

Abstract
The mammalian host responds to infection with Borrelia spirochetes through a highly orchestrated immune defense involving innate and adaptive effector functions aimed toward limiting pathogen burdens, minimizing tissue injury, and preventing subsequent reinfection. The evolutionary adaptation of Borrelia spirochetes to their reservoir mammalian hosts may allow for its persistence despite this immune defense. This review summarizes our current understanding of the host immune response to B. burgdorferi sensu lato, the most widely studied Borrelia spp. and etiologic agent of Lyme borreliosis. Pertinent literature will be reviewed with emphasis on in vitro, ex vivo and animal studies that influenced our understanding of both the earliest responses to B. burgdorferi as it enters the mammalian host and those that evolve as spirochetes disseminate and establish infection in multiple tissues. Our focus is on the immune response of inbred mice, the most commonly studied animal model of B. burgdorferi infection and surrogate for one of this pathogen’s principle natural reservoir hosts, the white-footed deer mouse. Comparison will be made to the immune responses of humans with Lyme borreliosis. Our goal is to provide an understanding of the dynamics of the mammalian immune response during infection with B. burgdorferi and its relation to the outcomes in reservoir (mouse) and non-reservoir (human) hosts.
Introduction
The mammalian immune system is critical for host defense against infection and for establishing memory responses that can respond rapidly to prevent or limit re-infection (Medzhitov, 2008). Sensor cells located in barrier tissues throughout the body play important roles in distinguishing pathogens as foreign (Murphy and Weaver, 2017). These cells orchestrate immune responses that evolutionarily have been selected as effective at controlling the perceived threat, either by its elimination or by limiting the pathogen burden and adverse effects on the host. Regulation of the inflammatory response is key to preventing excessive tissue injury as a consequence of this defense and restoring tissue homeostasis once the pathogen is controlled (Medzhitov, 2008; Feehan and Gilroy, 2019).
The goal of this review is to provide an understanding of the dynamics of the mammalian immune response during infection with Borrelia burgdorferi and how this response affects pathogen elimination and persistence. When visualized in tissues, B. burgdorferi is extracellular and has a predilection for extracellular matrix and connective tissue at all stages of infection (Duray and Steere, 1988; Barthold et al., 1990; Barthold et al., 1993). Although B. burgdorferi has been sighted rarely inside cells, both ex vivo and in vitro studies of its genome and its mechanisms for immune evasion (reviewed in Radolf and Samuels, 2021), which are directed toward subverting phagocyte, antibody, and complement-mediated destruction, point over-whelmingly toward its residing in the host extracellularly (Hyde, 2017; Lin et al., 2020). B. burgdorferi has evolved to persist for extended periods of time in its reservoir hosts without associated inflammatory pathologies despite engagement of both innate and adaptive immunity (Moody et al., 1994; Baum et al., 2012). The adaptive immune response, while unable to eliminate all spirochetes from the host, is capable of protecting the host from re-infection with autologous strains (Bockenstedt et al., 1997; Nadelman et al., 2012; Khatchikian et al., 2014; Jacquet et al., 2015). The factors that allow for this state of “concomitant immunity” await a better understanding of the genetic and antigenic composition of spirochetes that persist within the immunocompetent vertebrate host.
Much of what has been learned about the innate response has been derived from in vitro studies of the interactions of cultured B. burgdorferi and its components with cells or cell lines. While providing much insight into the capacity of mammalian immune cells to recognize and respond to B. burgdorferi components, the ability of B. burgdorferi to rapidly change its gene expression program depending on its environment (culture, tick, or mammal) (Radolf and Samuels, 2021) limits the extrapolation of in vitro studies to the unique in vivo microenvironments in which B. burgdorferi resides in the mammal (Radolf et al., 2012). A further distinction is made between B. burgdorferi infection in inbred mice, a surrogate reservoir host, and the clinical pathology known as “Lyme disease” or “Lyme borreliosis” resulting from B. burgdorferi infection of non-reservoir hosts, including humans. Studies in inbred mice provide important insights into immune components that are critical not only for limiting pathogen burden, resolving inflammation and preventing infection with B. burgdorferi, but also for identifying the spirochete’s key immune evasion strategies. Mice, however, do not develop the pathology characteristic of Lyme borreliosis in humans (Duray and Steere, 1988; Barthold et al., 1992a; Steere et al., 2016) even though in vitro studies suggest that similar immune pathways are engaged by mouse and human cells exposed to B. burgdorferi and its components (discussed herein). Recent studies in humans have used advanced technologies to probe host responses in blood and tissues, with a goal toward understanding how these responses result in clinical pathology. In this article, we review the innate and acquired host responses to B. burgdorferi that control pathogen burden, prevent reinfection and limit the extent of tissue injury and clinical disease, as well as those that may allow for B. burgdorferi persistence.
Establishment of infection: The role of the tick vector in B. burgdorferi infection
B. burgdorferi infection begins with introduction of spirochetes into the mammalian skin during tick feeding. The skin constitutes the first barrier to tick-borne pathogens, and breaching of this barrier at the tick feeding site creates a critical portal for passage of the pathogen from the tick to the bloodmeal host (Frischknecht, 2007). As with other vector-borne pathogens, B. burgdorferi benefits from mechanisms that Ixodes ticks have evolved to obtain the bloodmeal and to prevent host immunity to tick refeeding. Blood feeding of Ixodes ticks is a highly ordered process that occurs over 3–10 days, with the majority of the blood ingested during the final 24 hours of attachment (Kemp et al., 1982). With the initiation of feeding, B. burgdorferi replicates in the tick midgut and then migrates through the hemocoel to the salivary glands prior to deposition in the deep epidermis or dermis of the bloodmeal host (Dunham-Ems et al., 2009). During this time, tick saliva bathes the feeding pit with pharmacologically active substances that impair hemostasis, host immunity and wound healing, and which can secondarily facilitate pathogen transmission to the bloodmeal host (Ribeiro et al., 1985) (see also Radolf and Samuels, 2021). The composition of tick saliva changes during the course of tick feeding (Perner et al., 2018) and varies depending on the bloodmeal host species, possibly as a consequence of the vertebrate immune response (Narasimhan et al., 2019). Successful transmission of B. burgdorferi to the vertebrate host depends on its ability to exploit components of tick saliva as it moves from the tick salivary gland into the unique ecological niche of the tick feeding site (Nuttall and Labuda, 2004; Nuttall, 2019).
Several studies have shown that tick saliva can enhance infectivity of B. burgdorferi. Co-inoculation of B. burgdorferi with I. scapularis or I. ricinus salivary gland lysates into mice enhances pathogen burden in multiple organs in comparison to needle inoculation (Zeidner et al., 2002). This effect, however, is tick species- and spirochete strain-specific, as it was observed only when co-inoculating a spirochete strain isolated from the tick species from which the salivary gland lysates were obtained. Saliva and salivary gland extracts from I. ricinus promoted enhanced dissemination of B. afzelii when co-inoculated into mice, with an increase in pathogen burden in the skin and urinary bladder, but not the heart, by day 6 (Pechova et al., 2002; Horka et al., 2009). The early effect of tick saliva on pathogen survival is likely mediated by inhibition of cutaneous innate immune mechanisms. Consistent with this, I. ricinus salivary gland extracts impeded macrophage production of reactive oxygen species and killing of B. afzelii by mouse macrophages in vitro (Kuthejlova et al., 2001). A more recent study demonstrated that I. ricinus salivary gland extracts inhibited mast cell activation in vitro. While mast cell-deficient mice did not exhibit increased replication of B. burgdorferi in the skin, dissemination to the joints occurred more quickly (Bernard et al., 2017).
The factors within tick saliva that facilitate B. burgdorferi transmission and dissemination are only partially defined. Analyses of the I. scapularis and I. ricinus sialotranscriptomes have identified differentially expressed genes during tick feeding that encode proteins predicted to have a broad range of pharmacologic activities (Narasimhan et al., 2002; Ribeiro et al., 2006; Kotsyfakis et al., 2015; Perner et al., 2018). The timing for production of some of these molecules varies with duration of tick feeding (Perner et al., 2018). These molecules have been catalogued into functional groups, such as the basic tail family proteins (e.g. salp14) that impair blood clotting (Narasimhan et al., 2002; Ribeiro et al., 2006), the tissue factor pathway inhibitors ixolaris and penthalaris (Francischetti et al., 2002; Francischetti et al., 2004), vasodilators such as prostacyclin, prostaglandin E2, and adenosine (Ribeiro et al., 1988; Sa-Nunes et al., 2007), and inhibitors of angiogenesis (Francischetti et al., 2005). More relevant to the survival of B. burgdorferi in the skin are factors that retard local inflammation at the tick bite site, especially those that interfere with the activity of phagocytes, antibody and complement. Tick saliva contains an abundance of protease inhibitors, including sialostatin L, ILS919, and ISL 1373, that impair neutrophil functions such as chemotaxis, integrin expression, and reactive oxygen generation, respectively (Kotsyfakis et al., 2006) (Guo et al., 2009). Sialostatins also modulate mouse dendritic cell (DC) responses to B. burgdorferi in vitro, reducing DC maturation and IFN-β production, as well as DC production of the chemokines MIP-1α and IP-10 that recruit mononuclear cells, T cells and mast cells (Lieskovska et al., 2015). Other factors inhibit macrophage activation and interfere with the alternate pathway of complement activation, including salp20, Isac and Isac paralogs found in I. ricinus (Valenzuela et al., 2000; Daix et al., 2007; Tyson et al., 2007; Couvreur et al., 2008). Additionally, the TSLP1 family inhibits the lectin complement cascade (Schuijt et al., 2011; Wagemakers et al., 2016). RNAseq analysis of single I. ricinus salivary glands revealed that the most abundant transcripts expressed within the first 24 hours of tick feeding were secreted metalloproteases, members of the basic tail superfamily proteins, and anti-complement Isac paralogs, including IRAC1, IRACII and IxACB1–5 (Daix et al., 2007; Couvreur et al., 2008; Perner et al., 2018). The actions of these molecules at the tick feeding site interfere with the initial skin immune defense and may help pathogens like B. burgdorferi establish infection in the mammal.
Components of tick saliva modulate adaptive immunity, skewing T cell differentiation toward a TH2 phenotype through actions on DCs and T cells. This could facilitate B. burgdorferi infection by reducing the actions of TH1-associated cytokines on phagocytes. PGE2 in tick saliva increases IL-10 and reduces IL-12 and TNFα production by DCs, and suppresses DC-induced CD4+ T cell proliferation and IL-2 production in vitro (Sa-Nunes et al., 2007). Tick saliva also contains an IL-2 binding protein that inhibits T cell proliferation in vitro (Gillespie et al., 2001). Repeated tick infestation of C3H/HeN mice leads to reduced production of IL-2 and IFN-γ by splenocytes after concanavalin A stimulation and augmented production of IL-4 and IL-10 production. The increase in production of anti-inflammatory cytokines, especially IL-10, may help suppress phagocyte activation and clearance of B. burgdorferi. In support of this, treatment of C3H/HeJ mice with TNFα, or with IL-2 or IFN-γ for 10 days after onset of B. burgdorferi-infected tick feeding reduced mouse infection rates by 95% and 30–45%, respectively (Zeidner et al., 1996). Langerhans cells (LCs) are critical for tick-mediated suppression of the concanavalin A-induced TH1 response in lymph nodes draining the tick bite site, but their impact is attenuated when B. burgdorferi is introduced by feeding ticks (Vesely et al., 2009). Recently, a sphingomyelinase-like protein (IsSMase) has been identified in I. scapularis saliva that directly polarizes CD4+ T cells toward a TH2 phenotype and expression of IL-4 (Alarcon-Chaidez et al., 2009). B. burgdorferi also has exploited a tick salivary protein, salp15, to colonize the bloodmeal host (Ramamoorthi et al., 2005). The expression of this protein is upregulated in infected tick salivary glands and binds to outer surface protein C (OspC) on spirochetes as they enter the host. The binding of salp15 to OspC can shield B. burgdorferi s.s. from borrelicidal antibodies in vitro, suggesting that it could interfere with antibody-mediated killing in vivo. This effect appears to be species- specific as salp15 could not protect B. garinii or B. afzelii from being killed. Recombinant salp15 injected with spirochetes enhanced their survival in mice, and siRNA depletion of the protein in infected ticks reduced B. burgdorferi infectivity after vector transmission (Ramamoorthi et al., 2005). In addition, salp15 inhibited DC production of inflammatory cytokines through binding of DC-SIGN (Hovius et al., 2008), as well as T cell receptor signal transduction by binding to the T cell coreceptor CD4 (Garg et al., 2006; Juncadella et al., 2007). These diverse functions of salp15 not only aid B. burgdorferi infection and contribute to disease expression but also reduce the impact of host innate and adaptive immune responses induced by B. burgdorferi that could potentially interfere with tick feeding.
Initial host defense - the innate immune response
Both innate and acquired defenses control spirochetes in tissues, with the humoral response particularly important for lowering B. burgdorferi tissue burden, resolving disease and protecting from infection. Innate immune mechanisms are likely engaged to some extent at all stages of infection and wherever spirochetes are found in the mammal. Their greatest impact, however, would be during phases where B. burgdorferi initially enters the host, replicates and disseminates.
Early spirochetal dissemination into tissues and interactions with resident immune cells
During natural infection, ticks deposit B. burgdorferi within the epidermal or dermal layers of the skin, depending on the size/stage of the tick and the skin thickness of the new host. Studies of the earliest stages of tick-transmitted infection using intravital microscopy in living mice and GFP-expressing spirochetes have been unable to visualize spirochete deposition within the skin, consistent with the small numbers that are believed to be deposited in the feeding pit (Ohnishi et al., 2001; Bockenstedt et al., 2014). However, intradermal needle inoculation with a range of B. burgdorferi doses has been found to result in similar tissue burdens as achieved following tick transmission over time, with the absolute numbers varying depending on the mouse strain infected (Brown and Reiner, 1998b). After inoculation of cultured GFP-expressing spirochetes into mouse skin, intravital microscopy reveals that they immediately become motile within the skin, displaying a backward-forward motility along collagen fibers interspersed with longer runs, similar to patterns observed in tick-transmitted infection (Harman et al., 2012). Within 48 hours, they begin disseminating from the inoculation site through the dermis, particularly in areas possessing collagen (Barthold et al., 1991; Shih et al., 1992; Shih et al., 1993). During dissemination, spirochetes achieve velocities that average ~250 μm/min, while maintaining a primarily backward-forward motility interspersed with some longer runs in a single direction (Malawista and de Boisfleury Chevance, 2008; Harman et al., 2012). While some spirochetes access the bloodstream, the vast majority of bacteria appear to continue migrating through the dermis and lymphatic ducts (Tunev et al., 2011; Imai et al., 2013). After injection into one ear of a mouse, it consistently takes around 8 days for the spirochetes to reach the opposite ear, regardless of the inoculum dose; this also supports a mainly skin/lymphatics-based dissemination (Tunev et al., 2011). The first resident immune cells encountered during dissemination are LCs, which reside in the areas at the epidermal/dermal interface (Nestle et al., 2009), and constantly sample the environment via their long pseudopods that move at around 0.4 μm/min (Nguyen Hoang et al., 2014). Subsequently, the spirochetes come in contact with cells of the macrophage/DC lineage as they continue through the dermis (Nestle et al., 2009). These cells are capable of capturing and phagocytosing a subset of B. burgdorferi even though they move approximately 300-times slower than B. burgdorferi (Malawista and de Boisfleury Chevance, 2008). Within 4 hours of B. burgdorferi being injected into the skin, large numbers of neutrophils migrate specifically to the skin regions that contain spirochetes (Xu et al., 2007). These phagocytes travel at average speeds of 6 μm/min, which is around 40-times slower than spirochetes, capturing some of the disseminating B. burgdorferi (Park et al., 2018). The vast majority of spirochetes, however, continue to disseminate, with bacteria observed in the heart by ≤7 days and in the rear ankle joints by 7–10 days post-infection after inoculation into the back skin (Lazarus et al., 2006). The spirochetes can inhabit at least transiently almost any tissue in the host, including the meninges (Divan et al., 2018), residing in the extracellular spaces where they can interact with resident immune cells. During this persistent phase, B. burgdorferi may elicit inflammatory responses, though the exuberance of those responses is dependent largely on the host genetic make-up rather than the number of spirochetes that reside in those tissues (Ma et al., 1998; Brown et al., 1999; Miller et al., 2008b). Notably, the spirochetes continue to display the same backward-forward motility and velocities in skin tissues as demonstrated early in infection, even 2 years post-infection (Wooten et al., unpublished).
Aspects of the innate immune response appear to have the strongest effects on bacterial clearance.
A number of studies have helped delineate the relative importance of innate versus adaptive immunity in the control of B. burgdorferi numbers in host tissues. As outlined more below, SCID mice lacking B and T cells possessed similar spirochetal numbers as wild-type (WT) mice for several weeks post-infection. Over time, spirochetes reached higher numbers in several tissues of SCID mice compared to WT mice (Barthold et al., 1992b; Wang et al., 2005), with control of spirochete numbers mediated by B cells but not T cells (de Souza et al., 1993; Schaible et al., 1994). Mice lacking the pattern recognition receptor toll-like receptor 2 (TLR2) or myeloid differentiation antigen 88 (MyD88), the downstream signaling adaptor for numerous TLRs, including TLR2, quickly developed bacterial loads that were significantly higher than WT mice, and these differences were maintained even after the adaptive responses developed (Wooten et al., 2002b; Bolz et al., 2004) (Figure 1). Importantly, the antibody levels in these mice were similar to or significantly higher than those of infected WT mice, and passive transfer of their immune serum to naïve mice generated similar protection against B. burgdorferi challenge as immune serum from WT mice (Wooten et al., 2002b; Yoder et al., 2003; Bolz et al., 2004). These findings suggest that the absence of MyD88 reduces effector functions of the innate immune response important for controlling pathogen burden. Finally, massively parallel sequencing of a defined set of B. burgdorferi mutant strains following their transfer by needle-inoculation into WT and MyD88-deficient mice determined that the major bottleneck to infection occurs early at the site of infection, which is when innate immunity comprises the majority of immune responsiveness (Troy et al., 2013). Together with studies in SCID mice, these findings suggest that both the innate immune response and the production of antibodies are critical for controlling pathogen burden after B. burgdorferi infection in mice.
Figure 1.
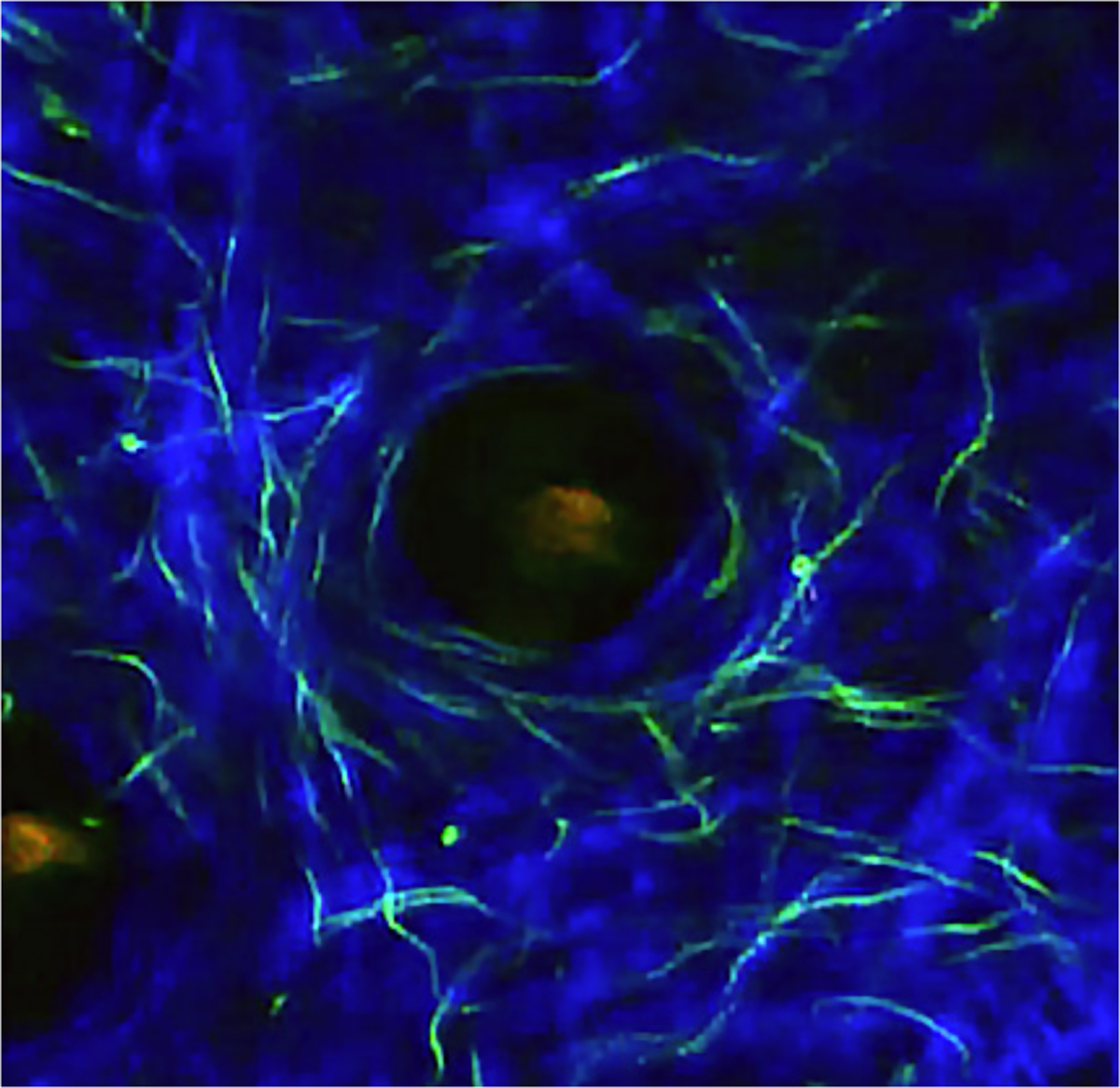
Image from a 2-photon intravital microscopy movie of GFP-expressing B. burgdorferi transformant 914 (derived from strain 297) in the ear skin of a 27-day infected C3H myD88−/− mouse. Infection was introduced by tick bite. Blue, second harmonics of skin collagen fibers. Note the numerous GFP spirochetes along the collagen fibers and surrounding the central hair follicle (See Bockenstedt, et al., 2012).
A caveat concerning most studies of B. burgdorferi interactions with immune cells is that they utilize assays performed in vitro or ex vivo, which are inadequate surrogates for the 3-dimensional tissues in which these spirochetal bacteria have evolved to reside. Some reasons for this include: 1) B. burgdorferi is a true obligate parasite that lacks many genes needed for growth outside a tick or vertebrate host; 2) B. burgdorferi is highly adaptable to the host or environment it encounters (i.e. vertebrate host vs. in vitro culture) and can rapidly change gene expression patterns; 3) we still do not know which gene products are expressed on the B. burgdorferi surface within a vertebrate host versus those expressed in an artificial environment; and 4) the relevance of B. burgdorferi motility and chemotaxis for immune evasion, particularly regarding the cellular responses, cannot be appreciated anywhere other than within the complex architecture of host tissues (Stevenson et al., 1995; Carroll et al., 1999; Charon and Goldstein, 2002; Rosa et al., 2005; Stewart et al., 2005). Thus, much of our understanding of how individual innate immune cell types respond to encounters with B. burgdorferi and/or its lipoproteins in vitro must be interpreted with caution. Some innate immune pathways have also been explored in mutant mice lacking relevant genes or after depletion of effector molecules. Table 1 summarizes those studies and the impact of interruption of specific innate immune pathways on pathogen burden, antibody responses and pathology.
Table 1.
Effect of Innate Immune Mediators on B. burgdorferi Infection
| Host gene or molecule affected | Pathogen burden relative to WT mice | Antibody (Ab) development | Disease development | References |
|---|---|---|---|---|
| CD1d/Jα18/Vα14 KO1 (no iNKT cells) | Increased | Increased | Increased arthritis, carditis | (Tupin et al., 2008; Olson et al., 2009; Lee et al., 2010; Lee et al., 2014) |
| Complement component C3 KO | Increased | Decreased | Increased | (Lawrenz et al., 2003) |
| E-selectin/P-selectin KO | No change | N/A | No change | (Seiler et al., 1998) |
| Gp91phox (NADPH oxidase subunit) KO | No change | N/A | No change | (Crandall et al., 2005) |
| IFN Type I KO or depleted | No change | No change | Decreased | (Hastey et al., 2014; Ma et al., 2014; Paquette et al., 2017) |
| IFNγ/IFNγR KO or depleted 3 | Increased | N/A | Decreased | (Brown and Reiner, 1999b; Olson et al., 2009; Sonderegger et al., 2012) |
| IL-4/IL-13 KO | No change | Shift from IgG1 to InG2b | No change | (Potter et al., 2000; Satoskar et al., 2000) |
| IL-10 KO | Decreased (skin, heart, joint) | Increased | Increased | (Brown et al., 1999; Lazarus et al., 2006; Sonderegger et al., 2012) |
| IL-12 depleted | Decreased (ear) | Decrease IgG2a only | Decreased arthritis | (Anguita et al., 1996; Anguita et al., 1997) |
| IL-17 KO | No change | No change | No change | (Lasky et al., 2015) |
| Mannose-binding lectin (MBL) KO | Increased early in skin; other tissues are WT | Increased | No change | (Coumou et al., 2019) |
| Myeloid-related protein (MRP) 8/14 KO | No change | No change | No change | (Mason et al., 2018) |
| MyD88 KO | Increased | No change | No change | (Bolz et al., 2004; Behera et al., 2006; Bockenstedt et al., 2006; Bolz et al., 2006) |
| Ncf1/p47phox (NADPH oxidase subunit) KO | No change | No change | No change | (Crandall et al., 2005) |
| Nitric oxide | No change | No change | No change | (Seiler et al., 1995; Brown and Reiner, 1999a) |
| Toll-like receptor 2 (TLR2) KO | Increased (ear, heart, joint, back skin) | Roughty similar to WT; lesser amounts of lipoprotein-specific Abs | No change | (Wooten et al., 2002; Yoder et al., 2003) |
KO – knock-out/gene deletion
N/A – Not assessed
Depleted – antibody-mediated depletion in mice
B. burgdorferi agonists and their innate pattern recognition receptors (PRRs)
B. burgdorferi populates the extracellular spaces of host tissues and, thus, is likely to initially interact with PRRs on cell surfaces. Early in vitro experiments indicated that B. burgdorferi is capable of stimulating a wide range of leukocytes and other host cells (Defosse and Johnson, 1992; Schoenfeld et al., 1992). Fractionation studies suggested that outer membrane lipoproteins were the primary pathogen-associated molecular patterns (PAMPs) through which innate immune cells sense B. burgdorferi. Prior to the identification of toll-like receptor (TLR) 4, the C3H/HeJ mouse line, a derivative of the C3H/HeN strain, was identified as resistant to septic shock after injection with lipopolysaccharide (LPS), but to be highly susceptible to infection with Gram-negative bacteria (Watson and Riblet, 1974; Weinstein et al., 1986); thus C3H/HeJ mice were presumed to lack an LPS receptor. Significantly, B. burgdorferi and/or their purified lipoproteins elicited similar responses by macrophages derived from either C3H/HeJ or C3H/HeN mice, and both mouse strains exhibited a similar course of infection with B. burgdorferi (Radolf et al., 1991; Yang et al., 1992; Barthold et al., 1993; Ma and Weis, 1993). This strongly suggested that B. burgdorferi lacked a classical LPS and that spirochetal lipoproteins activated macrophages through different innate receptors. The discovery of a family of TLRs (Medzhitov et al., 1997; Rock et al., 1998) set the stage for subsequent work that identified TLR2 to be the receptor for B. burgdorferi lipoproteins.
In addition to its surface PAMPs, intracellular PAMPs of B. burgdorferi are likely exposed when the bacterial cells are damaged or after their phagocytosis and degradation within certain host cell types. These PAMPs would then be recognized by PRRs residing within the endosomal/phagosomal compartment of phagocytes, where they can either amplify or suppress particular immune/inflammatory pathways (Cervantes et al., 2014). Evidence for the existence of pathways involving B. burgdorferi agonists and PRRs other than TLR2 have come from reports demonstrating that a greater complexity of responses are triggered in human and mouse mononuclear cells cultured with living B. burgdorferi than with purified lipoproteins or with sonicated preparations of spirochetes (Benhnia et al., 2005; Moore et al., 2007; Cruz et al., 2008; Lazarus et al., 2008; Dennis et al., 2009; Salazar et al., 2009) (Wooten et al., unpublished). The many immune molecules and pathways involved in the innate response to B. burgdorferi are described below.
CD14
The first receptor identified to mediate B. burgdorferi-associated inflammatory signaling was CD14. This receptor can be utilized by cells that either possess CD14 directly linked to their surface via a glycosylphosphatidylinositol linkage (mCD14) on myeloid lineage immune cells (e.g. macrophages, neutrophils, dendritic cells) or as a soluble protein (sCD14) in serum. Both macrophages (via mCD14) and endothelial cells (via sCD14) were activated by B. burgdorferi recombinant lipoproteins or lipopeptides using pathways accentuated by CD14 (Sellati et al., 1998; Wooten et al., 1998). Prototypic B. burgdorferi lipoproteins OspA and OspC were directly bound to CD14 in a similar fashion as LPS (Wooten et al., 1998). Activation of macrophages by lipoproteins occurred in the absence of CD14, albeit only at higher concentrations, indicating a role for CD14 in innate immune cell monitoring for bacterial lipid-containing pathogen-associated molecular patterns (PAMPs) in host tissues (Wooten et al., 1998; Hirschfeld et al., 1999). The mechanism differed from LPS-mediated interactions with CD14, in that lipoprotein activities mediated by CD14 occurred independently of LPS-binding protein activity (Sellati et al., 1998; Wooten et al., 1998). Studies using CD14-deficient mice indicated that B. burgdorferi interactions with CD14 in vivo were important for lowering bacterial numbers in multiple target tissues and diminishing B. burgdorferi-associated inflammatory pathologies (Benhnia et al., 2005). CD14-mediated signaling increased neutrophil-associated activities in B. burgdorferi-infected tissues while promoting matrix metalloprotease 9 (MMP9) activities and collagen remodeling (Zhao et al., 2007; Sahay et al., 2011). CD14 can promote B. burgdorferi phagocytosis through interactions with complement receptor 3 (CR3; CD11b) and lipid raft microdomains of macrophages completely independent of the TLR2-signaling pathway (Hawley et al., 2012). These findings suggest that CD14 acts as a scavenger receptor to target B. burgdorferi for enhanced phagocytosis and/or interacts with TLR2 (see below) to activate CD14-associated host cells.
TLR2
Early publications identified both TLR2 (Kirschning et al., 1998; Yang et al., 1998) and TLR4 (Poltorak et al., 1998) as the LPS-receptor. However, work by Hirschfeld et al indicated that the apparent LPS-mediated signaling via TLR2 was actually due to contaminating lipoproteins in the crude LPS preparations used in those studies (Hirschfeld et al., 2000), confirming that TLR4 was the actual LPS receptor. Transfection of five different TLRs individually into 293 and U373 cell lines lacking some or all TLRs identified TLR2 as a major receptor for B. burgdorferi-mediated signaling, as only cells expressing TLR2 alone could become activated in response to recombinant OspA and OspB lipoproteins, synthesized triacylated lipopeptides, or sonicated B. burgdorferi (Hirschfeld et al., 1999; Lien et al., 1999). TLR2-mediated signaling in response to lipoproteins also activated NF-κB in THP-1 cells, and could elicit apoptosis and/or respiratory burst-associated products in those cells (Aliprantis et al., 1999). TLR2-mediated signaling controlled B. burgdorferi numbers in vivo, as multiple target tissues from TLR2-deficient mouse lines possessed up to 100-fold more spirochetes compared to WT mice months post-infection (Wooten et al., 2002a). Interestingly, TLR2-deficient mice produced similar or significantly greater levels of B. burgdorferi-specific antibodies than WT mice, and these antibodies recognized a similar range of bacterial antigens, suggesting that TLR2-mediated innate responses were most important for controlling B. burgdorferi numbers but are similarly unable to clear the infection (Wooten et al., 2002a)(Wooten et al., unpublished). Immunization of TLR2-deficient and WT mice with rOspA elicited similar protection from challenge with B. burgdorferi, confirming that antibodies arising in the absence of TLR2 possessed indistinguishable protective capacities (Yoder et al., 2003). While TLR2-mediated signaling alone significantly contributes to the response to B. burgdorferi, the association of this receptor with TLR1 appears necessary for optimal production of antibodies against B. burgdorferi lipoproteins (Alexopoulou et al., 2002; Yoder et al., 2003). TLR2-activity is not required for phagocytosis of B. burgdorferi but does appear to be needed for sampling of spirochetal agonists after phagocytosis to allow optimal inflammatory signaling (Shin et al., 2008b; Salazar et al., 2009). In the case of human macrophages, phagocytosis of live B. burgdorferi elicits a more potent and varied upregulation of proinflammatory mediators compared to macrophages exposed to spirochetal lysates, suggesting that phagosomal signaling continues to be driven by subsequent exposure to TLR2-dependent and -independent B. burgdorferi agonists (Salazar et al., 2009). Interestingly, this TLR2-associated inflammatory signaling by murine macrophages is much stronger in response to killed B. burgdorferi or bacterial lysates compared to intact spirochetes, whereas human macrophages and DCs show much stronger signaling in response to live bacteria compared to bacterial lysates. The reason for these differences between mouse and human innate immune cell activation events is currently unknown (Lazarus et al., 2008; Salazar et al., 2009).
As noted above, B. burgdorferi infection of TLR2- or MyD88-deficient mice resulted in very high levels of bacteria in all target tissues tested compared to WT mice, despite similarity in their antibody responses (Bolz et al., 2004; Liu et al., 2004). Unlike TLR2-deficient macrophages, however, MyD88-deficient macrophages exhibited a defect in phagocytosis of B. burgdorferi and produced lower levels of inflammatory mediators in response to B. burgdorferi and its components than TLR2-deficient or WT macrophages (Shin et al., 2008b). These findings support a role for MyD88 in facilitating signals through innate receptors other than TLR2 after B. burgdorferi infection. Phagocytosis of B. burgdorferi also can be enhanced through signaling via the TIR-domain-containing adapter-inducing interferon-β (TRIF) adapter protein, which can be stimulated following TLR2 engagement (Shin et al., 2008b; Petnicki-Ocwieja et al., 2013; Petnicki-Ocwieja et al., 2015; Ullah et al., 2016). Together, these findings indicate that TLR2-signaling is critical for controlling B. burgdorferi numbers in tissues by affecting both innate and adaptive effector functions.
TLR1 and TLR6
TLR1 is expressed on the surface of many cell types and is known to dimerize with TLR2 to promote binding of bacterial triacylated lipoproteins, including those expressed by B. burgdorferi. TLR6 is also known to dimerize with TLR2 and to bind bacterial diacylated lipoproteins, which are presently mainly on Gram-positive bacteria. Thus, while one might predict that TLR1, but not TLR6, would have a major role in B. burgdorferi-signaling, there have been few studies assessing their function in controlling spirochete numbers independent of TLR2. TLR1 does appear necessary for optimal production of antibodies specific for B. burgdorferi lipoproteins (Alexopoulou et al., 2002). Co-cultures of B. burgdorferi lysates with different cell types isolated from the central nervous system elicited increased expression of TLR1 by astrocytes and microglia cells, but not neurons (Cassiani-Ingoni et al., 2006). In macrophages, TLR1 levels also were upregulated after exposure to B. burgdorferi (Izadi et al., 2007). Peritoneal macrophages from either TLR1- or TLR6-deficient mice exposed to B. burgdorferi in vitro produced a range of inflammatory cytokines at levels similar to those obtained from WT mice (Oosting et al., 2011a). Interestingly, splenocytes from TLR1-deficient mice produced significantly less IL-10 and significantly higher levels of IFNγ after exposure to B. burgdorferi in vitro compared to WT splenocytes. In contrast, TLR6-deficient mice exhibited a decreased IFNγ response. These findings indicate that the balance of TH1/TH2 associated cytokines is altered in the absence of TLR1 or TLR6 in mice. In contrast to TLR1-deficient mice, studies using PBMCs from human donors with polymorphisms in their TLR1 genes indicated that a subset of those mutations resulted in a significant inhibition of proinflammatory cytokines in response to B. burgdorferi in vitro (Oosting et al., 2011a), which could potentially impact the outcome of B. burgdorferi infection. Subsequent studies with a larger cohort of Lyme borreliosis patients identified a particular TLR1-polymorphism (i.e. TLR1-1805GG) that correlated with the development of persistent inflammatory arthritis after antibiotic treatment (post-antibiotic Lyme arthritis, discussed further below) (Strle et al., 2012). These patients developed higher levels of IFNγ and other inflammatory mediators than Lyme borreliosis patients who did not develop post-antibiotic Lyme arthritis, and PBMCs from these patients also produced higher levels of these mediators when exposed to B. burgdorferi in vitro. Together, these studies suggest that TLR1 can modulate inflammatory cytokine production in response to B. burgdorferi, and certain TLR1 polymorphisms may augment TH1-like inflammatory responses. While these effects may contribute to adverse outcomes from Lyme borreliosis, specific roles for TLR1 and TLR6 in controlling B. burgdorferi infection in vertebrate hosts have not yet been elucidated.
Other TLRs: TLR5/TLR7/TLR8/TLR9
TLR5 is a surface PRR that recognizes bacterial flagella. Although B. burgdorferi possess flagella, their location between the inner and outer membranes of the spirochete indicates they would not be exposed unless the spirochete has been damaged. While deficiency in TLR5 does not alter B. burgdorferi phagocytosis, it has been shown to affect production of inflammatory cytokines in macrophages and microglia. These observations suggest that phagocytosis and disruption of intact B. burgdorferi exposes flagella to TLR5 within endolysosomal compartments (Shin et al., 2008b; Parthasarathy and Philipp, 2015, 2017). However, macrophage responses to flagellin-deficient B. burgdorferi elicited similar levels of prototypic proinflammatory cytokines as produced in response to WT B. burgdorferi, indicating that TLR5 plays a minimal role in the inflammation caused by these spirochetes (Salazar et al., 2005). Disparate responses by certain TLRs are obtained depending on the host cell type studied. In contrast to human microglia, human monocytes stimulated with B. burgdorferi lysates or lipoproteins resulted in the upregulation of TLR2 and downregulation of TLR5, rendering the cells less responsive to TLR5 agonists (Cabral et al., 2006). TLR7 and TLR8 (PRRs for single-stranded RNA), as well as TLR9 (PRR for unmethylated CpG sequences in DNA), also respond to B. burgdorferi components that are likely exposed upon its degradation within host cells. Phagocytosis of B. burgdorferi has been shown to elicit production of Type I and III interferons from human PBMCs (Petzke et al., 2009; Love et al., 2014, 2015), monocytes, and DCs (Cervantes et al., 2011; Cervantes et al., 2013) via these TLR pathways, suggesting their involvement in the host response to B. burgdorferi infection. Interestingly, although type I IFN production was noted in B. burgdorferi-infected mice, production was shown to be independent of MyD88/TRIF, thus independent of TLR stimulation (Miller et al., 2008a; Hastey et al., 2014).
Nod-like receptors (NLRs)
Another group of intracellular PRRs is the NLRs that normally reside in the cytoplasm of leukocytes as well as other host cell types (Motta et al., 2015). Most of these possess a common C-terminal leucine-rich repeat segment that is associated with recognition of microbial agonists, as well as nucleotide-binding domains that allow protein-protein interactions. At least two different NLRs, NOD-1 and NOD-2, respond to muropeptides of bacterial peptidoglycan, with NOD-1 activated by muramyltripeptides of Gram-negative bacteria and NOD-2 by muramyl-dipeptides of Gram-positive bacteria. In those bacteria, inflammatory cytokines produced by macrophages are substantially reduced in the absence of NOD-2 but not NOD-1. B. burgdorferi produces a thin layer of peptidoglycan that elicits inflammatory events in mammals (Beck et al., 1990). As discussed elsewhere (Radolf and Samuels, 2021), peptidoglycan of Borrelia spirochetes has a unique muramyldipeptide and, unlike other bacteria, these spirochetes do not recycle their peptidoglycan fragments during cell wall turnover (Jutras et al., 2019). This shedding of a highly inflammatory B. burgdorferi component may contribute to the disproportionate host inflammatory reaction in infected tissues in comparison to the numbers of spirochetes present and may explain the more exuberant responses elicited from innate immune cells in vitro by sonicated/disrupted spirochetes versus intact viable organisms (Jutras et al., 2019).
NOD2 activation induces the proinflammatory cytokines IL-1β and IL-18. The receptors for both of these cytokines require MyD88 to transmit intracellular signals, suggesting that these cytokines could play an important role in host defense. Absence of IL-18, however, does not alter host susceptibility to B. burgdorferi infection and disease (Behera et al., 2006a). Secretion of IL-1β requires the intracellular assembly of the inflammasome, comprised of NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3), pro-caspase 1 and the adaptor apoptosis-associated speck-like protein ASC, through which caspase 1 cleaves pro-IL-β into its active form (Swanson et al., 2019). Upregulation of the inflammasome components occurs in response to perceived cell stress, including signals transmitted through pattern-associated molecular patterns (PAMPS; microbial agonists) and PRRs such as NOD2. Several in vitro studies have shown that B. burgdorferi induction of IL-1β from mouse and human macrophages requires caspase 1 and ASC (Liu et al., 2009; Oosting et al., 2011b). Despite these in vitro observations, in vivo studies have not supported a critical role for caspase 1-dependent inflammasome activation in host defense against B. burgdorferi (Liu et al., 2009). No significant difference in pathogen burden or arthritis severity was noted in mice deficient in either caspase 1 or ASC when infection was introduced by intradermal inoculation. However, absence of caspase 1 or ASC does attenuate arthritis induced by direct inoculation of cultured B. burgdorferi into the joint (Oosting et al., 2012). Surprisingly, joint inflammation was not dependent on NLRP3, the principal NLR involved in the caspase 1-inflammasome, suggesting the involvement of other pathways.
Cellular mediators of the innate immune response to B. burgdorferi
Langerhans cells (LCs).
LCs are a subset of myeloid cells that reside in the epidermis near the dermal interface (Kaplan, 2017). These cells are identified by the cell surface receptor Langerin (CD207), a PRR. They possess a full complement of PRRs that allow them to respond to pathogens encountered near the skin surface. Their activation leads to the upregulation of MHC class II and subsequent migration to the draining lymph nodes to present antigens to lymphocyte populations in those tissues. Electron microscopy studies identified B. burgdorferi associated with LCs in the epidermis of the skin lesion erythema migrans (EM), an early manifestation of Lyme borreliosis in humans (Hulinska et al., 1994). An ex vivo model using human skin tissues showed that LCs become activated by B. burgdorferi via a TLR2-mediated mechanism and subsequently migrate out of these tissues (Mason et al., 2016). A study of human skin incubated with B. burgdorferi in vitro found that, in contrast to other DC populations, LCs were not able to phagocytose Lyme Borrelia, (Filgueira et al., 1996). Increased numbers of LCs were observed in biopsies of acrodermatitis chronica atrophicans (ACA) lesions, a late skin manifestation of untreated Lyme borreliosis, and these cells demonstrated a decrease in MHC class II expression compared to LCs in normal skin (Silberer et al., 2000). Transfer of LC-enriched epidermal cell populations, pulsed in vitro with B. burgdorferi, into naïve mice elicited significant antibody production that protected recipients from challenge infection with tick-transmitted bacteria, suggesting that LCs play a significant role in B. burgdorferi-directed adaptive immune responses (Mbow et al., 1997). LCs also appear to be involved in the suppression of TH1 responses in regional lymph nodes during and after tick-feeding on mice, though the mechanisms for this activity are unknown (Vesely et al., 2009). Altogether, these findings suggest that LCs are not effectors that phagocytose B. burgdorferi. Instead, LCs efficiently sense spirochetal agonists in the skin and then activate adaptive immune responses in secondary lymphoid tissues.
Macrophages/Dendritic cells
Macrophages and dendritic cells (DCs) are tissue-resident professional phagocytes that can be generated from several tissues, though the majority represent a progression from myeloid progenitors in the bone marrow to blood-resident monocytes (Geissmann et al., 2010).Upon receiving maturation signals, monocytes leave the vasculature and migrate into tissues, where they mature into either macrophages or DCs. Macrophages and DCs are both professional antigen-presenting cells capable of initiating adaptive immune responses via antigen presentation by MHC class I or II. DCs are specialized for migration to the T cell zones of lymphatic tissues after phagocytosis of microbes where they present processed antigens to initiate T cell responses. In contrast, macrophages remain mostly in tissues at the site of infection where they phagocytose invading microbes and initiate localized inflammatory responses that attract additional leukocytes and soluble immune mediators. Currently, the literature on macrophage interactions with B. burgdorferi far outnumber papers on DCs, though both have similar effector responses (Chung et al., 2013).
In vitro studies indicate that macrophages and DCs can efficiently phagocytose and kill B. burgdorferi, and these activities are enhanced by opsonization via complement and/or spirochete-specific antibodies that bind to CR3 or Fc receptors, respectively (Benach et al., 1984a; Peterson et al., 1984; Montgomery et al., 1994; Filgueira et al., 1996; Cinco et al., 1997; Montgomery et al., 2002; Suhonen et al., 2002; Chung et al., 2013). Macrophage activation also occurs in response to B. burgdorferi recombinant lipoproteins via interactions with TLR2, producing a similar elicitation of activation markers as reported after exposure to intact spirochetes (Radolf et al., 1995; Hirschfeld et al., 1999; Wooten et al., 2002a; Chung et al., 2013). B. burgdorferi strains that have been passaged repeatedly in vitro are less infectious and more susceptible to phagocytosis by macrophages in vitro than low-passage strains, but both high and low passage strains equally elicit an oxidative burst after phagocytosis; the mechanisms behind these differences are unknown (Georgilis et al., 1991b).
Phagocytosis of B. burgdorferi also can be triggered by interaction of spirochetes with a number of surface PRRs, including integrin α3β1, MARCO, and TLRs, the latter capable of signaling via MyD88 and TRIF (Behera et al., 2006c; Behera et al., 2008; Petnicki-Ocwieja et al., 2013; Hawley et al., 2015; Killpack et al., 2017). Macrophages (and neutrophils) ingest B. burgdorferi primarily through both tube and coiling phagocytosis, in which a pseudopod of the cell wraps or “coils” around the entire length of the spirochete, resulting in its engulfment (Rittig et al., 1992; Montgomery and Malawista, 1996; Rittig et al., 1998; Linder et al., 2001; Naj et al., 2013; Naj and Linder, 2015) (Figures 2 and 3). Phagocytosis and degradation of B. burgdorferi releases intracellular PAMPs that macrophages then recognize via TLRs and other PRRs that are present in the phagolysosome and other endosomal compartments, as discussed above (Salazar et al., 2005; Shin et al., 2008b; Salazar et al., 2009; Cervantes et al., 2014; Petnicki-Ocwieja et al., 2015; Carreras-González et al., 2019). The cumulative effect of B. burgdorferi phagocytosis is a potent inflammatory response eliciting a wide range of pro-inflammatory mediators, including cytokines, chemokines, leukotrienes, and eicosanoids. These mediators prove to be a double-edged sword, in that they promote both spirochetal clearance, but also inflammatory pathology (Wang et al., 2008; Strle et al., 2009; Blaho et al., 2011; Pratt and Brown, 2014). This is reflected transiently in the joints and hearts of B. burgdorferi-infected mice, where macrophages have been shown to express phenotypic markers associated with both protective M1 and tissue remodeling M2 differentiation (Lasky et al., 2015b). Overall, these findings indicate a critical role for macrophages and DCs in initiating and mediating immune mechanisms necessary for controlling B. burgdorferi numbers in host tissues.
Figure 2.
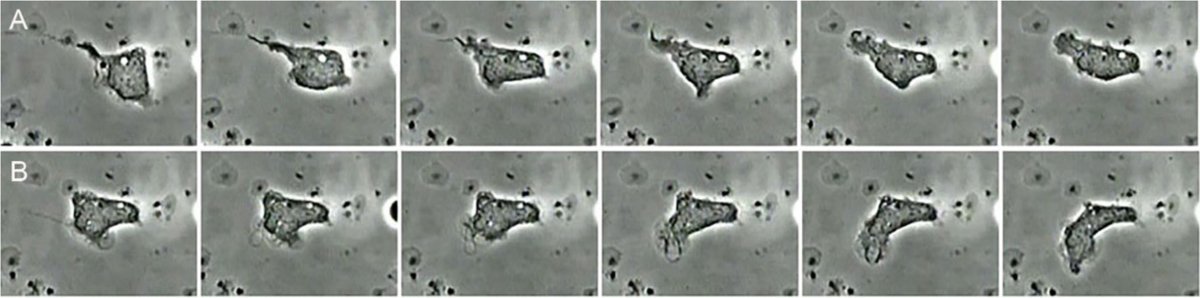
Serial images from a movie of a human neutrophil ingesting B. burgdorferi in vitro. Panel A shows engulfment of B. burgdorferi from one end by tube phagocytosis. Panel B shows the same neutrophil engulfing a spirochete initially attached at one end. The opposite end was subsequently captured, allowing the neutrophil to readily engulf the immobilized spirochete. From the unpublished archives of movies produced by Malawista (deceased) and de Boisfleury Chevance.
Figure 3.
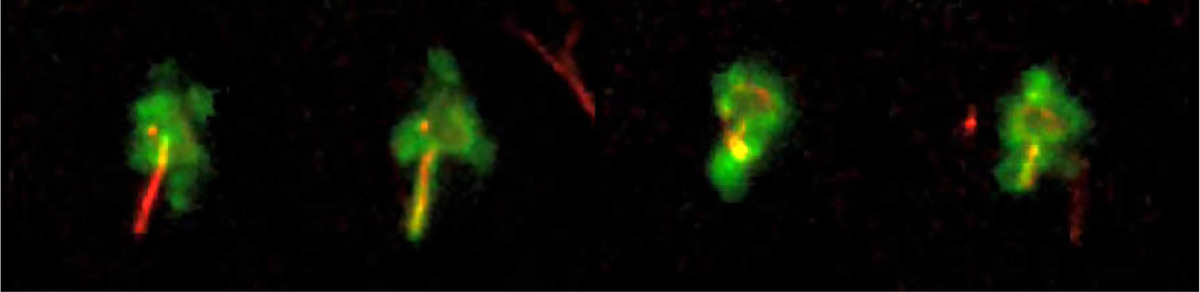
Phagocytosis of B. burgdorferi by a dermal professional antigen-presenting cell. Transgenic mice (I-Aβ-GFP expressing) possessing green MHC class II-expression were infected with DsRed-expressing B. burgdorferi intradermally into the ear. Intact ear tissues were imaged using intravital confocal microscopy on living mice at 24h post-infection. The four images were taken at 6-minute intervals (left-to-right). From the unpublished archives of movies produced by the R. Mark Wooten lab; images provided by John Presloid.
Neutrophils/Endothelial cells
Neutrophils are the most numerous phagocytic cells normally present in the circulation. When tissue-resident myeloid cells (e.g. macrophage and DCs) become activated, they secrete inflammatory mediators that activate endothelial cells lining local blood vessels. Activated endothelial cells upregulate the expression of adhesion molecules and chemokines that promote adherence of circulating immune cells and their subsequent migration from the bloodstream into the tissue. Chemotactic molecules secreted by activated tissue-resident immune cells create a gradient that directs leukocytes from the blood to the infected area, where they contribute to the phagocytosis and killing of extracellular pathogens. Since B. burgdorferi is known to persist long-term in the extracellular spaces in multiple tissues, endothelium and neutrophils must play an important role in controlling these infections.
In vitro studies using primary human umbilical vein endothelial cells (HUVECs) indicate they rapidly become activated after exposure to B. burgdorferi or their lipoproteins. Spirochetes bind to HUVECs via a mechanism involving fibronectin and then traverse the endothelial layers; this penetration was enhanced via binding of plasminogen (Szczepanski et al., 1990; Comstock and Thomas, 1991; Coleman et al., 1995). HUVECs co-cultured with B. burgdorferi or their lipoproteins elicit rapid NF-κB translocation to the nucleus, followed by upregulation of adhesion molecules critical for recruitment of leukocytes within 1–4 hours post-infection (Boggemeyer et al., 1994; Sellati et al., 1995; Wooten et al., 1996). Activation of HUVECs is greatly enhanced in the presence of sCD14 and activation is suppressed by B. burgdorferi-elicited IL-10 (Sellati et al., 1998; Wooten et al., 1998; Lisinski and Furie, 2002a). HUVEC secretion of pro-inflammatory cytokines and chemokines occurs within 4 hours of exposure, and neutrophils bind and traverse these endothelial layers by ≥4h post-exposure (Ma and Weis, 1993; Sellati et al., 1995; Sellati et al., 1996; Wooten et al., 1996; Ebnet et al., 1997).
In vitro studies indicate that neutrophils can efficiently phagocytose and kill B. burgdorferi, and these activities are enhanced in the presence of complement and/or spirochete-specific antibodies (Benach et al., 1984a; Peterson et al., 1984; Suhonen et al., 2000; Lusitani et al., 2002; Suhonen et al., 2002). Neutrophil activation also occurs in response to B. burgdorferi recombinant lipoproteins, producing a similar elicitation of activation markers as reported after exposure to intact spirochetes (Morrison et al., 1997; Cinco et al., 1998). Phagocytosis of B. burgdorferi by neutrophils occurs by tube phagocytosis in which pseudopods wrap around the entire 20–30μm length of these spirochetes to allow engulfment (Suhonen et al., 1998). Neutrophil-generated elastase, LL-37, and a number of other granular components can either directly kill B. burgdorferi or inhibit their growth in vitro (Garcia et al., 1998; Lusitani et al., 2002, 2003). Injection of B. burgdorferi or their lipoproteins into murine skin tissue results in a rapid influx of neutrophils (Norgard et al., 1995; Salazar et al., 2005). A number of chemokines and their receptors appear to be critical for recruitment of neutrophils into infected skin, heart, or joint tissues, including CXCR2 and KC (Georgilis et al., 1991a; Brown et al., 2003; Montgomery et al., 2007). Of significance, while neutrophils infiltrate B. burgdorferi-infected mouse skin efficiently within 6h, these numbers rapidly drop after 12h post-infection. Neutrophils are almost completely absent by day 7 post-infection, despite the continued presence of spirochetes (Xu et al., 2007). The downregulation of neutrophil influx may be a consequence of host adaptation of B. burgdorferi from the culture to the mammalian environment and appears to be important for the establishment of B. burgdorferi infection in this model. Of note, with tick-transmitted infection, inflammatory cells increased in numbers at the tick bite site during the first 3 days of tick attachment, but were excluded from the area immediately adjacent to the tick hypostome where spirochetes are deposited (Krause et al., 2009). Evidence that neutrophils can be deleterious to B. burgdorferi survival can be seen in studies using a B. burgdorferi transformant that expresses and secretes the murine neutrophil chemokine KC. Infection of mice with this transformant caused a significant increase and extended presence of neutrophils at the inoculation site, which correlated with a significantly increased clearance of bacteria in all tissues tested (Xu et al., 2007). Even though neutrophil presence highly correlated with more efficient control of B. burgdorferi numbers, mice deficient in genes that encode proteins within the NADPH oxidase complex (i.e. Ncf1 or Gp91phox) showed similar numbers of spirochetes in joints, hearts, and ear skin at 4 weeks post-infection as WT mice (Crandall et al., 2005). On the surface, this appears at odds with the finding that mouse strains that develop arthritis after B. burgdorferi infection exhibit a short-lived neutrophil-mediated inflammation in the joints (Barthold et al., 1990). Although quantification of B. burgdorferi in mouse tissues by PCR does not distinguish live from dead spirochetes, these findings suggest that either neutrophils are not important for controlling spirochete numbers in those tissues, or that other, NADPH oxidase-independent mechanisms may be engaged. Neutrophils responding to B. burgdorferi can generate extracellular traps (NETs) formed by the release of nuclear and cytosolic proteins and chromatin that ensnare and kill spirochetes (Menten-Dedoyart et al., 2012). The formation of NETs has been shown to occur through both NADPH oxidase-dependent and -independent mechanisms, suggesting a mechanism in addition to phagocytosis whereby neutrophils responding to B. burgdorferi in infected joints may kill spirochetes (Douda et al., 2015). Overall, the data suggest that neutrophils initially control B. burgdorferi numbers, but that there is a lack of sustained neutrophil infiltration into infected skin and perhaps other tissues. Identifying the mechanisms behind this lack of continued neutrophil activation will be important to understanding how these spirochetes can establish persistence.
Natural killer cells
Natural killer (NK) cells are derived from cells of the lymphocytic lineage during hematopoiesis but do not subsequently undergo antigen-receptor rearrangement compared to T and B cells (Abel et al., 2018). Instead, they act as cytotoxic innate immune cells with invariant receptors that recognize alterations in expression of major histocompatibility complex proteins on multiple types of host cells. These specificities of NK cell receptors allow recognition of host cells that have either become infected by different classes of microbial pathogens or that have been transformed to become tumorigenic, even in the absence of MHC expression. NK cell binding to these “altered” cells also promotes binding to Fas ligand.
The combined effects of these interactions lead to release of cytotoxic granules and formation of pores, leading to cell lysis and/or apoptosis. Within the host environment, their activities are enhanced by exposure to a subset of proinflammatory cytokines secreted by activated tissue-resident phagocytes, including IL-12 and IL-18, which promote their activation and secretion of IFNγ. This makes NK cells important for early detection of intracellular pathogens, such as viruses. The literature assessing the importance of NK cells in controlling B. burgdorferi during acute infection is limited. Patients with early or later stage manifestations of untreated Lyme borreliosis possessed significantly more CD16+ NK cells in their blood compared to patients that had been treated with antibiotics, and these cells expressed different cell markers associated with activation (Golightly et al., 1988). Interestingly, exposure of isolated NK cells to B. burgdorferi in vitro caused the suppression of their cytotoxicity. Crude preparations of murine primary NK cells from lymph nodes suggest they become activated in vitro after exposure to B. burgdorferi or their recombinant lipoproteins, as evidenced by production of IFNγ (Brown and Reiner, 1998a). Antibody-depletion of NK cells in mice did not appear to alter host control of B. burgdorferi numbers in multiple tissues, as measured by tissue outgrowth in BSK medium cultures (Barthold and de Souza, 1995; Brown and Reiner, 1998a). However, synovial tissues from Lyme borreliosis patients possess increased numbers of NK and T cells which secrete high levels of IFNγ, suggesting they do promote the inflammatory lesions associated with human Lyme arthritis and could contribute to spirochete killing (Katchar et al., 2013) (Jutras et al., 2019). NK cells also express inhibitory receptors and help to maintain self tolerance and potentially could contribute to resolution of inflammation (Pegram et al., 2011). Overall, while NK cells represent a resident immune cell type that can be activated in response to B. burgdorferi, their importance in controlling infection or mediating the pathology seen in Lyme borreliosis is still poorly understood.
Mast cells
Mast cells are tissue-resident innate immune cells that originate from hemopoietic stem cells (Frossi et al., 2017). They are primarily found in the connective tissues or mucosal surfaces near smooth muscle, glands, nerves, and blood vessels. When these cells become activated in response to their surface IgE molecules binding particular ligands, they degranulate to release a plethora of preformed cytokines, proteases, heparin, and multiple vasoactive materials. This results in rapid effects on proximal tissues, such as local blood flow, smooth muscle contraction, vessel permeability, and secretion activities. Because B. burgdorferi is introduced into and persists within skin tissue, it is possible that interactions with skin-resident mast cells might influence the development of B. burgdorferi induced inflammation. B. burgdorferi infection of gerbils resulted in increased numbers of mast cells in most tissues (Preac Mursic et al., 1990). Purified B. burgdorferi lipoproteins promoted the upregulation of CD28 on bone marrow-derived mast cells (Marietta et al., 1997), an important costimulatory receptor for T cells, and exposure of rodent mast cells to intact B. burgdorferi elicited degranulation and increased TNFα secretion in vitro (Talkington and Nickell, 1999). A more recent study found that exposure of C57BL/6 mouse mast cells to B. burgdorferi spirochetes in vitro resulted in degranulation and the production of the inflammatory cytokine IL-6 and upregulation of MCP1 mRNA; addition of I. ricinus salivary gland extract inhibited these responses (Bernard et al., 2017). Experiments in mast cell deficient (Kitwsh−/−) mice, however, revealed no significant impact on host resistance to infection, although B. burgdorferi was detected in the joints slightly earlier than in WT mice. Taken together, although early mast cell activation may occur in response to B. burgdorferi infection, there is limited data indicating these cells play a significant role in host defense or disease.
Microglia
B. burgdorferi are known to cause both peripheral and central nervous system pathology, collectively termed Lyme neuroborreliosis (Halperin, 2019). The central nervous system contains a diverse repertoire of myeloid cells, including microglia found in the brain parenchyma and non-parenchymal macrophages that can be found in the perivascular areas, the meninges and the choroid plexus (Prinz et al., 2011). To date, the responses of microglia are the only CNS macrophage type that has been studied in vitro. Microglia phagocytose invading pathogens and elicit inflammatory responses that can recruit a range of infiltrating immune cell types (Forrester et al., 2018). In vitro studies with murine microglial cell lines and primary murine or human microglia reveal that they are able to efficiently phagocytose B. burgdorferi, even in the absence of complement, and that spirochetes were efficiently trafficked into phagolysosomes and digested, followed by production of various proinflammatory mediators (Rasley et al., 2002; Kuhlow et al., 2005; Greenmyer et al., 2018). Phagocytosis of B. burgdorferi by primary microglia cells from rhesus monkeys correlates with increases in TLR1, TLR2, and TLR5 expression (Bernardino et al., 2008). Production of proinflammatory mediators by primary microglial cells in response to B. burgdorferi is augmented in the presence of NOD2 (Chauhan et al., 2009). Killed or damaged B. burgdorferi elicit a greater magnitude of inflammatory mediators compared to intact spirochetes, and these inflammatory responses are associated with increased apoptosis of bystander cells (Myers et al., 2009; Greenmyer et al., 2018). These findings, albeit derived from studies with microglia, suggest that CNS macrophages have the capacity to respond vigorously to B. burgdorferi and likely play a role in the host response in neuroborreliosis.
Soluble mediators that significantly affect B. burgdorferi infection
Interleukin-10
The production of pro-inflammatory cytokines is an important outcome of phagocyte-mediated killing of microbes due to their ability to amplify innate host defenses and to activate the adaptive immune response (Liu et al., 2004; Salazar et al., 2005; Moore et al., 2007; Cruz et al., 2008; Shin et al., 2008a; Salazar et al., 2009). These responses must eventually be suppressed, however, to prevent damage to the host tissues. One major anti-inflammatory cytokine that provides this function is IL-10, which is usually produced at later times after infection to downregulate the inflammatory responses and prevent excessive tissue injury. However, some pathogens are able to elicit early IL-10 production or encode an IL-10 homolog, either of which can prematurely shut down inflammatory/immune responses that are needed to effectively clear the infection. Thus, IL-10 production provides an important counterbalance to productive inflammatory responses and serves to modulate those responses to prevent damaging the host.
In vitro studies determined that B. burgdorferi and/or their lipoproteins potently elicit IL-10 production from macrophages and DCs (Chung et al., 2013). Similar trends in IL-10 production were observed in macrophages from mouse, human, and rhesus macaques incubated with B. burgdorferi and its lipoproteins, with optimal signaling dependent on both TLR2 and CD14 (Giambartolomei et al., 1998; Brown et al., 1999; Wooten et al., 2002b; Benhnia et al., 2005; Dennis et al., 2006). Interestingly, it was shown recently that living B. burgdorferi induce greater production of IL-10 by murine macrophages than do equivalent numbers of non-viable organisms (Lazarus et al., 2008). Addition of IL-10 to cell cultures suppresses pro-inflammatory cytokine production by macrophages and B cells in response to B. burgdorferi. The B. burgdorferi-induced production of IL-10 appears dependent on the host genetic make-up, and is greater in macrophages from mildly arthritic C57BL/6 mice than severely arthritic C3H mice (Brown et al., 1999; Lazarus et al., 2006). Kinetic studies indicated that IL-10 is elicited earlier in response to B. burgdorferi than the production of pro-inflammatory mediators, and correlated with prematurely suppressed pro-inflammatory responses (Lazarus et al., 2008; Chung et al., 2013). These findings are supported by the observation of greatly enhanced production of inflammatory mediators by macrophages and DCs from IL-10-deficient mice, as well as suppressed phagocytosis of B. burgdorferi and reduced expression of markers indicative of efficient activation of adaptive immune responses (Lazarus et al., 2008; Chung et al., 2013). Intravital confocal microscopy supports both the early kinetics of IL-10 production in infected mouse skin, with the majority of IL-10-producing cells falling within the phenotype of MHC class II-expressing cells, which are presumed to be macrophages/DCs (Wooten et al., unpublished).
The role of IL-10 in modulating arthritis severity in B. burgdorferi-infected mice is apparent, as infection of either IL-10−/−C57BL/6 mice or IL-10−/−C3H mice resulted in more severe disease than was observed in the appropriate WT strain (Brown et al., 1999) (Brown et al., 2008). The suppression of arthritis by IL-10 is not due to better control of spirochetes in tissues, as IL-10−/− mice harbored reduced numbers of bacteria in their tissues (Brown et al., 1999). IL-10 in infected joints was produced primarily by cells of the macrophage and CD4+ T cell lineage (Sonderegger et al., 2012b). The enhanced control of bacteria in vivo was dependent on innate host defenses, not to an observed heightened production of IgG (Lazarus et al., 2006). Thus, although release of the tight control of inflammation by the lack of IL-10 resulted in more severe arthritis, this was coupled with more effective control of the bacteria. The role of IL-10 in controlling arthritis development is not well understood, as regulation of IL-10 production is complex and depends on the cell type producing it (Moore et al., 2001). Mechanistic insight has come from in vitro studies in which SOCS1 and SOCS3 have been implicated in the IL-10-mediated suppression of B. burgdorferi-induced inflammatory responses in macrophages (Dennis et al., 2006), while others have found that IL-10 effects on the endothelium can influence inflammatory cell infiltration (Lisinski and Furie, 2002b). Apoptosis also was observed in human monocytes following treatment with lipoproteins or after phagocytosis of B. burgdorferi, suggesting another means of limiting inflammatory responses (Aliprantis et al., 1999; Cruz et al., 2008).
Complement system
The complement system refers to a group of 30+ proteins and their fragments that comprise an essential immune monitoring system to identify and control invading microbes, particularly those residing in extracellular spaces (Lin et al., 2020; Skare and Garcia, 2020). Activation can be initiated via binding of specific antibodies (i.e. classical pathway), binding by innate pattern recognition receptors such as mannose binding lectins (i.e. lectin-binding pathway), or by the continuous small-scale activation of complement component C3, which will bind surfaces and initiate activation unless inactivated by regulatory proteins (i.e. alternative pathway). Activation by any of these three pathways results in the formation of a C3-convertase which will generate activation of the enzymatically-amplified cascade of complement factors to result in a panoply of immune effects that can kill microbial invaders. One is the production of a membrane attack complex (MAC), which is a self-assembling complex that forms a pore in the microbial membrane, leading to a loss of membrane potential and/or lysis. Microbes that can resist this killing are known as serum-resistant, which is essential for establishing infection in most host tissues. Another mechanism for killing is via the effect of covalently bound fragments of the C3 protein (e.g. iC3b) on the microbial surface, which opsonizes the microbe to be recognized by phagocytes possessing complement receptors, which potently promotes phagocytosis and killing by those host cells. Thus, B. burgdorferi must possess a number of mechanisms to evade the killing mechanisms of complement to establish persistent infection (Radolf and Samuels, 2021).
Virtually all virulent B. burgdorferi sensu lato can grow/survive in the presence of human serum, a feature attributed to its ability to resist complement activation and complement-mediated lysis in the absence of specific antibodies (Kochi et al., 1991; Patarakul et al., 1999; Stevenson et al., 2002). This property is common to many human pathogens and is consistent with the need for this organism to survive extracellularly and disseminate hematogenously. Murine studies have described the involvement of a number of different complement components with B. burgdorferi during infection. In vitro studies indicated that B. burgdorferi can initiate complement activation via the classical and alternative pathways (Kochi and Johnson, 1988). While certain spirochete-specific antibodies have been reported to directly kill or inhibit growth of B. burgdorferi in vitro (Sadziene et al., 1993; Sadziene et al., 1994), in most cases complement is required for antibody-mediated killing of these bacteria (Kochi et al., 1993). Mice deficient in C3, which is central to all downstream complement-mediated immune mechanisms, harbored greater numbers of B. burgdorferi in some tissues and required five-fold fewer B. garinii spirochetes to establish infection (Lawrenz et al., 2003; Woodman et al., 2007). The fifth component of complement, C5, which is essential for formation of the membrane attack complex, is not required for antibody-mediated protection from infecting B. burgdorferi (Bockenstedt et al., 1993). Interestingly, serum-resistant B. burgdorferi strains express a CD59-like protein which can bind complement components C8 and C9, thus inhibiting assembly of the MAC (Pausa et al., 2003). B. burgdorferi sensu lato species also expresses three additional proteins (i.e. CspA, BGA66, and BGA71) that directly bind different components of the MAC, thus preventing the assembly of these lytic complexes (Hallstrom et al., 2013; Hammerschmidt et al., 2016). Together, these studies in mice suggest a major role for complement as an opsonin to promote clearance of the organism by phagocytes during early infection, rather than in direct lysis of the bacteria via the MAC, which is only seen at significant levels in the presence of B. burgdorferi-specific antibodies in vitro (Kochi et al., 1991).
B. burgdorferi is supremely adapted to evasion of complement mechanisms by producing a plethora of proteins capable of binding multiple host complement-regulatory proteins (Lin et al., 2020; Radolf and Samuels, 2021). These proteins include complement regulator-acquiring surface proteins (CRASPS) that bind factor H, OspC that binds C4b, BBK32 that binds the C1 complex and a 43 kD protein that binds the C4b binding protein C4bp. The combined effects of these interactions renders variable levels of resistance to clearance of spirochetes by the classical, alternative, and lectin-mediated pathways. Spirochete binding of host factor H prevents C3-convertases from accumulating on their surfaces, and thus renders them resistant to clearance by the alternative pathway. Overall, it is apparent that B. burgdorferi has invested much evolutionary currency to allow inactivation of complement immune activation. Collectively, these findings also suggest that therapies that can quell a subset of these activities might be able to allow the host innate responses to clear these infections. Recent examples of this are studies mutating the factor H-binding site of B. burgdorferi protein CspZ (i.e. CRASP-2) so that it is unable to bind factor H, and using this as a vaccine in mice (Marcinkiewicz et al., 2018; Marcinkiewicz et al., 2020). By preventing immediate complexing of this mutant protein with factor H within the mouse, antibodies are generated against multiple epitopes on the CspZ protein, which appears to confer resistance when challenged with B. burgdorferi.
Responses of acquired (adaptive) host defenses
T and B lymphocytes of the adaptive immune system and their secreted products are critical for controlling infections. These cells regulate anti-pathogen defenses following their highly antigen-specific, pathogen-induced activation, clonal expansion and eventual differentiation into various effector populations. These effectors control acute infections and generate memory responses that enable a host to more vigorously respond to repeat encounters with the same pathogen. The interaction of CD4 T cells with B cells is critical for the induction of optimal antibody responses, as well as the establishment of robust B cell memory. Experiments with SCID mice lacking T and B lymphocytes confirmed the importance of the adaptive immune system in controlling B. burgdorferi infections, as SCID mice cannot control B. burgdorferi spirochete burdens in blood and tissues and eventually succumb to uncontrolled B. burgdorferi expansion and the resulting inflammatory responses (Schaible et al., 1989; Schaible et al., 1990).
Yet, despite the fact that B. burgdorferi infection results in the generation of vigorous and specific antibody responses in their natural reservoir species, including small rodents such as Peromyscus leucopus and birds, infections are not cleared. Instead, as shown with studies on commonly used laboratory mouse strains, such as 129, BALB/c, C57BL/6 and C3H, tissue loads of B. burgdorferi are suppressed, but remain at levels that allow their acquisition by feeding ticks for many months. Clinical studies in dogs, horses, non-human primates and humans demonstrated that chronic, non-resolving infection can occur also in these incidental non-reservoir hosts (Appel et al., 1993; Barthold et al., 1993; Roberts et al., 1995; Straubinger et al., 1997; Radolf et al., 2012). Seroprevalence studies in humans as well as dogs residing in endemic areas have shown high exposure rates. Comparisons between IgG seroprevalence in individuals following a tick-bite and incidence of Lyme borreliosis suggests that only a fraction of exposed individuals who generate IgG will develop disease.(Fahrer et al., 1991; Rath et al., 1996; Huegli et al., 2011). Thus, while lifelong persistent infection in reservoir species is the likely outcome, in non-reservoir species outcomes vary from asymptomatic seroconversion to debilitating disease.
In persistent non-resolving infections of reservoir species, the clinical manifestations of disease are largely suppressed by the adaptive immune response. While spirochete burdens are reduced significantly, the pathogen is not eliminated. Importantly, the spirochete burden of disease-susceptible mice, such as the C3H mouse, which shows significant arthritis development after B. burgdorferi infection, is indistinguishable from that of disease-resistant laboratory mouse strains when using PCR to quantify Borrelia DNA (Barthold et al., 1993). Thus, disease associated with B. burgdorferi infection is not a manifestation of the pathogen causing damage, but rather the result of the inflammatory response to the pathogen. In C3H mice, for example, arthritis development is caused by a deficiency of lysosomal beta-glucoronidase. This leads to a failure to clear glycosaminoglycans in the joints of infected mice, which is a damage-associated molecular pattern (DAMP) that causes continued activation of inflammatory stimuli, resulting in tissue damage (Bramwell et al., 2014).
In sum, the adaptive immune response to B. burgdorferi infection is ineffective in clearing the pathogen from its natural reservoir species yet can protect the host against reinfection. In the infected host, it controls spirochete numbers, and thereby reduces the triggers of inflammatory responses that result in disease. In the clinical pathology characteristic of Lyme borreliosis, T cells may also become drivers of an ongoing inflammatory process. The features of adaptive immunity to B. burgdorferi, which have been studied mostly in mice, and which are summarized below, cannot be seen as reflecting an effective host defense that results in pathogen clearance. Rather, they represent the outcomes of a complex interplay of B. burgdorferi immune evasion strategies and the mammalian adaptive immune system that results in a state of concomitant immunity. This interplay allows B. burgdorferi to fulfill its complex lifecycle without inciting disease in the reservoir host.
B Cell Responses
B cells are critical for controlling B. burgdorferi tissue burden. Mice selectively lacking B cells (Igh6−/− mice) exhibited markedly elevated pathogen burdens in various tissues. B cells were also shown to be critical for preventing and/or resolving arthritis, carditis and myocarditis (McKisic et al., 2000). Their actions can be augmented by T cells, as carditis resolution was promoted by the adoptive transfer of IFNγ−producing T cells into T cell deficient mice, presumably through activation of macrophages, which are prominent in the diseased heart tissue (Bockenstedt et al., 2001). A role for antibodies as the main effectors produced by B cells was demonstrated in numerous passive transfer experiments, in which serum from previously infected mice or humans, but not non-infected controls, or the transfer of B and T cells, but not T cells alone, into SCID or T and B cell-deficient rag1−/− mice reduced pathogen burden and resolved arthritis (Schaible et al., 1989; Barthold et al., 1992b; McKisic and Barthold, 2000; Bockenstedt et al., 2001). Antibodies induced by B. burgdorferi infection are bactericidal, i.e. can kill the bacteria through both direct, complement-dependent and complement-independent spirochete lysis (LaRocca and Benach, 2008). They also can opsonize the spirochetes for uptake and degradation by macrophages and neutrophils following engagement of their surface-expressed FcγR or complement receptors (Benach et al., 1984b; Modolell et al., 1994).
Antigenic targets of the antibody response to B. burgdorferi infection.
Early clinical studies demonstrated strong serum antibody-antigen immune complex formation in patients with disseminated B. burgdorferi infection (Hardin et al., 1979; Hardin et al., 1984; Brunner and Sigal, 2001), indicating that robust antigen-specific humoral immune responses can be initiated among that group of patients. Indeed, strong early B cell responses have been associated recently with faster resolution of disease following antibiotic treatment (Blum et al., 2018). However, seroconversion takes time. Previous studies suggested that it is detectable in only about half of patients with a localized EM at the time of presentation (Aguero-Rosenfeld et al., 2005; Steere et al., 2008; Wormser et al., 2008; Pegalajar-Jurado et al., 2018; Nigrovic et al., 2019), although development of more sensitive modified-testing modalities have increased these numbers (Pegalajar-Jurado et al., 2018). Convalescent blood samples obtained after antibiotic treatment increases the percentage of patients testing positive for Lyme borreliosis. Lack of seroconversion has been observed in patients following rapid initiation of antibiotics, when the infection may have remained localized in the skin. In experimentally infected mice, seroconversion takes 10 −14 days, despite a robust and early B cell activation in the lymph nodes. Yet, by ELISPOT analysis, increased but transient local production of IgM and IgG was measurable in lymph nodes draining the site of infection by day 7. The data suggest that production of antibodies even in lymphoid tissues is not always reflected in the serum (Tunev et al., 2011; Hastey et al., 2012).
Comprehensive serological analysis of the immunogenic targets of B. burgdorferi-specific IgG from mice and humans, conducted with a genome-wide protein expression array, demonstrated the remarkable conservation of antibody targets between these species. Induction of antibodies was estimated to occur against ~15% of B. burgdorferi gene open reading frames (ORFs) (Barbour et al., 2008). This number is likely an underestimate, however, given the restrictions in this approach to screen effectively for all conformational epitopes. Nonetheless, the study provides the most comprehensive analysis yet on the antibody targets of B. burgdorferi. Previously identified surface-exposed lipoproteins such as OspC, p35, p37, p39, BmpA, BmpB and decorin-binding proteins (Dbp) A and B, which can induce antibodies with passive protective capacity (Fikrig et al., 1990; Fikrig et al., 1992; Fikrig et al., 1997b; Hanson et al., 1998; Pal et al., 2008), were strongly represented among B. burgdorferi peptide targets. In addition, the array analysis also identified strong IgG responses to previously unknown Borrelia proteins, including BBK07 among others, which were shown to be mostly absent from culture-grown B. burgdorferi but rapidly and strongly induced following mammalian infection (Barbour et al., 2008; Coleman et al., 2011). The ability of some of these antigens to elicit protective immunity after immunization appears to be B. burgdorferi strain-dependent. For example, neither OspC nor BmpA from B. burgdorferi N40 could elicit protective immunity, while they were protective to challenge with the B31 strain (Barthold et al., 1997; Bockenstedt et al., 1997).
The dynamic up- and down-regulation of surface proteins by B. burgdorferi affects the specificity of the antibody response induced to the infection and must be considered an immune evasion strategy. One prominent example is the expression of OspC, a protein required for mammalian infections (Grimm et al., 2004). Its expression is induced in the tick after initiation of the blood meal (Schwan et al., 1995; Pal et al., 2004), while OspA is downregulated (de Silva et al., 1996). Mutant B. burgdorferi with stabilized expression of OspC are rapidly cleared from the host (Embers et al., 2008). Once infection is established, however, expression of OspC is rapidly lost from B. burgdorferi in lymph nodes, where B cell responses are initiated (Skare et al., 2016). This suggests that the loss of this immunogenic surface protein reduces induction of OspC-specific antibodies, thereby avoiding antibody-mediated clearance during the earliest stages of B. burgdorferi infection, when Osp C is indispensable for the B. burgdorferi life-cycle (Tilly et al., 2006). Consistent with the expression pattern of OspC, IgG responses to this protein are overall low in infected mice compared to antibody responses to other surface proteins of B. burgdorferi (Barbour et al., 2008; Tunev et al., 2011).
Measurement of antibody responses to the surface-expressed VlsE protein of B. burgdorferi is used extensively for serological testing. The stalk region of this protein (the “C6” peptide) is highly invariable, immunogenic and appears to be constitutively expressed by B. burgdorferi. Most antibody responses to VlsE, however, are directed against the highly immunogenic protein head region, which is antibody accessible on the spirochete outer membrane (Eicken et al., 2002). That region undergoes extensive and continued antigenic variation, owing to the continued segmental recombination from a contiguous array of multiple vlsE silent-expression cassettes (Zhang et al., 1997; Zhang and Norris, 1998b, a; Bankhead and Chaconas, 2007; Coutte et al., 2009) (see also Radolf and Samuels, 2021). The process of antigenic variation is considered a prominent mechanism through which B. burgdorferi evades immune clearance, as it results in the generation of strong, but ultimately ineffective antibody responses that are induced to antigens expressed only for short periods of time (Norris, 2006; Bankhead and Chaconas, 2007; Coutte et al., 2009; Rogovskyy and Bankhead, 2013; Magunda and Bankhead, 2016). Thus, although a large number of antibodies against a variety of surface proteins of B. burgdorferi are induced, and the antibodies engage with and eliminate large numbers of spirochetes over the course of infection, the pathogen uses a multitude of immune evasion strategies to reduce the functionality of the antibodies.
Characteristics of the antibody response to B. burgdorferi Serum antibodies to B. burgdorferi are of the IgM, IgG and IgA isotypes in humans, with IgA responses most prevalent in early Lyme borreliosis in patients presenting with EM (Steere et al., 1979; D’Arco et al., 2017). IgM and IgG responses have been detected in the serum of 30–80% of patients at the initial visit (Aguero-Rosenfeld et al., 1996; Barbour et al., 2008; Tjernberg et al., 2009), which in the case of patients presenting with EM, is usually within the first 10–14 days after a tick bite. In experimentally-infected dogs, IgM responses were measurable as early as 7 days post infection and IgG by around day 10, similar to experimental infection of mice (Greene et al., 1988). As infection progresses beyond 6 weeks, IgM responses may remain elevated or may return to normal (Aguero-Rosenfeld et al., 1996). In untreated infection in humans, B. burgdorferi-specific IgG titers increase and are highest during the late phase of disease, when arthritis is present (Craft et al., 1986; Aguero-Rosenfeld et al., 2005). IgG titers may remain elevated for years after treatment despite clinical remission, or they may return to background levels following antibiotic treatment (Hammers-Berggren et al., 1994; Aguero-Rosenfeld et al., 1996; Nowakowski et al., 2003). Intrathecal IgM, IgG and IgA are also found in humans with neurological manifestations of the infection and are of high diagnostic value (Steere et al., 1990; Schutzer et al., 1997). Intrathecal antibodies to Borrelia are induced following direct intracerebral inoculation of spirochetes into mice but not when infection is introduced peripherally (outside the central nervous system) (Li et al., 2006).
A poorly-understood characteristic of the murine antibody response to B. burgdorferi, also reported for humans, is the continued presence of T-independent Borrelia-specific serum IgM, even months after initial infection (Steere, 2001; Liu et al., 2004; Gajovic et al., 2010; Hastey et al., 2020). Given that the half-life of serum IgM is short, on the order of 12 – 48 hours in mice, the continued presence of IgM in persistently-infected mice suggests either production of long-lived IgM secreting plasma cells or the continued de novo formation of plasmablasts. Because anti-Borrelia IgM responses are largely T-independent, the latter appears more plausible but would require the continued presence of Borrelia antigens. Of importance, the ratio of IgM and IgG therefore does not differ significantly between acute and persistent B. burgdorferi infection in mice. Ongoing IgM responses seem to control bacteremia, but do not appear to contribute to control of Borrelia tissue-burden, presumably because this very large molecule (850 kD) cannot gain access to tissues, except in situations of strong inflammation and associated vascular leakage (Blandino and Baumgarth, 2019; Hastey et al., 2020).
As stated above, hypergammaglobulinemia, often associated with chronic and persistent infections, and consisting of both Borrelia-specific and non-specific IgG, was found also in B. burgdorferi-infected mice (Hardin et al., 1979; Schutzer et al., 1990; Brunner and Sigal, 2001; Schutzer and Luan, 2003; Soulas et al., 2005). The mechanisms underlying the development of hypergammaglobulinemia, and the role this plays in effective immunity to B. burgdorferi is unknown. Studies on a chronic viral infection model in mice, the pathogen LCMV, suggested that hypergammaglobulinemia and strong production of self-reactive antibodies is dependent on CD4 T cell activation, and that this strong T cell induction paradoxically suppresses effective neutralizing antiviral antibody responses (Recher et al., 2004). Whether a similar effect might reduce the effectiveness of anti-Borrelia responses has not been tested.
B cell activation during B. burgdorferi infection
Following B. burgdorferi infection, B cells are activated and lymph nodes dramatically increase in size and cellularity, with much of the increase contributed by the accumulation of B cells (Sigal et al., 1988; Yang et al., 1992; Tunev et al., 2011). In contrast, B cell activation and induction of B. burgdorferi-specific antibody-secreting cells in the spleen are rarely observed in response to tick-borne infections or infection with host-adapted spirochetes via tissue-transplantation (Tunev et al., 2011). However, activation of marginal zone B cells resulting in secretion of IgM was reported in response to infection after intradermal inoculation of cultured B. burgdorferi, presumably in response to blood-borne spirochetes (Belperron et al., 2007; Malkiel et al., 2009). The extensive B cell accumulation observed in the lymph nodes was shown to depend on infection-induced, B cell extrinsic Type I IFN signaling, likely affecting lymph node stroma (Hastey et al., 2014). Co-cultures of B cells with culture-grown B. burgdorferi causes mitogenic B cell activation dependent largely on TLR2-mediated signals. However, the observed robust antibody response to B. burgdorferi infection of mice lacking either TLR2 or the adaptor protein MyD88, indicated that B cell activation and differentiation in vivo is not driven, or not driven solely, by these innate signals (Woods et al., 2008). Recent studies using transgenic mice expressing B cells specific for the hen egg lysozyme (HEL) demonstrated that HEL-reactive, Borrelia non-specific B cells are activated in vivo following infection (Soulas et al., 2005; Jung et al., 2016). These cells secreted IgM and could undergo class-switch recombination to IgG; however, the IgG-expressing cells were unable to differentiate into antibody-secreting cells. Thus, increases in transgene-induced, non-Borrelia-reactive antibodies following B. burgdorferi infection were largely of the IgM isotype, not considered to be pathogenic, and the HEL-specific B cells producing these antibodies contributed to the pool of activated B cells after infection (Soulas et al., 2005; Jung et al., 2016). This is consistent with another in vivo study, which concluded that much of the strong infection-induced IgG response in lymph nodes of B. burgdorferi-infected mice is Borrelia-specific, and, thus, the outcome of specific antigen-B cell interactions (Tunev et al., 2011). Paradoxically, mice lacking MyD88, the adaptor molecule of most TLRs, including TLR2, showed a dramatically enhanced hypergammaglobulinemia, which was shown to be a B cell-extrinsic effect (Woods et al., 2008), possibly triggered by the presence of increasing amounts of Borrelia antigens in these mice.
Some of the differences observed between B cell stimulation in vivo and in vitro may be explained by the extensive differences in Borrelia surface protein expression between culture-grown and mammalian-host adapted spirochetes (Samuels, 2011; Iyer and Schwartz, 2016), including expression of OspA, a mitogen for B cells (de Souza et al., 1992) that is rapidly downregulated after uptake of the bloodmeal and following transmission to the mammalian host (de Silva et al., 1996; Caimano et al., 2019). Thus, OspA is unlikely to drive early B cell responses in vivo, as it is not significantly expressed following infection of the mammalian host. (Schwan et al., 1995), consistent with a lack of antibody response development to OspA following natural infection or infection with host-adapted spirochetes. Taken together, B. burgdorferi infection typically results in the strong activation of lymph node B cells, including both Borrelia-specific and Borrelia non-specific, possibly self-reactive B cells. Their activation is affected by extrinsic and intrinsic inflammatory signals that act in concert with Borrelia antigens to drive strong B cell responses in the lymph nodes of infected mice. These responses result in strongly enhanced serum concentrations of both IgM and IgG that remain elevated throughout infection (Tran V.G. and Baumgarth, N., unpublished).
T-dependent follicular B cell responses to B. burgdorferi Follicular B cell responses are classified into either extrafollicular or germinal center (GC) B cell responses. Extrafollicular responses result in the rapid formation of foci containing short-lived plasmablasts in the T-B border and the medullary cord area of lymph nodes, and can be induced in either a T-dependent or T-independent manner. In contrast, GC responses are dependent on cognate CD4 T cell help from T follicular helper cells (TFH). GC responses are required for antibody somatic hyper-affinity maturation and result in the development of antibody-secreting long-lived plasma cells and recirculating memory B cells.
Despite the appreciated need for CD4 T cell help in the induction of strong B cell responses, antibodies that arise after B. burgdorferi infection in the absence of T cell help can support the resolution of arthritis and can, after passive transfer, protect mice from challenge infections (Fikrig et al., 1996; McKisic and Barthold, 2000). There are two possible explanations for these puzzling findings: First, most if not all T cell responses to B. burgdorferi are initiated in a T-independent manner through formation of extrafollicular foci; or second, CD4 T cell help is insufficient to enhance the functionality of the humoral response to B. burgdorferi. Support for induction of largely T-independent responses to B. burgdorferi was provided with studies in mice lacking CD40L, a critical co-stimulatory molecule expressed by T cells that induces Ig class-switch recombination and B cell activation (Fikrig et al., 1996). Antibodies generated to B. burgdorferi infection in these mice retained their ability to protect mice from a challenge infection. Similarly, serum from B. burgdorferi-infected mice lacking either all T cells, lacking CD4 T cells, or lacking MHCII, retained passive protective capacity (Fikrig et al., 1996; Fikrig et al., 1997a; McKisic and Barthold, 2000). Furthermore, strong extrafollicular plasmablast responses form in lymph nodes after B. burgdorferi infection (Tunev et al., 2011). However, IgG class-switching, with the exception of switching to IgG2b, was severely reduced in mice deficient in CD40L, MHCII, CD4 or all T cells (Fikrig et al., 1996; Fikrig et al., 1997a; McKisic and Barthold, 2000). Also, B. burgdorferi tissue burden was strongly increased in CD4 T cell-deficient mice (Elsner et al., 2015a); arthritis could not be resolved in SCID mice after passive-serum transfer (Fikrig et al., 1996; McKisic and Barthold, 2000; Elsner et al., 2015a); and carditis-resolution was delayed (Fikrig et al., 1997a). These data do not refute the notion that CD4 T cells are induced and likely important for the humoral response to B. burgdorferi. Rather, the passive protective capacity of antibody responses to B. burgdorferi is achieved even in the absence of IgG class switch recombination and the formation of T cell-dependent B cell responses. Thus, passive protection might not require antibody functionalities associated with antibody-tissue entry, which is needed for resolution of arthritis and carditis, as IgM is passively protective, or strongly hyper-affinity-matured antibodies, a process that requires T cell-dependent GC responses.
Taken together, these findings suggest that CD4 T cell helper responses to B. burgdorferi are insufficient to induce fully functional antibodies able to clear B. burgdorferi infection. In support of that finding, it was shown that functional GC responses do not develop after B. burgdorferi infection in mice (Hastey et al., 2012; Elsner et al., 2015a; Elsner et al., 2015b). Instead, they arise relatively late, around day 15 of infection, and then rapidly collapse by day 30, despite ongoing infection and despite the formation of CXCR5+ PD-1hi Bcl6+ CD4+ TFH (Hastey et al., 2012; Elsner et al., 2015a; Elsner et al., 2015b; Hastey et al., 2016). This lack of sustained GC causes a failure to sustain antibody affinity maturation, and a failure to induce memory B cells and long-lived plasma cells (Hastey et al., 2012; Elsner et al., 2015b). A failure to induce memory B cell responses may explain why, following antibiotic treatment, Borrelia-specific serum antibodies are often lost and, as shown for experimental mice, hosts can become susceptible to reinfection (Piesman et al., 1997; Elsner et al., 2015b). The lack of effective T-dependent B cell response induction may contribute to the inability of the host to clear B. burgdorferi infection. It will be important to determine the mechanisms underlying the inability of B. burgdorferi-infected mice to induce robust T-dependent B cell responses and to assess whether their induction could result in clearance of B. burgdorferi from the mammalian host.
Pre-existing and innate-like B cell immunity to B. burgdorferi
Humoral defense begins prior to deposition of spirochetes within the host, when ticks ingest interstitial fluid containing IgG or hemorrhaged blood, which contains all Ig subclasses, at the feeding site (Belperron and Bockenstedt, 2001). Infected ticks that feed on B cell-deficient mice (B6.Igh6−/−) harbor more spirochetes than those that feed on WT mice. Moreover, passive transfer of normal mouse serum into B cell-deficient mice reduces spirochete burden in feeding ticks. Pre-existing antibodies might be generated through long-lived plasma cells, or the presence of circulating IgG following recent exposure to B. burgdorferi. Even individuals never exposed to B. burgdorferi, however, harbor so-called “natural” antibodies, mostly of the IgM isotype, that exist in all individuals. Natural antibodies are secreted in mice by a distinct fetal-derived B cell subset termed B-1, which have the capacity to generate long-lived antigen-specific immunity in the absence of T cell help (Alugupalli, 2008). They also can reduce early pathogen burden to various pathogens, likely because of their ability to bind structures common to many pathogens, thus functioning similar to innate soluble pattern recognition receptors (Baumgarth et al., 2005; Baumgarth, 2011). The presence of natural B. burgdorferi-binding IgM, generated in the absence of any previous exposure, can complicate IgM-based serodiagnosis for Lyme borreliosis. Natural IgM is sufficient to cause B. burgdorferi reductions in the ticks during blood uptake, whereas IgG antibodies have no effect (Belperron and Bockenstedt, 2001). IgG can pass into the tick haemolymph, where it is bound by immunoglobulin-binding proteins and is excreted in the tick saliva back into the host, a mechanism by which the tick may interrupt the host immune response to tick feeding (Wang and Nuttall, 1999).
T cell responses
In contrast to the B cell’s ability to directly engage with Borrelia antigens, T cells require antigen-presentation by conventional or non-conventional MHC molecules. Conventional T cell activation entails the processing of protein antigens into peptides, followed by peptide loading into MHC I and II complexes, which allows their presentation by antigen-presenting cells (APC), such as dendritic cells, macrophages and B cells, to αβ T cell receptors (TCR) expressed on CD8 and CD4 T cells, respectively. The presentation of peptides by MHC II on B cells, following antigen binding and subsequent internalization by the BCR, is the hallmark of CD4 T-dependent B cell responses and results in the differentiation of CD4 T follicular helper cells (TFH) during B. burgdorferi infection. It also causes the activation of various CD4 and CD8 effector T cells (Teff), and populations of CD4 T regulatory (Treg) cells, the latter considered essential in controlling overshooting inflammation and tissue-damage, which are discussed below.
“Unconventional” antigen presentation involves the presentation of lipids and glycolipids to T cells (Park and Kim, 2018; Godfrey et al., 2019). These types of antigens cannot be presented on MHC I and MHC II, yet are common antigens expressed by B. burgdorferi. These antigens are processed for presentation by non-classical MHC Ib molecules such as CD1d, as well as Qa-I in mice, the ortholog of the non-polymorphic HLA-E of humans, which activate “natural killer” T cells (Park and Kim, 2018). Another non-classical MHC Ib molecule is MR1, which can bind to and present small bacterial metabolites and activates so-called Mucosa-Associated Invariant T cells (Godfrey et al., 2019). While CD1-restricted T cell activation has been shown for B. burgdorferi infection, activation of MAIT cells has not been described.
T cells utilizing the γδ TCR are another type of “unconventional” T cell reported to play a role in B. burgdorferi infections. Identifying the antigens recognized by the γδ TCR remains the subject of ongoing investigations. In contrast to αβ TCR and other non-conventional T cells, γδ TCR antigen-binding appears to be more akin to that of the BCR. About 0.5% of γδ T cells in mice can directly bind to the inducible non-classical MHC Ib molecule T10 and the related T22, which lack an antigen-binding groove (Crowley et al., 2000). Recognition of lipid antigens in CD1 has also been observed (Divan et al., 2015). However, they can also bind directly to protein antigens (phycoerythrin) as well as haptens (Zeng et al., 2012; Chien et al., 2014; D’Souza et al., 2019). A recent study, using a tetramerized human γδ TCR expressed by a T cell clone derived from the synovial fluid of a patient with chronic Lyme arthritis, reported expression of IL-1β-induced cell surface ligand(s) on activated monocytes and T cells, although their exact identity remains to be revealed (Collins et al., 2019).
Responses to B. burgdorferi by nonconventional T cells Antigens presented by CD1d to invariant TCR “natural killer” T (iNKT) cells, and by MR1 to MAIT cells, first demonstrated activation of T cell subsets with highly restricted TCR repertoires and are considered innate-like. The B. burgdorferi-derived glycolipid diacylglycerol (BbGL-II) was one of the first pathogen-derived antigens identified as being presented by CD1d to mouse and human invariant iNKT cells, causing their activation (Kinjo et al., 2006). iNKT cells were shown to be activated in response to the infection and to produce IFNγ (Tupin et al., 2008; Olson et al., 2009). Activation of iNKT cells appeared to be, at least in part, dependent on TLR2 signaling, as TLR2-deficient mice showed reduced accumulation of NKT cells in the arthritic joints, although their frequencies in the heart tissue was unchanged (Lasky et al., 2016). Production of IFNγ by iNKT cells may enhance macrophage-mediated B. burgdorferi uptake and thus could reduce Borrelia tissue burden; it can also drive the upregulation of CD1d as well as alter the chemokine milieu, thereby affecting the recruitment of macrophages and neutrophils (Sabino et al., 2011). However, IFNγ production by iNKT cells was not always observed (Whiteside et al., 2018). Lack of CD1d expression, and thus lack of iNKT cells, or deficiency in the major Vα14-expressing iNKT cell subset, resulted in enhanced arthritis development and increased spirochete tissue burden in mouse strains that are normally resistant to disease (C57BL/6 and BALB/c mice). Increased spirochete burden may underlie the observed increases in serum antibody titers against B. burgdorferi in these mice, including antibodies to the CD1d-ligand BbGLII (Kumar et al., 2000; Tupin et al., 2008). Together, the data suggest a protective role for CD1d-restricted iNKT cells; however, the mechanisms of that protective role remains to be revealed more fully.
In addition to CD1d-restricted invariant NKT cells, also termed “Type I NKT”, more recent studies using tetramerized CD1a, CD1b and CD1c from humans (mice only express CD1d), identified CD1-restricted αβ T cells with a diverse TCR repertoire (dNKT, or Type II NKT). These dNKT recognize antigens, including self-antigens, presented by CD1a, b or c, (Mori et al., 2016; Dhodapkar and Kumar, 2017). B. burgdorferi infection was shown to drive expression of CD1a-c on myeloid cells indirectly by stimulating the TLR2-induced secretion of IL-1β (Yakimchuk et al., 2011) and may drive the activation of CD1b-restricted NKT cells with self-antigen recognition potential in human patients (Reinink et al., 2019). The significance of these cells and their contributions to immunity to B. burgdorferi infection and/or infection-induced disease remain to be established.
T cells expressing the γδ TCR develop from the thymus throughout life, although their development includes early developmental waves that seed various tissues, including the skin, where they may interact with B. burgdorferi shortly after its release from the tick into the mammalian host (Divan et al., 2015). The main focus of studies on γδ T cells in infection has been on lymph tissue of mice and synovial fluid of patients with Lyme arthritis (Divan et al., 2015). Collectively, the results suggest that γδ T cells respond to cell stress and inflammatory responses triggered indirectly through TLR activation, as well as release of pro-inflammatory cytokines, such as IL-1β, that occurs during arthritis development. These signals seem to trigger the upregulation of γδ TCR ligands on numerous cell types, including monocytes, T cells, dendritic cells as well as fibroblasts (Collins et al., 2016; Collins et al., 2019). Similar to findings with CD1d-restricted iNKT cells, γδ T cells may contribute to production of IFNγ in tissues during B. burgdorferi infection (Bockenstedt et al., 2003).
Conventional T cell responses
The contributions of αβ T cells to host defense during B. burgdorferi infection remains incompletely defined. Studies with mice lacking all T cells (TCR β/δ −/−) or only αβ TCR-expressing cells, or with mice depleted of CD4 or CD8 T cells, demonstrated surprisingly modest effects of T cell-deficiency on the course of B. burgdorferi infection. These effects included some enhancements in spirochete tissue burden and increased arthritis severity (Keane-Myers and Nickell, 1995; Lim et al., 1995; Fikrig et al., 1997a; McKisic and Barthold, 2000; Elsner et al., 2015a). As discussed above, given that CD4 T cells are required for Ig class-switch recombination to most IgG subclasses, at least part of the effect on Borrelia-load can be explained by a failure to induce IgG in these animals. Antibody-independent mechanisms of host defense, however, cannot be ruled out (Keane-Myers and Nickell, 1995). These apparently modest effects of T cells on the course of B. burgdorferi infection are surprising, given the importance of CD4 T cells as crucial effectors and regulators of immune responses (Sakai et al., 2014; Tubo and Jenkins, 2014; Sallusto, 2016), and since effective B cell immunity usually requires T-dependent co-stimulatory signals that support Ig-class switch recombination, affinity-maturation, and the development of B cell memory and long-lived plasma cells. As outlined above, given that many of the features of a T-dependent B cell response are lacking in B. burgdorferi infected mice (Hastey et al., 2012; Hastey et al., 2014; Elsner et al., 2015a; Elsner et al., 2015b), together the data indicate that a fully-functional T cell response fails to develop or is suppressed during the infection, which could contribute to the development of Borrelia persistence.
Effector CD4 T cell responses are shaped by the inflammatory milieu in which they develop. Infections with extracellular pathogens, such as B. burgdorferi, are thought to be controlled by CD4 T cells polarized to express the transcriptional regulator RORγt and secreting the hallmark cytokine IL-17. Through secretion of cytokines, these cells regulate neutrophil accumulation at sites of infection, which then eliminate the pathogen via phagocytosis and enzyme-mediated killing. Exuberant and/or dysregulation of the pro-inflammatory IL-17-driven responses may also cause tissue damage (Chamoun et al., 2018). Remarkably, IL-17-deficiency on either the C57BL/6 or the C3H background did not significant impact B. burgdorferi tissue burden, course of disease, or the magnitude of the anti-B. burgdorferi antibody responses (Lasky et al., 2015a). As discussed in the innate immunity section of this review, the presence of B. burgdorferi in the skin of infected mice does not trigger continued accumulation of neutrophils or other effectors to the site of infection, further suggesting that TH17 responses are not significantly induced during B. burgdorferi infection in this model. However, the interpretation of the data is complicated by the fact that lack IL-17/RORγt can lead to reciprocal increases in T-bet expression and IFNγ production, which may mask some of the functions of IL-17 in B. burgdorferi infection by supporting a TH1-like pro-inflammatory response that contributes to Borrelia clearance.
Early studies suggested that induction of TH1 responses could drive the transient inflammatory pathology seen in mice infected with B. burgdorferi, and that a dominance of TH2 responses attenuated that response (Matyniak and Reiner, 1995). A pro-inflammatory role for IFNγ-producing T-bet+ CD4 TH1 cells in B. burgdorferi infection-induced inflammatory disease was suggested by the strong association between IFNγ−producing CD4 T cells with Lyme arthritis and carditis in mice and humans (Lim et al., 1995; Anguita et al., 1996; Gross et al., 1998b; McKisic et al., 2000). While adoptive transfer of T cells into B. burgdorferi-infected mice deficient in B cells led to significant inflammatory pathology, transfer of both B and T cells resolved inflammation, indicating that B cells are required for disease-ameliorating effects (McKisic et al., 2000). In another study, the transfer of IFNγ−producing CD4 T cells into T cell-deficient mice promoted carditis regression, not inflammation; this may be related to their effects on promoting macrophage-mediated clearance of B. burgdorferi at this site (Bockenstedt et al., 2001). Furthermore, treatment of mice with anti-IL-12 mAb, a cytokine that induces IFNγ production by T cells, reduced arthritis severity in C3H mice, but at the expense of an increased B. burgdorferi tissue-burden (Anguita et al., 1996). Others showed that the frequencies of activated CD44hi CD11ahi cells increased while total CD4 T cell numbers changed little in LN (or spleen) following B. burgdorferi infection (Hastey et al., 2012). No evidence of IFNγ production was found at those sites, despite the fact that B. burgdorferi-infection induces strong IFNβ responses, including in the LN (Miller et al., 2008a; Lochhead et al., 2012; Hastey et al., 2014; Ma et al., 2014). Moreover, C3H IFNγ−/− and DBA IL-4−/− mice developed arthritis of comparable severity and duration to their WT counterparts, as did disease-resistant 129/SvEv mice deficient in the IFNγ receptor, and C57BL/6 and BALB/c mice deficient in IL-4 or the IL-4 receptor alpha chain (Brown and Reiner, 1999; Potter et al., 2000; Glickstein et al., 2001).
Current studies on T cell responses to B. burgdorferi are hampered by inadequate tools to follow antigen-specific T cells. Recent evidence furthermore suggests that a significant proportion of T cells active in lymph nodes after B. burgdorferi infection do not react to Borrelia antigens (Whiteside et al., 2018). Mice deficient in the anti-inflammatory cytokine IL-10 develop robust arthritis on an otherwise disease-resistant background, which is associated with T cell infiltration of the synovium. A significant fraction of activated CD4 and CD8 T cells in the lymph nodes of these mice after B. burgdorferi infection were not induced by Borrelia-antigens but, rather, responded with strong “bystander” activation to inflammatory signals that required T cell-intrinsic TLR2-signaling. These bystander T cells, both CD4 and CD8, upregulated TLR2 in response to infection and secreted IFNγ, which supported inflammatory joint destruction (Whiteside et al., 2018). The data further highlight that control of the inflammatory milieu after B. burgdorferi infection can ameliorate the induction of potentially pathogenic T cell responses. They also highlight the fact that production of IFNγ is a double-edged sword, as it both supports reduction of B. burgdorferi tissue burden, but also drives inflammatory tissue destruction. Similarly, IL-10 is induced following B. burgdorferi infection (Sonderegger et al., 2012a) and appears to be a key regulatory factor that can reduce the amount of IFNγ produced following B. burgdorferi infection, and thereby the level of arthritis that develops in mice (Whiteside et al., 2018). However, as discussed above, greatly enhanced production of IL-10 may reduce macrophage activation and Borrelia clearance, which could cause secondary inflammatory responses due to excessive spirochete loads. In disease-resistant strains of mice, the balance between IFNγ and/or IL-17 and the production of IL-10 may support the failure to clear B. burgdorferi infections in the absence of overt manifestations of disease. In humans, who present with a spectrum of pathological manifestations following B. burgdorferi infections, a negative correlation was demonstrated between the levels of IL-10 produced by in vitro stimulated monocytes and reduced production of TNFα in response to TLR-signaling and disseminated Lyme borreliosis (Kisand et al., 2007).
Anti-inflammatory cytokines, such as IL-10 and TGFβ, are secreted in part by CD4+ CD25+ Foxp3+ Treg cells. The balance of Teff populations with Treg may contribute to the regulation of Lyme arthritis (Sakaguchi, 2004; Belkaid and Rouse, 2005). In support, Treg are found in the synovium of patients with chronic antibiotic-refractory Lyme arthritis, where they were presented in higher numbers compared to control patients with antibiotic-responsive arthritis. Moreover, in vitro inhibitory activity of Treg was reduced when taken from antibiotic-refractory Lyme arthritis compared to Treg isolated from those responding to antibiotic therapy (Shen et al., 2010; Vudattu et al., 2013). Activation of CD4+ Treg is observed also in the lymph nodes of B. burgdorferi-infected mice (Nardelli et al., 2004; Elsner et al., 2015a). Mice depleted of IFNγ and IL-17 developed no arthritis until mice were depleted also of CD25+ T cells (Nardelli et al., 2004). Furthermore, CD28−/− mice, which are deficient in natural Treg (Iliopoulou et al., 2007), had increased incidences, duration and severity of arthritis following B. burgdorferi infection in comparison to control mice. However, CD28−/− mice have impaired humoral immune responses (Shahinian et al., 1993) and lower pathogen-specific antibody titers after infection, which complicates interpretation of results.
Taken together, a diverse array of T cells is induced in response to Borrelia infection in both mice and humans. Production of proinflammatory cytokines such as TNFα and IFNγ supports the reduction in Borrelia tissue loads and inhibits the development of Borrelia-induced pathology, although it does not clear the infection. Conversely, induction of immune-regulatory cytokines, such as IL-10, may ameliorate manifestations of disease, while failing to support Borrelia-clearance. Altering the balance of pro- and anti-inflammatory stimuli to benefit stronger anti-pathogen responses can cause inflammatory disease, arthritis and carditis, either because B. burgdorferi infection is incompletely controlled, or because of the consequences of a dysregulated inflammatory response, as modeled in different strains of mice (Crandall et al., 2006; Ma et al., 2014; Paquette et al., 2017).
Host responses in human Lyme borreliosis
As discussed above, there are remarkable similarities between the immune molecules, cells, and signaling pathways that are engaged by B. burgdorferi and its components in mice and in humans. Without antibiotic treatment, spirochetes can persist in humans despite a panoply of immune responses, but most often these are associated with clinical disease. The outcomes in terms of pathology are quite different in untreated B. burgdorferi infection in mice and humans. Experimentally infected outbred Peromyscus leucopus, a main reservoir host for B. burgdorferi, exhibit no significant gross or microscopic pathology despite the presence of spirochetes in multiple tissues. An exception exists only when infection is introduced at a very young age (3 days), which can result in a transient arthritis (Moody et al., 1994). Inbred laboratory mice (Mus musculus) can exhibit inflammatory lesions in the hearts and joints, the appearance and severity of which are not only age- but also mouse strain-dependent (Barthold et al., 1990). In inbred mice, appearance and resolution of pathology follow the evolution of the immune responses to the spirochete as it multiplies in the skin and disseminates through the blood, lymphatics and tissues to establish foci of infection in other organ systems. The greatest burden of B. burgdorferi is detected during the period of peak disease, consistent with spirochete components driving the acute inflammatory response. Inbred mice also develop chronic inflammation in tissues, characterized by segmental perivascular and perineural infiltrates of lymphocytes and plasma cells that are often in association with visible spirochetes in adjacent tissues, although these pathologic changes do not give rise to clinical manifestations such as those seen in human Lyme borreliosis (Barthold et al., 1993).
B. burgdorferi infection of humans incites a broader range of tissue pathology, most often clinically apparent in the skin (EM, Borrelia lymphocytoma, and ACA), the heart (conduction system abnormalities and myocarditis), the nervous system (especially cranial nerve abnormalities, meningitis/meningo-encephalitis, and radiculoneuropathies) and in the joints (proliferative synovitis) (Steere et al., 2016) (see also Radolf and Samuels, 2021). These manifestations typically follow a characteristic timeline, with skin (except for ACA), heart and nervous system involvement in the first weeks to 1–2 months of infection, and arthritis and ACA occurring later. Clinical signs such as erythema migrans (EM) can remit without specific intervention, and others, like arthritis, can recur in the same or different site. Older studies of tissues from patients with Lyme borreliosis, many of whom had the infection for several years prior to diagnosis, have documented the extent of pathology that can occur in various organs during the course of untreated B. burgdorferi infection (Duray and Steere, 1988). It is important to note that lymphocytes dominate the cellular infiltrates of all human tissues examined by pathology, whereas innate immune cells are more prevalent in the transient inflammatory lesions in mice, with T and B lymphocytes infiltrating mouse organs as disease resolves. While these differences may reflect, in part, the duration of infection at which samples are evaluated for pathology, they also suggest that B. burgdorferi infection of reservoir hosts, such as mice, elicits an immune program much more permissive to silent persistent infection than the immune responses that arise in non-reservoir hosts exemplified by humans.
Host response in cutaneous manifestations of Lyme borreliosis
Experiments in laboratory mice have provided much information about the dynamics of the immune response to B. burgdorferi and its relationship to pathologic changes in the mouse, yet many features of human Lyme borreliosis remain to be explained. By the time patients present with clinical signs, cellular infiltrates in infected tissues reflect an adaptive immune response, with CD4+ T cells dominating the pathology (Steere et al., 2016). Typically, this presentation occurs within the first month of infection when one of the earliest clinical signs of Lyme borreliosis, the skin lesion EM, appears at the tick bite site (average 7–14 days after the bite). Not all humans present with EM, and mice do not develop EM after infection is introduced by tick bite or needle inoculation, even though the skin is a preferred site for B. burgdorferi infection and persistence (Barthold et al., 1992a). The reason for this is unclear, given that EM lesions are comprised of T lymphocytes, DCs, macrophages and rare plasma cells (Boer et al., 2007), and as described earlier, these cell subsets are engaged in mice within the first month of infection, when EM would be expected to appear. In humans, more spirochetes are found at the leading edge of the EM lesion than centrally, and smaller lesions harbor more spirochetes than larger ones (Liveris et al., 2002). As the size of the EM approximates the duration of infection, this suggests that the immune response is effective at reducing bacterial burdens in human skin, just as the bacterial burden at this site in mice declines with time, albeit without evidence of adaptive immune cell infiltration.
Characterization of EM by immunohistochemistry shows patchy perivascular and interstitial infiltrates, the density of which increase with the age of the lesion (Boer et al., 2007). It is presumed that skin-resident innate immune cells respond to the tick bite and B. burgdorferi deposited in the feeding pit as the effects of tick saliva wane, and their activation leads to release of inflammatory mediators that recruit additional immune cells to the infection site. In EM, perivascular infiltrates are dominated by CD4+ and CD8+ lymphocytes whereas interstitial infiltrates are comprised mainly of CD68+ histiocytes (Boer et al., 2007). CD1a+ DCs, which can drive TH1 differentiation, are less commonly found and are mainly perivascular. In one study, denser infiltrates had a greater abundance of B cells with rare CD138+ plasma cells, and this was associated with the age of the lesion (Boer et al., 2007). Flow cytometric analysis of dermal leukocytes extracted from EM lesions, using a blister technique, similarly revealed a predominance of T cells and also monocytes/macrophages and DCs, with occasional neutrophils (Salazar et al., 2003). T cells had an effector phenotype, and DCs had upregulated HLA-DR, CD14, and TLRs 1, 2 and 4 expression, consistent with a mature phenotype capable of presenting antigens to T cells. The dominant cytokines extracted from the lesions by the blister method were IL-6 and IFNγ, and the levels were higher in patients with isolated EM in comparison to those with multiple EM lesions. Responses in the skin are reflected, at least in part, in the blood. Analysis of cytokines and chemokines of patients who present with EM revealed elevation of innate cytokines CCL2, IL-6, IL-10, TNF and IL-1β, as well as the TH1-associated cytokines and chemokines CXCL0, CXCL10, IFNγ, and CCL19. In addition, marked elevated in TH17 cell associated cytokines IL-23, IL-27, IL-25, IL-22, IL17F, IL-21, and IL-17A has been observed (Strle et al., 2017).
The findings above are consistent with a much earlier study comparing expression of select genes in EM lesions versus those expressed in the late skin manifestation ACA (Mullegger et al., 2000). ACA typically occurs months to years after B. burgdorferi infection, most often on a distal extremity, and is associated with B. afzelii infection. Both EM and ACA exhibit mononuclear cell infiltrates, although they are patchy and found throughout the dermis in EM lesions, whereas infiltrates in ACA are denser and concentrated in the upper dermis. Cytokine mRNA expressed within EM lesions, as detected by in situ hybridization, revealed prominence of TNFα and IFNγ, as well as the anti-inflammatory cytokine IL-10, whereas ACA lesions showed only TNFα and IL-4. The differences in the dominant cytokines in EM vs ACA led to the notion that a strong IFNγ-producing T cell response that promoted macrophage activation was important for controlling B. burgdorferi burden and tissue dissemination, and that development of a TH2 response early may set the stage for the later complication of ACA. Whether the initial reaction to the tick bite, which as noted earlier might be modulated by the introduction of tick salivary proteins that can skew immune responses toward a TH2 phenotype, is permissive to ACA has not yet been explored. Transcriptomic analyses of RNA isolated from EM biopsies supported earlier findings of induction of innate and adaptive immune programs that favor TH1 differentiation and phagocyte activation (Marques et al., 2017). Of 254 genes differentially expressed in EM lesions versus healthy control skin, signatures for innate immune responses, cell migration and chemotaxis dominated among genes upregulated, whereas a prominent signature for tissue development and remodeling was found in those downregulated. In particular, two dominant clusters of induced genes were found: one with Type I and Type II IFN signaling, along with T cell chemoattractants CXCL9, CXCL10, and CXCL11; and the other related to innate immune cell functions, including phagosome formation, FcγR and TLRs 1 and 2. B cell chemoattractants, IFNγ, and genes involved in tryptophan catabolism, including the enzyme IDO1 (indoleamine 2,3-dioxygenase 1) were also found. Induction of CXCL9 and CXCL10 is consistent with the dominance of IFNγ−producing CD4+ TH1 cells in EM lesions that promote phagocyte activation, whereas CXCL11 promotes Treg formation. Phagocytic DC ingestion of spirochetes can lead to the induction of Type I IFNs in vitro, and this could drive the differentiation of IDO1-producing DCs that induce Treg cells, which can be found by immunohistochemistry in EM lesions. In addition to induction of Treg cells, tryptophan depletion can lead to suppression of the priming of CD8+ T cells, which are found in abundance in EM lesions. Taken together, these results suggest that the balance between proinflammatory responses and Treg cell induction may influence how permissive the host is to spirochete dissemination and ultimately to late complications of Lyme borreliosis.
Host response in the heart
Clinical involvement of the heart is rare in humans with Lyme borreliosis and most often occurs during the early, disseminated phase of B. burgdorferi infection. As a consequence, few studies have examined cardiac tissue specimens in Lyme carditis, which have largely been obtained either from endomyocardial biopsies or postmortem (Cary et al., 1990; Reimers et al., 1993; Tavora et al., 2008; Muehlenbachs et al., 2016). A recent autopsy case series evaluated the hearts of 5 patients who died of sudden cardiac death due to acute Lyme borreliosis (Muehlenbachs et al., 2016).The findings revealed significant inflammatory infiltrates in the atrioventricular and sinoatrial nodes, consistent with the clinical abnormalities typical of Lyme carditis, namely conduction system disease. There were also interstitial lymphoplasmacytic infiltrates in the myocardium in a curvilinear pattern with perivascular distribution extending from the endocardium to the epicardium, with infiltrates also involving the epicardial adipose tissue. Coronary arteries were not involved, nor was their evidence of vasculitis. Cellular infiltrates were predominantly T cells, histiocytes, and plasma cells, without significant recruitment of neutrophils or eosinophils. CD3+ T cells generally were in greater abundance than CD79a+ B cells, but lymphoid follicles were not observed. Cardiomyocyte complement components C4a and C9 were not detected. Cardiac tissue had increased collagen content and decorin was detected where collagen was present. These findings differ from the transient carditis seen in B. burgdorferi-infected mice, which arises in the absence of adaptive immune cells. In mice, inflammation is dominated by macrophages primarily located at the heart base, with T and B cells comprising less than 5% of the infiltrates (Ruderman et al., 1995). In all of the autopsy cases, spirochetes were detected in the heart tissues by Warthin-Starry stain and immunohistochemistry along the collagen fibers, consistent with their presence driving the inflammatory response. Despite involvement of the heart indicating disseminated B. burgdorferi infection, the only other site where spirochetes were found by immunohistochemistry was in the leptomeninges (2 patients) where they also were associated with mild perivascular inflammation. Spirochetes were not detected visually in other tissues available for analysis (lung, liver, kidney, spleen, prostate, nonlesional skin, and various soft tissues), including several that express decorin. It has been proposed from mouse studies that B. burgdorferi expression of DbpA is required for heart involvement and that absence of decorin diminishes the spirochete’s ability to colonize this site (Brown et al., 2001; Lin et al., 2014). The results of autopsy studies demonstrate that in humans, tissue expression of decorin is not sufficient to allow for spirochete colonization of a site, and the development of pathology leading to Lyme carditis suggests B. burgdorferi has a tropism for infection at this site.
Host response in neuroborreliosis
Neurologic disease is a third example of how mice and humans differ in the pathology that arises after B. burgdorferi infection. Early studies revealed that while spirochetes could be visualized in the mouse perineurium, there was no associated inflammation. They also could only be cultured from mouse brains early in infection, during periods when blood cultures were also positive (Barthold et al., 1992a). More recent immunohistochemical studies, however, revealed that while B. burgdorferi did not involve the brain parenchyma in mice, it was detected in the meninges, most often intra- and peri-vascular, in disseminated infection (Divan et al., 2018). In humans, Lyme neuroborreliosis can manifest as peripheral or central nervous system involvement, with meningoencephalitis, cranial nerve palsies and peripheral radiculoneuropathies most commonly seen (Halperin et al., 1988; Pachner and Steiner, 2007). A characteristic feature of Lyme neuroborreliosis is the immune cell composition of cerebrospinal fluid (CSF), which is notable for a lymphocytic infiltrate with elevated protein levels and antibodies to B. burgdorferi. Studies conducted primarily in Europe, where Lyme neuroborreliosis is more prevalent, have partially characterized the immune response in the CSF. Induction of matrix metalloproteases (MMPs) may permit B. burgdorferi invasion into neural tissue, and production of chemokines, including CXCL11 and CXCL13, contributes to cell recruitment. The CSF immune cells in Lyme neuroborreliosis are comprised of >60%T cells and ~15% B cells, with NK cells and monocytes representing a much smaller fraction of the cellular response (Kowarik et al., 2014). The relatively high frequency of B cells, which is much higher than in other bacterial or viral CNS diseases, may be explained by the fact that B. burgdorferi can induce human monocytes to produce the B cell chemoattractant CXCL13 in vitro, and monocytes with increased expression of CXCL13 are found in the CSF (Narayan et al., 2005; Rupprecht et al., 2007). B. burgdorferi-specific antibodies to a number of Borrelia antigens can be detected in the CSF. An elevated ratio of pathogen-specific IgG in the CSF compared to the peripheral blood is indicative of local antibody production. C1q, C4, C3 and C3a levels were elevated in the CSF, without associated findings in the plasma, suggesting that the inflammatory response is localized to the CSF (Rupprecht et al., 2007). In early neuroborreliosis, CD8+ T cells were the predominant T cells in CSF, which then gave way to a dominant CD4+ TH1 type response (Jacobsen et al., 2003; Lunemann et al., 2007). Analysis of the TH1 cells isolated from a single US patient with acute neuroborreliosis identified peptides of B. burgdorferi heat shock protein 90, 30S ribosomal protein, and proteins involved with porphrine and lipoprotein biosynthesis as relevant T cell antigens (Lunemann et al., 2007). Interestingly, a number of T cells had specificities for human peptide sequences, including CNPase, a structural protein involved in synaptic transmission, but whether these self-reactive T cells play a role in the immune response or contribute to clinical signs and symptoms of neuroborreliosis remains unknown.
More recently, Eckman and colleagues examined the cytokines and chemokines in US patients with Lyme neuroborreliosis using multiplex assays (Eckman et al., 2018). Their study confirmed the dominant increase in CXCL13 in CSF specimens and also reported elevated levels of the chemokines CXCL12, CXCL10, CXCL8, CXCL1 and eotaxin as well as the cytokines IL-12, IFNγ, IL-1α, IL-6, TNFα, IL-10, IL-1 receptor antagonist (IL-1RA) and granulocyte-monocyte colony stimulating factor (GM-CSF). The expression of many of these cytokines and chemokines can be induced when human microglia (the predominant phagocytic cell of the brain) or astrocytes (the primary glial cell forming the blood-brain barrier) are exposed to B. burgdorferi in vitro (Casselli et al., 2017; Greenmyer et al., 2018). No studies have specifically detailed responses in vitro of non-parenchymal macrophages (resident cells of the meninges) to B. burgdorferi. Transcriptomic studies of astrocytes exposed to B. burgdorferi in vitro also revealed changes in expression of 38 miRNAs, and for about half, expression of their respective gene targets was altered. Interestingly, the miRNAs expressed, which included miR145 and miR146b, were different from those identified as influencing Lyme arthritis, supporting the concept that in humans, B. burgdorferi elicits a tissue – specific immune response.
Host response in Lyme arthritis
Lyme arthritis is considered a late manifestation of Lyme borreliosis, occurring several months (average 6 months) after the onset of infection (Steere et al., 1987). The late appearance of arthritis relative to the onset of infection distinguishes the arthritis seen in B. burgdorferi-infected mice from the affliction in humans. Age plays a role in the clinical presentation, with young children often presenting with an acute mono- or pauciarticular inflammatory arthritis involving the knee or hip that resembles septic arthritis (Willis et al., 2003; Thompson et al., 2009; Baldwin et al., 2016), whereas Lyme arthritis in teens and adults usually presents with significant swelling in a joint without much pain (Steere et al., 2016). Adults often report no history of a clinical syndrome compatible with acute Lyme borreliosis in the year preceding the onset of arthritis. In mice, arthritis is polyarticular and is mediated by PMNs in the joint space and surrounding tissues. Although synovial fluid from Lyme arthritis patients similarly has a predominance of PMNs, synovial tissue is notable for infiltration with CD4+ T cells, macrophages, B cells and plasma cells (Duray and Steere, 1988; Steere and Angelis, 2006). The degree of synovial proliferation is dependent upon the number of episodes of arthritis and the duration of arthritis prior to treatment. In acute arthritis, B. burgdorferi DNA can be detected in the synovial fluid and tissue (Li et al., 2011), consistent with infection driving the host response. In untreated patients, rare spirochetes and globular antigen deposits have been observed in perivascular areas where lymphocytes are also present (Duray and Steere, 1988). The spontaneous remission of an episode of acute arthritis, which can last for only a few days or for several months, is presumed to be immune-mediated, but organisms can persist in the absence of antibiotic therapy and give rise to recurrent episodes of acute arthritis. In one study of patients who did not receive antibiotic therapy for EM, arthritis developed in more than half, but the number of patients with recurrent episodes of arthritis declined by 10–20% each year even in the absence of therapy, suggesting that either the inflammatory response was eventually down-regulated or spirochetes driving that response had been eliminated (Steere et al., 1987).
The late appearance of arthritis raises the question as to where spirochetes reside during the asymptomatic phase before arthritis onset. Two-photon intravital imaging of B. burgdorferi-infected mice has revealed spirochetes adjacent to cartilage and in the tendon insertion sites (entheses) around joints (Bockenstedt et al., 2012) (see also Radolf and Samuels, 2021). Entheses are relatively avascular areas rich in decorin, aggrecan, and other molecules B. burgdorferi is known to bind. Decorin in particular has been shown to be required for the development of B. burgdorferi-induced arthritis in mice (Brown et al., 2001). In both mice and humans, a specialized population of CD3+CD4−CD8−IL-23R+RORγt+ T cells has been identified in entheses that is believed to be involved in the pathogenesis of other forms of inflammatory arthritis through the production of IL-17 (Sherlock et al., 2012; Reinhardt et al., 2016; Cuthbert et al., 2017). These cells express PRRs and respond to local or systemic administration of IL-23. Interestingly, IL-23R+RORγt+ T cells are found at sites associated with mechanical stress, like the enthesis and the aorta, which also are areas associated with B. burgdorferi pathology. For other forms of arthritis in which IL-23R+RORγt+ T cells are believed to play a role, mechanical stress damaging the enthesis is thought to be an initiating event for an episode of joint inflammation. Adjacent synovial tissue cells, including macrophages and synovial intimal fibroblasts that normally contribute to entheseal nourishment and repair, drive the inflammatory response that involves the joint. Whether IL-23R+RORγt+ T cells are involved in the host response in Lyme borreliosis has not been explored.
The initiation of an episode of Lyme arthritis in humans likely involves exposure of synovial innate immune cells expressing TLRs to B. burgdorferi or its components, and the induction of MMPs from synovial cells and other cells within the joint. B. burgdorferi elicits MMPs and aggrecanases from human chondrocytes in vitro and MMP-2, MMP-3, and MMP-9 are found in synovial fluid (Hu et al., 2001; Behera et al., 2006b). As noted above, MMP-9 expression is also linked to the development of B. burgdorferi-induced arthritis in mice (Heilpern et al., 2009). Both αβ- and γδ T cells have been isolated and characterized from synovial fluid and tissues of patients with Lyme arthritis. αβ T cells include TH1, TH17, and TH2 subsets, and the dominance of TH1 cells correlates with larger effusions. Immunohistochemistry reveals upregulation of adhesion molecules (P-selectin, vascular adhesion protein-1, intercellular adhesion molecule-1 and vascular cell adhesion molecule-1) on synovial lining cells, endothelial cells, and cell infiltrates (Akin et al., 2001). Analysis of synovial fluid chemokines have detected macrophage and neutrophil chemoattractants, as well as TH1 chemoattractants CXCL9 and CXCL10, the TH17 cell chemoattractant CCL2, and the B cell chemoattractant CXCL13 (Shin et al., 2007). Both pro-inflammatory (IL-6, TNFα, IL-1β, IFNγ) and anti-inflammatory cytokines (IL-10, IL-5 and IL-4) have been reported (Shin et al., 2007). NapA has been identified as one B. burgdorferi component that drives joint inflammation in Lyme arthritis through the induction of IL-6, IL-1β, and TGFβ, and NapA-specific TH17 cells have been found in synovial fluid. A feature of Lyme arthritis is that in some patients, resolution of joint inflammation may take weeks to a few months after completion of antibiotics. The reason for the slow resolution of arthritis is not clear, although some studies suggest that adjunctive use of intra-articular steroids or oral non-steroidal anti-inflammatory agents may aid resolution. Recently B. burgdorferi peptidoglycan has been detected in the joints of patients with Lyme arthritis both before and after treatment and may provide a stimulus for continued inflammation even after antibiotics have killed viable organisms (Jutras et al., 2019).
Return to homeostasis and immunoregulation
The resolution of inflammation and return to tissue homeostasis after an immune challenge involves timely control of both innate and adaptive immune responses to prevent excessive tissue damage and limit energy consumption that occurs with on-going immune activation (Paludan et al., 2020). A return to a new immune homeostasis after pathogen encounter depends on the degree of perceived threat to the host. In B. burgdorferi reservoir hosts such as mice, that threshold appears to be low as mice exhibit minimal pathology despite persistence of spirochetes. The human encounter with B. burgdorferi, in contrast, results in a range of outcomes, from asymptomatic seroconversion to persistence of inflammatory sequelae after treatment, as for example in post-antibiotic Lyme arthritis (discussed below). These differences in outcomes likely relate to individual variation in host responses to infectious and immune challenges. A full understanding of how variations in human immunity contribute to the expression and outcomes of Lyme borreliosis awaits a more comprehensive understanding of the human immune system, its regulation and relation to other biological systems within the human host.
Regulation of innate and adaptive immune responses begins virtually immediately after their engagement (Vigano et al., 2012; Cui et al., 2014). Examples include the induction of IL-1 receptor antagonist after IL-1 receptor signaling, downregulation of cytokine receptor expression, modulation of signaling through induction of suppressor of cytokine signaling (SOCS), induction of CTLA4 after T cell activation, and actions of Treg cells. In an effort to assess the evolution of the immune response in Lyme borreliosis, Bouquet et al conducted a longitudinal transcriptome analysis of PBMCs isolated from whole blood samples of Lyme borreliosis patients presenting with EM and followed for 6 months after antibiotic treatment (Bouquet et al., 2016). Because a known signature of B. burgdorferi infection is Type I IFN response, a comparison was made between differentially expressed genes in Lyme borreliosis with those found in other infectious and inflammatory conditions, some of which are noted for Type I IFN induction. In the acute phase at study entry, the Lyme borreliosis samples revealed upregulation of gene sets common to influenza infection, another Type I interferon-associated pathogen, as well as sets shared with the Gram positive bacterium Streptococcus pneumoniae bacteremia. These gene sets included significant enrichment for transcripts encoding interferon signaling as well as PRR-associated innate immune responses. The elevation in these genes was protracted and detected even at 3 weeks after antibiotic treatment (~5 weeks after presentation). By 6 months, the number of differentially expressed genes (DEGs) had diminished and the pattern had changed, showing evolution of the response from genes primarily upregulated to those downregulated. At this stage, 60% of the DEGs were shared with those seen in systemic lupus erythematosus, a Type I IFN-associated autoimmune disease, and 30% with rheumatoid arthritis (other infectious diseases were not reported at this time point). The most downregulated gene transcript was EIF2, which encodes for eIF2, a global regulator necessary for the translation initiation of mRNAs and also downregulated in systemic lupus erythematosus. Although the study cohort included patients who had not yet resolved subjective symptoms such as fatigue at the 6 month timepoint, there was no difference in DEGs between those who reported return to their pre-infection clinical baseline and those who did not. Taken together, these findings confirmed a robust inflammatory transcriptome during the early phase of Lyme borreliosis that was sustained for a period after antibiotic therapy for the infection. The significance of a protracted inflammatory signature should be considered in the light of new studies revealing the impact of antibiotics on host immunity. Specifically, antibiotics have global effects on the host microbiome and can alter host immunity acutely in response to an immune challenge, enhancing inflammatory signatures in the blood (Hagan et al., 2019). Moreover, doxycycline (the most common antibiotic used to treat EM) not only inhibits MMPs but also interferes with M2 (anti-inflammatory) macrophage differentiation (He and Marneros, 2014). Both of these effects could lead to prolonged inflammatory signatures in the blood even though patients are clinically improving. The subsequent evolution of the immune response to one that shares some features with a Type I IFN-associated autoimmune disorder may represent engagement of common pathways aimed at reducing innate immune activation during the elimination of cellular and pathogen inflammatory debris and the energy expenditure of excessive activation of adaptive immunity.
Post-antibiotic Lyme arthritis – the importance of immune regulation
About 10% of patients with Lyme arthritis develop chronic synovitis, usually in a single joint, that persists despite antibiotic therapy. In an early study, arthritis often had been present for many months to years in patients prior to antibiotic institution, and even then, joint inflammation generally responded to this intervention (Steere et al., 1987). With time, a second subgroup of patients has been identified in whom arthritis does not resolve within >3 months of completion of antibiotic therapy, and often patients have received multiple antibiotic courses (Steere et al., 2006). Inflammation persists even though B. burgdorferi can no longer be detected in synovial fluid or synovial tissue (Nocton et al., 1994). Development of this “post-infectious” form of Lyme arthritis (now termed “post-antibiotic Lyme arthritis”) is associated with more invasive strains (RST1 type) of B. burgdorferi sensu strico, as well as genetic factors that may predispose to greater intensity and duration of inflammatory responses (Strle et al., 2011; Strle et al., 2012). Patients who develop post-antibiotic Lyme arthritis are more likely than those with antibiotic-responsive arthritis to bear rheumatoid arthritis-associated HLA-DRB1 molecules, especially HLA-DRB1*0401, HLA-DRB1*0101, and HLA-DRB1*0404 alleles. Because of the association of these HLA alleles with rheumatoid arthritis (Steere et al., 2006), much research has focused on autoreactivity in patients with post-antibiotic Lyme arthritis. A search for B. burgdorferi proteins that could incite autoreactivity through molecular mimicry initially identified an OspA peptide (aa163–175) as having some sequence similarity to human LFA-1, but subsequent studies have not supported its relevance to arthritis (Gross et al., 1998a; Trollmo et al., 2001). Linked T and B cell responses to several self-antigens have been identified subsequently, including endothelial cell growth factor (ECGF), MMP-10, apolipoprotein B 100, and annexin A2 (Drouin et al., 2013; Crowley et al., 2015; Crowley et al., 2016). Although antibodies to these proteins can be found in patients who resolve arthritis, they tend to be higher in those whose arthritis does not resolve.
These antibodies to self-antigens are generally of the IgG2 and IgG4 subclasses, whereas those directed against B. burgdorferi antigens are usually IgG1 and IgG3 (Sulka et al., 2018). Interestingly, the magnitude of IgG4 to some of these self-antigens correlates with the degree of obliterative vasculopathy and fibrosis characteristic of post-antibiotic Lyme arthritis (Londono et al., 2014; Sulka et al., 2018).
There is increasing evidence that a heightened and dysregulated immune response contributes to the magnitude and duration of Lyme arthritis. A retrospective analysis compared cytokine and chemokine expression in archived synovial fluid samples from patients who resolved arthritis versus those who developed post-antibiotic arthritis (Shin et al., 2007). Samples from patients in whom arthritis persisted after antibiotic therapy had higher levels of TNFα, IL-1β and IFNγ in synovial fluid and increased levels of T cell and monocyte chemoattractants (CXCL9, CXCL10 and CCL4), indicating a more exuberant inflammatory response. Most of the TH1 cytokines and chemokines declined over time in the post-antibiotic Lyme arthritis group once PCR for spirochete DNA became negative, although patients continued to have elevated levels in comparison to controls. Resolution of inflammation may in part be mediated by FoxP3 Treg cells, as post-antibiotic Lyme arthritis has been associated with low numbers of Treg cells in synovial fluid (Shen et al., 2010). The magnitude of the TH17 response has been correlated with the severity of joint inflammation and to the development of autoreactivity that may contribute to persistent synovitis (Strle et al., 2017). Genetic factors that could contribute to more severe inflammation include a SNP of the CD14 gene (CD14–550 C/T), which is related to the levels of soluble CD14 (Inoue et al., 2007); soluble CD14 has been detected at elevated levels in patients with a more severe inflammatory response to B. burgdorferi infection (Lin et al., 2000). In addition, a Toll-like receptor 1 (1805GG) polymorphism has also been associated with heightened TH1 inflammatory responses and is more prevalent among patients with post-antibiotic Lyme arthritis (Strle et al., 2012). Certain microRNAs that modulate inflammatory gene expression, including miR-155, miR-223 and miR-146a, are elevated in patients who experience persistent synovitis in comparison to those whose arthritis resolves with antibiotic treatment (Lochhead et al., 2017).
A study of the transcriptome of synovium from patients with post-antibiotic Lyme arthritis has shed light on how infection-induced inflammation can evolve to an immune-mediated chronic inflammation after the pathogen is eliminated (Lochhead et al., 2019a). Synovial tissue of patients with post-antibiotic Lyme arthritis exhibits heightened Type I and Type II IFN responses that correlate with the degree of inflammatory cytokine expression at the protein level, especially IFNγ, and are inversely correlated with genes involved in tissue repair. A dominance of T cells that produced IFNγ, including CD4, CD8 and double negative populations, as well as a smaller population of IFNγ-producing NK cells were found. Fibroblast-like synoviocytes, which have been implicated in the pathogenesis of rheumatoid arthritis, were a main source of IFN-inducible genes and when exposed to IFNγ in vitro, became hyper-responsive to B. burgdorferi (Lochhead et al., 2019b). Dysregulation of fibroblast-like synoviocytes may favor the continued recruitment of IFNγ-producing T cells into the synovium and give rise to many of the observed pathologic changes in post-antibiotic-Lyme arthritis. These findings underscore the importance of host regulation of both innate and acquired immune responses in order to return to a new state of tissue homeostasis after pathogen encounter, control and elimination.
Concluding Remarks
From the above, it is apparent that the immune response to B. burgdorferi is both complex and nuanced and involves many cell types, soluble molecules and varies based on the organ system affected by infection with this pathogen. While the immune response to B. burgdorferi reduces and controls spirochete numbers, and prevents or resolves manifestations of infection-induced disease, B. burgdorferi ultimately establishes persistent infection in many vertebrate reservoir hosts, enabling its re-acquisition by tick vectors and transmission to other hosts. Thus, co-evolutionary adaptation allows B. burgdorferi to maintain its essential enzootic cycle while helping the vertebrate host to avoid disease. Persistent infections by extracellular microbes are rare, more often seen following infections of viruses, such as CMV and EBV rather than large and complex bacteria that are constantly exposed to cells of the immune system. The complexity of how the balance between persistence and a disease-free state is achieved would be nearly impossible to maintain across the genetic diversity of the entire spectrum of hosts susceptible to infection with B. burgdorferi. Indeed, most infected vertebrates can either clear B. burgdorferi efficiently or develop some level of inflammatory disease, the latter seen following infections of canine, equine, and human hosts. A better understanding of the mechanisms enabling disease-free persistence of B. burgdorferi versus the mechanisms causing inflammation and disease are critical for enhancing our ability to develop therapeutics that more directly target the mechanisms rather than the manifestations of B. burgdorferi-induced disease. The recent advances in research technologies, including intravital microscopy, single-cell transcriptomics, proteomics and metabolomics, to name but a few, are likely to result in a more nuanced understanding of immune response regulation to infections with B. burgdorferi that may have relevance not only to human Lyme borreliosis but to infection with other Borrelia spp.
Acknowledgements
This work was supported by NIH U19AI089992 and the Harold W. Jockers Professorship to LKB, NIH R01AI121970 to RMW, and DoD TB170139 to NB
References
- Abel AM, Yang C, Thakar MS, and Malarkannan S (2018). Natural Killer Cells: Development, Maturation, and Clinical Utilization. Front Immunol 9, 1869. 10.3389/fimmu.2018.01869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguero-Rosenfeld ME, Nowakowski J, Bittker S, Cooper D, Nadelman RB, and Wormser GP (1996). Evolution of the serologic response to Borrelia burgdorferi in treated patients with culture-confirmed erythema migrans. J Clin Microbiol 34, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguero-Rosenfeld ME, Wang G, Schwartz I, and Wormser GP (2005). Diagnosis of Lyme borreliosis. Clin Microbiol Rev 18, 484–509. 10.1128/CMR.18.3.484-509.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akin E, Aversa J, and Steere AC (2001). Expression of adhesion molecules in synovia of patients with treatment-resistant Lyme arthritis. Infect Immun 69, 1774–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alarcon-Chaidez FJ, Boppana VD, Hagymasi AT, Adler AJ, and Wikel SK (2009). A novel sphingomyelinase-like enzyme in Ixodes scapularis tick saliva drives host CD4 T cells to express IL-4. Parasite Immunol 31, 210–219. 10.1111/j.1365-3024.2009.01095.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexopoulou L, Thomas V, Schnare M, Lobet Y, Anguita J, Schoen RT, Medzhitov R, Fikrig E, and Flavell RA (2002). Hyporesponsiveness to vaccination with Borrelia burgdorferi OspA in humans and in TLR1- and TLR2-deficient mice. Nat Med 8, 878–884. [DOI] [PubMed] [Google Scholar]
- Aliprantis AO, Yang RB, Mark MR, Suggett S, Devaux B, Radolf JD, Klimpel GR, Godowski P, and Zychlinsky A (1999). Cell activation and apoptosis by bacterial lipoproteins through toll-like receptor-2. Science 285, 736–739. [DOI] [PubMed] [Google Scholar]
- Alugupalli KR (2008). A distinct role for B1b lymphocytes in T cell-independent immunity. Curr Top Microbiol Immunol 319, 105–130. 10.1007/978-3-540-73900-5_5 [DOI] [PubMed] [Google Scholar]
- Anguita J, Persing DH, Rincon M, Barthold SW, and Fikrig E (1996). Effect of anti-interleukin 12 treatment on murine Lyme borreliosis. J Clin Invest 97, 1028–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appel MJ, Allan S, Jacobson RH, Lauderdale TL, Chang YF, Shin SJ, Thomford JW, Todhunter RJ, and Summers BA (1993). Experimental Lyme disease in dogs produces arthritis and persistent infection. J Infect Dis 167, 651–664. [DOI] [PubMed] [Google Scholar]
- Baldwin KD, Brusalis CM, Nduaguba AM, and Sankar WN (2016). Predictive Factors for Differentiating Between Septic Arthritis and Lyme Disease of the Knee in Children. J Bone Joint Surg Am 98, 721–728. 10.2106/JBJS.14.01331 [DOI] [PubMed] [Google Scholar]
- Bankhead T, and Chaconas G (2007). The role of VlsE antigenic variation in the Lyme disease spirochete: persistence through a mechanism that differs from other pathogens. Mol Microbiol 65, 1547–1558. https://doi.org/MMI5895 [pii] 10.1111/j.1365-2958.2007.05895.x [DOI] [PubMed] [Google Scholar]
- Barbour AG, Jasinskas A, Kayala MA, Davies DH, Steere AC, Baldi P, and Felgner PL (2008). A genome-wide proteome array reveals a limited set of immunogens in natural infections of humans and white-footed mice with Borrelia burgdorferi. Infect Immun 76, 3374–3389. https://doi.org/IAI.00048-08 [pii] 10.1128/IAI.00048-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barthold SW, Beck DS, Hansen GM, Terwilliger GA, and Moody KD (1990). Lyme borreliosis in selected strains and ages of laboratory mice. J Infect Dis 162, 133–138. [DOI] [PubMed] [Google Scholar]
- Barthold SW, and de Souza M (1995). Exacerbation of Lyme arthritis in beige mice. J Infect Dis 172, 778–784. [DOI] [PubMed] [Google Scholar]
- Barthold SW, de Souza M, Fikrig E, and Persing DH (1992a). Lyme borreliosis in the laboratory mouse. In Lyme Disease: Molecular and Immunologic Approaches, Schutzer SE, ed. (Plainview (New York): Cold Spring Harbor Laboratory Press; ), pp. 223–242. [Google Scholar]
- Barthold SW, de Souza MS, Janotka JL, Smith AL, and Persing DH (1993). Chronic Lyme borreliosis in the laboratory mouse. Am J Pathol 143, 959–971. [PMC free article] [PubMed] [Google Scholar]
- Barthold SW, Feng S, Bockenstedt LK, Fikrig E, and Feen K (1997). Protective and arthritis-resolving activity in sera of mice infected with Borrelia burgdorferi. Clin Infect Dis 25 Suppl 1, S9–17. [DOI] [PubMed] [Google Scholar]
- Barthold SW, Persing DH, Armstrong AL, and Peeples RA (1991). Kinetics of Borrelia burgdorferi dissemination and evolution of disease after intradermal inoculation of mice. Am J Pathol 139, 263–273. [PMC free article] [PubMed] [Google Scholar]
- Barthold SW, Sidman CL, and Smith AL (1992b). Lyme borreliosis in genetically resistant and susceptible mice with severe combined immunodeficiency. Am J Trop Med Hyg 47, 605–613. [DOI] [PubMed] [Google Scholar]
- Baum E, Hue F, and Barbour AG (2012). Experimental infections of the reservoir species Peromyscus leucopus with diverse strains of Borrelia burgdorferi, a Lyme disease agent. mBio 3, e00434–00412. 10.1128/mBio.00434-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgarth N (2011). The double life of a B-1 cell: self-reactivity selects for protective effector functions. Nat Rev Immunol 11, 34–46. 10.1038/nri2901 [DOI] [PubMed] [Google Scholar]
- Baumgarth N, Tung JW, and Herzenberg LA (2005). Inherent specificities in natural antibodies: a key to immune defense against pathogen invasion. Springer Semin Immunopathol 26, 347–362. 10.1007/s00281-004-0182-2 [DOI] [PubMed] [Google Scholar]
- Beck G, Benach JL, and Habicht GS (1990). Isolation, preliminary chemical characterization, and biological activity of Borrelia burgdorferi peptidoglycan. Biochem Biophys Res Commun 167, 89–95. 10.1016/0006-291x(90)91734-a [DOI] [PubMed] [Google Scholar]
- Behera AK, Durand E, Cugini C, Antonara S, Bourassa L, Hildebrand E, Hu LT, and Coburn J (2008). Borrelia burgdorferi BBB07 interaction with integrin alpha3beta1 stimulates production of pro-inflammatory mediators in primary human chondrocytes. Cell Microbiol 10, 320–331. 10.1111/j.1462-5822.2007.01043.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behera AK, Hildebrand E, Bronson RT, Perides G, Uematsu S, Akira S, and Hu LT (2006a). MyD88 deficiency results in tissue-specific changes in cytokine induction and inflammation in interleukin-18-independent mice infected with Borrelia burgdorferi. Infect Immun 74, 1462–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behera AK, Hildebrand E, Szafranski J, Hung HH, Grodzinsky AJ, Lafyatis R, Koch AE, Kalish R, Perides G, Steere AC, et al. (2006b). Role of aggrecanase 1 in Lyme arthritis. Arthritis Rheum 54, 3319–3329. [DOI] [PubMed] [Google Scholar]
- Behera AK, Hildebrand E, Uematsu S, Akira S, Coburn J, and Hu LT (2006c). Identification of a TLR-independent pathway for Borrelia burgdorferi-induced expression of matrix metalloproteinases and inflammatory mediators through binding to integrin alpha 3 beta 1. J Immunol 177, 657–664. [DOI] [PubMed] [Google Scholar]
- Belkaid Y, and Rouse BT (2005). Natural regulatory T cells in infectious disease. Nat Immunol 6, 353–360. [DOI] [PubMed] [Google Scholar]
- Belperron AA, and Bockenstedt LK (2001). Natural antibody affects survival of the spirochete Borrelia burgdorferi within feeding ticks. Infect Immun 69, 6456–6462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belperron AA, Dailey CM, Booth CJ, and Bockenstedt LK (2007). Marginal zone B-cell depletion impairs murine host defense against Borrelia burgdorferi infection. Infect Immun 75, 3354–3360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benach JL, Fleit HB, Habicht GS, Coleman JL, Bosler EM, and Lane BP (1984a). Interactions of phagocytes with the Lyme disease spirochete: role of the Fc receptor. J Infect Dis 150, 497–507. [DOI] [PubMed] [Google Scholar]
- Benach JL, Habicht GS, Gocinski BL, and Coleman JL (1984b). Phagocytic cell responses to in vivo and in vitro exposure to the Lyme disease spirochete. Yale J Biol Med 57, 599–605. [PMC free article] [PubMed] [Google Scholar]
- Benhnia MR, Wroblewski D, Akhtar MN, Patel RA, Lavezzi W, Gangloff SC, Goyert SM, Caimano MJ, Radolf JD, and Sellati TJ (2005). Signaling through CD14 attenuates the inflammatory response to Borrelia burgdorferi, the agent of Lyme disease. J Immunol 174, 1539–1548. [DOI] [PubMed] [Google Scholar]
- Bernard Q, Wang Z, Di Nardo A, and Boulanger N (2017). Interaction of primary mast cells with Borrelia burgdorferi (sensu stricto): role in transmission and dissemination in C57BL/6 mice. Parasit Vectors 10, 313. 10.1186/s13071-017-2243-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardino AL, Myers TA, Alvarez X, Hasegawa A, and Philipp MT (2008). Toll-like receptors: insights into their possible role in the pathogenesis of lyme neuroborreliosis. Infect Immun 76, 4385–4395. 10.1128/IAI.00394-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaho VA, Zhang Y, Hughes-Hanks JM, and Brown CR (2011). 5-Lipoxygenase-deficient mice infected with Borrelia burgdorferi develop persistent arthritis. J Immunol 186, 3076–3084. 10.4049/jimmunol.1003473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blandino R, and Baumgarth N (2019). Secreted IgM: New tricks for an old molecule. J Leukoc Biol 106, 1021–1034. 10.1002/jlb.3ri0519-161r [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum LK, Adamska JZ, Martin DS, Rebman AW, Elliott SE, Cao RRL, Embers ME, Aucott JN, Soloski MJ, and Robinson WH (2018). Robust B Cell Responses Predict Rapid Resolution of Lyme Disease. Front Immunol 9, 1634. 10.3389/fimmu.2018.01634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bockenstedt LK, Barthold S, Deponte K, Marcantonio N, and Kantor FS (1993). Borrelia burgdorferi infection and immunity in mice deficient in the fifth component of complement. Infect Immun 61, 2104–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bockenstedt LK, Gonzalez D, Mao J, Li M, Belperron AA, and Haberman A (2014). What ticks do under your skin: two-photon intravital imaging of Ixodes scapularis feeding in the presence of the Lyme disease spirochete. Yale J Biol Med 87, 3–13. [PMC free article] [PubMed] [Google Scholar]
- Bockenstedt LK, Gonzalez DG, Haberman AM, and Belperron AA (2012). Spirochete antigens persist near cartilage after murine Lyme borreliosis therapy. J Clin Invest 122, 2652–2660. 10.1172/JCI58813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bockenstedt LK, Hodzic E, Feng S, Bourrel KW, de Silva A, Montgomery RR, Fikrig E, Radolf JD, and Barthold SW (1997). Borrelia burgdorferi strain-specific Osp C-mediated immunity in mice. Infect Immun 65, 4661–4667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bockenstedt LK, Kang I, Chang C, Persing D, Hayday A, and Barthold SW (2001). CD4+ T helper 1 cells facilitate regression of murine Lyme carditis. Infect Immun 69, 5264–5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bockenstedt LK, Shanafelt MC, Belperron A, Mao J, and Barthold SW (2003). Humoral immunity reflects altered T helper cell bias in Borrelia burgdorferi-infected gamma delta T-cell-deficient mice. Infect Immun 71, 2938–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boer A, Bresch M, Dayrit J, and Falk TM (2007). Erythema migrans: a reassessment of diagnostic criteria for early cutaneous manifestations of borreliosis with particular emphasis on clonality investigations. Br J Dermatol 156, 1263–1271. [DOI] [PubMed] [Google Scholar]
- Boggemeyer E, Stehle T, Schaible UE, Hahne M, Vestweber D, and Simon MM (1994). Borrelia burgdorferi upregulates the adhesion molecules E-selectin, P-selectin, ICAM-1 and VCAM-1 on mouse endothelioma cells in vitro. Cell Adhes Commun 2, 145–157. [DOI] [PubMed] [Google Scholar]
- Bolz DD, Sundsbak RS, Ma Y, Akira S, Kirschning CJ, Zachary JF, Weis JH, and Weis JJ (2004). MyD88 plays a unique role in host defense but not arthritis development in Lyme disease. J Immunol 173, 2003–2010. [DOI] [PubMed] [Google Scholar]
- Bouquet J, Soloski MJ, Swei A, Cheadle C, Federman S, Billaud JN, Rebman AW, Kabre B, Halpert R, Boorgula M, et al. (2016). Longitudinal Transcriptome Analysis Reveals a Sustained Differential Gene Expression Signature in Patients Treated for Acute Lyme Disease. mBio 7, e00100–00116. 10.1128/mBio.00100-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramwell KK, Ma Y, Weis JH, Chen X, Zachary JF, Teuscher C, and Weis JJ (2014). Lysosomal beta-glucuronidase regulates Lyme and rheumatoid arthritis severity. J Clin Invest 124, 311–320. https://doi.org/72339 [pii] 10.1172/JCI72339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CR, Blaho VA, and Loiacono CM (2003). Susceptibility to experimental Lyme arthritis correlates with KC and monocyte chemoattractant protein-1 production in joints and requires neutrophil recruitment via CXCR2. J Immunol 171, 893–901. [DOI] [PubMed] [Google Scholar]
- Brown CR, Lai AY, Callen ST, Blaho VA, Hughes JM, and Mitchell WJ (2008). Adenoviral delivery of interleukin-10 fails to attenuate experimental Lyme disease. Infect Immun 76, 5500–5507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CR, and Reiner SL (1998a). Activation of natural killer cells in arthritis-susceptible but not arthritis-resistant mouse strains following Borrelia burgdorferi Infection. Infect Immun 66, 5208–5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CR, and Reiner SL (1998b). Clearance of Borrelia burgdorferi may not be required for resistance to experimental Lyme arthritis. Infect Immun 66, 2065–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CR, and Reiner SL (1999). Experimental Lyme arthritis in the absence of interleukin-4 or gamma interferon. Infect Immun 67, 3329–3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown EL, Wooten RM, Johnson BJ, Iozzo RV, Smith A, Dolan MC, Guo BP, Weis JJ, and Hook M (2001). Resistance to Lyme disease in decorin-deficient mice. J Clin Invest 107, 845–852. 10.1172/JCI11692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JP, Zachary JF, Teuscher C, Weis JJ, and Wooten RM (1999). Dual role of interleukin-10 in murine Lyme disease: Regulation of arthritis severity and host defense. Infect Immun 67, 5142–5150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner M, and Sigal LH (2001). Use of serum immune complexes in a new test that accurately confirms early Lyme disease and active infection with Borrelia burgdorferi. J Clin Microbiol 39, 3213–3221. 10.1128/jcm.39.9.3213-3221.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabral ES, Gelderblom H, Hornung RL, Munson PJ, Martin R, and Marques AR (2006). Borrelia burgdorferi lipoprotein-mediated TLR2 stimulation causes the down-regulation of TLR5 in human monocytes. J Infect Dis 193, 849–859. 10.1086/500467 [DOI] [PubMed] [Google Scholar]
- Caimano MJ, Groshong AM, Belperron A, Mao J, Hawley KL, Luthra A, Graham DE, Earnhart CG, Marconi RT, Bockenstedt LK, et al. (2019). The RpoS Gatekeeper in Borrelia burgdorferi: An Invariant Regulatory Scheme That Promotes Spirochete Persistence in Reservoir Hosts and Niche Diversity. Front Microbiol 10, 1923. 10.3389/fmicb.2019.01923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreras-González A, Barriales D, Palacios A, Montesinos-Robledo M, Navasa N, Azkargorta M, Peña-Cearra A, Tomás-Cortázar J, Escobes I, Pascual-Itoiz MA, et al. (2019). Regulation of macrophage activity by surface receptors contained within Borrelia burgdorferi-enriched phagosomal fractions. PLoS Pathog 15, e1008163. 10.1371/journal.ppat.1008163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll JA, Garon CF, and Schwan TG (1999). Effects of environmental pH on membrane proteins in Borrelia burgdorferi. Infect Immun 67, 3181–3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cary NR, Fox B, Wright DJ, Cutler SJ, Shapiro LM, and Grace AA (1990). Fatal Lyme carditis and endodermal heterotopia of the atrioventricular node. Postgrad Med J 66, 134–136. 10.1136/pgmj.66.772.134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casselli T, Qureshi H, Peterson E, Perley D, Blake E, Jokinen B, Abbas A, Nechaev S, Watt JA, Dhasarathy A, et al. (2017). MicroRNA and mRNA Transcriptome Profiling in Primary Human Astrocytes Infected with Borrelia burgdorferi. PLoS One 12, e0170961. 10.1371/journal.pone.0170961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassiani-Ingoni R, Cabral ES, Lunemann JD, Garza Z, Magnus T, Gelderblom H, Munson PJ, Marques A, and Martin R (2006). Borrelia burgdorferi Induces TLR1 and TLR2 in human microglia and peripheral blood monocytes but differentially regulates HLA-class II expression. J Neuropathol Exp Neurol 65, 540–548. 10.1097/00005072-200606000-00002 [DOI] [PubMed] [Google Scholar]
- Cervantes JL, Dunham-Ems SM, La Vake CJ, Petzke MM, Sahay B, Sellati TJ, Radolf JD, and Salazar JC (2011). Phagosomal signaling by Borrelia burgdorferi in human monocytes involves Toll-like receptor (TLR) 2 and TLR8 cooperativity and TLR8-mediated induction of IFN-beta. Proc Natl Acad Sci U S A 108, 3683–3688. 10.1073/pnas.1013776108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervantes JL, Hawley KL, Benjamin SJ, Weinerman B, Luu SM, and Salazar JC (2014). Phagosomal TLR signaling upon Borrelia burgdorferi infection. Front Cell Infect Microbiol 4, 55. 10.3389/fcimb.2014.00055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervantes JL, La Vake CJ, Weinerman B, Luu S, O’Connell C, Verardi PH, and Salazar JC (2013). Human TLR8 is activated upon recognition of Borrelia burgdorferi RNA in the phagosome of human monocytes. J Leukoc Biol 94, 1231–1241. 10.1189/jlb.0413206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamoun MN, Blumenthal A, Sullivan MJ, Schembri MA, and Ulett GC (2018). Bacterial pathogenesis and interleukin-17: interconnecting mechanisms of immune regulation, host genetics, and microbial virulence that influence severity of infection. Crit Rev Microbiol 44, 465–486. 10.1080/1040841X.2018.1426556 [DOI] [PubMed] [Google Scholar]
- Charon NW, and Goldstein SF (2002). Genetics of motility and chemotaxis of a fascinating group of bacteria: the spirochetes. Annu Rev Genet 36, 47–73. 10.1146/annurev.genet.36.041602.134359 041602.134359 [pii] [DOI] [PubMed] [Google Scholar]
- Chauhan VS, Sterka DG Jr., Furr SR, Young AB, and Marriott I (2009). NOD2 plays an important role in the inflammatory responses of microglia and astrocytes to bacterial CNS pathogens. Glia 57, 414–423. 10.1002/glia.20770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien YH, Meyer C, and Bonneville M (2014). Gammadelta T cells: first line of defense and beyond. Annu Rev Immunol 32, 121–155. 10.1146/annurev-immunol-032713-120216 [DOI] [PubMed] [Google Scholar]
- Chung Y, Zhang N, and Wooten RM (2013). Borrelia burgdorferi elicited-IL-10 suppresses the production of inflammatory mediators, phagocytosis, and expression of co-stimulatory receptors by murine macrophages and/or dendritic cells. PLoS One 8, e84980. 10.1371/journal.pone.0084980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cinco M, Murgia R, Perticarari S, and Presani G (1998). Surface receptors of neutrophils towards B. burgdorferi. Wien Klin Wochenschr 110, 866–869. [PubMed] [Google Scholar]
- Cinco M, Murgia R, Presani G, and Perticarari S (1997). Integrin CR3 mediates the binding of nonspecifically opsonized Borrelia burgdorferi to human phagocytes and mammalian cells. Infect Immun 65, 4784–4789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman AS, Rossmann E, Yang X, Song H, Lamichhane CM, Iyer R, Schwartz I, and Pal U (2011). BBK07 immunodominant peptides as serodiagnostic markers of Lyme disease. Clin Vaccine Immunol 18, 406–413. 10.1128/CVI.00461-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman JL, Sellati TJ, Testa JE, Kew RR, Furie MB, and Benach JL (1995). Borrelia burgdorferi binds plasminogen, resulting in enhanced penetration of endothelial monolayers. Infect Immun 63, 2478–2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins C, Lui Y, Santos AM, Ballif BA, Gogerly-Moragoda AM, Brouwer H, Ross R, Balagurunathan K, Sharma S, Wright GJ, et al. (2019). Detection of Cell Surface Ligands for Human Synovial gammadelta T Cells. J Immunol 203, 2369–2376. 10.4049/jimmunol.1900451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins CC, Bashant K, Erikson C, Thwe PM, Fortner KA, Wang H, Morita CT, and Budd RC (2016). Necroptosis of Dendritic Cells Promotes Activation of gammadelta T Cells. J Innate Immun 8, 479–492. 10.1159/000446498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comstock LE, and Thomas DD (1991). Characterization of Borrelia burgdorferi invasion of cultured endothelial cells. Microb Pathog 10, 137–148. [DOI] [PubMed] [Google Scholar]
- Coutte L, Botkin DJ, Gao L, and Norris SJ (2009). Detailed analysis of sequence changes occurring during vlsE antigenic variation in the mouse model of Borrelia burgdorferi infection. PLoS Pathog 5, e1000293. 10.1371/journal.ppat.1000293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couvreur B, Beaufays J, Charon C, Lahaye K, Gensale F, Denis V, Charloteaux B, Decrem Y, Prevot PP, Brossard M, et al. (2008). Variability and action mechanism of a family of anticomplement proteins in Ixodes ricinus. PLoS One 3, e1400. 10.1371/journal.pone.0001400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craft JE, Fischer DK, Shimamoto GT, and Steere AC (1986). Antigens of Borrelia burgdorferi recognized during Lyme disease. Appearance of a new immunoglobulin M response and expansion of the immunoglobulin G response late in the illness. J Clin Invest 78, 934–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crandall H, Dunn DM, Ma Y, Wooten RM, Zachary JF, Weis JH, Weiss RB, and Weis JJ (2006). Gene expression profiling reveals unique pathways associated with differential severity of Lyme arthritis. J Immunol 177, 7930–7942. 10.4049/jimmunol.177.11.7930 [DOI] [PubMed] [Google Scholar]
- Crandall H, Ma Y, Dunn DM, Sundsbak RS, Zachary JF, Olofsson P, Holmdahl R, Weis JH, Weiss RB, Teuscher C, et al. (2005). Bb2Bb3 regulation of murine Lyme arthritis is distinct from Ncf1 and independent of the phagocyte nicotinamide adenine dinucleotide phosphate oxidase. Am J Pathol 167, 775–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley JT, Drouin EE, Pianta A, Strle K, Wang Q, Costello CE, and Steere AC (2015). A Highly Expressed Human Protein, Apolipoprotein B-100, Serves as an Autoantigen in a Subgroup of Patients With Lyme Disease. J Infect Dis 212, 1841–1850. 10.1093/infdis/jiv310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley JT, Strle K, Drouin EE, Pianta A, Arvikar SL, Wang Q, Costello CE, and Steere AC (2016). Matrix metalloproteinase-10 is a target of T and B cell responses that correlate with synovial pathology in patients with antibiotic-refractory Lyme arthritis. J Autoimmun 69, 24–37. 10.1016/j.jaut.2016.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley MP, Fahrer AM, Baumgarth N, Hampl J, Gutgemann I, Teyton L, and Chien Y (2000). A population of murine gammadelta T cells that recognize an inducible MHC class Ib molecule. Science 287, 314–316. 10.1126/science.287.5451.314 [DOI] [PubMed] [Google Scholar]
- Cruz AR, Moore MW, La Vake CJ, Eggers CH, Salazar JC, and Radolf JD (2008). Phagocytosis of Borrelia burgdorferi, the Lyme disease spirochete, potentiates innate immune activation and induces apoptosis in human monocytes. Infect Immun 76, 56–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J, Chen Y, Wang HY, and Wang RF (2014). Mechanisms and pathways of innate immune activation and regulation in health and cancer. Hum Vaccin Immunother 10, 3270–3285. 10.4161/21645515.2014.979640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuthbert RJ, Fragkakis EM, Dunsmuir R, Li Z, Coles M, Marzo-Ortega H, Giannoudis PV, Jones E, El-Sherbiny YM, and McGonagle D (2017). Brief Report: Group 3 Innate Lymphoid Cells in Human Enthesis. Arthritis Rheumatol 69, 1816–1822. 10.1002/art.40150 [DOI] [PubMed] [Google Scholar]
- D’Arco C, Dattwyler RJ, and Arnaboldi PM (2017). Borrelia burgdorferi-specific IgA in Lyme Disease. EBioMedicine 19, 91–97. 10.1016/j.ebiom.2017.04.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Souza MP, Adams E, Altman JD, Birnbaum ME, Boggiano C, Casorati G, Chien YH, Conley A, Eckle SBG, Fruh K, et al. (2019). Casting a wider net: Immunosurveillance by nonclassical MHC molecules. PLoS Pathog 15, e1007567. 10.1371/journal.ppat.1007567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daix V, Schroeder H, Praet N, Georgin JP, Chiappino I, Gillet L, de Fays K, Decrem Y, Leboulle G, Godfroid E, et al. (2007). Ixodes ticks belonging to the Ixodes ricinus complex encode a family of anticomplement proteins. Insect Mol Biol 16, 155–166. 10.1111/j.1365-2583.2006.00710.x [DOI] [PubMed] [Google Scholar]
- de Silva AM, Telford SR 3rd, Brunet LR, Barthold SW, and Fikrig E (1996). Borrelia burgdorferi OspA is an arthropod-specific transmission-blocking Lyme disease vaccine. J Exp Med 183, 271–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Souza MS, Fikrig E, Smith AL, Flavell RA, and Barthold SW (1992). Nonspecific proliferative responses of murine lymphocytes to Borrelia burgdorferi antigens. J Infect Dis 165, 471–478. [DOI] [PubMed] [Google Scholar]
- de Souza MS, Smith AL, Beck DS, Terwilliger GA, Fikrig E, and Barthold SW (1993). Long-term study of cell-mediated responses to Borrelia burgdorferi in the laboratory mouse. Infect Immun 61, 1814–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Defosse DL, and Johnson RC (1992). In vitro and in vivo induction of tumor necrosis factor alpha by Borrelia burgdorferi. Infect Immun 60, 1109–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis VA, Dixit S, O’Brien SM, Alvarez X, Pahar B, and Philipp MT (2009). Live Borrelia burgdorferi spirochetes elicit inflammatory mediators from human monocytes via the Toll-like receptor signaling pathway. Infect Immun 77, 1238–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis VA, Jefferson A, Singh SR, Ganapamo F, and Philipp MT (2006). Interleukin-10 anti-inflammatory response to Borrelia burgdorferi, the agent of Lyme disease: a possible role for suppressors of cytokine signaling 1 and 3. Infect Immun 74, 5780–5789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhodapkar MV, and Kumar V (2017). Type II NKT Cells and Their Emerging Role in Health and Disease. J Immunol 198, 1015–1021. 10.4049/jimmunol.1601399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divan A, Budd RC, Tobin RP, and Newell-Rogers MK (2015). Gammadelta T Cells and dendritic cells in refractory Lyme arthritis. J Leukoc Biol 97, 653–663. 10.1189/jlb.2RU0714-343RR [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divan A, Casselli T, Narayanan SA, Mukherjee S, Zawieja DC, Watt JA, Brissette CA, and Newell-Rogers MK (2018). Borrelia burgdorferi adhere to blood vessels in the dura mater and are associated with increased meningeal T cells during murine disseminated borreliosis. PLoS One 13, e0196893. 10.1371/journal.pone.0196893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douda DN, Khan MA, Grasemann H, and Palaniyar N (2015). SK3 channel and mitochondrial ROS mediate NADPH oxidase-independent NETosis induced by calcium influx. Proc Natl Acad Sci U S A 112, 2817–2822. 10.1073/pnas.1414055112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drouin EE, Seward RJ, Strle K, McHugh G, Katchar K, Londono D, Yao C, Costello CE, and Steere AC (2013). A novel human autoantigen, endothelial cell growth factor, is a target of T and B cell responses in patients with Lyme disease. Arthritis Rheum 65, 186–196. 10.1002/art.37732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunham-Ems SM, Caimano MJ, Pal U, Wolgemuth CW, Eggers CH, Balic A, and Radolf JD (2009). Live imaging reveals a biphasic mode of dissemination of Borrelia burgdorferi within ticks. J Clin Invest 119, 3652–3665. 10.1172/JCI39401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duray PH, and Steere AC (1988). Clinical pathologic correlations of Lyme disease by stage. Ann N Y Acad Sci 539, 65–79. [DOI] [PubMed] [Google Scholar]
- Ebnet K, Brown KD, Siebenlist UK, Simon MM, and Shaw S (1997). Borrelia burgdorferi activates nuclear factor-kappa B and is a potent inducer of chemokine and adhesion molecule gene expression in endothelial cells and fibroblasts. J Immunol 158, 3285–3292. [PubMed] [Google Scholar]
- Eckman EA, Pacheco-Quinto J, Herdt AR, and Halperin JJ (2018). Neuroimmunomodulators in Neuroborreliosis and Lyme Encephalopathy. Clin Infect Dis 67, 80–88. 10.1093/cid/ciy019 [DOI] [PubMed] [Google Scholar]
- Eicken C, Sharma V, Klabunde T, Lawrenz MB, Hardham JM, Norris SJ, and Sacchettini JC (2002). Crystal structure of Lyme disease variable surface antigen VlsE of Borrelia burgdorferi. J Biol Chem 277, 21691–21696. 10.1074/jbc.M201547200 [DOI] [PubMed] [Google Scholar]
- Elsner RA, Hastey CJ, and Baumgarth N (2015a). CD4+ T cells promote antibody production but not sustained affinity maturation during Borrelia burgdorferi infection. Infect Immun 83, 48–56. https://doi.org/IAI.02471-14 [pii] 10.1128/IAI.02471-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsner RA, Hastey CJ, Olsen KJ, and Baumgarth N (2015b). Suppression of long-lived immunity following Borrelia burgdorferi induced Lyme disease. PLoS Pathog 11, e1004976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Embers ME, Alvarez X, Ooms T, and Philipp MT (2008). The failure of immune response evasion by linear plasmid 28–1-deficient Borrelia burgdorferi is attributable to persistent expression of an outer surface protein. Infect Immun 76, 3984–3991. https://doi.org/IAI.00387-08 [pii] 10.1128/IAI.00387-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahrer H, van der Linden SM, Sauvain MJ, Gern L, Zhioua E, and Aeschlimann A (1991). The prevalence and incidence of clinical and asymptomatic Lyme borreliosis in a population at risk. J Infect Dis 163, 305–310. 10.1093/infdis/163.2.305 [DOI] [PubMed] [Google Scholar]
- Feehan KT, and Gilroy DW (2019). Is Resolution the End of Inflammation? Trends Mol Med 25, 198–214. 10.1016/j.molmed.2019.01.006 [DOI] [PubMed] [Google Scholar]
- Fikrig E, Barthold SW, Chen M, Chang CH, and Flavell RA (1997a). Protective antibodies develop, and murine Lyme arthritis regresses, in the absence of MHC class II and CD4+ T cells. J Immunol 159, 5682–5686. [PubMed] [Google Scholar]
- Fikrig E, Barthold SW, Chen M, Grewal IS, Craft J, and Flavell RA (1996). Protective antibodies in murine Lyme disease arise independently of CD40 ligand. J Immunol 157, 1–3. [PubMed] [Google Scholar]
- Fikrig E, Barthold SW, Kantor FS, and Flavell RA (1990). Protection of mice against the Lyme disease agent by immunizing with recombinant OspA. Science 250, 553–556. [DOI] [PubMed] [Google Scholar]
- Fikrig E, Barthold SW, Marcantonio N, Deponte K, Kantor FS, and Flavell RA (1992). Roles of OspA, OspB, and flagellin in protective immunity to Lyme borreliosis in laboratory mice. Infect Immun 60, 657–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fikrig E, Barthold SW, Sun W, Feng W, Telford SR 3rd, and Flavell RA (1997b). Borrelia burgdorferi P35 and P37 proteins, expressed in vivo, elicit protective immunity. Immunity 6, 531–539. [DOI] [PubMed] [Google Scholar]
- Filgueira L, Nestle FO, Rittig M, Joller HI, and Groscurth P (1996). Human dendritic cells phagocytose and process Borrelia burgdorferi. J Immunol 157, 2998–3005. [PubMed] [Google Scholar]
- Forrester JV, McMenamin PG, and Dando SJ (2018). CNS infection and immune privilege. Nat Rev Neurosci 19, 655–671. 10.1038/s41583-018-0070-8 [DOI] [PubMed] [Google Scholar]
- Francischetti IM, Mather TN, and Ribeiro JM (2004). Penthalaris, a novel recombinant five-Kunitz tissue factor pathway inhibitor (TFPI) from the salivary gland of the tick vector of Lyme disease, Ixodes scapularis. Thromb Haemost 91, 886–898. [DOI] [PubMed] [Google Scholar]
- Francischetti IM, Mather TN, and Ribeiro JM (2005). Tick saliva is a potent inhibitor of endothelial cell proliferation and angiogenesis. Thromb Haemost 94, 167–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francischetti IM, Valenzuela JG, Andersen JF, Mather TN, and Ribeiro JM (2002). Ixolaris, a novel recombinant tissue factor pathway inhibitor (TFPI) from the salivary gland of the tick, Ixodes scapularis: identification of factor X and factor Xa as scaffolds for the inhibition of factor VIIa/tissue factor complex. Blood 99, 3602–3612. [DOI] [PubMed] [Google Scholar]
- Frischknecht F (2007). The skin as interface in the transmission of arthropod-borne pathogens. Cell Microbiol 9, 1630–1640. [DOI] [PubMed] [Google Scholar]
- Frossi B, Mion F, Tripodo C, Colombo MP, and Pucillo CE (2017). Rheostatic Functions of Mast Cells in the Control of Innate and Adaptive Immune Responses. Trends Immunol 38, 648–656. 10.1016/j.it.2017.04.001 [DOI] [PubMed] [Google Scholar]
- Gajovic O, Todorovic Z, Nesic L, and Lazic Z (2010). [Lyme borreliosis--diagnostic difficulties in interpreting serological results]. Med Pregl 63, 839–843. [DOI] [PubMed] [Google Scholar]
- Garcia R, Gusmani L, Murgia R, Guarnaccia C, Cinco M, and Rottini G (1998). Elastase is the only human neutrophil granule protein that alone is responsible for in vitro killing of Borrelia burgdorferi. Infect Immun 66, 1408–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg R, Juncadella IJ, Ramamoorthi N, Ashish, Ananthanarayanan SK, Thomas V, Rincon M, Krueger JK, Fikrig E, Yengo CM, et al. (2006). Cutting edge: CD4 is the receptor for the tick saliva immunosuppressor, Salp15. J Immunol 177, 6579–6583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, and Ley K (2010). Development of monocytes, macrophages, and dendritic cells. Science 327, 656–661. 10.1126/science.1178331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgilis K, Noring R, Steere AC, and Klempner MS (1991a). Neutrophil chemotactic factors in synovial fluids of patients with Lyme disease. Arthritis Rheum 34, 770–775. [DOI] [PubMed] [Google Scholar]
- Georgilis K, Steere AC, and Klempner MS (1991b). Infectivity of Borrelia burgdorferi correlates with resistance to elimination by phagocytic cells. J Infect Dis 163, 150–155. [DOI] [PubMed] [Google Scholar]
- Giambartolomei GH, Dennis VA, and Philipp MT (1998). Borrelia burgdorferi stimulates the production of interleukin-10 in peripheral blood mononuclear cells from uninfected humans and rhesus monkeys. Infect Immun 66, 2691–2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie RD, Dolan MC, Piesman J, and Titus RG (2001). Identification of an IL-2 binding protein in the saliva of the Lyme disease vector tick, Ixodes scapularis. J Immunol 166, 4319–4326. [DOI] [PubMed] [Google Scholar]
- Glickstein L, Edelstein M, and Dong JZ (2001). Gamma interferon is not required for arthritis resistance in the murine Lyme disease model. Infect Immun 69, 3737–3743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godfrey DI, Koay HF, McCluskey J, and Gherardin NA (2019). The biology and functional importance of MAIT cells. Nat Immunol 20, 1110–1128. 10.1038/s41590-019-0444-8 [DOI] [PubMed] [Google Scholar]
- Golightly M, Thomas J, Volkman D, and Dattwyler R (1988). Modulation of natural killer cell activity by Borrelia burgdorferi. Ann N Y Acad Sci 539, 103–111. 10.1111/j.1749-6632.1988.tb31843.x [DOI] [PubMed] [Google Scholar]
- Greene RT, Levine JF, Breitschwerdt EB, Walker RL, Berkhoff HA, Cullen J, and Nicholson WL (1988). Clinical and serologic evaluations of induced Borrelia burgdorferi infection in dogs. Am J Vet Res 49, 752–757. [PubMed] [Google Scholar]
- Greenmyer JR, Gaultney RA, Brissette CA, and Watt JA (2018). Primary Human Microglia Are Phagocytically Active and Respond to Borrelia burgdorferi With Upregulation of Chemokines and Cytokines. Front Microbiol 9, 811. 10.3389/fmicb.2018.00811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm D, Tilly K, Byram R, Stewart PE, Krum JG, Bueschel DM, Schwan TG, Policastro PF, Elias AF, and Rosa PA (2004). Outer-surface protein C of the Lyme disease spirochete: a protein induced in ticks for infection of mammals. Proc Natl Acad Sci U S A 101, 3142–3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross DM, Forsthuber T, Tary-Lehmann M, Etling C, Ito K, Nagy ZA, Field JA, Steere AC, and Huber BT (1998a). Identification of LFA-1 as a candidate autoantigen in treatment-resistant Lyme arthritis. Science 281, 703–706. 10.1126/science.281.5377.703 [DOI] [PubMed] [Google Scholar]
- Gross DM, Steere AC, and Huber BT (1998b). T helper 1 response is dominant and localized to the synovial fluid in patients with Lyme arthritis. J Immunol 160, 1022–1028. [PubMed] [Google Scholar]
- Guo X, Booth CJ, Paley MA, Wang X, DePonte K, Fikrig E, Narasimhan S, and Montgomery RR (2009). Inhibition of neutrophil function by two tick salivary proteins. Infect Immun 77, 2320–2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagan T, Cortese M, Rouphael N, Boudreau C, Linde C, Maddur MS, Das J, Wang H, Guthmiller J, Zheng NY, et al. (2019). Antibiotics-Driven Gut Microbiome Perturbation Alters Immunity to Vaccines in Humans. Cell 178, 1313–1328 e1313. 10.1016/j.cell.2019.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallstrom T, Siegel C, Morgelin M, Kraiczy P, Skerka C, and Zipfel PF (2013). CspA from Borrelia burgdorferi inhibits the terminal complement pathway. mBio 4. 10.1128/mBio.00481-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halperin JJ (2019). A Neurologist’s View of Lyme Disease and Other Tick-Borne Infections. Semin Neurol 39, 440–447. 10.1055/s-0039-1692143 [DOI] [PubMed] [Google Scholar]
- Halperin JJ, Pass HL, Anand AK, Luft BJ, Volkman DJ, and Dattwyler RJ (1988). Nervous system abnormalities in Lyme disease. Ann N Y Acad Sci 539, 24–34. [DOI] [PubMed] [Google Scholar]
- Hammers-Berggren S, Lebech AM, Karlsson M, Svenungsson B, Hansen K, and Stiernstedt G (1994). Serological follow-up after treatment of patients with erythema migrans and neuroborreliosis. J Clin Microbiol 32, 1519–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammerschmidt C, Klevenhaus Y, Koenigs A, Hallstrom T, Fingerle V, Skerka C, Pos KM, Zipfel PF, Wallich R, and Kraiczy P (2016). BGA66 and BGA71 facilitate complement resistance of Borrelia bavariensis by inhibiting assembly of the membrane attack complex. Mol Microbiol 99, 407–424. 10.1111/mmi.13239 [DOI] [PubMed] [Google Scholar]
- Hanson MS, Cassatt DR, Guo BP, Patel NK, McCarthy MP, Dorward DW, and Hook M (1998). Active and passive immunity against Borrelia burgdorferi decorin binding protein A (DbpA) protects against infection. Infect Immun 66, 2143–2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardin JA, Steere AC, and Malawista SE (1984). The pathogenesis of arthritis in Lyme disease: humoral immune responses and the role of intra-articular immune complexes. Yale J Biol Med 57, 589–593. [PMC free article] [PubMed] [Google Scholar]
- Hardin JA, Walker LC, Steere AC, Trumble TC, Tung KS, Williams RC Jr., Ruddy S, and Malawista SE (1979). Circulating immune complexes in Lyme arthritis. Detection by the 125I-C1q binding, C1q solid phase, and Raji cell assays. J Clin Invest 63, 468–477. 10.1172/JCI109324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harman MW, Dunham-Ems SM, Caimano MJ, Belperron AA, Bockenstedt LK, Fu HC, Radolf JD, and Wolgemuth CW (2012). The heterogeneous motility of the Lyme disease spirochete in gelatin mimics dissemination through tissue. Proc Natl Acad Sci U S A 109, 3059–3064. 10.1073/pnas.1114362109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastey CJ, Elsner RA, Barthold SW, and Baumgarth N (2012). Delays and diversions mark the development of B cell responses to Borrelia burgdorferi infection. J Immunol 188, 5612–5622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastey CJ, Elsner RA, Olsen KJ, Mundigl S, Barthold SW, and Baumgarth N (2020). Borrelia burgdorferi infection induces ongoing IgM production that controls bacteremia but not bacterial dissemination or tissue burden in mice. Submitted. [DOI] [PMC free article] [PubMed]
- Hastey CJ, Elsner RA, Olsen KJ, Tunev SS, Escobar ED, Barthold SW, and Baumgarth N (2016). Compartmentalization of humoral immunity to Borrelia burgdorferi infection. In preparation. [Google Scholar]
- Hastey CJ, Ochoa J, Olsen KJ, Barthold SW, and Baumgarth N (2014). MyD88- and TRIF-independent induction of type I interferon drives naive B cell accumulation but not loss of lymph node architecture in Lyme disease. Infect Immun 82, 1548–1558. https://doi.org/IAI.00969-13 [pii] 10.1128/IAI.00969-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley KL, Olson CM Jr., Carreras-González A, Navasa N, and Anguita J (2015). Serum C3 Enhances Complement Receptor 3-Mediated Phagocytosis of Borrelia burgdorferi. Int J Biol Sci 11, 1269–1271. 10.7150/ijbs.13395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley KL, Olson CM Jr., Iglesias-Pedraz JM, Navasa N, Cervantes JL, Caimano MJ, Izadi H, Ingalls RR, Pal U, Salazar JC, et al. (2012). CD14 cooperates with complement receptor 3 to mediate MyD88-independent phagocytosis of Borrelia burgdorferi. Proc Natl Acad Sci U S A 109, 1228–1232. 10.1073/pnas.1112078109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, and Marneros AG (2014). Doxycycline inhibits polarization of macrophages to the proangiogenic M2-type and subsequent neovascularization. J Biol Chem 289, 8019–8028. 10.1074/jbc.M113.535765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilpern AJ, Wertheim W, He J, Perides G, Bronson RT, and Hu LT (2009). Matrix metalloproteinase-9 plays a key role in Lyme arthritis but not dissemination of Borrelia burgdorferi. Infect Immun 77, 2643–2649. 10.1128/IAI.00214-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschfeld M, Kirschning CJ, Schwandner R, Wesche H, Weis JH, Wooten RM, and Weis JJ (1999). Cutting edge: inflammatory signaling by Borrelia burgdorferi lipoproteins is mediated by toll-like receptor 2. J Immunol 163, 2382–2386. [PubMed] [Google Scholar]
- Hirschfeld M, Ma Y, Weis JH, Vogel SN, and Weis JJ (2000). Repurification of LPS eliminates signaling through both human and murine Toll-like receptor 2. J Immunol 165, 618–622. [DOI] [PubMed] [Google Scholar]
- Horka H, Cerna-Kyckova K, Skallova A, and Kopecky J (2009). Tick saliva affects both proliferation and distribution of Borrelia burgdorferi spirochetes in mouse organs and increases transmission of spirochetes to ticks. Int J Med Microbiol 299, 373–380. 10.1016/j.ijmm.2008.10.009 [DOI] [PubMed] [Google Scholar]
- Hovius JW, de Jong MA, den Dunnen J, Litjens M, Fikrig E, van der Poll T, Gringhuis SI, and Geijtenbeek TB (2008). Salp15 binding to DC-SIGN inhibits cytokine expression by impairing both nucleosome remodeling and mRNA stabilization. PLoS Pathog 4, e31. 10.1371/journal.ppat.0040031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu LT, Eskildsen MA, Masgala C, Steere AC, Arner EC, Pratta MA, Grodzinsky AJ, Loening A, and Perides G (2001). Host metalloproteinases in Lyme arthritis. Arthritis Rheum 44, 1401–1410. [DOI] [PubMed] [Google Scholar]
- Huegli D, Moret J, Rais O, Moosmann Y, Erard P, Malinverni R, and Gern L (2011). Prospective study on the incidence of infection by Borrelia burgdorferi sensu lato after a tick bite in a highly endemic area of Switzerland. Ticks Tick Borne Dis 2, 129–136. 10.1016/j.ttbdis.2011.05.002 [DOI] [PubMed] [Google Scholar]
- Hulinska D, Bartak P, Hercogova J, Hancil J, Basta J, and Schramlova J (1994). Electron microscopy of Langerhans cells and Borrelia burgdorferi in Lyme disease patients. Zentralbl Bakteriol 280, 348–359. [DOI] [PubMed] [Google Scholar]
- Hyde JA (2017). Borrelia burgdorferi Keeps Moving and Carries on: A Review of Borrelial Dissemination and Invasion. Front Immunol 8, 114. 10.3389/fimmu.2017.00114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliopoulou BP, Alroy J, and Huber BT (2007). CD28 deficiency exacerbates joint inflammation upon Borrelia burgdorferi infection, resulting in the development of chronic Lyme arthritis. J Immunol 179, 8076–8082. [DOI] [PubMed] [Google Scholar]
- Imai DM, Samuels DS, Feng S, Hodzic E, Olsen K, and Barthold SW (2013). The early dissemination defect attributed to disruption of decorin-binding proteins is abolished in chronic murine Lyme borreliosis. Infect Immun 81, 1663–1673. 10.1128/IAI.01359-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue Y, Shimojo N, Suzuki Y, Campos Alberto EJ, Yamaide A, Suzuki S, Arima T, Matsuura T, Tomiita M, Aoyagi M, et al. (2007). CD14 –550 C/T, which is related to the serum level of soluble CD14, is associated with the development of respiratory syncytial virus bronchiolitis in the Japanese population. J Infect Dis 195, 1618–1624. [DOI] [PubMed] [Google Scholar]
- Iyer R, and Schwartz I (2016). Microarray-Based Comparative Genomic and Transcriptome Analysis of Borrelia burgdorferi. Microarrays (Basel) 5. 10.3390/microarrays5020009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izadi H, Motameni AT, Bates TC, Olivera ER, Villar-Suarez V, Joshi I, Garg R, Osborne BA, Davis RJ, Rincon M, et al. (2007). c-Jun N-terminal kinase 1 is required for Toll-like receptor 1 gene expression in macrophages. Infect Immun 75, 5027–5034. 10.1128/IAI.00492-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen M, Zhou D, Cepok S, Nessler S, Happel M, Stei S, Wilske B, Sommer N, and Hemmer B (2003). Clonal accumulation of activated CD8+ T cells in the central nervous system during the early phase of neuroborreliosis. J Infect Dis 187, 963–973. [DOI] [PubMed] [Google Scholar]
- Jacquet M, Durand J, Rais O, and Voordouw MJ (2015). Cross-reactive acquired immunity influences transmission success of the Lyme disease pathogen, Borrelia afzelii. Infect Genet Evol 36, 131–140. 10.1016/j.meegid.2015.09.012 [DOI] [PubMed] [Google Scholar]
- Juncadella IJ, Garg R, Ananthnarayanan SK, Yengo CM, and Anguita J (2007). T-cell signaling pathways inhibited by the tick saliva immunosuppressor, Salp15. FEMS Immunol Med Microbiol 49, 433–438. [DOI] [PubMed] [Google Scholar]
- Jung S, Schickel JN, Kern A, Knapp AM, Eftekhari P, Da Silva S, Jaulhac B, Brink R, Soulas-Sprauel P, Pasquali JL, et al. (2016). Chronic bacterial infection activates autoreactive B cells and induces isotype switching and autoantigen-driven mutations. Eur J Immunol 46, 131–146. 10.1002/eji.201545810 [DOI] [PubMed] [Google Scholar]
- Jutras BL, Lochhead RB, Kloos ZA, Biboy J, Strle K, Booth CJ, Govers SK, Gray J, Schumann P, Vollmer W, et al. (2019). Borrelia burgdorferi peptidoglycan is a persistent antigen in patients with Lyme arthritis. Proc Natl Acad Sci U S A 116, 13498–13507. 10.1073/pnas.1904170116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan DH (2017). Ontogeny and function of murine epidermal Langerhans cells. Nat Immunol 18, 1068–1075. 10.1038/ni.3815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katchar K, Drouin EE, and Steere AC (2013). Natural killer cells and natural killer T cells in Lyme arthritis. Arthritis Res Ther 15, R183. 10.1186/ar4373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane-Myers A, and Nickell SP (1995). T cell subset-dependent modulation of immunity to Borrelia burgdorferi in mice. J Immunol 154, 1770–1776. [PubMed] [Google Scholar]
- Kemp DH, Stone BF, and Binnington KC (1982). Tick attachment and feeding: Role of the mouthparts, feeding apparatus, salivary gland secretions and the host response. In Physiology of Ticks, Obenchain FD, and Galun R, eds. (Elmsford, NY: Pergamon Press; ), pp. 119–168. [Google Scholar]
- Khatchikian CE, Nadelman RB, Nowakowski J, Schwartz I, Wormser GP, and Brisson D (2014). Evidence for strain-specific immunity in patients treated for early Lyme disease. Infect Immun 82, 1408–1413. 10.1128/IAI.01451-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Killpack TL, Ballesteros M, Bunnell SC, Bedugnis A, Kobzik L, Hu LT, and Petnicki-Ocwieja T (2017). Phagocytic receptors activate Syk and Src signaling during Borrelia burgdorferi phagocytosis. Infect Immun 85. 10.1128/iai.00004-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinjo Y, Tupin E, Wu D, Fujio M, Garcia-Navarro R, Benhnia MR, Zajonc DM, Ben-Menachem G, Ainge GD, Painter GF, et al. (2006). Natural killer T cells recognize diacylglycerol antigens from pathogenic bacteria. Nat Immunol 7, 978–986. [DOI] [PubMed] [Google Scholar]
- Kirschning CJ, Wesche H, Merrill Ayres T, and Rothe M (1998). Human toll-like receptor 2 confers responsiveness to bacterial lipopolysaccharide. J Exp Med 188, 2091–2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisand KE, Prukk T, Kisand KV, Luus SM, Kalbe I, and Uibo R (2007). Propensity to excessive proinflammatory response in chronic Lyme borreliosis. APMIS 115, 134–141. 10.1111/j.1600-0463.2007.apm_538.x [DOI] [PubMed] [Google Scholar]
- Kochi SK, and Johnson RC (1988). Role of immunoglobulin G in killing of Borrelia burgdorferi by the classical complement pathway. Infect Immun 56, 314–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochi SK, Johnson RC, and Dalmasso AP (1991). Complement-mediated killing of the Lyme disease spirochete Borrelia burgdorferi. Role of antibody in formation of an effective membrane attack complex. J Immunol 146, 3964–3970. [PubMed] [Google Scholar]
- Kochi SK, Johnson RC, and Dalmasso AP (1993). Facilitation of complement-dependent killing of the Lyme disease spirochete, Borrelia burgdorferi, by specific immunoglobulin G Fab antibody fragments. Infect Immun 61, 2532–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotsyfakis M, Sa-Nunes A, Francischetti IM, Mather TN, Andersen JF, and Ribeiro JM (2006). Antiinflammatory and immunosuppressive activity of sialostatin L, a salivary cystatin from the tick Ixodes scapularis. J Biol Chem 281, 26298–26307. [DOI] [PubMed] [Google Scholar]
- Kotsyfakis M, Schwarz A, Erhart J, and Ribeiro JM (2015). Tissue- and time-dependent transcription in Ixodes ricinus salivary glands and midguts when blood feeding on the vertebrate host. Sci Rep 5, 9103. 10.1038/srep09103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowarik MC, Grummel V, Wemlinger S, Buck D, Weber MS, Berthele A, and Hemmer B (2014). Immune cell subtyping in the cerebrospinal fluid of patients with neurological diseases. J Neurol 261, 130–143. 10.1007/s00415-013-7145-2 [DOI] [PubMed] [Google Scholar]
- Krause PJ, Grant-Kels JM, Tahan SR, Dardick KR, Alarcon-Chaidez F, Bouchard K, Visini C, Deriso C, Foppa IM, and Wikel S (2009). Dermatologic changes induced by repeated Ixodes scapularis bites and implications for prevention of tick-borne infection. Vector Borne Zoonotic Dis 9, 603–610. 10.1089/vbz.2008.0091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlow CJ, Garcia-Monco JC, Coleman JL, and Benach JL (2005). Murine microglia are effective phagocytes for Borrelia burgdorferi. J Neuroimmunol 168, 183–187. [DOI] [PubMed] [Google Scholar]
- Kumar H, Belperron A, Barthold SW, and Bockenstedt LK (2000). Cutting edge: CD1d deficiency impairs murine host defense against the spirochete, Borrelia burgdorferi. J Immunol 165, 4797–4801. [DOI] [PubMed] [Google Scholar]
- Kuthejlova M, Kopecky J, Stepanova G, and Macela A (2001). Tick salivary gland extract inhibits killing of Borrelia afzelii spirochetes by mouse macrophages. Infect Immun 69, 575–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaRocca TJ, and Benach JL (2008). The important and diverse roles of antibodies in the host response to Borrelia infections. Curr Top Microbiol Immunol 319, 63–103. 10.1007/978-3-540-73900-5_4 [DOI] [PubMed] [Google Scholar]
- Lasky CE, Jamison KE, Sidelinger DR, Pratt CL, Zhang G, and Brown CR (2015a). Infection of Interleukin 17 Receptor A-Deficient C3H Mice with Borrelia burgdorferi Does Not Affect Their Development of Lyme Arthritis and Carditis. Infect Immun 83, 2882–2888. https://doi.org/IAI.00533-15 [pii] 10.1128/IAI.00533-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasky CE, Olson RM, and Brown CR (2015b). Macrophage polarization during murine Lyme borreliosis. Infect Immun 83, 2627–2635. 10.1128/iai.00369-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasky CE, Pratt CL, Hilliard KA, Jones JL, and Brown CR (2016). T Cells Exacerbate Lyme Borreliosis in TLR2-Deficient Mice. Front Immunol 7, 468. 10.3389/fimmu.2016.00468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrenz MB, Wooten RM, Zachary JF, Drouin SM, Weis JJ, Wetsel RA, and Norris SJ (2003). Effect of complement component C3 deficiency on experimental Lyme borreliosis in mice. Infect Immun 71, 4432–4440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarus JJ, Kay MA, McCarter AL, and Wooten RM (2008). Viable Borrelia burgdorferi enhances interleukin-10 production and suppresses activation of murine macrophages. Infect Immun 76, 1153–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarus JJ, Meadows MJ, Lintner RE, and Wooten RM (2006). IL-10 deficiency promotes increased Borrelia burgdorferi clearance predominantly through enhanced innate immune responses. J Immunol 177, 7076–7085. [DOI] [PubMed] [Google Scholar]
- Li L, Narayan K, Pak E, and Pachner AR (2006). Intrathecal antibody production in a mouse model of Lyme neuroborreliosis. J Neuroimmunol 173, 56–68. 10.1016/j.jneuroim.2005.11.019 [DOI] [PubMed] [Google Scholar]
- Li X, McHugh GA, Damle N, Sikand VK, Glickstein L, and Steere AC (2011). Burden and viability of Borrelia burgdorferi in skin and joints of patients with erythema migrans or lyme arthritis. Arthritis Rheum 63, 2238–2247. 10.1002/art.30384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lien E, Sellati TJ, Yoshimura A, Flo TH, Rawadi G, Finberg RW, Carroll JD, Espevik T, Ingalls RR, Radolf JD, et al. (1999). Toll-like receptor 2 functions as a pattern recognition receptor for diverse bacterial products. J Biol Chem 274, 33419–33425. 10.1074/jbc.274.47.33419 [DOI] [PubMed] [Google Scholar]
- Lieskovska J, Palenikova J, Langhansova H, Campos Chagas A, Calvo E, Kotsyfakis M, and Kopecky J (2015). Tick sialostatins L and L2 differentially influence dendritic cell responses to Borrelia spirochetes. Parasit Vectors 8, 275. 10.1186/s13071-015-0887-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim LC, England DM, DuChateau BK, Glowacki NJ, and Schell RF (1995). Borrelia burgdorferi-specific T lymphocytes induce severe destructive Lyme arthritis. Infect Immun 63, 1400–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin B, Noring R, Steere AC, Klempner MS, and Hu LT (2000). Soluble CD14 levels in the serum, synovial fluid, and cerebrospinal fluid of patients with various stages of Lyme disease. J Infect Dis 181, 1185–1188. [DOI] [PubMed] [Google Scholar]
- Lin YP, Benoit V, Yang X, Martinez-Herranz R, Pal U, and Leong JM (2014). Strain-specific variation of the decorin-binding adhesin DbpA influences the tissue tropism of the lyme disease spirochete. PLoS Pathog 10, e1004238. 10.1371/journal.ppat.1004238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YP, Diuk-Wasser MA, Stevenson B, and Kraiczy P (2020). Complement Evasion Contributes to Lyme Borreliae-Host Associations. Trends Parasitol. 10.1016/j.pt.2020.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder S, Heimerl C, Fingerle V, Aepfelbacher M, and Wilske B (2001). Coiling phagocytosis of Borrelia burgdorferi by primary human macrophages is controlled by CDC42Hs and Rac1 and involves recruitment of Wiskott-Aldrich syndrome protein and Arp2/3 complex. Infect Immun 69, 1739–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisinski TJ, and Furie MB (2002a). Interleukin-10 inhibits proinflammatory activation of endothelium in response to Borrelia burgdorferi or lipopolysaccharide but not interleukin-1β or tumor necrosis factor α. J Leukoc Biol 72, 503–511. [PubMed] [Google Scholar]
- Lisinski TJ, and Furie MB (2002b). Interleukin-10 inhibits proinflammatory activation of endothelium in response to Borrelia burgdorferi or lipopolysaccharide but not interleukin-1beta or tumor necrosis factor alpha. J Leukoc Biol 72, 503–511. [PubMed] [Google Scholar]
- Liu N, Belperron AA, Booth CJ, and Bockenstedt LK (2009). The caspase 1 inflammasome is not required for control of murine Lyme borreliosis. Infect Immun 77, 3320–3327. 10.1128/IAI.00100-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Montgomery RR, Barthold SW, and Bockenstedt LK (2004). Myeloid differentiation antigen 88 deficiency impairs pathogen clearance but does not alter inflammation in Borrelia burgdorferi-infected mice. Infect Immun 72, 3195–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liveris D, Wang G, Girao G, Byrne DW, Nowakowski J, McKenna D, Nadelman R, Wormser GP, and Schwartz I (2002). Quantitative detection of Borrelia burgdorferi in 2-millimeter skin samples of erythema migrans lesions: correlation of results with clinical and laboratory findings. J Clin Microbiol 40, 1249–1253. 10.1128/jcm.40.4.1249-1253.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochhead RB, Arvikar SL, Aversa JM, Sadreyev RI, Strle K, and Steere AC (2019a). Robust interferon signature and suppressed tissue repair gene expression in synovial tissue from patients with postinfectious, Borrelia burgdorferi-induced Lyme arthritis. Cell Microbiol 21, e12954. 10.1111/cmi.12954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochhead RB, Ordonez D, Arvikar SL, Aversa JM, Oh LS, Heyworth B, Sadreyev R, Steere AC, and Strle K (2019b). Interferon-gamma production in Lyme arthritis synovial tissue promotes differentiation of fibroblast-like synoviocytes into immune effector cells. Cell Microbiol 21, e12992. 10.1111/cmi.12992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochhead RB, Sonderegger FL, Ma Y, Brewster JE, Cornwall D, Maylor-Hagen H, Miller JC, Zachary JF, Weis JH, and Weis JJ (2012). Endothelial cells and fibroblasts amplify the arthritogenic type I IFN response in murine Lyme disease and are major sources of chemokines in Borrelia burgdorferi-infected joint tissue. J Immunol 189, 2488–2501. https://doi.org/jimmunol.1201095 [pii] 10.4049/jimmunol.1201095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochhead RB, Strle K, Kim ND, Kohler MJ, Arvikar SL, Aversa JM, and Steere AC (2017). MicroRNA Expression Shows Inflammatory Dysregulation and Tumor-Like Proliferative Responses in Joints of Patients With Postinfectious Lyme Arthritis. Arthritis Rheumatol 69, 1100–1110. 10.1002/art.40039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Londono D, Cadavid D, Drouin EE, Strle K, McHugh G, Aversa JM, and Steere AC (2014). Antibodies to endothelial cell growth factor and obliterative microvascular lesions in the synovium of patients with antibiotic-refractory Lyme arthritis. Arthritis Rheumatol 66, 2124–2133. 10.1002/art.38618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love AC, Schwartz I, and Petzke MM (2014). Borrelia burgdorferi RNA induces type I and III interferons via Toll-like receptor 7 and contributes to production of NF-kappaB-dependent cytokines. Infect Immun 82, 2405–2416. 10.1128/IAI.01617-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love AC, Schwartz I, and Petzke MM (2015). Induction of indoleamine 2,3-dioxygenase by Borrelia burgdorferi in human immune cells correlates with pathogenic potential. J Leukoc Biol 97, 379–390. 10.1189/jlb.4A0714-339R [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunemann JD, Gelderblom H, Sospedra M, Quandt JA, Pinilla C, Marques A, and Martin R (2007). Cerebrospinal fluid-infiltrating CD4+ T cells recognize Borrelia burgdorferi lysine-enriched protein domains and central nervous system autoantigens in early Lyme encephalitis. Infect Immun 75, 243–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusitani D, Malawista SE, and Montgomery RR (2002). Borrelia burgdorferi are susceptible to killing by a variety of human polymorphonuclear leukocyte components. J Infect Dis 185, 797–804. [DOI] [PubMed] [Google Scholar]
- Lusitani D, Malawista SE, and Montgomery RR (2003). Calprotectin, an abundant cytosolic protein from human polymorphonuclear leukocytes, inhibits the growth of Borrelia burgdorferi. Infect Immun 71, 4711–4716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Bramwell KK, Lochhead RB, Paquette JK, Zachary JF, Weis JH, Teuscher C, and Weis JJ (2014). Borrelia burgdorferi arthritis-associated locus Bbaa1 regulates Lyme arthritis and K/BxN serum transfer arthritis through intrinsic control of type I IFN production. J Immunol 193, 6050–6060. https://doi.org/jimmunol.1401746 [pii] 10.4049/jimmunol.1401746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Seiler KP, Eichwald EJ, Weis JH, Teuscher C, and Weis JJ (1998). Distinct characteristics of resistance to Borrelia burgdorferi-induced arthritis in C57BL/6N mice. Infect Immun 66, 161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, and Weis JJ (1993). Borrelia burgdorferi outer surface lipoproteins OspA and OspB possess B-cell mitogenic and cytokine-stimulatory properties. Infect Immun 61, 3843–3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magunda PR, and Bankhead T (2016). Investigating the potential role of non-vls genes on linear plasmid 28–1 in virulence and persistence by Borrelia burgdorferi. BMC Microbiol 16, 180. 10.1186/s12866-016-0806-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malawista SE, and de Boisfleury Chevance A (2008). Clocking the Lyme spirochete. PLoS One 3, e1633. 10.1371/journal.pone.0001633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malkiel S, Kuhlow CJ, Mena P, and Benach JL (2009). The loss and gain of marginal zone and peritoneal B cells is different in response to relapsing fever and Lyme disease Borrelia. J Immunol 182, 498–506. [DOI] [PubMed] [Google Scholar]
- Marcinkiewicz AL, Lieknina I, Kotelovica S, Yang X, Kraiczy P, Pal U, Lin YP, and Tars K (2018). Eliminating Factor H-Binding Activity of Borrelia burgdorferi CspZ Combined with Virus-Like Particle Conjugation Enhances Its Efficacy as a Lyme Disease Vaccine. Front Immunol 9, 181. 10.3389/fimmu.2018.00181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcinkiewicz AL, Lieknina I, Yang X, Lederman PL, Hart TM, Yates J, Chen WH, Bottazzi ME, Mantis NJ, Kraiczy P, et al. (2020). The Factor H-Binding Site of CspZ as a Protective Target against Multistrain, Tick-Transmitted Lyme Disease. Infect Immun 88. 10.1128/IAI.00956-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marietta EV, Weis JJ, and Weis JH (1997). CD28 expression by mouse mast cells is modulated by lipopolysaccharide and outer surface protein A lipoprotein from Borrelia burgdorferi. J Immunol 159, 2840–2848. [PubMed] [Google Scholar]
- Marques A, Schwartz I, Wormser GP, Wang Y, Hornung RL, Demirkale CY, Munson PJ, Turk SP, Williams C, Lee CR, et al. (2017). Transcriptome Assessment of Erythema Migrans Skin Lesions in Patients With Early Lyme Disease Reveals Predominant Interferon Signaling. J Infect Dis 217, 158–167. 10.1093/infdis/jix563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason LM, Wagemakers A, van ‘t Veer C, Oei A, van der Pot WJ, Ahmed K, van der Poll T, Geijtenbeek TB, and Hovius JW (2016). Borrelia burgdorferi Induces TLR2-Mediated Migration of Activated Dendritic Cells in an Ex Vivo Human Skin Model. PLoS One 11, e0164040. 10.1371/journal.pone.0164040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matyniak JE, and Reiner SL (1995). T helper phenotype and genetic susceptibility in experimental Lyme disease. J Exp Med 181, 1251–1254. 10.1084/jem.181.3.1251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbow ML, Zeidner N, Panella N, Titus RG, and Piesman J (1997). Borrelia burgdorferi-pulsed dendritic cells induce a protective immune response against tick-transmitted spirochetes. Infect Immun 65, 3386–3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKisic MD, and Barthold SW (2000). T-cell-independent responses to Borrelia burgdorferi are critical for protective immunity and resolution of lyme disease. Infect Immun 68, 5190–5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKisic MD, Redmond WL, and Barthold SW (2000). Cutting edge: T cell-mediated pathology in murine Lyme borreliosis. J Immunol 164, 6096–6099. [DOI] [PubMed] [Google Scholar]
- Medzhitov R (2008). Origin and physiological roles of inflammation. Nature 454, 428–435. 10.1038/nature07201 [DOI] [PubMed] [Google Scholar]
- Medzhitov R, Preston-Hurlburt P, and Janeway CA Jr. (1997). A human homologue of the Drosophila Toll protein signals activation of adaptive immunity [see comments]. Nature 388, 394–397. [DOI] [PubMed] [Google Scholar]
- Menten-Dedoyart C, Faccinetto C, Golovchenko M, Dupiereux I, Van Lerberghe PB, Dubois S, Desmet C, Elmoualij B, Baron F, Rudenko N, et al. (2012). Neutrophil extracellular traps entrap and kill Borrelia burgdorferi sensu stricto spirochetes and are not affected by Ixodes ricinus tick saliva. J Immunol 189, 5393–5401. 10.4049/jimmunol.1103771 [DOI] [PubMed] [Google Scholar]
- Miller JC, Ma Y, Bian J, Sheehan KC, Zachary JF, Weis JH, Schreiber RD, and Weis JJ (2008a). A critical role for type I IFN in arthritis development following Borrelia burgdorferi infection of mice. J Immunol 181, 8492–8503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JC, Ma Y, Crandall H, Wang X, and Weis JJ (2008b). Gene expression profiling provides insights into the pathways involved in inflammatory arthritis development: murine model of Lyme disease. Exp Mol Pathol 85, 20–27. 10.1016/j.yexmp.2008.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modolell M, Schaible UE, Rittig M, and Simon MM (1994). Killing of Borrelia burgdorferi by macrophages is dependent on oxygen radicals and nitric oxide and can be enhanced by antibodies to outer surface proteins of the spirochete. Immunol Lett 40, 139–146. 10.1016/0165-2478(94)90185-6 [DOI] [PubMed] [Google Scholar]
- Montgomery RR, Booth CJ, Wang X, Blaho VA, Malawista SE, and Brown CR (2007). Recruitment of macrophages and polymorphonuclear leukocytes in lyme carditis. Infect Immun 75, 613–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery RR, Lusitani D, Chevance Ad Ade B, and Malawista SE (2002). Human phagocytic cells in the early innate immune response to Borrelia burgdorferi. J Infect Dis 185, 1773–1779. [DOI] [PubMed] [Google Scholar]
- Montgomery RR, and Malawista SE (1996). Entry of Borrelia burgdorferi into macrophages is end-on and leads to degradation in lysosomes. Infect Immun 64, 2867–2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery RR, Nathanson MH, and Malawista SE (1994). Fc- and non-Fc-mediated phagocytosis of Borrelia burgdorferi by macrophages. J Infect Dis 170, 890–893. [DOI] [PubMed] [Google Scholar]
- Moody KD, Terwilliger GA, Hansen GM, and Barthold SW (1994). Experimental Borrelia burgdorferi infection in Peromyscus leucopus. J Wildl Dis 30, 155–161. 10.7589/0090-3558-30.2.155 [DOI] [PubMed] [Google Scholar]
- Moore KW, de Waal Malefyt R, Coffman RL, and O’Garra A (2001). Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol 19, 683–765. [DOI] [PubMed] [Google Scholar]
- Moore MW, Cruz AR, LaVake CJ, Marzo AL, Eggers CH, Salazar JC, and Radolf JD (2007). Phagocytosis of Borrelia burgdorferi and Treponema pallidum potentiates innate immune activation and induces gamma interferon production. Infect Immun 75, 2046–2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori L, Lepore M, and De Libero G (2016). The Immunology of CD1- and MR1-Restricted T Cells. Annu Rev Immunol 34, 479–510. 10.1146/annurev-immunol-032414-112008 [DOI] [PubMed] [Google Scholar]
- Morrison TB, Weis JH, and Weis JJ (1997). Borrelia burgdorferi outer surface protein A (OspA) activates and primes human neutrophils. J Immunology 158, 4838–4845. [PubMed] [Google Scholar]
- Motta V, Soares F, Sun T, and Philpott DJ (2015). NOD-like receptors: versatile cytosolic sentinels. Physiol Rev 95, 149–178. 10.1152/physrev.00009.2014 [DOI] [PubMed] [Google Scholar]
- Muehlenbachs A, Bollweg BC, Schulz TJ, Forrester JD, DeLeon Carnes M, Molins C, Ray GS, Cummings PM, Ritter JM, Blau DM, et al. (2016). Cardiac Tropism of Borrelia burgdorferi: An Autopsy Study of Sudden Cardiac Death Associated with Lyme Carditis. Am J Pathol 186, 1195–1205. 10.1016/j.ajpath.2015.12.027 [DOI] [PubMed] [Google Scholar]
- Mullegger RR, McHugh G, Ruthazer R, Binder B, Kerl H, and Steere AC (2000). Differential expression of cytokine mRNA in skin specimens from patients with erythema migrans or acrodermatitis chronica atrophicans. J Invest Dermatol 115, 1115–1123. 10.1046/j.1523-1747.2000.00198.x [DOI] [PubMed] [Google Scholar]
- Murphy K, and Weaver C (2017). Principles of innate immunity. In Janeway’s Immunobiology (New York, NY: Garland Science, Taylor & Francis Group, LLC; ), pp. 3–11. [Google Scholar]
- Myers TA, Kaushal D, and Philipp MT (2009). Microglia are mediators of Borrelia burgdorferi-induced apoptosis in SH-SY5Y neuronal cells. PLoS Pathog 5, e1000659. 10.1371/journal.ppat.1000659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadelman RB, Hanincova K, Mukherjee P, Liveris D, Nowakowski J, McKenna D, Brisson D, Cooper D, Bittker S, Madison G, et al. (2012). Differentiation of reinfection from relapse in recurrent Lyme disease. N Engl J Med 367, 1883–1890. 10.1056/NEJMoa1114362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naj X, Hoffmann AK, Himmel M, and Linder S (2013). The formins FMNL1 and mDia1 regulate coiling phagocytosis of Borrelia burgdorferi by primary human macrophages. Infect Immun 81, 1683–1695. 10.1128/iai.01411-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naj X, and Linder S (2015). ER-Coordinated Activities of Rab22a and Rab5a Drive Phagosomal Compaction and Intracellular Processing of Borrelia burgdorferi by Macrophages. Cell Rep 12, 1816–1830. 10.1016/j.celrep.2015.08.027 [DOI] [PubMed] [Google Scholar]
- Narasimhan S, Booth CJ, DePonte K, Wu MJ, Liang X, Mohanty S, Kantor F, and Fikrig E (2019). Host-specific expression of Ixodes scapularis salivary genes. Ticks Tick Borne Dis 10, 386–397. 10.1016/j.ttbdis.2018.12.001 [DOI] [PubMed] [Google Scholar]
- Narasimhan S, Santiago F, Koski RA, Brei B, Anderson JF, Fish D, and Fikrig E (2002). Examination of the Borrelia burgdorferi transcriptome in Ixodes scapularis during feeding. J Bacteriol 184, 3122–3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayan K, Dail D, Li L, Cadavid D, Amrute S, Fitzgerald-Bocarsly P, and Pachner AR (2005). The nervous system as ectopic germinal center: CXCL13 and IgG in lyme neuroborreliosis. Ann Neurol 57, 813–823. [DOI] [PubMed] [Google Scholar]
- Nardelli DT, Burchill MA, England DM, Torrealba J, Callister SM, and Schell RF (2004). Association of CD4+ CD25+ T cells with prevention of severe destructive arthritis in Borrelia burgdorferivaccinated and challenged gamma interferon-deficient mice treated with anti-interleukin-17 antibody. Clin Diagn Lab Immunol 11, 1075–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestle FO, Di Meglio P, Qin JZ, and Nickoloff BJ (2009). Skin immune sentinels in health and disease. Nat Rev Immunol 9, 679–691. https://doi.org/nri2622 [pii] 10.1038/nri2622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen Hoang AT, Chen P, Bjornfot S, Hogstrand K, Lock JG, Grandien A, Coles M, and Svensson M (2014). Technical advance: live-imaging analysis of human dendritic cell migrating behavior under the influence of immune-stimulating reagents in an organotypic model of lung. J Leukoc Biol 96, 481–489. 10.1189/jlb.3TA0513-303R [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigrovic LE, Lipsett SC, Molins CR, Wormser GP, Bennett JE, Garro AC, Levas MN, Balamuth F, Neville D, Lingampalli N, et al. (2019). Higher C6 enzyme immunoassay index values correlate with a diagnosis of noncutaneous Lyme disease. Diagn Microbiol Infect Dis 94, 160–164. 10.1016/j.diagmicrobio.2018.12.001 [DOI] [PubMed] [Google Scholar]
- Nocton JJ, Dressler F, Rutledge BJ, Rys PN, Persing DH, and Steere AC (1994). Detection of Borrelia burgdorferi DNA by polymerase chain reaction in synovial fluid from patients with Lyme arthritis. N Engl J Med 330, 229–234. [DOI] [PubMed] [Google Scholar]
- Norgard MV, Riley BS, Richardson JA, and Radolf JD (1995). Dermal inflammation elicited by synthetic analogs of Treponema pallidum and Borrelia burgdorferi lipoproteins. Infect Immun 63, 1507–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris SJ (2006). Antigenic variation with a twist--the Borrelia story. Mol Microbiol 60, 1319–1322. https://doi.org/MMI5204 [pii] 10.1111/j.1365-2958.2006.05204.x [DOI] [PubMed] [Google Scholar]
- Nowakowski J, Nadelman RB, Sell R, McKenna D, Cavaliere LF, Holmgren D, Gaidici A, and Wormser GP (2003). Long-term follow-up of patients with culture-confirmed Lyme disease. Am J Med 115, 91–96. https://doi.org/S0002934303003085 [pii] [DOI] [PubMed] [Google Scholar]
- Nuttall PA (2019). Tick saliva and its role in pathogen transmission. Wien Klin Wochenschr. 10.1007/s00508-019-1500-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuttall PA, and Labuda M (2004). Tick-host interactions: saliva-activated transmission. Parasitology 129 Suppl, S177–189. [DOI] [PubMed] [Google Scholar]
- Ohnishi J, Piesman J, and de Silva AM (2001). Antigenic and genetic heterogeneity of Borrelia burgdorferi populations transmitted by ticks. Proc Natl Acad Sci U S A 98, 670–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson CM Jr., Bates TC, Izadi H, Radolf JD, Huber SA, Boyson JE, and Anguita J (2009). Local production of IFN-gamma by invariant NKT cells modulates acute Lyme carditis. J Immunol 182, 3728–3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oosting M, Buffen K, Malireddi SR, Sturm P, Verschueren I, Koenders MI, van de Veerdonk FL, van der Meer JW, Netea MG, Kanneganti TD, et al. (2012). Murine Borrelia arthritis is highly dependent on ASC and caspase-1, but independent of NLRP3. Arthritis Res Ther 14, R247. 10.1186/ar4090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oosting M, Ter Hofstede H, Sturm P, Adema GJ, Kullberg BJ, van der Meer JW, Netea MG, and Joosten LA (2011a). TLR1/TLR2 heterodimers play an important role in the recognition of Borrelia spirochetes. PLoS One 6, e25998. 10.1371/journal.pone.0025998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oosting M, van de Veerdonk FL, Kanneganti TD, Sturm P, Verschueren I, Berende A, van der Meer JW, Kullberg BJ, Netea MG, and Joosten LA (2011b). Borrelia species induce inflammasome activation and IL-17 production through a caspase-1-dependent mechanism. Eur J Immunol 41, 172–181. 10.1002/eji.201040385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pachner AR, and Steiner I (2007). Lyme neuroborreliosis: infection, immunity, and inflammation. Lancet Neurol 6, 544–552. [DOI] [PubMed] [Google Scholar]
- Pal U, Wang P, Bao F, Yang X, Samanta S, Schoen R, Wormser GP, Schwartz I, and Fikrig E (2008). Borrelia burgdorferi basic membrane proteins A and B participate in the genesis of Lyme arthritis. J Exp Med 205, 133–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal U, Yang X, Chen M, Bockenstedt LK, Anderson JF, Flavell RA, Norgard MV, and Fikrig E (2004). OspC facilitates Borrelia burgdorferi invasion of Ixodes scapularis salivary glands. J Clin Invest 113, 220–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paludan SR, Pradeu T, Masters SL, and Mogensen TH (2020). Constitutive immune mechanisms: mediators of host defence and immune regulation. Nat Rev Immunol. 10.1038/s41577-020-0391-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paquette JK, Ma Y, Fisher C, Li J, Lee SB, Zachary JF, Kim YS, Teuscher C, and Weis JJ (2017). Genetic Control of Lyme Arthritis by Borrelia burgdorferi Arthritis-Associated Locus 1 Is Dependent on Localized Differential Production of IFN-beta and Requires Upregulation of Myostatin. J Immunol 199, 3525–3534. 10.4049/jimmunol.1701011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JS, and Kim JH (2018). Role of non-classical T cells in skin immunity. Mol Immunol 103, 286–292. 10.1016/j.molimm.2018.09.024 [DOI] [PubMed] [Google Scholar]
- Park SA, Choe YH, Park E, and Hyun YM (2018). Real-time dynamics of neutrophil clustering in response to phototoxicity-induced cell death and tissue damage in mouse ear dermis. Cell Adh Migr 12, 424–431. 10.1080/19336918.2018.1471322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parthasarathy G, and Philipp MT (2015). Inflammatory mediator release from primary rhesus microglia in response to Borrelia burgdorferi results from the activation of several receptors and pathways. J Neuroinflammation 12, 60. 10.1186/s12974-015-0274-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parthasarathy G, and Philipp MT (2017). Receptor tyrosine kinases play a significant role in human oligodendrocyte inflammation and cell death associated with the Lyme disease bacterium Borrelia burgdorferi. J Neuroinflammation 14, 110. 10.1186/s12974-017-0883-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patarakul K, Cole MF, and Hughes CA (1999). Complement resistance in Borrelia burgdorferi strain 297: outer membrane proteins prevent MAC formation at lysis susceptible sites. Microb Pathog 27, 25–41. [DOI] [PubMed] [Google Scholar]
- Pausa M, Pellis V, Cinco M, Giulianini PG, Presani G, Perticarari S, Murgia R, and Tedesco F (2003). Serum-resistant strains of Borrelia burgdorferi evade complement-mediated killing by expressing a CD59-like complement inhibitory molecule. J Immunol 170, 3214–3222. [DOI] [PubMed] [Google Scholar]
- Pechova J, Stepanova G, Kovar L, and Kopecky J (2002). Tick salivary gland extract-activated transmission of Borrelia afzelii spirochaetes. Folia Parasitol (Praha) 49, 153–159. [PubMed] [Google Scholar]
- Pegalajar-Jurado A, Schriefer ME, Welch RJ, Couturier MR, MacKenzie T, Clark RJ, Ashton LV, Delorey MJ, and Molins CR (2018). Evaluation of Modified Two-Tiered Testing Algorithms for Lyme Disease Laboratory Diagnosis Using Well-Characterized Serum Samples. J Clin Microbiol 56. 10.1128/JCM.01943-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pegram HJ, Andrews DM, Smyth MJ, Darcy PK, and Kershaw MH (2011). Activating and inhibitory receptors of natural killer cells. Immunol Cell Biol 89, 216–224. 10.1038/icb.2010.78 [DOI] [PubMed] [Google Scholar]
- Perner J, Kropackova S, Kopacek P, and Ribeiro JMC (2018). Sialome diversity of ticks revealed by RNAseq of single tick salivary glands. PLoS Negl Trop Dis 12, e0006410. 10.1371/journal.pntd.0006410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson PK, Clawson CC, Lee DA, Garlich DJ, Quie PG, and Johnson RC (1984). Human phagocyte interactions with the Lyme disease spirochete. Infect Immun 46, 608–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petnicki-Ocwieja T, Chung E, Acosta DI, Ramos LT, Shin OS, Ghosh S, Kobzik L, Li X, and Hu LT (2013). TRIF mediates Toll-like receptor 2-dependent inflammatory responses to Borrelia burgdorferi. Infect Immun 81, 402–410. 10.1128/iai.00890-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petnicki-Ocwieja T, Kern A, Killpack TL, Bunnell SC, and Hu LT (2015). Adaptor Protein-3-Mediated Trafficking of TLR2 Ligands Controls Specificity of Inflammatory Responses but Not Adaptor Complex Assembly. J Immunol 195, 4331–4340. 10.4049/jimmunol.1501268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petzke MM, Brooks A, Krupna MA, Mordue D, and Schwartz I (2009). Recognition of Borrelia burgdorferi, the Lyme disease spirochete, by TLR7 and TLR9 induces a type I IFN response by human immune cells. J Immunol 183, 5279–5292. 10.4049/jimmunol.0901390 [DOI] [PubMed] [Google Scholar]
- Piesman J, Dolan MC, Happ CM, Luft BJ, Rooney SE, Mather TN, and Golde WT (1997). Duration of immunity to reinfection with tick-transmitted Borrelia burgdorferi in naturally infected mice. Infect Immun 65, 4043–4047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poltorak A, He X, Smirnova I, Liu MY, Huffel CV, Du X, Birdwell D, Alejos E, Silva M, Galanos C, et al. (1998). Defective LPS Signaling in C3H/HeJ and C57BL/10ScCr Mice: Mutations in Tlr4 Gene. Science 282, 2085–2088. [DOI] [PubMed] [Google Scholar]
- Potter MR, Noben-Trauth N, Weis JH, Teuscher C, and Weis JJ (2000). Interleukin-4 (IL-4) and IL-13 signaling pathways do not regulate Borrelia burgdorferi-induced arthritis in mice: IgG1 is not required for host control of tissue spirochetes. Infect Immun 68, 5603–5609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt CL, and Brown CR (2014). The role of eicosanoids in experimental Lyme arthritis. Front Cell Infect Microbiol 4, 69. 10.3389/fcimb.2014.00069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preac Mursic V, Patsouris E, Wilske B, Reinhardt S, Gross B, and Mehraein P (1990). Persistence of Borrelia burgdorferi and histopathological alterations in experimentally infected animals. A comparison with histopathological findings in human Lyme disease. Infection 18, 332–341. [DOI] [PubMed] [Google Scholar]
- Prinz M, Priller J, Sisodia SS, and Ransohoff RM (2011). Heterogeneity of CNS myeloid cells and their roles in neurodegeneration. Nat Neurosci 14, 1227–1235. 10.1038/nn.2923 [DOI] [PubMed] [Google Scholar]
- Radolf JD, Arndt LL, Akins DR, Curetty LL, Levi ME, Shen Y, Davis LS, and Norgard MV (1995). Treponema pallidum and Borrelia burgdorferi lipoproteins and synthetic lipopeptides activate monocytes/macrophages. J Immunol 154, 2866–2877. [PubMed] [Google Scholar]
- Radolf JD, Caimano MJ, Stevenson B, and Hu LT (2012). Of ticks, mice and men: understanding the dual-host lifestyle of Lyme disease spirochaetes. Nat Rev Microbiol 10, 87–99. https://doi.org/nrmicro2714 [pii] 10.1038/nrmicro2714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radolf JD and Samuels DS (2021). Lyme Disease and Relapsing Fever Spirochetes: Genomics, Molecular Biology, Host Interactions and Disease Pathogenesis (Norfolk, UK: Caister Academic Press; ). 10.21775/9781913652616 [DOI] [Google Scholar]
- Radolf JD, Norgard MV, Brandt ME, Isaacs RD, Thompson PA, and Beutler B (1991). Lipoproteins of Borrelia burgdorferi and Treponema pallidum activate cachectin/tumor necrosis factor synthesis. Analysis using a CAT reporter construct. J Immunol 147, 1968–1974. [PubMed] [Google Scholar]
- Ramamoorthi N, Narasimhan S, Pal U, Bao F, Yang XF, Fish D, Anguita J, Norgard MV, Kantor FS, Anderson JF, et al. (2005). The Lyme disease agent exploits a tick protein to infect the mammalian host. Nature 436, 573–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasley A, Anguita J, and Marriott I (2002). Borrelia burgdorferi induces inflammatory mediator production by murine microglia. J Neuroimmunol 130, 22–31. 10.1016/s0165-5728(02)00187-x [DOI] [PubMed] [Google Scholar]
- Rath PM, Ibershoff B, Mohnhaupt A, Albig J, Eljaschewitsch B, Jurgens D, Horbach I, and Fehrenbach FJ (1996). Seroprevalence of Lyme borreliosis in forestry workers from Brandenburg, Germany. Eur J Clin Microbiol Infect Dis 15, 372–377. 10.1007/BF01690092 [DOI] [PubMed] [Google Scholar]
- Recher M, Lang KS, Hunziker L, Freigang S, Eschli B, Harris NL, Navarini A, Senn BM, Fink K, Lotscher M, et al. (2004). Deliberate removal of T cell help improves virus-neutralizing antibody production. Nat Immunol 5, 934–942. 10.1038/ni1102 [DOI] [PubMed] [Google Scholar]
- Reimers CD, de Koning J, Neubert U, Preac-Mursic V, Koster JG, Muller-Felber W, Pongratz DE, and Duray PH (1993). Borrelia burgdorferi myositis: report of eight patients. J Neurol 240, 278–283. 10.1007/BF00838161 [DOI] [PubMed] [Google Scholar]
- Reinhardt A, Yevsa T, Worbs T, Lienenklaus S, Sandrock I, Oberdorfer L, Korn T, Weiss S, Forster R, and Prinz I (2016). Interleukin-23-Dependent gamma/delta T Cells Produce Interleukin-17 and Accumulate in the Enthesis, Aortic Valve, and Ciliary Body in Mice. Arthritis Rheumatol 68, 2476–2486. 10.1002/art.39732 [DOI] [PubMed] [Google Scholar]
- Reinink P, Souter MNT, Cheng TY, van Gorkom T, Lenz S, Kubler-Kielb J, Strle K, Kremer K, Thijsen SFT, Steere AC, et al. (2019). CD1b presents self and Borrelia burgdorferi diacylglycerols to human T cells. Eur J Immunol 49, 737–746. 10.1002/eji.201847949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro JM, Alarcon-Chaidez F, Francischetti IM, Mans BJ, Mather TN, Valenzuela JG, and Wikel SK (2006). An annotated catalog of salivary gland transcripts from Ixodes scapularis ticks. Insect Biochem Mol Biol 36, 111–129. [DOI] [PubMed] [Google Scholar]
- Ribeiro JM, Makoul GT, Levine J, Robinson DR, and Spielman A (1985). Antihemostatic, antiinflammatory, and immunosuppressive properties of the saliva of a tick, Ixodes dammini. J Exp Med 161, 332–344. 10.1084/jem.161.2.332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro JM, Makoul GT, and Robinson DR (1988). Ixodes dammini: evidence for salivary prostacyclin secretion. J Parasitol 74, 1068–1069. [PubMed] [Google Scholar]
- Rittig MG, Jagoda JC, Wilske B, Murgia R, Cinco M, Repp R, Burmester GR, and Krause A (1998). Coiling phagocytosis discriminates between different spirochetes and is enhanced by phorbol myristate acetate and granulocyte-macrophage colony-stimulating factor. Infect Immun 66, 627–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rittig MG, Krause A, Haupl T, Schaible UE, Modolell M, Kramer MD, Lutjen-Drecoll E, Simon MM, and Burmester GR (1992). Coiling phagocytosis is the preferential phagocytic mechanism for Borrelia burgdorferi. Infect Immun 60, 4205–4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts ED, Bohm RF, Cogswell FB, NorbertLanners H, Lowrie RC, Povinelli L, Piesman J, and Philipp MT (1995). Chronic Lyme disease in the rhesus monkey. Lab Invest 72, 146–160. [PubMed] [Google Scholar]
- Rock FL, Hardiman G, Timans JC, Kastelein RA, and Bazan JF (1998). A family of human receptors structurally related to Drosophila Toll. Proc Natl Acad Sci U S A 95, 588–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogovskyy AS, and Bankhead T (2013). Variable VlsE is critical for host reinfection by the Lyme disease spirochete. PLoS One 8, e61226. 10.1371/journal.pone.0061226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosa PA, Tilly K, and Stewart PE (2005). The burgeoning molecular genetics of the Lyme disease spirochaete. Nat Rev Microbiol 3, 129–143. [DOI] [PubMed] [Google Scholar]
- Ruderman EM, Kerr JS, Telford SR 3rd, Spielman A, Glimcher LH, and Gravallese EM (1995). Early murine Lyme carditis has a macrophage predominance and is independent of major histocompatibility complex class II-CD4+ T cell interactions. J Infect Dis 171, 362–370. [DOI] [PubMed] [Google Scholar]
- Rupprecht TA, Kirschning CJ, Popp B, Kastenbauer S, Fingerle V, Pfister HW, and Koedel U (2007). Borrelia garinii induces CXCL13 production in human monocytes through Toll-like receptor 2. Infect Immun 75, 4351–4356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sa-Nunes A, Bafica A, Lucas DA, Conrads TP, Veenstra TD, Andersen JF, Mather TN, Ribeiro JM, and Francischetti IM (2007). Prostaglandin E2 is a major inhibitor of dendritic cell maturation and function in Ixodes scapularis saliva. J Immunol 179, 1497–1505. [DOI] [PubMed] [Google Scholar]
- Sabino GJ, Hwang SJ, McAllister SC, Mena P, and Furie MB (2011). Interferon-gamma influences the composition of leukocytic infiltrates in murine Lyme carditis. Am J Pathol 179, 1917–1928. 10.1016/j.ajpath.2011.06.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadziene A, Jonsson M, Bergstrom S, Bright RK, Kennedy RC, and Barbour AG (1994). A bactericidal antibody to Borrelia burgdorferi is directed against a variable region of the OspB protein. Infect Immun 62, 2037–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadziene A, Thompson PA, and Barbour AG (1993). In vitro inhibition of Borrelia burgdorferi growth by antibodies. J Infect Dis 167, 165–172. [DOI] [PubMed] [Google Scholar]
- Sahay B, Singh A, Gnanamani A, Patsey RL, Blalock JE, and Sellati TJ (2011). CD14 signaling reciprocally controls collagen deposition and turnover to regulate the development of lyme arthritis. Am J Pathol 178, 724–734. 10.1016/j.ajpath.2010.10.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi S (2004). Naturally arising CD4+ regulatory T cells for immunologic self-tolerance and negative control of immune responses. Annu Rev Immunol 22, 531–562. [DOI] [PubMed] [Google Scholar]
- Sakai S, Mayer-Barber KD, and Barber DL (2014). Defining features of protective CD4 T cell responses to Mycobacterium tuberculosis. Curr Opin Immunol 29, 137–142. https://doi.org/S0952-7915(14)00084-3 [pii] 10.1016/j.coi.2014.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar JC, Duhnam-Ems S, La Vake C, Cruz AR, Moore MW, Caimano MJ, Velez-Climent L, Shupe J, Krueger W, and Radolf JD (2009). Activation of human monocytes by live Borrelia burgdorferi generates TLR2-dependent and -independent responses which include induction of IFNβ. PLoS Pathog 5, e1000444. 10.1371/journal.ppat.1000444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar JC, Pope CD, Moore MW, Pope J, Kiely TG, and Radolf JD (2005). Lipoprotein-dependent and -independent immune responses to spirochetal infection. Clin Diagn Lab Immunol 12, 949–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar JC, Pope CD, Sellati TJ, Feder HM Jr., Kiely TG, Dardick KR, Buckman RL, Moore MW, Caimano MJ, Pope JG, et al. (2003). Coevolution of markers of innate and adaptive immunity in skin and peripheral blood of patients with erythema migrans. J Immunol 171, 2660–2670. [DOI] [PubMed] [Google Scholar]
- Sallusto F (2016). Heterogeneity of Human CD4(+) T Cells Against Microbes. Annu Rev Immunol 34, 317–334. 10.1146/annurev-immunol-032414-112056 [DOI] [PubMed] [Google Scholar]
- Samuels DS (2011). Gene regulation in Borrelia burgdorferi. Annu Rev Microbiol 65, 479–499. 10.1146/annurev.micro.112408.134040 [DOI] [PubMed] [Google Scholar]
- Schaible UE, Kramer MD, Museteanu C, Zimmer G, Mossmann H, and Simon M (1990). Lyme borreliosis in the severe combined immunodeficiency (scid) mouse manifests predominantly in the joints, heart and liver. Amer J Pathol 137, 811–820. [PMC free article] [PubMed] [Google Scholar]
- Schaible UE, Kramer MD, Museteanu C, Zimmer G, Mossmann H, and Simon MM (1989). The severe combined immunodeficiency (scid) mouse. A laboratory model for the analysis of Lyme arthritis and carditis. J Exp Med 170, 1427–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaible UE, Wallich R, Kramer MD, Nerz G, Stehle T, Museteanu C, and Simon MM (1994). Protection against Borrelia burgdorferi infection in SCID mice is conferred by presensitized spleen cells and partially by B but not T cells alone. Int Immunol 6, 671–681. [DOI] [PubMed] [Google Scholar]
- Schoenfeld R, Araneo B, Ma Y, Yang LM, and Weis JJ (1992). Demonstration of a B-lymphocyte mitogen produced by the Lyme disease pathogen, Borrelia burgdorferi. Infect Immun 60, 455–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuijt TJ, Coumou J, Narasimhan S, Dai J, Deponte K, Wouters D, Brouwer M, Oei A, Roelofs JJ, van Dam AP, et al. (2011). A tick mannose-binding lectin inhibitor interferes with the vertebrate complement cascade to enhance transmission of the Lyme disease agent. Cell Host Microbe 10, 136–146. 10.1016/j.chom.2011.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutzer SE, Coyle PK, Belman AL, Golightly MG, and Drulle J (1990). Sequestration of antibody to Borrelia burgdorferi in immune complexes in seronegative Lyme disease. Lancet 335, 312–315. 10.1016/0140-6736(90)90606-6 [DOI] [PubMed] [Google Scholar]
- Schutzer SE, Coyle PK, Krupp LB, Deng Z, Belman AL, Dattwyler R, and Luft BJ (1997). Simultaneous expression of Borrelia OspA and OspC and IgM response in cerebrospinal fluid in early neurologic Lyme disease. J Clin Invest 100, 763–767. 10.1172/JCI119589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutzer SE, and Luan J (2003). Early OspA immune complex formation in animal models of Lyme disease. J Mol Microbiol Biotechnol 5, 167–171. 10.1159/000070267 [DOI] [PubMed] [Google Scholar]
- Schwan TG, Piesman J, Golde WT, Dolan MC, and Rosa PA (1995). Induction of an outer surface protein on Borrelia burgdorferi during tick feeding. Proc Natl Acad Sci U S A 92, 2909–2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellati TJ, Abrescia LD, Radolf JD, and Furie MB (1996). Outer surface lipoproteins of Borrelia burgdorferi activate vascular endothelium in vitro. Infect Immun 64, 3180–3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellati TJ, Bouis DA, Kitchens RL, Darveau RP, Pugin J, Ulevitch RJ, Gangloff SC, Goyert SM, Norgard MV, and Radolf JD (1998). Treponema pallidum and Borrelia burgdorferi lipoproteins and synthetic lipopeptides activate monocytic cells via a CD14-dependent pathway distinct from that used by lipopolysaccharide. J Immunol 160, 5455–5464. [PubMed] [Google Scholar]
- Sellati TJ, Burns MJ, Ficazzola MA, and Furie MB (1995). Borrelia burgdorferi upregulates expression of adhesion molecules on endothelial cells and promotes transendothelial migration of neutrophils in vitro. Infection and Immunity 63, 4439–4447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahinian A, Pfeffer K, Lee KP, Kundig TM, Kishihara K, Wakeham A, Kawai K, Ohashi PS, Thompson CB, and Mak TW (1993). Differential T cell costimulatory requirements in CD28-deficient mice. Science 261, 609–612. [DOI] [PubMed] [Google Scholar]
- Shen S, Shin JJ, Strle K, McHugh G, Li X, Glickstein LJ, Drouin EE, and Steere AC (2010). Treg cell numbers and function in patients with antibiotic-refractory or antibiotic-responsive Lyme arthritis. Arthritis Rheum 62, 2127–2137. 10.1002/art.27468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherlock JP, Joyce-Shaikh B, Turner SP, Chao CC, Sathe M, Grein J, Gorman DM, Bowman EP, McClanahan TK, Yearley JH, et al. (2012). IL-23 induces spondyloarthropathy by acting on ROR-gammat+ CD3+CD4-CD8- entheseal resident T cells. Nat Med 18, 1069–1076. 10.1038/nm.2817 [DOI] [PubMed] [Google Scholar]
- Shih CM, Pollack RJ, Telford SR 3rd, and Spielman A (1992). Delayed dissemination of Lyme disease spirochetes from the site of deposition in the skin of mice. J Infect Dis 166, 827–831. [DOI] [PubMed] [Google Scholar]
- Shih CM, Telford SR 3rd, Pollack RJ, and Spielman A (1993). Rapid dissemination by the agent of Lyme disease in hosts that permit fulminating infection. Infect Immun 61, 2396–2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JJ, Glickstein LJ, and Steere AC (2007). High levels of inflammatory chemokines and cytokines in joint fluid and synovial tissue throughout the course of antibiotic-refractory Lyme arthritis. Arthritis Rheum 56, 1325–1335. [DOI] [PubMed] [Google Scholar]
- Shin OS, Isberg R, Akira S, Uematsu S, Behera AK, and Hu LT (2008a). Distinct roles for MyD88, TLR2, TLR5, and TLR9 in phagocytosis of Borrelia burgdorferi and inflammatory signaling. Infect Immun 76, 2341–2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin OS, Isberg RR, Akira S, Uematsu S, Behera AK, and Hu LT (2008b). Distinct roles for MyD88 and Toll-like receptors 2, 5, and 9 in phagocytosis of Borrelia burgdorferi and cytokine induction. Infect Immun 76, 2341–2351. 10.1128/IAI.01600-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigal LH, Steere AC, and Dwyer JM (1988). In vivo and in vitro evidence of B cell hyperactivity during Lyme disease. J Rheumatol 15, 648–654. [PubMed] [Google Scholar]
- Silberer M, Koszik F, Stingl G, and Aberer E (2000). Downregulation of class II molecules on epidermal Langerhans cells in Lyme borreliosis. Br J Dermatol 143, 786–794. https://doi.org/bjd3776 [pii] [DOI] [PubMed] [Google Scholar]
- Skare JT, and Garcia BL (2020). Complement Evasion by Lyme Disease Spirochetes. Trends Microbiol. 10.1016/j.tim.2020.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skare JT, Shaw DK, Trzeciakowski JP, and Hyde JA (2016). In Vivo Imaging Demonstrates That Borrelia burgdorferi ospC Is Uniquely Expressed Temporally and Spatially throughout Experimental Infection. PLoS One 11, e0162501. 10.1371/journal.pone.0162501PONE-D-16-28292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonderegger FL, Ma Y, Maylor-Hagan H, Brewster J, Huang X, Spangrude GJ, Zachary JF, Weis JH, and Weis JJ (2012a). Localized production of IL-10 suppresses early inflammatory cell infiltration and subsequent development of IFN-gamma-mediated Lyme arthritis. J Immunol 188, 1381–1393. 10.4049/jimmunol.1102359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonderegger FL, Ma Y, Maylor-Hagan H, Brewster J, Huang X, Spangrude GJ, Zachary JF, Weis JH, and Weis JJ (2012b). Localized production of IL-10 suppresses early inflammatory cell infiltration and subsequent development of IFNγ-mediated Lyme arthritis. J Immunol 188, 1381–1393. https://doi.org/jimmunol.1102359 [pii] 10.4049/jimmunol.1102359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soulas P, Woods A, Jaulhac B, Knapp AM, Pasquali JL, Martin T, and Korganow AS (2005). Autoantigen, innate immunity, and T cells cooperate to break B cell tolerance during bacterial infection. J Clin Invest 115, 2257–2267. 10.1172/JCI24646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steere AC (2001). Lyme disease. N Engl J Med 345, 115–125. 10.1056/NEJM200107123450207 [DOI] [PubMed] [Google Scholar]
- Steere AC, and Angelis SM (2006). Therapy for Lyme arthritis: strategies for the treatment of antibiotic-refractory arthritis. Arthritis Rheum 54, 3079–3086. [DOI] [PubMed] [Google Scholar]
- Steere AC, Berardi VP, Weeks KE, Logigian EL, and Ackermann R (1990). Evaluation of the intrathecal antibody response to Borrelia burgdorferi as a diagnostic test for Lyme neuroborreliosis. J Infect Dis 161, 1203–1209. 10.1093/infdis/161.6.1203 [DOI] [PubMed] [Google Scholar]
- Steere AC, Hardin JA, Ruddy S, Mummaw JG, and Malawista SE (1979). Lyme arthritis: correlation of serum and cryoglobulin IgM with activity, and serum IgG with remission. Arthritis Rheum 22, 471–483. 10.1002/art.1780220506 [DOI] [PubMed] [Google Scholar]
- Steere AC, Klitz W, Drouin EE, Falk BA, Kwok WW, Nepom GT, and Baxter-Lowe LA (2006). Antibiotic-refractory Lyme arthritis is associated with HLA-DR molecules that bind a Borrelia burgdorferi peptide. J Exp Med 203, 961–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steere AC, McHugh G, Damle N, and Sikand VK (2008). Prospective study of serologic tests for Lyme disease. Clin Infect Dis 47, 188–195. 10.1086/589242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steere AC, Schoen RT, and Taylor E (1987). The clinical evolution of Lyme arthritis. Ann Intern Med 107, 725–731. [DOI] [PubMed] [Google Scholar]
- Steere AC, Strle F, Wormser GP, Hu LT, Branda JA, Hovius JW, Li X, and Mead PS (2016). Lyme borreliosis. Nat Rev Dis Primers 2, 16090. 10.1038/nrdp.2016.90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson B, El-Hage N, Hines MA, Miller JC, and Babb K (2002). Differential binding of host complement inhibitor factor H by Borrelia burgdorferi Erp surface proteins: a possible mechanism underlying the expansive host range of Lyme disease spirochetes. Infect Immun 70, 491–497. 10.1128/iai.70.2.491-497.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson B, Schwan TG, and Rosa PA (1995). Temperature-related differential expression of antigens in the Lyme disease spirochete, Borrelia burgdorferi. Infect Immun 63, 4535–4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart PE, Byram R, Grimm D, Tilly K, and Rosa PA (2005). The plasmids of Borrelia burgdorferi: essential genetic elements of a pathogen. Plasmid 53, 1–13. [DOI] [PubMed] [Google Scholar]
- Straubinger RK, Summers BA, Chang YF, and Appel MJG (1997). Persistence of Borrelia burgdorferi in experimentally infected dogs after antibiotic treatment. J Clin Microbiol 35, 111–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strle K, Drouin EE, Shen S, Khoury JEI, McHugh G, Ruzic-Sabljic E, Strle F, and Steere AC (2009). Borrelia burgdorferi Stimulates Macrophages to Secrete Higher Levels of Cytokines and Chemokines than Borrelia afzelii or Borrelia garinii. J Infect Dis 200, 1936–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strle K, Jones KL, Drouin EE, Li X, and Steere AC (2011). Borrelia burgdorferi RST1 (OspC type A) genotype is associated with greater inflammation and more severe Lyme disease. Am J Pathol 178, 2726–2739. 10.1016/j.ajpath.2011.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strle K, Shin JJ, Glickstein LJ, and Steere AC (2012). Association of a Toll-like receptor 1 polymorphism with heightened Th1 inflammatory responses and antibiotic-refractory Lyme arthritis. Arthritis Rheum 64, 1497–1507. 10.1002/art.34383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strle K, Sulka KB, Pianta A, Crowley JT, Arvikar SL, Anselmo A, Sadreyev R, and Steere AC (2017). T-Helper 17 Cell Cytokine Responses in Lyme Disease Correlate With Borrelia burgdorferi Antibodies During Early Infection and With Autoantibodies Late in the Illness in Patients With Antibiotic-Refractory Lyme Arthritis. Clin Infect Dis 64, 930–938. 10.1093/cid/cix002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhonen J, Hartiala K, Tuominen-Gustafsson H, and Viljanen MK (2000). Borrelia burgdorferi--induced oxidative burst, calcium mobilization, and phagocytosis of human neutrophils are complement dependent. J Infect Dis 181, 195–202. [DOI] [PubMed] [Google Scholar]
- Suhonen J, Hartiala K, Tuominen-Gustafsson H, and Viljanen MK (2002). Sublethal concentrations of complement can effectively opsonize Borrelia burgdorferi. Scand J Immunol 56, 554–560. 10.1046/j.1365-3083.2002.01171.x [DOI] [PubMed] [Google Scholar]
- Suhonen J, Hartiala K, and Viljanen MK (1998). Tube phagocytosis, a novel way for neutrophils to phagocytize Borrelia burgdorferi. Infect Immun 66, 3433–3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulka KB, Strle K, Crowley JT, Lochhead RB, Anthony R, and Steere AC (2018). Correlation of Lyme Disease-Associated IgG4 Autoantibodies With Synovial Pathology in Antibiotic-Refractory Lyme Arthritis. Arthritis Rheumatol 70, 1835–1846. 10.1002/art.40566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson KV, Deng M, and Ting JP (2019). The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol 19, 477–489. 10.1038/s41577-019-0165-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczepanski A, Furie MB, Benach JL, Lane BP, and Fleit HB (1990). Interaction between Borrelia burgdorferi and endothelium in vitro. J Clin Invest 85, 1637–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talkington J, and Nickell SP (1999). Borrelia burgdorferi spirochetes induce mast cell activation and cytokine release. Infect Immun 67, 1107–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavora F, Burke A, Li L, Franks TJ, and Virmani R (2008). Postmortem confirmation of Lyme carditis with polymerase chain reaction. Cardiovasc Pathol 17, 103–107. 10.1016/j.carpath.2007.03.004 [DOI] [PubMed] [Google Scholar]
- Thompson A, Mannix R, and Bachur R (2009). Acute pediatric monoarticular arthritis: distinguishing Lyme arthritis from other etiologies. Pediatrics 123, 959–965. 10.1542/peds.2008-1511 [DOI] [PubMed] [Google Scholar]
- Tilly K, Krum JG, Bestor A, Jewett MW, Grimm D, Bueschel D, Byram R, Dorward D, Vanraden MJ, Stewart P, et al. (2006). Borrelia burgdorferi OspC protein required exclusively in a crucial early stage of mammalian infection. Infect Immun 74, 3554–3564. 10.1128/IAI.01950-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjernberg I, Sillanpaa H, Seppala I, Eliasson I, Forsberg P, and Lahdenne P (2009). Antibody responses to Borrelia IR(6) peptide variants and the C6 peptide in Swedish patients with erythema migrans. Int J Med Microbiol 299, 439–446. 10.1016/j.ijmm.2008.10.006 [DOI] [PubMed] [Google Scholar]
- Trollmo C, Meyer AL, Steere AC, Hafler DA, and Huber BT (2001). Molecular mimicry in Lyme arthritis demonstrated at the single cell level: LFA-1 alpha L is a partial agonist for outer surface protein A-reactive T cells. J Immunol 166, 5286–5291. 10.4049/jimmunol.166.8.5286 [DOI] [PubMed] [Google Scholar]
- Troy EB, Lin T, Gao L, Lazinski DW, Camilli A, Norris SJ, and Hu LT (2013). Understanding barriers to Borrelia burgdorferi dissemination during infection using massively parallel sequencing. Infect Immun 81, 2347–2357. 10.1128/IAI.00266-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubo NJ, and Jenkins MK (2014). CD4+ T Cells: guardians of the phagosome. Clin Microbiol Rev 27, 200–213. https://doi.org/27/2/200 [pii] 10.1128/CMR.00097-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tunev SS, Hastey CJ, Hodzic E, Feng S, Barthold SW, and Baumgarth N (2011). Lymphoadenopathy during Lyme borreliosis is caused by spirochete migration-induced specific B cell activation. PLoS Pathog 7, e1002066. 10.1371/journal.ppat.1002066PPATHOGENS-D-10-00234 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tupin E, Benhnia MR, Kinjo Y, Patsey R, Lena CJ, Haller MC, Caimano MJ, Imamura M, Wong CH, Crotty S, et al. (2008). NKT cells prevent chronic joint inflammation after infection with Borrelia burgdorferi. Proc Natl Acad Sci U S A 105, 19863–19868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyson K, Elkins C, Patterson H, Fikrig E, and de Silva A (2007). Biochemical and functional characterization of Salp20, an Ixodes scapularis tick salivary protein that inhibits the complement pathway. Insect Mol Biol 16, 469–479. [DOI] [PubMed] [Google Scholar]
- Ullah MO, Sweet MJ, Mansell A, Kellie S, and Kobe B (2016). TRIF-dependent TLR signaling, its functions in host defense and inflammation, and its potential as a therapeutic target. J Leukoc Biol 100, 27–45. 10.1189/jlb.2RI1115-531R [DOI] [PubMed] [Google Scholar]
- Valenzuela JG, Charlab R, Mather TN, and Ribeiro JM (2000). Purification, cloning, and expression of a novel salivary anticomplement protein from the tick, Ixodes scapularis. J Biol Chem 275, 18717–18723. [DOI] [PubMed] [Google Scholar]
- Vesely DL, Fish D, Shlomchik MJ, Kaplan DH, and Bockenstedt LK (2009). Langerhans cell deficiency impairs Ixodes scapularis suppression of Th1 responses in mice. Infect Immun 77, 1881–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigano S, Perreau M, Pantaleo G, and Harari A (2012). Positive and negative regulation of cellular immune responses in physiologic conditions and diseases. Clin Dev Immunol 2012, 485781. 10.1155/2012/485781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vudattu NK, Strle K, Steere AC, and Drouin EE (2013). Dysregulation of CD4+CD25(high) T cells in the synovial fluid of patients with antibiotic-refractory Lyme arthritis. Arthritis Rheum 65, 1643–1653. 10.1002/art.37910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagemakers A, Coumou J, Schuijt TJ, Oei A, Nijhof AM, van ‘t Veer C, van der Poll T, Bins AD, and Hovius JW (2016). An Ixodes ricinus Tick Salivary Lectin Pathway Inhibitor Protects Borrelia burgdorferi sensu lato from Human Complement. Vector Borne Zoonotic Dis 16, 223–228. 10.1089/vbz.2015.1901 [DOI] [PubMed] [Google Scholar]
- Wang G, Petzke MM, Iyer R, Wu H, and Schwartz I (2008). Pattern of proinflammatory cytokine induction in RAW264.7 mouse macrophages is identical for virulent and attenuated Borrelia burgdorferi. J Immunol 180, 8306–8315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, and Nuttall PA (1999). Immunoglobulin-binding proteins in ticks: new target for vaccine development against a blood-feeding parasite. Cell Mol Life Sci 56, 286–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Ma Y, Weis JH, Zachary JF, Kirschning CJ, and Weis JJ (2005). Relative contributions of innate and acquired host responses to bacterial control and arthritis development in Lyme disease. Infect Immun 73, 657–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson J, and Riblet R (1974). Genetic control of responses to bacterial lipopolysaccharides in mice. I. Evidence for a single gene that influences mitogenic and immunogenic respones to lipopolysaccharides. J Exp Med 140, 1147–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein DL, Lissner CR, Swanson RN, and O’Brien AD (1986). Macrophage defect and inflammatory cell recruitment dysfunction in Salmonella-susceptible C3H/HeJ mice. Cell Immunol 102, 68–77. [DOI] [PubMed] [Google Scholar]
- Whiteside SK, Snook JP, Ma Y, Sonderegger FL, Fisher C, Petersen C, Zachary JF, Round JL, Williams MA, and Weis JJ (2018). IL-10 Deficiency Reveals a Role for TLR2-Dependent Bystander Activation of T Cells in Lyme Arthritis. J Immunol 200, 1457–1470. 10.4049/jimmunol.1701248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis AA, Widmann RF, Flynn JM, Green DW, and Onel KB (2003). Lyme arthritis presenting as acute septic arthritis in children. J Pediatr Orthop 23, 114–118. [PubMed] [Google Scholar]
- Woodman ME, Cooley AE, Miller JC, Lazarus JJ, Tucker K, Bykowski T, Botto M, Hellwage J, Wooten RM, and Stevenson B (2007). Borrelia burgdorferi binding of host complement regulator factor H is not required for efficient mammalian infection. Infect Immun 75, 3131–3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods A, Soulas-Sprauel P, Jaulhac B, Arditi B, Knapp AM, Pasquali JL, Korganow AS, and Martin T (2008). MyD88 negatively controls hypergammaglobulinemia with autoantibody production during bacterial infection. Infect Immun 76, 1657–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wooten RM, Ma Y, Yoder RA, Brown JP, Weis JH, Zachary JF, Kirschning CJ, and Weis JJ (2002a). Toll-like receptor 2 is required for innate, but not acquired, host defense to Borrelia burgdorferi. J Immunol 168, 348–355. [DOI] [PubMed] [Google Scholar]
- Wooten RM, Ma Y, Yoder RA, Brown JP, Weis JH, Zachary JF, Kirschning CJ, and Weis JJ (2002b). Toll-like receptor 2 plays a pivotal role in host defense and inflammatory response to Borrelia burgdorferi. Vector Borne Zoonotic Dis 2, 275–278. [DOI] [PubMed] [Google Scholar]
- Wooten RM, Modur VR, McIntyre TM, and Weis JJ (1996). Borrelia burgdorferi outer membrane protein A induces nuclear translocation of nuclear factor-kB and inflammatory activation in human endothelial cells. J Immunol 157, 4584–4590. [PubMed] [Google Scholar]
- Wooten RM, Morrison TB, Weis JH, Wright SD, Thieringer R, and Weis JJ (1998). The role of CD14 in signaling mediated by outer membrane lipoproteins of Borrelia burgdorferi. J Immunol 160, 5485–5492. [PubMed] [Google Scholar]
- Wormser GP, Nowakowski J, Nadelman RB, Visintainer P, Levin A, and Aguero-Rosenfeld ME (2008). Impact of clinical variables on Borrelia burgdorferi-specific antibody seropositivity in acute-phase sera from patients in North America with culture-confirmed early Lyme disease. Clin Vaccine Immunol 15, 1519–1522. 10.1128/CVI.00109-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Seemanapalli SV, Reif KE, Brown CR, and Liang FT (2007). Increasing the recruitment of neutrophils to the site of infection dramatically attenuates Borrelia burgdorferi infectivity. J Immunol 178, 5109–5115. [DOI] [PubMed] [Google Scholar]
- Yakimchuk K, Roura-Mir C, Magalhaes KG, de Jong A, Kasmar AG, Granter SR, Budd R, Steere A, Pena-Cruz V, Kirschning C, et al. (2011). Borrelia burgdorferi infection regulates CD1 expression in human cells and tissues via IL1-beta. Eur J Immunol 41, 694–705. 10.1002/eji.201040808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Ma Y, Schoenfeld R, Griffiths M, Eichwald E, Araneo B, and Weis JJ (1992). Evidence for B-lymphocyte mitogen activity in Borrelia burgdorferi-infected mice. Infect Immun 60, 3033–3041. 10.1128/IAI.60.8.3033-3041.1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang RB, Mark MR, Gray A, Huang A, Xie MH, Zhang M, Goddard A, Wood WI, Gurney AL, and Godowski PJ (1998). Toll-like receptor-2 mediates lipopolysaccharide-induced cellular signalling. Nature 395, 284–288. [DOI] [PubMed] [Google Scholar]
- Yoder A, Wang X, Ma Y, Philipp MT, Heilbrun M, Weis JH, Kirschning CJ, Wooten RM, and Weis JJ (2003). Tripalmitoyl-S-glyceryl-cysteine-dependent OspA vaccination of toll-like receptor 2-deficient mice results in effective protection from Borrelia burgdorferi challenge. Infect Immun 71, 3894–3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeidner N, Dreitz M, Belasco D, and Fish D (1996). Suppression of acute Ixodes scapularis-induced Borrelia burgdorferi infection using tumor necrosis factor-alpha, interleukin-2, and interferon-gamma. J Infect Dis 173, 187–195. [DOI] [PubMed] [Google Scholar]
- Zeidner NS, Schneider BS, Nuncio MS, Gern L, and Piesman J (2002). Coinoculation of Borrelia spp. with tick salivary gland lysate enhances spirochete load in mice and is tick species-specific. J Parasitol 88, 1276–1278. [DOI] [PubMed] [Google Scholar]
- Zeng X, Wei YL, Huang J, Newell EW, Yu H, Kidd BA, Kuhns MS, Waters RW, Davis MM, Weaver CT, et al. (2012). Gammadelta T cells recognize a microbial encoded B cell antigen to initiate a rapid antigen-specific interleukin-17 response. Immunity 37, 524–534. 10.1016/j.immuni.2012.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JR, Hardham JM, Barbour AG, and Norris SJ (1997). Antigenic variation in Lyme disease Borreliae by promiscuous recombination of VMP-like sequence cassettes. Cell 89, 275–285. https://doi.org/S0092-8674(00)80206-8 [pii] [DOI] [PubMed] [Google Scholar]
- Zhang JR, and Norris SJ (1998a). Genetic variation of the Borrelia burgdorferi gene vlsE involves cassette-specific, segmental gene conversion. Infect Immun 66, 3698–3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JR, and Norris SJ (1998b). Kinetics and in vivo induction of genetic variation of vlsE in Borrelia burgdorferi. Infect Immun 66, 3689–3697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Fleming R, McCloud B, and Klempner MS (2007). CD14 mediates cross talk between mononuclear cells and fibroblasts for upregulation of matrix metalloproteinase 9 by Borrelia burgdorferi. Infect Immun 75, 3062–3069. 10.1128/IAI.00202-07 [DOI] [PMC free article] [PubMed] [Google Scholar]