

Summary
Today’s genomics workflows typically require alignment to a reference sequence, which limits discovery. We introduce a unifying paradigm, SPLASH (Statistically Primary aLignment Agnostic Sequence Homing), which directly analyzes raw sequencing data, using a statistical test to detect a signature of regulation: sample-specific sequence variation. SPLASH detects many types of variation and can be efficiently run at scale. We show that SPLASH identifies complex mutation patterns in SARS-CoV-2, discovers regulated RNA isoforms at the single cell level, documents the vast sequence diversity of adaptive immune receptors, and uncovers biology in non-model organisms undocumented in their reference genomes: geographic and seasonal variation and diatom association in eelgrass, an oceanic plant impacted by climate change, and tissue-specific transcripts in octopus. SPLASH is a unifying approach to genomic analysis that enables expansive discovery without metadata or references.
eTOC/In Brief:
Genomics workflows typically first map reads onto a reference genome as the foundation for downstream analyses. However, this poses severe limitations for biological discovery when references are incomplete or nonexistent and even for intensely studied genomes with rich population-level diversity. SPLASH is a highly efficient framework for statistics-driven analysis of sequence variation directly from raw sequencing data, overcoming previous limitations.
Graphical Abstract
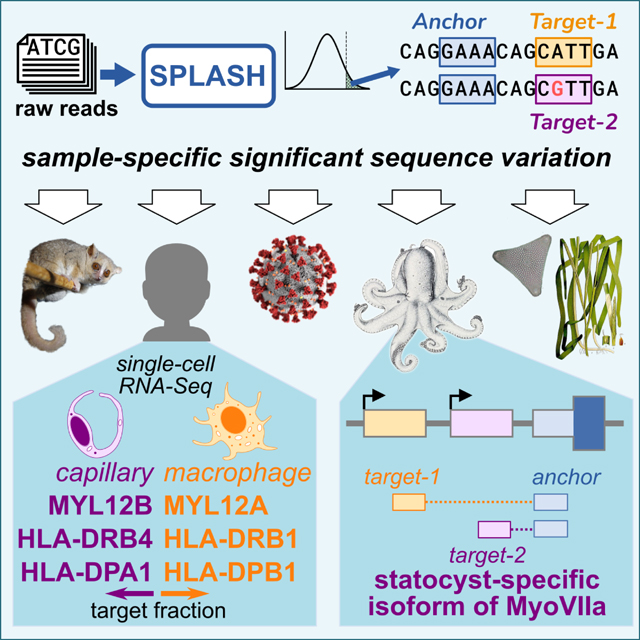
Introduction
Genomics is now foundational to biology, ecology and medicine, and as sequencing databases grow, so too does the opportunity to leverage them for discovery. How can this data best be analyzed to reveal regulation and function? Traditionally, bioinformatic pipelines start by assigning genomic positions to reads via alignment to a reference genome, an approach with many limitations. For less studied organisms, references can be partially misassembled, incomplete, or nonexistent. Even in the intensely studied human genome, it was found that under-studied populations have large amounts of sequence missing from the current reference1; such blindspots may exacerbate health disparities. Reference-based methods are not well-suited to deal with paralogs and repetitive elements (which comprise ~54% of the human genome2), so many analyses simply ignore them. They are also poorly suited for diseases such as cancer that are almost defined by their deviations from the reference, and vary even within a single tumor. Additionally, the enormous diversity of viral and microbial genomes and their constant adaptation3,4 makes it infeasible to define a complete set of references. Practically, alignment to references is computationally intensive, limiting the scale of genomic inference.
When dealing with genomic data, precise statistical analysis is critical. However, alignment-based methods are complex and difficult to model statistically; even seemingly simple tasks such as calling allele-specific expression can be fraught with statistical imprecision introduced during alignment5. Permutation-based methods are not a panacea; in addition to being slow, they can give 10-fold underestimates of the false discovery rate6,7.
Addressing these concerns has led us to a simple unifying paradigm for statistically detecting signals of biological interest directly from raw sequencing data without using a reference genome, which we call SPLASH (Statistically Primary aLignment Agnostic Sequence Homing). It relies on a simple formalization of sequence variation (short stretches of varying sequences, “targets”, adjacent to short stretches of constant sequences, “anchors”). SPLASH applies to myriad biological questions that can be framed as asking how sequence distributions vary within a set of samples. In the results below, we provide a snapshot of SPLASH’s wide possibilities for discovery, encompassing viral strain variation, single-cell level alternative isoforms, and antigen receptor diversity in human samples. We also show that SPLASH is easily applied to less studied organisms: lemur, octopus, and eelgrass. This demonstrates SPLASH’s potential to discover meaningful sequence variation without the aid of reference genomes, across many biological questions and organisms.
Results
SPLASH is a -mer based, statistics-first approach to identify sample-dependent sequence variation
The goal of SPLASH is to detect sequence variation between a set of samples. SPLASH uses a specific conception of variation based on -mers, or subsequences of length , in sequencing reads. This framework leads to a simple but powerful statistical test that identifies variation that is differentially distributed among the samples.
SPLASH only requires that each sample is represented by a separate file of sequencing data (FASTQ). The definition of a “sample” is dictated by the biological question: samples can denote different cells, different tissues, different individuals, or different mixed populations (metagenomics). Samples might differ by conditions as well – different times or treatments, and also in other features (cell-type, geographic location, etc.); we refer to these generically as “metadata”.
SPLASH characterizes variation using -mer pairs called “anchors” and “targets” (Figure 1A) ( by default, but is adjustable). Every -mer in the data is an anchor; each -mer a fixed offset downstream (, which may be zero) from a given anchor is one of its targets. Note that targets are always defined relative to an anchor. Anchors with more than one target can report on most sequence variation of interest: from changes at a single position to alternative splicing and isoform usage, gene rearrangements, and more.
Figure 1. Overview of SPLASH.

A. An anchor is a sequence of length (-mer) in a read; its target is the -mer that follows it after a fixed offset of length . An anchor may occur with different targets, which can capture many types of variation; examples are depicted schematically, with the anchor as a blue box and the targets as orange or violet boxes.
B. SPLASH compiles a table for each anchor, where the columns are samples, the rows are targets, and the entries are the respective occurrence counts. SPLASH tests multiple random splits of the samples, calculating a test statistic that measures the deviations between each sample’s target distribution and the average target distribution over all samples, searching for the most discriminating split. For the best split identified, SPLASH reports a -value bound.
C. Alignment-based pipelines are limited by the need for a reference genome, compute-intensive, and difficult to model statistically due to their complexity. SPLASH detects variation directly from raw reads with rigorous statistics, is compute-efficient, and detects many kinds of variation at once.
D. Some considerations of when SPLASH may be most useful, which reflect its design characteristics.
The SPLASH process is depicted in Figure 1B (detailed in STAR Methods, Figure S1). Conceptually, SPLASH steps through all positions in all reads of all samples, counting all anchor-target pairs. (To decrease compute time, SPLASH can analyze anchors at a subset of read positions, e.g., every fifth position, as used in this work.) SPLASH compiles a counts table for each anchor, with a column for each sample and row for each target; each entry is the count of a given target in a given sample (a contingency table). This requires only one pass through the raw sequences and does not involve reference alignment, so it is computationally efficient. Importantly, we have developed a highly flexible test statistic that captures the desire to find relatively discrete groups of samples with differing variation, and controls false positives even for low numbers of observations; it admits a closed form -value bound, which is thus fast to compute (unlike resampling methods). For each anchor, SPLASH calculates a -value bound for the null hypothesis that the observed target frequencies in samples all come from the same distribution, i.e., that there is no underlying variation of targets between samples. A low -value for an anchor implies that samples differ significantly in which targets they contain.
While SPLASH can use sample metadata (running in a “supervised” mode), SPLASH does not require such information. Indeed, for all the results presented here, SPLASH was run in its default unsupervised mode. For each anchor, SPLASH tries many random splits of the samples and the targets, retains the one which minimizes the -value, and reports the corresponding effect size for this grouping (next paragraph). This process can detect patterned target variation among the samples, if it exists (Figure S3A, STAR Methods).
SPLASH also calculates for each anchor an “effect size” that ranges from 0 to 1, with 0 meaning that the target distribution is the same between the two groups, and 1 meaning that the targets found between the two groups are disjoint. Effect size does not require that target distributions of all samples within a group are similar, just that they are different from the other group; thus, effect size can be high even when there are more than two natural groups. Anchors with large effect sizes have target variation that is more discrete across the samples, and are more promising for further biological investigation.
To interpret SPLASH’s findings, it can be useful to have longer sequence context than just the anchor and target. Thus, SPLASH also generates a “consensus” for each anchor, in each sample. Consensuses are longer sequences assembled from the raw reads of a given sample, looking at every occurrence of an anchor and extending base by base as long as the reads show a consensus (see STAR Methods). Mapping consensuses to protein sequences can identify protein domains – a powerful, reference-free attribution of biological function. Consensuses can also be aligned to sequence databases or reference genomes; aligning only the consensus sequences for significant anchors reduces the typical computational load by over 500-fold compared to usual approaches that align all reads (Figure S3B).
Figure 1C diagrams some of the differences between traditional alignment-first approaches and SPLASH; Figure 1D outlines some use cases for SPLASH. These guided us in our initial explorations with SPLASH, which are described below. While SPLASH has some adjustable parameters, we did not attempt to tune these (indeed, SPLASH seems robust to a range of parameters, STAR Methods); all analyses were run with the same settings, in unsupervised mode (blind to metadata). Despite this, we found that SPLASH performed well across a variety of datasets, and in all cases found significant patterns of sequence variation (-values for anchors, and binomial -values for target-fraction plots, are given in STAR Methods).
SPLASH identifies strain-defining and other mutations in SARS-CoV-2 de novo
Viral genomes have high mutation rates, at the extremes forming quasispecies3. The emergence of SARS-CoV-2 was followed by multiple surges caused by variant strains, over the course of just two years. This is an ideal setting for the application of SPLASH: viruses are always mutating, but out of a sea of mutations, scientists, clinicians and public health officials want to identify those showing consistent and significant variation.
We applied SPLASH to two SARS-CoV-2 datasets, both viral amplicon Illumina sequencing of nasopharyngeal swabs from infection-positive patients, taken from times when the dominant strains were Delta or Omicron. The samples from South Africa8 (Nov to Dec-2021) represent the rapid rise of Omicron during its first outbreak. The samples from France9 (Dec-2021 to Feb-2022) represent cases of co-infection by more than one strain; the study authors provided as metadata the assignment of the primary and secondary viral strains for each case (Table S1) (though not used by SPLASH).
SPLASH finds many significant anchors with low -values (<0.05) and high effect-sizes (>0.5) directly from sequencing reads (250 for South Africa dataset, 252 for France; Table S2). High effect sizes are expected for anchors whose targets partition samples by strain. To test if SPLASH recovers strain-defining and other variation, we examined the subset of significant anchors that perfectly map to a reference strain (Original, Delta, Omicron BA.1 or BA.2; defining mutations taken from CoVariants.org10), and call an anchor “strain-defining” if it has at least two targets (by definition different) with >5% abundance, at least one of which perfectly matches to a reference strain. We compare to a control set of anchors, those that are most abundant across all the reads. In the South Africa dataset, 98% (126/128) of SPLASH anchors that mapped perfectly were strain-defining, vs. 7/201 (3.5%) in the control set (hypergeometric -value <1.7E-79). In the France dataset, 100% (39/39) of SPLASH anchors were strain-defining, vs. 8.4% (21/250) of control anchors (hypergeometric -value <2.6E-33). Nearly all the control anchors have only a single abundant target. Thus SPLASH, though blind to strain reference sequences and sample metadata, detects strain differences with high precision.
Figure 2 shows exemplary strain-defining mutations identified by SPLASH in the Spike protein (S gene). Figure 2A shows an anchor that distinguishes at the major lineage level: one target has no mutations and is consistent with Delta; the other target has the mutation K417N, found in all Omicron strains (but not in Delta or Original). Target fractions across samples are consistent with the strain assignment metadata. Figure 2B shows an anchor that discriminates sub-lineages: one target has no mutation, consistent with Delta; the second target has the 3-nt deletion NL211I and the 9-nt insertion R214REPE which are Omicron BA.1 specific; the third target has the mutation V213G which is Omicron BA.2 specific. Figure 2C shows an anchor that detects emergent mutations not in our references. One target has a pair of mutations, N679K and P681H, found in all Omicron strains. The other targets all have P681R, a Delta-specific mutation, but two targets additionally encode Q677H (by different mutations). Q677 mutations have arisen independently multiple times in different lineages11,12, and Q677H in several strain backgrounds enhanced infectivity, syncytia formation, and resistance to neutralizing antibodies in pseudotype assay13.
Figure 2. SPLASH identifies strain-defining and other variation in SARS-Cov2.

In A-C, sets of targets that distinguish SARS-CoV-2 strains are shown; all are in the spike protein (S) gene. Each heatmap has columns for different samples (patients) and rows for different targets; the coloring indicates the fraction of that target observed in that patient. Summary anchor counts are given for rows and columns. Also shown is a map of the categorical metadata of what strains (primary and secondary) were identified in each patient in the original study; this data was not used by SPLASH, but there is evident agreement between the heatmap and the metadata strain assignment. Binomial -value bounds were computed per STAR Methods.
A. Distinguishing at the major strain level: target 1 (no mutation) matches Delta; target 2 contains K417N, found in all Omicron (both BA.1 and BA.2 sub-strains); two patients co-infected with Delta and Omicron show both targets. ().
B. Distinguishing at the sub-strain level: target 1 (no mutation) matches Delta; target 2 with V213G is specific for BA.2; target 3 with a deletion (NL211I) and insertion (R214REPE) is specific for BA.1. ()
C. Distinguishing non-strain related mutations: target 1 has P681R, Delta specific; targets 2 and 3 encode Q677H (by different mutations) and P681R; target 4 has N679K and P681H, Omicron specific. ()
D. Protein domain profiling in SARS-CoV-2. The top four and bottom four Pfam protein domain hits are shown. S1 Receptor binding domain (RBD) and S2 domain show high variation by SPLASH in both datasets. Other Pfam abbreviations: bCoV = beta-coronavirus; CoV = coronavirus, nucleocap = nucleocapsid N = N-terminal domain, SARS = Severe acute respiratory syndrome coronavirus, M, NS7A, NSP1, NSP8, 3b, NSP10 = viral proteins.
SPLASH results can also be analyzed completely without a reference genome by examining their coding potential.The consensus sequences are translated in silico to amino acid sequences (in all six reading frames) and matched against a database of protein domain models such as Pfam14. Protein profiles that are more frequently associated with significant SPLASH anchors, compared to control anchors, are candidates for proteins with important patterns of variation. The distribution of protein domains for SPLASH anchors was statistically different from controls (chi-squared test -values: France <3.7E-7, South Africa <2.5E-39) (Figure 2D). The top four protein domains in both datasets were beta-coronavirus receptor-binding domain (RBD; within S1 region of spike protein), coronavirus S2 domain (within spike protein), coronavirus M protein, and coronavirus ORF7a protein. By contrast, the bottom four domains for each dataset were completely different from each other. Protein profiling pinpoints domains undergoing high variation; for example, in the South Africa dataset the spike S2 domain had 23 SPLASH vs 3 control hits, (corrected hypergeometric -value). The spike protein is the major site of antigenic variation in coronaviruses, as it is a principal focus of the immune response; the RBD is well known as a target for natural and therapeutic neutralizing antibodies15, but in addition about 50% of natural anti-spike antibodies are directed against the S2 domain16.
We also carried out SPLASH protein domain profiling on an unrelated virus, rotavirus17. The domains enriched over controls were rotavirus VP3 and NSP3 proteins (Figure S2). These two proteins have roles in blocking host innate immunity18. Thus, variation in viral protein domains interacting with the immune system may be a recurring theme in SPLASH protein profiling of viral strains.
In summary, SPLASH finds patterns of variation in SARS-CoV-2, including those characterizing strains, without requiring reference sequences or metadata; the methodology should be generally applicable to other viruses. More broadly, SPLASH may be useful in surveillance for new strains or even completely new pathogens, and to cluster patients directly from raw sequencing samples.
SPLASH identifies regulated expression of paralogs and HLA in single cell RNA-seq
Current approaches to single-cell transcriptomics are reference-based and specialized; we sought to see if SPLASH’s unifying methodology could also be applied to single cell sequencing data generated with the Smart-Seq2 (SS2) protocol19, which provides broad transcript coverage.
Our first testbed was human macrophage and capillary cells from the Human Lung Cell Atlas20 (Table S2 and S3), as it was recently established that these cell types have regulated alternative splicing in MYL6, a light chain subunit of myosin motor protein, which serves as a positive control21. As expected, among SPLASH’s significant anchors are ones reporting on MYL6 alternative splicing (exon skipping or inclusion) (Figure S4). Interestingly, other SPLASH top anchors also involved myosin light chains, MYL12A and MYL12B, two paralogs with highly similar coding regions (95% nucleotide, 98% amino acid identity for human). Nevertheless, SPLASH finds targets that specifically distinguish them, showing that macrophages express more MYL12A, while capillary cells express more MYL12B, reproducible in two individuals (Figure 3A). Little is known about these genes, but they show differential expression in rat tissues22, and there is evolutionary conservation in mammals, birds, and reptiles of adjacent MYL12 paralogs within a syntenic region (e.g. human; rat22; Gallus gallus, NCBI Gene IDs 396284 and 770011; Chelonia mydas, Gene IDs 102938771 and 102937279). Besides the small number of amino acid differences between the paralogs, there may also be an important functional role for nucleotide sequence differences, as has been demonstrated for another pair of highly similar cytoskeletal paralogs, beta- and gamma-actin23.
Figure 3. Cell-type specific expression of paralogs and HLA from single-cell data.

Figures A-C show spread plots, each dot representing the relative isoform expression in a single cell; bar marks average relative expression across all cells.
A. Human MYL12A and MYL12B lie adjacent on chromosome 18, a region syntenic in mammals, chickens, and reptiles. The sequence alignment shows the two genes are very similar in the coding region, and marks the anchor and two targets for individual P2 (P3 has a distinct anchor). Macrophages express relatively more MYL12A and capillary cells more MYL12B, consistent in two individuals.
B. The HLA-DRB locus occurs as several different haplotypes, all containing DRB1 but differing in paralog (DRB3, DRB4, DRB5, or none; hg38 reference has DRB5). The anchor and its targets lie in the 3’ untranslated regions of DRB1 and DRB4; the two individuals have distinct alleles at both DRB1 and DRB4. Macrophages express mainly DRB1 and capillary cells mainly DRB4.
C. HLA-DPA1 and HLA-DPB1 overlap in head-to-head arrangement as shown in the UCSC Genome Browser diagram, which also shows the BLAT mapping of the anchor consensuses for DPA1 and DPB1, which lie on opposite strands. This is also depicted in the alignment; the targets are best assigned to opposite strands. An anchor simultaneously reporting on DPA1 and DPB1 was only found for individual P3; its targets show that macrophages exclusively express DPB1, while capillary cells mainly express DPA1.
D. The polymorphic HLA-B gene contains many SPLASH anchors from T cells, as depicted in the UCSC Browser diagram. The hg38 reference is the B*07:02 allele, whereas this individual matches best to B*08 and B*51 (consensus sequences 1 and 2). We investigated one HLA-B anchor, which lies in exon 2. In the alignment, differences from hg38 in the anchor and lookahead region are in lowercase. In the scatterplot, cells show a wide range of allele expression ratios, some expressing a single allele. Dashed lines mark a 98% confidence interval for the binomial distribution based on the population average expression (confidence depends on anchor counts); the observed data deviates significantly (binomial ), and some cells express almost exclusively one allele.
In the same data, SPLASH finds cell type-specific expression of genes in the major histocompatibility complex (MHC), known as HLA in humans. HLA is the most polymorphic region in the genome, with the most known disease associations; the polymorphism of HLA class I and class II proteins is intimately tied to their function in antigen presentation for adaptive immunity24. Due to the high levels of polymorphism, the region is challenging to represent in a reference genome and for alignment pipelines. Five major haplotypes have been identified at the HLA-DRB locus: all contain DRB1, but some contain a second functional paralog, either DRB3, DRB4, or DRB525. DRB1 is highly polymorphic (3516 alleles, in March 2023); the paralogs somewhat less so, e.g. DRB4 (236 alleles)26. SPLASH identifies an anchor with targets that distinguish between the highly similar 3’ untranslated regions (UTRs) of HLA-DRB1 and HLA-DRB4 (class II beta-chains) (Figure 3B). Macrophages express mainly DRB1, while capillary cells mainly express DRB4. This pattern is found in two individuals, who carry different alleles at DRB1 and DRB4. Macrophages are “professional” antigen-presenting cells and constitutively express HLA class II. Not all endothelial cells express class II, however most human capillary cells do27; endothelial MHC expression may be strongly cytokine-dependent28. Thus, class II expression in macrophages and capillary cells is likely to be regulated differently.
In one individual (P3), SPLASH finds a remarkable anchor whose two targets report on HLA-DPA1 and HLA-DPB1 (class II alpha and beta chains, respectively), unique among HLA genes in being organized head-to-head and transcribed in opposite directions. The anchor lies in sequence shared by DPB1 and a specific isoform of DPA1, while the targets lie in exons exclusive to each; SPLASH consensus sequences confirm opposite directionality as they bridge splice junctions. Macrophages in this individual express exclusively DPB1, while capillary cells express mainly DPA1 (Figure 3C). This pattern may be haplotype-specific, as we did not find a similar anchor for another individual (P2).
A final example is SPLASH’s detection of allele-specific expression of HLA-B in T cells (from a different dataset, see next section). The class I gene HLA-B is the most polymorphic of all HLA genes (9274 alleles)26, and HLA-B is the gene with the most anchors found by SPLASH in human T cells (Figure 3D). Since these T cells all come from one individual, this indicates substantial variation in expression of this individual’s two HLA-B alleles at the single-cell level (Figure 3D). Different T cells express a wide range of ratios of the two alleles, some cells expressing both alleles, but others expressing almost entirely one allele or the other (well outside the 98% confidence interval of what is expected by the average ratio). This is in keeping with a preprint that found allele-specific expression of HLA class I genes in normal breast epithelial cells29.
Overall, SPLASH finds multiple types of variation regulated at the single cell level, including paralogs, splicing, and alleles. This gives a glimpse into the complexity of HLA haplotype- and cell type-specific expression patterns, raising the possibility that disease-related HLA alleles might be expressed differently in key cell types compared to other alleles.
SPLASH identifies B and T cell receptor diversity in human and lemur single cell RNA-seq
Adaptive immune receptors for B cells (immunoglobulin or Ig), and T cells (T cell receptor or TCR) are generated combinatorially through V(D)J recombination, and Ig is further diversified through somatic hypermutation. Rearranged sequences are absent from germline reference genomes and cannot be cataloged comprehensively due to their huge potential diversity, empirically estimated at 1010-1011 for Ig heavy chains30. These genomic loci currently require manual curation due to their complexity and repetitive structure, so few species have high quality annotations. Existing methods to assign V(D)J rearrangements in single cells31 depend critically on annotations and so may have blindspots. Since SPLASH is designed to identify sequence diversity without a reference, we predicted that it would identify adaptive immune receptors de novo.
We ran SPLASH on 50 naive human B cells from peripheral blood of one individual, and separately on 128 CD4+ human T cells of another individual, taken from Tabula Sapiens, a large multi-organ dataset32 (Table S2 and S4). As a first reference-free pass, protein profiling found that the domains enriched in SPLASH anchors in B cells were Ig V-set and C1-set (variable-like and constant-like domains); these two domains were also matches in T cells (attributable to TCR) (Figure 4A). Mapping transcript gene-names to SPLASH anchors gives a similar picture: Ig light chain genes (both kappa and lambda) were strongly hit for B cells; HLA-B (discussed above) and TCR genes (both alpha and beta) were most prominent for T cells. These domains are not found among the control anchors (Figure 4A). Significant anchors were also found in Ig heavy chains, though fewer than in light chains. Ig/TCR anchors characteristically have a high diversity of targets (“target entropy”, a measure reported by SPLASH), and could be identified on that basis rather than requiring reference mapping. This is expected for clonally diverse receptors, and is evident in the clonotypic pattern (each cell expressing only its specific target) seen in heatmaps (Figure 4B).
Figure 4. B and T cell receptor diversity from human and lemur single cell data.

A. The “transcript mapping” plots show the number of anchors that align to a given gene name, for SPLASH on y-axis and Controls on x-axis, with immune receptor genes highlighted in red. For B cells, Ig genes (kappa = IGK and lambda = IGL) predominate among SPLASH anchors, but are not found at all in Control anchors. For T cells, TCR genes (alpha = TRA, beta = TRB) predominate, and are not found in Controls. The inset histograms show that immunoglobulin-type “V-set” and “C1-set” are among the top protein domain annotations identified by Pfam on anchor consensuses (for B cells, the top four and bottom four domains are shown; for T cells, all domains are shown). Mobile element activity is suggested by Pfam domains Tnp_22_dsRBD (“L1 transposable element dsRBD-like domain”) in B cells and RVT_1 (“Reverse transcriptase”) in T cells.
B. Targets associated with Ig/TCR anchors are clonotypically expressed, in both human and lemur: heatmaps show that most targets (rows) are expressed only in a single cell (columns). Target sequences are shown as bp color-maps (rows are targets, matching the heatmap; columns are bp positions, colored by base), for quick visualization of sequence diversity. Lemur NKT cells show shared TCR usage – see top two rows; the shared target sequence is different in the two individuals.
To showcase SPLASH’s utility for non-model organisms, we ran SPLASH on mouse lemur (Microcebus murinus) samples. Mouse lemurs are primates that diverged from humans 60–75 million years ago, and have potential as a model organism33. The lemur reference genome is incompletely annotated, especially at loci such as Ig and TCR. While the human reference does not suffice for alignment-first analysis of mouse lemur transcriptomes, we find that it is a reasonable approximation for interpreting SPLASH outputs; this may generalize to other organisms where a related, better-curated reference exists. From Tabula Microcebus, a multi-organ mouse lemur dataset34, we analyzed 111 natural killer T (NKT) cells from spleen; and separately, 289 B cells, also from spleen (Table S2 and S4). In both analyses, the cells came from two different individuals; for this reason, SPLASH also discovered numerous allelic differences between individuals, such as in COX2 (Figure S5A).
Our main focus was on adaptive immune receptors; similar to the human analyses, we found that SPLASH’s lemur anchors in B and NKT cells included Ig C1-set and V-set domains by protein profiling and Ig/TCR gene-names by transcript mapping (data not shown). As expected, SPLASH targets for lemur Ig heavy chain are predominantly clonotypically expressed (Figure 4B). Lemur NKT cells provide an interesting counterpoint. While there is some clonotypic diversity, a number of cells share TCR-alpha sequences; notably, the shared target is different between the two individuals (bottom-right heatmap, top row vs. second row). We selected NKT cells for analysis without foreknowledge of their properties. However, it is known in humans and mice that an NKT subset expresses an “invariant” TCR-alpha chain; NKT cells bridge between adaptive and innate immunity35. For Tabula Microcebus, NKT cells were operationally defined as co-expressing CD3E and KLRB1 (CD161)34; in this cell-type, SPLASH also finds shared usage in TCR-beta and TCR-gamma (Figure S5C and D).
To test if SPLASH can find diversity missed by standard methods, we analyzed a subset of 35 lemur B cells for which Ig light chain variable regions could not be assigned by the program BASIC36. BASIC assigns V-D-J regions based on curated human Ig sequences; although it was able to assign the large majority of lemur B cells, there was a subset for which it failed. We used SPLASH to find evidence of a light chain variable region in one of the 35 cells (STAR Methods); we were able to reconstruct the full variable region from reads (Data S1). In two cells, there were hits to the surrogate light chain (IGLL1/IGLL5 or λ5), which associates with Ig heavy chain when there is not yet a rearranged light chain37 (Data S1). This is proof-of-principle that SPLASH provides insights on data beyond traditional methods. In more recent work we have built on this capability (J. Salzman et al., unpublished work).
SPLASH applied to non-model organisms: octopus and eelgrass
To further explore SPLASH’s generality, we applied it to two understudied organisms: octopus and eelgrass. Octopuses have the most complex sensory and nervous systems among invertebrates, and are unusual in having high levels of RNA editing38. The marine angiosperm Zostera marina, or eelgrass, is the most widely distributed seagrass, and its adaptation to varying conditions, especially in the face of climate change, is of great interest and is only beginning to be explored at the genomic level39,40.
We focused narrowly on anchors where no more than one of its abundant targets mapped to the respective reference (STAR Methods), that is, where reference-based methods could not have detected variation. Hence, in these analyses we ignored many interesting findings of SPLASH that are reference-consistent.
For octopus, we analyzed an RNA-Seq dataset of Octopus bimaculoides41, encompassing a variety of tissues from a single individual (N. Bellono, personal communication). We examined several anchors with high effect sizes and BLAST hits to the closely related species Octopus sinensis (STAR Methods; Table S5). An anchor was found in O. sinensis myosin-VIIa, known as MYO7A in humans; MYO7A mutations cause Usher syndrome, leading to deafness and blindness42. Target 1 corresponds to the annotated first exon of O. sinensis myosin-VIIa, while target 2 represents an alternative first exon (not annotated in either species) expressed only in statocyst tissue (Figure 5A). The annotated O. bimaculoides myosin-VIIa gene is likely incomplete; it includes neither target, yet both target sequences are found upstream in the reference genome; also, the anchor is missing from the genome (Data S2). The O. sinensis myosin-VIIa gene is likely misassembled in a different way (Data S2). The statocyst-specific expression of an alternative first exon is intriguing given MYO7A’s association with Usher syndrome and deafness, as the octopus statocyst is a sensory organ for sound and balance43,44, suggesting homologous gene function.
Figure 5. Discovery of regulated variation in non-model organisms: octopus and eelgrass.

A. SPLASH identified alternative transcripts in the O. bimaculoides Myo-VIIa motor protein that are expressed mutually exclusively; the target 2 isoform is only found in statocyst. The transcripts have different first exons; the start codon lies in the shared second exon. The anchor and exon 2 are missing from the O. bimaculoides genome assembly, but are in the closely related O. sinensis genome. Targets 1 and 2 are in both genomes, but the statocyst-specific transcript is not annotated. The O. sinensis assembly has the Myo-VIIa gene in two inverted pieces (broken at the point marked by ‘??’ in protein domain schematic; Data S2).
B. The top four and bottom four domains identified by protein domain profiling in eelgrass (Z. marina) are plotted. The SPLASH domains chlorophyll A-B binding protein (Chloroa_b-bind) and silicon transporter (Silic_transp) derive from diatoms, based on BLAST protein alignment (Figure 7; Table S6). The other two top SPLASH domains, actin and ubiquitin, derive neither from diatoms nor eelgrass, so may be from other epiphytic species.
C. A Chloroa_b-bind anchor, identified by BLASTP as “fucoxanthin chlorophyll a/c protein” from diatoms (Figure 7C), has several differentially abundant targets. The most common target (top row) is mainly found in France/June samples; three targets that encode the same protein sequence (middle) are found in France/December samples; and one target (bottom row) is only in Norway/December samples.
D. An anchor in the eelgrass photosynthetic gene NdhL has four major targets. Targets 1–3 are allelic coding variants. Target 4 represents intron retention and gives a shortened protein. The scatterplot shows that Norway samples of June vs. December (red vs. green) are perfectly segregated by the fraction of target 4 (intron retention). A similar but less marked trend is seen for France samples of June vs. December (yellow vs. blue) – at the right edge, fraction target 4 values are collapsed to one dimension, with averages marked by bars.
Other SPLASH anchor-targets did not map to the O. bimaculoides genome yet did BLAST to 3’ UTRs of O. sinensis transcripts, including carboxypeptidase D, Upf2, and netrin receptor/DCC (Figure 6, Table S5). For all three, each target is expressed exclusively in some tissues and not others. Although O. bimaculoides has annotated transcripts for these genes, in all three cases the 3’ UTR region is missing or likely incorrect in the O. bimaculoides genome assembly. For two of the genes, the target variation may represent differential expression of alleles: a 13 nt deletion in carboxypeptidase D, and a short CAG repeat polymorphism in nonsense-mediated decay gene Upf2 (Figure 6A and B). For netrin receptor/DCC, involved in axon guidance and apoptosis, the variation SPLASH detects could be allelic but is also consistent with A-to-I RNA editing (Figure 6C). Our focus here on non-mapping anchor-targets excluded many more examples of regulated variation, including potential RNA editing in numerous octopus genes (data not shown).
Figure 6. O. bimaculoides 3’ UTR anchors show tissue-specific expression, related to Figure 5.

In the heatmaps, the parenthetical numbers are summed anchor counts.
A. Carboxypeptidase D (CPD). The anchor and targets align to the 3’ UTR of the O. sinensis CPD mRNA (XM_029795433.2), but are not in the O. bimaculoides genome assembly. The NCBI Browser screenshot at lower-right shows that the 3’ UTR of the O. bimaculoides CPD gene (LOC106880679, Ch.25) is entirely missing from the assembly: immediately after the coding region, a run of Ns begins (red box). Target 2 is identical to O. sinensis except for two mismatches; target 1 has a 12-nt deletion relative to target 2. Target 1 is only expressed in dissociated cells from sucker rims, and at a low level in one olfactory organ sample. All other tissues express only target 2.
B. Upf2 (regulator of nonsense transcripts 2). The alignment of Upf2 mRNAs from O. bimaculoides (XM_014915650.2) and O. sinensis (XM_036513028.1) shows that they diverge just before the stop codon, with unrelated 3’ UTRs. Our O. bimaculoides anchor-targets map only to O. sinensis Upf2 but not to the O. bimaculoides genome. The alignment also shows the downstream portion of the O. sinensis 3’ UTR where the anchor-targets map. Target 1 and 2 have six and five tandem CAG repeats, respectively. Target 1 is expressed in dissociated cells from sucker rims, and in olfactory organ; the other tissues express target 2.
C. Netrin receptor/DCC. Alignment of the O. bimaculoides genome (gene LOC106883766) and O. sinensis mRNA (XM_036508072.1) shows that the two diverge shortly after the stop codon. The O. bimaculoides gene ends in dinucleotide repeats just before the genome becomes a run of Ns. Our O. bimaculoides anchor-targets map to the O. sinensis netrin receptor 3’ UTRbut not to the O. bimaculoides genome. The targets differ at a single nucleotide: target 1 and 2 have G and A, respectively; O. sinensis has a G in this position. If the O. bimaculoides genome encodes A, then target 1 is consistent with A-to-I RNA editing (inosine read as G during reverse transcription). The majority of tissues express target 2 only, while target 1 is only expressed in dissociated cells of sucker rims.
We also applied SPLASH to RNA-Seq data from eelgrass (Zostera marina), collected in two locations, Montpellier, France (Mediterranean climate) and Rovika, Norway (near-arctic climate), in two seasons (winter and summer), and during day and night40. Considering each anchor with its most abundant target, a large number (14,680, 5.7%) did not align to eelgrass references (Table S6). A high-level view is provided by protein profiling: the top hits were chlorophyll A-B binding protein domain, Actin, Ubiquitin, and Silicon transporter (Figure 5B). BLASTP of some of the translated sequences finds that these have their best hits to a variety of organisms other than eelgrass, notably diatoms. Though a surprise to us, it has long been known that eelgrass is extensively colonized by epiphytes45, of which diatoms predominate46 and may provide as much as 71–83% of the primary production by the community47. For the most enriched protein domain, we investigated an anchor with high effect size whose consensus matches “fucoxanthin chlorophyll a/c protein” (FCP) in several diatoms, for example, Phaeodactylum tricornutum (95% amino acid identity, 81% nucleotide identity; Figure 7C). Given that the matches are imperfect, the true species of origin may not be in the NCBI database. FCPs function as light-harvesting antennae for photosynthesis48. This anchor has several targets whose abundance varies by location and time of year: target 1 is predominant in France in June; targets 3, 4, 5, which share the same amino acid sequence, together dominate in France in December; target 6 is predominant in Norway in December (Figure 5C). These targets could represent different diatom species or intra-species allelic variants. The abundance of this anchor (irrespective of target) is lower in Night samples (Figure 7C), indicating circadian regulation of this diatom photosynthetic gene. Other anchors mapping to diatoms, such as ferredoxin and high mobility group box-containing protein, also show targets that segregate by location, France vs. Norway (Figure 7A and B).
Figure 7. Diatom anchors in eelgrass samples show variation with location/season or Day vs Night, related to Figure 5.

A. HMG (high mobility group) box domain. The two targets show several nucleotide differences that result in two coding differences. The translation of the consensus sequence has its best two BLASTP matches to HMG box proteins from diatom species, shown in the inset. Target 1 is found only in Norway/December samples, while target 2 is found only in France/June samples.
B. Ferredoxin. The two targets show a silent single nucleotide polymorphism. The translation of the consensus sequence has its best BLASTP matches to ferredoxin from several diatom species, the top two are shown in the inset. Target 1 is found only in France/June samples, while target 2 is found only in Norway/December samples.
C. Fucoxanthin chlorophyll a/c protein (FCP). This anchor and its targets are also presented in Figure 5C. At left, the translated consensus sequence has its best protein BLAST matches to several diatom species, two are shown in the inset. The amino acid identity for Phaeodactylum tricornutum is 42/44 (95%). The consensus also BLASTs to the P. tricornutum genome, nucleotide identity 107/132 (81%) (not shown). At right, histogram shows total anchor counts for Night are ~60% lower than for Day, indicating circadian regulation of this gene. All are samples from France in December (where this anchor had both Day and Night representation).
One anchor and its targets, although mapping to the eelgrass genome, does report on noteworthy variation. It is in the NdhL subunit of chloroplast NADPH dehydrogenase complex (NDH). Of its four most abundant target sequences, three are within exon 3 and are SNP coding variants. The fourth represents retention of the intron following exon 3 (Figure 5D), altering the second transmembrane segment and terminating translation soon after (Data S2). The intron retention variant (target 4) is highest in winter (Figure 5D): for Norway, December (green) vs June (red) samples completely segregate by target 4 expression; the France samples show overlap, but on average December (blue) is higher than June (yellow). Figure 5D also illustrates other patterns: Norway samples do not express target 2 (instead they express target 1 and 3; data not shown); France samples have either a high fraction of target 2, or moderate (the latter also express target 1; data not shown). NDH is involved in cyclic electron transport49 and modulation of NDH function may affect photosynthetic efficiency and oxidative stress under varying light conditions50.
The above work with octopus and eelgrass are early forays, but show that SPLASH can discover regulated RNA splicing and isoforms, and bring to light allelic variation and communities of associated organisms. SPLASH’s statistics-first and reference-free methodology provides an unbiased approach to discovery, which can be augmented by protein profiling and the use of sequence databases beyond solely genomic references.
Discussion
Genomic analysis today is performed with complex computational workflows that are highly problem-specific and reference-dependent. Here we present a unifying statistics-first framework, SPLASH, which identifies sample-specific sequence variation directly from raw reads using a statistical test.
We provide a snapshot of SPLASH’s discoveries in disparate genomics subfields. When run on SARS-CoV-2 patient samples, without strain metadata or reference genomes, SPLASH finds many anchors capturing strain defining and emerging mutations. Using reference-free protein domain analysis, SPLASH identifies the spike protein as highly enriched for sequence variation. This points to broad potential for SPLASH in viral and other genomic surveillance.
In single-cell sequencing data, SPLASH is able to identify differential expression between highly similar genes, including myosin light chains MYL12A and B and several different HLA genes (traditionally difficult to analyze as they are highly polymorphic). SPLASH analysis was conducted in unsupervised mode, yet many of its significant anchors show cell-type regulation between macrophages and capillary endothelial cells. This testifies to the power of SPLASH’s unique statistical approach. When applied to B and T cells of both human and mouse lemur, SPLASH automatically identifies antigen receptor genes as exhibiting the most diverse variation. Post-facto analysis for lemur was performed using only an approximate genomic reference (human) that diverged from lemur ~60 million years ago.
To examine SPLASH’s ability to find variation not present in reference genomes, we applied it to two diverse organisms: octopus and eelgrass. In octopus, we identified several tissue-regulated isoforms not in the reference, in particular one in myosin-VIIa that is only expressed in statocyst. In the eelgrass dataset, SPLASH uncovered many sequences from epiphytic diatoms, with variation correlating with geography and season. This highlights the enormous potential in already existing datasets, and the need for tools like SPLASH to better explore them.
SPLASH should be of general interest to most genomic analyses. Users can easily run SPLASH on their own samples (FASTQ files): we provide it as a containerized Nextflow pipeline to minimize installation issues; it is lightweight, and can be run on a laptop (STAR Methods). The default parameters work well across all tested datasets (and SPLASH is robust to a range of parameters, STAR Methods). SPLASH outputs a list of significant anchors and targets; these results are a large data reduction and distillation of the variation present in the samples, and there are many ways they can be used. If metadata is available, it can be correlated with anchor-targets generated in unsupervised mode; alternatively, one can use metadata to supervise SPLASH analysis. If a reference genome is available, SPLASH can use it to align anchors and targets and provide gene names. SPLASH provides a number of metrics, such as a -value bound, effect size, target entropy, and average target similarity, which can be used to filter the anchor list. Another avenue for analyzing SPLASH’s results is BLAST of anchor-targets or consensus sequences against the NCBI databases and protein domain profiling with databases like Pfam, helpful especially when there is no reference genome or it is incomplete. Ultimately, users will bring their own domain expertise to bear in deciding how to best utilize SPLASH results.
Even in areas where there are existing pipelines, for example in differential alternative splicing, or antigen receptor identification, SPLASH provides a different approach and may well give additional insights. SPLASH scales to allow discovery to keep pace with the ever-increasing sequence data from the world at large, in particular microbes and metagenomic communities; recent collaborative work has further increased the computational efficiency of SPLASH51. SPLASH provides an expansive paradigm, and could be applied to a wide range of “omics” modalities, from DNA and protein sequencing to Hi-C and spatial transcriptomics, and more (STAR Methods). The statistical ideas underlying SPLASH are also expansive: anchor-target pairs can be generalized to tensors, and higher-dimensional relations between anchors, targets, and samples can be studied; other functions for splitting and hashing targets and samples can be considered, to optimize statistical power52.
In summary, SPLASH shifts from the “reference-first” approach to “statistics-first”, performing statistical hypothesis tests on raw sequencing data. By this design, SPLASH is highly computationally efficient. References are valuable for interpretation; however, the filtering of data by reference alignment introduces quantification biases and blindspots. SPLASH promises data-driven biological study with scope and power previously impossible.
Limitations of the study
SPLASH can be applied to problems across diverse fields which are of great current importance (STAR Methods), including those previously discussed. Naturally, some problems are not directly amenable to SPLASH analysis as formulated here. The most clear are cases where quantification of sample-specific RNA or DNA abundance alone is desired (e.g., differential gene expression analysis). Additionally, SPLASH is currently unable to distinguish which biological mechanism underlies the called variation, and work in progress seeks to address this.
STAR★Methods
RESOURCE AVAILABILITY
Lead contact
Correspondence and requests for materials should be addressed to the lead contact, Julia Salzman (julia.salzman@stanford.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table.
All original code has been deposited at Zenodo/Github and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| SARS-CoV-2 France | Bal 20229 | NCBI: SRP365166 |
| SARS-CoV-2 South Africa | Viana 20228 | NCBI: SRP348159 |
| Rotavirus | Simsek 202117 | NCBI: SRP328899 |
| Human Lung Cell Atlas | Travaglini 201920 | European Genome- phenome Archive: EGAS00001004344 |
| Tabula Sapiens | Tabula Sapiens Consortium 202232 | https://tabula-sapiens-portal.ds.czbiohub.org/ |
| Tabula Microcebus | The Tabula Microcebus Consortium 202134 |
https://tabula-microcebus.ds.czbiohub.org |
| Octopus bimaculoides RNA-Seq data | van Giesen 202041 | NCBI: SRP327909 |
| Zostera marina RNA-Seq data | Jueterbock 202140 | NCBI: SRP327909 |
| Pfam database (Pfam-A) | Mistry 202114 | https://www.ebi.ac.uk/interpro/download/pfam/ |
| Software and algorithms | ||
| SPLASH (commit 1b73949) | This study | https://doi.org/10.5281/zenodo.8271159 https://github.com/salzmanlab/nomad |
| FASTP v0.23.2 (installed with bioconda, 2/15/23) |
Chen 201853 | https://github.com/OpenGene/fastp |
| STAR | Dobin 201354 | https://github.com/alexdobin/STAR |
| bowtie2 | Langmead 200955 | https://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| bedtools | Quinlan 201056 | https://github.com/arq5x/bedtools |
| seqkit (installed with bioconda) | Shen 201657 | https://bioinf.shenwei.me/seqkit/ |
| HMMer3 | Eddy 201158 | http://hmmer.org/ |
| BLAST | Camacho 200959 | https://blast.ncbi.nlm.nih.gov/Blast.cgi |
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
We note that all datasets used are publicly available. We provide relevant details taken from the original papers. Because this work was a study of existing data and because the method can be used on any sample size above two, we did not perform sample size estimation (all samples in existing data were used). Also, samples were not explicitly allocated to experimental groups, rather the SPLASH procedure described in this manuscript uses random allocation and testing to find significant groupings.
Human samples
South Africa SARS-CoV-2 samples,
Sequencing was done on randomly selected nasopharyngeal and oropharyngeal swab samples from routine diagnostic SARS-CoV-2 PCR testing from public and private laboratories in South Africa and Botswana; requirement for participant consent was waived by the Research Ethics Committees. The large majority of samples were sequenced using Oxford Nanopore, but we only analyzed the samples sequenced using the Illumina COVIDseq assay; the paper does not explain how samples were chosen for Illumina sequencing. The paper does not give a breakdown of samples by age, sex, location, or ethnicity. 8
France SARS-CoV-2 samples,
The clinical samples presented in the paper are those that had evidence of co-infection by more than one strain of SARS-CoV-2; they were taken from a broader sequencing effort that included hospitalized patients and health care workers at the university hospital of Lyon (National Reference Center of Respiratory Viruses of Hospices Civils de Lyon (HCL)); and randomly selected positive samples nationwide from all diagnostic laboratories in France (EMERGEN consortium). The study was approved by the HCL ethics committee. We used all the samples in our analysis. Table 1 of the original paper gives some demographic information: among 15 outpatients with Delta/Omicron coinfection, median age was 31.13, 10 were male; 21 outpatients with BA.1/BA.2, median age was 22.36, 6 were male; 13 hospitalized with Delta/Omicron, median age 66.61, 7 were male; 3 hospitalized with BA.1/BA.2, median age 29.03, 2 were male.9
HLCA samples,
Normal lung tissues (approximately 5 cm3) were obtained from uninvolved regions of patients undergoing lobectomy for focal lung tumors; informed consent was obtained. Patient 2 was a 46-year-old male, non-smoker with a right middle lobe (RML) endobronchial carcinoid, who underwent surgical resection of the right upper and middle lobes; two blocks of tissue were selected from mid-bronchial region (medial 2) and periphery (distal 2) of right upper lobe (RUL). Patient 3 was a 51-year-old female, non-smoker with mild adult-onset asthma and a left lower lobe (LLL) endobronchial typical carcinoid, who underwent LLL lobectomy; three tissue blocks were resected from the bronchus (proximal 3), mid-bronchial (medial 2), and periphery (distal 3) of the LLL.20
Tabula Sapiens samples,
Donated tissues were procured in the Northern California through collaboration with the federally mandated organization Donor Network West (DNW, San Ramon, CA, USA); the research protocol was approved by the relevant boards of DNW and Stanford University. Donor TSP1 was a 59-year-old female with BMI of 23 and a history of stroke. She was found down with slurred speech. Donor TSP2 was a 61-year-old female with BMI of 41 and a history of deep vein thrombosis, hypertension, hypersensitive lung disease, type 2 diabetes, an infected knee replacement, and recent bout of the flu. She reported being short of breath and later went into respiratory arrest.32
Mouse lemur samples
Microcebus murinus gray mouse lemurs originated from the closed captive breeding colony at the Muséum National d’Histoire Naturelle in Brunoy, France, and transferred to the University of Texas (Austin) and then Stanford University. Mouse lemurs were housed indoors in an AAALAC-accredited facility in a temperature (24°C) and light-controlled environment (daily 14:10 h and 10:14 h light:dark alternating every 6 months) with perches and nest boxes, and were fed fresh fruits and vegetables, crushed primate chow plus live insect larvae. Animals in declining health that did not respond to standard therapy were euthanized by pentobarbital overdose under isoflurane anesthesia. Organs and tissues were removed and divided by a veterinary pathologist. We used data from two individuals: L2, 10 year old female; and L4, 11 year old male.34
Octopus samples
Adult female California two-spot octopuses (Octopus bimaculoides) were wild-caught (Aquatic Research Consultants, San Pedro, CA), fed daily with fiddler crabs (Uca pugnax, Northeast Brine Shrimp, Oak Hill, FL), and kept on a 12hr light/dark cycle in natural sea water. Sensory cells were isolated from suckers following tissue extraction and sucker epithelium dissection from animals that were sedated using step-wise increases in ethanol (ending at 3%). Animal protocols were approved by the Harvard University Animal Care and Use Committee.41
Eelgrass (Zostera marina) samples
Eelgrass shoots were sampled in Norway (Røvik, 67°16′06.2” N, 15°15′38.4″ E) and France (Sète, Thau Lagoon, 43°25′08.0” N, 3°40′03.9″ E). The youngest two shoots of each of six plants were collected at each site at noon and the following midnight around summer solstice (June 21) and winter solstice (December 21) 2017.40
METHOD DETAILS
SPLASH overview
Full details of SPLASH usage and outputs can be found at https://github.com/salzman-lab/nomad. Briefly, it takes as input a set of FASTQ sequencing data files (one per sample). SPLASH has several tunable parameters (anchor and target length, lookahead, minimum count thresholds, and more). SPLASH’s default settings work well in practice, and SPLASH’s inference is robust to these choices (see below). The standard output includes a table of anchors, targets, -values, etc., a table of “consensus” sequences (see below), and a table of “element annotations” (see below). SPLASH can also perform alignments with bowtie2 and STAR, to generate “genome annotations” and splice junction annotations (see below).
The code used in this work is available as a fully-containerized Nextflow pipeline60 at https://github.com/salzman-lab/nomad, commit 1b73949. The GitHub repository also contains the sample sheets for all analyses, including individual sample SRA accession numbers; as well as scripts for supplemental analysis. See Quantification And Statistical Analysis, below, for explanation of SPLASH -value and effect size computations.
SPLASH anchor preprocessing and parameter choices
Anchors and targets are defined as sequences of length (-mers) positioned at an offset apart, where is the length of the first read processed in the dataset, and is rounded to the nearest integer. If and , then . For a fixed number of anchor-target pairs, under alternatives such as differential exon skipping, larger choices of have provably higher power than smaller choices, following the style of analysis in [Salzman 2011]61. is typically long enough to be assigned a unique position in a genome while having a low probability of containing a sequencing error. Anchor sequences can be extracted as adjacent, disjoint sequences or as tiled sequences that begin at a fixed step size, to reduce computational burden. For this manuscript, SPLASH was run with default parameters: with 1M reads per FASTQ file, anchor sequences tiled by 5 bp, and . For HLCA datasets, both read 1 and read 2 were used; for other datasets, only read 1. Extracted anchor and target sequences are then counted for each sample with the UNIX command, `sort | uniq -c`, and anchor-target counts are then collected across all samples for restratification by the anchor sequence. This stratification step allows for user control over parallelization. To reduce the number of hypotheses tested and required to correct for, we discard anchors that have only one unique target, anchors that appear in only 1 sample, and (anchor, sample) pairs that have fewer than 6 counts. Then, we retain only anchors having more than 30 total counts after the above thresholds were applied. This approach efficiently constructs sample by target counts tables for each anchor.
SPLASH is robust to these parameter choices. We give examples of how choices of , , and tiling length impact results in France SARS-CoV-2 data as follows, showing that SPLASH yields similar results for a range of parameter choices. Default parameters shown in bold: we tested = [25, 27, 30]; Tile = [3, 5, 7]; Lookahead = [0, 15, 23]. For , 94.4% of anchors with default parameters contain at least one of the K=25 anchors as a substring. For , 93.8% of anchors with contain at least one of the anchors with default parameters a substring. For tile size of 3, 85% of the anchors from the default run can be found in the significant anchors of tile size of 3. For tile size of 7, 85% of the anchors from the default run can be found in the significant anchors of tile size of 5. For lookahead distance of 0, 37% of the anchors from the default run can be found in the significant anchors of tile size of 3; for lookahead distance of 15, 76% of the anchors from the default run can be found in the significant anchors. Overall, as tile size decreases, anchor calls increase (4715, 5522, 7891 for [7, 5, 3] respectively). As varies, anchor calls stay essentially the same (5875, 5522, and 5958 for = [25, 27, 30] respectively). Finally, for lookahead distance, the total number of calls decrease as lookahead distance increases (13239, 8295, 5522 for = [0, 15, 23] respectively).
Consensus sequences
For each significant anchor, a per sample consensus sequence is built for the sequence downstream of the anchor. A separate consensus is built for each sample by aggregating all reads from this sample that contain the given anchor. Then, SPLASH constructs the consensus as the plurality vote of all these reads; concretely, the consensus at base pair is the plurality vote of all reads that contain the anchor, base pairs after the anchor appears in the read (a read does not vote for consensus base if it has terminated within base pairs after the anchor appeared). The consensus base as well as the fraction agreement with this base among the reads is recorded. Some empirical behavior of consensuses is shown in Figure S4.
The consensus sequences can be used for splice site discovery as well as other applications, such as identifying point mutations and highly diversifying sequences, e.g. V(D)J rearrangements. The statistical properties of consensus building make it an appealing candidate for use in short read sequencing62, and may have information theoretic justification in de novo assembly63.
To provide intuition regarding the error correcting capabilities of the consensus, consider a simple probabilistic model where our reads from a sample all come from the same underlying sequence. In this case, under the substitution only error model, we have that the probability that our consensus for reads makes a mistake at a given location under independent sequencing error rate (substitution only) is at most
We can see that even for , this probability is less than 1.3E-7 for a given base pair, which we can union-bound over the length of the consensus to yield a vanishingly small probability of error. Thus, for a properly aligned read, if a base pair differs between the consensus and reference it is almost certainly a SNP.
Element annotations
To identify false positive sequences or contextualize mobile genetic elements, anchors and targets are aligned with bowtie2 to a set of indices, corresponding to databases of sequencing artifacts, transposable elements, and mobile genetic elements64. In these alignments, using bowtie2, the best hit is reported, relative to an order of priority. The references used are: UniVec, Illumina adapters, grass carp (Ctenopharyngodon idella genome, GCA_019924925)65, Escherichia phage phiX174, Rfam66, Dfam67, TnCentral68, ACLAME69, ICEberg70, CRISPR direct repeats71, ITSoneDB72, ITS273; and also the reference genome of interest for the study. (Grass carp was used as a control as it contains many artifactual Illumina adapters.) To perform these annotations, bowtie2 indices were built from the respective reference FASTAs, using bowtie2-build with default parameters. Anchors and targets were then aligned to each index, using bowtie2-align with default parameters. For each sequence, we report the alignment to the reference and the position of that alignment for each reference in the prespecified set. Anchors and targets, and their respective element annotations, are reported in the element annotation summary files.
Genome annotations
Anchor, target, and consensus sequences can be aligned by SPLASH to reference genomes and transcriptomes, to provide information about the location of sequences relative to genomic elements. All alignments reported are run in two modes in parallel: bowtie2 end-to-end mode (the bowtie2 default parameters) and bowtie2 local mode (`-local`, in addition to the bowtie2 default parameters). To report alignments to the transcriptome, the sequences are aligned to the reference transcriptome with bowtie2, with `-k 1`, in addition to the above parameters, to report a maximum of one alignment per sequence. If there is a transcriptome alignment, we report the alignment to the reference and the MAPQ score of the alignment. To report alignments to the genome, the sequences are aligned to the reference genome, with the same parameters above. If there is a genome alignment, we report the alignment to the reference, the strand of the alignment, and the alignment MAPQ score.
Splice junction calls
To identify exon coordinates for reporting annotations in this manuscript, consensus sequences are mapped with STAR aligner (default settings)54. Gapped alignments are extracted and their coordinates are annotated with known splice junction coordinates using ‘bedtools bamtobed --split’; each resulting contiguously mapping segment is called a “called exon”. From each consensus sequence, called exons are generated as start and end sites of each contiguously mapped sequence in the spliced alignment. These ‘called exons’ are then stratified as start sites and end sites. Note that the extremal positions of all called exons would not be expected to coincide with a splice boundary; “called exon” boundaries would coincide with an exon boundary if they are completely internal to the set of called exon coordinates. Each start and end site of each called exon is intersected with an annotation file of known exon coordinates; it receives a value of 0 if the site is annotated, and 1 if it is annotated as alternative. The original consensus sequence and the reported alignment of the consensus sequence are also reported. Gene names for each consensus are assigned by bedtools intersect with gene annotations (hg38 RefSeq for human data by default), possibly resulting in multiple gene names per consensus.
SPLASH protein domain profiles
Custom scripts were used to generate protein domain profiles. For each set of enriched anchors, homology-based annotation was attempted against an annotated protein database, Pfam14. For each dataset, up to 1000 of the most significant anchors (-value < 0.01) were retained for the following analysis: we first generated a substring of each downstream consensus by appending each consensus nucleotide assuming both conditions were met: a minimum observation count of 10 and a minimum agreement fraction of 0.8, until whichever metric first exhibited two consecutive failures at which point no further nucleotide was added. A limit of 1000 anchors was used due to computational constraints from HMMer3 (see below). Anchors that did not have any consensus nucleotides appended were kept as is. An extended anchor was generated for each experiment in which an anchor was found. Each extended anchor was then stored in a final concatenated multi FASTA file with unique seqID headers for each experiment’s extended anchors.
To assess these extended anchors for protein homology, this concatenated FASTA file was then translated in all six frames with the standard translation table using seqkit57 prior to using hmmsearch from the HMMer3 package74 to assess resulting amino acid sequences against the Pfam35 profile Hidden Markov Model (pHMM) database. The resulting ‘raw’ .tblout outputs were then processed, keeping the best hit (based on E-value) per each initial anchor, and any hits with an E-value better than 0.01 were parsed into an *_nomad.Pfam (or *_control.Pfam) file used for subsequent plotting.
All hits to the Pfam database were then binned at different E-value orders of magnitude. In each case, control assessments were performed by repeating the extension and homology searches against an equivalent number of control anchors (see below). Protein domains are ranked in the plot by the difference between SPLASH anchor hits and control anchor hits (for hits with E-value ≤ 1e-02). The number of matched anchors used for SPLASH and control analysis per dataset were as follows: 201 high effect size (.5) anchors in SARS-CoV-2 from South Africa, 252 high effect size (.5) anchors in SARS-CoV-2 from France; 1000 anchors (no effect size filter) were used for rotavirus, human T cells, human B cells, Microcebus natural killer T cells, and Microcebus B cells. We note that while the number of input anchors for SPLASH and control sets are matched, it is possible to have more control protein domains in the resulting barplots, as only high E-value hits to Pfam are reported in the visualizations. Domain profiling summaries are in Data S3.
A hypergeometric test was used to give -values for protein domain analysis. For a given domain, we construct the 2×2 contingency table, where the first row is the number of SPLASH hits for this domain, followed by the total number of SPLASH hits not in this domain. The second row is the mirror of this for control, where the first entry is the number of control hits for this domain, followed by the total number of control hits not in this domain. A one-sided -value is computed using Fisher’s exact test, which is identically a hypergeometric test. We apply Bonferroni correction for the total number of protein domains expressed by either SPLASH or control, to yield the stated -values.
Lastly it is worth noting that while only counts of the best scoring Pfam hits were assessed in this study, other information is also produced by HMMer3. In particular, relative alignment positions are given for each hit which could be used to more finely pinpoint the precise locus at which sequence variation is detected.
Control analyses
To construct control anchor lists based on abundance, we considered all anchors input to SPLASH and counted their abundance, collapsing counts across targets. That is, an anchor receives a count determined by the number of times it appears at an offset of 5 in the read up to position R - max(0,R/2–2*k) where R is the length of the read, summed over all targets. The 1000 most abundant anchors were output as the control set. For analysis comparing control to SPLASH anchors, min( |SPLASH anchor list|, 1000) most abundant anchors from the control set were used and the same number of SPLASH anchors were used, sorted by -value.
Generation of contingency table heatmaps.
To plot the anchor-target heatmaps, we exclude targets with low counts. Concretely, we by default filter out targets that occur fewer than 5 times, have less than 5% of the total counts of that anchor, and retain at most the top 10 targets, while ensuring that at least 2 targets are plotted. Then, all samples with fewer than 5 counts are discarded. For clarity of presentation, we include or remove rows corresponding to additional targets based on biological relevance.
SARS-CoV-2 analysis
SARS-CoV-2 data was downloaded from the NCBI: France9 (SRP365166) and South Africa8 (SRP348159). Sample metadata for the France dataset was provided by the authors via personal communication, and consists of their calls of the primary and secondary infecting strains for each patient sample. We note that sample ‘WTA-022002271301_S1’ appeared to be mislabelled, appearing in the metadata file but not in the NCBI sample list. Conversely, the sample ‘Pl924-022002271301_S1472’ appears in the NCBI sample list, but not in the metadata file. Thus, we associate these labels to each other, to obtain metadata labeling for all 106 samples. We do not have information regarding which samples are replicates. We provide the NCBI sample list and the strain metadata file in Table S1.
The SARS-CoV-2 datasets used in this manuscript were analyzed with SPLASH’s unsupervised mode (no sample metadata provided). To identify high effect size anchors, a threshold of èffect_size_randCjs` > 0.5 was used (Table S2).
For the purposes of strain-defining mutation analysis, we manually constructed “archetype” genome sequences for variant strains Delta, Omicron BA.1, and Omicron BA.2 by editing the Original (Wuhan) reference NC_045512.2 to contain all (and only) the defining mutations specified at CoVariants.org10; these are provided in Table S1.
To determine what SPLASH calls (and control anchors) were strain defining we perform the following. To generate SPLASH’s calls, we filter for anchors that are significant (with a BY corrected -value less than .05) and have large effect size (> .5), yielding a list of SPLASH-called anchors. Control anchors are generated by taking the anchors with the highest counts. For each of these anchors we construct their target × sample contingency table, first filtering out all anchors with fewer than 30 counts, only 1 unique target, or only 1 unique sample, and filtering out all samples with 5 or fewer counts. Then, we discard all targets that constitute less than 5% of the remaining counts for that anchor. The remaining anchors and targets are then bowtie aligned to an index comprised of the Original, Delta, Omicron BA.1, and Omicron BA.2 archetype genomes. For this alignment, options `-a -v 0` were used. Then, for each set of anchors (SPLASH calls, and controls), the list is filtered to only anchors that align perfectly to at least one of the reference assemblies, further requiring that each anchor have at least one target that aligns perfectly to a reference assembly. Then for each anchor, we declare it to be strain defining if, for any of the reference assemblies, it has at least one target that maps to it and one target that does not.
Identifying cell-type specific isoforms in single-cell data (lung macrophages and capillary cells)
The human lung scRNA-seq data used here (HLCA SS2)20 is accessible through the European Genome-phenome Archive (accession number: EGAS00001004344); FASTQ files from donor 1 (P2) and donor 2 (P3) generated with the Smart-seq2 protocol were used. In the analysis of HLCA SS2 data, we utilize “isoform detection conditions” for alternative isoform detection. These conditions select for (anchor, target) pairs that map exclusively to the human genome, anchors with at least one split-mapping consensus sequence, mu_lev > 5, and ; mu_lev is the average target distance from the most abundant target as measured by Levenshtein distance, is the total number of counts in the anchor’s contingency table. To identify anchors and targets that map exclusively to the human genome, we included anchors and targets that had exactly one element annotation, where that one element annotation must be grch38_1kgmaj. To identify anchors with at least one split-mapping consensus, we selected anchors that had at least one consensus sequence with at least 2 called exons. The conditions on Levenshtein distance, designed to require significant across-target sequence variation, significantly reduced anchors analyzed (excluding many SNP-like effects). We further restricted to anchors with , to account for the lower numbers in macrophage cells; note that the user can choose to use a lower requirement, based on input data. These isoform detection parameters were used to identify the SS2 examples discussed in this manuscript. For HLA discussion, gene names were called using consensus_gene_mode.
While here we focus on anchors that have aligned to the human genome, we note in passing that SPLASH makes many predictions of cell-type specific RNA expression that include sequences that map to repetitive elements or do not map to the human reference: for individual P2 (respectively P3), 53% (61%), of 4010 (4603) anchors map to the human genome and no other reference; 35% (30%) map to both the Rfam and human genome; 6% (7%) have no map to any reference used for annotation which includes repetitive and mobile elements. As an example, 9 and 18 such anchors (individual P2 and P3, respectively) BLAST to MHC alleles in the NCBI database.
Immune single-cell analysis
To study human B and T cells, we utilize Tabula Sapiens data (Smart-seq2)32, downloaded from https://tabula-sapiens-portal.ds.czbiohub.org/; B cells were used from donor 1 (TSP1) and CD4+ T cells from donor 2 (TSP2). Mouse lemur single-cell RNA-seq data used in this study was generated as part of the Tabula Microcebus consortium34; the FASTQ files were downloaded from https://tabula-microcebus.ds.czbiohub.org. B cells and natural killer T cells were analyzed separately; both were from spleen and were a mixture of individuals L2 and L4. To determine the most frequent transcriptome annotation for a dataset, all significant anchors were mapped to the human transcriptome (GRCh38, Gencode) with bowtie2, using default parameters and `-k 1` to report at most one alignment per anchor (Table S4). Then, the bowtie2 transcript hits are aggregated by counting over anchors. The transcript hits with the highest counts over all anchors were reported. Protein domain profiling was performed as described above.
SPLASH comparison to BASIC analysis in lemur spleen B cells
As part of the Tabula Microcebus consortium34, mouse lemur B cells were annotated with BASIC36 to identify Ig variable domains. However, BASIC was unable to identify the light chain variable domain in 35 cells. We used a simple approach to see if SPLASH could identify variable domains in these uncalled B cells. We checked the SPLASH genome annotations for these cells for anchors mapping to human “IGL” or “IGK” genes; there were only five such anchors, all to IGL, and these were found in only eight cells. For those eight cells, we retrieved the SPLASH consensus sequences for these anchors, which ranged from 2–5 per cell. Where consensuses for a cell overlapped, one was chosen, and these were submitted to BLAST against the nr/nt database. Many hits were to “immunoglobulin lambda-like polypeptide” 1 or 5 (IGLL), surrogate light chain genes that contain sequence similar to lambda J and C regions (as well as a unique N-terminal region) and so could mimic alignment to a true lambda variable domain. Therefore, BLAST alignments were checked to see whether the match could be assigned to V, J, C, or IGLL-unique regions. 4 cells matched C-region, 2 matched IGLL-unique region, and 2 had sequence beyond J-region (presumed V-region). For the latter two, we attempted to extend the consensus further into the V-region by `grep` in raw reads; one could not be extended as it only had adaptors adjacent to its J-region sequence. The other consensus was extended through the full V-region, and its sequence is given in Data S1, along with the IGLL-unique matches.
SPLASH for Zostera marina (eelgrass) and Octopus bimaculoides
Data was downloaded from SRP327909 (eelgrass40), and SRP278619 (Octopus41), using nf-core fetchngs run in default mode75 and preprocessed with fastp53 run in default mode to mitigate false positive calls due to adapter concatenation. An updated version of SPLASH51 (R-SPLASH v0.3.9, commit 5dafdc8) was run with gap length=0, anchor_unique_targets_threshold=1, anchor_count_threshold=50, anchor_samples_threshold=1, anchor_sample_counts_threshold=5, and excluding anchor-targets containing poly A / C / G / T run of length 8. 500 pairs of random and were chosen. fastp v0.3.9 was installed on 2/23/23 using bioconda.
The top 10 anchor-targets for each anchor were selected from the SPLASH calls if they had homopolymer length ≤ 5, effect size > 0.1, and corrected -value < 0.01. Element annotations were run using SPLASH commit ID 728066b; anchor-targets mapping to UniVec, Illumina adapters, SARS-CoV-2, or grass carp were removed. Anchor-targets were aligned with STAR 2.7.5 to a reference index generated from either the O. bimaculoides reference genome76 (NC_068981.1) and transcriptome41; or the Z. marina nuclear genome77 (v3.1, https://phytozome-next.jgi.doe.gov/info/Zmarina_v3_1) and mitochondrial and chloroplast genomes78 (NC_035345.1 and NC_036014.1, respectively).
For protein domain profiling, anchor-targets with no element annotation were in silico translated in all six frames and submitted to HMMer search of the Pfam database. For Figure 5B (eelgrass), anchors were ordered by descending number of observations; the top 200,000 were concatenated with their targets and submitted to element annotations, and unannotated anchor-targets were submitted to Pfam; these anchor-targets are defined as controls. For each anchor, we retain its best Pfam hit and full sequence E-value. Then for each Pfam domain, we tally (separately for SPLASH and control) the number of anchors that hit it with a full-sequence E-value ≤1e-02 and ≤1e-06.
For BLAST analysis, we selected anchors with no more than 1 target mapping with either STAR to the reference genome or Bowtie2 element annotations, thus cases where no sample-specific variation would be detected if a reference genome were used. In eelgrass, targets were selected if their fraction exceeded 0.5 and their anchor’s effect size exceeded 0.9; in octopus, targets were selected if their fraction exceeded 0.3 and if their anchor’s effect size exceeded 0.8. The 1808 anchor-target pairs satisfying these criteria in octopus were submitted to BLAST with parameters:
-db nt -evalue 0.1 -task blastn -dust no -word_size 24 -reward 1 -penalty −3 - max_target_seqs 4
BLAST hits were merged into SPLASH output, with an indicator variable for whether the sequence was queried. For octopus anchor-targets, 1061/1808 had a BLAST hit (max E-value 0.028). There were 288 hits to octopus, of which 281 were annotated as from O. sinensis and 7 from O. bimaculoides. Selected sequences mapping to O. sinensis were further analyzed. For eelgrass, 1606/4081 had a BLAST hit (max E-value 0.028).
SPLASH output, merged with Pfam and BLAST analyses, are in Table S5 and Table S6 (octopus and eelgrass, respectively).
SPLASH can detect myriad genomic events
In this work we focused our experimental results on identifying changes in viral strains and specific examples of RNA-seq analysis. SPLASH’s probabilistic formulation extends much further however, and subsumes a broad range of problems. Many other tasks, some described below, can also be framed under this unifying probabilistic formulation. Thus, SPLASH provides an efficient and general solution to disparate problems in genomics. We outline examples of SPLASH’s predicted applications in various biological contexts, highlighting the anchors that would be flagged as significant:
- RNA splicing, even if not alternative or regulated, can be detected by comparing DNA-seq and RNA-seq
- Examples of predicted significant anchors: sequences upstream of spliced or edited sequences including circular, linear, or gene fusions
- RNA editing can be detected by comparing RNA-seq and DNA-seq
- Examples of predicted significant anchors: sequences preceding edited sites
- Liquid biopsy – reference free detection of SNPs, centromeric and telomeric expansions with mutations
- Examples of predicted significant anchors: sequences in telomeres (resp. centromeres) preceding telomeric (resp. centromeric) sequence variants or chromosomal ends (telomeres) in cancer-specific chromosomal fragments
- Detecting MHC allelic diversity
- Examples of predicted significant anchors: sequences flanking MHC allelic variants
- Detecting disease-specific or person-specific mutations and structural variation in DNA
- Examples of predicted significant anchors: sequences preceding structural variants or mutations
- Cancer genomics e.g. BCR-ABL fusions
- Examples of predicted significant anchors: sequences preceding fusion breakpoints
- Transposon or retrotransposon insertions or mobile DNA/RNA
- Examples of predicted significant anchors: (retro)transposon arms or boundaries of mobile elements
- Adaptation
- Examples of predicted significant anchors: sequences flanking regions of DNA with time-dependent variation
- Novel virus’ and bacteria; emerging resistance to human immunity or drugs
- Examples of predicted significant anchors: sequences flanking rapidly evolving or recombined RNA/DNA
- Alternative 3’ UTR use
- Examples of predicted significant anchors: 3’ sequences with targets including both the poly(A) or poly(U), or adapters in cases of libraries prepared by adapter ligation versus downstream transcript sequence
- Hi-C or any proximity ligation
- Examples of predicted significant anchors: for Hi-C, DNA sequences with differential proximity to genomic loci as a function of sample; similarly, for other proximity ligation anchors would be predicted when the represented element has differential localization with other elements
- Finding combinatorially controlled genes e.g. V(D)J
- Examples of predicted significant anchors sequences in the constant, D, J, or V domains
SPLASH can use alternative anchor, target and consensus construction
SPLASH can function on any biological sequence and does not need anchor-target pairs to take the form of gapped -mers, and can take very general forms. For example, one could consider schemes that respect triplet codons: where are bases in the anchor and are bases in the target, this would focus specifically on variation in the wobble position, the fastest to diverge; similar schemes might be appropriate for mechanisms with known patterns of diversity, such as diversity generating retroelements79. X and Y could also be amino acid sequences or other discrete variables considered in molecular biology. Much more general forms of anchor-target pairs (or tensors) can be defined and analyzed, including other univariate or multivariate hash functions on targets or sample identity. SPLASH can also be further developed to analyze higher dimensional relationships between anchors, where statistical inference can be performed on tensors across anchors, targets, and samples. Similarly, hash functions can be optimized under natural maximization criterion, which is the subject of concurrent work. The hash functions can also be generalized to yield new statistics, optimizing power against different alternatives.
Computational benchmarking for SPLASH
SPLASH is computationally much more efficient than other approaches, due to its use of k-mers rather than reference alignment, and its closed-form statistics obviating compute-intensive significance testing. SPLASH is implemented as a fully containerized and parallelized workflow that requires only the FASTQ read files and no parameter tuning by the user. We ran SPLASH on a 2015 Intel laptop with an Intel® Core™ i7-6500U CPU @ 2.50GHz processor, generating significance calls for single cell RNA-seq totaling over 10 million reads in only 1 hour 45 min. When performed on a compute cluster, the same analysis is completed in an average of 22.8 minutes with 750 MB of memory for 10 million reads.
Because code was run on a server with dynamic memory, we report summary statistics as follows. For the steps parallelized by FASTQ file, such as anchor and target retrieval, total time for dataset run, as reported by Nextflow, was parsed per cell. Thus, the average time per cell is reported. For the steps parallelized by 64 files (-value calculations), total extracted times were summed and divided by number of cells. For steps that consisted of aggregating files, total run time was divided by number of cells. Thus, the total time and memory should be multiplied by the total number of cells to achieve an estimate of the pipeline time for this dataset.
Laptop specs:
An Intel® Core™ i7-6500U CPU @ 2.50GHz (launched in 2015)
2 cores, total of 4 threads, 3 of which SPLASH was allowed to use.
8 GB DDR3 RAM
SODIMM DDR3 Synchronous 1600 MHz (0.6 ns)
Laptop analysis dataset:
Ten B and T cells from donor 2 blood sequenced by Smart-Seq2 were used for the laptop benchmarking. These files totalled 43,870,027 reads, averaging 4.3M reads per cell. The fastq files for the Tabula Sapiens data were downloaded from https://tabula-sapiens-portal.ds.czbiohub.org/. Files used:
TSP2_Blood_NA_SS2_B114581_B133053_Lymphocytes_A13_S73_R1_001.fastq.gz
TSP2_Blood_NA_SS2_B114581_B133053_Lymphocytes_A18_S78_R1_001.fastq.gz
TSP2_Blood_NA_SS2_B114581_B133053_Lymphocytes_A19_S79_R1_001.fastq.gz
TSP2_Blood_NA_SS2_B114581_B133053_Lymphocytes_A21_S81_R1_001.fastq.gz
TSP2_Blood_NA_SS2_B114581_B133053_Lymphocytes_A3_S63_R1_001.fastq.gz
TSP2_Blood_NA_SS2_B114581_B133053_Lymphocytes_A5_S65_R1_001.fastq.gz
TSP2_Blood_NA_SS2_B114581_B133053_Lymphocytes_A6_S66_R1_001.fastq.gz
TSP2_Blood_NA_SS2_B114581_B133053_Lymphocytes_A8_S68_R1_001.fastq.gz
TSP2_Blood_NA_SS2_B114581_B133053_Lymphocytes_A9_S69_R1_001.fastq.gz
TSP2_Blood_NA_SS2_B114581_B133053_Lymphocytes_B10_S94_R1_001.fastq.gz
Anchor and target sequences, with -values and binomial -values
Targets are numbered by decreasing abundance, unless otherwise stated.
q-values are the BY-corrected p-values output by SPLASH, as detailed in Quantification And Statistical Analysis, below.
Binomial p-value calculations are described above, and are with respect to target 1, unless otherwise stated.
SARS-CoV-2 mutation K417N (Figure 2A)
-value: 9.4e-05
binomial -value: 6.4e-07
>anchor ATTCATTTGTAATTAGAGGTGATGAAG >target_1_Delta ACTGGAAAGATTGCTGATTATAATTAT >target_2_K417N_Omicron ACTGGAAATATTGCTGATTATAATTAT
SARS-CoV-2 mutations V213G, NL211I, R214REPE (Figure 2B)
-value: 8.3e-08
binomial -value: 1e-13
>anchor TTTAAGAATATTGATGGTTATTTTAAA >target_1_Delta TAATTTAGTGCGTGATCTCCCTCAGGG >target_2_V213G_BA.2 TAATTTAGGGCGTGATCTCCCTCAGGG >target_3_NL211I-R214REPE_BA.1 TATAGTGCGTGAGCCAGAAGATCTCCC
SARS-CoV-2 mutations P681R, N679K, P681H (Figure 2C)
-value: 1.2e-04
binomial -value: 4.9e-12
(reverse-complements are shown in Figure 1C)
>anchor GTGACATAGTGTAGGCAATGATGGATT >target_1_P681R_Delta (abundance order = 1) CGACGAGAATTAGTCTGAGTCTGATAA >target_2_P681R-Q677H (abundance order = 3) CGACGAGAATTAGTATGAGTCTGATAA >target_3_P681R-Q677H (abundance order = 4) CGACGAGAATTAGTGTGAGTCTGATAA >target_4_N679K-P681H_Omicron (abundance order = 2) CGATGAGACTTAGTCTGAGTCTGATAA
MYL12A / MYL12B (Figure 3A, S4B)
P2 -value: 2.5e-08
P2 binomial -value: 9.9e-37 (with respect to target 2)
P3 -value: 2.3E-42
P3 binomial -value: 2.2e-45 (with respect to target 1)
>P2_anchor AAGAGGCCTTCAACATGATTGATCAGA >P2_target_2_MYL12A TTCATTGGGGAAGAATCCAACTGATGA >P2_target_1_MYL12B TTCTCTAGGGAAGAATCCCACTGATGC >P2_consensus_MYL12A_macrophage ACAGAGATGGTTTCATCGACAAGGAAGATTTGCATGATATGCTTGCTTCATTGGGGAAGAATCCAACTGA TGAGTATCTAGATGCCATGATGAATGAGGCTCCAGGCCCCATCAATT >P2_consensus_MYL12B_capillary ACAGAGATGGCTTCATCGACAAGGAAGATTTGCATGATATGCTTGCTTCTCTAGGGAAGAATCCCACTGA TGCATACCTTGATGCCATGATGAATGAGGCCCCAGGGCCCATCA >P3_anchor AAGAGGCCTTCAACATGATTGATCAGA >P3_target_1_MYL12A GAAGATTTGCATGATATGCTTGCTTCA >P3_target_2_MYL12B GAAGATTTGCATGATATGCTTGCTTCT >p3_consensus_MYL12A_macrophage ACAGAGATGGTTTCATCGACAAGGAAGATTTGCATGATATGCTTGCTTCATTGGGGAAGAATCCAACTGA TGAGTATCTAGATGCCATGATGAATGAGGCTCCAGGCC >p3_cons_MYL12B_capillary ACAGAGATGGCTTCATCGACAAGGAAGATTTGCATGATATGCTTGCTTCTCTAGGGAAGAATCCCACTGA TGCATACCTTGATGCCATGATGAATGAGGCCCCAGGGCCCATCAATTT
HLA-DRB1 / HLA-DRB4 (Figure 3B)
P2 -value: 4.0e-10
P2 binomial -value: 2e-17
P3 -value: 1.2e-4
P2 binomial -value: 1.6e-08
(reverse-complements are shown in Figure 3B)
>P2_anchor GGAAGCCACAAGGGAGGACATTTTCTG >P2_target_1_DRB1 GTGGAAGAATAACTGCCAAGCAGGAAA >P2_target_2_DRB4 GGAAGAATAAGAGCCAAGTGGGAAAGC >P2_consensus_DRB1_macrophage GGAAGCCACAAGGGAGGACATTTTCTG CAGTTGCCGAACCAGTAGCAACCAGGTCCTGAGAAAGCCCTCTCTTGTGGAAGAATAACTGCCAAGCAGG AAAGCTTTTCATTCTGCAAAGCTGGGACAGAAGGTTCTTCCTTGAATGT >P2_consensus_DRB4_capillary CAGAGTTGCTGAACCAGTAACAACCTGGTCCTGACAAAGCTCTTGTGGAAGAATAAGAGCCAAGTGGGAA AGCTTTTCATCTTGCAAAGCTGGGGCAGAAGGTTCTTCCTTGAATGT >P3_anchor (same sequence as P2_anchor) GGAAGCCACAAGGGAGGACATTTTCTG >P3_target_1_DRB1 AGGTCCTGAGAAAGCCCTCTCTTGTGG >P3_target_3_DRB4 CCTGGTCCTGACAAAGCTCTTGTGGAA >P3_consensus_DRB1_macrophage CAGTTGCTGAACCAGTAGCAACCAGGTCCTGAGAAAGCCCTCTCTTGTGGAAGAATAACAGCCAGGAGGG AAAGCTTTTCATCCTGCAAAGCTGGGGCAGAAAGTTCTTCT >P3_consensus_DRB4_capillary GGAAGCCACAAGGGAGGACATTTTCTG CAGAGTTGCTGAACCAGTAACAACCTGGTCCTGACAAAGCTCTTGTGGAAGAATAAGAGCCAAGTGGGAA AGCCTTTCATCTTGCAAAGCTGGGGCAGAAGGTTCTTCCTTGA
HLA-DPA1 / HLA-DPB1 (Figure 3C, S4C)
P3 -value: 7.9e-22
P3 binomial -value: 9.15e-18
(anchor as given here is sense strand for DPA1, antisense strand for DPB1)
>P3_anchor AGATGTATCTCTCCAGGAAGCGCTGTG >P3_target_1_DPA1 TGCCGTCCCTGGAAAAGGTGAATCCCA >P3_target_2_DPB1 TGCCGTCCCTGGAAAAGGTAATTCTCT >P3_consensus_DPB1_macrophage TCCCATTAAACGCGTAGCATTCCTGCCGTCCCTGGAAAAGGTAATTCTCTGGAGTGGCCCTGCCCTGGAC CACAGATGTGAGCAGCACCATCAGTAACGCCGTCAGAGCCACT >P3_consensus_DPA1_capillary TCCCATTAAACGCGTAGCATTCCTGCCGTCCCTGGAAAAGGTGAATCCCAGCCATGCTGATTCCTCTCCA CCCATTTCCAGTGCTAGAGGCCCACAGTTTCAGTCTCATCTGC
HLA-B (Figure 3D, S4D)
-value: 2.7e-05
binomial -value: 1.7e-25
>anchor TTGGGACCGGAACACACAGATCTTCAA >target_1_HLA-B AGAGCCTGCGGAACCTGCGCGGCTACT >target_2_HLA-B AGAACCTGCGGATCGCGCTCCGCTACT >consensus_1_HLA-B TTGGGACCGGAACACACAGATCTTCAAGACCAACACACAGACTGACCGAGAGAGCCTGCGGAACCTGCGC GGCTACTACAACCAGAGCGAGGCCGGGTC >consensus_2_HLA-B TTGGGACCGGAACACACAGATCTTCAAGACCAACACACAGACTTACCGAGAGAACCTGCGGATCGCGCTC CGCTACTACAACCAGAGCGAGGCCGGGTC
human Ig-kappa C-region (Figure 4B)
-value = 1.6E-35
>anchor TGGCGGGAAGATGAAGACAGATGGTGC >Targ0 GCTTGGTCCCCTGGCCAAAAGTCCCGG >Targ1 GCTTGGTCCCCTGGCCAAAAGGGCTAC >Targ2 GCTTGGTCCCCTGGCCAAAAGTGTACG >Targ3 CCTTGGTCCCTCCGCCGAAAGAAGGTG >Targ4 GCTTGGTCCCCTGGCCAAAAGTGTCGT >Targ5 GCTTGGTCCCCTGGCCAAAAGTGCCCG >Targ6 CTTTGGTCCCAGGGCCGAAAGTGAATA >Targ7 CCTTGGTCCCTTGGCCGAACGTCCACC
human TCR-alpha C-region (Figure 4B)
-value = 3.4E-5
>anchor GTACACGGCAGGGTCAGGGTTCTGGAT >Targ1 TGCCTTTGCCGAAGTTGAGTGCATACC >Targ2 TCCCTGATCCAAAGATTATCTTGGAAG >Targ3 TGCCTGTCCCAAAGGTGAGTTTGTTTC >Targ4 TCCCAGCGCCCCAGATTAACTGATAGT >Targ5 TCCCCCTTGCAAAGAGCAGCTTCTGGC >Targ6 TTCCTCCTCCAAAAGTTAGCTTGTTGC >Targ7 TCCCTGTCCCAAAATAGAACTGGTTAC >Targ8 TTCCTCTTCCAAAGTATAGCCTCCCCA >Targ9 TTCCCTTTCCAAAGACCAGCTTTTCAG >Targ10 TTCCCTGTCCGAAGATAAGCTTTCCTC >Targ11 TCCCTGCTCCAAAGCGCATGTCATTGT >Targ12 TTCCCTTCCCAAAGATCAGAGCAGTTC >Targ13 TCCCAGATCCAAAGTAAAATTTGTTGA >Targ14 TCCCTTGCCCAAAGATTAGTTTGCCTG >Targ15 TTCCTCTTCCAAATGTAGGTATGTAGC >Targ16 TTCCATCTCCAAACATGAGTCTGGCAT >Targ17 TTCCACTCCCAAAAGTAAGTGCTCTCC >Targ18 TTCCTTTTCCAAATGTCAGTTTATAGT >Targ19 TGCCTGTTCCAAAGATGTATTTGTAGG >Targ20 TTCCAGTTCCAAAGGTAACTTTCTGGT >Targ21 TCCCTTGTCCAAATGTCAGCTTTCCAT >Targ22 TCCCCTTCCCGAAAGTGAGTTGGTAAC >Targ23 TGCCAGTTCCAAAGATGAGCTTGTTTG
lemur Ig-heavy V-region (Figure 4B)
-value = 1.3E-11
>anchor AGCCTGGGGGGTCCCTGAGACTCTCCT >Targ0 AGTGACTACTACATGAGCTGGGTCCGC >Targ1 AGCAGCTATGGGATGAACTGGGTCCGC >Targ2 AGCAACTACTGGATGAGCTGGGTCCGC >Targ3 AAGAACTATGAGATAAACTGGGTCCGC >Targ4 AGCAGCTACTACATGCACTGGGTCCGC >Targ5 AGCAGCTACGATATGAACTGGGTCCGA >Targ6 AGTGACTACTACATGAACTGGGTCCGC >Targ7 AGCAGCCATGGAATGCACTGGGTCCGC >Targ8 AGCAGCTACGATATGAACTGGGTCCGC >Targ9 AGCAGCTATGATATGCATTGGGTCCGC >Targ10 AGTGACCACCACATGAGCTGGGTCCGC >Targ11 GATGACTACCTCATGCACTGGATCCGC >Targ12 AGCAGCTATGCCATGAGCTGGGTCCGC >Targ13 AGTAGTTACTGGATGAACTGGGTCCGC >Targ14 GATTACTATGGCATGAACTGGGTCCGC >Targ15 ACCAATTTTGGGATGAACTGGGTCCGC >Targ16 AGCAGCTATGGGATGCACTGGGTCCGC >Targ17 ACCAGTTATGGGATGAACTGGGTCCGC
lemur TCR-alpha C-region (Figure 4B)
-value = 4.1E-7
>anchor TCAGCTGGTACACGGCGGGGTCAGGGT >Targ0 AGTCTGGTCCCTGCTCCAAAGCGCAGA >Targ1 AGCCTGGTCCCTGCTCCAAAAATCAAC >Targ2 AGCAGAGTGCCAGTCCCAAAGATGAGC >Targ3 ACGGTGGTTCCTTTCCCAAAGATCAAC >Targ4 AGTTGGGTGCCAGTTCCAAACACGGGT >Targ5 AACTGGGTCCCGGATCCAAAGGTCAGT >Targ6 AGTTGTGTCCCTTTTCCAAAGGTGACT >Targ7 AGTTTGGTCCCAGATCCAAAGTAAAAT >Targ8 AATCTGGTCCCAGTCCCAAAGATGAGC >Targ9 AGTCTGGTCCCTGATCCAAAGATTAGC
Octopus bimaculoides Myo-VIIa (Figure 5A)
-value = 4.0e-03
(reverse-complements shown in Figure 5A)
>anchor CCATTTTTGCTTTTTGTTTAAAATCCA >target_1 ATTATATCACAAGTTATAAGGCATGCC >target_2 ATTATATCTTAATAAATGGATACACTA
fucoxanthin chlorophyll a/c protein, diatom (Figure 5C)
-value = 6.0e-08
(reverse-complements shown in Figure 5C)
>anchor AAGTATCCAACAACGGCAAGCATGGAG >target_1 (abundance order = 1) ATACGTCCGTGCTTGAGCTCGACAAAT >target_2 (abundance order = 6) ATACGGCCGTGCTTGAGCTCGACAAAT >target_3 (abundance order = 2) ATACGTCCGTGCTTGATCTCGACGTAT >target_4 (abundance order = 4) ATACGTCCGTGCTTGATCTCAACGTAT >target_5 (abundance order = 5) ATACGTCCGTGCTTGATCTCGACGTAC >target_6 (abundance order = 3) ACACGTCCATGCTTAATTTCGACATAT
Zostera marina NADPH quinone oxidoreductase subunit L (NdhL) (Figure 5D)
-value = 6.5e-56
(reverse-complements shown in Figure 5D)
>anchor AATCGAAGCCAATTCATGATGATAGGC >target1 GGCATGATAAGGAAGTAGAAGAAAGCA >target2 GGCATGATAAGGAAGTAGAAGAAAACA >target3 GGCATGACAAGGAAGTAGAAGAAAGCA >target4 TTCGATCATGCAGTTCAATCAATGATC
human MYL6 (Figure S4A)
>P2_anchor AAGGTCCTCAGCCATTCAGCACCATGC >P2_consensus1_macrophage GGACGAGCTCTTCATAGTTGATACAACCATTGCTGTCCTCATGCCCTGCCACCAGCATCTCTACTTCTTC CTCTGTCATCTTCTCACCCAGTGTGACAAGAACATGCCGGATTTC >P2_consensus2_capillary GGACGAGCTCCGCCCCATGGGCCCGTCACCCCGACAGGATATGCCTCACAAACGCTTCATAGTTGATACA ACCATTGCTGTCCTCATGCCCTGCCACCAGCATCTCTACTTCTTCC >P2_target1 TGCCACCAGCATCTCTACTTCTTCCTC >P2_target2 CACAAACGCTTCATAGTTGATACAACC >P3_anchor (same as P2) AAGGTCCTCAGCCATTCAGCACCATGC >P3_consensus_macrophage AAGGTCCTCAGCCATTCAGCACCATGCGGACGAGCTCTTCATAGTTGATACAACCATTGCTGTCCTCATG CCCTGCCACCAGCATCTCTACTTCTTCCTCTGTCATCTTCTCACCCAGTGTGACAAGAACATGCCGGA >P3_consensus_capillary AAGGTCCTCAGCCATTCAGCACCATGCGGACGAGCTCCGCCCCATGGGCCCGTCACCCCGACAGGATATG CCTCACAAACGCTTCATAGTTGATACAACCATTGCTGTCCTCATGCCCTGCCACCAGCATCTCTACTTCT TCCT >P3_target1 CAACCATTGCTGTCCTCATGCCCTGCC >P3_target2 CGTCACCCCGACAGGATATGCCTCACA
mouse lemur COX2 (cytochrome c oxidase subunit II) (Figure S5A)
(reverse-complements are shown in Figure S5A)
>anchor ATTTAGGCGCCCTGGGATAGCATCTGT >target_1 TTCATGAATGTAGTACGTCTTCTGAAG >target_2 TTCATGAATGTAATACGTCTTCTGAAG
lemur IGLC3 with 97 targets (Figure S5B)
>anchor ACCGAGGGGGCGGCCTTGGGCTGACCT >Targ0 GCCGAACACCCCAGTGCCACCACTCCT >Targ1 GCCGAAGATATGACCACTCAGGCTGTC >Targ2 GCCGAACACATGATTGTAGCTGCCATC >Targ3 GCCGAATACATTAACACCACTGTTGTC >Targ4 GCCGAACACATAACCATATGAATCACC >Targ5 GCCGAACACACCACCACTGCTGTCCCC >Targ6 GCCGAACACATTAACACCACCGTCCCA >Targ7 GCCGAATACAGCACTGTTGTGCCACAC >Targ8 GCCGAAGATATAAGTGTTCCTGCCCGC >Targ9 GCCGAACACACCAACACCACTGCTGTC >Targ10 GCCGAACACACCAACACCAGTTTCCCA >Targ11 GCCGAAGATAACACCACTGTTGTCCCA >Targ12 GCCGAACACACTGTAGCTGCCATCATA >Targ13 GCCGAACACATAACCATATGAACCACC >Targ14 GCCGAAGATATACTGAATGCTGCTCCC >Targ15 GCCGAAGATATAAGTATTAGAGCTGCC >Targ16 GCCGAACACCCGAGCATCAAGACTGCT >Targ17 GCCGAATACATAAGCACTCAGGCTTTT >Targ18 GCCGAACACCCGACCATTCAGGCTGCT >Targ19 GCCGAATACATAAGTGCCACTGTTGGC >Targ20 GCCGAAGATATACGCACTCAGGCTACT >Targ21 GCCGAACACCTGACCACTCAGGCTACT >Targ22 GCCGAACACACCAACACCACTGTTGTC >Targ23 GCCGAACACCCAACTAGCACTGGCATC >Targ24 GCCGAACACACCAGCACGTAGGCTGCT >Targ25 GCCGAACACATGACCACTCAGGCTACT >Targ26 GCCGAACACATGAGCACTCAGGCTTCT >Targ27 GCCGAACACCCGACTGTAGCTGCCATC >Targ28 GCCGAAGATATTAACACCACTGTTGTC >Targ29 GCCGAAGATATCACTCAGGCTACTGTC >Targ30 GCCGAACACCCAACTCTTAGAGCTGCC >Targ31 GCCGAACACATCAGCACTGTTGTGCCA >Targ32 GCCGAACACAAGATTGTAGCTGCCATC >Targ33 GCCGAACACATAACTCTTAGAGCTGCC >Targ34 GCCGAACACCCCAGTGCCACCACTCTT >Targ35 GCCGAACACATCACCACTCAGGCTACT >Targ36 GCCGAACACCCTGCTGTCATAGGACTG >Targ37 GCCGAACACCCAATTAACACCACTGCT >Targ38 GCCGAACACCCAAGCATCAAGACTGGT >Targ39 GCCGAACACACGAGCATCAAGACTGCT >Targ40 GCCGAACACCCAACCATATGAATCACC >Targ41 GCCGAACACACCATGACCACTCAGGCT >Targ42 GCCGAACACACCATAGTTTCCATAACC >Targ43 GCCGAACACCGCATTAAGACTGCTGTC >Targ44 GCCGAAGATATACTGGTTGCTGAACCA >Targ45 GCCGAACACACCATGAGTACCAGTGCT >Targ46 GCCGAATACATGACCACTCAGGCTGTC >Targ47 GCCGAACACACCATCAAGACTGCTGTC >Targ48 GCCGAAGATATAAGTGCCGCTGCCCGC >Targ49 GCCGAACACATGACCACTCAGGCTTCT >Targ50 GCCGAACACACCAGCATCAAGACTGCT >Targ51 GCCGAAGATATAAGTGTTGCTGCCCGC >Targ52 GCCGAACACCCAAGCATCAAGACTGCT >Targ53 GCCGAACACACCATGACTCAGGCTGCT >Targ54 GCCGAACACCCAAACACCACTGTTGTC >Targ55 GCCGAACACATGAGCACTCAGGCTACT >Targ56 GCCGAACAGACCACTCAGGCTACTATC >Targ57 GCCGAAGATATACCCATATGAACCACC >Targ58 GCCGAAGATATGACCACTCAGGCTACT >Targ59 GCCGAACACCCAACCATATGAACCACC >Targ60 GCCGAATACATAATTGTAGCTGTCATC >Targ61 GCCGAACACACCACCACTCAGGCTGTC >Targ62 GCCGAACACAAAATTAACACCACTGCT >Targ63 GCCGAACACAGCACGCAGACTGCTGTC >Targ64 GCCGAACACCCAAGTGCCGCTGCCCGC >Targ65 GCCGAACACCCAGCACTGTTGTGCCAC >Targ66 GCCGAAAACATAAGTCTTAGACCTGCC >Targ67 GCCGAAGATATACGTATCAAGACTGCT >Targ68 GCCGAAGATATTGTTTTCACTAACCCA >Targ69 GCCGAAGATAGCACTGTTGTGCCACAC >Targ70 GCCGAACACACGAGCACCCAGACTACT >Targ71 GCCGAATACATGACCATTCAGGCTGCT >Targ72 GCCGAATATATAACTCTTAGAACTGCC >Targ73 GCCGAACACAAAACGGTTGCTGAACCA >Targ74 GCCGAACATCCAACTCTTAGAGCTGCC >Targ75 GCCGAACACCCAAGTCTTAGAGCTGCC >Targ76 GCCGAACACATGACTGTAGCTGTCATC >Targ77 GCCGAACACCCAATGGTTGCTGAACCA >Targ78 GCCGAACACCCAAAGTGCCGCTGCCCG >Targ79 GCCGAACACACCAGTCTTAGAGCTGCC >Targ80 GCCGAAGATATTAACACCAGTTTCCCA >Targ81 GCCGAACACACTGTAGCTGTCATCATA >Targ82 GCCGAATACAAATGGTTGCTGAACCAC >Targ83 GCCGAACACCCTATTAACACCACTGCT >Targ84 GCCGAACACAGCATCAAGACTGCTGTC >Targ85 GCCGAATACATAATCAAGACTGCTGTC >Targ86 GCCGAACACACCACTCAGGCTACTATC >Targ87 GCCGAAGATAGCATGAGTACCAGTATT >Targ88 GCCGAAGATAAGACCACTCAGGCTACT >Targ89 GCCGAACACAATAGCTGCCATCATAAG >Targ90 GCCGAACACCTGATTGTAGCTGTCATC >Targ91 GCCGAACACAAGACTAACACTGTCATC >Targ92 CAGAGGCCTGTGTCCACCTGGGGAGCC >Targ93 GCCGAACACACCTAGAGCTGCCATTCC >Targ94 GCCGAATACATTAACACCACTGCTGTC >Targ95 GCCGAATACATAATTGTAGCTGCCATC >Targ96 GCCGAAGACAAACATCGACTGAGGCTC
lemur TCR-beta J-region (Figure S5C)
>anchor CCGGGTCCCTGGCCCGAAGAACTGCTC >Targ0 TGCCGCTGCAGATGTAGACGCCGCTGT >Targ1 CGCAGAGATACAGGGCCGAGTCCCCCA >Targ2 TGGCACAGAGGTACGTGGCGGAGTCTT >Targ3 TGCTGGCACAGAGGTACGTGGCAGAGT >Targ4 AGAGGAACAGGGCCGAGTCCCCCAGCG
lemur TCR-gamma V-region (Figure S5D)
>anchor ACCCTCACCATTCACAATGTAGAGAAA >Targ0 TGCCCGTGAACTCTTCAGTAATGGAAC >Targ1 TGCCTCCTGGGAGTCTAGGAAACTCTT >Targ2 TGCCTCCTGGGACTGACGACTTACCAA >Targ3 TGCCTCCTGGGAGTTGAATTTTTATAG >Targ4 TGCCTCCTGGGAGTTGCACAGTGTCAC >Targ5 GCCCGTGAACTCTTCAGTAATGGAACA >Targ6 TGCCTCCTGGGAGTCGCTCTCTAATAT >Targ7 TGCCTCCTGGGAGTTGCACAGAAGATT
Octopus bimaculoides carboxypeptidase D (Figure S6A)
(reverse-complements are shown in Figure S6A)
>anchor GGAATTAGAAGAAAAATCTATTATGAA >target_1 AAATGTTTAGGCCAATATCTAAAGGCA >target_2 AAATGTTTAGGAAAAATTTTCTGCCAA
Octopus bimaculoides Upf2 (regulator of nonsense transcripts 2) (Figure S6B)
(reverse-complements are shown in Figure S6B)
>anchor GTATTGCACTGCATTGTACTGCACTGT >target_1 CGCTGCTGCTGCTGCTGCTGCCAATTG >target_2 CGCTGCTGCTGCTGCTGCCAATTGCCT
Octopus bimaculoides netrin receptor / DCC (Figure S6C)
(reverse-complements are shown in Figure S6C)
>anchor TCTATTACAGCTATCATCAATACACTT >target_1 TTGGATGTCTTCGTGTTCTCACTGCAG >target_2 TTGGATGTCTTTGTGTTCTCACTGCAG
HMG-box (diatom) (Figure S7A)
(reverse-complements are shown in Figure S7A)
>anchor TGCGGTCCTTGAATTCTTGCTTCTCTT >target_1 TATCCGAAAGAGCCCTCCACATTTCAC >target_2 CGTCCGTCAGAGCTCTCCACATTTCTC
ferredoxin (diatom) (Figure S7B)
(reverse-complements are shown in Figure S7B)
>anchor ACGGCACGAGTAGGGAAGTTCAATTCC >target_1 GGCTTCTTCAGCAGCGTCGACAATGAA >target_2 GGCTTCTTCGGCAGCGTCGACAATGAA
Open-source figure attributions
Person graphic, by Tanguy Krl: https://thenounproject.com/icon/person-1218528
Virus graphic, by Nuttapon Pohnprompratahn: https://thenounproject.com/icon/virus-2198681.
Gears graphic, by Tresnatiq: https://thenounproject.com/icon/gears-1088494.
Clock graphic, by sudan: https://thenounproject.com/icon/clock-5677937.
Book graphic, by Arjan: https://thenounproject.com/icon/open-book-1361747.
SARS-CoV-2 virion, by Centers for Disease Control and Prevention (CDC): https://commons.wikimedia.org/wiki/File:SARS-CoV-2_without_background.png Octopus bimaculatus, by United States Fish Commission (1910): https://commons.wikimedia.org/wiki/File:Octopus_bimaculatus1.jpg
Diatom, by George Swann: https://commons.wikimedia.org/wiki/File:Diatom_4.png
Zostera marina, by Carl Axel Magnus Lindman: https://commons.wikimedia.org/wiki/File:491_Zostera_marina.jpg
Gray mouse lemur, by Gabriella Skollar: https://commons.wikimedia.org/wiki/File:Gray_Mouse_Lemur_1.JPG
QUANTIFICATION AND STATISTICAL ANALYSIS
SPLASH -values
SPLASH’s analysis is based on a new statistical method for analyzing contingency tables, to reject the null hypothesis that row (target) counts are drawn from the same distribution for all samples. Despite its rich history, the field of statistical inference for contingency tables still has many open problems80. The field’s primary focus has been on either small contingency tables (2×2, e.g. Fisher’s exact test81), high counts settings where a chi-square test yields asymptotically valid -values, or computationally intensive Markov-Chain Monte-Carlo (MCMC) methods. While contingency tables have been widely analyzed in the statistics community80,82,83, to our knowledge no existing tests provide closed form, finite-sample valid statistical inference with desired power for the application at hand. In this work, we develop a flexible statistical test that provides closed form -value bounds, meaning that no permutation, resampling or numerical solutions to complex likelihoods are required for significance tests. In subsequent work, more sophisticated (optimization-based) approaches to computing improved and have been developed, leveraging a linear algebraic perspective on the test statistic52.
We develop a test statistic S that has power to detect many forms of sample-dependent sequence variation and is designed to have low power when there are a few outlying samples with low counts. Considering the target × sample contingency table, we first randomly draw a vector , which maps each target independently to {0,1} equiprobably. We then compute the mean value of targets with respect to this function. Next, we compute the mean target value within each sample with respect to . Then, an anchor-sample score is constructed for sample , , as a scaled version of the difference between these two (scaled by the square root of the total counts for this sample). Finally, the test statistic is computed as the weighted sum of these , with weights (which denote class-identity in the two-group case with metadata and are chosen randomly without metadata, see below). In the below equations, denotes the sequence of the -th target observed for the -th sample (for analysis purposes, we assume that the targets are drawn in a random order), is the number of observations in this sample, and denotes the total number of observations of this anchor.
Statistically valid -value bounds (non-asymptotic, providing control of Type I error for finite number of observations) are computed as:
by applying Hoeffding’s inequality on these sums of independent random variables (under the null). We provide a graphical description of this procedure in (Figure S1A), and details the derivation below.
First, observe that has mean 0 under the null hypothesis. This allows us to bound the probability that the random variable is larger than our observed anchor statistic as follows. Since and are fixed, and are independent of the data, we have that since are bounded between 0 and 1 we can apply Hoeffding’s inequality for bounded random variables. Defining as the expectation with respect to the common underlying distribution of (unknown), we center our random variables by subtracting the sample mean , our estimate of the true mean . Standard bounds can now be applied to decompose this deviation probability into two intuitive and standard terms, which we then bound with Hoeffding’s inequality for bounded random variables:
- the probability that the statistic , constructed with unavailable knowledge of the true
, is large - the probability that is far from .
The bound is optimized to within a factor of 2 by equating the two terms, with a as stated.
This statistic is computed for different random choices of , and in the case where sample group metadata is not used or available, jointly for each of the random choices of (each entry drawn independently as ±1), here with and . The choice of and that minimize the -value bound are reported, and are used for computing the -value bound for this anchor. To control Type I error we apply Bonferroni correction over the multiple hypotheses tested (just when sample metadata is used and randomization on is not performed). Then, to determine the significant anchors, we apply Benjamini-Yekutieli (BY) correction84 to the list of -value bounds (one for each anchor), yielding valid FDR controlled -values reported throughout the manuscript implemented with the statsmodels.api.stats.multipletests functionality in python. Additional theoretical properties of this statistic, and an improved -value bound, have been developed in follow-up work52.
SPLASH effect size
SPLASH provides a measure of effect size when the ’s used are ±1, to allow for prioritization of anchors with large inter-sample differences in target distributions (as the -value bound is heavily impacted by the number of observations). Effect size is calculated based on the split and vector that yield the smallest SPLASH -value bound. Fixing these, the effect size is the absolute value of the difference between the mean target value (with respect to ) across those samples with denoted , and the mean target value (with respect to ) across those samples with denoted .
The effect size should be thought of under the alternative hypothesis where the columns follow multinomial distributions with probability vector or probability vector , depending on the group identity . The effect size we compute can be thought of in this scenario as measuring the difference between the expectation of under and . In the case of maximizing the effect size over all possible {0,1}-valued , the effect size will be equal to the total variation distance between the empirical distributions of the group and . Thus, the effect size will be 1 if and only if the two sample groups partition targets into 2 disjoint sets on which the function takes opposite values, as to be expected from the total variation distance interpretation (Figure S1B). This will place a value of 1 on targets where the empirical frequency of the +1 group is larger than that of the −1 group . Since and are probability distributions, this ends up being exactly the total variation distance between them (i.e. half the vector distance).
Binomial p-value bound computation for plots depicting target fraction abundance
We provide -values to quantify the visually striking nature of the plots depicting fraction abundance per a specific target (target 1 by default). Under a null model, where all samples are expressing this target with the same probability, the number of times each sample expresses target 1 is binomial(,), for common . As seen from the plots, many samples exhibit highly deviating occurrences (number of observations of target 1 that are far from the expected . The -values we provide to this effect are not used in any SPLASH discovery or analysis, and are just used to quantify the visuals.
-values are constructed as follows: first, we compute p, the average occurrence of target 1 for this anchor (sum of counts of observations of target 1 divided by the total number of observations). Then, for all possible , we compute 1% and 99% quantiles (confidence bounds) for a binomial distribution with trials and heads probability p. If the fraction of target 1 in each sample was independent of sample identity, and were indeed binomially distributed, then each sample would have at least a 98% probability of falling within this confidence interval. Thus, we compute our test statistic as the number of samples that fall outside of the [1,99] quantiles, and compute as our -value the probability that a binomial random variable Bin(, )≥ , where with = number of samples and .
While intuitive, the above analysis is loose. Firstly, since binomials are discrete distributions, we will rarely be able to compute exact 1% and 99% quantiles. Thus, the probability that for any given a sample will fall outside of the [1,99] quantiles, which we denote , is almost always substantially less than .02. The true distribution of X is then poisson binomial, with this vector of probabilities (all at most .02), one for each sample. However, as this -value is numerically difficult to compute, we bound this -value as the probability that Bin(, ), where = number of samples with , where .
Supplementary Material
Data S1.
Data S2.
A. -value computation for SPLASH for user specified vectors and . Contingency table transposed for visual convenience (rows are samples and columns are targets). Starting with a counts matrix, SPLASH utilizes the vector to map targets to values within [0,1]. The mean with respect to is taken over the targets in each sample to yield , and an estimate for the mean over all target observations of is taken, yielding . The anchor-sample scores are then constructed as the difference between the row mean and the overall mean , scaled by (the number of observations in sample ). These anchor-sample scores are weighted by and summed to yield the anchor statistic . Finally, a -value bound is computed utilizing classical concentration inequalities, which we correct for multiple hypothesis testing (with dependence) by constructing -values using Benjamini-Yekutieli, a variant of Benjamini Hochberg testing which corrects for arbitrary dependence.
B. Effect size computation for SPLASH. Reported effect size is calculated based on the split and vector that yielded the most significant SPLASH -value. Fixing these, the effect size is computed as the difference between the mean across targets (with respect to ) across those samples with , and the mean across targets (with respect to ) across those samples with . This should be thought of as studying an alternative where samples from have targets that are independent and identically distributed with mean (under ) of , and samples with have targets that are independent and identically distributed with mean (under ) of . The total effect size is estimated as , which relates to the total variation distance between the two target distributions (STAR Methods).
We performed SPLASH protein domain profiling in a dataset of virally enriched samples from breakthrough infections in patients that had been vaccinated against rotavirus; nearly all did have rotavirus infection, but some also had other coinfections17. The top domains were rotavirus VP3 (Rotavirus_VP3, 76 SPLASH hits vs 9 control hits) followed by rotavirus NSP3 (Rota_NSP3, 87 SPLASH vs 35 control hits), indicating that these rotavirus genes have sequences that vary significantly among these patients; they have roles in blocking host innate immunity18, and so may be under selective pressure for variation.
A. SPLASH’s unsupervised approach (selection of random c vectors) yields good performance, and is able to recapitulate results obtained by the supervised method. We validate this on the HLCA dataset, where given metadata indicates whether a sample is from macrophage or capillary cells, which the supervised approach utilizes. Of the 3336 anchors (848 genes) called by the supervised approach in individual P2 (BY correction, alpha=.05), 71% of the genes called were also called by SPLASH’s unsupervised ’s (6407 called by anchors by random ’s, 1503 genes). Left plot indicates for each gene (dot) how many times it was called by samplesheet ’s vs random ’s. Red dots indicate those genes not called by random c’s. The right plot shows the fraction of genes that are called at least times by samplesheet ’s that are also called by random ’s. For (i.e. all genes hit by > 2 anchors), random ’s call >92% of those genes called by samplesheet ’s.
B. If reference alignment is desired, aligning only SPLASH results (consensuses) reduces the amount of data for alignment by >500-fold, compared to aligning all the raw sequencing reads; this is consistent in two different datasets. (MiB = mebibyte = 1,048,576 bytes)
A. MYL6 SPLASH results from two individuals (P2 and P3) are shown. For all the plots, columns represent different samples (single cells). The heatmap with rows labeled by target sequences is the sample×target contingency table, entries indicate the respective target fraction for a given sample; -ticks give the anchor counts in that sample; -ticks give the target counts across all samples. The top two plots show ’s; “samplesheet ” are the cell-type labels provided in the original study (pink = macrophage, blue = capillary), “ found by random” are SPLASH’s unsupervised assignments. The two bottom plots relate to the consensus sequences. The lower left plot depicts the nucleotide sequence using colors for the bases. Each column corresponds to the consensus sequence for the sample of the same column above it in the contingency table. The rows are base pair positions along each consensus. These consensus sequences are variable length, and a value of −1 (yellow color) on the bottom of a sequence indicates that the consensus has ended. The bottom right plot shows the fraction agreement per nucleotide within a sample with its consensus sequence. For samples where only one isoform / SNP is expressed the consensus stays near 100%, while for samples with a diverse set of targets the consensus is less uniform.
B. Macrophages tend to express the exon-skipping isoform, as shown by spread plots, with each dot representing the relative isoform expression in a single cell; a bar marks the average relative expression across all the cells.
C. UCSC Genome Browser diagram, showing BLAT mapping of macrophage and capillary consensus sequences, which fully cover inclusion or exclusion of the short alternative exon.
For panels B-D, target × sample heatmaps are shown on the left (samples are individual cells), and bp color-maps on the right. The small numbers at the sides of the heatmaps are summed target counts (over rows or columns).
A. COX2. SPLASH detects numerous anchors that have target fractions that correspond precisely to the identities of the two lemur individuals represented in the set of cells analyzed. One example is an anchor in COX2. The alignment of two consensuses (both from NKT cells) shows that targets 1 and 2 differ at a single silent position (C vs T) and another silent SNP is also present outside the target region (G vs A); these positions are highlighted in red. Consensuses 1 and 2 align perfectly to different lemur mitochondrial genome accessions (NC_028718.1 and KR911907.1). Targets 1 and 2 are found exclusively in individuals L4 and L2, respectively.
B. Ig-lambda C-region. Depicted is a lemur anchor that maps to the 5’ end of the human Ig-lambda C3 segment. It has 97 different targets, which lie within the hypervariable CDR3 region. Nearly all targets are expressed clonotypically (i.e., cells do not share targets).
C. Ig-beta J-region. We analyzed natural killer T (NKT) cells from lemur. In humans and mice, a subset of NKT cells express stereotypical or shared TCR genes; Figure 4B shows an example anchor in TCR-alpha. Here we show an anchor that maps to a human TCR-beta J-region (J2-1); targets reside in the V-region. A number of the NKT cells express a shared TCR-beta target (seen in the top row of the heatmap). All of these cells derive from individual L4.
D. Ig-gamma V-region. There is evidence for shared TCR-gamma sequences in NKT cells as well. This anchor maps to a human TCR-gamma V-region (TRGV9); the targets lie at the V-J junction. Individual L4 has two different shared targets (rows 1 and 3) while individual L2 shows three cells expressing a shared target (row 2). Other NKT cells express unique targets, however.
Table S1. SARS-CoV-2 metadata and strain genome sequences, related to Figure 2.
Table S3. Macrophage and capillary single-cell output tables, related to Figure 3.
Table S4. Human and lemur immune single-cell output tables, related to Figure 4.
Highlights.
Paradigm for statistical detection of sample-dependent variation in sequencing data
Computationally efficient, widely applicable, without need for reference genomes
Finds viral strain mutations; cell-type specific isoforms; Ig and TCR diversity
Finds octopus tissue isoforms; eelgrass/diatom seasonal, geographic variation
Acknowledgements
We thank Elisabeth Meyer for edits and figure help; Arjun Rustagi for virology discussion; Roozbeh Dehghannasiri for single-cell data selection; the Tabula Microcebus Consortium (Camille Ezran and Hannah Frank) for data sharing and discussion of antigen receptor detection; Jessica Klein for figure help; Michael Swift for discussion of antigen receptor detection; Robert Bierman for figure help and laptop timing; Sebastian Deorwicz and Marek Kokot for early access to code; Andy Fire, Aaron Straight, Catherine Blish, Peter Kim, Alex Starr, and Julia Olivieri for feedback and comments.
Funding
J.S. is supported by the Stanford University Discovery Innovation Award, National Institute of General Medical Sciences grant R35 GM139517 and the National Science Foundation Faculty Early Career Development Program Award MCB1552196. T.Z.B. is supported by the National Science Foundation Graduate Research Fellowship Program and the Stanford Graduate Fellowship.
Inclusion and Diversity
We support inclusive, diverse, and equitable conduct of research.
Footnotes
Declaration of Interests
K.C., T.Z.B. and J.S. are inventors on provisional patents related to this work. The authors declare no other competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sherman RM, Forman J, Antonescu V, Puiu D, Daya M, Rafaels N, Boorgula MP, Chavan S, Vergara C, Ortega VE, et al. (2019). Assembly of a pan-genome from deep sequencing of 910 humans of African descent. Nat. Genet 51, 30–35. 10.1038/s41588-018-0273-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nurk S, Koren S, Rhie A, Rautiainen M, Bzikadze AV, Mikheenko A, Vollger MR, Altemose N, Uralsky L, Gershman A, et al. (2022). The complete sequence of a human genome. Science 376, 44–53. 10.1126/science.abj6987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Domingo E, and Perales C. (2019). Viral quasispecies. PLoS Genet. 15, e1008271. 10.1371/journal.pgen.1008271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tettelin H, and Medini D. eds. (2020). The pangenome: diversity, dynamics and evolution of genomes (Springer; ) 10.1007/978-3-030-38281-0. [DOI] [PubMed] [Google Scholar]
- 5.Castel SE, Levy-Moonshine A, Mohammadi P, Banks E, and Lappalainen T. (2015). Tools and best practices for data processing in allelic expression analysis. Genome Biol. 16, 195. 10.1186/s13059-015-0762-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Romano Y, Sesia M, and Candès E. (2019). Deep Knockoffs. J. Am. Stat. Assoc, 1–27. 10.1080/01621459.2019.1660174.34012183 [DOI]
- 7.Chung E, and Romano JP (2013). Exact and asymptotically robust permutation tests. Ann. Statist 41, 484–507. 10.1214/13-AOS1090. [DOI] [Google Scholar]
- 8.Viana R, Moyo S, Amoako DG, Tegally H, Scheepers C, Althaus CL, Anyaneji UJ, Bester PA, Boni MF, Chand M, et al. (2022). Rapid epidemic expansion of the SARS-CoV-2 Omicron variant in southern Africa. Nature 603, 679–686. 10.1038/s41586-022-04411-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bal A, Simon B, Destras G, Chalvignac R, Semanas Q, Oblette A, Quéromès G, Fanget R, Regue H, Morfin F, et al. (2022). Detection and prevalence of SARS-CoV-2 co-infections during the Omicron variant circulation in France. Nat. Commun 13, 6316. 10.1038/s41467-022-33910-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hodcroft EB (2021). CoVariants: SARS-CoV-2 Mutations and Variants of Interest. https://covariants.org/faq#what-are-defining-mutations.
- 11.Hodcroft EB, Domman DB, Snyder DJ, Oguntuyo KY, Van Diest M, Densmore KH, Schwalm KC, Femling J, Carroll JL, Scott RS, et al. (2021). Emergence in late 2020 of multiple lineages of SARS-CoV-2 Spike protein variants affecting amino acid position 677. medRxiv. 10.1101/2021.02.12.21251658. [DOI]
- 12.Colson P, Delerce J, Burel E, Beye M, Fournier P-E, Levasseur A, Lagier J-C, and Raoult D. (2022). Occurrence of a substitution or deletion of SARS-CoV-2 spike amino acid 677 in various lineages in Marseille, France. Virus Genes 58, 53–58. 10.1007/s11262-021-01877-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zeng C, Evans JP, Faraone JN, Qu P, Zheng Y-M, Saif L, Oltz EM, Lozanski G, Gumina RJ, and Liu S-L (2021). Neutralization of SARS-CoV-2 Variants of Concern Harboring Q677H. MBio 12, e0251021. 10.1128/mBio.02510-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mistry J, Chuguransky S, Williams L, Qureshi M, Salazar GA, Sonnhammer ELL, Tosatto SCE, Paladin L, Raj S, Richardson LJ, et al. (2021). Pfam: The protein families database in 2021. Nucleic Acids Res. 49, D412–D419. 10.1093/nar/gkaa913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qi H, Liu B, Wang X, and Zhang L. (2022). The humoral response and antibodies against SARS-CoV-2 infection. Nat. Immunol 23, 1008–1020. 10.1038/s41590-022-01248-5. [DOI] [PubMed] [Google Scholar]
- 16.Voss WN, Hou YJ, Johnson NV, Delidakis G, Kim JE, Javanmardi K, Horton AP, Bartzoka F, Paresi CJ, Tanno Y, et al. (2021). Prevalent, protective, and convergent IgG recognition of SARS-CoV-2 non-RBD spike epitopes. Science 372, 1108–1112. 10.1126/science.abg5268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Simsek C, Bloemen M, Jansen D, Beller L, Descheemaeker P, Reynders M, Van Ranst M, and Matthijnssens J. (2021). High prevalence of coinfecting enteropathogens in suspected rotavirus vaccine breakthrough cases. J. Clin. Microbiol 59, e0123621. 10.1128/JCM.01236-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Antia A, Pinski AN, and Ding S. (2022). Re-Examining Rotavirus Innate Immune Evasion: Potential Applications of the Reverse Genetics System. MBio 13, e0130822. 10.1128/mbio.01308-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Picelli S, Faridani OR, Björklund AK, Winberg G, Sagasser S, and Sandberg R. (2014). Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc 9, 171–181. 10.1038/nprot.2014.006. [DOI] [PubMed] [Google Scholar]
- 20.Travaglini KJ, Nabhan AN, Penland L, Sinha R, Gillich A, Sit RV, Chang S, Conley SD, Mori Y, Seita J, et al. (2020). A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature 587, 619–625. 10.1038/s41586-020-2922-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Olivieri JE, Dehghannasiri R, Wang PL, Jang S, de Morree A, Tan SY, Ming J, Ruohao Wu A, Tabula Sapiens Consortium, Quake, S.R., et al. (2021). RNA splicing programs define tissue compartments and cell types at single-cell resolution. eLife 10. 10.7554/eLife.70692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grant JW, Taubman MB, Church SL, Johnson RL, and Nadal-Ginard B. (1990). Mammalian nonsarcomeric myosin regulatory light chains are encoded by two differentially regulated and linked genes. J. Cell Biol 111, 1127–1135. 10.1083/jcb.111.3.1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vedula P, Kurosaka S, Leu NA, Wolf YI, Shabalina SA, Wang J, Sterling S, Dong DW, and Kashina A. (2017). Diverse functions of homologous actin isoforms are defined by their nucleotide, rather than their amino acid sequence. eLife 6, e31661. 10.7554/eLife.31661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Trowsdale J, and Knight JC (2013). Major histocompatibility complex genomics and human disease. Annu. Rev. Genomics Hum. Genet 14, 301–323. 10.1146/annurev-genom-091212-153455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Andersson G, Svensson AC, Setterblad N, and Rask L. (1998). Retroelements in the human MHC class II region. Trends Genet. 14, 109–114. 10.1016/s0168-9525(97)01359-0. [DOI] [PubMed] [Google Scholar]
- 26.hla.alleles.org (2023). Numbers of HLA Alleles. HLA Informatics Group, Anthony Nolan Research Institute. https://hla.alleles.org/nomenclature/stats.html. [Google Scholar]
- 27.Daar AS, Fuggle SV, Fabre JW, Ting A, and Morris PJ (1984). The detailed distribution of MHC Class II antigens in normal human organs. Transplantation 38, 293–298. 10.1097/00007890-198409000-00019. [DOI] [PubMed] [Google Scholar]
- 28.Pober JS, Merola J, Liu R, and Manes TD (2017). Antigen presentation by vascular cells. Front. Immunol 8, 1907. 10.3389/fimmu.2017.01907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tian R, Zhu H, Pang Z, Tian Y, and Liang C. (2019). Extraordinary diversity of HLA class I gene expression in single cells contribute to the plasticity and adaptability of human immune system. BioRxiv. 10.1101/725119. [DOI]
- 30.Briney B, Inderbitzin A, Joyce C, and Burton DR (2019). Commonality despite exceptional diversity in the baseline human antibody repertoire. Nature 566, 393–397. 10.1038/s41586-019-0879-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Teraguchi S, Saputri DS, Llamas-Covarrubias MA, Davila A, Diez D, Nazlica SA, Rozewicki J, Ismanto HS, Wilamowski J, Xie J, et al. (2020). Methods for sequence and structural analysis of B and T cell receptor repertoires. Comput. Struct. Biotechnol. J 18, 2000–2011. 10.1016/j.csbj.2020.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tabula Sapiens Consortium RC, Karkanias J, Krasnow MA, Pisco AO, Quake SR, Salzman J, Yosef N, Bulthaup B, Brown P, et al. (2022). The Tabula Sapiens: A multiple-organ, single-cell transcriptomic atlas of humans. Science 376, eabl4896. 10.1126/science.abl4896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ezran C, Karanewsky CJ, Pendleton JL, Sholtz A, Krasnow MR, Willick J, Razafindrakoto A, Zohdy S, Albertelli MA, and Krasnow MA (2017). The mouse lemur, a genetic model organism for primate biology, behavior, and health. Genetics 206, 651–664. 10.1534/genetics.116.199448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.The Tabula Microcebus Consortium, Ezran C, Liu S, Chang S, Ming J, Botvinnik O, Penland L, Tarashansky A, de Morree A, Travaglini KJ, et al. (2021). Tabula Microcebus: A transcriptomic cell atlas of mouse lemur, an emerging primate model organism. BioRxiv. 10.1101/2021.12.12.469460. [DOI] [Google Scholar]
- 35.Heller NM, Berga-Bolanos R, Naler L, and Sen JM (2018). Natural killer T (NKT) cells in mice and men. In Signaling mechanisms regulating T cell diversity and function, Soboloff J. and Kappes DJ, eds. (CRC Press/Taylor & Francis; ). 10.1201/9781315371689-8. [DOI] [PubMed] [Google Scholar]
- 36.Canzar S, Neu KE, Tang Q, Wilson PC, and Khan AA (2017). BASIC: BCR assembly from single cells. Bioinformatics 33, 425–427. 10.1093/bioinformatics/btw631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Übelhart R, Werner M, and Jumaa H. (2016). Assembly and Function of the Precursor B-Cell Receptor. Curr. Top. Microbiol. Immunol 393, 3–25. 10.1007/82_2015_475. [DOI] [PubMed] [Google Scholar]
- 38.Liscovitch-Brauer N, Alon S, Porath HT, Elstein B, Unger R, Ziv T, Admon A, Levanon EY, Rosenthal JJC, and Eisenberg E. (2017). Trade-off between Transcriptome Plasticity and Genome Evolution in Cephalopods. Cell 169, 191–202.e11. 10.1016/j.cell.2017.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Röhr ME, Holmer M, Baum JK, Björk M, Chin D, Chalifour L, Cimon S, Cusson M, Dahl M, Deyanova D, et al. (2018). Blue carbon storage capacity of temperate eelgrass (Zostera marina) meadows. Global Biogeochem. Cycles 32, 1457–1475. 10.1029/2018GB005941. [DOI] [Google Scholar]
- 40.Jueterbock A, Duarte B, Coyer J, Olsen JL, Kopp MEL, Smolina I, Arnaud-Haond S, Hu Z-M, and Hoarau G. (2021). Adaptation of temperate seagrass to arctic light relies on seasonal acclimatization of carbon capture and metabolism. Front. Plant Sci 12, 745855. 10.3389/fpls.2021.745855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van Giesen L, Kilian PB, Allard CAH, and Bellono NW (2020). Molecular basis of chemotactile sensation in octopus. Cell 183, 594–604.e14. 10.1016/j.cell.2020.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu L, Pan L, Wei Z, and Zhang M. (2011). Structure of MyTH4-FERM domains in myosin VIIa tail bound to cargo. Science 331, 757–760. 10.1126/science.1198848. [DOI] [PubMed] [Google Scholar]
- 43.Solé M, Lenoir M, Durfort M, López-Bejar M, Lombarte A, and André M. (2013). Ultrastructural damage of Loligo vulgaris and Illex coindetii statocysts after low frequency sound exposure. PLoS ONE 8, e78825. 10.1371/journal.pone.0078825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Y, Shi F, Song J, Zhang X, and Yu S. (2015). Hearing characteristics of cephalopods: modeling and environmental impact study. Integr. Zool 10, 141–151. 10.1111/1749-4877.12104. [DOI] [PubMed] [Google Scholar]
- 45.Borowitzka MA, Lavery PS, and van Keulen M. (2006). Epiphytes of Seagrasses. In Seagrasses: Biology, Ecology and Conservation, Larkum AWD, Orth RJ, and Duarte CM, eds. (Springer; Netherlands: ), pp. 441–461. 10.1007/978-1-4020-2983-7_19. [DOI] [Google Scholar]
- 46.Prazukin AV, Lee RI, Firsov YK, and Kapranov SV (2022). Vertical distribution of epiphytic diatoms in relation to the eelgrass Zostera noltii canopy biomass and height. Aquatic Botany 176, 103466. 10.1016/j.aquabot.2021.103466. [DOI] [Google Scholar]
- 47.Cox TE, Cebrian J, Tabor M, West L, and Krause JW (2020). Do diatoms dominate benthic production in shallow systems? A case study from a mixed seagrass bed. Limnol. Oceanogr 5, 425–434. 10.1002/lol2.10167. [DOI] [Google Scholar]
- 48.Xu C, Pi X, Huang Y, Han G, Chen X, Qin X, Huang G, Zhao S, Yang Y, Kuang T, et al. (2020). Structural basis for energy transfer in a huge diatom PSI-FCPI supercomplex. Nat. Commun 11, 5081. 10.1038/s41467-020-18867-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Laughlin TG, Savage DF, and Davies KM (2020). Recent advances on the structure and function of NDH-1: The complex I of oxygenic photosynthesis. Biochim. Biophys. Acta Bioenerg 1861, 148254. 10.1016/j.bbabio.2020.148254. [DOI] [PubMed] [Google Scholar]
- 50.Ma M, Liu Y, Bai C, and Yong JWH (2021). The significance of chloroplast NAD(P)H dehydrogenase complex and its dependent cyclic electron transport in photosynthesis. Front. Plant Sci 12, 661863. 10.3389/fpls.2021.661863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kokot M, Dehghannasiri R, Baharav T, Salzman J, and Deorowicz S. (2023). SPLASH2 provides ultra-efficient, scalable, and unsupervised discovery on raw sequencing reads. BioRxiv. 10.1101/2023.03.17.533189. [DOI] [PubMed]
- 52.Baharav TZ, Tse D, and Salzman J. (2023). An interpretable, finite sample valid alternative to Pearson’s X2 for scientific discovery. BioRxiv. 10.1101/2023.03.16.533008. [DOI] [PMC free article] [PubMed]
- 53.Chen S, Zhou Y, Chen Y, and Gu J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. 10.1093/bioinformatics/bty560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shen W, Le S, Li Y, and Hu F. (2016). SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS ONE 11, e0163962. 10.1371/journal.pone.0163962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Eddy SR (2011). Accelerated profile HMM searches. PLoS Comput. Biol 7, e1002195. 10.1371/journal.pcbi.1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, and Madden TL (2009). BLAST+: architecture and applications. BMC Bioinformatics 10, 421. 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Di Tommaso P, Chatzou M, Floden EW, Barja PP, Palumbo E, and Notredame C. (2017). Nextflow enables reproducible computational workflows. Nat. Biotechnol 35, 316–319. 10.1038/nbt.3820. [DOI] [PubMed] [Google Scholar]
- 61.Salzman J, Jiang H, and Wong WH (2011). Statistical Modeling of RNA-Seq Data. Stat. Sci 26. 10.1214/10-STS343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ståhlberg A, Krzyzanowski PM, Jackson JB, Egyud M, Stein L, and Godfrey TE (2016). Simple, multiplexed, PCR-based barcoding of DNA enables sensitive mutation detection in liquid biopsies using sequencing. Nucleic Acids Res. 44, e105. 10.1093/nar/gkw224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Motahari A, Ramchandran K, Tse D, and Ma N. (2013). Optimal DNA shotgun sequencing: Noisy reads are as good as noiseless reads. In 2013 IEEE International Symposium on Information Theory (IEEE), pp. 1640–1644. 10.1109/ISIT.2013.6620505. [DOI] [Google Scholar]
- 64.Abante J, Wang PL, and Salzman J. (2022). DIVE: a reference-free statistical approach to diversity-generating and mobile genetic element discovery. BioRxiv. 10.1101/2022.06.13.495703. [DOI] [PMC free article] [PubMed]
- 65.Wu C-S, Ma Z-Y, Zheng G-D, Zou S-M, Zhang X-J, and Zhang Y-A (2022). Chromosome-level genome assembly of grass carp (Ctenopharyngodon idella) provides insights into its genome evolution. BMC Genomics 23, 271. 10.1186/s12864-022-08503-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kalvari I, Nawrocki EP, Ontiveros-Palacios N, Argasinska J, Lamkiewicz K, Marz M, Griffiths-Jones S, Toffano-Nioche C, Gautheret D, Weinberg Z, et al. (2021). Rfam 14: expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res. 49, D192–D200. 10.1093/nar/gkaa1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Storer J, Hubley R, Rosen J, Wheeler TJ, and Smit AF (2021). The Dfam community resource of transposable element families, sequence models, and genome annotations. Mob. DNA 12, 2. 10.1186/s13100-020-00230-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ross K, Varani AM, Snesrud E, Huang H, Alvarenga DO, Zhang J, Wu C, McGann P, and Chandler M. (2021). Tncentral: a prokaryotic transposable element database and web portal for transposon analysis. MBio 12, e0206021. 10.1128/mBio.02060-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Leplae R, Hebrant A, Wodak SJ, and Toussaint A. (2004). ACLAME: a CLAssification of Mobile genetic Elements. Nucleic Acids Res. 32, D45–9. 10.1093/nar/gkh084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bi D, Xu Z, Harrison EM, Tai C, Wei Y, He X, Jia S, Deng Z, Rajakumar K, and Ou H-Y (2012). ICEberg: a web-based resource for integrative and conjugative elements found in Bacteria. Nucleic Acids Res. 40, D621–6. 10.1093/nar/gkr846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Couvin D, Bernheim A, Toffano-Nioche C, Touchon M, Michalik J, Néron B, Rocha EPC, Vergnaud G, Gautheret D, and Pourcel C. (2018). CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 46, W246–W251. 10.1093/nar/gky425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Santamaria M, Fosso B, Licciulli F, Balech B, Larini I, Grillo G, De Caro G, Liuni S, and Pesole G. (2018). ITSoneDB: a comprehensive collection of eukaryotic ribosomal RNA Internal Transcribed Spacer 1 (ITS1) sequences. Nucleic Acids Res. 46, D127–D132. 10.1093/nar/gkx855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Selig C, Wolf M, Müller T, Dandekar T, and Schultz J. (2008). The ITS2 Database II: homology modelling RNA structure for molecular systematics. Nucleic Acids Res. 36, D377–80. 10.1093/nar/gkm827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Johnson LS, Eddy SR, and Portugaly E. (2010). Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinformatics 11, 431. 10.1186/1471-2105-11-431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ewels PA, Peltzer A, Fillinger S, Patel H, Alneberg J, Wilm A, Garcia MU, Di Tommaso P, and Nahnsen S. (2020). The nf-core framework for community-curated bioinformatics pipelines. Nat. Biotechnol 38, 276–278. 10.1038/s41587-020-0439-x. [DOI] [PubMed] [Google Scholar]
- 76.Albertin CB, Simakov O, Mitros T, Wang ZY, Pungor JR, Edsinger-Gonzales E, Brenner S, Ragsdale CW, and Rokhsar DS (2015). The octopus genome and the evolution of cephalopod neural and morphological novelties. Nature 524, 220–224. 10.1038/nature14668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ma X, Olsen JL, Reusch TBH, Procaccini G, Kudrna D, Williams M, Grimwood J, Rajasekar S, Jenkins J, Schmutz J, et al. (2021). Improved chromosome-level genome assembly and annotation of the seagrass, Zostera marina (eelgrass). F1000Res. 10, 289. 10.12688/f1000research.38156.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Olsen JL, Rouzé P, Verhelst B, Lin Y-C, Bayer T, Collen J, Dattolo E, De Paoli E, Dittami S, Maumus F, et al. (2016). The genome of the seagrass Zostera marina reveals angiosperm adaptation to the sea. Nature 530, 331–335. 10.1038/nature16548. [DOI] [PubMed] [Google Scholar]
- 79.Medhekar B, and Miller JF (2007). Diversity-generating retroelements. Curr. Opin. Microbiol 10, 388–395. 10.1016/j.mib.2007.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Agresti A. (1992). A Survey of Exact Inference for Contingency Tables. Stat. Sci 7, 131–153. 10.1214/ss/1177011454. [DOI] [Google Scholar]
- 81.Fisher RA (1922). On the Interpretation of X2 from Contingency Tables, and the Calculation of P. Journal of the Royal Statistical Society 85, 87–94. 10.2307/2340521. [DOI] [Google Scholar]
- 82.Diaconis P, and Sturmfels B. (1998). Algebraic algorithms for sampling from conditional distributions. Ann. Statist. 26 10.1214/aos/1030563990. [DOI]
- 83.Chen Y, Diaconis P, Holmes SP, and Liu JS (2005). Sequential Monte Carlo methods for statistical analysis of tables. J. Am. Stat. Assoc 100, 109–120. 10.1198/016214504000001303. [DOI] [Google Scholar]
- 84.Benjamini Y, and Yekutieli D. (2001). The control of the false discovery rate in multiple testing under dependency. Ann. Statist 29, 1165–1188. 10.1214/aos/1013699998. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1.
Data S2.
A. -value computation for SPLASH for user specified vectors and . Contingency table transposed for visual convenience (rows are samples and columns are targets). Starting with a counts matrix, SPLASH utilizes the vector to map targets to values within [0,1]. The mean with respect to is taken over the targets in each sample to yield , and an estimate for the mean over all target observations of is taken, yielding . The anchor-sample scores are then constructed as the difference between the row mean and the overall mean , scaled by (the number of observations in sample ). These anchor-sample scores are weighted by and summed to yield the anchor statistic . Finally, a -value bound is computed utilizing classical concentration inequalities, which we correct for multiple hypothesis testing (with dependence) by constructing -values using Benjamini-Yekutieli, a variant of Benjamini Hochberg testing which corrects for arbitrary dependence.
B. Effect size computation for SPLASH. Reported effect size is calculated based on the split and vector that yielded the most significant SPLASH -value. Fixing these, the effect size is computed as the difference between the mean across targets (with respect to ) across those samples with , and the mean across targets (with respect to ) across those samples with . This should be thought of as studying an alternative where samples from have targets that are independent and identically distributed with mean (under ) of , and samples with have targets that are independent and identically distributed with mean (under ) of . The total effect size is estimated as , which relates to the total variation distance between the two target distributions (STAR Methods).
We performed SPLASH protein domain profiling in a dataset of virally enriched samples from breakthrough infections in patients that had been vaccinated against rotavirus; nearly all did have rotavirus infection, but some also had other coinfections17. The top domains were rotavirus VP3 (Rotavirus_VP3, 76 SPLASH hits vs 9 control hits) followed by rotavirus NSP3 (Rota_NSP3, 87 SPLASH vs 35 control hits), indicating that these rotavirus genes have sequences that vary significantly among these patients; they have roles in blocking host innate immunity18, and so may be under selective pressure for variation.
A. SPLASH’s unsupervised approach (selection of random c vectors) yields good performance, and is able to recapitulate results obtained by the supervised method. We validate this on the HLCA dataset, where given metadata indicates whether a sample is from macrophage or capillary cells, which the supervised approach utilizes. Of the 3336 anchors (848 genes) called by the supervised approach in individual P2 (BY correction, alpha=.05), 71% of the genes called were also called by SPLASH’s unsupervised ’s (6407 called by anchors by random ’s, 1503 genes). Left plot indicates for each gene (dot) how many times it was called by samplesheet ’s vs random ’s. Red dots indicate those genes not called by random c’s. The right plot shows the fraction of genes that are called at least times by samplesheet ’s that are also called by random ’s. For (i.e. all genes hit by > 2 anchors), random ’s call >92% of those genes called by samplesheet ’s.
B. If reference alignment is desired, aligning only SPLASH results (consensuses) reduces the amount of data for alignment by >500-fold, compared to aligning all the raw sequencing reads; this is consistent in two different datasets. (MiB = mebibyte = 1,048,576 bytes)
A. MYL6 SPLASH results from two individuals (P2 and P3) are shown. For all the plots, columns represent different samples (single cells). The heatmap with rows labeled by target sequences is the sample×target contingency table, entries indicate the respective target fraction for a given sample; -ticks give the anchor counts in that sample; -ticks give the target counts across all samples. The top two plots show ’s; “samplesheet ” are the cell-type labels provided in the original study (pink = macrophage, blue = capillary), “ found by random” are SPLASH’s unsupervised assignments. The two bottom plots relate to the consensus sequences. The lower left plot depicts the nucleotide sequence using colors for the bases. Each column corresponds to the consensus sequence for the sample of the same column above it in the contingency table. The rows are base pair positions along each consensus. These consensus sequences are variable length, and a value of −1 (yellow color) on the bottom of a sequence indicates that the consensus has ended. The bottom right plot shows the fraction agreement per nucleotide within a sample with its consensus sequence. For samples where only one isoform / SNP is expressed the consensus stays near 100%, while for samples with a diverse set of targets the consensus is less uniform.
B. Macrophages tend to express the exon-skipping isoform, as shown by spread plots, with each dot representing the relative isoform expression in a single cell; a bar marks the average relative expression across all the cells.
C. UCSC Genome Browser diagram, showing BLAT mapping of macrophage and capillary consensus sequences, which fully cover inclusion or exclusion of the short alternative exon.
For panels B-D, target × sample heatmaps are shown on the left (samples are individual cells), and bp color-maps on the right. The small numbers at the sides of the heatmaps are summed target counts (over rows or columns).
A. COX2. SPLASH detects numerous anchors that have target fractions that correspond precisely to the identities of the two lemur individuals represented in the set of cells analyzed. One example is an anchor in COX2. The alignment of two consensuses (both from NKT cells) shows that targets 1 and 2 differ at a single silent position (C vs T) and another silent SNP is also present outside the target region (G vs A); these positions are highlighted in red. Consensuses 1 and 2 align perfectly to different lemur mitochondrial genome accessions (NC_028718.1 and KR911907.1). Targets 1 and 2 are found exclusively in individuals L4 and L2, respectively.
B. Ig-lambda C-region. Depicted is a lemur anchor that maps to the 5’ end of the human Ig-lambda C3 segment. It has 97 different targets, which lie within the hypervariable CDR3 region. Nearly all targets are expressed clonotypically (i.e., cells do not share targets).
C. Ig-beta J-region. We analyzed natural killer T (NKT) cells from lemur. In humans and mice, a subset of NKT cells express stereotypical or shared TCR genes; Figure 4B shows an example anchor in TCR-alpha. Here we show an anchor that maps to a human TCR-beta J-region (J2-1); targets reside in the V-region. A number of the NKT cells express a shared TCR-beta target (seen in the top row of the heatmap). All of these cells derive from individual L4.
D. Ig-gamma V-region. There is evidence for shared TCR-gamma sequences in NKT cells as well. This anchor maps to a human TCR-gamma V-region (TRGV9); the targets lie at the V-J junction. Individual L4 has two different shared targets (rows 1 and 3) while individual L2 shows three cells expressing a shared target (row 2). Other NKT cells express unique targets, however.
Table S1. SARS-CoV-2 metadata and strain genome sequences, related to Figure 2.
Table S3. Macrophage and capillary single-cell output tables, related to Figure 3.
Table S4. Human and lemur immune single-cell output tables, related to Figure 4.
Data Availability Statement
This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table.
All original code has been deposited at Zenodo/Github and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| SARS-CoV-2 France | Bal 20229 | NCBI: SRP365166 |
| SARS-CoV-2 South Africa | Viana 20228 | NCBI: SRP348159 |
| Rotavirus | Simsek 202117 | NCBI: SRP328899 |
| Human Lung Cell Atlas | Travaglini 201920 | European Genome- phenome Archive: EGAS00001004344 |
| Tabula Sapiens | Tabula Sapiens Consortium 202232 | https://tabula-sapiens-portal.ds.czbiohub.org/ |
| Tabula Microcebus | The Tabula Microcebus Consortium 202134 |
https://tabula-microcebus.ds.czbiohub.org |
| Octopus bimaculoides RNA-Seq data | van Giesen 202041 | NCBI: SRP327909 |
| Zostera marina RNA-Seq data | Jueterbock 202140 | NCBI: SRP327909 |
| Pfam database (Pfam-A) | Mistry 202114 | https://www.ebi.ac.uk/interpro/download/pfam/ |
| Software and algorithms | ||
| SPLASH (commit 1b73949) | This study | https://doi.org/10.5281/zenodo.8271159 https://github.com/salzmanlab/nomad |
| FASTP v0.23.2 (installed with bioconda, 2/15/23) |
Chen 201853 | https://github.com/OpenGene/fastp |
| STAR | Dobin 201354 | https://github.com/alexdobin/STAR |
| bowtie2 | Langmead 200955 | https://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| bedtools | Quinlan 201056 | https://github.com/arq5x/bedtools |
| seqkit (installed with bioconda) | Shen 201657 | https://bioinf.shenwei.me/seqkit/ |
| HMMer3 | Eddy 201158 | http://hmmer.org/ |
| BLAST | Camacho 200959 | https://blast.ncbi.nlm.nih.gov/Blast.cgi |