

Abstract
Activating cyclic GMP-AMP synthase-stimulator of interferon genes (cGAS-STING) holds great potential for cancer immunotherapy by eliciting type-I interferon (IFN-I) responses. Yet, current approaches to cGAS-STING activation rely on STING agonists, which suffer from difficult formulation, poor pharmacokinetics, and marginal clinical therapeutic efficacy. Here, we report nature-inspired oligonucleotide, Svg3, as a cGAS agonist for cGAS-STING activation in tumor combination immunotherapy. The hairpin-shaped Svg3 strongly binds to cGAS and enhances phase separation to form Svg3-cGAS liquid-like droplets. This results in cGAS-specific immunoactivation and robust IFN-I responses. Remarkably, Svg3 outperforms several state-of-the-art STING agonists in murine and human cells/tissues. Nanoparticle-delivered Svg3 reduces tumor immunosuppression and potentiates immune checkpoint blockade therapeutic efficacy of multiple syngeneic tumor models in wild-type mice, but in neither cGas−/− nor Sting−/− mice. Overall, these results demonstrate the great potential of Svg3 as a cGAS agonistic oligonucleotide for cancer combination immunotherapy.
Keywords: MT: Novel therapeutic targets and biomarker development Special Issue, cGAS, oligonucleotide therapeutics, immunostimulants, cancer vaccines, combination immunotherapy
Graphical abstract
Zhu and colleagues reported an oligonucleotide-based cGAS agonist to activate the cGAS-STING pathway and type-I interferon immune responses in both mice and human cells, and showed the potential for cancer combination immunotherapy.
Introduction
Immune checkpoint blockade (ICB) immunotherapy has improved the treatment outcomes of a growing number of cancers. However, most cancer patients have not benefited from current ICB, partly due to an immunosuppressive tumor microenvironment (TME), a lack of pre-existing antitumor immune cells, and immune-related adverse events.1 Immunostimulants and cancer antigen-specific vaccines hold a great potential to overcome these challenges and therefore maximize the clinical benefit of ICB for cancer treatment.
As pattern recognition receptors (PRRs), cGAS and STING are emerging targets for the development of immunostimulants in cancer immunotherapy. Indeed, the cGAS-STING immunostimulatory pathway is involved in various diseases such as cancer,2,3 autoimmune diseases,4,5 and senescence.6,7 Specifically, cytosolic long double-stranded DNA (dsDNA) activates cytosolic cGAS to synthesize 2′3′-cyclic GMP-AMP (2′3′-cGAMP) from endogenous ATP and GTP. 2′3′-cGAMP binds to and activates STING on endoplasmic reticulum membrane, resulting in IFN regulatory factor 3 (IRF3) phosphorylation and nuclear factor κB activation, and eventually IFN-I responses.8 IFN-I are critical cytokines for antigen presentation and T cell activation, making cGAS-STING activation appealing to elicit antitumor T cell responses, which are pivotal for cancer immunotherapy.9,10 Specifically, cGAS-STING activation upregulates the expression of proinflammatory chemokines and co-stimulatory molecules, which together promote antigen presentation and T cell priming, resulting in antigen-specific T cell responses; moreover, tumor IFN-I responses promote tumor infiltration of antitumor immune cells.11 This makes it attractive to activate the cGAS-STING pathway for cancer combination therapy.12,13
To this end, STING agonists, ranging from natural or analog cyclic dinucleotides (CDNs) to synthetic compounds, have been tested preclinically and clinically.14 However, current small-molecule STING agonists are associated with various limitations. Natural CDNs are susceptible to nuclease degradation (e.g., ectonucleotide pyrophosphatase/phosphodiesterase 1 [ENPP1]),15 which demands complex stability-enhancer modifications of CDNs to enhance their biostability. Furthermore, CDNs are very hydrophilic and small (∼700 Da) with negative electrostatic charge. This makes it difficult for current drug delivery technologies to efficiently deliver CDNs into target tissues, cells, and cytosol, where STING resides.16 As a result, the antitumor responses and tumor therapeutic efficacy of CDNs, such as a modified CDN ADU-S100 tested in a phase I clinical trial, has been marginal.17 Non-CDN small molecules are another class of STING agonists under development. However, DMXAA, a preclinical STING agonist, failed to benefit cancer therapy in a phase III clinical trial due to its selective activation of murine STING, but not human STING.18 More recent non-CDN STING agonists, such as diABZI, showed promising preclinical tumor therapeutic efficacy,19 although their clinical efficacy and safety remain to be evaluated. Finally, STING agonists can be subject to the restriction of human alleles.16 Overall, these challenges call for innovative approaches to drug development for cGAS-STING activation.
To this end, here, we report the engineering of oligonucleotide agonists for cGAS as a novel approach to cGAS-STING immunostimulation for combination cancer immunotherapy. cGAS can be activated by natural dsDNA in a sequence-independent and length-dependent manner. Specifically, cGAS activation requires relatively long dsDNA with length >45 base pairs (bp) to form ladder-like enzymatically active cGAS dimers.20,21 Interestingly, shorter dsDNA with guanosine (G)-rich overhangs also activate cGAS.22 Inspired by this, here, by structural engineering and screening a series of hairpin-shaped single-stranded DNA (ssDNA), we identified Svg3 as a potent cGAS agonist for versatile applications in combination cancer immunotherapy (Figure 1). Svg3 has a core structure of a hairpin with a GGG triplet in each of the four overhangs adjacent to the hairpin stem. Svg3 was easily synthesized on automated DNA synthesizers, and was efficiently formulated in well-established nucleic acid nanocarriers, such as liposomes and lipid nanoparticles, which allow efficient tissue, cell, and cytosolic delivery of Svg3. Svg3 elicited cGAS-dependent and cGAS-selective IFN-I responses, with undetectable inflammasome activation and pyroptosis, which could also be activated by long dsDNA. Svg3 outperformed several current state-of-the-art STING agonists to induce potent IFN-I responses in a dose-dependent manner in both murine and human immune cells and cancer cells, as well as human tumor tissues. Intratumoral (i.t.) administration of Svg3-loaded nanoparticles significantly reduced TME immunosuppression. In multiple poorly immunogenic syngeneic murine tumor models, i.t. administration of Svg3 nanoparticles significantly enhanced the therapeutic efficacies of ICB. Impressively, Svg3 outperformed several state-of-the-art STING agonists for tumor therapy. Such tumor therapeutic efficacy is cGAS and STING dependent, as verified in cGAS or STING knock out (KO) mice. Collectively, these results demonstrated the great potential of Svg3 as a novel, potent, and versatile immunostimulant for combination cancer immunotherapy.
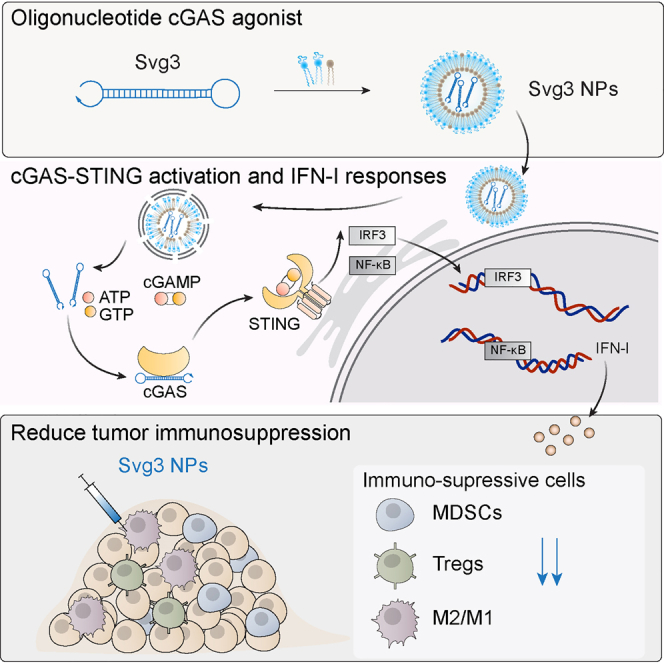
Figure 1.

Schematic illustration of an oligonucleotide-based cGAS agonist for cancer immunotherapy
A hairpin-shaped oligonucleotide, named Svg3, is engineered to selectively activate cGAS, and thereby eliciting type I IFN responses in mouse and human cells. Svg3 is readily loaded into well-established nanocarriers for efficient delivery into cells and, upon endosome escape, into cytosol where Svg3 binds to cytosolic cGAS. This results in cGAS-Svg3 phase separation and the formation of cGAS-Svg3 liquid-like droplets, in which cGAS is activated by Svg3 to trigger activation of the cGAS-STING signaling pathway and IFN-I responses. In the tumor microenvironment, Svg3-loaded nanoparticles (NPs) reduced tumor immunosuppression and enhanced antitumor immunity. Intratumoral vaccination of Svg3 NPs dramatically potentiated the tumor immunotherapeutic efficacy of ICB in multiple syngeneic murine tumor models.
Results
Identification of cGAS-agonistic Svg3 by oligonucleotide engineering and screening
Inspired by the ability of natural dsDNA to activate cGAS, we attempted to develop oligonucleotide therapeutics as cGAS agonists for application in combination cancer immunotherapy. Relative to dsDNA, ssDNA is expected to benefit from the simplicity of pharmaceutical manufacturing and quality control. Thus, to select potent cGAS-activating ssDNA, we designed a series of hairpin-structured ssDNA oligonucleotides (Figure 2A) by engineering the hairpin stem length and overhang length, as well as consecutive G numbers and positions in overhangs. These ssDNAs were transfected into RAW 264.7 murine macrophages for 24 h, followed by measuring IFN-I response. As a result, oligonucleotide with dsDNA stem length <16 bp was insufficient to triggering IFN-I response unless consecutive Gs were added in the overhangs (Figure S1A). Elongating the dsDNA stem from 10 to 24 bp, which was expected to enhance their cGAS binding affinity, indeed promoted their IFN-I responses, which plateaued at a stem length of 21 bp (Figure 2B). Further elongating the loop or adding G in the overhangs had minimal effect on IFN-I responses (Figures 2C and S1B). Svg3 showed a strong binding affinity with cGAS, with a Kd value of 262 ± 14 nM as measured by microscale thermophoresis (MST) (Figures 2D). As a result, mixing Svg3 with human cGAS protein resulted in the phase separation of cGAS-Svg3 and the formation of cGAS-Svg3 liquid-like droplets (Figure 2E). Moreover, in mouse bone marrow-derived macrophages and bone marrow-derived dendritic cells, as low as 25 nM Svg3 elicited significant IFN-β production (treatment: 24 h) (Figure 2F). Taken together, Svg3 was the oligonucleotide that elicited the strongest IFN-I responses and was therefore selected for further studies. Svg3 was predicted to form a hairpin secondary structure with a 21-bp dsDNA stem, a 9-nucleotide loop, and a GGG triplet adjacent to the stem in each of its four overhangs. Mutating the consecutive G to cytosines (C) in Svg3 overhangs dramatically reduced the IFN-I response (Figure S1C), validating their critical roles for cGAS activation. Svg3 elicited comparably potent IFN-I responses relative to interferon stimulatory DNA (ISD), a benchmark cGAS-activating 45-bp dsDNA (Figure 2G). We further validated that Svg3 treatment in RAW 264.7 macrophages resulted in efficient 2′3′-cGAMP production (Figure 2H) for at least 8 h, which outperformed ISD control, whose 2′3′-cGAMP production dramatically reduced to basal level 6 h after treatment. The efficient 2′3′-cGAMP production enabled by Svg3 is consistent with its ability to elicit potent IFN-I responses.
Figure 2.

Identification of Svg3 as a potent cGAS agonist by oligonucleotide engineering and screening
(A) Schematic structures of ssDNA oligonucleotides. (B) Elongating dsDNA stem length from 10 to 24 bp elevated IFN-I response in RAW-ISG macrophages, which plateaued at 21 bp in the stem. (C) Optimization of the loop sizes and G numbers in hairpin overhangs for cGAS-mediated IFN-I responses in RAW-ISG macrophages. (D) Svg3 showed a strong binding affinity with human cGAS as measured by MST. (E) The binding of cGAS with Svg3 induced cGAS-Svg3 phase separation to form liquid-like droplets. Alexa Fluor 488-labeled Svg3 was mixed with human cGAS for 30 min. (F) Svg3 elicited strong IFN-I responses in murine bone marrow-derived macrophages (BMDMs) and bone marrow-derived dendritic cells (BMDCs). (G) Svg3 elicited comparably potent IFN-I responses relative to ISD in RAW-ISG macrophages. (H) 2′3′-cGAMP production by RAW 264.7 cells upon treatment with Svg3 or ISD (100 nM) as a control for 4–8 h (n = 3). 2′3′-cGAMP concentration in cell lysis was measured by ELISA. Unless denoted otherwise, 25 nM DNA was transfected into cells by Lipofectamine 2000, followed by incubation for 24 h in cell culture medium before IFN-I measurement. Data: mean ± SD. p values were determined by t test. ns, not significant; p > 0.05, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Oligonucleotide therapeutics are subject to nuclease degradation that limits their biostability and therapeutic efficacy. We verified that Svg3 has decent biostability as shown by the integrity of Svg3 after incubation in PBS and cell culture medium containing 10% fetal bovine serum (FBS) (37°C, 24 h), respectively (Figure S1D). We attempted to further prolong the biostability of Svg3 by three approaches. First, we ligated the two terminals of Svg3 ssDNA into circular Svg3 that was expected to avoid exonuclease degradation; however, this did not significantly improve its ability to elicit IFN-I in RAW 264.7 macrophage (Figure S1E). Second, a phosphonothioate backbone, which is widely used in oligonucleotide therapeutics to resist nuclease degradation, in the dsDNA stem of Svg3 restricted Svg3’s ability to elicit IFN-I response in RAW 264.7 macrophages (Figure S1F). Third, adding nuclease-resistant inverted dT on the 3′ end of Svg3 did not improve the long-term IFN-I response in RAW 264.7 macrophages either (Figure S1F). Overall, although further comprehensive testing of modifications may further enhance Svg3 biostability, unmodified Svg3 has already shown great biostability and potent cGAS activation and was used in the following studies.
To elicit IFN-I responses for cancer immunotherapy in vivo, Svg3 is expected to be delivered to immune cells or tumor cells in tumor or lymphoid tissues (e.g., lymph nodes). For tissue and cell delivery of Svg3 in vivo, we employed liposomes, one of the most successful drug delivery systems thus far for oligonucleotide therapeutics and vaccines. Svg3 was efficiently loaded into liposomes (Figures S2A and S2B). Nanoparticles dramatically enhanced the cell uptake of Cy3-labeled Svg3 in RAW 264.7 macrophages (Figure S2C), followed by endosome escape of Svg3 to reach the cytosol, which allows Svg3 to activate cytosolic cGAS (Figure S2D). Moreover, liposomes prolonged the tumor retention of i.t. injected Svg3 in 4T1 mammary carcinoma in syngeneic BALB/c mice for at least 7 days (Figures S2E and S2F). These results demonstrate the ability of liposomes to prolong the tissue retention and facilitate cell uptake and cytosolic delivery of Svg3.
Svg3 is a cGAS-specific agonist
A variety of cytosolic nucleic acid sensors can be activated to elicit IFN-I responses. Specifically, aside from cGAS, cytosolic DNA sensors such as absent in melanoma 2 (AIM2), DEAD box helicase 41 (DDX41), interferon gammainducible protein (IFI16), Z-DNA binding protein (ZBP1), RNA polymerase III, and LRR binding FLII interacting protein 1 (LRRFIP1) also elicit IFN-I response.23 Moreover, DDX41, IFI16, and ZBP1 induce IFN-I responses in a cGAS-independent and STING-dependent manner.24 To study the cGAS specificity of Svg3-induced IFN-I responses, first, by RNA sequencing (RNA-seq), we analyzed the transcriptomic changes in RAW 264.7 macrophages transfected with Svg3, with blank Lipofectamine 2000 as a control. RNA-seq results verified that Svg3 induced a series of interferon-stimulated genes (ISGs), including Ifna2, Ifnb1, and Ifit1 (Figure 3A). Interestingly, Svg3 did not significantly upregulate the expression of Il-1β (encoding interleukin-1β [IL-1β]) and Il-18 (Figure 3A). This rules out AIM2 activation by Svg3, which would otherwise trigger confounding inflammasome responses, including IL-1β and IL-18 production. Macrophage IL-1β contributes to tumorigenesis25 and overexpressed IL-18 is related to tumor progression.26 This is further supported by the undetectable IL-1β and IL-18 production by Svg3 in RAW 264.7 macrophages (Figure 3B). Next, using RAW 264.7 macrophages with cGAS KO (RAW-Lucia ISG-KO-cGAS), we then verified that the Svg3-elicited IFN-I responses were cGAS dependent (Figure 3C), which also showed dose-dependent manner and lower half-maximal effective concentration (EC50) compared with STING agonists (Figure 3D). Taken together, these results demonstrate that Svg3 elicited cGAS-specific IFN-I responses.
Figure 3.

Svg3 specifically activates cGAS to induce potent IFN-I response
(A) Volcano plot of gene expression in RAW 264.7 macrophages treated with liposomal Svg3 (100 nM Svg3, 6 h). Blank liposome was used as a control. (B) IL-1β and IL-18 productions in RAW 264.7 macrophages treated with Svg3 (100 nM, 24 h). (C) IFN-I responses elicited by Svg3 (25 nM, 24 h) in RAW-ISG cells with wild-type cGAS (cGAS-WT) and cGAS-KO. (D) Svg3 was benchmarked against several state-of-the-art STING agonists to elicit IFN-I responses in murine RAW-ISG macrophages (n = 3). Svg3 outperformed these STING agonists to elicit IFN-I responses. Marked on the right of the legends are EC50 values, indicating that Svg3 showed 13.6×, 6.4×, and 43× lower EC50 values than ADU-S100, diABZI, and 2′3′-cGAMP, respectively, in RAW-ISG cells. Data: mean ± SD. p values were determined by t test. ns, not significant; p > 0.05, ∗p < 0.05.
Svg3 elicited potent IFN-I responses in human cells and human tumor tissues
We envision that local activation of intratumoral cGAS would promote multifaceted antitumor innate and adaptive immunity. To this end, clinical gene transcript analysis showed high transcript levels of cGAS and STING, respectively, in many types of human tumors such as breast cancer, melanoma, and head and neck cancer (Figure S3). This suggests the clinical potential of Svg3 to activate cGAS-STING in the corresponding types of human tumors. The inter-species differences in the immune system represents a barrier for the clinical translation of preclinically tested immunotherapeutics and vaccines. For example, due to slight structural differences of human and murine STINGs, a preclinically promising murine STING agonist, DMXAA, failed in a phase III clinical trial.18 Furthermore, there are structural differences between mouse cGAS (m-cGAS) and human cGAS (h-cGAS). Relative to m-cGAS, h-cGAS shows reduced binding ability for short dsDNAs (<45 bp).21 Therefore, it is critical to validate the ability of cGAS agonists to activate h-cGAS and elicit IFN-I responses in human systems. To this end, we first verified that Svg3 elicited potent IFN-I responses, with an EC50 as low as 15 nM in THP-1 human monocytes (Figure 4A). By benchmarking Svg3 against STING agonist 2′3′-cGAMP, Svg3 outperformed 2′3′-cGAMP to elicit IFN-I responses in THP-1 human monocytes (Figure 4A). Finally, we tested the ability of liposomal Svg3 to elicit IFN-I responses in cultured surgically collected human head and neck squamous cell carcinoma tissues. We treated these tissues with liposomal Svg3 (0.1 nmol Svg3) for 24 h, followed by quantifying the IFN-I response-associated gene transcripts by quantitative PCR (qPCR). As a result, Svg3 significantly upregulated IFN-I genes in these tissues (Figure 4B). These results highlight the potential of Svg3 for clinical translation in human patients.
Figure 4.

Svg3 elicited potent IFN-I responses in human monocytes and ex vivo human tumor tissues
(A) Svg3 elicited potent IFN-I responses that outperformed STING agonist 2′3′-cGAMP in human THP-I monocytes. Marked on the right of treatment legends are EC50 values. Svg3 and 2′3′-cGAMP were transfected using Lipofectamine 2000. (B) Relative gene expression showed that Svg3 elicited IFN-I responses in ex vivo head and neck squamous cell carcinoma tissues. Liposomal Svg3 (Svg3: 0.1 nmol) were injected directly into cultured tumor tissues (cohort study, n = 3) cultured in medium (1 mL). Twenty-four hours later, the mRNA levels of related cytokines were determined by qPCR. Data: mean ± SD. p values were determined by t test. ns, not significant; p > 0.05, ∗p < 0.05.
Svg3 reduced TME immunosuppression
TME is primarily where tumor cells suppress antitumor immunity. TME immunosuppression inhibits the tumor infiltration of antitumor immune cells and suppresses the antitumor immunity of intratumoral immune cells. Overcoming TME immunosuppression represents a hallmark to tumor immunotherapy, including the combination of immunostimulants with ICB such as αPD-1. Therefore, we studied TME immunomodulation by Svg3 that elicited potent antitumor IFN-I responses, alone or combined with αPD-1 (intraperitoneal [i.p.] injection) that can reinvigorate antitumor immune cells exhausted by prolonged exposure of inflammatory responses. Motivated by the long tumor retention of liposomal Svg3, we used liposomal Svg3 using 4T1 mammary carcinoma as a model in syngeneic BALB/c mice. Tumor (ca. 50 mm3)-bearing mice were treated with liposomal Svg3 (i.t. injection) and/or αPD-1 (i.p. injection) every 3 days for 3 times. Three days after the last treatment, tumors were excised to analyze the immune cell composition among all CD45+ leukocytes by flow cytometry. Myeloid-derived suppressor cells (MDSCs) and regulatory T cells (Tregs) are two of the most dominant immunosuppressive cell subsets in TME. Liposomal Svg3, especially when combined with αPD-1, significantly reduced the frequencies of MDSCs and CD4+Foxp3+CD25+ Tregs (Figure 5B and 5C). Meanwhile, liposomal Svg3, alone or combined with αPD-1, promoted the tumor infiltration of CD8+ T cells, and enhanced the ratio of CD8+ T cells over CD4+Foxp3+CD25+ T cells (Figures 5D–5F), both of which are expected to promote tumor therapeutic efficacy and predict tumor therapeutic efficacy. Moreover, liposomal Svg3 + αPD-1 enhanced the frequency of natural killer (NK) cells in TME and promoted the polarization of regulatory M2-like macrophage to proinflammatory M1-like macrophages, as measured M1/M2 ratio (Figure 5G and 5H). None of these treatments significantly impacted the densities of TME DCs (Figure S4). Taken together, liposomal Svg3, especially when combined with αPD-1, reduced multi-tier immunosuppressive cell densities and enhanced the antitumor immune cell densities in TME, which are expected to benefit tumor immunotherapy.
Figure 5.

Liposomal Svg3 reduced TME immunosuppression
(A) Timeline of TME immune cell subset analysis in a 4T1 tumor xenograft model treated with liposomal Svg3 (i.t.), alone or combined with αPD-1 (i.p.) as shown above. Tumors were s.c. injected on the flank of BALB/c mice (n = 3–5). Tumors were harvested on day 9 for single-cell analysis by flow cytometry. (B) Percentage of MDSCs among CD45+ cells in as-treated 4T1 tumors. (C) Ratio of CD4+Foxp3+CD25+ Tregs over all CD4+ T cells in as-treated 4T1 tumor. (D) Representative flow cytometric graphs of CD8+CD3+ T cells in as-treated 4T1 tumors. (E) Percentage of CD8+ T cells among CD45+ cells in as-treated 4T1 tumors. (F) Ratio of CD8+ T cells over CD4+ T cells in 4T1 tumors. (G) Densities of NK cells among CD45+ cells in as-treated 4T1 tumors. (H) Ratio of M1-like macrophages over M2-like macrophages in as-treated 4T1 tumors. Data: mean ± SEM. p values were determined by t test. ns, not significant; p > 0.05, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Combination of Svg3 and αPD-1 for robust tumor immunotherapy
The abilities of Svg3 to elicit IFN-I responses and reduce TME immunosuppression make this immunostimulant appealing for ICB combination immunotherapy of cancer. Specifically, we test this for Svg3 as both a therapeutic immunostimulant and an adjuvant for cancer antigen vaccines. We first evaluated Svg3 as an immunostimulant for the ICB combination immunotherapy of multiple poorly immunogenic murine tumor models in syngeneic mice, including 4T1 mammary carcinoma in BALB/c mice, and B16 melanoma and MOC2 oral squamous cell carcinoma in C57BL/6 mice. Worth noting, upon Svg3 treatment, the IFN-β production levels by these tumor cells varied significantly (Figure S5). To investigate the therapeutic efficacy of Svg3 in vivo, B16F10 tumor cells were subcutaneously (s.c.) inoculated in the right flank of syngeneic C57BL/6 mice. When tumors were approximately 50 mm3, mice started to be treated with liposomal Svg3 (i.t., 5 × 3 nmol) and αPD-1 (i.p., 5 × 100 μg) every 3 days for 5 times. While liposomal Svg3 monotherapy inhibited tumor progression, especially in B16F10 tumors, the combination of liposomal Svg3 with αPD-1 dramatically enhanced the tumor therapeutic efficacy in all these tumor models (Figures 6B–6F). Consistently, liposomal Svg3 + αPD-1 significantly prolonged mouse survival (Figure 6E). Worth noting, none of these treatments caused any significant reduction of mouse body weight (Figure S6), suggesting their potential of great safety (Figure 6G). Meanwhile, in naive C57BL/6 mice, we studied the systemic innate immune responses elicited by Svg3 by measuring a panel of serum chemokines and cytokines by Luminex. As a result, a single dose of s.c. administered liposomal Svg3 significantly elevated the serum levels of proinflammatory TNF-α and T cell-recruiting chemokine CXCL-10, but not IL-6 (Figure 6H). The increased expression of anti-inflammatory cytokine or chemokine IL-13 and eotaxin may also indicate rapid activation of CD4+ T cells upon exposure to Svg3.
Figure 6.

Svg3 improved ICB therapeutic efficacy in multiple poorly immunogenic tumor models
(A) Timeline of tumor therapy studies in mice. When tumors were approximately 50 mm3, mice were treated with liposomal Svg3 (i.t., 5 × 3 nmol) and αPD-1 (i.p., 5 × 100 μg) every 3 days for 5 times. (B) Average 4T1 tumor growth curves. p values were determined at day 34. (C) Individual 4T1 tumor growth curves. (D) Average B16F10 tumor growth curves. p values were determined at day 19. (E) Meier-Kaplan survival curves of B16F10 tumor-bearing mice after the above treatments. (F) Average MOC2 tumor growth curves. p values were determined at day 23. (G) Body weights of MOC2 tumor-bearing mice after the above treatments. Data: mean ± SEM. p values were determined by one-way ANOVA or t test. ns, not significance; ∗p < 0.05, ∗∗p < 0.01. (H) Luminex analysis of serum cytokines and chemokines 24 h after mice were s.c. treated with liposomal Svg3. Control, PBS.
We further investigated the roles of cGAS and STING in Svg3-mediated antitumor therapy using cGAS KO mice (B6(C)-Cgastm1d(EUCOMM)Hmgu/J or cGas−/−) and Goldenticket STING KO mice (C57BL/6J-Sting1gt/J or Stinggt/gt), respectively. Mice with s.c. B16F10 tumors (50 mm3) were again treated as above with five doses of liposomal Svg3, with liposomal STING agonist 2′3′-cGAMP as a control, alone or combined with αPD-1. In cGas−/− mice, B16F10 tumors, Svg3 lost its ability to improve the tumor therapeutic efficacy of αPD-1, suggesting that cGAS is essential for the tumor therapeutic efficacy of Svg3 (Figure 7A). In contrast, 2′3′-cGAMP significantly enhanced the ability of αPD-1 to retard tumor progression (Figure 7A), verifying the intact STING and downstream immunostimulatory signal pathway in this mouse model. Moreover, in Stinggt/gt mice, neither Svg3 nor 2′3′-cGAMP significantly potentiated the tumor therapeutic efficacy of αPD-1 (Figure 7B). This suggests that STING is essential for the tumor therapeutic efficacy of Svg3. Collectively, these results demonstrate the cGAS-STING-specific activation by Svg3 to potentiate the tumor therapeutic efficacy of ICB for combination immunotherapy.
Figure 7.

cGAS and STING are essential for Svg3 to potentiate ICB tumor immunotherapy in mice
(A) B16F10 tumor growth curves in cGas−/− mice treated with liposomal Svg3, with liposomal 2′3′-cGAMP as a control, alone or combined with αPD-1. Svg3, but not 2′3′-cGAMP, lost the ability to potentiate the tumor therapeutic efficacy of αPD-1. p values were determined by t test on day 19 (2′3′-cGAMP + αPD-1 vs. PBS) and day 22 (αPD-1 or Svg3 + αPD-1 vs. 2′3′-cGAMP + αPD-1). (B) B16F10 tumor grow curves in Stinggt/gt mice treated as above. Neither Svg3 nor 2′3′-cGAMP significantly potentiated the tumor therapeutic efficacy of αPD-1. Arrows mark treatments. Liposomal Svg3 and 2′3′-cGAMP, i.t., 5 × 3 nmol; αPD-1, i.p., 5 × 100 μg. Data: mean ± SEM. p values were determined by t test on day 19. ns, not significant; p > 0.05, ∗p < 0.05, ∗∗∗p < 0.001.
Discussion
Cancer immunotherapy, which leverages the host immune system to treat cancer, has significantly improved the treatment outcomes for many types of cancer. ICB is one of the current mainstream approaches to cancer immunotherapy. However, most cancer patients do not benefit from current ICB, due to a lack of pre-existing antitumor immune cells and immune checkpoints, as well as TME immunosuppression, among others. Moreover, some patients suffer from immune-related adverse events caused by the imbalanced immune homeostasis. This demands innovative approaches, such as cancer therapeutic vaccines, for combination immunotherapy with ICB to maximize the therapeutic potential of ICB. Cancer therapeutic vaccines can reduce systemic and tumor immunosuppression, elicit and augment antitumor innate and adaptive immunity, upregulate the expression of immune checkpoints thereby sensitizing them for ICB, and promote the tumor infiltration of antitumor cells and molecules. Immunostimulants, including PRR agonists, are extensively used in therapeutic vaccines as either non-tumor-specific innate immunostimulants or adjuvants for tumor-specific antigens. The cGAS-STING immunostimulation pathway plays a critical role in innate immunity that mediates anti-cancer therapies, including ICB. Upon dsDNA binding, cGAS is activated to synthesize cGAMP, which then stimulates STING to trigger antitumor IFN-I responses. Current approaches to the development of cancer therapeutic cGAS-STING-activating immunostimulants have been almost exclusively targeting STING. However, current STING agonists have shown limited tumor therapeutic efficacy in the clinic, largely due to their hydrophilicity, often negative electrostatic charges, and susceptibility to enzymatic degradation, which results in poor bioavailability. For example, i.t. administration of a CDN, MK-1454, showed minimal therapeutic efficacy in patients with solid tumors or lymphomas.27 In addition, a phase I clinical trial data of i.t.-administered STING agonist ADU-S100 in combination with αPD-1 (spartalizumab) showed modest clinical benefit.17
Compared with current small-molecule-based STING agonists, cGAS agonistic oligonucleotide therapeutics may have the following advantages: (1) reproducible and economic production using existing automated cGMP manufacturing facilities, (2) advanced drug delivery systems that dramatically improve the pharmacokinetics and therapeutic efficacy of oligonucleotides, (3) well-established oligonucleotide chemistry that may further improve the biostability, pharmacokinetics, safety, and therapeutic efficacy of oligonucleotide therapeutics, and (4) cGAS agonists can bypass the complications associated with the allele selectivity of STING in humans. Here, by structural optimization, we develop a cGAS-agonistic oligonucleotide, Svg3, for versatile applications in ICB combination cancer immunotherapy. Svg3 specifically activated cGAS, eliciting potent IFN-I responses not only in murine cells but also in human cells and tissues, which are pivotal for its future clinical translation. Remarkably, Svg3 outperformed several state-of-the-art STING agonists to elicit IFN-I responses in murine and human immune cells. Moreover, Svg3 did not significantly elevate AIM2-associated Il18 or Il1b expression or elicit detectable IL-18 or IL-1β production in RAW 264.7 macrophages. This rules out the possibility for Svg3 to activate AIM2, another cytosolic dsDNA sensor that can induce confounding inflammasome activation in these cells.
Svg3 was easily formulated in well-established liposomes, which improved the intracellular delivery, endosome escape, and tissue retention of Svg3 in tumors (for i.t. Svg3 administration). I.t. Svg3 nanoparticle administration reduced multifaceted immunosuppression and enhanced antitumor immunity in tumors. As a result, Svg3 significantly improved the therapeutic efficacy of ICB in multiple syngeneic tumor models in wild-type mice but not in cGas−/− or Stinggt/gt mice.
Further comprehensive optimization of Svg3 may further improve its cGAS activation efficacy. Future structural and biochemical studies of cGAS binding and activation by Svg3 may reveal the underlying mechanism of action and provide insight for its preclinical and clinical development. Moreover, future studies will explore the systemic delivery of Svg3 for the treatment of surgically inaccessible tumors and metastatic tumors, in which i.t. administration may find limited applicability. Systemic delivery of immunostimulants would likely elevate the systemic cytokine levels, which may lead to immune toxicity in a scenario such as a cytokine storm. Therefore, it will be important to optimize Svg3 formulation to have a balanced efficacy and safety. To this end, recent research has demonstrated the feasibility of systemic delivery of vaccines such as STING agonists and TLR7/8 agonist-adjuvanted nanovaccines.28,29 Overall, oligonucleotide-based cGAS agonists represent a promising approach as immunostimulant therapeutics and vaccine adjuvants for cancer combination immunotherapy.
Materials and methods
Cell culture
DC2.4 cells, 4T1 cells, and TC-1 were cultured in RPMI 1640 medium. RAW-ISG cells were obtained from InvivoGen and cultured in DMEM medium containing 100 μg/mL Normocin and 200 μg/mL Zeocin. MOC2 cells were cultured in DMEM/F12 medium. RAW 264.7 cells and B16F10 cells were cultured in DMEM medium. B3Z cells and THP-1 cells were cultured with RPMI 1640 medium supplemented with 2 mM L-glutamine, 1 mM sodium pyruvate, and 50 μM 2-mercaptoethanol. All media were supplemented with 10% FBS and 0.1% penicillin and streptomycin. All cells were cultured in a humidified atmosphere (5% CO2, 37°C) in a Biosafety Level II incubator.
IFN-I expression
IFN-I cytokine levels were measured by ELISA in RAW 264.7 cells and by using an IFN reporter cell in THP-1 cells. RAW 264.7 cells were seeded in 96-well plate at a density of 5 × 104 cells/well and incubated overnight for experiment. Svg3 was transfected into cells using Lipofectamine reagent at a final concentration of 25 nM and incubated at 37°C and 5% CO2 for 24 h. The medium was collected to test the concentration of IFN-β. THP-1 cells were seeded at densities of 1 × 104 cells/well in a 96-well plate. Cells were treated with Svg3 or cGAMP, respectively, for 24 h. At the same time, HEK-Blue IFN-α/β cells (InvivoGen) were seeded at densities of 1 × 104 cells/well in a 96-well plate. Then THP-1 cell supernatant was added to HEK-Blue IFN-α/β cells for 24 h. Cell supernatant (50 μL)was collected from each sample and added to 150 μL of QUANTI-Blue SEAP detection medium (InvivoGen) and incubated for 2 h at 37°C. SEAP activity was assessed by measuring the absorbance at 630 nm on a plate reader.
ISG expression
RAW-Lucia ISG cells, which were generated from a RAW 264.7 cell line by stable integration of an IRF-inducible Lucia luciferase reporter construct, were used to evaluate cGAS activation. To validate the role of cGAS in Svg3 stimulation, cGAS KO RAW-Lucia ISG cells were used at the same Svg3 treatment condition. For comparison of Svg3 and STING agonists, Svg3 and STING agonists including 2′3′-cGAMP, ADU-S100, or diABZI were transfected at different concentrations, respectively, using Lipofectamine reagent. The expression of IRF-induced Lucia luciferase in the culture medium was measured using QUANTI-Luc following the manufacturer’s instructions.
2′3′-cGAMP production by Svg3-activated cGAS
RAW 264.7 macrophages were seeded in 12-well plates and incubated overnight. Svg3 or ISD was transfected using Lipofectamine 2000 at a final concentration of 100 nM. The cells were collected at different time points after transfection, and lysed using RIPA lysis buffer containing protease inhibitors and EDTA on ice for 15 min. The concentrations of 2′3′-cGAMP were measured using 2′3′-cyclic cGAMP ELISA kit (Invitrogen) following the manufacturer’s instructions.
Svg3 binding affinity with cGAS
Cy5-labeled Svg3 (Cy5-Svg3) was used to measure the binding affinity with cGAS by MST, as described previously.30 In brief, the concentration of Cy5-Svg3 was determined to ensure the fluorescence intensity was between 800 and 1,000. cGAS was diluted using Tris buffer containing 0.05% Tween 20. Cy5-Svg3 of a series of different final concentrations was added into cGAS solutions and incubated for 15 min at room temperature. Then 20-μL samples were loaded into standard treated capillaries for measurements on a Monolith NT Automated (Nanotemper).
Co-stimulatory factor expression
RAW 264.7 macrophages and DC2.4 DCs were respectively seeded in 12-well plates and incubated overnight for experiment. Svg3 was transfected using Lipofectamine reagent at a final concentration of 25 nM. After treatment for 24 h, the cells were collected and stained with anti-mouse CD86-PerCp, anti-mouse CD80-Alexa Fluor 647, anti-mouse CD40-FITC, or anti-mouse I-A/I-E (MHC II)-PE, respectively, for flow cytometry (BD Canto).
B3Z T cell activation
B3Z is an SIINFEKL-specific CD8+ T cell hybridoma that can be activated by recognizing the SIINFEKL/H-2Kb complex.31 Activated B3Z cells produce β-galactosidase (β-gal), which can hydrolyze the substrate of chorophenol red-β-D-galactopyranoside (CPRG) into a red product. To evaluate B3Z activation, DC2.4 cells were seeded in 12-well plates and treated with lipo-transfected Svg3 with ovalbumin or free ovalbumin for 24 h. After incubation, the medium was removed and B3Z cells were cocultured for another 24 h. Then cells were lysed for 4 h at 37°C with lysis buffer (PBS with 100 mM 2-mercaptoethanol, 9 mM MgCl2, 0.2% Triton X-100, and 0.15 mM CPRG). The reaction was stopped by 1 M sodium carbonate. The magnitude of antigen priming was evaluated through absorbance measurements (λ = 570 nm).
RNA-seq transcriptomic analysis of Svg3-treated mouse macrophage
RAW 264.7 macrophages were seeded in 6-well plates and treated with lipo-transfected Svg3 at a final concentration of 25 nM. The control group was Lipofectamine without Svg3. After 6 h incubation, the culture medium was removed, and the cells were collected for RNA extraction and sequencing. An average of 30 million paired end reads of length 150 bp were generated for each sample. The quality of RNA-seq reads was assessed with FastQC v.0.11.9. The reads were aligned using STAR aligner32 v.2.7.6a to reference genome GRCm38. Raw gene counts of mapped reads were aggregated using featureCounts.33 The differential gene expression analysis was performed with Bioconductor package DESeq2 v.1.30.034 using the normalized and filtered counts per gene from the RNA-seq data.
qPCR
RAW 264.7 macrophages were seeded in 12-well plates and transfected with DNA at a final concentration of 25 nM. After incubation for 24 h, the cells were collected, and the RNA were extracted using RNA extraction kit (Invitrogen) following the manufacturer’s protocol. RNA was reverse-transcribed to cDNA using a High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher). qPCR was carried out using SYBR Green Master Mix (Applied Biosystems) on a QuantStudio 3 system (Applied Biosystems).
IFN responses in human tumor tissue
Human head and neck cancer tissues were provided by VCU Massey Cancer Center Tissue and Data Acquisition and Analysis Core. Fresh tumor tissues were evenly cut into cubes and randomly assigned into groups. The resulting tissues were cultured in 1 mL complete DMEM medium in six-well plates. Svg3 (0.1 nmol) loaded in liposomes was injected into the above tissues using syringe needles. After 24 h incubation in a tissue culture incubator, the tumor tissue RNA was extracted using RNA extraction kit (Invitrogen). RNA was reverse-transcribed to cDNA using a High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher). qPCR was carried out using SYBR Green Master Mix (Applied Biosystems) on a QuantStudio 3 system (Applied Biosystems).
Nanoparticle preparation
Liposomes: Svg3 was formulated into liposomes using the thin film hydration method as reported previously.35 In brief, DOTAP, cholesterol, DOPE, and DSPE-PEG2000 were dissolved in chloroform and mixed in a round-bottom bottle. The thin lipid film was formed by the removal of chloroform using a rotary evaporator. Svg3 aqueous solution then was added and vortexed vigorously for 30 min. The N/P ratio for lipids and DNA was 10. The crude liposomes were extruded using extruder (Avanti). Liposomes were diluted in 1× DPBS and measured using a Zetasizer (Malvern Panalytical) for size and zeta potential measurement.
Cell uptake of liposomal Svg3
Fluorescent-labeled Svg3 was used to evaluate the cellular uptake after being formulated into liposome using the above method. RAW 264.7 cells were seeded in six-well plates and cultured overnight for further experiment. Cy3-labeled Svg3 liposomes were diluted using serum-free medium and incubated at a final concentration of 100 nM per well. The medium was discarded, and cells were washed with 1× DPBS and then collected for flow cytometry (BD FACSCanto).
Animal studies
C57BL/6 mice and BALB/c mice (6–8 weeks old) were purchased from Charles River Laboratory. B6(C)-Cgastm1d(EUCOMM)Hmgu/J and C57BL/6J-Sting1gt/J mice were ordered from Jackson Laboratory. All animals were maintained at the animal facilities of Virginia Commonwealth University under specific pathogen-free conditions and treated in accordance with the regulations and guidelines of the Institutional Animal Care and Use Committee. All animal experiments were approved by the Virginia Commonwealth University (VCU) Institutional Animal Care and Use Committee.
IVIS imaging of Svg3 nanoparticle retention in tumor
For 4T1 tumor accumulation imaging, IR800-labeled Svg3 liposomes were i.t. injected to 4T1 tumor-bearing mice. The tumor retention effect of the IR800-Svg3 liposomes in tumors was visualized with the IVIS optical system for 7 days. The excitation wavelength was 780 nm and the emission wavelength was 794 nm.
Systemic cytokine and chemokine expression by Luminex
C57BL/6 mice were s.c. injected with Svg3 liposomes (3 nmol Svg3) or blank liposomes at the tail base. After 24 h, blood was collected and centrifuged for 5 min at 2,000 rpm to collect serum. Serum cytokines and chemokines were measured by Luminex at the University of Virginia Flow Cytometry Core.
Tumor therapy
Tumor therapy using i.t.-administered liposomal Svg3 was studied in three murine syngeneic tumor models: 4T1 mammary carcinoma in BALB/c mice, B16F10 melanoma, and MOC2 oral cancer in C57BL/6 mice, as well as B16F10 melanoma in cGas−/− mice (B6(C)-Cgastm1d(EUCOMM)Hmgu/J) and Goldenticket Stinggt/gt mice (C57BL/6J-Sting1gt/J) mice. Tumors were, respectively, established by s.c. injections of tumor cells (5 × 105) into the mouse right flank. Tumor volumes and body weights were monitored. Treatment was initiated when the average tumor volumes reached ∼50 mm.3 Liposomal Svg3 was i.t. injected at a dose of 3 nmol/mouse every 3 days for 5 times. For ICB, αPD-1 was intraperitoneally injected every 3 days for 5 times. Mouse humane endpoint was defined when the body weight dropped by 20% or tumor size reached 2,000 mm3.
TME immune analysis
4T1 tumors were established s.c. in BALB/c mice, as described previously, and treated i.t. with PBS, αPD-1, liposomal Svg3, and liposomal Svg3 + αPD-1, respectively, at days 0, 3, and 6. On day 9, tumor tissues were harvested and digested with collagenase A and DNase at 37°C for 40 min. Digestion mixtures were quenched by adding 10% FBS and samples were filtered through 40-μm cell strainers (Falcon). Cells were stained for 20–30 min in the dark on ice with the conjugated antibodies as follows (BioLegend unless denoted otherwise) following the manufacturer’s recommended concentrations. Myeloid panel: CD45-BV421, CD11b-PE-Cy5, CD11c-Alexa Fluor 594, CD86-PE, Ly-6G/Ly-6C-Alexa Fluor 488, F4/80-FITC, CD206-BV605. Lymphoid cell panel: CD45-BV421, CD3-Alexa Fluor 488, CD4-PerCp-Cy5, CD8α-APC-Cy7, NK-1.1-APC, FoxP3-Alexa Fluor 647. Cells were then fixed with BD Cytofix (BD Biosciences), followed by flow cytometry on a BD LSR Fortessa flow cytometer (BD Biosciences) and data analysis using FlowJo software (TreeStar) using gating trees shown in Figures S7 and S8.
Statistical analysis
Data are shown as mean ± standard deviation (SD) and mean ± standard error of the mean (SEM). Statistical significance was determined by t test or ANOVA in the experiment (Prism 9). ns, not significant; p > 0.05, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Data and code availability
All the data related with this study are available within the paper or can be obtained from the authors on request.
Acknowledgments
G.Z. acknowledges funding support from a American Cancer Society Research Scholar Grant (RSG-22-055-01-IBCD), the NIH (R01CA266981, R01AI168684, R35GM143014, and R21NS114455), DoD CDMRP Breast Cancer Breakthrough Award Level II (BC210931/P1), a METAvivor Early Career Investigator Award, the VCU Commercialization Fund, a University of Michigan Rogel Cancer Center Discovery Award, and the University of Michigan startup fund. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Microscopy was performed at the VCU Microscopy Facility, supported in part by funding from NINDS Center Core Grant 5 P30 NS047463 and, in part, by funding from the NCI Cancer Center Support Grant P30 CA016059. Bioinformatics analyses were provided by VCU Massey Cancer Center Bioinformatics Shared Resource; flow cytometry was performed at the VCU Massey Cancer Center Flow Cytometry Shared Resource; and human tumor tissues were provided by the VCU Massey Cancer Center Tissue and Data Acquisition and Analysis Core; these facilities are all supported, in part, with funding from NIH-NCI Cancer Center Support Grant P30 CA016059. We thank NIH Tetramer Core for providing tetramer reagents, and thank Prof. Xiang-Yang Wang at VCU for providing MOC2 cells.
Author contributions
Conceptualization, S.Z. and G.Z.; in vitro study, S.Z., T.S., F.C., and S.A.; animal work, S.Z., T.S., F.C., J.C., and X.L.; data analysis, S.Z., B.Z., J.Z., and G.Z.; writing, S.Z. and G.Z.
Declaration of interests
G.Z. and S.Z. were listed as inventors in a related patent application.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2024.102126.
Supplemental information
References
- 1.Sharma P., Allison J.P. Immune checkpoint therapy: Forging ahead. Sci. Transl. Med. 2022;14:eadf2947. doi: 10.1126/scitranslmed.adf2947. [DOI] [PubMed] [Google Scholar]
- 2.Kwon J., Bakhoum S.F. Cancer Discov; 2019. The Cytosolic DNA-Sensing cGAS–STING Pathway in Cancer; pp. 2159–8290. CD-19-0761v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schadt L., Sparano C., Schweiger N.A., Silina K., Cecconi V., Lucchiari G., Yagita H., Guggisberg E., Saba S., Nascakova Z., et al. Cancer-Cell-Intrinsic cGAS Expression Mediates Tumor Immunogenicity. Cell Rep. 2019;29:1236–1248.e7. doi: 10.1016/j.celrep.2019.09.065. [DOI] [PubMed] [Google Scholar]
- 4.Gao D., Li T., Li X.-D., Chen X., Li Q.-Z., Wight-Carter M., Chen Z.J. Activation of cyclic GMP-AMP synthase by self-DNA causes autoimmune diseases. Proc. Natl. Acad. Sci. USA. 2015;112:E5699–E5705. doi: 10.1073/pnas.1516465112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gray E.E., Treuting P.M., Woodward J.J., Stetson D.B. Cutting Edge: cGAS Is Required for Lethal Autoimmune Disease in the Trex1-Deficient Mouse Model of Aicardi–Goutières Syndrome. J. Immunol. 2015;195:1939–1943. doi: 10.4049/jimmunol.1500969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Glück S., Guey B., Gulen M.F., Wolter K., Kang T.-W., Schmacke N.A., Bridgeman A., Rehwinkel J., Zender L., Ablasser A. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat. Cell Biol. 2017;19:1061–1070. doi: 10.1038/ncb3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang H., Wang H., Ren J., Chen Q., Chen Z.J. cGAS is essential for cellular senescence. Proc. Natl. Acad. Sci. USA. 2017;114:E4612–E4620. doi: 10.1073/pnas.1705499114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cai X., Chiu Y.-H., Chen Z.J. The cGAS-cGAMP-STING Pathway of Cytosolic DNA Sensing and Signaling. Mol. Cell. 2014;54:289–296. doi: 10.1016/j.molcel.2014.03.040. [DOI] [PubMed] [Google Scholar]
- 9.Chen J., Cao Y., Markelc B., Kaeppler J., Vermeer J.A., Muschel R.J. Type I IFN protects cancer cells from CD8+ T cell–mediated cytotoxicity after radiation. J. Clin. Invest. 2019;129:4224–4238. doi: 10.1172/JCI127458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duong E., Fessenden T.B., Lutz E., Dinter T., Yim L., Blatt S., Bhutkar A., Wittrup K.D., Spranger S. Type I interferon activates MHC class I-dressed CD11b+ conventional dendritic cells to promote protective anti-tumor CD8+ T cell immunity. Immunity. 2022;55:308–323.e9. doi: 10.1016/j.immuni.2021.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang X., Bai X.C., Chen Z.J. Structures and Mechanisms in the cGAS-STING Innate Immunity Pathway. Immunity. 2020;53:43–53. doi: 10.1016/j.immuni.2020.05.013. [DOI] [PubMed] [Google Scholar]
- 12.Wang H., Hu S., Chen X., Shi H., Chen C., Sun L., Chen Z.J. cGAS is essential for the antitumor effect of immune checkpoint blockade. Proc. Natl. Acad. Sci. USA. 2017;114:1637–1642. doi: 10.1073/pnas.1621363114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fu J., Kanne D.B., Leong M., Glickman L.H., McWhirter S.M., Lemmens E., Mechette K., Leong J.J., Lauer P., Liu W., et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci. Transl. Med. 2015;7:283ra52. doi: 10.1126/scitranslmed.aaa4306. 283ra52-283ra52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Le Naour J., Zitvogel L., Galluzzi L., Vacchelli E., Kroemer G. Trial watch: STING agonists in cancer therapy. OncoImmunology. 2020;9:1777624. doi: 10.1080/2162402X.2020.1777624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chang D., Whiteley A.T., Bugda Gwilt K., Lencer W.I., Mekalanos J.J., Thiagarajah J.R. Extracellular cyclic dinucleotides induce polarized responses in barrier epithelial cells by adenosine signaling. Proc. Natl. Acad. Sci. USA. 2020;117:27502–27508. doi: 10.1073/pnas.2015919117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Motedayen Aval L., Pease J.E., Sharma R., Pinato D.J. Challenges and Opportunities in the Clinical Development of STING Agonists for Cancer Immunotherapy. JCM. 2020;9:3323. doi: 10.3390/jcm9103323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meric-Bernstam F., Sandhu S.K., Hamid O., Spreafico A., Kasper S., Dummer R., Shimizu T., Steeghs N., Lewis N., Talluto C.C., et al. Phase Ib study of MIW815 (ADU-S100) in combination with spartalizumab (PDR001) in patients (pts) with advanced/metastatic solid tumors or lymphomas. J. Clin. Orthod. 2019;37:2507. [Google Scholar]
- 18.Conlon J., Burdette D.L., Sharma S., Bhat N., Thompson M., Jiang Z., Rathinam V.A.K., Monks B., Jin T., Xiao T.S., et al. Mouse, but not Human STING, Binds and Signals in Response to the Vascular Disrupting Agent 5,6-Dimethylxanthenone-4-Acetic Acid. J. Immunol. 2013;190:5216–5225. doi: 10.4049/jimmunol.1300097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ramanjulu J.M., Pesiridis G.S., Yang J., Concha N., Singhaus R., Zhang S.-Y., Tran J.-L., Moore P., Lehmann S., Eberl H.C., et al. Design of amidobenzimidazole STING receptor agonists with systemic activity. Nature. 2018;564:439–443. doi: 10.1038/s41586-018-0705-y. [DOI] [PubMed] [Google Scholar]
- 20.Andreeva L., Hiller B., Kostrewa D., Lässig C., de Oliveira Mann C.C., Jan Drexler D., Maiser A., Gaidt M., Leonhardt H., Hornung V., Hopfner K.P. cGAS senses long and HMGB/TFAM-bound U-turn DNA by forming protein–DNA ladders. Nature. 2017;549:394–398. doi: 10.1038/nature23890. [DOI] [PubMed] [Google Scholar]
- 21.Du M., Chen Z.J. DNA-induced liquid phase condensation of cGAS activates innate immune signaling. Science. 2018;361:704–709. doi: 10.1126/science.aat1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Herzner A.-M., Hagmann C.A., Goldeck M., Wolter S., Kübler K., Wittmann S., Gramberg T., Andreeva L., Hopfner K.-P., Mertens C., et al. Sequence-specific activation of the DNA sensor cGAS by Y-form DNA structures as found in primary HIV-1 cDNA. Nat. Immunol. 2015;16:1025–1033. doi: 10.1038/ni.3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Keating S.E., Baran M., Bowie A.G. Cytosolic DNA sensors regulating type I interferon induction. Trends Immunol. 2011;32:574–581. doi: 10.1016/j.it.2011.08.004. [DOI] [PubMed] [Google Scholar]
- 24.Saeed A.F.U.H., Ruan X., Guan H., Su J., Ouyang S. Regulation of cGAS-Mediated Immune Responses and Immunotherapy. Adv. Sci. 2020;7:1902599. doi: 10.1002/advs.201902599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chew Z.H., Cui J., Sachaphibulkij K., Tan I., Kar S., Koh K.K., Singh K., Lim H.M., Lee S.C., Kumar A.P., et al. Macrophage IL-1β contributes to tumorigenesis through paracrine AIM2 inflammasome activation in the tumor microenvironment. Front. Immunol. 2023;14:1211730. doi: 10.3389/fimmu.2023.1211730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feng X., Zhang Z., Sun P., Song G., Wang L., Sun Z., Yuan N., Wang Q., Lun L. Interleukin-18 Is a Prognostic Marker and Plays a Tumor Suppressive Role in Colon Cancer. Dis. Markers. 2020;2020:1–9. doi: 10.1155/2020/6439614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gogoi H., Mansouri S., Jin L. The Age of Cyclic Dinucleotide Vaccine Adjuvants. Vaccines. 2020;8:453. doi: 10.3390/vaccines8030453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shae D., Becker K.W., Christov P., Yun D.S., Lytton-Jean A.K.R., Sevimli S., Ascano M., Kelley M., Johnson D.B., Balko J.M., Wilson J.T. Endosomolytic polymersomes increase the activity of cyclic dinucleotide STING agonists to enhance cancer immunotherapy. Nat. Nanotechnol. 2019;14:269–278. doi: 10.1038/s41565-018-0342-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baharom F., Ramirez-Valdez R.A., Tobin K.K.S., Yamane H., Dutertre C.-A., Khalilnezhad A., Reynoso G.V., Coble V.L., Lynn G.M., Mulè M.P., et al. Intravenous nanoparticle vaccination generates stem-like TCF1+ neoantigen-specific CD8+ T cells. Nat. Immunol. 2021;22:41–52. doi: 10.1038/s41590-020-00810-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tao J., Zhang X.-W., Jin J., Du X.-X., Lian T., Yang J., Zhou X., Jiang Z., Su X.-D. Nonspecific DNA Binding of cGAS N Terminus Promotes cGAS Activation. J. Immunol. 2017;198:3627–3636. doi: 10.4049/jimmunol.1601909. [DOI] [PubMed] [Google Scholar]
- 31.Wang P., Zhao P., Dong S., Xu T., He X., Chen M. An Albumin-binding Polypeptide Both Targets Cytotoxic T Lymphocyte Vaccines to Lymph Nodes and Boosts Vaccine Presentation by Dendritic Cells. Theranostics. 2018;8:223–236. doi: 10.7150/thno.21691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liao Y., Smyth G.K., Shi W. FeatureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 34.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550–621. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang H. In: Liposomes Methods in Molecular Biology. D’Souza G.G.M., editor. Springer New York; 2017. Thin-Film Hydration Followed by Extrusion Method for Liposome Preparation; pp. 17–22. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the data related with this study are available within the paper or can be obtained from the authors on request.