

Abstract
LYS006 is a novel, highly potent and selective, new‐generation leukotriene A4 hydrolase (LTA4H) inhibitor in clinical development for the treatment of neutrophil‐driven inflammatory diseases. We describe the complex pharmacokinetic to pharmacodynamic (PD) relationship in blood, plasma, and skin of LYS006‐treated nonclinical species and healthy human participants. In a randomized first in human study, participants were exposed to single ascending doses up to 100 mg and multiple ascending doses up to 80 mg b.i.d.. LYS006 showed rapid absorption, overall dose proportional plasma exposure and nonlinear blood to plasma distribution caused by saturable target binding. The compound efficiently inhibited LTB4 production in human blood and skin blister cells, leading to greater than 90% predose target inhibition from day 1 after treatment initiation at doses of 20 mg b.i.d. and above. Slow re‐distribution from target expressing cells resulted in a long terminal half‐life and a long‐lasting PD effect in ex vivo stimulated blood and skin cells despite low plasma exposures. LYS006 was well‐tolerated and demonstrated a favorable safety profile up to highest doses tested, without any dose‐limiting toxicity. This supported further clinical development in phase II studies in predominantly neutrophil‐driven inflammatory conditions, such as hidradenitis suppurativa, inflammatory acne, and ulcerative colitis.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Leukotriene A4 Hydrolase (LTA4H) inhibitors have been investigated by the pharmaceutical industry for the treatment of neutrophil‐driven inflammatory diseases. To date, no LTA4H inhibitors have reached the market, yet.
WHAT QUESTION DID THIS STUDY ADDRESS?
This study investigated the human pharmacokinetic (PK) to pharmacodynamic (PD) relationship and clinical safety of the potent and highly selective LTA4H inhibitor LYS006.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
This is the first disclosure of the PK/PD relationship and favorable clinical safety of the new generation LTA4H inhibitor LYS006, which demonstrates efficient and long‐lasting reduction of inflammatory leukotriene B4 production in blood and skin blister cells (PD) at low plasma exposures of compound (PK). Results of this first‐in‐human (FIH) study supported the further clinical development of LYS006 in phase II clinical trials.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
LYS006 is a reversible, competitive inhibitor of LTA4H. Its PK/PD characteristics, however, are reminiscent of covalent inhibitors that demonstrate long‐lasting target engagement, despite short overall systemic residence time and plasma exposure caused by the saturable binding and slow‐redistribution of LYS006 from target‐expressing cells. The study highlights the importance of in‐depth plasma/blood/target tissue PK/PD assessments in FIH trials for phase II dose selection.
INTRODUCTION
The effective treatment of a large spectrum of acute and chronic inflammatory diseases remains a high unmet medical need. Thus, drugs modulating key inflammatory pathways that are not yet targeted by available drugs could offer novel treatment options. Leukotriene B4 (LTB4) is a pro‐inflammatory lipid mediator and an important chemoattractant for numerous immune cells. 1 , 2 It is among the most potent chemotactic agents for neutrophils and elevated levels in tissue can lead to activation and swarming of neutrophils and release of downstream pro‐inflammatory and chemotactic mediators. 3 , 4 , 5 LTB4 has been shown to initiate and perpetuate inflammation and its persistent elevation contributes to the pathology of numerous preclinical models of acute and chronic inflammation, autoimmunity, infection, allergy, fibrosis and metabolic disease. 6 To date, no drug that specifically interferes with the biological function of LTB4 has reached the market, apart from 5‐Lipoxygenase (5‐LO) inhibitor zileuton (Zyflo), which additionally blocks the biosynthesis of cysteinyl leukotrienes, and other 5‐LO dependent lipid mediators, such as lipoxins (LXA4) and resolvins. 7 As a first‐in‐class treatment, zileuton is an approved drug for asthma despite its low potency, need for four‐times daily dosing in its original form, and the fact that it elevates liver transaminases. 8 Leukotriene A4 Hydrolase (LTA4H) is the final and rate limiting enzyme in the biosynthesis of LTB4. 9 Several inhibitors of LTA4H, which selectively inhibit the enzyme and change the balance in pro‐inflammatory LTB4 and anti‐inflammatory LXA4, have reached clinical development and were tested in phase I studies. 10 Prior to LYS006, however, only two compounds were tested in phase II clinical trials in patients: JNJ‐40929837 in patients with asthma 11 and CTX4430 (acebilustat) in patients with cystic fibrosis. 12 Neither compound progressed to the market due to lack of sufficient efficacy in the chosen target indications.
LYS006 is a potent and highly selective novel inhibitor of LTA4H. Preclinical studies demonstrated its excellent cellular potency across species with a human whole blood 90% inhibitory concentration (IC90) of ~57 ng/mL and a pronounced pharmacodynamic (PD) effect in rodent PD models. It demonstrated high selectivity in comprehensive preclinical off‐target profiling, and a favorable tolerability and safety profile in toxicology studies in two preclinical species. 13 Its preclinical and clinical pharmacokinetic (PK) profile is characterized by target mediated drug disposition (TMDD) leading to nonlinearity at lower exposures and a concentration dependent blood to plasma distribution due to saturable binding to target expressing blood cells, such as erythrocytes, monocytes, and neutrophils. 13 , 14 , 15 Clinical PK demonstrated a time to maximum plasma concentration (T max) of 1–2 h, rapid, predominantly renal‐mediated elimination of compound from plasma and a long‐terminal half‐life due to slow re‐distribution of LYS006 from target expressing cells. 15
Here, we describe the detailed PK/PD relationship of LYS006 in preclinical species and its translation to a randomized first‐in‐human (FIH) clinical study. The primary objective of this study was to investigate the safety and tolerability of single and multiple ascending oral doses of LYS006. The secondary objective was to investigate the PK of LYS006 in plasma, blood, and skin as well as PD (e.g., LTB4 release) in ex vivo stimulated blood or cantharidin‐induced skin blister cells after single and multiple doses of LYS006.
METHODS
Preclinical PK/PD studies
All animal studies were conducted according to the Swiss federal law for animal protection and animal protocols were approved by the authority of the Basel‐Stadt Cantonal Veterinary Office, Switzerland.
Rat blood/plasma distribution study
For the rat blood/plasma distribution study, ~9‐week‐old male Wistar rats (n = 3) were dosed p.o. with 3 mg/kg [3H]‐labeled LYS006 by gavage. Rats had access to food on all study days. Blood was collected from the vena cava after 4, 48, 72, 96, and 168 h, blood aliquots were frozen, and plasma was generated via centrifugation and frozen for later analysis by liquid scintillation counting. Determination of LYS006 in rat blood and plasma was performed after protein precipitation by liquid chromatography tandem mass spectrometry (LC–MS/MS).
Dog PK/PD study
Fasted, ~2‐year‐old male beagle dogs (n = 3) per group were dosed p.o. with 0.5 mg/kg or 3 mg/kg LYS006 or vehicle control by oral gavage once daily for 5 consecutive days (at 0, 24, 48, 72, and 96 h). Blood was collected via the vena cephalica at the following timepoints: 0.5, 1, 3, 5, 8, and 24 h (at trough before the second dose), 48 h, 72 h (at trough before third and fourth doses), 96 h (at trough before fifth dose), 98, 101, 104, 120, 144, 168, and 192 h post‐first dose. An aliquot was frozen for LYS006 quantification in blood, another aliquot was centrifuged for separation of plasma. LYS006 content in blood and plasma samples was quantified by LC–MS/MS.
For the PD readout, heparinized blood was diluted 1:3 with RPMI medium and stimulated with 10 μg/mL ionophore A23187 for 15 min. Levels of induced LTB4 was determined by enzyme‐linked immunosorbent assay (Assay Design) according to the manufacturer's manual.
Clinical PK/PD studies
Study population
The study was conducted according to the ethical principles of the Declaration of Helsinki. The study protocol and all amendments were reviewed by the Independent Ethics Committee for the study center. Informed consent was obtained from each participant in writing before any assessment was performed.
A total of 121 healthy adult men and women between 18 and 45 years of age were recruited to the study. Study participants were in good health as determined by their medical history, physical examination, vital signs, electrocardiogram (ECG), and laboratory tests at screening and required to weigh at least 50 kg with a body mass index within the range of 18–30 kg/m2. Participants were excluded if they had used other investigational drugs at the time of enrollment, or within five half‐lives of enrollment, or within 30 days. They were also excluded if they had used other prescription drugs within 4 weeks prior to study, or shown a history of clinically significant ECG abnormalities, long QT syndrome, or arrythmias. Furthermore, women of childbearing potential, including pregnant or nursing women, were excluded from the study.
Study design
This was a single‐center, randomized, participant‐blinded and investigator‐blinded, placebo‐controlled FIH study in 121 healthy male and female adults to assess the safety, tolerability, PK, and PD properties of LYS006 in plasma, blood, and skin. The study consisted of three parts: part 1 (single ascending dose [SAD]); part 2 food effect (FE), and part 3 (multiple ascending dose [MAD]; Figure 1). For part 1 and part 3, the study participants were included in a screening period for up to 28 days before baseline, a baseline visit, a treatment period, and a follow‐up period. As part 2 was a crossover design, each participant had two baseline visits after screening, and two treatment periods. In the SAD and MAD part, a complete and blinded safety assessment of all exposed participants was conducted by the investigator and the sponsor, before escalating to the next higher dose level. Arrows in Figure 1 indicate the progression from cohort to cohort. After the fourth cohort in the SAD part, not only the fifth cohort in the SAD part was initiated but in parallel also the FE part and the first cohort in the MAD cohort were initiated.
FIGURE 1.
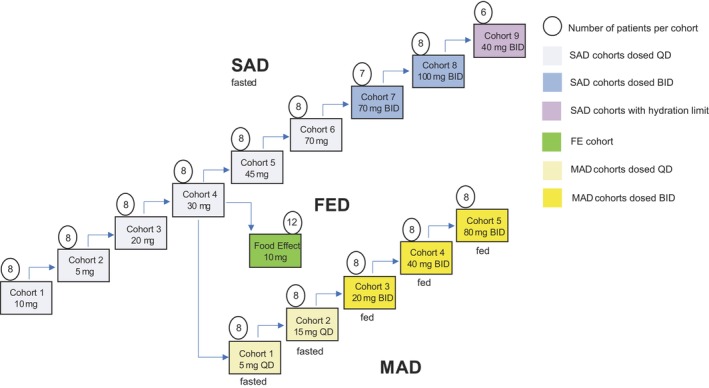
Study design of the first‐in‐human assessment of safety, PK and PD effects of LYS006. SAD part depicted in blue, FE arm depicted in green, and MAD part depicted in yellow. Numbers in circles indicate number of participants per cohort. Arrows indicate the sequence of the dosing in the different cohorts. After the fourth SAD cohort, the fifth SAD cohort, and, in addition, also the FE cohort and the first cohort in the MAD part were initiated. Cohorts with b.i.d. dosing received two equal doses per day separated by 12 h, in the SAD part for 1 day, and in the MAD part for 12 consecutive days. FE, food effect; MAD, multiple ascending dose; PD, pharmacodynamic; PK, pharmacokinetic; SAD, single ascending dose.
Sixty‐nine healthy participants were randomized to the nine cohorts of the SAD part, each cohort consisting of eight participants (6:2 ratio of active:placebo), except for cohort 7 with seven and cohort 9 with six participants. Eligible volunteers received either a single dose (cohorts 1–6) or two equal doses separated by 12 h (cohorts 7–9), as outlined in the study design (Figure 1). In the first cohort, two sentinel participants in a 1:1 ratio (active:placebo) were dosed first and a review of the safety data available from the first 72 h after dosing was performed prior to dosing the remaining six participants of the cohort, which occurred at least 72 h after dosing of the first participants. Due to higher than predicted exposures in the first SAD cohort (10 mg), the dose in the second cohort was reduced to 5 mg, before it was then further escalated in cohorts 3–8.
The FE cohort consisted of 12 patients who received 10 mg LYS006 open label in a two‐way crossover design in a 1:1 ratio starting either with fasted or fed conditions, respectively. The two treatment periods were separated by a washout period of at least 14 days.
Forty healthy participants were randomized to the five cohorts of the MAD part to receive LYS006 or placebo in a 6:2 ratio for 12 consecutive days. The first two cohorts were dosed under fasted conditions (5 and 15 mg q.d.), the remaining three under fed conditions, but b.i.d. The cohort 1 started after SAD cohort four results were known (as indicated by the arrows in Figure 1). In each cohort, a sentinel group of two participants, with one participant receiving active treatment, were dosed at the same time. A safety review of all the available safety parameters from the first 96 h was performed prior to dosing of the remaining six participants, which occurred no sooner than 168 ± 2 hours (i.e., 7 days) after dosing of the first two participants. PK, PD, and safety assessments were also performed during the follow‐up period and at end of study (EoS).
To assess the PD effect of LYS006 on a potential target organ, the skin, in this case, cantharidin skin blisters were induced on the arm of participants in selected cohorts of the MAD part by topical application of cantharidin. Blister fluid containing immune cells was collected 24 h after cantharidin application.
LYS006 1, 5, and 50 mg were provided as hard gelatin capsules, and their matching placebo was prepared by Novartis and supplied as open bulk to the unblinded site pharmacist. Participants, investigator staff, personnel performing assessments, and data analysts remained blinded to study treatments, except for the food effect part which was open label. Randomization of participants was done by Novartis Drug Supply Management using a validated automated system for random assignment of treatment arms.
Assessments
Pharmacokinetics
A detailed description of the sample collection and analysis of the PK samples of this study is described by Poller et al. 15
Blood pharmacodynamics
PD assessment was done via ex vivo stimulation of whole blood with ionophore A23187 for 30 min, which stimulates LTB4 production. Inhibition of LTA4H in treated participants at the time of blood collection can be assessed via LTB4 generation in stimulated blood pre‐ and postdose. In the SAD part, blood for PD assessment was collected before treatment start and 2, 4, 8, 24, 48, and 336 h postdose. In the MAD part, blood for PD assessment was collected before treatment start and 1, 2, 3, 8, 10, 11, 14, and 25 days postdose at predose timepoints (trough level PD). Levels of LTB4 were measured by LC–MS/MS and are given as percent reduction from predose levels.
Cantharidin skin blister induction and pharmacodynamics
In order to assess the PD effect of LYS006 in the skin, cantharidin skin blisters were induced by topical application of a solution of 0.1% cantharidin on a filter disc 24 h before blister fluid aspiration in participants at different dose levels of the MAD part of the study (placebo, 15 mg q.d., 20 mg b.i.d., 40 mg b.i.d., and 80 mg b.i.d.) of the MAD part of the study at day 8 of treatment. Blisters form within 24 h after application and contain next to blister fluid large numbers of immune cells, in particular neutrophils and macrophages. 16 Blister fluid was aspirated on day 9, predose, and cells were centrifuged for subsequent stimulation with ionophore A23187 for 30 min. Levels of LTB4 were measured by LC–MS/MS and are given as percent reduction from predose levels.
Safety
Safety assessments consisted of collecting adverse events (AEs), serious AEs (SAEs), liver safety monitoring, and renal safety monitoring. The laboratory evaluations, such as hematology and coagulation tests, clinical chemistry, urinalysis, special clinical laboratory evaluations, and crystalluria were done in regular intervals. Other safety assessments included physical examination, vital signs, height and weight, ECG monitoring, 24 h Holter ECG monitoring, pregnancy, and fertility assessments. The safety review of sentinel participants that was performed before proceeding to the dosing of the remaining participants of the cohort contained at least vital signs, ECGs, AEs, and laboratory safety.
RESULTS
Preclinical PK/PD
As we described previously, LYS006 exhibits a concentration dependent blood plasma distribution (Cb/Cp) in mice caused by TMDD, which is not observed in LTA4H knockout (KO) animals. 13 This concentration‐dependent Cb/Cp distribution could nicely be illustrated in a rat absorption, distribution, metabolism, and excretion (ADME) study with [3H]‐labeled LYS006 (Figure 2a). Four hours after oral administration of 3 mg/kg [3H]LYS006, leading to blood concentrations above 1000 ng/mL, a nearly equal distribution of compound was found in blood and plasma. Whereas LYS006 concentrations decreased steadily over time in plasma due to elimination processes, it persisted much longer in blood cells resulting in a steadily increasing Cb/Cp of greater than 20 until day 7 post oral administration. The slow redistribution of compound from blood and tissue into plasma also resulted in a long terminal plasma half‐life. Leukocytes express all 5‐LO pathway enzymes and are therefore the main source of LTB4 production in blood. Hence, we wanted to investigate whether the longer persistence of compound in LTA4H carrying blood cells would affect the PD effect of LYS006. We chose dogs for these extended PK/PD studies. A low dose of 0.5 mg/kg LYS006 p.o. led to a rapid absorption of the compound and distribution into blood cells with a maximum plasma concentration (C max) of 5.7 ng/mL and a blood C max of 46 ng/mL. Compound levels in plasma quickly dropped to around 3 ng/mL at trough plasma concentration (C trough) but stayed nearly stable in blood with a C trough of 33 ng/mL (Figure 2b). Blood and plasma C trough levels remained at similar levels after subsequent once daily doses of compound on days 2–4 and decreased slowly after the final dose (given at 96 h [day 5]). LTB4 release in ex vivo stimulated blood was reduced by ~60% (at peak) after the first dose of 0.5 mg/kg LYS006 (Figure 2c). After the fifth dose (peak concentration of 5.5 ng/mL plasma and 55 ng/mL blood), the PD effect reached 86% LTB4 inhibition, as also reflected by the higher blood exposure at peak of day 5. After the final dose, LTB4 inhibition only slowly returned to baseline levels in line with the long residence time of LYS006 in blood cells. After the first dose of 3 mg/kg, LYS006 peak levels reached 36 ng/mL in plasma and 71 ng/mL in blood 1 h after p.o. administration. This translated to a LTB4 inhibition of 93% 3 h after the initial dose. Plasma levels rapidly dropped to 5.3 ng/mL at C trough but only moderately declined in blood to 49 ng/mL (Figure 2b). Inhibition of LTB4 24 h after dosing was at 67% and showed similar trough level inhibition in subsequent days. At peak of the fifth dose, plasma C max was 50 ng/mL and blood C max was 89 ng/mL, translating to a the nearly complete PD effect of greater than 98% LTB4 inhibition (Figure 2c,d). Inhibition of LTB4 slowly declined after the last dose and reached predose levels 4 days after final dose despite persistent blood compound concentrations of around 30 ng/mL (Figure 2d).
FIGURE 2.
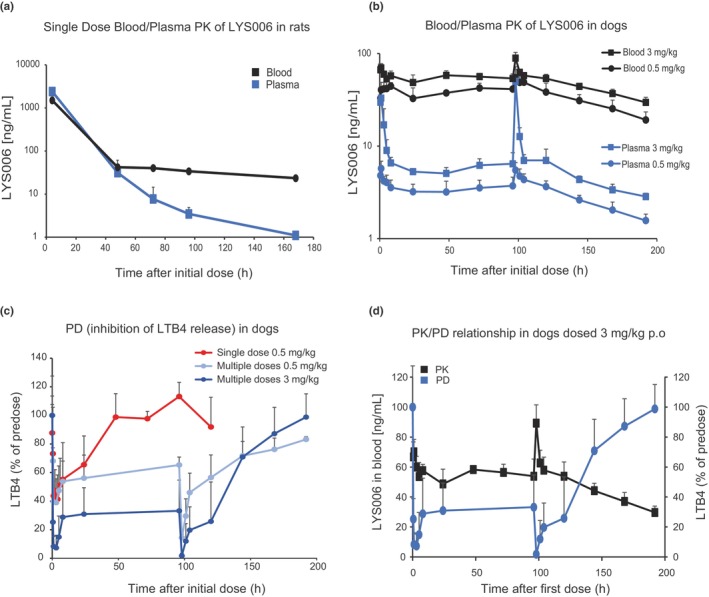
Characterization of the blood/plasma PK and PD relationship of LYS006 in preclinical species. (a) Male Wistar rats (n = 3) were dosed once with 3 mg/kg [3H]‐labeled LYS006 p.o. and distribution of compound between blood and plasma was determined by LC–MS/MS over time. Sampling was done at 4, 48, 72, 96, and 168 h postdose. Depicted are mean concentration‐time curves ±SD. (b) Fasted male beagle dogs (n = 3) were dosed once daily p.o. with 0.5 and 3 mg/kg LYS006 for 5 consecutive days. Concentration‐time curves (mean ± SD) of LYS006 in blood and plasma were determined by LC–MS/MS. Sampling was done at 0.5, 1, 3, 5, 8, and 24 h (at trough before the second dose), 48 h and 72 h (at trough before the third and fourth dose), and 96 h (at trough before the fifth dose), 98, 101, 104, 120, 144, 168, and 192 h post first dose. (c) LTB4 release of ionophore stimulated dog whole blood was determined at different timepoints after a single dose of 0.5 mg/kg LYS006 (red line). LTB4 release was also determined at the above‐mentioned timepoints after multiple doses of a once daily dosing regimen of 0.5 mg/kg (light blue) and 3 mg/kg (dark blue). LTB4 levels were determined by ELISA and given as percent LTB4 in relation to predose levels. (d) Overlay of PK (black curve) and PD (blue curve) of the 3 mg/kg p.o. dose group. Shown are means ± SD. ELISA, enzyme‐linked immunosorbent assay; LC–MS/MS, liquid chromatography tandem mass spectrometry; PD, pharmacodynamic; PK, pharmacokinetic.
Clinical study baseline demographics
A total of 121 healthy adult men and women between 18 and 45 years of age were recruited to the study. Sixty‐nine patients were randomized to the SAD cohorts, 40 to the MAD cohorts, and 12 participated in the FE cohort. Baseline demographics of study participants in the different arms of the study are summarized in Table 1.
TABLE 1.
Baseline demographics by cohorts.
| SAD cohorts | MAD cohorts | FE cohorts | |||
|---|---|---|---|---|---|
| Placebo, N = 18, n (%) | LYS006, N = 51, n (%) | Placebo, N = 10, n (%) | LYS006, N = 30, n (%) | LYS006, N = 12, n (%) | |
| Age in years, mean (SD) | 32.9 (6.62) | 33.1 (7.22) | 39.7 (6.36) | 34.2 (6.63) | 35.1 (7.87) |
| Males, n (%) | 17 (94.4) | 48 (94.1) | 9 (90) | 29 (96.7) | 11 (91.7) |
| Race, n (%) | |||||
| White | 17 (94.4) | 47 (92.2) | 10 (100) | 30 (100) | 11 (91.7) |
| American Indian or Alaska Native | 0 (0) | 1 (2) | 0 (0) | 0 (0) | 1 (8.3) |
| Black or African American | 1 (5.6) | 3 (5.9) | 0 (0) | 0 (0) | 0 (0) |
| Ethnicity, n (%) | |||||
| Non‐Hispanic or Latino | 18 (100) | 47 (92.2) | 10 (100) | 29 (96.7) | 11 (91.7) |
| Hispanic or Latino | 0 (0) | 1 (2) | 0 (0) | 1 (3.3) | 1 (8.3) |
| Not reported | 0 (0) | 3 (5.9) | 0 (0) | 0 (0) | 0 (0) |
| Weight in kg, mean (SD) | 84.1 (11.16) | 76.4 (9.22) | 78.8 (5.34) | 76.8 (11.27) | 78.0 (13.22) |
| Height in cm, mean (SD) | 182.8 (8.31) | 178.0 (8.04) | 174.5 (7.47) | 177.1 (6.73) | 175.9 (10.30) |
| BMI, kg/m2, mean (SD) | 25.3 (2.73) | 24.1 (2.33) | 25.6 (1.17) | 24.2 (2.86) | 25.2 (2.19) |
Abbreviations: BMI, body mass index; FE, food effect; MAD, multiple ascending dose; SAD, single ascending dose.
Clinical PK/PD during SAD
We first investigated the clinical PK/PD of LYS006 in human subjects after single ascending oral dose administrations. Similarly, as described above for rat and dog PK/PD profiles, LYS006 rapidly distributed to human blood cells after a single oral administration, resulting in high Cb/Cp ratios at low concentrations observed at C trough (Figure 3a). After single dose administration, a fast absorption was followed by rapidly decreasing plasma concentrations to below 10 ng/mL at 12 h for all dose groups and then slowly decreased further due to slow re‐distribution from blood and tissue to plasma (Figure 3a). Blood concentrations were on par with plasma concentrations for exposures above ~60 ng/mL but decreased to only about 30–50 ng/mL 12 h after administration. Fourteen days after the initial dose, blood concentrations were still at around 20 ng/mL for all dose groups. At the lowest tested dose of 5 mg q.d., ~70% reduction in LTB4 production could be achieved at peak (Figure 3b). Target engagement slowly declined and returned to placebo levels ~48 h after dosing. At doses of greater than or equal to 20 mg, a peak inhibition of greater than 95% was achieved. Despite rapidly decreasing plasma concentrations, a PD effect of greater than 80% was maintained 24 h after dosing for all doses of 20 mg and above, in line with the persistent blood exposures of LYS006. After a single dose of 70 mg, nearly complete (>95%) LTA4H inhibition was maintained for 24 h, but levels of inhibition returned to placebo levels at the 72 h timepoint despite maintenance of compound levels of around 30 ng/mL in blood.
FIGURE 3.
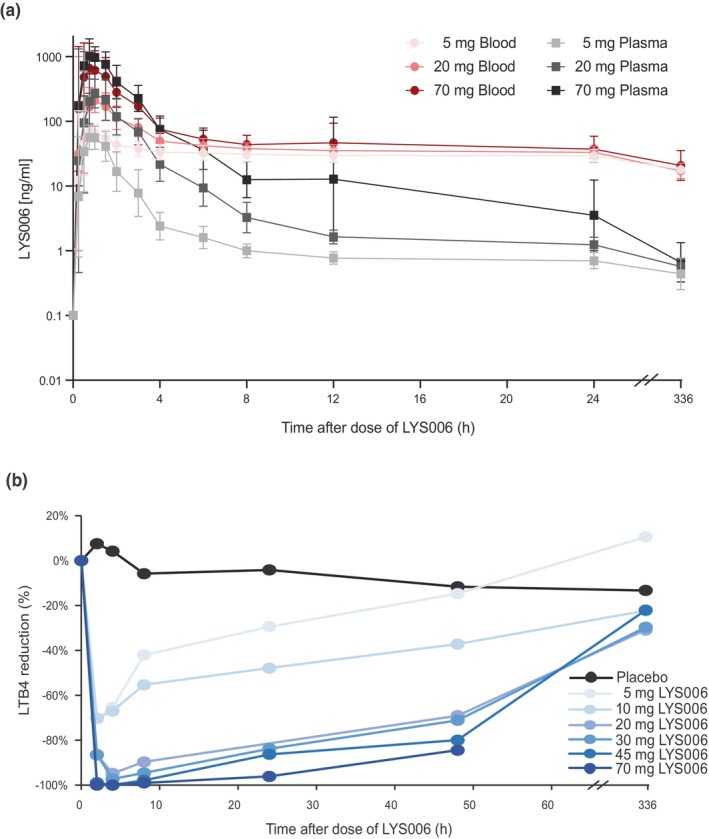
PK/PD profile of the SAD part. (a) Blood (red lines) and plasma (gray lines) concentrations of LYS006 after a single low (5 mg), a medium (20 mg), and a high (70 mg) dose of LYS006 during the SAD part of the study. Profiles of the first 24 h after dosing and of final timepoint 14 days after dosing are shown as arithmetic means ± errors. (b) Blood PDs of all single dose cohorts of the SAD part are shown for up to 48 h and at end of study after single ascending doses of LYS006. Reduction of LTB4 (in percent) versus pretreatment in ex vivo stimulated blood is shown as mean. PD, pharmacodynamic; PK, pharmacokinetic; SAD, single ascending dose.
Clinical PK/PD during MAD
In the MAD part of the study, predose (C trough) exposures showed high Cb/Cp levels for low doses and q.d. dosing regimens (5 and 15 mg q.d.). For the higher doses and b.i.d. dosing regimens Cb/Cp at C trough were between 2 (20 mg b.i.d.) and 1 (80 mg b.i.d.). After the end‐of‐treatment (EoT; last dose was given in the morning of day 12, for b.i.d. regimens only a single dose was given on day 12), we observed a rapid decline of plasma levels to ~1–2 ng/mL where they remained nearly stable. Blood exposures only marginally dropped and remained at around 30–40 ng/mL (Figure 4a). For the lowest dose of 5 mg q.d., we observed a slowly increasing PD effect until day 8 (Figure 4b). In addition, C trough plasma levels slowly increased visibly for this dose during the first 3 days. Target saturation was more quickly achieved with the 15 mg q.d. dose, reaching a reduction in LTB4 production of ~90% at C trough, 2 days after the initial dose which was maintained until EoT. For the b.i.d. dosing regimens (20–80 mg b.i.d.), a greater than 96% enzyme inhibition was already achieved at C trough of day 1 and complete inhibition of close to 100% (undetectable LTB4 after ex vivo stimulation) was maintained until EoT. At EoS assessment (day 25 after initial dose), also the PD effect of the high dose groups had returned to placebo levels. When we plotted C trough level plasma (Figure 4c) or blood (Figure 4d) exposures versus LTB4 inhibition, the C trough blood exposures leading to half‐maximal inhibitory concentration (IC50) or IC90 were largely in agreement with the potencies of LYS006 determined in human whole blood in vitro assays (IC50 = 21 ng/mL, IC90 = 57 ng/mL). 13
FIGURE 4.
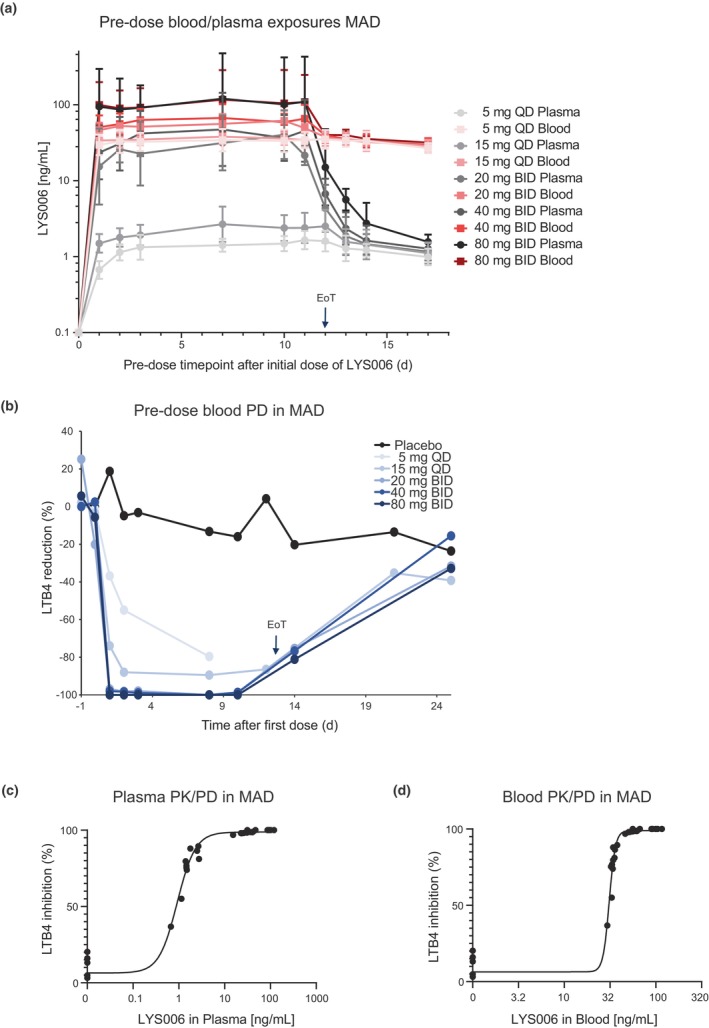
PK/PD profile of the MAD part. (a) Blood (red lines) and plasma (gray lines) predose concentrations of LYS006 at indicated timepoints after once‐daily doses of 5 mg and 15 mg q.d. or twice‐daily doses of 20 mg, 40 mg or 80 mg b.i.d. of LYS006. Profiles during the 12 days of repeated dosing and after EoT are depicted as arithmetic means ± errors. (b) PD effect of LYS006 in all MAD cohorts are shown at indicated days during repeated treatment and after EoT. PD effect is depicted at the predose timepoint of each day as LTB4 reduction versus pre‐treatment in percent (shown are means). (c) Mean predose plasma levels from all MAD cohorts are plotted versus mean predose LTB4 inhibition levels to depict a plasma PK/PD response curve. (d) Mean predose blood levels from all MAD cohorts are plotted versus mean predose LTB4 inhibition levels to depict a blood PK/PD response curve. EoT, end‐of‐treatment; MAD, multiple ascending dose; PD, pharmacodynamic; PK, pharmacokinetic.
PD in blood and skin and efficacious dose selection
The lowest dose of LYS006 to maintain full target engagement was the 20 mg b.i.d. dose. Overlay of the blood/plasma trough level PK and PD curves at this MAD dose, indicated that 96% (day 1) to 100% (day 8) inhibition of LTB4 release in blood was achieved at mean blood C trough exposures between 46 ng/mL (day 1), to 62 ng/mL (day 10; Figure 5a). Mean plasma C trough levels ranged from 15 ng/mL (day 1) to 40 ng/mL (day 10) at this dose. After EoT, a rapid decline in plasma levels to 1.5 ng/mL 48 h after the last dose but only moderate reduction in blood levels to 32 ng/mL maintained a PD effect of 75% at this timepoint (day 14). Given that LTA4H inhibition was considered an interesting target for the treatment of neutrophil‐driven inflammatory conditions of the skin, we were interested in assessing the degree of enzyme inhibition in immune cells of the skin. To this end, we induced cantharidin skin blisters in participants during the MAD phase. Despite high variability in the assessment of target inhibition in blister‐derived immune cells, it became evident that a dose‐dependent increase in LTA4H inhibition was also present in skin blister cells (Figure 5b). At a dose of 20 mg b.i.d., a median inhibition of 90% of predose LTB4 was achieved.
FIGURE 5.
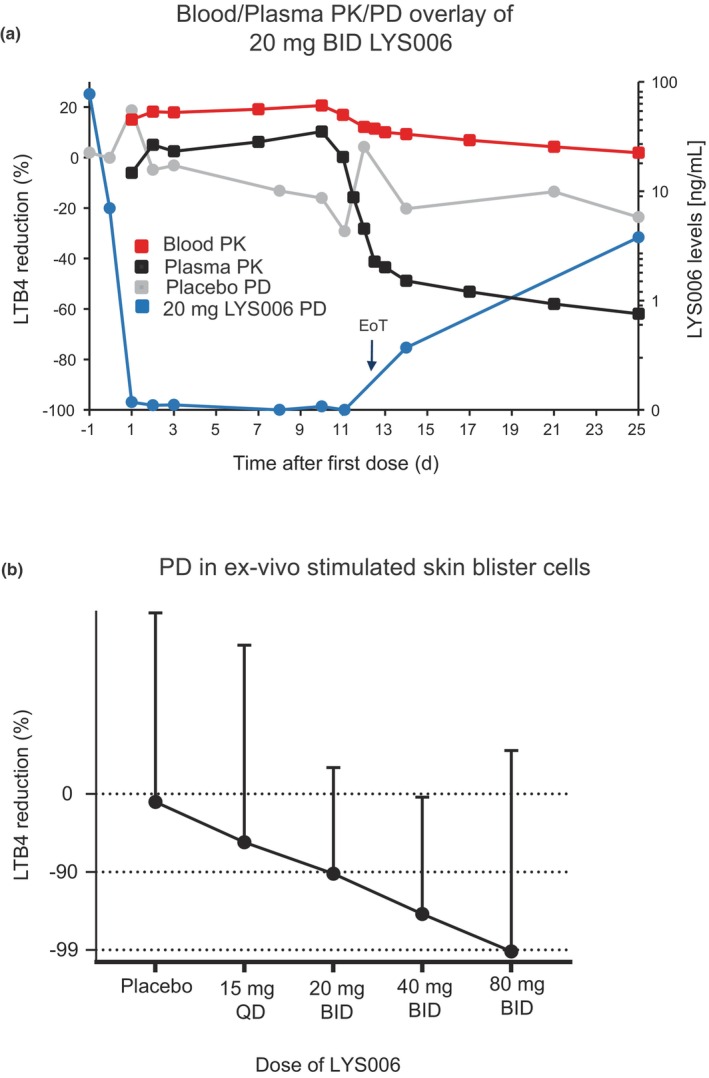
Blood and skin blister cell PD effect of LYS006 in MAD. (a) Overlays of blood PK (red curve), plasma PK (black curve), and PD effects of placebo (gray curve) and 20 mg b.i.d. LYS006 (blue curve) during repeated dose treatment and after EoT. PD effect is shown as mean LTB4 reduction in percentage versus pretreatment (left axis) and PK as mean LYS006 concentrations in blood and plasma (right axis) at indicated days at predose timepoints. (b) PD effect in skin blister cells is depicted as percent LTB4 reduction in ex vivo stimulated skin blister cells in placebo, 15 mg q.d. and 20, 40, and 80 mg b.i.d. treated patients. Shown are median values + errors. EoT, end‐of‐treatment; MAD, multiple ascending dose; PD, pharmacodynamic; PK, pharmacokinetic.
Safety and tolerability
Generally, LYS006 was well‐tolerated by all study participants and no deaths, no SAEs and no AEs leading to treatment discontinuation occurred in the active treatment arms in the SAD, FE, and MAD parts of the study. AEs and their breakdown by Medical Dictionary for regulatory activities preferred term and their severity are provided in Table 2.
TABLE 2.
Incidences of AEs by preferred term and their severity and causality for all doses within different cohorts.
| SAD cohorts | MAD cohorts | FE cohorts | |||
|---|---|---|---|---|---|
| Placebo, N = 18, n (%) | LYS006, N = 51, n (%) | Placebo, N = 10, n (%) | LYS006, N = 30, n (%) | LYS006, N = 12, n (%) | |
| Subjects with at least one AE | 8 (44.4) | 19 (37.3) | 8 (80.0) | 23 (76.7) | 8 (66.6) |
| Abdominal discomfort or pain | 1 (5.6) | 4 (7.8) | 1 (10) | 2 (6.7) | 2 (16.7) |
| Diarrhea | 3 (16.7) | 2 (3.9) | 1 (10) | 1 (3.3) | 0 (0) |
| Headache | 1 (5.6) | 3 (5.9) | 2 (20) | 8 (26.7) | 1 (8.3) |
| Lipase elevation | 0 (0) | 2 (3.9) | 1 (10) | 3 (10) | 0 (0) |
| Blood CPK elevation | 0 (0) | 0 (0) | 1 (10) | 5 (16.7) | 0 (0) |
| Amylase increase | 0 (0) | 0 (0) | 1 (10) | 2 (6.7) | 0 (0) |
| Back pain | 0 (0) | 1 (2) | 1 (10) | 2 (6.7) | 0 (0) |
| Dizziness | 0 (0) | 1 (2) | 1 (10) | 2 (6.7) | 0 (0) |
| Breast pain | 0 (0) | 0 (0) | 2 (20) | 0 (0) | 0 (0) |
| Nausea | 1 (5.6) | 0 (0) | 0 (0) | 2 (6.7) | 0 (0) |
| Pruritus | 0 (0) | 2 (3.9) | 1 (10) | 0 (0) | 0 (0) |
| Dry skin | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 2 (16.7) |
| Viral respiratory infection | 0 (0) | 0 (0) | 0 (0) | 2 (6.7) | 3 (25) |
| Feeling cold | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 2 (16.7) |
| AE severity | |||||
| Mild | 8 (44.4) | 18 (35.3) | 7 (70.0) | 22 (73.3) | 8 (66.6) |
| Moderate | 1 (5.6) | 2 (3.9) | 1 (10.0) | 4 (13.3) | 0 (0) |
| Severe | 0 (0) | 1 (2) | 1 (10.0) | 4 (13.3) | 0 (0) |
| Study drug‐related AEs | 0 (0) | 4 (7.8) | 1 (10) | 5 (16.7) | 0 (0) |
| SAEs | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| AEs leading to discontinuation | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Deaths | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
Abbreviations: AE, adverse event; CPK, blood creatine phosphokinase; FE, food effect; MAD, multiple ascending dose; SAD, single ascending dose; SAE, serious adverse event.
In the SAD part, AEs were reported in 19 out of all 51 treated LYS006 participants (37.3%) compared to eight out of 18 placebo participants (44.4%). The events (in the LYS006 and placebo‐treated participants) were of mild, moderate, and severe intensity for 26, 3, and 1 participant(s), respectively, and resolved without sequelae. The most‐commonly reported AEs were abdominal discomfort or pain in four (7.8%) LYS006 treated participants (compared to 1 [5.6%] for placebo), headache in three (5.9%) LYS006 treated participants (compared to 1 (5.6%) for placebo), and diarrhea in two (3.9%) LYS006 treated participants (compared to 3 (16.7%) in placebo). No dose dependency was observed. The only AE of severe intensity (grade 3 according to Common Terminology Criteria for Adverse Events version 4.03) was a lipase increase (≥2 × upper limit of normal [ULN]) without associated clinical sign in a participant who received the lowest single dose of LYS006 tested (5 mg) at day 8 after dosing.
During the open‐label two way crossover FE part, eight (66.7%) out of 12 patients receiving 10 mg LYS006 under fasted or fed conditions, reported at least one AE. All events were of mild intensity. The most commonly reported AEs were viral upper respiratory tract infection in three (25.0%) participants, abdominal pain in two (16.7%) participants, dry skin in two (16.7%) participants, and feeling cold in two (16.7%) participants.
In the MAD part, AEs were reported in 23 out of 30 LYS006‐treated participants (76.7%) compared to eight out of 10 placebo‐treated participants (80.0%). The events (in both LYS006 and placebo group) were of mild, moderate, and severe intensity for 29, five, and five participant(s), respectively, and resolved without sequelae. The most commonly reported AE was headache in 10 participants (8 [26.7%] in LYS006 group, 2 [20%] in placebo). In clinical chemistry assessments, the most frequent AEs were creatine phosphokinase (CPK) elevation in six participants (5 [16.7%] in the LYS006 group and 1 [10%] in the placebo group) and lipase elevation in four participants (3 [10%] in the LYS006 group and 1 [10%] in the placebo group). These laboratory elevations were not associated with concomitant clinical signs or symptoms, did not occur in a dose‐dependent fashion, and resolved spontaneously. AEs of severe intensity (grade 3) were detected in four LYS006‐treated participants (13.3%) and one placebo‐treated participant (10%). In the placebo group, the AE of severe intensity was related to a lipase elevation (≥2 × ULN). Two lipase elevations of severe intensity were also present in the LYS006 group. They were transient and rapidly returned to normal levels under continued treatment. The other two AEs of severe intensity in the LYS006 group were related to CPK elevations. These were as well transient and returned to normal levels under continued treatment.
In addition to the safety and tolerability assessments described above and due to the predominant renal clearance mechanisms of LYS006 (90% of parent compound is excreted into the urine), as described before, 15 we included a close monitoring of urine concentrations and close meshed assessment of urinary crystals. No crystalluria was observed in any patient having received LYS006 up to maximum tested supratherapeutic doses in SAD (2 × 100 mg) and MAD (80 mg b.i.d.), even when tested in a cohort undergoing restricted fluid intake (hydration limitation, cohort 9).
DISCUSSION
Prior to LYS006, four different LTA4H inhibitors had been tested in FIH studies: DeCode's DG‐051, Boehringer Ingelheim's BI691751, Janssen's JNJ‐40929837, and Celtaxys' Acebilustat, foreseen for the treatment of cardiovascular diseases, asthma, and cystic fibrosis. 10 Although detailed clinical PK/PD had only been published for JNJ‐40929837 11 and acebilustat, 17 , 18 high level information about the degree of target engagement and PK is also available for DG‐051 and BI691751. DG‐051 showed the lowest potency of these molecules in vitro 13 and it had been reported to reach LTB4 reductions in ex vivo stimulated blood of up to 70% after 7 days of once daily treatment. 19 For BI691751, neither chemical structure nor in vitro potency or clinical PD effects are disclosed, but a very long half‐life of 377 h in blood was determined clinically. 19 JNJ‐40929837, a highly potent LTA4H inhibitor in in vitro cellular assays, showed a reduction of LTB4 levels of greater than 95% over a 24 h period at doses of 100 mg and greater than 90% up to 98% in ex vivo stimulated blood of patients with asthma at trough and 92% reduction in sputum LTB4. 11 For acebilustat, a maximum PD effect of 89% reduction of LTB4 at peak in ex vivo stimulated blood at the highest tested dose of 200 mg q.d. was reported, with lower levels of LTB4 reduction in the blood of patients with cystic fibrosis (50%–70% at the 100 mg q.d. dose) and even less so in the sputum of these patients (20%–30% reduction at the 100 mg dose). 17 , 18
LYS006 is a highly potent and selective inhibitor of LTA4H that shows interesting characteristics in in vitro cellular assays as well as its preclinical and clinical PK/PD profile. As we demonstrated earlier, LYS006 shows a time dependency in its in vitro cellular potency, reaching maximum inhibition only after extended incubation times of greater than or equal to 4 h, suggesting a slow distribution of the compound into target cells expressing LTA4H. Its human whole blood IC90 was determined to be ~57 ng/mL. 13 Its preclinical and clinical PKs were characterized by rapid absorption, followed by a fast initial decline in plasma concentrations and a long terminal plasma and blood half‐life. The PK profile was linear for higher doses but nonlinear for lower doses, demonstrating a high Cb/Cp ratio at lower exposures in rats, dogs, and human participants. Saturable target binding impacting distribution (TMDD) is responsible for the nonlinear PK of LYS006, as we had previously demonstrated in LTA4H KO mice. 13 A key element of this TMDD appears to be a slow re‐distribution of LYS006 from cells expressing the target into the extracellular space, which is responsible for the long persistence of LYS006 in blood and other target‐expressing tissues such as the skin. It also explains the strong and long‐lasting PD effect of the compound at low plasma exposures.
In the SAD part of the FIH study, a single dose of 20 mg already achieved a PD effect of 95% 4 h after dosing, which was maintained above 80% 24 h after dosing despite plasma exposures of only 1.2 ng/mL. The reason for this sustained PD effect was the retention of LYS006 in blood cells at ~33 ng/mL 24 h after the single dose which ensured continued target engagement. The difference in blood levels between the low 5 mg dose and the high 70 mg dose was nearly dose proportional at T max but became very small at the 24 h timepoint (29.4 ng/mL vs. 37.1 ng/mL). Yet, this difference resulted in a strongly distinct PD effect of 29% versus 96% reduction of LTB4 levels, causing the very steep dose response curve between blood exposure and PD we see for LYS006. The steep dose response curve is explained by a combination of compound distribution and target binding. In the MAD part, we observed the trough level PD effects to slowly increase over time for the low dose 5 mg q.d. regimen, indicating a slowly increasing target saturation in blood cells at the low dose. At the 15 mg q.d. dose, target saturation was achieved already after two doses and trough level PD effect of ~90% was achieved at plasma levels of ~2 ng/mL and blood levels of ~35 ng/mL. Similarly, as observed during the SAD part, also in the MAD part, mean blood compound levels were very similar between the different dose groups 24 h after the last dose (EoT at day 12) ranging between 35 and 40 ng/mL from the 5 mg q.d. to the 80 mg b.i.d. dose. The very similar blood levels across the different dose groups are explained by the fact that only cells expressing LTA4H retained the compound 24 h after the last dose, whereas excess compound is cleared from plasma via the kidneys. The LYS006 blood concentration sufficient to saturate ~90% of LTA4H is between 35 to 40 ng/mL. Two interesting observations were made: (i) despite retention of compound in target expressing cells, complete target inhibition to undetectable levels of LTB4 at trough could only be achieved with a b.i.d. regimen. This likely has to do with the short half‐life of neutrophils in human blood, which is less than 1 day. 20 Therefore, new LTA4H expressing cells arrive in circulation capable of producing LTB4. The LTA4H expressed by these cells cannot be saturated due to the low LYS006 plasma concentrations a few hours after compound administration, leading to detectable levels of LTB4 upon ex vivo stimulation. (ii) A few days after EoT, PD values had returned to predose levels, even though considerable concentrations of LYS006 could still be detected in blood that only very slowly decreased. This is likely explained by the longer half‐life of LTA4H expressing erythrocytes. In contrast to neutrophils, erythrocytes have a half‐life of ~120 days. 21 Although they strongly express LTA4H, they are missing the upstream enzymatic machinery and, hence, cannot generate LTB4. Erythrocyte half‐life may therefore explain the presence of detectable levels of LYS006 in blood 2 weeks after EoT, without affecting the capacity of blood leukocytes to generate LTB4.
The PK/PD of LYS006 is therefore complex and influenced by slow compound distribution into and out of LTA4H expressing cells, rapid clearance from plasma, cell turnover, and enzyme turnover kinetics.
To achieve LTB4 reduction in blood to nearly undetectable levels at trough and to levels below 10% in skin blister immune cells, a dose of 20 mg b.i.d. was sufficient. We therefore defined this dose as the lowest fully efficacious dose to completely suppress LTB4 in blood (>99% at trough) and target tissue immune cells (>90% at trough) and chose it for subsequent proof of concept studies in inflammatory diseases of the skin. At this dose and dosing regimen, trough plasma levels (12 h after dosing) of 30 to 40 ng/mL and blood levels between 50 and 60 ng/mL of LYS006 were maintained at steady‐state.
Although LYS006 is a reversible, competitive inhibitor of LTA4H, its PK/PD characteristics are reminiscent of covalent inhibitors that demonstrate a long‐lasting target engagement and PD effect, despite short overall systemic residence time. 22 This represents a desired drug profile as strong and long‐lasting PD effects can be achieved with low and short overall systemic exposures, limiting potential off‐target effects. In agreement with this, LYS006 was well‐tolerated up to supratherapeutic doses of 80 mg b.i.d. in the MAD part and did not induce any clinical or laboratory safety signals of concern in healthy human participants. No dose‐limiting toxicity was observed, no SAEs and no AEs leading to study discontinuation were reported.
Overall, LYS006 demonstrated a favorable safety profile and a nearly complete PD effect in blood and skin blister cells with low systemic exposures. Its level of target engagement at trough is superior to other LTA4H inhibitors, clinically tested to date, suggesting this molecule may be the best‐in‐class LTA4H inhibitor tested in humans so far. As dosing in this FIH study was limited to 12 days, no long‐term safety in humans can be inferred from these data. Based on the favorable PK/PD and safety/tolerability results of this study and favorable long‐term preclinical toxicology data, LYS006 was moved to phase II efficacy studies in patients suffering from chronic inflammatory conditions, such as inflammatory acne, hidradenitis suppurativa, and ulcerative colitis.
AUTHOR CONTRIBUTIONS
T.A.R. and C.L. wrote the manuscript. T.A.R., D.P., C.L., H.M.W., K.K., H.O., and N.Z. designed the research. B.V.H., I.S., P.J., and B.P. performed the research. M.F., C.S., I.S., D.P., H.M.W., T.A.R., G.T., and C.M. analyzed the data.
FUNDING INFORMATION
The study was funded by Novartis Pharma AG, Switzerland.
CONFLICT OF INTEREST STATEMENT
We disclose that all authors associated with Novartis Biomedical Research or Novartis Pharma AG are current or former employees of and hold company stocks or stock options with Novartis Pharma AG, the company that is developing LYS006 as pharmacological treatment in inflammatory diseases. All other authors declared no competing interests for this work.
ACKNOWLEDGMENTS
The authors thank Solen Pichereau for PK/PD modeling and translation of preclinical to clinical data, Martina Sütterlin‐Hachmann for conducting the rat ADME study, Esther van de Kerhoff for serving as study director, Esther Scheidegger for conducting the dog study, Thierry Thevenin for ex vivo PD analysis of LTB4 by LC–MS/MS in FIH, and Justin McMullen for analysis of LTB4 in blister fluid cells.
Loesche C, Picard D, Van Hoorick B, et al. LTA4H inhibitor LYS006: Clinical PK/PD and safety in a randomized phase I clinical trial. Clin Transl Sci. 2024;17:e13724. doi: 10.1111/cts.13724
REFERENCES
- 1. Ford‐Hutchinson AW, Bray MA, Doig MV, Shipley ME, Smith MJ. Leukotriene B, a potent chemokinetic and aggregating substance released from polymorphonuclear leukocytes. Nature. 1980;286(5770):264‐265. [DOI] [PubMed] [Google Scholar]
- 2. Peters‐Golden M, Henderson WR Jr. Leukotrienes. N Engl J Med. 2007;357(18):1841‐1854. [DOI] [PubMed] [Google Scholar]
- 3. Afonso PV, Janka‐Junttila M, Lee YJ, et al. LTB4 is a signal‐relay molecule during neutrophil chemotaxis. Dev Cell. 2012;22(5):1079‐1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lammermann T, Afonso PV, Angermann BR, et al. Neutrophil swarms require LTB4 and integrins at sites of cell death in vivo. Nature. 2013;498(7454):371‐375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sadik CD, Luster AD. Lipid‐cytokine‐chemokine cascades orchestrate leukocyte recruitment in inflammation. J Leukoc Biol. 2012;91(2):207‐215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. He R, Chen Y, Cai Q. The role of the LTB4‐BLT1 axis in health and disease. Pharmacol Res. 2020;158:104857. [DOI] [PubMed] [Google Scholar]
- 7. Rao NL, Dunford PJ, Xue X, et al. Anti‐inflammatory activity of a potent, selective leukotriene A4 hydrolase inhibitor in comparison with the 5‐lipoxygenase inhibitor zileuton. J Pharmacol Exp Ther. 2007;321(3):1154‐1160. [DOI] [PubMed] [Google Scholar]
- 8. Wenzel SE, Kamada AK. Zileuton: the first 5‐lipoxygenase inhibitor for the treatment of asthma. Ann Pharmacother. 1996;30(7–8):858‐864. [DOI] [PubMed] [Google Scholar]
- 9. Haeggstrom JZ. Leukotriene A4 hydrolase and the committed step in leukotriene B4 biosynthesis. Clin Rev Allergy Immunol. 1999;17(1–2):111‐131. [DOI] [PubMed] [Google Scholar]
- 10. Rohn TA, Numao S, Otto H, Loesche C, Thoma G. Drug discovery strategies for novel leukotriene A4 hydrolase inhibitors. Expert Opin Drug Discovery. 2021;16(12):1483‐1495. [DOI] [PubMed] [Google Scholar]
- 11. Barchuk W, Lambert J, Fuhr R, et al. Effects of JNJ‐40929837, a leukotriene A4 hydrolase inhibitor, in a bronchial allergen challenge model of asthma. Pulm Pharmacol Ther. 2014;29(1):15‐23. [DOI] [PubMed] [Google Scholar]
- 12. Elborn JS, Konstan MW, Taylor‐Cousar JL, et al. Empire‐CF study: a phase 2 clinical trial of leukotriene A4 hydrolase inhibitor acebilustat in adult subjects with cystic fibrosis. J Cyst Fibros. 2021;20(6):1026‐1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Markert C, Thoma G, Srinivas H, et al. Discovery of LYS006, a potent and highly selective inhibitor of leukotriene A(4) hydrolase. J Med Chem. 2021;64(4):1889‐1903. [DOI] [PubMed] [Google Scholar]
- 14. McGee J, Fitzpatrick F. Enzymatic hydration of leukotriene A4. Purification and characterization of a novel epoxide hydrolase from human erythrocytes. J Biol Chem. 1985;260(23):12832‐12837. [PubMed] [Google Scholar]
- 15. Poller B, Pearson D, Leuthold LA, et al. Human pharmacokinetics of LYS006, an Oral leukotriene A4 hydrolase inhibitor displaying target‐mediated drug disposition. Drug Metab Dispos. 2022;50(12):1472‐1482. [DOI] [PubMed] [Google Scholar]
- 16. Day RM, Harbord M, Forbes A, Segal AW. Cantharidin blisters: a technique for investigating leukocyte trafficking and cytokine production at sites of inflammation in humans. J Immunol Methods. 2001;257(1–2):213‐220. [DOI] [PubMed] [Google Scholar]
- 17. Elborn JS, Bhatt L, Grosswald R, Ahuja S, Springman EB. Phase I studies of acebilustat: pharmacokinetics, pharmacodynamics, food effect, and CYP3A induction. Clin Transl Sci. 2017;10(1):20‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Elborn JS, Horsley A, MacGregor G, et al. Phase I studies of acebilustat: biomarker response and safety in patients with cystic fibrosis. Clin Transl Sci. 2017;10(1):28‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bhatt L, Roinestad K, Van T, Springman EB. Recent advances in clinical development of leukotriene B4 pathway drugs. Semin Immunol. 2017;33:65‐73. [DOI] [PubMed] [Google Scholar]
- 20. Lahoz‐Beneytez J, Elemans M, Zhang Y, et al. Human neutrophil kinetics: modeling of stable isotope labeling data supports short blood neutrophil half‐lives. Blood. 2016;127(26):3431‐3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Thiagarajan P, Parker CJ, Prchal JT. How do red blood cells die? Front Physiol. 2021;12:655393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kaul M, End P, Cabanski M, et al. Remibrutinib (LOU064): a selective potent oral BTK inhibitor with promising clinical safety and pharmacodynamics in a randomized phase I trial. Clin Transl Sci. 2021;14(5):1756‐1768. [DOI] [PMC free article] [PubMed] [Google Scholar]