

Abstract
The highly conserved MicroRNA-9 (miR-9) family consists of three members. We discovered that miR-9-1 deletion reduced mature miR-9 expression, causing 43% of the mice to display smaller size and postweaning lethality. MiR-9-1-deficient mice with growth defects experienced severe lymphopenia, but other blood cells were unaffected. The lymphopenia wasn’t due to defects in hematopoietic progenitors, as mutant bone marrow (BM) cells underwent normal lymphopoiesis after transplantation into wild-type recipients. Additionally, miR-9-1-deficient mice exhibited impaired osteoblastic bone formation, as mutant mesenchymal stem cells (MSCs) failed to differentiate into osteoblastic cells (OBs). RNA sequencing revealed reduced expression of master transcription factors for osteoblastic differentiation, Runt-related transcription factor 2 (Runx2) and Osterix (Osx), and genes related to collagen formation, extracellular matrix organization, and cell adhesion, in miR-9-1-deficient MSCs. Follistatin (Fst), an antagonist of bone morphogenetic proteins (BMPs), was found to be a direct target of miR-9-1. Its deficiency led to the up-regulation of Fst, inhibiting BMP signaling in MSCs, and reducing IL-7 and IGF-1. Thus, miR-9-1 controls osteoblastic regulation of lymphopoiesis by targeting the Fst/BMP/Smad signaling axis.
Keywords: Lymphopoiesis, Osteoblastic niche, MiR-9-1, Fst, BMP signaling
INTRODUCTION
HSC self-renewal and differentiation are tightly regulated by both hematopoietic cell intrinsic factors and by the soluble factors and extracellular matrix produced by bone marrow (BM) stromal cells such as mesenchymal progenitors (MSCs), osteoblasts (OBs), endothelial cells (ECs), peripheral nerves, fibroblasts, macrophages and adipocytes 1–6. The combination of stromal cells and the factors they produce help to determine the type of blood cells produced within specific microdomains, or niches.
B-cell development is especially dependent on the osteoblastic (endosteal) niche, which is notable for the presence of bone-forming OBs and osteogenic MSCs. OBs are critical for the support of HSC differentiation, including B-cell commitment, and conditional deletion of OBs severely impairs B-cell lymphopoiesis 7, 8. OBs also regulate HSC maintenance through Jagged/Notch and WNT signaling, and mediate HSC self-renewal and survival by producing CXCL12, osteopontin and thrombopoietin 9–15. OBs are derived from MSCs and osteoblastic differentiation of MSCs is regulated by bone morphogenic protein-2 (BMP-2) and BMP-6 16. Moreover, MSCs themselves are a critical component of the endosteal BM niche 17. MSCs express high levels of stem cell factor (SCF), vascular cell adhesion molecule 1 (VCAM1), CXCL12, IL-7 and IGF-1, all of which are important for supporting HSCs and lymphopoiesis 17–20.
A heretofore unexplored potential regulator of the lymphopoietic BM niche is the miR-9 family of microRNAs (miRNA). Overexpression of miRNA-9 in BM promotes myelopoiesis and suppresses lymphopoiesis, a pattern that has been observed in other models of endosteal niche dysregulation 21. MicroRNAs (miRNAs) are short noncoding RNAs of 20–22 nucleotides that silence gene expression by guiding Argonaute (AGO) proteins to the complementary sequences in the 3’-untranslated region (3’UTR) of mRNAs to destabilize target transcripts or inhibit protein translation 22, 23. MiRNAs are transcribed as longer primary transcripts and are processed into mature miRNAs by the RNaseIII enzyme, Dicer 24. MiRNAs are able to regulate multiple biological processes, including blood cell production, by targeting the expression of specific genes 25. In the BM, miRNAs play an important role in controlling the number of HSCs/progenitors and in directing cell differentiation during hematopoiesis. 26 MiR-181 regulates T cell selection 27, miR-150 controls B cell differentiation and modulates megakaryocyte specification 28–30, and miR-223 mediates granulocyte formation and function 31, 32.
MiR-9 is a highly conserved family of miRNAs which includes three members, miR-9-1, miR-9-2 and miR-9-3 33, 34. These three microRNAs are encoded by unique genes located on different chromosomes, the transcripts of which are processed into the same mature miR-9 sequence 33. In mice, miR-9-2/3 double deficiency leads to postnatal lethality, although miR-9-2 or miR-9-3 single-deficient mice are viable 33. The function of MiR-9 miRNAs has primarily been studied in the brain, where their expression is enriched. MiR-9 miRNAs regulate neurogenesis by promoting or suppressing the proliferation of neural progenitor cells 33, 35.
Here, we used CRISPR/Cas9-meidated genome editing to generate miR-9-1-deficient mice and observed a reduction in the overall expression of mature miR-9 in these mice. MiR-9-1-deficient mice were small and displayed incomplete penetrance of postweaning lethality. These mutant mice exhibited a reduction in CLPs and severely impaired lymphopoiesis, but normal erythropoiesis, thrombopoiesis and myelopoiesis. The reduced lymphopoiesis in miR-9-1-deficient mice was due to the impaired osteoblastic niche rather than intrinsic HSC defects. MiR-9-1-deficient MSCs failed to differentiate into OBs. Additionally, miR-9 targeted the BMP antagonist Fst, and miR-9-1 deficiency led to Fst up-regulation, which inhibited BMP/Smad signaling.
MATERIALS AND METHODS
Mice
MiR-9-1−/− mice were generated by the CRISPR-Cas9-mediated genome editing strategy. 36 Briefly, the Cas9 protein and the miR-9-1 sgRNA were co-injected into the zygotes from C57BL/6. The sequences of the miR-9-1 sgRNA were as follows: sg-miR-9-1RNA1: TGGTTATCT AGCTGTATGAG, sg-miR-9-1RNA2: GCGGGGTTGGTTGTTATCTT. Mutant mice were first maintained in the Animal Center of Fujian Normal University. Mutant mice were transferred to and maintained in the Biological Resource Center at the Medical College of Wisconsin (MCW). Experimental and control mice were 8–12 weeks old. Randomization was applied to allocate the experimental and control mice into different groups. The investigator was not blinded to the group allocation. All animal research and care procedures were conducted in accordance with the Guide for the Care and Use of Laboratory Animals and approved by the MCW Institutional Animal Care and Use Committee. The program is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care.
Flow cytometry analysis and cell sorting
Flow cytometric analysis was performed as previously described 37. Briefly, single-cell suspensions from the spleen and BM were treated with Gey’s solution to lyse RBCs and resuspended in PBS with 2% FBS. Cells were stained with a combination of fluorescence-conjugated antibodies. Allophycocyanin-conjugated anti-c-Kit, anti-Mac-1, anti-B220, anti-IgM, and anti-CD4; PE-Cy7-conjugated anti-Sca-1, anti-IL7R, anti-CD24, anti-CD25, and anti-CD23; PE-conjugated anti-B220, anti-CD4, anti-CD8, anti-Ter119, anti-Mac-1, anti-Gr-1, anti-CD45.2, and anti-IL7R; FITC-conjugated anti-CD34, anti-B220, anti-streptavidin and anti-Sca-1; Biotin-conjugated anti-CD135 and anti-FcγR; eFluor450-conjugated anti-CD45.2; eFluor710-conjugated anti-IgM and anti-Flt-3, and Percpcy5.5-conjugated anti-streptavidin were purchased from eBioscience. PE-conjugated anti-CD43, anti-CD21, anti-CD25, anti-IgD, and anti-NK1.1; and FITC-conjugated anti-CD8 were purchased from BD. APC-Cy7-conjugated anti-B220 and CD45.2 were purchased from BioLegend. Samples were applied to a flow cytometer (LSR II, Becton Dickinson). Data were collected and analyzed using FACSDiva software (Becton Dickinson) or FlowJo software (Tree Star).
Flow cytometric analysis of MSCs and OBs was performed as previously described. 38 Briefly, BM cells were flushed out using PBS with 2% FBS. Bones were minced with scissors and crushed bones were washed with HBSS until the bone chips were white. Endosteal stromal cells were released by digestion for 40 min at 37°C at 180 rpm with 200 μg/ml Liberase (Roche) and 200 μg/ml DNase I (Roche) dissolved in HBSS. The released endosteal cells were washed with HBSS + 2% FBS and residual bone material was removed by filtering through a 45 μm filter. Cells were stained with the indicated antibodies. PE-conjugated anti-CD4, anti-CD8, anti-B220, anti-Mac-1, anti-Gr-1 and anti-Ter119, PE-Cy7-conjugated anti-Sca-1, Percpcy5.5-conjugated anti-CD45.2, and FITC-conjugated anti-streptavidin were purchased from eBioscience. Alexa Fluor 647-conjugated anti-CD31 was purchased from BioLegend and Biotin-conjugated anti-CD51 was purchased from BD. Cells were sorted on a Melody after DAPI exclusion of dead cells. MSCs and OBs were sorted into Trizol (Invitrogen) for RNA preparation.
BM transplantation
BM transplantation was performed as previously described 37. Briefly, wild-type B6 SJL (CD45.1+) mice were irradiated with 1000 rads and injected with 2 × 106 total BM cells from wild-type or miR-9-1-deficient mice (CD45.2+) via tail vein. Recipients were analyzed 8 or 12 weeks after BM transplantation.
Competitive repopulation assay
BM cells (1 × 106) from wild-type or miR-9-1-deficient mice (CD45.2+) were mixed at a 1:1 ratio with competitor BM cells from wild-type B6 SJL mice (CD45.1+) and then transplanted into lethally irradiated (1,000 rads) wild-type B6 SJL mice by tail vein injection. The recipients were analyzed 6–8 weeks after BM transplantation.
In vitro osteoclast differentiation
In vitro osteoclast differentiation was performed as previously described 39. Briefly, BM cells isolated from the long bones of 6- to 12-week-old mice were cultured in α-MEM with 10% FBS and murine M-CSF (10 ng/ml) for 72 h. The nonadherent BM-derived macrophages (BMMs) were cultured with glutathione S-transferase-RANKL (100 ng/ml) and M-CSF (10 ng/ml) for 6 days. Osteoclast differentiation was detected by tartrate-resistant acid phosphatase (TRAP) staining according to the manufacturer’s instruction (Sigma). Cells that stained positively for TRAP were counted as osteoclasts.
In vitro osteoblastic differentiation
MSCs were isolated from BM cells as previously described 40. Briefly, BM cells from of 6- to 12-week-old mice were cultured in tissue culture flasks in alpha-MEM at 37°C for 48 hours and then non-adherent cells were removed. Adherent cells were collected as MSCs and cultured in α-MEM with 10% FBS. The CFU-F assay was used to quantify MSCs 41. 2 × 106 BM cells were plated in each well of a 6-well plate, cultured for 2 weeks, fixed in 4% PFA and stained with 0.5% crystal violet, followed by counting the stained colonies.
In vitro osteoblastic differentiation was performed as previously described. 41 Briefly, MSCs were cultured in osteogenic medium (10% FBS with 50 μg/mL ascorbic acid, 10 nM dexamethasone and 10 mM b-glycerophosphate). On day 7, cells were subjected to alkaline phosphatase (ALP) staining using ALP kit (SLCB3831; Sigma-Aldrich). Briefly, cells were fixed with Citrate-Acetone-Formaldehyde Fixative for 1 min after washing with deionized water. Sodium nitrite solution (100 μl) and fast red violet (FRV)-alkaline solution (100 μl) were mixed at room temperature for 2 min. The mixed solution was added to 4.5 ml of deionized water to prepare diazonium salt solution and then 100 μl of AS-BI alkaline solution was added to the final solution, in which cells were incubated in the dark at room temperature for 15 min. Cells were washed with deionized water twice and air-dried. The ALP-positive area indicating osteogenesis was calculated using ImageJ software. On day 21, the mineralization assay was performed by Alizarin red staining. Cells were fixed with Citrate-Acetone-Formaldehyde Fixative for 1 min after washing with deionized water. Next, cells were stained with 40 mM Alizarin red S (pH 4.2, Sigma) for 10 min in the dark and distained with 10% cetylpyridinium chloride. The calcium concentration was determined by absorbance measurements at 570 nm.
Microcomputed tomography (Micro-CT) analysis
Mouse femurs were cleaned of soft tissues, placed in 10% neutral formalin and stored at 4°C. The femurs were transferred to 70% alcohol 24 hours before used for Micro-CT scanning and then scanned by MICRO-CT SCANNER (Version2.6.5, SKYSCAN-1076, Bruker Skyscan). The scanning parameters were as follow: pixel size = (9.485 μm), source voltage = (70 kV), and source current = (141 μA). Scanning images were reconstructed with software Mimics (version10.01) and analyzed with software NRecon (Version1.4.4) and CTAn (Version1.7).
Quantitative real-time PCR (qRT-PCR) analysis
OBs and MSCs were sorted from wild-type and miR-9-1-deficient mice. Total RNAs were isolated from the cells and then first-strand cDNAs were synthesized with the Sensiscript RT kit (QIAGEN) using random primers (Invitrogen) according to the manufacturer’s protocol. Specific primer sequences are listed in Table S1. Gene expression level was normalized to actin abundance. qRT-PCR was performed in triplicate at 50°C for 2 min and then 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min using the 7500 Real-Time system (Applied Biosystems) in 10μl reaction volumes containing cDNA, primers, and iQ SYBR Green Supermix (Bio-Rad Laboratories). Data were analyzed using the 7500 system analyzing software and relative expression was calculated using the ΔΔCt method. Mature miR-9 was detected by using TaqMan miR-9 assay and U6 assay as normalization control (Applied Biosystems).
Osteoblastic activity and bone resorption measurement
Osteoblastic activity was assessed by measuring the serum levels of osteocalcin (OCN) using MyBioSource ELISA kit. Bone resorption was assessed by measuring the C-terminal telopeptides of collagen type I fragments (CTX) in mouse serum using MyBioSource ELISA kit.
Western blot analysis
MSCs were isolated from BM cells as described in the “In vitro osteoblastic differentiation” section above. MSCs were maintained in growth culture α-MEM and then were lysed in RIPA buffer (50 mM Tris pH7.4, 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS) in the presence of protease inhibitors. Total protein levels were determined by the bicinchoninic acid (BCA) protein assay kit (Pierce, Rockford, IL, USA). Cell lysates were subjected to Western blot analysis with the indicated antibodies.
3’-UTR luciferase reporter assay
The luciferase report assay was performed as previously described 42. Briefly, the wild-type Fst 3’UTR luciferase reporter vector was constructed by amplifying the mouse Fst 3’-UTR and cloning it into the NotI and XhoI sites of the psiCHECK-2 vector (Promega). The mutant Fst 3’-UTR was generated from the wild-type construct through the overlap extension PCR. Primary miRNA-9 was amplified and cloned into the NotI and XhoI sites of pcDNA3.0 vector. 293T cells were co-transfected with 100 ng luciferase reporter plasmid and 100 ng pCDNA3.0-miR-9 plasmid using X-tremeGENE HP DNA Transfection Reagent (Roche) according to the manufacturer’s instructions. After 48 h, lysates of 293T cells were harvested using lysis buffer (Promega), and luciferase activities were measured using the Dual-Luciferase reporter assay (Promega) according to the manufacturer’s instructions. Data were normalized for transfection efficiency by dividing Renilla luciferase activity with that of Firefly luciferase.
Overexpression of Fst
MSCs were transduced with 2 μg of pCMV3-mFst-HA (MG50035-CY, Sino Biological) or control plasmid using the X-tremeGENE HP DNA Transfection Reagent (Roche). The cells were collected for RNA extraction 48 h after transfection.
Bone histomorphometry analysis
Femurs were fixed in vitro with 1% paraformaldehyde-lysine-periodate solution, and frozen in OCT (Tissue Tek) after passage through sucrose gradient solution. 7 μm-thick cryostat sections were stained with hematoxylin and eosin, and Von Kossa staining kit (StatLab).
High-throughput RNA-seq and transcriptome analysis
High-throughput RNA-seq and transcriptome analysis were performed as previously described 37. MSCs and OBs were sorted from wild-type and miR-9-1-deficient mice, lysed in TRIzol (LifeTechnologies), and mRNAs were then isolated using NEBNext poly(A) mRNA Magnetic Isolation Module (New England Biolabs). The libraries were constructed using NEBNext Ultra RNA library Prep kit for Illumina (New England Biolabs) and quantified using Kapa Library Quantification Kit (KapaBiosystems) and QubitFluorometer (ThermoFisher). The average library size was measured using D1000 ScreenTape system (Agilent). The libraries (total 1.75 pmol) were sequenced on Illumina NextSeq 500 with NextSeq 500/550 v2 kit. Raw sequencing data was de-multiplexed, reads were aligned to the Mus musculus mm10 genome and counts for each gene were quantified using Basepair software (https://www.basepairtech.com). Read count normalization and differential expression analysis were conducted using DESeq2 v1.24.0 43. The Wald test was used to determine whether fold changes were significantly different from zero. Data were transformed using the regularized logarithm transformation for heatmaps 43. Batch effects were removed using the removeBatchEffect function from the limma v3.40.6 R package 44. Pre-ranked gene set enrichment analyses (GSEA) were conducted using shrunken fold-changes and clusterProfiler v3.12.0 45. Pathway databases, such as KEGG 46 and Reactome 47, were used. The Benjamini-Hochberg method was used to adjust p-values for false-discovery in both differential expression and GSEA analyses 48. GSEA was performed using GSEA software (https://www.gsea-msigdb.org). The gene sets for GSEA analysis were from the Molecular Signatures Database (MSigDB). GO analysis was performed with the DAVID online tool (https://david.ncifcrf.gov). Top GO categories were selected according to the adjusted P values. The gene sets related to BMP stimulation were from the genes that were upregulated by BMP2 stimulation in C2C12 cells (BMP_STIMULATE_VS _CONTROL_UP, 400_UP) 49.
Statistical analysis
The numbers of independent of experiments and the group size of experimental and control mice are indicated in the figure legends. No statistical method was used to predetermine sample size. Kaplan–Meier survival analysis was performed and survival differences between groups were assessed with the Log-rank test, assuming significance at p < 0.05. All statistical analysis was performed with the two-tailed unpaired Student t test, assuming significance at p < 0.05.
RESULTS
Small and underweight miR-9-1-deficient mice with incomplete penetrance
In both mouse and human, miR-9 is encoded by three genes, miR-9-1, miR-9-2 and miR-9-3 33, 34. To study the role of miR-9 in hematopoiesis, we examined the relative expression of primary miR-9-1, miR-9-2 and miR-9-3 in different subpopulations of hematopoietic cells. qRT-PCR revealed that miR-9-1 and miR-9-2 were expressed in HSCs and MPPs with relatively higher expression in CLPs (Supplementary Fig. S1A). Both miR-9-1 and miR-9-2 were expressed at very low levels in CMPs, MEPs and GMPs (Supplementary Fig. S1A). Similarly, both miR-9-1 and miR-9-2 were barely expressed in more mature hematopoietic subpopulations, such as CD4 T cells, CD8 T cells, NK cells, granulocytes, macrophages and dendritic cells (Supplementary Fig. S1B). However, miR-9-1 and miR-9-2 were expressed at low levels in B cells at the different developmental stages (Supplementary Fig. S1C). Of note, miR-9-3 was barely expressed in hematopoietic cells (Supplementary Fig. S1A–C).
To study the role of miR-9 in hematopoiesis, miR-9-1-deficient mice were generated by using the CRISPR-Cas9-mediated genome editing strategy technology (Supplementary Fig. S1D). We deleted 16 bp from the mature sequence of miR-9-1 without altering the sequences for miR-9-2 and miR-9-3 (Supplementary Fig. S1E). qRT-PCR analysis demonstrated that deletion of these base pairs from miR-9-1 (hereafter referred to as miR-9-1-deficient mice) reduced the expression of total mature miR-9 in BM cells and splenocytes relative to wild-type mice (Fig. 1A). About 43% of miR-9-1-deficient mice were smaller in size compared to gender- and age-matched wild-type controls (Fig. 1B). The smaller mutant mice displayed postweaning lethality within 12 weeks of age (Fig. 1C) whereas all the normal-sized mutants survived during this period (Supplementary Fig. S2A). The miR-9-1-deficient mice with growth defects had normal numbers of red blood cells and platelets, but markedly reduced white blood cells in the blood (Supplementary Fig. S2B). Consistently, the numbers of total BM cells, splenocytes and thymocytes were markedly decreased in growth-retarded miR-9-1-deficient relative to wild-type mice (Fig. 1D). Therefore, miR-9-1 deficiency is sufficient to cause growth defect and reduce white blood cells but not red blood cells and platelets with incomplete penetrance.
Fig. 1. Deficiency of miR-9-1 reduces overall mature miR-9 expression and causes growth defect and white blood cell reduction with incomplete penetrance.
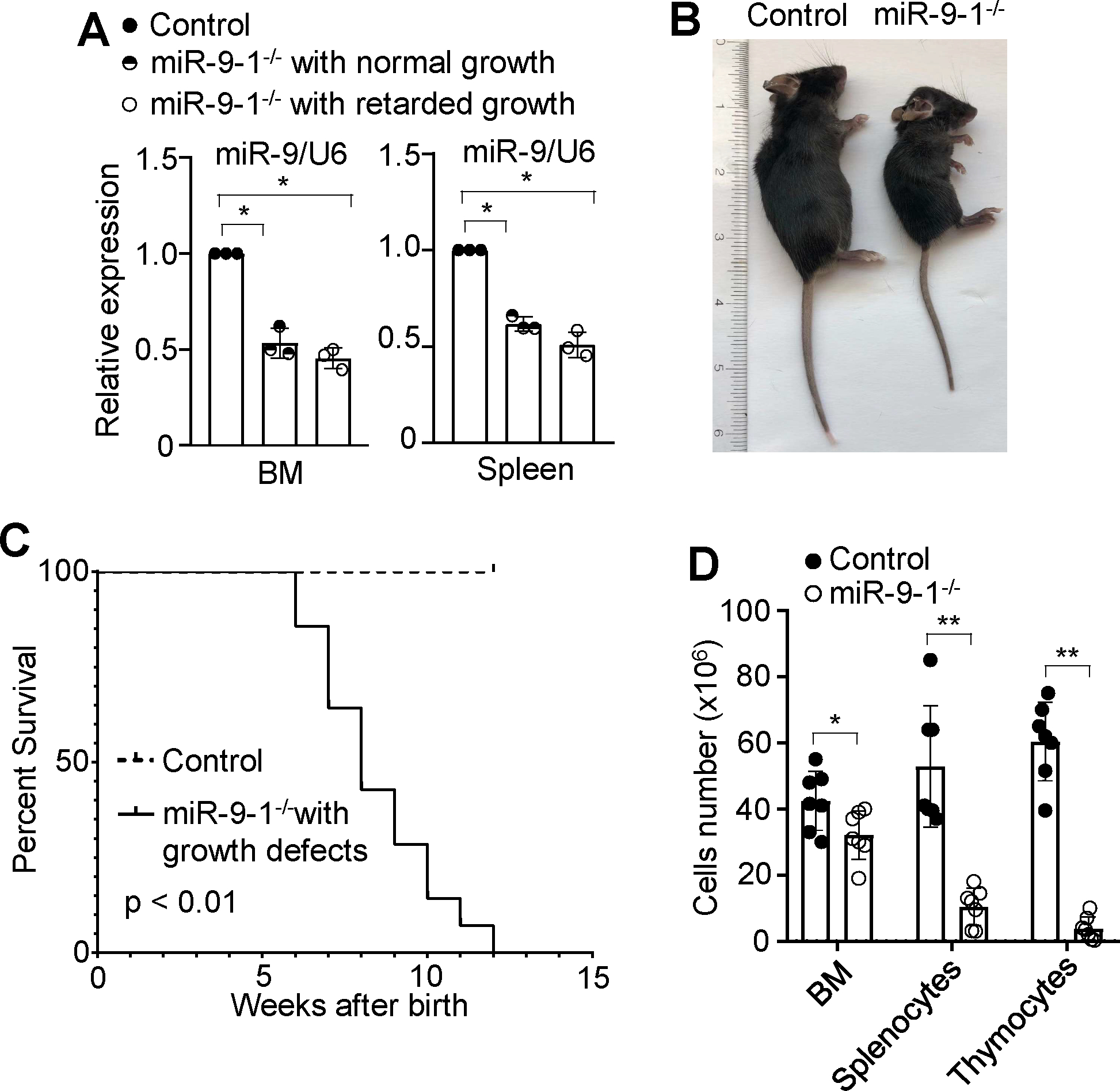
A Reduction in overall mature miR-9 expression in the BM and splenic cells from miR-9-1-deficient mice. The levels of mature miR-9 expression in BM and splenic cells from miR-9-1-deficient (miR-9-1−/−) mice with normal or retarded growth were quantified by qRT-PCR as a fraction of the corresponding wild-type control cells, which was set as 1. B Growth defect of miR-9-1-deficient mice. A representative image of 8-week-old growth-retarded male miR-9-1−/− mice and controls is shown. C Cumulative survival curve of miR-9-1-deficient mice with retarded growth. D Numbers of total BM cells, splenocytes and thymocytes in growth-retarded miR-9-1-deficient and control mice. Data shown are representative of or obtained from 3 independent experiments (A), and 9 (B, C) and 7 (D) pairs of wild-type control and growth-retarded miR-9-1−/− mice. Mean ± SD is shown. *, p < 0.05; **, p < 0.01.
Lack of miR-9-1 impairs lymphopoiesis
To further study the effect of miR-9-1 deficiency on white blood cell development, we first examined the B cell development in growth-retarded miR-9-1-deficient mice. The number and percentage of total B cells (B220+) in BM and spleen were markedly reduced in miR-9-1-deficient relative to wild-type mice (Fig. 2A). In addition, the populations of pro/pre- (B220+IgM−) and immature (B220+IgM+) but not mature (B220hiIgM+) B cells in BM were dramatically reduced in miR-9-1-deficient relative to wild-type controls (Fig. 2B). Within the pro/pre-B cell (B220+IgM−) population, the numbers and percentages of pro-(B220+CD43+) and pre-(B220+CD25+) B cells were reduced in miR-9-1-deficient relative to wild-type controls (Supplementary Fig. S2C). Thus, miR-9-1 deficiency impairs early B cell development.
Fig. 2. Lack of miR-9-1 impairs lymphopoiesis.
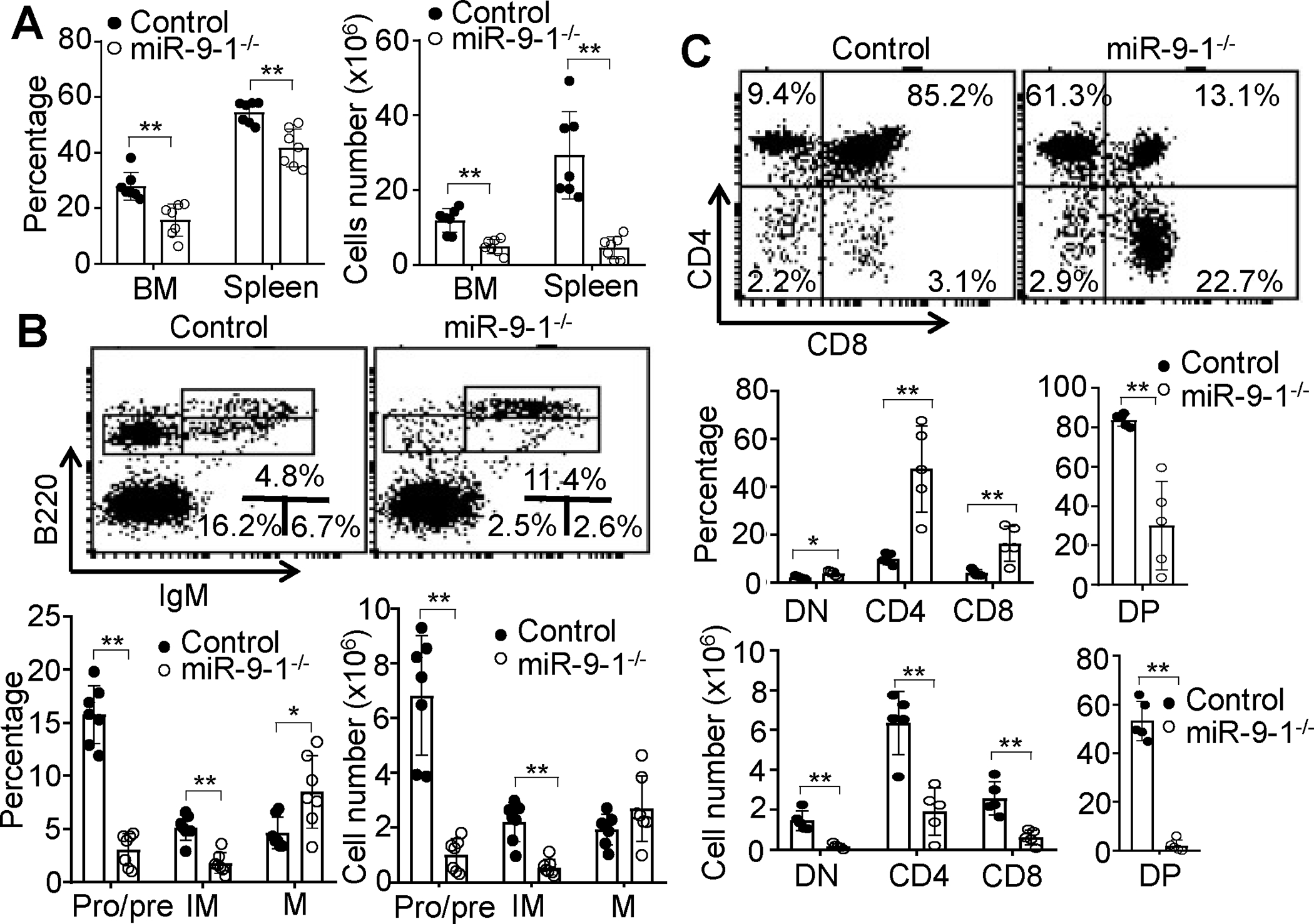
A Reduction in total B cells in the BM and spleen of miR-9-1-deficient mice. BM cells or splenocytes from growth-retarded miR-9-1−/− and wild-type control mice were stained with anti-B220. Percentages and numbers of B220+ B cells in the BM and spleen are shown, and percentages indicate cells in the gated live cells. B Reduction in pro/pre- and immature B cells in the BM of miR-9-1-deficient mice. BM cells from growth-retarded miR-9-1−/− and control mice were stained with anti-B220 and anti-IgM. Percentages and numbers of pro/pre-, immature and mature B cells in the BM are shown, and percentages indicate cells in the gated live cells. C Reduction in DN, DP and SP T cells in the thymus of miR-9-1-deficient mice. Thymocytes from growth-retarded miR-9-1−/− and control mice were stained with anti-CD4 and anti-CD8. Percentages and numbers of DN, DP and SP T cells in the thymus are shown, and percentages indicate cells in the gated live cells. Data shown are representative of or obtained from 7 (A, B) and 5 (C) control and growth-retarded miR-9-1−/− mice. Each dot represents an individual mouse. Mean ± SD is shown. *, p < 0.05; **, p < 0.01.
The effect of miR-9-1 deficiency on lymphocyte maturation was also examined. The percentages of transitional 1 (T1) (IgMhiIgDlo) was reduced, while the percentages of follicular (FO) (IgMloIgDhi) and marginal zone (MZ) (CD21hiCD23lo) mature B cells were increased in growth-retarded miR-9-1-deficient relative to control mice (Supplementary Fig. S2D). The numbers of T1, T2, FO and MZ B cells were all reduced in miR-9-1-deficient relative to control mice (Supplementary Fig. S2D). Thus, miR-9-1 deficiency impairs B cell maturation.
Early T cell development was also examined in growth-retarded miR-9-1-deficient mice. The percentages of DN (CD4−CD8−) and SP (CD4+CD8− and CD4−CD8+) thymocytes were increased, while DP (CD4+CD8+) thymocytes were decreased in miR-9-1-deficient mice relative to control mice (Fig. 2C). The absolute numbers of DN, DP, and SP thymocytes were markedly reduced due to the reduction in total thymocytes in miR-9-1-deficient mice (Fig. 2C). Moreover, the percentages of CD4+ and CD8+ T cells in the spleen were increased in miR-9-1-deficient mice. The absolute number of each subpopulation was decreased in miR-9-1-deficient relative to control mice (Supplementary Fig. S2E). Thus, miR-9-1 deficiency impairs T cell development and maturation. In contrast, the percentage and absolute number of myeloid cells were comparable between miR-9-1-deficient and control mice (Supplementary Fig. S2F). It is noteworthy that while mature miR-9 levels in BM cells and splenocytes were reduced to a similar level in miR-9-1-deficient mice with and without growth defects (Fig. 1A), the development of B and T cells was normal in miR-9-1-deficient mice without growth defects (Supplementary Fig. S3). Taken together, miR-9-1 deficiency impairs lymphopoiesis but not erythropoiesis, thrombopoiesis and myelopoiesis with incomplete penetrance.
Reduced hematopoietic progenitors, especially CLPs, in miR-9-1-deficient mice
To study the underlying mechanism by which miR-9-1 deficiency impairs lymphopoiesis, we examined hematopoietic stem cells and progenitors in growth-retarded miR-9-1-deficient mice. FACS analysis of the BM cells demonstrated that both the percentage and number of the Lin−IL7R−c-Kit+Sca1+ (LSK) population was reduced in miR-9-1-deficient relative to wild-type mice (Fig. 3A). The percentage and absolute number of the Lin−IL7R−c-Kit+Sca1− (LK) population were comparable between in miR-9-1-deficient and control mice (Fig. 3A). The LSK population comprises LT-HSC (CD34−CD135−), ST-HSC (CD34+CD135−) and MPP (CD34+CD135+). The percentages of LT-HSCs, ST-HSCs and MPPs were increased, decreased and not changed, respectively, in miR-9-1-deficient relative to control mice (Fig. 3B). Nonetheless, the absolute numbers of ST-HSCs and MPPs but not LT-HSC subpopulations were reduced in miR-9-1-deficient mice (Fig. 3B). Within the LK population, the percentage and number of CMPs (CD34+FcγRlo) but not GMPs (CD34+FcγRhi) or MEPs (CD34−FcγRlo) were decreased in miR-9-1-deficient relative to control mice (Fig. 3C). Moreover, the percentage and absolute number of CLPs (Lin−IL7R+c-KitmedSca1medCD135+) was reduced in miR-9-1-deficient relative to control mice (Fig. 3D). Taken together, the data demonstrate that miR-9-1 deficiency reduces hematopoietic progenitors, especially CLPs, but has no effect on GMPs and MEPs.
Fig. 3. MiR-9-1 deficiency reduces hematopoietic progenitors, especially CLPs.
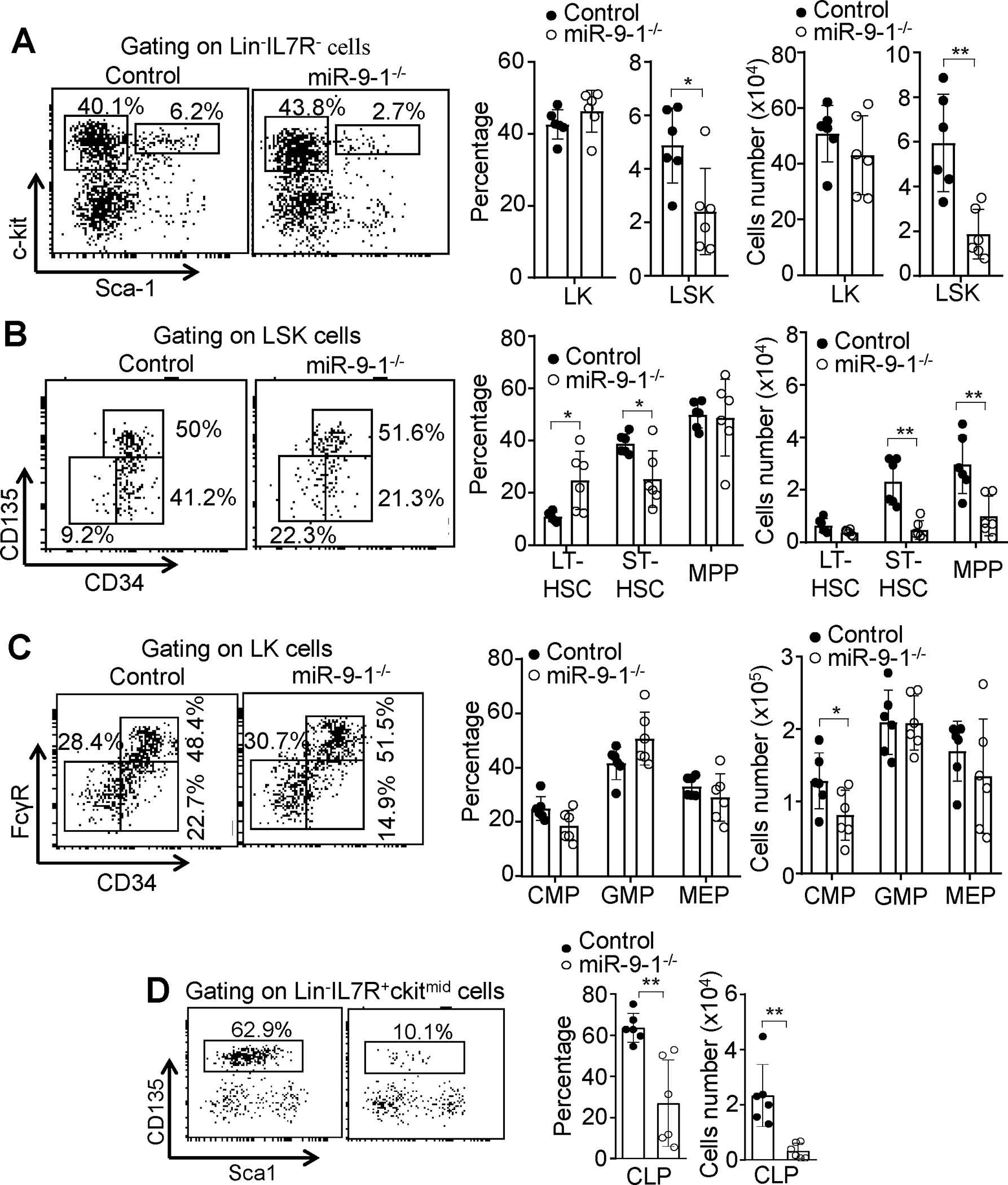
A Reduced LSK population in miR-9-1-deficient mice. BM cells from growth-retarded miR-9-1−/− and control mice were stained with the anti-lineage cocktail (Mac-1, Gr-1, B220, CD4, CD8, and Ter-119), IL-7R, c-Kit, and Sca-1 antibodies. Percentages and numbers of LSK and LK cells in the BM are shown, and percentages indicate cells in the gated lin−IL7R− population. B Normal LT-HSC but reduced ST-HSC and MPP subpopulations in miR-9-1-deficient mice. BM cells from growth-retarded miR-9-1−/− and control mice were stained with the anti-lineage cocktail, IL7R, c-Kit, Sca-1, CD34, and CD135 antibodies. Percentages and numbers of LT-HSC (CD34−CD135−), ST-HSC (CD34+CD135−) and MPP (CD34+CD135+) subpopulations in the BM are shown, and percentages indicate cells in the gated LSK population. C Reduced CMP and normal GMP and MEP populations in miR-9-1-deficient mice. BM cells from growth-retarded miR-9-1−/− and control mice were stained with the anti-lineage cocktail, IL7R, c-Kit, Sca-1, CD34 and FcγR antibodies. Percentages and numbers of CMP (CD34+FcγRlo), GMP (CD34+FcγRhi) and MEP (CD34−FcγRlo) subpopulations in the BM are shown, and percentages indicate cells in the gated LK population. D Reduced CLP populations in miR-9-1-deficient mice. BM cells from growth-retarded miR-9-1−/− and control mice were stained with the anti-lineage cocktail, IL-7R, c-Kit, Sca-1 and CD135 antibodies. Percentages and numbers of CLP subpopulations in the BM are shown, and percentages indicate CLP in the gated Lin−IL7R+ckitmid population. Data shown are representative of or obtained from 6 control and growth-retarded miR-9-1−/− mice. Each dot represents an individual mouse. Mean ± SD is shown. *, p < 0.05; **, p < 0.01.
BM niche-dependent impairment of lymphopoiesis in miR-9-1-deficient mice
To determine whether the defective lymphopoiesis in miR-9-1-deficient mice is the result of an intrinsic abnormality, we transplanted BM cells from CD45.2 growth-retarded miR-9-1-deficient or control mice into lethally irradiated congenic wild-type CD45.1 recipients. Eight weeks after transplantation, BM cells, splenocytes and thymocytes from the recipients that received the BM of miR-9-1-deficient or control mice were largely CD45.2+ (Supplementary Fig. S4A). In contrast to untransplanted miR-9-1-deficient mice (Supplementary Fig. S2B), wild-type recipients that received miR-9-1-deficient BM cells had equivalent numbers of white blood cells to those that received control BM cells (Supplementary Fig. S4B). Similar to miR-9-1-deficient mice (Supplementary Fig. S2B), recipients of miR-9-1-deficient BM cells possessed normal numbers of red blood cells and platelet numbers (Supplementary Fig. S4B). In addition, the recipients that received miR-9-1-deficient relative to wild-type BM cells displayed normal numbers of total BM cells, splenocytes and thymocytes (Fig. 4A). Moreover, the recipients that received miR-9-1-deficient or wild-type BM cells had comparable percentages and numbers of CD45.2+ LSK and LK cells (Supplementary Fig. S4C). Within the CD45.2+ LSK population, the recipients that received miR-9-1-deficient BM cells had normal percentages and numbers of LT-HSCs, ST-HSCs and MPPs (Fig. 4B). Within the CD45.2+ LK population, the recipients that received miR-9-1-deficient BM cells also had normal percentages and numbers of GMPs, CMPs and MEPs (Supplementary Fig. S4D). Further, the recipients that received miR-9-1-deficient BM cells exhibited normal percentages and numbers of Mac-1+Gr-1+ myeloid cells (Supplementary Fig. S4E). Thus, BM transplantation restores the populations of miR-9-1-deficient hematopoietic progenitors, including white blood cells and myeloid cells.
Fig. 4. Restored the hematopoietic progenitor populations and lymphopoiesis of miR-9-1-deficient BM cells after transplantation.
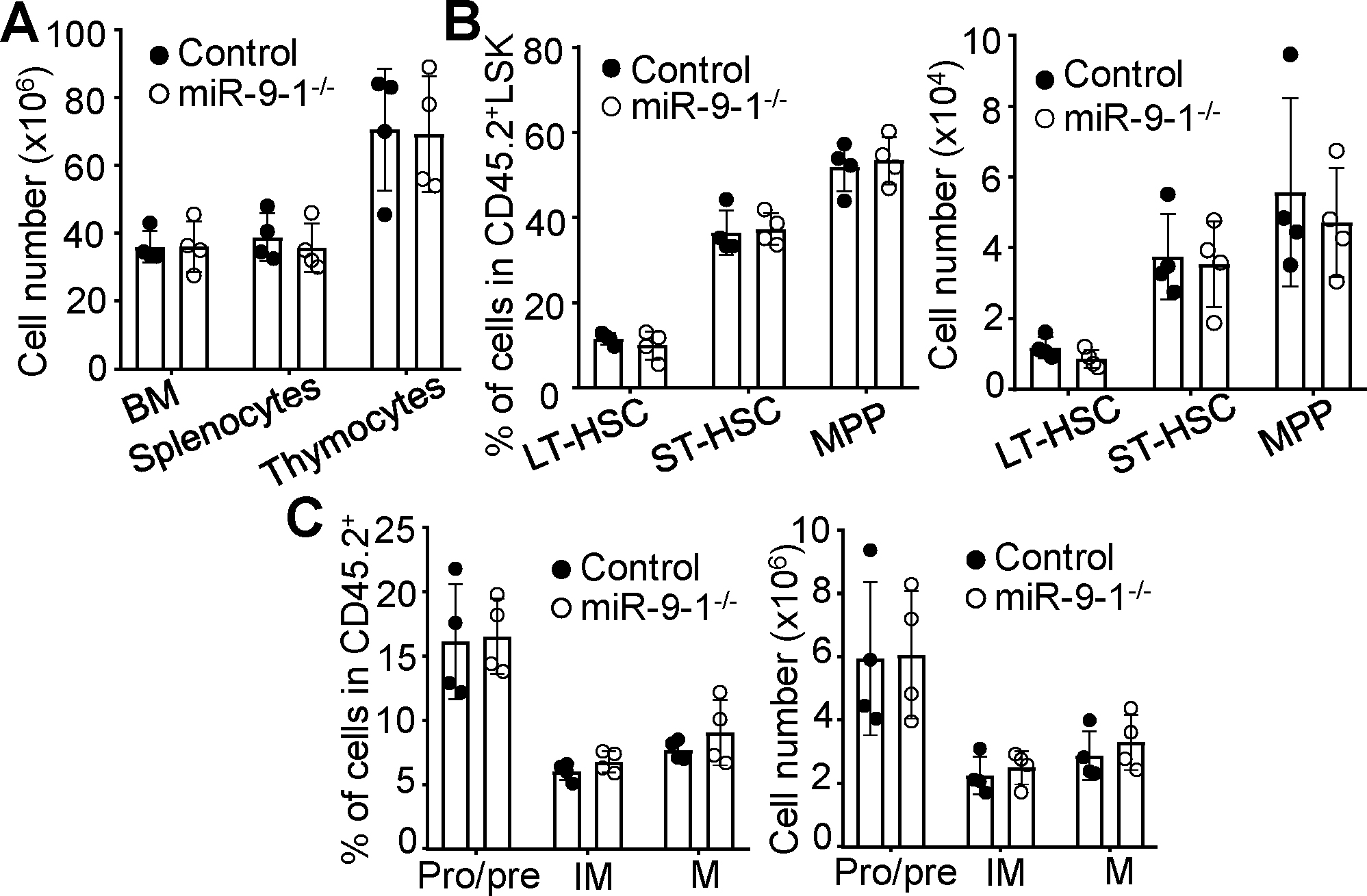
BM cells from growth-retarded miR-9-1−/− or wild-type control mice (CD45.2+) were transplanted into lethally irradiated CD45.1+ recipients. About eight weeks after transplantation, the recipients were analyzed. A The numbers of total BM cells, splenocytes and thymocytes the recipients that received BM cells from miR-9-1−/− and controls mice. B The percentages and numbers of various CD45.2+ hematopoietic progenitors in the BM of the recipients that received BM cells from miR-9-1−/− and controls mice. Percentages indicate LT-HSC, ST-HSC and MPP in the gated CD45.2+Lin−IL-7R−LSK population. C The percentages and numbers of B cells at the different developmental stages in the BM of the recipients that received BM cells from miR-9-1−/− and controls mice. Percentages indicate pro/pre-, immature (IM) and mature (M) B cells in the gated CD45.2+ population. Data shown are obtained from 4 recipients that received control or miR-9-1−/− BM cells. Each dot represents an individual mouse. Mean ± SD is shown. *, p < 0.05; **, p < 0.01.
In terms of B cell development, the recipients that received miR-9-1-deficient relative to wild-type BM cells acquired normal percentages and numbers of CD45.2+ pro/pre-, immature and mature B cells in the BM (Fig. 4C and Supplementary Fig. S4F) and CD45.2+ T1, T2, FO and MZ B cells in the spleen (Supplementary Fig. S4G, S4H). In terms of T cell development, the recipients that received miR-9-1-deficient relative to wild-type BM cells displayed normal percentages and numbers of CD4+ and CD8+ splenic T cells (Supplementary Fig. S4I) and DN, DP, CD4 and CD8 thymocytes (Supplementary Fig. S4J). It is noteworthy that, twelve weeks after transplantation, BM transplantation effectively restored the populations of miR-9-1-deficient lymphoid and myeloid progenitors, and promoted lymphocyte development (Supplementary Fig. S5), which aligns with the findings observed eight weeks post-transplantation (Supplementary Fig. S4). In conclusion, BM transplantation successfully restores the populations of miR-9-1-deficient lymphoid progenitors and facilitates the development of B and T cells.
The rescued engrafting capacity of miR-9-1-deficient hematopoietic progenitors was further confirmed by competitive repopulation BM transplantation. In competition with wild-type CD45.1+ hematopoietic progenitors, CD45.2+ BM cells from growth-retarded miR-9-1-deficient or control mice had comparable of chimerism in the progenitor cells, including LSK/LK, HSC, MPP, GMP, CMP, MEP and CLP (Supplementary Fig. S4K). We also observed that miR-9-1-deficient and control donor cells competed effectively with the competitor cells in BM and spleen, including B and T cells as well as Mac-1+Gr-1+ myeloid cells (Supplementary Fig. S4L). Thus, the impaired hematopoiesis and lymphopoiesis observed in miR-9-1-deficient mice are not hematopoietic cell autonomous but caused by alteration in the BM stromal microenvironment.
Impaired bone formation in miR-9-1-deficient mice
To study the function of miR-9-1 in regulating the formation of the BM niche, we first utilized high-resolution micro-computed tomography (μ-CT) to detect bone structures. Consistent with the small size of miR-9-1-deficient mice (Fig. 1 B and C), μ-CT analysis showed that the femurs of the growth-retarded mutant mice exhibited lower trabecular bone mass (Fig. 5A). The ratio of trabecular bone volume to tissue volume (BV/TV) in miR-9-1-deficient mice was 50% lower than that in the control mice (Fig. 5B). The decreased bone volumes were correlated with decreased trabecular number (Tb.N) and increased trabecular separation (Tb.Sp) in miR-9-1-deficient relative to control mice (Fig. 5B). Trabecular thickness (Tb.Th) was not significantly changed in miR-9-1-deficient mice (Fig. 5B). H&E staining showed that miR-9-1-deficient mice formed much less bone tissue when compared with control mice (Fig. 5C). Thus, these results demonstrate that miR-9-1 is required for bone formation.
Fig. 5. MiR-9-1 is required for osteoblastic differentiation.
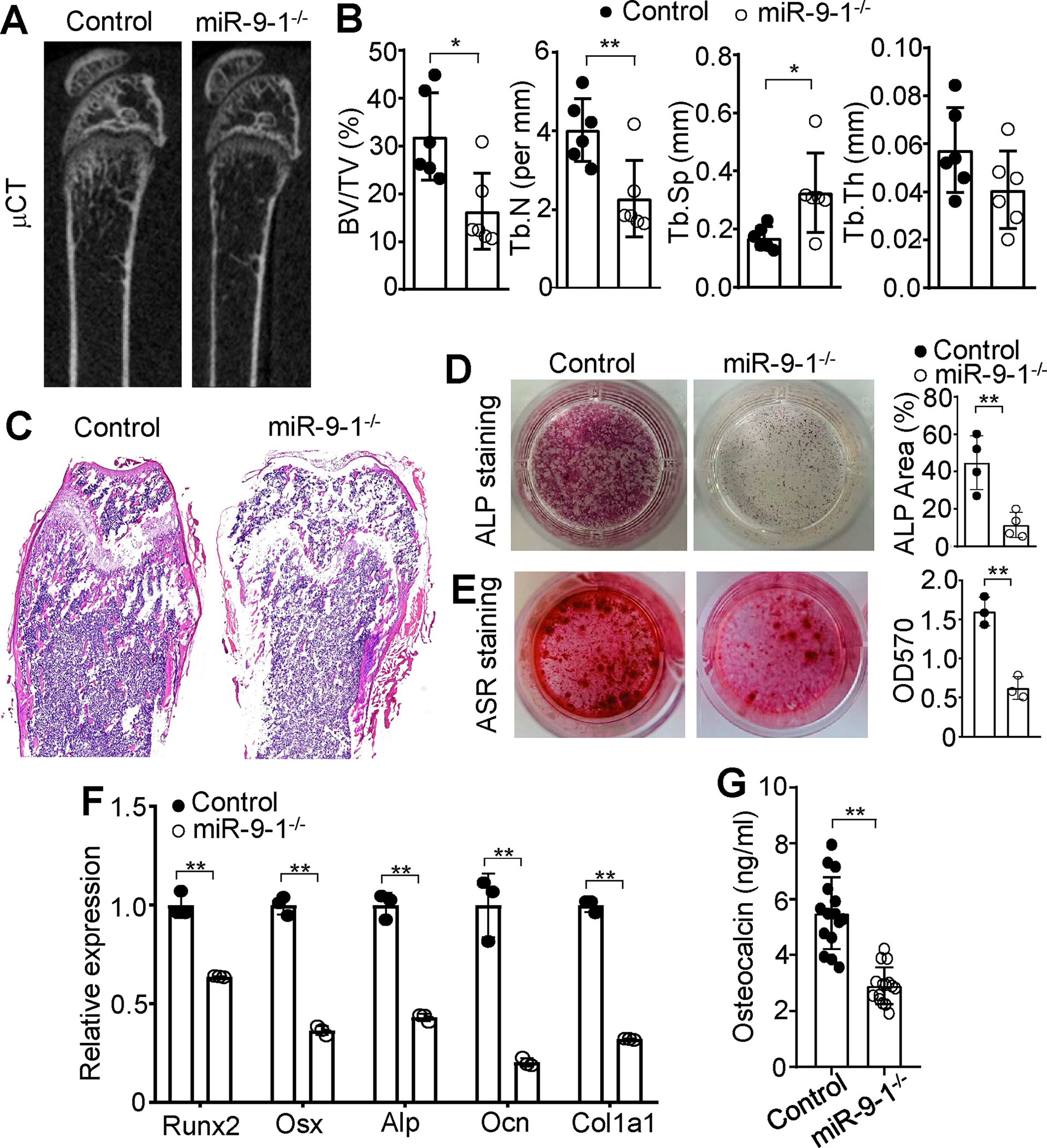
A μCT images of distal femurs from miR-9-1−/− and control mice. Distal femurs from 6–12 weeks growth-retarded miR-9-1−/− and control mice were subjected to μCT analysis and hematoxylin and eosin (H&E) staining. B Quantitative μCT analysis of distal femurs from miR-9-1−/− and control mice. Bone volume per tissue volume (BV/TV), trabecular number (Tb.N), trabecular seperation (Th.Sp) and trabecular thickness (Tb.Th) are shown. C H&E-stained sections of femurs from miR-9-1−/− and control mice. D ALP staining of miR-9-1-deficient MSCs after osteogenic differentiation. MiR-9-1−/− and control MSCs were cultured in osteogenic media for 7 days, and ALP staining (left) and quantification (right) were performed. E ARS staining of miR-9-1-deficient MSCs after osteoblastic differentiation. MiR-9-1−/− and control MSCs were cultured in osteogenic media for 21 days, and ARS staining and quantification were performed. F The relative mRNA levels of Runx2, Osx, Alp, Ocn and Col1α1 in miR-9-1-deficient MSCs. MiR-9-1−/− and control MSCs were cultured in osteogenic media for 7 days. Then, the mRNA levels of Runx2, Osx, Alp, Ocn and Col1α1 in miR-9-1−/− MSCs were quantified by qRT-PCR as a fraction of the corresponding control cells, average of which was set as 1. G Serum level of OCN in miR-9-1-deficient mice. Serum levels of OCN in 6–12 weeks old miR-9-1−/− and control mice were determined by the ELISA analysis. Data shown are representative of or obtained from 6 (A, B), 3 (C, E, F), 4 (D) and 15 (G) miR-9-1−/− and control mice. Each dot represents one biological replicate or an individual mouse. Mean ± SD is shown. *, p < 0.05; **, p < 0.01.
Requirement of miR-9-1 for osteoblastic but not osteoclastic differentiation
Bone development and growth is a continuous process that is the result of the balance between bone resorption by osteoclasts and bone formation by OBs 50. We first examined the role of miR-9-1 in osteoclastogenesis by performing in vitro osteoclast differentiation of miR-9-1-deficient BM-derived macrophages (BMMs). RANKL and M-CSF-induced in vitro osteoclast differentiation was normal in BMMs derived from growth-retarded miR-9-1-deficient relative to control mice (Supplementary Fig. S6A). To confirm that growth-retarded miR-9-1-deficient mice have normal osteoclasts in vivo, we compared the serum level of cross-linked C-telopeptide of type I collagen (CTX), an indicator of osteoclast activity in vivo 51, which were comparable in both growth-retarded miR-9-1-deficient and control mice (Supplementary Fig. S6B), indicating normal osteoclast activity in the mutant mice. Thus, these data demonstrate that miR-9-1 is not required for osteoclastogenesis.
We further investigated the role of miR-9-1 in osteoblastic differentiation by examining in vitro osteoblastogenesis of mutant MSCs. Primary Lin−CD45−CD31−CD51+Sca-1+ MSCs and Lin−CD45−CD31−CD51+Sca-1− OBs were isolated from the BM of control and miR-9-1-deficient mice. Analysis using qRT-PCR analysis demonstrated a marked reduction in the overall levels of miR-9 in CD51+Sca1+ and CD51+Sca1− cells from miR-9-1-deficient mice, both with normal and retarded growth, when compared to wild-type mice (Supplementary Fig. S6C). The colony-forming unit fibroblast (CFU-F) assay showed that miR-9-1-deficient MSCs displayed a slightly increased number of colonies compared to control cells (Supplementary Fig. S6D), indicating the unimpaired cell viability or growth rate of mutant MSCs. In contrast, MSCs from miR-9-1-deficient mice with retarded, but not normal, growth exhibited markedly decreased alkaline phosphatase (ALP) activity (Fig. 5D and Supplementary Fig. S6E, S6F) and mineralization visualized by Alizarin Red S (ARS) staining after osteoblastic differentiation (Fig. 5E), showing that miR-9-1 is important for osteoblastic differentiation. Consistently, the mRNA levels of the master transcription factor for osteogenesis, Runx2, and the osteoblast-specific transcription factor, Osx 52, 53, were markedly reduced in miR-9-1-deficient relative to control BM-derived MSCs (Fig. 5F). The markers of osteoblastic differentiation, Alp, osteocalcin (Ocn) and type I collagen (Col1α1) 54, were also clearly reduced in miR-9-1-deficient MSCs (Fig. 5F). In addition, the serum levels of the osteogenesis marker OCN was significantly decreased in miR-9-1 deficiency mice (Fig. 5G), demonstrating impaired OB function in the mutant mice. Taken together, these data demonstrate that miR-9-1 plays a critical role in osteoblastic but not osteoclastic differentiation.
Impaired lymphopoiesis and osteoblastogenesis were observed only in miR-9-1-deficient mice with retarded growth. To determine whether the phenotypes were solely due to the loss of miR-9-1 and not secondary to developmental, growth, or other genetic defects, we analyzed a second independent colony of miR-9-1-deficient mice with a 21bp deletion in the mature sequence of miR-9-1 (Supplementary Fig. S7A), which differed from the 16bp deletion in the original first colony. MiR-9-1-deficient mice from this second colony exhibited the same phenotypes as those from the first colony. Specifically, 48% of miR-9-1-deficient mice from the second colony exhibited small body size, lymphopenia, and impaired osteoblastogenesis compared to wild-type controls (Supplementary Fig. S7B–G). These findings suggest that the observed defects in miR-9-1-deficient mice are not due to secondary consequences of developmental or growth defects, or other genetic anomalies.
Regulation of the gene network of osteoblastic differentiation by miR-9-1
To understand the molecular mechanism by which miR-9-1 regulates osteoblastic differentiation, we performed a high-throughput RNA sequencing analysis of global gene expression patterns in miR-9-1-deficient MSCs and OBs to identify the genes regulated by miR-9-1. Endothelia cells (ECs), MSCs and OBs within the endosteal stromal compartment can be identified as Lin−CD45−CD31+Sca-1+, Lin−CD45−CD31−CD51+Sca-1+, Lin−CD45−CD31−CD51+Sca-1−, respectively (Supplementary Fig. S8A) 38. We examined the expression levels of miR-9-1, miR-9-2 and miR-9-3 in these cells and found that miR-9 was expressed in these BM niche cells, with the primary isoforms being miR-9-1 and miR-9-2 in CD51+Sca1+ MSCs and CD51+Sca1− OBs, and miR-9-1 in CD31+Sca1+ endothelial cells (Supplementary Fig. S8B). LepR+ MSCs have been reported to be important BM niche cells for lymphopoiesis 19, 55. Therefore, we also examined the expression levels of miR-9-1, miR-9-2 and miR-9-3 in Lin−CD45−CD31−CD51+Sca1+LepR+ MSCs and found that miR-9-1 and miR-9-2 were primary isoforms in LepR+ MSCs (Supplementary Fig. S8B). Differential gene expression analysis revealed that sorted CD51+Sca1+ MSCs from miR-9-1-deficient mice with retarded growth exhibited a marked change of the gene expression profile compared to wild-type control CD51+Sca1+ MSCs with a total of 1156 genes differentially expressed (Fig. 6A). Osteoblastic differentiation of MSCs is a multi-step process involving osteogenic lineage commitment and differentiation, extracellular matrix production and matrix mineralization, collagen secretion, and cell adhesion molecule upregulation. Analysis of individual genes demonstrated that osteoblast-specific genes, including Runx2, Sp7 (Osx) and Alp 52–54, 56, were markedly downregulated in miR-9-1−/− relative to control CD51+Sca1+ MSCs (Fig. 6B). The genes associated with bone formation, such as Col1α1 and Bglap (Ocn) 54, 56, were reduced in miR-9-1−/− CD51+Sca1+ MSCs (Fig. 6B). The genes related to bone extracellular matrix synthesis, including DMP1, Osteonectin (Sparc) and Keratocan (Kera) 57–60, were also decreased in miR-9-1−/− CD51+Sca1+ MSCs (Fig. 6B). In contrast, the genes important for tissue remodeling, such as matrix metallopeptidases (Mmp) and ADAM metallopeptidase (Adamts5) 60, 61, were increased in miR-9-1−/− relative to control CD51+Sca1+ MSCs (Fig. 6B). In addition, many genes related to extracellular organization were also strongly downregulated in miR-9-1−/− CD51+Sca1+ MSCs (Fig. 6B). A number of cell adhesion molecule genes 60 were also reduced in miR-9-1−/− relative to wild-type CD51+Sca1+ MSCs (Fig. 6B). Of note, the reduction of Runx2, Osx, Alp, Ocn and Col1α1 in directly sorted miR-9-1−/− CD51+Sca1− OBs was confirmed by qRT-PCR (Supplementary Fig. S8C). These gene expression profile changes are consistent with the observation that miR-9-1-deficient MSCs had impaired osteoblastic differentiation (Fig. 5D–G).
Fig. 6. MiR-9-1 regulates the expression of the genes associated with osteoblastic differentiation and function in MSCs.
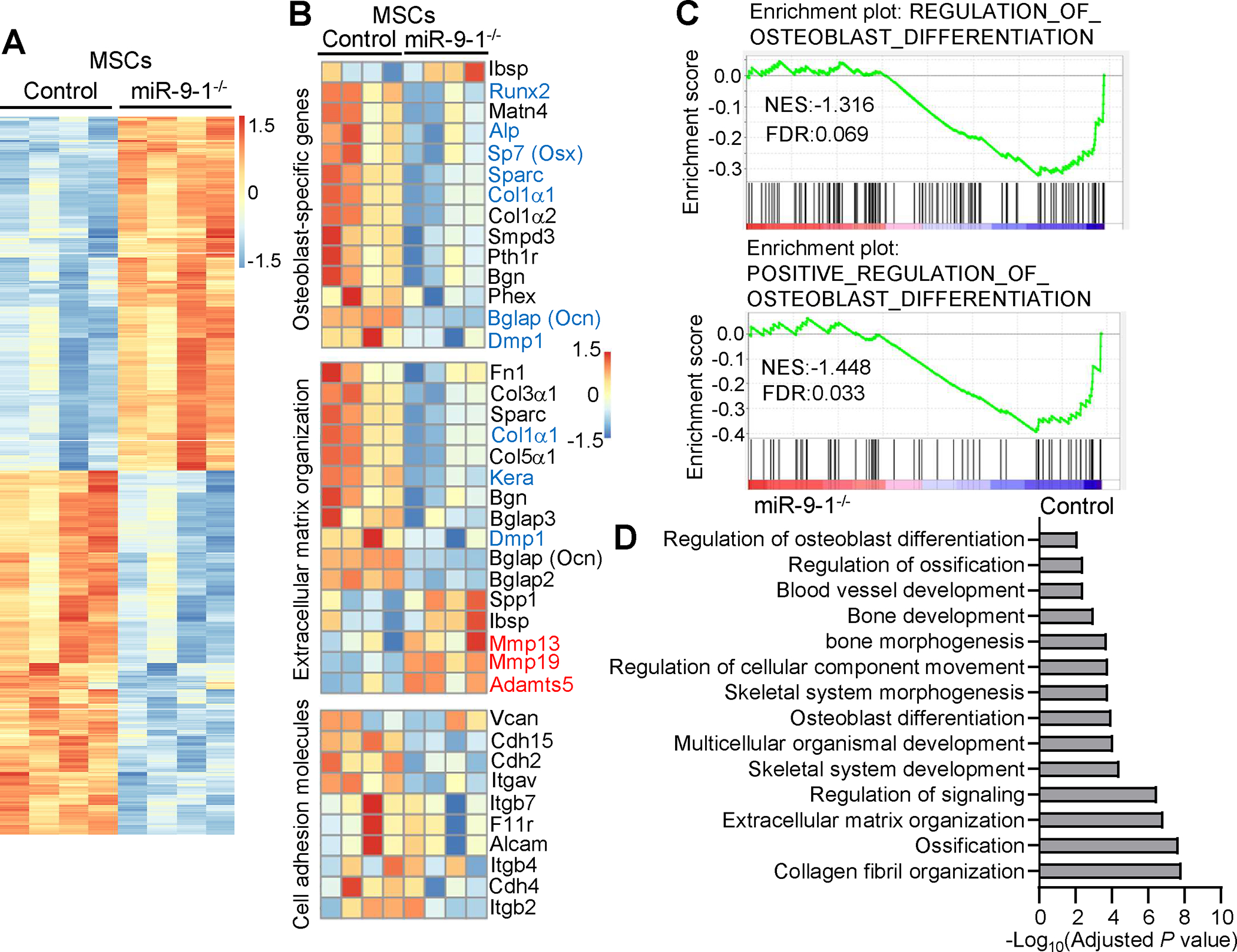
MSCs (Lin−CD45−CD31−CD51+Sca-1+) were sorted from growth-retarded miR-9-1−/− and wild-type control mice and directly subjected to RNA-seq. A Heatmap of differentially expressed genes between miR-9-1−/− and control MSCs. Genes with an adjusted p-value < 0.05 found by DESeq2 were assigned as differentially expressed. B Heatmap of differentially expressed osteoblast-specific, extracellular organization and cell adhesion molecule genes in miR-9-1−/− relative control MSCs. C GSEA plots of osteoblast differentiation gene signatures in miR-9-1−/− and control MSCs. D GO enrichment analysis of downregulated genes in miR-9-1−/− relative to control MSCs. Bar plots show negative log10 of p-values for GO enrichment analysis of the biological pathways in control relative to miR-9-1−/− MSCs. Data shown is obtained from 4 growth-retarded miR-9-1−/− and wild-type mice.
Comparative gene set enrichment analysis (GSEA) of osteoblastic differentiation signatures showed that the sets of genes associated with osteoblastic differentiation were enriched in control relative to miR-9-1-deficient CD51+Sca1+ MSCs (Fig. 6C). In contrast, the sets of genes associated with the differentiation of myocytes, chondrocytes and adipocytes were not significantly changed in miR-9-1-deficient relative to control CD51+Sca1+ MSCs (Supplementary Fig. S8D). Consistent with the GSEA analysis, the ability of CD51+Sca1+ MSCs from miR-9-1-deficient mice with retarded growth to differentiate into adipocytes was normal relative to wild-type CD51+Sca1+ MSCs (Supplementary Fig. S8E–F). GSEA also found that the gene sets associated with bone development and mineralization were reduced in miR-9-1-deficient relative to control CD51+Sca1+ MSCs (Supplementary Fig. S8G). Gene ontology (GO) analysis identified that the genes associated with osteoblastic differentiation, bone development and ossification, extracellular matrix organization, and collagen fibril organization were downregulated in miR-9-1−/− relative to control CD51+Sca1+ MSCs (Fig. 6D). Further, pathway enrichment analysis using the REACTOME database revealed marked reduction of the pathways associated with collagen formation, extracellular matrix organization and cell adhesion molecules in miR-9-1−/− relative to control CD51+Sca1+ MSCs (Supplementary Fig. S8H). Consistent with the RNA-seq data, the defect in bone formation in miR-9-1-deficient mice was observed by Von Kossa staining of sections across the femurs (Supplementary Fig. S8I). Taken together, these data demonstrate that miR-9-1 regulates genes associated with osteoblast differentiation and function of MSCs.
Similarly, OBs (Lin−CD45−CD31−CD51+Sca-1−) were sorted out from the endosteal stromal compartment (Supplementary Fig. S8A), and qRT-PCR analysis showed that the OB-specific-genes, such as Runx2, Col1a1 and Ocn, were highly expressed in sorted CD51+Sca-1− OBs relative to the Lin−CD45−CD31−CD51−Sca-1− control population (Supplementary Fig. S9A), confirming the OB identity of the sorted cells. Of note, qRT-PCR analysis demonstrated that miR-9-1 deficiency markedly reduced the total level of mature miR-9 in mutant relative to wild-type CD51+Sca-1− OBs (Supplementary Fig. S9B), demonstrating the reduction of the total mature miR-9 level in OBs by miR-9-1 deficiency. Following RNA-seq, differential gene expression analysis revealed that miR-9-1-deficient CD51+Sca-1− OBs displayed a total of 290 genes differentially expressed compared to wild-type control CD51+Sca-1− OBs (Supplementary Fig. S9C). Individual gene analysis demonstrated that osteoblast-specific genes, including Runx2, Sp7 (Osx) and Alpl, Dmp1, Ocn and Col1a1, were downregulated in miR-9-1−/− relative to control CD51+Sca-1− OBs (Supplementary Fig. S9D). The genes associated to extracellular matrix organization, such as Kera and Sparc, were also decreased whereas the genes important for tissue remodeling, such as Mmp13, Mmp19 and Adamts5, were increased in miR-9-1−/− relative to control CD51+Sca-1− OBs (Supplementary Fig. S9D). Cell adhesion molecule genes, including integrin (Itg) and cadherin (Cdh), were also reduced in miR-9-1−/− relative to wild-type CD51+Sca-1− OBs (Supplementary Fig. S9D). GO analysis revealed that the gene sets associated with extracellular matrix organization and bone development and ossification were downregulated in miR-9-1−/− relative to control CD51+Sca-1− OBs (Supplementary Fig. S9E). Taken together, these data further support the notion that miR-9-1 deficiency reduces the expression of multiple OB-specific genes and impairs osteoblastic differentiation.
The numbers of OB (CD51+Sca1−) cells, but not MSCs (CD51+Sca1+) or ECs (CD31+Sca1+), were reduced in the stromal cells from growth-retarded miR-9-1-deficient relative to wild-type mice (Supplementary Fig. S9F). Apart from osteoblasts, CD51+Sca1− cells are known to contain various heterogeneous BM niche subtypes 62 that can be identified by the expression of NG2 63, CD200 64, and Leptin receptor (LepR) 65. We investigated whether the altered heterogeneity within CD51+Sca1− cells in miR-9-1-deficient mice contributes to the reduced expression of OB-specific genes. To do so, we examined the niche subtypes within CD51+Sca1− cells. The subpopulations of NG2+, CD200+ and LepR+ cells within the CD51+Sca1− cells were comparable between growth-retarded miR-9-1-deficient and wild-type mice (Supplementary Fig. S9H–I). Therefore, the findings suggest that the reduced expression of OB-specific genes in miR-9-1-deficient CD51+Sca1− cells is not due to altered cell heterogeneity of CD51+Sca1− cells in mutant mice.
Directly targeting Fst by miR-9-1 to regulate BMP/Smad signaling
To further investigate the molecular mechanism by which miR-9-1 regulates osteoblastic differentiation, we aimed to identify the genes directly regulated by miR-9-1. MiRNAs usually guide AGO proteins to the complementary sequences in the 3’UTR of mRNAs to destabilize target transcripts 22, 23. Theoretically, expression of genes that are direct targets of miR-9-1 should be up-regulated in miR-9-1-deficient relative to control cells. Based on the criteria of adjusted p value < 0.05, the Venn diagram showed 569 and 200 genes were up-regulated in CD51+Sca1+ MSCs and CD51+Sca-1− OBs from growth-retarded miR-9-1-deficient mice, respectively, and there were 109 genes were increased in both mutant CD51+Sca1+ MSCs and CD51+Sca-1− OBs (Fig. 7A). Although the mature miRNA could be generated from both the 5’ and 3’ arms of the single miRNA hairpin precursor as miRNA-5p and miRNA-3p respectively, miR-5p (the guide strand) is preferentially loaded onto AGO2 and is functionally relevant while miR-3p (the passenger strand) is rapidly degraded 22, 23. The 3’-UTR of mRNAs generally possesses the miR target sites that are complementary to the miR-5p seed region, which is the key criterion for target-site prediction. Thus, we used the online database miRDB (http://mirdb.org) 66 to predict miR-9-5p target mRNAs and found 1068 potential target genes of miR-9-5p (Fig. 7A). Importantly, the Venn diagram showed 5 genes that were up-regulated in miR-9-1-deficient CD51+Sca1+ MSCs and CD51+Sca-1− OBs and possessed the miR-9-5p target site, including Fst, the negative regulator of BMP activity 67 (Fig. 7A). Indeed, differential gene expression analysis demonstrated that the expression of the 5 genes, including Fst, was markedly increased in miR-9-1−/− relative to control CD51+Sca1+ MSCs and CD51+Sca-1− OBs (Fig. 7B). The higher expression of Fst in miR-9-1-deficient CD51+Sca1+ MSCs and CD51+Sca-1− OBs was confirmed by qRT-PCR (Fig. 7C). Thus, these data indicate that miR-9-1 targets the Fst gene to reduce its mRNA level.
Fig. 7. MiR-9-5p directly targets Fst to regulate BMP signaling in MSCs.
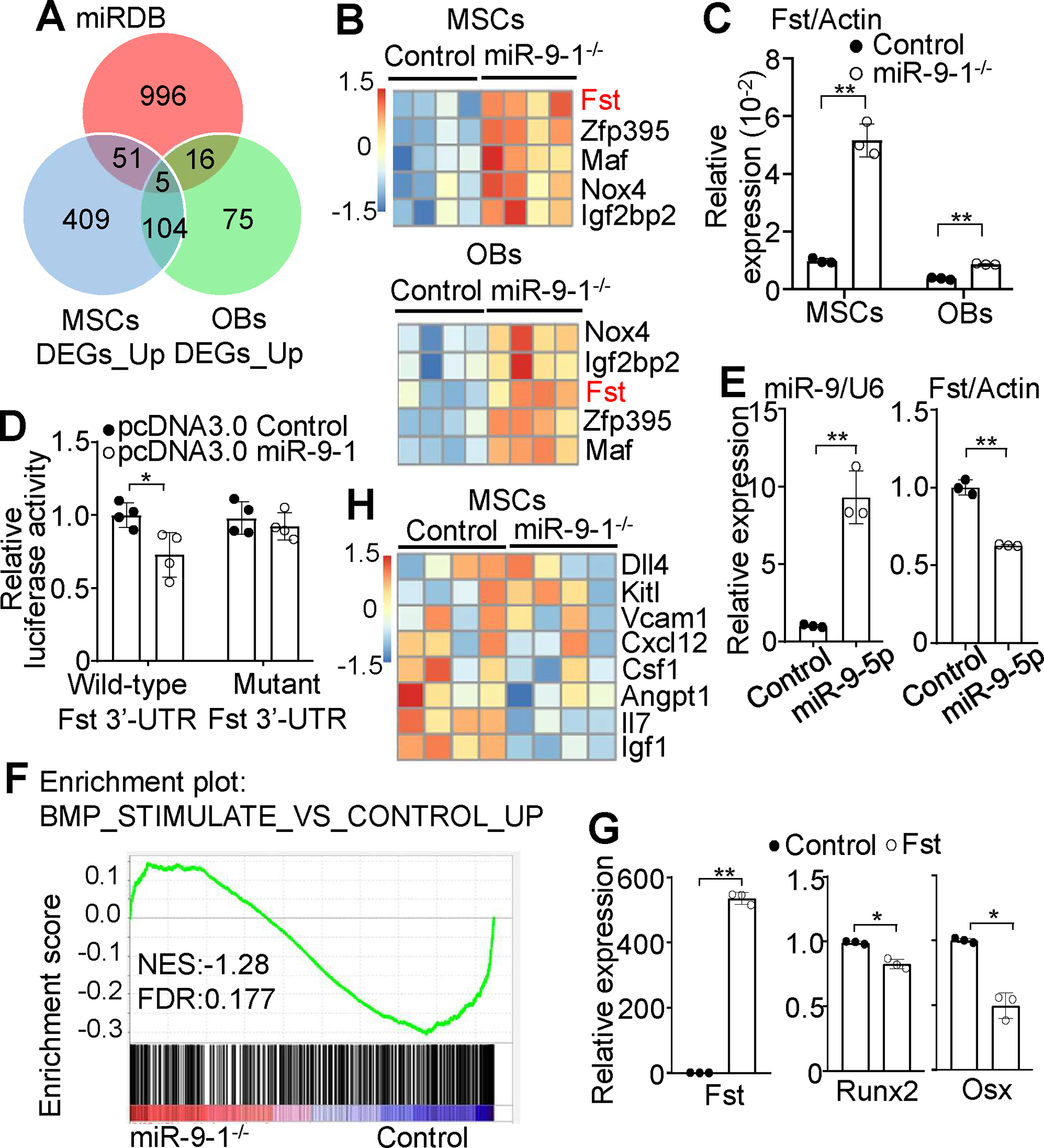
A Venn diagram showing the overlap among genes with miR-9-binding sites predicted by miRDB in the 3’-UTR and genes upregulated in miR-9-1-deficient relative to control MSCs and OBs. B Differential gene expression analysis of RNA-seq data showing increased levels of Fst mRNA in miR-9-1−/− relative to control MSCs and OBs. C qRT–PCR confirming increased levels of Fst mRNA in miR-9-1−/− relative to control MSCs and OBs. The relative mRNA levels of Fst in miR-9-1−/− and control MSCs and OBs were normalized to Actin. D Suppression of the Renilla luciferase activity of the reporter with wild-type 3’-UTR of Fst but not mutant by miR-9-5p. The level of luciferase activity in 293T cells transfected with the luciferase reporter vector containing the wild-type or mutant 3′-UTR of Fst with miR-9-5p was quantified by the luciferase assay as a fraction of the corresponding control cells transfected with the luciferase reporter vector containing the wild-type 3′-UTR of Fst with empty vector, which was set as 1. E Reduced level of endogenous Fst mRNA in primary MSCs by miR-9-5p overexpression. The expression levels of miR-9-5p and Fst in MSCs transfected with empty pcDNA3.0 vector (control) or miR-9-5p expression pcDNA3.0 (miR-9-5p) was determined by qRT-PCR with U6 small nuclear RNA or Actin as the internal control, respectively. F Downregulation of the genes related to BMP stimulation in miR-9-1-deficient MSCs. Comparative GSEA of the BMP stimulation-related gene signatures in sorted primary miR-9-1-deficient and control CD51+Sca1+ MSCs. G Effect of Fst overexpression on Runx2 and Osx expression. The relative expression levels of Fst, Runx2 and Osx were compared between wild-type MSCs overexpressing Fst (Fst) and wild-type MSCs transfected with empty vector (control) by qRT-PCR analysis. H Differential gene expression analysis of RNA-seq data showing decreased levels of IL-7 and IGF-1 mRNA in miR-9-1−/− relative to control MSCs. Data shown are representative of or obtained from 4 (A, B, F, H) and 3 (E, G) miR-9-1−/− and control mice, and representative of or obtained from 4 independent experiments (C, D). Each dot represents one biological replicate or an individual mouse. Mean ± SD is shown. *, p < 0.05; **, p < 0.01.
To validate that miR-9-5p directly targeted Fst mRNA, we performed a luciferase reporter assay. The wild-type 3’-UTR of mouse Fst mRNA was cloned downstream of Renilla luciferase gene in psi-CHECK2 luciferase reporter vector whereas the mutant 3’-UTR of mouse Fst mRNA with mutated the seed-matched sequence, the predicted binding site of miR-9-1, was cloned downstream of Renilla luciferase gene in psi-CHECK2 luciferase reporter vector as negative control (Supplementary Fig. S10A). The luciferase activity in 293T cells transfected with the luciferase reporter vector containing the wild-type 3′-UTR of Fst was repressed by co-transfection with miR-9-5p (Fig. 7D). In contrast, co-transfection with miR-9-5p failed to repress the luciferase activity in 293T cells transfected with the luciferase reporter vector containing the mutant 3′-UTR of Fst (Fig. 7D), showing that miR-9-5p directly targets Fst mRNA. In addition, overexpression of miR-9 reduced the levels of Fst mRNA and protein in primary BM-derived MSCs (Fig. 7E and Supplementary Fig. S10B), demonstrating that miR-9 negatively regulates the expression level of endogenous Fst. It’s noteworthy that Fst showed relatively high expression in CD51+Sca1+ MSCs, but relatively low expression in CD51+Sca1− OBs, with no expression in HSCs (Supplementary Fig. S10C). These findings suggest that miR-9 functions in MSCs by inhibiting the production of Fst.
Fst has been shown to interact with BMP, negatively regulates BMP/Smad signaling, and inhibits BMP activity 67. Consistently, GSEA found that the genes related to BMP stimulation were reduced in freshly sorted primary miR-9-1-deficient relative to wild-type CD51+Sca1+ MSCs (Fig. 7F), demonstrating that the BMP/Smad signaling is reduced by miR-9-1 deficiency in vivo. In addition, the level of p-smad1/5/8, an indicator of BMP signaling, was reduced in primary miR-9-1-deficient relative to control BM-derived MSCs (Supplementary Fig. S10D), biochemically confirming that the BMP/Smad signaling is reduced by miR-9-1 deficiency. Moreover, overexpression of Fst in primary BM-derived MSCs decreased the expression levels of the master transcription factor for osteogenesis, Runx2, and the osteoblast-specific transcription factor, Osx (Fig. 7G), confirming Fst as a negative regulator of osteoblastic differentiation. MSCs are the main source of SCF, CXCL12, IL-7 and IGF-1 that sustain HSCs and lymphopoiesis 19, 20, 55, 68. BMP induces the expression of IGF-1 69. Differential gene expression analysis of hematopoietic and lymphopoietic growth factors/cytokines produced by MSCs found that the IGF-1 and IL-7 transcript level was significantly decreased in mutant relative to wild-type CD51+Sca1+ MSCs (Fig. 7H). The specific reduction of the IGF-1 and IL-7 transcripts and preserved expression of both CXCL12 and CSF1 transcripts in miR-9-1−/− MSCs was confirmed by qRT-PCR (Supplementary Fig. S10E).
It has been reported that LepR+ MSCs with osteogenic and adipogenic properties secrete IL-7 and support B cell development, whereas OBs do not contribute to this process 19, 55. In addition, LepR+ peri-sinusoidal stromal cells are the primary source of IL-7 for early B cell development 68. Therefore, we examined whether miR-9-1 deficiency affects the expression of IL-7 in miR-9-1-deficient LepR+ cells. Lin−CD45−CD31−CD51+LepR+ cells were isolated from wild-type and miR-9-1-deficient mice with normal or retarded growth, and qRT-PCR analysis showed that the expression of IL-7 was significantly reduced in LepR+ cells isolated from miR-9-1-deficient mice with retarded growth, but not in those with normal growth, when compared to wild-type mice (Supplementary Fig. S10F and S11). Moreover, the expression level of CXCL12, which is known to be highly expressed by most IL-7+ cells in BM 19, was also significantly reduced in LepR+ cells from miR-9-1-deficient mice with retarded growth, but not in those with normal growth (Supplementary Fig. S10F and S11). However, the levels of Runx2, Osx and Fst were comparable between miR-9-1-deficient mice with retarded growth and wild-type control mice (Supplementary Fig. S10F and S11), indicating that the effect of miR-9-1 deficiency on Runx2, Osx and Fst could not be detected in LepR+ cells, which are a highly heterogenous population 70. In addition, the levels of IGF-1 and Csf1 were similar among miR-9-1-deficient mice with retarded and normal growth and wild-type control mice (Supplementary Fig. S10F and S11). Therefore, our findings suggest that miR-9-1 deficiency impairs the expression of IL-7 and CXCL12 in LepR+ cells, but its impact on Runx2, Osx and Fst cannot be detected in these cells.
Taken together, these findings demonstrate that miR-9-1 directly downregulates Fst, a negative regulator of BMP activity, and miR-9-1 deficiency augments Fst and thus reduces BMP signaling and impairs osteoblastic differentiation, including IL-7, possibly IGF-1 and CXCL-12 production, in CD51+Sca1+ MSCs (Supplementary Fig. S12). Targeting the Fst/BMP/Smad signaling axis is at least one mechanism by which miR-9-1 controls osteoblastic regulation of lymphopoiesis (Supplementary Fig. S12).
DISCUSSION
The miR-9 family of miRNAs consists of three members in both mice and humans, namely miR-9-1, miR-9-2 and miR-9-3. Each member is encoded by a unique gene located on different chromosomes 33, 34. It is worth noting that all three miR-9s are processed into the same mature miR-9 sequence 33, indicating a potentially redundant role of the three members. Indeed, miR-9-2 or miR-9-3 single-deficient mice are viable whereas miR-9-2/3 double-deficient mice are postnatally lethal 33. While miR-9 has been shown to be enriched in brain and regulates neurogenesis 35, we identified distinct expression patterns of the three members in the hematopoietic system. Specifically, miR-9-1 and miR-9-2 but not miR-9-3 were expressed in hematopoietic cells. Both miR-9-1 and miR-9-2 were expressed in HSCs, MPPs and CLPs but at very low levels in CMPs, MEPs, GMPs, and more mature hematopoietic subpopulations. Furthermore, we found that miR-9-1 and miR-9-2 were the main isoforms highly expressed in MSCs, LepR+ MSCs and osteoblasts.
Importantly, miR-9-1 deficiency significantly reduced the overall level of miR-9 in mutant mice, and this reduction was sufficient to cause a global growth defect and impaired lymphopoiesis, underscoring the importance of miR-9 gene dosage. However, miR-9-1-deficient mice displayed growth defect and impaired B-cell lymphopoiesis and osteoblastic differentiation with incomplete penetrance. Notably, mutant mice with normal growth exhibited normal lymphopoiesis and osteoblastic differentiation. Additionally, RNA-seq analysis revealed that the gene expression profile in LepR+ MSCs from miR-9-1-deficient mice with normal growth resembled that of MSCs from wild-type mice. We confirmed a 16bp deletion in the mature sequence of miR-9-1 in miR-9-1-deficient mice with normal growth through genomic DNA sequencing. The levels of total mature miR-9 were reduced in BM cells, splenocytes, CD51+Sca1+ MSCs, and CD51+Sca1− OBs of miR-9-1-deficient mice with normal growth to a similar extent as in miR-9-1-deficient mice with retarded growth. Importantly, we generated a second colony of miR-9-1-deficient mice and found that mutant mice from the second colony exhibited the same phenotypes as those from the first colony. These data support the idea that the incomplete penetrance of the observed defects in miR-9-1-deficient mice is not due to secondary consequences of developmental/growth defects or other genetic defects. It is worth noting that the genetic background can often impact the phenotypic outcome of a gene mutation. In our study, we generated and maintained miR-9-1-deficient mice on a C57BL/6 background. Although the underlying molecular mechanism is not yet clear, it is possible that the combination of the subtle genetic background variations and the dosage sensitivity of miR-9-1 gene contributes to the incomplete penetrance of the miR-9-1 deletion.
Our findings indicate that the impaired hematopoiesis, especially lymphopoiesis, resulting from miR-9-1 deficiency was not intrinsic to hematopoietic cells, but rather a consequence of a BM niche defect. The primary isoforms expressed in both MSCs and OBs were miR-9-1 and miR-9-2, while Fst was highly expressed in MSCs and OBs. In contrast, Fst was barely detectable in HSCs. Our results suggest that miR-9 is produced in MSCs, where it inhibits Fst production. Notably, BM transplantation restored the populations of miR-9-1-deficient lymphoid progenitors, as well as B and T cell development, indicating that miR-9 is produced and functions within the BM niche MSCs. Additionally, we demonstrated that the absence of miR-9-1 inhibited MSC function. The osteoblastic (endosteal) and perivascular niches have distinct subsets of MSCs and MSC-progeny that differentially regulate the maintenance and differentiation of HSCs and subsequent hematopoiesis 1. The osteoblastic niche consists of MSCs and MSC-progeny, including osteoprogenitor cells, OBs, mature OBs, osteocytes and osteoclasts 71. We observed a decrease in OBs but no MSCs in miR-9-1-deficient mice with growth retardation. Through RNA-seq and qRT-PCR analyses, we identified a reduction in the expression of the master transcription factors for osteoblastic differentiation, Runx2 and Osx, along with genes associated with collagen formation, extracellular matrix organization, and cell adhesion in miR-9-1-deficient MSCs. These findings clearly demonstrate the critical role of miR-9-1 in the early stages of osteoblastic differentiation, commencing from the MSC stage. Notably, within the osteoblastic niche, MSCs, osteo-progenitors, and OBs direct HSC differentiation, and the elimination of OBs results in the loss of HSC quiescence, suppresses lymphopoiesis, and facilitates myelopoiesis 7, 72. OBs specifically promote B lymphopoiesis by supporting lymphoid commitment of HSCs and subsequent B cell differentiation 8, 65, 73. Therefore, blockade of lymphopoiesis but not erythropoiesis, thrombopoiesis or myelopoiesis by miR-9-1 deficiency is likely due to the loss of MSC function and OB generation, and the resulting impairment of the osteoblastic niche in the growth-retarded mutant mice. The perivascular niche, which consists of endothelial cells, Schwann cells and MSCs, sustains HSC maintenance 1, 74. Peri-arteriolar MSCs express CXCL12, SCF, Nestin (Nes), LepR, NG2 and PRX1, and depletion of MSCs results in a reduction of HSCs 17, 74. The reduction of LT-HSCs, ST-HSCs and MPPs in growth-retarded miR-9-1-deficient mice is likely due to the dysfunction of MSCs within the perivascular niche and the resultant early hematopoietic phenotype.
MiRNAs typically guide AGO proteins to the complementary sequence of the miR-5p (the guide strand) seed region at the 3’-UTR of their direct target mRNAs, resulting in the subsequent degradation of the target transcripts 22, 23. We identified five miR-9-1direct target genes whose expression levels were increased in both miR-9-1-deficient MSCs and OBs and that possessed the miR-9-5p target sites at their 3’-UTRs. One of the 5 genes is Fst, which strongly and directly interacts with multiple BMPs, negatively regulates BMP/Smad signaling and inhibits BMP activities 67. BMPs are essential for osteoblast differentiation and bone formation 75, 76. It is worth noting that the Fst/BMP signaling pathway is just one of several targets through which miR-9 may mediate lymphopoiesis. There are likely other target pathways involved in these cellular processes. In addition to Fst, there are other antagonists of BMPs, such as noggin (Nog) and gremlin (Grem1) 77. Previous studies have demonstrated that Nog and Fst have redundant roles in inhibiting BMP function. Accordingly, the deficiency of Fst alone does not appear to significantly impact BMP-mediated skeleton development 78. It is probable that the regulation of these processes by miR-9 involves multiple target pathways, where Fst plays an important role but is not the sole determinant.
Our biochemical analysis and high-throughput RNA-seq data clearly demonstrated that BMP-regulated Smad activation and gene expression were reduced by miR-9-1 deficiency, supporting the notion that the up-regulation of Fst suppresses BMP activities and subsequently reduces the production of growth factors and cytokines, such as IL-7 and IGF-1 by CD51+Sca1+ MSCs and IL-7 and CXCL12 by LepR+ cells. This reduction of growth factors and cytokines leads to decreased numbers of CLPs and possible impaired functions of mutant CLPs. The dramatic decrease in B cell progenitors and total thymocyte numbers, including both DN and DPs, is consistent with the reduction of CLP numbers and impaired functions. The shift in the ratio between DNs and DPs may be due to the impaired function of mutant CLPs. The impaired lymphopoiesis in miR-9-1-deficient mice can be attributed, at least in part, to the reduction of IL-7, IGF-1 and CXCL12 in BM niche cells through the miR-9/Fst/BMP signaling axis. However, the exact mechanism underlying the change in the ratio between DNs and DPs is yet to be determined. Further, we found that the overexpression of Fst decreased the expression levels of the master transcription factor for osteogenesis, Runx2, and the osteoblast-specific transcription factor, Osx, in MSCs and OBs. These findings demonstrate that targeting Fst and thus controlling BMP activity is at least one important molecular mechanism by which miR-9-1 regulates MSC function and osteoblast differentiation.
OBs have been shown to produce various growth factors and cytokines, such as SCF, CXCL12, and IL-7 8, 65, 73. CD51+Sca1− OBs are known to harbor multiple heterogeneous BM niche subtypes that can be identified by the expression of NG2, CD200, and LepR 62–65. Meanwhile, MSCs are a critical source of SCF, CXCL12, IL-7, and IGF-1 20, 74, which are crucial for supporting HSCs and lymphopoiesis 19, 20, 55, 68. Deletion of CXCL12 in osteoblasts has been found to decrease CLPs and early lymphoid progenitors 65. Our differential gene expression analysis found that these growth factors/cytokines were mainly produced by CD51+Sca1+ MSCs, and that the levels of IL-7 and IGF-1 were evidently decreased in miR-9-deficient CD51+Sca1+ MSCs. IL-7 is essential for both early T and B cell development, and its deficiency severely impairs lymphopoiesis 79. Recent studies have also shown that IGF-1 is an important MSC-derived growth factor that prevents HSC from aging and supports B lymphopoiesis 20, 80. Notably, BMP is able to induce the expression of IGF-1 in osteoblast lineage cells 69. Our results demonstrated that the levels of IL-7 and CXCL12 were significantly reduced in miR-9-1-deficient LepR+ cells compared to wild-type mice. However, the impact of miR-9-1 deficiency on the levels of Runx2, Osx, Fst and IGF-1 could not be detected in LepR+ cells, which are known to be a highly heterogenous population 70. Overall, these results demonstrate that miR-9-1 directly downregulates Fst, thereby enhancing BMP activity to partially promote the production of growth factors and cytokines, such as IL-7 and IGF-1, in CD51+Sca1+ MSCs. Consequently, the miR-9-1 deficiency impairs lymphopoiesis partly due to the reduction of IL-7 and IGF-1 in BM niche MSCs through the miR-9/Fst/BMP signaling axis. The administration of IL-7, IGF-1 or CXCL12 holds significant promise as a solution to rescue impaired lymphopoiesis. This avenue of research is highly intriguing and warrants further exploration. By delving into interventions involving the administration of IL-7, IGF-1 or CXCL12, we have the opportunity to gain novel insights and make substantial contributions to future research endeavors. The findings from our studies have provided a solid foundation for future investigations in this field.
Supplementary Material
ACKNOWLEDGMENTS
The authors acknowledge Robert Burns and Shikan Zheng for their assistance with the analysis of the high-throughput sequence data. This work is supported in part by NIH grants AI079087 (D.W.), HL130724 (D.W.), and HL148120 (R.W.). Y.Z. (Yongwei Zheng) is supported by The Elizabeth Elser Doolit - the Postdoctoral Fellowship. Y.Z. (Yongguang Zhang) receives stipend from Fujian Normal University, China.
Footnotes
COMPETING INTERESTS
The authors have no financial conflicts of interest.
ADDITIONAL INFORMATION
All data needed to evaluate the conclusions in the article are present in the article or the Supplementary Materials.
REFERENCES
- 1.Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature 2014. Jan 16; 505(7483): 327–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yin T, Li L. The stem cell niches in bone. The Journal of clinical investigation 2006. May; 116(5): 1195–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell 2008. Feb 22; 132(4): 631–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kiel MJ, Morrison SJ. Maintaining hematopoietic stem cells in the vascular niche. Immunity 2006. Dec; 25(6): 862–864. [DOI] [PubMed] [Google Scholar]
- 5.Wang LD, Wagers AJ. Dynamic niches in the origination and differentiation of haematopoietic stem cells. Nature reviews Molecular cell biology 2011. Sep 2; 12(10): 643–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perry JM, Li L. Disrupting the stem cell niche: good seeds in bad soil. Cell 2007. Jun 15; 129(6): 1045–1047. [DOI] [PubMed] [Google Scholar]
- 7.Visnjic D, Kalajzic Z, Rowe DW, Katavic V, Lorenzo J, Aguila HL. Hematopoiesis is severely altered in mice with an induced osteoblast deficiency. Blood 2004. May 1; 103(9): 3258–3264. [DOI] [PubMed] [Google Scholar]
- 8.Zhu J, Garrett R, Jung Y, Zhang Y, Kim N, Wang J, et al. Osteoblasts support B-lymphocyte commitment and differentiation from hematopoietic stem cells. Blood 2007. May 1; 109(9): 3706–3712. [DOI] [PubMed] [Google Scholar]
- 9.Arai F, Hirao A, Ohmura M, Sato H, Matsuoka S, Takubo K, et al. Tie2/angiopoietin-1 signaling regulates hematopoietic stem cell quiescence in the bone marrow niche. Cell 2004. Jul 23; 118(2): 149–161. [DOI] [PubMed] [Google Scholar]
- 10.Taichman RS. Blood and bone: two tissues whose fates are intertwined to create the hematopoietic stem-cell niche. Blood 2005. Apr 1; 105(7): 2631–2639. [DOI] [PubMed] [Google Scholar]
- 11.Stier S, Ko Y, Forkert R, Lutz C, Neuhaus T, Grunewald E, et al. Osteopontin is a hematopoietic stem cell niche component that negatively regulates stem cell pool size. The Journal of experimental medicine 2005. Jun 6; 201(11): 1781–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoshihara H, Arai F, Hosokawa K, Hagiwara T, Takubo K, Nakamura Y, et al. Thrombopoietin/MPL signaling regulates hematopoietic stem cell quiescence and interaction with the osteoblastic niche. Cell stem cell 2007. Dec 13; 1(6): 685–697. [DOI] [PubMed] [Google Scholar]
- 13.Nilsson SK, Johnston HM, Whitty GA, Williams B, Webb RJ, Denhardt DT, et al. Osteopontin, a key component of the hematopoietic stem cell niche and regulator of primitive hematopoietic progenitor cells. Blood 2005. Aug 15; 106(4): 1232–1239. [DOI] [PubMed] [Google Scholar]
- 14.Fleming HE, Janzen V, Lo Celso C, Guo J, Leahy KM, Kronenberg HM, et al. Wnt signaling in the niche enforces hematopoietic stem cell quiescence and is necessary to preserve self-renewal in vivo. Cell stem cell 2008. Mar 6; 2(3): 274–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Calvi LM, Adams GB, Weibrecht KW, Weber JM, Olson DP, Knight MC, et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 2003. Oct 23; 425(6960): 841–846. [DOI] [PubMed] [Google Scholar]
- 16.Jung Y, Song J, Shiozawa Y, Wang J, Wang Z, Williams B, et al. Hematopoietic stem cells regulate mesenchymal stromal cell induction into osteoblasts thereby participating in the formation of the stem cell niche. Stem Cells 2008. Aug; 26(8): 2042–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mendez-Ferrer S, Michurina TV, Ferraro F, Mazloom AR, Macarthur BD, Lira SA, et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 2010. Aug 12; 466(7308): 829–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Greenbaum A, Hsu YM, Day RB, Schuettpelz LG, Christopher MJ, Borgerding JN, et al. CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature 2013. Mar 14; 495(7440): 227–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cordeiro Gomes A, Hara T, Lim VY, Herndler-Brandstetter D, Nevius E, Sugiyama T, et al. Hematopoietic Stem Cell Niches Produce Lineage-Instructive Signals to Control Multipotent Progenitor Differentiation. Immunity 2016. Dec 20; 45(6): 1219–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu VW, Lymperi S, Oki T, Jones A, Swiatek P, Vasic R, et al. Distinctive Mesenchymal-Parenchymal Cell Pairings Govern B Cell Differentiation in the Bone Marrow. Stem Cell Reports 2016. Aug 9; 7(2): 220–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Senyuk V, Zhang Y, Liu Y, Ming M, Premanand K, Zhou L, et al. Critical role of miR-9 in myelopoiesis and EVI1-induced leukemogenesis. Proceedings of the National Academy of Sciences of the United States of America 2013. Apr 2; 110(14): 5594–5599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gebert LFR, MacRae IJ. Regulation of microRNA function in animals. Nature reviews Molecular cell biology 2019. Jan; 20(1): 21–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jonas S, Izaurralde E. Towards a molecular understanding of microRNA-mediated gene silencing. Nature reviews Genetics 2015. Jul; 16(7): 421–433. [DOI] [PubMed] [Google Scholar]
- 24.Bernstein E, Caudy AA, Hammond SM, Hannon GJ. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 2001. Jan 18; 409(6818): 363–366. [DOI] [PubMed] [Google Scholar]
- 25.Bartel DP. Metazoan MicroRNAs. Cell 2018. Mar 22; 173(1): 20–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guo S, Lu J, Schlanger R, Zhang H, Wang JY, Fox MC, et al. MicroRNA miR-125a controls hematopoietic stem cell number. Proceedings of the National Academy of Sciences of the United States of America 2010. Aug 10; 107(32): 14229–14234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li QJ, Chau J, Ebert PJ, Sylvester G, Min H, Liu G, et al. miR-181a is an intrinsic modulator of T cell sensitivity and selection. Cell 2007. Apr 6; 129(1): 147–161. [DOI] [PubMed] [Google Scholar]
- 28.Xiao C, Calado DP, Galler G, Thai TH, Patterson HC, Wang J, et al. MiR-150 controls B cell differentiation by targeting the transcription factor c-Myb. Cell 2007. Oct 5; 131(1): 146–159. [DOI] [PubMed] [Google Scholar]
- 29.Zhou B, Wang S, Mayr C, Bartel DP, Lodish HF. miR-150, a microRNA expressed in mature B and T cells, blocks early B cell development when expressed prematurely. Proceedings of the National Academy of Sciences of the United States of America 2007. Apr 24; 104(17): 7080–7085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu J, Guo S, Ebert BL, Zhang H, Peng X, Bosco J, et al. MicroRNA-mediated control of cell fate in megakaryocyte-erythrocyte progenitors. Dev Cell 2008. Jun; 14(6): 843–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fazi F, Rosa A, Fatica A, Gelmetti V, De Marchis ML, Nervi C, et al. A minicircuitry comprised of microRNA-223 and transcription factors NFI-A and C/EBPalpha regulates human granulopoiesis. Cell 2005. Dec 2; 123(5): 819–831. [DOI] [PubMed] [Google Scholar]
- 32.Johnnidis JB, Harris MH, Wheeler RT, Stehling-Sun S, Lam MH, Kirak O, et al. Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature 2008. Feb 28; 451(7182): 1125–1129. [DOI] [PubMed] [Google Scholar]
- 33.Shibata M, Nakao H, Kiyonari H, Abe T, Aizawa S. MicroRNA-9 regulates neurogenesis in mouse telencephalon by targeting multiple transcription factors. J Neurosci 2011. Mar 2; 31(9): 3407–3422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yuva-Aydemir Y, Simkin A, Gascon E, Gao FB. MicroRNA-9: functional evolution of a conserved small regulatory RNA. RNA Biol 2011. Jul-Aug; 8(4): 557–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krichevsky AM, Sonntag KC, Isacson O, Kosik KS. Specific microRNAs modulate embryonic stem cell-derived neurogenesis. Stem Cells 2006. Apr; 24(4): 857–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 2013. May 9; 153(4): 910–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu M, Chen Y, Zeng H, Zheng Y, Fu G, Zhu W, et al. PLCgamma-dependent mTOR signalling controls IL-7-mediated early B cell development. Nature communications 2017. Nov 13; 8(1): 1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Winkler IG, Sims NA, Pettit AR, Barbier V, Nowlan B, Helwani F, et al. Bone marrow macrophages maintain hematopoietic stem cell (HSC) niches and their depletion mobilizes HSCs. Blood 2010. Dec 2; 116(23): 4815–4828. [DOI] [PubMed] [Google Scholar]
- 39.Chen Y, Wang X, Di L, Fu G, Chen Y, Bai L, et al. Phospholipase Cgamma2 mediates RANKL-stimulated lymph node organogenesis and osteoclastogenesis. The Journal of biological chemistry 2008. Oct 24; 283(43): 29593–29601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Soleimani M, Nadri S. A protocol for isolation and culture of mesenchymal stem cells from mouse bone marrow. Nature protocols 2009; 4(1): 102–106. [DOI] [PubMed] [Google Scholar]
- 41.Liu Z, Yao X, Yan G, Xu Y, Yan J, Zou W, et al. Mediator MED23 cooperates with RUNX2 to drive osteoblast differentiation and bone development. Nature communications 2016. Apr 1; 7: 11149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tian J, Rui K, Tang X, Ma J, Wang Y, Tian X, et al. MicroRNA-9 Regulates the Differentiation and Function of Myeloid-Derived Suppressor Cells via Targeting Runx1. J Immunol 2015. Aug 1; 195(3): 1301–1311. [DOI] [PubMed] [Google Scholar]
- 43.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology 2014; 15(12): 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015. Apr 20; 43(7): e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 2012. May; 16(5): 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 2000. Jan 1; 28(1): 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fabregat A, Jupe S, Matthews L, Sidiropoulos K, Gillespie M, Garapati P, et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res 2018. Jan 4; 46(D1): D649–D655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society Series B (Methodological) 1995; 57(1): 289–300. [Google Scholar]
- 49.Wei Q, Holle A, Li J, Posa F, Biagioni F, Croci O, et al. BMP-2 Signaling and Mechanotransduction Synergize to Drive Osteogenic Differentiation via YAP/TAZ. Adv Sci (Weinh) 2020. Aug; 7(15): 1902931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cao X Targeting osteoclast-osteoblast communication. Nature medicine 2011. Nov 7; 17(11): 1344–1346. [DOI] [PubMed] [Google Scholar]
- 51.Vasikaran S, Eastell R, Bruyere O, Foldes AJ, Garnero P, Griesmacher A, et al. Markers of bone turnover for the prediction of fracture risk and monitoring of osteoporosis treatment: a need for international reference standards. Osteoporos Int 2011. Feb; 22(2): 391–420. [DOI] [PubMed] [Google Scholar]
- 52.Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell 1997. May 30; 89(5): 755–764. [DOI] [PubMed] [Google Scholar]
- 53.Nakashima K, Zhou X, Kunkel G, Zhang Z, Deng JM, Behringer RR, et al. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell 2002. Jan 11; 108(1): 17–29. [DOI] [PubMed] [Google Scholar]
- 54.Owen TA, Aronow M, Shalhoub V, Barone LM, Wilming L, Tassinari MS, et al. Progressive development of the rat osteoblast phenotype in vitro: reciprocal relationships in expression of genes associated with osteoblast proliferation and differentiation during formation of the bone extracellular matrix. J Cell Physiol 1990. Jun; 143(3): 420–430. [DOI] [PubMed] [Google Scholar]
- 55.Zhou BO, Yue R, Murphy MM, Peyer JG, Morrison SJ. Leptin-receptor-expressing mesenchymal stromal cells represent the main source of bone formed by adult bone marrow. Cell stem cell 2014. Aug 7; 15(2): 154–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nishikawa K, Nakashima T, Takeda S, Isogai M, Hamada M, Kimura A, et al. Maf promotes osteoblast differentiation in mice by mediating the age-related switch in mesenchymal cell differentiation. The Journal of clinical investigation 2010. Oct; 120(10): 3455–3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Narayanan K, Ramachandran A, Hao J, He G, Park KW, Cho M, et al. Dual functional roles of dentin matrix protein 1. Implications in biomineralization and gene transcription by activation of intracellular Ca2+ store. The Journal of biological chemistry 2003. May 9; 278(19): 17500–17508. [DOI] [PubMed] [Google Scholar]
- 58.Rosset EM, Bradshaw AD. SPARC/osteonectin in mineralized tissue. Matrix Biol 2016. May-Jul; 52–54: 78–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Igwe JC, Gao Q, Kizivat T, Kao WW, Kalajzic I. Keratocan is expressed by osteoblasts and can modulate osteogenic differentiation. Connect Tissue Res 2011. Oct; 52(5): 401–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schepers K, Pietras EM, Reynaud D, Flach J, Binnewies M, Garg T, et al. Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell stem cell 2013. Sep 5; 13(3): 285–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Krane SM, Inada M. Matrix metalloproteinases and bone. Bone 2008. Jul; 43(1): 7–18. [DOI] [PubMed] [Google Scholar]
- 62.Green AC, Tjin G, Lee SC, Chalk AM, Straszkowski L, Kwang D, et al. The characterization of distinct populations of murine skeletal cells that have different roles in B lymphopoiesis. Blood 2021. Jul 29; 138(4): 304–317. [DOI] [PubMed] [Google Scholar]
- 63.Kunisaki Y, Bruns I, Scheiermann C, Ahmed J, Pinho S, Zhang D, et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature 2013. Oct 31; 502(7473): 637–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Menon S, Salhotra A, Shailendra S, Tevlin R, Ransom RC, Januszyk M, et al. Skeletal stem and progenitor cells maintain cranial suture patency and prevent craniosynostosis. Nature communications 2021. Jul 30; 12(1): 4640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ding L, Morrison SJ. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature 2013. Mar 14; 495(7440): 231–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen Y, Wang X. miRDB: an online database for prediction of functional microRNA targets. Nucleic Acids Res 2020. Jan 8; 48(D1): D127–D131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Iemura S, Yamamoto TS, Takagi C, Uchiyama H, Natsume T, Shimasaki S, et al. Direct binding of follistatin to a complex of bone-morphogenetic protein and its receptor inhibits ventral and epidermal cell fates in early Xenopus embryo. Proceedings of the National Academy of Sciences of the United States of America 1998. Aug 4; 95(16): 9337–9342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Balzano M, De Grandis M, Vu Manh TP, Chasson L, Bardin F, Farina A, et al. Nidogen-1 Contributes to the Interaction Network Involved in Pro-B Cell Retention in the Peri-sinusoidal Hematopoietic Stem Cell Niche. Cell reports 2019. Mar 19; 26(12): 3257–3271 e3258. [DOI] [PubMed] [Google Scholar]
- 69.Yeh LC, Adamo ML, Kitten AM, Olson MS, Lee JC. Osteogenic protein-1-mediated insulin-like growth factor gene expression in primary cultures of rat osteoblastic cells. Endocrinology 1996. May; 137(5): 1921–1931. [DOI] [PubMed] [Google Scholar]
- 70.Mo C, Guo J, Qin J, Zhang X, Sun Y, Wei H, et al. Single-cell transcriptomics of LepR-positive skeletal cells reveals heterogeneous stress-dependent stem and progenitor pools. EMBO J 2022. Feb 15; 41(4): e108415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Le PM, Andreeff M, Battula VL. Osteogenic niche in the regulation of normal hematopoiesis and leukemogenesis. Haematologica 2018. Dec; 103(12): 1945–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bowers M, Zhang B, Ho Y, Agarwal P, Chen CC, Bhatia R. Osteoblast ablation reduces normal long-term hematopoietic stem cell self-renewal but accelerates leukemia development. Blood 2015. Apr 23; 125(17): 2678–2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wu JY, Purton LE, Rodda SJ, Chen M, Weinstein LS, McMahon AP, et al. Osteoblastic regulation of B lymphopoiesis is mediated by Gs{alpha}-dependent signaling pathways. Proceedings of the National Academy of Sciences of the United States of America 2008. Nov 4; 105(44): 16976–16981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wei Q, Frenette PS. Niches for Hematopoietic Stem Cells and Their Progeny. Immunity 2018. Apr 17; 48(4): 632–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yamaguchi A, Katagiri T, Ikeda T, Wozney JM, Rosen V, Wang EA, et al. Recombinant human bone morphogenetic protein-2 stimulates osteoblastic maturation and inhibits myogenic differentiation in vitro. J Cell Biol 1991. May; 113(3): 681–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sampath TK, Maliakal JC, Hauschka PV, Jones WK, Sasak H, Tucker RF, et al. Recombinant human osteogenic protein-1 (hOP-1) induces new bone formation in vivo with a specific activity comparable with natural bovine osteogenic protein and stimulates osteoblast proliferation and differentiation in vitro. The Journal of biological chemistry 1992. Oct 5; 267(28): 20352–20362. [PubMed] [Google Scholar]
- 77.Stafford DA, Brunet LJ, Khokha MK, Economides AN, Harland RM. Cooperative activity of noggin and gremlin 1 in axial skeleton development. Development 2011. Mar; 138(5): 1005–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Stafford DA, Monica SD, Harland RM. Follistatin interacts with Noggin in the development of the axial skeleton. Mech Dev 2014. Feb; 131: 78–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.von Freeden-Jeffry U, Vieira P, Lucian LA, McNeil T, Burdach SE, Murray R. Lymphopenia in interleukin (IL)-7 gene-deleted mice identifies IL-7 as a nonredundant cytokine. The Journal of experimental medicine 1995. Apr 1; 181(4): 1519–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Young K, Eudy E, Bell R, Loberg MA, Stearns T, Sharma D, et al. Decline in IGF1 in the bone marrow microenvironment initiates hematopoietic stem cell aging. Cell stem cell 2021. Aug 5; 28(8): 1473–1482 e1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.