

SUMMARY
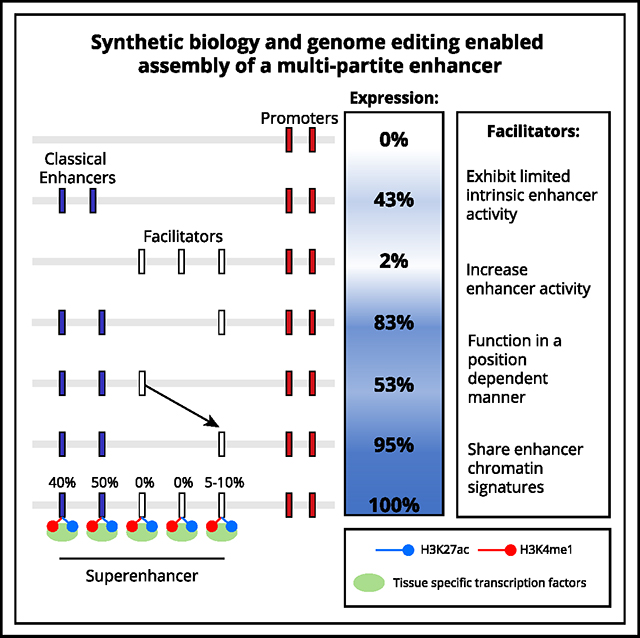
Super-enhancers are compound regulatory elements that control expression of key cell identity genes. They recruit high levels of tissue-specific transcription factors and co-activators such as the Mediator complex and contact target gene promoters with high frequency. Most super-enhancers contain multiple constituent regulatory elements, but it is unclear whether these elements have distinct roles in activating target gene expression. Here, by rebuilding the endogenous multipartite α-globin super-enhancer, we show that it contains bioinformatically equivalent but functionally distinct element types: classical enhancers and facilitator elements. Facilitators have no intrinsic enhancer activity, yet in their absence, classical enhancers are unable to fully upregulate their target genes. Without facilitators, classical enhancers exhibit reduced Mediator recruitment, enhancer RNA transcription, and enhancer-promoter interactions. Facilitators are interchangeable but display functional hierarchy based on their position within a multipartite enhancer. Facilitators thus play an important role in potentiating the activity of classical enhancers and ensuring robust activation of target genes.
In brief
De novo construction of a multipartite enhancer unveils a new class of regulatory element referred to as “facilitators.” Facilitators lack intrinsic enhancer activity but enhance the activity of classical enhancers in a position-dependent manner.
Graphical abstract
INTRODUCTION
Enhancers are regions of DNA that recruit transcription factors (TFs) and activate expression of target genes in a cell-type-specific manner.1–3 Generally, enhancers are classified bioinformatically based on chromatin accessibility, TF occupancy, and enrichment for particular histone modifications including H3K4Me1 and H3K27Ac. The term super-enhancer (SE) has been coined to describe clusters of elements bearing the bioinformatic signature of enhancers, which are enriched for particularly high levels of H3K27Ac, TF recruitment, and recruitment of transcriptional co-activators such as the Mediator complex.4 SEs are often regulators of cell identity genes and are frequently mutated in association with complex traits and genetic diseases.5–9
Despite extensive analysis, it remains unclear whether SEs are merely groups of independent classical enhancers or whether they are cooperatives containing functionally distinct element types.10–14 SEs may not all be mechanistically the same. This has become a highly debatable topic as the genetic dissection of SEs is challenging, given that many studied loci regulate genes that control complex transcriptional and epigenetic programs. Disruption of such pathways makes it difficult to separate changes in SE-regulated gene expression from associated changes in cell lineage and differentiation. Most previous studies analyzing SEs have drawn conclusions from deleting just one or two constituent elements, and many studies have relied on artificial reporter-based assays divorced from their functionally relevant chromatin contexts.15 To date, a number of studies have dissected SEs16–22 with conflicting conclusions. More rigorous analysis is essential to determine their true nature.
The mouse α-globin SE (α-SE) is made up of five constituent elements (R1, R2, R3, Rm, and R4); it lies together with the duplicated α-globin genes in a well-defined 65 kb sub-topologically associating domain (sub-TAD) and upregulates α-globin gene expression in terminally differentiating red blood cells. The α-SE is an ideal model for detailed genetic analysis, as its perturbation has no effect on cell identity or differentiation.17 Previous dissection of the endogenous α-SE, removing each of its constituents individually or in selective pairs, suggested that it is a cluster of five independent elements: two classical enhancers (R1 and R2) capable of significantly upregulating gene expression, and three inactive elements (R3, Rm, and R4), albeit conserved over ~70 million years of evolution23, bearing the bioinformatic signature of enhancers but with little/no ability to activate transcription in all previously defined assays of enhancer function.17 These deletions shed light on the necessity of each individual element within the otherwise intact SE, but they did not report on each element’s sufficiency or the functional relationships between the five constituent elements.
Here, we engineered the endogenous mouse α-SE to enable us to rebuild the native α-SE from the bottom up, generating all informative element combinations. Coupling efficient whole locus genome editing24,25 with a recently developed embryoid body (EB)-based in vitro differentiation of mouse embryonic stem cells (mESCs) and erythroid purification system26 has allowed us to unpick the complex relationships between the constituents of a well-characterized model of a mammalian SE. We show in EB-derived erythroid cells that the α-SE is comprised of two functionally distinct element types: classical enhancers R1 and R2 and facilitators R3, Rm, and R4. The three facilitators have little or no intrinsic enhancer activity, but in their absence, the two classical enhancers are unable to effectively upregulate α-globin expression. Furthermore, we present an in vivo mouse model lacking all but the strongest classical enhancer at the endogenous α-globin locus to demonstrate that without facilitator elements, classical enhancers cannot recruit high levels of Mediator, transcribe high levels of enhancer RNA, or interact with their target gene promoters with high frequency. Importantly, the relative effect of facilitators appears to depend on their position within their composite SE. Review of diverse, albeit incomplete, previous analyses of SEs16,17,19,20,22 suggest that facilitators, as described here, may be relatively common elements in multipartite enhancers. We propose that facilitators, such as R3, Rm, and R4, are a novel form of regulatory element important for potentiating the activity of classical enhancers.
RESULTS
Engineering the mouse α-globin cluster as a test bed for elements of the SE
Engineering several independent mutations in a single allele using conventional editing is time-consuming and complicated, requiring multiple steps to ensure that all mutations are precise and present in cis to one another. Multiple editing steps in a single cell line can also introduce “off-target” effects, which may compromise the ability of the model mESC to divide and differentiate normally into erythroid cells or to generate a subsequent mouse model.
To overcome these issues, we used a recently developed protocol for de novo assembly of large DNA fragments (Figure S1A)27 to design and synthesize 86 kb alleles containing either the wild-type (WT) α-globin sub-TAD or individually designed variants of the allele. First, we designed an allele in which only R2 remained, with R1, R3, Rm, and R4 deleted (R2-only). These two synthetic alleles (WT and R2-only) were each integrated using recombinase-mediated genomic replacement (RMGR)25 into mESCs in which one copy of the entire α-globin locus had already been deleted (Figure 1A). The resultant hemizygous cells therefore contained only one (synthetically derived) α-globin allele, which allowed genomic analysis to be conducted specifically on each newly synthesized locus. A third genetic model was made by deleting the remaining R2 element from R2-only mESCs using a CRISPR-Cas9 approach, creating an enhancerless model in which all elements of the α-SE had been removed from the locus (Δα-SE) (Figure 1A). We then used an EB-based in vitro differentiation of mESCs and an erythroid purification system26 to analyze hemizygous WT, Δα-SE, and R2-only erythroid cells.
Figure 1. Generation of an enhancerless (Δα-SE) in vitro mouse model and an R2-only in vivo mouse model to test the sufficiency of the R2 enhancer element.
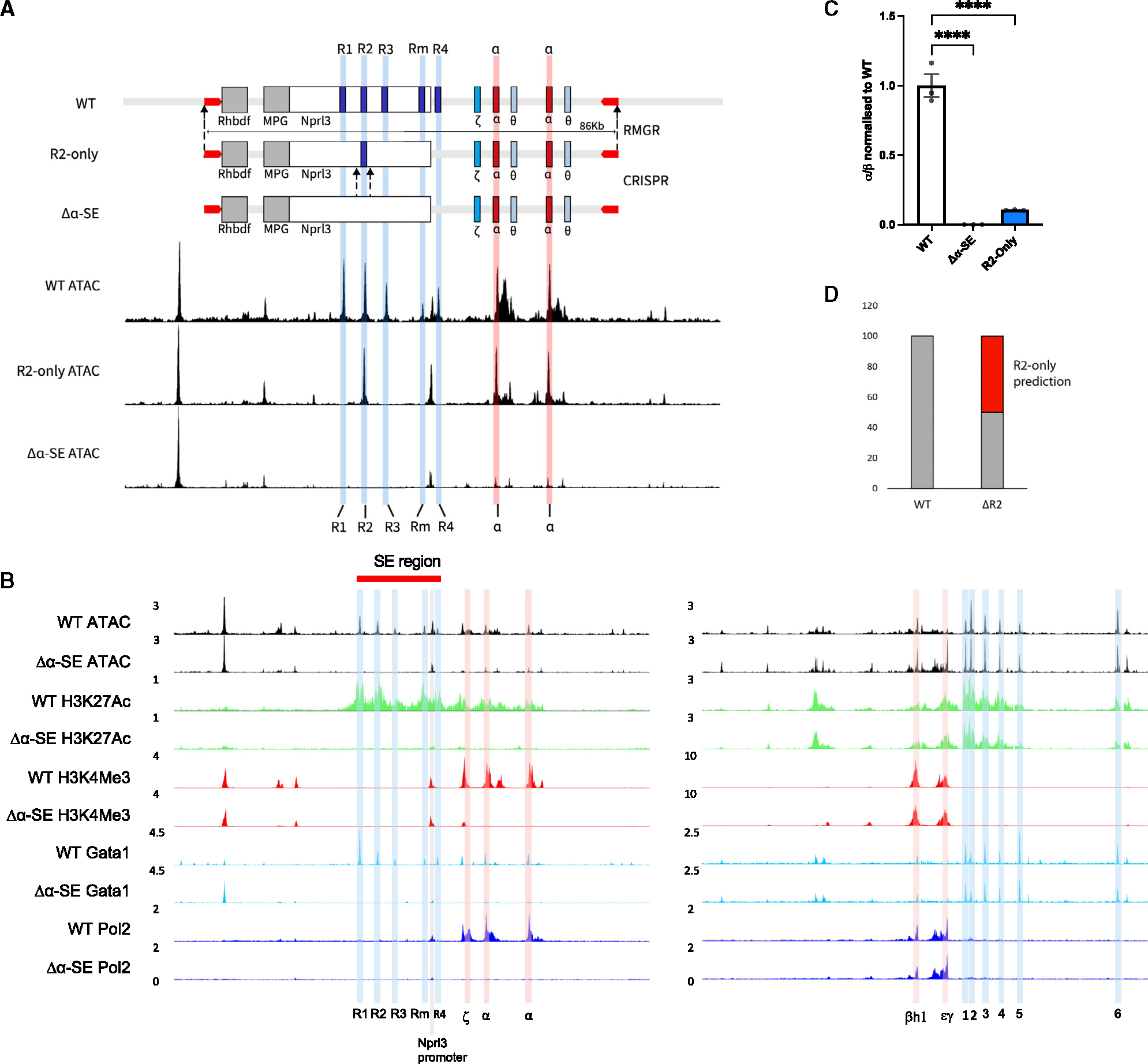
(A) A graphical representation of the design of the Δα-SE and R2-only α-globin loci. R2-only locus was synthesized, assembled into a bacterial artificial chromosome (BAC), and delivered into the wild-type (WT) α-globin locus through recombination-mediated genomic replacement (RMGR; STAR Methods). Δα-SE was generated by CRISPR deletion of R2 from the R2-only hemizygous mESCs. UCSC tracks represent cpm (count per million) normalized ATAC-seq data from WT (top), R2-only FL erythroid cells (middle), and Δα-SE EB-derived erythroid cells (bottom).
(B) ATAC-seq from WT and Δα-SE EB-derived erythroid cells (top) and ChIPmentation for H3K27Ac, H3K4Me3, GATA1, and Pol II (bottom; n ≥ 2). Left, α-globin locus with highlighted regulatory elements (R1–R4, blue) and promoters (Hba-x or ζ corresponding to the embryonic α-globin and Hba-a1, Hba-a2, or α corresponding to the adult α-globin expressed in EB-derived primitive erythroid cells, red; Nprl3 promoter, pink); right, β-globin locus with highlighted regulatory elements (HS1–HS6, blue) and promoters (Hbb-h1 or βh1 and Hbb-y or εγ corresponding to the embryonic β-globin expressed in EB-derived primitive erythroid cells, red). All tracks are cpm normalized.
(C) α-globin gene expression in WT, Δα-SE, and R2-only EB-derived erythroid cells (n ≥ 3) assayed by RT-qPCR. Expression normalized to β-globin and displayed as a proportion of WT expression. Dots, biological replicates; error bars, Standard Error. Statistical analysis was performed using one-way ANOVA with a Tukey post-hoc test: ****p ≤ 0.00001.
(D) Prediction of R2-only α-globin gene expression, calculated by subtracting ΔR2 α-globin expression from WT and the observed R2-only expression levels in (C).
See also Figures S1 and S3.
Gene expression in the absence of all enhancers
Upon deletion of all five elements of the α-SE (Δα-SE), EB-derived erythroid cells display an almost complete loss of α-globin expression (>99.9% loss), and all chromatin marks normally associated with the SE elements are lost (Figures 1B, 1C, and S3A). Very small ATAC-seq peaks persist over the α-globin promoters, but they are no longer bound by GATA1 or Pol II nor marked by H3K4Me3 (Figure 1B). In the absence of the enhancers, H3K27Ac is almost completely lost from the entire locus, with only a very small peak associated with the embryonic ζ-globin gene remaining (Figure 1B). In summary, the Δα-SE model provides a well-characterized baseline for studying the role of the SE elements individually and in combination during erythropoiesis.
A single enhancer-driven α-globin locus (R2-only) is associated with severe downregulation of α-globin expression and embryonic lethality
To determine the individual contribution of the R2 enhancer element to α-globin expression, we compared the structure and function of the enhancerless locus (Δα-SE) with the R2-only locus, in which R2 is present at its normal position in the absence of R1, R3, Rm, and R4. In this case, all enhancer activity comes from R2 alone (Figure 1A). Previously, we showed that deleting R2 from the otherwise intact α-SE (ΔR2) causes a 50% reduction in α-globin transcription compared to WT.17 We therefore predicted that R2-only erythroid cells would produce 50% α-globin expression (Figure 1D). Unexpectedly, EB-derived R2-only cells expressed only 10% α-globin, 5-fold less than predicted (Figure 1C).
To investigate the R2-only phenotype further, we generated an R2-only mouse model in which the endogenous α-SE is replaced with an SE containing R2 but not R1, R3, Rm, or R4. Previous ΔR2 mice displayed a 50% reduction in α-globin expression and no significant changes in red cell parameters;17 we therefore predicted that R2-only mice would present with a similar gene expression and hematological phenotype (Figure 1D). In contrast to ΔR2 mice, R2-only mice were largely non-viable. In 16 heterozygote crosses harvested at embryonic days (E)9.5, E10.5, E12.5, E14.5, and E17.5, the Mendelian ratio of WT:heterozygotes:homozygotes was as expected (Table S1); however, homozygous R2-only embryos were visibly smaller and paler than their WT and heterozygous littermates (Figure 2A). We obtained only one surviving homozygote with anemia and severe splenomegaly, which died prematurely at 7 weeks.
Figure 2. R2-only mice exhibit severely attenuated α-globin expression and are non-viable.
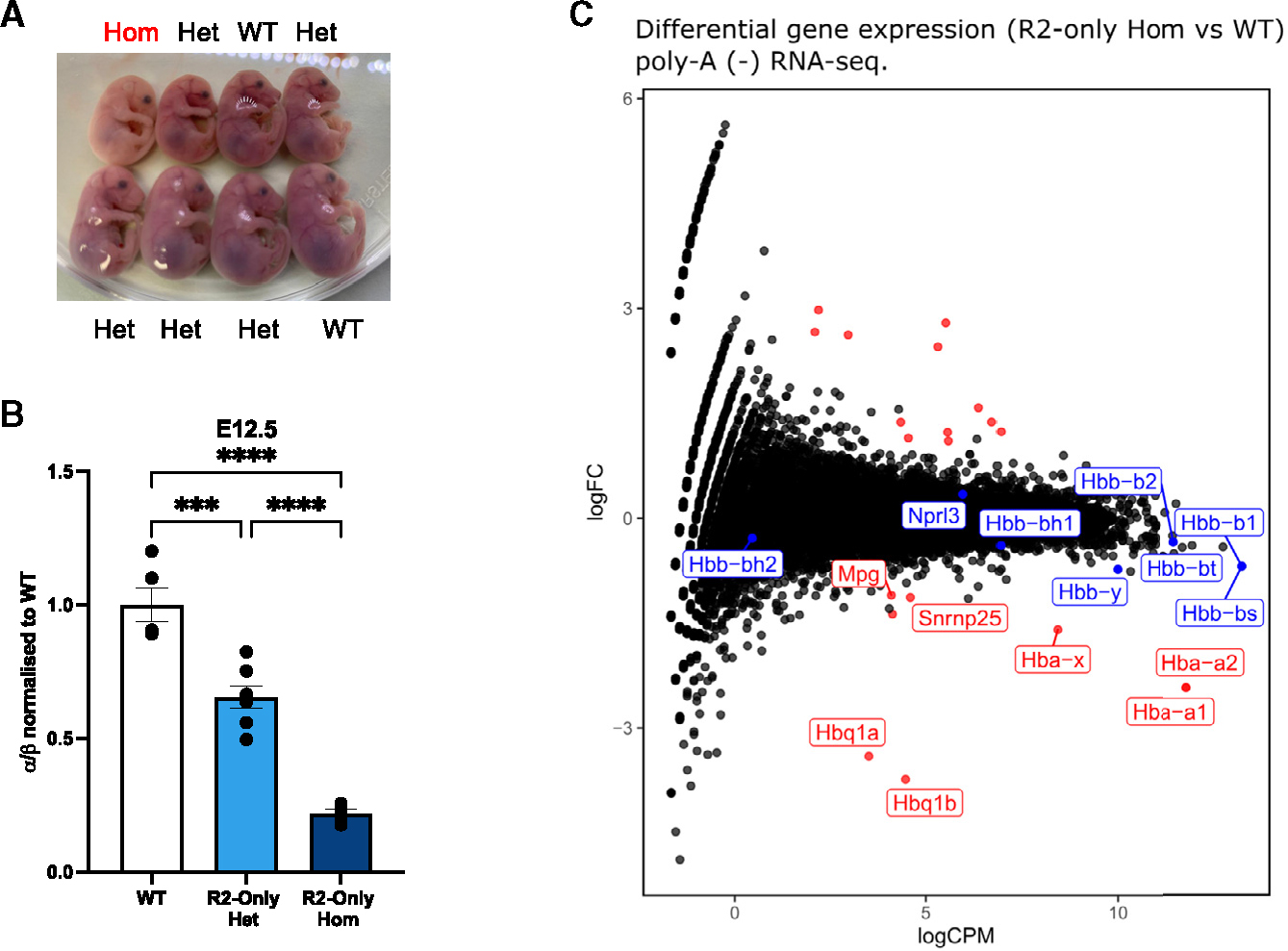
(A) Representative image of R2-only litter from heterozygote crossings. Pregnant female was sacrificed at embryonic day E17.5 and fetuses extracted. Hom, homozygotes; Het, heterozygotes.
(B) RT-qPCR comparing α-globin expression in FL erythroid cells from WT, R2-only heterozygous, and R2-only homozygous E12.5 littermates. Expression normalized to β-globin and displayed as a proportion of WT expression. Dots, biological replicates; error bars, Standard Error. Statistical analysis was performed using one-way ANOVA with a Tukey post-hoc test: ***p ≤ 0.0001, ****p ≤ 0.00001.
(C) Poly-A minus RNA-seq comparing gene expression in FL erythroid cells from WT (n = 2) and R2-only homozygous (n = 3) littermates. Red dots, differentially expressed (>2-fold); blue dots, informative erythroid genes (non-differentially expressed); black dots, no statistically significant change in expression (<2-fold). As well as reductions in transcription of the α-like globins, expression of Snrnp25 and Mpg (two genes lying upstream of the 5′ boundary of the α-globin sub-TAD) was reduced.
See also Figures S2 and S3 and Table S1.
The severe anemia in R2-only homozygotes suggested that removing R1, R3, Rm, and R4 had compromised α-globin expression more than expected. We first wanted to exclude that any phenotype we observe is a reflection of impaired erythropoiesis and a readout of non-equivalent cell populations. We assessed the differentiation state of the erythroid populations derived from WT, R2-only heterozygotes, and R2-only homozygotes by isolating E12.5 fetal livers (FLs) (the definitive erythroid compartment at this developmental stage) and performing fluorescence-activated cell sorting (FACS) analysis using standard erythroid immunophenotyping surface markers (CD71 and Ter119); this showed that the erythroid populations are not affected by the R2-only α-globin genotype and that the populations are comparable (Figure S2A). We further excluded impaired erythropoiesis by a more in-depth investigation of the genome-wide open chromatin; we performed a principal component analysis (PCA) on ATAC-seq peaks generated from WT- and R2-only-derived FL erythroid cells and included other FL erythroid cell populations, spleen-derived erythroid cells, and mESCs. The PCA confirmed that R2-only FL erythroid cells were developmentally equivalent to all the other WT FL cell populations (Figure S2B). To assess α-globin transcription, we performed RT-qPCR on the same FL erythroid cells. R2-only erythroid cells expressed only 15% α-globin compared to WT littermates, rather than the predicted 50% (Figure 2B). A similar downregulation of α-globin expression was observed at all developmental stages from E9.5–E17.5 (Figure S3B). Poly-A minus RNA-seq on FL erythroid cells from three R2-only and two WT littermates confirmed the RT-qPCR results and showed that expression of various erythroid and developmental markers was unaffected in R2-only FL erythroid cells (Figure 2C). Interestingly, the level of steady-state RNA from the Nprl3 gene, in which 4 of the α-SE elements are embedded, appears to be unaffected in the R2-only erythroid cells. However, analysis of nascent expression from its promoter may be influenced by the α-SE (Figures 2C and S3C and unpublished observation). Downregulation of two genes upstream of the α-globin locus was also observed (Figure 2C); Snrnp25 and Mpg genes are housekeeping genes and only seem to be affected in the erythroid cells, linking their downregulation in the R2-only homozygote mice to the defective α-globin erythroid-specific enhancer cluster (Figure S3C).
The R2 element retains its enhancer identity in the absence of other SE elements but fails to recruit high levels of co-activators
To determine if R2 retains characteristics generally associated with an active enhancer, after removing R1, R3, Rm, and R4, we examined the chromatin accessibility and epigenetic status of the R2-only α-globin locus. ATAC-seq revealed that R2 and both α-globin promoters remain accessible in R2-only FL erythroid cells and that R1, R3, Rm and R4 were by far the most differentially accessible regions genome-wide when R2-only FL erythroid cell data were compared to those of WT (Figure 3A). ChIPmentation experiments in the R2-only model using antibodies against H3K4Me1, H3K4Me3, and H3K27Ac showed that R2 and the α-globin promoters are marked by active enhancer- (H3K4Me1 and H3K27Ac) and promoter-associated (H3K4Me3 and H3K27Ac) histone modifications, respectively, albeit to a severely compromised level compared to those in WT cells (Figure S4). Furthermore, enhancers recruit high levels of tissue-specific TFs. Erythroid-specific TFs (e.g., Gata1 and Nf-e2) occupied both R2 and the α-globin promoters to an equivalent degree in R2-only and WT FL erythroid cells (Figure 3B). We conclude that in the absence of other elements in the α-SE, R2 retains its identity as an enhancer, recruiting transcription factors and creating a region of open chromatin.
Figure 3. R2 retains the hallmarks of an active enhancer in R2-only erythroid cells, but co-activator recruitment is significantly reduced.
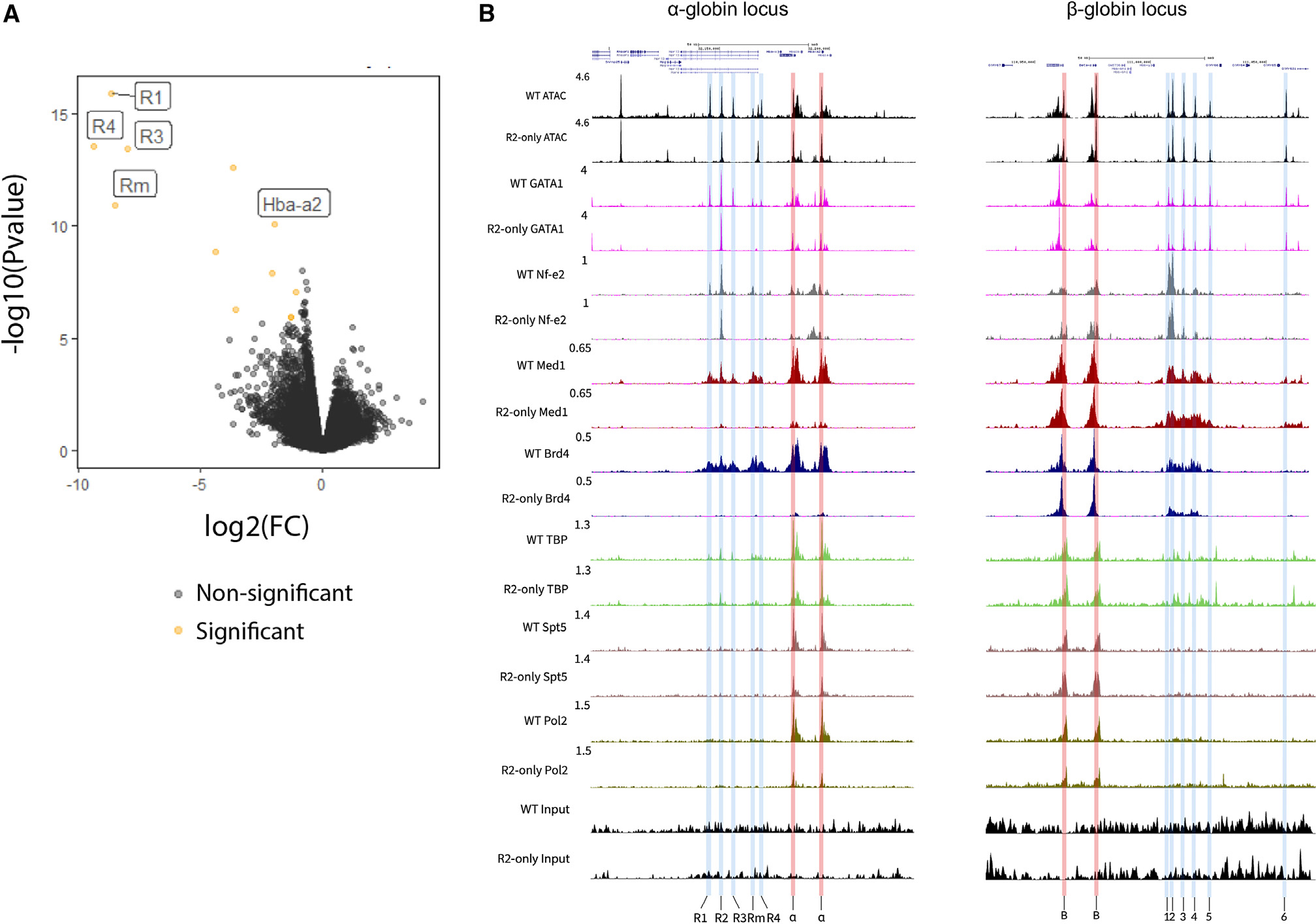
(A) Genome-wide differential accessibility assessed through ATAC-seq in FL erythroid cells from WT (n = 3) and R2-only homozygous (n = 3) littermates. Yellow, differentially accessible; black, non-significant.
(B) UCSC tracks represents data from WT (n = 3) and R2-only homozygous (n = 3) FL erythroid cells at the α-globin locus, starting at the top and continuing down: ATAC-seq (black); ChIPmentation for GATA1; Nf-e2; Med1; Brd4; TBP (n = 2); Spt5 (n = 2); Pol II; input ChIPmentation in WT (n = 2) and R2-only homozygous (n = 2) FL erythroid cells. Tracks = merged biological replicates. All tracks are cpm normalized. Left, α-globin locus with highlighted regulatory elements (R1–R4 in blue) and promoters (corresponding to Hba-a1 and Hba-a2, the adult α-globin expressed in definitive FL erythroid cells, red), coordinates = chr11: 32,090,000–32,235,000 (mm9); right, β-globin locus with highlighted regulatory elements (HS1–HS6 in blue) and promoters (corresponding to Hbb-b1 and Hbb-b2, the adult β-globin expressed in definitive FL erythroid cells, pink), shown as an unaffected control, coordinates = chr7: 110,936,000–111,070,000 (mm9).
See also Figure S4.
SEs are, in part, defined by the extent to which they recruit high levels of transcriptional co-activators.4 To investigate R2’s capacity to recruit co-activators in the absence of the other four α-SE constituents, we performed ChIPmentation with antibodies against Med1, a member of the Mediator complex, and bromodomain-containing protein 4 (Brd4), a transcriptional and epigenetic regulator. WT FL erythroid cells recruit high levels of Med1 and Brd4 to the α-SE and α-globin promoters, but in R2-only FL erythroid cells, recruitment of both factors was severely reduced (Figure 3B). Mediator plays a central role in Pol II recruitment and stability at the promoter; therefore, we asked whether reduced Med1 occupancy at the α-globin promoters correlates with changes to the formation of the preinitiation complex. We performed ChIPmentation experiments with antibodies against TATA binding protein (TBP) and Pol II. There was no change in TBP recruitment in R2-only erythroid cells, consistent with its autonomous DNA-binding activity; however, there was a substantial reduction in Pol II occupancy at both α-globin promoters (Figure 3B).
R2 eRNA transcription is reduced in the absence of R1, R3, Rm, and R4
Enhancers are actively transcribed, producing bidirectional transcripts of varying lengths.28 Enhancer transcription appears to be related to enhancer activity; whether enhancer RNAs (eRNAs) have any function, or whether the relationship between transcription and enhancer strength is merely correlative, remains unclear.29 To explore eRNA transcription from the R2 element, we analyzed the poly-A minus RNA-seq data. Because R2 is located in an intron of the Nprl3 gene, which is active in erythroid cells and transcribed on the negative strand, we had to restrict our investigation of R2 eRNA transcription to the positive strand. In WT FL erythroid cells, we found clear transcripts originating from all five α-SE constituents (Figure S5), whereas in R2-only cells, only the R2 enhancer showed any evidence of transcription.
To compare R2 eRNA transcription in WT and R2-only cells quantitatively, we performed a “virtual qPCR,” normalizing levels of R2 eRNA to eRNA originating from the β-globin HS2 enhancer, a member of the β-globin locus control region (LCR) and a well-characterized SE. This revealed a ~3-fold reduction in R2 eRNA transcription in R2-only cells compared to WT (Figure 4A).
Figure 4. R2 eRNA transcription and R2-α-promoter interaction frequency are reduced in R2-only erythroid cells.
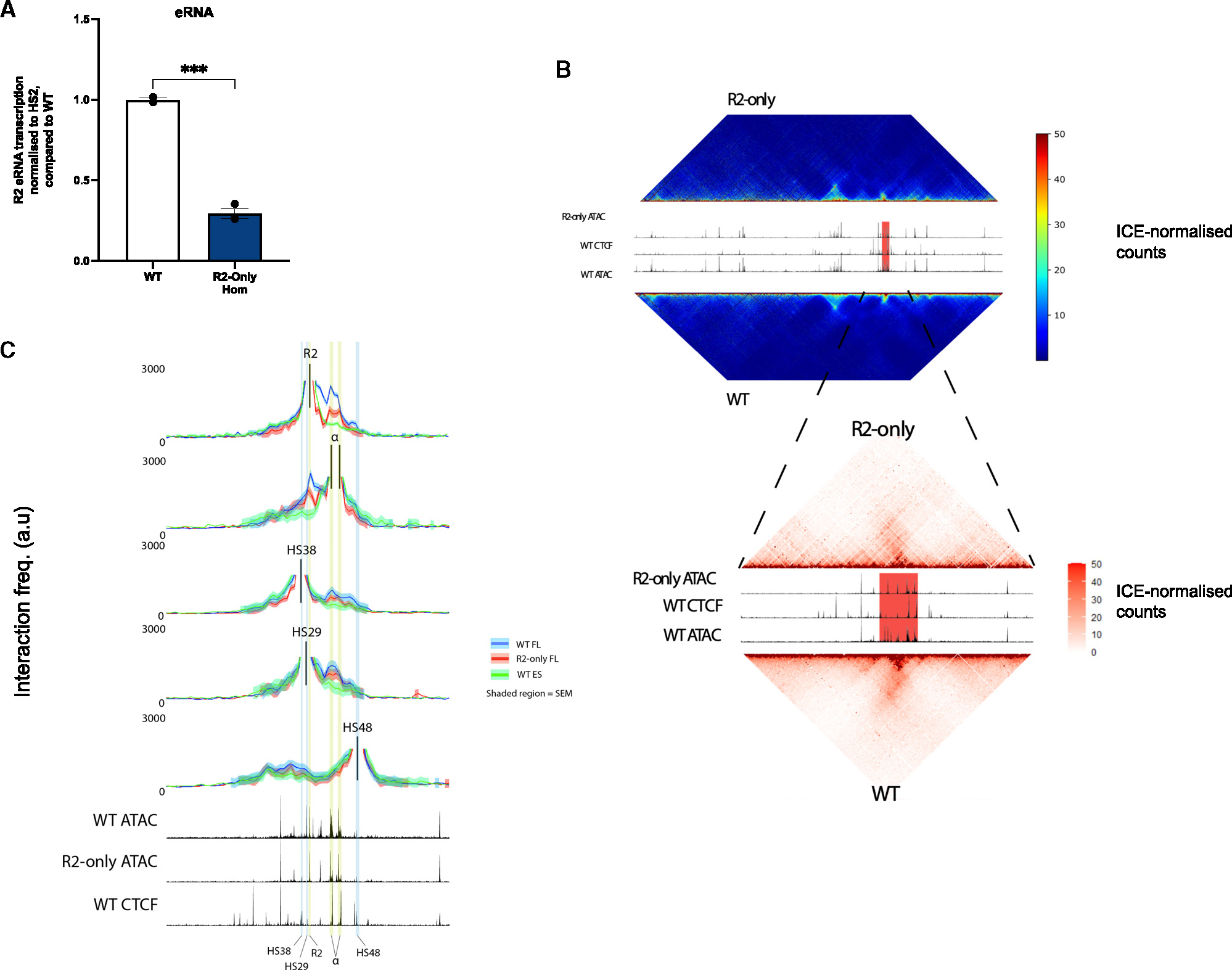
(A) Virtual qPCR conducted on poly-A minus RNA-seq, comparing R2 eRNA expression in WT (n = 2) and R2-only homozygous (n = 3) FL erythroid cells. Transcripts originating from the R2 enhancer were normalized to those originating from the HS2 enhancer within the β-globin LCR and displayed as a proportion of WT expression. Dots, biological replicates; error bars, Standard Error. Statistical analysis was performed using one-way ANOVA with a Tukey post-hoc test: ***p ≤ 0.0001.
(B) Tiled-C heatmaps comparing all-vs-all interaction frequency throughout the α-globin locus in R2-only homozygous (n = 3) and WT (n = 2) FL erythroid cells. Upper, coordinates = chr11: 29,900,000–33,230,000 (mm9). Lower, zoomed heatmap over the α-locus; coordinates = chr11: 31,900,000–32,400,000 (mm9). Heatmaps, merged biological replicates. Bins, 2,000 bp; UCSC tracks (top to bottom), R2-only homozygous ATAC-seq; WT CTCF ChIP-seq; WT ATAC-seq. Highlighted red region, α-locus; color scale, iterative correction and eigenvector decomposition (ICE)-normalized counts.
(C) Virtual capture plots, pairwise interactions throughout the zoomed tiled locus (chr11: 31,900,000–32,400,000 [mm9]) in which viewpoints participate. Viewpoints (top to bottom): R2 enhancer; α-promoters (combined as almost identical in sequence); CTCF sites HS38, HS29, HS48. FL erythroid cells are represented as blue, WT (n = 2); red, homozygous R2-only (n = 3); green, mESC cells (n = 3).
See also Figure S5.
Enhancer-promoter interaction is compromised in the R2-only locus in the absence of the other α-SE constituents
Numerous publications have demonstrated high-frequency interactions between SEs and their cognate target genes.11,30–34 These interactions appear crucial for effective upregulation of the target gene, although the mechanism(s) facilitating interactions between an SE and its target gene and the spatiotemporal relationship between interaction and activation remain unclear. Indeed, previous chromatin conformation capture (3C)-based studies have shown that in erythroid cells, the α-SE constituents, particularly R1 and R2, interact frequently with the α-globin promoters.17,35–40 To investigate whether R2’s ability to contact the α-globin promoters is affected in the R2-only locus in erythroid cells, we performed tiled-C, a low-input, high-resolution 3C-based technique that allows comparison of “all-vs-all” pairwise chromatin interactions at a specific genomic locus (in this case, 3.3 Mb surrounding α-globin).41
Chromatin interaction heat maps suggested a reduction in the overall frequency of pairwise interactions throughout the α-globin sub-TAD in R2-only FL erythroid cells (Figure 4B). To quantitatively assess the reduction, we generated five virtual capture plots, examining all pairwise chromatin interactions throughout the tiled region in which individual informative “viewpoints” participate: three CTCF sites (two flanking the α-globin sub-TAD and one situated between the R1 and R2 enhancers), the R2 enhancer, and the α-globin promoters. Since the two α-globin promoters are identical in sequence except for a single SNP, we considered both as one viewpoint. As well as generating virtual capture plots from WT and R2-only erythroid cells, we re-analyzed a previously published WT mESC tiled-C dataset41 to serve as a non-erythroid control. Chromatin interactions between each CTCF site (HS38 and HS29 upstream of the locus and HS48 downstream) and the surrounding DNA were unperturbed in R2-only FL erythroid cells, demonstrating that the 65 kb α-globin sub-TAD still forms in the absence of the R1, R3, Rm, and R4 elements (Figure 4C). However, interrogation of R2’s chromatin interaction profile revealed a striking reduction in interaction frequency between R2 and the α-globin promoters, which was corroborated by reciprocal virtual capture from the promoters themselves (Figure 4C). Although the frequency of these interactions was reduced in R2-only FL erythroid cells, it was still significantly higher than that in the WT mESC baseline.
The α-SE constituents are not equivalent and perform two distinct functions
In the R2-only mouse model, the R2 enhancer retains many characteristics of an active enhancer; however, its ability to recruit co-activators, interact with its target gene promoters, produce bidirectional eRNA transcripts, and upregulate α-globin expression are all severely attenuated. We next set out to rebuild the α-SE in various configurations to determine the role of each of the other SE elements. Because rebuilding the SE entailed the generation of numerous genetic models—too many to reasonably study in mice—we returned to the orthogonal in vitro EB-based mESC erythroid differentiation system.26
Our initial conclusion that the α-SE combines additively was based on deleting individual elements from an otherwise intact SE.17 Therefore, to revalidate these findings, we reconstituted those same deletions in hemizygous mESCs. Analysis of chromatin accessibility and α-globin gene expression in EB-derived erythroid cells was entirely consistent with our previous findings (Figures 5A, 5C, and S6A, yellow bars). Individual deletion of R1 (R2R3RmR4) or R2 (R1R3RmR4) significantly reduced α-globin expression, and deleting both R1 and R2 (R3RmR4), leaving only the R3, Rm, and R4 elements, reduced expression to ~2% of WT. Meanwhile, deleting R3 (R1R2RmR4) or Rm (R1R2R3R4) alone had no discernible effect on gene expression, and deleting R4 (R1R2R3Rm) led to a small (~15%) but statistically significant reduction in α-globin expression (Figures 5A and S6A, green bars).
Figure 5. The R1 and R2 enhancers rely on R3, Rm, and R4 in order to exert their full potential.
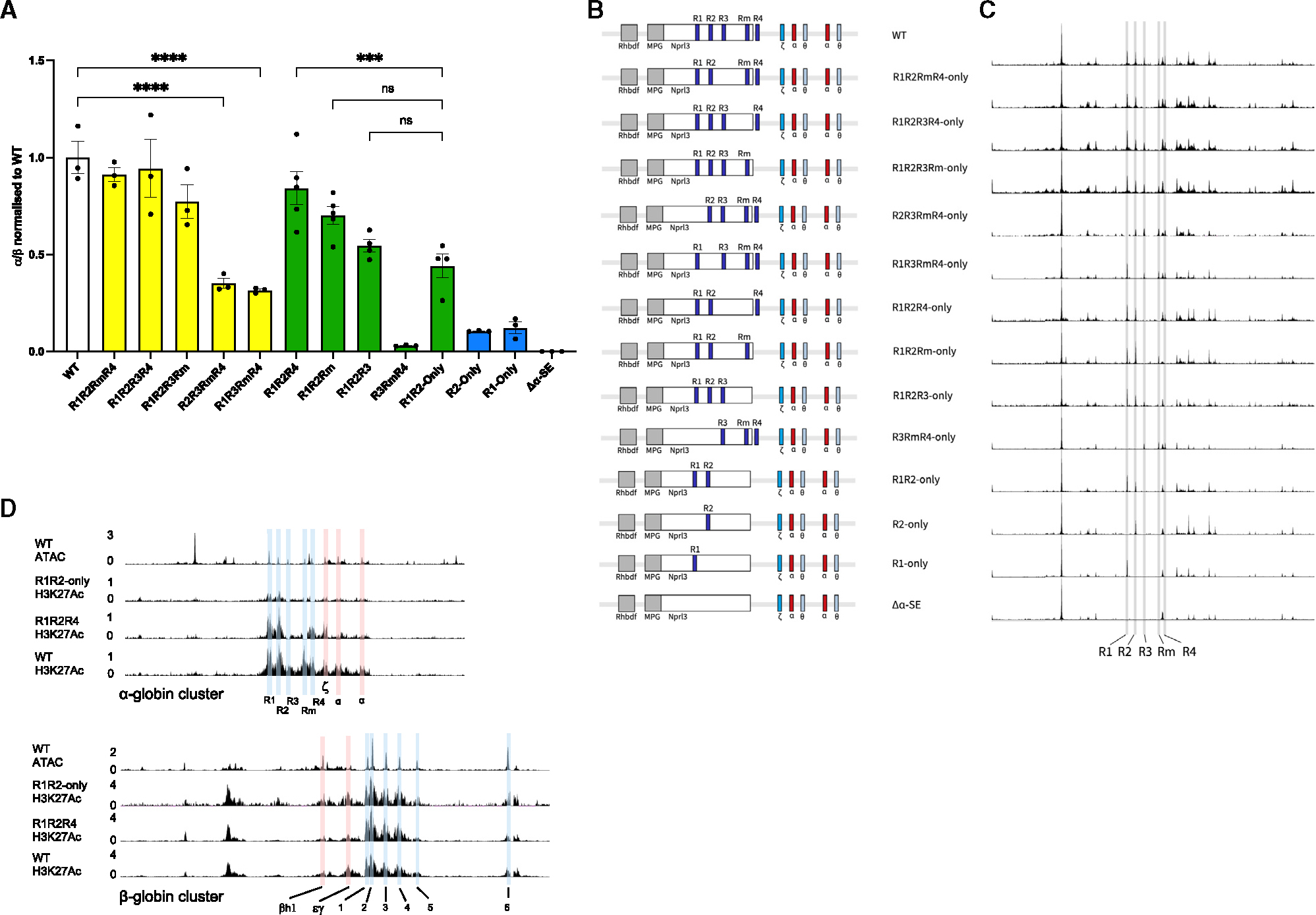
(A) α-globin gene expression from EB-derived erythroid cells (n ≥ 3) assayed by RT-qPCR. Expression normalized to β-globin and displayed as a proportion of WT expression. Dots, biological replicates; error bars, Standard Error. Yellow bars represent α-SE single deletion models that recapitulate the published results obtained in primary mouse erythroid cells in WT, R2R3RmR4 (equivalent to ΔR1), R1R3RmR4 (equivalent to ΔR2), R1R2RmR4 (equivalent to ΔR3), R1R2R3R4 (equivalent to ΔRm), and R1R2R3Rm (equivalent to ΔR4). Data are also shown for the enhancerless model, Δα-SE, and the single enhancer driven models, R1-only and R2-only (blue bars). R1R2-only model is shown along with the rescue models where each of the elements R3, Rm, and R4 (green bars) are assessed for their potential to restore the R1R2-only α-globin expression. Note that these three elements combined in the absence of R1 and R2 have no activation potential (R3RmR4, green bar). Statistical analysis was performed using one-way ANOVA with a Tukey post-hoc test: ****p ≤ 0.00001, ***p ≤ 0.0001.
(B) Graphical representation of the bottom-up approach of α-SE constituent enhancer functional dissection: fourteen genetic models combine various elements to rebuild the α-SE in hemizygous mESCs. All models screened by PCR, Sanger sequencing, and ATAC-seq.
(C) ATAC-seq in EB-derived erythroid cells corresponding the enhancer titration models (n ≥ 3) in (B). Tracks = merged biological replicates. Gray bars highlight the five elements (R1, R2, R3, Rm, and R4) at the α-globin locus.
(D) ATAC-seq in EB-derived WT erythroid cells (n = 3, merged; top). H3K27Ac ChIPmentation in R1R2-only (n = 1), R1R2R4 (n = 1), and WT (n = 1) EB-derived erythroid cells. Top panel, α-globin locus (regulatory elements and promoters highlighted and annotated as in Figure 1B); bottom panel, β-globin locus (regulatory elements and promoters highlighted and annotated as in Figure 1B). All tracks are cpm normalized.
See also Figure S6.
Next, we investigated whether reinserting the α-SE’s second major activator, R1, into the enhancerless Δα-SE locus would be sufficient to restore high levels of α-globin transcription. Similar to R2-only, EB-derived R1-only erythroid cells only expressed 10% α-globin compared to WT (Figures 5A and S6A, blue bars). Unexpectedly, even a model harboring both major activators in their native positions (R1R2-only) was incapable of restoring high levels of α-globin transcription (Figures 5A and S6A, green bars).
The R3, Rm, and R4 elements display little or no inherent conventional enhancer activity, but they appear to still be necessary for full α-SE activity. We called these elements “facilitators.” To investigate how R3, Rm, and R4 complement the activity of R1 and R2, we generated an “enhancer titration series,” sequentially rebuilding the native α-SE from the deficient R1R2-only model to WT and generating all R3/Rm/R4 permutations (Figure 5B).
We generated at least three separately targeted clones for each model and verified the integrity of each one using PCR and Sanger sequencing. To confirm that the newly designed models do not inadvertently create a sequence with potential function, we used the JASPAR42,43 and Sasquatch44 in silico tools to screen for predicted changes in TF motifs and DNA accessibility at the deletion and insertion sites. Using ATAC-seq, we show that in each model, the chromatin associated with the appropriate elements becomes accessible in erythroid cells, and there were no unexpected changes in accessibility throughout the remainder of the locus (Figure 5C).
To evaluate the ability of the facilitators (R3, Rm, and R4) to potentiate the classical enhancers (R1 and R2), we reinserted them individually and in combination into an allele containing just R1 and R2 (R1R2-only). Reinserting the R3 element into the R1R2-only background only rescued gene expression by ~10% (not statistically significant), whereas reinsertion of Rm or R4 had a more significant effect, increasing expression from R1R2-only (50%) by 50% and 80%, respectively (Figures 5A and S6A, green bars). Reinsertion of the R4 element was accompanied by a large increase in H3K27Ac over the R1 and R2 elements (Figure 5D), suggesting that R4’s main role is to facilitate the full activity of the two classical enhancers.
Reinsertion of the R3 element upregulated α-globin transcription to approximately the same degree in the presence of Rm, R4, or both (Figure S6B). Meanwhile, reintroducing Rm into a cluster containing R4 (e.g., inserting Rm into R1R2R4 cells) only raised expression by 5%–10% (Figure S6B). In the presence of R4, Rm seems redundant. Likewise, the positive effect of reinserting R4 into a locus already containing Rm (e.g., reinserting R4 into R1R2Rm cells) was less than reinserting R4 into a locus containing only R1, R2, and/or R3 (Figure S6B). Therefore, in their native context, Rm and R4, but not R3, appear to be at least partially redundant in their ability to facilitate the function of the classical enhancers R1 and R2, with R4 having a stronger effect than Rm.
R4’s rescue potential is dependent on its position
To investigate the cause of R4’s superior rescue potential, we reanalyzed an existing DNase-seq dataset and conducted FIMO (MEME-suite) motif analysis on the five α-SE elements. Unsurprisingly, R1 and R2 contained the highest density of TF motifs and the most complex DNase foot-printing signals (Figure 6A). However, motif analysis demonstrated that R3 contains more erythroid TF motifs (by absolute number and motif diversity) than Rm and R4 combined, which was supported by R3’s richer DNase foot-printing signal compared to Rm and R4 (Figure 6A). Inspection of Gata1, Nf-e2, and Tal1 ChIP-seq and ChIPmentation tracks from a number of WT erythroid tissues further supported the results of our motif analysis (data not shown). It is possible that R4 recruits other unknown factors, but our data suggest that the relative rescue capacities of R3, Rm, and R4 are not simply encoded in their relative capacities to recruit transcription factors.
Figure 6. The relative activity of R3, Rm, and R4 is primarily encoded in their positions.
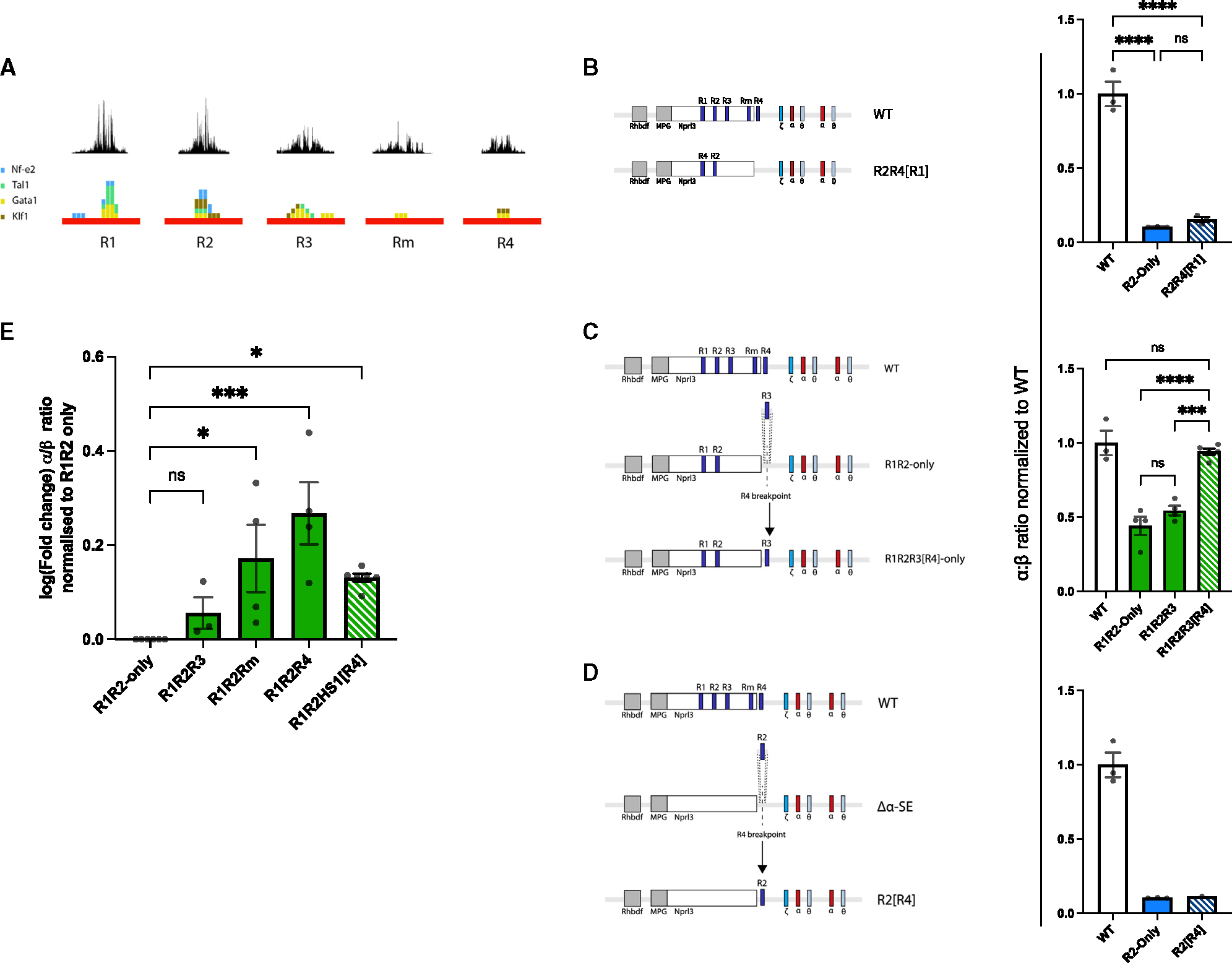
(A) DNase foot-printing over each of the α-SE constituents (top). FIMO motif analysis conducted on each α-SE constituent, searching for occurrences of Gata1 (yellow), Nf-e2 (blue), Tal1 (green), and Klf1 (brown) motifs (bottom).
(B) Schematic of the R2R4[R1] model and α-globin gene expression in WT, R2-only, and R2R4[R1] EB-derived erythroid cells (n ≥ 3) assayed by RT-qPCR. Expression normalized to β-globin and displayed as a proportion of WT expression. Dots, biological replicates; error bars, Standard Error. Statistical analysis was performed using one-way ANOVA with a Tukey post-hoc test: ****p ≤ 0.00001, ns: non-significant.
(C) Schematic of the R1R2R3[R4] model and α-globin gene expression in WT, R1R2-only, R1R2R3, and R1R2R3[R4] EB-derived erythroid cells (n ≥ 3) assayed by RT-qPCR. Expression normalized to β-globin and displayed as a proportion of WT expression. Dots, biological replicates; error bars, Standard Error. Statistical analysis was performed using one-way ANOVA with a Tukey post-hoc test: ****p ≤ 0.00001, ***p ≤ 0.0001, ns: non-significant.
(D) Schematic of the R2[R4] model and α-globin gene expression in WT (n = 3), R2-only (n = 3) and R2[R4] (n = 1) EB-derived erythroid cells assayed by RT-qPCR. Expression normalized to β-globin and displayed as a proportion of WT expression. Dots, biological replicates; error bars, Standard Error.
(E) Fold increase in α-globin expression over that calculated in R1R2-only erythroid cells. Expression normalized to β-globin and shown for the R1R2-only rescue by α-globin facilitators R3, Rm, and R4 as well as HS1, a β-globin identified facilitator. Dots, biological replicates; error bars, Standard Error. Statistical analysis was performed using one-way ANOVA with a Dunnett’s correction test: ***p ≤ 0.0001, *p ≤ 0.01, ns: non-significant.
See also Figure S7.
Rescue potential of R3, Rm, and R4 inversely correlates with distance to the α-globin promoters (Figures 5A and S6A, green bars). We therefore asked whether each element’s ability to bolster transcription depends more on its sequence or its proximity to the α-globin promoters. To test whether R4’s sequence is sufficient to rescue expression, we modified the R2-only model by reinserting R4; however, rather than placing R4 in its native position (close to the α-globin promoters), we reinserted it in the position of R1 (the element located furthest from the promoters; R2R4[R1]) and showed accessible chromatin at the reinsertion site (Figures 6B and S7). EB-derived R2R4[R1] erythroid cells only expressed 12% α-globin, a level comparable to that in R2-only erythroid cells, suggesting that R4’s rescue capacity is not exclusively based on its sequence (Figure 6B).
Next, to test the importance of element positioning, we modified the R1R2-only model by inserting R3 in the position of R4 (R1R2R3[R4]) and showed accessible chromatin at the reinsertion site (Figures 6C and S7). Moving R3 closer to the α-globin promoters in this model had a dramatic effect, increasing gene expression by >85% compared to the ~12% rescue of the R1R2-only model driven by R3 in its native position (R1R2R3; Figure 6C). Together, this strongly indicates that R4’s position, rather than its sequence, underpins its potency in rescuing gene expression.
R2-only FL erythroid cells exhibited reduced interaction frequency between R2 and the α-globin promoters (Figure 4C), and it seems that R4’s position close to the α-globin promoters is important for facilitating full R1 and R2 enhancer activity. We therefore speculated that R4 might play a role in increasing interaction frequency between the α-SE and promoters. To test whether the R2-only transcriptional deficit could be rescued by simply reducing the linear distance between R2 and its cognate promoters, we modified the Δα-SE model by inserting R2 at the position of R4 (R2[R4]) and showed accessible chromatin at the reinsertion site (Figures 6D and S7). Surprisingly, moving R2 closer to the α-globin promoters had no positive effect on gene expression (Figure 6D). This demonstrates that the physical linear proximity of R2 to the α-globin promoter was insufficient to restore R2’s full activity.
Identification of facilitators in other multipartite enhancers
An important question is whether other multipartite enhancer clusters contain facilitator elements as defined here, i.e., elements within an enhancer cluster that have the chromatin signature of enhancers, are poor in tissue-specific transcription factor binding sites, harbor little or no intrinsic enhancer activity when tested in reporter assays, and are necessary for the full activity of canonical enhancers within the cluster. To identify these elements requires a thorough epigenetic, genetic, and functional dissection of a cluster. To date, erythroid SEs have not been studied at the required depth to reveal these elements, but the β-globin LCR has been fully characterized, comparable to the α-globin cluster. The β-globin LCR includes six regulatory elements (HS1–6) and has been classified as a super-enhancer.17 When examined closely, HS1 seems a good candidate to test as a facilitator: when tested individually, HS1 has no intrinsic enhancer activity in embryonic, fetal, or adult erythropoiesis;45 deletion of HS1 from the SE results in substantial sensitivity to position effects in transgenic mice;46 and the predominant TF binding sites in HS1 are just two GATA binding elements.47 Here, we have taken HS1 and placed it in the position of the α-globin facilitator (R4) and found that despite its lack of intrinsic enhancer activity, like the α-globin facilitators, it shows a significant rescue potential of the R1R2 allele, observed as a 36% increase in the α-globin expression (Figure 6E), albeit lower than the native facilitators’ rescue effects (Rm ~50% and R4 >80%). HS1 fulfills our definition of a facilitator, exhibiting the hallmarks of such elements at the α-globin locus despite being transported from an independent erythroid-specific SE.
DISCUSSION
Since the seminal description of an enhancer element in 198148 and the first report that followed two years later of what was effectively an enhancer cluster,49 there has been an immense amount of research into what enhancers are, how they work, and how they influence development and disease. Despite the fact that enhancer clusters have been studied for over forty years, we are yet to understand many of the basic principles governing their activity, from the manner(s) by which cluster constituents cooperate with one another to the biochemical processes increasing target gene expression.
Over the years, many groups have reported different “flavors” of biologically significant enhancer clusters, among them locus control regions,50 shadow enhancers,51 regulatory archipelagos,52 Greek islands,53 stretch enhancers,54 and super-enhancers.4 Many enhancer clusters satisfy the criteria of multiple classes. To simplify our analyses, we focused on studying the functional characteristics of super-enhancers, selecting this particular class due to their clear bioinformatic definition (using the ROSE algorithm) and the fact that the field has widely adopted the “super-enhancer” nomenclature. Even with this definition, we do not assume that the underlying mechanism of their action is always the same, as discussed in Blobel et al.10
SEs are defined by high levels of enhancer-associated H3K27Ac, high levels of TF and Mediator occupancy, and the limited genomic distances between their constituents.4 Numerous publications have demonstrated that SEs activate high levels of gene expression with a tendency to regulate lineage-specific genes.4,11,14,18 Despite this, it remains unclear whether there is a functional distinction separating SEs from clusters of regular enhancer elements. Perhaps the key question is whether SEs are clusters of independent elements combining in an additive fashion or cohesive units exhibiting activities greater than the sum of their parts.10,11 A number of groups have dissected SEs, yielding various conclusions, from additive16 to super-additive,22 redundant,19 synergistic,21 and hierarchical20 cooperation. Similar to the variation in TF cooperation and sequence grammar manifested within individual enhancers, SE cooperation may well be variable, with subsets of SEs combining additively and others non-additively. The majority of SE dissection studies have been hampered by incomplete dissection of the clusters under investigation. For example, although dissection of the β-globin SE suggested that it combines additively, the degree to which the cluster was dissected was comparable to our previous study at the α-globin SE.16,17 As we have shown here, this level of dissection is insufficient to unequivocally conclude whether a cluster combines additively or non-additively. Regardless, previous studies deleting the entire β-globin SE showed that the β-globin genes remain accessible and marked by histone acetylation;55–57 this is contrary to what we see upon deleting the entire α-SE, suggesting that the effect SEs have on their target genes is likely to be somewhat variable between loci. Other SE dissection studies have been confounded by disruption of clusters that regulate pleiotropic transcription factors or co-factors that influence cell fate, rendering it impossible to control whether WT and manipulated models are equivalent in their developmental stage and cell type.
Here, we have comprehensively dissected the tractable α-globin SE in situ to investigate how its five constituent elements cooperate. The α-SE is an ideal genetic model for this study; the SE and the TAD in which it is contained have been extensively characterized,58 and the SE is activated exclusively during terminal erythroid differentiation, meaning its manipulation has no effect on cell fate or in non-erythroid cells. Previous dissection of the α-SE suggested that its five constituents combine additively as independent elements,17 a conclusion drawn through generating a series of mouse models harboring single and selective pairwise element deletions from an otherwise intact SE. Our present work further evaluates this conclusion and demonstrates unequivocally that R2 requires (a subset of) the other four α-SE constituents to achieve its full enhancer potential. Despite maintaining the biochemical signature of an active enhancer, R2 by itself is not sufficient to upregulate high levels of α-globin expression, exhibiting very low levels of co-activator recruitment, reduced interactions with its target genes’ promoters, and lower levels of eRNA transcription.
Rebuilding the α-SE demonstrated that our previous single deletion models were simply inadequate to fully resolve the cooperation between the five α-SE constituents. This serves as a cautionary tale and clearly shows that extensive genetic dissection and synthetic rebuilding is essential to fully understand how an enhancer cluster operates. Combinatorial reconstruction of the α-SE exposed a complex network of functional interactions between its constituents: R1 and R2 cooperate synergistically, each upregulating gene expression 100-fold alone versus 450-fold when combined, whereas the R3, Rm, and R4 elements display no intrinsic enhancer activity in the models tested here; instead, they facilitate the activities of R1 and R2. The three “facilitator” elements display a hierarchy wherein R4 is the most potent facilitator and R3 the least potent. Whereas R3 facilitates the activities of R1 and R2 to a similar degree regardless of Rm/R4 coincidence, Rm and R4 function in a context-dependent manner, each partially redundant to the other.
Is there evidence for similar facilitator elements elsewhere in the genome? Interestingly, Sahu and colleagues recently used a STARR-seq method to show that four out of the five MYC SE constituents have no detectable enhancer activity in HepG2 cells.59 From this and other observations in their report, the authors conclude that unlike classical enhancers, elements they refer to as “chromatin-dependent enhancers” do not strongly transactivate a heterologous promoter but act to increase gene expression via chromatin modification or structural changes in higher-order chromatin.59 Similarly, Hnisz and colleagues previously showed that the E8 enhancer within the Pri-miR-290–295 has very little enhancer activity measured by luciferase assay; nevertheless, deletion of E8 caused a large decrease in Pri-miR-290–295 expression.18 E8 is the SE constituent located most proximally to Pri-miR-290–295. These findings are similar to what we see at R4, namely, an SE constituent that lacks enhancer activity, is important for overall SE function, and is located between its target gene and all other SE constituents. Finally, as shown here, the HS1 element of the β-globin LCR, which has no intrinsic enhancer activity, acts to facilitate the α-SE when placed in the position of R4. Together, these findings suggest that facilitators could be a common feature of SEs.
The mechanism(s) by which facilitators augment SE activity remain elusive. It is possible that facilitators act by providing cooperativity for the recruitment or stability of TFs or co-factors analogous to the mechanism proposed in the enhanceosome model for individual enhancer elements.60,61 By analogy, it is possible that appropriately located enhancers and facilitators combine to create a distinct three-dimensional structure. From this point of view, it is interesting that the hierarchy of facilitators is encoded in their positions more than their sequences. The position dependence of facilitators is particularly interesting in light of recent work reporting functional directionality as a property of SEs.62 Moreover, moving R2 closer to the α-globin promoters had no effect on gene expression, suggesting that facilitators do not solely act to increase enhancer-promoter interaction frequency; nevertheless, this does not preclude them playing a role in forming or stabilizing specific three-dimensional structures. A recent study in Drosophila has identified what may be similar elements, which are not enhancers but are thought to facilitate interactions between regulatory elements by tethering them together.63
A number of studies have suggested that enhancer clusters, including SEs, may act cooperatively to form foci containing high concentrations of tissue-specific TFs, co-activators such as the Mediator complex, and Pol II.11 The biochemical processes leading to formation of such subnuclear structures are debated, but current theories propose liquid-liquid phase separation18,64–66 and some form of TF trapping22,67 or engagement of TFs in multivalent interactions independent of assembly into phase-separated liquid-like droplets.68 All of these proposals require recruitment of a critical mass of TFs within a given three-dimensional space. It is feasible that R4 could rescue gene expression by increasing the density of TF binding sites at a particularly influential position along the chromatin fiber; subsequent recruitment of co-activators such as the Mediator complex could then be instructive for establishing a regulatory hub. Though speculative, this explanation is consistent both with R3’s ability to rescue transcription when transplanted to the position of R4 in the R1R2R3[R4] model and with R2’s continued insufficiency in the R2[R4] model.
In summary, our findings demonstrate that SEs can constitute complex cohesive networks of regulatory elements, displaying simultaneous additive, redundant, and synergistic cooperation. We present evidence that SEs can act as cohorts of functionally distinct elements, including classical enhancers, responsible for activating a target gene’s expression, and facilitators, which in some way augment activator function. Without facilitators, we see severely attenuated coactivator recruitment, enhancer-promoter interaction frequency, and eRNA transcription. Most importantly, we rigorously show that an SE can manifest emergent properties resulting from the interaction of classical enhancers and what we define here as facilitators.
Limitations of the study
Although it seems likely that facilitators will be a common feature of multipartite enhancer clusters, this remains to be further tested. The fact that observations on gene regulation made at the globin gene clusters have always illustrated general principles is encouraging. At present, the ability to identify facilitators genome-wide is limited by the lack of a distinguishing signature for such elements and the extensive genetic engineering required to analyze each element. Both of these points are being addressed by constructing a screening system. Although there are clues to the mechanisms by which facilitators might potentiate the activity of classical enhancers, this has not been formally addressed in this study and will be the focus of future investigations.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Mira Kassouf (mira.kassouf@imm.ox.ac.uk).
Materials availability
Materials associated with the paper (the R2-only mouse model, the engineered mouse ESCs) are available upon request.
Data and code availability
All data generated for this study are included in this published article and its supplementary information. Standardized data types (ChIP-seq, ATAC-seq RNA-seq and Tiled Capture-C data, raw data and processed files) are publicly accessible in the Gene Expression Omnibus (GEO) under accession numbers GEO: GSE220463.
This paper does not report original code. Codes used in the analysis of this manuscript are referenced.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODELS AND STUDY PARTICIPANT DETAILS
Mouse model generation
All mouse work was performed in accordance with UK Home office regulations, under the appropriate animal licenses. Mouse model generation and animal husbandry was conducted by the Mouse Transgenics Core Facility at the Weatherall Institute of Molecular Medicine. R2-only BAC-integrated mESCs were karyotyped, microinjected into C57BL/6 blastocysts and implanted into pseudopregnant C57BL/6 females. Three resulting chimeric males were back-crossed with WT C57BL/6 females. Between three litters from one chimera, 10 pups were identified with germline transmission, as assessed by agouti coat color derived from the E14 (E14-TG2a.IV) mESC background. Of these 10, five pups were confirmed to be heterozygous for the Hprt+ R2-only modification by PCR-based genotyping. Heterozygotes were crossed to a Flp-expressing line to promote recombinase-mediated excision of the Hprt cassette. Hprt- R2-only heterozygotes were then back-crossed and inter-crossed to establish a Flp-negative heterozygous line and set up timed matings for embryo dissection and tissue harvesting and experimentation.
Genotyping was performed on material from non-erythroid tissue: embryonic material (dissected embryos), ear notches (live pups) or brain (dead pups). DNA from ear notches and embryos was prepared using the DIRECTPCR-EAR PEQGOLD reagent (VWR) with 50 μg/mL Proteinase K treatment at 55°C overnight (Thermo Fisher). After Proteinase K inactivation for 5 min at 95°C, samples were spun at maximum speed for 3 min and supernatant added directly to standard PCR reactions using IMMOLASE DNA Polymerase (Bioline) supplemented with 1M betaine. Brain tissue was lysed overnight in a buffer of 50 μM Tris pH 8.0, 100 mM EDTA, 100 mM NaCl and 1% SDS with 50 mg/mL Proteinase K, then DNA was obtained by standard phenol-chloroform extraction and ethanol precipitation. Genotyping was performed using PCR primers detailed in Table S3.
Timed-heterozygote crosses: R2-only homozygotes were not viable as shown by the Mendelian ratio in Table S1. Therefore, all analyses were restricted to embryonic timepoints. Pregnant mice were sacrificed at embryonic days E8.5, E9.5, E10.5, E12.5, E14.5, or E17.5 post coitum. Embryos were dissected from the pregnant females and tissue was taken for genotyping by PCR. Erythropoietic cells/compartments were then isolated for analysis, blinded to the genotype. E8.5–10.5 embryos were deposited in heparinised PBS and primitive erythroid cells drained from the embryos were aspirated into fresh tubes for processing. Fetal livers (FL, the definitive erythroid compartment) were isolated from E12.5-E17.5 embryos. FL were mechanically disaggregated to a single cell suspension in FACS buffer, and filtered through pre-separation filters; brain tissue was stored for genotyping by PCR and gene expression analysis (RT-qPCR). Erythroid cells were processed for analysis by RT-qPCR/RNA-seq, ATAC-seq, ChIP/ChIPmentation and 3C-based methods on the day of harvest (see below). FACS analysis following staining for the CD71 and Ter119 cell surface markers in E12.5 FL cells, revealed that WT and R2-only FL are composed of ~95% CD71+/Ter119+ erythroid cells, indicating that no further selection (beyond mechanical disaggregation, and filtration through pre-separation filters (miltenyibiotec) was required.
Genetically engineered mouse embryonic stem cell lines and in vitro erythroid differentiation system
E14-TG2a.IV (E14) mESCs, or genetic models derived from these cells, were cultured in gelatinised plates using standard methods69,70: cells were maintained in ES-complete medium, a GMEM-based medium supplemented amongst other standard tissue culture media reagents with FBS and Leukemia inhibitory factor (LIF).
An in vitro Embryoid Body (EB)-based mESCs differentiation system was used to generate erythroid cells.26 Briefly, 24–48 h pre-differentiation, mESCs cells were induced for differentiation by passaging into base media (Iscove’s modified Dulbecco’s medium (IMDM), 1.4×10–4 M monothioglycerol (Sigma-Aldrich) and 50 U/ml penicillin-streptomycin (Thermo Fisher)) supplemented with 15% heat-inactivated FBS and 1000 U/ml LIF. For embryoid body (EB) generation, cells were disaggregated by trypsinisation and quenched in base media (as above) supplemented with 10% ΔFCS. Differentiation media was prepared fresh on the day of differentiation by supplementing base media (as above) with 15% ΔFCS, 5% protein-free hybridoma medium (PFHMII) (Thermo Fisher), 2 mM L-glutamine (Thermo Fisher), 50 μg/mL L-ascorbic acid (Sigma Aldrich), 3×10−4 M monothioglycerol and 300 μg/mL human transferrin (Sigma Aldrich). Cells were plated in triple vent petri dishes (Thermo Fisher) at 1–2 ×103 cells in 10 mL differentiation media. EBs were left to differentiate for up to seven days without disruption except for gentle manual shaking every few days to disrupt cell sticking.
After seven days of differentiation, EBs were disaggregated into single cell suspension, through incubation in 0.25% trypsin for ~3 min, and then quenched with FCS-containing media. The bulk population was analyzed for erythroid differentiation by immune-phenotyping using two erythroid surface markers, the transferrin receptor CD71 and Ter119. cells were labeled for CD71 in staining buffer for 20 min at 4°C, rolling, then washed by adding staining buffer (1 mL per 107 cells) and spinning. After supernatant removal, cells were incubated with MACS anti-FITC separation microbeads (Miltenyli; 10 μL per 107 cells) in ice-cold separation buffer (PBS plus 0.5% bovine serum albumin (BSA) and 2 mM EDTA; 90 μL per 107 cells) for 15 min at 4°C, rolling, and washed by adding separation buffer (1 mL per 107 cells) and spinning.
Bead-labelled cells were resuspended in 500 μL cold separation buffer and added to a pre-equilibrated LS column (following manufacturer’s instructions). The negative fraction was washed through with two flushes of 3 mL cold separation buffer and the positive fraction collected by forcing cells from the column in 5 mL separation buffer. After spinning and supernatant removal, cells were resuspended in staining buffer as needed for downstream processing. Population purity and selection efficiency were determined by flow cytometry. All antibodies and reagents are listed in Key Resources Table (KRT).
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
| H3K27ac | Abcam | cat# ab4729, RRID:AB_2118291 |
| H3K4me1 | Abcam | cat# ab8895, RRID:AB_306847 |
| H3K4me3 | Abcam | cat# ab8580, RRID:AB_306649 |
| Gatal | Abcam | cat# ab11852, RRID:AB_298635 |
| Rad21 | Abcam | cat# ab992, RRID:AB_2176601 |
| Polli | Santa Cruz Biotechnology | cat# sc-899, RRID:AB_632359 |
| Nf-e2 | Santa Cruz Biotechnology | cat# sc-22827, RRID:AB_2152924 |
| Medi | Bethyl | cat# IHC-00149, RRID:AB_2144026, Discontinued |
| Brd4 | Bethyl | cat# IHC-00396, RRID:AB_1604188, |
| CTCF | Cambridge Bioscience, supplier: Active Motif | cat# 61311, RRID:AB_2614975 |
| CD71-FITC | eBioscience | cat# 11–0711-85, RRID:AB_465125 |
| ter119-PE | BD Pharmingen | cat # 553673, RRID:AB_394986 |
| Anti-FITC magnetic microbeads | Miltenyi | cat# 130–048-701, RRID:AB_244371 |
|
| ||
| Biological Samples | ||
|
| ||
| mouse embryonic blood | this paper | N/A |
| mouse adult blood | this paper | N/A |
| mouse adult brain | this paper | N/A |
| mouse fetal liver cells | this paper | N/A |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| PFHM II | Gibco | cat# 12040077 |
| L-Ascorbic Acid | Sigma | cat# A4544–25G |
| Transferrin | Merck | cat# 10652202001 |
| LIF | Cell Bioscience Cell Guidance Systems (supplier) | cat# GFM200–100 |
| FCS used in mESC cultures | Gibco | cat# 10270–106 |
| Cesium chloride powder | Merck | cat# C4036–100g |
| UltraPure Ethidium bromide | Life | cat# 15585011 |
| OptiMEM medium | Thermofisher Scientific | cat# 31985062 |
| DpnII enzyme for Tiled-C library prep | New England Biolabs | cat# R0543M |
| T4 DNA Ligase, HC (30 U/uL)-5,000 units | Life Technologies Ltd | cat# EL0013 |
| Immolase DNA Polymerase | Bioline | cat# BIO-21046 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Tapestation High-Sensitivity D1000 reagents | Agilent | cat# 5067–5585 |
| Tapestation High-Sensitivity D1000 screentapes | Agilent | cat# 5067–5584 |
| Tapestation D1000 ScreeTape | Agilent | cat# 5067–5582 |
| Tapestation D1000 reagents | Agilent | cat# 5067–5583 |
| RNA Screentape | Agilent | cat# 5067–5576 |
| Qubit dsDNA BR Assay Kit | Invitrogen | cat# Q32850 |
| Qubit dsDNA HS Assay Kit | Invitrogen | cat# Q32851 |
| Qubit BR RNA Assay Kit | Invitrogen | cat# Q10211 |
| Qubit HS RNA Assay Kit | Invitrogen | cat# Q32855 |
| KAPA Quantification kit | KAPA | cat# KK4824 |
| Illumina Tagment DNA Enzyme and Buffer Large Kit | Illumina | cat# 20034198 |
| NEBNext High-Fidelity 2x PCR Master Mix | NEB | cat# M0541 |
| NextSeq® 500/550 High Output Kit v2.5(75 cycles) | illumina | cat# 20024906 |
| NEBNext Ultra II Directional RNA Library Prep Kit | NEB | cat# E7760 |
| NEBNext rRNA Depletion Kit (Human/ Mouse/Rat) | NEB | cat# E6310 |
| NEBNext® Ultra™ II DNA Library Prep with Sample Purification Beads | NEB | cat# E7103S |
| Applied BiosystemsTaqMan Universal PCR Master Mix - 5mL | Thermofisher Scientific | cat# 4304437 |
| NEBNext Multiplex Oligos for Illumina | NEB | cat# E7335/E7500 |
| SuperScript™ III First-Strand Synthesis SuperMix for qRT-PCR | Thermofisher Scientific | cat# 11752050 |
| Lipofectamine™ LTX Reagent with PLUS™ Reagent | Thermofisher Scientific | cat# 15338100 |
| Hba-a1/2 (FAM-MGB) | Thermofisher Scientific | Assay ID Mm02580841_g1 |
| Hbb-b (FAM-MGB) | Thermofisher Scientific | Assay ID Mm01611268_g1 |
| Hbb-y (FAM-MGB) | Thermofisher Scientific | Assay ID Mm00433936_g1 |
| Hba-x (FAM-MGB) | Thermofisher Scientific | Assay ID Mm00439255_m1 |
| Snrnp25 (FAM-MGB) | Thermofisher Scientific | Assay ID Mm00547218_m1 |
| Hbb-bh1 (FAM-MGB) | Thermofisher Scientific | Assay ID Mm00433932_g1 |
| Mpg (FAM-MGB) | Thermofisher Scientific | Assay ID Mm00447872_m1 |
| Nprl3 (FAM-MGB) | Thermofisher Scientific | Assay ID Mm01193449_m1 |
| Rhbdf1 (FAM-MGB) | Thermofisher Scientific | Assay ID Mm00711711_m1 |
| RPS18 (FAM-MGB) | Thermofisher Scientific | Assay ID Mm02601777_g1 |
| LS selection columns | Miltenyi 130–042-401 | cat# 130–042-401 |
| Direct-zol MicroPrep Kit | Zymo Research | cat# R2060 |
| AMPure XP Beads | Beckman Coulter | cat# A63881 |
| Nextera XT Library Preparation kit | illumina | cat# FC-131–1024 |
| Dynabeads M-280 Streptavidin | ThermoFisher | cat# 11205D |
| NEBNext Poly(A) mRNA Magnetic Isolation Module | NEB | cat# E7490S |
|
| ||
| Deposited data | ||
|
| ||
| ChIP-seq, ATAC-seq, RNA-seq and NG Capture-C data (sequence reads and processed files) | This paper, Gene Expression Omnibus | GEO: GSE220463 |
|
| ||
| Experimental models: cell lines | ||
|
| ||
| 1- mDist mouse ESC line (RMGR ready) | modified in Prof. Doug Higgs Lab | DOI: https://doi.org/10.1016/j.cell.2006.11.044 |
| 2- R2-only mouse ESC line (RMGR ready) | This paper, derived from mDist mouse ESC line, heterozygote for the R2-only BAC inserted allele. Used to generate the R2- only mouse model | N/A |
| 3- Hemizygous mDist mouse ESC line (RMGR-ready on the undeleted allele) | This paper, used as Wildtype and base line for other genetically modified clones | N/A |
| 4- Hemizygous mDist mouse ESC line (RMGR-ready and and R2-only on the undeleted allele) | This paper, used as R2-only mESC clone and base line for other genetically modified clones | N/A |
| 5- R1-only mESC | This paper, derived from mESC #4 in this list | N/A |
| 6- R1R2-only mESC | This paper, derived from mESC #4 in this list | N/A |
| 7- R1R2R3-only mESC | This paper, derived from mESC #3 in this list | N/A |
| 8- R1R2R3Rm-only mESC | This paper, derived from mESC #3 in this list | N/A |
| 9- R1R2R3R4-only mESC | This paper, derived from mESC #3 in this list | N/A |
| 10- R1R3RmR4-only mESC | This paper, derived from mESC #3 in this list | N/A |
| 11- R2R3RmR4-only mESC | This paper, derived from mESC #3 in this list | N/A |
| 12- RmR3R4-only mESC | This paper, derived from mESC #3 in this list | N/A |
| 13- R1R2Rm-only mESC | This paper, derived from mESC #3 in this list | N/A |
| 14- R1R2R4-only mESC | This paper, derived from mESC #3 in this list | N/A |
| 15- R1R2R4-only mESC | This paper, derived from mESC #3 in this list | N/A |
| 16- R2R4[R1] mESC | This paper, derived from mESC #4 in this list | N/A |
| 17- R2[R4] mESC | This paper, derived from mESC #4 in this list | N/A |
| 18- R1R2R3[R4] mESC | This paper, derived from mESC #4 in this list | N/A |
| 19- R1R2HS1[R4] mESC | This paper, derived from mESC #4 in this list | N/A |
| 20- Δα-SE mESC | This paper, derived from mESC #4 in this list | N/A |
|
| ||
| Experimental models: organisms/strains | ||
|
| ||
| mouse R2-only model WT, Heterozygotes, Homozygotes analyzed from the same colony | This paper, derived by microinjecting mDist-R2-only mouse ESCs into blastocysts from C57BL/6 mice and implanted into pseudopregnant females. | N/A |
|
| ||
| Oligonucleotides | ||
| Guide RNA sequences | This paper, designed and provided by the Genome Engineering Facility at the Weatherall Institute of Molecular Medicine by Dr Philip Hublitz; see Table S2 | N/A |
| PCR primers for genome engineering screening | This paper, designed and provided by the Genome Engineering Facility at the Weatherall Institute of Molecular Medicine by Dr Philip Hublitz; see Table S3 | N/A |
| Tiled-C Capture oligonucleotides | STAR Methods | DOI: https://doi.org/10.1038/s41467-020-16598-7 |
|
| ||
| Recombinant DNA | ||
|
| ||
| pSpCas9(BB)-2A-GFP (pX458) vector | Addgene plasmid | http://n2t.net/addgene:48138;RRID:Addgene_48138 |
| pSpCas9(BB)-2A-Ruby (pX458) | This paper, modified version of the pX458-GFP vector, Provided by the Genome Engineering Facility at the Weatherall Institute of Molecular Medicine by Dr Philip Hublitz | N/A |
| HDR donor vectors | this paper, GeneArt Gene Synthesis custom design and in-house cloning | N/A |
| pCAGGS-Cre-IRESpuro plasmid | This paper, provided by the Genome Engineering Facility at the Weatherall Institute of Molecular Medicine | DOI: https://doi.org/10.1038/sj.onc.1205530 |
| Flippase (Flp)-expressing vector | This paper, provided by the Genome Engineering Facility at the Weatherall Institute of Molecular Medicine | https://doi.org/10.1002/gene.1076 |
|
| ||
| Software and algorithms | ||
|
| ||
| CRISPOR | https://doi.org/10.1093/nar/gky354 | http://crispor.tefor.net/ |
| BreakingCas | https://doi.org/10.1093/nar/gkw407 | https://bioinfogp.cnb.csic.es/tools/breakingcas/ |
| ggplot2 | https://doi.org/10.1007/978-3-319-24277-4 | https://ggplot2.tidyverse.org/ |
| bowtie2 | https://doi.org/10.1038/nmeth.1923 | https://github.com/BenLangmead/bowtie2 |
| SAMtools | https://doi.org/10.1093/bioinformatics/btp352 | http://samtools.sourceforge.net |
| deepTools (bamcoverage, FilteredRNAstrand) | deeptools.ie-freiburg.mpg.de | https://doi.org/10.1093/nar/gkw257 |
| MACS2 | https://doi.org/10.1186/GB-2008-9-9-R137 | https://hbctraining.github.io/Intro-to-ChIPseq/lessons/05_peak_calling_macs.html |
| DESeq2 | https://genomebiology.biomedcentral.com/articles/10.1186/s13059-014-0550-8 | http://www.bioconductor.org/packages/release/bioc/html/DESeq2.html |
| MEME suite | https://doi.org/10.1093/NAR/GKV416 | http://meme-suite.org |
| Diffbind, rgl, magick for Principal Component Analysis (PCA) | RStudio | http://bioconductor.org/packages/release/bioc/vignettes/DiffBind/inst/doc/DiffBind.pdf |
| HiC-Pro | https://doi.org/10.1186/S13059-015-0831-X | http://github.com/nservant/HiC-Pro |
| star alignment tool | https://doi.org/10.1093/BIOINFORMATICS/BTS635 | http://code.google.com/p/rna-star/ |
| Rsubread featurecounts | https://doi.org/10.1093/nar/gkz114 | http://www.bioconductor.org |
| edgeR expression differential analysis | RStudio | https://www.bioconductor.org/packages/devel/bioc/vignettes/edgeR/inst/doc/edgeRUsersGuide.pdf |
| bedtools | https://doi.org/10.1093/BIOINFORMATICS/BTQ033 | http://code.google.com/p/bedtools |
| JASPAR | https://doi.org/10.1093/nar/gkab1113 | https://jaspar.genereg.net/ |
| SASQUATCH | https://doi.org/10.1101/gr.220202.117 | https://github.com/Hughes-Genome-Group/sasquatch |
| Plotgardner | https://doi.org/10.1093/bioinformatics/btac057 | https://github.com/PhanstielLab/plotgardener |
|
| ||
| Other | ||
|
| ||
| Beckman Coulter OptiSealpolypropylene Centrifuge tube 56 tubes and plugs (13 × 48mm) | Beckman Coulter | cat# 361621 |
METHOD DETAILS
Synthetic BAC generation
Two ‘RMGR-ready’ versions of the α-globin locus, the first encoding the five enhancer elements and the second deleting all but the R2 element, were constructed. A previously constructed BAC spanning the α-globin locus plus RMGR parts25 (RP23–46918; BACPAC Resources Center, Children’s Hospital Oakland Research Institute71; ) was used as template to generate PCR amplicons with 50–200 base pairs of overlapping sequence for yeast homologous recombination. Lox sites were integrated 85 kb apart, flanking the α-globin regulatory region (85,145 bp; Chr11:32,115,389–32,200,533). Gblocks (IDT) or fusion PCR products were used to provide homology with non-overlapping adjacent segments (e.g., enhancer deletions, vector-adjacent amplicons). A variant of the eSwAP-In method24 was used to produce the two constructs, which were sequence-verified using illumina short-read sequencing (Figure S1A). BACs containing synthetic versions of the mouse α-globin locus were received as transformed bacteria samples and grown up in LB under selection with kanamycin (20 μg/mL) and ampicillin (50 μg/mL). 1 L overnight cultures were cleared for cell debris using Plasmid Maxi Kit reagents P1–3 (Qiagen) as instructed for low-copy plasmids, with precipitated material removed by filtration. DNA was precipitated from the cleared supernatant using 1:1 isopropanol and washed with 70% ethanol, then purified by cesium chloride centrifugation. BAC preparations were quantified by the Qubit dsDNA Broad-Range Assay (Thermo Fisher) and checked for sequence integrity by restriction digest and visualisation on a 1% agarose gel.
BAC transfection
RMGR-competent mESCs (mDist cells derived from E14-TG2a.IV (E14) mESCs25) were co-transfected by lipofection (Lipofectamine LTX reagent) with Purified BAC DNA and a pCAGGS-Cre-IRESpuro plasmid.72 Transfections were performed in 6-well format by plating freshly trypsinised mESCs in 2 mL culture media supplemented with the transfection mix prepared to manufacturer’s instructions: 5 μL LTX reagent, 2 μg plasmid DNA (1.5 μg BAC plus 0.5 μg pCAGGS-Cre-IRESpuro), 2 μL PLUS reagent and 250 μL Opti-MEM (Thermo Fisher). Media was changed after 24 h to remove the lipofection reagents and selection for complementation of the Hprt gene was started after an additional 24 h with 1x HAT supplement (0.1 mM hypoxathine, 0.4 mM aminopterin, 0.016 mM thymidine). Selection was continued for up to two weeks, at which point surviving colonies were picked into individual wells and HAT selection replaced with HT (0.1 mM hypoxanthine, 0.016 mM thymidine) recovery media. Cells were selected for Hprt complementation, and the Hprt gene later removed by transfection with a transient flippase-expressing plasmid.73 Cells were screened by selection with 6-thioguanine (6-TG) and PCR (for genotyping primer refer to KRT).
In the case of the R2-only model, the structural integrity of the locus was checked with linked-read library preparation (10x Genomics) followed by illumina sequencing, all performed at the Oxford Genomics Center, Wellcome Center for Human Genetics (Oxford, UK). (Figure S1B).
CRISPR-Cas9 editing for the generation of mouse ESC genetic models
All clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 targeting strategies were designed, prepared and tested by Dr Philip Hublitz and colleagues in the Genome Engineering Core Facility at the Weatherall Institute of Molecular Medicine. Guide RNAs were designed using the CRISPOR and BreakingCas online gRNA design tools (refer to KRT). Candidates with the fewest predicted off-targets were selected and further screened for their effectiveness, using an in vitro surveyor assay (according to the manufacturer, IDT). For guide RNA sequences see Table S3.
mDist (RMGR-ready) mESCs targeted with WT and R2-only BAC DNA generated two cell-lines which were then modified into hemizygous ‘RMGR-ready’ WT and R2-only mESCs respectively. These two cell-lines were used as the base cell lines for the rest of mESC models reported in this paper. To generate the hemizygous lines, gRNAs (cloned into pSpCas9 (BB)-2A-GFP (pX458) vector, a gift from Feng Zhang (Addgene plasmid: #48138), or pX458-ruby74) were designed such as a 117kb region that encompasses the full RMGR-ready region of the α-globin locus (86Kb) is removed in addition to flanking sequences, deleting in the process all the CTCF sites that contribute to the formation of the α-globin sub-TAD. Screening for this deletion was done using various PCR strategies (for sequences see Table S2) and Sanger sequencing that tested the junction created by the deletion and for the presence of an intact RMGR locus. For enhancer deletions, gRNAs were designed flanking the targeted enhancer, and cloned into pSpCas9 (BB)-2A-GFP (pX458) vector, a gift from Feng Zhang (Addgene plasmid: #48138), or pX458-ruby.74 Hemizygous WT mESCs were co-transfected, by lipofection, with the appropriate 5′ targeting vector (expressing GFP) and 3′-targeting vector (expressing mRuby), and 24–36 h later, GFP-mRuby co-fluorescent cells were FACS sorted into individual wells of a 96 well plate. Individual clones were grown in each well for 8–10 days without disruption. When colonies were visible in each well, cells were split into two plates: one for screening, and the other for analysis/freezing. Clones were screened for successful enhancer deletion by the appropriate PCR strategy; this entailed PCR amplification using primers flanking each deleted element (sequences in Table S3), such that a successfully deleted allele would produce a smaller product than a WT allele. Clones were then screened by Sanger sequencing and ATAC-seq.
To produce R2[R4], R1R2R3[R4], R2R4[R1] and R1R2HS1[R4] mESC models, existing hemizygous Δα-SE, R2-only, or R1R2-only mESC models were re-targeted for the desired outcome. Each new model was generated using a single round of targeting – either through insertion of the R2 element at the position of R4 in Δα-SE cells, insertion of the R4 element in the position of R1 in R2-only cells or insertion of the R3 or HS1 element in the position of R4 in R1R2-only cells. Homology directed repair (HDR) donors were designed encoding the R2, R4, R3, and HS1 elements flanked by 500bp homology arms, homologous to the native position of the desired insertion site. A Sal1 restriction enzyme recognition site was inserted at the 5′ of the R2 enhancer in the initial HDR donor, and an Mlu1 site at the 3′ of the element. This enabled efficient restriction-ligation exchange of the R2 element within the HDR donor with the R3 and HS1 elements. The HDR donor construct was ordered as a GeneART Gene synthesis custom design. The HDR donor was also designed to inactivate the protospacer adjacent motif. R2[R4], R1R2R3[R4] and R1R2HS1[R4] donors were screened with Sasquatch 43and JASPAR42,43 to ensure no novel accessibility sites or motifs were predicted at the newly created junctions,43,44 prior to synthesis and transfection. The donor vector sequence was also verified using Plasmidsaurus.
Δα-SE, R2-only and R1R2-only cells were transfected, by lipofection, with pX458 vectors (expressing gRNA targeting desired insertion position for various insertions) and the appropriate HDR donor using a 3:1 HDR donor:guide plasmid(s) ratio by mass. 24–36 h post-transfection, GFP positive cells were FACS sorted into single wells in a 96-well format, and screened as described above but with the size and sequence of the inserted fragments taken into account.
68,69,25RNA extraction and RT-PCR
On the day of cell harvest, samples of 105-2×106 cells (primary mouse cells, or CD71+ mES cell-derived models) were lysed in TRI Reagent (Sigma) and immediately frozen at −80°C. RNA was extracted using a Direct-zol MicroPrep kit (Zymo Research) with a 45 min DNase step on-column at room temperature. RNA quality was assessed by Agilent TapeStation instrument, using RNA ScreenTape (Agilent). Only samples with an RNA integrity score of at least 8 were taken forwards for subsequent analysis. The extracted RNA was reverse transcribed using Superscript III First-Strand Synthesis SuperMix (Life Technologies) including an RNase step to degrade remaining template molecules.
Real-time PCR (RT-PCR) was performed with Taqman probes (refer to KRT for the assays) to analyze gene expression in each model. Results were normalised to RPS18 or the relevant β-globin genes as an erythroid-specific highly expressed unaffected control gene. Reverse transcriptase (RT-) enzyme negative controls were included to confirm that DNase treatment was complete. Analysis steps including all statistical tests (ANOVA) and graphical plotting were conducted in RStudio. The R package ggplot2 was used to generate and render each plot (refer to KRT for all software).
NGS assays
ATAC-seq:
Assay for transposase-accessible chromatin (ATAC)-seq was performed on ~7×104 cells, using the illumina Tagment DNA enzyme and buffer kit (illumina), as previously described.17,75 Briefly, cells were lysed in a gentle NP-40 containing lysis buffer, and resuspended in Tn5 buffer with illumina adaptor-loaded Tn5 enzyme. Cells were incubated for 30 min at 37°C, and then tagmented DNA was purified using AMPure XP beads (mybeckman), before indexing with Nextera indexing primers (illumina). Indexed ATAC samples were assessed by tape station, using a High Sensitivity (HS) D1000 ScreenTape (Agilent).
ChIPmentation experiments were performed as previously described,76 with few modifications. On the day of cell harvest, aliquots of 1×105-1×106 cells (primary mouse cells, or CD71+ mouse ES cell-derived models) were either single-fixed with 1% formaldehyde for 10 min, followed by quenching with 125mM glycine, or double-fixed with 2mM disuccinimidyl glutarate (DSG) for 50 min, followed by 1% formaldehyde for 10 min, before quenching with 125mM Glycine. Single-fixed samples were ultimately used for ChIPmentation experiments assaying histone modifications; double-fixed samples were used for experiments assaying transcription factor occupancy (for antibodies refer to KRT).
Cells were spun down and washed with PBS, before being snap frozen. Fixed aliquots were stored at −80°C. Cell pellets were lysed in 0.5% SDS lysis buffer and sonicated, using a Covaris ME220 sonicator, to fragment DNA to an average fragment length of ~200–300bp. Sonicated chromatin was analyzed by Tapestation, using a D1000 or D1000 HS ScreenTape (Agilent). SDS in the lysis buffer was neutralised with 1% Triton X-, and the sonicate was incubated overnight with a mix of protein A and G dynabeads (Thermofisher) and the appropriate antibody (KRT). The following morning, chromatin-bound beads were washed three times using a low salt, a high salt and a LiCl-containing wash buffer, followed by tagmentation of the immunoprecipitated chromatin with sequencing adaptor-loaded tn5. Samples were indexed, using Nextera indices (illumina) and NEBNext 2X High fidelity mastermix.
Tiled-C, a high-resolution Chromosome Conformation Capture (3C) method that produces contact matrices of selected regions of interest was conducted as previously described.41 On the day of harvest, aliquots of 5×105 cells (primary mouse cells, or CD71+ mouse ES cell-derived models) were fixed with 2% formaldehyde for 10 min, before quenching with 125mM Glycine.
Cells were spun down, washed with PBS, and the pellet suspended in a mild NP-40-containing lysis buffer. Samples were then snap frozen and stored at −80°C. Cells in lysis buffer were thawed and spun down, before resuspension in restriction enzyme buffer mix. An appropriate volume of DpnII was added, and samples were incubated overnight at 37°C. Fresh aliquots of DpnII were added the following morning and afternoon. The DpnII was heat inactivated, and proximal DpnII-digested “sticky ends” were ligated using T4 ligase. Digested-re-ligated DNA was extracted using XP AMPure beads (mybeckman) and sonicated using a Covaris ME220 sonicator. Sonicated chromatin was analyzed by Tapestation, using a D1000 or D1000 HS screen tape (Agilent). The resultant fragments were indexed using the NEBNext Ultra II library preparation kit (New England BioLabs). Fragments corresponding to the region of interest (chr11:29902951–33226736) were enriched using oligo capture with biotinylated oligos (for oligo information, check KRT) complementary to every DpnII fragment within the tiled region, before streptavidin pulldown using Dynabeads M-280 Streptavidin (ThermoFisher).
RNA-seq:
on the day of harvest, aliquots of 5×105 cells (primary mouse cells, or CD71+ mouse ES cell-derived models) and processed as mentioned above.
Poly-A positive and negative RNA-seq was performed on 5×105 cells, using the NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (New England BioLabs). Ribosomal RNA was depleted using the NEBNext rRNA Depletion Kit, and then the poly-A positive and negative fractions were separated using the NEBNext Poly(A) mRNA Magnetic Isolation Module.
Sequencing and bioinformatic analysis
All NGS sequencing was performed using TG NSQ 500/550 Hi Output v2.5 (75 CYS) kits (illumina); these kits are paired-end sequencing kits which produce two 40 base pair reads, corresponding to the 5′ and the 3′ of the fragment being sequenced. Generally, ~25–40 million reads were desirable for each ATAC or ChIPmentation sample, ~10–20 million for each RNA-seq sample, and ~5–10 million for each Tiled-C sample, although actual sequencing depth was variable.
ATAC and ChIPmentation
The quality of the FASTQ files from ATAC-seq and ChIPmentation were assessed using FASTQC, and the reads aligned to the mm9 mouse genome, using bowtie2. Non-aligning reads were trimmed using Cutadapt trimgalore and then realigned to the mm9 genome using bowtie2.77 All reads which still failed to align were extracted, and flashed using FLASH, before realignment to the mm9 genome using bowtie2. All of the files containing successfully aligning reads were concatenated, and aligned to the mm9 genome together using bowtie2. Resultant SAM files were filtered, sorted, and PCR duplicates removed, using SAMtools (samtools view, sort, and rmdup, respectively). The resultant BAM file was indexed using SAMtools index, and converted to a bigwig file using deepTools bamcoverage.78 Each bigwig was visualised using the University of California Santa Cruz (UCSC) genome browser, and traces corresponding to regions of interest were downloaded from here. Peaks were called in each sample using MACS279 with default parameters, and differential accessibility/binding analysis was conducted using Bioconductor DESeq2 in RStudio.80 Generation of consensus peak files from multiple biological replicates was performed using bedtools intersect, and analysis of overlapping peaks/peak distances was performed using bedtools intersect and bedtools closest.81 Motif analysis was performed using the MEME suite82 (meme-chip for de novo motif analysis and fimo for finding occurrences of known motifs), using HOCOMOCO mouse position weight matrices. Principal component analysis was performed on ATAC samples, using the DiffBind, rgl and magick packages in RStudio.
Tiled-C
Samples were analyzed using the HiC-Pro pipeline,83 using the capture Hi-C workflow (aligning the data to the mm9 genome). To avoid interaction bias between regions within and outside of the tiled region, all data mapping to the tiled region was extracted and the remaining data discarded from subsequent analysis steps. Interaction matrices were ICE-normalised using HiC-Pro, and heatmaps generated for visualisation using ggplot2 in RStudio. Virtual capture plots were generated by extracting all entries within the tiled-C matrix in which a specific viewpoint of interest participates, and interaction scores normalised by dividing interaction scores by the total number of interactions within the tiled region. Virtual capture plots were produced for visualisation using ggplot2 in RStudio. Loess (local regression) smoothing (span = 0.05) was used reduce noise in the virtual capture-C plots. This effectively runs multiple local regressions for each datapoint along the x axis, with the span variable dictating the proportion of data taken into account when performing each regression. By re-plotting each datapoint based on the local regression prediction, therefore taking into consideration bins either side of the processed bin, I could reduce the noise in each plot. Because loess smoothing gives dramatically more weight to the values lying closest (along x) to the processed datapoint (weighting α (1-(distance/maximum distance)3),3 it allows one to smooth the data with little information loss.
RNA-seq
RNA-seq data was aligned to the mm9 genome, using star.84 The resultant SAM files were then filtered and sorted using SAMtools (samtools view and sort, respectively). The resultant BAM files were indexed using SAMtools index, and directional, rpkm normalised bigwigs generated using deepTools bamcoverage, with the filteredRNAstrand flag enabled. Each sample bigwig was visualised using the University of California Santa Cruz (UCSC) genome browser, and traces corresponding to regions of interest downloaded from here. Read coverage over each gene in the mm9 genome was calculated using Rsubread featurecounts,85 and differential expression analysis performed using edgeR in RStudio Y.
Plots were generated using ggplot2 in Rstudio. Principal component analysis was performed on RNA-seq samples, using the DiffBind, rgl and magick packages in RStudio. To compare enhancer RNA transcription in WT and R2-only cells, levels of poly-A negative RNA over the R1, R2, R3, Rm and R4 enhancers were visually assessed on the UCSC genome browser; however, this was only possible on the + strand, as the Nprl3 gene, in which the R1, R2 and R3 enhancers are located, is transcribed on the – strand. To compare R2 enhancer RNA transcription quantitatively, a virtual qPCR was performed, by normalizing the number of reads mapping to the R2 enhancer in each sample to the number of reads mapping to the HS2 enhancer of the β-globin LCR or the RPS18 gene in the same sample. Levels of the normalised enhancer RNA transcription in WT and R2-only samples were then compared and the results plotted, using ggplot2 in Rstudio.
QUANTIFICATION AND STATISTICAL ANALYSIS
One-way ANOVA with Tukey multiple comparisons of means with 95% family-wise confidence level was applied to all the expression analysis of all the genetic models generated in this paper. The statistical details of experiments can be found in the figure legends and depicted graphically in figures, including the statistical tests used, exact value of n, what n represents (e.g., number of animals, number of samples used). For the bioinformatic analyses, statistical solutions for differential detection of reads/peaks are included in various packages such a Bioconductor DESeq2, DiffBind, and EdgeR in Rstudio.
Supplementary Material
Highlights.
Large synthetic alleles allow de novo assembly of multipartite enhancers
Mouse α-globin super-enhancer contains classical enhancers and facilitators
Facilitators have no inherent enhancer activity but potentiate classical enhancers
Newly identified facilitators act in a position-dependent manner
ACKNOWLEDGMENTS
The authors would like to express their gratitude to all colleagues who contributed to this work, in particular Jackie Sloane-Stanley and the MRC Weatherall Institute of Molecular Medicine Transgenics Core Facility, Dr Philip Hublitz and the MRC Weatherall Institute of Molecular Medicine Genome Engineering Facility, and the MRC Weatherall Institute of Molecular Medicine Flow Cytometry Facility. The main contributing authors are funded by the Wellcome Trust (219979/Z/19/Z to J.W.B., 109097/Z/15/Z to H.F., 222843/Z/21/Z to L.C., and 215111/Z/18/Z to R.S.), the Chinese Academy of Medical Sciences (CAMS) Innovation Fund for Medical Science (CIFMS), China (grant 2018-I2M-2-002), and the UKRI Medical Research Council (MRC) (MR/T014067/1). This research was supported in part by National Institutes of Health (NIH/NHGRI) CEGS grant RM1HG009491 to J.D.B. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research or within their geographical location.
Footnotes
DECLARATION OF INTERESTS
J.D.B. is a Founder and Director of CDI Labs, Inc.; a Founder of and consultant to Neochromosome, Inc.; a Founder, SAB member of, and consultant to ReOpen Diagnostics and Logomix, Inc., LLC; and serves or served on the Scientific Advisory Board of the following: Sangamo, Inc., Modern Meadow, Inc., Rome Therapeutics, Inc., Sample6, Inc., Tessera Therapeutics, Inc., and the Wyss Institute. All other authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.cell.2023.11.030.
REFERENCES
- 1.Long HK, Prescott SL, and Wysocka J (2016). Ever-Changing Landscapes: Transcriptional Enhancers in Development and Evolution. Cell 167, 1170–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shlyueva D, Stampfel G, and Stark A (2014). Transcriptional enhancers: from properties to genome-wide predictions. Nat. Rev. Genet. 15, 272–286. [DOI] [PubMed] [Google Scholar]
- 3.Spitz F, and Furlong EEM (2012). Transcription factors: from enhancer binding to developmental control. Nat. Rev. Genet. 13, 613–626. [DOI] [PubMed] [Google Scholar]
- 4.Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, and Young RA (2013). Master Transcription Factors and Mediator Establish Super-Enhancers at Key Cell Identity Genes. Cell 153, 307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dębek S, and Juszczyński P (2022). Super enhancers as master gene regulators in the pathogenesis of hematologic malignancies. Biochim. Biophys. Acta. Rev. Cancer 1877, 188697. [DOI] [PubMed] [Google Scholar]
- 6.Harteveld CL, and Higgs DR (2010). α-Thalassaemia. Orphanet J. Rare Dis. 5, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Higgs DR, Engel JD, and Stamatoyannopoulos G (2012). Thalassaemia. Lancet 379, 373–383. [DOI] [PubMed] [Google Scholar]
- 8.Tang F, Yang Z, Tan Y, and Li Y (2020). Super-enhancer function and its application in cancer targeted therapy. npj Precis. Oncol. 4, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamagata K, Nakayamada S, and Tanaka Y (2020). Critical roles of super-enhancers in the pathogenesis of autoimmune diseases. Inflamm. Regen. 40, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blobel GA, Higgs DR, Mitchell JA, Notani D, and Young RA (2021). Testing the super-enhancer concept. Nat. Rev. Genet. 22, 749–755. [DOI] [PubMed] [Google Scholar]
- 11.Grosveld F, van Staalduinen J, and Stadhouders R (2021). Transcriptional Regulation by (Super)Enhancers: From Discovery to Mechanisms. Annu. Rev. Genom. Hum. Genet. 22, 127–146. [DOI] [PubMed] [Google Scholar]
- 12.Moorthy SD, Davidson S, Shchuka VM, Singh G, Malek-Gilani N, Langroudi L, Martchenko A, So V, Macpherson NN, and Mitchell JA (2017). Enhancers and super-enhancers have an equivalent regulatory role in embryonic stem cells through regulation of single or multiple genes. Genome Res. 27, 246–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pott S, and Lieb JD (2015). What are super-enhancers? Nat. Genet. 47, 8–12. [DOI] [PubMed] [Google Scholar]
- 14.Wang X, Cairns MJ, and Yan J (2019). Super-enhancers in transcriptional regulation and genome organization. Nucleic Acids Res. 47, 11481–11496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Catarino RR, and Stark A (2018). Assessing Sufficiency and Necessity of Enhancer Activities for Gene Expression and the Mechanisms of Transcription Activation. Genes Dev. 32, 202–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bender MA, Ragoczy T, Lee J, Byron R, Telling A, Dean A, and Groudine M (2012). The hypersensitive sites of the murine β-globin locus control region act independently to affect nuclear localization and transcriptional elongation. Blood 119, 3820–3827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hay D, Hughes JR, Babbs C, Davies JOJ, Graham BJ, Hanssen L, Kassouf MT, Marieke Oudelaar AM, Sharpe JA, Suciu MC, et al. (2016). Genetic dissection of the α-globin super-enhancer in vivo. Nat. Genet. 48, 895–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hnisz D, Schuijers J, Lin CY, Weintraub AS, Abraham BJ, Lee TI, Bradner JE, and Young RA (2015). Convergence of Developmental and Oncogenic Signaling Pathways at Transcriptional Super-Enhancers. Mol. Cell 58, 362–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hörnblad A, Bastide S, Langenfeld K, Langa F, and Spitz F (2021). Dissection of the Fgf8 regulatory landscape by in vivo CRISPR-editing reveals extensive intra- and inter-enhancer redundancy. Nat. Commun. 12, 439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang J, Li K, Cai W, Liu X, Zhang Y, Orkin SH, Xu J, and Yuan G-C (2018). Dissecting super-enhancer hierarchy based on chromatin interactions. Nat. Commun. 9, 943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shin HY, Willi M, HyunYoo K, Zeng X, Wang C, Metser G, and Hennighausen L (2016). Hierarchy within the mammary STAT5-driven Wap super-enhancer. Nat. Genet. 48, 904–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thomas HF, Kotova E, Jayaram S, Pilz A, Romeike M, Lackner A, Penz T, Bock C, Leeb M, Halbritter F, et al. (2021). Temporal dissection of an enhancer cluster reveals distinct temporal and functional contributions of individual elements. Mol. Cell 81, 969–982.e13. [DOI] [PubMed] [Google Scholar]
- 23.Hughes Jim R., Cheng Jan-Fang, Ventress Nicki, Prabhakar Shyam, Clark Kevin, Anguita Eduardo, Marco De Gobbi, Pieter de Jong, Eddy Rubin, and Douglas R. Higgs. Proc Natl Acad Sci U S A. 2005. Jul 12; 102(28): 9830–9835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mitchell LA, McCulloch LH, Pinglay S, Berger H, Bosco N, Brosh R, Bulajić M, Huang E, Hogan MS, Martin JA, et al. (2021). De novo assembly and delivery to mouse cells of a 101 kb functional human gene. Genetics 218, iyab038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wallace HAC, Marques-Kranc F, Richardson M, Luna-Crespo F, Sharpe JA, Hughes J, Wood WG, Higgs DR, and Smith AJH (2007). Manipulating the Mouse Genome to Engineer Precise Functional Syntenic Replacements with Human Sequence. Cell 128, 197–209. [DOI] [PubMed] [Google Scholar]
- 26.Francis HS, Harold CL, Beagrie RA, King AJ, Gosden ME, Blayney JW, Jeziorska DM, Babbs C, Higgs DR, and Kassouf MT (2022). Scalable in vitro production of defined mouse erythroblasts. PLoS One 17, e0261950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mitchell LA, and Boeke JD (2014). Circular permutation of a synthetic eukaryotic chromosome with the telomerator. Proc. Natl. Acad. Sci. USA 111, 17003–17010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sartorelli V, and Lauberth SM (2020). Enhancer RNAs are an important regulatory layer of the epigenome. Nat. Struct. Mol. Biol. 27, 521–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arnold PR, Wells AD, and Li XC (2019). Diversity and Emerging Roles of Enhancer RNA in Regulation of Gene Expression and Cell Fate. Front. Cell Dev. Biol. 7, 377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Allahyar A, Vermeulen C, Bouwman BAM, Krijger PHL, Verstegen MJAM, Geeven G, van Kranenburg M, Pieterse M, Straver R, Haarhuis JHI, et al. (2018). Enhancer hubs and loop collisions identified from single-allele topologies. Nat. Genet. 50, 1151–1160. [DOI] [PubMed] [Google Scholar]
- 31.Beagrie RA, Scialdone A, Schueler M, Kraemer DCA, Chotalia M, Xie SQ, Barbieri M, de Santiago I, Lavitas L-M, Branco MR, et al. (2017). Complex multi-enhancer contacts captured by genome architecture mapping. Nature 543, 519–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ing-Simmons E, Seitan VC, Faure AJ, Flicek P, Carroll T, Dekker J, Fisher AG, Lenhard B, and Merkenschlager M (2015). Spatial enhancer clustering and regulation of enhancer-proximal genes by cohesin. Genome Res. 25, 504–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li Y, Hu M, and Shen Y (2018). Gene regulation in the 3D genome. Hum. Mol. Genet. 27, R228–R233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oudelaar AM, and Higgs DR (2021). The relationship between genome structure and function. Nat. Rev. Genet. 22, 154–168. [DOI] [PubMed] [Google Scholar]
- 35.Hanssen LLP, Kassouf MT, Oudelaar AM, Biggs D, Preece C, Downes DJ, Gosden M, Sharpe JA, Sloane-Stanley JA, Hughes JR, et al. (2017). Tissue-specific CTCF/Cohesin-mediated chromatin architecture delimits enhancer interactions and function in vivo. Nat. Cell Biol. 19, 952–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hua P, Badat M, Hanssen LLP, Hentges LD, Crump N, Downes DJ, Jeziorska DM, Oudelaar AM, Schwessinger R, Taylor S, et al. (2021). Defining Genome Architecture at Base-Pair Resolution. Nature 595, 125–129. [DOI] [PubMed] [Google Scholar]
- 37.Hughes JR, Roberts N, McGowan S, Hay D, Giannoulatou E, Lynch M, De Gobbi M, Taylor S, Gibbons R, and Higgs DR (2014). Analysis of hundreds of cis-regulatory landscapes at high resolution in a single, high-throughput experiment. Nat. Genet. 46, 205–212. [DOI] [PubMed] [Google Scholar]
- 38.King AJ, Songdej D, Downes DJ, Beagrie RA, Liu S, Buckley M, Hua P, Suciu MC, Marieke Oudelaar A, Hanssen LLP, et al. (2021). Reactivation of a developmentally silenced embryonic globin gene. Nat. Commun. 12, 4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oudelaar AM, Davies JOJ, Hanssen LLP, Telenius JM, Schwessinger R, Liu Y, Brown JM, Downes DJ, Chiariello AM, Bianco S, et al. (2018). Single-Allele Chromatin Interactions Identify Regulatory Hubs in Dynamic Compartmentalized Domains. Nat. Genet. 50, 1744–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oudelaar AM, Harrold CL, Hanssen LLP, Telenius JM, Higgs DR, and Hughes JR (2019). A revised model for promoter competition based on multi-way chromatin interactions at the α-globin locus. Nat. Commun. 10, 5412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oudelaar AM, Beagrie RA, Gosden M, de Ornellas S, Georgiades E, Kerry J, Hidalgo D, Carrelha J, Shivalingam A, El-Sagheer AH, et al. (2020). Dynamics of the 4D Genome during in Vivo Lineage Specification and Differentiation. Nat. Commun. 11, 2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Castro-Mondragon JA, Riudavets-Puig R, Rauluseviciute I, Lemma RB, Turchi L, Blanc-Mathieu R, Lucas J, Boddie P, Khan A, Manosalva Pérez N, et al. (2022). JASPAR 2022: the 9th release of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 50, D165–D173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fornes O, Castro-Mondragon JA, Khan A, van der Lee R, Zhang X, Richmond PA, Modi BP, Correard S, Gheorghe M, Baranašić D, et al. (2020). JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 48, D87–D92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schwessinger R, Suciu MC, McGowan SJ, Telenius J, Taylor S, Higgs DR, and Hughes JR (2017). Sasquatch: predicting the impact of regulatory SNPs on transcription factor binding from cell- and tissue-specific DNase footprints. Genome Res. 27, 1730–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fraser P, Pruzina S, Antoniou M, and Grosveld F (1993). Each hypersensitive site of the human beta-globin locus control region confers a different developmental pattern of expression on the globin genes. Genes Dev. 7, 106–113. [DOI] [PubMed] [Google Scholar]
- 46.Milot E, Strouboulis J, Trimborn T, Wijgerde M, de Boer E, Langeveld A, Tan-Un K, Vergeer W, Yannoutsos N, Grosveld F, and Fraser P (1996). Heterochromatin Effects on the Frequency and Duration of LCR-Mediated Gene Transcription. Cell 87, 105–114. [DOI] [PubMed] [Google Scholar]
- 47.Hardison R, Slightom JL, Gumucio DL, Goodman M, Stojanovic N, and Miller W (1997). Locus control regions of mammalian β-globin gene clusters: combining phylogenetic analyses and experimental results to gain functional insights. Gene 205, 73–94. [DOI] [PubMed] [Google Scholar]
- 48.Banerji J, Rusconi S, and Schaffner W (1981). Expression of a β-globin gene is enhanced by remote SV40 DNA sequences. Cell 27, 299–308. [DOI] [PubMed] [Google Scholar]
- 49.Mercola M, Wang X-F, Olsen J, and Calame K (1983). Transcriptional Enhancer Elements in the Mouse Immunoglobulin Heavy Chain Locus. Science 221, 663–665. [DOI] [PubMed] [Google Scholar]
- 50.Grosveld F, van Assendelft GB, Greaves DR, and Kollias G (1987). Position-independent, high-level expression of the human β-globin gene in transgenic mice. Cell 51, 975–985. [DOI] [PubMed] [Google Scholar]
- 51.Hong J-W, Hendrix DA, and Levine MS (2008). Shadow Enhancers as a Source of Evolutionary Novelty. Science 321, 1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Montavon T, Soshnikova N, Mascrez B, Joye E, Thevenet L, Splinter E, de Laat W, Spitz F, and Duboule D (2011). A Regulatory Archipelago Controls Hox Genes Transcription in Digits. Cell 147, 1132–1145. [DOI] [PubMed] [Google Scholar]
- 53.Markenscoff-Papadimitriou E, Allen WE, Colquitt BM, Goh T, Murphy KK, Monahan K, Mosley CP, Ahituv N, and Lomvardas S (2014). Enhancer Interaction Networks as a Means for Singular Olfactory Receptor Expression. Cell 159, 543–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Parker SCJ, Stitzel ML, Taylor DL, Orozco JM, Erdos MR, Akiyama JA, van Bueren KL, Chines PS, Narisu N, et al. ; NISC Comparative Sequencing Program (2013). Chromatin stretch enhancer states drive cell-specific gene regulation and harbor human disease risk variants. Proc. Natl. Acad. Sci. USA 110, 17921–17926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bender MA, Bulger M, Close J, and Groudine M (2000). β-globin Gene Switching and DNase I Sensitivity of the Endogenous β-globin Locus in Mice Do Not Require the Locus Control Region. Mol. Cell 5, 387–393. [DOI] [PubMed] [Google Scholar]
- 56.Epner E, Reik A, Cimbora D, Telling A, Bender MA, Fiering S, Enver T, Martin DI, Kennedy M, Keller G, and Groudine M (1998). The β-Globin LCR Is Not Necessary for an Open Chromatin Structure or Developmentally Regulated Transcription of the Native Mouse β-Globin Locus. Mol. Cell 2, 447–455. [DOI] [PubMed] [Google Scholar]
- 57.Schübeler D, Groudine M, and Bender MA (2001). The murine β-globin locus control region regulates the rate of transcription but not the hyperacetylation of histones at the active genes. Proc. Natl. Acad. Sci. USA 98, 11432–11437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Oudelaar AM, Beagrie RA, Kassouf MT, and Higgs DR (2021). The mouse alpha-globin cluster: a paradigm for studying genome regulation and organization. Curr. Opin. Genet. Dev. 67, 18–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sahu B, Hartonen T, Pihlajamaa P, Wei B, Dave K, Zhu F, Kaasinen E, Lidschreiber K, Lidschreiber M, Daub CO, et al. (2022). Sequence determinants of human gene regulatory elements. Nat. Genet. 54, 283–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Thanos D, and Maniatis T (1995). Virus induction of human IFNβ gene expression requires the assembly of an enhanceosome. Cell 83, 1091–1100. [DOI] [PubMed] [Google Scholar]
- 61.Merika M, and Thanos D (2001). Curr. Opin. Genet. Dev. 11, 205–208. [DOI] [PubMed] [Google Scholar]
- 62.Kassouf MT, Francis HS, Gosden M, Suciu MC, Downes DJ, Harrold C, Larke M, Oudelaar M, Cornell L, Blayney J, et al. (2022). Multipartite Super-Enhancers Function in an Orientation-Dependent Manner. Preprint at bioRxiv.. 10.1101/2022.07.14.499999. [DOI] [Google Scholar]
- 63.Levo M, Raimundo J, Bing XY, Sisco Z, Batut PJ, Ryabichko S, Gregor T, and Levine MS (2022). Transcriptional coupling of distant regulatory genes in living embryos. Nature 605, 754–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Boija A, Klein IA, Sabari BR, Dall’Agnese A, Coffey EL, Zamudio AV, Li CH, Shrinivas K, Manteiga JC, Hannett NM, et al. (2018). Transcription Factors Activate Genes through the Phase-Separation Capacity of Their Activation Domains. Cell 175, 1842–1855.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sabari BR, Dall’Agnese A, Boija A, Klein IA, Coffey EL, Shrinivas K, Abraham BJ, Hannett NM, Zamudio AV, Manteiga JC, et al. (2018). Coactivator condensation at super-enhancers links phase separation and gene control. Science 361, eaar3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gurumurthy A, Shen Y, Gunn EM, and Bungert J (2019). Phase Separation and Transcription Regulation: Are Super-Enhancers and Locus Control Regions Primary Sites of Transcription Complex Assembly? Bio-essays 41, 1800164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sigova AA, Abraham BJ, Ji X, Molinie B, Hannett NM, Guo YE, Jangi M, Giallourakis CC, Sharp PA, and Young RA (2015). Transcription factor trapping by RNA in gene regulatory elements. Science 350, 978–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Trojanowski J, Frank L, Rademacher A, Mücke N, Grigaitis P, and Rippe K (2022). Transcription activation is enhanced by multivalent interactions independent of phase separation. Mol. Cell 82, 1878–1893.e10. [DOI] [PubMed] [Google Scholar]
- 69.Jackson M, Taylor AH, Jones EA, and Forrester LM (2010). Mouse Cell Culture, Methods and Protocols. Methods Mol. Biol. 633, 1–18. [DOI] [PubMed] [Google Scholar]
- 70.Smith AG (1991). Culture and differentiation of embryonic stem cells. J. Tissue Cult. Methods 13, 89–94. [Google Scholar]
- 71.Osoegawa K, Tateno M, Woon PY, Frengen E, Mammoser AG, Catanese JJ, Hayashizaki Y, and de Jong PJ (2000). Bacterial artificial chromosome libraries for mouse sequencing and functional analysis. Genome Res. 10, 116–128. [PMC free article] [PubMed] [Google Scholar]
- 72.Smith AJH, Xian J, Richardson M, Johnstone KA, and Rabbitts PH (2002). Cre-loxP chromosome engineering of a targeted deletion in the mouse corresponding to the 3p21.3 region of homozygous loss in human tumours. Oncogene 21, 4521–4529. [DOI] [PubMed] [Google Scholar]
- 73.Schaft J, Ashery-Padan R, van der Hoeven F, Gruss P, and Stewart AF (2001). Efficient FLP recombination in mouse ES cells and oocytes. genesis 31, 6–10. [DOI] [PubMed] [Google Scholar]
- 74.Kredel S, Oswald F, Nienhaus K, Deuschle K, Röcker C, Wolff M, Heilker R, Nienhaus GU, and Wiedenmann J (2009). mRuby, a Bright Monomeric Red Fluorescent Protein for Labeling of Subcellular Structures. PLoS One 4, e4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Buenrostro JD, Wu B, Chang HY, and Greenleaf WJ (2015). ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr. Protoc. Mol. Biol. 109, 21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schmidl C, Rendeiro AF, Sheffield NC, and Bock C (2015). ChIPmentation: fast, robust, low-input ChIP-seq for histones and transcription factors. Nat. Methods 12, 963–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based Analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bailey TL, Johnson J, Grant CE, and Noble WS (2015). The MEME Suite. Nucleic Acids Res. 43, W39–W49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Servant N, Varoquaux N, Lajoie BR, Viara E, Chen C-J, Vert J-P, Heard E, Dekker J, and Barillot E (2015). HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Liao Y, Smyth GK, and Shi W (2019). The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 47, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated for this study are included in this published article and its supplementary information. Standardized data types (ChIP-seq, ATAC-seq RNA-seq and Tiled Capture-C data, raw data and processed files) are publicly accessible in the Gene Expression Omnibus (GEO) under accession numbers GEO: GSE220463.
This paper does not report original code. Codes used in the analysis of this manuscript are referenced.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.