

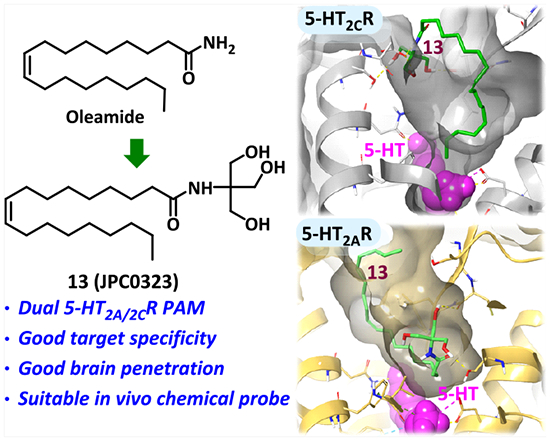
Abstract
The serotonin 5-HT2A receptor (5-HT2AR) and 5-HT2CR localize to the brain and share overlapping signal transduction facets that contribute to their roles in cognition, mood, learning, and memory. Achieving selective targeting of these receptors is challenged by the similarity in their 5-HT orthosteric binding pockets. A fragment-based discovery approach was employed to design and synthesize novel oleamide analogues as selective 5-HT2CR or dual 5-HT2CR/5-HT2AR positive allosteric modulators (PAMs). Compound 13 (JPC0323) exhibited on-target properties, acceptable plasma exposure and brain penetration, as well as negligible displacement to orthosteric sites of ~50 GPCRs and transporters. Furthermore, compound 13 suppressed novelty-induced locomotor activity in a 5-HT2CR-dependent manner, suggesting 5-HT2CR PAM, but not 5-HT2AR, activity at the level of the whole organism at the employed doses of 13. We discovered new selective 5-HT2CR PAMs and first-in-class 5-HT2CR/5-HT2AR dual PAMs that broaden the pharmacological toolbox to explore the biology of these vital receptors.
Graphical Abstract
INTRODUCTION
Fourteen serotonin (5-HT) receptors are identified with 1 ionotropic receptor (5-HT3R) and 13 class A G protein-coupled receptors (GPCRs) designated as 5-HT1–7R based on structural and pharmacological criteria.1,2 Serotonin 5-HT2 receptors (5-HT2Rs) are a pharmacologically important 5-HT receptor family that includes the three subtypes 5-HT2AR, 5-HT2BR, and 5-HT2CR, which share approximately 80% sequence homology in the transmembrane (TM) ligand-binding regions. Among them, 5-HT2CR and 5-HT2AR have generated immense interest from pharmacologists and medicinal chemists in recent decades.2–6 5-HT2AR and 5-HT2CR are broadly distributed in the mammalian central nervous system (CNS) and mediate various brain functions including cognition, mood, learning, and memory.2,7–9
The three 5-HT2R subtypes (5-HT2AR, 5-HT2BR, and 5-HT2CR) display similar molecular structures with a highly conserved endogenous agonist (5-HT) binding site and intersecting signal transduction pathways and pharmacology.10 Traditional agonists target the orthosteric ligand binding site within the seven-transmembrane bundle (7TM) of the receptor that is highly conserved, challenging the goal to achieve individual subtype selectivity. Allosteric modulators targeting a spatially and topographically distinct site provide a novel pharmacological paradigm for GPCR drug discovery.11–15 The design of allosteric modulators that selectively target 5-HT2R subtypes is a viable and practical approach for avoiding ligand binding to other 5-HT receptors as well as the serotonin reuptake transporter and is especially critical for avoiding 5-HT2BR stimulation, which is known to be associated with cardiac valvulopathy and pulmonary hypertension adverse effects.16
The goal to develop 5-HT2R allosteric modulators is a recent endeavor with a focus on 5-HT2CR positive allosteric modulators (PAMs);17–21 as depicted in Figure 1, the complex natural product derivative compound 1 (PNU-69176E) was the first reported 5-HT2CR PAM and was characterized by a stereo-dependent functional activity profile (Figure 1).22,23 We demonstrated two medicinal chemistry design iterations toward the optimization of 1 (PNU-69176E). Given the poor drug-likeness of 1, we focused on the α-d-galactopyrano-side fragment, termed the polar head (PH), and the undecyl substituent at the 4-position of the piperidine, which is termed the lipophilic tail (LT). These efforts provided efficacious 5-HT2CR PAMs 2 (CYD-1–79) and 3 (CTW0415).18,20 These 5-HT2CR PAMs displayed improved pharmacokinetic (PK) profiles and demonstrated in vivo activity in preclinical animal models. Recent discovery efforts by others produced the N-benzyl-indole 4 (VA012) and piperazine-linked phenyl cyclopropyl methanone 5 as 5-HT2cR PAMs.17,19 Among them, compound 5 also exhibited 5-HT2BR negative allosteric modulation (NAM) activity.19
Figure 1.

Chemical structures of representative small molecule 5-HT2R allosteric modulators 1–5. Compounds 1–4 are 5-HT2CR PAMs. Compound 5 is a 5-HT2CR PAM and 5-HT2BR NAM.
Compound 6 (oleamide, (Z)-9-octadecenamide; Figure 2), an endogenous fatty acid amide, was identified in the cerebrospinal fluid of sleep-deprived cats as well as human plasma.24,25 Compound 6 is implicated in several biological and behavioral phenomena such as sleep induction, feeding regulation, and hypothermia;26–29 6 was noted to act nonselectively as an agonist or allosteric modulator at 5-HT1AR, 5-HT2AR, and 5-HT2CR and an inhibitor at 5-HT7R as well as at other receptor systems.26,27,29–34 The activity of 6 in these systems is structurally sensitive, and only modest chemical modifications have been attempted thus far.31,33,35
Figure 2.

Design strategy for novel 5-HT2CR and/or dual 5-HT2CR/5-HT2AR PAMs. (A) The minimized three-dimensional structures of compound 2 (CYD-1–79, blue dashes) and 7 (red dashes) are presented in an overlay. Energy minimization was carried out using the Schrödinger Drug Discovery Suite (Schrödinger Release 2015–4: LigPrep, version 3.6, Schrödinger, LLC, New York, NY, 2015). (B) The overlay poses of compounds 2 (CYD-1–79) (red) and 7 (orange) docked to the 5-HT2CR X-ray crystal structure (PDB ID: 6BQG) are illustrated as a ribbon representation, and compounds are shown in stick representation. (C) The current structural modification strategy is presented with new probes designed by hybridization of compounds 6 (oleamide) and 2 (CYD-1–79).
Compound 6 shares the common feature of a long LT and a terminal PH with the 5-HT2CR PAM 2 (CYD-1–79).18 Although the LT of 6 is longer than that of PAM 2 (18- vs 15-carbon tail, respectively), an energy minimization overlay of 2 and 7 (the analogue of 6 modified by coupling 6 with a privileged 1,2-diol PH fragment) shows that, because of the cis conformation of the double bond, the tail lengths are approximately the same in maximal length (17.98 vs 17.78 Å; Figure 2A). Molecular docking of 2 and 7 to the 5-HT2CR (PDB ID: 6BQG) X-ray crystal structure demonstrates a similar binding pose and directionality of both the LT and PH, as expected (Figure 2B).18,20 We previously demonstrated that the proper size and volume of the LT and hydroxyl-rich PH are important to the activity of 5-HT2cR PAMs,18,20 which together contribute to the formation of hydrophobic and hydrogen bond interactions with the receptor extracellular loop 2 (ECL2) and TM helices. Given the similarity in the structural scaffolds of compounds 2 and 7, along with the molecular docking predictions, we reasoned that these two molecules may potentially act at the same allosteric site on the 5-HT2CR and potentiate 5-HT signaling as a PAM. Additionally, we rationalized that the substitution of 4-alkylpiperidine-2-carboxamide within the oleamide scaffold to remove the multiple chiral centers of 5-HT2CR PAMs compounds 2 and 3 would reduce the chemical complexity of the PAMs and improve the drug-likeness. Herein, we describe our recent progress in the design, synthesis, and pharmacological evaluation of a novel series of structurally optimized analogues by chemical hybridization of compounds 2 and 6, providing PAMs of 5-HT2R subtypes. This series of 5-HT2CR PAMs and dual 5-HT2CR/5-HT2AR PAMs exhibited in vitro functional activity and are valuable chemical probes for exploring the mechanisms underlying 5-HT2CR and 5-HT2AR allosteric modulation (Figure 2C).
RESULTS AND DISCUSSION
Chemistry and Cellular Signaling Analyses.
The synthesis of new oleamide analogues 7–30 with simplified PHs is depicted in Scheme 1. An effective and convenient one-step coupling of oleic acid, a long-chain unsaturated omega-9 fatty acid, with various aminol alcohol analogues, amino acid residues, or monoamine-like fragments was conducted via adapting the common condensation reagent N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate (HBTU) or 1-ethyl-(3-dimethylaminopropyl)-carbodiimide hydrochloride (EDCI) in combination with 1-hydroxybenzotriazole and the organic base N,N-diisopropylethylamine (DIPEA). Compounds 16–19 underwent saponification according to standard protocols to yield the corresponding carboxylic acid compounds 20–23. Compounds 7–30 were accomplished in 42–94% yields and then subjected to in vitro functional assessment.
Scheme 1.

Synthesis of Oleamide Analogues 7–30
Agonist binding to 5-HT2CR and 5-HT2AR recruits phospholipase Cβ (PLCβ) via Gαq/11 proteins and in turn elicits the efflux of intracellular calcium (Cai2+).36 We employed a fluorescence-based assay to measure 5-HT-evoked Cai2+ levels as a measure of receptor activity in Chinese hamster ovary (CHO) cells stably transfected with the unedited (INI) isoform of human (h) h5-HT2CR (h5-HT2CR-CHO cells), h5-HT2AR (h5-HT2AR-CHO cells), or h5-HT2BR (h5-HT2BR-CHO cells).18 20 The capacity of 5-HT to promote Cai2+ release (Emax) was established and set as the 100% response.
Previous studies demonstrated that the 5-HT2CR PAM 2 at the concentration of 1 nM evokes ~23% upward regulation of 5-HT-evoked Cai2+ release.18 Thus, compounds were screened at 1 nM in the presence of an increasing concentration of 5-HT to assess the enhancement of 5-HT-induced Cai2+ release (5-HT Emax). At 1 nM, none of the tested compounds exhibited intrinsic agonist activity to induce Cai2+ release in h5-HT2CR-CHO, h5-HT2AR-CHO, or h5-HT2BR-CHO cells when administered 15 min prior to the addition of 5-HT (for details, see Figure S1, sections A–Y; Figure S2, sections A–J; and Figure S3, sections A–F).
Privileged PH fragments are instrumental in forming crucial interactions with the ECL2 and certain TM helices of the receptor, as noted previously.18,20 The new series of oleamide analogues 7–30 with varied PHs were first screened in vitro in h5-HT2CR-CHO cells (Table 1). Oleamide (6) exhibited moderate enhancement (~11%) of 5-HT-evoked Cai2+ release under the test conditions, whereas 7, which possesses the privileged 1,2-diol PH as 2 and 3, did not significantly increase 5-HT2CR-evoked Cai2+ (Table 1; Figure S1). Exclusion of the middle hydroxyl group of 7 afforded the less-bulky oleyl propanolamide 8 that potentiated 5-HT-evoked Cai2+ release (Table 1; Figure 3A). Compound 9 with ethanolamide PH maintained 5-HT2CR PAM activity (Table 1; Figure 4A).
Table 1.
Effects of Oleamide Analogues 7–30 (1 nM) on 5-HT-Induced Cai2+ Release in h5-HT2CR-CHO Cells
| Compound | PH | Emax RFU (%5-HT)a | Compound | PH | Emax RFU (%5-HT)a |
|---|---|---|---|---|---|
| 5-HT | 100.0% | 5-HT | 100.0% | ||
| 6 | H | 111.4 ± 4.3b | 19 |

|
125.4 ± 5.2b |
| 7 |

|
108.0 ± 6.7 | |||
| 8 |
|
126.3 ± 8.5b | 20 |

|
111.9 ± 4.4 |
| 9 |
|
124.0 ± 6.8b | 21 |

|
117.8 ± 6.9 |
| 10 |

|
100.1 ± 8.8 | 22 |

|
106.6 ± 9.0 |
| 11 |
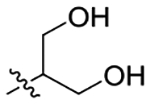
|
143.9 ± 14.1b | 23 |

|
117.4 ± 12.4 |
| 12 |

|
126.0 ± 6.9b | 24 |

|
111.9 ± 11.8 |
| 13 |

|
113.2 ± 3.7b | 25 |

|
122.8 ± 6.7b |
| 14 |
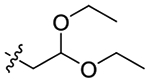
|
111.1 ± 5.4 | 26 |

|
116.0 ± 18.1 |
| 15 |
|
106.2 ± 2.1b | 27 |

|
120.6 ± 15.5 |
| 16 |

|
109.5 ± 1.6b | 28 |

|
121.4 ± 5.2b |
| 17 |

|
126.8 ± 11.4 | 29 |
|
118.5 ± 6.4 |
| 18 |

|
127.8 ± 15.3 | 30 |

|
92.9 ± 1.3b |
Addition of the synthetic compound (1 nM) occurred 15 min prior to assessment of Cai2+ release evoked by increasing concentrations of 5-HT (vehicle, 10−11 to 10−5 M) in h5-HT2CR-CHO cells. Data are presented as maximal 5-HT-induced Cai2+ release (Emax).
p < 0.05. Comparisons between means for Emax were conducted with an unpaired t test with Welch’s correction (GraphPad Prism). All statistical analyses were conducted with an experiment-wise error rate of α = 0.05.
Figure 3.

Concentration–response curves for compounds 8, 12, 15, 16, and 25 for 5-HT-induced Cai2+ release in (A) h5-HT2CR-CHO cells or (B) h5-HT2AR-CHO cells. Representative curves demonstrate the concentration–response curve for 5-HT in the absence (black circles) and in the presence (closed triangles) of the test compounds, vehicle (open circle), and test compound assessed alone (open triangle). The maximum 5-HT-induced Cai2+ release in the absence of the test compounds was set as 100%, and the Emax values of the test compounds are listed in Tables 1 and 2.
Figure 4.

Concentration–response curve of compounds 9, 11, 13, 19, and 28 for 5-HT-induced Cai2+ release in live (A) h5-HT2CR-CHO cells or (B) h5-HT2AR-CHO cells. Representative curves demonstrate test compounds against concentration–response curves for 5-HT in the absence (black circles) and in the presence (closed triangles), vehicle (open circle), and vehicle in the presence of test compound (open triangle). The maximum 5-HT-induced Cai2+ release in the absence of the test compounds was set as 100%, and the Emax values of the test compounds are listed in Tables 1 and 2.
Because compound 2 exhibited a stereo conformation preference for 5-HT2CR PAM activity, compound 10, furnished by introducing an S configurational 1,2-diol to the amide, was then explored and identified as inactive (Table 1), suggesting that the privileged 1,2-diol PH of 2 and 3 is not favorable for the oleamide derivatives to produce 5-HT2CR PAM activity. Compound 11, which has a 1,3-diol moiety vs a 1,2-diol PH, promoted a ~44% increase in 5-HT2CR-evoked Cai2+ (Table 1; Figure 4A). Compound 12, which incorporates a phenyl into the diol PH of 11, maintained 5-HT2CR PAM activity (Table 1; Figure 3A), as does compound 13 (Table 1; Figure 4A) with one more hydroxymethyl group to 11. Compound 14, which includes the hydroxyl group with ether, did not evoke PAM activity, most likely due to the loss of the terminal H-bond donor and the bulkier volume of the PH (Table 1). Meanwhile, lengthening the PH of 9 by etherification with another ethanol fragment was less favorable (15, Figure 3A). These findings illustrate that the hydroxyl containing moiety is important for 5-HT2CR allosteric potentiation, and a proper size of the PH is required for 5-HT2CR PAM activity.
We then synthesized the polar head with other chemical elements, including chiral amino acids (16–23), terminal phenol (24 and 26), catechol (25 and 27), and morpholino (28) or amino (29–30) substituted alkylamines (Table 1). As the introduction of a hydroxyl-containing moiety is beneficial for 5-HT2CR PAM efficacy, hydroxyl or terminal phenol containing chiral amino acids were applied by condensation of the methyl ester of threonine, serine, and tyrosine, affording compounds 16–18. Among them, compound 16 (Table 1; Figure 3A) demonstrated 5-HT2CR PAM activity. Interestingly, compound 19 (Table 1; Figure 4A), incorporating the 5-HT indole-like tryptophan methyl ester in the terminal position of the PH, resulted in a 5-HT-evoked Emax of 125.4% (Figure 4A). Saponification of the methyl esters provided free acids 20–23 potentially capable of additional H-bonding and salt interactions with the target receptor. However, these carbonyl acid derivatives 20–23 at 1 nM did not potentiate 5-HT efficacy (Table 1). Furthermore, both 4-aminoalkyl phenolic and catechol moieties were explored as potential PHs (24–27). Terminal phenol compound 25 with a 4-aminomethyl catechol PH promoted 5-HT2CR PAM activity (Table 1; Figure 3A), whereas other terminal phenol compounds with less phenolic hydroxyl or more carbon spaced chain did not display 5-HT2CR PAM efficacy (the 4-aminoalkyl phenolic 24, 26, and two-carbon spaced catechol 27; Table 1). Intriguingly, when aliphatic amines were employed as PHs, the two-carbon spaced morpholino compound (28) displayed 5-HT2CR PAM activity (Table 1; Figure 4A), whereas the n-butylamine compound (29) with a bulky amine terminus did not induce a 5-HT2CR PAM effect (Table 1). To our surprise, when 4-(aminomethyl)piperidine was incorporated at the end of oleamide, a comparable ~10% decrease in 5-HT2CR-mediated Cai2+ release was observed, which may signal the potential for 30 to function as a negative allosteric modulator (NAM) (Table 1).
Oleamide (6) itself has a complex pharmacological profile,26,27,29–34 and a subset of the 10 oleamide-like compounds characterized as 5-HT2CR PAMs (Table 1) was further evaluated in the in vitro Cai2+ efflux assay using h5-HT2AR-CHO cells (Table 2). After screening this subset of 10 analogues at 1 nM in the h5-HT2AR-CHO cells, we identified compounds with two pharmacological profiles. Compounds 8, 12, 15, 16, and 25 were identified as 5-HT2CR PAMs (Table 1; Figure 3A) and exhibited no efficacy as allosteric modulators of the 5-HT2AR (Table 2; Figure 3B). However, compounds 9, 11, 13, 19, and 28 were identified as 5-HT2CR PAMs (Table 1; Figure 4A) and also exhibited efficacy as 5-HT2AR PAMs (Table 2; Figure 4B). Thus, we differentiated 5-HT2CR PAMs (8, 12, 15, 16, and 25) and dual 5-HT2CR/5-HT2AR PAMs (9, 11, 13, 19, and 28) within this current series of oleamide-like compounds.
Table 2.
Effects of Identified Oleamide-like 5-HT2CR PAMs (1 nM) on 5-HT-Induced Cai2+ Release in h5-HT2AR-CHO Cells
| Compound | PH | Emax RFU (%5-HT)a |
|---|---|---|
| 5-HT | 100.0% | |
| 8 |
|
99.2 ± 2.6 |
| 9 |
|
121.0 ± 3.7b |
| 11 |

|
119.0 ± 3.2b |
| 12 |

|
98.8 ± 3.8 |
| 13 |

|
132.4 ± 6.1b |
| 15 |
|
99.6 ± 0.6 |
| 16 |

|
106.0 ± 5.3 |
| 19 |

|
109.2 ± 3.5b |
| 25 |

|
102.5 ± 2.7 |
| 28 |

|
105.7 ± 2.0b |
Addition of the synthetic compound (1 nM) occurred 15 min prior to assessment of Cai2+ release evoked by increasing concentrations of 5-HT (vehicle, 10−11 to 10−5 M) in h5-HT2AR-CHO cells. Data are presented as maximal 5-HT-induced Cai2+ release (Emax).
p < 0.05. Comparisons between means for Emax were conducted with an unpaired t test with Welch’s correction (GraphPad Prism). All statistical analyses were conducted with an experiment-wise error rate of α = 0.05.
Compound 9, which possesses an ethanol PH, displayed 5-HT2AR PAM activity (Table 2; Figure 4B). Compounds 11 and 13, introducing a second and a third methanol to the PH of 9, respectively, maintained 5-HT2AR allosteric effects (Table 2; Figure 4B). This result suggests that the incorporation of the two-carbon spaced hydroxyl moiety is favorable for producing 5-HT2AR PAM activity. Compound 19 bearing a methyl tryptophan as PH and compound 28 with a two-carbon linked morpholino PH also act as 5-HT2AR PAMs (Table 2; Figure 4B). Lengthening the linker length of 9 by one more carbon (8), etherification with another ethanol fragment (15), or retaining the 1,3-diol moiety of 11 with the addition of a phenyl (12) did not result in 5-HT2AR PAM activity (Table 2; Figure 4A). Compounds 16 and 25 with longer PHs as methyl threonine and 4-aminomethyl catechol did not result in 5-HT2AR PAM activity (Table 2; Figure 3B). These findings indicate that the structure of 5-HT2AR PAMs is more sensitive to the length and volume change of PH than 5-HT2CR PAMs (Tables 1 and 2). Representative 5-HT2CR PAMs 8, 12, and 25 and dual 5-HT2CR/5-HT2AR PAMs 9, 11, and 13 were next assessed in the in vitro functional assay in h5-HT2BR-CHO cells (Table 3; Figure S3). None of the compounds altered 5-HT2BR-evoked Cai2+ alone or in the presence of 5-HT (Table 3).
Table 3.
Effects of Representative 5-HT2CR PAMs and Dual 5-HT2CR/5-HT2AR PAMs (1 nM) on 5-HT-Induced Cai2+ Release in h5-HT2BR-CHO Cellsb
| Compound | PH | Emax RFU (%5-HT)a |
|---|---|---|
| 8 |
|
95.6 ± 4.6 |
| 9 |
|
106.1 ± 7.5 |
| 11 |

|
97.2 ± 9.2 |
| 12 |

|
92.4 ± 3.2 |
| 13 |

|
98.4 ± 4.6 |
| 25 |

|
100.6 ± 4.4 |
Addition of the synthetic compound (1 nM) occurred 15 min prior to assessment of Cai2+ release evoked by increasing concentrations of 5-HT (vehicle, 10−11 to 10−5 M) in h5-HT2BR-CHO cells. Data are presented as maximal 5-HT-induced Cai2+ release (Emax).
p < 0.05. Comparisons between means for Emax were conducted with an unpaired t test with Welch’s correction (GraphPad Prism). All statistical analyses were conducted with an experiment-wise error rate of α = 0.05.
Molecular Docking to 5-HT2CR and 5-HT2AR.
The molecular docking of the receptor–ligand interaction provides useful information for structure-based drug design. To obtain comprehensive docking poses of these PAMs at the 5-HT2CR and 5-HT2AR allosteric sites, we employed the Schrödinger Drug Discovery Suite for modeling the 5-HT2CR and 5-HT2AR and selected PAM molecules by utilizing recently reported X-ray crystal structures (see SI for details on docking protocol).37,38 Docking studies for representative molecules 8, 9, 11, 12, 13, and 25 to the 5-HT2CR followed the same protocol we reported previously.18,20 Briefly, we preprocessed and optimized the 5-HT2CR X-ray crystal structure (PDB: 6BQG), except for using the newly released OPLS4 force field in Maestro 12.7, to provide the docking model for 5-HT2CR PAMs herein.37,39 The docking model for 5-HT2AR PAMs was obtained by utilizing the 5-HT2AR X-ray crystal structure (PDB: 6WGT), which is in complex with d-lysergic acid diethylamide (LSD).38 The structure was preprocessed and optimized with the Schrödinger protein preparation wizard, including filling loops with missing residues, replacing stabilizing mutations, and removing stabilization domains not part of the receptor. Subsequently, we ran the Schrödinger Induced Fit Docking (IFD) protocol to replace LSD with 5-HT in the orthosteric binding site to provide a suitable canvas upon which to dock our molecules. Selected compounds 9, 11, and 13 were then docked into the 5-HT2AR–5-HT complex model using the Schrödinger Glide software platform, employing extra-precision (XP), standard-precision (SP), and IFD docking protocols to obtain reliable and reproducible poses. The representative pose for each molecule was selected from a top-scoring, enriched cluster, along with the biological and chemical rationality evaluation (e.g., interactions at the binding site and physicochemical properties).
Each of the agonists and oleamide-like PAM compounds modeled was prepared with the Maestro LigPrep module, ensuring biologically accurate ionization states and conformations with the OPLS4 force field. As the global overview of the docking poses shows in Figure 5A,B, the six oleamide-like compounds—8, 9, 11, 12, 13, 25—occupy a similar docking cavity for both 5-HT2CR and 5-HT2AR, distinct from the 5-HT binding pocket and differentiated by a somewhat more restricted, smaller volume in the 5-HT2AR. The six compounds localize in a spatially distinct allosteric binding pocket near the top of the 7TM bundle formed by helices I–III, V–VII, and ECL2 and directly above the orthosteric binding site. Consistent with our previous docking studies,20 the six molecules exhibited the flat stretch pose over the binding cavity with the PH responsible for key hydrogen bonding interactions to ECL2 and TM helix residues and the long LT oriented within a spatially constrained hydrophobic pocket, generating beneficial hydrophobic interactions (Figure 5A,B). The calculated distance between the 5-HT indole N-atom and the nearest carbon in LT of oleamide-derived compounds in both 5-HT2Rs was carefully measured. For the 5-HT2CR, the average distance between the six compounds and 5-HT was calculated to be 8.59 Å, and for the 5-HT2AR, this average distance for the three compounds modeled was 8.13 Å. This measurement indicates an occupancy deeper into the TM bundle for the new 5-HT2CR PAMs compared to the previous PAM 3 into the hydrophobic pocket (measured at 13.5 Å between 5-HT and 3), along with a tighter packed profile.20 Moreover, as shown in Figure 5C,D, the dual 5-HT2CR/5-HT2AR PAMs reach deeper into the more compact binding pocket of 5-HT2AR, displaying a slightly closer distance to the orthosteric ligand 5-HT (8.13 Å) compared to the allosteric site in 5-HT2CR. This may be the reason that 5-HT2AR PAMs are more sensitive to the length and volume than 5-HT2CR PAMs. This feature may be related to distinctions in the sequence of the ECL2 between the two receptors; the 5-HT2CR structure has a VAL208 in the same spatial position that the 5-HT2AR contains the more voluminous LEU229 (Figure 5C,D).
Figure 5.
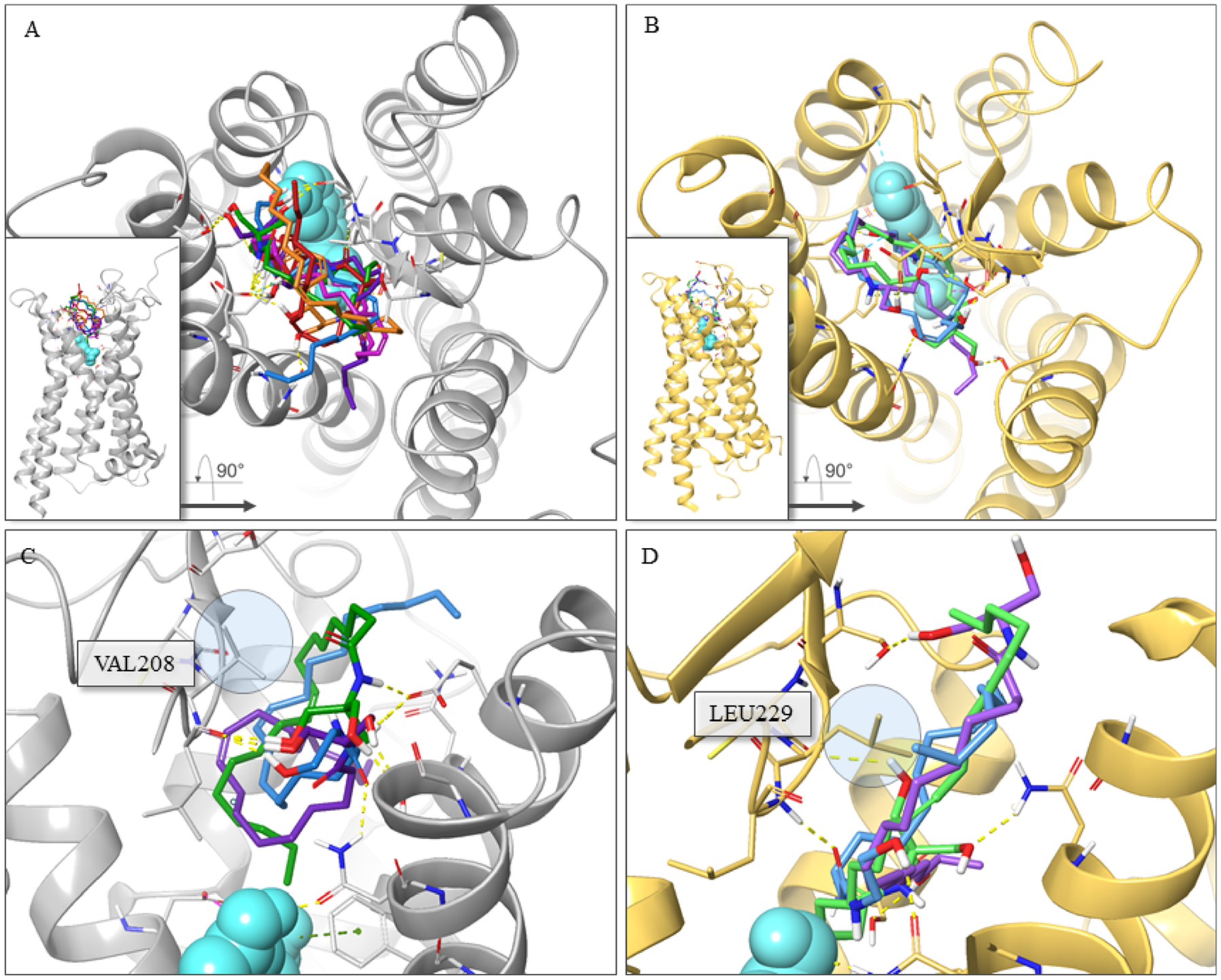
Extracellular overview of the crystal structure of 5-HT2CR (PDB ID: 6BQG) or 5-HT2AR (PDB ID: 6WGT)-PAMs complex with serotonin (teal) binding in the orthosteric site. (A) Global overview of 5-HT2CR with representative 5-HT2CR PAMs 8 (magenta), 12 (orange), and 25 (red) and dual 5-HT2CR/5-HT2AR PAMs 9 (blue), 11 (purple), and 13 (green). (B) Global overview of 5-HT2AR with representative dual 5-HT2CR/5-HT2AR PAMs 9 (blue), 11 (purple), and 13 (green). (C) Docking poses of dual 5-HT2CR/5-HT2AR PAMs 9 (blue), 11 (purple), and 13 (green) in 5-HT2CR. (D) Docking poses of dual 5-HT2CR/5-HT2AR PAMs 9 (blue), 11 (purple), and 13 (green) in 5-HT2AR. The 5-HT2CR is gray colored, and the 5-HT2AR is gold colored. PAM compounds are shown in stick representation. Hydrogen bonds are in yellow dotted lines.
Figure 6 illustrates the detailed docking pose of dual 5-HT2CR/5-HT2AR PAMs 9 and 13. The amide carbonyl and terminal hydroxyl groups of 9 accept two hydrogen binding interactions with residues ASN331 (TMVI) and LEU209 (ECL2) in the 5-HT2CR–5-HT complex, respectively (Figure 6A). Identical interactions with ASN343 (TMVI) and LEU228 (ECL2) are observed for the 5-HT2AR–5-HT model (Figure 6B). The LT moiety of our oleamide-like compounds had been difficult to precisely dock into our previous allosteric model for 5-HT2CR due to its lack of functional groups and overall flexibility.18,20 However, with new structures and model improvements, the hydrophobic cavities created by the active-state receptor provide useful constraints for modeling the LT orientation. This is evident in the comparison between pose differences for compound 13 at the two receptors (Figure 6C,D). The binding pocket tunnel is created in 5-HT2AR by LEU229 compared to the less imposing VAL208 in 5-HT2CR in the corresponding ECL2 location. This spatial difference is likely to be advantageous for designing new 5-HT2AR- or 5-HT2CR-selective allosteric modulators. An additional difference includes a flipped orientation of ASN331 (5-HT2CR) and ASN343 (5-HT2AR) in the TMVI helix between the two 5-HT2Rs (Figure 6A,B). The docking study for compound 13 also demonstrates the relative openness of the 5-HT2CR allosteric pocket in comparison (Figure 6E,F). The binding poses are essentially flipped primarily because of crowding in 5-HT2AR by LEU229 (5-HT2CR has VAL208) as denoted by the bar (5-HT2CR) and arrow (5-HT2AR). Importantly, in the docking poses for both the 5-HT2AR and 5-HT2CR, compound 13 interacts via hydrogen bonds with residues in the ECL2 and additional residues in the TM helices. Specifically, the amide and terminal hydroxyl groups form interactions with residues LEU209 (ECL2), GLU347 (TMVII), and SER334 (TMVI) in 5-HT2CR and residues LEU229 (ECL2), ASN363 (TMVII), and SER131 (TMII) in 5-HT2AR. This work suggests that the stabilization of the ECL2 of both 5-HT2Rs by bridging interactions between key LEU209 (5-HT2CR) and LEU229 (5-HT2AR) residues toward adjacent TM helix residues results in a conformational state by which positive allosteric modulation is achieved. Further ECL2 stabilization by filling the resultant hydrophobic allosteric pocket with a complimentary LT moiety likely plays a role in the observed PAM activity. In addition, we compared the 5-HT2BR structure (PDB ID: 4IB4) with the 5-HT2CR and 5-HT2AR X-ray crystal structures by carrying out a superimposition and pairwise sequence alignment. Structural analysis of these receptors shows that the 5-HT2CR/5-HT2AR PAM 9 and 13 binding site in the corresponding area of 5-HT2BR presents structural distinctions. Whereas the key residues of ECL2 remain almost the same among three structures (ECL2: 2A_LEU229/2B_LEU209/2C_LEU209), the key residues on TMVI, TMVII, and TMII that are forming H-bonds with the amide and terminal hydroxy groups of ligands are substantially different from those on 5-HT2BR (TMVI: 2A_ALA346/2B_LEU347/2C_SER334; TMVII: 2A_GLY359/2B_GLN359/2C_GLU347; TMVII: 2A_ASN363/2B_GLU363/2C_ASN351; TMII: 2A_SER131/2B_ALA111/2C_SER110). Ligand-mediated stabilization does not appear to be effectively achieved between ECL2 and the adjacent TM helix for 5-HT2BR due to structural differences, which might contribute to the selectivity of compound 13.
Figure 6.

Detailed interaction diagram of dual 5-HT2CR/5-HT2AR PAMs 9 and 13 to the 5-HT2CR (PDB ID: 6BQG) or 5-HT2AR (PDB ID: 6WGT) X-ray crystal structure. Detailed interaction diagram of 9 (blue) to the (A) 5-HT2CR X-ray crystal structure or (B) 5-HT2AR X-ray crystal structure. Top view of 13 (green) to the (C) 5-HT2CR X-ray crystal structure or (D) 5-HT2AR X-ray crystal structure. Side view of 13 (green) to the (E) 5-HT2CR X-ray crystal structure or (F) 5-HT2AR X-ray crystal structure. The 5-HT2CR is gray colored, and the 5-HT2AR is gold colored. PAM compounds are shown in stick representation. Hydrogen bonds are noted in yellow dotted lines.
In Vitro Radioligand Binding Displacement Study.
Considering the dual in vitro activity at the 5-HT2CR and 5-HT2AR, compounds 11 and 13 were selected as representative tool compounds out of the active analogues for further pharmacological evaluation. To explore the off-target profile of the two compounds, we utilized the National Institute of Mental Health (NIMH) Psychoactive Drug Screening Program (PDSP) to assess a broad-panel of GPCRs and monoamine transporters (Table 4; Table S1).40 Generally, compounds 11 and 13 exhibited no off-target effects for most of the receptors and monoamine transporters evaluated, except that 11 exhibited a Ki of 0.46 μM at 5-HT2CR (Table S1). Compound 13 did not displace 5-HT2AR or 5-HT2CR binding, suggesting an important feature for selectivity of a 5-HT2A/2CR PAM. Both compounds 11 and 13 did not displace binding to the human ether-a-go-go-related gene (hERG) potassium channel, suggesting a low risk of cardiac adverse events.41 Compound 13 featured micromolar displacement at the H3 receptor (Ki = 5.3 μM) and σ2 receptor (Ki = 3.6 μM) (Table S1). Nonetheless, the off-target profile of compound 13 is considered promising, and compound 13 was selected as a representative dual PAM for further evaluation (for the full panel results, see Table S1).
Table 4.
Displacement of Radioligand Binding by Compound 13 in a Broad Panel of Receptors and Transporters
| receptor/transporter | radioligand | % inhibitiona | Ki (μM)b |
|---|---|---|---|
| 5-HT1A | [3H]-8-OH-DPAT | 13.99 | NT |
| 5-HT1B | [3H]-GR125743 | −5.9 | NT |
| 5-HT1D | [3H]-GR125743 | −6.35 | NT |
| 5-HT1E | [3H]-5-HT | 6.48 | NT |
| 5-HT2A | [3H]-ketanserin | 2.03 | NT |
| 5-HT2B | [3H]-LSD | 33.25 | NT |
| 5-HT2C | [3H]-mesulergine | 44.64 | NT |
| 5-HT3 | [3H]-LY278584 | 5.63 | NT |
| 5-HT5A | [3h]-LSD | −7.85 | NT |
| 5-HT6 | [3H]-LSD | −8.61 | NT |
| 5-HT7 | [3H]-LSD | −8.95 | NT |
| D1 | [3H]-SCH23390 | 22.64 | NT |
| D2 | [3H]-N-methylspiperone | 2.44 | NT |
| D3 | [3H]-N-methylspiperone | −6.88 | NT |
| D4 | [3H]-N-methylspiperone | 19.02 | NT |
| D5 | [3H]-SCH23390 | 56.32 | >10Avg |
| DAT | [3H]-WIN35428 | 4.01 | NT |
| SERT | [3H]-citalopram | −20.8 | NT |
| NET | [3H]-nisoxetine | −0.8 | NT |
| α 2A | [3H]-rauwolscine | 28.38 | NT |
| α 2B | [3H]-rauwolscine | 14.82 | NT |
| α 2C | [3H]-rauwolscine | 20.26 | NT |
| HERG | [3H]-dofetilide | 1.6 | NT |
The binding replacement test was conducted at 10 μM of compound 13. A binding inhibition result >50% was considered reliable replacement of target receptor radioligand by the test compound.
The Ki value was calculated via a nonlinear regression analysis of radioligand competition isotherms for ligand binding inhibition >50%. NT = not tested, Avg = average Ki from repeated experiments.
We calculated the central nervous system (CNS) multi-parameter optimization (MPO) value for compound 13.42 The CNS MPO was adopted to increase the odds of prospectively designing CNS-targeted molecules that achieve CNS exposure.42,43 The calculated score for compound 13 is 3.3 out of a collective score range from 0 to 6. Of note, for this series of molecules, the predictive nature is inherently limited by a lower number of structurally comparable molecules in the training data set. The literature suggests that a higher MPO value is desirable for the CNS drugs (Table S2).
Toxicity Profile and Pharmacokinetics (PK) Profile of Compound 13.
In silico toxicity predictions were conducted to evaluate compound 13 in various toxicity end points such as acute toxicity, hepatotoxicity, cytotoxicity, carcinogenicity, mutagenicity, immunotoxicity, adverse outcome (ProTox-II) pathways, and toxicity targets (Table 5; Table S3). The results suggest that compound 13 exhibits a profile consistent with a low expectation of adverse drug reactions or toxic effects and was ranked in a nontoxic class VI (LD50 > 5000 mg/kg). A study of CYP450 inhibition was then carried out in human liver microsomes to assess the inhibitory potential of compound 13 (10 μM) against CYP450 isoforms (Table 5). Compound 13 displayed ≤20% inhibition of CYP3A4, CYP1A2, CYP2C8, CYP2C19, CYP2D6, and CYP2C9 and ~50% inhibition of CYP2B6.
Table 5.
Toxicity Profile and Human Liver Microsome P450 Inhibition Profile for 13a
| In silico toxicity profile | ||||||||
|---|---|---|---|---|---|---|---|---|
| predicted LD50 | 10,000 mg/kg | |||||||
| toxicity classb | 6 (low) | |||||||
| prediction accuracy | 70.97% | |||||||
| targetc | prediction (probability) | targetc | prediction (probability) | |||||
| hepatotoxicity | inactive (0.84) | AR-LBD | inactive (0.98) | |||||
| carcinogenicity | inactive (0.59) | aromatase | inactive (0.99) | |||||
| immunotoxicity | inactive (0.96) | PPAR-gamma | inactive (0.98) | |||||
| mutagenicity | inactive (0.89) | Nrf2/ARE | inactive (0.94) | |||||
| cytotoxicity | inactive (0.82) | HSE | inactive (0.94) | |||||
| aryl hydrocarbon receptor | inactive (0.98) | MMP | inactive (0.95) | |||||
| estrogen receptor α | inactive (0.89) | phosphoprotein p53 | inactive (0.96) | |||||
| ER-LBD | inactive (0.97) | ATAD5 | inactive (0.99) | |||||
| androgen receptor | inactive (0.98) | |||||||
| Human liver microsome P450 inhibition profile | ||||||||
| P450 isoforms | CYP3A4d | CYP3A4e | CYP1A2 | CYP2B6 | CYP2C8 | CYP2C9 | CYP2C19 | CYP2D6 |
| inhibitory % | 18.72 | 11.11 | 3.4 | 53.31 | 11.87 | 21.2 | 18.34 | 10.13 |
For detailed in silico toxicity prediction profile (ProTox-II), see SI Table S3 (more information at http://tox.charite.de/protox II/).44 Cytochrome P450 enzymatic inhibition assays for compound 13 performed at 10 μM and represented as percent inhibition.
Toxicity class ranks from 1 to 6: 1 = high, 6 = low.
ER-LBD = estrogen receptor ligand binding domain; AR-LBD = androgen receptor ligand binding domain; PPAR-gamma = peroxisome proliferator activated receptor gamma; Nrf2/ARE = nuclear factor (erythroid-derived 2)-like 2/antioxidant responsive element; HSE = heat shock factor response element; MMP = mitochondrial membrane potential; ATAD5 = ATPase family AAA domain-containing protein 5.
CYP3A4 (midazolam).
CYP3A4 (testosterone).
A membrane permeability evaluation of compound 13 was carried out in hMDRI-MDCKII cells to investigate potential CNS permeability and drug efflux (Table 6). Compound 13 demonstrated a moderate permeability and a low efflux ratio of 0.6 and 0.4, respectively, in the absence or presence of a P-glycoprotein (P-gp) inhibitor, indicating that 13 is not a predicted substrate for P-gp. Compound 13 displayed a kinetic solubility in the PBS buffer (pH = 7.2) of 48.55 μg/mL. The rate of disappearance of 13 following incubation with rat or human liver microsomes was monitored to determine the in vitro intrinsic clearance; 13 showed a higher clearance rate than prior 5-HT2CR PAMs.18,20 Nonetheless, in vivo PK evaluation in male Sprague–Dawley rats after a single dose of 10 mg/kg by intraperitoneal (ip) or 20 mg/kg by oral (po) administration was performed to assess the drug-like properties and in vivo probe potential of 13. As summarized in Table 7, 10 mg/kg of 13 administered ip (T1/2 = 2.41 ± 1.73 h) or 20 mg/kg of 13 administered po (T1/2 = 2.14 ± 0.18 h) resulted in a similar plasma exposure (AUC0–inf; ip: 1885 ± 232 ng·h·mL−1; po: 615 ± 94 ng·h·mL−1) to 5-HT2CR PAM 2 (AUC0–inf; intravenous: 939 ± 108 ng·h·mL−1; po: 737 ± 56 ng·h·mL−1), although it was slightly inferior to 5-HT2CR PAM 3.18,20 Compound 13 exhibited brain/plasma (B/P) ratios of 0.589 (15 min) and 2.05 (1 h) after intraperitoneal administration, significantly higher than 0.3, which was reported as the cutoff to classify CNS drugs. These data suggest that 13 has a comparable plasma exposure to that of PAM 2 and good brain permeability as a suitable in vivo pharmacological tool compound for further in vivo animal studies.
Table 6.
In Vitro PK Data for Compound 13
| MDCK-MDR1 permeability | without P-gp inhibitor: PappA → B = 5.86 × 10−6 cm/s; PappB → A = 3.23 × 10−6 cm/s; efflux ratio: 0.6 |
| with P-gp inhibitor:PappA → B = 10.64 × 10−6 cm/s; PappB → A = 4.77 × 10−6 cm/s; efflux ratio: 0.4 | |
| kinetic solubility (PBS pH = 7.2) | 48.55 μg/mL |
| liver microsomal clearance | CLr = 174.66 μL/min/mg |
| CLh = 148.85 μL/min/mg |
Table 7.
In Vivo PK and Brain Penetrability for Compound 13a
| In vivo PK profile | ||||
|---|---|---|---|---|
| dose (mg/kg) | T1/2 (h) | Tmax (h) | Cmax (ng/mL) | AUC0–inf (ng·h·mL−1) |
| 10, ip | 2.41 ± 1.73 | 0.5 | 878 ± 236 | 1885 ± 232 |
| 20, po | 2.14 ± 0.18 | 0.667 ± 0.289 | 181 ± 36 | 615 ± 94 |
| Brain penetration analysis | ||||
| dose (mg/kg) | time (h) | brain conc (ng/g) | plasma conc (ng/mL) | brain/plasma ratio |
| 10, ip | 0.25 | 378 ± 58.8 | 642 ± 133 | 0.589 |
| 10, ip | 1.0 | 1120 ± 65 | 547 ± 18.3 | 2.05 |
T1/2, half-life; Tmax, time of maximum concentration; Cmax, maximum concentration; AUC0–inf, area under the plasma concentration–time curve; time, hours after dose for brain collection; brain conc, averaged concentration of 13 in tissue sample. Experiments were studied in biological triplicates, and data values are shown as the mean ± SEM (± standard error of the mean) from male Sprague–Dawley rats. Vehicle, 10% dimethyl sulfoxide (DMSO)/90% 2-hydroxypropyl-β-cyclodextrin (HP-β-CD).
Effects of Compound 13 on Spontaneous Locomotor Activity.
Locomotor activity in an unfamiliar environment is frequently measured to assess novelty-evoked exploratory behavior that predicts vulnerability to maladaptive psychiatric-like states (e.g., anxiety, initiation of problematic drug use) in rodent models.45–49 Ligands that activate the 5-HT2CR or augment its functional efficacy are well-described to dose-dependently suppress novelty-induced motor activity in rodents.17,18,50,51 Preferential 5-HT2AR agonists are noted to increase or decrease novelty-induced motor activity in rodents depending on the dose administered.50,52 We surmised that 13, a proposed dual 5-HT2CR/5-HT2AR PAM in vitro, would impact novelty-induced motor activity in vivo through enhancement of endogenous 5-HT actions at 5-HT2CR and/or 5-HT2AR. Thus, in the present experiment, we tested the hypothesis that 13 would dose-dependently alter novelty-induced motor activity in male Sprague–Dawley rats. We assessed the effects of 13 (3, 10, or 30 mg/kg ip) on total ambulations over a 90 min session (mean ± S.E.M) (Figure 7A).18,53,54 A main effect of treatment (F3,44 = 7.826, p = 0.0003) was observed for total ambulations; a priori comparisons revealed that 30 mg/kg of 13 significantly reduced total ambulations vs vehicle (p < 0.0001).
Figure 7.

Impact of compound 13 on novelty-induced locomotor activity in Sprague–Dawley rats. (A) Male rats (n = 12/treatment) naïve to the activity monitors were injected with vehicle (ip) or compound 13 (3, 10, or 30 mg/kg, ip) 30 min prior to the start of the 90 min locomotor assessment. The mean total ambulations (±SEM) for the 90 min session are plotted with open circles representing individual rats (*p < 0.05 vs Veh). (B) In a separate cohort (n = 11–12/treatment), Veh, the 5-HT2CR antagonist SB242084 (0.5 mg/kg, ip), or the 5-HT2AR antagonist M100907 (0.1 mg/kg, ip) plus Veh or 13 (30 mg/kg) was administered 30 min prior to the 90 min locomotor assessment. Average activity (mean ± SEM) is shown for each treatment group and compared to animals receiving Veh + Cmpd 13 (*p < 0.05). The average basal activity (Veh + Veh; dashed line) is shown for reference. Please see Experimental Section for Veh descriptions.
A separate cohort of rats was then employed to assess the impact of the selective 5-HT2AR (M100907, 0.1 mg/kg, ip) or the 5-HT2CR antagonist (SB242084, 0.5 mg/kg, ip) on the suppressive motor effects of 13 to illuminate 5-HT2CR- and 5-HT2AR-specific contributions. The employed doses of both 5-HT2R antagonists were selected based on their lack of effect on novelty-evoked motor activity in naïve male rats (data not shown).55,56 Total ambulations in a novel environment for the 90 min session are presented (mean ± SEM) (Figure 7B). Veh + Cmpd 13 (30 mg/kg ip) reduced activity (~34%) relative to Veh + Veh baseline (dashed line) as seen in Figure 7A (~31%). A main effect of treatment (F2,32 = 8.971, p = 0.0008) was observed for ambulations across the entire 90 min session (Figure 7B). SB242084 (0.5 mg/kg) pretreatment reversed 13-evoked suppression of motor activity (p = 0.0039 vs Veh + Cmpd 13), whereas M100907 (0.1 mg/kg) treatment was ineffective (p = 0.7664 vs Veh + Cmpd 13). This behavioral profile has been described for previously identified 5-HT2CR PAMs, suggesting that 13 is a preferential 5-HT2CR PAM in vivo.18
CONCLUSIONS
We report the molecular design, chemical synthesis, and pharmacological evaluation of a novel series of oleamide-based analogues as novel selective 5-HT2R PAMs in vitro. Several compounds (8, 12, 15, 16, and 25) were identified as selective 5-HT2CR PAMs, whereas 9, 11, 13, 19, and 28 were characterized as dual 5-HT2CR/5-HT2AR PAMs. Further in vitro evaluation of representative 5-HT2CR PAMs and dual 5-HT2CR/5-HT2AR PAMs suggested acceptable 5-HT subtype selectivity with no efficacy at 5-HT2BR. Molecular docking studies identified a spatially distinct allosteric binding site for 5-HT2CR and 5-HT2AR differentiated by a narrower pocket with a reduced volume for 5-HT2AR vs 5-HT2CR, providing a clue for the discovery of subtype selective allosteric 5-HT2CR and 5-HT2AR modulators. Compound 13 (JPC0323) showed negligible displacement of the orthosteric binding sites of roughly 50 GPCRs and transporters, including 5-HT2AR and 5-HT2CR. Therefore, 13 is a first-in-class dual 5-HT2CR/5-HT2AR PAM selected from an extended medicinal chemistry endeavor. Collectively, 13 exhibited a suitable CNS MPO value, acceptable plasma exposure at 10 mg/kg (ip), and decent brain penetrability and behavioral efficacy in a spontaneous locomotor activity assay. These cumulative findings suggest that 13 maintains favorable drug-like properties for utility as an in vivo probe of allosteric modulation of the 5-HT2CR/5-HT2AR.
Compound 13 suppressed spontaneous ambulations in a 5-HT2CR-, but not 5-HT2AR-, dependent manner, suggesting that 13 demonstrates a preferential 5-HT2CR PAM profile in vivo. However, these observations do not definitively demonstrate a lack of efficacy of 13 to engage 5-HT2AR signaling. Both 5-HT2CR agonists and PAMs reliably blunt horizontal activity in rodents, whereas the impact of 5-HT2AR activation on motor activity is ligand- and dose-dependent.17,18,50−52 Further, 5-HT2CR agonists block the 5-HT2AR-mediated head twitch response in rodents,57 whereas selective 5-HT2CR antagonists can unmask 5-HT2AR activity produced by nonselective 5-HT2CR agonists.58 These data as well as cellular studies suggest that functional cross talk between the 5-HT2AR and 5-HT2CR is biologically meaningful.6,59–61 Stimulation of the 5-HT2AR is necessary for the powerful effects of psychedelics to alter perception and cognition.62,63 Our in vivo results support the involvement of the 5-HT2CR, but not the 5-HT2AR, in the behavioral effects of compound 13, and future studies are required to characterize the effects of 13 in 5-HT2AR-dependent preclinical models. PAM actions at 5-HT2AR would distinguish from full 5-HT2AR agonists that are hallucinogenic and may provide an alternative, potentially safer therapeutic approach in this light.62,63 5-HT2CR agonists and 5-HT2AR agonists have been proposed for the treatment of anxiety, obsessive–compulsive, and substance use disorders.62–65 As a dual 5-HT2CR/5-HT2AR PAM, compound 13 would be expected to amplify signaling at the 5-HT2CR and 5-HT2AR, with a reduced likelihood of agonist-induced adverse side effects, such as 5-HT2AR agonist-evoked hallucinations.
Our phased success in the chemical modification of oleamide enriches the 5-HT2R allosteric modulatory library with a novel series of structurally diversified scaffolds. This series will facilitate pharmacological investigations to examine 5-HT2CR and 5-HT2AR allosteric modulation to promote the development of novel therapeutics for chronic diseases with impaired serotonergic function. Further chemical optimization studies focusing on modifying the long lipophilic tail based on these identified chemical leads will improve the overall physicochemical and pharmacokinetic profiles and provide optimized drug-like compounds for further in vitro cellular and in vivo analysis.
EXPERIMENTAL SECTION
Chemistry.
General.
All commercially available reaction reagents and solvents were reagent grade and used directly. Preparative column chromatography was carried out using silica gel 60, particle size 0.063–0.200 mm (70–230 mesh, flash). Analytical TLC was performed employing silica gel 60 F254 plates (Merck, Darmstadt). NMR spectra were recorded on a Bruker-600 (1H, 600 MHz; 13C, 150 MHz) spectrometer or Bruker-300 (1H, 300 MHz; 13C, 75 MHz). 1H and 13C NMR spectra were recorded with tetramethylsilane (TMS) as an internal reference. Chemical shifts were presented in ppm, and J values were expressed in Hz. Melting points were obtained on a Thermo Scientific electrothermal digital melting point apparatus. High-resolution mass spectra (HRMS) were acquired with a Thermo Fisher LTQ Orbitrap Elite mass spectrometer. Parameters were as follows: the nano ESI spray voltage was 1.8 kV, capillary temperature was 275 °C, resolution was 60,000, and ionization was achieved by positive mode. Purity of final compounds was carried out on a Shimadzu HPLC system (model CBM-20A LC-20 AD SPD-20A UV/vis) with analytical conditions as follows: Waters μBondapak C18 (300 × 3.9 mm), flow rate 0.5 mL/min, UV detection at 254 and 210 nm, and linear gradient from 30% acetonitrile in water (0.1% TFA) to 100% acetonitrile (0.1% TFA) in 20 min followed by 30 min of the last-named solvent. All newly synthesized compounds were characterized with 1H NMR, 13C NMR, HRMS, and HPLC analysis. All biologically evaluated compounds are >95% pure.
General Procedure for the Synthesis of Oleamide Analogues 7–30.
To a solution of oleic acid (1.0 equiv) in dichloromethane (2 mL) was added HBTU (1.3 equiv) or EDCI (1.5 equiv) in combination with 1-hydroxybenzotriazole (1.5 equiv) and stirred under room temperature; then alcohol analogues, amino acid analogues, and monoamine-like fragments (1.1 equiv) along with DIPEA (2.5 equiv) were added to the solution. The reaction mixture was stirred for another 8 h, and a TLC plate was used to detect the reaction with the potassium permanganate chromogenic agent. After the completion of reaction, saturated ammonium chloride aqueous solution (10 mL) was titrated to the solution to quench the reaction, and then the mixture system was extracted with ethyl acetate (20 mL × 3), and washed with water and brine, dried over anhydrous Na2SO4, and filtered. The organic solvent was concentrated under reduced pressure and purified on a silica gel column (DCM/MeOH = 99:1), which afforded the desired products 7–30.
N-(2,3-Dihydroxypropyl)oleamide (7).
Compound 7 (57 mg, 80%) was prepared from oleic acid (0.20 mmol) following the general synthetic procedure for 7–30 as a white wax-like material. 1H NMR (300 MHz, CDCl3) δ 6.13 (s, 1H), 5.36 (h, J = 4.0 Hz, 2H), 3.77 (q, J = 5.2 Hz, 1H), 3.57 (t, J = 4.1 Hz, 2H), 3.42 (q, J = 5.8 Hz, 2H), 2.23 (t, J = 7.6 Hz, 2H), 2.02 (q, J = 6.2 Hz, 4H), 1.64 (t, J = 7.4 Hz, 2H), 1.30 (d, J = 10.6 Hz, 20H), 0.89 (t, J = 6.4 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 175.3, 130.0, 129.7, 71.2, 63.6, 42.2, 36.6, 31.9, 29.8, 29.7, 29.5, 29.3, 29.2, 29.1, 27.22, 27.16, 25.7, 22.7, 14.1. HRMS (ESI) calcd for C21H41NO3 [M + H]+ 356.3159; found 356.3158.
N-(3-Hydroxypropyl)oleamide (8).
Compound 8 (25 mg, 52%) was prepared from oleic acid (0.14 mmol) following the general synthetic procedure for 7–30 as a white solid; mp 63.0–63.5 °C. 1H NMR (300 MHz, CDCl3) δ 5.91 (s, 1H), 5.43–5.30 (m, 2H), 3.64 (t, J = 5.6 Hz, 2H), 3.46–3.37 (m, 2H), 2.20 (t, J = 7.6 Hz, 2H), 2.02 (q, J = 6.2 Hz, 4H), 1.66 (dt, J = 14.6, 6.9 Hz, 4H), 1.42–1.18 (m, 20H), 0.95–0.82 (m, 3H). 13C NMR (75 MHz, CDCl3/MeOD) δ 175.1, 129.9, 129.6, 59.1, 38.5, 36.5, 36.1, 36.0, 31.9, 31.8, 29.7, 29.6, 29.4, 29.23, 29.19, 29.1, 27.13, 27.10, 25.8, 22.6, 14.0. HRMS (ESI) calcd for C21H41NO2 [M + H]+ 340.3210; found 340.3352.
N-(2-Hydroxyethyl)oleamide (9).
Compound 9 (52 mg, 71%) was prepared from oleic acid (0.27 mmol) following the general synthetic procedure for 7–30 as a white solid; mp 63.0–63.5 °C. 1H NMR (300 MHz, CDCl3) δ 6.34 (s, 1H), 5.41–5.26 (m, 2H), 3.70 (t, J = 4.9 Hz, 2H), 3.56 (s, 1H), 3.40 (dd, J = 10.2, 5.4 Hz, 2H), 2.24–2.15 (m, 2H), 2.06–1.95 (m, 4H), 1.73–1.51 (m, 2H), 1.29 (d, J = 10.0 Hz, 20H), 0.88 (t, J = 6.7 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 174.6, 130.0, 129.7, 62.1, 42.4, 36.6, 31.9, 29.8, 29.7, 29.5, 29.31, 29.28, 29.2, 27.21, 27.17, 25.7, 22.7, 14.1. HRMS (ESI) calcd for C20H40NO2 [M + H]+ 326.3054; found 326.3570.
(S)-N-(2,3-Dihydroxypropyl)oleamide (10).
Compound 10 (32 mg, 60%) was prepared from oleic acid (0.15 mmol) following the general synthetic procedure for 7–30 as a white wax-like material. 1H NMR (300 MHz, CDCl3) δ 6.44 (t, J = 6.1 Hz, 1H), 5.41–5.29 (m, 2H), 3.98 (bs, 1H), 3.85 (s, 1H), 3.76 (t, J = 5.2 Hz, 1H), 3.55 (bs, 2H), 3.40 (tq, J = 14.1, 8.2, 6.8 Hz, 2H), 2.22 (t, J = 7.6 Hz, 2H), 2.02 (q, J = 6.4 Hz, 4H), 1.77–1.54 (m, 2H), 1.40–1.19 (m, 20H), 0.93–0.84 (m, 3H). 13C NMR (75 MHz, CDCl3) δ 175.4, 130.0, 129.7, 71.1, 63.6, 42.1, 36.6, 31.9, 29.8, 29.7, 29.5, 29.32, 29.29, 29.2, 27.23, 27.18, 25.7, 22.7, 14.1. HRMS (ESI) calcd for C21H41NO3 [M + H]+ 356.3159; found 356.3154.
N-(1,3-Dihydroxypropan-2-yl)oleamide (11).
Compound 11 (58 mg, 60%) was prepared from oleic acid (0.27 mmol) following the general synthetic procedure for 7–30 as a whitish wax. 1H NMR (300 MHz, CDCl3) δ 6.69 (d, J = 7.9 Hz, 1H), 5.46–5.16 (m, 2H), 3.97–3.77 (m, 1H), 3.65 (ddd, J = 28.2, 11.3, 4.8 Hz, 4H), 2.86 (s, 2H), 2.26–2.09 (m, 2H), 2.07–1.85 (m, 4H), 1.71–1.50 (m, 2H), 1.26 (d, J = 9.6 Hz, 20H), 0.86 (t, J = 6.6 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 175.0, 130.0, 129.7, 61.7, 52.4, 52.3, 36.6, 36.5, 31.8, 29.71, 29.69, 29.5, 29.3, 29.2, 29.1, 27.2, 27.1, 25.7, 22.6, 14.0. HRMS (ESI) calcd for C21H41NO3 [M + H]+ 356.3159; found 356.3150.
N-((2S)-1,3-Dihydroxy-1-phenylpropan-2-yl)oleamide (12).
Compound 12 (61 mg, 94%) was prepared from oleic acid (0.15 mmol) following the general synthetic procedure for 7–30 as an off-white wax-like material. 1H NMR (300 MHz, CDCl3/CD3OD) δ 7.41–7.14 (m, 5H), 6.68–6.77 (d, J = 8.4 Hz, 1H), 5.39–5.25 (m, 2H), 4.97 (d, J = 3.5 Hz, 1H), 4.08–3.99 (m, 1H), 3.89 (d, J = 2.5 Hz, 2H), 3.63 (qd, J = 11.1, 5.8 Hz, 2H), 2.08 (t, J = 7.3 Hz, 2H), 1.99 (q, J = 7.2 Hz, 4H), 1.44 (p, J = 7.4 Hz, 2H), 1.26 (d, J = 9.4 Hz, 20H), 0.92–0.78 (m, 3H). 13C NMR (75 MHz, CDCl3/CD3OD) δ 175.0, 141.5, 129.9, 129.7, 128.1, 127.4, 125.7, 71.8, 62.2, 56.4, 56.3, 36.5, 36.4, 31.8, 29.69, 29.66, 29.4, 29.24, 29.19, 29.1, 29.0, 27.1, 25.7, 22.6, 14.0. HRMS (ESI) calcd for C27H45NO3 [M + H]+ 432.3472; found 432.3465.
N-(1,3-Dihydroxy-2-(hydroxymethyl)propan-2-yl)oleamide (13).
Compound 13 (73 mg, 94%) was prepared from oleic acid (0.20 mmol) following the general synthetic procedure for 7–30 as a white wax. 1H NMR (300 MHz, CDCl3) δ 6.51 (s, 1H), 5.36 (s, 2H), 3.60 (s, 6H), 2.24 (t, J = 7.6 Hz, 2H), 2.02 (d, J = 6.1 Hz, 4H), 1.62 (s, 2H), 1.30 (d, J = 11.4 Hz, 20H), 0.90 (d, J = 6.0 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 175.3, 130.0, 129.7, 61.8, 61.6, 37.0, 31.9, 29.7, 29.5, 29.3, 29.2, 29.1, 27.2, 25.8, 22.7, 14.1. HRMS (ESI) calcd for C22H44NO4 [M + H]+ 386.3265; found 386.3262.
N-(2,2-Diethoxyethyl)oleamide (14).
Compound 14 (56 mg, 70%) was prepared from oleic acid (0.20 mmol) following the general synthetic procedure for 7–30 as a colorless oil. 1H NMR (300 MHz, CDCl3) δ 5.78–5.65 (m, 1H), 5.41–5.25 (m, 2H), 4.50 (t, J = 5.2 Hz, 1H), 3.70 (dq, J = 9.6, 7.1 Hz, 2H), 3.53 (dq, J = 15.9, 6.8 Hz, 2H), 3.38 (t, J = 5.5 Hz, 2H), 2.17 (t, J = 7.6 Hz, 2H), 2.00 (q, J = 6.2 Hz, 4H), 1.62 (t, J = 7.4 Hz, 2H), 1.42–1.14 (m, 26H), 0.88 (t, J = 6.5 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 173.2, 130.0, 129.7, 100.8, 62.8, 41.9, 36.7, 31.9, 29.73, 29.68, 29.5, 29.3, 29.2, 29.1, 27.2, 27.1, 25.7, 22.6, 15.3, 14.1. HRMS (ESI) calcd for C24H47NO3Na [M + Na]+ 420.3448; found 420.3446.
N-(2-(2-Hydroxyethoxy)ethyl)oleamide (15).
Compound 15 (62 mg, 83%) was prepared from oleic acid (0.20 mmol) following the general synthetic procedure for 7–30 as a colorless oil. 1H NMR (300 MHz, CDCl3) δ 6.21–6.03 (m, 1H), 5.33 (q, J = 6.3 Hz, 2H), 3.82–3.69 (m, 2H), 3.57 (q, J = 4.4 Hz, 4H), 3.46 (q, J = 5.4 Hz, 2H), 2.59 (s, 1H), 2.18 (t, J = 7.6 Hz, 2H), 2.01 (q, J = 6.4 Hz, 4H), 1.63 (t, J = 7.4 Hz, 2H), 1.29 (d, J = 9.6 Hz, 20H), 0.88 (t, J = 6.3 Hz, 3H).13C NMR (75 MHz, CDCl3) δ 173.5, 130.0, 129.7, 72.2, 70.0, 61.7, 39.2, 36.7, 31.9, 29.7, 29.5, 29.3, 29.1, 27.1, 25.7, 22.6, 14.1. HRMS (ESI) calcd for C22H44NO3 [M + H]+ 370.3316; found 370.3314.
Methyl Oleoyl-l-allothreoninate (16).
Compound 16 (113 mg, 68%) was prepared from oleic acid (0.42 mmol) following the general synthetic procedure for 7–30 as a white solid; mp 63.0–63.5 °C. 1H NMR (300 MHz, CDCl3) δ 6.67–6.32 (m, 1H), 5.47–5.10 (m, 2H), 4.69–4.48 (m, 1H), 4.33 (s, 1H), 3.74 (s, 3H), 3.26 (s, 1H), 2.27 (t, J = 7.6 Hz, 2H), 2.11–1.89 (m, 4H), 1.79–1.51 (m, 2H), 1.28 (d, J = 11.6 Hz, 20H), 1.19 (d, J = 6.4 Hz, 3H), 0.87 (t, J = 6.6 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 174.1, 171.7, 130.0, 129.7, 67.8, 57.3, 52.4, 38.6, 36.5, 31.9, 29.74, 29.71, 29.5, 29.29, 29.27, 29.24, 29.15, 27.19, 27.16, 25.7, 22.6, 20.0, 14.1. HRMS (ESI) calcd for C23H43NO4 [M + H]+ 398.3265; found 398.3453.
Methyl Oleoyl-l-serinate (17).
Compound 17 (100 mg, 62%) was prepared from oleic acid (0.42 mmol) following the general synthetic procedure for 7–30 as a colorless oil. 1H NMR (300 MHz, CDCl3) δ 6.62 (s, 1H), 5.44–5.18 (m, 2H), 4.77–4.54 (m, 1H), 4.08–3.82 (m, 2H), 3.78 (s, 3H), 3.36 (s, 1H), 2.33–2.15 (m, 2H), 2.12–1.91 (m, 4H), 1.78–1.51 (m, 2H), 1.28 (d, J = 10.1 Hz, 20H), 0.88 (t, J = 6.6 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 173.9, 171.1, 130.0, 129.7, 63.2, 54.6, 52.7, 36.4, 31.9, 29.8, 29.7, 29.5, 29.30, 29.26, 29.2, 29.1, 27.21, 27.17, 25.6, 22.7, 14.1. HRMS (ESI) calcd for C22H41NO4 [M + H]+ 384.3108; found 384.3457.
Methyl Oleoyl-l-tyrosinate (18).
Compound 18 (143 mg, 74%) was prepared from oleic acid (0.42 mmol) following the general synthetic procedure for 7–30 as a white solid; mp 71.5–72.3 °C. 1H NMR (300 MHz, CDCl3) δ 7.16 (s, 1H), 6.95 (d, J = 8.5 Hz, 2H), 6.75 (d, J = 8.5 Hz, 2H), 6.05 (d, J = 8.0 Hz, 1H), 5.47–5.23 (m, 2H), 4.90 (dt, J = 8.0, 6.0 Hz, 1H), 3.75 (s, 3H), 3.04 (ddd, J = 30.8, 14.0, 5.9 Hz, 2H), 2.29–2.11 (m, 2H), 2.12–1.90 (m, 4H), 1.71–1.48 (m, 2H), 1.28 (s, 20H), 0.89 (t, J = 6.7 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 173.5, 172.5, 155.7, 130.2, 130.0, 129.8, 126.9, 115.6, 53.2, 52.4, 37.3, 36.6, 31.9, 29.8, 29.7, 29.5, 29.3, 29.2, 29.1, 27.23, 27.19, 25.6, 22.7, 14.1. HRMS (ESI) calcd for C28H45NO4 [M + H]+ 460.3421; found 460.3436.
Methyl Oleoyl-l-tryptophanate (19).
Compound 19 (152 mg, 75%) was prepared from oleic acid (0.42 mmol) following the general synthetic procedure for 7–30 as a yellow oil. 1H NMR (300 MHz, CDCl3) δ 8.43 (s, 1H), 7.56 (d, J = 7.8 Hz, 1H), 7.37 (d, J = 8.0 Hz, 1H), 7.17 (dtd, J = 14.8, 7.1, 1.1 Hz, 2H), 6.98 (d, J = 2.4 Hz, 1H), 6.03 (d, J = 7.8 Hz, 1H), 5.49–5.24 (m, 2H), 5.00 (dt, J = 7.9, 5.4 Hz, 1H), 3.71 (s, 3H), 3.34 (dd, J = 5.3, 1.4 Hz, 2H), 2.22–2.11 (m, 2H), 2.03 (dd, J = 7.8, 4.7 Hz, 4H), 1.69–1.50 (m, 2H), 1.30 (s, 20H), 0.91 (t, J = 6.7 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 172.9, 172.6, 136.2, 130.0, 129.8, 127.7, 122.7, 122.2, 119.6, 118.5, 111.3, 110.0, 52.9, 52.3, 36.6, 31.9, 29.8, 29.7, 29.5, 29.34, 29.26, 29.2, 29.1, 27.7, 27.3, 27.2, 25.5, 22.7, 14.1. HRMS (ESI) calcd for C30H46N2O3 [M + H]+ 483.3581; found 483.3417.
Oleoyl-l-allothreonine (20).
Solid LiOH monohydrate (16.8 mg, 0.4 mmol) was added to a solution of 16 (40 mg, 0.10 mmol) in THF/H2O 3:1 (2 mL) at rt. The reaction mixture was stirred for 48 h and determined complete by TLC. The reaction mixture was neutralized with HCl and extracted with EtOAc (3 × 10 mL). The combined organic extracts were washed with brine (5 mL) and concentrated under reduced pressure to afford 20 (25 mg, 66%) as a colorless gel. 1H NMR (600 MHz, CDCl3) δ 6.99–6.71 (bs, 2H), 5.53–5.11 (m, 2H), 4.52 (d, J = 6.9 Hz, 1H), 4.42 (s, 1H), 2.40–2.25 (m, 2H), 2.10–1.89 (m, 4H), 1.65 (s, 2H), 1.30 (d, J = 18.5 Hz, 20H), 1.22 (d, J = 5.2 Hz, 3H), 0.90 (t, J = 6.9 Hz, 3H). 13C NMR (150 MHz, CDCl3) δ 175.4, 174.3, 130.0, 129.6, 67.5, 57.7, 36.4, 31.9, 29.8, 29.6, 29.35, 29.32, 29.2, 27.24, 27.21, 25.8, 22.7, 19.4, 14.1. HRMS (ESI) calcd for C22H41NO4 [M + H]+ 384.3108; found 384.3166.
Oleoyl-l-serine (21).
Compound 21 (20 mg, 54%) was prepared from 17 by a procedure similar to that used to prepare compound 20, as a white wax-like material. 1H NMR (300 MHz, CDCl3) δ 5.31 (td, J = 4.7, 2.1 Hz, 2H), 4.52 (t, J = 3.8 Hz, 1H), 3.95 (dd, J = 11.6, 3.9 Hz, 1H), 3.80 (dd, J = 11.5, 3.7 Hz, 1H), 3.61 (s, 3H), 2.25 (dt, J = 10.4, 7.5 Hz, 2H), 1.98 (q, J = 6.3 Hz, 4H), 1.71–1.50 (m, 2H), 1.38–1.18 (m, 20H), 0.94–0.78 (m, 3H). 13C NMR (75 MHz, CDCl3/MeOD) δ 174.4, 172.7, 130.0, 129.7, 62.6, 36.3, 31.9, 29.71, 29.68, 29.5, 29.3, 29.24, 29.20, 29.1, 27.2, 25.5, 22.6, 14.0. HRMS (ESI) calcd for C21H39NO4 [M + H]+ 370.2952; found 370.2995.
Oleoyl-l-tyrosine (22).
Compound 22 (40 mg, 90%) was prepared from 18 by a procedure similar to that used to prepare compound 20, as a white solid; mp 170.0–170.5 °C. 1H NMR (600 MHz, CDCl3) δ 6.90 (d, J = 8.0 Hz, 2H), 6.62 (d, J = 8.0 Hz, 2H), 5.41–5.11 (m, 2H), 4.38 (s, 1H), 3.94–3.59 (bs, 1H), 3.02–2.91 (m, 1H), 2.90–2.74 (m, 1H), 2.04–1.93 (m, 6H), 1.44 (d, J = 6.2 Hz, 2H), 1.36–1.10 (m, 20H), 0.85 (t, J = 7.0 Hz, 3H). 13C NMR (150 MHz, CDCl3/MeOD) δ 177.9, 174.3, 155.3, 130.2, 129.9, 129.7, 128.3, 115.3, 56.1, 37.0, 36.4, 31.9, 29.7, 29.5, 29.30, 29.27, 29.2, 27.2, 25.7, 22.6, 14.0. HRMS (ESI) calcd for C27H43NO4 [M + H]+ 446.3265; found 446.3216.
Oleoyl-l-tryptophan (23).
Compound 23 (22 mg, 42%) was prepared from 19 by a procedure similar to that used to prepare compound 20, as a white wax-like material. 1H NMR (300 MHz, CDCl3) δ 8.42 (s, 1H), 7.57 (d, J = 7.8 Hz, 1H), 7.32 (d, J = 8.0 Hz, 1H), 7.19 (t, J = 7.2 Hz, 1H), 7.11 (t, J = 7.2 Hz, 1H), 6.97 (s, 1H), 6.18 (d, J = 7.6 Hz, 1H), 5.46–5.26 (m, 2H), 4.92 (dd, J = 12.4, 5.4 Hz, 1H), 3.48–3.09 (m, 2H), 2.12–1.93 (m, 6H), 1.57–1.41 (m, 2H), 1.29 (s, 15H), 1.20 (s, 5H), 0.90 (t, J = 6.7 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 175.5, 174.3, 136.1, 130.0, 129.8, 127.8, 123.3, 122.1, 119.7, 118.4, 111.5, 109.5, 53.6, 36.4, 31.9, 29.8, 29.7, 29.6, 29.4, 29.3, 29.2, 29.1, 27.3, 27.2, 27.0, 25.4, 22.7, 14.1. HRMS (ESI) calcd for C29H44N2O3 [M + H]+ 469.3425; found 469.3500.
N-(4-Hydroxybenzyl)oleamide (24).
Compound 24 (40 mg, 69%) was prepared from oleic acid (0.15 mmol) following the general synthetic procedure for 7–30 as a white solid; mp 71.5–72.3 °C. 1H NMR (300 MHz, CDCl3) δ 7.85–7.67 (m, 1H), 7.14 (d, J = 8.5 Hz, 2H), 6.86 (d, J = 8.5 Hz, 2H), 6.03 (t, J = 5.5 Hz, 1H), 5.61–5.25 (m, 2H), 4.39 (d, J = 5.6 Hz, 2H), 2.37–2.18 (m, 2H), 2.06 (dd, J = 7.9, 4.2 Hz, 4H), 1.79–1.61 (m, 2H), 1.34 (s, 20H), 0.95 (t, J = 6.7 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 173.8, 156.2, 130.0, 129.7, 129.20, 129.17, 115.8, 43.4, 36.8, 31.9, 29.8, 29.7, 29.5, 29.32, 29.25, 29.2, 29.1, 27.23, 27.18, 25.8, 22.7, 14.1. HRMS (ESI) calcd for C25H41NO2 [M + H]+ 388.3210; found 388.3480.
N-(3,4-Dihydroxybenzyl)oleamide (25).
Compound 25 (30 mg, 50%) was prepared from oleic acid (0.15 mmol) following the general synthetic procedure for 7–30 as a colorless oil. 1H NMR (300 MHz, CDCl3) δ 6.93–6.79 (m, 2H), 6.67 (dd, J = 8.1, 1.9 Hz, 1H), 6.17 (t, J = 5.7 Hz, 1H), 5.59–5.24 (m, 2H), 4.34 (d, J = 5.8 Hz, 2H), 2.34–2.21 (m, 2H), 2.15–1.95 (m, 4H), 1.81–1.55 (m, 2H), 1.34 (s, 20H), 0.95 (t, J = 6.7 Hz, 3H).13C NMR (75 MHz, CDCl3) δ 174.4, 144.6, 144.3, 130.0, 129.8, 129.7, 119.7, 115.1, 114.9, 43.6, 36.8, 31.9, 29.8, 29.7, 29.5, 29.3, 29.22, 29.19, 29.1, 27.23, 27.17, 25.8, 22.7, 14.1. HRMS (ESI) calcd for C25H41NO3 [M + H]+ 404.3159; found 404.3338.
N-(4-Hydroxyphenethyl)oleamide (26).
Compound 26 (24 mg, 42%) was prepared from oleic acid (0.14 mmol) following the general synthetic procedure for 7–30 as a white wax-like material. 1H NMR (300 MHz, CDCl3) δ 7.72 (s, 1H), 7.04 (d, J = 8.5 Hz, 2H), 6.94–6.74 (m, 2H), 5.83–5.68 (m, 1H), 5.49–5.25 (m, 2H), 3.52 (q, J = 6.9 Hz, 2H), 2.77 (t, J = 7.0 Hz, 2H), 2.25–2.14 (m, 2H), 2.13–1.97 (m, 4H), 1.77–1.53 (m, 2H), 1.32 (s, 20H), 0.93 (t, J = 6.7 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 174.0, 155.4, 130.0, 129.7, 129.71, 129.66, 115.7, 41.0, 36.8, 34.8, 31.9, 29.8, 29.7, 29.5, 29.3, 29.2, 29.1, 27.23, 27.18, 25.8, 22.7, 14.1. HRMS (ESI) calcd for C26H43NO2 [M + H]+ 402.3367; found 402.3293.
N-(3,4-Dihydroxyphenethyl)oleamide (27).
Compound 27 (45 mg, 68%) was prepared from oleic acid (0.14 mmol) following the general synthetic procedure for 7–30 as a white solid; mp 69.1–71.0 °C. 1H NMR (300 MHz, CDCl3) δ 7.94 (s, 1H), 6.85 (d, J = 8.0 Hz, 1H), 6.79 (d, J = 2.0 Hz, 1H), 6.59 (dd, J = 8.0, 2.0 Hz, 1H), 5.86 (t, J = 5.1 Hz, 1H), 5.49–5.25 (m, 2H), 3.51 (dd, J = 13.1, 6.9 Hz, 2H), 2.72 (t, J = 7.1 Hz, 2H), 2.29–2.14 (m, 2H), 2.14–1.97 (m, 4H), 1.75–1.52 (m, 2H), 1.32 (s, 20H), 0.93 (t, J = 6.7 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 174.6, 144.5, 143.3, 130.4, 130.0, 129.7, 120.4, 115.5, 115.3, 41.0, 36.8, 34.9, 31.9, 29.8, 29.7, 29.5, 29.33, 29.31, 29.21, 29.18, 29.1, 27.23, 27.18, 25.8, 22.7, 14.1. HRMS (ESI) calcd for C26H43NO3 [M + H]+ 418.3316; found 418.3624.
N-(2-Morpholinoethyl)oleamide (28).
Compound 28 (71 mg, 67%) was prepared from oleic acid (0.27 mmol) following the general synthetic procedure for 7–30 as an off-white wax-like material. 1H NMR (300 MHz, CDCl3) δ 5.99 (s, 1H), 5.44–55.26 (m, 2H), 3.78–3.64 (m, 4H), 3.37 (q, J = 6.0 Hz, 2H), 2.57–2.41 (m, 6H), 2.19 (t, J = 6.0 Hz, 2H), 2.08–1.94 (m, 4H), 1.73–1.54 (m, 2H), 1.30 (d, J = 11.9 Hz, 20H), 0.89 (t, J = 6.7 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 173.2, 130.0, 129.7, 66.9, 57.1, 53.3, 36.8, 35.5, 31.9, 29.8, 29.7, 29.5, 29.32, 29.29, 29.2, 27.22, 27.17, 25.8, 22.7, 14.1. HRMS (ESI) calcd for C24H46N2O2 [M + H]+ 395.3632; found 395.3628.
tert-Butyl (4-Oleamidobutyl)carbamate (29).
Compound 29 (73 mg, 81%) was prepared from oleic acid (0.20 mmol) following the general synthetic procedure for 7–30 as a colorless oil. 1H NMR (300 MHz, CDCl3) δ 5.78 (s, 1H), 5.33 (d, J = 5.3 Hz, 2H), 4.66 (s, 1H), 3.26 (q, J = 6.2 Hz, 2H), 3.13 (d, J = 6.4 Hz, 2H), 2.16 (t, J = 7.6 Hz, 2H), 2.08–1.94 (m, 4H), 1.62 (t, J = 7.5 Hz, 2H), 1.51 (d, J = 4.5 Hz, 4H), 1.44 (s, 9H), 1.28 (d, J = 9.2 Hz, 20H), 0.93–0.83 (m, 3H). 13C NMR (75 MHz, CDCl3) δ 173.2, 156.1, 130.0, 129.7, 40.1, 39.0, 36.8, 31.9, 29.74, 29.70, 29.5, 29.29, 29.25, 29.1, 28.4, 27.6, 27.20, 27.16, 26.7, 25.8, 22.7, 14.1. HRMS (ESI) calcd for C27H53N2O3 [M + H]+ 453.4051; found 453.4059.
tert-Butyl 4-(Oleamidomethyl)piperidine-1-carboxylate (30).
Compound 30 (81 mg, 85%) was prepared from oleic acid (0.20 mmol) following the general synthetic procedure for 7–30 as a colorless oil. 1H NMR (300 MHz, CDCl3) δ 5.59 (t, J = 6.1 Hz, 1H), 5.34 (td, J = 7.5, 4.7 Hz, 2H), 4.20–4.02 (m, 2H), 3.15 (s, 2H), 2.68 (t, J = 12.8 Hz, 2H), 2.18 (t, J = 7.6 Hz, 2H), 2.02 (q, J = 6.7 Hz, 4H), 1.65 (tt, J = 8.3, 4.3 Hz, 5H), 1.46 (s, 9H), 1.29 (d, J = 9.9 Hz, 20H), 1.19–1.09 (m, 2H), 0.89 (t, J = 6.5 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 173.3, 154.8, 130.0, 129.7, 79.4, 44.8, 43.6, 36.9, 36.4, 31.9, 29.8, 29.7, 29.5, 29.3, 29.24, 29.17, 29.13, 29.10, 28.4, 27.21, 27.16, 25.8, 22.7, 14.1. HRMS (ESI) calcd for C29H55N2O3 [M + H]+ 479.4207; found 479.4216.
In Vitro Pharmacology.
Intracellular Calcium (Cai2+) Release Assay in h5-HT2R-CHO Cells.
The assays were conducted with Chinese hamster ovary (CHO) cells stably transfected with the human unedited (INI) h5-HT2CR (h5-HT2CR-CHO cells), human h5-HT2AR (h5-HT2AR-CHO cells), or h5-HT5-HT2BR5-HT2BR-CHO cells, which were generous gifts from Drs. Kelly A. Berg and William P. Clarke (University of Texas Health Science Center, San Antonio). The h5-HT2BR (5-HT2BR1/5-HT2BR; h5-HT2BR-CHO) cells were purchased from GenScript, Piscataway, NJ. The cellular growth environment was as follows: 37 °C, 5% CO2, and 85% relative humidity. The h5-HT2CR-CHO and h5-HT2AR-CHO cells were cultured in a GlutaMax-MEM medium (Invitrogen, Carlsbad, CA) containing 5% fetal bovine serum (Atlanta Biologicals, Atlanta, GA) and 100 μg/mL hygromycin (Mediatech, Manassas, VA). The h5-HT2BR-CHO cells were cultured in Ham’s F12 media supplemented with 10% FBS and 200 μg/mL Zeocin (Thermo Fisher Scientific, Carlsbad, CA). All cells were passaged when they reached 80% confluency.
The Cai2+ release assay was performed according to our previous publications, with minor modifications.18,20,23 Specifically, cells (150 μL; passages 9–15) were plated in serum-replete medium at a density of 14,000–16,000 (FlexStation 3; Molecular Devices) or 30,000 cells/well (FLIPRTETRA; Molecular Devices) in black-wall 96-well culture plates with optically clear flat bottoms. After ~24 h, the medium was replaced with serum-free (SF) GlutaMax-MEM medium (h5-HT2CR-CHO and h5-HT2AR-CHO cells) or serum free HAM’s F12 (h5-HT2BR-CHO cells) supplemented with 20 nM to 100 μM putrescine (Sigma-Aldrich, St. Louis, MO), 20 nM to 100 μM progesterone (Sigma-Aldrich), and 1:100 ITS (1000 mg/L human recombinant insulin, 550 mg/L human recombinant transferrin, 0.67 mg/L selenious acid; Corning Inc., Corning, NY) (SF+ medium). After an incubation for another 3 h, SF+ medium for the h5-HT2CR-CHO and h5-HT2AR-CHO cells was replaced with 40 μL of Hank’s balanced saline solution (HBSS; without CaCl2 or MgCl2, pH 7.4) plus 40 μL of Calcium 6 dye solution (FLIPR No-wash kit, Molecular Devices, Sunnyvale CA) supplemented with 2.5 mM of water-soluble probenecid (Sigma-Aldrich), and then the plate was incubated with dye solution in the dark for 2 h at 37 °C followed by 15 min at room temperature. For h5-HT2BR-CHO cells, Calcium 6 dye was incubated in the presence of serum-free Ham’s F12 medium supplemented with progesterone, putrescine, and ITS as described above. The drug was diluted at 5× concentration in 1× HBSS, and controls contained the same final concentration of diluent. The delivery of the compound (20 μL/well) was 15 min prior to the addition of 5-HT (10 pM to 100 μM; 25 μL/well), and a baseline was established for each well before the addition of the compound and 5-HT. The fluorescence readings were then adopted to evaluate the allosteric modulation of 5-HT-induced Cai2+ release. FlexStation 3 (Molecular Device) or FLIPRTETRA (80–130 gain, 80% intensity, 0.3 s exposure) was used to measure fluorescence. For FlexStation 3, a 17 s baseline was established before the compound was added, and fluorescence was recorded every 1.7 s thereafter for 240 s. The maximum peak height of each well was determined by SoftMax software (Pro 5.4.5). For FLIPRTETRA, a 10 s baseline was established before adding the compound, and then the fluorescence was recorded every 1 s for 120 s after the compound or 360 s after 5-HT. The maximum peak height of each well was determined by ScreenWorks 4.0 software. After the final reading, the cells were fixed in 2% paraformaldehyde (Sigma) overnight.
A four-parameter nonlinear regression analysis (GraphPad Prism 7 or 9) was used to determine the 5-HT-induced Cai2+ maximum release (Emax) in the presence of the test compound and calculated from four to six biological replicates, with each biological replicate performed in technical triplicates. The Emax of the test compound plus 5-HT was normalized to the Emax of 5-HT alone. Subsequently, Welch’s unpaired t test (GraphPad prism) was used for post hoc comparison of the Emax means. All statistical analyses were performed with an experimental error rate of α = 0.05. All treatment assignments were blinded to investigators who performed in vitro assays and end point statistical analyses.
In Vivo PK and Brain Penetration Analyses.
Male Sprague–Dawley rats (n = 3/treatment group; Beijing Vital River Laboratory, Animal Technology Co., Ltd., Beijing, China) weighing 200–250 g at the beginning of the experiment were housed three per cage in a pathogen-free, temperature-controlled (20–26 °C), and humidity-controlled (40–70%) environment with a 12 h light–dark cycle and ad libitum access to food and filtered water. Rats were randomly assigned to treatment groups. Vehicle [10% dimethyl sulfoxide (DMSO) and 90% 2-hydroxypropyl-β-cyclodextrin (HP-β-CD); Cyclodextrin Technologies Development, Inc., High Springs, FL, USA] or compound 13 dissolved in vehicle was administered to rats ip at 10 mg/kg or po at 20 mg/kg. Blood samples (300 μL) were collected from the jugular vein before dosing and at 0.08, 0.25, 0.5, 1.0, 2.0, 4.0, 8.0, and 24 h postdosing for ip administration and 0.25, 0.5, 1.0, 2.0, 4.0, 8.0, and 24 h postdosing for po administration. The blood samples were placed in heparinized tubes and centrifuged at 6000 rpm for 5 min at 4 °C. Brain samples were collected at 0.25 and 1 h postdosing. All samples were stored at −20 °C. The concentration of 13 in each sample was analyzed by Sundia MediTech Co., Ltd. The study and the related standard operating procedures were reviewed and approved by the Bioduro-Sundia Institutional Animal Care and Use Committee. The Bioduro-Sundia animal facility is approved with yearly inspection by the Shanghai Laboratory Animal Management Committee. The PK parameters of compound 13 were calculated according to a noncompartmental model using WinNonlin 8.1 (Pharsight Corporation, ver 5.3, Mountain View, CA, USA). The peak concentration (Cmax) and time of peak concentration (Tmax) were directly obtained from the plasma concentration–time profile. The elimination rate constant (λ) was obtained by the least-squares fitted terminal log-linear portion of the slope of the plasma concentration–time profile. The elimination half-life (T1/2) was evaluated according to 0.693/λ. The area under the plasma concentration–time curve from 0 to time t (AUC0–t) was evaluated using the linear trapezoidal rule and further extrapolated to infinity (AUC0–inf) following the equation AUC0–inf = AUC0–t + Clast/λ. The PK parameters and brain concentrations are presented as mean ± SEM.
Effects of Compound 13 on Spontaneous Locomotor Activity.
Animals.
A total of 96 male Sprague–Dawley rats (Envigo RMS, LLC; Indianapolis, IN) weighing 175–199 g at the start of experiments were used. Rats were housed two per cage and allowed to acclimate for 7 days in a colony room at a constant temperature (21–23 °C) and humidity (45–50%) on a 12 h light–dark cycle (lights on 0600–1800 h). Food and water were available ad libitum, and rats were handled for 7 days prior to the start of behavioral testing. All experiments were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (2011) and with the approval of the UTMB Institutional Animal Care and Use Committee. All efforts were made to minimize animal suffering, to reduce the number of animals used, and to utilize alternatives to in vivo techniques, when available.
Drugs.
SB242084 (6-chloro-5-methyl-1[[2-(2-methylpyrid-3-yloxy)pyrid-5-yl]carbamoyl]indoline dihydrochloride; Cayman Chemical, Ann Arbor, MI) was dissolved in 0.9% NaCl containing 10 mM citric acid (Sigma Chemical Co.) and 8% HP-β-CD (Trappsol, Cyclodextrin Technologies Development Inc., High Springs, FL) with the final pH of the solution adjusted to 5.6. M100907 ((R)-(2,3-dimethoxyphenyl)-[1-[2-(4-fluorphenyl)ethyl]-piperidin-4-yl]methanol) (synthesized by Kenner Rice, National Institute on Drug Abuse, Bethesda, MD) was dissolved in 1% Tween-80 in 0.9% NaCl. Compound 13 was dissolved in 4% Tween 80 and 1% DMSO in 0.9% NaCl. To facilitate dissolution, prepared doses of compound 13 were briefly sonicated and maintained at ~37 °C until injection. SB242084 and M100907 were administered ip at a volume of 1 mL/kg. Because of solubility limits, compound 13 was injected at a volume of 2 mL/kg to achieve the desired dose.
Locomotor Activity Assessments.
Locomotor activity was monitored and quantified under low light conditions using a modified open field activity system (San Diego Instruments, San Diego, CA) as previously described.18 Clear plexiglass chambers (40 × 40 × 40 cm3) were surrounded by a 4 × 4 photobeam matrix positioned 4 cm from the chamber floor. Total ambulations were quantified as the sum of consecutive photobeam breaks that occurred within the inner central 16 × 16 cm2 perimeter and in the surrounding outer peripheral 16 × 16 cm2 perimeter of the activity monitor.
A between-subjects design was implemented to evaluate the dose–effect relationship of compound 13 on spontaneous motor activity. On test day, rats (n = 48) were administered vehicle (4% Tween 80 and 1% DMSO in 0.9% NaCl, 2 mL/kg, ip) or compound 13 (3, 10, or 30 mg/kg, 2 mL/kg, ip) 30 min prior to placement in activity monitors; ambulations and vertical activity were assessed for 90 min.
In a separate cohort, a between-subjects design was employed to ascertain 5-HT2R subtypes mediating locomotor suppression induced by compound 13. Rats (n = 48) underwent a 7 day drug washout period before the start of experiment ii. On test day, rats were administered the 5-HT2AR-selective antagonist M10090766 (0.1 mg/kg, 1 mL/kg, ip), the 5-HT2CR-selective antagonist SB24208467 (0.5 mg/kg, 1 mL/kg, ip), or saline (1 mL/kg, ip) immediately followed by administration of vehicle (4% Tween 80 and 1% DMSO in 0.9% NaCl, 2 mL/kg, ip) or compound 13 (30 mg/kg, 2 mL/kg, ip) 30 min prior to placement in activity monitors; ambulations and vertical activity were assessed for 90 min. One animal was removed from the analysis because of a photobeam failure in its activity monitor.
Statistical Analyses.
Locomotor activity data are presented as mean ambulations (± SEM) totaled over the 90 min session for both the compound 13 dose–response curve and combination treatments with a 5-HT2AR or 5-HT2CR antagonist. The main effect of treatment on total ambulations was analyzed with a one-way analysis of variance (ANOVA); predetermined comparisons between treatment groups were made with Dunnett’s procedure. All statistical analyses were conducted with an experiment-wise error rate of α = 0.05.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants R21 MH093844 (J.Z./K.A.C.), R01 DA038446 (J.Z./K.A.C.), K05 DA020087 (K.A.C.), P30 DA028821 (K.A.C.), T32 DA007287 (C.T.W., E.A.W., C.R.M., A.A.B.), F31 DA038922 (C.T.W.), and F31 DA045511 (E.A.W.) from the National Institutes of Health, the John D. Stobo, M.D. Distinguished Chair Endowment Fund (J.Z.), the Edith & Robert Zinn Chair in Drug Discovery Endowment Fund (J.Z.), and the Center for Addiction Sciences and Therapeutics at UTMB. We thank Drs. Lawrence C. Sowers, Jason Herring, and Tianzhi Wang for the NMR spectroscopy assistance. Receptor binding profiles and agonist functional data were generously conducted and provided by the National Institute of Mental Health Psychoactive Drug Screening Program (PDSP), Contract #HHSN-271-2013-00017-C (NIMH PDSP). We thank Drs. Joanna M. Miszkiel and Claudia A. Soto for their initial technical and conceptual input.
ABBREVIATIONS
- 5-HT
serotonin
- 5-HT2AR
5-HT2A receptor
- 5-HT2BR
5-HT2B receptor
- 5-HT2CR
5-HT2C receptor
- 5-HT2Rs
serotonin 5-HT2 receptors
- PAMs
positive allosteric modulators
- CNS
central nervous system
- GPCRs
G protein-coupled receptors
- LSD
d-lysergic acid diethylamide
- LT
lipophilic tail
- PH
polar head
- ECL2
extracellular loop 2
- 7TM
seven-transmembrane bundle
- SAR
structure–activity relationship
- HBTU
N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate
- EDCI
1-ethyl-(3-dimethylaminopropyl)carbodiimide hydrochloride
- DIPEA
N,N-diisopropylethylamine
- TFA
trifluoroacetic acid
- PTLC
preparative thin layer chromatographic
- PLCβ
phospholipase Cβ
- Cai2+
intracellular calcium
- CHO
Chinese hamster ovary
- E max
maximum 5-HT-induced Cai2+ release
- NAM
negative allosteric modulator
- NIMH
National Institute of Mental Health
- PDSP
psychoactive drug screening program
- MPO
multiparameter optimization
- P-gp
P-glycoprotein
- PK
pharmacokinetics
- IFD
Induced Fit Docking
- XP
extra-precision
- SP
standard-precision
- ECL
extracellular loop
- TM
transmembrane helix
- hERG
human ether-a-go-go-related gene
- T 1/2
half-life
- T max
time of maximum concentration
- C max
maximum concentration
- ip
intraperitoneal
- AUC0–inf
area under the concentration–time curves from time zero to infinity
- B/P
brain/plasma
- DMSO
dimethyl sulfoxide
- HP-β-CD
2-hydroxypropyl-β-cyclodextrin
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.3c00908
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.3c00908.
Radioligand binding data, CNS-MPO value calculation, cellular assay data, docking protocol, in silico toxicity prediction, and 1H and 13C NMR spectra as well as HPLC analysis for the compounds described in this paper. Authors will release the atomic coordinates upon article publication. (PDF)
Molecular formula strings (CSV)
5HT2CR-Cmpd7-Cmpd2 (PDB)
6BQG_5HT2CR merged ligands (PDB)
6WGT_5HT2AR merged ligands (PDB)
5HT-6BQG-Cmpd9 (PDB)
5HT-6BQG-Cmpd13 (PDB)
5HT-6WGT-Cmpd9 (PDB)
5HT-6WGT-Cmpd13 (PDB)
Notes
The authors declare the following competing financial interest(s): Dr. Cunningham is a consultant for Delix Therapeutics which is unrelated to this manuscript. Dr. Zhou is partially supported by MapLight Therapeutics, Inc. through an industry-funded collaborative research project that is unrelated to this manuscript.
NOTE ADDED AFTER ASAP PUBLICATION
On July 27, 2023, in the Experimental Section, the name for compound 13 was corrected to read N-(1,3-Dihydroxy-2-(hydroxymethyl)propan-2-yl)oleamide.
Contributor Information
Jianping Chen, Center for Addiction Sciences and Therapeutics and Chemical Biology Program and Department of Pharmacology and Toxicology, University of Texas Medical Branch (UTMB), Galveston, Texas 77555, United States.
Erik J. Garcia, Center for Addiction Sciences and Therapeutics, University of Texas Medical Branch (UTMB), Galveston, Texas 77555, United States;.
Christina R. Merritt, Center for Addiction Sciences and Therapeutics, University of Texas Medical Branch (UTMB), Galveston, Texas 77555, United States
Joshua C. Zamora, Center for Addiction Sciences and Therapeutics, University of Texas Medical Branch (UTMB), Galveston, Texas 77555, United States
Andrew A. Bolinger, Center for Addiction Sciences and Therapeutics and Chemical Biology Program and Department of Pharmacology and Toxicology, University of Texas Medical Branch (UTMB), Galveston, Texas 77555, United States
Konrad Pazdrak, Center for Addiction Sciences and Therapeutics, University of Texas Medical Branch (UTMB), Galveston, Texas 77555, United States.
Susan J. Stafford, Center for Addiction Sciences and Therapeutics, University of Texas Medical Branch (UTMB), Galveston, Texas 77555, United States
Randy C. Mifflin, Center for Addiction Sciences and Therapeutics, University of Texas Medical Branch (UTMB), Galveston, Texas 77555, United States
Eric A. Wold, Center for Addiction Sciences and Therapeutics and Chemical Biology Program and Department of Pharmacology and Toxicology, University of Texas Medical Branch (UTMB), Galveston, Texas 77555, United States
Christopher T. Wild, Center for Addiction Sciences and Therapeutics and Chemical Biology Program and Department of Pharmacology and Toxicology, University of Texas Medical Branch (UTMB), Galveston, Texas 77555, United States
Haiying Chen, Center for Addiction Sciences and Therapeutics and Chemical Biology Program and Department of Pharmacology and Toxicology, University of Texas Medical Branch (UTMB), Galveston, Texas 77555, United States.
Noelle C. Anastasio, Center for Addiction Sciences and Therapeutics and Chemical Biology Program and Department of Pharmacology and Toxicology, University of Texas Medical Branch (UTMB), Galveston, Texas 77555, United States;.
Kathryn A. Cunningham, Center for Addiction Sciences and Therapeutics and Chemical Biology Program and Department of Pharmacology and Toxicology, University of Texas Medical Branch (UTMB), Galveston, Texas 77555, United States;.
Jia Zhou, Center for Addiction Sciences and Therapeutics and Chemical Biology Program and Department of Pharmacology and Toxicology, University of Texas Medical Branch (UTMB), Galveston, Texas 77555, United States;.
REFERENCES
- (1).Hynie S 5-hydroxytryptamine (serotonin) receptors–nomenclature and classification of types and subtypes. Cesk. Fysiol 1995, 44, 135–138. [PubMed] [Google Scholar]
- (2).Wold EA; Wild CT; Cunningham KA; Zhou J Targeting the 5-HT2C Receptor in Biological Context and the Current State of 5-HT2C Receptor Ligand Development. Curr. Top. Med. Chem 2019, 19, 1381–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Barnes NM; Ahern GP; Becamel C; Bockaert J; Camilleri M; Chaumont-Dubel S; Claeysen S; Cunningham KA; Fone KC; Gershon M; Di Giovanni G International Union of Basic 956 and Clinical Pharmacology. CX. Classification of Receptors for 5-hydroxytryptamine; 957 Pharmacology and Function. Pharmacol. Rev 2021, 73, 310–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Soogrim V; Ruberto VL; Murrough J; Jha MK Spotlight on Pimavanserin Tartrate and Its Therapeutic Potential in the Treatment of Major Depressive Disorder: The Evidence to Date. Drug Des. Devel. Ther 2021, Volume 15, 151–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Georgescu T; Lyons D; Heisler LK Role of serotonin in body weight, insulin secretion and glycaemic control. J. Neuroendocrinol 2021, 33, No. e12960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Anastasio NC; Stutz SJ; Fink LH; Swinford-Jackson SE; Sears RM; DiLeone RJ; Rice KC; Moeller FG; Cunningham KA Serotonin (5-HT) 5-HT2A Receptor (5-HT2AR):5-HT2CR Imbalance in Medial Prefrontal Cortex Associates with Motor Impulsivity. ACS Chem. Neurosci 2015, 6, 1248–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Zhang G; Stackman RW Jr. The role of serotonin 5-HT2A receptors in memory and cognition. Front. Pharmacol 2015, 6, 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Cunningham KA; Bubar MJ; Anastasio NC The serotonin 5-HT2C receptor in medial prefrontal cortex exerts rheostatic control over the motivational salience of cocaine-associated cues: new observations from preclinical animal research. Neuropsychopharmacology 2010, 35, 2319–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Zhou J; Cunningham KA Positive-allosteric modulation of the 5-HT(2C) receptor: implications for neuropsychopharmacology and neurotherapeutics. Neuropsychopharmacol 2019, 44, 230–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Barnes NM; Sharp T A review of central 5-HT receptors and their function. Neuropharmacology 1999, 38, 1083–1152. [DOI] [PubMed] [Google Scholar]
- (11).Jeffrey Conn P; Christopoulos A; Lindsley CW Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat. Rev. Drug Discovery 2009, 8, 41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Lu S; Li S; Zhang J Harnessing allostery: a novel approach to drug discovery. Med. Res. Rev 2014, 34, 1242–1285. [DOI] [PubMed] [Google Scholar]
- (13).Wild C; Cunningham KA; Zhou J Allosteric Modulation of G Protein-Coupled Receptors: An Emerging Approach of Drug Discovery. Austin. J. Pharmacol. Ther 2014, 2, 1101. [PMC free article] [PubMed] [Google Scholar]
- (14).Wold EA; Zhou J GPCR Allosteric Modulators: Mechanistic Advantages and Therapeutic Applications. Curr. Top. Med. Chem 2018, 18, 2002–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Wold EA; Chen J; Cunningham KA; Zhou J Allosteric Modulation of Class A GPCRs: Targets, Agents, and Emerging Concepts. J. Med. Chem 2019, 62, 88–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Fitzgerald LW; Burn TC; Brown BS; Patterson JP; Corjay MH; Valentine PA; Sun JH; Link JR; Abbaszade I; Hollis JM; Largent BL; Hartig PR; Hollis GF; Meunier PC; Robichaud AJ; Robertson DW Possible role of valvular serotonin 5-HT(2B) receptors in the cardiopathy associated with fenfluramine. Mol. Pharmacol 2000, 57, 75–81. [PubMed] [Google Scholar]
- (17).García-Cárceles J; Decara JM; Vázquez-Villa H; Rodríguez R; Codesido E; Cruces J; Brea J; Loza MI; Alén F; Botta J; McCormick PJ; Ballesteros JA; Benhamú B; Rodríguez de Fonseca F; López-Rodríguez ML A Positive Allosteric Modulator of the Serotonin 5-HT(2C) Receptor for Obesity. J. Med. Chem 2017, 60, 9575–9584. [DOI] [PubMed] [Google Scholar]
- (18).Wild CT; Miszkiel JM; Wold EA; Soto CA; Ding C; Hartley RM; White MA; Anastasio NC; Cunningham KA; Zhou J Design, Synthesis, and Characterization of 4-Undecylpiperidine-2-carboxamides as Positive Allosteric Modulators of the Serotonin (5-HT) 5-HT(2C) Receptor. J. Med. Chem 2019, 62, 288–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Singh K; Sona C; Ojha V; Singh M; Mishra A; Kumar A; Siddiqi MI; Tripathi RP; Yadav PN Identification of dual role of piperazine-linked phenyl cyclopropyl methanone as positive allosteric modulator of 5-HT(2C) and negative allosteric modulator of 5-HT(2B) receptors. Eur. J. Med. Chem 2019, 164, 499–516. [DOI] [PubMed] [Google Scholar]
- (20).Wold EA; Garcia EJ; Wild CT; Miszkiel JM; Soto CA; Chen J; Pazdrak K; Fox RG; Anastasio NC; Cunningham KA; Zhou J Discovery of 4-Phenylpiperidine-2-Carboxamide Analogues as Serotonin 5-HT2C Receptor-Positive Allosteric Modulators with Enhanced Drug-like Properties. J. Med. Chem 2020, 63, 7529–7544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Heng HL; Chee CF; Thy CK; Tee JT; Chin SP; Herr DR; Buckle MJ; Paterson IC; Doughty SW; Abd Rahman N; Chung LY In vitro functional evaluation of isolaureline, dicentrine and glaucine enantiomers at 5-HT(2) and α(1) receptors. Chem. Biol. Drug Des 2019, 93, 132–138. [DOI] [PubMed] [Google Scholar]
- (22).Im WB; Chio CL; Alberts GL; Dinh DM Positive allosteric modulator of the human 5-HT2C receptor. Mol. Pharmacol 2003, 64, 78–84. [DOI] [PubMed] [Google Scholar]
- (23).Ding C; Bremer NM; Smith TD; Seitz PK; Anastasio NC; Cunningham KA; Zhou J Exploration of synthetic approaches and pharmacological evaluation of PNU-69176E and its stereoisomer as 5-HT2C receptor allosteric modulators. ACS Chem. Neurosci 2012, 3, 538–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Cravatt BF; Prospero-Garcia O; Siuzdak G; Gilula NB; Henriksen SJ; Boger DL; Lerner RA Chemical characterization of a family of brain lipids that induce sleep. Science 1995, 268, 1506–1509. [DOI] [PubMed] [Google Scholar]
- (25).Arafat ES; Trimble JW; Andersen RN; Dass C; Desiderio DM Identification of fatty acid amides in human plasma. Life Sci 1989, 45, 1679–1687. [DOI] [PubMed] [Google Scholar]
- (26).Hedlund PB; Danielson PE; Thomas EA; Slanina K; Carson MJ; Sutcliffe JG No hypothermic response to serotonin in 5-HT7 receptor knockout mice. Proc. Natl. Acad. Sci. U. S. A 2003, 100, 1375–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Soria-Gómez E; Márquez-Diosdado MI; Montes-Rodríguez CJ; Estrada-González V; Prospéro-García O Oleamide administered into the nucleus accumbens shell regulates feeding behaviour via CB1 and 5-HT2C receptors. Int. J. Neuropsychopharmacol 2010, 13, 1247–1254. [DOI] [PubMed] [Google Scholar]
- (28).Prospéro-García O; Amancio-Belmont O; Becerril Meléndez AL; Ruiz-Contreras AE; Méndez-Díaz M Endocannabinoids and sleep. Neurosci. Biobehav. Rev 2016, 71, 671–679. [DOI] [PubMed] [Google Scholar]
- (29).Méndez-Díaz M; Amancio-Belmont O; Estrada-González V; Ruiz-Contreras AE; Prospéro-García O CB1R mediates oleamide’s reward while 5HT2cR mediates aversion in the nucleus accumbens shell of rats. Neurosci. Lett 2019, 706, 189–193. [DOI] [PubMed] [Google Scholar]
- (30).Thomas EA; Carson MJ; Neal MJ; Sutcliffe G Unique allosteric regulation of 5-hydroxytryptamine receptor-mediated signal transduction by oleamide. Proc. Natl. Acad. Sci. U. S. A 1997, 94, 14115–14119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Boger DL; Patterson JE; Jin Q Structural requirements for 5-HT2A and 5-HT1A serotonin receptor potentiation by the biologically active lipid oleamide. Proc. Natl. Acad. Sci. U. S. A 1998, 95, 4102–4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Cheer JF; Cadogan A-K; Marsden CA; Fone KCF; Kendall DA Modification of 5-HT2 receptor mediated behaviour in the rat by oleamide and the role of cannabinoid receptors. Neuropharmacology 1999, 38, 533–541. [DOI] [PubMed] [Google Scholar]
- (33).Hedlund PB; Carson MJ; Sutcliffe JG; Thomas EA Allosteric regulation by oleamide of the binding properties of 5-hydroxytryptamine7 receptors. Biochem. Pharmacol 1999, 58, 1807–1813. [DOI] [PubMed] [Google Scholar]
- (34).Fedorova I; Hashimoto A; Fecik RA; Hedrick MP; Hanus LO; Boger DL; Rice KC; Basile AS Behavioral Evidence for the Interaction of Oleamide with Multiple Neurotransmitter Systems. J. Pharmacol. Exp. Ther 2001, 299, 332–342. [PubMed] [Google Scholar]
- (35).Mueller GP; Driscoll WJ Biosynthesis of oleamide. Vitam. Horm 2009, 81, 55–78. [DOI] [PubMed] [Google Scholar]
- (36).Millan MJ; Marin P; Bockaert J Mannoury la Cour, C. Signaling at G-protein-coupled serotonin receptors: recent advances and future research directions. Trends Pharmacol. Sci 2008, 29, 454–464. [DOI] [PubMed] [Google Scholar]
- (37).Peng Y; McCorvy JD; Harpsøe K; Lansu K; Yuan S; Popov P; Qu L; Pu M; Che T; Nikolajsen LF; Huang XP; Wu Y; Shen L; Bjørn-Yoshimoto WE; Ding K; Wacker D; Han GW; Cheng J; Katritch V; Jensen AA; Hanson MA; Zhao S; Gloriam DE; Roth BL; Stevens RC; Liu ZJ 5-HT(2C) Receptor Structures Reveal the Structural Basis of GPCR Polypharmacology. Cell 2018, 172, 719–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Kim K; Che T; Panova O; DiBerto JF; Lyu J; Krumm BE; Wacker D; Robertson MJ; Seven AB; Nichols DE; et al. Structure of a Hallucinogen-Activated Gq-Coupled 5-HT2A Serotonin Receptor. Cell 2020, 182, 1574–1588.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Lu C; Wu C; Ghoreishi D; Chen W; Wang L; Damm W; Ross GA; Dahlgren MK; Russell E; Von Bargen CD; et al. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory Comput 2021, 17, 4291–4300. [DOI] [PubMed] [Google Scholar]
- (40).Besnard J; Ruda GF; Setola V; Abecassis K; Rodriguiz RM; Huang X-P; Norval S; Sassano MF; Shin AI; Webster LA; et al. Automated design of ligands to polypharmacological profiles. Nature 2012, 492, 215–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Garrido A; Lepailleur A; Mignani SM; Dallemagne P; Rochais C hERG toxicity assessment: Useful guidelines for drug design. Eur. J. Med. Chem 2020, 195, No. 112290. [DOI] [PubMed] [Google Scholar]
- (42).Wager TT; Hou X; Verhoest PR; Villalobos A Central Nervous System Multiparameter Optimization Desirability: Application in Drug Discovery. ACS Chem. Neurosci 2016, 7, 767–775. [DOI] [PubMed] [Google Scholar]
- (43).Wager TT; Hou X; Verhoest PR; Villalobos A Moving beyond Rules: The Development of a Central Nervous System Multiparameter Optimization (CNS MPO) Approach To Enable Alignment of Druglike Properties. ACS Chem. Neurosci 2010, 1, 435–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Banerjee P; Eckert AO; Schrey AK; Preissner R ProTox-II: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res 2018, 46, 257–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Slosky LM; Pires A; Bai Y; Clark NB; Hauser ER; Gross JD; Porkka F; Zhou Y; Chen X; Pogorelov VM; et al. Establishment of multi-stage intravenous self-administration paradigms in mice. Sci. Rep 2022, 12, 21422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Mitchell JM; Cunningham CL; Mark GP Locomotor activity predicts acquisition of self-administration behavior but not cocaine intake. Behav. Neurosci 2005, 119, 464–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).White DA; Kalinichev M; Holtzman SG Locomotor response to novelty as a predictor of reactivity to aversive stimuli in the rat. Brain Res 2007, 1149, 141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Sell SL; Dillon AM; Cunningham KA; Thomas ML Estrous cycle influence on individual differences in the response to novelty and cocaine in female rats. Behav. Brain Res 2005, 161, 69–74. [DOI] [PubMed] [Google Scholar]
- (49).Piazza PV; Deminière JM; Le Moal M; Simon H Factors that predict individual vulnerability to amphetamine self-administration. Science 1989, 245, 1511–1513. [DOI] [PubMed] [Google Scholar]
- (50).Halberstadt AL; van der Heijden I; Ruderman MA; Risbrough VB; Gingrich JA; Geyer MA; Powell SB 5-HT(2A) and 5-HT(2C) receptors exert opposing effects on locomotor activity in mice. Neuropsychopharmacol 2009, 34, 1958–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Filip M; Bubar MJ; Cunningham KA Contribution of serotonin (5-hydroxytryptamine; 5-HT) 5-HT2 receptor subtypes to the hyperlocomotor effects of cocaine: acute and chronic pharmacological analyses. J. Pharmacol. Exp. Ther 2004, 310, 1246–1254. [DOI] [PubMed] [Google Scholar]
- (52).Bishop C; Tessmer JL; Ullrich T; Rice KC; Walker PD Serotonin 5-HT2A receptors underlie increased motor behaviors induced in dopamine-depleted rats by intrastriatal 5-HT2A/2C agonism. J. Pharmacol. Exp. Ther 2004, 310, 687–694. [DOI] [PubMed] [Google Scholar]
- (53).Soto CA; Du HC; Fox RG; Yang T; Hooson J; Anastasio NC; Gilbertson SR; Cunningham KA In Vivo and In Vitro Analyses of Novel Peptidomimetic Disruptors for the Serotonin 5-HT(2C) Receptor Interaction With Phosphatase and Tensin Homolog. Front. Pharmacol 2019, 10, 907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Cunningham KA; Fox RG; Anastasio NC; Bubar MJ; Stutz SJ; Moeller FG; Gilbertson SR; Rosenzweig-Lipson S Selective serotonin 5-HT(2C) receptor activation suppresses the reinforcing efficacy of cocaine and sucrose but differentially affects the incentive-salience value of cocaine- vs. sucrose-associated cues. Neuropharmacology 2011, 61, 513–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Higgins GA; Ouagazzal AM; Grottick AJ Influence of the 5-HT(2C) receptor antagonist SB242,084 on behaviour produced by the 5-HT(2) agonist Ro60–0175 and the indirect 5-HT agonist dexfenfluramine. Br. J. Pharmacol 2001, 133, 459–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Higgins GA; Enderlin M; Haman M; Fletcher PJ The 5-HT2A receptor antagonist M100,907 attenuates motor and ‘impulsive-type’ behaviours produced by NMDA receptor antagonism. Psychopharmacol 2003, 170, 309–319. [DOI] [PubMed] [Google Scholar]
- (57).Fantegrossi WE; Simoneau J; Cohen MS; Zimmerman SM; Henson CM; Rice KC; Woods JH Interaction of 5-HT2A and 5-HT2C receptors in R(−)-2,5-dimethoxy-4-iodoamphetamine-elicited head twitch behavior in mice. J. Pharmacol. Exp. Ther 2010, 335, 728–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Vickers SP; Easton N; Malcolm CS; Allen NH; Porter RH; Bickerdike MJ; Kennett GA Modulation of 5-HT(2A) receptor-mediated head-twitch behaviour in the rat by 5-HT(2C) receptor agonists. Pharmacol. Biochem. Behav 2001, 69, 643–652. [DOI] [PubMed] [Google Scholar]
- (59).Felsing DE; Anastasio NC; Miszkiel JM; Gilbertson SR; Allen JA; Cunningham KA Biophysical validation of serotonin 5-HT2A and 5-HT2C receptor interaction. PLoS One 2018, 13, No. e0203137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Price AE; Sholler DJ; Stutz SJ; Anastasio NC; Cunningham KA Endogenous Serotonin 5-HT(2A) and 5-HT(2C) Receptors Associate in the Medial Prefrontal Cortex. ACS Chem. Neurosci 2019, 10, 3241–3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Moutkine I; Quentin E; Guiard BP; Maroteaux L; Doly S Heterodimers of serotonin receptor subtypes 2 are driven by 5-HT(2C) protomers. J. Biol. Chem 2017, 292, 6352–6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Roth BL; Gumpper RH Psychedelics as Transformative Therapeutics. Am. J. Psychiatry 2023, 180, 340–347. [DOI] [PubMed] [Google Scholar]
- (63).Vargas MV; Meyer R; Avanes AA; Rus M; Olson DE Psychedelics and Other Psychoplastogens for Treating Mental Illness. Front. Psychiatry 2021, 12, No. 727117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Chagraoui A; Thibaut F; Skiba M; Thuillez C; Bourin M 5-HT2C receptors in psychiatric disorders: A review. Prog. Neuropsychopharmacol. Biol. Psychiatry 2016, 66, 120–135. [DOI] [PubMed] [Google Scholar]
- (65).Howell LL; Cunningham KA Serotonin 5-HT2 receptor interactions with dopamine function: implications for therapeutics in cocaine use disorder. Pharmacol. Rev 2015, 67, 176–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Kehne JH; Baron BM; Carr AA; Chaney SF; Elands J; Feldman DJ; Frank RA; van Giersbergen PL; McCloskey TC; Johnson MP; McCarty D; Poirot M; Senyah Y; Siegel BW; Widmaier C Preclinical characterization of the potential of the putative atypical antipsychotic MDL 100,907 as a potent 5-HT2A antagonist with a favorable CNS safety profile. J. Pharmacol. Exp. Ther 1996, 277, 968–981. [PubMed] [Google Scholar]
- (67).Kennett GA; Wood MD; Bright F; Trail B; Riley G; Holland V; Avenell KY; Stean T; Upton N; Bromidge S; Forbes IT SB 242084, a selective and brain penetrant 5-HT2C receptor antagonist. Neuropharmacology 1997, 36, 609–620. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.