.221 Excessive TGF-β signaling is detrimental to joint degeneration. Notably, CED patients or mouse models carrying gain-of-function mutations of TGFB1 are prone to developing OA.221,222 Suppression of TGF-β signaling by deleting Tgfbr2 in nestin-positive MSCs ameliorates the development of OA after anterior cruciate ligament transection (ACLT) compared to a control.221 The contradictory roles of TGF-β in OA have been linked to the opposite regulatory functions of its type I receptors, ALK1 and ALK5, to transduce signals to SMAD1/5/8 and SMAD2/3, respectively, in chondrocytes.20,21 ALK1 signaling is destructive by inducing the expression of matrix-degrading enzyme MMP-13. In contrast, ALK5 signaling is protective by inducing the expression of matrix proteins aggrecan, type II collagen, and PRG4 (proteoglycan 4).207,223 In addition, ALK1 opposes TGF-β-ALK5-induced phosphorylation of SMAD3 and inhibits the expression of chondrogenic genes induced by TGF-β, including fibronectin and type II collagen.20 Furthermore, ALK1/ALK5 ratio is increased in aging and osteoarthritic cartilage in mice.223 Disturbed balance between ALK1 and ALK5 signalings might contribute to articular cartilage degeneration.223 In addition, TGF-β signaling also promotes the clustering of nestin-positive MSCs, leading to the formation of marrow osteoid islets accompanied by high levels of angiogenesis to deteriorate OA condition.221
Abnormal BMP activation is associated with OA since BMP accelerates chondrocyte terminal differentiation.199 Recently, Occhetta et al. 224 found that selective inhibition of BMP signaling helps control differentiation of MSCs into chondrocytes at precisely the stage as those in articular cartilage. As cultured chondrocytes usually undergo terminal differentiation, this finding indicates that targeting BMP signaling provides a strategy for cartilage regeneration. BMP activity also needs to be inhibited spatially in vivo during development or in postnatal cartilage to prevent further chondrocyte differentiation as well as the overexpression of its antagonists, such as Gremlin.224
Regulation of TGF-β and BMP signaling in bone
TGF-β and BMP signaling is regulated at multiple levels from extracellular space to nucleus (Figs. 1 and 2). Extracellularly, matrix proteins such as FBNs and collagens control the latency of TGF-βs and BMPs; metalloproteinases contribute to the release and activation of TGF-β and BMP peptides; antagonists interrupt the binding of TGF-β and BMP ligands to their receptors. At the cell membranes, co-receptors such as β-glycan and endoglin (ENG) facilitate the ligand–receptor interactions. In the cytoplasm, I-SMAD, ubiquitin ligases, and deubiquitinases regulate the activation and stability of SMAD complexes. Nuclear envelope proteins control the transport of SMAD complexes from cytoplasm to nucleus. Various transcription co-factors and epigenetic factors cooperate with SMAD complexes in the nucleus to regulate their transcription activity. Here, we will summarize how those regulators coordinate BMP and TGF-β signaling in bone and cartilage.
Latency and release control of the ligands
LTBPs interact with LAPs and active TGF-β peptides to form the LLC. LTBP is indispensable for the latency, correct folding, and secretion of TGF-β. It is also essential for storing TGF-β in the ECM through interactions with platform proteins. Currently, four different LTBPs (LTBP-1–4) have been identified.225 Among them, LTBP-3 is the most studied. Ltbp-3-null mice have impaired TGF-β signaling, exhibiting multiple skeletal malformations and an OA-like phenotype.226,227 Impaired TGF-β signaling in LTBP-3 null cells also reduced proliferation and osteogenic potential.228
The FBN microfibril network controls the latency of TGF-βs and BMPs by serving as their reservoir in the bone and cartilage matrix. The major component of the microfibril network, Fbn, binds the LLCs or CPLXs through its unique N-terminal region. Fbn-1 and -2 are both found to be expressed in the cancellous bone.229 In humans, mutations in FBN-1 and FBN-2 cause pleiotropic manifestations in Marfan syndrome (MIM #154700) and congenital contractural arachnodactyly (MIM #121050), respectively77,230 (Table 2). Fbn-1-null mice had systemic sclerosis due to abnormal activation of both TGF-β and BMP signalings.231 However, Fbn-2 deficiency in mice induced a low bone mass phenotype due to improper activation of TGF-β inhibiting osterix expression and increasing osteoblast-induced osteoclast formation.231,232 Microfibril-associated glycoprotein-1 (MAGP1) is another constitutive component in microfibril network.233,234 Magp1-null mice, resembling Fbn-2-null mice, developed progressive osteopenia due to abnormal activation of TGF-β.233,234
Type I collagens (COL1s), COL1A1 and COL1A2, also serve as reservoirs for TGF-βs in the bone matrix. Autosomal dominant mutations of COL1 in humans cause osteogenesis imperfecta (OI; MIM #259420), a bone dysplasia characterized by bone deformities, low bone mass, poor bone quality, frequent fractures, and high non-union rate (Table 2).235 Cartilage-associated protein (CRTAP) catalyzes the maturation of COL1 by 3-hydroxylation, and its mutations also cause OI. Both Col1a2G610c/+ and Crtap–/– mouse models recapitulated OI phenotypes due to excessive TGF-β signaling.236,237 Importantly, anti-TGF-β antibody 1D11 treatment both corrects the bone phenotype and improves fracture healing in the OI mouse model, highlighting the potential of targeting TGF-β signaling in treatment for OI.236,237
Heparin sulfate (HS) is abundant in the cartilage matrix and binds to latent TGF-βs and BMPs. EXT1 and EXT2 are Golgi-resident glycosyltransferases participating in the biosynthesis of HS.238 Mutations of EXT1 and EXT2 in humans cause hereditary multiple exostoses (MIM #133700, #133701), a human autosomal skeletal disorder characterized by the formation of cartilage-capped bony growths (osteochondroma) at the ends of the bones, due to excessive BMP signaling.238 Mouse models with CKO of Ext1 in cartilage tissue develop osteochondroma and enhanced chondrocyte hypertrophy due to increased BMP-SMAD activity.239–241
CTGF is a cartilage matrix protein bound to latent TGF-βs.242 The Ctgf-deficient mice developed more severe OA than control mice due to increased TGF-β-SMAD activity.242
A disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS) and MMPs help release active TGF-βs and BMPs from ECM via a proteolytic process. As reported, ADAMTSL2, ADAMTS17, and ADAMTS10 regulate skeletal development by activating TGF-βs or BMPs. Mutations of ADAMTSL2 in humans are associated with recessive geleophysic dysplasia (MIM #231050), characterized by short stature, short extremities, and skeletal abnormalities243 (Table 2). Delhon et al. 244 generated whole-body and chondrocyte-specific Adamtsl2-deficient mice, both of which displayed skeletal abnormalities reminiscent of the human phenotype due to impaired TGF-β signaling. Mutations of ADAMTS10 and ADAMTS17 in humans cause Weill-Marchesani syndrome (WMS; MIM# 277600, 608328) and WMS-like syndromes, characterized by short stature and brachydactyly245 (Table 2). Adamts17–/– mice recapitulated WMS phenotype with shortened growth plate due to impaired BMP activation.246 TGF-βs or BMPs are also activated by proteolytic processing of the PD by MMPs, including MMP-2, -9, and -13.11–13
Extracellular antagonists
Noggin is a twelve-membered cystine knot protein and a critical antagonist of BMP ligands in bone. The crystal structure of the BMP–Noggin binding complex has been previously determined, showing that Noggin acts by sequestering the ligand in an inactive state.247 Noggin has a similar expression pattern to BMPs in bone during prenatal and postnatal development.248,249 In animal and cell models, Noggin blocks osteoblast formation by inhibiting BMP activation. Administration of Noggin suppresses osteogenesis,250,251 and neutralizing Noggin promotes osteoblast differentiation.252 Mice with conditional overexpression of Noggin showed dramatic decreases in bone mineral density and bone formation rates.250,253 However, deletion of Noggin in mature osteoblasts resulted in more osteoclast formation and osteopenia.254 Whether the detrimental impact on bone is attributed to an excessive presence of BMP or whether Noggin plays a BMP-independent role in skeletal homeostasis remains uncertain. In humans, mutations of NOGGIN are associated with various ankylosis deformities, including proximal symphalangism (SYM1: MIM #185800), multiple synostosis syndrome (SYNS: MIM#186500), tarsal–carpal coalition syndrome (TCC; MIM#186570), and stapes ankylosis with broad thumb and toes (SABTT; MIM#184460) (Table 2).59 Noggin mutant cells from ankylosis patients outperformed healthy cohorts in osteogenic differentiation capacity due to enhanced BMP activity.255 Noggin prevents cranial suture closure by inhibiting BMP signaling during cranial bone development. Therefore, Noggin downregulation also contributes to syndromic craniosynostoses.256
Gremlin-1 and Gremlin-2 (Grem1 and Grem2) are DAN family proteins and extracellular antagonists of BMPs. The structure of the Grem2–GDF5 complex reveals that two Grem dimers bind perpendicularly to each ligand monomer as a stable aggregate-like structure, which is not observed in Noggin and FS.257 Suppression of Grem promotes osteogenesis in vivo and in vitro due to sensitization of BMP signaling.258 Consistently, the osteoblast-specific Grem1 CKO mice are osteosclerotic, and osteoblast-specific Grem1-overexpressing mice are osteopenic.258,259 Grem1 expression also defines a population of skeletal stem cells in the bone marrow required for both bone remodeling and fracture repair, as reported by Worthley et al. 260 Grem1+ stem cells can self-renew and differentiate into osteoblasts, chondrocytes, and reticular marrow stromal cells while lacking the capacity to develop into adipocytes.260
FS binds and neutralizes several different members in the TGF-β superfamily, including BMPs, Activin A, GDF11, and myostatin/GDF8. Among them, BMP promotes osteogenesis, myostatin and Activin A are negative regulators of bone mass, and the role of GDF11 in bone homeostasis is controversial.6,261,262 So far, FS was mostly reported to play anti-osteogenic roles.262,263 FS restricts BMP2 action in osteoblastogenesis in vitro, and mice overexpressing FS exhibited spontaneous bone fractures.262,263
Chordin is another well-established BMP antagonist and has a role in early embryonic neural development. Very few studies characterized the role of chordin in bone, showing that the expression of chordin is inversely related to osteoblast and chondrocyte differentiation.264,265
Co-receptors
β-glycan, also regarded as the TGF-β type III receptor, acts as a membrane-anchored proteoglycan to enhance TGF-β association with the TGFBR2–TGFBR1 complex, but its soluble form may also associate with TGF-βs, activins, or BMPs to inhibit signal transduction. β-glycan is expressed in osteoblasts and promotes osteogenesis in vivo and in vitro.266,267 β-glycan-knockout embryos displayed reduced vascular and osteoblast differentiation.267
Neuropilins (Nrps) interact with TGFBR1 to promote downstream signaling. Nrp2 is expressed in both osteoblasts and osteoclasts, and Nrp2-knockout mice had increased osteoclast number, decreased osteoblast number, and low bone mass.268 While Nrps also bind and transduce signals downstream of semaphorins, how much its role in bone homeostasis is attributed to TGF-β signaling is still unclear.
ENG may bind to BMP ligands or BMPR2 receptors to facilitate signal transduction while associating with TGF-β1 or TGF-β3 for signaling through ALK3. ENG enhances BMP2-induced osteogenesis of periodontal ligament (PDL) cells in osteoblasts.269 ENG also acts as a co-receptor for BMP9 and BMP10 to induce osteogenesis in conjunction with ALK1.15,270 ENG is also expressed in human chondrocytes, and its expression increases in the chondrocytes of OA patients.271,272 Yet its function in chondrocytes remains controversial.271–273 ENG enhances Smad1/5 signaling and inhibits Smad2/3 activation to promote cartilage matrix protein production.271 However, knockdown of ENG also impaired cartilaginous tissue formation.274
Neogenin binds to BMP receptors.275 Neogenin-null mice have impaired limb development and endochondral ossification due to decreased BMP-SMAD signaling and Runx2 expression.276
Regulation machinery in the cytoplasm
I-SMADs Smad6 and Smad7
In the cytoplasm, the signaling is mainly negatively regulated by I-Smads (Smad6 and Smad7). I-SMADs inhibit the receptor-mediated activation of R-Smads through several mechanisms including interfering with type I receptor–R-Smad interaction, recruiting ubiquitin ligases to induce type I receptor or R-SMAD protein degradation, and interfering with the formation of R-SMAD–Co-SMAD complex.277 Therefore, the inhibitory functions of I-SMADs largely depend on their direct interactions with the type I receptors or R-SMADs. I-SMADs bind R-SMADs or receptors through their C-terminal MH2 domains, which show high similarity between SMAD6 and SMAD7. However, their N-terminal Leu-rich motifs (LRMs) have a low similarity rate of 36.7%, laying down the structural basis for their functional differences. SMAD6 prefers to inhibit BMP signaling, whereas SMAD7 inhibits TGF-β and BMP signaling.277 Like Noggin, SMAD6 mutations in humans also cause craniosynostosis due to the augmentation of BMP signaling.278 Smad6 transgene blocked BMP activation and led to osteopenia and dwarfism in mice.279 Smad6-null mice exhibited axial and appendicular skeletal development defects, with an expanded hypertrophic zone attributed to increased BMP responsiveness.280 Smad6 also recruits Smurf1 to ubiquitinate and degrade Runx2 to inhibit osteoblast differentiation.281 In contrast, SMAD7 might be anabolic for bone. Partial loss of Smad7 decreased bone formation and increased bone resorption.282 Smad7 overexpression impacts both early and late stages of chondrocyte differentiation due to downregulation of both BMP and TGF-β signalings.283
E3 ubiquitin ligases
I-SMAD recruits ubiquitin ligases to degrade target proteins, mainly the neural precursor cell expressed developmentally downregulated 4 (NEDD4) subfamily of HECT (homologous to the E6-accessory protein) E3 ubiquitin ligases, such as Smurf1, Smurf2, and Nedd4.
Smurf1, together with Smad6, catalyzes the poly-ubiquitination and degradation of multiple targets with an osteogenic function, such as SMAD-1, -5 and -8, MEKK2, and Runx2.281,284–287 Therefore, Smurf1 has an anti-osteogenic function. Double overexpression of Smad6 and Smurf1 delayed ossification more severely than Smad6 overexpression alone.279 The Smurf1 transgenic mice also had significantly reduced bone formation, while Smurf1-null mice had increased bone mass.279,285,286 A chalcone derivative inhibiting Smurf1 activity promotes local spinal fusion and systematic bone formation in mice, indicating that targeting Smurf1 is a potential treatment for bone healing.288
Pleckstrin homology domain-containing family O member 1 (PLEKHO1) associates with Smurf1 to promote the ubiquitination of Smad1/5 to inhibit BMP signaling and bone formation.289 Furthermore, the expression of PLEKHO1 increased during aging, indicating its involvement in aging-related bone loss.289
Smurf2 is a negative regulator of BMP and TGF-β signaling. Smurf2 is detrimental to cartilage homeostasis by antagonizing TGF-β signaling. Smurf2 overexpression promotes chondrocyte maturation, causing spontaneous OA and accelerated age-related IVD degeneration.290–292 Smurf2 deficiency protects both young and aged mice from surgically-induced OA.293 Smurf2 negatively regulates BMP signaling to inhibit osteogenesis.294 Smurf2 was proposed to induce degradation of the TGF-β receptors, Smad2, and Smad3. However, neither of those proteins increased in Smurf2-null mice.295 Instead, mono-ubiquitination of SMAD3 was reduced to favor SMAD complex formation in the absence of Smurf2, which mediates the interaction between SMAD3 and vitamin D receptor to modulate RANKL production and osteoclast formation.295,296
Nedd4 regulates the degradation of Smad1 to antagonize BMP signaling and inhibit bone formation.297,298 Nedd4 overexpression in osteoblasts increases bone mass, and Nedd4 deletion in osteoblasts reduces bone formation.298
Deubiquitination
Deubiquitylating enzyme USP15 stabilizes ALK3 to enhance BMP activation and osteoblast differentiation.299 USP15 also inhibits OA progression by deubiquitinating ERK2 and enhancing ERK2-induced TGF-β/SMAD2 signaling.300
Osteogenic LIM mineralization protein (LMP-1) antagonizes SMAD ubiquitination to promote TGF-β and BMP activation. LMP-1 interacts with Smurf1 to prevent Smad-1 and -5 ubiquitination, and interacts with Jab1 to prevent Smad7-induced Smad-4 and -5 ubiquitination.301
Valosin-containing protein (VCP)/p97, together with its adaptor nuclear protein localization 4 (NPL4), interacts explicitly with Smurf1 and delivers the ubiquitinated Smurf1 for degradation. Mutation of VCP/p97 causes rare forms of Paget’s disease of bone (PDB)-like syndromes by increasing BMP activity.302
COP9 signalosome is a protein complex with isopeptidase activity responsible for the deneddylation of RING ubiquitin ligases (CRL) by catalyzing the hydrolysis of NEDD protein CRL. Jab1, also known as Csn5/Cops5, is a crucial subunit of the COP9 signalosome. Jab1 deletion in preosteoblast reduced the response to TGF-β and BMP signaling, impairing osteoblast differentiation and reducing the trabecular bone number.303
Phosphatases and kinases
TGF-β and BMP receptor activity is also regulated by phosphorylation and dephosphorylation. Endosome-associated FYVE-domain protein (endofin), previously implicated in regulating membrane trafficking, also recruits protein phosphatase 1 catalytic subunit (PP1c) to exert a negative regulative effect on BMP signaling by dephosphorylating the BMP type I receptor.304 A single point mutation of endofin (F872A) disrupts endofin–PP1c interaction and sensitizes BMP signaling to increase osteogenesis in vitro and in vivo.304 Casein kinase II (CK2) phosphorylates the ALK3 receptor to block its activity, reducing BMP2’s osteogenic effects on osteoblasts in patients with osteoporosis.305
Regulation in the nucleus
Nuclear envelope proteins
Transport of the SMAD complex into the nucleus is controlled by the nuclear pore complex (NPC), comprising multiple copies of ~30 different proteins located on the nuclear envelope. As the boundary between the cell nucleus and cytoplasm, the nuclear envelope comprises a double-membrane sheet, the inner nuclear membrane (INM) and the outer nuclear membrane (ONM). LEM domain containing 3 (LEMD3), an INM protein and transmembrane protein 53 (TMEM53) have been reported to regulate bone BMP and TGF-β signaling. Loss of function of LEMD3 results in unique sclerosing bone disease spectrums, osteopoikilosis (MIM #166700), melorheostosis (MIM #155950) and Buschke-Ollendorff syndrome (BOS; MIM #166700) (Table 2).306,307 LEMD3 has been shown to antagonize BMP and TGF-β by interacting with SMAD-1, -2, -5, and -9. TMEM53 inhibits BMP signaling in osteoblast lineage cells by blocking cytoplasm-nucleus translocation of SMAD1/5/8 specifically.308 In humans, TMEM53 was identified as a susceptibility gene for osteoporosis in several studies,309,310 and was recently associated with a previously unknown type of sclerosing bone disease (Table 2).308
Transcription repressors
Ski is a nuclear proto-oncogene protein homolog of the avian sarcoma viral (v-ski) oncogene and is a repressor of TGF-β and BMP signaling by inhibiting the transcription activity of SMAD complex.311 It also recruits histone deacetylases HDAC4 and HDAC5 as co-repressors.312 SKI mutations in humans cause Shprintzen-Goldberg syndrome (GOSHS; MIM #182212),313 which share multiple skeletal anomalies with Marfan syndrome caused by mutations of FBN-1 (Table 2). Both diseases are associated with enhanced TGF-β and BMP signaling.
SnoN, a Ski proto-oncogene homolog, also interacts with the SMAD complex. A negative feedback mechanism, regulated by SnoN, can be evoked by TGF-β to oppose BMP signaling in chondrocytes and osteoblasts.314,315 SnoN and Ski might have different functions since they are differently recruited by Smad2 and Smad3.89
Nkx3.2 is a transcriptional repressor expressed in the sclerotome and developing cartilage, where it activates the chondrocyte differentiation program via a BMP-dependent manner. Mechanistically, Nkx3.2 forms a complex with histone deacetylase 1 (HDAC1) and Smad-1 and -4 in a BMP-dependent manner through its homeodomain and NK domain to repress gene expression cooperatively.316
Tob is a member of the emerging family of anti-proliferative proteins and negatively regulates BMP signaling in osteoblasts by directly interacting with Smad-1, -5, and -8 in the nucleus. Tob-null mice have a greater bone mass due to an increased number of osteoblasts.317
FOXC1 could repress the transcriptional activity of SMAD-1 and -5 to modulate the expression of BMP-responsive genes to prevent osteoblast differentiation.318
Transcription co-factors
Runx2 is a critical transcription factor in promoting osteoblast differentiation and chondrocyte hypertrophy. Runx2 is physically and functionally associated with Smad proteins in osteoblasts and chondrocytes.319,320 Javed et al. 320 reported that BMP-induced osteogenesis is blunted in Runx2-null cells, and Runx2 with mutations in Smad-interacting domain (HTY (426–428)) is only marginally functional in promoting osteoblast differentiation at early stages.
TCF4 and β-catenin are the transcription factors activated by canonical WNT signaling and are anabolic for osteogenesis. They form a complex with Smad proteins on the promoter of osteoblastic genes and recruit co-activators such as CBP or p300, cooperatively regulating the expression of early osteoblast genes such as Dlx5, Msx2, Runx2, and osterix.139
Sox9 is the key chondrogenic transcription factor. Sox9 interacts with Smad2/3 on the Col2 enhancer region in a TGF-β-dependent manner and recruits co-activators such as CBP or p300 to promote transcription.321
c-Fos, a key osteoclastic transcription factor, interacts directly with SMAD-2 and -3 to promote osteoclast diffrentiation.38
Lysine demethylase 4B (KDM4B), a histone demethylase whose expression is induced by TGF-β, potentiates TGF-β-mediated chondrogenesis of human MSCs in a positive feedback loop.322 Mechanistically, KDM4B removes the silencing H3K9me3 marks on the SOX9 promoter to facilitate SMAD3 binding and transcription.322
Conclusion and Perspectives
BMP and TGF-β signaling is essential in embryonic skeleton development and postnatal bone and cartilage homeostasis. Dysregulated TGF-β and BMP signaling causes numerous hereditary skeletal diseases in humans. For example, excessive TGF-β signaling in humans due to TGFB1, SMAD3, or MAP2K1 gene mutations leads to a spectrum of sclerosis symptoms. Excessive TGF-β activation is also associated with OI. NOGGIN, SMAD6, or ALK2 mutations augment BMP signaling to cause craniosynostosis or HO. Mutations of FBN-1/2 or SKI enhance both TGF-β and BMP signalings to cause similar skeletal anomalies in humans. Mutations of ADAMTS block TGF-β and BMP activation and lead to short stature anomalies. Moreover, genome-wide association studies have identified several genes in TGF-β and BMP signaling associated with bone density. Most phenotypes were recapitulated in genetic mouse models carrying those disease-associated mutations, which provide disease models for pathomechanism studies and drug screening. Targeting TGF-β and BMP signaling effectively cures their associated skeletal disorders in diseased mouse models and clinical trials, such as OI and HO.
TGF-βs and BMPs belong to the same family, share structural similarities, and transduce signals through both SMAD-dependent and -independent pathways. However, they recruit different receptors to activate independent sets of SMAD proteins, laying down the molecular basis for their diverse functions. For example, BMPs, but not TGF-βs, are essential for limb bud outgrowth. In chondrocytes, BMPs promote differentiation at all stages; in contrast, TGF-β promotes chondrocyte early development but antagonizes its hypertrophy. BMPs promote osteoblast and osteoclast differentiation and are applied to improve fracture healing. Meanwhile, TGF-β signaling plays dual roles in osteoblast and osteoclast formation. Moreover, BMP and TGF-β also play opposite roles in articular cartilage homeostasis.
Genetics and molecular biology studies have advanced our understanding of the function and dynamic regulations of BMP and TGF-β signaling in the skeleton. However, more precise knowledge is still in demand and might promote the development of effective therapeutic strategies to treat related skeletal disorders. Future directions may lie in answering the following questions:
Why do BMP and TGF-β signalings have dynamic functions? As reviewed here, this question could be partially answered by the diversity of ligand–receptor combinations and the complex intracellular regulatory network that causes the dynamic readout of the TGF-β and BMP signaling. In particular, the BMP signaling pathway comprises multiple ligands and receptors that interact promiscuously with one another. A series of works from Dr. Michael B. Elowitz’s group demonstrated that the promiscuous ligand–receptor interaction systems of BMP signaling are critical for its dynamic regulations.323–325 Their work elucidated how the BMP pathway processes multi-ligand inputs using a repertoire of computational mechanisms, including ratiometric sensing, balance detection, and imbalance detection. Since cells have different expression patterns of receptors and ligands, the promiscuous interaction system allows a small number of ligands, acting in combinations, to address the issue of a larger number of individual cell types.
What transcriptional mechanism operates to bring about the diversity of transcriptional outcomes that arise in different cell types in response to the same ligand? Answering this question would require using state-of-the-art techniques such as ChIP-seq, co-IP/MS, ATAC-seq, and CUT&Tag-seq. DNA and histone modification status varies in different cell types and might alter the affinity of SMAD complex binding with the chromosomes. Therefore, analyzing the epigenetic marks on the transcription factor binding sequences would help answer this question. Characterization of the receptor–ligand interaction mode and chromatin status in specific cell contexts might also explain why TGF-β signaling has stage-dependent functions in most skeletal cells.
How to circumvent the side effects of BMPs and TGF-βs when applying them in clinical settings? Excessive BMP and TGF-β signaling is associated with multiple anomalies in bone tissues. Thus, further study and intervention are needed to prevent those side effects when applying BMPs and TGF-βs in clinical settings. Although TGF-β signaling maintains cartilage degeneration, hyperactivated TGF-β signaling aggravates OA. Despite its dual functions, TGF-β signaling is still proposed as a potential treatment to alleviate OA, although it needs more study to design the proper timing and dose for the treatment.
How to safely and effectively modulate BMP and TGF-β signaling in skeletal disorders caused by their dysfunctions? Targeting BMP and TGF-β signaling is proposed as the therapeutic strategy to treat OI, HO, or osteosclerosis disorders while effective treatment is still under development.
Acknowledgements
We apologize to the many researchers whose important primary papers could not be cited in the References due to space limitations. We thank Ms. Abigail McVicar for her excellent assistance with manuscript editing. We thank Figdraw for the support concerning the figure drawing. This work was supported by the National Institutes of Health (AR-070135 and AG-056438 to W.C., and AR075735, DE023813, and DE028264 to Y.P.L), the National Natural Science Foundation of China (81900806 and 32070814 to M.W), the Qizhen Grant of Zhejiang University (226-2022-00132 to M.W.).
Author contributions
M.W. and Y.P.L. discussed and conceived the framework of the manuscript. M.W., S.W., W.C. and Y.P.L. searched and collected papers related to the topic. M.W., S.W., W.C. and Y.P.L. drafted the manuscript. M.W. and W.C. drew the figures and tables. M.W., Y.P.L. and W.C. revised the manuscript.
Competing interests
The authors declare no competing interests.
Contributor Information
Mengrui Wu, Email: mengruiwu@zju.edu.cn.
Yi-Ping Li, Email: yli81@tulane.edu.
References
- 1.Moses HL, Roberts AB, Derynck R. The discovery and early days of TGF-β: A historical perspective. Cold Spring Harb. Perspect. Biol. 2016;8:a021865. doi: 10.1101/cshperspect.a021865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Katagiri T, Watabe T. Bone morphogenetic proteins. Cold Spring Harb. Perspect. Biol. 2016;8:a021899. doi: 10.1101/cshperspect.a021899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Derynck R, Budi EH. Specificity, versatility, and control of TGF-β family signaling. Sci. Signal. 2019;12:eaav5183. doi: 10.1126/scisignal.aav5183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mahmood A, Harkness L, Schrøder HD, Abdallah BM, Kassem M. Enhanced differentiation of human embryonic stem cells to mesenchymal progenitors by inhibition of TGF-beta/activin/nodal signaling using SB-431542. J. Bone Miner. Res. 2010;25:1216–1233. doi: 10.1002/jbmr.34. [DOI] [PubMed] [Google Scholar]
- 5.Cianciolo G, et al. The role of activin: the other side of chronic kidney disease-mineral bone disorder? Nephrol. Dial. Transplant. 2021;36:966–974. doi: 10.1093/ndt/gfaa203. [DOI] [PubMed] [Google Scholar]
- 6.Lee SJ, et al. Targeting myostatin/activin A protects against skeletal muscle and bone loss during spaceflight. Proc. Natl. Acad. Sci. USA. 2020;117:23942–23951. doi: 10.1073/pnas.2014716117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maridas, D. E., et al. Chapter 48 - Bone morphogenetic proteins. In: Bilezikian J. P., Martin T. J., Clemens T. L., Rosen C. J. eds. Principles of Bone Biology (Fourth Edition): Academic Press: 1189–1197 (2020).
- 8.Xu, X. & Cao, X. Chapter 47 - Transforming growth factor-β and skeletal homeostasis1. In: Bilezikian J. P., Martin T. J., Clemens T. L., Rosen C. J. eds. Principles of Bone Biology (Fourth Edition): Academic Press: 1153–1187 (2020).
- 9.Gipson GR, et al. Structural perspective of BMP ligands and signaling. Bone. 2020;140:115549–115549. doi: 10.1016/j.bone.2020.115549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shi M, et al. Latent TGF-β structure and activation. Nature. 2011;474:343–349. doi: 10.1038/nature10152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karsdal MA, et al. Matrix metalloproteinase-dependent activation of latent transforming growth factor-beta controls the conversion of osteoblasts into osteocytes by blocking osteoblast apoptosis. J. Biol. Chem. 2002;277:44061–44067. doi: 10.1074/jbc.M207205200. [DOI] [PubMed] [Google Scholar]
- 12.Dallas SL, Rosser JL, Mundy GR, Bonewald LF. Proteolysis of latent transforming growth factor-beta (TGF-beta)-binding protein-1 by osteoclasts. A cellular mechanism for release of TGF-beta from bone matrix. J. Biol. Chem. 2002;277:21352–21360. doi: 10.1074/jbc.M111663200. [DOI] [PubMed] [Google Scholar]
- 13.D’Angelo M, Billings PC, Pacifici M, Leboy PS, Kirsch T. Authentic matrix vesicles contain active metalloproteases (MMP). a role for matrix vesicle-associated MMP-13 in activation of transforming growth factor-beta. J. Biol. Chem. 2001;276:11347–11353. doi: 10.1074/jbc.M009725200. [DOI] [PubMed] [Google Scholar]
- 14.Wang J, et al. Atp6i deficient mouse model uncovers transforming growth factor-β1 /Smad2/3 as a key signaling pathway regulating odontoblast differentiation and tooth root formation. Int. J. Oral Sci. 2023;15:35. doi: 10.1038/s41368-023-00235-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Salmon RM, et al. Molecular basis of ALK1-mediated signalling by BMP9/BMP10 and their prodomain-bound forms. Nat. Commun. 2020;11:1621. doi: 10.1038/s41467-020-15425-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Neugebauer JM, et al. The prodomain of BMP4 is necessary and sufficient to generate stable BMP4/7 heterodimers with enhanced bioactivity in vivo. Proc. Natl. Acad. Sci. USA. 2015;112:E2307–E2316. doi: 10.1073/pnas.1501449112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sengle G, Ono RN, Sasaki T, Sakai LY. Prodomains of transforming growth factor beta (TGFbeta) superfamily members specify different functions: extracellular matrix interactions and growth factor bioavailability. J. Biol. Chem. 2011;286:5087–5099. doi: 10.1074/jbc.M110.188615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gregory KE, et al. The prodomain of BMP-7 targets the BMP-7 complex to the extracellular matrix. J. Biol. Chem. 2005;280:27970–27980. doi: 10.1074/jbc.M504270200. [DOI] [PubMed] [Google Scholar]
- 19.Martinez-Hackert E, Sundan A, Holien T. Receptor binding competition: A paradigm for regulating TGF-β family action. Cytokine Growth Factor Rev. 2021;57:39–54. doi: 10.1016/j.cytogfr.2020.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Finnson KW, Parker WL, ten Dijke P, Thorikay M, Philip A. ALK1 opposes ALK5/Smad3 signaling and expression of extracellular matrix components in human chondrocytes. J. Bone Miner. Res. 2008;23:896–906. doi: 10.1359/jbmr.080209. [DOI] [PubMed] [Google Scholar]
- 21.Goumans MJ, et al. Activin receptor-like kinase (ALK)1 is an antagonistic mediator of lateral TGFbeta/ALK5 signaling. Mol. Cell. 2003;12:817–828. doi: 10.1016/S1097-2765(03)00386-1. [DOI] [PubMed] [Google Scholar]
- 22.Mang T, et al. BMPR1A is necessary for chondrogenesis and osteogenesis, whereas BMPR1B prevents hypertrophic differentiation. J. Cell Sci. 2020;133:jcs246934. doi: 10.1242/jcs.246934. [DOI] [PubMed] [Google Scholar]
- 23.Zhu D, et al. BMP-9 regulates the osteoblastic differentiation and calcification of vascular smooth muscle cells through an ALK1 mediated pathway. J. Cell Mol. Med. 2015;19:165–174. doi: 10.1111/jcmm.12373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kokabu S, et al. BMP3 suppresses osteoblast differentiation of bone marrow stromal cells via interaction with Acvr2b. Mol. Endocrinol. 2012;26:87–94. doi: 10.1210/me.2011-1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yi SE, Daluiski A, Pederson R, Rosen V, Lyons KM. The type I BMP receptor BMPRIB is required for chondrogenesis in the mouse limb. Development. 2000;127:621–630. doi: 10.1242/dev.127.3.621. [DOI] [PubMed] [Google Scholar]
- 26.van Caam A, et al. The high affinity ALK1-ligand BMP9 induces a hypertrophy-like state in chondrocytes that is antagonized by TGFβ1. Osteoarthritis Cartilage. 2015;23:985–995. doi: 10.1016/j.joca.2015.02.007. [DOI] [PubMed] [Google Scholar]
- 27.Zhang D, et al. ALK2 functions as a BMP type I receptor and induces Indian hedgehog in chondrocytes during skeletal development. J. Bone Miner. Res. 2003;18:1593–1604. doi: 10.1359/jbmr.2003.18.9.1593. [DOI] [PubMed] [Google Scholar]
- 28.Daluiski A, et al. Bone morphogenetic protein-3 is a negative regulator of bone density. Nat. Genet. 2001;27:84–88. doi: 10.1038/83810. [DOI] [PubMed] [Google Scholar]
- 29.Macias MJ, Martin-Malpartida P, Massagué J. Structural determinants of Smad function in TGF-β signaling. Trends Biochem. Sci. 2015;40:296–308. doi: 10.1016/j.tibs.2015.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gámez B, Rodríguez-Carballo E, Graupera M, Rosa JL, Ventura F. Class I PI-3-kinase signaling is critical for bone formation through regulation of SMAD1 activity in osteoblasts. J. Bone Miner. Res. 2016;31:1617–1630. doi: 10.1002/jbmr.2819. [DOI] [PubMed] [Google Scholar]
- 31.Zhu Y, et al. Crosstalk between Smad2/3 and specific isoforms of ERK in TGF-β1-induced TIMP-3 expression in rat chondrocytes. PLoS Genet. 2017;21:1781–1790. doi: 10.1111/jcmm.13099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baron R, et al. Balancing BMP signaling through integrated inputs into the Smad1 linker. Nat. Commun. 2007;25:441–454. doi: 10.1016/j.molcel.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 33.Urata M, et al. A peptide that blocks the interaction of NF-κB p65 subunit with Smad4 enhances BMP2-induced osteogenesis. J. Cell. Physiol. 2018;233:7356–7366. doi: 10.1002/jcp.26571. [DOI] [PubMed] [Google Scholar]
- 34.Sun X, et al. TGF-β inhibits osteogenesis by upregulating the expression of ubiquitin ligase SMURF1 via MAPK-ERK signaling. J. Cell. Physiol. 2018;233:596–606. doi: 10.1002/jcp.25920. [DOI] [PubMed] [Google Scholar]
- 35.Kua HY, et al. c-Abl promotes osteoblast expansion by differentially regulating canonical and non-canonical BMP pathways and p16INK4a expression. Nat. Cell Biol. 2012;14:727–737. doi: 10.1038/ncb2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martin-Malpartida P, et al. Structural basis for genome wide recognition of 5-bp GC motifs by SMAD transcription factors. Nat. Commun. 2017;8:2070. doi: 10.1038/s41467-017-02054-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miyazono K, Maeda S, Imamura T. BMP receptor signaling: Transcriptional targets, regulation of signals, and signaling cross-talk. Cytokine Growth Factor Rev. 2005;16:251–263. doi: 10.1016/j.cytogfr.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 38.Omata Y, et al. Genomewide comprehensive analysis reveals critical cooperation between Smad and c-Fos in RANKL-induced osteoclastogenesis. J. Bone Miner. Res. 2015;30:869–877. doi: 10.1002/jbmr.2418. [DOI] [PubMed] [Google Scholar]
- 39.Yu S, et al. BMP2-dependent gene regulatory network analysis reveals Klf4 as a novel transcription factor of osteoblast differentiation. Cell Death Dis. 2021;12:197. doi: 10.1038/s41419-021-03480-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yan J, et al. Smad4 deficiency impairs chondrocyte hypertrophy via the Runx2 transcription factor in mouse skeletal development. J. Biol. Chem. 2018;293:9162–9175. doi: 10.1074/jbc.RA118.001825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Berendsen AD, Olsen BR. Bone development. Bone. 2015;80:14–18. doi: 10.1016/j.bone.2015.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Long F, Ornitz DM. Development of the endochondral skeleton. Cold Spring Harb. Perspect. Biol. 2013;5:a008334. doi: 10.1101/cshperspect.a008334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tang CY, et al. Runx1 up-regulates chondrocyte to osteoblast lineage commitment and promotes bone formation by enhancing both chondrogenesis and osteogenesis. Biochem. J. 2020;477:2421–2438. doi: 10.1042/BCJ20200036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tang J, et al. Runt-related transcription factor 1 is required for murine osteoblast differentiation and bone formation. J. Biol. Chem. 2020;295:11669–11681. doi: 10.1074/jbc.RA119.007896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tian F, et al. Core binding factor beta (Cbfβ) controls the balance of chondrocyte proliferation and differentiation by upregulating Indian hedgehog (Ihh) expression and inhibiting parathyroid hormone-related protein receptor (PPR) expression in postnatal cartilage and bone formation. J. Bone Miner. Res. 2014;29:1564–1574. doi: 10.1002/jbmr.2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu M, et al. Deletion of core-binding factor β (Cbfβ) in mesenchymal progenitor cells provides new insights into Cbfβ/Runxs complex function in cartilage and bone development. Bone. 2014;65:49–59. doi: 10.1016/j.bone.2014.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu M, et al. Chondrocyte-specific knockout of Cbfβ reveals the indispensable function of Cbfβ in chondrocyte maturation, growth plate development and trabecular bone formation in mice. Int. J. Biol. Sci. 2014;10:861–872. doi: 10.7150/ijbs.8521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yoon BS, et al. Bmpr1a and Bmpr1b have overlapping functions and are essential for chondrogenesis in vivo. Proc. Natl. Acad. Sci. USA. 2005;102:5062–5067. doi: 10.1073/pnas.0500031102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rigueur D, et al. The type I BMP receptor ACVR1/ALK2 is required for chondrogenesis during development. J. Bone Miner. Res. 2015;30:733–741. doi: 10.1002/jbmr.2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shu B, et al. BMP2, but not BMP4, is crucial for chondrocyte proliferation and maturation during endochondral bone development. J. Cell Sci. 2011;124:3428–3440. doi: 10.1242/jcs.083659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bandyopadhyay A, et al. Genetic analysis of the roles of BMP2, BMP4, and BMP7 in limb patterning and skeletogenesis. PLoS Genet. 2006;2:e216. doi: 10.1371/journal.pgen.0020216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Selever J, Liu W, Lu MF, Behringer RR, Martin JF. Bmp4 in limb bud mesoderm regulates digit pattern by controlling AER development. Dev. Biol. 2004;276:268–279. doi: 10.1016/j.ydbio.2004.08.024. [DOI] [PubMed] [Google Scholar]
- 53.Nissim S, Hasso SM, Fallon JF, Tabin CJ. Regulation of Gremlin expression in the posterior limb bud. Dev. Biol. 2006;299:12–21. doi: 10.1016/j.ydbio.2006.05.026. [DOI] [PubMed] [Google Scholar]
- 54.Maatouk DM, Choi KS, Bouldin CM, Harfe BD. In the limb AER Bmp2 and Bmp4 are required for dorsal-ventral patterning and interdigital cell death but not limb outgrowth. Dev. Biol. 2009;327:516–523. doi: 10.1016/j.ydbio.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 55.Choi KS, Lee C, Maatouk DM, Harfe BD. Bmp2, Bmp4 and Bmp7 are co-required in the mouse AER for normal digit patterning but not limb outgrowth. PLoS One. 2012;7:e37826. doi: 10.1371/journal.pone.0037826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pajni-Underwood S, Wilson CP, Elder C, Mishina Y, Lewandoski M. BMP signals control limb bud interdigital programmed cell death by regulating FGF signaling. Development. 2007;134:2359–2368. doi: 10.1242/dev.001677. [DOI] [PubMed] [Google Scholar]
- 57.Benazet JD, Zeller R. Dual requirement of ectodermal Smad4 during AER formation and termination of feedback signaling in mouse limb buds. Genesis. 2013;51:660–666. doi: 10.1002/dvg.22412. [DOI] [PubMed] [Google Scholar]
- 58.Pignatti E, Zeller R, Zuniga A. To BMP or not to BMP during vertebrate limb bud development. Semin. Cell Dev. Biol. 2014;32:119–127. doi: 10.1016/j.semcdb.2014.04.004. [DOI] [PubMed] [Google Scholar]
- 59.Takano K, et al. A novel nonsense mutation in the NOG gene causes familial NOG-related symphalangism spectrum disorder. Hum. Genome Var. 2016;3:16023. doi: 10.1038/hgv.2016.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Seemann P, et al. Activating and deactivating mutations in the receptor interaction site of GDF5 cause symphalangism or brachydactyly type A2. J. Clin. Invest. 2005;115:2373–2381. doi: 10.1172/JCI25118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dixon ME, Armstrong P, Stevens DB, Bamshad M. Identical mutations in NOG can cause either tarsal/carpal coalition syndrome or proximal symphalangism. Genet. Med. 2001;3:349–353. doi: 10.1097/00125817-200109000-00004. [DOI] [PubMed] [Google Scholar]
- 62.Lehmann K, et al. A new subtype of brachydactyly type B caused by point mutations in the bone morphogenetic protein antagonist NOGGIN. Am. J. Hum. Genet. 2007;81:88–396. doi: 10.1086/519697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dathe K, et al. Duplications involving a conserved regulatory element downstream of BMP2 are associated with brachydactyly type A2. Am. J. Hum. Genet. 2009;84:483–492. doi: 10.1016/j.ajhg.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lehmann K, et al. Mutations in bone morphogenetic protein receptor 1B cause brachydactyly type A2. Proc. Natl. Acad. Sci. USA. 2003;100:12277–12282. doi: 10.1073/pnas.2133476100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yoon BS, et al. BMPs regulate multiple aspects of growth-plate chondrogenesis through opposing actions on FGF pathways. Development. 2006;133:4667–4678. doi: 10.1242/dev.02680. [DOI] [PubMed] [Google Scholar]
- 66.Brunet LJ, McMahon JA, McMahon AP, Harland RM. Noggin, cartilage morphogenesis, and joint formation in the mammalian skeleton. Science. 1998;280:1455–1457. doi: 10.1126/science.280.5368.1455. [DOI] [PubMed] [Google Scholar]
- 67.McMahon JA, et al. Noggin-mediated antagonism of BMP signaling is required for growth and patterning of the neural tube and somite. Genes Dev. 1998;12:1438–1452. doi: 10.1101/gad.12.10.1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Keller B, et al. Interaction of TGFβ and BMP signaling pathways during chondrogenesis. PLoS One. 2011;6:e16421. doi: 10.1371/journal.pone.0016421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Retting KN, Song B, Yoon BS, Lyons KM. BMP canonical Smad signaling through Smad1 and Smad5 is required for endochondral bone formation. Development. 2009;136:1093–1104. doi: 10.1242/dev.029926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tsumaki N, et al. Role of CDMP-1 in skeletal morphogenesis: promotion of mesenchymal cell recruitment and chondrocyte differentiation. J. Cell Biol. 1999;144:161–173. doi: 10.1083/jcb.144.1.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gao L, et al. TAK1 regulates SOX9 expression in chondrocytes and is essential for postnatal development of the growth plate and articular cartilages. J. Cell Sci. 2013;126:5704–5713. doi: 10.1242/jcs.135483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bertoli-Avella AM, et al. Mutations in a TGF-β ligand, TGFB3, cause syndromic aortic aneurysms and dissections. J. Am. Coll. Cardiol. 2015;65:1324–1336. doi: 10.1016/j.jacc.2015.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lindsay ME, et al. Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat. Genet. 2012;44:922–927. doi: 10.1038/ng.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.van de Laar IM, et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat. Genet. 2011;43:121–126. doi: 10.1038/ng.744. [DOI] [PubMed] [Google Scholar]
- 75.Loeys BL, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat. Genet. 2005;37:275–281. doi: 10.1038/ng1511. [DOI] [PubMed] [Google Scholar]
- 76.Vandeloo B, et al. Spontaneous coronary artery dissection in a man with a novel missense mutation in SMAD2 treated by optical coherence tomography-guided percutaneous coronary intervention. JACC Cardiovasc. Interv. 2019;12:e45–e47. doi: 10.1016/j.jcin.2018.09.007. [DOI] [PubMed] [Google Scholar]
- 77.Milewicz DM, et al. Marfan syndrome. Nat. Rev. Dis. Primers. 2021;7:64. doi: 10.1038/s41572-021-00298-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Seo HS, Serra R. Deletion of Tgfbr2 in Prx1-cre expressing mesenchyme results in defects in development of the long bones and joints. Dev. Biol. 2007;310:304–316. doi: 10.1016/j.ydbio.2007.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Baffi MO, et al. Conditional deletion of the TGF-beta type II receptor in Col2a expressing cells results in defects in the axial skeleton without alterations in chondrocyte differentiation or embryonic development of long bones. Dev. Biol. 2004;276:124–142. doi: 10.1016/j.ydbio.2004.08.027. [DOI] [PubMed] [Google Scholar]
- 80.Sohn P, Cox M, Chen D, Serra R. Molecular profiling of the developing mouse axial skeleton: a role for Tgfbr2 in the development of the intervertebral disc. BMC Dev. Biol. 2010;10:29. doi: 10.1186/1471-213X-10-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Spagnoli A, et al. TGF-beta signaling is essential for joint morphogenesis. J. Cell Biol. 2007;177:1105–1117. doi: 10.1083/jcb.200611031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Longobardi L, et al. TGF-β type II receptor/MCP-5 axis: at the crossroad between joint and growth plate development. Cancers. 2012;23:71–81. doi: 10.1016/j.devcel.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Matsunobu T, et al. Critical roles of the TGF-beta type I receptor ALK5 in perichondrial formation and function, cartilage integrity, and osteoblast differentiation during growth plate development. Dev. Biol. 2009;332:325–338. doi: 10.1016/j.ydbio.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sueyoshi T, Yamamoto K, Akiyama H. Conditional deletion of Tgfbr2 in hypertrophic chondrocytes delays terminal chondrocyte differentiation. Matrix Biol. 2012;31:352–359. doi: 10.1016/j.matbio.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 85.Sanford LP, et al. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development. 1997;124:2659–2670. doi: 10.1242/dev.124.13.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Proetzel G, et al. Transforming growth factor-beta 3 is required for secondary palate fusion. Nat. Genet. 1995;11:409–414. doi: 10.1038/ng1295-409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kaartinen V, et al. Abnormal lung development and cleft palate in mice lacking TGF-beta 3 indicates defects of epithelial-mesenchymal interaction. Nat. Genet. 1995;11:415–421. doi: 10.1038/ng1295-415. [DOI] [PubMed] [Google Scholar]
- 88.Kulkarni AB, et al. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc. Natl. Acad. Sci. USA. 1993;90:770–774. doi: 10.1073/pnas.90.2.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang W, Song B, Anbarchian T, Shirazyan A, Sadik JE. Smad2 and Smad3 regulate chondrocyte proliferation and differentiation in the growth plate. PLoS Genet. 2016;12:e1006352. doi: 10.1371/journal.pgen.1006352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Edwards JR, Mundy GR. Advances in osteoclast biology: old findings and new insights from mouse models. Nat. Rev. Rheumatol. 2011;7:235–243. doi: 10.1038/nrrheum.2011.23. [DOI] [PubMed] [Google Scholar]
- 91.Long F. Building strong bones: molecular regulation of the osteoblast lineage. Nat. Rev. Mol. Cell Biol. 2011;13:27–38. doi: 10.1038/nrm3254. [DOI] [PubMed] [Google Scholar]
- 92.Chen W, et al. Cbfβ deletion in mice recapitulates cleidocranial dysplasia and reveals multiple functions of Cbfβ required for skeletal development. Proc. Natl. Acad. Sci. USA. 2014;111:8482–8487. doi: 10.1073/pnas.1310617111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wu M, et al. Cbfβ governs osteoblast-adipocyte lineage commitment through enhancing β-catenin signaling and suppressing adipogenesis gene expression. Proc. Natl. Acad. Sci. USA. 2017;114:10119–10124. doi: 10.1073/pnas.1619294114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tang CY, et al. Runx1 is a central regulator of osteogenesis for bone homeostasis by orchestrating BMP and WNT signaling pathways. PLoS Genet. 2021;17:e1009233. doi: 10.1371/journal.pgen.1009233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Delgado-Calle J, Bellido T. The osteocyte as a signaling cell. Physiol. Rev. 2022;102:379–410. doi: 10.1152/physrev.00043.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Langdahl BL, Carstens M, Stenkjaer L, Eriksen EF. Polymorphisms in the transforming growth factor beta 1 gene and osteoporosis. Bone. 2003;32:297–310. doi: 10.1016/S8756-3282(02)00971-7. [DOI] [PubMed] [Google Scholar]
- 97.Panach L, et al. Comparative transcriptome analysis identifies CARM1 and DNMT3A as genes associated with osteoporosis. Sci. Rep. 2020;10:16298. doi: 10.1038/s41598-020-72870-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gregson CL, et al. Genome-wide association study of extreme high bone mass: Contribution of common genetic variation to extreme BMD phenotypes and potential novel BMD-associated genes. Bone. 2018;114:62–71. doi: 10.1016/j.bone.2018.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pei YF, et al. Genome-wide association meta-analyses identified 1q43 and 2q32.2 for hip Ward’s triangle areal bone mineral density. Bone. 2016;91:1–10. doi: 10.1016/j.bone.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.He JW, Yue H, Hu WW, Hu YQ, Zhang ZL. Contribution of the sclerostin domain-containing protein 1 (SOSTDC1) gene to normal variation of peak bone mineral density in Chinese women and men. J. Bone Miner. Metab. 2011;29:571–581. doi: 10.1007/s00774-010-0253-5. [DOI] [PubMed] [Google Scholar]
- 101.Moffett SP, et al. Identification and association analysis of single nucleotide polymorphisms in the human noggin (NOG) gene and osteoporosis phenotypes. Bone. 2009;44:999–1002. doi: 10.1016/j.bone.2008.12.024. [DOI] [PubMed] [Google Scholar]
- 102.Wang H, et al. Association of bone morphogenetic protein-2 gene polymorphisms with susceptibility to ossification of the posterior longitudinal ligament of the spine and its severity in Chinese patients. Eur. Spine J. 2008;17:956–964. doi: 10.1007/s00586-008-0651-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lin GT, et al. SNP combinations in chromosome-wide genes are associated with bone mineral density in Taiwanese women. Chin. J. Physiol. 2008;51:32–41. [PubMed] [Google Scholar]
- 104.Medici M, et al. BMP-2 gene polymorphisms and osteoporosis: the Rotterdam Study. J. Bone Miner. Res. 2006;21:845–854. doi: 10.1359/jbmr.060306. [DOI] [PubMed] [Google Scholar]
- 105.Gregson CL, et al. A rare mutation in SMAD9 associated with high bone mass identifies the SMAD-dependent BMP signaling pathway as a potential anabolic target for osteoporosis. J. Bone Miner. Res. 2020;35:92–105. doi: 10.1002/jbmr.3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kim BJ, et al. Association of SMAD2 polymorphisms with bone mineral density in postmenopausal Korean women. Osteoporos. Int. 2011;22:2273–2282. doi: 10.1007/s00198-010-1450-8. [DOI] [PubMed] [Google Scholar]
- 107.Lowery JW, Rosen V. Bone morphogenetic protein-based therapeutic approaches. Cold Spring Harb. Perspect. Biol. 2018;10:a022327. doi: 10.1101/cshperspect.a022327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Begam H, Nandi SK, Kundu B, Chanda A. Strategies for delivering bone morphogenetic protein for bone healing. Mater. Sci. Eng. C Mater. Biol. Appl. 2017;70:856–869. doi: 10.1016/j.msec.2016.09.074. [DOI] [PubMed] [Google Scholar]
- 109.Bharadwaz A, Jayasuriya AC. Osteogenic differentiation cues of the bone morphogenetic protein-9 (BMP-9) and its recent advances in bone tissue regeneration. Mater. Sci. Eng. C Mater. Biol. Appl. 2021;120:111748. doi: 10.1016/j.msec.2020.111748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Eiraku N, et al. BMP9 directly induces rapid GSK3-β phosphorylation in a Wnt-independent manner through class I PI3K-Akt axis in osteoblasts. FASEB J. 2019;33:12124–12134. doi: 10.1096/fj.201900733RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Tsuji K, et al. BMP4 is dispensable for skeletogenesis and fracture-healing in the limb. J. Bone Joint Surg. Am. 2008;90:14–18. doi: 10.2106/JBJS.G.01109. [DOI] [PubMed] [Google Scholar]
- 112.Tsuji K, et al. BMP2 activity, although dispensable for bone formation, is required for the initiation of fracture healing. Nat. Genet. 2006;38:1424–1429. doi: 10.1038/ng1916. [DOI] [PubMed] [Google Scholar]
- 113.Hu Y. Acvr1 deletion in osteoblasts impaired mandibular bone mass through compromised osteoblast differentiation and enhanced sRANKL-induced osteoclastogenesis. J. Cell. Physiol. 2021;236:4580–4591. doi: 10.1002/jcp.30183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Yang C, Yang L, Wan M, Cao X. Generation of a mouse model with expression of bone morphogenetic protein type II receptor lacking the cytoplasmic domain in osteoblasts. Ann. N. Y. Acad. Sci. 2010;1192:286–291. doi: 10.1111/j.1749-6632.2009.05248.x. [DOI] [PubMed] [Google Scholar]
- 115.Gamer LW, Cox K, Carlo JM, Rosen V. Overexpression of BMP3 in the developing skeleton alters endochondral bone formation resulting in spontaneous rib fractures. Dev. Dyn. 2009;238:2374–2381. doi: 10.1002/dvdy.22048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Greenblatt MB, et al. The p38 MAPK pathway is essential for skeletogenesis and bone homeostasis in mice. J. Clin. Invest. 2010;120:2457–2473. doi: 10.1172/JCI42285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Thouverey C, Caverzasio J. The p38α MAPK positively regulates osteoblast function and postnatal bone acquisition. Cell Mol. Life Sci. 2012;69:3115–3125. doi: 10.1007/s00018-012-0983-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Salazar VS, et al. Embryonic ablation of osteoblast Smad4 interrupts matrix synthesis in response to canonical Wnt signaling and causes an osteogenesis-imperfecta-like phenotype. J. Cell Sci. 2013;126:4974–4984. doi: 10.1242/jcs.131953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Salazar VS, et al. Postnatal ablation of osteoblast Smad4 enhances proliferative responses to canonical Wnt signaling through interactions with β-catenin. J. Cell Mol. Med. 2013;126:5598–5609. doi: 10.1242/jcs.132233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zhang J, et al. The inhibition effects of insulin on BMP2-induced muscle heterotopic ossification. Biomaterials. 2014;35:9322–9331. doi: 10.1016/j.biomaterials.2014.07.056. [DOI] [PubMed] [Google Scholar]
- 121.Agarwal S, et al. Strategic targeting of multiple BMP receptors prevents trauma-induced heterotopic ossification. Mol. Ther. 2017;25:1974–1987. doi: 10.1016/j.ymthe.2017.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.van Dinther M, et al. ALK2 R206H mutation linked to fibrodysplasia ossificans progressiva confers constitutive activity to the BMP type I receptor and sensitizes mesenchymal cells to BMP-induced osteoblast differentiation and bone formation. J. Bone Miner. Res. 2010;25:1208–1215. doi: 10.1359/jbmr.091110. [DOI] [PubMed] [Google Scholar]
- 123.Fukuda T, et al. A unique mutation of ALK2, G356D, found in a patient with fibrodysplasia ossificans progressiva is a moderately activated BMP type I receptor. Biochem. Biophys. Res. Commun. 2008;377:905–909. doi: 10.1016/j.bbrc.2008.10.093. [DOI] [PubMed] [Google Scholar]
- 124.Hatsell SJ, et al. ACVR1R206H receptor mutation causes fibrodysplasia ossificans progressiva by imparting responsiveness to activin A. Sci. Transl. Med. 2015;7:303ra137. doi: 10.1126/scitranslmed.aac4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hino K, et al. Neofunction of ACVR1 in fibrodysplasia ossificans progressiva. Proc. Natl. Acad. Sci. USA. 2015;112:15438–15443. doi: 10.1073/pnas.1510540112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Billings PC, et al. Dysregulated BMP signaling and enhanced osteogenic differentiation of connective tissue progenitor cells from patients with fibrodysplasia ossificans progressiva (FOP) J. Bone Miner. Res. 2008;23:305–313. doi: 10.1359/jbmr.071030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Yu PB, et al. BMP type I receptor inhibition reduces heterotopic [corrected] ossification. Nat. Med. 2008;14:1363–1369. doi: 10.1038/nm.1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lees-Shepard JB, et al. Activin-dependent signaling in fibro/adipogenic progenitors causes fibrodysplasia ossificans progressiva. Nat. Commun. 2018;9:471. doi: 10.1038/s41467-018-02872-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Chakkalakal SA, et al. An Acvr1 R206H knock-in mouse has fibrodysplasia ossificans progressiva. J. Bone Miner. Res. 2012;27:1746–1756. doi: 10.1002/jbmr.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Fukuda T, et al. Generation of a mouse with conditionally activated signaling through the BMP receptor, ALK2. Genesis. 2016;44:159–167. doi: 10.1002/dvg.20201. [DOI] [PubMed] [Google Scholar]
- 131.Lodberg A. Principles of the activin receptor signaling pathway and its inhibition. Cytokine Growth Factor Rev. 2021;60:1–17. doi: 10.1016/j.cytogfr.2021.04.001. [DOI] [PubMed] [Google Scholar]
- 132.Shimono K, et al. Potent inhibition of heterotopic ossification by nuclear retinoic acid receptor-γ agonists. Nat. Med. 2011;17:454–460. doi: 10.1038/nm.2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Pignolo RJ, et al. Reduction of new heterotopic ossification (HO) in the open-label, phase 3 MOVE trial of palovarotene for fibrodysplasia ossificans progressiva (FOP) J. Bone Miner. Res. 2023;38:381–394. doi: 10.1002/jbmr.4762. [DOI] [PubMed] [Google Scholar]
- 134.Meng X, Wang H, Hao J. Recent progress in drug development for fibrodysplasia ossificans progressiva. Mol. Cell. Biochem. 2022;477:2327–2334. doi: 10.1007/s11010-022-04446-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Williams E, et al. Saracatinib is an efficacious clinical candidate for fibrodysplasia ossificans progressiva. JCI Insight. 2021;6:e95042. doi: 10.1172/jci.insight.95042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wang Q, et al. Bone morphogenetic protein 2 activates Smad6 gene transcription through bone-specific transcription factor Runx2. J. Biol. Chem. 2007;282:10742–10748. doi: 10.1074/jbc.M610997200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Jeon EJ, et al. Bone morphogenetic protein-2 stimulates Runx2 acetylation. J. Biol. Chem. 2006;281:16502–16511. doi: 10.1074/jbc.M512494200. [DOI] [PubMed] [Google Scholar]
- 138.Jun JH, et al. BMP2-activated Erk/MAP kinase stabilizes Runx2 by increasing p300 levels and histone acetyltransferase activity. J. Biol. Chem. 2010;285:36410–36419. doi: 10.1074/jbc.M110.142307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Rodríguez-Carballo E, et al. Conserved regulatory motifs in osteogenic gene promoters integrate cooperative effects of canonical Wnt and BMP pathways. J. Bone Miner. Res. 2011;26:718–729. doi: 10.1002/jbmr.260. [DOI] [PubMed] [Google Scholar]
- 140.Pawaputanon Na Mahasarakham C, et al. BMP-2 enhances Lgr4 gene expression in osteoblastic cells. J. Cell. Physiol. 2016;231:887–895. doi: 10.1002/jcp.25180. [DOI] [PubMed] [Google Scholar]
- 141.Ko FC, et al. Acute phosphate restriction impairs bone formation and increases marrow adipose tissue in growing mice. J. Bone Miner. Res. 2016;31:2204–2214. doi: 10.1002/jbmr.2891. [DOI] [PubMed] [Google Scholar]
- 142.Yang G, et al. BMP-2 induction of Dlx3 expression is mediated by p38/Smad5 signaling pathway in osteoblastic MC3T3-E1 cells. J. Cell. Physiol. 2014;229:943–954. doi: 10.1002/jcp.24525. [DOI] [PubMed] [Google Scholar]
- 143.Hopkins A, Mirzayans F, Berry F. Foxc1 expression in early osteogenic differentiation is regulated by BMP4-SMAD activity. J. Cell. Biochem. 2016;117:1707–1717. doi: 10.1002/jcb.25464. [DOI] [PubMed] [Google Scholar]
- 144.Ramazzotti G, et al. BMP-2 induced expression of PLCβ1 that is a positive regulator of osteoblast differentiation. J. Cell. Physiol. 2016;231:623–629. doi: 10.1002/jcp.25107. [DOI] [PubMed] [Google Scholar]
- 145.Guo Y, et al. BMP-IHH-mediated interplay between mesenchymal stem cells and osteoclasts supports calvarial bone homeostasis and repair. Bone Res. 2018;6:30. doi: 10.1038/s41413-018-0031-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Liu Z, et al. Molecules mimicking Smad1 interacting with Hox stimulate bone formation. J. Biol. Chem. 2004;279:11313–11319. doi: 10.1074/jbc.M312731200. [DOI] [PubMed] [Google Scholar]
- 147.Lim J, et al. Dual function of Bmpr1a signaling in restricting preosteoblast proliferation and stimulating osteoblast activity in mouse. Development. 2016;143:339–347. doi: 10.1242/dev.126227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Zhang H, et al. Loss of BMP signaling mediated by BMPR1A in osteoblasts leads to differential bone phenotypes in mice depending on anatomical location of the bones. Bone. 2020;137:115402. doi: 10.1016/j.bone.2020.115402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Zhang Y, et al. Loss of BMP signaling through BMPR1A in osteoblasts leads to greater collagen cross-link maturation and material-level mechanical properties in mouse femoral trabecular compartments. Bone. 2016;88:74–84. doi: 10.1016/j.bone.2016.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kamiya N, et al. Wnt inhibitors Dkk1 and Sost are downstream targets of BMP signaling through the type IA receptor (BMPRIA) in osteoblasts. J. Bone Miner. Res. 2010;25:200–210. doi: 10.1359/jbmr.090806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Liu Z, Tang Y, Qiu T, Cao X, Clemens TL. A dishevelled-1/Smad1 interaction couples WNT and bone morphogenetic protein signaling pathways in uncommitted bone marrow stromal cells. J. Biol. Chem. 2006;281:17156–17163. doi: 10.1074/jbc.M513812200. [DOI] [PubMed] [Google Scholar]
- 152.Tang Y, et al. TGF-beta1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat. Med. 2009;15:757–765. doi: 10.1038/nm.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Seo HS, Serra R. Tgfbr2 is required for development of the skull vault. Dev. Biol. 2009;334:481–490. doi: 10.1016/j.ydbio.2009.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Peters SB, Wang Y, Serra R. Tgfbr2 is required in osterix expressing cells for postnatal skeletal development. Bone. 2017;97:54–64. doi: 10.1016/j.bone.2016.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Corps K, Stanwick M, Rectenwald J, Kruggel A, Peters SB. Skeletal deformities in Osterix-Cre;Tgfbr2(f/f) mice may cause postnatal death. Genes. 2021;12:975. doi: 10.3390/genes12070975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Borton AJ, Frederick JP, Datto MB, Wang XF, Weinstein RS. The loss of Smad3 results in a lower rate of bone formation and osteopenia through dysregulation of osteoblast differentiation and apoptosis. J. Bone Miner. Res. 2001;16:1754–1764. doi: 10.1359/jbmr.2001.16.10.1754. [DOI] [PubMed] [Google Scholar]
- 157.Wang Y, Cox MK, Coricor G, MacDougall M, Serra R. Inactivation of Tgfbr2 in Osterix-Cre expressing dental mesenchyme disrupts molar root formation. Dev. Biol. 2013;382:27–37. doi: 10.1016/j.ydbio.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kinoshita A, et al. Domain-specific mutations in TGFB1 result in Camurati-Engelmann disease. Nat. Genet. 2000;26:19–20. doi: 10.1038/79128. [DOI] [PubMed] [Google Scholar]
- 159.Velchev JD, Verstraeten A, Loeys B. Hide and seek: Somatic SMAD3 mutations in melorheostosis. J. Exp. Med. 2020;217:e20200185. doi: 10.1084/jem.20200185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Kang H, et al. Somatic SMAD3-activating mutations cause melorheostosis by up-regulating the TGF-β/SMAD. Pathway. J. Exp. Med. 2020;217:e20191499. doi: 10.1084/jem.20191499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Kang H, et al. Somatic activating mutations in MAP2K1 cause melorheostosis. Nat. Commun. 2018;9:1390. doi: 10.1038/s41467-018-03720-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Nesti LJ, et al. TGF-beta1-stimulated osteoblasts require intracellular calcium signaling for enhanced alpha5 integrin expression. Ann. N. Y. Acad. Sci. 2002;961:178–182. doi: 10.1111/j.1749-6632.2002.tb03078.x. [DOI] [PubMed] [Google Scholar]
- 163.Arnott JA, et al. Molecular requirements for induction of CTGF expression by TGF-beta1 in primary osteoblasts. Bone. 2008;42:871–885. doi: 10.1016/j.bone.2008.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Li J, et al. Smad2 overexpression enhances Smad4 gene expression and suppresses CBFA1 gene expression in osteoblastic osteosarcoma ROS17/2.8 cells and primary rat calvaria cells. J. Biol. Chem. 1998;273:31009–31015. doi: 10.1074/jbc.273.47.31009. [DOI] [PubMed] [Google Scholar]
- 165.Lin HT, et al. Dynamic expression of SMAD3 is critical in osteoblast differentiation of PDMCs. Int. J. Mol. Med. 2019;43:1085–1093. doi: 10.3892/ijmm.2018.4001. [DOI] [PubMed] [Google Scholar]
- 166.Kang JS, Alliston T, Delston R, Derynck R. Repression of Runx2 function by TGF-beta through recruitment of class II histone deacetylases by Smad3. EMBO J. 2005;24:2543–2555. doi: 10.1038/sj.emboj.7600729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Hjelmeland AB, Schilling SH, Guo X, Quarles D, Wang XF. Loss of Smad3-mediated negative regulation of Runx2 activity leads to an alteration in cell fate determination. Mol. Cell. Biol. 2005;25:9460–9468. doi: 10.1128/MCB.25.21.9460-9468.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Lian N, et al. Transforming growth factor β suppresses osteoblast differentiation via the vimentin activating transcription factor 4 (ATF4) axis. J. Biol. Chem. 2012;287:35975–35984. doi: 10.1074/jbc.M112.372458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Ehnert S, et al. TGF-β(1) impairs mechanosensation of human osteoblasts via HDAC6-mediated shortening and distortion of primary cilia. J. Mol. Med. 2017;95:653–663. doi: 10.1007/s00109-017-1526-4. [DOI] [PubMed] [Google Scholar]
- 170.Nam B, Park H. TGFβ1 suppressed matrix mineralization of osteoblasts differentiation by regulating SMURF1-C/EBPβ-DKK1 axis. Int. J. Mol. Sci. 2020;21:9771. doi: 10.3390/ijms21249771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Ochiai H, et al. Inhibition of insulin-like growth factor-1 (IGF-1) expression by prolonged transforming growth factor-β1 (TGF-β1) administration suppresses osteoblast differentiation. J. Biol. Chem. 2012;287:22654–22661. doi: 10.1074/jbc.M111.279091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Qiu T, et al. TGF-beta type II receptor phosphorylates PTH receptor to integrate bone remodelling signalling. Nat. Cell Biol. 2010;12:224–234. doi: 10.1038/ncb2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Kwok S, Qin L, Partridge NC, Selvamurugan N. Parathyroid hormone stimulation and PKA signaling of latent transforming growth factor-beta binding protein-1 (LTBP-1) mRNA expression in osteoblastic cells. J. Cell. Biochem. 2005;95:1002–1011. doi: 10.1002/jcb.20453. [DOI] [PubMed] [Google Scholar]
- 174.Sowa H, et al. Parathyroid hormone-Smad3 axis exerts anti-apoptotic action and augments anabolic action of transforming growth factor beta in osteoblasts. J. Biol. Chem. 2003;278:52240–52252. doi: 10.1074/jbc.M302566200. [DOI] [PubMed] [Google Scholar]
- 175.Kamiya N, et al. Disruption of BMP signaling in osteoblasts through type IA receptor (BMPRIA) increases bone mass. J. Bone Miner. Res. 2008;23:2007–2017. doi: 10.1359/jbmr.080809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Kamiya N, et al. Targeted disruption of BMP signaling through type IA receptor (BMPR1A) in osteocyte suppresses SOST and RANKL, leading to dramatic increase in bone mass, bone mineral density and mechanical strength. Bone. 2016;91:53–63. doi: 10.1016/j.bone.2016.07.002. [DOI] [PubMed] [Google Scholar]
- 177.Tan X, et al. Smad4 is required for maintaining normal murine postnatal bone homeostasis. J. Cell Sci. 2007;120:2162–2170. doi: 10.1242/jcs.03466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Moon YJ, et al. Smad4 controls bone homeostasis through regulation of osteoblast/osteocyte viability. Exp. Mol. Med. 2016;48:e256. doi: 10.1038/emm.2016.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Wu H, et al. Inhibitory effects of combined bone morphogenetic protein 2, vascular endothelial growth factor, and basic fibroblast growth factor on osteoclast differentiation and activity. Tissue Eng. Part A. 2021;27:1387–1398. doi: 10.1089/ten.tea.2020.0325. [DOI] [PubMed] [Google Scholar]
- 180.Tasca A, et al. Smad1/5 and Smad4 expression are important for osteoclast differentiation. J. Cell. Biochem. 2015;116:1350–1360. doi: 10.1002/jcb.25092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Miao X, et al. Bone morphogenetic protein-2 promotes osteoclasts-mediated osteolysis via Smad1 and p65 signaling pathways. Spine. 2021;46:E234–E242. doi: 10.1097/BRS.0000000000003770. [DOI] [PubMed] [Google Scholar]
- 182.Omi M, Kaartinen V, Mishina Y. Activin A receptor type 1-mediated BMP signaling regulates RANKL-induced osteoclastogenesis via canonical SMAD-signaling pathway. J. Biol. Chem. 2019;294:17818–17836. doi: 10.1074/jbc.RA119.009521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Okamoto M, et al. Conditional deletion of Bmpr1a in differentiated osteoclasts increases osteoblastic bone formation, increasing volume of remodeling bone in mice. J. Bone Miner. Res. 2011;26:2511–2522. doi: 10.1002/jbmr.477. [DOI] [PubMed] [Google Scholar]
- 184.Crane JL, Cao X. Bone marrow mesenchymal stem cells and TGF-β signaling in bone remodeling. J. Clin. Invest. 2014;124:466–472. doi: 10.1172/JCI70050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Karsdal MA, et al. Transforming growth factor-beta controls human osteoclastogenesis through the p38 MAPK and regulation of RANK expression. J. Biol. Chem. 2003;278:44975–44987. doi: 10.1074/jbc.M303905200. [DOI] [PubMed] [Google Scholar]
- 186.Morita M, et al. Smad4 is required to inhibit osteoclastogenesis and maintain bone mass. Sci. Rep. 2016;6:35221. doi: 10.1038/srep35221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Pan W, et al. SIS3 suppresses osteoclastogenesis and ameliorates bone loss in ovariectomized mice by modulating Nox4-dependent reactive oxygen species. Biochem. Pharmacol. 2022;195:114846. doi: 10.1016/j.bcp.2021.114846. [DOI] [PubMed] [Google Scholar]
- 188.Houde N, Chamoux E, Bisson M, Roux S. Transforming growth factor-beta1 (TGF-beta1) induces human osteoclast apoptosis by up-regulating Bim. J. Biol. Chem. 2009;284:23397–23404. doi: 10.1074/jbc.M109.019372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Quinn JM, et al. Transforming growth factor beta affects osteoclast differentiation via direct and indirect actions. J. Bone Miner. Res. 2001;16:1787–1794. doi: 10.1359/jbmr.2001.16.10.1787. [DOI] [PubMed] [Google Scholar]
- 190.Dole NS, et al. Osteocyte-intrinsic TGF-β signaling regulates bone quality through perilacunar/canalicular remodeling. Cell Rep. 2017;21:2585–2596. doi: 10.1016/j.celrep.2017.10.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Dole NS, Yee CS, Mazur CM, Acevedo C, Alliston T. TGFβ regulation of perilacunar/canalicular remodeling is sexually dimorphic. J. Bone Miner. Res. 2020;35:1549–1561. doi: 10.1002/jbmr.4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Schurman CA, Verbruggen SW, Alliston T. Disrupted osteocyte connectivity and pericellular fluid flow in bone with aging and defective TGF-β signaling. Proc. Natl. Acad. Sci. USA. 2021;118:e2023999118. doi: 10.1073/pnas.2023999118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Liu W, et al. TGF-β1 facilitates cell-cell communication in osteocytes via connexin43- and pannexin1-dependent gap junctions. Cell Death Discov. 2019;5:141. doi: 10.1038/s41420-019-0221-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Jähn K, et al. Phenotype and viability of MLO-Y4 cells is maintained by TGFβ3 in a serum-dependent manner within a 3D-co-culture with MG-63 cells. Int. J. Mol. Sci. 2018;19:1932. doi: 10.3390/ijms19071932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Bailey KN, et al. Mechanosensitive control of articular cartilage and subchondral bone homeostasis in mice requires osteocytic transforming growth factor β signaling. Arthritis Rheumatol. 2021;73:414–425. doi: 10.1002/art.41548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Grol MW, Lee BH. Gene therapy for repair and regeneration of bone and cartilage. Curr. Opin. Pharmacol. 2018;40:59–66. doi: 10.1016/j.coph.2018.03.005. [DOI] [PubMed] [Google Scholar]
- 197.Patil AS, Sable RB, Kothari RM. An update on transforming growth factor-β (TGF-β): sources, types, functions and clinical applicability for cartilage/bone healing. J. Cell. Physiol. 2011;226:3094–3103. doi: 10.1002/jcp.22698. [DOI] [PubMed] [Google Scholar]
- 198.He Y, et al. Reduction of Smad2 caused by oxidative stress leads to necrotic death of hypertrophic chondrocytes associated with an endemic osteoarthritis. Rheumatology. 2021;61:440–451. doi: 10.1093/rheumatology/keab286. [DOI] [PubMed] [Google Scholar]
- 199.Li TF, et al. Smad3-deficient chondrocytes have enhanced BMP signaling and accelerated differentiation. J. Bone Miner. Res. 2006;21:4–16. doi: 10.1359/JBMR.050911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Motaung SCKM, Di Cesare PE, Hari Reddi A. Differential response of cartilage oligomeric matrix protein (COMP) to morphogens of bone morphogenetic protein/transforming growth factor-β family in the surface, middle and deep zones of articular cartilage. J. Tissue Eng. Regen. Med. 2011;5:e87–e96. doi: 10.1002/term.358. [DOI] [PubMed] [Google Scholar]
- 201.Niikura T, Reddi AH. Differential regulation of lubricin/superficial zone protein by transforming growth factor β/bone morphogenetic protein superfamily members in articular chondrocytes and synoviocytes. Arthritis Rheum. 2007;56:2312–2321. doi: 10.1002/art.22659. [DOI] [PubMed] [Google Scholar]
- 202.Malemud CJ, Killeen W, Hering TM, Purchio AF. Enhanced sulfated-proteoglycan core protein synthesis by incubation of rabbit chondrocytes with recombinant transforming growth factor-β1. J. Cell. Physiol. 1991;149:152–159. doi: 10.1002/jcp.1041490119. [DOI] [PubMed] [Google Scholar]
- 203.Takahashi N, et al. Elucidation of IL-1/TGF-β interactions in mouse chondrocyte cell line by genome-wide gene expression. Osteoarthritis Cartilage. 2005;13:426–438. doi: 10.1016/j.joca.2004.12.010. [DOI] [PubMed] [Google Scholar]
- 204.Wiegertjes R, et al. TGF-β dampens IL-6 signaling in articular chondrocytes by decreasing IL-6 receptor expression. Osteoarthritis Cartilage. 2019;27:1197–1207. doi: 10.1016/j.joca.2019.04.014. [DOI] [PubMed] [Google Scholar]
- 205.Cherian JJ, et al. Preliminary results of a phase II randomized study to determine the efficacy and safety of genetically engineered allogeneic human chondrocytes expressing TGF-β1 in patients with grade 3 chronic degenerative joint disease of the knee. Osteoarthritis Cartilage. 2015;23:2109–2118. doi: 10.1016/j.joca.2015.06.019. [DOI] [PubMed] [Google Scholar]
- 206.Ramaswamy G, Sohn P, Eberhardt A, Serra R. Altered responsiveness to TGF-β results in reduced Papss2 expression and alterations in the biomechanical properties of mouse articular cartilage. Arthritis Res. Ther. 2012;14:R49. doi: 10.1186/ar3762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.van Caam A, et al. TGFβ1-induced SMAD2/3 and SMAD1/5 phosphorylation are both ALK5-kinase-dependent in primary chondrocytes and mediated by TAK1 kinase activity. Arthritis Res. Ther. 2017;19:112. doi: 10.1186/s13075-017-1302-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Wang Q, Tan Q. Postnatal deletion of Alk5 gene in meniscal cartilage accelerates age-dependent meniscal degeneration in mice. J. Cell. Physiol. 2018;234:595–605. doi: 10.1002/jcp.26802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Shen J, et al. Deletion of the transforming growth factor β receptor type II gene in articular chondrocytes leads to a progressive osteoarthritis-like phenotype in mice. Arthritis Rheum. 2013;65:3107–3119. doi: 10.1002/art.38122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Namdari S, Wei L, Moore D, Chen Q. Reduced limb length and worsened osteoarthritis in adult mice after genetic inhibition of p38 MAP kinase activity in cartilage. Arthritis Rheum. 2008;58:3520–3529. doi: 10.1002/art.23999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Frazier K, et al. Inhibition of ALK5 signaling induces physeal dysplasia in rats. Toxicol. Pathol. 2007;35:284–295. doi: 10.1080/01926230701198469. [DOI] [PubMed] [Google Scholar]
- 212.Prasadam I, et al. Inhibition of p38 pathway leads to OA-like changes in a rat animal model. Rheumatology. 2012;51:813–823. doi: 10.1093/rheumatology/ker360. [DOI] [PubMed] [Google Scholar]
- 213.Wang C, Shen J, Ying J, Xiao D, O’Keefe RJ. FoxO1 is a crucial mediator of TGF-β/TAK1 signaling and protects against osteoarthritis by maintaining articular cartilage homeostasis. Proc. Natl. Acad. Sci. USA. 2020;117:30488–30497. doi: 10.1073/pnas.2017056117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Mori H, Izawa T, Tanaka E. Smad3 deficiency leads to mandibular condyle degradation via the sphingosine 1-phosphate (S1P)/S1P3 signaling axis. Am. J. Pathol. 2015;185:2742–2756. doi: 10.1016/j.ajpath.2015.06.015. [DOI] [PubMed] [Google Scholar]
- 215.Wang H, Zhang J, Sun Q, Yang X. Altered gene expression in articular chondrocytes of Smad3(ex8/ex8) mice, revealed by gene profiling using microarrays. J. Genet. Genomics. 2007;34:698–708. doi: 10.1016/S1673-8527(07)60079-4. [DOI] [PubMed] [Google Scholar]
- 216.Zhang Y, et al. Runx1 is a key regulator of articular cartilage homeostasis by orchestrating YAP, TGFβ, and Wnt signaling in articular cartilage formation and osteoarthritis. Bone Res. 2022;10:63. doi: 10.1038/s41413-022-00231-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Ueland T, et al. Increased serum and bone matrix levels of transforming growth factor {beta}1 in patients with GH deficiency in response to GH treatment. Eur. J. Endocrinol. 2011;165:393–400. doi: 10.1530/EJE-11-0442. [DOI] [PubMed] [Google Scholar]
- 218.Pombo-Suarez M, Castaño-Oreja MT, Calaza M, Gomez-Reino J, Gonzalez A. Differential upregulation of the three transforming growth factor beta isoforms in human osteoarthritic cartilage. Ann. Rheum. Dis. 2009;68:568–571. doi: 10.1136/ard.2008.090217. [DOI] [PubMed] [Google Scholar]
- 219.Blaney Davidson EN, Vitters EL, van der Kraan PM, van den Berg WB. Expression of transforming growth factor-beta (TGFbeta) and the TGFbeta signalling molecule SMAD-2P in spontaneous and instability-induced osteoarthritis: role in cartilage degradation, chondrogenesis and osteophyte formation. Ann. Rheum. Dis. 2006;65:1414–1421. doi: 10.1136/ard.2005.045971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.François RJ, Neure L, Sieper J, Braun J. Immunohistological examination of open sacroiliac biopsies of patients with ankylosing spondylitis: detection of tumour necrosis factor alpha in two patients with early disease and transforming growth factor beta in three more advanced cases. Ann. Rheum. Dis. 2006;65:713–720. doi: 10.1136/ard.2005.037465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Zhen G, et al. Inhibition of TGF-β signaling in mesenchymal stem cells of subchondral bone attenuates osteoarthritis. Nat. Med. 2013;19:704–712. doi: 10.1038/nm.3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Zheng L, et al. Aberrant activation of latent transforming growth factor-β initiates the onset of temporomandibular joint osteoarthritis. Bone Res. 2018;6:26. doi: 10.1038/s41413-018-0027-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Blaney Davidson EN, et al. Increase in ALK1/ALK5 ratio as a cause for elevated MMP-13 expression in osteoarthritis in humans and mice. J. Immunol. 2009;182:7937–7945. doi: 10.4049/jimmunol.0803991. [DOI] [PubMed] [Google Scholar]
- 224.Occhetta P, et al. Developmentally inspired programming of adult human mesenchymal stromal cells toward stable chondrogenesis. Proc. Natl. Acad. Sci. USA. 2018;115:4625–4630. doi: 10.1073/pnas.1720658115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Janssens K, ten Dijke P, Janssens S, Van Hul W. Transforming growth factor-β1 to the bone. Endocr. Rev. 2005;26:743–774. doi: 10.1210/er.2004-0001. [DOI] [PubMed] [Google Scholar]
- 226.Dabovic B, et al. Osteopetrosis-like phenotype in latent TGF-beta binding protein 3 deficient mice. Bone. 2005;37:25–31. doi: 10.1016/j.bone.2005.02.021. [DOI] [PubMed] [Google Scholar]
- 227.Dabovic B, et al. Bone defects in latent TGF-beta binding protein (Ltbp)-3 null mice; a role for Ltbp in TGF-beta presentation. J. Endocrinol. 2002;175:129–141. doi: 10.1677/joe.0.1750129. [DOI] [PubMed] [Google Scholar]
- 228.Koli K, Ryynänen MJ, Keski-Oja J. Latent TGF-beta binding proteins (LTBPs)-1 and -3 coordinate proliferation and osteogenic differentiation of human mesenchymal stem cells. Bone. 2008;43:679–688. doi: 10.1016/j.bone.2008.06.016. [DOI] [PubMed] [Google Scholar]
- 229.Kitahama S, et al. Expression of fibrillins and other microfibril-associated proteins in human bone and osteoblast-like cells. Bone. 2000;27:61–67. doi: 10.1016/S8756-3282(00)00292-1. [DOI] [PubMed] [Google Scholar]
- 230.Putnam EA, Zhang H, Ramirez F, Milewicz DM. Fibrillin-2 (FBN2) mutations result in the Marfan-like disorder, congenital contractural arachnodactyly. Nat. Genet. 1995;11:456–458. doi: 10.1038/ng1295-456. [DOI] [PubMed] [Google Scholar]
- 231.Nistala H, et al. Fibrillin-1 and -2 differentially modulate endogenous TGF-β and BMP bioavailability during bone formation. J. Cell Biol. 2010;190:1107–1121. doi: 10.1083/jcb.201003089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Nistala H, Lee-Arteaga S, Smaldone S, Siciliano G, Ramirez F. Extracellular microfibrils control osteoblast-supported osteoclastogenesis by restricting TGF{beta} stimulation of RANKL production. J. Biol. Chem. 2010;285:34126–34133. doi: 10.1074/jbc.M110.125328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Craft CS, et al. Oophorectomy-induced bone loss is attenuated in MAGP1-deficient mice. J. Cell. Biochem. 2012;113:93–99. doi: 10.1002/jcb.23331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Craft CS, et al. Microfibril-associated glycoprotein-1, an extracellular matrix regulator of bone remodeling. J. Biol. Chem. 2010;285:23858–23867. doi: 10.1074/jbc.M110.113019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Marini JC, et al. Osteogenesis imperfecta. Nat. Rev. Dis. Primers. 2017;3:17052. doi: 10.1038/nrdp.2017.52. [DOI] [PubMed] [Google Scholar]
- 236.Zieba J, et al. Fracture healing in collagen-related preclinical models of osteogenesis imperfecta. J. Bone Miner. Res. 2020;35:1132–1148. doi: 10.1002/jbmr.3979. [DOI] [PubMed] [Google Scholar]
- 237.Grafe I, et al. Excessive transforming growth factor-β signaling is a common mechanism in osteogenesis imperfecta. Nat. Med. 2014;20:670–675. doi: 10.1038/nm.3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Pacifici M. The pathogenic roles of heparan sulfate deficiency in hereditary multiple exostoses. Matrix Biol. 2018;71-72:28–39. doi: 10.1016/j.matbio.2017.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Kawashima K, et al. Heparan sulfate deficiency leads to hypertrophic chondrocytes by increasing bone morphogenetic protein signaling. Osteoarthritis Cartilage. 2020;28:1459–1470. doi: 10.1016/j.joca.2020.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Inubushi T, Nozawa S, Matsumoto K, Irie F, Yamaguchi Y. Aberrant perichondrial BMP signaling mediates multiple osteochondromagenesis in mice. JCI Insight. 2017;2:e90049. doi: 10.1172/jci.insight.90049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Inubushi T, Lemire I, Irie F, Yamaguchi Y. Palovarotene inhibits osteochondroma formation in a mouse model of multiple hereditary exostoses. J. Bone Miner. Res. 2018;33:658–666. doi: 10.1002/jbmr.3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Tang X, et al. Connective tissue growth factor contributes to joint homeostasis and osteoarthritis severity by controlling the matrix sequestration and activation of latent TGFβ. Ann. Rheum. Dis. 2018;77:1372–1380. doi: 10.1136/annrheumdis-2018-212964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Le Goff C, et al. ADAMTSL2 mutations in geleophysic dysplasia demonstrate a role for ADAMTS-like proteins in TGF-beta bioavailability regulation. Nat. Genet. 2008;40:1119–1123. doi: 10.1038/ng.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Delhon L, et al. Impairment of chondrogenesis and microfibrillar network in Adamtsl2 deficiency. FASEB J. 2019;33:2707–2718. doi: 10.1096/fj.201800753RR. [DOI] [PubMed] [Google Scholar]
- 245.Morales J, et al. Homozygous mutations in ADAMTS10 and ADAMTS17 cause lenticular myopia, ectopia lentis, glaucoma, spherophakia, and short stature. Am. J. Hum. Genet. 2009;85:558–568. doi: 10.1016/j.ajhg.2009.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Oichi T, et al. Adamts17 is involved in skeletogenesis through modulation of BMP-Smad1/5/8 pathway. Cell Mol. Life Sci. 2019;76:4795–4809. doi: 10.1007/s00018-019-03188-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Groppe J, et al. Structural basis of BMP signaling inhibition by Noggin, a novel twelve-membered cystine knot protein. J. Bone Joint Surg. Am. 2003;85-A:52–58. doi: 10.2106/00004623-200300003-00010. [DOI] [PubMed] [Google Scholar]
- 248.Pregizer SK, Mortlock DP. Dynamics and cellular localization of Bmp2, Bmp4, and Noggin transcription in the postnatal mouse skeleton. J. Bone Miner. Res. 2015;30:64–70. doi: 10.1002/jbmr.2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Yoshimura Y, et al. Colocalization of noggin and bone morphogenetic protein-4 during fracture healing. J. Bone Miner. Res. 2001;16:876–884. doi: 10.1359/jbmr.2001.16.5.876. [DOI] [PubMed] [Google Scholar]
- 250.Wu XB, et al. Impaired osteoblastic differentiation, reduced bone formation, and severe osteoporosis in noggin-overexpressing mice. J. Clin. Invest. 2003;112:924–934. doi: 10.1172/JCI15543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Iwata T, et al. Noggin blocks osteoinductive activity of porcine enamel extracts. J. Dent. Res. 2002;81:387–391. doi: 10.1177/0810387. [DOI] [PubMed] [Google Scholar]
- 252.Wan DC, et al. Noggin suppression enhances in vitro osteogenesis and accelerates in vivo bone formation. J. Biol. Chem. 2007;282:26450–26459. doi: 10.1074/jbc.M703282200. [DOI] [PubMed] [Google Scholar]
- 253.Devlin RD, et al. Skeletal overexpression of noggin results in osteopenia and reduced bone formation. Endocrinology. 2003;144:1972–1978. doi: 10.1210/en.2002-220918. [DOI] [PubMed] [Google Scholar]
- 254.Canalis E, Brunet LJ, Parker K, Zanotti S. Conditional inactivation of noggin in the postnatal skeleton causes osteopenia. Endocrinology. 2012;153:1616–1626. doi: 10.1210/en.2011-1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Xie Z, et al. Imbalance between bone morphogenetic protein 2 and noggin induces abnormal osteogenic differentiation of mesenchymal stem cells in ankylosing spondylitis. Arthritis Rheumatol. 2016;68:430–440. doi: 10.1002/art.39433. [DOI] [PubMed] [Google Scholar]
- 256.Warren SM, Brunet LJ, Harland RM, Economides AN, Longaker MT. The BMP antagonist noggin regulates cranial suture fusion. Nature. 2003;422:625–629. doi: 10.1038/nature01545. [DOI] [PubMed] [Google Scholar]
- 257.Nolan K, et al. Structure of Gremlin-2 in complex with GDF5 gives insight into DAN-family-mediated BMP antagonism. Cell Rep. 2016;16:2077–2086. doi: 10.1016/j.celrep.2016.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Gazzerro E, et al. Conditional deletion of gremlin causes a transient increase in bone formation and bone mass. J. Biol. Chem. 2007;282:31549–31557. doi: 10.1074/jbc.M701317200. [DOI] [PubMed] [Google Scholar]
- 259.Gazzerro E, et al. Skeletal overexpression of gremlin impairs bone formation and causes osteopenia. Endocrinology. 2005;146:655–665. doi: 10.1210/en.2004-0766. [DOI] [PubMed] [Google Scholar]
- 260.Worthley DL, et al. Gremlin 1 identifies a skeletal stem cell with bone, cartilage, and reticular stromal potential. Cell. 2015;160:269–284. doi: 10.1016/j.cell.2014.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Eijken M, et al. The activin A-follistatin system: potent regulator of human extracellular matrix mineralization. FASEB J. 2007;21:2949–2960. doi: 10.1096/fj.07-8080com. [DOI] [PubMed] [Google Scholar]
- 262.Suh J, et al. GDF11 promotes osteogenesis as opposed to MSTN, and follistatin, a MSTN/GDF11 inhibitor, increases muscle mass but weakens bone. Proc. Natl. Acad. Sci. USA. 2020;117:4910–4920. doi: 10.1073/pnas.1916034117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Abe Y, Abe T, Aida Y, Hara Y, Maeda K. Follistatin restricts bone morphogenetic protein (BMP)-2 action on the differentiation of osteoblasts in fetal rat mandibular cells. Proc. Natl. Acad. Sci. USA. 2004;19:1302–1307. doi: 10.1359/JBMR.040408. [DOI] [PubMed] [Google Scholar]
- 264.Zhang D, et al. A role for the BMP antagonist chordin in endochondral ossification. J. Bone Miner. Res. 2002;17:293–300. doi: 10.1359/jbmr.2002.17.2.293. [DOI] [PubMed] [Google Scholar]
- 265.Petryk A, et al. Twisted gastrulation and chordin inhibit differentiation and mineralization in MC3T3-E1 osteoblast-like cells. Bone. 2005;36:617–626. doi: 10.1016/j.bone.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 266.Cook LM, et al. Betaglycan drives the mesenchymal stromal cell osteogenic program and prostate cancer-induced osteogenesis. Oncogene. 2019;38:6959–6969. doi: 10.1038/s41388-019-0913-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Hill CR, Jacobs BH, Brown CB, Barnett JV, Goudy SL. Type III transforming growth factor beta receptor regulates vascular and osteoblast development during palatogenesis. Dev. Dyn. 2015;244:122–133. doi: 10.1002/dvdy.24225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Verlinden L, et al. Nrp2 deficiency leads to trabecular bone loss and is accompanied by enhanced osteoclast and reduced osteoblast numbers. Bone. 2013;55:465–475. doi: 10.1016/j.bone.2013.03.023. [DOI] [PubMed] [Google Scholar]
- 269.Ishibashi O, et al. Endoglin is involved in BMP-2-induced osteogenic differentiation of periodontal ligament cells through a pathway independent of Smad-1/5/8 phosphorylation. J. Cell. Physiol. 2010;222:465–473. doi: 10.1002/jcp.21968. [DOI] [PubMed] [Google Scholar]
- 270.Lawera A, et al. Role of soluble endoglin in BMP9 signaling. Proc. Natl. Acad. Sci. USA. 2019;116:17800–17808. doi: 10.1073/pnas.1816661116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Finnson KW, et al. Endoglin differentially regulates TGF-β-induced Smad2/3 and Smad1/5 signalling and its expression correlates with extracellular matrix production and cellular differentiation state in human chondrocytes. Osteoarthritis Cartilage. 2010;18:1518–1527. doi: 10.1016/j.joca.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 272.Parker WL, Goldring MB, Philip A. Endoglin is expressed on human chondrocytes and forms a heteromeric complex with betaglycan in a ligand and type II TGFbeta receptor independent manner. J. Bone Miner. Res. 2003;18:289–302. doi: 10.1359/jbmr.2003.18.2.289. [DOI] [PubMed] [Google Scholar]
- 273.Alzahrani A, et al. Endoglin haploinsufficiency is associated with differential regulation of extracellular matrix production during skin fibrosis and cartilage repair in mice. J. Cell Commun. Signal. 2018;12:379–388. doi: 10.1007/s12079-018-0461-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Bianchi VJ, Parsons M, Backstein D, Kandel RA. Endoglin level is critical for cartilage tissue formation in vitro by passaged human chondrocytes. Tissue Eng. Part A. 2021;27:1140–1150. doi: 10.1089/ten.tea.2020.0120. [DOI] [PubMed] [Google Scholar]
- 275.Hagihara M, et al. Neogenin, a receptor for bone morphogenetic proteins. J. Biol. Chem. 2011;286:5157–5165. doi: 10.1074/jbc.M110.180919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Zhou Z, et al. Neogenin regulation of BMP-induced canonical Smad signaling and endochondral bone formation. Dev. Cell. 2010;19:90–102. doi: 10.1016/j.devcel.2010.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Miyazawa K, Miyazono K. Regulation of TGF-β family signaling by inhibitory Smads. Cold Spring Harb. Perspect. Biol. 2017;9:a022095. doi: 10.1101/cshperspect.a022095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Timberlake AT, et al. Two locus inheritance of non-syndromic midline craniosynostosis via rare SMAD6 and common BMP2 alleles. Elife. 2016;5:e20125. doi: 10.7554/eLife.20125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Horiki M, et al. Smad6/Smurf1 overexpression in cartilage delays chondrocyte hypertrophy and causes dwarfism with osteopenia. J. Cell Biol. 2004;165:433–445. doi: 10.1083/jcb.200311015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Estrada KD, Retting KN, Chin AM, Lyons KM. Smad6 is essential to limit BMP signaling during cartilage development. J. Bone Miner. Res. 2011;26:2498–2510. doi: 10.1002/jbmr.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Shen R, et al. Smad6 interacts with Runx2 and mediates Smad ubiquitin regulatory factor 1-induced Runx2 degradation. J. Biol. Chem. 2006;281:3569–3576. doi: 10.1074/jbc.M506761200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Li N, et al. Partial loss of Smad7 function impairs bone remodeling, osteogenesis and enhances osteoclastogenesis in mice. Bone. 2014;67:46–55. doi: 10.1016/j.bone.2014.06.033. [DOI] [PubMed] [Google Scholar]
- 283.Iwai T, Murai J, Yoshikawa H, Tsumaki N. Smad7 inhibits chondrocyte differentiation at multiple steps during endochondral bone formation and down-regulates p38 MAPK pathways. J. Biol. Chem. 2008;283:27154–27164. doi: 10.1074/jbc.M801175200. [DOI] [PubMed] [Google Scholar]
- 284.Zhao M, Qiao M, Oyajobi BO, Mundy GR, Chen D. E3 ubiquitin ligase Smurf1 mediates core-binding factor alpha1/Runx2 degradation and plays a specific role in osteoblast differentiation. J. Biol. Chem. 2003;278:27939–27944. doi: 10.1074/jbc.M304132200. [DOI] [PubMed] [Google Scholar]
- 285.Zhao M, et al. Smurf1 inhibits osteoblast differentiation and bone formation in vitro and in vivo. Int. J. Mol. Sci. 2004;279:12854–12859. doi: 10.1074/jbc.M313294200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Yamashita M, et al. Ubiquitin ligase Smurf1 controls osteoblast activity and bone homeostasis by targeting MEKK2 for degradation. Cell. 2005;121:101–113. doi: 10.1016/j.cell.2005.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Sapkota G, et al. signaling through integrated inputs into the Smad1 linker. Mol. Cell. 2007;25:441–454. doi: 10.1016/j.molcel.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 288.Liang C, et al. Inhibition of osteoblastic Smurf1 promotes bone formation in mouse models of distinctive age-related osteoporosis. Nat. Commun. 2018;9:3428. doi: 10.1038/s41467-018-05974-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Liu J, et al. Increased PLEKHO1 within osteoblasts suppresses Smad-dependent BMP signaling to inhibit bone formation during aging. Aging Cell. 2017;16:360–376. doi: 10.1111/acel.12566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Wu Q, et al. Regulation of embryonic endochondral ossification by Smurf2. J. Orthop. Res. 2008;26:704–712. doi: 10.1002/jor.20563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Wu Q, et al. Induction of an osteoarthritis-like phenotype and degradation of phosphorylated Smad3 by Smurf2 in transgenic mice. Arthritis Rheum. 2008;58:3132–3144. doi: 10.1002/art.23946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Wu Q, Huang JH. Ectopic expression of Smurf2 and acceleration of age-related intervertebral disc degeneration in a mouse model. J. Neurosurg. Spine. 2017;27:116–126. doi: 10.3171/2016.11.SPINE16901. [DOI] [PubMed] [Google Scholar]
- 293.Huang H, Veien ES, Zhang H, Ayers DC, Song J. Skeletal characterization of Smurf2-deficient mice and in vitro analysis of Smurf2-deficient chondrocytes. PLoS One. 2016;11:e0148088. doi: 10.1371/journal.pone.0148088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Kushioka J, Kaito T. A novel negative regulatory mechanism of Smurf2 in BMP/Smad signaling in bone. Bone Res. 2020;8:41. doi: 10.1038/s41413-020-00115-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Tang LY, et al. Ablation of Smurf2 reveals an inhibition in TGF-β signalling through multiple mono-ubiquitination of Smad3. EMBO J. 2011;30:4777–4789. doi: 10.1038/emboj.2011.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Xu Z, et al. SMURF2 regulates bone homeostasis by disrupting SMAD3 interaction with vitamin D receptor in osteoblasts. Nat. Commun. 2017;8:14570. doi: 10.1038/ncomms14570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Kim BG, Lee JH, Yasuda J, Ryoo HM, Cho JY. Phospho-Smad1 modulation by nedd4 E3 ligase in BMP/TGF-β signaling. J. Bone Miner. Res. 2011;26:1411–1424. doi: 10.1002/jbmr.348. [DOI] [PubMed] [Google Scholar]
- 298.Jeon SA, Lee JH, Kim DW, Cho JY. E3-ubiquitin ligase NEDD4 enhances bone formation by removing TGFβ1-induced pSMAD1 in immature osteoblast. Bone. 2018;116:248–258. doi: 10.1016/j.bone.2018.08.012. [DOI] [PubMed] [Google Scholar]
- 299.Herhaus L, et al. USP15 targets ALK3/BMPR1A for deubiquitylation to enhance bone morphogenetic protein signalling. Open Biol. 2014;4:140065. doi: 10.1098/rsob.140065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Wang W, Zhu Y, Sun Z, Jin C, Wang X. Positive feedback regulation between USP15 and ERK2 inhibits osteoarthritis progression through TGF-β/SMAD2 signaling. Arthritis Res. Ther. 2021;23:84. doi: 10.1186/s13075-021-02456-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Sangadala S, et al. Characterization of a unique motif in LIM mineralization protein-1 that interacts with jun activation-domain-binding protein 1. Mol. Cell. Biochem. 2014;385:145–157. doi: 10.1007/s11010-013-1823-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Li H, et al. VCP/p97 increases BMP signaling by accelerating ubiquitin ligase Smurf1 degradation. FASEB J. 2019;33:2928–2943. doi: 10.1096/fj.201801173R. [DOI] [PubMed] [Google Scholar]
- 303.Samsa WE, et al. The master developmental regulator Jab1/Cops5/Csn5 is essential for proper bone growth and survival in mice. Bone. 2021;143:115733. doi: 10.1016/j.bone.2020.115733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Zhang F, et al. Sustained BMP signaling in osteoblasts stimulates bone formation by promoting angiogenesis and osteoblast differentiation. J. Bone Miner. Res. 2009;24:1224–1233. doi: 10.1359/jbmr.090204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Durbano HW, et al. Aberrant BMP2 signaling in patients diagnosed with osteoporosis. Int. J. Mol. Sci. 2020;21:6909. doi: 10.3390/ijms21186909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Mumm S, et al. Deactivating germline mutations in LEMD3 cause osteopoikilosis and Buschke-Ollendorff Syndrome, but not sporadic melorheostosis. J. Bone Miner. Res. 2007;22:243–250. doi: 10.1359/jbmr.061102. [DOI] [PubMed] [Google Scholar]
- 307.Hellemans J, et al. Loss-of-function mutations in LEMD3 result in osteopoikilosis, Buschke-Ollendorff syndrome and melorheostosis. Nat. Genet. 2004;36:1213–1218. doi: 10.1038/ng1453. [DOI] [PubMed] [Google Scholar]
- 308.Guo L, et al. Deficiency of TMEM53 causes a previously unknown sclerosing bone disorder by dysregulation of BMP-SMAD signaling. Nat. Commun. 2021;12:2046. doi: 10.1038/s41467-021-22340-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Mullin BH, et al. Genome-wide association study meta-analysis for quantitative ultrasound parameters of bone identifies five novel loci for broadband ultrasound attenuation. Hum. Mol. Genet. 2017;26:2791–2802. doi: 10.1093/hmg/ddx174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Moayyeri A, et al. Genetic determinants of heel bone properties: genome-wide association meta-analysis and replication in the GEFOS/GENOMOS consortium. Hum. Mol. Genet. 2014;23:3054–3068. doi: 10.1093/hmg/ddt675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Luo K. Negative regulation of BMP signaling by the ski oncoprotein. J. Bone Joint Surg. Am. 2003;85-A:39–43. doi: 10.2106/00004623-200300003-00008. [DOI] [PubMed] [Google Scholar]
- 312.Kim KO, et al. Ski inhibits TGF-β/phospho-Smad3 signaling and accelerates hypertrophic differentiation in chondrocytes. J. Cell. Biochem. 2012;113:2156–2166. doi: 10.1002/jcb.24089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Doyle AJ, et al. Mutations in the TGF-β repressor SKI cause Shprintzen-Goldberg syndrome with aortic aneurysm. Nat. Genet. 2012;44:1249–1254. doi: 10.1038/ng.2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Kawamura I, et al. SnoN suppresses maturation of chondrocytes by mediating signal cross-talk between transforming growth factor-β and bone morphogenetic protein pathways. J. Biol. Chem. 2012;287:29101–29113. doi: 10.1074/jbc.M112.349415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Ehnert S, et al. Transforming growth factor β1 inhibits bone morphogenic protein (BMP)-2 and BMP-7 signaling via upregulation of Ski-related novel protein N (SnoN): possible mechanism for the failure of BMP therapy? BMC Med. 2012;10:101. doi: 10.1186/1741-7015-10-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Kim DW, Lassar AB. Smad-dependent recruitment of a histone deacetylase/Sin3A complex modulates the bone morphogenetic protein-dependent transcriptional repressor activity of Nkx3.2. Mol. Cell. Biol. 2003;23:8704–8717. doi: 10.1128/MCB.23.23.8704-8717.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Yoshida Y, et al. Negative regulation of BMP/Smad signaling by Tob in osteoblasts. Cell. 2000;103:1085–1097. doi: 10.1016/S0092-8674(00)00211-7. [DOI] [PubMed] [Google Scholar]
- 318.Caddy JC, Luoma LM, Berry FB. FOXC1 negatively regulates BMP-SMAD activity and Id1 expression during osteoblast differentiation. J. Cell. Biochem. 2020;121:3266–3277. doi: 10.1002/jcb.29595. [DOI] [PubMed] [Google Scholar]
- 319.Leboy P, et al. Smad-Runx interactions during chondrocyte maturation. J. Bone Joint Surg. Am. 2001;83-A:S15–S22. [PubMed] [Google Scholar]
- 320.Javed A, et al. Structural coupling of Smad and Runx2 for execution of the BMP2 osteogenic signal. J. Biol. Chem. 2008;283:8412–8422. doi: 10.1074/jbc.M705578200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Furumatsu T, Tsuda M, Taniguchi N, Tajima Y, Asahara H. Smad3 induces chondrogenesis through the activation of SOX9 via CREB-binding protein/p300 recruitment. J. Biol. Chem. 2005;280:8343–8350. doi: 10.1074/jbc.M413913200. [DOI] [PubMed] [Google Scholar]
- 322.Lee HL, Yu B, Deng P, Wang CY, Hong C. Transforming growth factor-β-induced KDM4B promotes chondrogenic differentiation of human mesenchymal stem cells. Stem Cells. 2016;34:711–719. doi: 10.1002/stem.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Su CJ, et al. Ligand-receptor promiscuity enables cellular addressing. Cell Syst. 2022;13:408–425.e12. doi: 10.1016/j.cels.2022.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Klumpe HE, et al. The context-dependent, combinatorial logic of BMP signaling. Cell Syst. 2022;13:388–407.e10. doi: 10.1016/j.cels.2022.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Antebi YE, et al. Combinatorial signal perception in the BMP pathway. Cell. 2017;170:1184–1196.e24. doi: 10.1016/j.cell.2017.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Luo G, et al. BMP-7 is an inducer of nephrogenesis, and is also required for eye development and skeletal patterning. Genes Dev. 1995;9:2808–2820. doi: 10.1101/gad.9.22.2808. [DOI] [PubMed] [Google Scholar]
- 327.Storm EE, et al. Limb alterations in brachypodism mice due to mutations in a new member of the TGF beta-superfamily. Nature. 1994;368:639–643. doi: 10.1038/368639a0. [DOI] [PubMed] [Google Scholar]
- 328.Coleman CM, Scheremeta BH, Boyce AT, Mauck RL, Tuan RS. Delayed fracture healing in growth differentiation factor 5-deficient mice: a pilot study. Clin. Orthop. Relat. Res. 2011;469:2915–2924. doi: 10.1007/s11999-011-1912-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Daans M, Luyten FP, Lories RJ. GDF5 deficiency in mice is associated with instability-driven joint damage, gait and subchondral bone changes. Ann. Rheum. Dis. 2011;70:208–213. doi: 10.1136/ard.2010.134619. [DOI] [PubMed] [Google Scholar]
- 330.Rountree RB, et al. BMP receptor signaling is required for postnatal maintenance of articular cartilage. PLoS Biol. 2004;2:e355. doi: 10.1371/journal.pbio.0020355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Shi C, et al. Deletion of BMP receptor type IB decreased bone mass in association with compromised osteoblastic differentiation of bone marrow mesenchymal progenitors. Sci. Rep. 2016;6:24256. doi: 10.1038/srep24256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Zhao M, et al. Bone morphogenetic protein receptor signaling is necessary for normal murine postnatal bone formation. J. Cell Biol. 2002;157:1049–1060. doi: 10.1083/jcb.200109012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Wang Q, et al. Cartilage-specific deletion of Alk5 gene results in a progressive osteoarthritis-like phenotype in mice. Osteoarthritis Cartilage. 2017;25:1868–1879. doi: 10.1016/j.joca.2017.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Abou-Ezzi G, et al. TGF-β signaling plays an essential role in the lineage specification of mesenchymal stem/progenitor cells in fetal bone marrow. Stem Cell Rep. 2019;13:48–60. doi: 10.1016/j.stemcr.2019.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Lowery JW, et al. Loss of BMPR2 leads to high bone mass due to increased osteoblast activity. J. Cell Sci. 2015;128:1308–1315. doi: 10.1242/jcs.156737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Goh BC, et al. Activin receptor type 2A (ACVR2A) functions directly in osteoblasts as a negative regulator of bone mass. J. Biol. Chem. 2017;292:13809–13822. doi: 10.1074/jbc.M117.782128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Wang M, et al. Smad1 plays an essential role in bone development and postnatal bone formation. Osteoarthritis Cartilage. 2011;19:751–762. doi: 10.1016/j.joca.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Gunnell LM, et al. TAK1 regulates cartilage and joint development via the MAPK and BMP signaling pathways. J. Bone Miner. Res. 2010;25:1784–1797. doi: 10.1002/jbmr.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Estrada KD, et al. Smad7 regulates terminal maturation of chondrocytes in the growth plate. Dev. Biol. 2013;382:375–384. doi: 10.1016/j.ydbio.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]