

Abstract
Activation of the androgen receptor (AR) and AR-driven transcriptional programs is central to the pathophysiology of prostate cancer. Despite successful translational efforts in targeting AR, therapeutic resistance often occurs as a result of molecular alterations in the androgen signaling axis. The efficacy of next-generation AR-directed therapies for castration-resistant prostate cancer has provided crucial clinical validation for the continued dependence on AR signaling and introduced a range of new treatment options for men with both castration-resistant and castration-sensitive disease. Despite this, however, metastatic prostate cancer largely remains an incurable disease, highlighting the need to better understand the diverse mechanisms by which tumors thwart AR-directed therapies, which may inform new therapeutic avenues. In this review, we revisit concepts in AR signaling and current understandings of AR signaling-dependent resistance mechanisms as well as the next frontier of AR targeting in prostate cancer.
Androgen signaling axis targeting in prostate cancer and molecular basis of response and resistance
INTRODUCTION
Androgens are essential hormones in the maintenance of normal male physiology and sex differentiation, including in the prostate.1,2 Activation of the androgen receptor (AR) is a hallmark of prostate cancer, in which AR-driven transcriptional programs can instigate and support tumor growth. Recognition of this dependence dates back to the original observations made by Charles Huggins and Clarence Hodges in the 1940s that surgical castration induced tumor regression, thus proving the pathophysiologic reliance on androgens and pioneering the therapeutic targeting of AR in this sex-biased disease.3,4 For this seminal discovery, Huggins was awarded the Nobel Prize in Physiology or Medicine in 1966.
CONTEXT
Key Objective
What are important molecular and metabolic variants in the androgen signaling axis that can arise in the context of therapeutic resistance in prostate cancer?
Knowledge Generated
The molecular landscape of therapeutic resistance to androgen receptor (AR)–directed therapies in prostate cancer is characterized by multiple changes converging on the androgen signaling axis, which include perturbations to AR, to androgen biosynthesis, or to downstream AR-regulated oncogenic pathways. Understanding these different mechanisms may inform investigations into novel treatment approaches for prostate cancer.
Relevance (M.A. Carducci)
-
AR biology, signaling, and targeting has been the mainstay of prostate cancer therapy, yet new knowledge impacts how we monitor current approaches and develop new strategies in order to tailor and improve outcomes of men with advance prostate cancer. This review places the current biology and evolving treatment approaches/combinations in a practical and clinical focus.*
*Relevance section written by JCO Associate Editor Michael A. Carducci, MD, FACP, FASCO.
Although targeting AR in prostate cancer has been a translational success story, resistance inevitably arises, often driven by molecular alterations in the androgen signaling axis. With the advent of multiple effective next-generation AR-directed therapies, the landscape of resistance mechanisms has become increasingly diverse and complex. Furthermore, the reality that metastatic prostate cancer largely remains an incurable malignancy despite targeting the apparent Achilles heel of this disease indicates an unmet need to understand how tumors evade AR-directed therapies. In this review, we will revisit concepts in AR signaling, with a perspective focused on the next frontier of AR targeting in prostate cancer.
OVERVIEW OF AR ACTION
The AR is a ligand-dependent transcription factor and member of the steroid receptor family consisting of the estrogen receptor, progesterone receptor (PR), glucocorticoid receptor (GR), and mineralocorticoid receptor (MR).5 These steroid receptors share varying degrees of homology but are functionally distinct, with unique actions dictated primarily by the specificity of cognate ligand binding and differential transcriptional programs.6 Under physiological conditions, the principal androgenic ligands for AR are testosterone and its more potent 5α-reduced derivative, dihydrotestosterone (DHT).7 In the absence of ligand, the inactive AR generally resides within the cellular cytoplasm bound by chaperone proteins. Androgen binding triggers conformational changes that promote AR nuclear translocation, homodimerization, binding to DNA at androgen response elements, and direct transcriptional activation of target genes (Fig 1).6,8,9
FIG 1.

Androgen signaling in prostate cancer is highlighted by multiple receptor and pre/postreceptor mechanisms that serve as targets for different therapeutic approaches. Androgen biosynthesis is tightly regulated by the hypothalamic-pituitary-gonadal and hypothalamic-pituitary-adrenal axes, which govern the production of gonadal and adrenal androgens that serve as precursors for DHT, the principal AR ligand in the prostate (pre--receptor activity). On ligand binding, AR translocates from the cytoplasm to the nucleus to bind to DNA as a homodimer, permitting transactivation of target genes and pathways (postreceptor activity). Examples of different clinically approved as well as investigational inhibitors are highlighted. ACTH, adrenocorticotropic hormone; AR, androgen receptor; ARE, androgen response element; DHT, dihydrotestosterone; FSH, follicle-stimulating hormone; GnRH, gonadotropin-releasing hormone; HSP, heat shock protein; LH, luteinizing hormone; NTD, N-terminal domain; PROTACs, proteolysis-targeting chimeras; PSA, prostate-specific antigen; SARDs, selective androgen receptor degraders.
THERAPEUTIC TARGETING OF AR IN PROSTATE CANCER—A HISTORICAL PERSPECTIVE
Since the initial demonstration by Huggins and Hodges, depletion of gonadal testosterone by surgical or medical castration (also referred to as androgen deprivation therapy [ADT]) has remained a mainstay of therapy for prostate cancer. Evidence for AR activation in promoting prostate cancer even in early stages of disease derives from two key observations. First, prostate-specific antigen (PSA) is frequently elevated at diagnosis and often rises to herald disease progression. The KLK3 gene encoding PSA is a direct transcriptional target of AR.10 Second, AR-driven transcription can be hijacked by genomic rearrangements that fuse regulatory elements from AR target genes with proto-oncogene gene bodies, thereby coupling physiologic AR signaling with dysregulated oncogenic pathways.11 The prototypical example is TMPRSS2-ETS fusions, which are among the most common and earliest genomic aberrations found in primary prostate cancer and precancerous lesions.12,13 However, despite this seemingly vital role of AR signaling, alterations in AR are rare in primary prostate cancer and untreated metastatic disease, indicating that AR signaling is necessary but not sufficient alone to drive early tumor development.14-19
Although initially effective, the response to ADT is invariably followed by recurrence of castration-resistant prostate cancer (CRPC). For decades, the prevailing dogma was that CRPC represented an androgen-independent state, although it is now appreciated that inappropriate restoration of the androgen signaling axis occurs in most cases of CRPC to drive disease progression. This is supported by the fact that AR is frequently overexpressed in CRPC, with a rise in PSA usually accompanying its onset. In early investigations, it became clear that AR overexpression could promote tumor proliferation in response to castrate levels of androgens,20-22 the clinical importance of which is underscored by the high frequency of AR gene amplification in tumors after hormonal therapy.16-18 Importantly, despite castrate levels of serum testosterone, tissue depletion of androgens is incomplete after ADT.23-26 This is likely due to intratumoral androgen production from alternative steroidal precursors such as adrenal androgens.24,27,28 The recognition of diverse resistance mechanisms involving AR has been the impetus for designing more potent AR signaling inhibitors (Fig 2).20,30
FIG 2.
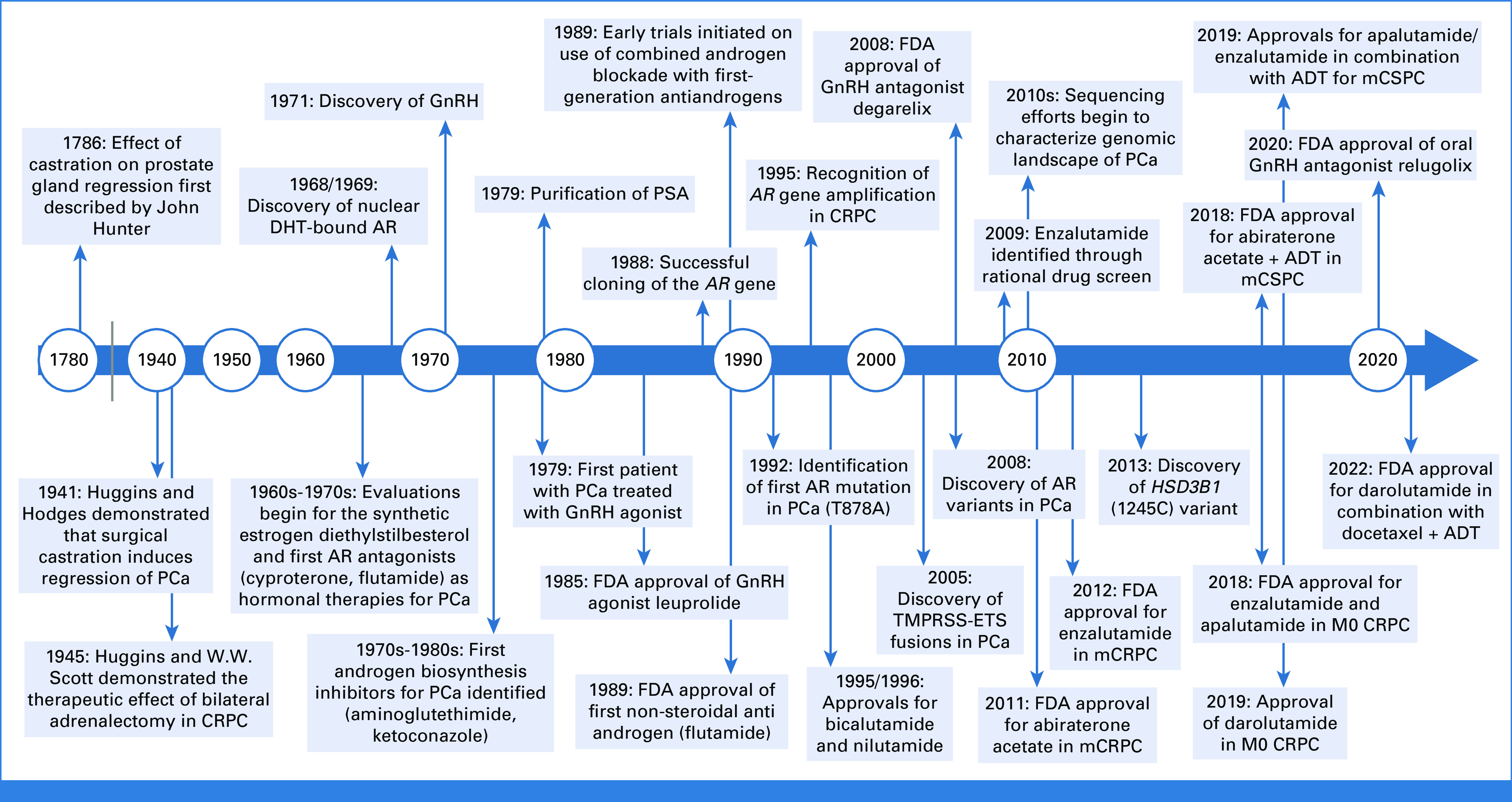
A timeline of key translational discoveries and therapeutic innovations in the treatment of PCa. Illustration was created with BioRender.29 ADT, androgen deprivation therapy; AR, androgen receptor; CRPC, castration-resistant prostate cancer; DHT, dihydrotestosterone; FDA, US Food and Drug Administration; GnRH, gonadotropin-releasing hormone; mCSPC, metastatic castration-sensitive prostate cancer; PCa, prostate cancer; PSA, prostate-specific antigen.
A pivotal milestone in the development of AR-targeting agents was achieved with enzalutamide (formerly MDV3100), a second-generation competitive antagonist that binds to AR with 5-8 fold greater affinity than bicalutamide.30 In parallel, abiraterone was also developed as an irreversible inhibitor of CYP17A1—the enzyme that converts pregnenolone to dehydroepiandrosterone (DHEA), a precursor for potent androgen biosynthesis (Fig 3).33 Both agents changed the treatment landscape for prostate cancer, demonstrating for the first time an overall survival benefit with retargeting AR in metastatic CRPC after progression on chemotherapy.34,35 This provided critical clinical validation that CRPC continues to rely on AR, which then spurred the development of other second-generation AR antagonists, such as apalutamide and darolutamide, as well as the intensification of AR blockade in earlier disease stages, including nonmetastatic CRPC.36-39 Additionally, the possibility of incomplete AR signaling suppression with ADT even before the clinical onset of CRPC provided rationale for combining ADT with second-generation AR-directed therapies.40-45 Multiple phase III trials have now confirmed a clear survival benefit to these approaches, which have become standard of care. Yet, despite these translational successes, a minority of patients experience primary resistance, and most men will unfortunately experience disease progression after treatment with these agents. Nevertheless, one lesson learned from prior successes is that the pathophysiology of prostate cancer remains deeply tied to the molecular regulation of AR.
FIG 3.
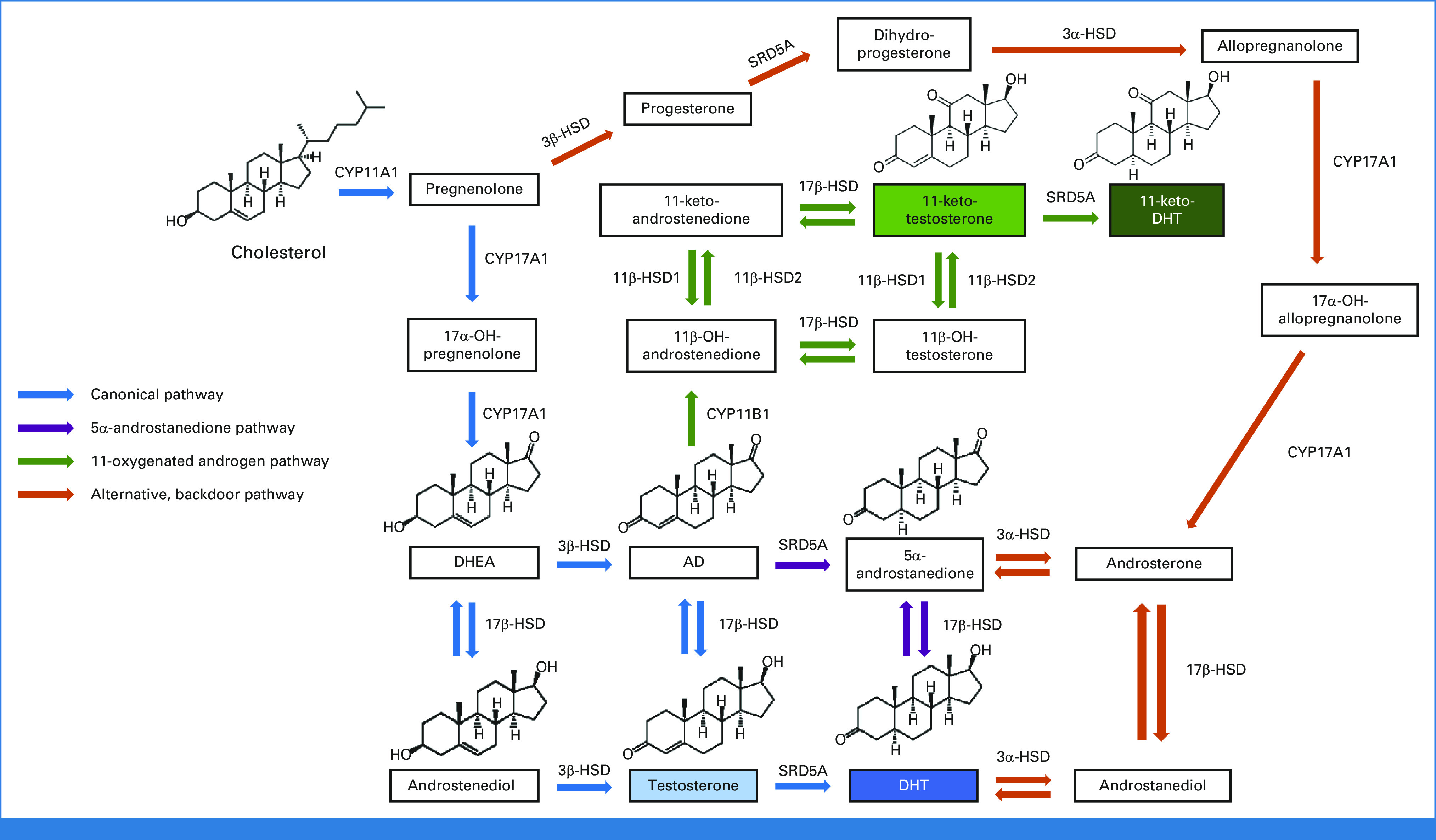
Several key pathways of steroidogenesis contributing to androgen biosynthesis, which include the canonical pathway of DHT synthesis through testosterone (blue) as described in normal physiology, as well as the 5α-androstanedione (5α-dione) pathway, which is the primary route of DHT biosynthesis from adrenal precursors in prostate cancer.31,32 More recently, the 11-oxygenated androgen pathway has gained increasing recognition for generation of 11-keto-testosterone and 11-keto-DHT, which can serve as bona fide AR agonists in prostate cancer (green). In addition, multiple other potentially relevant pathways exist, including the alternative, backdoor pathway (orange). Of note, this simplified schematic is not comprehensive in depicting all possible pathways, including those through 17α-OH progesterone derivatives as intermediates. AD, androstenedione; AR, androgen receptor; DHEA, dehydroepiandrosterone; DHT, dihydrotestosterone; HSD, hydroxysteroid dehydrogenase.
MOLECULAR MECHANISMS OF RESISTANCE TO AR TARGETING IN PROSTATE CANCER
Most commonly, CRPC overcomes AR signaling inhibition by reactivating the androgen signaling axis through various genetic and epigenetic alterations while a minority of cases can develop epigenetic alterations that bypass a requirement for AR signaling. Common alterations within the androgen signaling axis include AR overexpression and gene amplification, ligand-binding domain (LBD) mutations, structural rearrangements, constitutively active AR variants (AR-Vs), and alterations in pathways of androgen biosynthesis. Although alterations in AR are uncommon in primary disease, they become highly prevalent in CRPC, as is evident from multiple large-scale tissue genomic sequencing studies.15-18,46,47 Serial sampling by plasma cell-free DNA in patients on second-generation AR-directed therapies likewise confirms that the genetic alterations arising in the setting of treatment largely converge on AR with evolving changes seen in gene copy number and structural rearrangements.48
The increasing emergence of AR-negative prostate cancers is likely an outcome of selective pressure from intensive AR suppression with second-generation AR-directed agents, forcing the reprogramming of tumor cells to survive via AR-independent pathways.49 A subset of these tumors exhibit markers of neuroendocrine differentiation and may morphologically resemble small cell carcinoma, despite arising originally from adenocarcinoma.49,50 A rise in the incidence of treatment-related AR-negative prostate cancers presents a unique challenge from a treatment perspective and has spurred interest in targeting AR-independent pathways or restoring AR expression in these tumors.51 Taken together, these findings strongly suggest that AR is a master regulator of prostatic differentiation and lineage-dependent survival pathways that are subsequently usurped by prostate cancer—such that resistance to potent AR signaling blockade necessitates augmentation of AR signaling or a switch to AR-independent programs.52
Below, we have organized AR resistance mechanisms into two groups: (1) those which directly perturb the AR protein and (2) those that influence either the availability of steroid ligands for AR or modify AR binding/actions (later designated as prereceptor and postreceptor, respectively; Fig 1). Understanding which of these mechanisms are operative in individual tumors could inform which strategies may be most effective to overcome treatment resistance. However, one challenge is that the varied resistance mechanisms to AR-directed therapies are not necessarily mutually exclusive. Nevertheless, comprehensive characterization of these mechanisms should hopefully clarify and refine the molecular taxonomy of treatment-resistant disease.
AR STRUCTURE/FUNCTION
The AR gene comprises eight exons spanning 183 kb of the X chromosome at Xq11-12 and encodes a 110 kDa protein that is approximately 919 amino acids (Fig 4). Notably, AR gene amplification (commonly by tandem duplication) is the most frequent molecular alteration in CRPC, occurring in about approximately 60%-70% of cases.46 Furthermore, the amplicon often encompasses both the AR gene body as well as an enhancer site approximately 650 kb centromeric to AR.53,54 However, in approximately 10%-15% of cases, amplification of this enhancer can occur independently of the AR gene body, which drives AR overexpression similar to AR gene body amplification.54 Interestingly, this region displays the epigenetic hallmarks of a developmental enhancer that is potentially reactivated in CRPC.53 Similar to gene amplification, structural rearrangements in AR are also common in CRPC, occurring in approximately 13%-33% of patients before abiraterone or enzalutamide treatment and increasing in frequency to approximately 25%-50% after treatment.55,56 These structural rearrangements can occur concomitantly with or independent of AR amplification and can give rise to diverse AR variant proteins with constitutive activity.55,56 Both AR gene amplification and structural rearrangements have been implicated in driving resistance to enzalutamide and abiraterone.48,57,58
FIG 4.

The AR gene locus and protein. The mRNA transcript encoding the full-length AR protein encompasses eight exons, which consists of four major functional protein domains. AR mutations most frequently occur in the LBD. Several AR-Vs including AR-V7, AR-V9, and AR-V12/ARv567es are depicted as well, including their corresponding exons. AR, androgen receptor; AR-Vs, AR variants; DBD, DNA-binding domain; LBD, ligand-binding domain; NTD, N-terminal domain; UTR, untranslated region.
Like other steroid nuclear receptors, AR comprises four major structural domains: an N-terminal domain (NTD), a DNA-binding domain (DBD), a hinge region, and a C-terminally positioned LBD (Fig 4).6 The DBD and LBD are the most highly conserved across different species and share significant homology with other steroid receptors while the NTD is unique, possibly reflecting its specificity in AR function.9 The NTD (exon 1) harbors a strong transcriptional activation element termed AF-1, which is the primary effector of transactivation.59-61 Loss of the LBD manifests with constitutive activity, indicating its basal repressive role on the NTD.62,63 In vitro studies have also suggested critical interactions between the N-terminus and C-terminus in AR transactivation, although more contemporary in vivo work suggests that this property may be dispensible.64 In recent years, various constitutively active AR-Vs have been characterized that lack the LBD and may command AR programs in a ligand-independent manner.65-68 Importantly, the majority of current AR-directed agents either directly interact with or require a functioning LBD and thus do not act on AR-Vs. Accordingly, development of NTD inhibitors has been an attractive concept, although the intrinsic structural disorder of this domain is a crucial biophysical property for transcriptional activity that also presents an inherent challenge for the design of inhibitors.69 EPI-506 is a bisphenol-like compound that was developed as a covalent NTD inhibitor and recently tested in a phase I study of patients with mCRPC resistant to second-generation AR-directed therapies.70 EPI-506 achieved only minor PSA declines, a finding that later attributed to poor bioavailability.70,71 EPI-7386 is a successor drug with greater metabolic stability and more potent activity, which is currently undergoing investigation.72 Notably, although these agents target the NTD, they may have broader, less specific actions that also contribute to their therapeutic effect.73
The DBD (exons 2-3) is the most conserved region of AR, which is perhaps unsurprising given the critical interactions with DNA required for gene expression.9 The first zinc finger of the DBD makes base-specific contacts within the major groove of DNA via a conserved series of amino acids known as the P(roximal)-box, whereas the second zinc finger mediates receptor dimerization via a similarly conserved D(istal)-box.6 Remarkably, both the P-box and D-box residues as well as the consensus hexameric repeat sequence of DNA recognized by AR are also shared by GR, PR, and MR. This may explain some degree of overlap between the genomic binding sites of AR and GR, which is pertinent in CRPC, as GR appears to be upregulated in certain enzalutamide-resistant tumors, leading to GR binding to and transcriptionally regulating a subset of AR target genes.74,75
The DBD and LBD are joined by a flexible hinge region (exons 3-4). The hinge region contains target sites for post-translational modifications and a bipartite nuclear localization signal that orchestrates nuclear import.6 The LBD (exons 4-8) governs ligand-dependent AR activity, illustrated by the fact that deletion of this region renders the AR constitutively active and unresponsive to androgens.62 In prostate cancer, the LBD is the most frequent site of gain-of-function mutations. In addition to the ligand-binding pocket, the LBD also contains an AF-2 element which enables interaction with AR coregulators, a nuclear export signal that excludes unliganded AR from the nucleus, and an allosteric BF-3 regulatory site.76-78 These sites are potential targets for noncompetitive inhibitors, which remain an active area of investigation.79-81
AR MUTATIONS
AR mutations detected in prostate cancer typically arise after exposure to antiandrogens. The majority of these are gain-of-function missense mutations concentrated within the LBD that enable receptor promiscuity and inappropriate activation by a broad range of noncanonical ligand partners or even antagonists. Contemporary next-generation sequencing methods have shown that four hotspot LBD mutations (L702H, W742C/L, H875Y, and T878A/S) encompass a significant number of cases, together being found in approximately 10%-25% of CRPC.16-18,47,82 The first AR mutation described in prostate cancer was T878A, identified initially in the LNCaP cell line (derived from a man with CRPC) after the observation that hydroxyflutamide was an agonist in this model and later confirmed in a patient with CRPC.83-85 In vitro functional characterization of T878A/S and H875Y revealed that these mutations confer increased AR activation in response to various noncanonical steroidal ligands such as progesterone, estradiol, and DHEA, as well as to antiandrogens such as flutamide.86-89 Similarly, AR W742C/L can be activated by bicalutamide.90,91 The ability of these mutations to grant modest agonist potential to antagonists is the purported mechanism of antiandrogen withdrawal syndrome, a phenomenon initially described with first-generation antiandrogens, wherein discontinuation of therapy leads to PSA declines.90,92,93
Several mutations, including a more recently described F877L mutation, have potential to promote resistance to second-generation AR-directed therapies.94-97 In vitro studies suggest that F877L can confer agonist activity to enzalutamide and apalutamide (previously ARN-509), although this mutation occurs infrequently in patients overall and does not appear to be enriched by treatment.98 Notably, in contrast to enzalutamide and apalutamide, darolutamide (previously ODM-201) bears a distinct chemical structure with inhibitory activity even in enzalutamide-resistant models that harbor AR F877L or other resistance mutations.99 However, the optimal sequencing of treatment with AR-directed agents including darolutamide in the context of AR mutations remains to be determined.
AR T878A, H875Y, and L702H mutations have similarly been observed in patients experiencing disease progression on abiraterone.57,58,100 These mutations may hinder efficacy by enabling AR to be activated by noncanonical ligands such as progesterone and other steroids synthesized upstream of CYP17A1, which are thus not suppressed by abiraterone. This has prompted interest in the development of steroid biosynthesis inhibitors that target enzymatic steps upstream of CYP17A1 (discussed below). Of note, the L702H mutation, alone or in combination with T878A, appears to be activated by glucocorticoids, which is a largely unavoidable obstacle given that abiraterone requires concurrent glucocorticoid administration to prevent mineralocorticoid excess. Furthermore, given their steroidal structure, abiraterone and its metabolites can also directly bind to AR to influence AR activity, which could explain some degree of cross-resistance between AR antagonists and abiraterone.101-104
AR-Vs
A number of AR-Vs have been described which lack the LBD and can thus maintain AR signaling in a constitutive, ligand-independent manner. AR-Vs generally arise via alternative RNA splicing of intronic sequences or through structural rearrangements in the AR gene which promote altered RNA splicing patterns. To date, more than 20 AR-Vs have been identified.65-68 Among those arising from alternative RNA splicing of intronic sequences, AR-V7 appears to be the most abundant in CRPC and is encoded by splicing of AR exons 1-3, followed by a cryptic exon CE3. In vitro, AR-V7 has been shown to either homodimerize or heterodimerize with full-length AR (AR-FL) to mediate gene transcription.105-107 Expression of AR-V7 increases after ADT and correlates strikingly with inferior clinical outcomes after enzalutamide and abiraterone therapy, which has now been validated across multiple cohorts.108-112 In light of this, AR-V7 may serve as a useful predictive biomarker, although how this dictates alternative treatment selection and timing remains an area of active investigation.108-110 Other AR-Vs detected in prostate cancer tissues include AR-V9, which is similarly encoded by RNA splicing of AR exons 1-3 followed by a cryptic exon CE5, as well as AR-V12 (also referred to as ARv567es), arising from structural rearrangement and skipping of exons 5, 6, and 7 (Fig 4). Like AR-V7, detection of AR-V9 in CRPC biopsies may predict for resistance to abiraterone.113 However, given that expression of AR-V7 and AR-V9 generally mirrors that of AR-FL, ongoing and unresolved questions remain regarding whether AR-Vs drive CRPC independent of AR-FL, as well as if AR-Vs activate similar or different transcriptional programs compared with AR-FL (with data to support both conclusions).105-107,114 The identification of certain AR gene structural rearrangements in CRPC tissues that block expression of AR-FL while promoting AR-Vs indicates that, in specific circumstances, AR-Vs could drive therapeutic resistance.55,56
Given the multiple resistance mechanisms which circumvent effective targeting of the LBD, there has been interest in alternative approaches to AR signaling inhibition. In addition to the aforementioned NTD inhibitors, proteolysis-targeting chimeras (PROTACs) and selective androgen receptor degraders (SARDs) have recently emerged as novel and promising therapeutic strategies. PROTACs are heterobifunctional molecules consisting of two ligands connected by a central linker; one ligand binds to AR while the other recruits an E3 ubiquitin ligase to facilitate ubiquitination and proteasome-mediated degradation. ARV-110 is a first-in-class PROTAC, for which phase I/II data were recently reported and appears to show encouraging clinical activity among patients with heavily pretreated mCRPC, particularly among those with detectable T878A/S and H875Y mutations.115 Similarly, several SARDs have also shown activity in preclinical models for CRPC, including against AR-V7, and could represent a new class of AR-directed therapies.116,117
INTRATUMORAL ANDROGEN BIOSYNTHESIS (prereceptor mechanisms)
The biosynthesis of all steroid hormones begins with 27-carbon cholesterol, which can undergo stepwise enzymatic modification, first to downstream 21-carbon steroids (progestins), followed by further conversion to 19-carbon androgens. In men, the major circulating androgens in serum are testosterone and DHEA, predominantly produced by the testes and adrenal glands, respectively.118 As ADT does not influence the production of extragonadal androgens, CRPC can engage in intracrine androgen biosynthesis via alternative androgenic precursors to maintain AR signaling despite castrate serum levels of testosterone. Importantly, adrenal-derived DHEA can be readily metabolized to DHT (the principal AR ligand in the prostate) via a limited repertoire of enzymes expressed within prostatic tissue (Fig 3).119 These steroidogenic enzymes are frequently upregulated in CRPC to enable more efficient androgen biosynthesis of AR ligands.25,28
Multiple biosynthetic pathways converge on DHT as the final active metabolite (Fig 3).120 Although targeting of these enzymes is appealing given their requirement for androgen production, one consideration for therapeutic development is that inhibiting enzymes more proximally in the pathway may inadvertently disrupt synthesis of other physiologically indispensable steroids (such as mineralocorticoids and glucocorticoids), while blocking more distal enzymes spares the generation of upstream metabolites and creates opportunities for escape mechanisms. For instance, despite inhibition of CYP17A1 (17α-hydroxylase/17,20-lyase) activity, abiraterone does not prevent the generation of progestins that can activate AR in the context of specific AR mutations.100 Although there has been interest in inhibiting CYP11A1 upstream to overcome this issue, this maneuver mandates glucocorticoid and mineralocorticoid replacement therapy. In phase I/II trials, the first-in-class CYP11A1 inhibitor ODM-208 was more effective in patients with detectable AR LBD mutations in achieving PSA declines but was associated with grade 3 adrenal insufficiency at higher doses.121,122 Thus, striking a balance between blockade of AR mutants while allowing for physiologic glucocorticoid/mineralocorticoid signaling appears to be a challenge with CYP11A1 inhibition.
Immediately downstream to CYP17A1 is 3β-hydroxysteroid dehydrogenase (3β-HSD), which catalyzes the rate-limiting step in the conversion of DHEA to androstenedione (AD). A germline variant in 3β-HSD1 (encoded by HSD3B1, the predominant isoenzyme expressed in the prostate) renders this enzyme resistant to ubiquitin-mediated degradation and increases protein stability, with resultant increased metabolic flux of DHEA to downstream androgens.123 Inheritance of HSD3B1 (1245C), the adrenal-permissive allele that encodes for the more stable form of 3β-HSD1, has been associated with rapid onset of resistance to ADT and poorer clinical outcomes in CRPC, which has been independently validated across several different cohorts.124-128 In addition, CRPC tumors from patients who are germline heterozygotes can acquire a second somatic mutation or undergo loss of heterozygosity.123 Recent evidence also indicates that 3β-HSD1 activity may require phosphorylation by the tyrosine kinase BMX, a finding that could present novel therapeutic avenues to modulate androgen biosynthesis.129
Conversion of AD to DHT requires two final reactions mediated by 17β-hydroxysteroid dehydrogenase and 5α-reductase family enzymes. In prostate cancer, AKR1C3 (type 5 17β-hydroxysteroid dehydrogenase) is overexpressed in response to ADT.28,130,131 However, development of potent, selective AKR1C3 inhibitors is challenging given the sequence similarity of AKR1C3 to several other enzymes within this family, of which inhibition could lead to potentially undesirable effects.132 Furthermore, although 5α-reductase inhibitors are routinely used in the treatment of benign prostatic conditions, their role in prostate cancer is less clear, especially as blockade can result in unintended upstream accumulation of testosterone to potentially rescue AR activity.133,134
Beyond these well-described pathways, other alternative, underappreciated biosynthetic pathways likely also exist that are relevant in prostate cancer (Fig 3).120 For instance, C19 steroid 11β-OH derivatives of AD can be metabolized by CRPC into 11-keto-testosterone/11-keto-dihydroteststerone, which can act as bona fide AR agonists (Fig 3).135,136 Aberrant cortisol metabolism via dysregulation of 11β-HSD2 may also promote upregulation of GR signaling to bypass AR and mediate enzalutamide resistance.137,138 Ultimately, effective therapeutic blockade of androgen biosynthesis requires understanding these different pathways and their contributions toward restoring or circumventing AR activity.
POSTRECEPTOR MECHANISMS
In light of multiple prereceptor and receptor-level resistance mechanisms that promote continued AR activity, a potentially favorable approach might be to target postreceptor mechanisms, which include AR binding and activation of specific downstream genes or oncogenic pathways modulated by AR. This requires a deep understanding of specific transcriptional programs directed by AR, as well as how AR-dependent transcription is regulated by a variety of coregulators.139 For example, the AR cistrome undergoes extensive reprogramming with malignant transformation and progression.140-142 The importance of molecular partners in this process is perhaps best exemplified by the high frequency of driver mutations in key proteins such as FOXA1 and SPOP, which have been shown to interface with AR signaling to promote prostate cancer.17,143,144 Similarly, structural variants such as TMPRSS2-ETS fusions can hijack AR-driven programs, which may be particularly relevant in early-onset disease.145 Some evidence also suggests that AR signaling itself can conversely provoke these nonrandom translocation events to promote carcinogenesis.146-148 Of note, although a majority of primary prostate cancers express AR and can be characterized by a taxonomy-defining alteration, approximately 30% lack a clear driver alteration, despite clinically resembling tumors with identifiable driver alterations. Indeed, a lack of well-defined molecular correlates for Gleason grade exists, and further understanding is thus required in terms of the processes that drive aggressive primary disease.14,149-151
In the context of various potential cellular functions of AR, there remains interest in how to exploit these functions therapeutically. For instance, it is well-recognized that AR can engage in cross-talk with oncogenic signaling pathways such as PI3K/AKT to facilitate tumor progression152,153 The relationship between AR and mediating DNA damage repair as well as the immune response has also prompted efforts to combine AR-directed therapies with other agents, such as PARP inhibitors or immunotherapy.148,154-157 AR target gene expression is also strongly affected by epigenetic processes, including histone acetylation and methylation, which can modify chromatin accessibility and AR binding.140-142 Bromodomain and extraterminal family proteins are epigenetic readers of acetylated histones that are targets for inhibitor design, given that they influence expression of prostate cancer oncogenes, including c-MYC.158-160 Epigenetic regulation can also contribute to enzalutamide resistance because of GR upregulation or other mechanisms that regulate endogenous repeat elements.161,162 More recently, it has become apparent that mechanisms operating in CRPC cells to restore AR activity may also manifest with divergent actions that might be therapeutically exploited.148,156,163-165 These mechanisms are perhaps the basis for the phenomenon of bipolar androgen therapy, in which high-dose testosterone can paradoxically induce clinical responses in a subset of patients.166 Although this is not an exhaustive review of the breadth of postreceptor mechanisms, it highlights a fundamental need to better understand the varied cellular functions of AR and how AR specifically orchestrates prostate cancer programs, which may yield new insights and directions in the treatment of prostate cancer.
CONCLUDING REMARKS
In conclusion, the field of prostate cancer has seen remarkable advances in the past several decades, driven in large part by our understandings of the androgen signaling axis. With this also comes a greater appreciation for the complex mechanisms employed by prostate cancers to thwart effective inhibition of AR signaling. Despite considerable progress in the development of effective next-generation AR-directed therapies, most patients will eventually develop resistance. However, recent and ongoing molecular investigations have led to unprecedented insights into AR structure and function, which has the potential to enhance therapeutic precision and galvanize newfound directions in the treatment of men with prostate cancer.
Scott M. Dehm
Consulting or Advisory Role: Celgene, Oncternal Therapeutics, Janssen Research & Development
Research Funding: Medivation/Astellas (Inst)
Patents, Royalties, Other Intellectual Property: Royalties from licensing genome-engineered prostate cancer cell lines
Travel, Accommodations, Expenses: Oncternal Therapeutics
No other potential conflicts of interest were reported.
Nima Sharifi
Research Funding: Astellas Pharma (Inst), Bristol Myers Squibb Foundation (Inst)
Patents, Royalties, Other Intellectual Property: A patent application has been filed by Cleveland Clinic for a method of steroid dependent disease treatment based on HSD3B1. N.S. is a co-inventor on this patent application.
SUPPORT
Supported by 2T32CA071345-21A1 (to C.D.) and R01CA261995, R01CA236780, R01CA172382, and R01CA249279 (to N.S.).
AUTHOR CONTRIBUTIONS
Conception and design: All authors
Financial support: Nima Sharifi
Collection and assembly of data: Charles Dai, Nima Sharifi
Data analysis and interpretation: All authors
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Targeting the Androgen Signaling Axis in Prostate Cancer
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/authors/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Scott M. Dehm
Consulting or Advisory Role: Celgene, Oncternal Therapeutics, Janssen Research & Development
Research Funding: Medivation/Astellas (Inst)
Patents, Royalties, Other Intellectual Property: Royalties from licensing genome-engineered prostate cancer cell lines
Travel, Accommodations, Expenses: Oncternal Therapeutics
No other potential conflicts of interest were reported.
Nima Sharifi
Research Funding: Astellas Pharma (Inst), Bristol Myers Squibb Foundation (Inst)
Patents, Royalties, Other Intellectual Property: A patent application has been filed by Cleveland Clinic for a method of steroid dependent disease treatment based on HSD3B1. N.S. is a co-inventor on this patent application.
REFERENCES
- 1. Wilson JD, George FW, Griffin JE. The hormonal control of sexual development. Science. 1981;211:1278–1284. doi: 10.1126/science.7010602. [DOI] [PubMed] [Google Scholar]
- 2. Quigley CA, De Bellis A, Marschke KB, et al. Androgen receptor defects: Historical, clinical, and molecular perspectives. Endocr Rev. 1995;16:271–321. doi: 10.1210/edrv-16-3-271. [DOI] [PubMed] [Google Scholar]
- 3. Huggins C, Hodges CV. Studies on prostatic cancer I The effect of castration, of estrogen and of androgen Injection on serum phosphatases in metastatic carcinoma of the prostate. Cancer Res. 1941;1:293–297. doi: 10.3322/canjclin.22.4.232. [DOI] [PubMed] [Google Scholar]
- 4. Huggins C, Stevens RE, Hodges CV. Studies on prostatic cancer: II The effects of castration on advanced carcinoma of the prostate gland. Arch Surg. 1941;43:209–223. [Google Scholar]
- 5. Evans RM. The steroid and thyroid hormone receptor superfamily. Science. 1988;240:889–895. doi: 10.1126/science.3283939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Claessens F, Denayer S, van Tilborgh N, et al. Diverse roles of androgen receptor (AR) domains in AR-mediated signaling. Nucl Recept Signal. 2008;6:e008. doi: 10.1621/nrs.06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Deslypere JP, Young M, Wilson JD, et al. Testosterone and 5 alpha-dihydrotestosterone interact differently with the androgen receptor to enhance transcription of the MMTV-CAT reporter gene. Mol Cell Endocrinol. 1992;88:15–22. doi: 10.1016/0303-7207(92)90004-p. [DOI] [PubMed] [Google Scholar]
- 8. Wasmuth EV, Broeck AV, LaClair JR, et al. Allosteric interactions prime androgen receptor dimerization and activation. Mol Cell. 2022;82:2021–2031.e5. doi: 10.1016/j.molcel.2022.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gelmann EP. Molecular biology of the androgen receptor. J Clin Oncol. 2002;20:3001–3015. doi: 10.1200/JCO.2002.10.018. [DOI] [PubMed] [Google Scholar]
- 10. Riegman PHJ, Vlietstra RJ, van der Korput JAGM, et al. The promoter of the prostate-specific antigen gene contains a functional androgen responsive element. Mol Endocrinol. 1991;5:1921–1930. doi: 10.1210/mend-5-12-1921. [DOI] [PubMed] [Google Scholar]
- 11. Rubin MA, Maher CA, Chinnaiyan AM. Common gene rearrangements in prostate cancer. J Clin Oncol. 2011;29:3659. doi: 10.1200/JCO.2011.35.1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tomlins SA, Rhodes DR, Perner S, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–648. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 13. Mosquera JM, Perner S, Genega EM, et al. Characterization of TMPRSS2-ERG fusion high-grade prostatic intraepithelial neoplasia and potential clinical implications. Clin Cancer Res. 2008;14:3380. doi: 10.1158/1078-0432.CCR-07-5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. The Cancer Genome Atlas Research Network The molecular taxonomy of primary prostate cancer. Cell. 2015;163:1011–1025. doi: 10.1016/j.cell.2015.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gundem G, van Loo P, Kremeyer B, et al. The evolutionary history of lethal metastatic prostate cancer. Nature. 2015;520:353–357. doi: 10.1038/nature14347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Taylor BS, Schultz N, Hieronymus H, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Grasso CS, Wu Y-M, Robinson DR, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–243. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Robinson D, Van Allen EM, Wu Y-M, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161:1215–1228. doi: 10.1016/j.cell.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fraser M, Sabelnykova VY, Yamaguchi TN, et al. Genomic hallmarks of localized, non-indolent prostate cancer. Nature. 2017;541:359–364. doi: 10.1038/nature20788. [DOI] [PubMed] [Google Scholar]
- 20. Chen CD, Welsbie DS, Tran C, et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10:33–39. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- 21. Waltering KK, Helenius MA, Sahu B, et al. Increased expression of androgen receptor sensitizes prostate cancer cells to low levels of androgens. Cancer Res. 2009;69:8141–8149. doi: 10.1158/0008-5472.CAN-09-0919. [DOI] [PubMed] [Google Scholar]
- 22. Visakorpi T, Hyytinen E, Koivisto P, et al. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet. 1995;9:401–406. doi: 10.1038/ng0495-401. [DOI] [PubMed] [Google Scholar]
- 23. Geller J, Albert J, Loza D, et al. DHT concentrations in human prostate cancer tissue. J Clin Endocrinol Metab. 1978;46:440–444. doi: 10.1210/jcem-46-3-440. [DOI] [PubMed] [Google Scholar]
- 24. Titus MA, Schell MJ, Lih FB, et al. Testosterone and dihydrotestosterone tissue levels in recurrent prostate cancer. Clin Cancer Res. 2005;11:4653–4657. doi: 10.1158/1078-0432.CCR-05-0525. [DOI] [PubMed] [Google Scholar]
- 25. Montgomery RB, Mostaghel EA, Vessella R, et al. Maintenance of intratumoral androgens in metastatic prostate cancer: A mechanism for castration-resistant tumor growth. Cancer Res. 2008;68:4447–4454. doi: 10.1158/0008-5472.CAN-08-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nishiyama T, Hashimoto Y, Takahashi K. The influence of androgen deprivation therapy on dihydrotestosterone levels in the prostatic tissue of patients with prostate cancer. Clin Cancer Res. 2004;10:7121–7126. doi: 10.1158/1078-0432.CCR-04-0913. [DOI] [PubMed] [Google Scholar]
- 27. Huggins C, Scott WW. Bilateral adrenalectomy in prostatic cancer: Clinical features and urinary excretion of 17-ketosteroids and estrogen. Ann Surg. 1945;122:1031–1041. [PMC free article] [PubMed] [Google Scholar]
- 28. Stanbrough M, Bubley GJ, Ross K, et al. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 2006;66:2815–2825. doi: 10.1158/0008-5472.CAN-05-4000. [DOI] [PubMed] [Google Scholar]
- 29. BioRender.
- 30. Tran C, Ouk S, Clegg NJ, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324:787–790. doi: 10.1126/science.1168175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chang K-H, Li R, Papari-Zareei M, et al. Dihydrotestosterone synthesis bypasses testosterone to drive castration-resistant prostate cancer. Proc Natl Acad Sci USA. 2011;108:13728–13733. doi: 10.1073/pnas.1107898108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dai C, Chung YM, Kovac E, et al. Direct metabolic interrogation of dihydrotestosterone biosynthesis from adrenal precursors in primary prostatectomy tissues. Clin Cancer Res. 2017;23:6351–6363. doi: 10.1158/1078-0432.CCR-17-1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Haidar S, Ehmer PB, Barassin S, et al. Effects of novel 17α-hydroxylase/C17, 20-lyase (P450 17, CYP 17) inhibitors on androgen biosynthesis in vitro and in vivo. J Steroid Biochem Mol Biol. 2003;84:555–562. doi: 10.1016/s0960-0760(03)00070-0. [DOI] [PubMed] [Google Scholar]
- 34. de Bono JS, Logothetis CJ, Molina A, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364:1995–2005. doi: 10.1056/NEJMoa1014618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Scher HI, Fizazi K, Saad F, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367:1187–1197. doi: 10.1056/NEJMoa1207506. [DOI] [PubMed] [Google Scholar]
- 36. Hussain M, Fizazi K, Saad F, et al. Enzalutamide in men with nonmetastatic, castration-resistant prostate cancer. N Engl J Med. 2018;378:2465–2474. doi: 10.1056/NEJMoa1800536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fizazi K, Shore N, Tammela TL, et al. Nonmetastatic, castration-resistant prostate cancer and survival with darolutamide. N Engl J Med. 2020;383:1040–1049. doi: 10.1056/NEJMoa2001342. [DOI] [PubMed] [Google Scholar]
- 38. Smith MR, Saad F, Chowdhury S, et al. Apalutamide and overall survival in prostate cancer. Eur Urol. 2021;79:150–158. doi: 10.1016/j.eururo.2020.08.011. [DOI] [PubMed] [Google Scholar]
- 39. Sternberg CN, Fizazi K, Saad F, et al. Enzalutamide and survival in nonmetastatic, castration-resistant prostate cancer. N Engl J Med. 2020;382:2197–2206. doi: 10.1056/NEJMoa2003892. [DOI] [PubMed] [Google Scholar]
- 40. Fizazi K, Tran NP, Fein L, et al. Abiraterone acetate plus prednisone in patients with newly diagnosed high-risk metastatic castration-sensitive prostate cancer (LATITUDE): Final overall survival analysis of a randomised, double-blind, phase 3 trial. Lancet Oncol. 2019;20:686–700. doi: 10.1016/S1470-2045(19)30082-8. [DOI] [PubMed] [Google Scholar]
- 41. James ND, de Bono JS, Spears MR, et al. Abiraterone for prostate cancer not previously treated with hormone therapy. N Engl J Med. 2017;377:338–351. doi: 10.1056/NEJMoa1702900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Smith MR, Hussain M, Saad F, et al. Darolutamide and survival in metastatic, hormone-sensitive prostate cancer. N Engl J Med. 2022;386:1132–1142. doi: 10.1056/NEJMoa2119115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Davis ID, Martin AJ, Zielinski RR, et al. Updated overall survival outcomes in ENZAMET (ANZUP 1304), an international, cooperative group trial of enzalutamide in metastatic hormone-sensitive prostate cancer (mHSPC) J Clin Oncol. 2022;40 doi: 10.1200/JCO.21.00941. suppl 17; abstr LBA5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chi KN, Chowdhury S, Bjartell A, et al. Apalutamide in patients with metastatic castration-sensitive prostate cancer: Final survival analysis of the randomized, double-blind, phase III TITAN study. J Clin Oncol. 2021;39:2294–2303. doi: 10.1200/JCO.20.03488. [DOI] [PubMed] [Google Scholar]
- 45. Armstrong AJ, Szmulewitz RZ, Petrylak DP, et al. ARCHES: A randomized, phase III study of androgen deprivation therapy with enzalutamide or placebo in men with metastatic hormone-sensitive prostate cancer. J Clin Oncol. 2019;37:2974–2986. doi: 10.1200/JCO.19.00799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Quigley DA, Dang HX, Zhao SG, et al. Genomic hallmarks and structural variation in metastatic prostate cancer. Cell. 2018;174:758–769.e9. doi: 10.1016/j.cell.2018.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Abida W, Cyrta J, Heller G, et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc Natl Acad Sci USA. 2019;166:11428–11436. doi: 10.1073/pnas.1902651116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Annala M, Taavitsainen S, Khalaf DJ, et al. Evolution of castration-resistant prostate cancer in ctDNA during sequential androgen receptor pathway inhibition. Clin Cancer Res. 2021;27:4610–4623. doi: 10.1158/1078-0432.CCR-21-1625. [DOI] [PubMed] [Google Scholar]
- 49. Beltran H, Prandi D, Mosquera JM, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med. 2016;22:298–305. doi: 10.1038/nm.4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Beltran H, Tagawa ST, Park K, et al. Challenges in recognizing treatment-related neuroendocrine prostate cancer. J Clin Oncol. 2012;30:e386–e389. doi: 10.1200/JCO.2011.41.5166. [DOI] [PubMed] [Google Scholar]
- 51. Puca L, Vlachostergios PJ, Beltran H. Neuroendocrine differentiation in prostate cancer: Emerging biology, models, and therapies. Cold Spring Harb Perspect Med. 2019;9:a030593. doi: 10.1101/cshperspect.a030593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Garraway LA, Sellers WR. Lineage dependency and lineage-survival oncogenes in human cancer. Nat Rev Cancer. 2006;6:593–602. doi: 10.1038/nrc1947. [DOI] [PubMed] [Google Scholar]
- 53. Takeda DY, Spisák S, Seo JH, et al. A somatically acquired enhancer of the androgen receptor is a noncoding driver in advanced prostate cancer. Cell. 2018;174:422. doi: 10.1016/j.cell.2018.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Viswanathan SR, Ha G, Hoff AM, et al. Structural alterations driving castration-resistant prostate cancer revealed by linked-read genome sequencing. Cell. 2018;174:433–447.e19. doi: 10.1016/j.cell.2018.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Henzler C, Li Y, Yang R, et al. Truncation and constitutive activation of the androgen receptor by diverse genomic rearrangements in prostate cancer. Nat Commun. 2016;7:1–12. doi: 10.1038/ncomms13668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Li Y, Yang R, Henzler CM, et al. Diverse AR gene rearrangements mediate resistance to androgen receptor inhibitors in metastatic prostate cancer. Clin Cancer Res. 2020;26:1965–1976. doi: 10.1158/1078-0432.CCR-19-3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Azad AA, Volik Sv, Wyatt AW, et al. Androgen receptor gene aberrations in circulating cell-free DNA: Biomarkers of therapeutic resistance in castration-resistant prostate cancer. Clin Cancer Res. 2015;21:2315–2324. doi: 10.1158/1078-0432.CCR-14-2666. [DOI] [PubMed] [Google Scholar]
- 58. Romanel A, Tandefelt DG, Conteduca V, et al. Plasma AR and abiraterone-resistant prostate cancer. Sci Transl Med. 2015;7:312re10. doi: 10.1126/scitranslmed.aac9511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Callewaert L, van Tilborgh N, Claessens F. Interplay between two hormone-independent activation domains in the androgen receptor. Cancer Res. 2006;66:543–553. doi: 10.1158/0008-5472.CAN-05-2389. [DOI] [PubMed] [Google Scholar]
- 60. Jenster G, van der Korput HA, Trapman J, et al. Identification of two transcription activation units in the N-terminal domain of the human androgen receptor. J Biol Chem. 1995;270:7341–7346. doi: 10.1074/jbc.270.13.7341. [DOI] [PubMed] [Google Scholar]
- 61. Yu X, Yi P, Hamilton RA, et al. Structural insights of transcriptionally active, full-length androgen receptor coactivator complexes. Mol Cell. 2020;79:812–823.e4. doi: 10.1016/j.molcel.2020.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Jenster G, van der Korput HA, van Vroonhoven C, et al. Domains of the human androgen receptor involved in steroid binding, transcriptional activation, and subcellular localization. Mol Endocrinol. 1991;5:1396–1404. doi: 10.1210/mend-5-10-1396. [DOI] [PubMed] [Google Scholar]
- 63. Simental JA, Sar M, Lane MV, et al. Transcriptional activation and nuclear targeting signals of the human androgen receptor. J Biol Chem. 1991;266:510–518. [PubMed] [Google Scholar]
- 64. el Kharraz S, Dubois V, Launonen KM, et al. N/C interactions are dispensable for normal in vivo functioning of the androgen receptor in male mice. Endocrinology. 2022;163:bqac104. doi: 10.1210/endocr/bqac104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Dehm SM, Schmidt LJ, Heemers HV, et al. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008;68:5469–5477. doi: 10.1158/0008-5472.CAN-08-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Guo Z, Yang X, Sun F, et al. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res. 2009;69:2305–2313. doi: 10.1158/0008-5472.CAN-08-3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hu R, Dunn TA, Wei S, et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009;69:16–22. doi: 10.1158/0008-5472.CAN-08-2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sun S, Sprenger CCT, Vessella RL, et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clin Invest. 2010;120:2715–2730. doi: 10.1172/JCI41824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zhu J, Salvatella X, Robustelli P. Small molecules targeting the disordered transactivation domain of the androgen receptor induce the formation of collapsed helical states. Nat Commun. 2022;13:1–15. doi: 10.1038/s41467-022-34077-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Maurice-Dror C, le Moigne R, Vaishampayan U, et al. A phase 1 study to assess the safety, pharmacokinetics, and anti-tumor activity of the androgen receptor n-terminal domain inhibitor epi-506 in patients with metastatic castration-resistant prostate cancer. Invest New Drugs. 2022;40:322–329. doi: 10.1007/s10637-021-01202-6. [DOI] [PubMed] [Google Scholar]
- 71. Le Moigne R, Zhou H-J, Obst JK, et al. Lessons learned from the metastatic castration-resistant prostate cancer phase I trial of EPI-506, a first-generation androgen receptor N-terminal domain inhibitor. J Clin Oncol. 2019;37 suppl 7; abstr 257. [Google Scholar]
- 72. Le Moigne R, Pearson P, Lauriault V, et al. Preclinical and clinical pharmacology of EPI-7386, an androgen receptor N-terminal domain inhibitor for castration-resistant prostate cancer. J Clin Oncol. 2021;39 suppl 6; abstr 119. [Google Scholar]
- 73. Brand LJ, Olson ME, Ravindranathan P, et al. EPI-001 is a selective peroxisome proliferator-activated receptor-gamma modulator with inhibitory effects on androgen receptor expression and activity in prostate cancer. Oncotarget. 2015;6:3811. doi: 10.18632/oncotarget.2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Arora VK, Schenkein E, Murali R, et al. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell. 2013;155:1309–1322. doi: 10.1016/j.cell.2013.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Isikbay M, Otto K, Kregel S, et al. Glucocorticoid receptor activity contributes to resistance to androgen-targeted therapy in prostate cancer. Horm Cancer. 2014;5:72–89. doi: 10.1007/s12672-014-0173-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Heery DM, Kalkhoven E, Hoare S, et al. A signature motif in transcriptional co-activators mediates binding to nuclear receptors. Nature. 1997;387:733–736. doi: 10.1038/42750. [DOI] [PubMed] [Google Scholar]
- 77. Bevan CL, Hoare S, Claessens F, et al. The AF1 and AF2 domains of the androgen receptor interact with distinct regions of SRC1. Mol Cell Biol. 1999;19:8383–8392. doi: 10.1128/mcb.19.12.8383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Estébanez-Perpiñá E, Arnold LA, Nguyen P, et al. A surface on the androgen receptor that allosterically regulates coactivator binding. Proc Natl Acad Sci USA. 2007;104:16074–16079. doi: 10.1073/pnas.0708036104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Lack NA, Axerio-Cilies P, Tavassoli P, et al. Targeting the binding function 3 (BF3) site of the human androgen receptor through virtual screening. J Med Chem. 2011;54:8563–8573. doi: 10.1021/jm201098n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Tan ME, Li J, Xu HE, et al. Androgen receptor: Structure, role in prostate cancer and drug discovery. Acta Pharmacol Sin. 2014;36:3–23. doi: 10.1038/aps.2014.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Lallous N, Leblanc E, Munuganti RSN, et al. Targeting binding function-3 of the androgen receptor blocks its co-chaperone interactions, nuclear translocation, and activation. Mol Cancer Ther. 2016;15:2936–2945. doi: 10.1158/1535-7163.MCT-16-0354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Ledet EM, Lilly MB, Sonpavde G, et al. Comprehensive analysis of AR alterations in circulating tumor DNA from patients with advanced prostate cancer. Oncologist. 2020;25:327–333. doi: 10.1634/theoncologist.2019-0115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Suzuki H, Sato N, Watabe Y, et al. Androgen receptor gene mutations in human prostate cancer. J Steroid Biochem Mol Biol. 1993;46:759–765. doi: 10.1016/0960-0760(93)90316-o. [DOI] [PubMed] [Google Scholar]
- 84. Veldscholte J, Ris-Stalpers C, Kuiper GG, et al. A mutation in the ligand binding domain of the androgen receptor of human LNCaP cells affects steroid binding characteristics and response to anti-androgens. Biochem Biophys Res Commun. 1990;173:534–540. doi: 10.1016/s0006-291x(05)80067-1. [DOI] [PubMed] [Google Scholar]
- 85. Wilding G, Chen M, Gelmann EP. Aberrant response in vitro of hormone-responsive prostate cancer cells to antiandrogens. Prostate. 1989;14:103–115. doi: 10.1002/pros.2990140204. [DOI] [PubMed] [Google Scholar]
- 86. Veldscholte J, Berrevoets CA, Ris-Stalpers C, et al. The androgen receptor in LNCaP cells contains a mutation in the ligand binding domain which affects steroid binding characteristics and response to antiandrogens. J Steroid Biochem Mol Biol. 1992;41:665–669. doi: 10.1016/0960-0760(92)90401-4. [DOI] [PubMed] [Google Scholar]
- 87. Taplin ME, Bubley GJ, Shuster TD, et al. Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. N Engl J Med. 1995;332:1393–1398. doi: 10.1056/NEJM199505253322101. [DOI] [PubMed] [Google Scholar]
- 88. Tan J, Sharief Y, Hamil KG, et al. Dehydroepiandrosterone activates mutant androgen receptors expressed in the androgen-dependent human prostate cancer xenograft CWR22 and LNCaP cells. Mol Endocrinol. 1997;11:450–459. doi: 10.1210/mend.11.4.9906. [DOI] [PubMed] [Google Scholar]
- 89. Taplin ME, Bubley GJ, Ko YJ, et al. Selection for androgen receptor mutations in prostate cancers treated with androgen antagonist. Cancer Res. 1999;59:2511–2515. [PubMed] [Google Scholar]
- 90. Hara T, Miyazaki J, Araki H, et al. Novel mutations of androgen receptor: A possible mechanism of bicalutamide withdrawal syndrome. Cancer Res. 2003;63:149–153. [PubMed] [Google Scholar]
- 91. Yoshida T, Kinoshita H, Segawa T, et al. Antiandrogen bicalutamide promotes tumor growth in a novel androgen-dependent prostate cancer xenograft model derived from a bicalutamide-treated patient. Cancer Res. 2005;65:9611–9616. doi: 10.1158/0008-5472.CAN-05-0817. [DOI] [PubMed] [Google Scholar]
- 92. Scher HI, Kelly WK. Flutamide withdrawal syndrome: Its impact on clinical trials in hormone-refractory prostate cancer. J Clin Oncol. 1993;11:1566–1572. doi: 10.1200/JCO.1993.11.8.1566. [DOI] [PubMed] [Google Scholar]
- 93. Sartor AO, Tangen CM, Hussain MHA, et al. Antiandrogen withdrawal in castrate-refractory prostate cancer: A Southwest Oncology Group trial (SWOG 9426) Cancer. 2008;112:2393–2400. doi: 10.1002/cncr.23473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Joseph JD, Lu N, Qian J, et al. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discov. 2013;3:1020–1029. doi: 10.1158/2159-8290.CD-13-0226. [DOI] [PubMed] [Google Scholar]
- 95. Balbas MD, Evans MJ, Hosfield DJ, et al. Overcoming mutation-based resistance to antiandrogens with rational drug design. Elife. 2013;2:e00499. doi: 10.7554/eLife.00499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Korpal M, Korn JM, Gao X, et al. An F876L mutation in androgen receptor confers genetic and phenotypic resistance to MDV3100 (enzalutamide) Cancer Discov. 2013;3:1030–1043. doi: 10.1158/2159-8290.CD-13-0142. [DOI] [PubMed] [Google Scholar]
- 97. Lallous N, Volik Sv, Awrey S, et al. Functional analysis of androgen receptor mutations that confer anti-androgen resistance identified in circulating cell-free DNA from prostate cancer patients. Genome Biol. 2016;17:10. doi: 10.1186/s13059-015-0864-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Rathkopf DE, Smith MR, Ryan CJ, et al. Androgen receptor mutations in patients with castration-resistant prostate cancer treated with apalutamide. Ann Oncol. 2017;28:2264–2271. doi: 10.1093/annonc/mdx283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Borgmann H, Lallous N, Ozistanbullu D, et al. Moving towards precision urologic oncology: Targeting enzalutamide-resistant prostate cancer and mutated forms of the androgen receptor using the novel inhibitor darolutamide (ODM-201) Eur Urol. 2018;73:4–8. doi: 10.1016/j.eururo.2017.08.012. [DOI] [PubMed] [Google Scholar]
- 100. Chen EJ, Sowalsky AG, Gao S, et al. Abiraterone treatment in castration-resistant prostate cancer selects for progesterone responsive mutant androgen receptors. Clin Cancer Res. 2015;21:1273–1280. doi: 10.1158/1078-0432.CCR-14-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Richards J, Lim AC, Hay CW, et al. Interactions of abiraterone, eplerenone, and prednisolone with wild-type and mutant androgen receptor: A rationale for increasing abiraterone exposure or combining with MDV3100. Cancer Res. 2012;72:2176–2182. doi: 10.1158/0008-5472.CAN-11-3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Li Z, Bishop AC, Alyamani M, et al. Conversion of abiraterone to D4A drives anti-tumour activity in prostate cancer. Nature. 2015;523:347–351. doi: 10.1038/nature14406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Li Z, Alyamani M, Li J, et al. Redirecting abiraterone metabolism to fine-tune prostate cancer anti-androgen therapy. Nature. 2016;533:547–551. doi: 10.1038/nature17954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Khalaf DJ, Annala M, Taavitsainen S, et al. Optimal sequencing of enzalutamide and abiraterone acetate plus prednisone in metastatic castration-resistant prostate cancer: A multicentre, randomised, open-label, phase 2, crossover trial. Lancet Oncol. 2019;20:1730–1739. doi: 10.1016/S1470-2045(19)30688-6. [DOI] [PubMed] [Google Scholar]
- 105. Cao B, Qi Y, Zhang G, et al. Androgen receptor splice variants activating the full-length receptor in mediating resistance to androgen-directed therapy. Oncotarget. 2014;5:1646–1656. doi: 10.18632/oncotarget.1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Xu D, Zhan Y, Qi Y, et al. Androgen receptor splice variants dimerize to transactivate target genes. Cancer Res. 2015;75:3663–3671. doi: 10.1158/0008-5472.CAN-15-0381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Watson PA, Chen YF, Balbas MD, et al. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc Natl Acad Sci USA. 2010;107:16759–16765. doi: 10.1073/pnas.1012443107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Antonarakis ES, Lu C, Luber B, et al. Androgen receptor splice variant 7 and efficacy of taxane chemotherapy in patients with metastatic castration-resistant prostate cancer. JAMA Oncol. 2015;1:582–591. doi: 10.1001/jamaoncol.2015.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Armstrong AJ, Halabi S, Luo J, et al. Prospective multicenter validation of androgen receptor splice variant 7 and hormone therapy resistance in high-risk castration-resistant prostate cancer: The PROPHECY study. J Clin Oncol. 2019;37:1120–1129. doi: 10.1200/JCO.18.01731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Scher HI, Lu D, Schreiber NA, et al. Association of AR-V7 on circulating tumor cells as a treatment-specific biomarker with outcomes and survival in castration-resistant prostate cancer. JAMA Oncol. 2016;2:1441–1449. doi: 10.1001/jamaoncol.2016.1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Antonarakis ES, Lu C, Luber B, et al. Clinical significance of androgen receptor splice variant-7 mRNA detection in circulating tumor cells of men with metastatic castration-resistant prostate cancer treated with first- and second-line abiraterone and enzalutamide. J Clin Oncol. 2017;35:2149–2156. doi: 10.1200/JCO.2016.70.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Sharp A, Coleman I, Yuan W, et al. Androgen receptor splice variant-7 expression emerges with castration resistance in prostate cancer. J Clin Invest. 2019;129:192–208. doi: 10.1172/JCI122819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Kohli M, Ho Y, Hillman DW, et al. Androgen receptor variant AR-V9 is coexpressed with AR-V7 in prostate cancer metastases and predicts abiraterone resistance. Clin Cancer Res. 2017;23:4704–4715. doi: 10.1158/1078-0432.CCR-17-0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Sperger JM, Emamekhoo H, McKay RR, et al. Prospective evaluation of clinical outcomes using a multiplex liquid biopsy targeting diverse resistance mechanisms in metastatic prostate cancer. J Clin Oncol. 2021;39:2926–2937. doi: 10.1200/JCO.21.00169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Gao X, III HAB, Vuky J, et al. Phase 1/2 study of ARV-110, an androgen receptor (AR) PROTAC degrader, in metastatic castration-resistant prostate cancer (mCRPC) J Clin Oncol. 2022;40:17. [Google Scholar]
- 116. Ponnusamy S, He Y, Hwang DJ, et al. Orally bioavailable androgen receptor degrader, potential next-generation therapeutic for enzalutamide-resistant prostate cancer. Clin Cancer Res. 2019;25:6764–6780. doi: 10.1158/1078-0432.CCR-19-1458. [DOI] [PubMed] [Google Scholar]
- 117. Mohler ML, Sikdar A, Ponnusamy S, et al. An overview of next-generation androgen receptor-targeted therapeutics in development for the treatment of prostate cancer. Int J Mol Sci. 2021;22:1–20. doi: 10.3390/ijms22042124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Bélanger A, Candas B, Dupont A, et al. Changes in serum concentrations of conjugated and unconjugated steroids in 40- to 80-year-old men. J Clin Endocrinol Metab. 1994;79:1086–1090. doi: 10.1210/jcem.79.4.7962278. [DOI] [PubMed] [Google Scholar]
- 119. Wilson EM, French FS. Binding properties of androgen receptors Evidence for identical receptors in rat testis, epididymis, and prostate. J Biol Chem. 1976;251:5620–5629. [PubMed] [Google Scholar]
- 120. Dai C, Heemers H, Sharifi N. Androgen signaling in prostate cancer. Cold Spring Harb Perspect Med. 2017;7:a030452. doi: 10.1101/cshperspect.a030452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Bernard-Tessier A, Utriainen T, Cook N, et al. Impact of activating androgen receptor (AR) mutations on AR sensitivity to alternative ligands and response to ODM-208, a selective, first-in-class CYP11A1 inhibitor, in patients with advanced metastatic castration-resistant prostate cancer (mCRPC) J Clin Oncol. 2022;40:5057. [Google Scholar]
- 122. Fizazi K, Cook N, Barthélémy P, et al. Phase 1 results of the ODM-208 first-in-human phase 1-2 trial in patients with metastatic castration-resistant prostate cancer (CYPIDES) J Clin Oncol. 2022;40:18. [Google Scholar]
- 123. Chang K-H, Li R, Kuri B, et al. A gain-of-function mutation in DHT synthesis in castration-resistant prostate cancer. Cell. 2013;154:1074–1084. doi: 10.1016/j.cell.2013.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Hearn JWD, Sweeney CJ, Almassi N, et al. HSD3B1 genotype and clinical outcomes in metastatic castration-sensitive prostate cancer. JAMA Oncol. 2020;6:e196496. doi: 10.1001/jamaoncol.2019.6496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Hearn JW, AbuAli G, Reichard CA, et al. HSD3B1 and resistance to androgen deprivation therapy in prostate cancer: A multi-cohort study. Lancet Oncol. 2016;17:1435–1444. doi: 10.1016/S1470-2045(16)30227-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Agarwal N, Hahn AW, Gill DM, et al. Independent validation of effect of HSD3B1 genotype on response to androgen-deprivation therapy in prostate cancer. JAMA Oncol. 2017;3:856–857. doi: 10.1001/jamaoncol.2017.0147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Khalaf DJ, Aragón IM, Annala M, et al. HSD3B1 (1245A>C) germline variant and clinical outcomes in metastatic castration-resistant prostate cancer patients treated with abiraterone and enzalutamide: Results from two prospective studies. Ann Oncol. 2020;31:1186–1197. doi: 10.1016/j.annonc.2020.06.006. [DOI] [PubMed] [Google Scholar]
- 128. Lu C, Terbuch A, Dolling D, et al. Treatment with abiraterone and enzalutamide does not overcome poor outcome from metastatic castration-resistant prostate cancer in men with the germline homozygous HSD3B1 c1245C genotype. Ann Oncol. 2020;31:1178–1185. doi: 10.1016/j.annonc.2020.04.473. [DOI] [PubMed] [Google Scholar]
- 129. Li X, Berk M, Goins C, et al. BMX controls 3βHSD1 and sex steroid biosynthesis in cancer. J Clin Invest. 2023;133:e163498. doi: 10.1172/JCI163498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Mitsiades N, Sung CC, Schultz N, et al. Distinct patterns of dysregulated expression of enzymes involved in androgen synthesis and metabolism in metastatic prostate cancer tumors. Cancer Res. 2012;72:6142. doi: 10.1158/0008-5472.CAN-12-1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Penning TM. AKR1C3 (type 5 17β-hydroxysteroid dehydrogenase/prostaglandin F synthase): Roles in malignancy and endocrine disorders. Mol Cell Endocrinol. 2019;489:82–91. doi: 10.1016/j.mce.2018.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Penning TM. Aldo-keto reductase (AKR) 1C3 inhibitors: A patent review. Expert Opin Ther Pat. 2017;27:1329. doi: 10.1080/13543776.2017.1379503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Shah SK, Trump DL, Sartor O, et al. Phase II study of Dutasteride for recurrent prostate cancer during androgen deprivation therapy. J Urol. 2009;181:621–626. doi: 10.1016/j.juro.2008.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Rittmaster R, Hahn RG, Ray P, et al. Effect of dutasteride on intraprostatic androgen levels in men with benign prostatic hyperplasia or prostate cancer. Urology. 2008;72:808–812. doi: 10.1016/j.urology.2008.06.032. [DOI] [PubMed] [Google Scholar]
- 135. Storbeck KH, Bloem LM, Africander D, et al. 11β-Hydroxydihydrotestosterone and 11-ketodihydrotestosterone, novel C19 steroids with androgenic activity: A putative role in castration resistant prostate cancer? Mol Cell Endocrinol. 2013;377:135–146. doi: 10.1016/j.mce.2013.07.006. [DOI] [PubMed] [Google Scholar]
- 136. du Toit T, Bloem LM, Quanson JL, et al. Profiling adrenal 11β-hydroxyandrostenedione metabolites in prostate cancer cells, tissue and plasma: UPC2-MS/MS quantification of 11β-hydroxytestosterone, 11keto-testosterone and 11keto-dihydrotestosterone. J Steroid Biochem Mol Biol. 2017;166:54–67. doi: 10.1016/j.jsbmb.2016.06.009. [DOI] [PubMed] [Google Scholar]
- 137. Li J, Alyamani M, Zhang A, et al. Aberrant corticosteroid metabolism in tumor cells enables GR takeover in enzalutamide resistant prostate cancer. Elife. 2017;6:e20183. doi: 10.7554/eLife.20183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Li J, Berk M, Alyamani M, et al. Hexose-6-phosphate dehydrogenase blockade reverses prostate cancer drug resistance in xenograft models by glucocorticoid inactivation. Sci Transl Med. 2021;13:eabe8226. doi: 10.1126/scitranslmed.abe8226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Heemers HV, Tindall DJ. Androgen receptor (AR) coregulators: A diversity of functions converging on and regulating the AR transcriptional complex. Endocr Rev. 2007;28:778–808. doi: 10.1210/er.2007-0019. [DOI] [PubMed] [Google Scholar]
- 140. Wang Q, Li W, Zhang Y, et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell. 2009;138:245–256. doi: 10.1016/j.cell.2009.04.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Sharma NL, Massie CE, Ramos-Montoya A, et al. The androgen receptor induces a distinct transcriptional program in castration-resistant prostate cancer in man. Cancer Cell. 2013;23:35–47. doi: 10.1016/j.ccr.2012.11.010. [DOI] [PubMed] [Google Scholar]
- 142. Pomerantz MM, Li F, Takeda DY, et al. The androgen receptor cistrome is extensively reprogrammed in human prostate tumorigenesis. Nat Genet. 2015;47:1346–1351. doi: 10.1038/ng.3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Barbieri CE, Baca SC, Lawrence MS, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet. 2012;44:685–689. doi: 10.1038/ng.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Wang D, Garcia-Bassets I, Benner C, et al. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature. 2011;474:390–394. doi: 10.1038/nature10006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Weischenfeldt J, Simon R, Feuerbach L, et al. Integrative genomic analyses reveal an androgen-driven somatic alteration landscape in early-onset prostate cancer. Cancer Cell. 2013;23:159–170. doi: 10.1016/j.ccr.2013.01.002. [DOI] [PubMed] [Google Scholar]
- 146. Lin C, Yang L, Tanasa B, et al. Nuclear receptor-induced chromosomal proximity and DNA breaks underlie specific translocations in cancer. Cell. 2009;139:1069–1083. doi: 10.1016/j.cell.2009.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Mani R-S, Tomlins SA, Callahan K, et al. Induced chromosomal proximity and gene fusions in prostate cancer. Science. 2009;326:1230. doi: 10.1126/science.1178124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Haffner MC, Aryee MJ, Toubaji A, et al. Androgen-induced TOP2B-mediated double-strand breaks and prostate cancer gene rearrangements. Nat Genet. 2010;42:668–675. doi: 10.1038/ng.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Chodak GW, Kranc DM, Puy LA, et al. Nuclear localization of androgen receptor in heterogeneous samples of normal, hyperplastic and neoplastic human prostate. J Urol. 1992;147:798–803. doi: 10.1016/s0022-5347(17)37389-5. [DOI] [PubMed] [Google Scholar]
- 150. Ruizeveld De Winter JA, Janssen PJA, Sleddens HMEB, et al. Androgen receptor status in localized and locally progressive hormone refractory human prostate cancer. Am J Pathol. 1994;144:735. [PMC free article] [PubMed] [Google Scholar]
- 151. Yoshikawa H, Ikeuchi T, Kai Y. Immunohistochemical study of androgen receptor in adenocarcinoma of the human prostatic cancer [in Japanese] Nihon Hinyokika Gakkai Zasshi. 1996;87:956–963. doi: 10.5980/jpnjurol1989.87.956. [DOI] [PubMed] [Google Scholar]
- 152. Mulholland DJ, Tran LM, Li Y, et al. Cell autonomous role of PTEN in regulating castration-resistant prostate cancer growth. Cancer Cell. 2011;19:792–804. doi: 10.1016/j.ccr.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Carver BS, Chapinski C, Wongvipat J, et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell. 2011;19:575–586. doi: 10.1016/j.ccr.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Goodwin JF, Schiewer MJ, Dean JL, et al. A hormone-DNA repair circuit governs the response to genotoxic insult. Cancer Discov. 2013;3:1254–1271. doi: 10.1158/2159-8290.CD-13-0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Polkinghorn WR, Parker JS, Lee MX, et al. Androgen receptor signaling regulates DNA repair in prostate cancers. Cancer Discov. 2013;3:1245–1253. doi: 10.1158/2159-8290.CD-13-0172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Chatterjee P, Schweizer MT, Lucas JM, et al. Supraphysiological androgens suppress prostate cancer growth through androgen receptor–mediated DNA damage. J Clin Invest. 2019;129:4245. doi: 10.1172/JCI127613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Guan X, Polesso F, Wang C, et al. Androgen receptor activity in T cells limits checkpoint blockade efficacy. Nature. 2022;606:791–796. doi: 10.1038/s41586-022-04522-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Wyce A, Degenhardt Y, Bai Y, et al. Inhibition of BET bromodomain proteins as a therapeutic approach in prostate cancer. Oncotarget. 2013;4:2419. doi: 10.18632/oncotarget.1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Coleman DJ, Gao L, Schwartzman J, et al. Maintenance of MYC expression promotes de novo resistance to BET bromodomain inhibition in castration-resistant prostate cancer. Sci Rep. 2019;9:3823. doi: 10.1038/s41598-019-40518-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Delmore JE, Issa GC, Lemieux ME, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Shah N, Wang P, Wongvipat J, et al. Regulation of the glucocorticoid receptor via a BET-dependent enhancer drives antiandrogen resistance in prostate cancer. Elife. 2017;6:e27861. doi: 10.7554/eLife.27861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Baratchian M, Tiwari R, Khalighi S, et al. H3K9 methylation drives resistance to androgen receptor–antagonist therapy in prostate cancer. Proc Natl Acad Sci USA. 2022;119:e2114324119. doi: 10.1073/pnas.2114324119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Litvinov Iv, vander Griend DJ, Antony L, et al. Androgen receptor as a licensing factor for DNA replication in androgen-sensitive prostate cancer cells. Proc Natl Acad Sci USA. 2006;103:15085–15090. doi: 10.1073/pnas.0603057103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Sena LA, Kumar R, Sanin DE, et al. Androgen receptor activity in prostate cancer dictates efficacy of bipolar androgen therapy through MYC. J Clin Invest. 2022;132:e162396. doi: 10.1172/JCI162396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Kumar R, Mendonca J, Owoyemi O, et al. Supraphysiologic testosterone induces ferroptosis and activates immune pathways through nucleophagy in prostate cancer. Cancer Res. 2021;81:5948–5962. doi: 10.1158/0008-5472.CAN-20-3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Denmeade SR, Wang H, Agarwal N, et al. TRANSFORMER: A randomized phase II study comparing bipolar androgen therapy versus enzalutamide in asymptomatic men with castration-resistant metastatic prostate cancer. J Clin Oncol. 2021;39:1371–1382. doi: 10.1200/JCO.20.02759. [DOI] [PMC free article] [PubMed] [Google Scholar]