

Abstract
Genetics and omics studies of Alzheimer’s disease and other dementia subtypes enhance our understanding of underlying mechanisms and pathways that can be targeted. We identified key remaining challenges: First, can we enhance genetic studies to address missing heritability? Can we identify reproducible omics signatures that differentiate between dementia subtypes? Can high-dimensional omics data identify improved biomarkers? How can genetics inform our understanding of causal status of dementia risk factors? And which biological processes are altered by dementia-related genetic variation? Artificial intelligence (AI) and machine learning approaches give us powerful new tools in helping us to tackle these challenges, and we review possible solutions and examples of best practice. However, their limitations also need to be considered, as well as the need for coordinated multidisciplinary research and diverse deeply phenotyped cohorts. Ultimately AI approaches improve our ability to interrogate genetics and omics data for precision dementia medicine.
Keywords: Dementia, genetics, omics, artificial intelligence, machine learning, etiology, biomarkers, pathology, causality, risk factors, disease pathways
1. Introduction
Dementia results from a variety of heterogeneous pathologies, such as Alzheimer’s disease (AD), Parkinson’s disease dementia (PDD), Dementia with Lewy bodies (DLB), Frontotemporal Dementia (FTD), and cerebrovascular disease [1]. The number of people living with dementia worldwide is around 45 million and, as life expectancy increases and populations age, this number is expected to increase [2]. Genome-wide association studies (GWAS) have led to the identification of an increasing number of genetic loci associated with the risk of dementias and related neurodegenerative diseases in older adults, primarily of European ancestry [3][4][5][6][7][8][9][10]. However, even with established bonafide associations, the task of characterizing variants and genes in the context of complex disease molecular pathophysiology, as well as its interacting genes and pathways, remains a daunting challenge [11].
Recent progress in cutting-edge genetic and omics technologies, such as epigenomics, transcriptomics, proteomics and metabolomics, which refer to the comprehensive assessment of a set of specific types of biological molecules, allied with emerging computational methods, hold promise of faster discoveries. However, because of the large number of associations investigated in most omics scale studies, it is necessary to have large sample sizes collected in a consistent manner. Scaling up multidisciplinary dementia studies, such as those using omics approaches, comes with challenges and implies the need of coordinated efforts from clinicians, basic and computational scientists. Appropriate funding and infrastructures capable of dealing with large numbers of biological samples and big data are also needed.
As the omics field continues to expand in dementia research, artificial intelligence (AI)-powered technologies, and in particular machine learning (ML) and deep learning (DL), are well-suited for the detection of undiscovered patterns in high-dimensional data and advance dementia research in unprecedented ways (Figure 1). COVID-19 demonstrated that progress can rapidly be made towards tackling a disease when certain scientific practices are altered [12]. Coordinated action across interested parties can result in extraordinary progress within short periods of time. Significant progress could be made rapidly in dementia research if interested parties were able to organize such that we could tackle the systemic problems that hold back the field, some of which are discussed below.
Figure 1. Illustration of multiple aspects of dementia research that can be enhanced by the use of appropriate genetics and omics data allied with the implementation of artificial intelligence approaches.
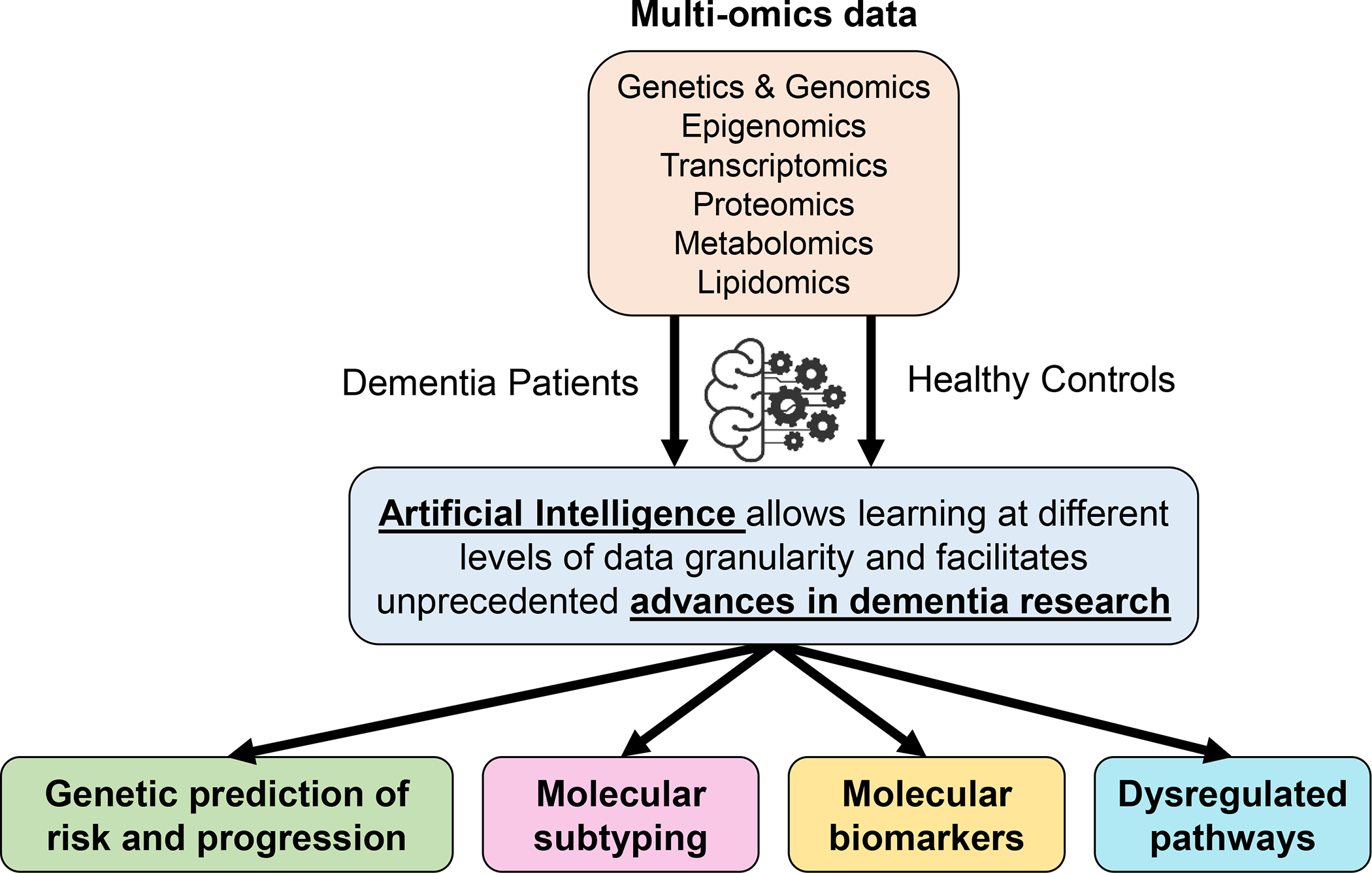
Here we identify and discuss five unresolved key questions in dementia research, which could be addressed using omics combined with advanced AI approaches: 1) How can we enhance genetic studies to inform our understanding of dementia risk? 2) Can we find reproducible omics brain signatures that differentiate between dementia subtypes? 3) Can high-dimensional omics data identify improved molecular biomarkers for dementia compared to single marker approaches? 4) How do we use genetics to inform our understanding of causal risk factors? And 5) Which biological processes are altered by genetic risk for dementia-related diseases? Tackling these questions is crucial to improving our understanding of dementia, and involves coordinating a multitude of players whose expertise go well beyond omics. It also involves improving the availability of bioresources and clinical data as well as developing analytical tools and ML algorithms to deal with high-dimensional and heterogeneous data. We note some of the challenges which must be surmounted to answer these questions within the next decade. In each instance, we highlight possible solutions and exemplar projects and communities, who have set good examples that can be used to improve our performance as a dementia research community.
This review is one of a series of eight articles in a Special Issue on ‘Artificial Intelligence for Alzheimer’s Disease and Related Dementias’ published in Alzheimer’s & Dementia. Together, this series provides a comprehensive overview of current applications of AI to dementia, and future opportunities for innovation to accelerate research. Each review focuses on a different area of dementia research, including experimental models [this issue], drug discovery and trials optimization [this issue], genetics and omics (this article), biomarkers [this issue], neuroimaging [this issue], prevention [this issue], applied models and digital health [this issue], and methods optimization [this issue].
2. Key Challenges
2.1. How can we enhance genetic studies to inform our understanding of dementia risk?
2.1.1. State of the science
The majority of GWAS rely upon logistic or linear regression-based approaches to test for associations between individual genetic variants (single nucleotide polymorphisms; SNPs) and a binary or continuous outcome [13][14]. This process is repeated until an estimate of association has been generated separately for each genetic variant. Then p-values are used to gauge whether any of these individual associations are strong enough to be considered genome-wide significant when correcting for multiple testing (a conventional threshold for ‘hits’ is 5 × 10−8) [15]. After a GWAS has been conducted it is often then possible to construct a polygenic risk score (PRS) by summing the value for each genetic variant weighted by the effect size from the initial GWAS [16]. PRS have important applications as research tools, in clinical trials and in clinical practice, as they can facilitate causal inference modeling and genetic risk stratification on an individual level. Despite twin study heritability estimates of around 60–80% for AD [17], recent SNP-based estimates of common variant heritability of AD from GWAS and PRS are much lower (up to 20%) [18], suggesting that much of the genetic contribution to dementia risk remains unexplained. Other approaches are needed to uncover this missing heritability by integrating multi-omics or non-linear modeling.
2.1.2. What problems need addressing?
The diagnosis of dementia and its subtypes is imprecise [19]. Current GWAS are based on cases for whom diagnosis of a specific dementia subtype has been largely made based upon clinical signs and symptoms. Thus, although current dementia GWAS are likely to be enriched for pathology related to the dementia subtype of interest, they will inevitably also contain other dementia subtypes and pathologies in their cases. This is problematic since etiology and risk factors are likely to differ for each dementia subtype, so genetic markers with small effect sizes that are specific to a single dementia subtype will be harder to detect than generalized dementia pathways.
There is currently a marked lack of diversity within dementia genetics studies, with GWAS discovery being largely confined to the genetics of AD in non-Hispanic White adults of European ancestry. Although some small GWAS have been conducted in non-European samples [20][21][22][23], have measured non-AD dementias [6][9][10], and incorporated dementia-related intermediate quantitative phenotypes or endophenotypes (such as amyloid-beta and cerebral small vessel disease) [24][25][26], these studies are largely underpowered. Certain ancestries remain understudied, for example South Asians despite representing around a quarter of the total global population. Without enhancing diversity in GWAS, or developing appropriate reference panels and genotyping chips, we are unable to construct PRS for all ancestral groups. This perpetuates ethnic bias in future research and clinical practice. We need better methods that can leverage diversity when evaluating risk. Not only from the standpoint of genetics, but integrating multimodal data that may interact with genetic or epigenetic factors as part of comprehensive risk assessment and risk prediction.
The study of both coding and non-coding rare/structural variants associated with dementia risk needs to be further pursued through short- and long-read sequencing technologies, which are thought to be important contributors to missing heritability in dementia [27]. Under the hood, long-read sequencing is powered by DL, using GPU-powered alignment algorithms to better characterize the genome. Other potential reasons for missing heritability include unmeasured interactions between genes (epistasis) and failing to account for correlations between genetic variants due to population structure, dynastic effects, assortative mating or functional relationships [28].
2.1.3. Possible solutions
Perhaps the simplest way to enhance future GWAS is to further increase sample sizes and the diversity of these samples. This has been the main strategy so far, and has been reasonably successful in identifying additional genetic variants and, to a lesser degree, improving the phenotypic variance explained. It is reasonable to assume that by further increasing sample sizes (essentially more of the same) further discoveries will be made. Increasing sample sizes considerably will involve enhancing existing research studies or establishing new studies. It is also important to consider the existence of different dementia subtypes and how to distinguish them. It may be possible to take advantage of existing well characterized samples that have not previously been genotyped due to resource limitations, such as gold standard post-mortem brain bank material with linked clinical data. That said, the cost of new studies which include clinical characterization is likely to remain high, and the number of existing samples is finite, raising practical concerns. Although there is no theoretical upper limit, in practice a predictive accuracy plateau in part limited by heritability is often reached, beyond which additional training data is not helpful. Given the large amount of missing heritability remaining, it is likely that increasing sample sizes may be needed but will not be sufficient in future GWAS, and alternative approaches will be required [29].
Leveraging population diversity, rather than omitting it, can both improve statistical power and better detect causal variants. For example, a transfer learning approach was used to enhance the findings from a modestly sized GWAS in a Japanese population using summary statistics from a larger European ancestry GWAS [21]. Conversely, trans-ancestry cohorts can also be used to improve genetic variant discovery and localization in European ancestry GWAS. Transfer learning heuristics can also potentially be employed with different rates across global and local admixture levels in some populations for higher accuracy.
As an alternative to the standard linear approaches employed in traditional GWAS, advanced ML approaches may offer various benefits [30] (Table 1), including the ability to: 1) capture main genetic effects more accurately; 2) capture multi-scale, non-linear epistatic interactions overlooked when investigating genetic variants individually; 3) better handle trans-ethnic variation; 4) flexibly integrate multimodal (e.g. neuroimaging, clinical biomarkers) and/or multi-omics data; and 5) accurately predict multiple outcomes, such as subtraits, symptoms, and endophenotypes, at once. For example, a gradient tree boosting method followed by an adaptive iterative genetic variant search was used to capture complex non-linear epistatic interactions and select interacting genetic variants with high predictiveness for breast cancer [31]. Similarly, improvements have been observed by applying DL to predict survival in age-related macular degeneration [32] and reduce multiple testing burden [33]. The tool DeepWAS [34] was used to identify genetic variants associated with multiple sclerosis and major depressive disorder while simultaneously predicting their cell-type-specific regulatory effects using multi-omics data integration. DeepNull [35] is a DL-based tool that models non-linear associations between the phenotype and non-genetic covariates. This improved GWAS hits detection by 6% and phenotypic prediction by 23% on average across 10 different UK Biobank traits, while also substantially reducing the false positive rate. Despite these advances, few attempts have so far been made to apply these techniques to dementia. While early attempts to apply ML-based methods to improve AD risk variant prediction have yet to find substantial improvements over traditional GWAS, the cohorts in which these models have been applied are extremely underpowered [36][37], leaving ample opportunities to fully leverage ML-based methods on large-scale genomic data [38].
Table 1.
Examples of artificial intelligence methods to potentially address current challenges in the study of dementia genetics and omics.
| Challenge | Use of AI/ML/DL |
|---|---|
| Multi-scale or non-linear epistatic interactions are overlooked when investigating genetic variants individually through GWAS | • ML accurately predicts multiple outcomes at a time • Tree-based methods can be used to capture complex non-linear epistatic interactions and select interacting genetic variants |
| GWAS are limited by genetic detection of genome-wide hits | • DL models can deal with non-linear associations between the phenotype and non-genetic covariates to improve GWAS hits detection |
| GWAS are limited by European ancestry based research | • ML models in some cases are better to incorporate trans-ethnic variation and implement transfer learning |
| Cell-type effects and specific pathologies are difficult to reproducibly categorize | • DL can predict cell-type-specific regulatory effects using multi-omics data integration substantially reducing the false positive rate • DL and computer vision can be used for generating harmonized digital pathology datasets |
| PRS are limited by predictive accuracy and hampered by heritability | • Novel DL-based model that does not only rely on the addictive effect of risk SNPs, may outperform more traditional PRS models across a variety of disease phenotypes |
| Causal inferences are often underpowered and limited in scope | • DeepMR [41] approaches integrate ML with MR by using multi-task DL models to learn the relationship between different sets of genomic marks associated with a pathway or phenotype of interest and then uses MR to examine causal relationships between them. |
AI - artificial intelligence; ML - machine learning; DL - deep learning; GWAS - Genome-wide association studies; PRS - polygenic risk score; MR - Mendelian Randomization; DeepMR - Deep Mendelian Randomization.
These ML approaches may provide the key to the development of PRS with greater predictive accuracy and specificity [39]. However, the degree of improvement offered by ML methods may be partly dependent on the complexity and inter-individual heterogeneity of the genetic architecture underlying the disease of interest. For instance, DeepPRS [40], a novel DL-based model that does not only rely on the additive effect of risk SNPs, outperformed more traditional PRS models across a variety of disease phenotypes, including AD. Thus we anticipate further improvements in these approaches will unlock some of the unexplained heritability observed in prior GWAS, enhancing future research, trials, and clinical practice.
2.1.4. Examples of best practice
The Global Parkinson’s Genetics Program (GP2) [41] is in the process of collecting 100,000 European Parkinson’s Disease cases, and a further 50,000 cases from under-represented populations around the world. They are primarily achieving this through collaborations and partnerships with researchers and organizations in other countries across the world, highlighting that large collaborative efforts are crucial for success.
Recent work in multi-ancestry PRS is a good first step in the right direction [42], but with larger sample sizes of participant level data, a ML approach could perform well. Lake and colleagues leverage genetically quantified admixture and random effects models in a population with complex substructures using both random-effects derived risk scores and a risk heuristic that leverages the rates of genetic admixture to build a better predictive model [22].
2.2. Can we find reproducible omics brain signatures that differentiate between dementia subtypes?
2.2.1. State of the science
Omics technologies have been increasingly applied to human brain samples from individuals with dementia and related neurodegenerative conditions [43][44][45][46]. Similarly to the GWAS described in the previous section, the largest brain omics studies have focused exclusively on AD. For example, a meta-analysis of the AD human brain transcriptome [47], which using gene expression data from over 2,000 samples identified 30 coexpression modules as the major source of AD transcriptional perturbations. Additionally, a meta-analysis of AD epigenome-wide association studies [48], using DNA methylation data from over 2,000 individuals identified 334 differentially methylated positions associated with AD neuropathology across cortical regions. Yet, robust disease-specific omics signatures or signatures shared across diseases are lacking. Neurodegenerative diseases are heterogeneous entities and there is extensive clinical, pathological and genetic overlap [49]. Co-pathologies alongside a dominant condition are frequent (e.g. presence of Lewy bodies in AD patients) [50]. Cross disease/pathology studies are starting to emerge, for example addressing epigenetic changes across neurodegenerative diseases [51][52], and disentangling amyloid-β and tau-pathology-associated transcriptomic profiles in AD [53]. However, to find distinguishing molecular signatures we require large well-powered trans-diagnostic cohorts, with a range of primary co-pathologies, and to develop powerful unsupervised ML methods to cluster omics data [54]. Although the increasing availability of single-disease datasets has opened the way to meta-analysis and multiple-cohort reanalysis [55][56][57][58][59][60], much more is needed to assess which mechanisms are conserved across pathologies and which are disease-specific.
2.2.2. What problems need addressing?
It is yet to be understood how and why selective vulnerability occurs in different brain regions and cell types across different neurodegenerative diseases. However, findings from omics studies are often not replicable at the gene/effect level even within a single disease. How then can replicability be enhanced? Several issues need to be addressed: First, studies are often undertaken in small cohorts, which lack statistical power to detect significant molecular changes, and may reflect sampling bias and disease heterogeneity [59]. Availability of brain tissue, especially for rare diseases and for matched cognitively normal controls [61], is a limiting factor. Second, phenotype definitions are not unified. The dominant pathology (e.g. AD or Parkinson’s disease) is often used as the label, but variable degrees of co-pathologies impact molecular signatures. Instead, multiple pathologies could be combined as a quantitative ‘polypathology score’. Third, hemispheric asymmetry in neuronal processes is a fundamental feature of the human brain and drives symptom lateralization (e.g. Parkinson’s disease and FTD), which is reflected molecularly [62][63]. This interferes with histopathology to omics comparisons, mostly investigated in opposite hemispheres [62]. Fourth, genetic variability between individuals is often not accounted for in omics studies. Fifth, there is considerable heterogeneity across studies including differences in brain regions, brain cell type compositions, protocols and platforms to generate the molecular data, and analytic pipelines used. Sixth, the influence of confounding factors, such as batch effects, postmortem interval or RNA/DNA quality, can vary substantially between brain banks due to distinct standard procedures [64][65][66].
2.2.3. Possible solutions
Achieving well-powered cohorts will require an escalation in brain donations, especially for control brains. With appropriate funding of brain banks, or through encouraging and funding brain collection in large-scale population studies, this could be achieved. The adoption of standardized procedures across brain banks is crucial to ensure preservation of appropriate and comparable quality tissue for molecular analyses, and allow seamless integration of samples from different banks. Furthermore, omics studies require deep clinical and pathological phenotyping to reduce heterogeneity and to account for covariates in subsequent data analyses.
The ML paradigm may be useful in multiple ways for the identification of reliable and discriminatory brain omics signatures. There is a clear need to integrate omics data generated for samples both from different brain regions and different cohorts, thus enabling the latent space modeling of multimodal brain omics [67], different brain regions, different cell types [68][69] and different neurodegenerative phenotypes or diseases. This latent space will allow the uniform treatment of samples and a seamless creation of ML models for downstream tasks, such as diagnosis or interpretation.
Multi-omics data in well characterized pathology samples will allow us to refine dementia subtyping. AI can play a huge role in this. DL and computer vision can be used for generating harmonized digital pathology datasets [70]. These datasets and samples can then be input into the pipeline for omics characterization. Data from such pathology-based omics studies will be harmonized across sites using a number of unsupervised learning methods. At its core, single cell resolution using tools like scVI [71] rely on ML to annotate and quantify cellular components of multi-omics datasets which can then be used for multimodal subtyping at the intersection of genomics and pathology.
2.2.4. Examples of best practice
ML approaches applied to dementia brain omics data, such as epigenomics, transcriptomics and proteomics data, have started to emerge and illustrate the promise of using such methods to maximize findings from existing data. Huang and colleagues have recently developed EWASplus, a computational method that uses a supervised ML strategy to extend EWAS coverage to the entire genome [38], and implicates additional epigenetic loci for AD that are not found using array-based AD EWASs. Wang and colleagues implemented a DL method that analyzes RNA-seq data from brain donors to characterize post-mortem brain transcriptome signatures associated with amyloid-β plaques, tau neurofibrillary tangles and clinical severity in multiple AD and related dementia populations [58]. In the proteomics space, Tasaki and colleagues applied a deep neural network approach to predict protein abundance from mRNA expression, in an attempt to track the early protein drivers of AD and related dementia subtypes [72]. These approaches demonstrate how such methodologies can be used to identify potential early protein drivers and possible drug targets for preventing or treating AD and related dementias.
2.3. Can high-dimensional omics data identify improved molecular biomarkers for dementia compared to single marker approaches?
2.3.1. State of the science
Technological advances and large, shared, international datasets allow a new approach to understanding diseases including biomarker identification. Single molecule assays such as Simoa, allow accurate measurement of plasma proteins [73]. Notably, plasma neurofilament light (NfL) has been comprehensively shown by many research groups to be substantially increased in a diverse array of neurological brain conditions when compared with age-matched controls, leading to the proposal of NfL being the first established blood-biomarker for neurological and cognitive decline [74]. Targeted biomarkers such as NfL have begun to be translated into clinical settings but the use of multi-omics data has so far been limited. However, omics modalities present opportunities for the identification and application of new biomarkers. For example, most dementias appear to have a considerable polygenic component, which present potential as multi-assay risk biomarkers. Genome sequences comprising petabytes of data can be resolved to common single nucleotide variation, rare variants, and structural variants all with potential as markers of disease risk. RNA expression data is currently used in biomarker discovery though not yet achieving the accuracy of blood proteins in disease prediction [75][76].
DNA methylation data can provide a route to identify non-recorded environmental exposures through imputation of these risk factors from published predictors [77]. This strategy could help validate epidemiological reports of environmental risk factors and help stratify patients across diagnostic boundaries, which may provide stimuli for additional analyses and clinical follow-up [78]. Genes where DNA methylation is altered by specific environmental factors could identify molecular pathways of relevance across dementias. In addition to markers of aging, they have also been used as predictors of cognitive function [79]. However, before these markers can be translated to the clinic they would need to demonstrate stringent accuracy in independent validation cohorts.
While these multimodal datasets described above can contribute to biomarker discovery, many diagnostics companies and regulatory bodies prefer a single readout approach. This is contrary to the basic concept that multimodal data can more accurately reflect complex biological systems.
2.3.2. What problems need addressing?
The development of large harmonized omics datasets is challenging. The first challenge relates to the issue of data quality: high dimensional omics data are acquired from different sources, in distinct formats and over multiple sites, and accompanied by patient medical records. As errors may occur during measurement or processing (i.e. batch effects), they risk potentially compromising the reproducibility and the usability of the generated data. The second challenge is of a computational nature: the preliminary analyses of multi-omics data require a data harmonization process and the development of integration, clustering, functional characterization and visualization tools. Beyond this step, one of the goals in the biomarker study is the inference and the prediction of biological systems [80]. The statistical method traditionally deployed in the inference requires explicit assumptions, which are not necessarily intuitive in the large omics dataset [81]. Finally, given dimensionality constraints posed by integrating large multiple omics datasets, the computational burden and storage space requirements can be limiting. The last challenge is to make these datasets sharable and accessible to a large community [82]. The development of a large omics dataset therefore requires establishing standardized protocols for the acquisition, transfer and analysis of clinical and omics data that can be used by the scientific research community.
At its core, the issues with multimodal datasets needed for building the next generation of complex biomarkers is both a wide data and sparsity problem. Studies are simply not large enough, similar enough, or data easily accessible enough to identify better biomarkers which have clinical relevance.
2.3.3. Possible solutions
Recently, ML approaches have made considerable advances in genomics, multi-omics, biomedicine, and data-driven therapeutics discovery [83][84][85][39]. Application of DL approaches on large scale omics datasets allows researchers to detect new disease relationships with the data. Translating these discoveries into multi-panel tests will be key in applying potential biomarkers. As the costs of omics assays continue to drop, the standard use of high-throughput DNA, RNA, protein and metabolomics biomarkers in the clinic need to become a reality. Large-scale sequencing initiatives that focus on the genomic underpinnings of neurodegenerative diseases [41][86][87][88][89][90] will aid in the development of more targeted and cost-effective tests such as PRSs and metabolite panels [91]. Collectively, these initiatives will enable many opportunities for biomarker identification, validation in both diagnosis and early disease detection, as well as raise important ethical and technical challenges.
In its simplest terms, information theory dictates that adding impactful and independent features to a model should improve its predictability, although limiting analyses to such features may be difficult due to wide data issues in genomics. In ML, facing high dimensionality problems where the number of features is much greater than the number of samples is relatively frequent. That is why the problem of feature selection has worsened in recent decades [92][93]. In addition, techniques such as federated learning [94] are likely to be useful in analyzing biomarkers across datasets that cannot be combined for ethical or practical reasons safely.
2.3.4. Examples of best practice
Analyzing datasets from independent cohorts and then combining them in a meta-analysis can improve statistical power and the ability to detect significant associations. For example, a meta-analysis of 569 lipidomics species measured in the Australian Imaging, Biomarkers and Lifestyle (AIBL) cohort and the Alzheimer’s Disease Neuroimaging Initiative (ADNI) cohort identified multiple lipids from several species predictive of prevalent and incident AD [95]. Within cohort integration of data modalities can also yield novel disease markers, for example, co-expression networks of metabolite and gene expression data from the ADNI cohort identified new metabolite candidate markers [96]. The European Medical Information Framework Alzheimer’s Disease (EMIF-AD) project (http://www.emif.eu/emif-ad-2/), set up a pan-European platform for large-scale research on biomarkers and risk factors for neurodegenerative disorders. The EMIF-AD Multimodal Biomarker Discovery study harmonized and pooled clinical data from 11 cohort studies and samples from CSF, plasma, DNA, and MRI scans were centrally analyzed using different omics techniques (proteomics, metabolomics, and genomics) and integrated analysis has demonstrated the power of such approaches. The Accelerating Medicines Partnership - Alzheimer’s Disease (AMP-AD) (https://www.nia.nih.gov/research/amp-ad) allows researchers to access multiple cohorts via a single platform. It is a partnership between government, industry, and nonprofit organizations to transform the current model for developing new diagnostics and treatments for AD. The sharing of multi-omics datasets through this centralized data infrastructure, the AD Knowledge Portal, enables integrative and collaborative analyses to more easily and effectively advance biomarker identification and replication. Improved standardization and harmonization of multi-omics data across silos will benefit the field in the future. In addition, combining multi-omics and clinical data with wearable or other streaming data may yield exciting results such as has been seen in the Parkinson’s disease field by Rune Labs’ AppleWatch app (https://www.accessdata.fda.gov/cdrh_docs/pdf21/K213519.pdf).
2.4. How do we use genetics to inform our understanding of causal risk factors?
2.4.1. State of the science
It was recently estimated that reducing modifiable risk factors could prevent around 40% of all-cause dementia cases [97]. However, the evidence-base for most hypothesized risk factors being causal is weak, with conflicting findings across studies depending on study design, time of risk factor measurement, type of outcome, sample size and study population [97][98]. Many studies are prone to bias by unmeasured or residual confounding, reverse causation due to dementia’s long latency period, and survival bias. Traditionally, randomized controlled trials (RCTs) have been necessary to confirm causal pathways between a risk factor and an outcome. However, these are notoriously challenging for dementia research because it would require monitoring participants over many decades due to the long and ill-defined prodromal period of dementia. In addition, it would be impractical or unethical to conduct an RCT of harmful risk factors such as air pollution and traumatic brain injury. These limitations make it difficult to ascertain which risk factors would be the most useful to target in interventions, and at what point in life such interventions would be most efficacious.
Mendelian randomization (MR) gives us a strong foundation to interrogate the causal status of risk factors. MR overcomes several limitations inherent to observational research, whilst utilizing more easily accessible cross-sectional rather than prospective data [99]. MR uses genetic variants as instrumental variables (IVs) for risk factors in what has been dubbed a natural RCT. Because an individual’s genome is assigned randomly at conception, it is largely independent of confounding factors that often cause bias in observational research. The genome also cannot be modified by subsequent disease, making bias due to reverse causation unlikely. MR is a widely used method and can be a useful tool for understanding the etiology of risk factors [100][101][102][103][104], but it also has limitations that should be carefully considered [105][106]. Despite the clear advantages of MR studies few other methods have been developed that can explore the causal relationships between risk factors and dementia-related outcomes.
2.4.2. What problems need addressing?
There are several common problems that can impact causal inference if they are not duly addressed and can lead to unreliable conclusions being made. Power is problematic in many MR studies examining causality of risk factors on dementia [100]. Confidence intervals are often wide, so meaningful effects in either direction cannot be excluded. This is often the case for risk factors that are difficult to measure (e.g. sleep disturbance and physical inactivity) [107][108]. Weak instruments (i.e. those with an F-statistic <10) can introduce bias [109]. Examples of strong instruments that have been used in MR of dementia risk include plasma glucose [110], educational attainment and intelligence [111], type-2 diabetes mellitus and glycated hemoglobin (HbA1c) [112], but these only represent a small fraction of dementia risk factors.
Collider bias can also be introduced into causal analyses when an included sample suffers from selection bias, for example due to differential patterns of survival associated with the risk factor of interest [113]. Individuals need to live long enough to obtain a dementia diagnosis so observed causal effects of any risk factor associated with premature mortality (e.g. smoking) on dementia risk are likely biased [114]. Very few studies attempt to identify and, if necessary, correct for survival bias, despite it being demonstrated to produce spurious protective effects in MR studies of causal risk factors for AD and Parkinson’s disease [115][116]. Causal analyses may also be biased by population effects that confound the relationship between the genetic instrument and outcome variable (violating the ‘independence’ MR assumption [117]). Certain dementia risk factors, such as educational attainment, have been shown to be highly influenced by assortative mating (i.e. non-random mating) within populations [117], but this has not yet been systematically assessed in studies of dementia risk factors, so we do not know the extent to which current causal estimates are being biased by these population effects.
Confounding due to horizontal pleiotropy is especially problematic in MR studies that measure the causal association between a complex risk factor (i.e. a phenotype that is highly polygenic) and an outcome. It is becoming increasingly apparent that many SNPs in the genome causally influence multiple traits, making the ‘exclusion restriction’ MR assumption (i.e. that the only path between the genetic instrument and the outcome is via the exposure) less likely to be upheld. In addition, even though many dementia risk factors are genetically inter-correlated [118] and co-occurrence of multiple risk factors within an individual increases dementia risk more than being exposed to a single risk factor [119], most studies only measure the causality of one risk factor on dementia. By only measuring bivariate relationships, we are likely overlooking synergistic effects or overlapping causal pathways between dementia risk factors, reducing our ability to identify shared biological pathways that are especially central in raising dementia risk and to characterize the patterns of pleiotropic effects between risk factors. There are methods to disentangle this such as genomic or transcriptomic structural equation modeling-SEM [120][121], but they require well-powered GWAS, which are not available for all risk factors.
Aside from MR, few causal modeling methods have been developed for use with genetic data. Even in cases where new causal methods have been proposed, such as Bayesian network analysis (BN)[122], latent causal variable analysis (LCV) [123] and the multi-SNP mediation intersection-union test (SMUT)[124], these have not yet been applied in dementia risk factor research and there is a noticeable lack of causal ML modeling in the genomics field.
2.4.3. Possible solutions
One of the key ways that AI methods could be harnessed to improve causal analyses in dementia research is to use ML/DL to strengthen genetic instruments for MR. Traditionally, instruments are created from GWAS summary statistics that are measured using logistic regression and defined p-value thresholds, whereas COMBI [28] and DeepCOMBI [33] use Support Vector Machines (SVM) and deep neural networks, respectively, to identify SNPs related to a phenotype. Particularly, DeepCOMBI has been shown to replicate known disease loci, as well as identify novel ones. DeepMR integrates ML with MR by using multi-task DL models to initially learn the relationship between different sets of genomic marks (e.g. chromatin marks) associated with a pathway or phenotype of interest and then uses MR to examine causal relationships between them [125], which could help to identify more functionally relevant SNPs for inclusion in the exposure instrumental variable.
Existing methods that quantify and correct for known sources of bias should also be routinely implemented. Automated AI methods could help support this, for example MR-MoE (MR-Mixture of Experts), which is an ML framework that applies random forest learning algorithms to MR results to identify the method for your analysis that is least likely to be biased by horizontal pleiotropy [126].
Several of the associations between dementia and its risk factors are likely non-linear. For example, the association between sleep duration and dementia is likely to be U-shaped: both too little and too much sleep have been associated with increased dementia risk [97][127][128]. In this instance, sleep duration is a categorical discrete rather than a truly continuous phenotype, and its genetic instruments are weak in comparison with other risk factors [110]. Non-linear MR accounts for non-linearity between continuous exposures and outcomes [129] but it has scarcely been applied to MR studies of dementia risk. One recent study used non-linear MR to assess the causal influence of sleep duration on dementia-related cognitive outcomes [130]. Thus, to use MR to understand non-linear relationships between risk factors and dementia, we should focus future GWAS efforts on improving the modeling of continuous risk factors in situations where observational evidence suggests that there is a non-linear causal relationship with dementia.
Room for future improvement includes the potential leveraging of tree-based, boosted, bagged or other ML algorithms to create interpretable model cascades of causal risk. This could increase the value of previous MR studies while at the same time addressing their shortcoming of generally focusing on only a single exposure at a time. AI has the power to model multiple potentially connected causal risk factors at scale.
2.4.4. Examples of best practice
Recently, a multivariate GWAS was performed using random forest regression to predict causal SNPs for 56 neuroimaging phenotypes, which identified the APOE SNP rs429358 as the top locus as well as additional lead SNPs that mapped to genes relevant to brain disorders, which were not identified by traditional linear regression methods [131]. Another study introduced the MR-based Structure Learning (MRSL) algorithm, which used graph theory combined with multivariable MR to uncover causal and mediating pathways between 44 diseases and 26 biomarkers using publicly available GWAS summary statistics [132]. Together, these results highlight the potential benefits of utilizing ML-based multivariate approaches to model the genetics underlying inter-correlated risk factor traits when performing causal analyses in dementia research.
Noyce and colleagues previously assessed the impact of survival bias on estimates of the causal effect of BMI on Parkinson’s disease [116]. They performed simulations to estimate the likely effect that their MR analysis would show if survival bias was present, when assuming that BMI was not truly related to Parkinson’s disease. The objective was to see if the likely magnitude of the survival bias was large enough to explain the MR results estimated from the real data. They demonstrated that the seemingly protective effect of higher BMI on Parkinson’s disease risk was likely due to survival bias related to increased frailty in people with lower BMI, rather than being the true causal driver. Since effects from survival bias are likely to be especially important for causal analysis of risk factors in dementia research it is crucial that we start to consistently test for this and other common forms of bias in future studies to minimize the impact of spurious findings within our field.
2.5. Which biological processes are altered by genetic risk for dementia-related diseases?
2.5.1. State of the science
Highly penetrant variants in APP, PSEN1 or PSEN2 have pointed to a central role of amyloid-β in early-onset AD [133]. Separately, GWAS for late-onset AD identified several biological processes enriched for genes associated with disease risk, including amyloid-β processing, lipid metabolism, and immune responses [134][135]. Although most AD GWAS associations are non-coding, rare coding variants have implicated key microglial genes such as TREM2 and PLCG2 [135][136]. Follow-up experiments in cellular and animal models confirmed the effects of these genes on microglial activation and lipid processing [137][138]. Epigenomic maps from purified cell populations [139] or single cells [140] have localized non-coding AD risk variants to microglia-specific enhancers, regulating genes including BIN1 and RIN3. An alternative way of linking risk variants to genes is to identify quantitative trait loci (QTLs) that influence gene expression, followed by a test for statistical colocalization with nearby GWAS loci. A variation on the previously discussed topic of MR called SMR is often used to establish causal inferences for the function of these QTLs in the context of disease risk on a per gene level. Recent studies in purified microglia from living [141] or post-mortem [142][143] donors have nominated some AD and Parkinson’s disease risk genes, but so far they are underpowered relative to bulk brain datasets. Thus, while genetic studies of AD indicate a clear role of microglia [144][135][136][141][145], the roles of specific cell types are still being discovered in other neurodegenerative conditions, such as Parkinson’s disease [139][146] and amyotrophic lateral sclerosis [147].
2.5.2. What problems need addressing?
GWAS for different dementias have so far mainly used a case-control framework to identify genetic loci associated with a clinical diagnosis. However, this approach ignores the complexity of neuropathological changes that occur in patients, which usually predate clinical symptoms by years or decades, and which may involve multiple distinct pathologies [54][148]. The decoupling of genetic associations from specific pathologies makes it difficult to identify the most relevant cellular model for a given locus. In this absence, most cellular models have focused on a single cell type, and thereby fail to elucidate the probable interplay between different cell types that leads to neurodegeneration. Furthermore, identifying and validating the causal genes at GWAS loci continues to remain challenging, due to both the uncertainty in the specific causal variants and the cell types through which they act [149]. Additionally, GWAS loci may arise only in a specific cellular state, such as response to a pathology, as has been recently shown for the UNC13A amyotrophic lateral sclerosis/FTD locus [150][151]. As a result, the genes and biological processes that are identified as relevant have depended largely upon the prior hypotheses of investigators and on the cellular models and analysis methods that were used. Although the scale and resolution of single-cell transcriptomic and epigenomic datasets is increasing, there isn’t yet a robust and reproducible catalog of all cell types and cell states relevant to brain function and disease processes. Additionally, curated resources cataloging genes involved in many biological processes are often victims of bias due to publication and funding issues as well as reporting bias.
2.5.3. Possible solutions
New technologies have the potential to improve our understanding of neurodegenerative diseases, if applied systematically and at scale. Single-cell technologies are beginning to reveal the cell type diversity of the human brain [152], and to identify cell type-specific gene expression changes in disease [140,153]. The GTEx project [154] was transformative in describing gene regulation across human tissues, enabling others to link these genetic effects to human disease risks. However, its sampling of bulk tissues limits its use for understanding biological mechanisms. Single-cell technologies now make it possible to envision a cell type-specific gene regulatory atlas of the human brain. Such an atlas should be built in a robust way across multiple labs, and include both healthy and diseased donors of different ages.
We must also seek to recapitulate the spatial dimension of cell type localization and gene expression. Only by probing gene expression directly in a tissue section can we reliably establish organ-wide patterns of gene expression, reconstruct cell-cell interactions and assess how neuropathology affects local gene expression. Mouse models have highlighted how amyloid plaques influence oligodendrocyte and microglia gene expression across disease stages [155]. Going forward, a brain-wide, spatially-resolved gene expression atlas, possibly integrating splicing information [156], would be a rich complement to a standard gene regulatory atlas.
To understand the molecular mechanisms of neurodegenerative disease genetic associations, we need to perturb the function of candidate genes and measure their effects in relevant cellular models. However, an ad-hoc approach in the most accessible cell types will not lead to robust conclusions. With CRISPR-based tools these perturbations can be done at genome-wide scale, in specific cell types derived from human induced pluripotent stem cells (iPSCs), and with high-throughput phenotyping assays. As a community, we should coordinate to systematically investigate a broad set of candidate genes, across multiple cellular phenotypes and in a range of cellular models. Additionally, as part of therapeutic development, these perturbed screens will likely need to be carried out across networks upstream of known targets.
2.5.4. Examples of best practice
For psychiatric disease, the PsychENCODE project set an example by collecting multiple types of omic data from over a thousand post-mortem brains across three diseases and three brain regions [157][46,158]. Crucially, integrative analyses need to leverage these multiple omic layers to generate novel insights, as demonstrated in previous studies of bulk brain [46][159]. Recent studies have used scRNA-seq methods to examine specific brain regions in disease and control individuals for AD [153][160], amyotrophic lateral sclerosis and FTD [161], revealing cell type-specific effects of disease pathology. For all of these datasets and analyzes to be most useful, robust ML methods are needed to integrate distinct omics modalities and to ensure reproducible results. Promising approaches in this direction have recently been applied to large-scale single-cell data from mouse motor cortex [162], and the human immune system [163].
As genetic studies of dementias increase in size, so does the need to identify the causal genes at associated loci. New methods enable enhanced fine-mapping using functional genomic data (e.g. PolyFun [164]), and better prediction of enhancer-promoter connections (e.g. activity-by-contact score). One such example is the identification of USP6NL as the putative causal gene within the AD GWAS locus ‘ECHDC3’ by linking a functionally fine-mapped variant within a microglia enhancer with the USP6NL promoter [142]. This finding was further supported by strong colocalization between the GWAS-eQTL. This methodology has also been applied to Parkinson’s disease [165]. DL models have also shown dramatic improvements in predicting the effects of genetic variants on splicing, pathogenicity (coding variants), and gene expression. Along with experimental data, both variant effect predictions and fine-mapping data can be used as input to ML methods that directly predict the most likely causal genes at GWAS loci.
Beyond cellular maps and genetic associations, a systematic approach to model systems is needed. An NIH-funded project, the iPSC Neurodegenerative Disease Initiative (iNDI) [166], is creating more than 100 isogenic iPSC lines with mutations associated with dementias. How these are used to model neurodegeneration in specific derived cell types will be up to the creativity and vision of the research community.
CRISPR based studies and methods such as perturbSeq and CROPseq have pushed the boundaries of what can be assayed rapidly with edited cell lines [167]. These techniques are already being sought after by biotechs looking to quantify up and downstream effects of genetic and genomic therapeutic targets. Enough of this type of data, combined with DL to recognize patterns of functionally connected genes or graph-based network models could identify communities of risk factors that are functionally connected to disease risk [168]. These new communities could serve as less biased pathways derived from the appropriate tissues and cell types.
3. Limitations of AI and ML in the dementia omics field
High-throughput methods, such the full suite of omics platforms, including genomic, transcriptomic, epigenomic, proteomic, metabolomic, and related technologies, have inaugurated a new era of systems biology. This provides abundant and detailed data, which conventional analytical and statistical approaches are often not capable of dealing with. AI and ML algorithms, which are designed to automatically mine data for insights into complex relationships in these massive datasets, are still at its infancy in dementia genetics and omics research, and far from being explored at its full capacity. Despite major strengths and achievements so far, it is worth having in mind possible caveats of AI models in the omics field, including the following examples: 1) Interpretation (the black box), as often the complexity of certain models makes it difficult to understand the learned patterns and consequently it is challenging to infer the causal relationship between the data and an outcome; 2) “Curse” of dimensionality: omics datasets represent a huge number of variables and often a small number of samples, as mentioned in multiple sections of this paper; 3) Imbalanced classes: most models applied to omics data deal with disease classification problems (e.g. use of major pathology labels in the presence of co-pathologies, as mentioned in section 2.2); and 4) Heterogeneity and sparsity: data from omics applications is often heterogeneous and sparse since it comes from subgroups of the population (e.g. as highlighted in section 2.1), different platforms (e.g. multiple array and sequencing based platforms), multiple omics modalities (e.g. transcriptomics, epigenomics, proteomics) and is often resource intensive to generate. Many of these limitations, however, can be overcomed with improvements to data generation (e.g. larger more diverse harmonizable studies) and analysis (e.g. using dimensionality reduction strategies and interpretable ML approaches).
4. Concluding remarks
In conclusion, omics technologies, including genomics, epigenomics, transcriptomics, proteomics, and metabolomics, can provide increasingly comprehensive high-dimensional insights into the biological system of each individual when combined with AI approaches. This in turn can contribute immensely to a better understanding of AD and other forms of dementia, and to the development of personalized medicines. However, a number of thorny issues hamper the use of omics technologies and AI in dementia research. These include the need for better and more comprehensive and less biased genetics and omics dementia-related data resources, the development of improved AI algorithms, and the need for more collaborative multidisciplinary collaboration. Increased funding, a more coordinated collaborative global effort, and a greater number of diverse and deeply phenotyped cohorts, together with innovative AI methods have the potential to overcome these challenges and to increase the pace of discovery that we are able to achieve. Ultimately, this would have a major impact on our understanding of the underlying disease processes and help to improve the prevention, diagnosis, and treatment of dementia.
Supplementary Material
Highlights.
We have identified five key challenges in dementia genetics and omics studies
AI can enable detection of undiscovered patterns in dementia genetics and omics data
Enhanced and more diverse genetics and omics datasets are still needed
Multidisciplinary collaborative efforts using AI can boost dementia research
Acknowledgements
With thanks to the Deep Dementia Phenotyping (DEMON) Network State of the Science symposium participants (in alphabetical order): Peter Bagshaw, Robin Borchert, Magda Bucholc, James Duce, Charlotte James, David Llewellyn, Donald Lyall, Sarah Marzi, Danielle Newby, Neil Oxtoby, Janice Ranson, Tim Rittman, Nathan Skene, Eugene Tang, Michele Veldsman, Laura Winchester, Zhi Yao.
Funding
This paper was the product of a DEMON Network state of the science symposium entitled “Harnessing Data Science and AI in Dementia Research” funded by Alzheimer’s Research UK. CB is supported by Alzheimer’s Research UK (ARUK-RF2019B-005) and Multiple System Atrophy Trust. NS is supported by the UK Dementia Research Institute which receives its funding from UK DRI Ltd, funded by the UK Medical Research Council, Alzheimer’s Society and Alzheimer’s Research UK. NS also received funding from a UKRI Future Leaders Fellowship (MR/T04327X/1). EA is supported by MRC Skills Development Fellowship (MR/W011581/1) and UKRI Future Leaders Fellowship (MR/W011581/1). LW is supported Alzheimer’s Research UK. IFF is supported by the National Institute on Aging (RF1AG073593). MAN’s participation in this project was part of a competitive contract awarded to Data Tecnica International LLC by the National Institutes of Health to support open science research. JH is supported by the NIH National Institute of Neurological Disorders and Stroke (U54NS123743). SJM is funded by the Edmond and Lily Safra Early Career Fellowship Program and the UK Dementia Research Institute, which receives its funding from UK DRI Ltd, funded by the UK Medical Research Council, Alzheimer’s Society and Alzheimer’s Research UK. AAK is funded by ALS Association Milton Safenowitz Research Fellowship (grant number22-PDF-609. DOI:10.52546/pc.gr.150909), The Motor Neurone Disease Association (MNDA) Fellowship (Al Khleifat/Oct21/975-799), The Darby Rimmer Foundation, and The NIHR Maudsley Biomedical Research Centre. ELH is supported by the Alzheimer’s Society (AS-RF-21-017) and the Cambridge British Heart Foundation Centre of Research Excellence (RE/18/1/34212). VG is supported by Diabetes UK (15/0005250), British Heart Foundation (SP/16/6/32726) and Professor David Matthews Non-Clinical Fellowship from the Diabetes Research and Wellness Foundation (SCA/01/NCF/22). CS is supported by the UK Dementia Research Institute (UK DRI) funded by the Medical Research Council (MRC), Alzheimer’s Society and Alzheimer’s Research UK, and by the Ser Cymru II programme which is part-funded by Cardiff University and the European Regional Development Fund through the Welsh Government. SK is supported by a PhD studentship award from Alzheimer’s Society, UK (AS-PhD-19b-014) and the Ser Cymru II programme. JMR and DJL are supported by Alzheimer’s Research UK and the Alan Turing Institute/Engineering and Physical Sciences Research Council (EP/N510129/1). DJL also receives funding from the Medical Research Council (MR/X005674/1), National Institute for Health Research (NIHR) Applied Research Collaboration South West Peninsula, National Health and Medical Research Council (NHMRC), and National Institute on Aging/National Institutes of Health (RF1AG055654). This research was supported in part by the Intramural Research Program of the NIH, National Institute on Aging (NIA), National Institutes of Health, Department of Health and Human Services; project number ZO1 AG000535 and ZIA AG000949, as well as the National Institute of Neurological Disorders and Stroke (NINDS). The views expressed in this publication are those of the authors and not necessarily those of the NIHR, NHS, or UK Department of Health and Social Care.
Footnotes
Conflicts of interest
JS is an employee of Illumina Inc. MAN currently serves on the scientific advisory board for Character Biosciences Inc and Neuron 23 Inc. All other authors declare no competing interests.
References
- [1].Robinson L, Tang E, Taylor J-P. Dementia: timely diagnosis and early intervention. BMJ 2015;350:h3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].GBD 2016 Dementia Collaborators. Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol 2019;18:88–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Jansen IE, Savage JE, Watanabe K, Bryois J, Williams DM, Steinberg S, et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet 2019;51:404–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wightman DP, Jansen IE, Savage JE, Shadrin AA, Bahrami S, Holland D, et al. A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer’s disease. Nat Genet 2021;53:1276–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Nalls MA, Blauwendraat C, Vallerga CL, Heilbron K, Bandres-Ciga S, Chang D, et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol 2019;18:1091–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ferrari R, Hernandez DG, Nalls MA, Rohrer JD, Ramasamy A, Kwok JBJ, et al. Frontotemporal dementia and its subtypes: a genome-wide association study. Lancet Neurol 2014;13:686–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Guerreiro R, Ross OA, Kun-Rodrigues C, Hernandez DG, Orme T, Eicher JD, et al. Investigating the genetic architecture of dementia with Lewy bodies: a two-stage genome-wide association study. Lancet Neurol 2018;17:64–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bellenguez C, Küçükali F, Jansen IE, Kleineidam L, Moreno-Grau S, Amin N, et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat Genet 2022;54:412–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Chia R, Sabir MS, Bandres-Ciga S, Saez-Atienzar S, Reynolds RH, Gustavsson E, et al. Genome sequencing analysis identifies new loci associated with Lewy body dementia and provides insights into its genetic architecture. Nat Genet 2021;53:294–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Fongang B, Sargurupremraj M, Jian X, Mishra A, Damotte V, Bis JC, et al. A meta-analysis of genome-wide association studies identifies new genetic loci associated with all-cause and vascular dementia. bioRxiv 2022:2022.10.11.509802. 10.1101/2022.10.11.509802. [DOI] [Google Scholar]
- [11].Karczewski KJ, Snyder MP. Integrative omics for health and disease. Nat Rev Genet 2018;19:299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Park JJH, Mogg R, Smith GE, Nakimuli-Mpungu E, Jehan F, Rayner CR, et al. How COVID-19 has fundamentally changed clinical research in global health. The Lancet Global Health 2021;9:e711–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Staley JR, Jones E, Kaptoge S, Butterworth AS, Sweeting MJ, Wood AM, et al. A comparison of Cox and logistic regression for use in genome-wide association studies of cohort and case-cohort design. Eur J Hum Genet 2017;25:854–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Stranger BE, Stahl EA, Raj T. Progress and Promise of Genome-Wide Association Studies for Human Complex Trait Genetics. Genetics 2011;187:367–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Fadista J, Manning AK, Florez JC, Groop L. The (in)famous GWAS P-value threshold revisited and updated for low-frequency variants. Eur J Hum Genet 2016;24:1202–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Choi SW, Mak TS-H, O’Reilly PF. Tutorial: a guide to performing polygenic risk score analyses. Nat Protoc 2020;15:2759–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Gatz M, Reynolds CA, Fratiglioni L, Johansson B, Mortimer JA, Berg S, et al. Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry 2006;63:168–74. [DOI] [PubMed] [Google Scholar]
- [18].Fuente J de la, de la Fuente J, Grotzinger AD, Marioni RE, Nivard MG, Tucker-Drob EM. Integrated analysis of direct and proxy genome wide association studies highlights polygenicity of Alzheimer’s disease outside of the APOE region. PLOS Genetics 2022;18:e1010208. 10.1371/journal.pgen.1010208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ryan J, Fransquet P, Wrigglesworth J, Lacaze P. Phenotypic Heterogeneity in Dementia: A Challenge for Epidemiology and Biomarker Studies. Front Public Health 2018;6:181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kunkle BW, Schmidt M, Klein H-U, Naj AC, Hamilton-Nelson KL, Larson EB, et al. Novel Alzheimer Disease Risk Loci and Pathways in African American Individuals Using the African Genome Resources Panel: A Meta-analysis. JAMA Neurol 2021;78:102–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Shigemizu D, Mitsumori R, Akiyama S, Miyashita A, Morizono T, Higaki S, et al. Ethnic and trans-ethnic genome-wide association studies identify new loci influencing Japanese Alzheimer’s disease risk. Transl Psychiatry 2021;11:151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Lake J, Solsberg CW, Kim JJ, Acosta-Uribe J, Makarious MB, Li Z, et al. Multi-ancestry meta-analysis and fine-mapping in Alzheimer’s Disease. medRxiv 2022:2022.08.04.22278442. 10.1101/2022.08.04.22278442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Sherva R, Zhang R, Sahelijo N, Jun G, Anglin T, Chanfreau C, et al. African Ancestry GWAS of Dementia in a Large Military Cohort Identifies Significant Risk Loci. medRxiv 2022:2022.05.25.22275553. 10.1101/2022.05.25.22275553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Persyn E, Hanscombe KB, Howson JMM, Lewis CM, Traylor M, Markus HS. Genome-wide association study of MRI markers of cerebral small vessel disease in 42,310 participants. Nat Commun 2020;11:2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Yan Q, Nho K, Del-Aguila JL, Wang X, Risacher SL, Fan K-H, et al. Genome-wide association study of brain amyloid deposition as measured by Pittsburgh Compound-B (PiB)-PET imaging. Mol Psychiatry 2021;26:309–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Damotte V, van der Lee SJ, Chouraki V, Grenier-Boley B, Simino J, Adams H, et al. Plasma amyloid β levels are driven by genetic variants near APOE, BACE1, APP, PSEN2: A genome-wide association study in over 12,000 non-demented participants. Alzheimers Dement 2021;17:1663–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Vialle RA, de Paiva Lopes K, Bennett DA, Crary JF, Raj T. Integrating whole-genome sequencing with multi-omic data reveals the impact of structural variants on gene regulation in the human brain. Nat Neurosci 2022;25:504–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Mieth B, Kloft M, Rodríguez JA, Sonnenburg S, Vobruba R, Morcillo-Suárez C, et al. Combining Multiple Hypothesis Testing with Machine Learning Increases the Statistical Power of Genome-wide Association Studies. Sci Rep 2016;6:36671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Nicholls HL, John CR, Watson DS, Munroe PB, Barnes MR, Cabrera CP. Reaching the End-Game for GWAS: Machine Learning Approaches for the Prioritization of Complex Disease Loci. Front Genet 2020;11:350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Machine learning approaches to genome-wide association studies. Journal of King Saud University - Science 2022;34:101847. [Google Scholar]
- [31].Behravan H, Hartikainen JM, Tengström M, Pylkäs K, Winqvist R, Kosma V-M, et al. Machine learning identifies interacting genetic variants contributing to breast cancer risk: A case study in Finnish cases and controls. Sci Rep 2018;8:13149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Sun T, Wei Y, Chen W, Ding Y. Genome-wide association study-based deep learning for survival prediction. Stat Med 2020;39:4605–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Mieth B, Rozier A, Rodriguez JA, Höhne MMC, Görnitz N, Müller K-R. DeepCOMBI: explainable artificial intelligence for the analysis and discovery in genome-wide association studies. NAR Genom Bioinform 2021;3:lqab065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Arloth J, Eraslan G, Andlauer TFM, Martins J, Iurato S, Kühnel B, et al. DeepWAS: Multivariate genotype-phenotype associations by directly integrating regulatory information using deep learning. PLoS Comput Biol 2020;16:e1007616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].McCaw ZR, Colthurst T, Yun T, Furlotte NA, Carroll A, Alipanahi B, et al. DeepNull models non-linear covariate effects to improve phenotypic prediction and association power. Nat Commun 2022;13:241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].De Velasco Oriol J, Vallejo EE, Estrada K, Taméz Peña JG, Disease Neuroimaging Initiative TA. Benchmarking machine learning models for late-onset alzheimer’s disease prediction from genomic data. BMC Bioinformatics 2019;20:709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Jo T, Nho K, Bice P, Saykin AJ, Alzheimer’s Disease Neuroimaging Initiative. Deep learning-based identification of genetic variants: application to Alzheimer’s disease classification. Brief Bioinform 2022;23. 10.1093/bib/bbac022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Huang Y, Sun X, Jiang H, Yu S, Robins C, Armstrong MJ, et al. A machine learning approach to brain epigenetic analysis reveals kinases associated with Alzheimer’s disease. Nat Commun 2021;12:4472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ho DSW, Schierding W, Wake M, Saffery R, O’Sullivan J. Machine Learning SNP Based Prediction for Precision Medicine. Front Genet 2019;10:267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Peng J, Li J, Han R, Wang Y, Han L, Peng J, et al. A deep learning-based genome-wide polygenic risk score for common diseases identifies individuals with risk. bioRxiv 2021. 10.1101/2021.11.17.21265352. [DOI] [Google Scholar]
- [41].Program TGPG, The Global Parkinson’s Genetics Program. GP2 : The Global Parkinson’s Genetics Program. Movement Disorders 2021;36:842–51. 10.1002/mds.28494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Wang Y, Tsuo K, Kanai M, Neale BM, Martin AR. Challenges and Opportunities for Developing More Generalizable Polygenic Risk Scores. Annu Rev Biomed Data Sci 2022;5:293–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Yang C, Farias FHG, Ibanez L, Suhy A, Sadler B, Fernandez MV, et al. Genomic atlas of the proteome from brain, CSF and plasma prioritizes proteins implicated in neurological disorders. Nat Neurosci 2021;24:1302–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].De Jager PL, Srivastava G, Lunnon K, Burgess J, Schalkwyk LC, Yu L, et al. Alzheimer’s disease: early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat Neurosci 2014;17:1156–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Lunnon K, Smith R, Hannon E, De Jager PL, Srivastava G, Volta M, et al. Methylomic profiling implicates cortical deregulation of ANK1 in Alzheimer’s disease. Nat Neurosci 2014;17:1164–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Marzi SJ, Leung SK, Ribarska T, Hannon E, Smith AR, Pishva E, et al. A histone acetylome-wide association study of Alzheimer’s disease identifies disease-associated H3K27ac differences in the entorhinal cortex. Nat Neurosci 2018;21:1618–27. [DOI] [PubMed] [Google Scholar]
- [47].Wan Y-W, Al-Ouran R, Mangleburg CG, Perumal TM, Lee TV, Allison K, et al. Meta-Analysis of the Alzheimer’s Disease Human Brain Transcriptome and Functional Dissection in Mouse Models. Cell Rep 2020;32:107908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Shireby G, Dempster EL, Policicchio S, Smith RG, Pishva E, Chioza B, et al. DNA methylation signatures of Alzheimer’s disease neuropathology in the cortex are primarily driven by variation in non-neuronal cell-types. Nat Commun 2022;13:5620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Ahmed RM, Devenney EM, Irish M, Ittner A, Naismith S, Ittner LM, et al. Neuronal network disintegration: common pathways linking neurodegenerative diseases. J Neurol Neurosurg Psychiatry 2016;87:1234–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Robinson JL, Lee EB, Xie SX, Rennert L, Suh E, Bredenberg C, et al. Neurodegenerative disease concomitant proteinopathies are prevalent, age-related and APOE4-associated. Brain 2018;141:2181–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Sanchez-Mut JV, Heyn H, Vidal E, Moran S, Sayols S, Delgado-Morales R, et al. Human DNA methylomes of neurodegenerative diseases show common epigenomic patterns. Transl Psychiatry 2016;6:e718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Smith AR, Smith RG, Burrage J, Troakes C, Al-Sarraj S, Kalaria RN, et al. A cross-brain regions study of ANK1 DNA methylation in different neurodegenerative diseases. Neurobiol Aging 2019;74:70–6. [DOI] [PubMed] [Google Scholar]
- [53].Gerrits E, Brouwer N, Kooistra SM, Woodbury ME, Vermeiren Y, Lambourne M, et al. Distinct amyloid-β and tau-associated microglia profiles in Alzheimer’s disease. Acta Neuropathol 2021;141:681–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Cornblath EJ, Robinson JL, Irwin DJ, Lee EB, Lee VM-Y, Trojanowski JQ, et al. Defining and predicting transdiagnostic categories of neurodegenerative disease. Nat Biomed Eng 2020;4:787–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Smith RG, Pishva E, Shireby G, Smith AR, Roubroeks JAY, Hannon E, et al. A meta-analysis of epigenome-wide association studies in Alzheimer’s disease highlights novel differentially methylated loci across cortex. Nat Commun 2021;12:3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Noori A, Mezlini AM, Hyman BT, Serrano-Pozo A, Das S. Systematic review and meta-analysis of human transcriptomics reveals neuroinflammation, deficient energy metabolism, and proteostasis failure across neurodegeneration. Neurobiol Dis 2021;149:105225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Haytural H, Benfeitas R, Schedin-Weiss S, Bereczki E, Rezeli M, Unwin RD, et al. Insights into the changes in the proteome of Alzheimer disease elucidated by a meta-analysis. Sci Data 2021;8:312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Wang Q, Chen K, Su Y, Reiman EM, Dudley JT, Readhead B. Deep learning-based brain transcriptomic signatures associated with the neuropathological and clinical severity of Alzheimer’s disease. Brain Commun 2022;4:fcab293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Patel H, Dobson RJB, Newhouse SJ. A Meta-Analysis of Alzheimer’s Disease Brain Transcriptomic Data. J Alzheimers Dis 2019;68:1635–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Fodder K, Murthy M, Rizzu P, Toomey CE, Hasan R, Humphrey J, et al. Brain DNA methylomic analysis of frontotemporal lobar degeneration reveals OTUD4 and CEBPZ in shared dysregulated signatures across pathological subtypes. bioRxiv 2022:2022.10.21.513088. 10.1101/2022.10.21.513088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Samarasekera N, Al-Shahi Salman R, Huitinga I, Klioueva N, McLean CA, Kretzschmar H, et al. Brain banking for neurological disorders. Lancet Neurol 2013;12:1096–105. [DOI] [PubMed] [Google Scholar]
- [62].Hasan R, Humphrey J, Bettencourt C, Newcombe J, NYGC ALS Consortium, Lashley T, et al. Transcriptomic analysis of frontotemporal lobar degeneration with TDP-43 pathology reveals cellular alterations across multiple brain regions. Acta Neuropathol 2022;143:383–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Li P, Ensink E, Lang S, Marshall L, Schilthuis M, Lamp J, et al. Hemispheric asymmetry in the human brain and in Parkinson’s disease is linked to divergent epigenetic patterns in neurons. Genome Biol 2020;21:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Scholefield M, Church SJ, Xu J, Robinson AC, Gardiner NJ, Roncaroli F, et al. Effects of Alterations of Post-Mortem Delay and Other Tissue-Collection Variables on Metabolite Levels in Human and Rat Brain. Metabolites 2020;10. 10.3390/metabo10110438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Sjöholm LK, Ransome Y, Ekström TJ, Karlsson O. Evaluation of Post-Mortem Effects on Global Brain DNA Methylation and Hydroxymethylation. Basic Clin Pharmacol Toxicol 2018;122:208–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Clement C, Hill JM, Dua P, Culicchia F, Lukiw WJ. Analysis of RNA from Alzheimer’s Disease Post-mortem Brain Tissues. Mol Neurobiol 2016;53:1322–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Kang M, Ko E, Mersha TB. A roadmap for multi-omics data integration using deep learning. Brief Bioinform 2022;23. 10.1093/bib/bbab454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Li H, Brouwer CR, Luo W. A universal deep neural network for in-depth cleaning of single-cell RNA-Seq data. Nat Commun 2022;13:1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Lin E, Mukherjee S, Kannan S. A deep adversarial variational autoencoder model for dimensionality reduction in single-cell RNA sequencing analysis. BMC Bioinformatics 2020;21:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Cooper LAD, Carter AB, Farris AB, Wang F, Kong J, Gutman DA, et al. Digital Pathology: Data-Intensive Frontier in Medical Imaging: Health-information sharing, specifically of digital pathology, is the subject of this paper which discusses how sharing the rich images in pathology can stretch the capabilities of all otherwise well-practiced disciplines. Proc IEEE Inst Electr Electron Eng 2012;100:991–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Lopez R, Regier J, Cole MB, Jordan MI, Yosef N. Deep generative modeling for single-cell transcriptomics. Nat Methods 2018;15:1053–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Tasaki S, Xu J, Avey DR, Johnson L, Petyuk VA, Dawe RJ, et al. Inferring protein expression changes from mRNA in Alzheimer’s dementia using deep neural networks. Nat Commun 2022;13:655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Andreasson U, Blennow K, Zetterberg H. Update on ultrasensitive technologies to facilitate research on blood biomarkers for central nervous system disorders. Alzheimers Dement 2016;3:98–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Ashton NJ, Janelidze S, Al Khleifat A, Leuzy A, van der Ende EL, Karikari TK, et al. A multicentre validation study of the diagnostic value of plasma neurofilament light. Nat Commun 2021;12:3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Soleimani Zakeri NS, Pashazadeh S, MotieGhader H. Gene biomarker discovery at different stages of Alzheimer using gene co-expression network approach. Sci Rep 2020;10:12210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Shigemizu D, Mori T, Akiyama S, Higaki S, Watanabe H, Sakurai T, et al. Identification of potential blood biomarkers for early diagnosis of Alzheimer’s disease through RNA sequencing analysis. Alzheimers Res Ther 2020;12:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell 2013;49:359–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Lu AT, Hannon E, Levine ME, Crimmins EM, Lunnon K, Mill J, et al. Genetic architecture of epigenetic and neuronal ageing rates in human brain regions. Nat Commun 2017;8:15353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].McCartney DL, Hillary RF, Conole ELS, Banos DT, Gadd DA, Walker RM, et al. Blood-based epigenome-wide analyses of cognitive abilities. Genome Biol 2022;23:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Website n.d. 10.1098/rsif.2017.0387. [DOI]
- [81].Bzdok D, Altman N, Krzywinski M. Statistics versus machine learning. Nat Methods 2018;15:233–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Krassowski M, Das V, Sahu SK, Misra BB. State of the Field in Multi-Omics Research: From Computational Needs to Data Mining and Sharing. Front Genet 2020;11:610798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Eraslan G, Avsec Ž, Gagneur J, Theis FJ. Deep learning: new computational modelling techniques for genomics. Nat Rev Genet 2019;20:389–403. [DOI] [PubMed] [Google Scholar]
- [84].Zou J, Huss M, Abid A, Mohammadi P, Torkamani A, Telenti A. A primer on deep learning in genomics. Nat Genet 2019;51:12–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Jurtz VI, Johansen AR, Nielsen M, Almagro Armenteros JJ, Nielsen H, Sønderby CK, et al. An introduction to deep learning on biological sequence data: examples and solutions. Bioinformatics 2017;33:3685–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Beecham GW, Bis JC, Martin ER, Choi S-H, DeStefano AL, van Duijn CM, et al. The Alzheimer’s Disease Sequencing Project: Study design and sample selection. Neurol Genet 2017;3:e194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Iwaki H, Leonard HL, Makarious MB, Bookman M, Landin B, Vismer D, et al. Accelerating Medicines Partnership: Parkinson’s Disease. Genetic Resource. Mov Disord 2021;36:1795–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].The Foundational Data Initiative for Parkinson Disease: Enabling efficient translation from genetic maps to mechanism. Cell Genomics 2023;3:100261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Schekman R, Riley EA. Coordinating a new approach to basic research into Parkinson’s disease. Elife 2019;8. 10.7554/eLife.51167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Birkenbihl C, Westwood S, Shi L, Nevado-Holgado A, Westman E, Lovestone S, et al. ANMerge: A Comprehensive and Accessible Alzheimer’s Disease Patient-Level Dataset. J Alzheimers Dis 2021;79:423–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Badhwar A, McFall GP, Sapkota S, Black SE, Chertkow H, Duchesne S, et al. A multiomics approach to heterogeneity in Alzheimer’s disease: focused review and roadmap. Brain 2020;143:1315–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Friedman J, Hastie T, Tibshirani R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J Stat Softw 2010;33:1–22. [PMC free article] [PubMed] [Google Scholar]
- [93].Breiman L. Mach Learn 2001;45:5–32. [Google Scholar]
- [94].Konczyk J. Federated Learning with TensorFlow. 2019.
- [95].Huynh K, Lim WLF, Giles C, Jayawardana KS, Salim A, Mellett NA, et al. Concordant peripheral lipidome signatures in two large clinical studies of Alzheimer’s disease. Nat Commun 2020;11:5698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Saykin AJ, Shen L, Yao X, Kim S, Nho K, Risacher SL, et al. Genetic studies of quantitative MCI and AD phenotypes in ADNI: Progress, opportunities, and plans. Alzheimers Dement 2015;11:792–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Livingston G, Huntley J, Sommerlad A, Ames D, Ballard C, Banerjee S, et al. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet 2020;396:413–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Yu J-T, Xu W, Tan C-C, Andrieu S, Suckling J, Evangelou E, et al. Evidence-based prevention of Alzheimer’s disease: systematic review and meta-analysis of 243 observational prospective studies and 153 randomised controlled trials. J Neurol Neurosurg Psychiatry 2020;91:1201–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Bandres-Ciga S, Noyce AJ, Traynor BJ. Mendelian Randomization-A Journey From Obscurity to Center Stage With a Few Potholes Along the Way. JAMA Neurol 2020;77:7–8. [DOI] [PubMed] [Google Scholar]
- [100].Kuźma E, Hannon E, Zhou A, Lourida I, Bethel A, Levine DA, et al. Which Risk Factors Causally Influence Dementia? A Systematic Review of Mendelian Randomization Studies. J Alzheimers Dis 2018;64:181–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Andrews SJ, Fulton-Howard B, O’Reilly P, Marcora E, Goate AM, collaborators of the Alzheimer’s Disease Genetics Consortium. Causal Associations Between Modifiable Risk Factors and the Alzheimer’s Phenome. Ann Neurol 2021;89:54–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Larsson SC, Traylor M, Malik R, Dichgans M, Burgess S, Markus HS, et al. Modifiable pathways in Alzheimer’s disease: Mendelian randomisation analysis. BMJ 2017;359:j5375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Østergaard SD, Mukherjee S, Sharp SJ, Proitsi P, Lotta LA, Day F, et al. Associations between Potentially Modifiable Risk Factors and Alzheimer Disease: A Mendelian Randomization Study. PLoS Med 2015;12:e1001841; discussion e1001841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Malik R, Georgakis MK, Neitzel J, Rannikmäe K, Ewers M, Seshadri S, et al. Midlife vascular risk factors and risk of incident dementia: Longitudinal cohort and Mendelian randomization analyses in the UK Biobank. Alzheimers Dement 2021;17:1422–31. [DOI] [PubMed] [Google Scholar]
- [105].Gagliano Taliun SA, Evans DM. Ten simple rules for conducting a mendelian randomization study. PLoS Comput Biol 2021;17:e1009238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Davies NM, Holmes MV, Davey Smith G. Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. BMJ 2018;362:k601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Anderson EL, Richmond RC, Jones SE, Hemani G, Wade KH, Dashti HS, et al. Is disrupted sleep a risk factor for Alzheimer’s disease? Evidence from a two-sample Mendelian randomization analysis. Int J Epidemiol 2021;50:817–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Baumeister SE, Karch A, Bahls M, Teumer A, Leitzmann MF, Baurecht H. Physical activity and risk of Alzheimer disease: A 2-sample mendelian randomization study. Neurology 2020;95:e1897–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol 2015;44:512–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Benn M, Nordestgaard BG, Tybjærg-Hansen A, Frikke-Schmidt R. Impact of glucose on risk of dementia: Mendelian randomisation studies in 115,875 individuals. Diabetologia 2020;63:1151–61. [DOI] [PubMed] [Google Scholar]
- [111].Anderson EL, Howe LD, Wade KH, Ben-Shlomo Y, Hill WD, Deary IJ, et al. Education, intelligence and Alzheimer’s disease: evidence from a multivariable two-sample Mendelian randomization study. Int J Epidemiol 2020;49:1163–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Garfield V, Farmaki A-E, Fatemifar G, Eastwood SV, Mathur R, Rentsch CT, et al. Relationship Between Glycemia and Cognitive Function, Structural Brain Outcomes, and Dementia: A Mendelian Randomization Study in the UK Biobank. Diabetes 2021;70:2313–21. [DOI] [PubMed] [Google Scholar]
- [113].Smit RAJ, Trompet S, Dekkers OM, Jukema JW, le Cessie S. Survival Bias in Mendelian Randomization Studies: A Threat to Causal Inference. Epidemiology 2019;30:813–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Weuve J, Proust-Lima C, Power MC, Gross AL, Hofer SM, Thiébaut R, et al. Guidelines for reporting methodological challenges and evaluating potential bias in dementia research. Alzheimers Dement 2015;11:1098–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Desai R, John A, Saunders R, Marchant NL, Buckman JEJ, Charlesworth G, et al. Examining the Lancet Commission risk factors for dementia using Mendelian randomisation. BMJ Ment Health 2023;26. 10.1136/bmjment-2022-300555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Noyce AJ, Kia DA, Hemani G, Nicolas A, Price TR, De Pablo-Fernandez E, et al. Estimating the causal influence of body mass index on risk of Parkinson disease: A Mendelian randomisation study. PLoS Med 2017;14:e1002314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Torvik FA, Eilertsen EM, Hannigan LJ, Cheesman R, Howe LJ, Magnus P, et al. Modeling assortative mating and genetic similarities between partners, siblings, and in-laws. Nat Commun 2022;13:1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Foote IF, Jacobs BM, Mathlin G, Watson CJ, Bothongo PL, Waters S, et al. The shared genetic architecture of modifiable risk for Alzheimer’s disease: a genomic structural equation modelling study. Neurobiol Aging 2022;117:222–35. [DOI] [PubMed] [Google Scholar]
- [119].Peters R, Booth A, Rockwood K, Peters J, D’Este C, Anstey KJ. Combining modifiable risk factors and risk of dementia: a systematic review and meta-analysis. BMJ Open 2019;9:e022846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Grotzinger AD, Rhemtulla M, de Vlaming R, Ritchie SJ, Mallard TT, Hill WD, et al. Genomic structural equation modelling provides insights into the multivariate genetic architecture of complex traits. Nat Hum Behav 2019;3:513–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Grotzinger AD, de la Fuente J, Davies G, Nivard MG, Tucker-Drob EM. Transcriptome-wide and Stratified Genomic Structural Equation Modeling Identify Neurobiological Pathways Underlying General and Specific Cognitive Functions. medRxiv 2021:2021.04.30.21256409. 10.1101/2021.04.30.21256409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Howey R, Shin S-Y, Relton C, Davey Smith G, Cordell HJ. Bayesian network analysis incorporating genetic anchors complements conventional Mendelian randomization approaches for exploratory analysis of causal relationships in complex data. PLoS Genet 2020;16:e1008198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].O’Connor LJ, Price AL. Distinguishing genetic correlation from causation across 52 diseases and complex traits. Nat Genet 2018;50:1728–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Zhong W, Spracklen CN, Mohlke KL, Zheng X, Fine J, Li Y. Multi-SNP mediation intersection-union test. Bioinformatics 2019;35:4724–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Malina S, Cizin D, Knowles DA. Deep mendelian randomization: Investigating the causal knowledge of genomic deep learning models. PLoS Comput Biol 2022;18:e1009880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Hemani G, Bowden J, Haycock P, Zheng J, Davis O, Flach P, et al. Automating Mendelian randomization through machine learning to construct a putative causal map of the human phenome. bioRxiv 2017:173682. 10.1101/173682. [DOI] [Google Scholar]
- [127].Liang Y, Qu L-B, Liu H. Non-linear associations between sleep duration and the risks of mild cognitive impairment/dementia and cognitive decline: a dose-response meta-analysis of observational studies. Aging Clin Exp Res 2019;31:309–20. [DOI] [PubMed] [Google Scholar]
- [128].Wu L, Sun D, Tan Y. A systematic review and dose-response meta-analysis of sleep duration and the occurrence of cognitive disorders. Sleep Breath 2018;22:805–14. [DOI] [PubMed] [Google Scholar]
- [129].Staley JR, Burgess S. Semiparametric methods for estimation of a nonlinear exposure-outcome relationship using instrumental variables with application to Mendelian randomization. Genet Epidemiol 2017;41:341–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Henry A, Katsoulis M, Masi S, Fatemifar G, Denaxas S, Acosta D, et al. The relationship between sleep duration, cognition and dementia: a Mendelian randomization study. Int J Epidemiol 2019;48:849–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Malik MA, Lundervold AS, Michoel T. rfPhen2Gen: A machine learning based association study of brain imaging phenotypes to genotypes 2022.
- [132].Hou L, Geng Z, Shi X, Wang C, Li H, Xue F. MRSL: A phenome-wide causal discovery algorithm based on GWAS summary data. medRxiv 2022:2022.06.29.22277051. 10.1101/2022.06.29.22277051. [DOI] [Google Scholar]
- [133].Van Cauwenberghe C, Van Broeckhoven C, Sleegers K. The genetic landscape of Alzheimer disease: clinical implications and perspectives. Genet Med 2016;18:421–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Kunkle BW, Grenier-Boley B, Sims R, Bis JC, Damotte V, Naj AC, et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat Genet 2019;51:414–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Sims R, van der Lee SJ, Naj AC, Bellenguez C, Badarinarayan N, Jakobsdottir J, et al. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat Genet 2017;49:1373–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, et al. TREM2 variants in Alzheimer’s disease. N Engl J Med 2013;368:117–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Takalo M, Wittrahm R, Wefers B, Parhizkar S, Jokivarsi K, Kuulasmaa T, et al. The Alzheimer’s disease-associated protective Plcγ2-P522R variant promotes immune functions. Mol Neurodegener 2020;15:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Andreone BJ, Przybyla L, Llapashtica C, Rana A, Davis SS, van Lengerich B, et al. Alzheimer’s-associated PLCγ2 is a signaling node required for both TREM2 function and the inflammatory response in human microglia. Nat Neurosci 2020;23:927–38. [DOI] [PubMed] [Google Scholar]
- [139].Nott A, Holtman IR, Coufal NG, Schlachetzki JCM, Yu M, Hu R, et al. Brain cell type-specific enhancer-promoter interactome maps and diseaserisk association. Science 2019;366:1134–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Corces MR, Shcherbina A, Kundu S, Gloudemans MJ, Frésard L, Granja JM, et al. Single-cell epigenomic analyses implicate candidate causal variants at inherited risk loci for Alzheimer’s and Parkinson’s diseases. Nat Genet 2020;52:1158–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Young AMH, Kumasaka N, Calvert F, Hammond TR, Knights A, Panousis N, et al. A map of transcriptional heterogeneity and regulatory variation in human microglia. Nat Genet 2021;53:861–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Lopes K de P, Snijders GJL, Humphrey J, Allan A, Sneeboer MAM, Navarro E, et al. Genetic analysis of the human microglial transcriptome across brain regions, aging and disease pathologies. Nat Genet 2022;54:4–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Kosoy R, Fullard JF, Zeng B, Bendl J, Dong P, Rahman S, et al. Genetics of the human microglia regulome refines Alzheimer’s disease risk loci. Nat Genet 2022;54. 10.1038/s41588-022-01149-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Schwartzentruber J, Cooper S, Liu JZ, Barrio-Hernandez I, Bello E, Kumasaka N, et al. Genome-wide meta-analysis, fine-mapping and integrative prioritization implicate new Alzheimer’s disease risk genes. Nat Genet 2021;53:392–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Gjoneska E, Pfenning AR, Mathys H, Quon G, Kundaje A, Tsai L-H, et al. Conserved epigenomic signals in mice and humans reveal immune basis of Alzheimer’s disease. Nature 2015;518:365–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Reynolds RH, Botía J, Nalls MA, International Parkinson’s Disease Genomics Consortium (IPDGC), System Genomics of Parkinson’s Disease (SGPD), Hardy J, et al. Moving beyond neurons: the role of cell type-specific gene regulation in Parkinson’s disease heritability. NPJ Parkinsons Dis 2019;5:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].van Rheenen W, van der Spek RAA, Bakker MK, van Vugt JJFA, Hop PJ, Zwamborn RAJ, et al. Common and rare variant association analyses in amyotrophic lateral sclerosis identify 15 risk loci with distinct genetic architectures and neuron-specific biology. Nat Genet 2021;53:1636–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Young AL, Marinescu RV, Oxtoby NP, Bocchetta M, Yong K, Firth NC, et al. Uncovering the heterogeneity and temporal complexity of neurodegenerative diseases with Subtype and Stage Inference. Nat Commun 2018;9:4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Hutchinson A, Asimit J, Wallace C. Fine-mapping genetic associations. Hum Mol Genet 2020;29:R81–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Brown A-L, Wilkins OG, Keuss MJ, Hill SE, Zanovello M, Lee WC, et al. TDP-43 loss and ALS-risk SNPs drive mis-splicing and depletion of UNC13A. Nature 2022;603:131–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Ma XR, Prudencio M, Koike Y, Vatsavayai SC, Kim G, Harbinski F, et al. TDP-43 represses cryptic exon inclusion in the FTD-ALS gene UNC13A. Nature 2022;603:124–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Fujita M, Gao Z, Zeng L, McCabe C, White CC, Ng B, et al. Cell-subtype specific effects of genetic variation in the aging and Alzheimer cortex. bioRxiv 2022:2022.11.07.515446. 10.1101/2022.11.07.515446. [DOI] [Google Scholar]
- [153].Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019;570:332–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Consortium GTEx. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 2020;369:1318–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Chen W-T, Lu A, Craessaerts K, Pavie B, Sala Frigerio C, Corthout N, et al. Spatial Transcriptomics and In Situ Sequencing to Study Alzheimer’s Disease. Cell 2020;182:976–91.e19. [DOI] [PubMed] [Google Scholar]
- [156].Lebrigand K, Bergenstråhle J, Thrane K, Mollbrink A, Meletis K, Barbry P, et al. The spatial landscape of gene expression isoforms in tissue sections. bioRxiv 2022:2020.08.24.252296. 10.1101/2020.08.24.252296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].PsychENCODE Consortium, Akbarian S, Liu C, Knowles JA, Vaccarino FM, Farnham PJ, et al. The PsychENCODE project. Nat Neurosci 2015;18:1707–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Wang D, Liu S, Warrell J, Won H, Shi X, Navarro FCP, et al. Comprehensive functional genomic resource and integrative model for the human brain. Science 2018;362. 10.1126/science.aat8464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Hannon E, Spiers H, Viana J, Pidsley R, Burrage J, Murphy TM, et al. Methylation QTLs in the developing brain and their enrichment in schizophrenia risk loci. Nat Neurosci 2016;19:48–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Davila-Velderrain J, Mathys H, Mohammadi S, Ruzicka B, Jiang X, Ng A, et al. Single-cell anatomical analysis of human hippocampus and entorhinal cortex uncovers early-stage molecular pathology in Alzheimer’s disease. bioRxiv 2021:2021.07.01.450715. 10.1101/2021.07.01.450715. [DOI] [Google Scholar]
- [161].Sebastian Pineda S, Lee H, Fitzwalter BE, Mohammadi S, Pregent LJ, Gardashli ME, et al. Single-cell profiling of the human primary motor cortex in ALS and FTLD. bioRxiv 2021:2021.07.07.451374. 10.1101/2021.07.07.451374. [DOI] [Google Scholar]
- [162].Zhang M, Eichhorn SW, Zingg B, Yao Z, Cotter K, Zeng H, et al. Spatially resolved cell atlas of the mouse primary motor cortex by MERFISH. Nature 2021;598:137–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Hao Y, Hao S, Andersen-Nissen E, Mauck WM 3rd, Zheng S, Butler A, et al. Integrated analysis of multimodal single-cell data. Cell 2021;184:3573–87.e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Weissbrod O, Hormozdiari F, Benner C, Cui R, Ulirsch J, Gazal S, et al. Functionally informed fine-mapping and polygenic localization of complex trait heritability. Nat Genet 2020;52:1355–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Schilder BM, Raj T. Fine-mapping of Parkinson’s disease susceptibility loci identifies putative causal variants. Hum Mol Genet 2022;31:888–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Ramos DM, Skarnes WC, Singleton AB, Cookson MR, Ward ME. Tackling neurodegenerative diseases with genomic engineering: A new stem cell initiative from the NIH. Neuron 2021;109:1080–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Tian R, Abarientos A, Hong J, Hashemi SH, Yan R, Dräger N, et al. Genome-wide CRISPRi/a screens in human neurons link lysosomal failure to ferroptosis. Nat Neurosci 2021;24:1020–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Makarious MB, Leonard HL, Vitale D, Iwaki H, Sargent L, Dadu A, et al. Multi-modality machine learning predicting Parkinson’s disease. NPJ Parkinsons Dis 2022;8:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.