

Abstract
Aging, tau pathology, and chronic inflammation in the brain play crucial roles in synaptic loss, neurodegeneration, and cognitive decline in tauopathies, including Alzheimer's disease. Senescent cells accumulate in the aging brain, accelerate the aging process, and promote tauopathy progression through their abnormal inflammatory secretome known as the senescence‐associated secretory phenotype (SASP). Tau oligomers (TauO)—the most neurotoxic tau species—are known to induce senescence and the SASP, which subsequently promote neuropathology, inflammation, oxidative stress, synaptic dysfunction, neuronal death, and cognitive dysfunction. TauO, brain inflammation, and senescence are associated with heterogeneity in tauopathy progression and cognitive decline. However, the underlying mechanisms driving the disease heterogeneity remain largely unknown, impeding the development of therapies for tauopathies. Based on clinical and preclinical evidence, this review highlights the critical role of TauO and senescence in neurodegeneration. We discuss key knowledge gaps and potential strategies for targeting senescence and TauO to treat tauopathies.
Highlights
Senescence, oligomeric Tau (TauO), and brain inflammation accelerate the aging process and promote the progression of tauopathies, including Alzheimer's disease.
We discuss their role in contributing to heterogeneity in tauopathy and cognitive decline.
We highlight strategies to target senescence and TauO to treat tauopathies while addressing key knowledge gaps.
Keywords: aging, Alzheimer's disease, cognitive deficits, inflammation, senescence‐associated secretory phenotype, senescent cells, tau, tauopathies
1. INTRODUCTION
Chronic neuroinflammation, cognitive impairment, and the deposition of pathological tau in the brain are common features of aging‐related neurodegenerative tauopathies, including Alzheimer's disease (AD), for which no disease‐modifying treatments are available to date. 1 In AD, the accumulation of amyloid beta (Aβ) and tau aggregates are neurotoxic, and their increased accumulation disrupts the function and communication of neuronal and glial cells that leads to synaptic and neuronal loss. Chronic inflammation is another hallmark, often stemming from the malfunction of astrocytes and microglia—cells responsible for neuronal homeostasis and waste clearance in the brain. In AD, astrocytes and microglia struggle to support neuronal cells and remove waste, including Aβ and tau aggregates that contribute to neuronal dysfunction and persistent inflammation in the brain. Additionally, vascular factors also play a role in AD. Deposition of Aβ and tau aggregates in brain arteries, atherosclerosis, and mini‐strokes can reduce blood flow and oxygen to the brain and impair the integrity of blood‐brain barrier (BBB)—which protects the brain from harmful factors while selectively allowing transportation of vital factors. These impaired BBB and vascular functions consequently contribute to exacerbated inflammation and brain atrophy. Emerging evidence suggests that AD‐related brain pathology results from a complex interplay between Aβ and tau and several other factors. It appears that pathological tau accumulates in specific brain regions that are critically involved in memory, and the pathological tau spread throughout the brain following a stereotypical pattern that correlates with the clinical severity and dementia. 2 AD initially affects short‐term memory, learning, and spatial navigation, due to early neurodegeneration in the temporal lobe. However, in the end stages, the pathology spreads to other brain regions and causes various impairments in language, motor skills, and other abilities 2 AD is also classified as a neurodegenerative tauopathy characterized by accumulation of the microtubule‐associated protein tau into meta‐stable tau aggregates known as neurofibrillary tangles (NFTs). In AD patients, the clinical severity of dementia strongly correlates with the load of pathological tau aggregates. 3 , 4 Furthermore, pathological tau aggregates, in the absence of amyloid plaques, can induce neurotoxicity, chronic brain inflammation, and subsequent cognitive impairment in many primary human tauopathies, including Pick's disease (PiD), progressive supranuclear palsy (PSP), and corticobasal degeneration. 4 , 5 , 6 Despite NFTs being pathological hallmarks of AD and other human tauopathies, NFT‐containing neurons can survive for decades, 7 , 8 indicating that NFTs are neither obligatory nor sufficient to induce neuronal dysfunction and toxicity. In this context, our laboratory and others found that smaller, soluble tau oligomers (TauO) —not NFTs—are the most toxic tau species. 9 , 10 , 11 Neurons can extracellularly release TauO, which serve as “seeds” that promote spreading and propagation of tau pathology in a prion‐like fashion and induce cognitive deficits. 11 , 12 , 13 TauO are linked to Aβ and other pathological features of AD in several ways. Aβ can trigger the production of TauO by activating inflammatory pathways and impairing the clearance of tau. TauO can also enhance the toxicity of amyloid by facilitating its uptake and aggregation in neurons and it also promotes BBB damage, facilitating more inflammatory factors to enter the brain and worsen the neurodegeneration. TauO are also involved in synaptic dysfunction, which is a major cause of cognitive impairment in AD. 14 Additionally, we observed elevated levels of TauO in the brain, cerebrospinal fluid (CSF), and sera from AD patients with clinical severity, 9 , 15 , 16 , 17 , 18 , 19 , 20 suggesting that pathogenic TauO plays a crucial role in the etiology of AD and other tauopathies.
Aging is the primary risk factor for AD. AD affects one in every ten individuals aged 65 or older and its prevalence increases exponentially with advancing age. AD remains the fifth‐leading cause of death among individuals aged 65 or older. The Alzheimer's Association estimates that the number of AD cases in the US will increase from 6.7 million in 2023 to 12.7 million in 2060. 21 This will pose a huge burden on individuals, families, health care systems, and society. Current treatments for AD can only alleviate some of the symptoms, but they cannot stop or reverse the underlying brain damage caused by the disease. Therefore, there is an urgent need for new therapies that can modify the course of AD and prevent or delay its onset and progression. Recent studies in humans and animal models demonstrate that aging is associated with increased accumulation of senescent cells. Various endogenous and exogenous stressors (eg, pathological tau aggregates) can induce senescence in different types of brain cells. 22 , 23 , 24 , 25 Importantly, selective clearance of senescent cells extends the healthy lifespan in aged animals and also prevents AD‐associated brain pathology and cognitive decline, 22 , 23 , 24 , 25 suggesting that senescent cells drive aging and play a causal role in AD progression. These detrimental effects of senescent cells are mediated by an abnormal inflammatory secretome called the senescence‐associated secretory phenotype (SASP), which is linked to aging‐related chronic inflammation (ie, inflammaging). 26 , 27 , 28 Thus, TauO, along with senescence and inflammaging, have emerged as important potential therapeutic targets. These findings suggest that TauO, senescence, and inflammaging are crucial elements in AD and may cooperatively trigger the underlying complex pathomechanisms involved in AD progression and cognitive decline. 9 , 29 , 30 , 31 , 32 , 33
In this review, we provide insight into the pathomechanisms of TauO, senescence, and inflammaging and their subsequent impact on brain pathology and cognitive function in humans and animal models. We summarize mechanistic links that explain the involvement of TauO, senescence, and inflammaging in the progression and onset of tauopathies. Finally, we discuss potential therapeutic strategies for targeting senescence, inflammaging, and TauO for the future treatment of AD and related dementias.
2. CELLULAR SENESCENCE AS A COMPONENT OF AD PATHOLOGY
Population studies suggest that aging is the most significant risk factor for the progression and development of AD and neurodegenerative tauopathies. Lopez‐Otin et al. reviewed in detail the nine hallmarks of aging: cellular senescence, mitochondrial dysfunction, telomere attrition, genomic instability, epigenetic alterations, dysregulated nutrient sensing, loss of proteostasis, stem cell exhaustion, and altered intercellular communication. 34 These aging processes are critically involved in driving multiple neurodegenerative diseases, including AD and tauopathies. 35 Among these aging hallmarks, cellular senescence plays a crucial role in brain aging, neurodegeneration, and dementia 36 through the SASP, which represents a major source of inflammation. 37 Recent studies elegantly demonstrate that selective removal of senescent cells using genetic or pharmacological approaches extends the healthspan and lifespan of aged animals, 38 decreases chronic inflammation and brain pathology, and improves cognitive function, suggesting that cellular senescence contributes not only to aging but also to cognitive decline. In addition to several genetic and environmental factors, SASP or inflammaging may influence each other and increase the risk of AD (Figure 1).
FIGURE 1.
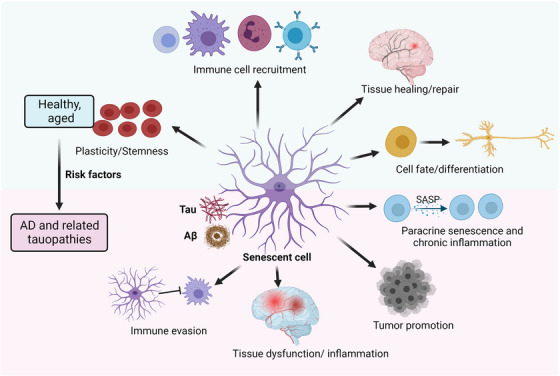
Cellular senescence as a component of healthy aging and AD. Recent studies suggest that increased inflammation in the brain of AD patients is driven by increased cellular senescence triggered by tau pathology and/or Aβ. Similarly, aging facilitates the accumulation of senescent cells, which contributes to chronic inflammation through SASP. Senescence‐induced inflammation may increase the risk of AD alongside additional genetic and/or environmental factors. Aβ, amyloid beta; AD, Alzheimer's disease; SASP, senescence‐associated secretory phenotype.
Cellular senescence was first discovered by Hayflick more than 60 years ago as a phenomenon of ceasing cell proliferation. 39 , 40 The concept of senescence was extended to incorporate premature senescence induced by oncogenic activation or exposure to cytotoxic agents. 41 , 42 Recently, cellular senescence has been redefined as a multi‐step and complex biological process involving irreversible cell cycle arrest, resistance to apoptosis, increased protein production, metabolic shifts (such as increased glycolysis, reduced fatty acid oxidation), enhanced reactive oxygen species (ROS) production, and acquisition of the SASP. Numerous processes, including DNA damage, telomer attrition, oncogenic mutation, oxidative stress, chronic inflammation/infection, and proteotoxic stress, are linked to or cause senescence, all of which also increase with aging. 43
RESEARCH IN CONTEXT
Systematic review: A literature review was performed by searching PubMed and meeting abstracts. An emerging body of evidence indicates a causal role of cellular senescence and inflammaging in neurodegenerative tauopathies including Alzheimer's disease (AD), suggesting great promise for therapeutically targeting senescence, tau oligomers, and brain inflammation. Relevant studies elucidating these links are discussed.
Interpretation: There are evident associations among brain inflammation, senescence, and tau oligomer pathology in AD progression and cognitive decline. Diverse and dynamic compositions of tau oligomeric strains, inflammatory secretome, and senescent cell types may influence the heterogeneity and rate of disease progression in tauopathies, suggesting the need for personalized and combination‐based therapeutic approaches.
Future directions: This review summarizes newly discovered cellular and molecular mechanisms in AD progression. Future studies should investigate the underlying mechanisms of diversity and dynamics of tau oligomeric strains, senescent cell types, and the inflammatory secretome.
Senescent cells exhibit permanent cell cycle arrest, resistance to apoptosis, and the DNA‐damage response (DDR) accompanied by a unique secretory phenotype that produces inflammatory and matrix‐degrading proteins known as the SASP. 44 Senescent cells show enlarged cell morphology and swollen lysosomal compartments, which allows detection of β‐galactosidase activity at a suboptimal pH of 6, commonly called senescence‐associated β‐galactosidase (SA‐β‐gal) activity. 45 Senescence can be triggered by various factors, including potential DDR inducers, oncogene activation and mutation, oxidative stress, metabolic insults, proteotoxic stress induced by Aβ and tau, and chronic inflammation associated with damage‐associated molecular patterns (DAMPs) and pathogen‐associated molecular patterns (Figure 2). Senescent cells evade apoptosis through the upregulation of proteins associated with the DDR (eg, γH2AX), anti‐apoptosis (eg, BCL‐XL/BCL‐W), and cell cycle arrest (eg, p16INK4A and p21Cip1). 45 , 46 , 47 Recent studies established the role of pathological tau and Aβ aggregates in triggering senescence in AD. TauO and Aβ aggregates and other stimuli predominantly activate DDR signaling, the main underlying cause of senescence. DDR triggers activation of several protein kinases, such as phosphorylated histone H2AX (γH2AX), apical kinases ataxia telangiectasia mutated (ATM) and ataxia telangiectasia and Rad3‐related protein (ATR). These signaling cascades initiate recruitment of DDR mediators, (eg, MDC1, 53BP1), activate CHK1 and CHK2 checkpoints, and instigate p53–p21Cip1 pathway activation, ultimately resulting in irreversible cell cycle arrest. 45 , 46 , 47 ,
FIGURE 2.
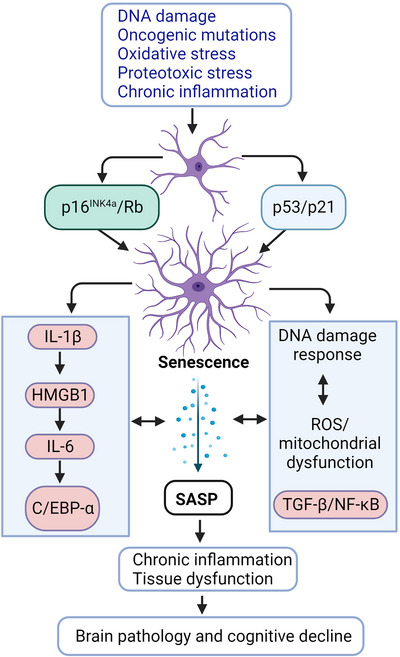
Several inducers alone or in combination can trigger senescence through p16INK4a/Rb and p53/p21 pathways. Senescence‐triggering signals may involve DNA damage (eg, telomere shortening), oncogenic mutation (eg, Myc, Ras), oxidative stress (eg, ROS), and proteotoxic stress (eg, Aβ and tau protein aggregation and protein misfolding). These senescence‐inducers contribute to chromatin remodeling and alterations in gene expression that underlie senescence‐associated cell cycle arrest and SASP. Intracellular autocrine signaling reinforces senescence and initiation of SASP. Senescence not only negatively impacts progenitor/stem cells but also contributes to tissue dysfunction, AD pathology, and subsequent cognitive decline through SASP, chronic inflammation, and loss of extracellular matrix. Aβ, amyloid beta; AD, Alzheimer's disease; ROS, reactive oxygen species; SASP, senescence‐associated secretory phenotype.
With advanced age, senescent cell loads increase in different tissues throughout the body, including the brain. 34 Senescent cells contribute to aging‐associated tissue deterioration and dysfunction and chronic human diseases. 26 , 48 , 49 , 50 Other main features of senescent cells involve chromatin remodeling due to loss of nuclear lamina protein lamin B1 and high‐mobility group box 1 (HMGB1), which is subsequently released into the extracellular microenvironment (Figure 2). As a result, the activation of innate immune responses leads to both acute and chronic inflammatory responses. Several recent high‐profile publications emphasize the targeting and clearance of tau and/or Aβ‐induced senescent cells as a new therapeutic approach to prevent neurodegeneration associated with AD. 22 , 24 , 25 Thus, in this review we examine the evidence linking senescence, inflammaging, and AD.
3. SASP: THE MAJOR SOURCE OF CHRONIC INFLAMMATION IN AGING
Although senescent cells account for only 4% to 15% of total tissue, they are a major contributor in the progression of aging‐related diseases. 51 Senescent cells are highly metabolically active and thus produce enormous amounts of SASP, which is one of the most important pathological factors inducing chronic inflammation during aging. 44 The SASP disrupts tissue homeostasis by inducing apoptosis or paracrine senescence in neighboring cells, thereby accelerating the process of biological aging. 52 The composition of the SASP is heterogeneous and dynamic and it varies depending on the cell type, tissue, and senescence inducer. 53 However, overall, the SASP performs three crucial functions. First, the SASP reinforces cell cycle arrest and perpetuation of its own phenotype via a positive feedforward mechanism, which explains the tumor‐suppressor activity of senescence. 54 , 55 Second, numerous components of the SASP transmit senescence to neighboring cells through a biochemical process called paracrine senescence. 56 , 57 Third, the SASP recruits immune cells to remove senescent cells from tissue and halt inflammation. Interestingly, although the immunosurveillance of senescent cells is advantageous in the context of cancer and embryonic development, 58 immunosurveillance of senescent cells in the aged brain may contribute to cognitive decline through natural killer cell‐mediated cytotoxicity and impaired neurogenesis. 59 Immune cell senescence impairs immune system function, reducing clearance of senescent cells. 60 , 61 Fundamentally, the SASP impairs tissue regenerative capacity through paracrine senescence in stem cells. 46 The SASP further exacerbates chronic inflammation and increases the risk of mortality and morbidity in older adults. 28 Increased levels of SASP factors are detected in human plasma in an age‐dependent manner, positively correlating with frailty and adverse postsurgical clinical outcomes. 62 More importantly, increased SASP levels are found in the blood and CSF of aged individuals and correlate with reduced neurogenesis in aging mice. Mimicking increased SASP in young adult mice impairs neurogenesis, hippocampal‐dependent learning, and memory. These observations suggest that the SASP from peripheral blood negatively impacts neurogenesis and cognitive functions. 63 In line with this evidence, a recent study using heterochronic blood exchange demonstrates that blood from aged mice induces cell and tissue senescence in young mice and treatment with senolytic drugs abrogates these progeronic actions of the SASP, indicating that SASP factors in the blood trigger rapid and robust spread of senescence. 64 Furthermore, clearance of senescent cells in aging mice attenuates the production of inflammatory mediators in circulation, underscoring that senescent cells are a key source of inflammation. 26 Conversely, the transplantation of a small number of senescent cells into young mice increases chronic inflammation, leads to persistent physical dysfunction, and decreases lifespan. 65 Therefore, the SASP is an important factor driving the biological aging process and the onset of chronic aging‐associated diseases, including neurodegenerative diseases. 66 SASP components have been shown to be induced by TauO and Aβ and to contribute to cognitive impairments in AD models or patients in various ways. Interleukin‐1β (IL‐1β) is a pro‐inflammatory cytokine that is part of the SASP and can be induced by Aβ and TauO in the brain. IL‐1β can activate microglia and astrocytes, which can further produce inflammatory mediators and exacerbate brain inflammation. IL‐1β can also impair synaptic plasticity and memory formation by affecting the expression and function of N‐methyl‐D‐aspartic acid (NMDA) receptors, which are important for learning and memory. 67 Recent studies have shown that IL‐1β production is regulated by the NLRP3 inflammasome, which is a multiprotein complex consisting of NLRP3, ASC, and caspase 1. The activation of the NLRP3 inflammasome drives tau pathology. 68 The knockout of ASC or NLRP3 ameliorates AD‐like brain pathology in animal models. 68 Tumor necrosis factor alpha (TNF‐α) is another pro‐inflammatory cytokine that is part of the SASP and can be induced by Aβ and TauO in the brain. TNF‐α can also activate microglia and astrocytes, and induce the production of ROS, which can damage neurons and synapses. TNF‐α can also promote the phosphorylation and aggregation of tau proteins, which can form neurotoxic oligomers and fibrils. TNF‐α can also impair synaptic transmission and plasticity by affecting the expression and function of AMPA receptors, which are important for synaptic strength and plasticity. 67 Transforming growth factor beta (TGF‐β) is another important component of Aβ and TauO induced‐SASP, and it can also enhance neuroinflammation, tau pathology, and neurodegeneration by activating the non‐Smad signaling pathway, which can induce oxidative stress, apoptosis, and autophagy impairment. 67
Most of the SASP proteins are evolutionarily conserved across cell types, but different cell types and tissues secrete distinct SASP compositions. 44 Further investigation is needed to determine whether different glial cells produce unique SASP components in AD pathogenesis and progression. The contribution of nonprotein SASP factors, such as bradykinins, ROS, or nucleotides remains poorly studied. It is worth noting that SASP composition may vary over time after senescence induction and depending on the stimulus or initiating mechanism, as observed in senescence induced by RAS, p16INK4a, and p53. 44 , 69 Therefore, SASP is dynamic and heterogeneous, with its composition, quantity, and extent depend on the specific cell type and inducer. In the context of brain cells, including neurons, astrocytes, microglia, oligodendrocytes, and endothelial cells, senescence phenotypes linked to Aβ or tau have been observed in the brains of animal models and human AD patients, indicating that SASP composition and dynamics may contribute to disease heterogeneity and the rate of cognitive decline. 70
4. HMGB1: AN INFLAMMATORY SASP COMPONENT AND PARACRINE SENESCENCE MEDIATOR
In this section, we will discuss the role of HMGB1 in senescence, inflammation, and AD pathogenesis. HMGB1 is a highly conserved non‐histone DNA‐binding protein belonging to the HMG protein family. It was named because of its rapid electrophoretic mobility. 71 In mammalian nuclei, HMGB1 is the most abundant non‐histone protein, with one HMGB1 molecule per 10 nucleosomes. 71 Despite its high abundance and conservation in nuclei, HMGB1 has been extensively studied as an extracellular inflammatory factor and DAMP and is hence known as an “alarmin.” 72 , 73 When there is an infection, injury, or Aβ or tau pathology, HMGB1 is actively secreted by activated immune cells or passively released by necrotic and damaged cells to serve as an alarmin. Extracellular HMGB1 interacts with toll‐like receptor 4 (TLR4) and receptor for advanced glycation end products (RAGE) receptors, triggering chronic inflammation. 74 , 75 This extracellular HMGB1 contributes to chronic inflammation, 72 , 73 which could slow or stop tissue regeneration and homeostasis, ultimately causing tissue deterioration. In the context of senescent cells, HMGB1 undergoes translocation from the nucleus to the cytosol, where it is released into the extracellular space to trigger inflammation via RAGE and TLR4 signaling pathways. HMGB1 serves as an early and central mediator of senescence phenotypes in both human and mouse tissue, and studies have shown that HMGB1 relocalization and secretion regulate the SASP and thereby contribute to paracrine senescence both in vitro and in vivo. 56 , 76 , 77 In proliferating cells, HMGB1 plays a crucial role in DNA repair, V(D)J recombination, and chromatin assembly. HMGB1‐deficient cells have fewer nucleosomes, making chromatin more susceptible to DNA damage, dysregulated transcription, and inflammatory response. The protein binds chromatin, bends and twists DNA, and facilitates the binding of transcription factors such as p53. 78 Neither p53 nor pRB is essential for SASP, and in fact p53 deficiency further amplifies SASP. 44 , 69 Upstream signaling molecules of p53 (such as H2AX, CHK2, NBS1, ATM) play an indispensable role in the SASP and also function as DDR proteins. 79 , 80 Recent studies have revealed that HMGB1 coordinates SASP‐associated chromatin folding and RNA homeostasis, thereby regulatiing autocrine and paracrine senescence. 81 The depletion of HMGB1 from the nuclei and its subsequent secretion is a hallmark of cellular senescence in mammalian cells. In contrast, continuously proliferating cancer cells exhibit higher levels of HMGB1 in nuclei than those in normal tissue, suggesting that loss of nuclear HMGB1 is a key event in the progression of the senescence pathway. HMGB1 binds and regulates a distinct set of mRNA transcripts that are implicated in senescence regulation. 81 Senescent cells in culture are also known to actively release HMGB1, and depletion or knockdown of HMGB1 induces senescence. Further, quantitative analysis of HMGB1 shows higher serum levels of the protein in aged mice than in younger mice, 76 indicating that depletion of nuclear HMGB1 and its active release is an important conserved feature of senescent cells in both mice and humans. 76 , 82
Additionally, we and others demonstrated that extracellular HMGB1 promotes paracrine senescence in neighboring cells and induces chronic tissue inflammation via the SASP, 56 , 57 suggesting that senescence can be transmitted through the SASP/HMGB1 axis. Schafer et al. investigated the correlation between SASP components in the blood and aging‐related dysfunction in humans. 62 They reported positive correlation between SASP components and age, frailty, and postsurgical health deficits, indicating that circulating levels of SASP are linked to advanced aging and detrimental health outcomes. These findings suggest that SASP could serve as a clinical biomarker and therapeutic target for advanced age‐related diseases. 83 , 84 Importantly, experimental evidence suggests that the presence of nuclear HMGB1 is necessary for survival, as HMGB1 knock‐out mice die shortly after birth due to severe hypoglycemia. 85 Loss of nuclear HMGB1 is also linked to organ failure in mice and humans. 86 We reported that in postmortem brain tissue from AD patients, translocation of HMGB1 from the nucleus to the cytoplasm is a molecular signature of senescence and brain inflammation. 57
HMGB1 is thought to be involved in the pathogenesis and progression of AD by promoting brain inflammation, Aβ and tau pathology, neurite degeneration, and cognitive impairment. HMGB1 mainly triggers paracrine senescence and inflammation via microglia and astrocytes. This chronic inflammation promotes tau phosphorylation and aggregation and increases their accumulation in the cytoplasm, leading to synaptic and neuronal dysfunction and death. 57 , 87 It is worth mentioning that elevated HMGB1 levels are found in the brain and CSF of AD patients and aged rodents and correlate with disease severity. 88 , 89 , 90 Importantly, exposure to pathological protein aggregates such as Aβ and TauO triggers HMGB1 release, chronic inflammation, and upregulation of RAGE and TLR4 signaling pathways. 57 Using a 5XFAD transgenic mouse model of AD, researchers have demonstrated that extracellular HMGB1 initiates neurite degeneration through the TLR4‐MARCKS pathway in early AD. 89 , 91 Furthermore, treatment with anti‐HMGB1 antibodies or HMGB1 release inhibitors decreases AD‐associated neuropathology, reduces the burden of senescent cells in the brain, alleviates neuroinflammation, and improves cognitive function in AD mouse models. 89 Notably, significant neuronal necrosis occurs at very early stages of presymptomatic AD; hence, HMGB1 levels are significantly elevated in clinically diagnosed presymptomatic AD but not in symptomatic advanced‐stage AD. 91 Upon neuronal necrosis, pathological Aβ and TauO are released into the extracellular space and become seeds for the propagation of AD neuropathology in a prion‐like fashion. Moreover, evidence from AD models have demonstrated that targeting HMGB1, RAGE, or TLR4 with antibodies, inhibitors, or antagonists can reduce neuroinflammation, Aβ and tau pathology, and improve cognitive function. 92 Together, these findings highlight a crucial contribution of HMGB1 to AD pathogenesis by triggering paracrine senescence, inflammation, tau pathology, and clinical severity. Therefore, targeting HMGB1, in combination with TauO, could hold promise as a therapeutic strategy for AD. Our lab recently has shown that inhibiting release of HMGB1 using ethyl pyruvate and glycyrrhizic acid alleviates tau oligomer‐induced senescence like phenotype, neuroinflammation and cognitive decline in tauopathy mice model. 54 Further research efforts are needed to discover new molecules that can effectively target HMGB1 or its endogenous regulators.
5. INFLAMMAGING: A KEY CONTRIBUTOR TO ACCELERATED AGING
Inflammaging refers to aging‐associated elevated levels of inflammatory markers in blood and tissues, which lead to chronic low‐grade inflammation. 93 It is an important pathological factor in several diseases, including AD and tauopathies that are highly prevalent in elderly individuals, contributing to disability and accelerated aging. Inflammaging serves as a biomarker of accelerated aging in humans and can worsen the natural age‐related tissue deterioration through paracrine mechanisms. 93 Inflammaging can drive tau pathology and neurodegeneration in AD by impairing the clearance of Aβ peptides, altering BBB integrity, and affecting the synaptic functions. 68 Levels of inflammatory cytokines, such as IL‐6, TNF‐α, and IL‐1β, increase along with immune system dysregulation (ie, loss of vaccine efficiency, increased morbidity in infection/injury, increased incidence of cancer and autoimmune diseases). 28 , 94 Studies demonstrate a positive correlation between levels of circulatory inflammatory molecules and the progression of neurodegenerative diseases. 95 , 96
Inflammaging can drive tau pathology and neurodegeneration in AD by several mechanisms. 68 , 97 One mechanism is that inflammaging impairs the clearance of Aβ and tau aggregates, which can trigger the activation of microglia and astrocytes, and induce the production of pro‐inflammatory cytokines and ROS. These inflammatory mediators can damage neurons and synapses, and also promote the tau phosphorylation and aggregation that ultimately impair axonal transport, synaptic function, and neuronal viability. 97 Another mechanism is that inflammaging impairs BBB integrity thereby increasing entry of peripheral inflammatory factors, such as cytokines, chemokines, and immune cells, into the brain. These factors can further amplify brain inflammation and exacerbate tau pathology and neurodegeneration in AD. A third mechanism is that inflammaging affects the balance between pre‐ and post‐synaptic tau levels, and impairs the interactions of tau with other synaptic proteins, such as Fyn kinase, PSD‐95, and NMDA receptors. 97 These alterations can impair synaptic transmission and plasticity, and lead to memory impairment. 97
SASP components, including C‐reactive protein and IL‐1 family cytokines, have been proposed as markers of disease progression in AD. 98 In the aging brain, neurotoxic misfolded protein aggregates, such as Aβ and tau, are recognized by microglia—the resident macrophages of the brain—and astrocytes—the neuronal supporting cells—through highly conserved pattern recognition receptors (eg, TLR4). This exacerbates neuronal toxicity, protein aggregation, synaptic dysfunction, and chronic brain inflammation, creating a feedforward loop that amplifies and sustains brain inflammation. This detrimental cycle further contributes to the structural and functional deterioration of the brain, fueling the progression of neurodegeneration. The chronology of AD pathology and inflammation is described as a wave model with multiple interconnected cellular and molecular processes that accelerate neurodegeneration (Figure 3).
FIGURE 3.
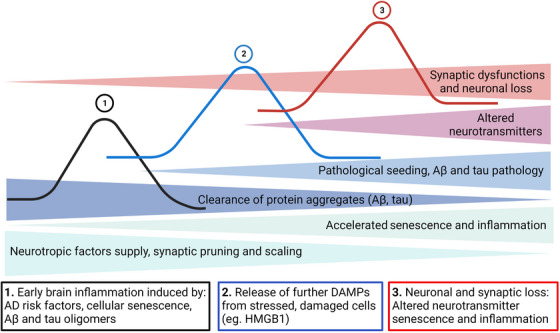
Three‐wave model showing chronology of pathological events in AD. Multiple intermingling cellular and molecular processes could influence the onset and progression of AD. For example, recognition of pathological protein aggregates (Aβ and tau oligomers) by glial cells is followed by induction of the inflammatory response and release of endogenous DAMPs (eg, HMGB1) that aggravate AD progression. Aβ and tau oligomers impair proteasome functions that compromise the clearance of protein aggregates and facilitate their release into the extracellular space, which causes pathological seeding and propagation of AD pathology and further fuels the inflammatory response. Chronic inflammatory responses, in turn, trigger neuronal dysfunction and death. Aβ, amyloid beta; AD, Alzheimer's disease; DAMPs, damage‐associated molecular patterns.
Aβ and tau exist in different aggregation stages, including monomeric, oligomeric, fibrils, and plaques/tangles, each with varying effects on glial activation and chronic brain inflammation. The impaired clearance of these pathological protein aggregates leads to the seeding and propagation of AD pathology. Glial activation triggers the release of inflammatory cytokines, such as IL‐1β, TNFα, and IL‐6, that can be found in the brain and CSF of AD patients. Overexpression of these inflammatory mediators activates neuronal signaling cascades that trigger activation of protein kinases like cyclin‐dependent kinase 5, along with deactivation of phosphatases like PP1. This results in tau phosphorylation and formation of neurotoxic oligomers and fibrils, further amplifying glial activation and brain inflammation. 99 The excessive release of neurotoxic inflammatory mediators negatively impacts neurotransmitter levels, leading to neuronal dysfunction and death (Figure 4), suggesting Aβ and tau pathology and inflammaging together contribute to AD progression. Therefore, modulating inflammaging/SASP may represent a promising therapeutic approach for AD and tauopathies.
FIGURE 4.
Schematic diagram of AD pathogenesis. Several risk factors, such as aging, genetics, sleep deprivation, stress, diet, and head injury, can influence AD progression via chronic immune activation and cellular senescence. This, in turn, leads to aging‐associated chronic inflammatory responses, also known as inflammaging, which allows penetration of inflammatory molecules (pathogen‐ and/or damage‐associated molecular patterns, eg, lipopolysaccharide and HMGB1) through the BBB via glial senescence. In the brain, risk factors and inflammation impact neuronal homeostasis and induce Aβ and tau aggregation to form oligomers that cause neuronal insults, which further fuels brain inflammation. Additionally, disruption of the BBB facilitates the entry of peripheral inflammatory molecules and cells into the brain, which promotes gliosis and glial senescence followed by chronic brain inflammation. These events lead to the progression of AD pathology, neuronal and synaptic dysfunction, and cognitive decline. Aβ, amyloid beta; AD, Alzheimer's disease; BBB, blood‐brain barrier; SASP, senescence‐associated secretory phenotype.
6. BRAIN INFLAMMATION INDUCED BY TAU, SENESCENCE, AND INFLAMMAGING CONTRIBUTES TO AD
Clinical AD symptoms and dementia typically manifest after a prolonged preclinical or prodromal AD stage, suggesting that identifying and targeting early pathogenic events could halt or delay the progression of AD. 31 Clinical evidence shows that TauO and brain inflammation also appear in the preclinical or prodromal AD stage, many decades prior to the onset of dementia. 100 This early brain inflammation may be exacerbated or driven by TauO and senescent cells in the brain and often linked to cognitive decline. Supporting this notion, one study reports that brain inflammation contributes to early memory loss, increases brain inflammation, and a higher load of senescent cells in the brain. 101 It establishes a connection between the other two key pathological features, Aβ and tau aggregates. 102 A growing body of evidence suggests that inflammation has an intricate relationship with tau pathology that contributes to AD pathology and cognitive decline. 103 Importantly, treatment with ibuprofen, a nonsteroidal anti‐inflammatory drug, markedly reduces brain inflammation, senescent cell load and improves cognitive function. These observations suggest that senescence‐induced chronic brain inflammation is an important pathological factor in aging‐related cognitive decline. 101 Brain inflammation can also trigger deposition of pathological amyloid proteins via upregulation of interferon‐induced transmembrane protein 3 (IFITM3). 104 Of note, IFITM3 mediates paracrine senescence through small extracellular vehicles released by senescent cells in the aging brain, 105 further supporting the crosstalk among amyloid protein deposition, chronic inflammation, and senescence in brain aging. These findings support the notion that inhibiting brain inflammation could halt or delay the progression from mild cognitive impairment (MCI) to dementia. The involvement of senescence and SASP in MCI and AD is further strengthened by experimental evidence that senolytic therapy provides beneficial effects in accelerated aging, 106 tauopathy, 22 and AD mouse models 25 by reducing brain inflammation. The ability of senolytic therapy to modulate the progression of AD is also being evaluated in a pilot clinical trial. 32 Besides aging, other important sources of brain inflammation are linked with AD risk factors. For example, traumatic brain injury (TBI) induces brain inflammation through acute and chronic alterations in the immune system and increased susceptibility to infections. 107 Importantly, TBI also triggers the formation of pathological TauO 108 , 109 and upregulation of senescence biomarkers in glial cells, thus establishing links among TBI, TauO, and cellular senescence. 108 , 110 However, further research is necessary to understand the underlying mechanisms of TBI‐induced senescence and inflammation and its role in AD pathogenesis.
Additionally, numerous factors from the periphery reach the brain as a result of aging‐associated vascular damage, representing another important source and stimulus of chronic brain inflammation. 111 For example, cerebral small vessel disease increases the risk of cognitive decline and dementia in AD. 112 Chronic inflammation‐induced disruption of the BBB is an early biomarker of human cognitive dysfunction and often leads to dementia, 113 and both Aβ deposits and tau pathology aggravate vascular damage. 114 , 115 Pericytes play a role in cerebral blood flow regulation and support BBB function, are important for maintaining brain homeostasis, 116 and their loss is known to influence AD‐associated neurodegeneration. 117 It has been argued that the accumulation of Aβ and tau are causally linked to pericyte dysfunction, which contributes to reduced cerebral blood flow in early AD. 114 , 118 Interestingly, pericytes communicate with neurons primarily through the chemokine ligand 2, a critical component of the SASP that mediates paracrine senescence. 56 Additionally, recent evidence suggests that obesity 119 and hyperinsulinemia 120 can also trigger senescence and chronic inflammation, impairing brain homeostasis.
Genome‐wide association studies have revealed that various polymorphic variants of genes implicated in inflammation serve as risk factor for AD, suggesting an important role of inflammation in AD. 121 Two important genes linked to AD risk are APOE and TREM2. 122 Studies using an AD mouse model along with postmortem analyses of human AD brain tissue indicate that activation of the TREM2‐APOE pathway triggers transition in microglia from a homeostatic phenotype to a disease‐associated microglia (DAM) phenotype characterized by an increased inflammatory response. 123 Interestingly, senescence and the SASP facilitate emergence of the DAM phenotype and contribute to AD pathology. 23 Together, these studies emphasize that brain cells undergo senescence upon exposure to TauO and Aβ and systemic inflammatory factors, thereby triggering further brain pathology, inflammatory responses, and cognitive decline.
7. EVIDENCE OF SENESCENCE IN DIFFERENT CELL TYPES IN THE AD BRAIN
Based on the current literature, evidence of cell types that undergo senescence in the AD brain remains sparse. In this section, we briefly discuss senescence in different types of brain cells in the context of AD.
7.1. Microglia
Microglia are the first responders and innate immune cells of the brain. They are involved in surveillance of the brain and actively eliminate cell debris and pathogenic molecules through phagocytosis, thus playing key roles in brain homeostasis. Numerous studies report aging‐related changes in microglia. Senescent microglia are linked to the spreading of pathological tau and neurodegeneration in AD. 124 Compared with the brains of non‐demented age‐matched control individuals, the number of senescent microglia is higher in the brains of patients with AD, dementia with Lewy bodies (DLB), and limbic‐predominant age‐related TDP‐43 encephalopathy. 125 Under pathological conditions, microglia show typical characteristics of senescence such as increased oxidative stress and nuclear chromatin alterations. 126 During the onset of Aβ and tau pathology, microglia show rapid proliferative responses and contribute to disease progression. 127 , 128 , 129 Interestingly, microglia that undergo rapid proliferation initially can later switch to a DAM phenotype, characterized by harmful inflammatory responses. 129 DAM also display typical characteristics of senescence such as increased SA‐β‐gal activity, telomer shortening, and transcriptional signatures. 23 These studies indicate that in the brains of individuals with AD, a subset of microglia could exhibit a senescent phenotype that drives inflammation and neurodegeneration.
7.2. Astrocytes
Astrocytes are highly abundant glial cells in the brain that provide metabolic, structural, and functional support to neurons. They are involved in regulating neuronal trophic support, ionic homeostasis, brain inflammation, and cognitive function. 130 Astrocytes also control oxidative stress, the release of growth factors, and contribute to the establishment and maintenance of BBB integrity. 131 The astrocytic end feet form an important part of the perivascular network and the glymphatic system, facilitating the exchange of interstitial fluid and CSF. 132 Due to their close proximity to neurons, astrocytes facilitate neurotransmission between multiple neurons via a tripartite synapse and are involved in neurotransmitter recycling. 133 The collective evidence underscores the essential role of astrocytes in supporting neuronal function.
Astrocytes in AD undergo senescence, playing a crucial role in the development and progression of AD pathology and cognitive decline. 22 , 49 , 57 , 134 , 135 Senescent astrocytes lose their neuroprotective functions and acquire neurotoxic functions, leading to neuronal dysfunction and neuropathology. Astrocyte senescence negatively impacts neurons through gain of the SASP or loss of metabolite transferring ability, which is crucial for neuronal support. Astrocyte senescence induces neurotoxic effects through the gain of inflammatory SASP and contributes to the etiology of neurodegenerative diseases, including AD, PSP, and PiD. 135 , 136 Senescent astrocytes are characterized by increased levels of glial fibrillary acidic protein and vimentin protein, upregulation of SA‐β‐gal activity, p16INK4a expression, SASP, and loss of nuclear lamin B1 and HMGB1. 70 , 137 A single‐nucleus RNA sequencing study using the 5XFAD AD mouse model revealed that senescent astrocytes are inflammatory disease‐associated astrocytes and show features of senescence and an inflammatory phenotype, which appears at the early stages of AD pathology, and their abundance increases with the progression of disease. 138 This study also demonstrates the presence of senescent astrocytes in aging human brains, suggesting a link among inflammatory disease‐associated astrocytes, aging, and AD pathology. Additionally, upon exposure to oxidative stress and X‐ray irradiation, primary human astrocytes undergo senescence in vitro and show senescence biomarkers such as SA β‐gal activity and increased levels of p16INK4A and p21. This senescence induction in astrocytes downregulates glutamate and potassium transporters, which leads to excitotoxicity and neuronal cell death. 139 , 140 Analysis of postmortem brain tissue confirms an exacerbated load of senescent astrocytes in human AD brains. 49 , 57 , 135 , 141
Bussian et al. found that the burden of senescent astrocytes (16%) is higher than that of senescent microglia (4%) in the brain of animals with tauopathy. 22 This study demonstrated that selective clearance of these senescent glial cells slows the progression of tauopathy and cognitive impairment, suggesting that senescent astrocytes play a key role in tauopathy progression and cognitive decline. 22 However, it is possible that microglia—as first responders—undergo senescence first, which then induces astrocyte senescence through paracrine mechanisms. Our recent study demonstrated that astrocytes internalize pathological TauO from the extracellular environment. TauO induces a senescence phenotype in primary astrocytes and triggers a chronic inflammatory response through secretion of IL‐1β, IL‐6, and TNF‐α. It is also noteworthy that TauO‐associated astrocytes show a senescence phenotype in human AD and frontotemporal dementia brains. 57 The accumulation of senescent cells is associated with neurovascular dysfunction, which manifests as cognitive decline. 142 These results suggest that the accumulation of TauO and senescent astrocytes in the brain, along with the deleterious consequences of SASP, contributes to the progression of AD pathology and cognitive decline. Hence, a combined approach targeting TauO, senescent astrocytes and/or SASP could effectively delay the progression of AD and cognitive decline.
7.3. Oligodendrocytes
Oligodendrocytes, the third major type of glial cells in the brain, originate from oligodendrocyte progenitor cells (OPCs). 143 Upon brain injury/pathology, OPCs migrate to the site of brain damage and differentiate into oligodendrocytes to facilitate the remyelination of damaged axons. 36 However, the proliferation of OPCs is impaired with advanced age, leading to failure in the remyelination capacity of oligodendrocytes that contributes to disease progression. 144 , 145 In this context, a recent study demonstrated that Aβ triggers a senescence phenotype in OPCs in the brains of AD patients as well as in an APP/PS1 transgenic mouse model of AD. 25 These observations are consistent with earlier reports suggesting that pathological conditions can promote OPC senescence. 146 Further investigations are required to determine whether OPC senescence is a specific response to Aβ or whether other pathological proteins like tau, α‐synuclein, and TDP‐43 can also induce OPC senescence.
In general, compared with microglia and astrocytes, the role of OPCs and oligodendrocytes in AD pathogenesis is not as well studied. Thus, investigating how OPCs and oligodendrocytes respond to AD pathology will greatly enhance our understanding of AD pathogenesis. A study by Zhang et al. demonstrated that senolytic treatment using the FDA‐approved small molecules dasatinib and quercetin (D+Q) prevents Aβ accumulation and OPC senescence, resulting in improved learning and memory and reduced inflammation. Notably, this study also revealed that D+Q can prevent Aβ plaque buildup prior to the appearance of senescence, suggesting a non‐targeted effect of D+Q. It is important to note that quercetin has been shown to prevent the release of HMGB1 and reduce inflammation, thereby improving cognitive function in various AD models. 147 , 148 , 149 Future investigations are required to determine the senolytic and non‐senolytic effects of D+Q in AD models and clinical trials. Collectively, these studies suggest that D+Q treatment may be an effective strategy for preventing or delaying AD pathogenesis by reducing Aβ accumulation, OPC senescence, and HMGB1‐mediated inflammation and paracrine senescence.
7.4. Neurons
Neurons are the fundamental cells responsible for transmitting and processing information in the brain. Upon exposure to toxic Aβ and TauO, most neurons die; however, some neurons abnormally re‐enter the cell cycle, which protects them from cell death. 150 This raises the possibility that during pathological conditions, neurons can undergo senescence. In this regard, the prevalence of senescent neurons is reported to increase with advanced age, indicating a potential role of neuronal senescence in brain aging and neurodegenerative diseases. 151 Interestingly, senescent cells are resistant to apoptosis, indicating that senescence can promote other mechanisms of neuronal death such as necroptosis, which is linked to neuronal loss in the AD brain. 152 Although neurons are considered terminally differentiated cells, whether they undergo senescence remains a debatable question. Studies have shown conflicting results; Bussain et al. demonstrated that neurons do not exhibit a senescence phenotype in a tauopathy mouse model. 22 On the other hand, Musi et al. showed that tau accumulation is linked to neuronal senescence in another tauopathy mouse model. 24 The evolutionary mechanism that favors senescence over apoptosis in neurons may have benefits in terms of preserving cell numbers in the brain. However, this could come at the cost of limited regenerative potential and the promotion of deleterious SASP factors. 153 Further research is needed to elucidate the role of neuronal senescence and its contribution to AD. Understanding the interplay between senescence, apoptosis, and other forms of neuronal cell death will provide valuable insights into the mechanisms underlying neurodegeneration and may guide the development of novel therapeutic strategies.
7.5. Endothelial cells
Vascular dysfunction, particularly endothelial dysfunction, plays an important role in AD pathogenesis and progression. Studies have shown that senescence induction in cerebromicrovascular endothelial cells leads to endothelial dysfunction and BBB disruption, which in turn promotes cognitive impairment. 154 A study by Bryant et al. demonstrated that compared with age‐matched control patients, B3 microvessels from AD patients have markedly elevated levels of senescence markers and adhesion proteins, which are specifically linked to tau pathology. 155 Cerebral small vessel disease significantly raises the risk of cognitive decline and the onset of dementia in AD cases. 112 Pericytes play a vital role in regulating cerebral blood flow and supporting the integrity of the BBB. 116 Their depletion has been associated with the progression of neurodegeneration linked to AD. 117 It has been proposed that the accumulation of both Aβ and tau are interconnected with pericyte malfunction, which contributes to the decline in cerebral blood flow seen in the early stages of AD. 114 , 118 Interestingly, pericytes communicate with neurons primarily through the chemokine ligand 2, a key player in the SASP that orchestrates paracrine senescence. 56 Furthermore, emerging research highlights that conditions such as obesity 109 and hyperinsulinemia 110 can also induce senescence and chronic inflammation, thus impairing the brain's equilibrium and homeostasis. 119 , 120 This finding suggests that pathological tau‐induced endothelial cell senescence could be involved in AD pathogenesis. Therefore, detailed investigations are needed to establish the role of endothelial senescence in AD.
8. IMPLICATIONS OF PATHOLOGICAL TAU STRAINS IN DISEASE HETEROGENEITY, DIVERSITY OF CLINICAL SYMPTOMS, NEUROPATHOLOGY, AND RATE OF COGNITIVE DECLINE
Although tau deposition is a common pathological hallmark in neurodegenerative tauopathies, cases exhibit clinical and neuropathological heterogeneity, leading to different rates of cognitive decline. 156 , 157 Advanced neuroimaging methods have enabled the detection of tau and amyloid‐beta (Aβ) deposition even in younger adults aged 30 to 49 years. 158 In the last decade, numerous studies have explored the notion that tau and other amyloid‐forming proteins contribute to AD in a prion‐like fashion. The groundbreaking discovery of neurodegenerative diseases mediated by the propagation of unique protein conformations, as exemplified by prion diseases, was awarded the 1997 Nobel Prize in Physiology or Medicine to Stanley Prusiner. In AD, pathological tau is released in the interstitial fluid by neurons, propagating and spreading tau pathology in a stereotypical anatomical pattern. 159 Extensive evidence from animal and cell models convincingly supports the prion‐like properties of tau, acting as a seed that disseminates tau pathology across the brain. 159 , 160 , 161 The spread of tau pathology in the brain positively correlates with the heterogeneity and severity of clinical symptoms. 3 , 4 Consequently, tau aggregates can form distinct prion‐like strains or conformations that influence disease phenotypes, symptoms, and neuropathology in neurodegenerative tauopathies. Recent high‐profile studies have revealed that despite a relatively uniform anatomical pattern of tauopathy progression, AD patients show significant heterogeneity in clinical symptoms, neuropathology, and rate of cognitive decline. 3 , 4 Importantly, patient‐to‐patient heterogeneity can be attributed to different seed competent TauO confirmations (ie, TauO strains). In AD patients, deposition of tau aggregates in the brain strongly correlates with neuronal loss, cognitive decline, and dementia. Notably, tau aggregation alone can cause cognitive decline. 3 , 4 Pathological tau deposits display distinct cell and brain region specificities and distribution patterns in various tauopathies. Biochemical and morphological differences suggest that pathological tau adopts disease‐specific structural conformations akin to prion strains. Recent advances in cryo‐electron microscopy have unveiled atomic structures of pathological tau filaments isolated from the brains of patients with different tauopathies. Each neurodegenerative tauopathy is characterized by a distinctive tau filament conformation highly conserved across patients with the same disease. 162 , 163 , 164 In vitro aggregation studies have also demonstrated the propagation of distinct tau strains from seeds in the brain. Furthermore, the injection of these tau strains in mouse brain recapitulates the unique tau pathology found in specific cell types and brain regions in human tauopathies. 165 The composition of tau isoforms, post‐translational modifications, and interactions with cofactors may influence formation of tau strains in the brain, ultimately contributing to diversity of clinical phenotype and the rate of cognitive decline. We speculate that tau strains differentially propagate in the brain in a cell type‐ and region‐specific manner, influencing the clinical and pathological heterogeneity observed in patients with tauopathies (Figure 5). Understanding tau strain formation and its impact on neuronal and glial cells will be critical to deciphering the molecular mechanisms that underlie the clinical and pathological diversity in human tauopathies. Notably, other neuropathological amyloid proteins such as Aβ and α‐synuclein also exhibit seeding activities. Moreover, Aβ, tau, and α‐synuclein interact with each other, leading to co‐pathologies and an accelerated neuropathological burden in the brain. In Aβ‐expressing mice, inoculation of human wild‐type tau protein downregulates genes involved in synaptic regulation as well as behavioral abnormalities, indicating that Aβ and tau cooperatively contribute to progression of synaptic and cognitive impairment. 166 A study by Narasimhan et al. demonstrated that different tau strains isolated from human tauopathy brains were associated with diverse tau neuropathology. This study revealed that an increase in the tau seeding activity of a particular TauO strain (with high seeding activity), but not others, correlated with worse clinical outcomes in AD patients, suggesting the existence of TauO strains in different individuals with typical AD. 167 These findings highlight the possibility that AD, like cancer, involves multiple molecular drivers of common disease phenotypes. They also underscore the potential for personalized therapeutic approaches tailored to target specific tau strains and halt the clinical progression of AD. 167
FIGURE 5.
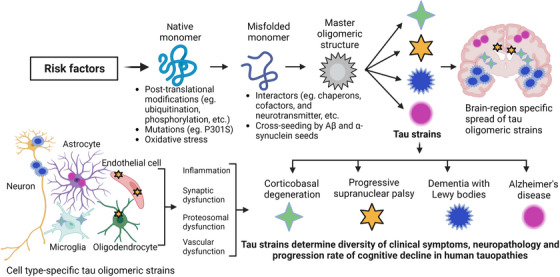
Schematic diagram showing potential mechanism for the formation of TauO strains and their implications in disease heterogeneity, diversity of clinical symptoms, neuropathology, and progression rate of cognitive decline in human tauopathies. Various risk factors (eg, aging, genetic background) induce post‐translational modifications (eg, ubiquitination, phosphorylation) and oxidative stress that triggers tau misfolding. This misfolded tau can interact with Aβ and α‐synuclein seeds, chaperons, co‐factors, and neurotransmitters, resulting in the formation of distinct misfolded monomers that aggregate and form different TauO strains. Distinct conformations of TauO strains lead to different seeding activities as well as brain region and cell type specificity. As a result, tau strains differentially induce inflammation, synaptic dysfunction, proteasomal impairment, and vascular dysfunction, which contributes to diversity in human tauopathies. Aβ, amyloid beta; TauO, tau oligomers.
Like tau, α‐synuclein and Aβ oligomers also exhibit distinct strains that influence disease phenotypes. 168 , 169 , 170 Intriguingly, after local and systemic injections, α‐synuclein strains can also induce distinct synucleinopathies with varying levels of toxicity and behavioral phenotypes. 171 Furthermore, cross‐seeding activities between aggregation‐prone proteins such as tau and α‐synuclein can lead to the formation of distinct amyloid strains. Given the coexistence of α‐synuclein and tau aggregates in Parkinson's disease and AD, it is plausible that they promote cross‐seeding of each other. In support of this notion, our previous study demonstrated that α‐synuclein oligomers induce a distinct toxic strain of TauO that evades fibril formation. Injection of α‐synuclein/TauO into the brains of a tauopathy mouse model accelerates endogenous TauO formation and concurrent neurodegeneration, thereby contributing to disease progression. 172 In support of this notion, the co‐occurrence of different disease‐associated protein aggregates is frequently observed in human neurodegenerative diseases. For instance, over 50% of AD patients exhibit Lewy bodies, while Aβ plaques and NFTs are commonly detected in Parkinson's disease and DLB brains. 173 Postmortem brain analyses of 161 clinically normal individuals revealed that 86% exhibited at least one pathology, with 63% displaying mixed pathologies. 174 Additionally, a meta‐analysis involving 4677 individuals reported that one‐third of those with intermediate‐to‐high AD neuropathology did not develop dementia during their lifetime. 175 Histological studies suggest that individuals with high AD neuropathology and normal cognition show resistance to synaptic degradation. In our recent study, we demonstrated that the functional integrity of synapses in cognitively normal individuals with moderate‐to‐high AD neuropathology is linked to an absence of synaptic TauO. 176 This study further supports the critical role of TauO in synaptic dysfunction and cognitive impairment in AD patients. Interactions of TauO with α‐synuclein, Aβ, and TDP43, leading to comorbidities and mixed neuropathology, often contribute to disease heterogeneity and differences in the progression rates of clinical symptoms in AD patients. The mechanisms underlying how these co‐pathologies contribute to disease progression and clinical heterogeneity remain open questions that warrant further investigation. Detailed discussions on the molecular interactions among Aβ, tau, and α‐synuclein and their contributions to neurodegeneration can be found in our recent review. 177 In this context, it is plausible that one form of protein aggregates may directly cross‐seed other amyloidogenic proteins, resulting in the generation of distinct strains that have common structural features but different seeding activities and toxicity. 178 , 179 It is likely that distinct strains of pathological tau and α‐synuclein exist in AD brains, contributing to the remarkable disease heterogeneity observed. This suggests the presence of a variety of dominant oligomeric strains across different proteinopathies, underpinning disease progression with various manifestations and subtypes. In this context, significant advancements have been made in the development of novel anti‐tau antibodies that specifically recognize disease‐relevant tau strains and conformations. These antibodies target unique conformational epitopes, independent of specific linear amino acid sequences, and exhibit preferences for different subsets of tau oligomers. 16 , 29 , 180 , 181 , 182 , 183 Furthermore, recent studies have introduced a rapid strain identification method called EMBER (excitation multiplexed bright emission recording). EMBER utilizes fluorescent excitation/emission spectra of amyloid‐binding dyes and employs machine learning techniques to identify distinct conformational strains in vitro and in transgenic mouse models. Notably, EMBER enables the swift identification of conformational differences in Aβ and tau deposits in human brain slices derived from various neurodegenerative diseases, including sporadic and familial AD, Down syndrome, as well as PiD. 184 These remarkable findings significantly contribute to our understanding of protein misfolding diseases and hold promising implications for future advancements in diagnostic and personalized therapeutic approaches in this field. Furthermore, it is hypothesized that TauO strains may differentially induce senescence and SASP in a cell type‐ and brain region‐specific manner, further contributing to the clinical heterogeneity and variable rates of disease progression. Therefore, future in‐depth investigations into tau strains within the context of the clinical disease heterogeneity observed in AD and related dementias are essential and warranted.
9. INTERVENTIONS TARGETING SENESCENCE, SASP, AND TAUO STRAINS
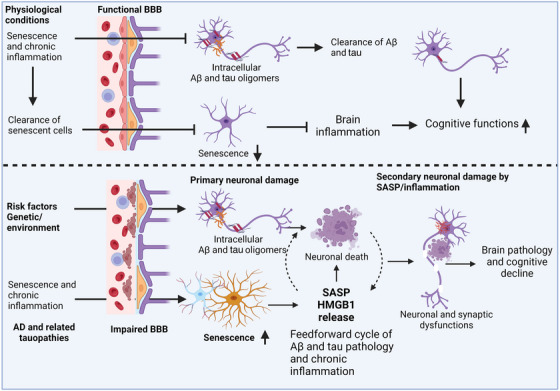
Despite the high prevalence of AD, no cure is currently available for this devastating disease. AD involves a complex interplay among different brain cell types along with factors from the periphery, which trigger multifaceted pathomechanisms including cellular senescence, chronic inflammation, mitochondrial dysfunction, impaired protein homeostasis, and compromised nuclear and BBB integrity, all of which are involved in disease progression. Developing therapies that target a single mechanism does not seem to be feasible and effective. Therapies that aim to decrease Aβ load have mostly had disappointing outcomes, although recent FDA approvals of anti‐Aβ antibodies such as lecanemab and aducanumab have provided some hope. Emerging evidence from clinical and preclinical studies suggests that senescence, chronic brain inflammation, and TauO play key roles in AD, appearing decades before onset of clinical symptoms and driving disease progression. In fact, there is a correlation between the buildup of senescent cells, inflammation, and the accumulation of aggregated tau in the progression of AD. This highlights that targeting one or a combination of these factors could be an effective way to develop therapeutic interventions. Several studies demonstrated that tau aggregates can trigger cellular senescence and inflammation, and eliminating senescent cells with senolytics reduces tau aggregation and inflammation and improves cognitive performance in vivo. 22 , 24 , 25 Human studies also revealed that senescence markers are associated with inflammation and tau pathology in the brain. Several studies have revealed that the gene CDKN2A (p16INK4a), responsible for regulating cell cycle and senescence, is highly elevated in the brains of individuals with AD as compared to age‐matched healthy individuals. This increase in p16INK4a is also found to be correlated with inflammation, the density of tau tangles, and Braak stage, which is a measure of tau pathology progression. 155 SASP components, which are secreted by senescent cells and can have pro‐inflammatory effects, have been found to be elevated in the CSF of AD patients when compared to controls. This elevation of SASP components in AD patients correlates with CSF tau levels, inflammation, and cognitive impairment. 17 , 20 , 63 , 99 In this section, we briefly discuss therapeutic strategies for AD that focus on senescence, SASP, and TauO.
9.1. Senotherapeutics: strategies for targeting senescence
A promising therapeutic approach for AD and other aging‐related diseases involves the use of senolytics, which selectively eliminate senescent cells through modulation of anti‐apoptotic pathways (eg, BCL‐2‐BCL‐xL, PI3K/AKT, MDM2‐p53‐p21Cip1‐serpine elements). Senolytic therapy has shown promising results in various animal models, including tauopathies. 22 , 25 , 57 , 185 Currently, around 20 clinical trials of senotherapies are ongoing, two of which (NCT04785300 and NCT04685590) are evaluating the therapeutic efficacy of D+Q in individuals with MCI or AD. 186 A phase I clinical trial demonstrates the elimination of senescent cells in humans with diabetic kidney disease using the combination of D+Q. 187 However, the elimination of senescent cells using senolytics may also have adverse side effects. 188 , 189 ,
FIGURE 6.
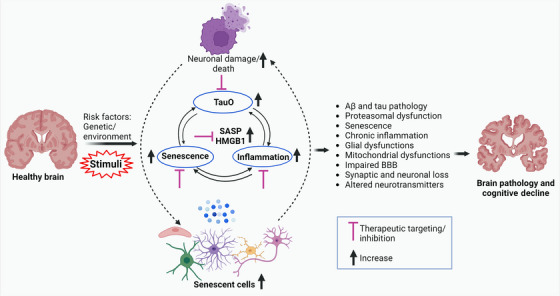
An overview diagram illustrating the role of senescence, brain inflammation, and TauO in AD and potential therapeutic targets. Various risk factors, including aging, can trigger senescence, inflammation, and TauO production, resulting in impaired synaptic and neuronal function loss, which are essential for learning and memory. This also induces the release of HMGB1 and initiates the SASP which lead to Aβ and tau pathology, proteasomal dysfunction, glial dysfunctions, mitochondrial dysfunctions, impaired BBB, synaptic and neuronal loss. Senescence, brain inflammation, and TauO can interact with each other and contribute to AD pathogenesis through several mechanisms. For example, TauO can trigger senescence and inflammation in the brain, which can impair the clearance of TauO and promote their aggregation and propagation. Senescence and inflammation can also impair the BBB functions, allowing more peripheral inflammatory factors to enter the brain and worsen neurodegeneration. Therefore, targeting senescence, inflammation, and TauO in combination may be a promising therapeutic strategy for AD. Aβ, amyloid beta; AD, Alzheimer's disease; BBB, blood‐brain barrier; SASP, senescence‐associated secretory phenotype; TauO, tau oligomers.
Alternatively, senomorphic compounds offer a different approach by alleviating senescence‐associated pathologies through the suppression of the SASP without killing senescent cells. Senomorphics interfere with SASP expression pathways (eg, p38MAPK, JAK/STAT, mTOR) and transcription factors (eg, NF‐κΒ, STAT3). 190 Neutralizing specific SASP factors with antibodies or small molecules is another strategy for mitigating the spread of senescence, SASP‐mediated chronic inflammation, and subsequent tissue damage. Several preclinical studies have confirmed the beneficial effects of senomorphic treatment in age‐related diseases. However, senomorphic treatment may require continuous administration and could potentially disrupt tissue homeostasis due to SASP blockade. 191 , 192 To date, several senomorphic agents (eg, RAD001, ruxolitinib, KU‐60019, loperamide, simvastatin, anakinra, canakinumab, etanercept, siltuximab, tocilizumab) evaluated in clinical and preclinical studies have been found to diminish SASP, prevent senescence progression, and improve physical function in naturally aged animals. 193 Nevertheless, further clinical studies are needed to establish the effectiveness of senotherapeutics in humans.
9.2. TauO strain‐specific immunotherapy
Developing effective tau immunotherapy has proven challenging in clinical trials, as many anti‐tau antibodies have yielded negative results. Antibodies such as semorinemab, bepranemab, E2814, JNJ‐63733657, Lu AF87908, APNmAb005, MK‐2214, PNT00, and PRX005 are currently being evaluated, but none have progressed to phase III trials. 194 Clinical trials for tilavonemab and gosuranemab, which target extracellular tau, have also not shown improvements in patients with AD and PSP. 195 Future research is needed to explore strategies like tau‐degrading intrabodies, single‐domain antibodies for intracellular tau, peptibodies for multi‐aggregate clearance, and brain shuttles to increase antibody bioavailability. 196 , 197 It is important to note that strain‐specific immunotherapy for tau is still in preclinical stages, and optimizing its efficacy is crucial to effectively target and clear distinct tau strains involved in disease heterogeneity and cognitive decline in tauopathies. 29 , 198
10. CONCLUDING REMARKS
This review provided valuable insights into the complex nature and heterogeneity of AD and its underlying mechanisms. We have explored the role of TauO, cellular senescence, and brain inflammation. It is evident that AD is a multifactorial disease involving the interplay of various cellular and molecular processes. The accumulation of TauO, accompanied by senescence and brain inflammation, contributes to neuronal dysfunction and cognitive decline (Figure 6). Microglia and astrocytes play critical roles in maintaining brain homeostasis, but their dysfunction through senescence can exacerbate neuroinflammation and neuronal damage. Looking ahead, therapeutic strategies for AD need to consider the multifaceted nature of the disease. Targeting Aβ or tau alone has shown limited success, highlighting the importance of exploring alternative approaches. Although therapeutic targeting of senescence, SASP, or pathological tau has shown promise in preclinical studies, tangible improvements in cognition have yet to be achieved in clinical trials. Interventions that show promise in mice often fail in clinical trials due to unique and complex aspects of human biology, unexpected side effects, and many other issues. By addressing multiple aspects of the disease pathology, combination therapy may offer a more comprehensive and effective approach to mitigate brain pathology and cognitive decline.
CONFLICT OF INTEREST STATEMENT
The authors declare that there are no conflicts of financial interest related to this publication. Author disclosures are available in the supporting information.
Supporting information
Supporting Information
ACKNOWLEDGMENTS
We thank Kayed lab members for their assistance. This work was supported by grants from the National Institutes of Health (AG054025, NS094557, AG072458, and T32 AG067952 to R.K.), an Alzheimer's Association Research Fellowship (AARF‐22‐967275 to S.G.), and a UTMB 2021 Claude D. Pepper OAIC Pilot grant (to S.G.).
Gaikwad S, Senapati S, Haque MA, Kayed R. Senescence, brain inflammation, and oligomeric tau drive cognitive decline in Alzheimer's disease: Evidence from clinical and preclinical studies. Alzheimer's Dement. 2024;20:709–727. 10.1002/alz.13490
REFERENCES
- 1. Guzman‐Martinez L, Maccioni RB, Andrade V, Navarrete LP, Pastor MG, Ramos‐Escobar N. Neuroinflammation as a common feature of neurodegenerative disorders. Front Pharmacol. 2019;10:1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kent SA, Spires‐Jones TL, Durrant CS. The physiological roles of tau and Abeta: implications for Alzheimer's disease pathology and therapeutics. Acta Neuropathol. 2020;140:417‐447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Arriagada PV, Growdon JH, Hedley‐Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology. 1992;42:631‐639. [DOI] [PubMed] [Google Scholar]
- 4. Giannakopoulos P, Herrmann FR, Bussiere T, et al. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer's disease. Neurology. 2003;60:1495‐1500. [DOI] [PubMed] [Google Scholar]
- 5. Gao YL, Wang N, Sun FR, Cao XP, Zhang W, Yu JT. Tau in neurodegenerative disease. Ann Transl Med. 2018;6:175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Iqbal K, Liu F, Gong CX, Alonso Adel C, Grundke‐Iqbal I. Mechanisms of tau‐induced neurodegeneration. Acta Neuropathol. 2009;118:53‐69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. de Calignon A, Spires‐Jones TL, Pitstick R, Carlson GA, Hyman BT. Tangle‐bearing neurons survive despite disruption of membrane integrity in a mouse model of tauopathy. J Neuropathol Exp Neurol. 2009;68:757‐761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Morsch R, Simon W, Coleman PD. Neurons may live for decades with neurofibrillary tangles. J Neuropathol Exp Neurol. 1999;58:188‐197. [DOI] [PubMed] [Google Scholar]
- 9. Lasagna‐Reeves CA, Castillo‐Carranza DL, Jackson GR, Kayed R. Tau oligomers as potential targets for immunotherapy for Alzheimer's disease and tauopathies. Curr Alzheimer Res. 2011;8:659‐665. [DOI] [PubMed] [Google Scholar]
- 10. Lasagna‐Reeves CA, Castillo‐Carranza DL, Sengupta U, Clos AL, Jackson GR, Kayed R. Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild‐type mice. Mol Neurodegener. 2011;6:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lasagna‐Reeves CA, Castillo‐Carranza DL, Sengupta U, et al. Alzheimer brain‐derived tau oligomers propagate pathology from endogenous tau. Sci Rep. 2012;2:700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Maeda S, Takashima A. Tau oligomers. Adv Exp Med Biol. 2019;1184:373‐380. [DOI] [PubMed] [Google Scholar]
- 13. Shafiei SS, Guerrero‐Munoz MJ, Castillo‐Carranza DL. Tau oligomers: cytotoxicity, propagation, and mitochondrial damage. Front Aging Neurosci. 2017;9:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Marcatti M, Fracassi A, Montalbano M, et al. Abeta/tau oligomer interplay at human synapses supports shifting therapeutic targets for Alzheimer's disease. Cell Mol Life Sci. 2022;79:222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Castillo‐Carranza DL, Nilson AN, Van Skike CE, et al. Cerebral microvascular accumulation of tau oligomers in Alzheimer's disease and related tauopathies. Aging Dis. 2017;8:257‐266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Castillo‐Carranza DL, Sengupta U, Guerrero‐Munoz MJ, et al. Passive immunization with tau oligomer monoclonal antibody reverses tauopathy phenotypes without affecting hyperphosphorylated neurofibrillary tangles. J Neurosci: the official journal of the Society for Neuroscience. 2014;34:4260‐4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kolarova M, Sengupta U, Bartos A, Ricny J, Kayed R. Tau oligomers in sera of patients with Alzheimer's disease and aged controls. J Alzheimers Dis. 2017;58:471‐478. [DOI] [PubMed] [Google Scholar]
- 18. Lasagna‐Reeves CA, Castillo‐Carranza DL, Sengupta U, et al. Identification of oligomers at early stages of tau aggregation in Alzheimer's disease. FASEB J. 2012;26:1946‐1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Niewiadomska G, Niewiadomski W, Steczkowska M, Gasiorowska A. Tau oligomers neurotoxicity. Life (Basel). 2021;11:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sengupta U, Portelius E, Hansson O, Farmer K, et al. Tau oligomers in cerebrospinal fluid in Alzheimer's disease. Ann Clin Transl Neurol. 2017;4:226‐235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. 2023 Alzheimer's disease facts and figures. Alzheimers Dement. 2023;19:1598‐1695. doi: 10.1002/alz.13016 [DOI] [PubMed] [Google Scholar]
- 22. Bussian TJ, Aziz A, Meyer CF, Swenson BL, van Deursen JM, Baker DJ. Clearance of senescent glial cells prevents tau‐dependent pathology and cognitive decline. Nature. 2018;562:578‐582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hu Y, Fryatt GL, Ghorbani M, et al. Replicative senescence dictates the emergence of disease‐associated microglia and contributes to Abeta pathology. Cell Rep. 2021;35:109228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Musi N, Valentine JM, Sickora KR, et al. Tau protein aggregation is associated with cellular senescence in the brain. Aging Cell. 2018;17:e12840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang P, Kishimoto Y, Grammatikakis I, et al. Senolytic therapy alleviates Abeta‐associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer's disease model. Nat Neurosci. 2019;22:719‐728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Baker DJ, Childs BG, Durik M, et al. Naturally occurring p16(Ink4a)‐positive cells shorten healthy lifespan. Nature. 2016;530:184‐189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Birch J, Gil J. Senescence and the SASP: many therapeutic avenues. Genes Dev. 2020;34:1565‐1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age‐associated diseases. J Gerontol A Biol Sci Med Sci. 2014;69(Suppl 1):S4‐S9. [DOI] [PubMed] [Google Scholar]
- 29. Bittar A, Al‐Lahham R, Bhatt N, et al. Passive immunotherapy targeting tau oligomeric strains reverses tauopathy phenotypes in aged human‐tau mice in a mouse model‐specific manner. J Alzheimers Dis. 2022;90:1103‐1122. [DOI] [PubMed] [Google Scholar]
- 30. Castillo‐Carranza DL, Guerrero‐Munoz MJ, Sengupta U, et al. Tau immunotherapy modulates both pathological tau and upstream amyloid pathology in an Alzheimer's disease mouse model. J. Neurosci: the official journal of the Society for Neuroscience. 2015;35:4857‐4868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. De Strooper B, Karran E. The cellular phase of Alzheimer's disease. Cell. 2016;164:603‐615. [DOI] [PubMed] [Google Scholar]
- 32. Gonzales MM, Garbarino VR, Marques Zilli E, et al. Senolytic therapy to modulate the progression of Alzheimer's disease (SToMP‐AD): a pilot clinical trial. J Prev Alzheimers Dis. 2022;9:22‐29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kosyreva AM, Sentyabreva AV, Tsvetkov IS, Makarova OV. Alzheimer's disease and inflammaging. Brain Sci. 2022;12:1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lopez‐Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194‐1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kennedy BK, Berger SL, Brunet A, et al. Geroscience: linking aging to chronic disease. Cell. 2014;159:709‐713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Baker DJ, Petersen RC. Cellular senescence in brain aging and neurodegenerative diseases: evidence and perspectives. J Clin Invest. 2018;128:1208‐1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. van Deursen JM. The role of senescent cells in ageing. Nature. 2014;509:439‐446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang L, Pitcher LE, Yousefzadeh MJ, Niedernhofer LJ, Robbins PD, Zhu Y. Cellular senescence: a key therapeutic target in aging and diseases. J Clin Invest. 2022:132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res. 1965;37:614‐636. [DOI] [PubMed] [Google Scholar]
- 40. Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585‐621. [DOI] [PubMed] [Google Scholar]
- 41. Gorgoulis V, Adams PD, Alimonti A, et al. Cellular senescence: defining a path forward. Cell. 2019;179:813‐827. [DOI] [PubMed] [Google Scholar]
- 42. Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593‐602. [DOI] [PubMed] [Google Scholar]
- 43. Martinez‐Cue C, Rueda N. Cellular senescence in neurodegenerative diseases. Front Cell Neurosci. 2020;14:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Coppe JP, Patil CK, Rodier F, et al. Senescence‐associated secretory phenotypes reveal cell‐nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853‐2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Neurohr GE, Terry RL, Lengefeld J, et al. excessive cell growth causes cytoplasm dilution and contributes to senescence. Cell. 2019;176:1083‐1097 e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chang J, Wang Y, Shao L, et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med. 2016;22:78‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hu L, Li H, Zi M, et al. Why senescent cells are resistant to apoptosis: an insight for senolytic development. Front Cell Dev Biol. 2022;10:822816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Baker DJ, Wijshake T, Tchkonia T, et al. Clearance of p16Ink4a‐positive senescent cells delays ageing‐associated disorders. Nature. 2011;479:232‐236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bhat R, Crowe EP, Bitto A, et al. Astrocyte senescence as a component of Alzheimer's disease. PLoS One. 2012;7:e45069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, van Deursen JM. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science. 2016;354:472‐477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Herbig U, Ferreira M, Condel L, Carey D, Sedivy JM. Cellular senescence in aging primates. Science. 2006;311:1257. [DOI] [PubMed] [Google Scholar]
- 52. Yousefzadeh MJ, Wilkinson JE, Hughes B, et al. Heterochronic parabiosis regulates the extent of cellular senescence in multiple tissues. Geroscience. 2020;42:951‐961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Basisty N, Kale A, Jeon OH, et al. A proteomic atlas of senescence‐associated secretomes for aging biomarker development. PLoS Biol. 2020;18:e3000599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Acosta JC, O'Loghlen A, Banito A, et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008;133:1006‐1018. [DOI] [PubMed] [Google Scholar]
- 55. Kuilman T, Michaloglou C, Vredeveld LC, et al. Oncogene‐induced senescence relayed by an interleukin‐dependent inflammatory network. Cell. 2008;133:1019‐1031. [DOI] [PubMed] [Google Scholar]
- 56. Acosta JC, Banito A, Wuestefeld T, et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol. 2013;15:978‐990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gaikwad S, Puangmalai N, Bittar A, et al. Tau oligomer induced HMGB1 release contributes to cellular senescence and neuropathology linked to Alzheimer's disease and frontotemporal dementia. Cell Rep. 2021; 36:109419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Storer M, Mas A, Robert‐Moreno A, et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell. 2013; 155:1119‐1130. [DOI] [PubMed] [Google Scholar]
- 59. Jin WN, Shi K, He W, et al. Neuroblast senescence in the aged brain augments natural killer cell cytotoxicity leading to impaired neurogenesis and cognition. Nat Neurosci. 2021; 24:61‐73. [DOI] [PubMed] [Google Scholar]
- 60. Ovadya Y, Landsberger T, Leins H, et al. Impaired immune surveillance accelerates accumulation of senescent cells and aging. Nat Commun. 2018; 9: 5435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yousefzadeh MJ, Flores RR, Zhu Y, et al. An aged immune system drives senescence and ageing of solid organs. Nature. 2021; 594: 100‐105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Schafer MJ, Zhang X, Kumar A, et al. The senescence‐associated secretome as an indicator of age and medical risk. JCI Insight. 2020;5:e133668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Villeda SA, Luo J, Mosher KI, et al. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature. 2011;477:90‐94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Jeon OH, Mehdipour M, Gil TH, et al. Systemic induction of senescence in young mice after single heterochronic blood exchange. Nat Metab. 2022;4:995‐1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Xu M, Pirtskhalava T, Farr JN, et al. Senolytics improve physical function and increase lifespan in old age. Nat Med. 2018;24:1246‐1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hou Y, Dan X, Babbar M, et al. Ageing as a risk factor for neurodegenerative disease. Nat Rev Neurol. 2019;15:565‐581. [DOI] [PubMed] [Google Scholar]
- 67. Guo T, Zhang D, Zeng Y, Huang TY, Xu H, Zhao Y. Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer's disease. Mol Neurodegener. 2020;15:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ising C, Venegas C, Zhang S, et al. NLRP3 inflammasome activation drives tau pathology. Nature. 2019;575:669‐673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Coppe JP, Rodier F, Patil CK, Freund A, Desprez PY, Campisi J. Tumor suppressor and aging biomarker p16(INK4a) induces cellular senescence without the associated inflammatory secretory phenotype. J Biol Chem. 2011;286:36396‐36403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Guerrero A, De Strooper B. Arancibia‐Carcamo IL. Cellular senescence at the crossroads of inflammation and Alzheimer's disease. Trends Neurosci. 2021;44:714‐727. [DOI] [PubMed] [Google Scholar]
- 71. Chikhirzhina E, Starkova T, Beljajev A, Polyanichko A, Tomilin A. Functional diversity of non‐histone chromosomal protein HmgB1. Int J Mol Sci. 2020;21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hudson BI, Lippman ME. Targeting RAGE signaling in inflammatory disease. Annu Rev Med. 2018;69:349‐364. [DOI] [PubMed] [Google Scholar]
- 73. Yang H, Wang H, Andersson U. Targeting inflammation driven by HMGB1. Front Immunol. 2020;11:484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Bonaldi T, Talamo F, Scaffidi P, et al. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 2003;22:5551‐5560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191‐195. [DOI] [PubMed] [Google Scholar]
- 76. Davalos AR, Kawahara M, Malhotra GK, et al. p53‐dependent release of alarmin HMGB1 is a central mediator of senescent phenotypes. J Cell Biol. 2013;201:613‐629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Salminen A, Kauppinen A, Kaarniranta K. Emerging role of NF‐kappaB signaling in the induction of senescence‐associated secretory phenotype (SASP). Cell Signal. 2012;24:835‐845. [DOI] [PubMed] [Google Scholar]
- 78. Rowell JP, Simpson KL, Stott K, Watson M, Thomas JO. HMGB1‐facilitated p53 DNA binding occurs via HMG‐Box/p53 transactivation domain interaction, regulated by the acidic tail. Structure. 2012;20:2014‐2024. [DOI] [PubMed] [Google Scholar]
- 79. Rodier F, Coppe JP, Patil CK, et al. Persistent DNA damage signalling triggers senescence‐associated inflammatory cytokine secretion. Nat Cell Biol. 2009;11:973‐979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Rodier F, Munoz DP, Teachenor R, et al. DNA‐SCARS: distinct nuclear structures that sustain damage‐induced senescence growth arrest and inflammatory cytokine secretion. J Cell Sci. 2011;124:68‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Sofiadis K, Josipovic N, Nikolic M, et al. HMGB1 coordinates SASP‐related chromatin folding and RNA homeostasis on the path to senescence. Mol Syst Biol. 2021;17:e9760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Gaikwad S, Kayed R. HMGB1‐mediated senescence and brain inflammation contributes to cognitive dysfunctions. Alzheimer's & Dementia. 2022;18:e068747. [Google Scholar]
- 83. Fagiolo U, Cossarizza A, Scala E, et al. Increased cytokine production in mononuclear cells of healthy elderly people. Eur J Immunol. 1993;23:2375‐2378. [DOI] [PubMed] [Google Scholar]
- 84. Franceschi C, Bonafe M, Valensin S, et al. Inflamm‐aging. an evolutionary perspective on immunosenescence. Ann N Y Acad Sci. 2000;908:244‐254. [DOI] [PubMed] [Google Scholar]
- 85. Calogero S, Grassi F, Aguzzi A, et al. The lack of chromosomal protein Hmg1 does not disrupt cell growth but causes lethal hypoglycaemia in newborn mice. Nat Genet. 1999;22:276‐280. [DOI] [PubMed] [Google Scholar]
- 86. Funayama A, Shishido T, Netsu S, et al. Cardiac nuclear high mobility group box 1 prevents the development of cardiac hypertrophy and heart failure. Cardiovasc Res. 2013;99:657‐664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Yin XY, Tang XH, Wang SX, et al. HMGB1 mediates synaptic loss and cognitive impairment in an animal model of sepsis‐associated encephalopathy. J Neuroinflammation. 2023;20:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Fonken LK, Frank MG, Kitt MM, et al. The alarmin hmgb1 mediates age‐induced neuroinflammatory priming. the journal of neuroscience: the official journal of the society for. Neuroscience. 2016;36:7946‐7956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Fujita K, Motoki K, Tagawa K, et al. HMGB1, a pathogenic molecule that induces neurite degeneration via TLR4‐MARCKS, is a potential therapeutic target for Alzheimer's disease. Sci Rep. 2016;6:31895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Paudel YN, Shaikh MF, Chakraborti A, et al. HMGB1: a common biomarker and potential target for TBI, neuroinflammation, epilepsy, and cognitive dysfunction. Front Neurosci. 2018;12:628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Tanaka H, Homma H, Fujita K, et al. YAP‐dependent necrosis occurs in early stages of Alzheimer's disease and regulates mouse model pathology. Nat Commun. 2020;11:507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Paudel YN, Angelopoulou E, Piperi C, Othman I, Aamir K, Shaikh MF. Impact of HMGB1, RAGE, and TLR4 in Alzheimer's disease (AD): from risk factors to therapeutic targeting. Cells. 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Ferrucci L, Fabbri E. Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat Rev Cardiol. 2018;15:505‐522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Pilling LC, Joehanes R, Melzer D, et al. Gene expression markers of age‐related inflammation in two human cohorts. Exp Gerontol. 2015;70:37‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Cao W, Zheng H. Peripheral immune system in aging and Alzheimer's disease. Mol Neurodegener. 2018;13:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Kim JW, Stewart R, Kang HJ, et al. Longitudinal associations between serum cytokine levels and dementia. Front Psychiatry. 2018;9:606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Bok E, Leem E, Lee BR, et al. Role of the lipid membrane and membrane proteins in tau pathology. Front Cell Dev Biol. 2021;9:653815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Fernandes A, Tabuas‐Pereira M, Duro D, et al. C‐reactive protein as a predictor of mild cognitive impairment conversion into Alzheimer's disease dementia. Exp Gerontol. 2020;138:111004. [DOI] [PubMed] [Google Scholar]
- 99. Morales I, Farias G, Maccioni RB. Neuroimmunomodulation in the pathogenesis of Alzheimer's disease. Neuroimmunomodulation. 2010;17:202‐204. [DOI] [PubMed] [Google Scholar]
- 100. Heneka MT, Carson MJ, El Khoury J, et al. Neuroinflammation in Alzheimer's disease. Lancet Neurol. 2015;14:388‐405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Fielder E, Tweedy C, Wilson C, et al. Anti‐inflammatory treatment rescues memory deficits during aging in NFKB1(‐/‐) mice. Aging Cell. 2020;19:e13188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Kinney JW, Bemiller SM, Murtishaw AS, Leisgang AM, Salazar AM, Lamb BT. Inflammation as a central mechanism in Alzheimer's disease. Alzheimers Dement (N Y). 2018;4:575‐590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Chen Y, Yu Y. Tau and neuroinflammation in Alzheimer's disease: interplay mechanisms and clinical translation. J Neuroinflammation. 2023;20:165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Hur JY, Frost GR, Wu X, et al. The innate immunity protein IFITM3 modulates gamma‐secretase in Alzheimer's disease. Nature. 2020;586:735‐740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Borghesan M, Fafian‐Labora J, Eleftheriadou O, et al. Small extracellular vesicles are key regulators of non‐cell autonomous intercellular communication in senescence via the interferon protein IFITM3. Cell Rep. 2019;27:3956‐3971 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Ogrodnik M, Evans SA, Fielder E, et al. Whole‐body senescent cell clearance alleviates age‐related brain inflammation and cognitive impairment in mice. Aging Cell. 2021;20:e13296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Faden AI, Barrett JP, Stoica BA, Henry RJ. Bidirectional brain‐systemic interactions and outcomes after TBI. Trends Neurosci. 2021;44:406‐418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Bittar A, Bhatt N, Hasan TF, et al. Neurotoxic tau oligomers after single versus repetitive mild traumatic brain injury. Brain communications. 2019;1:fcz004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Edwards G 3rd, Zhao J, Dash PK, Soto C, Moreno‐Gonzalez I. Traumatic brain injury induces tau aggregation and spreading. J Neurotrauma. 2020;37:80‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Ritzel RM, Doran SJ, Glaser EP, et al. Old age increases microglial senescence, exacerbates secondary neuroinflammation, and worsens neurological outcomes after acute traumatic brain injury in mice. Neurobiol Aging. 2019;77:194‐206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Pluvinage JV, Wyss‐Coray T. Systemic factors as mediators of brain homeostasis, ageing and neurodegeneration. Nat Rev Neurosci. 2020;21:93‐102. [DOI] [PubMed] [Google Scholar]
- 112. Pan Y, Li H, Wardlaw JM, Wang Y. A new dawn of preventing dementia by preventing cerebrovascular diseases. BMJ. 2020;371:m3692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Nation DA, Sweeney MD, Montagne A, et al. Blood‐brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat Med. 2019;25:270‐276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Bennett RE, Robbins AB, Hu M, et al. Tau induces blood vessel abnormalities and angiogenesis‐related gene expression in P301L transgenic mice and human Alzheimer's disease. Proc Natl Acad Sci U S A. 2018;115:E1289‐E1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Greenberg SM, Bacskai BJ, Hernandez‐Guillamon M, Pruzin J, Sperling R, van Veluw SJ. Cerebral amyloid angiopathy and Alzheimer disease—one peptide, two pathways. Nat Rev Neurol. 2020;16:30‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Nelson AR, Sagare MA, Wang Y, Kisler K, Zhao Z, Zlokovic BV. Channelrhodopsin excitation contracts brain pericytes and reduces blood flow in the aging mouse brain in vivo. Frontiers in aging neuroscience. 2020;12:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Sagare AP, Bell RD, Zhao Z, et al. Pericyte loss influences Alzheimer‐like neurodegeneration in mice. Nat Commun. 2013;4:2932. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 118. Nortley R, Korte N, Izquierdo P, et al. Amyloid beta oligomers constrict human capillaries in Alzheimer's disease via signaling to pericytes. Science. 2019:365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Ogrodnik M, Zhu Y, Langhi LGP, et al. Obesity‐induced cellular senescence drives anxiety and impairs neurogenesis. Cell Metab. 2019;29:1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Chow HM, Shi M, Cheng A, et al. Age‐related hyperinsulinemia leads to insulin resistance in neurons and cell‐cycle‐induced senescence. Nat Neurosci. 2019;22:1806‐1819. [DOI] [PubMed] [Google Scholar]
- 121. Efthymiou AG, Goate AM. Late onset Alzheimer's disease genetics implicates microglial pathways in disease risk. Mol Neurodegener. 2017;12:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Scheltens P, De Strooper B, Kivipelto M, et al. Alzheimer's disease. Lancet. 2021;397:1577‐1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Krasemann S, Madore C, Cialic R, et al. The TREM2‐APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity. 2017;47:566‐581 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Streit WJ, Braak H, Xue QS, Bechmann I. Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer's disease. Acta Neuropathol. 2009;118:475‐485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Shahidehpour RK, Higdon RE, Crawford NG, et al. Dystrophic microglia are associated with neurodegenerative disease and not healthy aging in the human brain. Neurobiol Aging. 2021;99:19‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Bisht K, Sharma KP, Lecours C, et al. Dark microglia: a new phenotype predominantly associated with pathological states. Glia. 2016;64:826‐839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Condello C, Yuan P, Schain A, Grutzendler J. Microglia constitute a barrier that prevents neurotoxic protofibrillar Abeta42 hotspots around plaques. Nat Commun. 2015;6:6176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Mancuso R, Fryatt G, Cleal M, et al. CSF1R inhibitor JNJ‐40346527 attenuates microglial proliferation and neurodegeneration in P301S mice. Brain. 2019;142:3243‐3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Mathys H, Adaikkan C, Gao F, et al. Temporal tracking of microglia activation in neurodegeneration at single‐cell resolution. Cell Rep. 2017;21:366‐380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Vasile F, Dossi E, Rouach N. Human astrocytes: structure and functions in the healthy brain. Brain Struct Funct. 2017;222:2017‐2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Fakhoury M. Microglia and astrocytes in Alzheimer's disease: implications for therapy. Curr Neuropharmacol. 2018;16:508‐518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Reid MJ, Beltran‐Lobo P, Johnson L, Perez‐Nievas BG, Noble W. Astrocytes in tauopathies. Front Neurol. 2020;11:572850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Verkhratsky A, Nedergaard M. Physiology of astroglia. Physiol Rev. 2018;98:239‐389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Blasko I, Stampfer‐Kountchev M, Robatscher P, Veerhuis R, Eikelenboom P, Grubeck‐Loebenstein B. How chronic inflammation can affect the brain and support the development of Alzheimer's disease in old age: the role of microglia and astrocytes. Aging Cell. 2004;3:169‐176. [DOI] [PubMed] [Google Scholar]
- 135. Cohen J, Torres C. Astrocyte senescence: evidence and significance. Aging Cell. 2019;18:e12937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Maciel‐Baron LA, Morales‐Rosales SL, Silva‐Palacios A, et al. The secretory phenotype of senescent astrocytes isolated from Wistar newborn rats changes with anti‐inflammatory drugs, but does not have a short‐term effect on neuronal mitochondrial potential. Biogerontology. 2018;19:415‐433. [DOI] [PubMed] [Google Scholar]
- 137. Muszynski P, Groblewska M, Kulczynska‐Przybik A, Kulakowska A, Mroczko B. YKL‐40 as a potential biomarker and a possible target in therapeutic strategies of Alzheimer's disease. Curr Neuropharmacol. 2017;15:906‐917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Habib N, McCabe C, Medina S, et al. Disease‐associated astrocytes in Alzheimer's disease and aging. Nat Neurosci. 2020;23:701‐706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Limbad C, Oron TR, Alimirah F, et al. Astrocyte senescence promotes glutamate toxicity in cortical neurons. PLoS One. 2020;15:e0227887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Zou Y, Zhang N, Ellerby LM, et al. Responses of human embryonic stem cells and their differentiated progeny to ionizing radiation. Biochem Biophys Res Commun. 2012;426:100‐105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Turnquist C, Horikawa I, Foran E, et al. p53 isoforms regulate astrocyte‐mediated neuroprotection and neurodegeneration. Cell Death Differ. 2016;23:1515‐1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Ungvari Z, Tarantini S, Hertelendy P, et al. Cerebromicrovascular dysfunction predicts cognitive decline and gait abnormalities in a mouse model of whole brain irradiation‐induced accelerated brain senescence. Geroscience. 2017;39:33‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Geha S, Pallud J, Junier MP, et al. NG2+/Olig2+ cells are the major cycle‐related cell population of the adult human normal brain. Brain Pathol. 2010;20:399‐411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Antel JP, Lin YH, Cui QL, et al. Immunology of oligodendrocyte precursor cells in vivo and in vitro. J Neuroimmunol. 2019;331:28‐35. [DOI] [PubMed] [Google Scholar]
- 145. Choi CI, Yoo KH, Hussaini SM, et al. The progeroid gene BubR1 regulates axon myelination and motor function. Aging (Albany NY). 2016;8:2667‐2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Tang DG, Tokumoto YM, Apperly JA, Lloyd AC, Raff MC. Lack of replicative senescence in cultured rat oligodendrocyte precursor cells. Science. 2001;291:868‐871. [DOI] [PubMed] [Google Scholar]
- 147. Cui W, Hu G, Peng J, Mu L, Liu J, Qiao L. Quercetin exerted protective effects in a rat model of sepsis via inhibition of reactive oxygen species (ROS) and downregulation of high mobility group box 1 (HMGB1) protein expression. Med Sci Monit. 2019;25:5795‐5800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Khan H, Ullah H, Aschner M, Cheang WS, Akkol EK. Neuroprotective effects of quercetin in Alzheimer's disease. Biomolecules. 2019;10:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Tang D, Kang R, Xiao W, et al. Quercetin prevents LPS‐induced high‐mobility group box 1 release and proinflammatory function. Am J Respir Cell Mol Biol. 2009;41:651‐660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Ippati S, Deng Y, van der Hoven J, et al. Rapid initiation of cell cycle reentry processes protects neurons from amyloid‐beta toxicity. Proc Natl Acad Sci U S A. 2021;118:e2011876118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Jurk D, Wang C, Miwa S, et al. Postmitotic neurons develop a p21‐dependent senescence‐like phenotype driven by a DNA damage response. Aging Cell. 2012;11:996‐1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Koper MJ, Van Schoor E, Ospitalieri S, et al. Necrosome complex detected in granulovacuolar degeneration is associated with neuronal loss in Alzheimer's disease. Acta Neuropathol. 2020;139:463‐484. [DOI] [PubMed] [Google Scholar]
- 153. Sapieha P, Mallette FA. Cellular senescence in postmitotic cells: beyond growth arrest. Trends Cell Biol. 2018;28:595‐607. [DOI] [PubMed] [Google Scholar]
- 154. Kiss T, Nyul‐Toth A, Balasubramanian P, et al. Single‐cell RNA sequencing identifies senescent cerebromicrovascular endothelial cells in the aged mouse brain. Geroscience. 2020;42:429‐444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Bryant AG, Hu M, Carlyle BC, et al. Cerebrovascular senescence is associated with tau pathology in Alzheimer's. Disease Front Neurol. 2020;11:575953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Dujardin S, Commins C, Lathuiliere A, et al. Tau molecular diversity contributes to clinical heterogeneity in Alzheimer's disease. Nat Med. 2020;26:1256‐1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Komarova NL, Thalhauser CJ. High degree of heterogeneity in Alzheimer's disease progression patterns. PLoS computational biology. 2011;7:e1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Bischof GN, Rodrigue KM, Kennedy KM, Devous MD Sr, Park DC. Amyloid deposition in younger adults is linked to episodic memory performance. Neurology. 2016;87:2562‐2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Sanders DW, Kaufman SK, DeVos SL, et al. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron. 2014;82:1271‐1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Colin M, Dujardin S, Schraen‐Maschke S, et al. From the prion‐like propagation hypothesis to therapeutic strategies of anti‐tau immunotherapy. Acta Neuropathol. 2020;139:3‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Gerson JE, Mudher A, Kayed R. Potential mechanisms and implications for the formation of tau oligomeric strains. Crit Rev Biochem Mol Biol. 2016;51:482‐496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Kametani F, Yoshida M, Matsubara T, et al. Comparison of common and disease‐specific post‐translational modifications of pathological tau associated with a wide range of tauopathies. Front Neurosci. 2020;14:581936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Li Y, Phoo WW, Loh YR, et al. Structural characterization of the linked NS2B‐NS3 protease of Zika virus. FEBS Lett. 2017;591:2338‐2347. [DOI] [PubMed] [Google Scholar]
- 164. Scheres SH, Zhang W, Falcon B, Goedert M. Cryo‐EM structures of tau filaments. Curr Opin Struct Biol. 2020;64:17‐25. [DOI] [PubMed] [Google Scholar]
- 165. Narasimhan S, Guo JL, Changolkar L, et al. Pathological tau strains from human brains recapitulate the diversity of tauopathies in nontransgenic mouse brain . J Neurosci : the official journal of the Society for Neuroscience. 2017;37:11406‐11423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Pickett EK, Herrmann AG, McQueen J, et al. Amyloid beta and tau cooperate to cause reversible behavioral and transcriptional deficits in a model of Alzheimer's disease. Cell Rep. 2019;29:3592‐3604 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Dujardin S, Commins C, Lathuiliere A, et al. Author correction: tau molecular diversity contributes to clinical heterogeneity in Alzheimer's disease. Nat Med. 2021;27:356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Glabe CG. Structural classification of toxic amyloid oligomers. J Biol Chem. 2008;283:29639‐29643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Malfertheiner K, Stefanova N, Heras‐Garvin A. The concept of alpha‐synuclein strains and how different conformations may explain distinct neurodegenerative disorders. Front Neurol. 2021;12:737195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Maxwell AM, Yuan P, Rivera BM, et al. Emergence of distinct and heterogeneous strains of amyloid beta with advanced Alzheimer's disease pathology in Down syndrome. Acta Neuropathol Commun. 2021;9:201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Peelaerts W, Bousset L, Van der Perren A, et al. alpha‐synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature. 2015;522:340‐344. [DOI] [PubMed] [Google Scholar]
- 172. Castillo‐Carranza DL, Guerrero‐Munoz MJ, Sengupta U, Gerson JE. Kayed R. Alpha‐synuclein oligomers induce a unique toxic tau strain. Biol Psychiatry. 2018;84:499‐508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Goedert M, Spillantini MG, Del Tredici K, Braak H. 100 years of Lewy pathology. Nat Rev Neurol. 2013;9:13‐24. [DOI] [PubMed] [Google Scholar]
- 174. Wennberg AM, Whitwell JL, Tosakulwong N, et al. The influence of tau, amyloid, alpha‐synuclein, TDP‐43, and vascular pathology in clinically normal elderly individuals. Neurobiol Aging. 2019;77:26‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Azarpazhooh MR, Avan A, Cipriano LE, et al. A third of community‐dwelling elderly with intermediate and high level of Alzheimer's neuropathologic changes are not demented: a meta‐analysis. Ageing Res Rev. 2020;58:101002. [DOI] [PubMed] [Google Scholar]
- 176. Singh A, Allen D, Fracassi A, et al. Functional integrity of synapses in the central nervous system of cognitively intact individuals with high Alzheimer's disease neuropathology is associated with absence of synaptic tau oligomers. J Alzheimers Dis. 2020;78:1661‐1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Sengupta U, Kayed R. Amyloid beta, tau, and alpha‐synuclein aggregates in the pathogenesis, prognosis, and therapeutics for neurodegenerative diseases. Prog Neurobiol. 2022;214:102270. [DOI] [PubMed] [Google Scholar]
- 178. Guo JL, Covell DJ, Daniels JP, et al. Distinct alpha‐synuclein strains differentially promote tau inclusions in neurons. Cell. 2013;154:103‐117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Kayed R, Head E, Thompson JL, et al. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486‐489. [DOI] [PubMed] [Google Scholar]
- 180. HITT BD, Gupta A, Singh R, et al. Anti‐tau antibodies targeting a conformation‐dependent epitope selectively bind seeds. bioRxiv. 2023:2023.05. 04.539475. [DOI] [PMC free article] [PubMed]
- 181. Hitt BD, Yang T, Beaver JD, et al. Conformation‐specific anti‐tau antibodies distinguish human tauopathies. Alzheimer's & Dementia. 2022;18:e066069. [Google Scholar]
- 182. Gibbons GS, Kim SJ, Wu Q, et al. Conformation‐selective tau monoclonal antibodies inhibit tau pathology in primary neurons and a mouse model of Alzheimer's disease. Mol Neurodegener. 2020;15:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Montalbano M, Majmundar L, Sengupta U, Fung L, Kayed R. Pathological tau signatures and nuclear alterations in neurons, astrocytes and microglia in Alzheimer's disease, progressive supranuclear palsy, and dementia with Lewy bodies. Brain Pathol. 2023;33:e13112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Yang H, Yuan P, Wu Y, et al. EMBER multidimensional spectral microscopy enables quantitative determination of disease‐ and cell‐specific amyloid strains. Proc Natl Acad Sci U S A. 2023;120:e2300769120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Currais A, Farrokhi C, Dargusch R, et al. Fisetin reduces the impact of aging on behavior and physiology in the rapidly aging SAMP8 mouse. J Gerontol A Biol Sci Med Sci. 2018;73:299‐307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Chaib S, Tchkonia T, Kirkland JL. Cellular senescence and senolytics: the path to the clinic. Nat Med. 2022;28:1556‐1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Hickson LJ, Langhi Prata LGP, Bobart SA, et al. Senolytics decrease senescent cells in humans: preliminary report from a clinical trial of dasatinib plus quercetin in individuals with diabetic kidney disease. EBioMedicine. 2019;47:446‐456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Demaria M, Ohtani N, Youssef SA, et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF‐AA. Dev Cell. 2014;31:722‐733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Grosse L, Wagner N, Emelyanov A, et al. Defined p16 (high) senescent cell types are indispensable for mouse healthspan. Cell Metab. 2020;32:87‐99 e6. [DOI] [PubMed] [Google Scholar]
- 190. Song S, Tchkonia T, Jiang J, Kirkland JL, Sun Y. Targeting senescent cells for a healthier aging: challenges and opportunities. Adv Sci (Weinh). 2020;7:2002611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. De Cecco M, Ito T, Petrashen AP, et al. L1 drives IFN in senescent cells and promotes age‐associated inflammation. Nature. 2019;566:73‐78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Xu M, Tchkonia T, Ding H, et al. JAK inhibition alleviates the cellular senescence‐associated secretory phenotype and frailty in old age. Proc Natl Acad Sci U S A. 2015;112:E6301‐E6310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Sun Y, Li Q, Kirkland JL. Targeting senescent cells for a healthier longevity: the roadmap for an era of global aging. Life Med. 2022;1:103‐119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Panza F, Dibello V, Sardone R, et al. Clinical development of passive tau‐based immunotherapeutics for treating primary and secondary tauopathies. Expert Opin Investig Drugs. 2023:1‐10. [DOI] [PubMed] [Google Scholar]
- 195. Ji C, Sigurdsson EM. Current status of clinical trials on tau immunotherapies. Drugs. 2021;81:1135‐1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Congdon EE, Pan R, Jiang Y, et al. Single domain antibodies targeting pathological tau protein: influence of four IgG subclasses on efficacy and toxicity. EBioMedicine. 2022;84:104249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Gallardo G, Wong CH, Ricardez SM, et al. Targeting tauopathy with engineered tau‐degrading intrabodies. Mol Neurodegener. 2019;14:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Bittar A, Sengupta U, Kayed R. Prospects for strain‐specific immunotherapy in Alzheimer's disease and tauopathies. NPJ Vaccines. 2018;3:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information