

Abstract
Obesity is a chronic, multifactorial, and morbid disease. In the United States, 69% of adults are overweight or have obesity and the global prevalence of obesity is increasing. Obesity is influenced by genetic, neurologic, metabolic, enteric, and behavioral processes. It remains a key modifiable risk factor for many comorbid diseases, including cardiovascular disease, diabetes mellitus, and cancer. While there are recent and significant advances in obesity therapy, including diets, lifestyle modifications, pharmacotherapies, endoscopic procedures, and bariatric surgeries, there is an immense need for a better understanding of the heterogeneity in obesity pathophysiology and outcomes. Here we review salient pathophysiology underlying the development and morbidity of obesity, as well as pathophysiologically based classification systems that inform current obesity management and may inform improved and individualized management in the future.
Keywords: Obesity, Bariatric, Obesity Comorbidities, Adipose
BACKGROUND
Obesity is a chronic and multifactorial disease and has become a worldwide epidemic with devastating health and economic consequences. In 2015, there were an estimated 603.7 million adults and 107.7 million children with obesity worldwide and the rate of obesity had doubled in nearly half of countries since 1980.1 Projections estimate that obesity will affect about 1 in every 2 adults in the United States by 2030.2 Further, in the United States alone, obesity costs the healthcare system an estimated $480 billion annually.3
The World Health Organization defines obesity as an excess of adiposity that increases health risks. Obesity is usually classified by a body mass index (BMI) greater than 30 kg/m.4 Obesity affects essentially every organ system in the body and obesity is a risk factor for many morbid conditions, the most impactful of which are type 2 diabetes (T2DM), hypertension (HTN), dyslipidemia, cardiovascular disease (CVD), and some cancers.5 Obesity also directly decreases life expectancy.6,7 Furthermore, it is one of the most severe risk factors for hospitalization and mortality from coronavirus disease 2019 (Covid-19).8 Although smoking remains the leading risk factor for global disease burden in several countries, high BMI became the leading risk in several parts of the world including Australasia and southern Latin America.9
Obesity outcomes and responses to weight loss interventions (i.e., lifestyle modifications, anti-obesity medications [AOM], endoscopic procedures, and bariatric surgery) vary significantly between individuals. Advances in our understanding of obesogenic processes, the pathophysiology of obesity related comorbidities, and how these processes vary between individuals help explain some of this heterogeneity. Recently developed physiologic and risk-based obesity categorizations serve as tools to classify individuals based on some of these differences, including the degree of central obesity, metabolic dysfunction, abnormalities in mechanical gastric functions, and satiation. These advances enhance the assessment of individual patient risk and have also allowed for the identification of variable obesogenic processes that may be therapeutic targets in afflicted groups. This review will discuss the pathophysiology and comorbidity of obesity, heterogeneity in etiology and outcomes, and impactful obesity classification systems.
METHODS
We searched PubMed and Google Scholar for articles published in English relating to the epidemiology, physiology and pathophysiology, and comorbidity of obesity. Our search focused on recent genetic and physiologic studies in human subjects and, where human trials were insufficient, animal models, as well as cross-sectional epidemiologic studies, systematic reviews and meta-analyses, randomized clinical trials (RCTs), and prospective and retrospective cohort studies. There was no specified range of publication dates to restrict this literature search though preference was given to studies published within the last 10 years.
OBESITY PATHOPHYSIOLOGY: ENERGY REGULATION
Energy homeostasis can be divided into energy intake and expenditure. A positive energy balance drives weight gain.10 Established genetic and environmental factors are associated with a positive energy balance and the development of obesity. Physiologically, energy regulation involves complex, reciprocal processes including enteric sensory-motor processes, peripheral hormonal signaling, and peripheral and central neurologic pathways. Understanding these obesity related traits and processes is crucial to a functional understanding of obesity itself.
Genetics
Obesity can be partially explained by a combination of genetic factors and environmental conditions. The hereditability of BMI is estimated between 40–70% based on twin, family, and adoption studies.11 However, single-gene mutation (monogenic) obesity is rare and occurs in less than 1% of cases12 Most of the single-gene mutations identified are connected to the leptin-melanocortin pathway and may be present in as many as 6% of individuals with severe, childhood onset obesity.13 Genome-wide association studies from large-scale cohorts and biobanks have identified numerous genetic variants associated with obesity-related metrics, behaviors, and processes, including BMI, food preference, energy regulation, lipid metabolism, and neurologic pathways involved in feeding.11 However, only about 5% of the variability in individual obesity measures can be attributed to these genes14–16
There are multiple apparent obesogenic gene-environment interactions that contribute to the hereditability of obesity. To date, more than 1,100 independent loci have been associated with a range of obesity traits. However, only 12 loci have been identified in gene-by-environment interaction analysis, and whose effects on obesity are attenuated or exacerbated by non-genetic factors, including physical activity, diet, and smoking.17 For example, increased physical activity or a balanced diet can reduce the influence of the obesity-associated (FTO) locus on obesity risk by 30–40%.18 Obesity hereditability is also influenced by epigenetics, which describes reversible DNA alterations that can affect gene expression regardless of nucleotide sequence changes.19 Epigenetic studies have demonstrated tissue-specific methylation pattern alterations associated with environmental and behavioral factors, including exercise, diet, weight gain, and bariatric surgery.11 Epigenome-wide association studies revealed a link between methylation modifications and obesity-related anthropometrics. However, association analyses suggest that these epigenetic changes are predominantly the consequence, as opposed to the cause, of elevated adiposity. Little is known about the mechanisms that cause these alterations.20
Environment
Obesogenic environmental factors, such as increased availability of palatable food, changes in the food supply, altered consumption patterns, decreased physical activity in both occupational and leisure settings, use of medications with the potential side effect of weight gain, and alterations in sleep patterns, have all contributed to and sustain this epidemic.15 There are also significant social factors that appear to influence obesity prevalence. Communities affected by poverty have significantly higher rates of obesity and obesity-related comorbidity. An analysis of the National Health Interview survey demonstrated that a greater burden of unfavorable social determinants of health related to financial stability, community cohesion, food security, educational attainment, and health care access, was associated with greater rates of obesity, with the most burdened quartile having 50% and 75% greater relative rates of BMI of 30–40 and > 40 kg/m2, respectively.21 Studies in pediatric populations show that non-Hispanic black and Hispanic children, children in households with a single parent or where the primary language is not English, and other factors were associated with increased rates of obesity.22 More research is needed to understand the etiology of these trends and identify ways to remedy the social inequities as well as their contribution to obesity.
Energy Balance
There are numerous reciprocal and redundant pathways that regulate food intake and energy expenditure. Communications between the brain and peripheral tissues, including adipose tissue and the gut, are at the center of these processes (Figure 1). Energy intake is regulated by enteric sensory pathways that result in vagal afferent signaling and endocrine cascades which activate, among other things, brain regions controlling appetite and feeding behavior. Energy expenditure is moderated by many of these same pathways. Resting metabolic rate accounts for around 70% of total energy expenditure and is closely correlated to fat free mass.23 The remainder of energy expenditure is attributable to the thermic effect of food and physical activity. Specific hormonal and neurologic pathways mediate these domains of energy expenditure 23, 24 and these pathways may be consequential in obesity.
Figure 1: Central and Peripheral Energy Regulation:
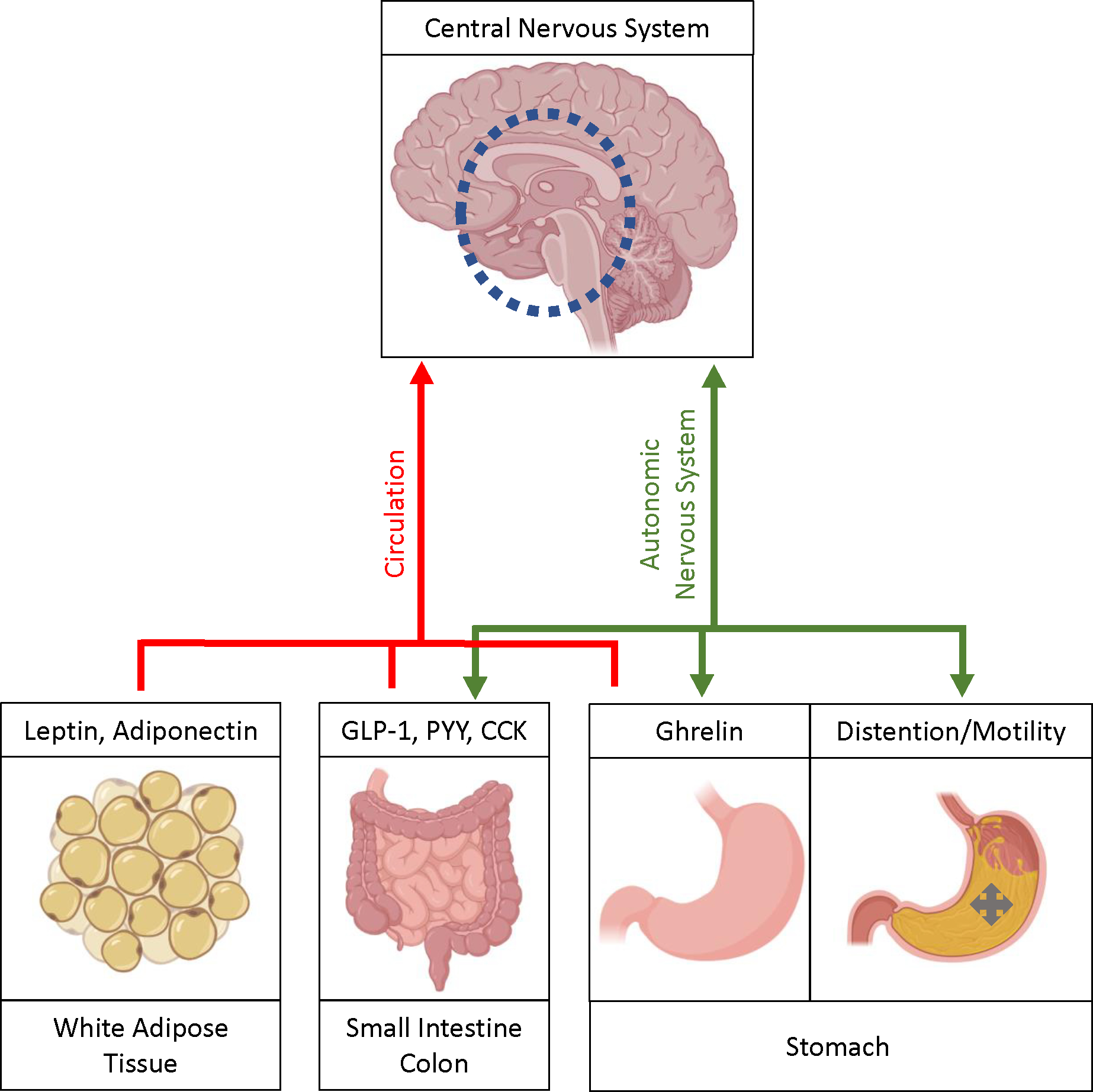
Important enteric sensory modalities detect nutrients and distention. This results in hormone secretion, including ghrelin, CCK, GLP-1, and PYY, and afferent autonomic signaling. Enteric-derived hormones act on autonomic nerve endings embedded in the lumen wall and result in signaling to the nucleus of the solitary tract in the brainstem via afferent vagal and splanchnic nerves. These hormones, along with leptin and adiponectin which are largely derived from adipose tissues in response to alterations in nutrient stores, may also enter the circulatory system and activate AGRP and POMC receptors at the arcuate nucleus. Both of these signaling modalities can result in anorexigenic and orexigenic signals to the periventricular nucleus and other brain regions, which subsequently communicate with other central nervous system domains that moderate feeding behaviors and physical activity. Gastric emptying, gastric accommodation, and other enteric secretory and motor functions are modified by the above hormones, intra-enteric neuronal signaling, and efferent autonomic signaling via efferent vagus nerve pathways, which reciprocally influence distention and nutrient signaling.
Peripheral Regulation
GASTRIC SENSORY FUNCTIONS
The stomach provides important information regarding the quantity of meal intake and controls the rate of introduction of meal contents to the small intestine, a mechanism known as gastric emptying (GE). The neurosensory capacity of the stomach involves a network of diverse cell and tissue types, including interstitial cells of Cajal, fibroblast like cells, smooth muscle layers, and the enteric nervous system.25 The vagal nerve detects distention of various structures located in the muscular layers and submucosa, including widely distributed afferent nerve endings, intra-ganglionic laminar endings, and intra-muscular arrays.26
Gastric distention results in the release of neurotransmitters, both via the intrinsic (enteric) and extrinsic (vagal) neural networks, that moderate meal-related gastric secretomotor processes and CNS signaling. Distention-related signals are primarily transmitted through afferent vagal nerves whereas intrinsic signalling within the enteric nervous system is mediated by interneurons which connect primary afferent neurons with motor and secretomotor neurons.25
These processes largely account for the measurable effect of distention on feeding behavior. Regardless of caloric content, distention is an important source of negative feedback in food intake regulation. Distention decreases appetite and food intake 27 and studies have shown a correlation between tolerated gastric volumes and weight.28
GASTRIC MOTOR
GE controls the transit of meal contents from the stomach to the intestine, thereby mediating gastric distention and the introduction of nutrients to the neurohormonally active intestinal lumen. This allows GE to modify important signaling processes, in particular vagal signaling and hormonal feedback, which, in turn, reciprocally influence GE. Several associations emphasize the role of GE in feeding behavior and energy regulation. In adults, fasting gastric volume and accelerated GE are associated with obesity 29 and with subsequent weight gain.30 Rapid GE has also been associated with obesity and alterations in satiety and increased hunger 31 and greater weight loss in bariatric endoscopic procedures.32
ENTERIC and ADIPOSE HORMONES
Upon entry of meal contents to the duodenum, various nutrients trigger enteroendocrine cell nutrient-specific receptors and result in the release of hormones that influence motility, including GE, and regulate food intake.33 Some of these hormones are appetite inducing (orexigenic), such as ghrelin, whereas others are satiety inducing (anorexigenic), such as peptide YY (PYY), glucagon-like peptide 1 (GLP-1), oxyntomodulin, cholecystokinin (CCK), and amylin, as has been extensively reviewed elsewhere.34 These peptides influence the function of neighboring cells, activate autonomic neural pathways, and enter circulation to act on distant sites.
These general processes are well represented by the specific action of GLP-1. GLP-1 is secreted by L-cell enteroendocrine cells in the small intestine and colon in response to intraluminal nutrients, in particular glucose and fat.35 GLP-1 further influences nutrient utilization, as do other incretin hormones, by promoting insulin secretion and inhibiting glucagon secretion.34 GLP-1 activates vagal afferent nerves and myenteric neurons which decrease GE rate 36 and have an anorexigenic effect through promotion of satiety and decreased energy intake.34 Other enteroendocrine produced peptides have similar effects, including CCK, which also activates vagal afferents, promotes meal termination, and slows GE.34 While circulating meal related peptides, such as amylin, are known to enter circulation and act directly on the CNS postrema and nucleus tractus solitarii (NTS) (i.e. not solely via vagal afferent signaling),37 it is less clear whether other major enteroendocrine hormones such as GLP-1 and CCK behave in a similar fashion.
Ghrelin is an orexigenic peptide hormone secreted by P/D1 enteroendocrine cells. It activates the vagus nerve and ultimately stimulates food intake and alterations in various gastrointestinal functions. In addition to altering energy intake, rat models show that injection with ghrelin results in a significant decrease in physical activity 38 and may mediate decreases in energy expenditure that can contribute to an energy surplus, weight gain and obesity.
It is unclear whether many hormones, such as PYY and CCK, directly contribute to obesity. Despite the clear meal-time related functions of PYY and CCK, studies show contradictory results with inconsistently lower baseline hormone levels and blunted post-prandial secretion in patients with obesity.39 Other hormones, like GLP-1, have a clear role in weight loss. Physiologic studies show that post-meal GLP-1 levels are blunted in individuals with obesity and essentially normalize following weight reduction,39 though these studies do not discern causation from correlation. However, drug trials consistently demonstrate that GLP-1 receptor agonism results in significant weight loss.40 GIP, like GLP-1, is an incretin hormone though it has previously been shown to have a minimal effect on weight.40 However, combination GIP and GLP-1 agonism results in robust weight loss, potentially related to GIP’s unique alterations in adipose cell lipid cycling 41 and attenuation of the nauseating effects of GLP-1 monotherapy.40 The effect of this combination emphasizes that these hormones function in dynamic biological systems. Additional physiologic studies are needed to clarify the role of meal-related peptides in feeding and energy regulation.
Another major modulator of energy regulation and potential driver of obesity is adipose tissue itself. Leptin and adiponectin are among the best understood and most impactful adipose-derived signals. Leptin is largely, though not solely, produced by adipocytes. Its production is closely correlated to white adipose mass.42 The physiological role of leptin is signaling nutritional status, particularly energy depletion. Accordingly, fasting and weight loss are associated with a drop in leptin levels. There are also human and animal models that demonstrate leptin’s ability to mediate neuroendocrine adjustments to decrease energy intake.43 Leptin modulates satiety and energy expenditure through binding receptors in the arcuate nucleus, hypothalamus, and brainstem. Homozygous loss-of-function mutations that decrease leptin synthesis, secretion, or biological activity or alter signaling through the leptin receptor cause severe obesity.44 Leptin also appears to alter energy expenditure. In rat models, disruption of leptin signaling pathways in the arcuate nucleus results in decreased thermogenesis. Stimulation of these pathways results in increased thermogenesis.45 This shows how leptin may oppose a positive energy balance and weight gain via both alterations in energy intake and expenditure.
The impact of leptin is empirically demonstrated in the weight loss and metabolic effects of exogenous leptin therapy 46 and the MC4R receptor agonist setmelanotide 47 in patients with severe hypoleptinemia or receptor deficiency. However, while leptin resistance and hyperleptinemia are present in the majority of patients with obesity, exogenous leptin does not promote significant weight loss in these patients based on existing studies.46 There is active research into other hormones implicated in these signaling pathways, such amylin, which may enhance leptin signaling, though the of the underlying physiology and clinical implications are still uncertain.46
Adiponectin is produced exclusively by adipocytes and plays a major role in weight homeostasis and metabolic functions, including insulin sensitivity, lipid regulation, and fatty acid oxidation. Lower plasma adiponectin concentrations are associated with obesity as well as visceral adipose burden.48 In fact, diet and weight loss are associated with increased adiponectin concentrations.49
Central Regulation
The CNS receives peripheral signals from afferent autonomic pathways (e.g., vagal afferents), peripherally secreted hormones (e.g., leptin, CCK, GLP-1, PYY3-36), and circulating nutrients (e.g., free fatty acids [FFA]). There are multiple brain regions implicated in integrating and processing these signals.
Many enteric hormonal and neuronal signals, such as distention and CCK, converge in the vagus afferent nerves which terminate at the NTS. The NTS projects to multiple important appetite centers, including the parabrachial nucleus, central amygdala, stria terminalis, and medulla.50 These brain centers moderate complex reward signaling pathways and feeding behaviors.
An overlapping, yet distinct, regulatory pathway involves the arcuate and paraventricular hypothalamic nuclei which contain orexigenic agouti-related peptide (AGRP) and anorexigenic proopiomelanocortin (POMC) expressing neurons. These are essentially opposing pathways that converge at melanocortin 4 receptor (MC4R) expressing cells in the paraventricular nucleus (PVN) of the hypothalamus, among other targets. Mice models show certain stimuli, including fasting 51 and ghrelin,52 result in stimulation of AGRP neurons. AGRP neurons project to the paraventricular hypothalamic nucleus, anterior bed nucleus of the stria terminalis, lateral hypothalamic area, and periventricular thalamus and antagonize MC4R actions.53 Targeted stimulation of AgRP neurons provokes instant and dramatic increase in feeding in mice.54
POMC neurons have an essentially opposite effect. Stimulation is mediated by hormones including leptin,55 whereas fasting decreases POMC expression in these brain regions.56 POMC neurons project to multiple regions and cell types and appear to stimulate MC4R expressing cells in the paraventricular nucleus which mediate satiety via subsequent projections, including to the parabrachial nucleus and subsequent reward centers33,57 POMC and AGRP pathways also reciprocally influence peripheral pathways involved in appetite and eating behavior, such as through direct projections to the dorsal vagal complex.58 This region controls vagal efferent output, which is known to regulate enteric functions such as GE. The dorsal vagal complex has a high density of MC4R and targeted stimulation of these receptors results in marked alterations to feeding in animal models.59,60 The impact of these CNS pathways is empirically evident in the robust weight loss and metabolic effects of therapeutics targeting the leptin-MC4R pathway as reviewed above. More research is needed on the role of these pathways in various forms of obesity.
Related neurologic pathways are also implicated in energy expenditure. Activation of orexin neurons in the lateral hypothalamus results in increased spontaneous physical activity and overall energy expenditure.61 Stimulation of NPY neurons in the arcuate nucleus alters signaling to the PVN and decreases sympathetic output to brown adipose tissue and decreases thermogenesis.62 While the full implications of these effects are not known, the involvement of these neurologic domains in physical activity and thermogenesis further supports the role of the gut-brain axis in controlling energy intake and expenditure.
OBESITY PATHOPHYSIOLOGY: MORBIDITY
The pathophysiological impact of positive energy balance, as described above, results in the excess of calories stored predominantly in adipose tissue. The excess of adiposity seen in obesity is associated with various metabolic, inflammatory, immune, and mechanical changes that mediate tissue injury (Figure 2) and obesity associated comorbidities (Figure 3). The most notable obesity related comorbidities are CVD, T2DM, and cancer, with the majority of obesity-related costs attributable to the former two.63 Here we review major obesity related pathophysiologic changes and impactful comorbidities associated with these changes.
Figure 2: Obesity-related tissue dysfunction.
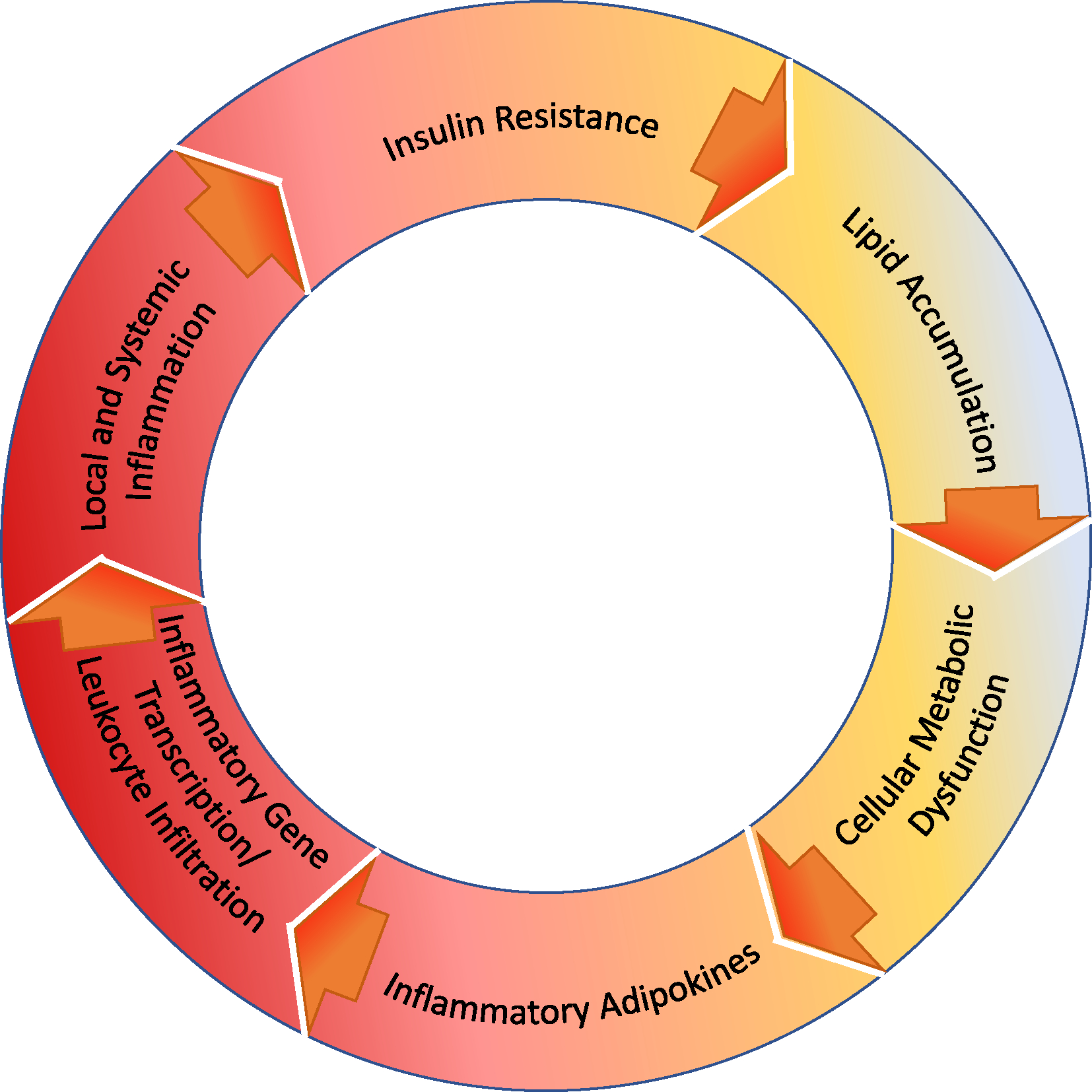
Obesity-associated lipid accumulation overwhelms lipid metabolic pathways. This dysfunction appears to promote endoplasmic reticulum and mitochondrial dysfunction and increase reactive oxidative species as well as FFA and FFA intermediates, such as ceramide. FFA intermediates activate various pro-inflammatory kinases resulting in pro-inflammatory adipokines and immune cell (macrophage, TH1) infiltration. FFA intermediates and the resulting inflammatory milieu also result in NF-κB activation and altered gene transcription. These changes also have direct and indirect effects on insulin signaling, such as through alterations in insulin receptor phosphorylation and function, and result in tissue injury, particularly in the case of ectopic fat accumulation in the liver and vascular structures. These numerous changes may all contribute to local and systemic inflammation.
Figure 3: Pathophysiology of predominant obesity-related comorbidity.
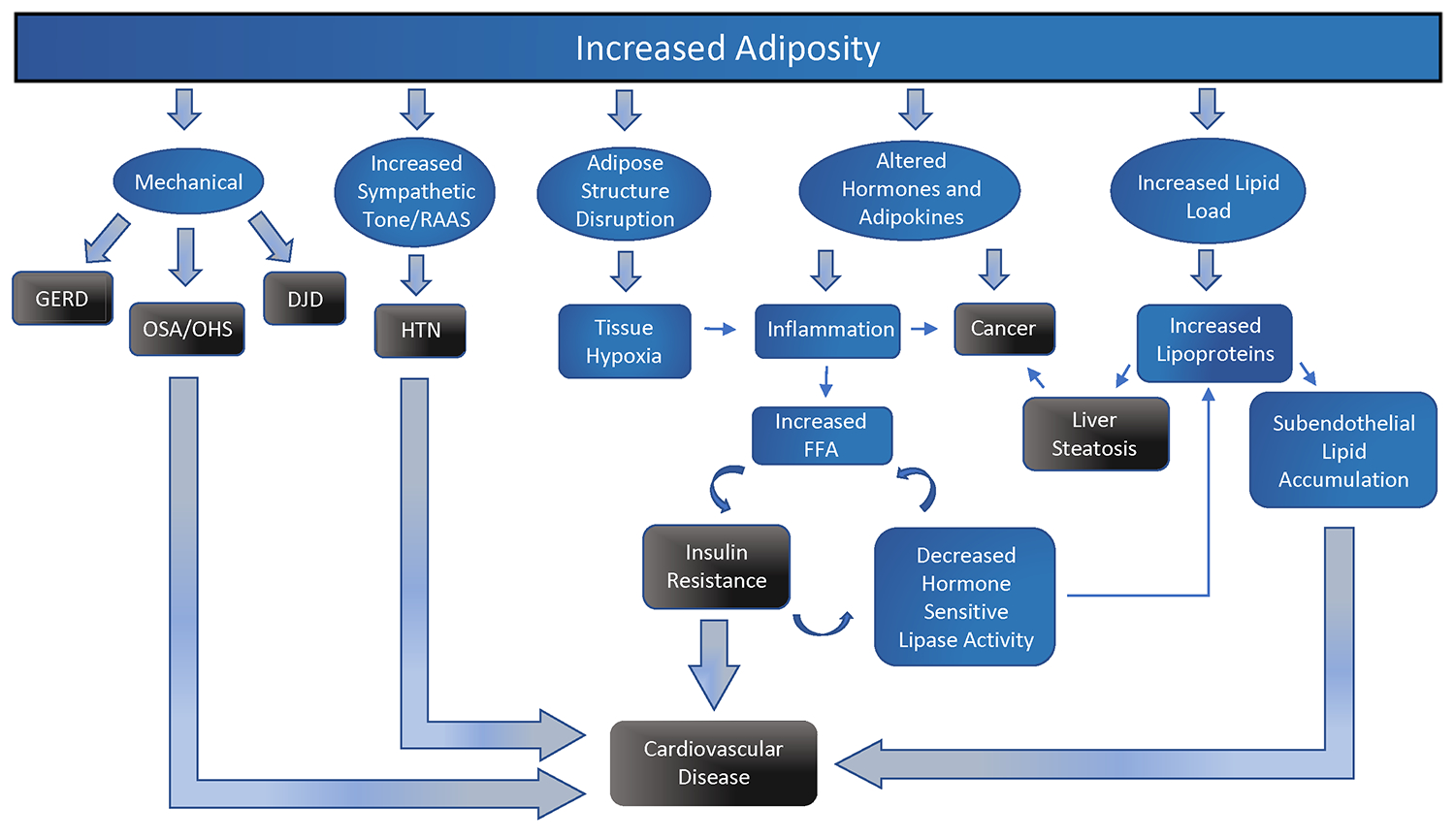
Increased adiposity contributes to various mechanical, sympathetic, cellular, and metabolic changes that contribute to chronic disease, most notably cancer, insulin resistance, and cardiovascular disease.
Dyslipidemia and Inflammation
Obesity results in systemic and local inflammation. Markers of inflammation are strongly associated with measures of adiposity and cardiometabolic risks.64 This inflammation may be largely caused by lipid pathway dysfunction and adipokine alterations which promote potent immune and inflammatory pathways. While the inciting processes remain somewhat theoretical, elevated intracellular lipid loads appear to overwhelm oxidative pathways and result in the build-up of fatty acids and fatty acid intermediates, such as ceramide,65 contributing to inflammation and metabolic dysregulation.66–68 Obesity is also associated with altered adipose structure and hypoxia, which may each further contribute to inflammatory and metabolic derangement.69 Studies have demonstrated increased adipose secretion of IL-6 and tumor necrosis factor-α (TNF-α) in patients with obesity related insulin resistance.70
Obesity is also marked by tissue specific immunogenic alterations. FFA and intermediates may trigger toll-like receptor 4 receptors and immune cell recruitment.71 Adipose tissue from patients with obesity is marked by both innate and adaptive immune responses, characterized by increased macrophage 72 and T-cell 73 infiltration and activity.
Insulin Resistance
This obesity-associated inflammatory milieu, marked by elevated inflammatory cytokines and immune cell activity, promotes insulin resistance via multiple mechanisms. Inflammatory signals, including TNF, result in altered activity of kinases involved in insulin-mediated glucose uptake 74 and inhibit insulin receptor signaling through altering receptor phosphorylation patterns.75 Elevated FFA intermediates are also associated with alterations to transcription via activation of NF-κB and protein kinase C, which could promote further inflammation and insulin receptor dysfunction.76 The role of inflammatory processes in insulin resistance is empirically supported by animal models which show that suppression or deficiency in specific pro-inflammatory kinase pathways protects against insulin resistance.77 Obesity related changes in insulin signaling occur diffusely, including in skeletal muscle and the liver, which are particularly impactful in glucose homeostasis. Studies have shown that hepatic levels of FFA intermediates, in particular diacylglycerol, are correlated with insulin resistance.78 insinuating similar nutrient mediated insulin signaling alterations as reviewed above. The nature of interactions between nutrients, kinases, and metabolic pathways and how these vary between tissue types are not fully understood.
Vascular Injury
The above mentioned inflammatory and metabolic changes occur locally and systematically to contribute to significant cardiovascular injury. Lipoproteins characteristically elevated in dyslipidemia and obesity, including LDL and chylomicrons, are known to migrate into the vascular sub-endothelium and promote an inflammatory immune reaction culminating in atherosclerosis.79 Lipid particles within the arterial intima of the vascular wall may be oxidized and contribute to endothelial cell activation, adhesion molecule expression, and sub-endothelial leukocyte accumulation.80 Lipid-laden macrophages, known as foam cells, mediate local injury and apoptosis, contributing to chemokine and antigenic build-up that may facilitate T-cell recruitment and plaque progression.80 The importance of these processes is empirically supported by decreased atherosclerosis in chemokine deficient animal models with absent immune cell recruitment.81
Cardiovascular Disease
The association between cardiovascular disease and obesity is largely driven by the above described metabolic, inflammatory, and vascular changes. Cardiovascular disease is the leading cause of death for adults in the United States. Studies consistently show the staggering burden of cardiovascular disease in patients with obesity. A large prospective study demonstrated that a BMI> 25 kg/m2 has a relative risk (RR) of 1.21 for men and 1.2 for women for cardiovascular disease.82 A large meta-analysis further showed that a BMI of ≥35 kg/m2 had an increased risk of death related to cardiovascular disease with a hazard ratio of 1.8 compared to individuals with a BMI of 22.5–24.9 kg/m2.7 The graded effect of BMI on CVD has been consistently replicated, including in a 2016 population-based cohort study which showed that a BMI increase of 4.8 kg/m2 was associated with an increased risk of stroke, coronary heart disease, and hypertension, as well as T2DM.83 T2DM plays a significant role in the association between obesity and CVD. A BMI> 40kg/m2 is associated with a drastically increased risk of T2DM (OR 7.37) 84 and CVD is the most notable complication of T2DM, though patients are also at risk of numerous other highly morbid complications, including renal and neurologic disease.85
Non-Alcoholic Fatty Liver Disease
The above metabolic and inflammatory changes also contribute to non-alcoholic fatty liver disease (NAFLD). NAFLD is defined by the build-up of fat in the liver that cannot be attributed to other causes and is perhaps the most obvious example of morbid ectopic fat. Several important metabolic alterations are associated and likely mediate these changes, including increased FFAs related to increased lipolysis and dietary fat, insulin signaling alterations resulting in impaired fatty acid oxidation and decreased lipid clearance, and de novo lipogenesis.86 Seventy-five percent of patients with obesity have NAFLD compared to 16% in control patients.87 NAFLD may be an asymptomatic feature of metabolic syndrome or progress to cirrhosis, end-stage liver disease, or death. Moreover, NAFLD has emerged as a dominant etiology of hepatocellular carcinoma (HCC) and is predicted to become the leading cause of liver transplantation in the United States.88
Cancer
Inflammatory and metabolic changes also appear to play a large role in the association between obesity and certain malignancies 89–95 (Table). Increased obesity related cancer risk may be further mediated by altered hormone profiles, oxidative stress, carcinogen metabolism, and tissue growth homeostasis.94 The significant morbidity of these various cancers is beyond the scope of this review.
Table: Obesity-associated cancer risk.
The above table reports comparison of mean (as RR unless otherwise specified) for various cancers in groups with and without obesity.
| Relative Risk | BMI (“Control”) kg/m2 | BMI (“Obesity”) kg/m2 | Reference | |
|---|---|---|---|---|
| Esophageal Cancer | 4.76a | <25 | >40 | 89 |
| Gastric Cardia Cancer | 1.82 | <25 | >30 | 90 |
| Pancreatic Cancer | 1.47 | 21–23 | >30 | 91 |
| Gallbladder Cancer | 1.62 | 18.5–24.9 | >30 | 92 |
| Extrahepatic Bile Duct Cancer | 1.48 | 18.5–24.9 | >30 | 92 |
| Colorectal Cancer | 1.19 | <25 | >30 | 93 |
| Endometrial Cancer | 3.6 | <23.5b | >30b | 94 |
| Breast Cancer (Receptor-positive, postmenopausal) | 1.39 | <25 | >30 | 95 |
Odds Ratio
Average BMI cut-off for included studies.
Mechanical Stress
Obesity is linked to anatomic changes that lead to a mechanical stress on multiple systems, including connective tissues, intraabdominal organs, and the upper airway. Increased adipose mass causes stress on musculoskeletal structures throughout the body, likely through synergistic alterations to skeletal alignment, muscular deconditioning, and mechanical load. The result is a significant increase in the risk of musculoskeletal injury and chronic pathology.96 Increased adipose mass increases intraabdominal and intrathoracic compressive forces, which are theorized to contribute to peri-renal compression and hypertension.15 These forces may also lead to alterations in upper airway structure and load which, in conjunction with decreased compensatory respiratory drive may contribute to altered respiratory physiology, decreased pulmonary function,97 and ventilatory abnormalities. Case-control studies demonstrate that the frequency and severity of sleep apnea increases with the severity of obesity.98 Obesity also predisposes to obesity hypoventilation syndrome (OHS) with cohort studies showing 31% of inpatients with a BMI >35 kg/m2 have hypercapnia without an apparent alternative etiology.99 These associations are further supported by the reversibility of lung dysfunction with weight loss, with systematic reviews showing that OSA improved in 77–99% of patients undergoing weight loss surgery.100
The above alterations to respiratory physiology, as well as changes in metabolism, hormone profiles, and immune cell function,8 may help explain the association between obesity and an increased incidence and severity of certain pulmonary infections. A meta-analysis of 75 studies demonstrated that among individuals with COVID-19 infections, obesity was associated with an increased likelihood of hospitalization (OR 2.13), admission to the ICU (OR 1.74), and death (OR 1.48).8 Similar trends have been apparent in other pandemic illnesses, including influenza.101 The risk with COVID-19 appears to be mitigated by weight loss after bariatric surgery based on case control studies.102
Given the devastating morbidity of obesity, it is necessary to acknowledge the role and impact of obesity treatment. Even modest weight loss reduces the risk and can even reverse many obesity-related comorbidities. Effective treatments for obesity have been comprehensively reviewed elsewhere 103–105 and are beyond the scope of this review.
OBESITY CLASSIFICATIONS
Obesity has traditionally been categorized by the degree of BMI elevation. A BMI of 25 to 30 kg/m2 is defined as overweight, whereas BMI of 30 to 35, 35–40, and > 40 kg/m2 are classified as class 1, 2, and 3 obesity, respectively.106 These classifications are used to guide screening recommendations 107 and inform guidelines for pharmacologic and surgical obesity treatments.108 However, BMI has limited utility in predicting obesity-related risks and treatment responses. Categorization systems that may allow for improved prognostication, as well as individualized management, include waist circumference, metabolic status, sarcopenia, and physiologic phenotype based on gastric motility, energy expenditure, psycho-behavioral traits, and satiation. Some of these systems are thoroughly validated and others, while promising, require further study to define their role clinically.
Metabolically “Healthy” and “Unhealthy” Obesity
Individuals with obesity and metabolic dysfunction, based on criteria such as dyslipidemia, hypertension, insulin resistance, and elevated inflammatory markers,109 are considered “metabolically unhealthy”. Around 30% of individuals with obesity are, alternatively, “metabolically healthy”.109 Studies show that, among individuals with obesity, metabolic dysfunction by these criteria is associated with future morbidity including cardiovascular disease.109 Obesity without metabolic derangement is still a significant risk factor for morbidity, particularly CVD, when compared to metabolically healthy individuals without obesity.110 These concepts are applied clinically by various classification systems, including the Edmonton Obesity Staging system (EOSS). EOSS stages are defined by the presence of BMI > 30 kg/m2 and metabolic alterations including fasting glucose, cholesterol, and hypertension. This system also incorporates chronic conditions including osteoarthritis, mood symptoms, and functional limitations. Longitudinal data shows that individuals with a higher EOSS stage are at elevated risk of mortality and cardiovascular mortality compared to normal weight and lower stage individuals.111
Central Adiposity
There is wide variation in the degree of central and visceral adiposity between individuals of similar BMI. Central obesity is associated with increased morbidity, including cardiovascular disease,112 metabolic syndrome, and insulin resistance.113 A meta-analysis comparing various metrics of obesity showed that metrics utilizing waist circumference predicted hypertension, T2DM, and dyslipidemia more accurately than BMI alone.114 Central obesity also predicts outcomes in acute illness and is even a significant risk factor for death in critically ill patients admitted to the ICU.115 Due to the strength of the associations between central obesity and negative health outcomes, many medical organizations have incorporated measurements of central obesity into their official obesity guidelines.116 There are clinical classification systems that account for both metabolic factors and cardiovascular events as well as waist circumference, such as the Cardiometabolic Disease Staging System (CDSS). A large cohort study showed that the risk of cardiovascular mortality was progressive with each CDSS stage, with hazard ratios ranging from 3.6 for stage 1 to 14.6 for stage 4.117
Sarcopenic Obesity
Sarcopenic obesity may occur with or without central adiposity and is defined by concurrent obesity and decreased muscle mass or function.118 The development of sarcopenic obesity is related to activity, hormonal, inflammatory, and myocellular changes. While study criteria for sarcopenic obesity vary and are typically based on muscle mass measurement or strength testing, studies estimate a prevalence from 2.1–11%. Regardless of the specific definition, it is clearly a highly morbid entity with markedly higher risk of dyslipidemia, insulin resistance, functional deficits, depression, and mortality compared to patients with sarcopenia or obesity alone.118
Energy Intake and Expenditure
The classification of obesity based on BMI, waist circumference, and metabolic dysfunction provides useful information regarding obesity associated morbidity. However, these paradigms do not address heterogeneity in the pathophysiologic drivers of obesity. Recent research reveals promising measurable traits associated with alterations in energy intake and expenditure allow for identification of the etiology of obesity on an individual level.
Energy Intake
Energy intake is driven by homeostatic processes that result in sensations of hunger and fullness, as well as hedonic processes which result in eating to cope with positive and negative emotions. Homeostatic processes are primarily mediated by sensory mechanisms integrated through the gut-brain axis and regulate hunger, satiation (perceived fullness at the end of a meal), and satiety (post-prandial processes that determine the return of hunger).119 Standardized metrics to assess both satiation (measured by caloric intake in standardized meal resulting in fullness) and satiety (measured by GE rates) have allowed for quantification of these processes, which are associated with obesity related traits and weight gain, as reviewed above (see GASTRIC MOTOR).
Energy Expenditure
A large body of research has revealed that energy expenditure is variably modified to promote and preserve obesity. Decreased energy stores result in metabolic and feeding changes that promote energy conservation and weight gain. Studies have consistently shown variability in individual energy expenditure reduction in response to both calorie restriction and weight-loss.120 Prospective studies have shown that disproportionately high energy conservation in response to such changes predicts future weight gain.120 Excessive energy conservation is durable and may result in decreased energy expenditure up to or beyond 6 years after weight-loss.121 Addressing this derangement in affected patients may allow for improved treatment of obesity.
Energy Intake and Expenditure Phenotypes
Most of the current severity-based classifications and staging systems fail to address the pathophysiological and etiological heterogeneity of obesity.122–124 However, a recent classification was proposed based on pathophysiological and behavioral phenotypes to stratify obesity based on homeostatic eating, hedonic eating behavior and abnormal energy expenditure.125 Consequently, four actionable phenotypes were established: abnormal satiation (measured by calories ingested to experience postprandial fullness), abnormal postprandial satiety (duration of fullness and measured by GE), emotional eating behavior (measured by questionnaires) and abnormal resting energy expenditure (measured by indirect calorimetry). This classification uses the 25th or 75th percentile cutoff for each measurement and applied separately for females and males.
This obesity-phenotype based classification aims to match a treatment to the underlying pathophysiological abnormality seen in patients with obesity. This classification guided an individualized selection of lifestyle interventions and diets for each phenotype, which showed significantly better weight loss outcomes compared to patients treated irrespective of these traits.126 In addition, in multiple post-hoc analyses of pilot studies using AOMs and bariatric endoscopic devices, obesity phenotypes demonstrated a better weight loss response to these interventions.125 For instance, phentermine-topiramate extended-release (ER) works better for the abnormal satiation phenotype in which the use of this medication was associated with reduced energy intake at an ad libitum meal (mean Δ 260 kcal, p=0.03) and delayed GE of solids (mean Δ GE T½ 19 min, p=0.06).12. Similarly, exenatide, a GLP-1 agonist, significantly slowed GE of solids (p<0.001) and reduced calorie intake at an ad libitum meal by an average of 130kcal compared to placebo. Similarly, liraglutide administered daily for 16 weeks delayed GE at 5 (p<0.0001) and 16 weeks (p=0.03) compared to placebo with a higher weight loss at 16 weeks (6.1 ± 2.8 kg) compared to 2.2 ± 5 kg in the control group (p<0.01). Interestingly, at 5 and 16 weeks, GE of solids correlated with differences in weight loss on liraglutide (p<0.02). Accelerated GE at baseline seen in patients with abnormal postprandial satiety was a predictor for a better weight loss response when GLP-1 receptor agonists are used.127
Based on the mechanisms of action of AOMs, Acosta et al. proposed a unique method to use AOM to target each obesity phenotype. For example, patients with abnormal satiation might have better weight loss outcomes with phentermine-topiramate ER as it modulates the adrenergic and GABAergic pathways. Similarly, GLP-1 receptor agonists might benefit patients with abnormal satiety phenotype who are characterized by accelerated GE by delaying GE and reducing inter-meal hunger. In addition, patients with abnormal emotional hunger phenotype who are characterized by negative mood eating and reward-seeking behaviors due to negative and positive emotions, might benefit from naltrexone/bupropion sustained-release, which modulates the dopamine and opioid pathways.125 In a 12-month, pragmatic, real-world trial with 312 patients, patients in the phenotype-guided group had 1.75-fold greater weight loss (15.9%) compared to 9.0% in the non-phenotype-guided group [difference −6.9% (95%CI −9.4 to −4.5), p<0.001]. The proportion of patients who lost >10% at 12 months was 79% in the phenotype-guided group compared to 34% in the non-phenotype-guided treatment group.125 Hence, targeting the pathophysiological and behavioral phenotypes may enhance weight loss.
Moreover, a phenotype-guided approach has demonstrated promising results in bariatric endoscopic trials. For example, baseline and post-procedural GE rates predicted efficacy and tolerability of space occupying intragastric balloons 128 while a greater decrease in maximum tolerated volume was correlated with better weight loss outcomes in patient undergoing endoscopic sleeve gastroplasty.129 Further research is needed to better understand these physiologic and etiologic traits which may improve individualized treatment of obesity.
CONCLUSION and FUTURE DIRECTIONS
Obesity has emerged as a devastating global epidemic. Significant advancements have been made in understanding the pathophysiology of obesity and obesity related conditions. However, defining obesity related characteristics that allow for accurate individualized prognosis and management has remained elusive. This review has summarized the established and ongoing research on the pathophysiology of obesity etiology and morbidity. We have also reviewed obesity classification systems that may allow for improved individualized management.
Article Highlights:
A concise review of the domains that influence and regulate feeding, energy balance, and weight.
A concise review of the pathophysiology of obesity.
A review of actionable obesity categorization systems and how these systems inform and improve the understanding of obesity pathophysiology and its treatment.
Acknowledgements
The images in figure 1 were created using BioRender software.
The authors received no financial or material support requiring disclosure for this project.
Abbreviations:
- AGRP
Agouti-Related Peptide
- AOM
anti-obesity medication
- BMI
Body Mass Index
- CCK
Cholecystokinin
- CDSS
Cardiometabolic Disease Staging System
- CVD
Cardiovascular Disease
- COVID-19
Coronavirus Disease 2019
- EOSS
Edmonton Obesity Staging system
- FFA
Free Fatty Acids
- FTO
Obesity-Associated Gene
- GE
Gastric Emptying
- GERD
Gastroesophageal Reflux Disease
- GIP
Gastric Inhibitory Polypeptide
- GLP-1
Glucagon-Peptide 1
- HCC
Hepatocellular Carcinoma
- HTN
Hypertension
- MC4R
Melanocortin 4 Receptor
- NAFLD
Non-Alcoholic Fatty Liver Disease
- NTS
Nucleus Tractus Solitarii
- OHS
Obesity Hypoventilation Syndrome
- OR
Odds Ratio
- OSA
Obstructive Sleep Apnea
- POMC
Proopiomelanocortin
- PVN
Paraventricular Nucleus
- PYY
Peptide YY
- RCTs
Randomized Clinical Trials
- RR
Relative Risk
- T2DM
Type 2 Diabetes Mellitus
- TNF-α
tumor necrosis factor-α
Footnotes
Disclosure: Dr. Acosta is a stockholder in Gila Therapeutics, Phenomix Sciences; he served as a consultant for Rhythm Pharmaceuticals, General Mills, Amgen, Bausch Health.
CRediT Form: Obesity: A Review of Pathophysiology and Classification
Bradley Busebee, M.D.: Conceptualization, Visualization, Writing-Original draft, review and editing
Wissam Ghusn, M.D.: Writing-Review and editing
Lizeth Cifuentes, M.D.: Writing-Review and editing
Andres Acosta, M.D., PhD: Supervision, Writing-Review and editing
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Afshin A, Forouzanfar MH, Reitsma MB, et al. Health Effects of Overweight and Obesity in 195 Countries over 25 Years. The New England journal of medicine. 2017;377:13–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ward ZJ, Bleich SN, Cradock AL, et al. Projected U.S. State-Level Prevalence of Adult Obesity and Severe Obesity. The New England journal of medicine. 2019;381:2440–2450. [DOI] [PubMed] [Google Scholar]
- 3.Waters H, Graf M. America’s obesity crisis. The Health and Economic Costs of Excess Weight. Santa Monica, California: Milken Institute. 2018. [Google Scholar]
- 4.WHO. Obesity. World Health Organization; 2022. [Google Scholar]
- 5.Pi-Sunyer FX. Comorbidities of overweight and obesity: current evidence and research issues. Medicine and science in sports and exercise. 1999;31:S602–608. [DOI] [PubMed] [Google Scholar]
- 6.Fontaine KR, Redden DT, Wang C, Westfall AO, Allison DB. Years of life lost due to obesity. Jama. 2003;289:187–193. [DOI] [PubMed] [Google Scholar]
- 7.Berrington de Gonzalez A, Hartge P, Cerhan JR, et al. Body-mass index and mortality among 1.46 million white adults. The New England journal of medicine. 2010;363:2211–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Popkin BM, Du S, Green WD, et al. Individuals with obesity and COVID-19: A global perspective on the epidemiology and biological relationships. 2020;21:e13128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lim SS, Vos T, Flaxman AD, et al. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2224–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Romieu I, Dossus L, Barquera S, et al. Energy balance and obesity: what are the main drivers? Cancer Causes Control. 2017;28:247–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bray MS, Loos RJ, McCaffery JM, et al. NIH working group report-using genomic information to guide weight management: From universal to precision treatment. Obesity (Silver Spring). 2016;24:14–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hurtado AM, Acosta A. Precision Medicine and Obesity. Gastroenterology clinics of North America. 2021;50:127–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Farooqi IS, Keogh JM, Yeo GS, Lank EJ, Cheetham T, O’Rahilly S. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. The New England journal of medicine. 2003;348:1085–1095. [DOI] [PubMed] [Google Scholar]
- 14.Pigeyre M, Yazdi FT, Kaur Y, Meyre DJCs. Recent progress in genetics, epigenetics and metagenomics unveils the pathophysiology of human obesity. 2016;130:943–986. [DOI] [PubMed] [Google Scholar]
- 15.Heymsfield SB, Wadden TA. Mechanisms, Pathophysiology, and Management of Obesity. The New England journal of medicine. 2017;376:254–266. [DOI] [PubMed] [Google Scholar]
- 16.Locke AE, Kahali B, Berndt SI, et al. Genetic studies of body mass index yield new insights for obesity biology. 2015;518:197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Loos RJF, Yeo GSH. The genetics of obesity: from discovery to biology. Nature reviews. Genetics. 2021:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qi Q, Chu AY, Kang JH, et al. Fried food consumption, genetic risk, and body mass index: gene-diet interaction analysis in three US cohort studies. BMJ (Clinical research ed.). 2014;348:g1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hamilton JP. Epigenetics: principles and practice. Dig Dis. 2011;29:130–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wahl S, Drong A, Lehne B, et al. Epigenome-wide association study of body mass index, and the adverse outcomes of adiposity. Nature. 2017;541:81–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Javed Z, Valero-Elizondo J, Maqsood MH, et al. Social determinants of health and obesity: Findings from a national study of US adults. 2022;30:491–502. [DOI] [PubMed] [Google Scholar]
- 22.Yusuf ZI, Dongarwar D, Yusuf RA, et al. Social determinants of overweight and obesity among children in the United States. 2020;9:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tran LT, Park S, Kim SK, et al. Hypothalamic control of energy expenditure and thermogenesis. 2022;54:358–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Westerterp KR. Control of Energy Expenditure in Humans. In: Feingold KR, Anawalt B, Boyce A, et al. , eds. Endotext. South Dartmouth (MA): MDText.com, Inc. [Google Scholar]
- 25.Goyal RK, Hirano IJNEJoM. The enteric nervous system. 1996;334:1106–1115. [DOI] [PubMed] [Google Scholar]
- 26.Brookes SJ, Spencer NJ, Costa M, Zagorodnyuk VPJNrG, hepatology. Extrinsic primary afferent signalling in the gut. 2013;10:286–296. [DOI] [PubMed] [Google Scholar]
- 27.Cannon WB. The mechanical factors of digestion: Longmans, Green & Company; 1911. [Google Scholar]
- 28.Geliebter A, Schachter S, Lohmann-Walter C, Feldman H, Hashim SAJTAjocn. Reduced stomach capacity in obese subjects after dieting. 1996;63:170–173. [DOI] [PubMed] [Google Scholar]
- 29.Acosta A, Camilleri M, Shin A, et al. Quantitative gastrointestinal and psychological traits associated with obesity and response to weight-loss therapy. Gastroenterology. 2015;148:537–546.e534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pajot G, Camilleri M, Calderon G, et al. Association between gastrointestinal phenotypes and weight gain in younger adults: a prospective 4-year cohort study. Int J Obes (Lond). 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gonzalez-Izundegui D, Campos A, Calderon G, et al. Association of gastric emptying with postprandial appetite and satiety sensations in obesity. Obesity (Silver Spring). 2021;29:1497–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vargas EJ, Bazerbachi F, Calderon G, et al. Changes in time of gastric emptying after surgical and endoscopic bariatrics and weight loss: a systematic review and meta-analysis. 2020;18:57–68. e55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Field BC, Chaudhri OB, Bloom SR. Bowels control brain: gut hormones and obesity. Nat Rev Endocrinol. 2010;6:444–453. [DOI] [PubMed] [Google Scholar]
- 34.Makaronidis JM, Batterham RLJCOiP. The role of gut hormones in the pathogenesis and management of obesity. 2019;12:1–11. [Google Scholar]
- 35.Matzinger D, Degen L, Drewe J, et al. The role of long chain fatty acids in regulating food intake and cholecystokinin release in humans. 2000;46:689–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Imeryüz Ne, Yeğen BC, Bozkurt A, et al. Glucagon-like peptide-1 inhibits gastric emptying via vagal afferent-mediated central mechanisms. 1997;273:G920–G927. [DOI] [PubMed] [Google Scholar]
- 37.Sexton P, Paxinos G, Kenney M, Wookey P, Beaumont KJN. In vitro autoradiographic localization of amylin binding sites in rat brain. 1994;62:553–567. [DOI] [PubMed] [Google Scholar]
- 38.Tang-Christensen M, Vrang N, Ortmann S, Bidlingmaier M, Horvath TL, Tschöp MJE. Central administration of ghrelin and agouti-related protein (83–132) increases food intake and decreases spontaneous locomotor activity in rats. 2004;145:4645–4652. [DOI] [PubMed] [Google Scholar]
- 39.Steinert RE, Feinle-Bisset C, Asarian L, Horowitz M, Beglinger C, Geary NJPr. Ghrelin, CCK, GLP-1, and PYY (3–36): secretory controls and physiological roles in eating and glycemia in health, obesity, and after RYGB. 2017;97:411–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hayes MR, Borner T, De Jonghe BC. The Role of GIP in the Regulation of GLP-1 Satiety and Nausea. Diabetes. 2021;70:1956–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Samms RJ, Coghlan MP, Sloop KWJTiE, Metabolism. How may GIP enhance the therapeutic efficacy of GLP-1? 2020;31:410–421. [DOI] [PubMed] [Google Scholar]
- 42.Considine RV, Sinha MK, Heiman ML, et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. The New England journal of medicine. 1996;334:292–295. [DOI] [PubMed] [Google Scholar]
- 43.Ahima RS, Prabakaran D, Mantzoros C, et al. Role of leptin in the neuroendocrine response to fasting. Nature. 1996;382:250–252. [DOI] [PubMed] [Google Scholar]
- 44.van der Klaauw AA, Farooqi IS. The hunger genes: pathways to obesity. Cell. 2015;161:119–132. [DOI] [PubMed] [Google Scholar]
- 45.Kong D, Tong Q, Ye C, et al. GABAergic RIP-Cre neurons in the arcuate nucleus selectively regulate energy expenditure. 2012;151:645–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Obradovic M, Sudar-Milovanovic E, Soskic S, et al. Leptin and obesity: role and clinical implication. 2021;12:585887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Collet TH, Dubern B, Mokrosinski J, et al. Evaluation of a melanocortin-4 receptor (MC4R) agonist (Setmelanotide) in MC4R deficiency. Molecular metabolism. 2017;6:1321–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kaser S, Tatarczyk T, Stadlmayr A, et al. Effect of obesity and insulin sensitivity on adiponectin isoform distribution. European journal of clinical investigation. 2008;38:827–834. [DOI] [PubMed] [Google Scholar]
- 49.Pasarica M, Tchoukalova YD, Heilbronn LK, et al. Differential effect of weight loss on adipocyte size subfractions in patients with type 2 diabetes. Obesity (Silver Spring). 2009;17:1976–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Andermann ML, Lowell BBJN. Toward a wiring diagram understanding of appetite control. 2017;95:757–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu T, Kong D, Shah BP, et al. Fasting activation of AgRP neurons requires NMDA receptors and involves spinogenesis and increased excitatory tone. 2012;73:511–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen SR, Chen H, Zhou JJ, et al. Ghrelin receptors mediate ghrelin-induced excitation of agouti-related protein/neuropeptide Y but not pro-opiomelanocortin neurons. Journal of neurochemistry. 2017;142:512–520. [DOI] [PubMed] [Google Scholar]
- 53.Heisler LK, Lam DD. An appetite for life: brain regulation of hunger and satiety. Current opinion in pharmacology. 2017;37:100–106. [DOI] [PubMed] [Google Scholar]
- 54.Aponte Y, Atasoy D, Sternson SMJNn. AGRP neurons are sufficient to orchestrate feeding behavior rapidly and without training. 2011;14:351–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cowley MA, Smart JL, Rubinstein M, et al. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. 2001;411:480–484. [DOI] [PubMed] [Google Scholar]
- 56.Mizuno TM, Kleopoulos SP, Bergen HT, Roberts JL, Priest CA, Mobbs CVJD. Hypothalamic pro-opiomelanocortin mRNA is reduced by fasting in ob/ob and db/db mice, but is stimulated by leptin. 1998;47:294–297. [DOI] [PubMed] [Google Scholar]
- 57.Garfield AS, Li C, Madara JC, et al. A neural basis for melanocortin-4 receptor–regulated appetite. 2015;18:863–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zheng H, Patterson LM, Phifer CB, Berthoud H-RJAJoP-R, Integrative, Physiology C. Brain stem melanocortinergic modulation of meal size and identification of hypothalamic POMC projections. 2005;289:R247–R258. [DOI] [PubMed] [Google Scholar]
- 59.Sivarao D, Krowicki Z, Hornby PJN, Society mtojotEGM. Role of GABAA receptors in rat hindbrain nuclei controlling gastric motor function. 1998;10:305–313. [DOI] [PubMed] [Google Scholar]
- 60.Camilleri M Peripheral mechanisms in appetite regulation. Gastroenterology. 2015;148:1219–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zink AN, Bunney PE, Holm AA, Billington CJ, Kotz CMJIJoO. Neuromodulation of orexin neurons reduces diet-induced adiposity. 2018;42:737–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shi Y-C, Lau J, Lin Z, et al. Arcuate NPY controls sympathetic output and BAT function via a relay of tyrosine hydroxylase neurons in the PVN. 2013;17:236–248. [DOI] [PubMed] [Google Scholar]
- 63.Thorpe KE, Yang Z, Long KM, Garvey WT. The impact of weight loss among seniors on Medicare spending. Health Econ Rev. 2013;3:7–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.de Rooij SR, Nijpels G, Nilsson PM, et al. Low-grade chronic inflammation in the relationship between insulin sensitivity and cardiovascular disease (RISC) population: associations with insulin resistance and cardiometabolic risk profile. Diabetes care. 2009;32:1295–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Summers SA. Ceramides in insulin resistance and lipotoxicity. Progress in lipid research. 2006;45:42–72. [DOI] [PubMed] [Google Scholar]
- 66.de Ferranti S, Mozaffarian D. The perfect storm: obesity, adipocyte dysfunction, and metabolic consequences. Clin Chem. 2008;54:945–955. [DOI] [PubMed] [Google Scholar]
- 67.Schenk S, Saberi M, Olefsky JM. Insulin sensitivity: modulation by nutrients and inflammation. The Journal of clinical investigation. 2008;118:2992–3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sathyanarayana P, Barthwal MK, Kundu CN, et al. Activation of the Drosophila MLK by ceramide reveals TNF-alpha and ceramide as agonists of mammalian MLK3. Molecular cell. 2002;10:1527–1533. [DOI] [PubMed] [Google Scholar]
- 69.Bays HE, González-Campoy JM, Bray GA, et al. Pathogenic potential of adipose tissue and metabolic consequences of adipocyte hypertrophy and increased visceral adiposity. Expert Rev Cardiovasc Ther. 2008;6:343–368. [DOI] [PubMed] [Google Scholar]
- 70.Kern PA, Ranganathan S, Li C, Wood L, Ranganathan G. Adipose tissue tumor necrosis factor and interleukin-6 expression in human obesity and insulin resistance. American journal of physiology. Endocrinology and metabolism. 2001;280:E745–751. [DOI] [PubMed] [Google Scholar]
- 71.Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. The Journal of clinical investigation. 2006;116:3015–3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Xu H, Barnes GT, Yang Q, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. The Journal of clinical investigation. 2003;112:1821–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rocha VZ, Folco EJ, Sukhova G, et al. Interferon-gamma, a Th1 cytokine, regulates fat inflammation: a role for adaptive immunity in obesity. Circulation research. 2008;103:467–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Stratford S, Dewald DB, Summers SAJBJ. Ceramide dissociates 3′-phosphoinositide production from pleckstrin homology domain translocation. 2001;354:359–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Aguirre V, Uchida T, Yenush L, Davis R, White MF. The c-Jun NH(2)-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307). The Journal of biological chemistry. 2000;275:9047–9054. [DOI] [PubMed] [Google Scholar]
- 76.Itani SI, Ruderman NB, Schmieder F, Boden G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IkappaB-alpha. Diabetes. 2002;51:2005–2011. [DOI] [PubMed] [Google Scholar]
- 77.Arkan MC, Hevener AL, Greten FR, et al. IKK-beta links inflammation to obesity-induced insulin resistance. Nature medicine. 2005;11:191–198. [DOI] [PubMed] [Google Scholar]
- 78.Magkos F, Su X, Bradley D, et al. Intrahepatic diacylglycerol content is associated with hepatic insulin resistance in obese subjects. 2012;142:1444–1446. e1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Klop B, Elte JW, Cabezas MC. Dyslipidemia in obesity: mechanisms and potential targets. Nutrients. 2013;5:1218–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rocha VZ, Libby P. Obesity, inflammation, and atherosclerosis. Nature reviews. Cardiology. 2009;6:399–409. [DOI] [PubMed] [Google Scholar]
- 81.Boring L, Gosling J, Cleary M, Charo IF. Decreased lesion formation in CCR2−/− mice reveals a role for chemokines in the initiation of atherosclerosis. Nature. 1998;394:894–897. [DOI] [PubMed] [Google Scholar]
- 82.Wilson PW, D’Agostino RB, Sullivan L, Parise H, Kannel WB. Overweight and obesity as determinants of cardiovascular risk: the Framingham experience. Arch Intern Med. 2002;162:1867–1872. [DOI] [PubMed] [Google Scholar]
- 83.Lyall DM, Celis-Morales C, Ward J, et al. Association of Body Mass Index With Cardiometabolic Disease in the UK Biobank: A Mendelian Randomization Study. JAMA cardiology. 2017;2:882–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mokdad AH, Ford ES, Bowman BA, et al. Prevalence of obesity, diabetes, and obesity-related health risk factors, 2001. 2003;289:76–79. [DOI] [PubMed] [Google Scholar]
- 85.Nathan DMJNEjom. Long-term complications of diabetes mellitus. 1993;328:1676–1685. [DOI] [PubMed] [Google Scholar]
- 86.Koo SH. Nonalcoholic fatty liver disease: molecular mechanisms for the hepatic steatosis. Clinical and molecular hepatology. 2013;19:210–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bellentani S, Marino M. Epidemiology and natural history of non-alcoholic fatty liver disease (NAFLD). Annals of hepatology. 2009;8 Suppl 1:S4–8. [PubMed] [Google Scholar]
- 88.Pais R, Barritt 4th AS, Calmus Y, et al. NAFLD and liver transplantation: Current burden and expected challenges. 2016;65:1245–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hoyo C, Cook MB, Kamangar F, et al. Body mass index in relation to oesophageal and oesophagogastric junction adenocarcinomas: a pooled analysis from the International BEACON Consortium. 2012;41:1706–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chen Y, Liu L, Wang X, et al. Body mass index and risk of gastric cancer: a meta-analysis of a population with more than ten million from 24 prospective studies. 2013;22:1395–1408. [DOI] [PubMed] [Google Scholar]
- 91.Genkinger JM, Spiegelman D, Anderson KE, et al. A pooled analysis of 14 cohort studies of anthropometric factors and pancreatic cancer risk. 2011;129:1708–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Li L, Gan Y, Li W, Wu C, Lu ZJO. Overweight, obesity and the risk of gallbladder and extrahepatic bile duct cancers: a meta-analysis of observational studies. 2016;24:1786–1802. [DOI] [PubMed] [Google Scholar]
- 93.Moghaddam AA, Woodward M, Huxley RJCe, biomarkers p. Obesity and risk of colorectal cancer: a meta-analysis of 31 studies with 70,000 events. 2007;16:2533–2547. [DOI] [PubMed] [Google Scholar]
- 94.Bianchini F, Kaaks R, Vainio H. Overweight, obesity, and cancer risk. The Lancet. Oncology. 2002;3:565–574. [DOI] [PubMed] [Google Scholar]
- 95.Munsell MF, Sprague BL, Berry DA, Chisholm G, Trentham-Dietz AJEr. Body mass index and breast cancer risk according to postmenopausal estrogen-progestin use and hormone receptor status. 2014;36:114–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wearing SC, Hennig EM, Byrne NM, Steele JR, Hills AP. Musculoskeletal disorders associated with obesity: a biomechanical perspective. Obesity reviews : an official journal of the International Association for the Study of Obesity. 2006;7:239–250. [DOI] [PubMed] [Google Scholar]
- 97.Chen Y, Rennie D, Cormier YF, Dosman J. Waist circumference is associated with pulmonary function in normal-weight, overweight, and obese subjects. The American Journal of Clinical Nutrition. 2007;85:35–39. [DOI] [PubMed] [Google Scholar]
- 98.Peppard PE, Young T, Palta M, Dempsey J, Skatrud JJJ. Longitudinal study of moderate weight change and sleep-disordered breathing. 2000;284:3015–3021. [DOI] [PubMed] [Google Scholar]
- 99.Nowbar S, Burkart KM, Gonzales R, et al. Obesity-associated hypoventilation in hospitalized patients: prevalence, effects, and outcome. 2004;116:1–7. [DOI] [PubMed] [Google Scholar]
- 100.Sarkhosh K, Switzer NJ, El-Hadi M, Birch DW, Shi X, Karmali SJOs. The impact of bariatric surgery on obstructive sleep apnea: a systematic review. 2013;23:414–423. [DOI] [PubMed] [Google Scholar]
- 101.Louie JK, Acosta M, Samuel MC, et al. A novel risk factor for a novel virus: obesity and 2009 pandemic influenza A (H1N1). Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2011;52:301–312. [DOI] [PubMed] [Google Scholar]
- 102.Aminian A, Tu C, Milinovich A, Wolski KE, Kattan MW, Nissen SE. Association of Weight Loss Achieved Through Metabolic Surgery With Risk and Severity of COVID-19 Infection. JAMA Surgery. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Grunvald E, Shah R, Hernaez R, et al. AGA Clinical Practice Guideline on Pharmacological Interventions for Adults With Obesity. Gastroenterology. 2022;163:1198–1225. [DOI] [PubMed] [Google Scholar]
- 104.Glass J, Chaudhry A, Zeeshan MS, Ramzan Z. New Era: Endoscopic treatment options in obesity-a paradigm shift. World journal of gastroenterology. 2019;25:4567–4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Arterburn DE, Telem DA, Kushner RF, Courcoulas AP. Benefits and Risks of Bariatric Surgery in Adults: A Review. Jama. 2020;324:879–887. [DOI] [PubMed] [Google Scholar]
- 106.CDC. Defining Adult Overweight and Obesity. 3/November/2023. [Google Scholar]
- 107.Force USPST. A and B Recommendations. 2023. [Google Scholar]
- 108.Acosta A, Streett S, Kroh MD, et al. White Paper AGA: POWER - Practice Guide on Obesity and Weight Management, Education, and Resources. Clin Gastroenterol Hepatol. 2017;15:631–649.e610. [DOI] [PubMed] [Google Scholar]
- 109.Wildman RP, Muntner P, Reynolds K, et al. The obese without cardiometabolic risk factor clustering and the normal weight with cardiometabolic risk factor clustering: prevalence and correlates of 2 phenotypes among the US population (NHANES 1999–2004). Arch Intern Med. 2008;168:1617–1624. [DOI] [PubMed] [Google Scholar]
- 110.Eckel N, Meidtner K, Kalle-Uhlmann T, Stefan N, Schulze MB. Metabolically healthy obesity and cardiovascular events: A systematic review and meta-analysis. Eur J Prev Cardiol. 2016;23:956–966. [DOI] [PubMed] [Google Scholar]
- 111.Kuk JL, Ardern CI, Church TS, et al. Edmonton Obesity Staging System: association with weight history and mortality risk. 2011;36:570–576. [DOI] [PubMed] [Google Scholar]
- 112.Rexrode KM, Carey VJ, Hennekens CH, et al. Abdominal adiposity and coronary heart disease in women. Jama. 1998;280:1843–1848. [DOI] [PubMed] [Google Scholar]
- 113.Shuster A, Patlas M, Pinthus JH, Mourtzakis M. The clinical importance of visceral adiposity: a critical review of methods for visceral adipose tissue analysis. Br J Radiol. 2012;85:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lee CM, Huxley RR, Wildman RP, Woodward M. Indices of abdominal obesity are better discriminators of cardiovascular risk factors than BMI: a meta-analysis. J Clin Epidemiol. 2008;61:646–653. [DOI] [PubMed] [Google Scholar]
- 115.Paolini JB, Mancini J, Genestal M, et al. Predictive value of abdominal obesity vs. body mass index for determining risk of intensive care unit mortality. Crit Care Med. 2010;38:1308–1314. [DOI] [PubMed] [Google Scholar]
- 116.Wharton S, Lau DC, Vallis M, et al. Obesity in adults: a clinical practice guideline. 2020;192:E875–E891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Guo F, Moellering DR, Garvey WTJO. The progression of cardiometabolic disease: validation of a new cardiometabolic disease staging system applicable to obesity. 2014;22:110–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Batsis JA, Villareal DTJNRE. Sarcopenic obesity in older adults: aetiology, epidemiology and treatment strategies. 2018;14:513–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Cummings DE, Overduin J. Gastrointestinal regulation of food intake. The Journal of clinical investigation. 2007;117:13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Reinhardt M, Thearle MS, Ibrahim M, et al. A Human Thrifty Phenotype Associated With Less Weight Loss During Caloric Restriction. Diabetes. 2015;64:2859–2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Fothergill E, Guo J, Howard L, et al. Persistent metabolic adaptation 6 years after “The Biggest Loser” competition. Obesity (Silver Spring). 2016;24:1612–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hebebrand J, Holm JC, Woodward E, et al. A Proposal of the European Association for the Study of Obesity to Improve the ICD-11 Diagnostic Criteria for Obesity Based on the Three Dimensions Etiology, Degree of Adiposity and Health Risk. Obes Facts. 2017;10:284–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mechanick JI, Hurley DL, Garvey WT. Adiposity-Based Chronic Disease as a New Diagnostic Term: The American Association of Clinical Endocrinologists and American College of Endocrinology Position Statement. Endocr Pract. 2017;23:372–378. [DOI] [PubMed] [Google Scholar]
- 124.Blundell JE, Dulloo AG, Salvador J, Fruhbeck G, BMI ESWGo. Beyond BMI--phenotyping the obesities. Obes Facts. 2014;7:322–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Acosta A, Camilleri M, Abu Dayyeh B, et al. Selection of Antiobesity Medications Based on Phenotypes Enhances Weight Loss: A Pragmatic Trial in an Obesity Clinic. Obesity (Silver Spring). 2021;29:662–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Cifuentes L, Ghusn W, Feris F, et al. Phenotype tailored lifestyle intervention on weight loss and cardiometabolic risk factors in adults with obesity: a single-centre, non-randomised, proof-of-concept study. eClinicalMedicine. 2023;58:101923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Maselli D, Atieh J, Clark MM, et al. Effects of liraglutide on gastrointestinal functions and weight in obesity: A randomized clinical and pharmacogenomic trial. Obesity (Silver Spring). 2022;30:1608–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lopez-Nava G, Jaruvongvanich V, Storm AC, et al. Personalization of Endoscopic Bariatric and Metabolic Therapies Based on Physiology: a Prospective Feasibility Study with a Single Fluid-Filled Intragastric Balloon. Obes Surg. 2020;30:3347–3353. [DOI] [PubMed] [Google Scholar]
- 129.Rapaka B, Maselli DB, Lopez-Nava G, et al. Effects on physiologic measures of appetite from intragastric balloon and endoscopic sleeve gastroplasty: results of a prospective study. Chin Med J (Engl). 2022;135:1234–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]