

Abstract
Recent advances in spatially resolved transcriptomics (SRT) have brought ever-increasing opportunities to characterize expression landscape in the context of tissue spatiality. Nevertheless, there still exist multiple challenges to accurately detect spatial functional regions in tissue. Here, we present a novel contrastive learning framework, SPAtially Contrastive variational AutoEncoder (SpaCAE), which contrasts transcriptomic signals of each spot and its spatial neighbors to achieve fine-grained tissue structures detection. By employing a graph embedding variational autoencoder and incorporating a deep contrastive strategy, SpaCAE achieves a balance between spatial local information and global information of expression, enabling effective learning of representations with spatial constraints. Particularly, SpaCAE provides a graph deconvolutional decoder to address the smoothing effect of local spatial structure on expression’s self-supervised learning, an aspect often overlooked by current graph neural networks. We demonstrated that SpaCAE could achieve effective performance on SRT data generated from multiple technologies for spatial domains identification and data denoising, making it a remarkable tool to obtain novel insights from SRT studies.
Keywords: spatially resolved transcriptomics, spatial domain identification, spatially contrastive learning, graph embedding variational autoencoder
INTRODUCTION
Understanding the intricate process underlying organ genesis and pathology necessitates a comprehensive study of in situ gene expression variations across different tissue regions. The recent emergence of spatially resolved transcriptomics (SRT) technologies has revolutionized scientific research in these domains. These innovative technologies enable the concurrent measurement of gene expression and their precise spatial locations, significantly advancing our capabilities in these fields [1, 2]. Sequencing-based SRT methods offer the potential for whole-genome-wide gene expression profiling [3, 4]. However, they are constrained by resolution, typically quantifying gene expression within small groups of cells known as spots, rather than at the single-cell level. For instance, the widely used 10x Visium technology can measure a spot encompassing approximately 10 cells, with a diameter of 55 μm. Recently, high-resolution SRT technologies, such as Slide-seq [5], Slide-seqV2 [6] and Stereo-seq [7], have emerged, offering near single-cell resolution measurements. These cutting-edge technologies, while promising, have presented a common challenge—the data they generate often exhibit dropouts and elevated noise levels. This challenge is particularly pronounced in large-scale high-resolution datasets, underscoring the need for robust computational methods to effectively address these complexities.
Spatial domain detection is a fundamental task in SRT data analysis. It involves identifying clusters of spots with similar gene expression patterns and spatial coherence. Such spatial domains often represent regions with shared biological functions, making them crucial for downstream analyses, including the identification of spatially variable genes (SVGs) and prognostic gene discovery in tumor tissues. To effectively address spatial domain detection, computational methods must simultaneously model gene expression data and their corresponding spatial dependencies. Previously proposed computational approaches for spatial domain detection have fallen into two primary categories: probabilistic models and deep learning models. Probabilistic models, such as hidden Markov random field (HMRF) [8], BayesSpace [4] and SpatialPCA [9], typically tailor probabilistic graphical models to accommodate spatial information. In contrast, deep learning models, including SpaGCN [10], STAGATE [11], DeepST [12], GraphST [13] and SpaceFlow [14], leverage graph convolutional modules and their variations to model spatial dependencies. The existing methods directly utilize sparse SRT data as input, limiting their ability to fully exploit the spatial dependence of gene expression and effectively mitigate the impact of high sparsity. Therefore, how to effectively use highly sparse SRT data with spatial context to explore the expression landscape in tissue is still a challenge.
Here, we develop a novel contrastive learning, SPAtially Contrastive variational AutoEncoder (SpaCAE), which discerns fine-grained tissue structures by contrasting the transcriptomic signals between each spot and its spatial neighborhoods. Specifically, SpaCAE generates spatially augmented expression of each spot by borrowing the shared information from spatially neighboring spots. Then, SpaCAE builds the graph convolutional encoder and graph deconvolutional decoder to learn effective representation with spatial constraint from the original and augmented expression data. Finally, SpaCAE provides a deep contrastive strategy to achieve a harmonious integration of spatial local information and global information during the process of representation learning. Especially, SpaCAE reduces the smoothing effect of local spatial structure on expression’s self-supervised learning by graph deconvolutional decoder, which is ignored by current graph neural networks. In this way, SpaCAE not only rectifies the low-quality gene expression by adopting the spatial dependence of gene expression but also balances spatial coherence and expression variability to improve the performance of spatial domains detection.
We demonstrate the superiority of SpaCAE by benchmarking it against seven state-of-the-art methods on a total of 16 SRT datasets generated by 10x Visium, Slide-seqV2 and ST technologies. For the SRT datasets with known ground truth annotations from 10x Visium and imaging-based platform, SpaCAE exhibits higher clustering accuracy than other methods. Applied to mouse hippocampus Slide-seqV2 data, SpaCAE reveals finer-grained anatomical regions for interpreting tissue functions. Employed in human breast cancer ST data, SpaCAE deciphers tumor heterogeneity and provides more biological insights on cancer-associated genes, providing valuable validation by the survival analysis of independent clinical data.
METHODS
Overview of SpaCAE
SpaCAE learns the representation with spatial constraint from multi-modal data by balancing the spatial dependence and variance of gene expression (Figure 1A–C). Specifically, SpaCAE employs the spatial dependence of gene expression to contrast the biological signals of each spot with those of its adjoining spatial neighbors during representation learning (Figure 1B).
Figure 1.

Schematic overview of SpaCAE augmentation-encoding-decoding (A–C) processes and potential biological applications of SpaCAE in downstream SRT analysis (D, E). (A) The three data sources for SpaCAE inputs are STR expression matrix, spatial coordinates and hematoxylin and eosin (H&E) staining image. (B) Weighted graph (i.e. 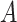 ) construction from spatial multi-modal data and spatial expression augmentation from aggregating expression information from neighborhood spots. SpaCAE integrates spatial information into gene expression to augment the shared expression between spots by aggregating the gene expression from their K spatial neighbors through weighted graph, which forms the spots’ spatial contrastive structure (e.g.
) construction from spatial multi-modal data and spatial expression augmentation from aggregating expression information from neighborhood spots. SpaCAE integrates spatial information into gene expression to augment the shared expression between spots by aggregating the gene expression from their K spatial neighbors through weighted graph, which forms the spots’ spatial contrastive structure (e.g. 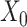 →
→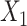 ). (C) SpaCAE model. SpaCAE uses robust graph convolutional encoder and deep contrastive variational autoencoder to generate the low-dimensional representation (i.e.
). (C) SpaCAE model. SpaCAE uses robust graph convolutional encoder and deep contrastive variational autoencoder to generate the low-dimensional representation (i.e. 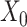 →
→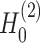 ,
, 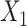 →
→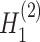 ). The input data are the original gene expression matrix (i.e.
). The input data are the original gene expression matrix (i.e. 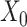 ), the augmented gene expression matrix (i.e.
), the augmented gene expression matrix (i.e. 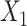 ) and weighted spatial graph
) and weighted spatial graph 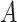 from (B). Based on the original and low-dimensional data, SpaCAE performs data reconstructions via graph deconvolutional decoder and deep contrastive variational autoencoder. SpaCAE iteratively learns the graph convolutional encoder
from (B). Based on the original and low-dimensional data, SpaCAE performs data reconstructions via graph deconvolutional decoder and deep contrastive variational autoencoder. SpaCAE iteratively learns the graph convolutional encoder 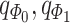 and graph deconvolutional decoder
and graph deconvolutional decoder 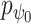 ,
, 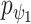 by minimizing the sum of reconstruction losses and contrastive losses (see Methods). When SpaCAE reaches convergence, the optimum is achieved for further downstream analyses. (D, E) Biological applications for SpaCAE including data denoising and spatial domain identification. (D) The reconstructed augmented spatial expression
by minimizing the sum of reconstruction losses and contrastive losses (see Methods). When SpaCAE reaches convergence, the optimum is achieved for further downstream analyses. (D, E) Biological applications for SpaCAE including data denoising and spatial domain identification. (D) The reconstructed augmented spatial expression 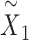 can be employed to denoise data. (E) The low-dimensional representation (i.e. (
can be employed to denoise data. (E) The low-dimensional representation (i.e. (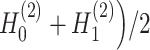 ) can be applied to detect spatial domains.
) can be applied to detect spatial domains.
SpaCAE first borrows the shared information from spatially neighboring spots to generate augmented expression of each spot based on the constructed graph 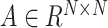 (i.e. original data
(i.e. original data 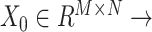 augmented data
augmented data 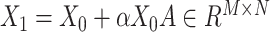 ,
, 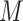 and
and 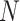 denote the number of genes and spots). Then, SpaCAE learns effective representation from the original data
denote the number of genes and spots). Then, SpaCAE learns effective representation from the original data 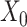 and the augmented data
and the augmented data 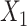 based on a novel graph embedding contrastive variational autoencoder (GC-VAE) model (Figure 1C). Note that SpaCAE leverages a graph convolutional network (GCN) encoder to ensure the learned representation with spatial constraint and provides a graph deconvolutional network (GDN) decoder to reduce the smoothing effect of local spatial structure during representation learning. To balance spatial local information and global information of expression from original data
based on a novel graph embedding contrastive variational autoencoder (GC-VAE) model (Figure 1C). Note that SpaCAE leverages a graph convolutional network (GCN) encoder to ensure the learned representation with spatial constraint and provides a graph deconvolutional network (GDN) decoder to reduce the smoothing effect of local spatial structure during representation learning. To balance spatial local information and global information of expression from original data 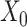 and the augmented data
and the augmented data 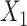 , SpaCAE proposes the deep contrastive strategy to contrast each hidden layer (i.e.
, SpaCAE proposes the deep contrastive strategy to contrast each hidden layer (i.e. 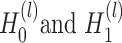 ,
, 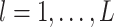 ,
, 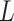 is the number layers) of GC-VAE.
is the number layers) of GC-VAE.
In this way, SpaCAE can learn an optimal latent representation (i.e. 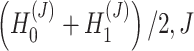 is the number of encoder layers) and reconstructed expression matrix
is the number of encoder layers) and reconstructed expression matrix 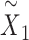 , which can be used for multiple downstream analytical tasks (Figure 1D and E). Matrix
, which can be used for multiple downstream analytical tasks (Figure 1D and E). Matrix 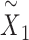 enables SpaCAE to denoise the expression profile to improve individual gene analysis (Figure 1D), e.g. SVGs or differentially expressed genes (DEGs) identification. The optimal
enables SpaCAE to denoise the expression profile to improve individual gene analysis (Figure 1D), e.g. SVGs or differentially expressed genes (DEGs) identification. The optimal 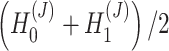 serves to detect spatial domains (Figure 1E) interoperating with Mclust [15].
serves to detect spatial domains (Figure 1E) interoperating with Mclust [15].
Building SpaCAE model
Constructing graph-structure data form spatial multi-modal data
SpaCAE converts spatial multi-modal data into a weighted graph 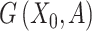 . Matrix
. Matrix 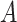 denotes the relationship between each spot and its spatial neighbors, which reflects spatially local structure of SRT.
denotes the relationship between each spot and its spatial neighbors, which reflects spatially local structure of SRT.
First, SpaCAE uses the strategy from SpaGCN [10] to calculate the Euclidean distance of each pair of spots based on the spatial coordinates and the corresponding histology image information and screens out the K-nearest spatial neighbors [16] of each spot (i.e. K default to 10 for 10x Visium datasets and 30 for Silde-seqV2 and Stereo-seq datasets).
Then, SpaCAE calculates the cosine distance of these neighboring spots based on PCA embedding (i.e. PCs) of expression (i.e. 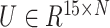 , 15 is the number of PCs from expression data) and exponentially transforms this distance to a similarity matrix (i.e.
, 15 is the number of PCs from expression data) and exponentially transforms this distance to a similarity matrix (i.e. 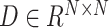 ) as
) as
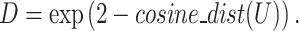 |
(1) |
The weight matrix 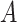 (i.e.
(i.e. 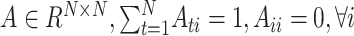 ) is the normalized form of the similarity matrix
) is the normalized form of the similarity matrix 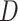 .
.
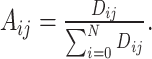 |
(2) |
Spatial expression augmentation
The gene expression of each spot is modified by incorporating shared information from its surrounding neighborhoods, and it is regarded as the augmented spatial expression of the corresponding spot. Using a weight matrix 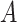 , the augmented expression matrix
, the augmented expression matrix 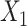 can be calculated specifically as follows:
can be calculated specifically as follows:
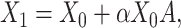 |
(3) |
where the tunable parameter 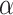 can be manually set according to the obtained outcomes, which controls the impact of spatial neighborhood similarity on spatial domains (Supplementary Figure 1).
can be manually set according to the obtained outcomes, which controls the impact of spatial neighborhood similarity on spatial domains (Supplementary Figure 1).
Graph embedding contrastive variational autoencoder
We build a GC-VAE to contrast the biological signals of each spot and its spatial neighbors for learning representation. There are three principal components: graph convolutional encoder, graph deconvolutional decoder and deep contrastive variational autoencoder (VAE). The main procedure can be stated as follows.
Graph convolutional encoder: we use the GCN as the encoder to learn representation with spatial constraint from the original and augmented expression data (i.e.
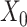 and
and 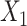 ). In general, graph convolutional operation can be interpreted as a special form of Laplacian smoothing [17]. From the spectral perspective, the tth layer of the graph convolutional encoder for
). In general, graph convolutional operation can be interpreted as a special form of Laplacian smoothing [17]. From the spectral perspective, the tth layer of the graph convolutional encoder for 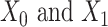 , denoted as
, denoted as 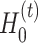 and
and 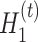 , is defined as
, is defined as
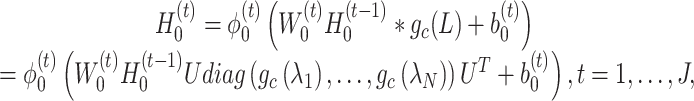 |
(4) |
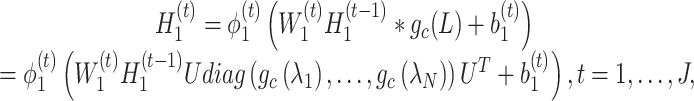 |
(5) |
where 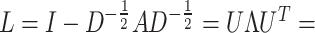
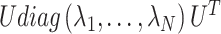 is the normalized Laplacian matrix and
is the normalized Laplacian matrix and 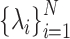 and
and 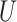 are the eigenvalues and eigenvectors of matrix
are the eigenvalues and eigenvectors of matrix 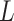 .
. 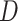 denotes the degree matrix.
denotes the degree matrix. 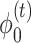 and
and 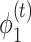 are the activation functions of the tth GCN encoder layer for
are the activation functions of the tth GCN encoder layer for 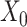 and
and 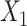 , respectively.
, respectively. 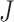 is the number of encoder layers. * denotes convolutional operator.
is the number of encoder layers. * denotes convolutional operator. 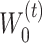 and
and 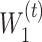 denote the weight matrix of encoder for
denote the weight matrix of encoder for 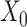 and
and 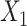 , respectively.
, respectively. 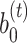 and
and 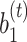 correspond to the bias term of VAE for
correspond to the bias term of VAE for 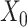 and
and 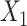 , respectively. We use the convolutional operator of GCN as the convolution operation of this model (i.e.
, respectively. We use the convolutional operator of GCN as the convolution operation of this model (i.e. 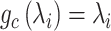 ). For convenience, we denote the gene expression matrix
). For convenience, we denote the gene expression matrix 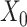 and
and 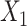 as
as 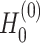 and
and 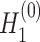 , respectively.
, respectively.
Graph deconvolutional decoder: although graph convolution can effectively learn representation with local spatial information, the resulting smoothness affects the expression data reconstruction and weakens the global information of the learnable representation from the gene expression data. To solve this problem, we propose the GDN as the decoder to attenuate the effect of graph convolution, enabling efficient representation to be learned from the expression. As an inverse to graph convolution, the kth layer (
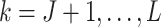 ,
, 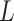 is the number of GC-VAE layers) of the graph deconvolution decoder for
is the number of GC-VAE layers) of the graph deconvolution decoder for 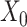 and
and 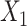 , denoted as
, denoted as 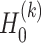 and
and 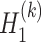 , is defined as
, is defined as
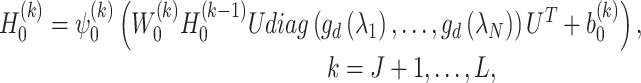 |
(6) |
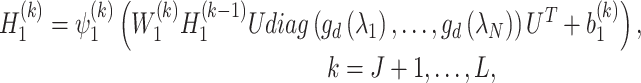 |
(7) |
where 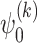 and
and 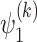 are the activation functions of the
are the activation functions of the 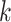 th GDN decoder layer for
th GDN decoder layer for 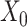 and
and 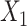 , respectively.
, respectively. 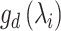 is the deconvolutional operator of GDN.
is the deconvolutional operator of GDN. 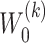 and
and 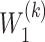 denote the weight matrix of decoder for
denote the weight matrix of decoder for 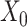 and
and 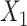 , respectively.
, respectively. 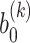 and
and 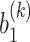 correspond to the bias term of VAE for
correspond to the bias term of VAE for 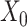 and
and 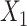 , respectively.
, respectively.
In general, the deconvolutional operator 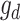 is the inverse function of
is the inverse function of 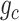 , e.g.
, e.g. 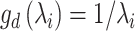 . Due to the randomness of the latent representation in GC-VAE, the general deconvolutional operator is not suitable for this model (Supplementary Note S1). Therefore, we adopt the following new deconvolution operators for the
. Due to the randomness of the latent representation in GC-VAE, the general deconvolutional operator is not suitable for this model (Supplementary Note S1). Therefore, we adopt the following new deconvolution operators for the 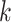 th layer decoder
th layer decoder 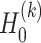 and
and 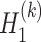 :
:
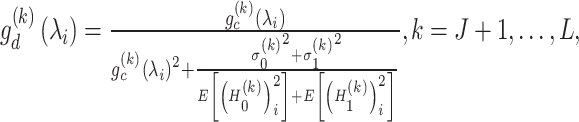 |
(8) |
where 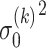 and
and 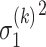 are the variance of the stochastic disturbance in
are the variance of the stochastic disturbance in 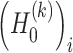 and
and 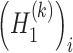 and
and 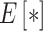 denotes mathematical expectation, respectively. The average values (i.e.
denotes mathematical expectation, respectively. The average values (i.e. 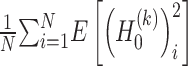 and
and 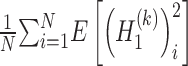 ) are used to approximate the value of
) are used to approximate the value of 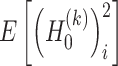 and
and 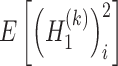 , respectively. Due to the following statistical formula
, respectively. Due to the following statistical formula
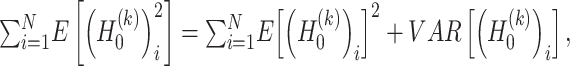 |
(9) |
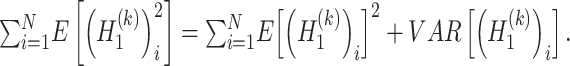 |
(10) |
The values of 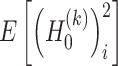 and
and 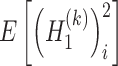 are estimated as follows:
are estimated as follows:
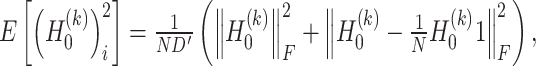 |
(11) |
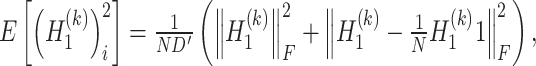 |
(12) |
where 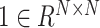 is the all ones matrix and
is the all ones matrix and 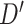 is the feature size of the
is the feature size of the 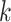 th layer of decoder. The variance
th layer of decoder. The variance 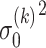 ,
, 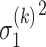 are estimated by considering their neighborhoods as
are estimated by considering their neighborhoods as
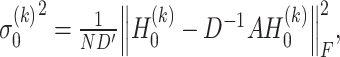 |
(13) |
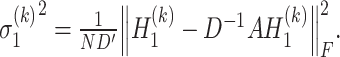 |
(14) |
Deep contrastive VAE: to balance spatial local information and global information from the original and augmented expression data, we present a deep contrastive strategy as the soft constraint during representation learning. The contrastive loss can be expressed as
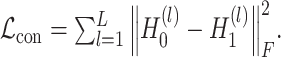 |
(15) |
Combined with the loss (i.e. 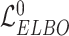 and
and 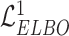 ) of VAE for the original data
) of VAE for the original data 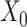 and the augmented expression data
and the augmented expression data 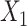 , the overall loss function of SpaCAE is denoted as follows:
, the overall loss function of SpaCAE is denoted as follows:
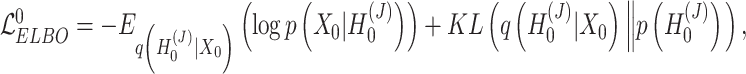 |
(16) |
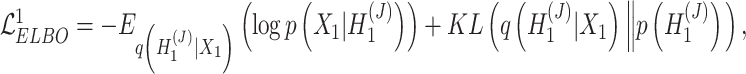 |
(17) |
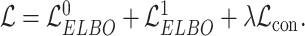 |
(18) |
where the tunable parameter 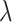 can be manually set and defaults to 1.
can be manually set and defaults to 1.
Hyperparameter tunings and implementations
In SpaCAE model, two crucial hyperparameters, namely, 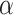 and
and 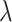 , significantly impact its performance. Parameter
, significantly impact its performance. Parameter 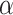 controls the proportion of the neighborhood to the center spot in the augmented gene expression
controls the proportion of the neighborhood to the center spot in the augmented gene expression 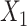 , thereby influencing the spatially local structures or similarities of the learned representation (Supplementary Figure 1). On the other hand, parameter
, thereby influencing the spatially local structures or similarities of the learned representation (Supplementary Figure 1). On the other hand, parameter 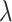 determines the strength of the contrastive loss, facilitating the model in learning consistent representations from both the original gene expression
determines the strength of the contrastive loss, facilitating the model in learning consistent representations from both the original gene expression 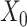 and the augmented gene expression
and the augmented gene expression 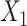 (Supplementary Figures 2 and 3). Increasing both parameters
(Supplementary Figures 2 and 3). Increasing both parameters 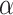 and
and 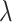 results in a final representation learned by SpaCAE with a stronger spatially local structure and improved spatial consistency of the identified spatial domains.
results in a final representation learned by SpaCAE with a stronger spatially local structure and improved spatial consistency of the identified spatial domains.
In real applications, a fixed value of 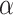 cannot work for all kinds of datasets because different datasets could have different sensitivities to the changes of
cannot work for all kinds of datasets because different datasets could have different sensitivities to the changes of 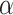 . We started to search
. We started to search 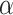 from a small value, and the default search list for
from a small value, and the default search list for 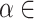 [0,2] in SpaCAE is {0.005, 0.01, 0.05, 0.1, 0.2, 0.3, …, 2}. For each fixed
[0,2] in SpaCAE is {0.005, 0.01, 0.05, 0.1, 0.2, 0.3, …, 2}. For each fixed 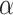 ,
, 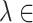 [0,5] is determined by grid search based on the cross-validation. The goal of SpaCAE is to identify the spatial regions that are most consistent with the pathological characteristics and physiological structure. Based on this motivation as a priori, we decide
[0,5] is determined by grid search based on the cross-validation. The goal of SpaCAE is to identify the spatial regions that are most consistent with the pathological characteristics and physiological structure. Based on this motivation as a priori, we decide 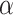 and
and 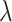 by maximizing unsupervised cluster evaluation measurements ARI or purity. In cases where datasets lack manual annotations but include reference diagrams, we provide a search range for parameters
by maximizing unsupervised cluster evaluation measurements ARI or purity. In cases where datasets lack manual annotations but include reference diagrams, we provide a search range for parameters 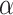 and
and 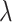 in the SRT data from different platforms. Based on these search range, the optimal combination of parameters
in the SRT data from different platforms. Based on these search range, the optimal combination of parameters 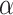 and
and 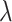 is determined based on the output results (Supplementary Table 1).
is determined based on the output results (Supplementary Table 1).
Data collection and general preprocessing
We used 16 datasets from four different SRT platforms including 12 10x Visium datasets, 2 imaging-based SRT slices, 1 Slide-seqV2 dataset and 1 ST [18] dataset. Based on SCANPY [19] Python package, we firstly selected top 2000 highly variable genes (HVGs). Then, we log-transformed and standardized the original gene expression data as the input of SpaCAE.
Spatial domain identification, gene expression denoising and survival analysis
We used the latent representation 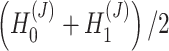 to identify spatial domains by Mclust algorithms. We used the reconstructed augmented expression
to identify spatial domains by Mclust algorithms. We used the reconstructed augmented expression 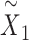 to denoise the SRT profiles. We performed Kaplan–Meier (KM) analysis and evaluated the prognostic significance of domain-specific marker genes using bulk expression profiling data with patient survival information in cancer study.
to denoise the SRT profiles. We performed Kaplan–Meier (KM) analysis and evaluated the prognostic significance of domain-specific marker genes using bulk expression profiling data with patient survival information in cancer study.
Performance evaluation
We describe below the metrics to evaluate the performance of SpaCAE. Details of the benchmarking approaches are provided in Supplementary Note S1.
Accuracy of spatial domains identification: Adjusted Rand index (ARI) [20–26] and cluster purity (i.e. Equation (19)) [4] are used to quantify the accuracy of identifying spatial domains.
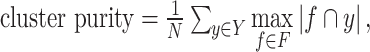 |
(19) |
where Y is the set of clusters or spatial domains; 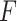 is the set of reference groups.
is the set of reference groups.
Spatial continuity and expression specificity of gene after denoising: the Moran’s I statistic is employed to assess the spatial autocorrelation of gene expression both before and after the denoising process. Additionally, we evaluate the specificity of gene expression before and after denoising by comparing the log2 fold change (Log2FC) values of the top marker genes associated with each spatial domain.
RESULTS
Benchmarking SpaCAE against existing spatial domain detection methods
To quantitatively evaluate the spatial domain identification ability of SpaCAE, we first applied SpaCAE onto 12 human dorsolateral prefrontal cortex (DLPFC) sections from 10x Visium. Maynard et al. [27] has manually annotated the cortical layers (L1-L6) and white matter (WM) of these slides. Regarding them as the ground truth, we compared SpaCAE with existing state-of-the-art methods (i.e. STAGATE [11], GraphST [13], BayesSpace [4], DeepST [12] and SpaGCN [10]) and graph autoencoder (GAE) [28] and VAE [29] in terms of ARI, and the results demonstrated that the spatial domains identified by SpaCAE were consistent with the manual annotation of DLPFC and the definition of cortical stratification in neuroscience.
For the boundary division of cortical layers, the ARI of SpaCAE was 0.601 ± 0.079, which was substantially higher than that of other methods (e.g. GraphST, ARI = 0.538 ± 0.084; STAGATE, ARI = 0.499 ± 0.106; DeepST, ARI = 0.472 ± 0.071; BayesSpace, ARI = 0.448 ± 0.076; SpaGCN, ARI = 0.413 ± 0.082, the difference between SpaCAE and each of the others is confirmed by the Wilcoxon rank-sum test, P < 0.05, as depicted in Figure 2B and Supplementary Figure 4). Taking slide 151675 (there are 3592 spots and 33 538 genes) as an example, we found that SpaCAE obtained the higher clustering accuracy with ARI = 0.686 (Figure 2A and D). Notably, domains identified by SpaCAE demonstrated smoother boundaries and less noisy.
Figure 2.

Comparative performance of SpaCAE to existing spatial and non-spatial methods on spatial domain identification. (A) DLPFC layers were annotated by Maynard et al. The ground truth of spots was mapped on their spatial position in slide 151675 (n = 3592 spots), which was separated into six cortical layers (L1–L6) and WM. Layers with annotations are provided on the remaining slides (Supplementary Figure 4). (B) Boxplots of the performance of SpaCAE and other algorithms for all 12 DLPFCs. The y-axis shows the ARI, which was used to compare the similarity of the predicted spatial layers and the manually annotated layers for each algorithm. (C) ARI boxplots of whether deep spatial contrast is used, whether spatial data augmentation is used and whether replace GCN/GDN with linear layer in SpaCAE are shown. (D) Identification of spatial domains by SpaCAE and existing state-of-the-art algorithms (GraphST, STAGATE, DeepST, BayesSpace, SpaGCN), VAE and GAE for slide 151675.
Specifically, we systematically evaluated the contribution of SpaCAE components to overall performance. First, we replaced graph convolution in encoder 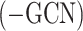 and graph deconvolution in decoder (
and graph deconvolution in decoder (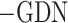 ) with linear layers on 12 DLPFCs and calculated their ARI values (Figure 2C).
) with linear layers on 12 DLPFCs and calculated their ARI values (Figure 2C). 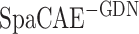 (ARI = 0.523 ± 0.097) obtained higher ARI values and better model robustness (
(ARI = 0.523 ± 0.097) obtained higher ARI values and better model robustness (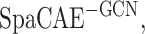 ARI = 0.362 ± 0.092) relatively, but it was still inferior to the complete SpaCAE (ARI = 0.601 ± 0.079). On the one hand, the resolved relative spatial locations of spots provide valuable information about neighboring context, and the graph convolution contributed to representations with local information constraints; on the other hand, the corresponding graph deconvolution compensated for the global information reduction caused by local smoothness of graph convolution exactly. However, in the absence of graph convolution, the bias could be further propagated to local areas through graph deconvolution networks. Considering the fact that the deep contrast strategy is what differentiates SpaCAE from other spatial algorithms, we ran SpaCAE without 1st and 3rd contrast operations [Figure 2C, –SC (h1/h3, i.e. spatial contrast), ARI = 0.506 ± 0.097] and without entire deep spatial contrast [Figure 2C, −SC(h1/h2/h3), ARI = 0.432 ± 0.120]. Deep contrast could significantly improve SpaCAE performance. To quantitatively assess the contribution of the augmentation matrix
ARI = 0.362 ± 0.092) relatively, but it was still inferior to the complete SpaCAE (ARI = 0.601 ± 0.079). On the one hand, the resolved relative spatial locations of spots provide valuable information about neighboring context, and the graph convolution contributed to representations with local information constraints; on the other hand, the corresponding graph deconvolution compensated for the global information reduction caused by local smoothness of graph convolution exactly. However, in the absence of graph convolution, the bias could be further propagated to local areas through graph deconvolution networks. Considering the fact that the deep contrast strategy is what differentiates SpaCAE from other spatial algorithms, we ran SpaCAE without 1st and 3rd contrast operations [Figure 2C, –SC (h1/h3, i.e. spatial contrast), ARI = 0.506 ± 0.097] and without entire deep spatial contrast [Figure 2C, −SC(h1/h2/h3), ARI = 0.432 ± 0.120]. Deep contrast could significantly improve SpaCAE performance. To quantitatively assess the contribution of the augmentation matrix 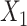 on SpaCAE performances, we ran VAE with matrix
on SpaCAE performances, we ran VAE with matrix 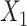 [Figure 2C, VAE (with DA, i.e. data augmented), ARI = 0.377 ± 0.069] and with matrix
[Figure 2C, VAE (with DA, i.e. data augmented), ARI = 0.377 ± 0.069] and with matrix 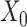 [Figure 2C, VAE (without DA), ARI = 0.190 ± 0.128]. The results show that matrix
[Figure 2C, VAE (without DA), ARI = 0.190 ± 0.128]. The results show that matrix 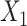 does improve the performance of clustering results than matrix
does improve the performance of clustering results than matrix 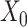 , but it is still inferior to the representation of SpaCAE. Thus, spatial data augmentation could significantly reduce noise interference and clarify spatial patterns to improve clustering performance. These results indicated these components all play crucial roles in accurately identifying tissue spatial structure.
, but it is still inferior to the representation of SpaCAE. Thus, spatial data augmentation could significantly reduce noise interference and clarify spatial patterns to improve clustering performance. These results indicated these components all play crucial roles in accurately identifying tissue spatial structure.
SpaCAE detects tissue functional regions in single-cell resolution SRT data
SpaCAE could work well on various SRT data independent of platforms. Aside from the 10x Visium platform, we then tested that SpaCAE on the osmFISH data from mouse somatosensory cortex includes 5328 positions and 33 genes at a coverage of 162 UMIs per bead, which profiles the typical anatomical multi-layered structure of the cortex [30]. Strikingly, SpaCAE presented the highest ARI than the competing methods over the slice (SpaCAE, ARI = 0.466; DeepST, ARI = 0.425; SpaGCN, ARI = 0.406; BayesSpace, ARI = 0.383; SpatialPCA, ARI = 0.316; STAGATE, ARI = 0.227) and relatively less noise points (the difference between SpaCAE and each of the others is confirmed by the Wilcoxon rank-sum test based on random seed settings, P < 0.01, as depicted in Figure 3A and B and Supplementary Figure 5).
Figure 3.

SpaCAE improves spatial domain recognition for high-resolution ST data. (A) Mouse somatosensory cortex osmFISH data (n = 5328 spots) was manually annotated into 12 categories. (B) Cluster assignments of somatosensory cortex layers generated by SpaCAE and five spatial algorithms (DeepST, SpaGCN, BayesSpace, SpatialPCA and STAGATE). (C) The ground truth of imaging-based molecular MIBI-TOF data (n = 1023 spots). (D) Spatial subdomains detected by above five methods and comparison of ARI.
Similarly, SpaCAE was applied to another imaging-based molecular multiplexed ion beam imaging by time of flight (MIBI-TOF) data, which imaged 36 labeled antibodies with histochemical staining and endogenous elements [12]. SpaCAE revealed partial regional continuity and local element fusion on the imaging results, which was almost compatible with original annotation (SpaCAE, ARI = 0.357; SpaGCN, ARI = 0.336; DeepST, ARI = 0.330; BayesSpace, ARI = 0.328; SpatialPCA, ARI = 0.206; STAGATE, ARI = 0.200, the difference between SpaCAE and each of the others is confirmed by the Wilcoxon rank-sum test based on random seed settings, P < 0.01, as depicted in Figure 3C and D and Supplementary Figure 5). Overall, SpaCAE also demonstrated better performance on spatial functional regions for the single-cell resolution SRT data.
SpaCAE reveals the finer-grained anatomical regions on mouse hippocampus Slide-seqV2 data
SpaCAE enables to identify spatial domains of tissue structure from SRT data of different spatial resolution. We next evaluated the performance of SpaCAE on a mouse hippocampus Slide-seqV2 dataset (41 786 locations), which has a higher resolution than the 10x Visium platform [6]. Slide-seqV2 spatial expression profiles have more spots (>10 000 per slice), but higher dropouts, leading to greater challenges to identify tissue structure efficiently and greater time and computational consumption in domain detection.
Considering the anatomical diagram from Allen Mouse Brain Atlas [31] as the illustrative reference, we compared SpaCAE with several methods to validate the performance of SpaCAE on a high-resolution and large-scale dataset. It was revealed that SpaCAE and STAGATE shows better regional continuity than other involved methods. In terms of fine-grained hippocampus structure, such as CA2, CA3, ventricle, dorsal part of the lateral geniculate complex (LGD) and ventral posteromedial nucleus of the thalamus (VPM) in the thalamus region, SpaCAE detected the corresponding regions with high consistency with Allen’ s reference (Figure 4A).
Figure 4.

SpaCAE identifies tissue structures and functional genes in mouse hippocampus Slide-seqV2 (n = 41 786 spots) data. (A) The corresponding anatomical definitions obtained from the Allen Mouse Brain Atlas (first image in A) is shown as reference. The identified spatial domains by all the involved approaches are illustrated on the spatial coordinates and distinguished using different colors without anatomical correspondence. The fine anatomical regions, for example, CA3, thalamus (LGD + VPM), CA2 and ventricle sections (second image in A) that SpaCAE detected while other methods did not are marked by black circles on computational results. (B) Individual loadings of each spatial domain identified by SpaCAE (top), the expression of domain-specific differential genes (middle) and the corresponding ISH (bottom).
Key domains were further separated and validated individually from the perspective of genetic landscape. The expressions of many known gene markers also verified the cluster partition of SpaCAE. Concerning the regions identified only by SpaCAE, for CA2 and CA3 areas firstly, we discovered that the domain locations demonstrated high correspondence with the expression profiles of the marker genes (i.e. S100b for CA2 [32] and Prkcd for CA3 [33]) and independent ISH (in situ hybridization) image (Figure 4B). Then for the ventricle area, the shape of the detected domain was consistent with that of the region where the marker gene (i.e. Adck4 [34]) expressed at a prominently high level. Furthermore, SpaCAE successfully identified the LGD and VPM regions in the thalamus, which were not captured by other methods, and Tnnt1 displayed specific spatial gene expression patterns in these areas [35] (Figure 4B). These results indicated that SpaCAE could dissect spatial heterogeneity and further uncover spatial expression patterns; meanwhile, SpaCAE could be well adapted to large-scale and high-resolution datasets.
SpaCAE provides more biological insights into human breast cancer heterogeneity on ST data
We tested the HER2-positive breast tumor data from the ST [18] technology, containing 613 locations and 15 030 genes to demonstrate the capacity of SpaCAE on cancer tissue. The tumor section was examined and annotated by a pathologist [36]. To accurately evaluate the effectiveness of SpaCAE, we took the histopathological image and annotations as reference and used clustering purity as quantitative assessment criteria.
Clusters based on SpaCAE categorized the tissue into seven spatial domains, consistent with the regional annotations and were more accurate (purity = 0.648) than that by other five spatial clustering algorithms (BayesSpace, purity = 0.600; GraphST, purity = 0.577; SpatialPCA, purity = 0.575; STAGATE, purity = 0.569; SpaGCN, purity = 0.564; DeepST, purity = 0.447, the difference between SpaCAE and each of the others is confirmed by the Wilcoxon rank-sum test based on random seed settings, P < 0.01, as depicted in Figure 5D). Regions of carcinoma in situ and infiltrating carcinoma identified by the model were the most similar to that of the pathological tissue sections (Figure 5A and B and Supplementary Figure 6).
Figure 5.

SpaCAE provides more biological insights into tumor heterogeneity in human HER2 breast cancer ST data (n = 613 spots). (A) H&E staining image (left) shows distinct tissue regions annotated by a pathologist in the original study. The annotated tissue regions include invasive cancer, fat tissue, fibrous tissue, normal breast glands, in situ cancer/DCIS and immune cells. (B) The comparison of spatial domain identification on breast cancer slice with domains = 7, the cluster purity is used to compare the similarity between the identified spatial domains and the ground truth annotations. (C) The change of gene differential expression and spatial autocorrelation patterns before and after data denoising. Since the second domain detected by SpaCAE was spatially corresponding to the undetermined tag of the real tissue slice and there were only two differential genes detected in the second domain against others, it was of little biological significance to discuss noise reduction in this spatial domain and the column of the second domain was removed from the boxplot. FC: fold change of gene expression. (D) Spatial expression of selected domain marker genes before (left) and after (middle) data denoising and the change of Moran’s I. Survival analysis (right) of the genes (CD24, HPS90AB1) associated with breast cancer.
On account that the data sparsity may hinder downstream analysis, we studied whether SpaCAE could provide more insights into the underlying tumor heterogeneity. Firstly, SpaCAE was employed to denoise the low-quality matrix and recover gene spatial expression patterns. According to the augmented gene expression, the DEGs) in each domain were identified. To evaluate the effectiveness of SpaCAE in denoising, we demonstrated the changes of Log2FC between original and denoised gene expression profile through SpaCAE and STAGATE, respectively, as to the changes of Moran’s I (Figure 5C and Supplementary Figure 7). The greater change of the two indicators before and after denoising, the stronger denoising ability.
Comprehensively, the comparison highlighted the remarkable improvement of biological specificity brought by SpaCAE denoising across the detected and annotated domains (Figure 5C and Supplementary Figures 7–9). Particularly, some DEGs expression (e.g. CD24 and HSP90AB1), which have been verified to be the potential prognostic risk factor for HER2-positive breast tumor, appeared more spatial specificity on cancerous areas (Figure 5D). Specifically, breast tumor expression of CD24 promotes immune escape by avoiding clearance of macrophages through its association with Siglec-10, a inhibitory receptor expressed on tumor-associated macrophages [37]. HSP90AB1 is one of the heat shock proteins (HSPs) 90 isoforms [38, 39]. High-level expression of HSP90AB1 was driven by chromosome coding region amplifications and were independent factors that led to death from breast cancer among patients with HER2−/ER+ subtypes [39]. Collectively, these results exhibited the ability of SpaCAE to diminish noise and augment spatial expression patterns.
DISCUSSION
Recent breakthroughs in SRT technologies provide new perspectives to understand the heterogeneities of tissues and cancer under spatial context. The key issue of deciphering tissue spatial functional regions is to mitigate the high sparsity and noise of SRT data. However, the current methods commonly utilize high-sparse SRT data as input and modeling spatial constraints solely based on meta-genes, resulting in these approaches failing to effectively mitigate the impact of high sparsity by fully leveraging the spatial dependence of gene expression. In this work, SpaCAE was designed as a novel spatially contrastive learning based on the spatial dependence of gene expression. SpaCAE contrasts the transcriptomic signals of each spot and those of its spatial neighbors to learn effective representation with spatial constraints by building the graph convolutional encoder, graph deconvolutional decoder and deep contrastive strategy, which are then used for accurate detecting spatial domain and denoising gene expression. The superiority of SpaCAE was verified on spatial domains detection and data denoising across multiple datasets of various technologies, i.e. 10x Visium, Slide-seqV2, ST. SpaCAE revealed the tissue heterogeneity of tumor and brain and the corresponding domain-specific genes. These findings suggest that SpaCAE is a powerful tool for identifying complex spatial expression landscape in SRT study.
The superiority of SpaCAE is attributed to its distinctive design, which involves contrasting the transcriptomic signals of each spot and its spatial neighbors. Different from the current existing methods, SpaCAE uses the gene expression with spatial dependence as one of the model inputs, which avoids the high sparsity affecting the performance of the method caused by only using the original expression modeling. To achieve this purpose, SpaCAE not only generates spatially strengthened expression of each spot but also builds the graph convolutional encoder and graph deconvolutional decoder to learn effective representation with spatial constraints from the original and augmented expression data. Additionally, SpaCAE provides a deep contrastive strategy to balance spatial local information and global information during representation learning. Thus, SpaCAE corrects the low-quality gene expression by borrowing the spatial dependence of gene expression before modeling and learns the representation with balancing spatial coherence and expression variance to improve the performance of spatial domains detection.
The spatially contrastive strategy introduced by SpaCAE presents a novel approach for studying SRT data. However, this strategy is currently limited to spatial domain identification and lacks the capability to delve into the molecular mechanisms underlying tissue structure. This limitation of SpaCAE hinders its application in conducting in-depth analysis for biological interpretations. Meanwhile, gene co-expression networks provide a way to enhance SRT data. Therefore, drawing on the current gene regulatory networks inference methods [40–42], the next crucial work is to effectively infer gene regulatory networks through spatial contrast modeling and investigate spatially specific regulatory factors.
Key Points
SpaCAE is a spatially contrastive variational autoencoder framework that effectively identifies spatial domains and denoises highly sparse SRT data, providing a novel contrastive learning model to facilitate the sharing of neighboring bio-signals and learn spatially constrained representations.
SpaCAE presents a tunable deep contrastive strategy to achieve a harmonious integration of spatial local and global information and utilizes graph convolutional encoder and graph deconvolutional decoder to capitalize on spatial information.
SpaCAE is a remarkable tool to provide novel insights in SRT studies and can be scaled for diverse SRT platforms.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the anonymous reviewers for useful suggestions. Q.S. and C.Z. conceived and designed the framework and the experiments. Y.H. performed the experiments. Y.H. and C.Z. analyzed the data and wrote the paper. Y.H., Q.S., K.X., H.Y., X.L. and C.Z. revised the manuscript.
Author Biographies
Yaofeng Hu is a master student in Key Laboratory of Systems Health Science of Zhejiang Province, Hangzhou Institute for Advanced Study, University of Chinese Academy of Sciences, Chinese Academy of Sciences, Hangzhou 310024, China. Her current research interests include single-cell transcriptomics and spatial transcriptomics.
Kai Xiao is a master student in State Key Laboratory of Cell Biology, Shanghai Institute of Biochemistry and Cell Biology, Center for Excellence in Molecular Cell Science, Chinese Academy of Sciences, Shanghai 200031, China. His current research interests include single-cell transcriptomics and spatial transcriptomics.
Hengyu Yang is a master student in Key Laboratory of Systems Health Science of Zhejiang Province, Hangzhou Institute for Advanced Study, University of Chinese Academy of Sciences, Chinese Academy of Sciences, Hangzhou 310024, China. His current research interests include deep learning and algorithm design for the biological fields.
Xiaoping Liu is a professor in Key Laboratory of Systems Health Science of Zhejiang Province, School of Life Science, Hangzhou Institute for Advanced Study, University of Chinese Academy of Sciences, Hangzhou 310024, China. He studies the fields of computational systems biology and bioinformatics.
Chuanchao Zhang received the PhD from Wuhan University, Wuhan, China, in 2017. He is currently an assistant research fellow in Key Laboratory of Systems Health Science of Zhejiang Province, Hangzhou Institute for Advanced Study, University of Chinese Academy of Sciences, Chinese Academy of Sciences, Hangzhou 310024, China. His current research interests include machine learning, deep learning, single-cell transcriptomics and spatial transcriptomics.
Qianqian Shi received the PhD from Shanghai Institute of Biological Sciences, University of Chinese Academy of Sciences, Chinese Academy of Sciences, China, in 2017. She is currently an associate professor at College of Informatics, Huazhong Agricultural University, Wuhan, China. Her current research interests include machine learning, deep learning, network biology, computational biology, single-cell transcriptomics and spatial transcriptomics.
Contributor Information
Yaofeng Hu, Key Laboratory of Systems Health Science of Zhejiang Province, School of Life Science, Hangzhou Institute for Advanced Study, Hangzhou 310024; University of Chinese Academy of Sciences, China.
Kai Xiao, State Key Laboratory of Cell Biology, Shanghai Institute of Biochemistry and Cell Biology, Center for Excellence in Molecular Cell Science, Chinese Academy of Sciences, Shanghai 200031, China; University of Chinese Academy of Sciences, Beijing 100049, China.
Hengyu Yang, Key Laboratory of Systems Health Science of Zhejiang Province, School of Life Science, Hangzhou Institute for Advanced Study, Hangzhou 310024; University of Chinese Academy of Sciences, China.
Xiaoping Liu, Key Laboratory of Systems Health Science of Zhejiang Province, School of Life Science, Hangzhou Institute for Advanced Study, Hangzhou 310024; University of Chinese Academy of Sciences, China.
Chuanchao Zhang, Key Laboratory of Systems Health Science of Zhejiang Province, School of Life Science, Hangzhou Institute for Advanced Study, Hangzhou 310024; University of Chinese Academy of Sciences, China.
Qianqian Shi, Hubei Engineering Technology Research Center of Agricultural Big Data, Huazhong Agricultural University, Wuhan 430070, Hubei, China; Hubei Key Laboratory of Agricultural Bioinformatics, College of Informatics, Huazhong Agricultural University, Wuhan 430070, China.
FUNDING
This work is supported by National Key Research and Development Program of China (2022YFA1004800), National Natural Science Foundation of China (Grant No. 62202120), the R&D project of Pazhou Lab (Huangpu) under Grant 2023K0602, Zhejiang Provincial Natural Science Foundation of China under Grant No. LZ22C060001, the research funds of Hangzhou Institute for advanced study, UCAS (No. 2022ZZ01013, 2022ZZ01016, 2023HIAS-Y024) and the open project of BGI-Shenzhen, Shenzhen 518000, China.
DATA AND CODE AVAILABILITY
The human dorsolateral prefrontal cortex (DLPFC) datasets are available in the spatialLIBD package at http://spatial.libd.org/spatialLIBD. The MIBI-TOF dataset is available at https://github.com/scverse/squidpy. The osmFISH dataset of mouse somatosensory cortex is available at https://github.com/drieslab/spatial-datasets. Mouse hippocampus data by Slide-seq V2 is available at https://singlecell.broadinstitute.org/single_cell/study/SCP815. The HER2-positive breast tumor data from ST platform is available at https://github.com/almaan/her2st. Python source code of SpaCAE, under the open-source BSD 3-Clause license, is available at https://github.com/HYF01/SpaCAE.
References
- 1. Larsson L, Frisén J, Lundeberg J. Spatially resolved transcriptomics adds a new dimension to genomics. Nat Methods 2021;18(1):15–8. [DOI] [PubMed] [Google Scholar]
- 2. Marx V. Method of the year: spatially resolved transcriptomics. Nat Methods 2021;18(1):9–14. [DOI] [PubMed] [Google Scholar]
- 3. Zhang C, Li X, Huang W, et al. Spatially aware self-representation learning for tissue structure characterization and spatial functional genes identification. Brief Bioinform 2023;24(4):bbad197. [DOI] [PubMed] [Google Scholar]
- 4. Zhao E, Stone MR, Ren X, et al. Spatial transcriptomics at subspot resolution with BayesSpace. Nat Biotechnol 2021;39(11):1375–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rodriques SG, Stickels RR, Goeva A, et al. Slide-seq: a scalable technology for measuring genome-wide expression at high spatial resolution. Science 2019;363(6434):1463–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Stickels RR, Murray E, Kumar P, et al. Highly sensitive spatial transcriptomics at near-cellular resolution with Slide-seqV2. Nat Biotechnol 2021;39(3):313–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wei X, Fu S, Li H, et al. Single-cell stereo-seq reveals induced progenitor cells involved in axolotl brain regeneration. Science 2022;377(6610):eabp9444. [DOI] [PubMed] [Google Scholar]
- 8. Dries R, Zhu Q, Dong R, et al. Giotto: a toolbox for integrative analysis and visualization of spatial expression data. Genome Biol 2021;22:1–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shang L, Zhou X. Spatially aware dimension reduction for spatial transcriptomics. Nat Commun 2022;13(1):7203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hu J, Li X, Coleman K, et al. SpaGCN: integrating gene expression, spatial location and histology to identify spatial domains and spatially variable genes by graph convolutional network. Nat Methods 2021;18(11):1342–51. [DOI] [PubMed] [Google Scholar]
- 11. Dong K, Zhang S. Deciphering spatial domains from spatially resolved transcriptomics with an adaptive graph attention auto-encoder. Nat Commun 2022;13(1):1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xu C, Jin X, Wei S, et al. DeepST: identifying spatial domains in spatial transcriptomics by deep learning. Nucleic Acids Res 2022;50(22):e131–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Long Y, Ang KS, Li M, et al. Spatially informed clustering, integration, and deconvolution of spatial transcriptomics with GraphST. Nat Commun 2023;14(1):1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ren H, Walker BL, Cang Z, Nie Q. Identifying multicellular spatiotemporal organization of cells with SpaceFlow. Nat Commun 2022;13(1):4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Scrucca L, et al. Model-Based Clustering, Classification, and Density Estimation Using mclust in R. Chapman and Hall/CRC, 2023. [Google Scholar]
- 16. Cover T, Hart P. Nearest neighbor pattern classification. IEEE Trans Inf Theory 1967;13(1):21–7. [Google Scholar]
- 17. Cheng J, Li M, Li J, et al. Wiener Graph Deconvolutional Network Improves Graph Self-Supervised Learning. Proc AAAI Conf Artif Intell 2023;37:7131–39. [Google Scholar]
- 18. Ståhl PL, Salmén F, Vickovic S, et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 2016;353(6294):78–82. [DOI] [PubMed] [Google Scholar]
- 19. Wolf FA, Angerer P, Theis FJ. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol 2018;19:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shi Q, Li X, Peng Q, et al. scDA: single cell discriminant analysis for single-cell RNA sequencing data. Comput Struct Biotechnol J 2021;19:3234–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hubert L, Arabie P. Comparing partitions. J Classif 1985;2(1):193–218. [Google Scholar]
- 22. Chuanchao Zhang, Juan Liu, Qianqian Shi, et al. Integration of multiple heterogeneous omics data. 2016 IEEE Int Conf Bioinforma Biomed BIBM 2016;564–69.
- 23. Shi Q, Zhang C, Peng M, et al. Pattern fusion analysis by adaptive alignment of multiple heterogeneous omics data. Bioinformatics 2017;33(17):2706–14. [DOI] [PubMed] [Google Scholar]
- 24. Zhang C, Liu J, Shi Q, et al. Differential function analysis: identifying structure and activation variations in dysregulated pathways. Sci China Inf Sci 2017;60(1):12108. [Google Scholar]
- 25. Zhang C, Liu J, Shi Q, et al. Comparative network stratification analysis for identifying functional interpretable network biomarkers. BMC Bioinformatics 2017;18:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xu J, Zhang A, Liu F, et al. CIForm as a transformer-based model for cell-type annotation of large-scale single-cell RNA-seq data. Brief Bioinform 2023;24:bbad195. [DOI] [PubMed] [Google Scholar]
- 27. Maynard KR, Collado-Torres L, Weber LM, et al. Transcriptome-scale spatial gene expression in the human dorsolateral prefrontal cortex. Nat Neurosci 2021;24(3):425–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kipf TN, Welling M. Variational graph auto-encoders. In: Gal Y, et al. eds. Neural Information Processing Systems Workshop on Bayesian Deep Learning. Curran Associates, Inc.; 2016.
- 29. Kingma DP, Welling M. An introduction to variational autoencoders. Found Trends Mach Learn 2019;12(4):307–92. [Google Scholar]
- 30. Codeluppi S, Borm LE, Zeisel A, et al. Spatial organization of the somatosensory cortex revealed by osmFISH. Nat Methods 2018;15(11):932–5. [DOI] [PubMed] [Google Scholar]
- 31. Sunkin SM, Ng L, Lau C, et al. Allen brain atlas: an integrated spatio-temporal portal for exploring the central nervous system. Nucleic Acids Res 2012;41(D1):D996–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xiong Z-G, O'Hanlon D, Becker LE, et al. Enhanced calcium transients in glial cells in neonatal cerebellar cultures derived from S100B null mice. Exp Cell Res 2000;257(2):281–9. [DOI] [PubMed] [Google Scholar]
- 33. Brewer GJ, Boehler MD, Leondopulos S, et al. Toward a self-wired active reconstruction of the hippocampal trisynaptic loop: DG-CA3. Front Neural Circuits 2013;7:165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Alvi NH, Turkstani BA, Ashi AS, et al. COQ8B-related steroid-resistant nephrotic syndrome in Saudi Arabia: a case report. Cureus 2022;14(11):e31922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Peng H, Xie P, Liu L, et al. Morphological diversity of single neurons in molecularly defined cell types. Nature 2021;598(7879):174–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Andersson A, Larsson L, Stenbeck L, et al. Spatial deconvolution of HER2-positive breast cancer delineates tumor-associated cell type interactions. Nat Commun 2021;12(1):6012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Barkal AA, Brewer RE, Markovic M, et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature 2019;572(7769):392–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ozgur A, Tutar L, Tutar Y. Regulation of heat shock proteins by miRNAs in human breast cancer. Microrna 2014;3(2):118–35. [DOI] [PubMed] [Google Scholar]
- 39. Cheng Q, Chang JT, Geradts J, et al. Amplification and high-level expression of heat shock protein 90 marks aggressive phenotypes of human epidermal growth factor receptor 2 negative breast cancer. Breast Cancer Res 2012;14:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xu J, Zhang A, Liu F, Zhang X. STGRNS: an interpretable transformer-based method for inferring gene regulatory networks from single-cell transcriptomic data. Bioinformatics 2023;39(4):btad165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Peng H, Xu J, Liu K, et al. EIEPCF: accurate inference of functional gene regulatory networks by eliminating indirect effects from confounding factors. Brief Funct Genomics 2023;elad040. [DOI] [PubMed] [Google Scholar]
- 42. Wang L, Trasanidis N, Wu T, et al. Dictys: dynamic gene regulatory network dissects developmental continuum with single-cell multiomics. Nat Methods 2023;20(9):1368–78. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The human dorsolateral prefrontal cortex (DLPFC) datasets are available in the spatialLIBD package at http://spatial.libd.org/spatialLIBD. The MIBI-TOF dataset is available at https://github.com/scverse/squidpy. The osmFISH dataset of mouse somatosensory cortex is available at https://github.com/drieslab/spatial-datasets. Mouse hippocampus data by Slide-seq V2 is available at https://singlecell.broadinstitute.org/single_cell/study/SCP815. The HER2-positive breast tumor data from ST platform is available at https://github.com/almaan/her2st. Python source code of SpaCAE, under the open-source BSD 3-Clause license, is available at https://github.com/HYF01/SpaCAE.