

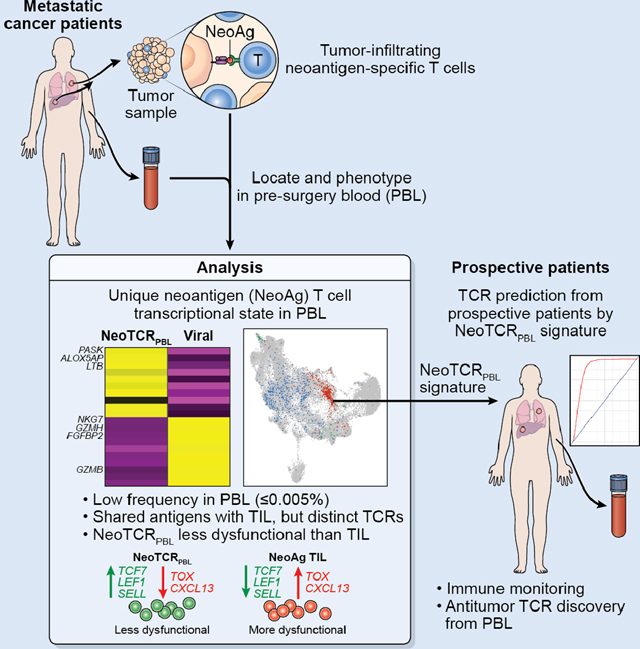
SUMMARY
Circulating T cells from peripheral blood (PBL) can provide a rich and non-invasive source for antitumor T cells. By single-cell transcriptomic profiling of 36 neoantigen-specific T cell clones from 6 metastatic cancer patients, we report the transcriptional and cell surface signatures of antitumor PBL-derived CD8+ T cells (NeoTCRPBL). Comparison of TIL- and PBL-neoantigen-specific T cells revealed that NeoTCRPBL T cells are low in frequency and display less-dysfunctional memory phenotypes relative to their TIL counterparts. Analysis of 100 antitumor TCR clonotypes indicates that most NeoTCRPBL populations target the same neoantigens as TILs. However, NeoTCRPBL TCR repertoire is only partially shared with TIL. Prediction and testing of NeoTCRPBL signature-derived TCRs from PBL of 6 prospective patients demonstrate high enrichment of clonotypes targeting tumor mutations, a viral oncogene, and patient-derived tumor. Thus, the NeoTCRPBL signature provides an alternative source for identifying antitumor T cells from PBL of cancer patients, enabling immune monitoring and immunotherapies.
Keywords: Blood, CD8+ T cells, neoantigens, adoptive cell transfer, T cell receptors
eTOC blurb
Yossef et al. report that antitumor T cells in the blood of metastatic cancer patients exhibit a distinct transcriptional profile. They further show that this gene signature can be leveraged to predict and identify antitumor T cell receptors, providing a noninvasive source to develop T cell therapies against cancer.
Graphical Abstract
INTRODUCTION
Immunotherapies such as adoptive cell therapy (ACT), and immune checkpoint blockade therapy (ICB), represent promising approaches to treat metastatic human solid epithelial cancers 1–4. While immune infiltration of tumors varies depending on stage and histology5, the vast majority of metastatic human tumors still retain antitumor CD8+ and CD4+ T cell immune recognition of tumor antigens and epitopes encoded by somatic tumor mutations (neoantigens), suggesting an ongoing antitumor immune response even in tumors conventionally thought of as immunologically “cold”6–9.
Identification of antitumor T cells, their antigenic specificities, and their cognate T cell receptors (TCRs) have provided crucial insights into antitumor T cell immunity and led to the design of next-generation engineered cell therapies. Tumor-infiltrating lymphocytes (TIL) from surgically resected tumors represent an enriched source for isolating and identifying antitumor T cells 10. In vitro cultured TIL can be assessed for functional recognition of tumor-specific antigens, and their TCRs can be isolated using cell surface phenotypic markers of exhaustion6,9,11,12. However, these markers are often non-specific as their expression on bystander tumor-irrelevant TIL corrupts the accurate identification of antitumor TIL that also suffer from functional impairment13. Alternatively, fluorescent, metal-labeled, or DNA-barcoded multimers of human leukocyte antigen (HLA) proteins loaded with predicted neopeptides can be used to identify neoantigen-reactive CD8+ T cells from TIL and blood13–17; however, this approach requires a priori knowledge of candidate HLA-binding peptides based on prediction algorithms tailored to the patients’ HLA alleles.
Recent advances utilizing single-cell transcriptomic (scRNA) profiling of antitumor, neoantigen-specific TIL have provided unprecedented insights into the phenotypes, differentiation states, and TCR-repertoire of tumor-reactive human TIL18. These studies have identified the shared expression of antitumor T cell dysfunction programs across human cancers, and have together highlighted the predominance of dysfunctional TIL within metastatic tumor deposits19–25. Additionally, these studies have also demonstrated that identification of anti-tumor TCRs based only on the transcriptomic states of TILs, is possible20,22. However, such TIL profiling requires the invasive surgical resection of tumors from patients followed by the separation of TIL from tumors before phenotypic analyses.
Circulating blood represents an alternative source for tumor-specific T cells and their TCRs, circumventing the need for invasive surgery and growth of TIL. From an immune monitoring perspective, identifying and studying antitumor T cell states in the circulation can provide insights into the breadth and landscape of antitumor T cell immunity in patients with metastatic cancer. Importantly, it is currently unknown if the antigenic repertoire, TCR repertoire, and the phenotypic states of antitumor T cells vary between tumor and blood in humans. Recent murine studies on antitumor T cells from circulating blood have suggested that there is a clonal and phenotypic overlap with TIL, although their relevance to metastatic human cancers remains unclear 26,27. Defining the cellular states and targets of circulating blood-derived neoantigen-specific T cells within PBL of heavily pretreated metastatic cancer patients, however, is inherently challenging due to their extremely low frequencies 28–30.
RESULTS
A tetramer based single-cell transcriptomic pipeline identifies pre-surgery circulating neoantigen-specific T cell phenotypes
Here we sought to define the phenotypic states of circulating anti-tumor reactive T cells in a cohort of metastatic cancer patients largely composed of patients with solid epithelial cancers who had not received prior ICB (Table S1A). To avoid in vitro growth of antitumor T cells that might alter their native phenotypes, and to distinguish their phenotypes from irrelevant T cells, we devised a PBL-neoantigen T cell discovery strategy wherein we located bonafide neoantigen-specific CD8+ clonotypes identified from patient TIL within their PBL compartment by tetramer-enrichment via fluorescent activated cell sorting (FACS) (Fig. 1A). We then mixed the known proportion of neoantigen tetramer+ PBL T cells back into CD8+ tetramer-negative PBL T cells, followed by single-cell transcriptome (scRNA) and TCR (scTCR) sequencing analysis (Fig. 1A–D).
Figure 1. Tetramer enrichment and scRNAseq of circulating neoantigen-reactive CD8+ T cells.
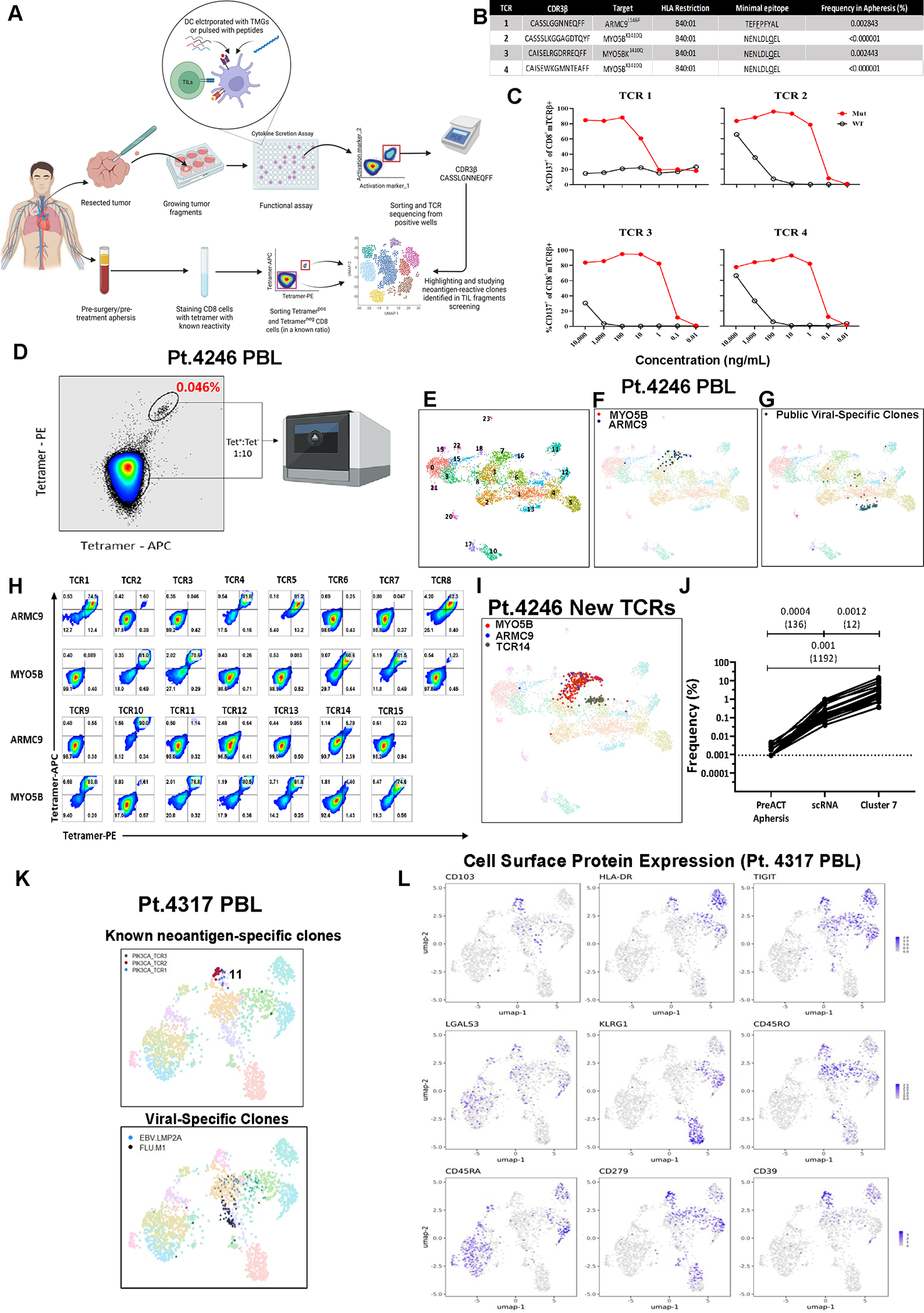
(A) Graphical pipeline summary of discovery, tetramer-enrichment and phenotypic analysis of neoantigen-reactive circulating CD8+ cells (prepared using BioRender.com). (B) Known neoantigen-reactive TCR CDR3β sequences, specificities, HLA of restriction, minimal epitope, and their frequencies in the pre-surgery blood of metastatic colon cancer patient (Pt. 4246). (C) CD137 expression of health-donor PBL TCR-transduced PBLs following overnight co-culture with 4246 dendritic cells (DCs) pulsed with various dilutions of their cognate minimal mutated or wild-type peptides. (D) Tetramer-enrichment sort of circulating neoantigen-reactive T cells spiked back into bulk CD8+ population at a 1:10 ratio. (E) UMAP projection of the single-cell transcriptome of Pt.4246 PBL. (F) Previously known neoantigen-reactive T cells (against ARMC9L146F or MYO5BK1410Q) highlighted in Pt.4246 PBL (G) Public viral-targeting TCRs highlighted (H) Healthy donor PBLs virally-transduced with candidate TCRs from cluster 7 (C7) stained with ARMC9L146F or MYO5BK1410Q fluorescent tetramers. (I) Highlighting all T cell clones expressing neoantigen-reactive TCRs. (J) Summary of enrichment of neoantigen-reactive clones in the pre-enrichment PBL, scRNA after enrichment, and specifically in cluster 7. (K) UMAP projection of scRNAseq of PIK3CAP449T:HLA-A*02:01-tetramer-enriched T cells from the peripheral blood sample of a metastatic cancer patient (Pt. 4317). (L) Expression of cell surface feature-barcoded CITE-Seq antibody staining intensities of protein markers. Also see Fig. S1–3 and Table S1.
We first employed this strategy on PBL obtained from a colorectal cancer patient (pt.4246) where we had previously identified 4 neoantigen-specific TCRs by screening the patient’s in vitro cultured TIL against tumor-derived mutations using tandem minigene and mutant peptide-pool screening approach we had previously developed (Fig. 1B–C, Fig. S1A–B) 6,9. We FACS-sorted 2000 PBL-derived CD8+ HLA-B*40:01 tetramer-enriched ARMC9L1410Q and MYO5BK1010Q NeoTCRPBL CD8+ T cells and spiked them into 18000 CD8+ tetramer negative T cells and performed single-cell transcriptome (scRNA) and TCR (scTCR) sequencing (Fig. 1D). Unsupervised transcriptomic clustering of 5054 combined PBL CD8+ T cells followed by uniform manifold approximation and projection (UMAP) analysis defined 24 cell states comprising 1770 distinct clonotypes. Transcriptomic states of PBL-derived CD8+ T cells ranged from less-differentiated naive T cells (cluster 0) to differentiated T cell states, such as effector-like (cluster 1) and effector memory (cluster 2), to highly differentiated cytotoxic T cells (cluster 4) (Fig. 1E, Fig. S1C, Table S1B). We mapped the 4 known neoantigen-specific TCR clonotypes identified from TIL within the PBL (0.77% of total T cells) by mapping their TCR identity to their cell state as described previously20,22. The majority (69.23%) of the known MYO5BK1010Q and ARMC9L1410Q- specific T cells were observed within cluster 7, while 25.6% of these neoantigen-specific T cells were found distributed between clusters 3 and 16 (Fig. 1F, Fig. S1D). Cluster 7 had a unique transcriptional profile expressing cell signaling and T cell activation genes such as COTL1, PASK, ALOX5AP, HLA-DRB1, HLA-DPA1, as well as memory, quiescence markers such as SELL (CD62L), LTB, KLF2, LGALS3 but relatively low expression of genes encoding cytotoxic molecules PRF1, GZMH, GNLY (Fig. S1C, Table S1B). In contrast, 74 CD8+ virus-specific T cells expressing 6 public viral TCR clonotypes targeting cytomegalovirus (CMV), Epstein-Barr virus (EBV), and Influenza A virus (Flu) predominantly mapped to cluster 13 (72%), distinct from those comprising neoantigen-specific T cells (Fig. 1G).
Since the known neoantigen-specific T cell clonotypes only represented 9.75% of cells in cluster 7, we reasoned that cluster 7 might contain additional unidentified ARMC9 or MYO5B-reactive neoantigen-specific TCR clonotypes enriched by our tetramer sorting (Fig. 1D). To test this hypothesis, we utilized a transcriptome based method that we had previously shown to predict novel antitumor, neoantigen-specific TCR clonotypes using transcriptomic states of uncultured TIL20,22,23. We screened PBL that were transduced with retroviral constructs encoding fourteen dominant TCR clonotypes from PBL-derived cluster 7, and one clonally expanded dominant clone that was also enriched in cluster 6 (TCR14) and performed neoantigen-specific T cell screening (Fig. 1H, Fig. S1D–F). Neoantigen-specific tetramer staining, and functional screening demonstrated that 14/15 TCRs derived from cluster 7 displayed high neoantigen-specificity with varying functional TCR avidities (Fig. 1H, Fig. S1E–F). Similar to the clonal distribution of known neoantigen-TCR clonotypes, 64.7% of newly discovered ARMC9L1410Q- and MYO5BK1010Q clonotype expressing T cells were found in cluster 7, with 31.7% found in cluster 3, and cluster 16 (Fig.1I, Fig.S1D). Notably, TCR14 stained tetramer-negative suggesting that clonally expanded tumor irrelevant bystander T cells are abundant in cluster 6 and in the PBL (Fig. 1I, Fig. S1D). Combined analysis of all 18 neoantigen-specific PBL clonotypes from pt.4246 indicated a >1100-fold enrichment of neoantigen-specific TCR clonotypes within cluster 7 (P = 0.001, paired T-test compared to bulk PBL, Fig. 1J). We performed a similar scRNA analysis on neoantigen-specific (CMTR1K601T) tetramer-enriched CD8+ T cells doped into CD8+ PBL from a second metastatic colon cancer patient pt.4287 (Fig. S2A–B). Among the 3358 CD8+ T cells that were analyzed, the 2 known CMTR1K601T neoantigen-TCR clonotypes comprised 0.42% of all cells (Fig. S2C). All neoantigen-reactive T cells (100%) were observed in cluster 4, distinct from transcriptomic states of known public viral TCR clonotype expressing T cells (Fig. S2D). These results support our neoantigen-PBL T cell phenotypic profiling approach and demonstrate that neoantigen-specific CD8+ T cells from circulating blood exist in unique transcriptional states.
PBL-derived circulating neoantigen-specific T cells express unique cell surface protein markers
We attempted to circumvent the need for HLA-specific tetramers by comprehensively profiling circulating neoantigen-reactive CD8+ T cells based on the expression of cell-surface markers. We, therefore, performed Cellular Indexing of Transcriptomes and Epitopes by Sequencing (CITE-Seq, Table S1C) on neoantigen-specific HLA A*02:01-restricted PIK3CAP449T CD8+ T cells we had identified by in vitro TIL screening from a metastatic rectal cancer patient TIL (pt.4317, Table S1D), along with Flu M1(58–66) and EBV LMP2A(426–434) viral-tetramer T cells (Fig. 1K, Fig. S2E–G, Table S1E) 22. The majority of the 3 PIK3CAP449T PBL TCR clonotypes converged within transcriptomic cluster 11 (median > 66.67%), while Flu and EBV tetramer+ T cells were predominantly distributed between transcriptomic clusters 1, 4, and 9 (Fig. 1K, Fig. S2F–G). Analysis of protein expression of PIK3CAP449T T cells within cluster 11 indicated relatively high levels of CD45RO and lower levels of CD45RA cell-surface protein expression (Fig. 1L, Fig. S2I), indicative of a memory phenotype, as has been suggested by other studies, as well as scRNA results from pt.4246 (Fig. S1C) 28. PIK3CAP449T T cells within cluster 11 also had high cell surface protein expression of the HLA-DRA activation marker, the tissue-residency marker CD103, and TIGIT, PD-1, and CD39, which have all been associated with dysfunctional T cells and enriched for neoantigen-specific TIL (Fig. 1L, Fig. S2I) 13,20,22,23,25. Based on these results, we developed a tetramer-agnostic neoantigen T cell enrichment strategy by FACS-sorting CD8+CD45RO+HLA-DRAhi-CD39+, CD103+ or CD39+CD103+ PBL-T cells from 3 colorectal cancer patients (pt.4382, pt.4422, pt.4324) and doping the sorted cells back into bulk CD8+ T cells followed by scRNA-scTCR analysis (Fig. S3A–B). Neoantigen-specific T cell clones, previously identified in cultured TIL from circulating PBL of 3 patients, were enriched in specific CD8+ T cell transcriptional states, similar to those seen in tetramer-enriched samples (Fig. S3C–F). These data suggest that circulating neoantigen-reactive CD8+ T cells also possess a unique cell-surface marker profile that can be leveraged to enrich antitumor T cells without the need for prior knowledge of patients’ HLA haplotypes or mutations present in the tumor.
Circulating neoantigen-specific CD8+ T cell phenotypic states are shared among patients
To gain a granular understanding of the phenotypes of circulating neoantigen-specific CD8+ T cells, we performed a combined analysis of the transcriptomic states of 36 neoantigen-specific CD8+ NeoTCRPBL clones from all 6 patients (Fig. 2A, Table S1D). The dominant transcriptional program of circulating T cells was shared among patients, with a median frequency of 75% percent of all 36 NeoTCRPBL clones from these 6 patients observed in cluster 9, and 11.76% found within cluster 3 (Fig. 2A–B, Table S1D). NeoTCRPBL clones from each of the 6 patients were specifically enriched within cluster 9 (Fig. S4A). In contrast, 39 bystander viral T cell clones defined either by high confidence public TCR clonotypes, or by viral antigen-specific tetramer, or functional peptide recognition (Table S1E, STAR methods) in these six patient PBL were distributed across multiple distinct phenotypic states (clusters 1, 2, 3, 4, 5 and 7 in Fig. 2B) and were largely low frequency within the dominant neoantigen-specific transcriptional state C9 (mean 0.7% of all viral TCR clones, Fig. 2B).
Figure 2: Transcriptional program of circulating neoantigen-reactive CD8+ T cells.
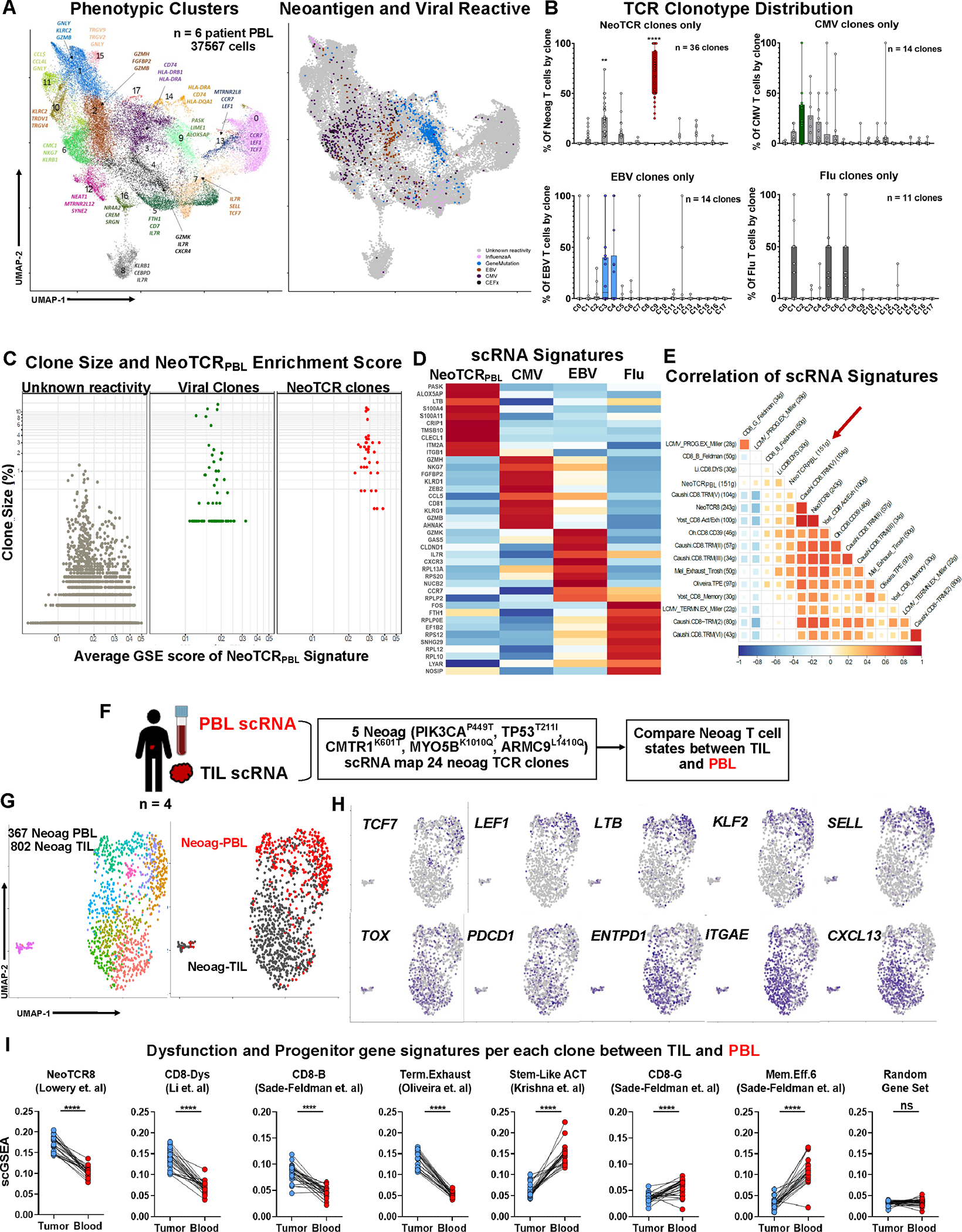
(A) Transcriptional clustering and UMAP projection of circulating CD8+ cells from six metastatic cancer patients’ blood samples (Left) and Projection of neoantigen- and viral-reactive clones on the UMAP space (right). (B) Frequency of neoantigen- and viral-reactive clones within UMAP clusters. **P < 0.01, ****P < 0.001 by Two-way ANOVA adjusted by Bonferroni multiple corrections (C) Comparison of clonal frequency and average NeoTCRPBL signature score by scGSEA within neoantigen-, viral-specific clones, and rest of clones with unknown-reactivities. Welch Two Sample t-test of clone size between viral and NeoTCRPBL clones P = 0.227. (D) Heatmap of top 10 differentially expressed genes between Neoantigen-, CMV, EBV-, and InfluenzaA-reactive T cells from the six patient PBL. (E) Pearson correlation between public TIL gene-signatures and NeoTCRPBL. (F) Schematic representing the combined TIL + PBL neoantigen T cell phenotypic states analysis within each patient. (G) UMAP displaying the combined analysis of neoantigen-specific T cells (24 total neoantigen T cell clones) from each of the 4 patients (left panel), and segregation of transcriptomic states based on their TIL compartment (Neoag-TIL) or peripheral blood compartment (Neoag-PBL) (right panel). (H) Gene expression of candidate memory progenitor genes (top panel), and tissue-residency, dysfunctional T cell genes (bottom panel) across the combined TIL-PBL UMAP. (I) Average gene signature scores (scGSEA) scores of immunotherapy response and non-response associated gene signatures that indicate T cell dysfunction, stem-like progenitor states within each individual neoantigen-reactive clone (n = 24) compared between its TIL compartment and PBL compartment from all 4 patients. Random gene set of 50 genes are displayed as control gene signature. ****P < 0.0001 by Paired T-test per each neoantigen T cell clonotype. Also see Fig S.4–5 and Table S1–2.
The C9 transcriptional program remained intact over a wide range of resolution parameters used for single-cell transcriptomic analysis (Fig. S4B), suggesting a distinct phenotypic profile of circulating antitumor neoantigen-specific T-cell clones. The combined scRNA analysis demonstrated that cells within C9 had relatively lower expression of genes encoding cytotoxic molecules (GZMB, GNLY), but had higher expression of genes involved in T cell activation (HLA-DRB, ALOX5AP, COTL1), memory quiescence (LEF1, KLF2, LTB, SELL), and tissue-residency (ZNF683, CXCR6, ITGAE) suggesting that antitumor T cells in the circulation likely home to, or egress from tumoral sites (Fig. S4C, Table S2). Reflecting cell-surface protein expression of CD39, circulating neoantigen T cells had transcriptional expression of inhibitory markers PDCD1, CTLA4, TIGIT (Table S2, Fig. S4C). Cluster 9 also had lower expression of cytotoxic effector molecules such as GZMA, GZMH, GZMK, GZMB when compared to cluster 3 which contained a minor proportion of neoantigen-specific T cells (Table S2C). Pathway analysis of the differentially expressed genes within the C9 transcriptional state indicated positive enrichment of pathways associated with PI3K/AKT signaling, mTOR signaling and cytoskeleton, integrin signaling pathways, and negative association with EIF2 signaling and Granzyme A signaling pathways suggesting lower T cell effector function (Fig. S5A).
While there was variation in the expression of genes within C9 between the 6 patient PBL (Fig. S5B), given the high enrichment of neoantigen-specific T cell clones from each of the patients within cluster 9 (Fig. S4A), we reasoned that a transcriptional signature of 151 differentially upregulated genes in C9 derived from the combined analysis of the 36 clones might encompass a common gene signature of circulating antitumor T cells (cut-off of Log2FC > 0.5, termed NeoTCRPBL signature, Supplementary Materials and Methods) (Table S2D). Single-cell gene signature analysis (scGSEA) demonstrated that NeoTCRPBL T cells exhibited phenotypic states distinct from acute (Flu) or latent/chronic states of infection (EBV, CMV) (p < 2.2e-16, Welch’s T-Test, Fig. 2A–D), despite displaying clonal expansion comparable to that of bystander virus-specific T cells in the circulation (p = 0.227, Welch’s T-Test, Fig. 2C). Importantly, NeoTCRPBL T cells could be distinguished from EBV- and CMV-specific T cells by relatively low expression of genes encoding cytotoxic programs (NKG7, GZMH, GZMK, and KLRG1, Fig. 2D). Despite shared expression of some memory genes, NeoTCRPBL T cells could be distinguished from Flu-specific T cells by their lower expression of IL7R and TCF7, as shown in recent TIL studies (Fig. 2D) 20,21. Correlation analyses of the NeoTCRPBL transcriptional program revealed a positive correlation with metastatic TIL dysfunction signatures previously described by us and others (e.g. NeoTCR8, Yost-Exh) and with resident-memory (Caushi TRM) and progenitor-exhausted (Oliveira TPE) TIL signatures (Fig. 2E) 19–22,31. These results suggest that circulating neoantigen-specific T cells exhibit transcriptional identities that were likely originated from tumor sites, with memory states reflecting prior antigen encounter, and are distinct from different classes of viral T cell clonotypes.
Circulating neoantigen-specific CD8+ T cells exhibit less-dysfunctional transcriptional states compared to tumor resident neoantigen-specific TIL
We next investigated whether, within the same patient, circulating PBL-derived antitumor neoantigen-specific T cells maintained less-dysfunctional memory progenitor states relative to their uncultured TIL counterparts resected from tumor specimens. To this end, from 4 patients (pt.4317, pt.4324, pt. 4246, pt.4287), we performed paired scRNA analysis on 1169 neoantigen-specific CD8+ T cells derived from PBL and TIL after normalization (Fig. 2F–G, STAR methods). We then compared the phenotypic states of neoantigen-specific T cell clones found within circulating blood to the same T cell clones found within TIL that had previously been reported to possess unique transcriptomic signatures (Fig. 2F)19,20,22,25,32. Neoantigen-specific T cells from these four patients separated into distinct phenotypic clusters that were predominantly associated with either their TIL or PBL identities (Fig. 2G, right panel), despite the shared tumor-relevant gene expression programs observed between the two sources (as shown by correlation analyses in Fig. 2E). Relative to NeoTCR T cells from within TIL, NeoTCRPBL T cells displayed higher gene expression of canonical stemness markers TCF7, KLF2, LTB, SELL (CD62L) and LEF1, with relatively lower transcriptional expression of canonical dysfunction markers TOX, ENTPD1 (CD39) and CXCL13 (Fig. 2H). Importantly, at the clonotype level, relative to the same clones within the TIL at that time point, NeoTCRPBL T cells scored higher for less-dysfunctional transcriptional gene signatures associated with immune checkpoint blockade-therapy response (CD8-G, Sade-Feldman et al. 2018), and TIL-ACT response associated stemlike signatures (Stem-like ACT, Krishna et al, 2020) (Fig. 2I, P < 0.0001, paired T-Test, n = 24 NeoTCR clones). Additionally, relative to neoantigen-specific T cell clones within TIL, NeoTCRPBL clones within the circulation also scored lower for immunotherapy non-responsive signatures such as CD8-B, and terminally-dysfunctional TIL signatures (NeoTCR8, Lowery et al, Term.Exhaust, Oliveira et al gene signatures) (Fig. 2I, P < 0.0001, paired T-Test, n = 24 NeoTCR clones). Importantly, no such differences between the same clones within TIL and PBL were observed when a random gene set was used as a control comparator for these gene signature analysis (Fig. 2I). These data indicate an intra-patient site-dependent phenotypic heterogeneity within tumor-specific T cell clones and suggest that circulating blood-derived neoantigen T cells, despite sharing TIL dysfunction programs, consistently display a less-dysfunctional tissue-resident memory-profile associated with better immunotherapy responses.
NeoTCRPBL transcriptomic signatures can prospectively identify neoantigen-specific TCR clonotypes
We then evaluated canonical T cell subset markers, the previously reported PD1 and the NeoTCRPBL cell-surface markers identified in this study for their ability to capture and enrich neoantigen T cell clonotypes from pt.4246 PBL 28,33,34 (Fig. 3A, Fig. S6A–D). While previously reported central memory markers and cell surface PD1 enriched for known neoantigen TCR clonotypes from PBL, NeoTCRPBL marker-based FACS-sorting resulted in a >2100-fold enrichment of known neoantigen-specific TCR clonotypes (P =0.0002) whereas FACS-sorting by PD-1 alone resulted in a 33-fold enrichment of these clonotypes (P=0.0002, Fig. 3A). We therefore hypothesized that the NeoTCRPBL cell-surface markers followed by gene-signature analysis identified can be used to prospectively isolate neoantigen-specific circulating T cells from patients with varying tumor histologies. We then FACS-sorted circulating CD8+ CD45RO+HLA-DRhiCD39+CD103+ T cells from 3 prospective patients with metastatic breast cancer (pt.4180), colon cancer (pt.4359), and melanoma (pt.3791), spiked into bulk PBL CD8+ T cells followed by scRNA-scTCR analysis (Fig. 3B–C, Fig. S6E–H, Fig. S7). We identified putative NeoTCRPBL signature expressing clones by scGSEA from each PBL sample, using AUCell (see STAR methods) and reconstructed the top expanded TCR clonotypes for prospective experimental testing against candidate tumor neoepitopes (Fig. 3C, Fig.S6F, Fig. S7B).
Figure 3: Prospective prediction of neoantigen-reactive circulating CD8+ T cells patient PBL.
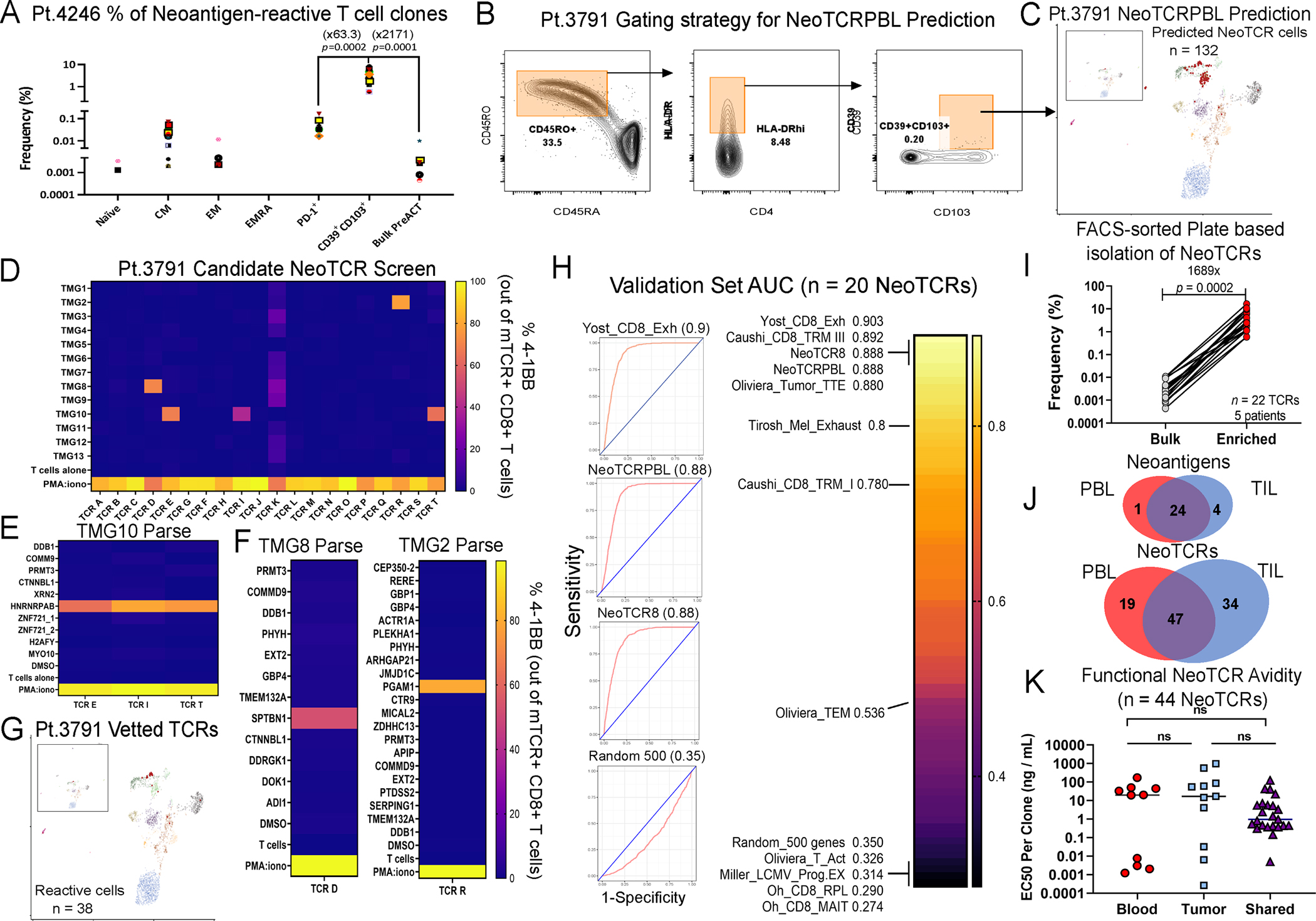
(A) Frequency of known neoantigen-reactive T cell clones in FACS-sorted T cell subsets from pt.4246 PBL. Fold change of the mean frequencies of neoantigen-reactive T cell clones between indicated groups is presented in parentheses. T-test p-value is shown between the groups. Each colored shape represents a different neoantigen TCR clone from pt.4246 PBL. (B) FACS-sorting enrichment gating of circulating CD8+ T cells from pt.3791 for scRNAseq. 3,290 CD39+CD103+ were sorted and mixed with 9500 bulk CD8+ T cells from PBL. (C) Clustering and projection of predicted neoantigen-reactive T cells from PBL, based on AUCell (red). (D) Frequency of TCR-transduced CD8+ cells expressing CD137 following co-culture with imDCs electroporated with patient’s TMGs (D) or mutated peptides for the corresponding TMG. (E-F) Deconvolution of TMG hits to identify specific neoantigens recognized by patient NeoTCRPBL. (G) Summary back-projection of experimentally vetted NeoTCRPBL cells on pt.3791 PBL UMAP. (H) Summarized mean AUC scores of ROC analysis comparing NeoTCRPBL signature and published TIL gene-signatures for prediction of neoantigen-reactive T cells from three validation set samples (pt.3791, pt.4180, pt.4359). Random 500 gene set is shown as control (I) FACS-based enrichment and identification of neoantigen-reactive clones within circulating lymphocytes from PBL of 5 prospective patients. Neoantigen-reactive clone frequency was compared between bulk lymphocytes and within enriched sorted populations (based on Table S1). Numbers represent fold enrichment and p-value of Paired T-test. (J) Summary of the landscape of neoantigen-reactive TCR clonotypes and their cognate neoantigens shared between TIL and PBL (identified by NeoTCRPBL signature or FACS-sorting) from all patients (total of 100 TCRs from TIL and PBL). (K). Functional avidity of 44 NeoTCR clones that were either found only in the PBL compartment (Blood), TIL compartment (Tumor), or shared between PBL and TIL (Shared). Data shown is the half-maximal reactivity for each TCR clone assessed by titrating the reactivity across a wide range of cognate mutated neopeptide concentrations relative to the wildtype peptide pulsed on autologous APCs. Also see Fig. S6–8 and Table S1.
From PBL of metastatic melanoma patient pt.3791, we evaluated 20 candidate NeoTCRPBL clonotypes and identified 3 TCRs targeting neoantigen HNRNRPABG87E, 1 TCR targeting SPTBN1A837V, and 1 TCR targeting PGAM1A237T neoantigens (Fig. 3D–G). Three of these positive TCRs were novel and had not been identified in conventional TIL screening. The frequencies of all neoantigen-reactive TCRs in bulk unenriched circulating T cells were below the limit of detection (<0.002%, Fig. 3G) (Table S1F). From PBL sample obtained from a triple-negative breast cancer patient pt.4180, we tested 22 candidate NeoTCRPBL clonotypes for tumor-reactivity and identified 5 TCRs targeting TTLL12K306 neoantigen, 2 TCRs targeting CCT8R317M and 1 TCR targeting NUP93 neoantigen (Fig. S6G–H, Table S1F). The cumulative frequency of the 8 positive TCRs prior to enrichment was ≤ 0.05% among circulating lymphocytes, with 5 TCR clonotypes being novel and undetected within TIL fragments (Table S1F). From PBL sample obtained from a metastatic colorectal cancer patient pt.4359, we tested 23 candidate NeoTCRPBL TCR clonotypes and identified 6 TCRs targeting tumor neoantigen EIF2AG186A, 4 of which were novel, and 1 novel TCR targeting RPS13R99Q neoantigen (Fig. S7C–D). Only one clone (TCR U) was detected within T cells in the PBL by bulk TCR-sequencing without enrichment (0.00238%, Table S1G). From the 3 prospective patient PBL samples, 965 total TCR candidates (including singletons clonotypes) fulfilled the criteria of clonal expansion or high NeoTCRPBL signature scores (STAR methods). We experimentally evaluated 65 TCR clonotype candidates to identify 20 neoantigen-reactive TCRs (an estimated 30.7%) suggesting that the NeoTCRPBL signature can successfully prospectively identify antitumor TCRs from very low circulating frequencies (Table S1G).
Sensitivity and Specificity of NeoTCRPBL gene signature in identifying antitumor TCRs
To assess the sensitivity and specificity of NeoTCRPBL gene signatures and other TIL signatures to identify circulating antitumor TCRs from patient PBL, we performed receiver operating characteristic (ROC) analysis using scGSEA scores on scRNA-scTCR data from the 3 validation samples (STAR Methods). We found that NeoTCRPBL gene signature along with TIL exhaustion signatures previously described by us and others (NeoTCR8, Oliveira_Tumor_TTE, Yost_CD8_Exh, Caushi_CD8_TRM III) to have high sensitivity and specificity (area under the curve (AUC) > 0.88) in identifying neoantigen-specific T cells from circulating blood (Fig. 3H) 20–22,31. In contrast, gene sets composed of random genes, gene signatures describing effector memory, activation, and proliferation states of T cells performed poorly in predicting experimentally vetted NeoTCRPBL clonotypes (Fig. 3H, Fig. S8A top panel). Notably, a “circulating TIL” signature identified in a previous study based on bulk TCR clonotypic overlap between PBL and TIL27, as well as a combination of previously described TIL neoantigen-specific gene markers using ENTPD1 (CD39) and ITGAE (CD103)35, demonstrated lower sensitivity and specificity relative to NeoTCRPBL signature in predicting CD8+ NeoTCRs from PBL (AUCs = 0.64 and 0.69 respectively) in the prospective cohort (Fig. S8A bottom panel), indicating the importance of utilizing transcriptomic signatures trained specifically on experimentally vetted and confirmed antitumor T cells for TCR prediction. Finally, in this validation cohort, NeoTCRPBL scores exhibited high sensitivity and specificity for only predicting neoantigen-specific clonotypes (Fig. 3H), but not tumor-irrelevant bystander public viral clonotypes (n = 9 viral clonotypes, AUC = 0.377, Fig. S8B).
We compared and contrasted the NeoTCRPBL signature to the 5 top TIL gene signatures described in Figure 3H. Interestingly, while genes were shared between each of the different transcriptional programs, no single gene was shared across all 5 gene signatures, suggesting that TIL and PBL transcriptional states are likely picking up distinct aspects of antitumor T cell dysfunction or tissue residence (Fig. S8C). This is further evidenced by the high sensitivity and specificity performance of genes that were not shared with TIL and were unique only to the NeoTCRPBL gene signature (NeoTCRPBL-unique, 111/151 distinct genes not shared with previously studied TIL signatures). The ROC analysis of this gene set with 111 unique genes resulted in a comparable AUC score (0.864) with the original 151 gene signature (0.888) (Fig. S8D, left panel). ROC analysis of the top 50 genes within NeoTCRPBL gene signature also performed comparably as the original and NeoTCRPBL-unique gene signatures in predicting prospective neoantigen TCR clonotypes (AUC=0.88, Fig. S8D, right panel). Taken together, these data demonstrate that transcriptional states from tumor and circulation can be used to identify neoantigen-specific TCR clonotypes with high accuracy.
NeoTCRPBL-based cell surface markers can prospectively capture neoantigen-specific TCR clonotypes
We further developed a simple FACS-based plate-sorting TCR-identification strategy based on a nested CDR3ɑ and CDR3β PCR amplification strategy that we had previously developed by single-cell sorting of PBL CD8+ T cells expressing CD45RO+HLA-DRhi and CD39+CD103+ from 8 metastatic cancer patients (Table S1H) followed by experimental testing against all patient tumor mutations. We identified reactive TCR clonotypes in 6/8 patients with a median frequency of 15.4% in the sorted T cells with a fold enrichment of >1650 compared to frequencies of bulk circulating lymphocytes (p=0.0002, paired T-test, Fig. 3I), including one TCR clone targeting the human papillomavirus 16 E4 antigen from an anal squamous cell carcinoma patient, and 3 TCRs targeting patient-derived tumor material (4421 and 4323 organoid reactive TCRs in Table S1H)22. Interestingly, in the two patients (4428, 4429) without detectable NeoTCRPBL clones in the PBL, no neoantigen-specific clonotypes were detected by TIL screening (Table S1H) suggesting high concordance of peripheral T cell immunity with that of tumoral immunity. We further evaluated the frequency of the NeoTCRPBL cell surface phenotypic subset in the PBL from 11 metastatic epithelial cancer patients and age-matched healthy donors (n=10, Fig. S8E). The frequency of CD8+CD45RO+HLA-DRAhi-CD39+CD103+ T cells in PBL is low (<0.5% out of live CD8+ T cells) and not significantly different between PBL of metastatic cancer patients and age-matched healthy donor PBL (Fig. S8E), suggesting the phenotype is not exclusive to patients with metastatic solid cancers.
Frequency, specificity, and avidity comparison of NeoTCR clonotypes between TIL and PBL
Based on a combined analysis of the 72 total NeoTCRPBL clonotypes assessed in this study with available pre-treatment blood availability, we estimate a median frequency of ≤ 0.001–0.005% for each NeoTCR clone in the pre-surgery circulating peripheral blood without prior enrichment (Table S1I), which is significantly lower than estimates from within the tumor microenvironment (median NeoTCR clonotype frequency of 0.25% out of live intratumoral T cells, estimated from Lowery et al, Science, 2022)22. Next, we conducted a comprehensive analysis to compare and contrast the extent of overlap of the 29 neoantigen targets and 100 antitumor TCR clonotypes between the PBL and TIL compartments from all the patient samples utilized in this study. NeoTCRs found within TIL and PBL targeted both clonal and subclonal neoantigens from within the patient’s resected tumors with no obvious differences in the type of candidate antigen targeted between TCR clones from each of the compartments (Table S1I). Interestingly, we observed that ~82% of neoantigens were targeted by T cells from both TIL and PBL compartments (Fig. 3J, top panel). However, 47% of neoantigen-specific TCR clonotypes found in circulating blood were also found in the TIL (Fig. 3J, bottom panel). These data indicate that while the antitumor TCR repertoire is only partially shared between tumor and blood, the neoantigenic landscape of tumors recognized by antitumor T cells is likely similar between tumor and in the circulation.
Finally, given emerging data suggesting that TCR avidities might impact T cell phenotypic states36,37, we experimentally assessed the functional TCR avidities of 44 NeoTCRs from 4 patients on autologous antigen-presenting cells presenting a wide range of concentrations of the cognate neoepitopes (Fig. 3K). We found that NeoTCRs unique to TIL or PBL (n=21 TCRs), as well as those shared between the two compartments (n=23 NeoTCRs) had a wide range of functional avidities to their mutant neoepitopes, with no significant avidity differences across the 3 different groups (Fig. 3K), and thus the phenotypic states of circulating T cells are not fully explained by differences in functional TCR avidities to their mutated neoepitopes.
DISCUSSION
Adoptive cell therapy targeting private neoantigens as well as shared driver neoantigen targets can mediate durable tumor regressions7,8,12. Identifying antitumor TCRs from a minimally invasive source such as peripheral blood represents an important avenue for engineered cell therapies against cancer. In this study, we have identified cell surface and transcriptional phenotypes of antitumor, neoantigen-specific CD8+ T cells in the peripheral blood of metastatic cancer patients. Our results provide the first-in-human deep transcriptional profiling of pre-surgery circulating antitumor neoantigen-specific CD8+ T cells that supports findings from prior murine T cell phenotyping efforts 26,27. The success of NeoTCRPBL gene signature in prospectively identifying antitumor TCR clonotypes in PBL of patients with differing tumor histologies suggests that this T cell transcriptomic program is highly conserved in humans with metastatic cancers (Fig. 3). At present it is unclear if the approximately 1 in 3 prospectively tested TCRs found to be neoantigen-reactive represents all possible antitumor targets at the time point the apheresis was obtained from the patient. This is further evidenced by our identification of tumor-material reactive “orphan” TCRs as well as HPV16-E4-directed TCRs within the NeoTCRPBL compartment (Table S1H). Since we did not test candidate TCRs against tumor-overexpressed targets, nonmutated antigens, alternative mutations, and patient-derived autologous tumor material in all the patient samples, we anticipate that there are additional antitumor TCR clonotypes in the circulating blood that we did not identify in this study.
Several lines of evidence suggest that the NeoTCRPBL gene signature is a unique transcriptional program distinct from those of virus-specific T cells, likely reflecting the functions of tumor resident T cells leaving the tumor microenvironment after antigen encounter (Fig.1, Fig.2). First, NeoTCRPBL gene expression program shares markers of TIL activation and dysfunction (~9.8% overlap, Table S2), including HLA-DR, PDCD1, TIGIT, TOX, along with cell surface protein expression of CD39 (ENTPD1), HLA-DR, and CD103 (ITGAE) 13,35. Yet, they largely lacked expression of CXCL13, GZMB, CXCR6, and other genes expressed in terminally dysfunctional neoantigen-specific metastatic TIL gene programs (e.g. NeoTCR8) 20,22–24. Second, the NeoTCRPBL gene signature also had expression of canonical genes involved in T cell quiescence (LTB, LEF1), tissue-resident memory (ZNF683), CD62L (SELL), and a cell surface phenotype of CD45RO+ CD45RA− together suggesting antigenic recall and progenitor potential (Fig. 1, Table S2). Third, the NeoTCRPBL gene expression signature showed high positive correlation with both TIL dysfunctional gene signatures, as well as progenitor-exhausted, tissue-resident memory TIL signatures, suggesting that these cells likely possess an intermediate level of dysfunction (Fig.2E) 19–22. Lastly, comparing the same neoantigen-specific TCR clonotypes within a patient, circulating CD8+ T cells scored higher for immunotherapy-associated stem-like progenitor T cell signatures (CD8-G, Krishna-ACT-Stem-like), and lower for terminal exhaustion signatures (CD8-B, Oliveria Term-Exhaust), relative to tumor-resident T cells.
Although the impact of ICB was not examined in the current study, our findings indicate that T cells possessing the NeoTCRPBL signature may play a role in ICB-driven antitumor immune response as suggested by a recent study in head and neck carcinomas wherein circulating KLRG1−PD1+ resident-memory phenotypes in the blood were found to be potent antitumor immune responders unleashed by ICB 38. Since terminal dysfunction within neoantigen-specific TIL represent a major hurdle for cellular immunotherapies25, our study also raises the possibility that circulating PBL might represent an alternative source of less dysfunctional antitumor T cells that can be leveraged for ACT. However, such low-frequency clones from the circulating blood still need to be expanded to large numbers prior to adoptive cell transfer. Thus, future studies that compare frequency and phenotypic differences between in vitro expanded antitumor T cell clones isolated from TIL and PBL for ACT are warranted.
While some genes comprising the NeoTCRPBL phenotypic state have been described before in the context of T cell intracellular signaling (PASK)39, cytotoxicity (ITGB1)40, and memory (LTB)41, the NeoTCRPBL signature is also comprised of less well-described genes in the context of antitumor T cell phenotypic states in humans such as TMSB10, S100A4, S100A11, and CLEC1A (Fig. 2D, Fig. S4). Some of these genes are reported to be involved in actin, cytoskeletal remodeling, and cell migration, differentiation in context of T cells and other cell types42–44, while CLEC1A has been reported on myeloid cell compartment45. While it is conceivable that NeoTCRPBL states reflect antitumor T cells in transit between tissue-resident tumor and secondary lymphoid organs, the specific roles of many of these genes in antitumor T cells warrant testing and assessment in future studies.
The NeoTCRPBL signature is highly enriched for circulating neoantigen-specific T cell clonotypes that were generally of very low frequency in blood (≤0.001–0.002%, Table S1). This is expected since there are a paucity of chronic tumor neoantigen sources in the blood as opposed to neoantigen-density rich tumor sites; however, it is unclear if the low-frequency NeoTCRPBL phenotypes within age-matched donors without detectable cancer (Fig. S8E) represent active tumor immune surveillance in the circulation or if they comprise tumor-irrelevant clones in patients without apparent disease. In our prospective NeoTCR validation from 6 patients by single-cell gene signature or FACS-sorting methods, despite screening for all candidate tumor mutations for NeoTCRPBL identification, we largely defined the same private and driver neoantigens targeted by TIL clonotypes (Fig. 3J). Thus, despite the redundancy of a diverse antitumor TCR repertoire, which is only partially shared between tumor and blood (Fig. 3J), the neoantigenic landscape of tumors recognized by antitumor T cells is similar between tumor and in the circulation. These results are consistent with our prior efforts in defining immunogenic neoantigens in epithelial cancers as well as a recent study conducted on melanoma patients treated with immune checkpoint blockade both of which demonstrate that a limited number of immunodominant mutations are recurrently recognized by a diverse T cell repertoire in patients with metastatic cancers6,46. A recent study described the first-in-human CRISPR-edited neoantigen-directed TCR therapy for solid tumors by identifying from PBL via neuropeptide-loaded multimers showed minimal clinical benefit in patients47. Since our study demonstrates that utilizing the NeoTCRPBL phenotype identifies vast numbers of antitumor TCRs targeting the same tumor-relevant neoantigens as those found within TIL compartment (Fig. 3J), we propose that the NeoTCRPBL signature can aid the identification of tumor-relevant TCRs to develop such PBL-derived TCR-engineered cell therapies against metastatic tumors.
Finally, we leveraged our CITE-seq and transcriptomic results to develop a strategy for prospective prediction of antitumor TCRs in the blood. In our prospective cohort, we demonstrated that scGSEA analysis of CD45RA−CD45RO+HLA-DRhiCD39+CD103+ enriched samples from three patient samples using NeoTCRPBL gene-signature was successful in predicting neoantigen-reactive TCR with high sensitivity and specificity. We also developed a FACS-based plate FACS-sorting method for TCR discovery that validated the high dimensional transcriptomic analysis. Our study opens the possibility that circulating blood may serve as an alternative for the isolation and engineering of TCR-based cell therapies as well as for immune monitoring in the context of immunotherapies. In vitro expanded PBL using the markers identified in this study and TCRs isolated from the blood using the NeoTCRPBL gene program circumvents the need for in vitro TIL growth, thus expanding the potential to treat patients without any viable or with minimal tumor material, or whom tumor-resection would entail a significant risk of morbidity or mortality. While the immunological cause and mechanistic basis resulting in circulating antitumor neoantigen-specific T cells and the roles of many of the genes expressed in the blood-derived T cell transcriptomic program remain to be explored in future studies, we believe that the NeoTCRPBL signature provides a rapid, and minimally invasive avenue to isolate, study, and utilize circulating antitumor T cells and their TCR clonotypes for cancer immunotherapies.
Limitations of this study
In this study, although we demonstrate that circulating antitumor, neoantigen-specific CD8+ T cells are highly enriched in a distinct transcriptional and cell surface phenotypic state, we cannot rule out the possibility that other non-tumor-reactive T cells might also exist in this phenotypic population, or the possibility that antitumor T cells that were not found within TIL exist in a different phenotype in circulating PBL. Addressing this possibility requires the testing of all T cell clones within this phenotypic population as well as other transcriptional clusters against the entire HLA-I self, and nonself-peptidome, which remains technologically challenging. Additionally, due to resource limitations, we did not reconstruct and prospectively screen all the 965 potential TCR clonotypes including unexpanded singletons that fit the NeoTCRPBL signature criteria (Table S1), which might over-or underestimate the true frequency of antitumor TCR clonotypes in the circulation within the NeoTCRPBL compartment. While we validated NeoTCRPBL signatures on various tumor histologies, the number and histology of patient samples used as a validation cohort in this study might also be a potential limitation to extrapolate the findings from this study to multiple tumor types. Thus, we cannot rule out the possibility that immunotherapy-sensitive tumor types might harbor NeoTCRPBL clonotypes in phenotypes that are different from those derived from largely immunotherapy non-responsive epithelial tumors described here. These remain to be assessed in future studies. Finally, the antibody combination of our CITE-seq antibody panel was limited at the time of this study. Previously reported alternative surface proteins such as PD-1 34, in addition to cell surface markers that correspond to high transcriptomic expression in NeoTCRPBL signature (Table S2D) such as CD62L, AQP3, CD29 (ITGB4), CD52, CD55 and others for a more accurate and efficient isolation of circulating neoantigen-specific T cells and TCRs remain to be evaluated in future efforts.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Steven A. Rosenberg (sar@nih.gov).
Materials availability
We will share expression plasmids and unique reagents upon request and signing of an MTA in compliance with National Cancer Institute regulations.
Data and code availability
Raw transcriptomic and V(D)J data scRNA-seq sequencing data is available through dbGaP accession # ID phs003064.v1.p1. All software used in this study are freely available. Any additional information required to analyze the data in this study will be available upon request.
EXPERIMENTAL MODEL AND SUBJECTS DETAILS
Patient samples
Samples were collected from patients enrolled on Surgery Branch treatment protocol NCT00068003. Apheresis samples, used to identify circulating neoantigen-reactive CD8+ T cells, were collected under NCT00001823 protocol. Metastatic tumor specimens were obtained from patients enrolled in NCT01174121 protocol. Clinical protocols were reviewed and approved by National Cancer Institute (NCI) Institutional Review Board (IRB). In accordance with the Declaration of Helsinki, informed consent was reviewed, signed, and documented.
METHOD DETAILS
Fluorescence-Activated Cell Sorting (FACS) and Flow Cytometry
Peripheral blood samples obtained by apheresis were thawed into T cell culture media 50/50 media supplemented with DNaseI (StemCell, #7900), washed, and cultured for 2 hours at 37°C 5%CO2. Non-adherent cells were collected, washed, and rested overnight at 37°C in 50/50 media w/o cytokines. Adherent cells were used to generate immature dendritic cells (imDCs), as previously described 6,48. On the following day, non-adherent cells were filtered through 70μm filters to exclude cell clumps and cells were then negatively enriched for CD8+ (StemCell, #17953). For tetramer-based sorting, cells were stained with APC- and PE-conjugated UV-peptide exchange tetramers49, anti-CD4 (BioLegend, Clone SK3), and DAPI (BioLegend). For Pt.4287, HLA-B*49:01 tetramers were used to sort known reactive T cells against CMTR1K601T, ST7E171A, and MRRFG198C and HLA-A*01:01 for reactivity against SUPT3HS32C. 1500 tetramerpos cells were sorted with each tetramer, however no known reactive TCRs against mutated ST7, MRRF, and SUPT3H were seen in the scRNA-seq.
For CITE-seq staining in Pt.4317, we use used a panel of 20 TotalseqC barcoded antibodies and 2 TotalSeqC barcoded streptavidin-PE (BioLegend), that were used to conjugate HLA-A*02:01 FluM and EBV LMP2A tetramers (Table S1). 280 PIK3CAP449T-HLA-A*02:01+, 270 EBV LMP2-HLA-A*02:01+ (CLGGLLTMV) and 266 FluM-HLA-A*02:01+ (GILGFVFTL) cells were sorted and spiked into bulk CD8+ cells. Subsequently, cells were washed and stained with a mixture of TotalSeqC antibodies with T cell relevance that were available at the time of the study (listed in Table S1C) for 30min, on ice. Cells were then washed twice before scRNA-seq.
For surface marker-based sorting, cells were stained with anti-CD45RO (BD Biosciences, Clone UCHL1), anti-CD45RA (BD Biosciences, Clone HI100), anti-HLA-DR (BD Biosciences, Clone L243), anti-CD103 (BioLegend, Clone Ber-ACT8) and anti-CD39 (BD Biosciences, Clone TU66, BioLegend, Clone A1) antibodies. Cells were sorted using custom-made BD FACSAriaII, SONY SH800S, or SONY MA900 cell sorters.
Whole-Exome Sequencing (WES) and bulk RNAseq
To identify mutated genes, WES and RNA-seq for tumor specimens were performed in Surgery Branch Genomics Core with Illumina NextSeq®550, as previously described 6. For normal tissue sequencing, we used autologous peripheral blood cells. Sequences were aligned to hg38, and following data cleanup, putative mutations were called based on: (1) tumor/normal sequencing coverage >10, (2) variant allele frequency ≥ 7%, and (3) variant reads ≥ 4. For RNA-seq, STAR was used for alignment to hg19 and duplicates were marked using Picard’s MarkDuplicates. Final bam files and variants were called with Varscan2.
Single Cell Transcriptomics Analysis
Single cell samples were processed using Cell Ranger software 50 version 5.0.1 & version 3.1.0 for gene expression & VDJ analysis respectively. Cell ranger makefastq was used to demultiplex sequencer files. Cell Ranger count & VDJ were used under default conditions to generate h5 files which were used as input to Seurat gene expression pipeline. High quality cells with at least 250 detected genes, less than 20% mitochondrial RNA content and unique molecular identifier (UMIs) greater than or equal 500 were retained for downstream analysis. Low expressed genes, with total UMI count across all cells less than 4 and TR[AB]V & TR[AB]J genes were eliminated from the dataset to minimize the noise and bias due to TRAV/TRBV in gene-expression based UMAP cell clustering.
For Discovery set: QC passed single cell data of discovery patients, 4324, 4382, 4317, 4246, 4287 & 4422 were first normalized separately using Seurat SCTransform function using “glmGamPoi” package and then integrated using Seurat’s IntegrateData function 51. UMAP coordinates, neighbors and clusters were predicted using “PCA” parameters. Stable cells cluster were found at 0.5 resolution. Each cluster specific markers were identified by using FindAllMarkers function in Seurat with min.pct and log2fc threshold set to 0.25. VDJ dataset for these patients were merged into single dataset with one V, CDR3 & J entry for each TCRα or TCRβ chains per cell-barcode. In case of cells with multiple TCRα or TCRβ chains, CDR3 with the highest UMI was considered. Cells with TCR showing in-vitro antigen reactivity are annotated as “NeoTCRPBL”. All other TCRs which matched the public VDJ-viral database (https://vdjdb.cdr3.net/search)52 were annotated as “Public_viral”. For NeoTCRPBL gene-signature we concatenated a list of top expressed genes in cluster 9 that showed differentially expression of a log2FC ≥ 0.5. (151 genes). We refer this set of markers as “NeoTCRPBL” gene signature.
For Prospective patients: To evaluate the discriminatory power of NeoTCRPBL gene signature to predict neoantigen-reactive clones within sorted sample, we estimated enrichment of NeoTCRPBL geneset in three prospective patients, 4180, 3791, 4359 using AUCell R package and putative neoantigen-reactive TCRs were constructed based on the scGSEA score of the cells 53,54. In brief, TCRs were listed and sorted based on the median NeoTCRPBL score and expansion of the T cell clones in descending order. clones with a median NeoTCRPBL score higher than AUC were reconstructed and tested. Approximately 20–23 TCR clonotypes from expanded T cell clones with median NeoTCRPBL score higher than average AUCell NeoTCRPBL score for the patient sample PBL were selected for reconstruction and testing. Clones that had a similar cell number that scored higher than AUC were prioritized by the average score of cells of this clone.
Clone size and NeoTCRPBL enrichment calculation
We labeled TCRs from PBL samples of patients who showed positive reactivity in in-vitro assays as “Neoantigen reactive”. TCRs that matched CDR3 in the VDJ-viral database (https://vdjdb.cdr3.net/search) were labeled as “Viral”. The remaining TCRs were marked as “Unknown”. We calculated the total clones, ClonesT (unique cell-barcodes) and clones per clonotype, ClonesTRA-TRB (ClonesTRA-TRB) for each class. Then, we determined the average geneset enrichment score (GSE) of the “NeoTCRPBL (151g)” signature for clones within a clonotype. The frequency of a clonotype was calculated by dividing the total clones of that clonotype by the total clones in the clonotype class.
Signature Correlation Analysis
scRNA-Seq data of all patients was checked for quality and assembled into a single Seurat object. Gene set enrichment analysis (GSEA) was performed on the discovery set for the NeoTCRPBL gene set and other public gene-signatures using AUCell R package 55. Then, we computed Pearson correlation between NeoTCRPBL and other signatures to measure phenotypic similarity between them.
Analysis of Tumor and Blood
We integrated single cell sequencing data of only neoantigen-reactive T cells in PBL and TIL of four patients, 4317,4324,4246,4287 using harmony (0.1.0). Then we performed transcriptomic clustering of the integrated data using Seurat (4.0.2) FindClusters pipeline using standard parameters. We performed an unsupervised and unbiased single cell clustering using UMAP algorithm from Seurat (4.0.2) single cell analysis package. We performed AUCell analysis as previously described to compute the scGSEA score of various signatures on the TIL, PBL neoantigen-specific T cells. We then calculated the median score of each gene signature per each neoantigen clonotype from PBL or TIL for clonotype comparison data shown in Fig. 2I.
IPA Pathway analysis
Gene pathway analysis was performed on differentially expressed genes in cluster 9 / NeoTCRBlood signature using QIAGEN’s Ingenuity® Pathway Analysis (IPA®, QIAGEN Redwood City, www.qiagen.com/ingenuity) tools [1]. Log_pvalue is a measure of the significant enrichment of a pathway and zscore indicates whether the pathway was activated (positive zscore) or inhibted (negative zscore).
AUC-ROC analysis for prospective patient group
In the three prospective patients, we found 511 cells with TCRs that showed positive reactivity in in-vitro antigen specificity assay. These cells were annotated as true positives (labeled 1). For the remaining cells we applied a ‘Closed-world’ assumption, i.e., every unannotated cell is considered negative (labeled 0). To build the ROC curve for NeoTCRPBL signature, we ranked cells by their enrichment score and recorded the true positive rate and false positive rate after each cell. These values were used as the x and y values when plotting ROC curve and this curve is summarized to AUC score. AUC score ranges from 0 to 1. AUC score 1 shows perfect predictive value and any value greater than greater than 0.5 shows better than random chance.
Public Viral TCR Analysis
For the projection and analysis of T cell clones targeting common viruses, we used a table of CDR3β targeting InfluenzaA, CMV, EBV, and HSV-2 from VDJdb database (https://vdjdb.cdr3.net/) 52. Cells in patients’ samples that expressed TCRs found in the table with a confidence score ≥2 were designated as viral-targeting cells.
CDR3β Survey and Deep Sequencing
CDR3β and TRBV sequencing were performed on genomic DNA from cell pellets, ranging from 5×104 to 5×106, by immunoSEQ® (Adaptive Biotechnology, Seattle WA). Analyses were carried using immunoSEQ ANALYZER 3.0 (Adaptive Biotechnology, Seattle WA). Only productive CDR3β rearrangements were used for calculations of TCR frequencies.
TIL fragment screening for detection and isolation of neoantigen-reactive T cells
TIL fragments screening was performed as part of NCT00068003 clinical trials in intent to treat patients with metastatic cancer with in vitro-expanded TIL fragments that showed neoantigen-reactivity or TCR-transduced PBLs expressing neoantigen-reactive TCRs, as previously described3,6. Briefly, TIL fragments were co-cultured with autologous imDCs pulsed with peptides encompassing patients tumor’s mutation flanked by 12mer from both ends or electroporated with a string of in vitro transcribed (IVT) tandem-minigenes (TMGs) encompassing the mutations, as previously described9. Following co-culture, IFNγ secretion was measured by ELISpot or ELISA and expression of T-cell activation markers was measured by flow-cytometry6. To sequence the neoantigen-reactive TCR(s), T cells upregulating the activation marker 4–1BB (CD137) were FACS-sorted and scPCR was performed using TCRα and TCRβ specific primers, as previously described56.
Construction, Cloning, and Retroviral Transduction of T Cell Receptors
To test receptors for their anti-tumor reactivity, TRBV-CDR3β-TRBJ, and TRAV-CDR3α-TRAJ were fused to modified murine TRBC and TRAC chains, respectively. TRB and TRA were synthesized as a single-chain separated by a furin SGSG P2A linker and cloned into pMSGV-1 (GenScript, Piscataway NJ). Retroviral supernatants were produced using HEK-293GP packaging line, as previously described57. Briefly, 0.7–1×106 cells per well were plated in 6-wells poly-D-Lysine-coated plates and co-transfected with 2 μg/well TCR-encoding p-MSGV-1 and 1.4 μg/well envelope-encoding pRD114 plasmids using Lipofectamine 2000 (Invitrogen, Cat. 11668–019), and supernatants were collected 48–72hrs following transfection. Next, supernatants were plated in non-tissue culture treated-plates pre-coated with 10–20 μg/mL retronectin (Takara, T100B) and centrifuged at 2000xg, 32°C for 2hrs. Subsequently, supernatants were discarded and 1–2×106 stimulated healthy donor PBLs (0.5×106 cells/mL) were centrifuged onto the retrovirus-coated plated at 350×g for 10min. Transduced cells were removed and transferred into tissue-treated plates after 24 hours and grown in rhIL-2-containing media for 10–14 days before screening.
Quantification and Statistical Analysis
For cluster-based scRNA analysis, the FindAllMarkers function (using default values) within Seurat was used to identify differentially expressed genes between clusters with min.pct and log2fc threshold set to 0.25. Genes with an adjusted p-value of < 0.05 were included as DEGs. The frequency and enrichment of neoantigen-specific NeoTCRPBL clones were compared between bulk, and specific scRNA clusters, and select population-based enrichment by Paired T-test with a p-value significance level of < 0.05. NeoTCRPBL distribution of cells across clusters was compared by Two-way analysis of variance (ANOVA) adjusted by Bonferroni correction for multiple comparisons. For cluster enrichment and signature score per clone between population calculations, paired T-tests were performed and Bonferroni correction for multiple comparisons test was performed. For signature score and gene SCT counts comparisons between tumor and blood Wilcoxon rank-sum test was performed.
Supplementary Material
Table S1: Patient demography, Pt.4246 differential gene-analysis, CITEseq antibodies, and T-cell receptors used in this study. (A) Patients’ demographics, related to Figure 1, 2, and 3. (B) Differentially expressed genes from each cluster in pt.4246 within patient PBL (related to Figure 1E–G). (C) TotalSeqC antibodies used in pt.4317 sample labeling and analysis (related to Figure 1K–L). (D) Discovery set - pre-surgery frequencies of archival antitumor, neoantigen-specific TCR clonotypes (related to Figure 2A–B). (E) Bystander viral-reactive clones used for analysis in discovery set (related to Figure 2A–B). (F) Experimentally tested TCRs in prospective patient sample PBL along with their NeoTCRPBL signature scores, clone size, and AUCell criteria (related to Figure 3D, Fig. S6, Fig. S7). (G) Prospective samples - Pre-surgery frequencies of antitumor, neoantigen-specific TCR clonotypes (related to Figure 3E–F, Fig. S6, Fig. S7). (H) FACS based samples - Pre-surgery frequencies of antitumor, neoantigen-specific TCR clonotypes (related to Figure 3I). (I) Clonality status of neoantigens analyzed in this study (related to Figure 3J–K)
Table S2: Transcriptomic analysis of circulating anti-tumor T cells and NeoTCRPBL gene-signature, related to Figure 2 and 3. (A) Differentially expressed genes analysis of circulating T cells from 6 patients, related to Figure 2. (B) Differentially expressed genes by cluster, related to Figure 2. (C) Differentially expressed genes between clusters 3 and 9, related to Figure 2. (D) NeoTCRPBL gene-signature, related to Figure 2 and 3. (E) Top differentially 30 upregulated and downregulated genes, related to Figure 2 and 3. (F) Differentially expressed genes between NeoTCRPBL and virus-specific T cells, related to Figure 2.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-human CD45RO APC | BD Biosciences | Cat#559865; AB_398673 |
| Anti-human CD45RA PcrCP5.5 | BD Biosciences | Cat#563429; AB_2738199 |
| Anti-human HLA-DR APC-Cy7 | BD Biosciences | Cat#641393; AB_1645739 |
| Anti-human CD39 FITC | BD Biosciences | Cat#561444; AB_10896292 |
| Anti-human CD39 FITC | Biolegend | Cat#328206; AB_940425 |
| Anti-human CD103 PE | Biolegend | Cat#350206; AB_10641843 |
| Anti-human CD4 PE-Cy7 | BD Biosciences | Cat#348789; AB_400379 |
| Anti-human CD8 PE-Cy7 | BD Biosciences | Cat#335787; AB_399966 |
| Anti-mTCRp PE | BD Biosciences | Cat#553172; AB_394684 |
| Anti-human CD3 APC-Cy7 | BD Biosciences | Cat#341090; AB_400214 |
| Anti-human CD137 (4–1BB) APC | BD Biosciences | Cat#550890; AB_398477 |
| TotalSeqC Anti-human LGALS3 | Biolegend | Cat#125423; AB_2819851 |
| TotalSeqC Anti-human KLRG1 | Biolegend | Cat#138433; AB_2800649 |
| TotalSeqC Anti-human CD28 | Biolegend | Cat#302963; AB_2800751 |
| TotalSeqC Anti-human CD29 | Biolegend | Cat#303029; AB_10752594 |
| TotalSeqC Anti-human CD45RA | Biolegend | Cat#304163; AB_2800764 |
| TotalSeqC Anti-human CD45RO | Biolegend | Cat#304259; AB_2800766 |
| TotalSeqC Anti-human CD62L | Biolegend | Cat#304851; AB_2800770 |
| TotalSeqC Anti-human CD94 | Biolegend | Cat#305523; AB_2814143 |
| TotalSeqC Anti-human HLA-DR | Biolegend | Cat#307663; AB_2800795 |
| TotalSeqC Anti-human CD39 | Biolegend | Cat#328237; AB_2800853 |
| TotalSeqC Anti-human CD274 (PD-L1) | Biolegend | Cat#329751; AB_2800860 |
| TotalSeqC Anti-human CD279 (PD-1) | Biolegend | Cat#329963; AB_2800862 |
| TotalSeqC Anti-human TIM-3 | Biolegend | Cat#345049; AB_2800925 |
| TotalSeqC Anti-human CD103 | Biolegend | Cat#350233; AB_2800933 |
| TotalSeqC Anti-human CD127 (IL7R) | Biolegend | Cat#351356; AB_2800937 |
| TotalSeqC Anti-human CD197 (CCR7) | Biolegend | Cat#353251; AB_2800943 |
| TotalSeqC Anti-human CD183 (CXCR3) | Biolegend | Cat#353747; AB_2800949 |
| TotalSeqC Anti-human CD357 (GITR) | Biolegend | Cat#371227; AB_2810583 |
| TotalSeqC Anti-human TIGIT | Biolegend | Cat#372729; AB_2801021 |
| TotalSeqC Anti-human LAG-3 | Biolegend | Cat#369335; AB_2814327 |
| TotalSeqC PE Strepavidin (EBV LMP2A tetramer) | Biolegend | Cat#405265 |
| TotalSeqC PE Strepavidin (Influenza M1 tetramer) | Biolegend | Cat#405263 |
|
| ||
| Biological samples | ||
|
| ||
| Patient peripheral blood samples | This Paper | n/a |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Tandem mini genes | Genscript | custom made |
| HPLC peptides | Genscript | custom made |
| Crude peptides | Genscript and inhouse | custom made |
| Custom-made photo-cleavable HLA monomers | Gift from Pia Kvistborg | custom made |
|
| ||
| Critical commercial assays | ||
|
| ||
| Chromium Next GEM Single Cell 5’ Kit v2 | 10xGenomics | Cat#1000263 |
| Chromium Single Cell Human TCR Amplification Kit | 10xGenomics | Cat#1000252 |
| Chromium Next GEM Chip K Single Cell kit | 10xGenomics | Cat#1000287 |
| Library Construction Kit | 10xGenomics | Cat#1000190 |
| 10x Chromium controller system | 10xGenomics | Cat#120223 |
| NextSeq 2000 | Illumina | Cat#20038897 |
|
| ||
| Deposited data | ||
|
| ||
| Human GI cancer patient 4246 PBL scRNAseq dataset | This paper | dbGaP: phs003064.v1.p1 |
| Human breast cancer patient 4180 PBL scRNAseq dataset | This paper | dbGaP: phs003064.v1.p1 |
| Human GI cancer patient 4287 PBL scRNAseq dataset | This paper | dbGaP: phs003064.v1.p1 |
| Human GI cancer patient 4324 PBL scRNAseq dataset | This paper | dbGaP: phs003064.v1.p1 |
| Human GI cancer patient 4422 PBL scRNAseq dataset | This paper | dbGaP: phs003064.v1.p1 |
| Human GI cancer patient 4317 PBL scRNAseq dataset | This paper | dbGaP: phs003064.v1.p1 |
| Human GI cancer patient 4382 PBL scRNAseq dataset | This paper | dbGaP: phs003064.v1.p1 |
| Human melanoma cancer patient 3791 PBL scRNAseq dataset | This paper | dbGaP: phs003064.v1.p1 |
| Human GI cancer patient 4359 PBL scRNAseq dataset | This paper | dbGaP: phs003064.v1.p1 |
|
| ||
| Software and algorithms | ||
|
| ||
| CellRanger | 10xGenomics | https://support.10xgenomics.com/single-cell-gene-expression/software |
| Seurat (4.0.2) | Satija Laboratory | https://github.com/satijalab/seurat |
| R (4.0.4) | R project | https://www.r-project.org/ |
| FlowJo v10.08 | Tree Star | https://www.flowjo.com/ |
| GraphPad Prism 9.0 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
Highlights.
Circulating anti-tumor T cells (NeoTCRPBL) express a unique transcriptional signature
NeoTCRPBL are low in frequency and are less dysfunctional compared to neoantigen-TIL
NeoTCRPBL transcriptional signature predicts circulating anti-tumor T cell receptors
NeoTCRPBL provide a non-invasive source for anti-tumor T cells and their receptors
ACKNOWLEDGMENTS
We thank Abraham Sachs, Zhili Zheng, Zhiya Yu, and Biman C. Paria for their technical support. We thank NIH Integrated Data Analysis Platform (NIDAP) for their computational support. We also thank Pia Kvitsborg, from the Netherlands Cancer Institute, for providing the photo-cleavable HLA monomers. This work utilized the computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov).
INCLUSION AND DIVERSITY
One or more of the authors of this paper self-identifies as underrepresented ethnic minority in their field of research or within their geographical location. One or more of the authors self-identifies as a member of the LGBTQIA+ community.
Footnotes
DECLARATION OF INTEREST
R.Y., A.R.C., S.K., F.J.L. P.F.R, and S.A.R. are listed on a patent application (PCT/US2021/023225) that covers the use of NeoTCRPBL signature to identify antitumor TCRs. The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Pardoll DM (2012). The blockade of immune checkpoints in cancer immunotherapy. Nature Reviews Cancer 12, 252–264. 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosenberg SA, and Restifo NP (2015). Adoptive cell transfer as personalized immunotherapy for human cancer. Science 348, 62–68. 10.1126/science.aaa4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, Royal RE, Topalian SL, Kammula US, Restifo NP, et al. (2006). Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 314, 126–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.June CH, and Sadelain M (2018). Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 379, 64–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang T-H, Porta-Pardo E, Gao GF, Plaisier CL, Eddy JA, et al. (2019). The Immune Landscape of Cancer. Immunity 51, 411–412. [DOI] [PubMed] [Google Scholar]
- 6.Parkhurst MR, Robbins PF, Tran E, Prickett TD, Gartner JJ, Jia L, Ivey G, Li YF, El-Gamil M, Lalani A, et al. (2019). Unique Neoantigens Arise from Somatic Mutations in Patients with Gastrointestinal Cancers. Cancer Discov. 9, 1022–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tran E, Robbins PF, Lu Y-C, Prickett TD, Gartner JJ, Jia L, Pasetto A, Zheng Z, Ray S, Groh EM, et al. (2016). T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N. Engl. J. Med. 375, 2255–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zacharakis N, Huq LM, Seitter SJ, Kim SP, Gartner JJ, Sindiri S, Hill VK, Li YF, Paria BC, Ray S, et al. (2022). Breast Cancers Are Immunogenic: Immunologic Analyses and a Phase II Pilot Clinical Trial Using Mutation-Reactive Autologous Lymphocytes. Journal of Clinical Oncology. 10.1200/jco.21.02170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robbins PF, Lu Y-C, El-Gamil M, Li YF, Gross C, Gartner J, Lin JC, Teer JK, Cliften P, Tycksen E, et al. (2013). Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat. Med. 19, 747–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rosenberg SA, Spiess P, and Lafreniere R (1986). A New Approach to the Adoptive Immunotherapy of Cancer with Tumor-Infiltrating Lymphocytes. Science 233, 1318–1321. 10.1126/science.3489291. [DOI] [PubMed] [Google Scholar]
- 11.Gros A, Robbins PF, Yao X, Li YF, Turcotte S, Tran E, Wunderlich JR, Mixon A, Farid S, Dudley ME, et al. (2014). PD-1 identifies the patient-specific CD8+ tumor-reactive repertoire infiltrating human tumors. J. Clin. Invest. 124, 2246–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tran E, Ahmadzadeh M, Lu Y-C, Gros A, Turcotte S, Robbins PF, Gartner JJ, Zheng Z, Li YF, Ray S, et al. (2015). Immunogenicity of somatic mutations in human gastrointestinal cancers. Science 350, 1387–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simoni Y, Becht E, Fehlings M, Loh CY, Koo S-L, Teng KWW, Yeong JPS, Nahar R, Zhang T, Kared H, et al. (2018). Bystander CD8 T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 557, 575–579. [DOI] [PubMed] [Google Scholar]
- 14.Lee PP, Yee C, Savage PA, Fong L, Brockstedt D, Weber JS, Johnson D, Swetter S, Thompson J, Greenberg PD, et al. (1999). Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients. Nat. Med. 5, 677–685. [DOI] [PubMed] [Google Scholar]
- 15.Bentzen AK, Marquard AM, Lyngaa R, Saini SK, Ramskov S, Donia M, Such L, Furness AJS, McGranahan N, Rosenthal R, et al. (2016). Large-scale detection of antigen-specific T cells using peptide-MHC-I multimers labeled with DNA barcodes. Nat. Biotechnol. 34, 1037–1045. [DOI] [PubMed] [Google Scholar]
- 16.Holm JS, Funt SA, Borch A, Munk KK, Bjerregaard A-M, Reading JL, Maher C, Regazzi A, Wong P, Al-Ahmadie H, et al. (2022). Neoantigen-specific CD8 T cell responses in the peripheral blood following PD-L1 blockade might predict therapy outcome in metastatic urothelial carcinoma. Nat. Commun. 13, 1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cohen CJ, Gartner JJ, Horovitz-Fried M, Shamalov K, Trebska-McGowan K, Bliskovsky VV, Parkhurst MR, Ankri C, Prickett TD, Crystal JS, et al. (2015). Isolation of neoantigen-specific T cells from tumor and peripheral lymphocytes. J. Clin. Invest. 125, 3981–3991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van der Leun AM, Thommen DS, and Schumacher TN (2020). CD8 T cell states in human cancer: insights from single-cell analysis. Nat. Rev. Cancer 20, 218–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li H, van der Leun AM, Yofe I, Lubling Y, Gelbard-Solodkin D, van Akkooi ACJ, van den Braber M, Rozeman EA, Haanen JBA, Blank CU, et al. (2020). Dysfunctional CD8 T Cells Form a Proliferative, Dynamically Regulated Compartment within Human Melanoma. Cell 181, 747. 10.1016/j.cell.2020.04.017. [DOI] [PubMed] [Google Scholar]
- 20.Oliveira G, Stromhaug K, Klaeger S, Kula T, Frederick DT, Le PM, Forman J, Huang T, Li S, Zhang W, et al. (2021). Phenotype, specificity and avidity of antitumour CD8 T cells in melanoma. Nature 596, 119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Caushi JX, Zhang J, Ji Z, Vaghasia A, Zhang B, Hsiue EH-C, Mog BJ, Hou W, Justesen S, Blosser R, et al. (2021). Transcriptional programs of neoantigen-specific TIL in anti-PD-1-treated lung cancers. Nature 596, 126–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lowery FJ, Krishna S, Yossef R, Parikh NB, Chatani PD, Zacharakis N, Parkhurst MR, Levin N, Sindiri S, Sachs A, et al. (2022). Molecular signatures of antitumor neoantigen-reactive T cells from metastatic human cancers. Science 375, 877–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hanada K-I, Zhao C, Gil-Hoyos R, Gartner JJ, Chow-Parmer C, Lowery FJ, Krishna S, Prickett TD, Kivitz S, Parkhurst MR, et al. (2022). A phenotypic signature that identifies neoantigen-reactive T cells in fresh human lung cancers. Cancer Cell 40, 479–493.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zheng C, Fass JN, Shih Y-P, Gunderson AJ, Sanjuan Silva N, Huang H, Bernard BM, Rajamanickam V, Slagel J, Bifulco CB, et al. (2022). Transcriptomic profiles of neoantigen-reactive T cells in human gastrointestinal cancers. Cancer Cell 40, 410–423.e7. [DOI] [PubMed] [Google Scholar]
- 25.Krishna S, Lowery FJ, Copeland AR, Bahadiroglu E, Mukherjee R, Jia L, Anibal JT, Sachs A, Adebola SO, Gurusamy D, et al. (2020). Stem-like CD8 T cells mediate response of adoptive cell immunotherapy against human cancer. Science 370, 1328–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pauken KE, Shahid O, Lagattuta KA, Mahuron KM, Luber JM, Lowe MM, Huang L, Delaney C, Long JM, Fung ME, et al. (2021). Single-cell analyses identify circulating anti-tumor CD8 T cells and markers for their enrichment. J. Exp. Med. 218. 10.1084/jem.20200920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lucca LE, Axisa P-P, Lu B, Harnett B, Jessel S, Zhang L, Raddassi K, Zhang L, Olino K, Clune J, et al. (2021). Circulating clonally expanded T cells reflect functions of tumor-infiltrating T cells. J. Exp. Med. 218. 10.1084/jem.20200921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cafri G, Yossef R, Pasetto A, Deniger DC, Lu Y-C, Parkhurst M, Gartner JJ, Jia L, Ray S, Ngo LT, et al. (2019). Memory T cells targeting oncogenic mutations detected in peripheral blood of epithelial cancer patients. Nat. Commun. 10, 449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Malekzadeh P, Yossef R, Cafri G, Paria BC, Lowery FJ, Jafferji M, Good ML, Sachs A, Copeland AR, Kim SP, et al. (2020). Antigen Experienced T Cells from Peripheral Blood Recognize p53 Neoantigens. Clin. Cancer Res. 26, 1267–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Strønen E, Toebes M, Kelderman S, van Buuren MM, Yang W, van Rooij N, Donia M, Böschen M-L, Lund-Johansen F, Olweus J, et al. (2016). Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. Science 352, 1337–1341. [DOI] [PubMed] [Google Scholar]
- 31.Yost KE, Satpathy AT, Wells DK, Qi Y, Wang C, Kageyama R, McNamara KL, Granja JM, Sarin KY, Brown RA, et al. (2019). Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat. Med. 25, 1251–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, Lieb DJ, Chen JH, Frederick DT, Barzily-Rokni M, et al. (2019). Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell 176, 404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gros A, Parkhurst MR, Tran E, Pasetto A, Robbins PF, Ilyas S, Prickett TD, Gartner JJ, Crystal JS, Roberts IM, et al. (2016). Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat. Med. 22, 433–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gros A, Tran E, Parkhurst MR, Ilyas S, Pasetto A, Groh EM, Robbins PF, Yossef R, Garcia-Garijo A, Fajardo CA, et al. (2019). Recognition of human gastrointestinal cancer neoantigens by circulating PD-1+ lymphocytes. J. Clin. Invest. 129, 4992–5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Duhen T, Duhen R, Montler R, Moses J, Moudgil T, de Miranda NF, Goodall CP, Blair TC, Fox BA, McDermott JE, et al. (2018). Co-expression of CD39 and CD103 identifies tumor-reactive CD8 T cells in human solid tumors. Nat. Commun. 9, 2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Purcarea A, Jarosch S, Barton J, Grassmann S, Pachmayr L, D’Ippolito E, Hammel M, Hochholzer A, Wagner KI, van den Berg JH, et al. (2022). Signatures of recent activation identify a circulating T cell compartment containing tumor-specific antigen receptors with high avidity. Sci Immunol 7, eabm2077. [DOI] [PubMed] [Google Scholar]
- 37.Shakiba M, Zumbo P, Espinosa-Carrasco G, Menocal L, Dündar F, Carson SE, Bruno EM, Sanchez-Rivera FJ, Lowe SW, Camara S, et al. (2022). TCR signal strength defines distinct mechanisms of T cell dysfunction and cancer evasion. J. Exp. Med. 219. 10.1084/jem.20201966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Luoma AM, Suo S, Wang Y, Gunasti L, Porter CBM, Nabilsi N, Tadros J, Ferretti AP, Liao S, Gurer C, et al. (2022). Tissue-resident memory and circulating T cells are early responders to pre-surgical cancer immunotherapy. Cell. 10.1016/j.cell.2022.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Duraiswamy J, Ibegbu CC, Masopust D, Miller JD, Araki K, Doho GH, Tata P, Gupta S, Zilliox MJ, Nakaya HI, et al. (2011). Phenotype, function, and gene expression profiles of programmed death-1(hi) CD8 T cells in healthy human adults. J. Immunol. 186, 4200–4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nicolet BP, Guislain A, and Wolkers MC (2021). CD29 Enriches for Cytotoxic Human CD4 T Cells. J. Immunol. 207, 2966–2975. [DOI] [PubMed] [Google Scholar]
- 41.Wang X, Shen X, Chen S, Liu H, Hong N, Zhong H, Chen X, and Jin W (2022). Reinvestigation of Classic T Cell Subsets and Identification of Novel Cell Subpopulations by Single-Cell RNA Sequencing. J. Immunol. 208, 396–406. [DOI] [PubMed] [Google Scholar]
- 42.Maelan AE, Rasmussen TK, and Larsson L-I (2007). Localization of thymosin beta10 in breast cancer cells: relationship to actin cytoskeletal remodeling and cell motility. Histochem. Cell Biol. 127, 109–113. [DOI] [PubMed] [Google Scholar]
- 43.Shankar J, Messenberg A, Chan J, Underhill TM, Foster LJ, and Nabi IR (2010). Pseudopodial actin dynamics control epithelial-mesenchymal transition in metastatic cancer cells. Cancer Res. 70, 3780–3790. [DOI] [PubMed] [Google Scholar]
- 44.Brisslert M, Bian L, Svensson MND, Santos RF, Jonsson I-M, Barsukov I, Erlandsson M, Andersson K, Carmo AM, and Bokarewa MI (2014). S100A4 regulates the Src-tyrosine kinase dependent differentiation of Th17 cells in rheumatoid arthritis. Biochim. Biophys. Acta 1842, 2049–2059. [DOI] [PubMed] [Google Scholar]
- 45.Drouin M, Saenz J, Gauttier V, Evrard B, Teppaz G, Pengam S, Mary C, Desselle A, Thepenier V, Wilhelm E, et al. (2022). CLEC-1 is a death sensor that limits antigen cross-presentation by dendritic cells and represents a target for cancer immunotherapy. Sci Adv 8, eabo7621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Puig-Saus C, Sennino B, Peng S, Wang CL, Pan Z, Yuen B, Purandare B, An D, Quach BB, Nguyen D, et al. (2023). Neoantigen-targeted CD8 T cell responses with PD-1 blockade therapy. Nature 615, 697–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Foy SP, Jacoby K, Bota DA, Hunter T, Pan Z, Stawiski E, Ma Y, Lu W, Peng S, Wang CL, et al. (2023). Non-viral precision T cell receptor replacement for personalized cell therapy. Nature 615, 687–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tran E, Ahmadzadeh M, Lu Y-C, Gros A, Turcotte S, Robbins PF, Gartner JJ, Zheng Z, Li YF, Ray S, et al. (2015). Immunogenicity of somatic mutations in human gastrointestinal cancers. Science 350, 1387–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Toebes M, Coccoris M, Bins A, Rodenko B, Gomez R, Nieuwkoop NJ, van de Kasteele W, Rimmelzwaan GF, Haanen JBAG, Ovaa H, et al. (2006). Design and use of conditional MHC class I ligands. Nat. Med. 12, 246–251. [DOI] [PubMed] [Google Scholar]
- 50.Zheng GXY, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J, et al. (2017). Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 8, 14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shugay M, Bagaev DV, Zvyagin IV, Vroomans RM, Crawford JC, Dolton G, Komech EA, Sycheva AL, Koneva AE, Egorov ES, et al. (2018). VDJdb: a curated database of T-cell receptor sequences with known antigen specificity. Nucleic Acids Res. 46, D419–D427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Matsumoto H, Kiryu H, Furusawa C, Ko MSH, Ko SBH, Gouda N, Hayashi T, and Nikaido I (2017). SCODE: an efficient regulatory network inference algorithm from single-cell RNA-Seq during differentiation. Bioinformatics 33, 2314–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lowery FJ, Krishna S, Yossef R, Parikh NB, Chatani PD, Zacharakis N, Parkhurst MR, Levin N, Sindiri S, Sachs A, et al. (2022). Molecular signatures of antitumor neoantigen-reactive T cells from metastatic human cancers. Science 375, 877–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Aibar S, González-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, Rambow F, Marine J-C, Geurts P, Aerts J, et al. (2017). SCENIC: single-cell regulatory network inference and clustering. Nat. Methods 14, 1083–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pasetto A, Gros A, Robbins PF, Deniger DC, Prickett TD, Matus-Nicodemos R, Douek DC, Howie B, Robins H, Parkhurst MR, et al. (2016). Tumor- and Neoantigen-Reactive T-cell Receptors Can Be Identified Based on Their Frequency in Fresh Tumor. Cancer Immunol Res 4, 734–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pasetto A, Gros A, Robbins PF, Deniger DC, Prickett TD, Matus-Nicodemos R, Douek DC, Howie B, Robins H, Parkhurst MR, et al. (2016). Tumor- and Neoantigen-Reactive T-cell Receptors Can Be Identified Based on Their Frequency in Fresh Tumor. Cancer Immunol Res 4, 734–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Patient demography, Pt.4246 differential gene-analysis, CITEseq antibodies, and T-cell receptors used in this study. (A) Patients’ demographics, related to Figure 1, 2, and 3. (B) Differentially expressed genes from each cluster in pt.4246 within patient PBL (related to Figure 1E–G). (C) TotalSeqC antibodies used in pt.4317 sample labeling and analysis (related to Figure 1K–L). (D) Discovery set - pre-surgery frequencies of archival antitumor, neoantigen-specific TCR clonotypes (related to Figure 2A–B). (E) Bystander viral-reactive clones used for analysis in discovery set (related to Figure 2A–B). (F) Experimentally tested TCRs in prospective patient sample PBL along with their NeoTCRPBL signature scores, clone size, and AUCell criteria (related to Figure 3D, Fig. S6, Fig. S7). (G) Prospective samples - Pre-surgery frequencies of antitumor, neoantigen-specific TCR clonotypes (related to Figure 3E–F, Fig. S6, Fig. S7). (H) FACS based samples - Pre-surgery frequencies of antitumor, neoantigen-specific TCR clonotypes (related to Figure 3I). (I) Clonality status of neoantigens analyzed in this study (related to Figure 3J–K)
Table S2: Transcriptomic analysis of circulating anti-tumor T cells and NeoTCRPBL gene-signature, related to Figure 2 and 3. (A) Differentially expressed genes analysis of circulating T cells from 6 patients, related to Figure 2. (B) Differentially expressed genes by cluster, related to Figure 2. (C) Differentially expressed genes between clusters 3 and 9, related to Figure 2. (D) NeoTCRPBL gene-signature, related to Figure 2 and 3. (E) Top differentially 30 upregulated and downregulated genes, related to Figure 2 and 3. (F) Differentially expressed genes between NeoTCRPBL and virus-specific T cells, related to Figure 2.
Data Availability Statement
Raw transcriptomic and V(D)J data scRNA-seq sequencing data is available through dbGaP accession # ID phs003064.v1.p1. All software used in this study are freely available. Any additional information required to analyze the data in this study will be available upon request.