

The late Michael Walker had just installed a triple quadrupole mass spectrometer (HPLC/MS/MS) in 2001 when I joined his lab (HB). He had successfully measured Anandamide using a single quad (LC/MS; [1]); however, the amount of material needed to get a reading and the reliability of the accuracy was limiting, so he focused his resources towards obtaining this cutting-edge instrument. The triple quad technology generates a more exacting mass fingerprint of the molecule by measuring the parent mass, dissociating the molecule, and then measuring the fragments of that collision. In 2017 we published a review [2] outlining some of the different types of mass spectrometric (MS) techniques used for endocannabinoid analysis (e.g. isotope dilution gas chromatography, electrospray ionization, tandem mass spectrometry), so an overview of these will not be provided here. Suffice to say that each of these mass spectrometric techniques has been used to quantify endocannabinoids (eCBs) and related lipids, so the process of building MS methods is an issue of manufacture and instrument type and will be regarded, only in the context of this chapter, as equivalent. Even with the level of molecular fingerprinting accuracy of an instrument like the triple quadrupole mass spectrometer, if one is not getting the most optimized sample to the detector in a way that this improved technology can be of use, then advancements can be stymied. Here, our focus is on review and discussion of sample preparation methodologies used to isolate the eCB, Anandamide (N-arachidonoyl ethanolamine; AEA) and its close congeners (N-acyl ethanolamines; NAEs; Fig. 1) and its structural congeners (a.k.a. lipo amino acids, lipoamines, N-acyl amides; [3–12]; Fig. 2) in biological fluids. Most of our focus will be on the analysis of these lipids in plasma/serum, but we will also discuss how the same techniques can be used for analysis of saliva and breast milk.
Figure 1.
Examples of N-acyl ethanolamines (NAEs).
Figure 2.
Examples of Lipoamines (a.k.a, lipo amino acids, N-acyl amides).
Sample preparation prior to introduction to mass spectrometric detectors is critical for optimizing eCB measurements
Excellent reviews have outlined the history, chemistry, and physics of lipidomics techniques and there use in biological systems [13–16]. There are entire subfields of science dedicated to understanding the relationships of chemistry and physics that allow us to accurately measure molecules and lipidomics is a further specialization of this intersection. The use of some type of “liquid-liquid extraction” (LLE) extraction typically using chloroform as the initial solvent is often considered the standard protocol for the most general lipid extraction that will capture the most different types of lipid species [13,16]. However, multiple recent studies have also highlighted the importance of the choice of the initial solvent choice for lipid extraction in the pursuit of more targeted lipidomics analyses [17–19]. These studies highlight the importance in recognizing that if the goal is to measure only specific types of lipids (e.g. eCBs and related lipids), a general type of LLE preparation may provide you with an enriched sample of many lipids; however, by doing so, it has the potential to diminish the likelihood of enriching the sample of specific lipids being identified for study. The question remains as to whether using an all-inclusive LLE to extract “all” lipids could actually functionally drown out the signal of the least abundant species, like eCBs even with advanced MS techniques?
With an increase in the ability for sample preparation optimization techniques with an aim to measure specific types of lipid species, this also allows for a wider range of lipids that can be analyzed within that subspecies. AEA and the NAEs (Fig. 1) are arguably the most abundant members of their class as evidenced by their relative levels throughout the body [6,20–26]. It is also easily argued that it is this abundance that made it possible to isolate and characterize these lipids decades ago [27]. Optimized lipidomics techniques coupled to MS analysis has provided the opportunity for the scientific community to move beyond simply measuring only these abundant lipids within biological fluids and towards being able to analyze a much broader arrays of lipids within the class (Fig. 2; [2,4,6,8,21–26,28–33]). To do so, we must recognize which of the techniques will best allow for this broader range of lipids to be analyzed.
Tables 1–3 contains summaries of lipid extraction methods in published reports used to isolate AEA and, in some cases, related lipids in biological fluids such as plasma, serum, saliva, and breast milk [34–55]. Some of these studies also measured lipids in other tissues; however, the focus here is on the methods used for biological fluids. The methodological details are as close to verbatim as possible to maintain the integrity of the details. Only minor edits have been made to clarify abbreviations and jargon. This is not meant to be an exhaustive list of methodologies or published data on biological fluid analysis of eCBs and related lipids, but it does provide a representative overview of the current techniques. There was an attempt to highlight the most recent publications in the field and have, therefore, also indicated when those papers referenced earlier work if the methods were not provided in the most recent publication. In these cases, information from the referenced article is provided. The species of lipids analyzed are also listed and illustrates that most authors only report a few lipids even though the process used would isolate hundreds or more.
Table 1. Summaries of sample preparations that rely on lipid-lipid extractions (LLE) to enrich lipids prior to MS analysis.
See “List of Abbreviations” for a key to those reagents abbreviated. Comments in italics were not in the original manuscripts but are clarifications by the authors here. Only the first author and publication year are listed in the table, though the full reference is available at the end of the Chapter.
| Author, year | Sample type | eCB Lipids Measured | Methods that use LLE plus evaporation/reconstitution as the primary means of lipid enrichment |
|---|---|---|---|
| Ney, 2019 | Saliva, plasma | 2-AG, AEA | Saliva and plasma samples were frozen at −20C until processed. After thawing then centrifugation, 400μL of saliva supernatant was added to 1600μL of 1:1 methanol: acetone. 400μL of plasma was added to 1mL of ethyl acetate: cyclohexane 1:1. 20ng/mL of d5–2-AG and d4-AEA were added as internal standards and solutions vortexed. Saliva was frozen (−20°C, 1h) and then both saliva and plasma were centrifuged at 4°C. Each was evaporated under nitrogen and reconstituted with either 150μL methanol for saliva or 100μL acetonitrile for plasma. Samples were evaporated again and reconstituted in 50% MeOH for saliva or acetonitrile for plasma to a final volume of 30μL. |
| Ulu, 2021 | Plasma | AEA, DHEA, OEA 2-AG, 2-DG, | To analyze plasma eCBs, they collected blood in K2EDTA tubes, purified lipids as reported in Argueta, 2017. |
| Argueta, 2017 | Plasma | 2-AG, AEA | Collected blood via cardiac puncture and kept it in EDTA tubes on ice, then centrifuged 1,500xG, 10min at 4°C to separate plasma and stored at −80°C. Plasma (0.1mL*) was combined with 0.9% saline (0.9 ml) then added to 2mL chloroform. The organic phase was separated on open-bed silica gel chromatography, the resulting eluate dried under nitrogen, resuspended in 0.1mL MeOH: chloroform (9:1), which was used for MS analysis. *amount of plasma used was inferred by the statement that “0.1 mL plasma at the expense of saline” in reference to the 1ml volume of saline. Otherwise, no clear statement on the amount of plasma per sample was used. |
| Martin-Perez, 2021 | Plasma | 2-AG, AEA, POEA | Collected blood in K2-EDTA tubes, centrifuged at 1,700xG, 15min, 4°C, then stored at −80°C. Plasma (0.5mL) was spiked with deuterium labeled standards in 0.5mL of 0.1M ammonium acetate buffer. Tert-butyl methyl ether (6.0mL) was added to the spiked plasma solution and the organic layer separated then evaporated at 39°C under nitrogen. Sample was reconstituted in 100μL water: acetonitrile (1:9, v/v) that contained 0.1% formic acid, and used for MS analysis. |
| Sempio, 2021 | Plasma | NAEs, 2-AG, DH-g-LEA, NADA, 2-AGE, ODA | 200μL of plasma was spiked with deuterium-labeled standards and added to 800μL acetonitrile. The solution was mixed via vortex for 10min, then centrifuged (25,000xG, 4°C, 10min). 750μL of the supernatant was added to 450μL of 0.1% formic acid in water and mixed via vortex. 400μL of the solution was used for MS analysis. |
| De Icco, 2021 | Plasma | AEA, PEA | 500μL of plasma was mixed with 1mL of methanol + d4-AEA and d4-PEA and added to 2mL of chloroform, centrifuged at 3000xRPM (no g indicated) for 15min at 4°C. The organic phase was transferred to glass vials, and the aqueous fraction extracted again with chloroform (1 mL) and 500μL phosphate buffered saline. The 2 resulting organic phases were combined, dried under N2, and the residues dissolved in 2mL chloroform. Silica Gel G columns were used to fraction the extracts, eluting into 2mL chloroform/MeOH (9:1), then 2mL chloroform/MeOH (8:2). The solutions were evaporated under nitrogen and reconstituted in 75μL of 9:1 chloroform/MeOH to be analyzed via MS. |
| Manca, 2021 | Plasma | NAEs 2-acyl glycerols | Same protocol as Turcotte 2020 to extract lipids (below) |
| Turcotte, 2020 | Plasma | 2-acyl glycerols LPA; free fatty acids including AA; AEA | 200μL plasma was added to 300μL TRIS buffer, then added 2mL toluene + internal standards. Solution was mixed via vortex and then separated via centrifuge at 4,000xG for 5min. Samples were then placed in an ethanol-dry-ice bath (−80°C) to freeze the aqueous phase (bottom). The organic phase (top) was then collected and evaporated to dryness under a stream of nitrogen. Samples were reconstituted in 25μL of HPLC solvent A (H2O with 0.05% acetic acid and 1 mM NH4+) and 25μL of solvent B (MeCN/H2O, 95/5, v/v, with 0.05% acetic acid and 1 mM NH4+). A 25μL aliquot was used for MS analysis. |
| Brunk-horst Kanaan, 2021 | Plasma | AEA, 2-AG, 1-AG, OEA, PEA | Blood was collected in K+-EDTA tubes on ice, centrifuged at 3000RCF (no G provided), for 4°C, 10min, then plasma stored at −80°C until processing. Plasma was thawed and internal standard added. Ethyl acetate/hexane was added to the plasma solution as 9:1 (v:v). Organic phase was removed, evaporated, then reconstituted in acetonitrile for MS analysis. |
| Kirkwood, 2016 | Serum | AEA, OEA, PEA, 2/1-AG | Collected blood was “spun” to separate serum, then frozen in aliquots at −80°C. Sample preparation protocol was adapted from Zoerner, 2012. 50μL of serum was added to 1mL of toluene + 100pg/mL internal standards, mixed via vortex for 15min, medium speed at room temperature. 400μL of water + 3% formic acid was added and mixed via vortex for 5min. Samples were held at −80°C for 2hr, then moved to 4°C to thaw and centrifuged at 3,000xG for 15min at 4°C. Organic phase was evaporated under nitrogen, resuspended in 40μL of 50/50 acetonitrile/MeOH, then mixed via vortex for 2min. 60μL water + 0.2% formic acid was added and mixed via vortex for 2min and then used for MS analysis. |
| Siebers, 2021 | Plasma | AEA, 2-AG, PEA, AA | Collected blood in a tube coated with EDTA and centrifuged 10min, 2000xG, at 4°C then stored at −85°Cs. eCBs were measured from 100μL plasma. Detailed methods were not provided and authors referenced Post 2020, Lerner 2019, and Lerner 2018. |
| Post et al 2020 | N/A | N/A | This reference is to a “Jove” video presentation that is still inaccessible with a notice that “Video is coming soon” in a Jan 2022 search, so it was not accessible to evaluate. |
| Lerner, 2019 | Brain, plasma | 2-AG, AEA, free fatty acids including AA, LA, and EPA, | 40μL of plasma was combined with 1000μL MTBE/methanol (10:3), spiking solution, and 250 μL water+ 5μM THL/URB597 and 10ug/mL BHT. The authors then reference the methods of Lerner 2016; however, there was no “Lerner 2016” that matched the reference; however, this was likely due to an online publication date, as there is a Lerner 2017 that matches the title. (see below). |
| Lerner, 2017 | Brain, liver, plasma | NAEs including OEA, PEA, AEA; 2-AG; AA; eicosanoids | Blood was collected in chilled 500μL EDTA tubes containing 10μM indomethacin and 25 μM tetradydrolipstatin/URB, and centrifuged 2,000xG, 4°C 10min to separate plasma which was removed and stored at −80°C. To extract lipids they used 100μL of plasma + 1200μL ethylacetate/n-hexane (9:1) + internal standards + 600μL of 0.1M formic acid. They vortexed for 2 min at 4°C, then centrifuged 13,000xRPM for 20min at 4°C, froze them for 10 min, then evaporated under nitrogen at 37°C and reconstituted in 50μL ACN/water (1:1). |
Table 3. Summaries of publications with ambiguous or missing details on sample preparation prior to MS analysis.
See “List of Abbreviations” for a key to those reagents abbreviated. Comments in italics were not in the original manuscripts but are clarifications by the authors here. Only the first author and publication year are listed in the table, though the full reference is available at the end of the Chapter.
| Author, year | Sample type | Measuring | Methods where the sample preparation was unclear/unknown |
|---|---|---|---|
| Diaz-Rua, 2021 | Brain, plasma | AEA, 2-AG | The methods described for eCB analysis only detail the MS parameters. There is no description of any aspect of sample preparation in this study and references 2 previous studies. 1) Diaz-Rua et al 2020a, which does not describe any aspect of sample preparation and only describe MS parameters. 2) Diaz-Rua et al 2020b, which, likewise, does not describe any aspect of sample preparation. |
| Manca, 2020 | Plasma | Free fatty acids, 2-AG, 2-PG, 2-LG, 2-OG, AEA, DHEA, OEA, PEA | An “aliquot” (no specific volume) of plasma was used eCB analysis. d8-AEA, d5–2-AG, d2-OEA, d4-PEA, d3-SEA were added as internal standards to the plasma aliquots. There is no further information on sample preparation. It is possible that the samples were used in this state with no further preparation for MS analysis. |
Methodologies highlighted in Table 1 are from publications that use some version of an LLE technique that relies upon the chemistry of lipophilicity wherein those compounds that are most like the organic solvent would remain within that solvent and those that are more polar would be retained in the more aqueous phase. LLE typically requires that the organic layer be evaporated and reconstituted in a solvent that is more amenable to chromatographic and mass spectrometric techniques. The evaporation and reconstitution steps are also the primary way in which this method can concentrate the sample to be able to, at least theoretically, increase the signal at whatever detector is be employed (e.g. MS, MS/MS). While a detailed discussion of the chemistry and physics of signal suppression in MS detectors is beyond the scope of this chapter, it is important to state that this phenomenon is an issue that can cause a sample to have a false negative for a compound of interest or dramatically increase variability with a highly enriched sample. There are excellent reviews that discuss this phenomenon [56,57], in brief, signal suppression refers to the loss of MS “signal”, which is the ability to measure a specific lipid within a complex matrix, due to the interference by all the other lipids in the matrix. Therefore, signal suppression very often happens when a sample consists of a wide range of lipids from a sample that was extracted using a general lipid extraction (e.g. LLE alone) and then further concentrated. Some of this can be overcome with chromatography as part of the analysis technique and more sensitive MS/MS methods; however, arguably the most effective way is to have a pre-separation of lipid classes prior to introduction to the HPLC/MS system [57].
A means to overcome signal suppression that is often observed with LLE alone is to partially purify the sample matrix through separation with solid phase extraction (SPE) columns prior to MS analysis [56,57]. Multiple laboratories, including the Bradshaw lab, use SPEs in sample preparation for eCB and related lipid analyses and these methodologies are highlighted in Table 2. This is not a new technique and the Balvers 2009 paper [35], was added to illustrate that this technique has been used for at least few decades for biological fluids. Of interest, is that many of these methodologies that use SPE also use an evaporation and reconstitution step to further enrich the sample, though what this means for signal suppression is an open question. The methodology in the Bradshaw lab highlighted in Bashashati et al, 2021 (Table 2,[36], does not employ an evaporation step and the signal to noise level is robust ([36], Figure 3). Likewise, evaporation is not used in the methods described by Luque-Cordoba et al. 2018 [58], Biernacki, 2021 [37], and Gaisberger, 2021 [59]. In addition to a reduction in signal suppression, an argument for the use of SPE without the evaporation/reconstitution steps is that the loss of lipids via oxidation, degradation, or adherence to the vial that does occur with this step is eliminated. Taken together, the use of SPE to drive targeted lipidomics allows for an enhanced ability to measure compounds of interest in a smaller amount of starting material with a greater signal to noise and less signal suppression.
Table 2. Summaries of sample preparations that rely on Solid Phase Extraction partial purification techniques to enrich specific lipid species prior to MS analysis.
See “List of Abbreviations” for a key to those reagents abbreviated. Comments in italics were not in the original manuscripts but are clarifications by the authors here. Only the first author and publication year are listed in the table, though the full
| Author, year | Sample type | Measuring | Methods that use SPE columns to partially purify and enrich samples with specific lipid classes |
|---|---|---|---|
| Balvers, 2009 | Plasma | 2-AG, 2-AG ether, AEA, DEA, NADA, DLE, NAGly, O-AEA, OLDA, OEA, PEA, SEA | Collected blood in K3EDTA tubes on ice, centrifuged at 2,000xG, 15min, 4°C, added phenylmethanesulfonyl fluoride (PMSF) to plasma then stored at −80°C. 1mL plasma was added to 4mL acetonitrile + d8–2-AG, d8-AEA, d8-NADA, d8-NAGly + 100μM PMSF, then centrifuged at 3,000xG for 5min, and decanted into 15mL water + 0.133% trifluoro acetic acid (TFA). Partial purification was performed on C8-SPE columns. Columns were initially washed with 1mL methanol, then 1mL water, sample was loaded, washed with 2mL of 20% acetonitrile/ 80% water / 0.1% TFA, and eluted with 2mL of 80% acetonitrile/20% water/0.1% TFA. This elution was evaporated with a vacuum and stored at −80°C until analysis. At the time of MA analysis, sample was reconstituted the in 100μL acetonitrile/0.1% TFA spiked with d4-PEA. 5μL was used for MS analysis. |
| Taglia-monte, 2021 | Plasma | 2-AG, AEA, OEA, LEA, PEA | Blood was collected by venipuncture in tubes containing EDTA and kept on ice. Plasma samples were centrifuged 3,000RPM (information on g not provided), 15min, 4°C within 15min of collection, then stored at −80 °C. On day of processing, samples were thawed at 4°C and then kept on ice. 500μL of plasma was diluted 1:2 in distilled water + 50μL of 200ηg/mL d8-AEA. Samples were vortexed, then centrifuged at 21,1000xG, 5min, 4°C. Supernatant partially purified on HLB Oasis solid phase extraction columns that had been conditioned with 1mL MeOH and 1mL water, were washed with 1mL 40% MeOH, and eluted with 1mL of acetonitrile. The eluate was dried under nitrogen, reconstituted in 100μL of acetonitrile: water (50:50), which was used for MS analysis. |
| Gaisber-ger, 2021 | Plasma | AEA | Collected blood in serum- and plasma-specific tubes, then froze and stored at −80°C until processing. Samples were thawed and prior to partial purification via C18 solid phase extraction column separation. Columns were conditioned with 1mL acetonitrile (0.1% formic acid), and 1mL water (0.1% formic acid). 500μL of plasma spiked with 1μL of 1ηg/mL d4-AEA was added to the column. The column was washed with 1mL of 20% acetonitrile (0.1% formic acid), then eluted with 400μL of acetonitrile (0.1% formic acid), which was used for MS analysis. |
| Biernacki, 2021 | Plasma | AEA, 2-AG | Blood was collected in tubes with EDTA and butylhydroxytoluene, centrifuged at 3,000xG 4°C to collect plasma. Luque-Cordoba 2018 is referenced the as protocol for sample preparation and MS analysis. |
| Luque-Cordoba, 2018 | Serum | DHEA, AEA, NAGly, NADA, 2-AG, PEA, DEA, OEA, 2-AG ether, PALDA, OLDA/ODA, SEA, STEARDA | Samples were thawed on ice then centrifuged at 20,200×g for 10 min, then filtrated by a nylon filter with a 0.2 μm pore size. 200μL of the filtrated sample is introduced in an opaque vial with a glass insert and located in the autosampler of the SPE (C18 solid phase extraction) system, which is maintained at 4°C. SPE column is conditioned with 10mL of methanol, then 1mL of methanol, followed by two solvation steps each with 1 mL of 25% (v/v) ACN in water containing 0.7% of formic acid. The 100μL sample loop is filled with the analytical sample that is then loaded in the sorbent cartridge with 1.5mL of solvation solution. In this process, the analytes are retained in the sorbent and the sample matrix removed to the waste. Then, the analytes are eluted with the chromatographic mobile phase (elution time 90 s), and the cartridge is cleaned by five rinsing steps with 5 mL of water, methanol, water, 30% (v/v) ACN in water, and water. |
| Bashashati et al, 2021 | Plasma | AEA, OEA, LEA, DEA, SEA, PEA, 2-AG, 2-PG, 2-LG, 2-OG, AA, LA, OA | Plasma was aliquoted into 150μL volumes for each participant and then added to 2mL of (HPLC)-grade methanol. Solutions were spiked with 500 picomoles deuterium labeled N-arachidonoyl glycine (d8-NAGly; Cayman Chemical) and centrifuged at 19,000 × g for 20 minutes at 24°C. Supernatants were diluted with HPLC water to make a 15% supernatant solution. Lipid extractions were performed using C18 solid-phase extraction columns. SPE columns were conditioned with 5 mL HPLC methanol followed by 2.5 mL HPLC water. Then, the supernatant/water solution was loaded onto the column, followed by 2.5 mL HPLC water. A series of 5 elutions with 1.5 mL 40%, 60%, 75%, 85%, and 100% methanol were collected for MS analysis. #Extractions and MS analysis performed in Bradshaw lab. |
| Gaitan, 2018 | Milk | AA, DHA, EPA, AEA, PEA, OEA, DHEA, EPEA, EEA, 2-acyl glycerols | Participants expressed milk at home and kept in the fridge for a maximum of 24h until transferred to lab on ice. In lab, samples were heated with a 37°C water bath, gently mixed, and aliquoted into 15mL to store at −80°C. To process, samples were thawed in 37°C water bath, vortexed 10sec and then added to an un-specified volume of acetonitrile + PBS + internal standards then centrifuged at 14,000xG, 4°C for 5min. The supernatant was mixed with 4 volumes of 5% phosphoric acid before partial purification on solid phase extraction columns (OASIS HLB reverse phase cartridges). SPEs were conditioned with MeOH and water. Sample loaded, they washed with 40% MeOH, then eluted with acetonitrile. The elution was the evaporated under nitrogen, reconstituted in EtOH, vortexed, sonicated, and centrifuged prior to MS analysis. |
| Bruun, 2018 | Milk | OEA, SEA, PEA | Milk was collected in lab, aliquoted to 10mL, and stored at 5°C for a maximum of 3 days. The sample was then centrifuged3,600RPM, 21°C, 5min, the fat, skimmed and solid fractions separated, and stored at −80°C. To extract lipids, they centrifuged to remove particles, added 20μL of a 20ηg/mL d4-OEA, d3-SEA, d4-PEA, and then passed them through solid phase extraction columns. They washed with 5% MeOH, 0.1% acetic acid, then eluted into 2mL of acetonitrile and 2mL MeOH. Then they dried w/speed vacuum, reconstituted in 100μL MeOH, added 10μL recovery (0.025ug/mL 12-[[cyclohexylamino)carbonyl amino] dodecanoic acid), and centrifuged again using an Eppendorf tube filter prior to MS analysis. |
Fig. 3. Comparisons of AEA method used on plasma between Sciex API 7500, Sciex API 6000, and API 3000.
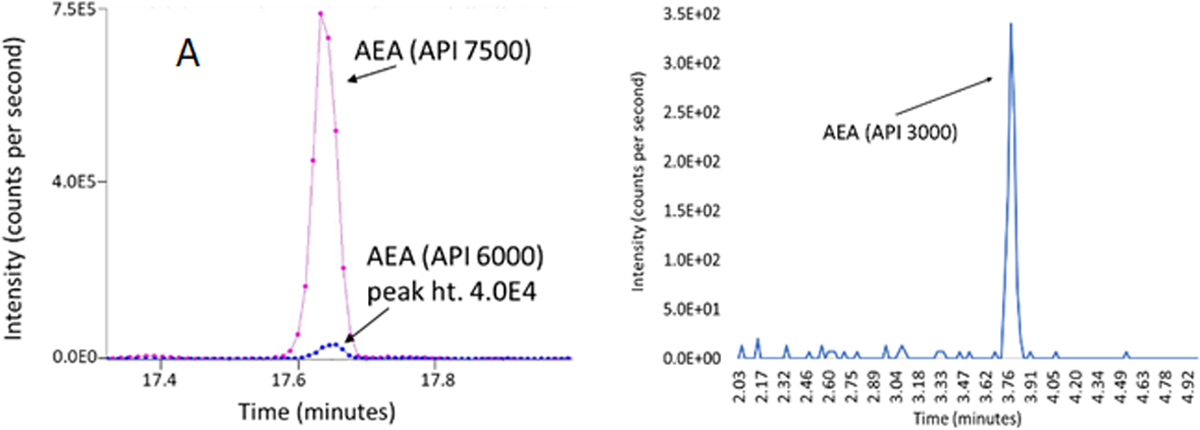
A) Data are from equivalent amounts of NIST human plasma (100μl) dissolved in methanol, supernatant dried, and reconstituted in 50 μl in 50:50 water: methanol and 50 μl injected on a C18 Phenomenex analytical column. The pink line represents the output from the API 7500 and the blue line is the API 6000. B) Data are from a 20 μl injection on a Zorbex C18 column from 25 μl of plasma partially purified on C18 solid phase extraction column and eluted in 1.5ml 100% methanol. The identical parent and fragment pairs [H+]348/62 in positive ion mode were used for analysis in each instrument.
A final grouping of methodological summaries shown in Table 3 is for those publications in which the sample preparation methods are not stated or are vague to the point of being unknown. This is an overall issue for both reviewers and authors. There is no suggestion of subterfuge on the part of the authors, but merely a lack of attention to detail for an aspect of the methodology that can be thought of so standardized as to lack the need for attention. Tables 1 and 2 illustrate that no two labs are using the same sample preparation technique, so this sentiment could not be further from the reality of the situation. There is quite literally a lack of any standardization of sample preparation across labs that investigate endogenous levels of eCB related lipids; therefore, the need to explain how the samples were prepared is important.
Additional factors to consider for optimization of eCB measurement in biological fluids
While a detailed discussion of advances in MS technology is not appropriate here, it is important to state that the evolution of MS technologies has driven an advent of no sample preparation prior to MS analysis in some cases. There is an assumption based on the lack of any discussion of sample preparation beyond the addition of internal standards in the Manca et al 2020 report [45] that plasma samples were directly analyzed via HPLC/MS/MS with no sample preparation. Figure 3A shows data from promotional materials for the API 6000 and the API 7500 from Applied Biosystems in which AEA is analyzed from 100μl of plasma that was dissolved in methanol (unknown amount) and reconstituted in 50μl of 50:50 methanol:water and then all 50μl injected for HLPC/MS/MS analysis. One aspect of the data was to show that the improved technology of the API 7500 is superior to the API 6000; however, another is to demonstrate that one should not need to have much sample preparation to detect AEA. As mentioned previously, AEA is a relatively abundant lipid in biological systems, and it is important to note that the sample is 100μl. In Figure 3B, we show that with an API 3000 (~20 years older than the API 7500), we can use 25μl of plasma and the SPE-dependent sample preparation describe in Table 2 and our previous report [36] and have a clear AEA signal as well as be able to measure multiple other lipids (Figure 4). One can only speculate what one could measure by combining the power of SPE partial purification with these improved MS/MS techniques.
Figure 4. Change in percent recovery of lipid analytes in plasma as a function of initial methanolic extraction solution.
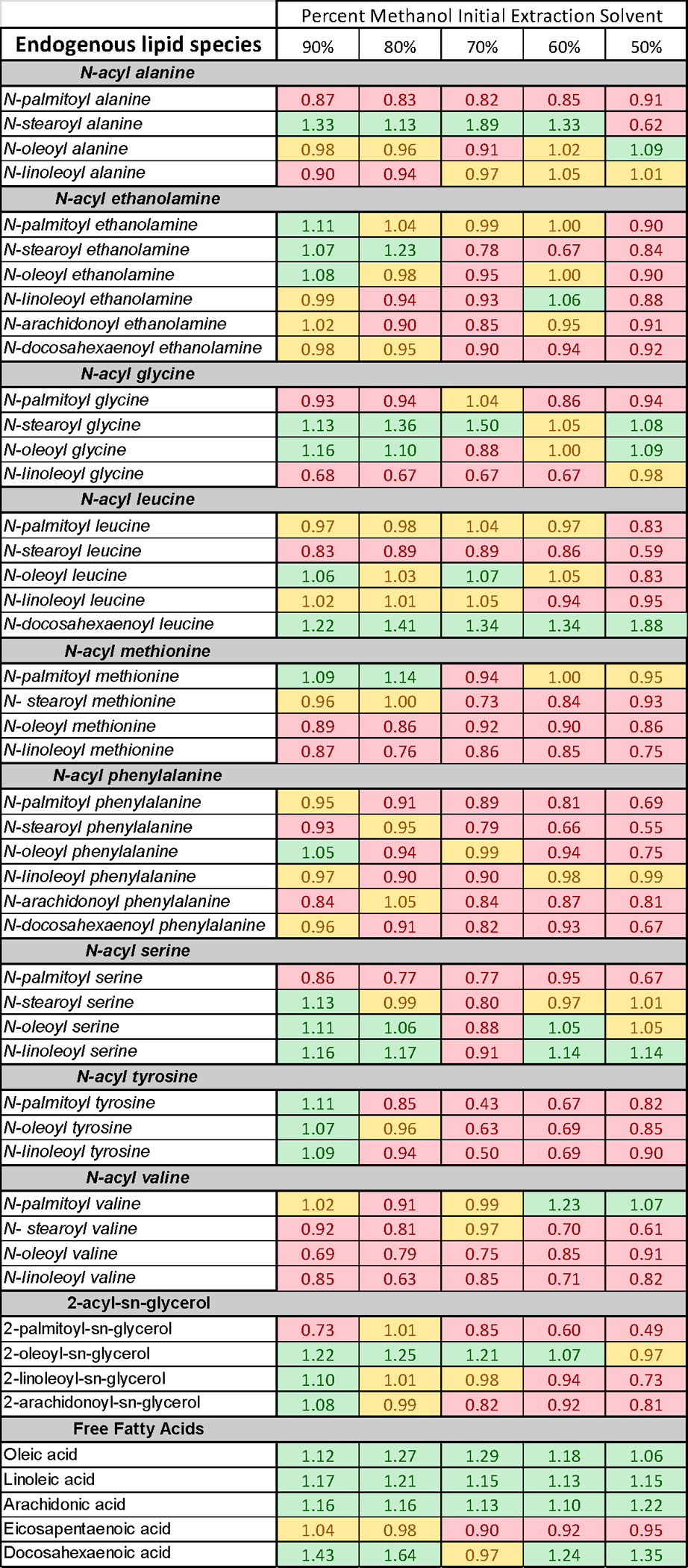
See text for methodological and analytical details.
If, as we argue, one of the keys to optimization for the measurement of AEA and related lipids in plasma and other biological fluids is to work towards enriching the sample with fewer lipids, would changes in the amount of initial methanolic solvent coupled to SPE preparation be a primary way to achieve this end? Is it possible to refine this process by reducing the lipid noise further by the introduction of water into the initial solvent extractions step and not separating the organic phase? Here, we show data of a lipidomics screen of a range of eCB endogenous analogs and free fatty acids from plasma with changes in the percentage of the initial methanolic extraction. Plasma samples were pooled, mixed, and separated into 75μl aliquots. The only exception to the protocol for sample preparation for MS analysis previous described by the Bradshaw [36] was that the percent of methanol as the initial extraction solvent was either 100% (as used in the original protocol), 90%, 80%, 70%, 60%, and 50%. N=6 for each treatment group. To determine if changing the initial extraction solvent would change the recovery of endogenous analytes, fold differences from the mean were calculated from the standard protocol solvent (e.g. 100% methanol) for each analyte measured in plasma in all 6 samples for each solvent group. 80 lipid analytes were assayed and 49 had measurable chromatographic peaks in all 6 samples in each group (Figure 4). Fold change was separated into 3 categories. Category A (shown in green in Fig. 4) had a greater than or equal to 1.05-fold increase; category B (shown in yellow in Fig. 4) had a fold change between 0.95 and 1.049; and finally, category C (shown in red) had a fold change equal or less than 0.949. The actual fold changes are shown for each analyte in Fig. 4 in the cell that corresponds to the specific lipid and the initial staring solvent. These data show that 23 of the 49 lipids analyzed were in category A with an initial methanolic percentage of 90; 13 with 80%; 8 with 70%, 10 with 60%, and 8 with 50%. It is important to note that the lipid species most effected by the change in initial solvent percentage were free fatty acids and that other endogenous AEA analogs were differentially affected such the same 8 lipids were not increased in both 80% and 50%. Likewise, the largest deficit in recovery was measured using 50% methanol as the initial solvent with 32 of the 49 lipids having lower recovery. These data are provided to illustrate the point that the staring extraction solvent influences the lipids that can be measured and that it is not possible to have a “one size fits” all approach to sample preparation for MS analysis of lipids.
Is a standardized sample preparation for eCB and related lipids possible to achieve? Is it even important?
As we journeyed through the methodological landscape of our peers to gather information for this Chapter it became evident that there are many ways to achieve a similar end when it comes to analyzing eCBs and related lipids. That the individuality of sample preparation techniques is quite literally as different as the individuals who developed and published them is both a source of consternation and delight. In our own lab as our scientific questions take us to situations that require samples that are smaller in size than our previous limits (e.g. primary cell culture, capillary blood draws, 1mm brain tissue punches), the need to couple SPE with evaporation becomes more apparent. Having moved away from the evaporation technique over a decade ago, it is challenging to think of this is a viable option; however, evaluating the data from peers who can combine the techniques brings into focus the need to continue to evaluate the types of sample preparation to match the needs of the study. Is it a hope that those who use only LLE analysis and evaluate only a small number of eCB-class lipids will consider using SPE sample preparation and provide the scientific community with much needed data on how their disease or behavioral model of choice is driving changes in a broader array of lipids? Yes, of course. So, does this mean that there is a recommendation for everyone to move towards a unified sample preparation with SPE for each preparation? Actually, no. An argument against standardization is that the richness gained by labs continuing to develop novel analytical techniques would be lost if there was a push for all techniques to be standardized. It can be difficult enough to maintain the systems in place to accurately measure eCBs and related lipids, so advocating for a change in this neither equitable nor realistic. The only argument towards any standardization is a drive towards clearer methodological details within publications. Many of the summaries in Tables 1–3 required deep dives into multiple aspects of the publication and to dig back into multiple previous publications, sometimes to no avail. Sample preparation is the cornerstone to all levels of endogenous lipid analysis and should not simply be relegated to the oft used phrase “as previously described”. Even small changes in methodologies can have larger effects. When we finally zeroed in on the combination of LLE and SPE coupled to HPLC/MS/MS that allowed us to identify many previously unidentified lipo amino acids (sometimes in the upper attomole range of sensitivity;[3–5,31,60]) the late Michael Walker was looking through the data and exclaimed, “You can measure everything and see nothing!” The beauty is that the “nothing” (e.g. low-abundance lipids) we were seeing has now been shown to have many different biological effects in multiple disease and genetic KO models [6,20–26,29,31,36], with the scientific community still only scratching the surface on understanding what they do. Having the methodological capability to continue to “see nothing” is reliant on our ability to learn from each other and use the best sample preparation methodology for the question and the tissue type. Optimization processes can be tailored to fit the tissue, and biological fluids are specialized tissue that deserve a specialized approach. It is the hope of the authors that the overviews provided here will spark new thoughts and ideas for how to best approach this goal in future experiments and discoveries.
Abbreviations
- ACN
Acetonitrile
- MeOH
methanol
- EtOH
ethanol
- 2-AG
2-arachidonoyl glycerol
- 2-AG ether; 2-AGE
2-arachidonoyl glycerol ether
- 2-AG
1-arachidonoyl glycerol
- 2-PG
2-palmitoyl glycerol
- 2-LG
2-linoleoyl glycerol
- 2-OG
2-oleoyl glycerol
- 2-DG
2-docosahexaenoyl glycerol
- NAE
N-acyl ethanolamine
- AEA
N-arachidonoyl ethanolamine
- PEA
N-palmitoyl ethanolamine
- POEA
palmitoleoylethanolamide
- SEA
N-stearoyl ethanolamine
- OEA
N-oleoyl ethanolamine
- LEA
N-linoleoyl ethanolamine
- DHEA
N-docosahexaenoyl ethanolamine
- EPEA
N-eicosapentaenoyl ethanolamine
- O-AEA
O-arachidonoyl ethanolamide
- DLE; DH-g-LEA
dihomo-ɣ-linolenoyl ethanolamide
- DEA
docosatetraenoyl ethanolamide
- EEA
eicosenoyl ethanolamide
- SA
stearic acid
- LA
linoleic acid
- AA
arachidonic acid
- OA
oleic acid
- EPA
eicosapentaenoic acid
- DHA
docosahexaenoic acid
- NADA
N-arachidonoyl dopamine
- OLDA; ODA
N-oleoyl dopamine
- STEARDA
N-stearoyl dopamine
- PALDA
N-palmitoyl dopamine
- NAGly
N-arachidonoyl glycine
- OLA
oleamide
- PBS
phosphate buffer saline
- PMSF
phenylmethanesulfonyl fluoride
- TFA
trifluoro acetic acid
- SPE
solid phase extraction
- EDTA
ethylenediaminetetraacetic acid
- MTBE
methyl tertbutyl ether
References
- 1.Walker JM, Huang SM, Strangman NM, Tsou K, and Sanudo-Pena MC, Pain modulation by release of the endogenous cannabinoid anandamide. Proc Natl Acad Sci U S A, 1999. 96(21): p. 12198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Piscitelli F and Bradshaw HB, Endocannabinoid Analytical Methodologies: Techniques That Drive Discoveries That Drive Techniques. Adv Pharmacol, 2017. 80: p. 1–30. [DOI] [PubMed] [Google Scholar]
- 3.Bradshaw HB and Walker JM, The expanding field of cannabimimetic and related lipid mediators. Br J Pharmacol, 2005. 144(4): p. 459–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rimmerman N, Bradshaw HB, Hughes HV, Chen JS, Hu SS, McHugh D, Vefring E, Jahnsen JA, Thompson EL, Masuda K, Cravatt BF, Burstein S, Vasko MR, Prieto AL, O’Dell DK, and Walker JM, N-palmitoyl glycine, a novel endogenous lipid that acts as a modulator of calcium influx and nitric oxide production in sensory neurons. Mol Pharmacol, 2008. 74(1): p. 213–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bradshaw HB, Rimmerman N, Hu SS, Burstein S, and Walker JM, Novel endogenous N-acyl glycines identification and characterization. Vitam Horm, 2009. 81: p. 191–205. [DOI] [PubMed] [Google Scholar]
- 6.Raboune S, Stuart JM, Leishman E, Takacs SM, Rhodes B, Basnet A, Jameyfield E, McHugh D, Widlanski T, and Bradshaw HB, Novel endogenous N-acyl amides activate TRPV1–4 receptors, BV-2 microglia, and are regulated in brain in an acute model of inflammation. Front Cell Neurosci, 2014. 8: p. 195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Y, Plastina P, Vincken JP, Jansen R, Balvers M, Ten Klooster JP, Gruppen H, Witkamp R, and Meijerink J, N-Docosahexaenoyl Dopamine, an Endocannabinoid-like Conjugate of Dopamine and the n-3 Fatty Acid Docosahexaenoic Acid, Attenuates Lipopolysaccharide-Induced Activation of Microglia and Macrophages via COX-2. ACS Chem Neurosci, 2017. 8(3): p. 548–557. [DOI] [PubMed] [Google Scholar]
- 8.Wu J, Zhu C, Yang L, Wang Z, Wang L, Wang S, Gao P, Zhang Y, Jiang Q, Zhu X, and Shu G, N-Oleoylglycine-Induced Hyperphagia Is Associated with the Activation of Agouti-Related Protein (AgRP) Neuron by Cannabinoid Receptor Type 1 (CB1R). J Agric Food Chem, 2017. 65(5): p. 1051–1057. [DOI] [PubMed] [Google Scholar]
- 9.Epand RF, Infante MR, Flanagan TD, and Epand RM, Properties of lipoamino acids incorporated into membrane bilayers. Biochim Biophys Acta, 1998. 1373(1): p. 67–75. [DOI] [PubMed] [Google Scholar]
- 10.Console-Bram L, Ciuciu SM, Zhao P, Zipkin RE, Brailoiu E, and Abood ME, N-arachidonoyl glycine, another endogenous agonist of GPR55. Biochem Biophys Res Commun, 2017. 490(4): p. 1389–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Macfarlane MG, Phosphatidylglycerols and lipoamino acids. Adv Lipid Res, 1964. 2: p. 91–125. [DOI] [PubMed] [Google Scholar]
- 12.Pignatello R, Jansen G, Kathmann I, Puglisi G, and Toth I, Lipoamino acid conjugates of methotrexate with antitumor activity. J Pharm Sci, 1998. 87(3): p. 367–71. [DOI] [PubMed] [Google Scholar]
- 13.Hu T and Zhang JL, Mass-spectrometry-based lipidomics. J Sep Sci, 2018. 41(1): p. 351–372. [DOI] [PubMed] [Google Scholar]
- 14.Hyotylainen T, Ahonen L, Poho P, and Oresic M, Lipidomics in biomedical research-practical considerations. Biochim Biophys Acta Mol Cell Biol Lipids, 2017. 1862(8): p. 800–803. [DOI] [PubMed] [Google Scholar]
- 15.Wood PL, Mass spectrometry strategies for clinical metabolomics and lipidomics in psychiatry, neurology, and neuro-oncology. Neuropsychopharmacology, 2014. 39(1): p. 24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu T, Hu C, Xuan Q, and Xu G, Recent advances in analytical strategies for mass spectrometry-based lipidomics. Anal Chim Acta, 2020. 1137: p. 156–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reis A, Rudnitskaya A, Blackburn GJ, Mohd Fauzi N, Pitt AR, and Spickett CM, A comparison of five lipid extraction solvent systems for lipidomic studies of human LDL. J Lipid Res, 2013. 54(7): p. 1812–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iriondo A, Tainta M, Saldias J, Arriba M, Ochoa B, Goni FM, Martinez-Lage P, and Abad-Garcia B, Isopropanol extraction for cerebrospinal fluid lipidomic profiling analysis. Talanta, 2019. 195: p. 619–627. [DOI] [PubMed] [Google Scholar]
- 19.Ulmer CZ, Jones CM, Yost RA, Garrett TJ, and Bowden JA, Optimization of Folch, Bligh-Dyer, and Matyash sample-to-extraction solvent ratios for human plasma-based lipidomics studies. Anal Chim Acta, 2018. 1037: p. 351–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ho JM, Bergeon Burns CM, Rendon NM, Rosvall KA, Bradshaw HB, Ketterson ED, and Demas GE, Lipid signaling and fat storage in the dark-eyed junco. Gen Comp Endocrinol, 2017. 247: p. 166–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leishman E, Kunkler PE, Hurley JH, Miller S, and Bradshaw HB, Bioactive Lipids in Cancer, Inflammation and Related Diseases : Acute and Chronic Mild Traumatic Brain Injury Differentially Changes Levels of Bioactive Lipids in the CNS Associated with Headache. Adv Exp Med Biol, 2019. 1161: p. 193–217. [DOI] [PubMed] [Google Scholar]
- 22.Leishman E, Kunkler PE, Manchanda M, Sangani K, Stuart JM, Oxford GS, Hurley JH, and Bradshaw HB, Environmental Toxin Acrolein Alters Levels of Endogenous Lipids, Including TRP Agonists: A Potential Mechanism for Headache Driven by TRPA1 Activation. Neurobiol Pain, 2017. 1: p. 28–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leishman E, Mackie K, and Bradshaw HB, Elevated Levels of Arachidonic Acid-Derived Lipids Including Prostaglandins and Endocannabinoids Are Present Throughout ABHD12 Knockout Brains: Novel Insights Into the Neurodegenerative Phenotype. Front Mol Neurosci, 2019. 12: p. 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sałaga M, Mokrowiecka A, Zakrzewski PK, Cygankiewicz A, Leishman E, Sobczak M, Zatorski H, Małecka-Panas E, Kordek R, Storr M, Krajewska WM, Bradshaw HB, and Fichna J, Experimental colitis in mice is attenuated by changes in the levels of endocannabinoid metabolites induced by selective inhibition of fatty acid amide hydrolase (FAAH). J Crohns Colitis, 2014. 8(9): p. 998–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun X, Tavenier A, Deng W, Leishman E, Bradshaw HB, and Dey SK, Metformin attenuates susceptibility to inflammation-induced preterm birth in mice with higher endocannabinoid levels. Biol Reprod, 2018. 98(2): p. 208–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Toguri JT, Leishman E, Szczesniak AM, Laprairie RB, Oehler O, Straiker AJ, Kelly MEM, and Bradshaw HB, Inflammation and CB2 signaling drive novel changes in the ocular lipidome and regulate immune cell activity in the eye. Prostaglandins Other Lipid Mediat, 2018. 139: p. 54–62. [DOI] [PubMed] [Google Scholar]
- 27.Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, and Mechoulam R, Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science, 1992. 258(5090): p. 1946–9. [DOI] [PubMed] [Google Scholar]
- 28.McHugh D, Hu SS, Rimmerman N, Juknat A, Vogel Z, Walker JM, and Bradshaw HB, N-arachidonoyl glycine, an abundant endogenous lipid, potently drives directed cellular migration through GPR18, the putative abnormal cannabidiol receptor. BMC Neurosci, 2010. 11: p. 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brierley DI, Harman JR, Giallourou N, Leishman E, Roashan AE, Mellows BAD, Bradshaw HB, Swann JR, Patel K, Whalley BJ, and Williams CM, Chemotherapy-induced cachexia dysregulates hypothalamic and systemic lipoamines and is attenuated by cannabigerol. J Cachexia Sarcopenia Muscle, 2019. 10(4): p. 844–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Milman G, Maor Y, Abu-Lafi S, Horowitz M, Gallily R, Batkai S, Mo FM, Offertaler L, Pacher P, Kunos G, and Mechoulam R, N-arachidonoyl L-serine, an endocannabinoid-like brain constituent with vasodilatory properties. Proc Natl Acad Sci U S A, 2006. 103(7): p. 2428–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smoum R, Bar A, Tan B, Milman G, Attar-Namdar M, Ofek O, Stuart JM, Bajayo A, Tam J, Kram V, O’Dell D, Walker MJ, Bradshaw HB, Bab I, and Mechoulam R, Oleoyl serine, an endogenous N-acyl amide, modulates bone remodeling and mass. Proc Natl Acad Sci U S A, 2010. 107(41): p. 17710–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cohen-Yeshurun A, Willner D, Trembovler V, Alexandrovich A, Mechoulam R, Shohami E, and Leker RR, N-arachidonoyl-L-serine (AraS) possesses proneurogenic properties in vitro and in vivo after traumatic brain injury. J Cereb Blood Flow Metab, 2013. 33(8): p. 1242–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Greco R, Demartini C, Zanaboni AM, Tumelero E, Reggiani A, Misto A, Piomelli D, and Tassorelli C, FAAH inhibition as a preventive treatment for migraine: A pre-clinical study. Neurobiol Dis, 2020. 134: p. 104624. [DOI] [PubMed] [Google Scholar]
- 34.Argueta DA and DiPatrizio NV, Peripheral endocannabinoid signaling controls hyperphagia in western diet-induced obesity. Physiol Behav, 2017. 171: p. 32–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Balvers MG, Verhoeckx KC, and Witkamp RF, Development and validation of a quantitative method for the determination of 12 endocannabinoids and related compounds in human plasma using liquid chromatography-tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci, 2009. 877(14–15): p. 1583–90. [DOI] [PubMed] [Google Scholar]
- 36.Bashashati M, Leishman E, Bradshaw H, Sigaroodi S, Tatro E, Bright T, McCallum R, and Sarosiek I, Plasma endocannabinoids and cannabimimetic fatty acid derivatives are altered in gastroparesis: A sex- and subtype-dependent observation. Neurogastroenterol Motil, 2021. 33(1): p. e13961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Biernacki M, Brzóska MM, Markowska A, Gałażyn-Sidorczuk M, Cylwik B, Gęgotek A, and Skrzydlewska E, Oxidative Stress and Its Consequences in the Blood of Rats Irradiated with UV: Protective Effect of Cannabidiol. Antioxidants (Basel), 2021. 10(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.De Icco R, Greco R, Demartini C, Vergobbi P, Zanaboni A, Tumelero E, Reggiani A, Realini N, Sances G, Grillo V, Allena M, and Tassorelli C, Spinal nociceptive sensitization and plasma palmitoylethanolamide levels during experimentally induced migraine attacks. Pain, 2021. 162(9): p. 2376–2385. presentations from Eli-Lilly and Novartis. C. Tassorelli received honoraria for participation in advisory boards or for oral presentations from Allergan, ElectroCore, Eli-Lilly, Novartis, and Teva. C. Tassorelli has no ownership interest and does not own stocks of any pharmaceutical company. C. Tassorelli serves as Chief Section Editor of Frontiers in Neurology—Section Headache Medicine and Facial Pain and on the editorial board of The Journal of Headache and Pain. The remaining authors have no conflicts of interest to declare. Sponsorships or competing interests that may be relevant to content are disclosed at the end of this article. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Díaz-Rúa A, Chivite M, Comesaña S, Velasco C, Soengas JL, and Conde-Sieira M, Central administration of endocannabinoids exerts bimodal effects in food intake of rainbow trout. Horm Behav, 2021. 134: p. 105021. [DOI] [PubMed] [Google Scholar]
- 40.Díaz-Rúa A, Chivite M, Velasco C, Comesaña S, Soengas JL, and Conde-Sieira M, Periprandial response of central cannabinoid system to different feeding conditions in rainbow trout Oncorhynchus mykiss. Nutr Neurosci, 2020: p. 1–12. [DOI] [PubMed] [Google Scholar]
- 41.Ney L, Stone C, Nichols D, Felmingham K, Bruno R, and Matthews A, Endocannabinoid reactivity to acute stress: Investigation of the relationship between salivary and plasma levels. Biol Psychol, 2021. 159: p. 108022. [DOI] [PubMed] [Google Scholar]
- 42.Ulu A, Burr A, Heires AJ, Pavlik J, Larsen T, Perez PA, Bravo C, DiPatrizio NV, Baack M, Romberger DJ, and Nordgren TM, A high docosahexaenoic acid diet alters lung inflammation and recovery following repetitive exposure to aqueous organic dust extracts. J Nutr Biochem, 2021. 97: p. 108797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Martín-Pérez C, Contreras-Rodríguez O, Pastor A, Christensen E, Andrews ZB, de la Torre R, and Verdejo-García A, Endocannabinoid signaling of homeostatic status modulates functional connectivity in reward and salience networks. Psychopharmacology (Berl), 2021. [DOI] [PubMed] [Google Scholar]
- 44.Sempio C, Klawitter J, Jackson M, Freni F, Shillingburg R, Hutchison K, Bidwell LC, Christians U, and Klawitter J, Analysis of 14 endocannabinoids and endocannabinoid congeners in human plasma using column switching high-performance atmospheric pressure chemical ionization liquid chromatography-mass spectrometry. Anal Bioanal Chem, 2021. 413(12): p. 3381–3392. [DOI] [PubMed] [Google Scholar]
- 45.Manca C, Lacroix S, Pérusse F, Flamand N, Chagnon Y, Drapeau V, Tremblay A, Di Marzo V, and Silvestri C, Oral Capsaicinoid Administration Alters the Plasma Endocannabinoidome and Fecal Microbiota of Reproductive-Aged Women Living with Overweight and Obesity. Biomedicines, 2021. 9(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Turcotte C, Archambault AS, Dumais É, Martin C, Blanchet MR, Bissonnette E, Ohashi N, Yamamoto K, Itoh T, Laviolette M, Veilleux A, Boulet LP, Di Marzo V, and Flamand N, Endocannabinoid hydrolysis inhibition unmasks that unsaturated fatty acids induce a robust biosynthesis of 2-arachidonoyl-glycerol and its congeners in human myeloid leukocytes. Faseb j, 2020. 34(3): p. 4253–4265. [DOI] [PubMed] [Google Scholar]
- 47.Brunkhorst-Kanaan N, Trautmann S, Schreiber Y, Thomas D, Kittel-Schneider S, Gurke R, Geisslinger G, Reif A, and Tegeder I, Sphingolipid and Endocannabinoid Profiles in Adult Attention Deficit Hyperactivity Disorder. Biomedicines, 2021. 9(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kirkwood JS, Broeckling CD, Donahue S, and Prenni JE, A novel microflow LC-MS method for the quantitation of endocannabinoids in serum. J Chromatogr B Analyt Technol Biomed Life Sci, 2016. 1033–1034: p. 271–277. [DOI] [PubMed] [Google Scholar]
- 49.Siebers M, Biedermann SV, Bindila L, Lutz B, and Fuss J, Exercise-induced euphoria and anxiolysis do not depend on endogenous opioids in humans. Psychoneuroendocrinology, 2021. 126: p. 105173. [DOI] [PubMed] [Google Scholar]
- 50.Lerner R, Post JM, Ellis SR, Vos DRN, Heeren RMA, Lutz B, and Bindila L, Simultaneous lipidomic and transcriptomic profiling in mouse brain punches of acute epileptic seizure model compared to controls. J Lipid Res, 2018. 59(2): p. 283–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lerner R, Post J, Loch S, Lutz B, and Bindila L, Targeting brain and peripheral plasticity of the lipidome in acute kainic acid-induced epileptic seizures in mice via quantitative mass spectrometry. Biochim Biophys Acta Mol Cell Biol Lipids, 2017. 1862(2): p. 255–267. [DOI] [PubMed] [Google Scholar]
- 52.Tagliamonte S, Laiola M, Ferracane R, Vitale M, Gallo MA, Meslier V, Pons N, Ercolini D, and Vitaglione P, Mediterranean diet consumption affects the endocannabinoid system in overweight and obese subjects: possible links with gut microbiome, insulin resistance and inflammation. Eur J Nutr, 2021. 60(7): p. 3703–3716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gaitán AV, Wood JT, Solomons NW, Donohue JA, Ji L, Liu Y, Nikas SP, Zhang F, Allen LH, Makriyannis A, and Lammi-Keefe CJ, Endocannabinoid Metabolome Characterization of Milk from Guatemalan Women Living in the Western Highlands. Curr Dev Nutr, 2019. 3(6): p. nzz018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gaitán AV, Wood JT, Zhang F, Makriyannis A, and Lammi-Keefe CJ, Endocannabinoid Metabolome Characterization of Transitional and Mature Human Milk. Nutrients, 2018. 10(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bruun S, Gouveia-Figueira S, Domellöf M, Husby S, Neergaard Jacobsen L, Michaelsen KF, Fowler CJ, and Zachariassen G, Satiety Factors Oleoylethanolamide, Stearoylethanolamide, and Palmitoylethanolamide in Mother’s Milk Are Strongly Associated with Infant Weight at Four Months of Age-Data from the Odense Child Cohort. Nutrients, 2018. 10(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Annesley TM, Ion suppression in mass spectrometry. Clin Chem, 2003. 49(7): p. 1041–4. [DOI] [PubMed] [Google Scholar]
- 57.Guan Z, Discovering novel brain lipids by liquid chromatography/tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci, 2009. 877(26): p. 2814–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Luque-Córdoba D, Calderón-Santiago M, Luque de Castro MD, and Priego-Capote F, Study of sample preparation for determination of endocannabinoids and analogous compounds in human serum by LC-MS/MS in MRM mode. Talanta, 2018. 185: p. 602–610. [DOI] [PubMed] [Google Scholar]
- 59.Gaisberger M, Fuchs J, Riedl M, Edtinger S, Reischl R, Grasmann G, Hölzl B, Landauer F, Dobias H, Eckstein F, Offenbächer M, Ritter M, and Winklmayr M, Endogenous anandamide and self-reported pain are significantly reduced after a 2-week multimodal treatment with and without radon therapy in patients with knee osteoarthritis: a pilot study. Int J Biometeorol, 2021. 65(7): p. 1151–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tan B, O’Dell DK, Yu YW, Monn MF, Hughes HV, Burstein S, and Walker JM, Identification of endogenous acyl amino acids based on a targeted lipidomics approach. J Lipid Res, 2010. 51(1): p. 112–9. [DOI] [PMC free article] [PubMed] [Google Scholar]