

ABSTRACT
Efficacy of cancer immunotherapies relies on correct recognition of tumor antigens by lymphocytes, eliciting thus functional responses capable of eliminating tumor cells. Therefore, important efforts have been carried out in antigen identification, with the aim of understanding mechanisms of response to immunotherapy and to design safer and more efficient strategies. In addition to classical tumor-associated antigens identified during the last decades, implementation of next-generation sequencing methodologies is enabling the identification of neoantigens (neoAgs) arising from mutations, leading to the development of new neoAg-directed therapies. Moreover, there are numerous non-classical tumor antigens originated from other sources and identified by new methodologies. Here, we review the relevance of neoAgs in different immunotherapies and the results obtained by applying neoAg-based strategies. In addition, the different types of non-classical tumor antigens and the best approaches for their identification are described. This will help to increase the spectrum of targetable molecules useful in cancer immunotherapies.
KEYWORDS: Tumor neoantigens, mutations, non-classical tumor antigens, ribosome profiling, immunopeptidomics
Introduction
A main feature of the immune system is its ability to discriminate between self and non-self to protect the organism from exogenous microbial pathogens. This exquisite capacity relies on the presence of lymphocyte receptors that specifically recognize a wide array of antigen molecules. In the case of tumor cells, although it took many years, it was finally demonstrated that, despite being own cells, they express antigenic molecules than can be recognized by lymphocytes and that, in some cases, led to control of tumor growth.1 Malignant transformation of tumor cells arises from a series of mutations, which ultimately lead to the generation of proteins with new sequences, changes in the expression of non-mutated proteins and even in post-translational modifications of some of these proteins. These changes in the repertoire of molecules expressed by tumor cells increase the antigenicity of tumor cells and allow the activation of lymphocytes capable of recognizing and destroying tumor cells.2 B lymphocytes, by means of their surface immunoglobulins, recognize tumor antigens expressed on the cell membrane in their native conformation. On the other side, by using their antigen-specific T cell receptor (TCR), T cells recognize processed antigens presented as short peptides by MHC class I and class II molecules, leading to the activation of CD8 and CD4 T lymphocytes, respectively. Although B cell-mediated immunity is important in the antitumor response, and antibodies and antibody-derivatives (e.g. CAR-T therapies) have been successfully used in tumor immunotherapy, and due to the relevance of cellular immunity, this review will focus on antigens presented by MHC molecules for T lymphocyte recognition. Lymphocyte recognition of tumor antigens determines the interactions between tumor cells and the immune system, modulates tumor growth, sculpting the tumor antigen landscape and shaping the effect of immune-based therapies.3 Therefore, identification of these antigen molecules expressed by tumor cells has been of paramount importance, not only to study and understand tumor/immune interactions, but to unravel mechanisms of response to therapies as well as to design new immunotherapies. For these reasons, during the last decades, with the implementation of new methodologies, a plethora of tumor antigens has been identified, and different studies have been carried out to characterize their expression pattern, considering differences between healthy vs tumor tissue, frequency of expression among individuals with a type of tumor, as well as their expression by different tumor types.4,5 These features may determine their immunogenicity, as expression in healthy cells promotes immune tolerance mechanisms and potential applicability. From initially identified antigens using cDNA libraries to the newly discovered antigens by means of next-generation sequencing (NGS) technologies and immunopeptidomics, a number of antigens with different expression patterns and immunogenicity levels have been described. They include overexpressed, differentiation and oncofetal antigens, among the different types of classical tumor-associated antigens,4 as well as those more tumor-specific antigens, such as antigens derived from viral sequences.6 Belonging to this last group of tumor-specific antigens, neoantigens (neoAgs) generated from mutations have emerged during the last years as a category of tumor antigens with important implications in cancer immunotherapy. These mutations or alterations, which result in new amino acid sequences or in post-translational modifications of original sequences, generate new epitopes recognizable by lymphocytes. In this review, we will first focus on mutated neoAgs and second, in a new category of non-classical antigens. As opposed to classical antigens, this last group usually includes proteins that have not been annotated in databases and can be added to the already available list of tumor antigens. Precise knowledge of these antigens, their expression, immunogenicity and how these features impact tumor growth and response to therapies, may help to develop better immunotherapies.
Neoantigens originated from mutations
Types of mutations responsible for neoantigen generation
NeoAgs are generated through different mechanisms, such as non-synonymous single-nucleotide variants (ns-SNVs), insertions and deletions (INDELs) and gene fusions (Figure 1a).7 Mutated peptides resulting from the processing of these new sequences must bind to MHC molecules and be presented on the cell surface, so that they can be finally recognized by those T cell receptors (TCR) that have escaped central tolerance mechanisms.7,8 Lack of tolerance against these sequences is a common event, since they are not expressed by healthy cells, with the exception of neoAgs generated from mutations originating familial cancers.
Figure 1.
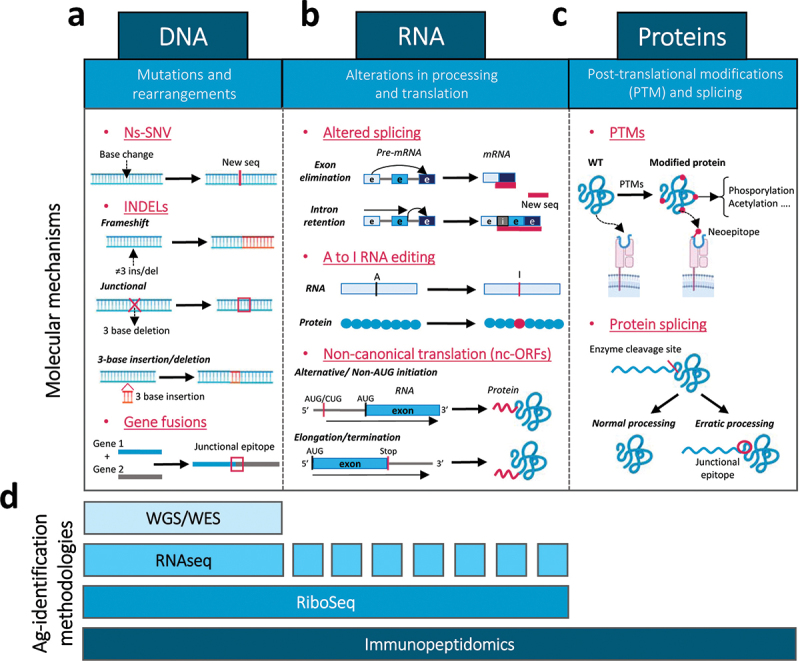
Molecular mechanisms responsible for the generation of tumor antigens at different cellular processes: methodologies to identify these antigens. New tumor antigens and epitopes are generated by different molecular mechanisms: (a) mutations and re-arrangements at the DNA level, including ns-SNVs, INDELs and gene fusions; (b) alterations in RNA processing and translation, such as altered splicing, A to I RNA editing and processes of non-canonical translation due to alternative initiation, elongation or termination; and (c) post-translational protein modification and splicing. (d) Several antigen identification methodologies can detect these new antigens or neoepitopes, including WGS/WES for DNA alterations, RiboSeq and RNASeq for DNA and RNA alterations (although not all RNA-associated alterations can be detected by RNASeq) and immunopeptidomics, which can detect any protein change originated from DNA, RNA or protein modification events.
Ns-SNVs-derived NeoAgs
Among the different neoAgs, the most commonly studied are those originated from ns-SNVs, since in the majority of cases, and in different tumor types, the majority of neoAgs-specific T cells found recognized ns-SNVs.9 However, the identification of higher rates of ns-SNV-specific T cells may be biased, since methods for identification of ns-SNVs are usually more developed and reliable than those used for identification of INDELs or gene fusion antigens.7,10,11 Also, ns-SNVs are not submitted to non-sense mediated decay (NMD) mechanisms that eliminate transcripts with early stop codons usually generated after frameshifts.12 Generation of neoAgs, including those derived from ns-SNVs, may occur by two different mechanisms: 1) by enhancing binding of the mutated peptide to MHC molecules; 2) by generating a different amino acid at TCR contact sites (Figure 2). By making visible a peptide that was not presented on its original sequence or by generating a new sequence to be recognized by a new set of TCR, these mutations play a significant role in tumor antigenicity, as they can induce highly tumor-specific T cell responses, with a lower risk of cross-reactivity with normal cells. Nevertheless, the number of immunogenic antigens derived from ns-SNVs is low compared to the total identified ns-SNVs, which indicates that only a small proportion of mutated sequences are finally true neoAgs. Systematic studies analyzing immunogenicity of mutation-harboring peptides by using T cell stimulation assays or MHC-multimer technology have demonstrated that in different tumors (e.g. ovarian, gastrointestinal and melanoma cancers),13–15 only a small proportion of peptides are immunogenic and generate T cell responses. In some cases, this may be related to amino acid changes that do not fulfil one of the two criteria for effective recognition by T cells indicated above. However, in other cases, the immunosuppressive tumor microenvironment may preclude priming of neoAg-specific immunity despite the presence of peptide structural features compatible with immunogenicity.
Figure 2.
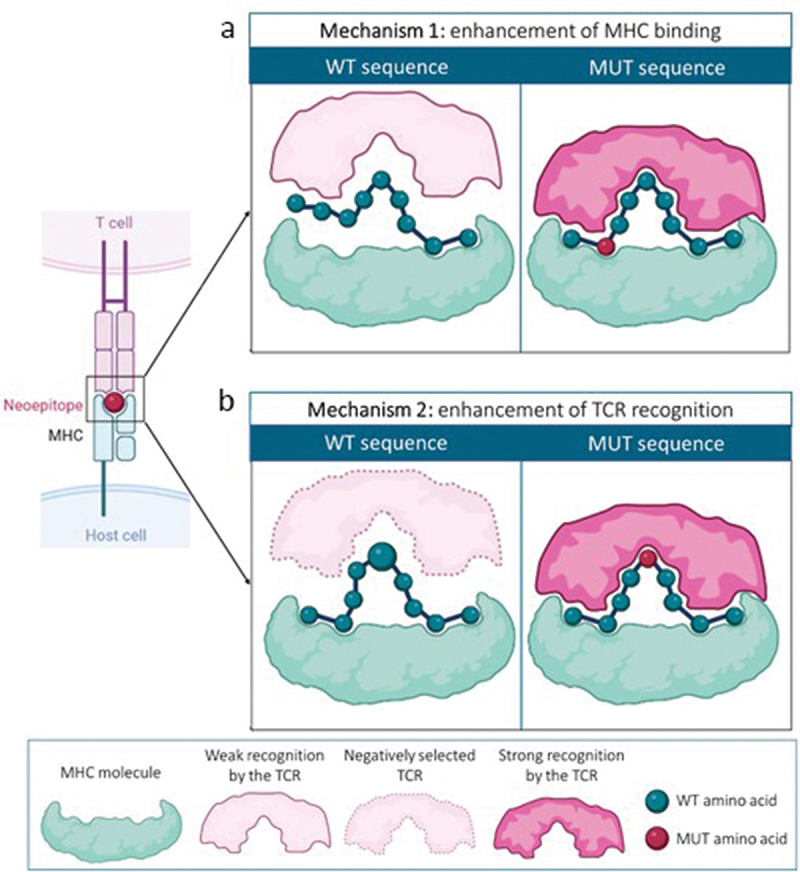
Neoantigen generation mechanisms. (a) neoantigens can be generated by a change of an amino acid located at an MHC-anchoring position, enhancing binding to MHC molecules and favoring a strong recognition by a TCR. (b) in other cases, the mutation occurs at an amino acid located at a TCR recognition site. The mutated amino acid has to differ sufficiently from the WT version to allow recognition of this neoepitope by a TCR that has escaped central tolerance mechanisms.
Most ns-SNVs-derived neoAgs are patient-specific, although some shared neoAgs have been described among patients (e.g. KRASG12D in patients with pancreatic and colorectal cancer).16,17 Shared neoAgs are considered an interesting target for “off-the-shelf” vaccination and other immunotherapeutic approaches, such as those based on transgenic TCRs. The challenge faced in this case is that shared neoAgs are less prevalent with respect to the total neoAg load identified in each patient and importantly, neoAgs are HLA-restricted, indicating that immunogenicity of a mutated peptide may depend on the HLA molecules expressed by the patient.18–20 Interestingly, a recent study using a high-throughput discovery platform has identified a considerable number of neoAgs shared among patients and tumor types.21 On the other side, the fact that the majority of neoAgs are private supposes a change of the cancer treatment paradigm into the development of personalized therapies and vaccines.18 For this matter, standardized neoAg identification protocols and vaccine production have to be established for large-scale applications.
INDEL- and gene fusion-derived NeoAgs
NeoAgs derived from INDELs and gene fusions may differ more from the original sequence than those generated by a single amino acid change, as is the case of ns-SNVs. These changes, especially those leading to frameshifted sequences, can generate new ORFs encoding sequences with a higher number of amino acid changes, increasing the possibility of generating neoAgs and resulting in a higher immunogenicity. A pan-cancer study analyzing the load of ns-SNVs and INDELs and the possibility of neoAg generation from these mutations revealed different levels of INDELs across cancer types. Interestingly, despite a higher number of ns-SNVs and their corresponding putative neoAgs, the number of neoAgs per mutation was clearly higher for INDELs. With regard to the clinical significance of these mutations, the INDEL count was significantly associated with response to checkpoint inhibitor immunotherapy in several cohorts of melanoma patients.9 Similar results have also been reported in other patient groups,22,23 suggesting the relevance of the immunogenic potential of INDELs. In addition to these studies reporting the INDEL and the corresponding putative neoAg load, some initial studies also demonstrated T cell responses specific for INDELs corresponding to TGFBRII or CDKN2A.24,25 Finally, in a more recent study carried out in patients with kidney cancer, the presence of INDEL-specific T cells was demonstrated by using the MHC multimer technology, which corresponded to the 21% of the recognized neoAgs.26
Similar to INDELs, fusion-derived neoAgs have to be considered as an additional source of tumor antigens.27 The high level of chromosomal instability observed in cancer results in chromosomal translocations or inversions that can cause gene fusions and the formation of hybrid proteins. Some of them, such as the BCR-ABL fusion protein, are oncogenic and result in specific epitopes from the fusion region that can be recognized by T cells.7,28 However, the number of well-described gene fusions is low, and the fusion region may vary between patients,29 hindering clinical translation. Nevertheless, a pan-cancer study of fusion-derived neoAgs revealed that these neoAgs tend to have a higher immunogenic potential, as compared with neoAgs derived from ns-SNVs or INDELs, although the burden of these neoAgs is not related with immunotherapy outcome.29 Therefore, these mutated sequences can generate an additional set of potential neoAgs with clinical relevance.19
Identification of mutation-derived NeoAgs
Advances in NGS technologies and bioinformatic tools have enabled the identification of mutations and prediction of mutation-derived neoAgs, allowing the development of different therapeutic strategies focused on neoAgs as targets.30,31 For this purpose, different algorithms have been developed and are currently applied. Briefly, neoAg identification requires first whole genome/exome sequencing of tumor and healthy tissue, alignment to a reference genome and, after eliminating germline polymorphisms, identification of mutations by using variant caller tools (e.g. VARSCAN, SomaticSniper, Mutect2, etc).32–37 This results in a list of mutations that can be useful for tumor mutational burden (TMB) calculation, with potential applicability for clinical purposes. To predict neoAgs within the list of mutations, most algorithms rely on HLA binding prediction algorithms (reviewed in Xie et al.19). Sequences of mutated peptides are assayed against the panel of HLA alleles expressed by the patient, previously identified from sequencing data. In addition to HLA binding, parameters such as dissimilarity with the self-proteome, similarity with epitopes present in pathogens or the presence of flanking amino acids favoring antigen processing, have also been considered by some algorithms.38–41 Together with these intrinsic immune-related properties of putative neoAgs, other factors such as tumor heterogeneity play an important role. Indeed, differences between clonal and subclonal neoAgs may determine their relevance in tumor rejection and edition, dictating the interactions between the tumor and immune system.42 Finally, if available and as an additional filter for neoAg identification, gene expression obtained by RNASeq can be used to identify those putatively expressed neoAgs.30 In this respect, it has been recently reported that many published mutated neoAgs are also expressed (at the RNA level) in normal tissues and cannot be considered as true neoantigens, suggesting the need of using adequate tools to identify tumor-specific neoAgs.43 Overall, this strategy bears important limitations, such as the suboptimal prediction capacity of some algorithms (e.g. for less common HLA alleles), the inability to identify post-translational modifications that affect immunogenicity or the identification of peptides generated from splicing of proteins.44
An alternative less used approach, not relying on gene sequencing, is immunopeptidomics, based on proteomic technologies. In this case, tumor tissue is lysed and peptide/HLA complexes are purified by using immunoprecipitation with anti-HLA antibodies, and after peptide elution, they are identified by mass spectrometry analysis, by matching peptide sequences with those found in databases or obtained from NGS data using patient tumor cells.45–48 The main advantage of this methodology is that it can identify peptides derived from genes with confirmed expression and translation. Since they are eluted from HLA molecules, identified peptides fulfil the requirements for antigen processing and binding to HLA molecules, not relying on prediction algorithms. On the other side, the amount of biological material required for these studies is usually higher than that used for gene sequencing, although in recent years these methodologies are being improved.49,50
However, in the absence of additional assays analyzing the presence of neoAg-specific T cells, none of the identification strategies mentioned above provide any information about neoAg immunogenicity. To gain insight on this, different assays have been carried out. A classical assay involves stimulation of patient T cells with peptides or tandem minigenes containing the whole set of predicted/identified neoAgs to detect T cell activation.51 In this context, besides to classical techniques measuring cytokine production or upregulation of activation markers, immunogenicity of neoAgs has been tested by using a fast, easy and very efficient method called MANAFEST, which detects changes in the frequencies of T cell clonotypes.52 Alternatively, fluorochrome-tagged HLA multimers loaded with the selected neoAgs are used to label T cells and identify those with neoAg specificity.53 In this last case, any T cell specific for the neoAg of interest can be detected, irrespective of their functional capacity. Since in Ag-based stimulation assays, nonfunctional T cells are not detected, one cannot exclude that the lack of response is due to the poor/null immunogenicity of the neoAgs selected or to the immunosuppressive tumor microenvironment (TME) and highly differentiated and exhausted state of T cells observed in many patients. These methods have been used not only to confirm immunogenicity of neoAgs predicted by HLA binding algorithms or identified by immunopeptidomics but also in unbiased screening procedures, considering all peptides with mutations, irrespective of the HLA of the patient.54 Besides the enhanced complexity of these technologies, availability of lymphocytes from the patients is necessary, making difficult in some cases the characterization of neoAg immunogenicity. Alternative approaches to overcome this include the use of blood from HLA-matched healthy donors (with a better lymphocyte availability and lacking tumor-induced immunosuppression) to perform in vitro priming assays,55 and in vivo experiments using transgenic mice expressing the HLA alleles of interest.56,57 Taken as a whole, new methodologies have been developed to identify neoAgs generated by different types of mutations, helping to characterize their immunogenicity.
Impact of specific genomic alterations on the immune response to neoantigens
Several studies have found an association of high TMB with increased immune infiltration and response to immunotherapy.58 However, the utility of TMB as a predictive biomarker has not been fully demonstrated across all cancers.58,59 Cancer types where TMB does not predict response generally show no relationship between neoAg load and T-cell infiltration. There are some genetic alterations, i.e. deletion and loss-of-function mutations in HLA and B2M genes, that impair antigen presentation,60–63 and T-cell recruitment into tumors. There are also some genetic variants that play an important role in the evasion of antitumor immunity. One of the first pathways to be related to poorly inflamed tumors, despite containing a high number of mutations, was the WNT/β-catenin signaling pathway.64 Activation of this pathway inhibits the recruitment of dendritic cells of the Batf3 lineage, thus decreasing the infiltration of CD8+ T cells, through the negative regulation of CC chemokine ligand 4 (CCL4).65 Various gene mutations are involved in the activation of the WNT/β-catenin pathway. Thus, in addition to gene copy number changes, somatic mutations involving activation (exon 3) of CTNNB1 or loss of function of genes such as APC, APC2, AXIN1 and AXIN2, contribute to its activation.66 Another example is mutations in the RNF43 gene, which are common in high microsatellite instability (MSI-H) tumors. It has been recently observed that frameshift mutations in RNF43 implying loss of functions are associated with non-inflamed TME by activating the WNT/β-catenin signaling pathway.67
Inactivating mutations in Janus kinases (JAK1 or JAK2) produce defective IFNγ signaling with multiple effects, including altered antigen presentation and failed recruitment of T cells into the tumor.68–70 On the other hand, loss-of-function mutations of the PTEN gene cause increased activation of the phosphatidylinositol 3-kinase (PI3K)-AKT pathway and is also significantly associated with a non-inflamed tumor phenotype.71 Mutations in RHOA72 and hepatic serine/threonine kinase B1 (LKB1; also known as STK11)73,74 have also been associated with an immunosuppressive TME and reduction in tumor-infiltrating lymphocytes. Inactivating mutations of the Trp53 gene have also been associated with low immune infiltration of tumors.75 Notably, TP53-mutated tumor cells lack the production of key chemokines necessary for the recruitment of NK cells and T cells to TME.76 In glioma, mutations in the IDH1 and IDH2 isocitrate dehydrogenase genes that confer the ability to convert α-ketoglutaramate into the oncometabolite R-2-hydroxyglutaramate77 are also associated with low-infiltrated tumors.78 Accumulation of R-2-hydroxyglutaramate limits intratumoral production of CXCL9 and CXCL10 chemokines, resulting in decreased T-cell recruitment.78
Taking these findings into account, we must evaluate not only the number of neoantigenic mutations but also the presence of other genetic alterations that may play an important role in the evasion of antitumor immunity.
NeoAgs and efficacy of cancer immunotherapies
Since immune responses against tumor cells constitute the basis of immunotherapies, tumor antigens recognized by therapy-boosted cells play a prominent role.18,79,80 There are immunotherapeutic strategies that rely on antigen-agnostic mechanisms, including immune checkpoint inhibitors (ICI), IL-2 administration, adoptive transfer of autologous tumor-infiltrating lymphocytes (TILs) or the use of oncolytic viruses. However, despite this, all of them stimulate T cells that comprise a pool of cells with specificity for tumor antigens. On the other side, strategies like vaccines and transfer of TCR- or CAR-transduced T cells fully depend on the knowledge of the antigen to be targeted. Therefore, either for immunotherapy design, to understand their efficacy or for patient selection, many studies have focused on the analyses of immunotherapy-targeted antigens, characterizing their abundance, properties, specificity/similarity with self-proteins, etc. In this regard, neoAgs have also been considered as an important factor in the efficacy of some immunotherapies.
Immune checkpoint inhibitors and NeoAgs
Immune checkpoint inhibitors (ICI) have become the most common cancer immunotherapy used in the last decade. This therapy is based on the administration of antibodies blocking signaling pathways triggered by T lymphocyte negative receptors and denominated immune checkpoints.81,82 Although many molecules have been described in this group, so far most clinical applications involve blockade of PD-1/PD-L1 and CTLA-4-derived pathways. Inhibition of these negative signals rescues functional properties of tumor-specific T lymphocytes, rendering them capable of eliminating tumor cells. Since not all patients respond to ICI, important efforts have been carried out to identify biomarkers of response for patient selection.83 Among them, TMB has emerged as an important feature. Initial studies carried out in highly mutated tumors like melanoma84,85 and lung cancer86 demonstrated an association between TMB and response to ICI. Later, this was extended to other tumors, including those with MSI-H and/or mismatch repair deficiency (dMMR).87,88 By considering a set of patients with tumors bearing different TMB levels, Yarchoan et al. reported a positive correlation between TMB and response rate to PD-1/PD-L1 blockade,89 and this sensitivity to ICI increases when focusing on clonal neoAgs.42 As indicated above, the mechanistic explanation postulates that tumors with a higher TMB would present increased neoAg levels, resulting in more “strange” or immunogenic tumors. Consequently, this would favor lymphocyte priming and tumor infiltration, deriving a higher possibility of being rescued by ICI. Although this correlation is true for heavily mutated tumors, where the high number of mutations may generate a sufficient number of neoAgs, it does not fit equally well for tumors with low or moderate TMB. Interestingly, for these tumors, neoAg load may behave as a better biomarker than TMB to predict response to ICI. In this regard, neoAg load, but not TMB, accurately predicts treatment efficacy in melanoma, lung cancer and gynecological tumors, whereas it has a poorer predictive capacity in urinary system and liver tumors.90 Association between neoAg load and response to ICI can be observed not only when considering the classical mutations originated from SNV but also from frameshift mutations, known to generate a higher number of neoAgs per mutation.9,91
However, most of these studies were inferred on associations between TMB or neoAg load with T cell immunity and concomitant response to ICI, without any experimental demonstration of the presumed molecular and cellular mechanisms. In a study across different cancers of patients where high TMB does not predict response to ICI, there is no association between neoAg load and CD8 T cell infiltration,92 indicating the relevance of these mechanisms mainly when infiltrating T cells are neoAg-specific. More recently, Puig-Saus et al. have demonstrated the existence of neoAg-specific T cells in patients responding to antiPD-1, at superior levels to those found in non-responder patients.93 Interestingly, the T cell responses to mutational neoAgs were not linearly correlated with the number of mutations. However, in another study in patients with metastatic urothelial carcinoma, early expansion of neoAg-specific T cells associated with the response to ICI.94
Adoptive T-cell therapy and neoAgs
Treatment based on the passive administration of antitumor T cells is called adoptive T-cell therapy (ACT). ACT using TILs has been applyied for years in different tumors, with pioneer studies in melanoma. In a randomized prospective study, TIL therapy demonstrated objective responses in 54% of melanoma patients, including 24% complete responses, which in most cases were durable.95 The lack of toxicity to normal tissues in these trials suggested that the transferred TILs targeted molecules unique to cancer cells. Indeed, a series of retrospective studies revealed that infused TILs frequently targeted mutation-derived neoAgs.51–96–99
Although TIL therapy has been shown to be very effective in treating melanoma, this has not been the case for other tumors. Interestingly, in tumors other than melanoma, the conventional procedure to expand TILs does not yield tumor-reactive TIL products with the same efficiency.100 Among the possible reasons for this low reactivity are the lower number of mutations of these tumor types, and consequently, the lower frequency of mutation-reactive TILs. For some years now, a series of methods have been developed to detect somatic mutation-reactive autologous T cells and specifically increase their frequency in the infusion product.101 A series of clinical case reports provided the first evidence that enriching TILs in neoAg-specific T cells can mediate durable tumor regressions without causing toxicity in patients with non-melanoma epithelial cancers.17,102,103 An illustrative case is that of a patient with refractory hormone receptor-positive breast cancer who was treated with an autologous TIL product enriched in T cell clones recognizing four different neoAgs (SLC3A2, KIAA0368, CADPS2 and CTSB), representing the 23% of the infused product. Treatment with this TIL product in combination with pembrolizumab produced complete and long-lasting regression of multiple metastases.103
However, the low presence of neoAg-specific TILs and their highly differentiation and exhaustion state in the tumor sample may make it difficult to obtain infusion products sufficiently enriched in neoAg-reactive cells to have therapeutic efficacy.104 Further application of ACT led to the development of techniques to genetically modify normal peripheral blood T cells to express a receptor capable of recognizing the tumor, providing them with antitumor activity.105 Genetic modification of normal peripheral blood T cells with an exogenous TCR specific for a neoAg (neoTCR) could be an interesting alternative to TILs targeting somatic mutations. The fact that neoAg are highly tumor-specific antigens would mitigate the danger of off-target toxicities observed with TCRs directed at self-antigens.106
Among the different types of neoAgs, those derived from driver mutations are attractive targets for TCR therapy. This enables the development of off-the-shelf neoTCRs that could be used to treat a larger number of cancer patients who express the mutation and the adequate HLA restriction. This is the case of KRAS, an oncogene that is frequently mutated in pancreatic and colorectal cancer. Recently, a patient with progressive metastatic pancreatic cancer showed objective response after treatment with a mixture of autologous T cells that had been genetically modified to express two HLA-C * 08:02-restricted TCRs targeting the KRAS G12D driver mutation,17 expressed by the patient’s tumors.107 Additional TCRs targeting KRAS G12D and other hot-spot KRAS mutants restricted by different HLA molecules have been identified from TIL cultures108–110 that could be used to extend TCR therapy against mutant KRAS to a larger number of patients. TCR libraries against other driver mutations, such as TP53, BRAF, PIK3CA and EGFR, may be promising.
However, most neoAgs result from random passenger mutations, behaving as patient-specific or ‘private’ neoAgs. TILs specific to these mutations have been found in various types of cancer.51,96,97,102 Although private neoAgs may be subjected to clonal heterogeneity, an important mechanism of resistance to immunotherapy, TIL therapy directed against this type of neoAgs has been able to generate prolonged responses,102,103 deserving thus to be considered as a potential target of ACT. However, while private neoAgs are feasible targets for TIL therapy, they pose a significant challenge for TCR therapy because they require customization. Recently, a first-in-human Phase I clinical trial (NCT03970382) has demonstrated the feasibility of manufacturing multiple neoTCR engineered T cells using non-viral precision genome-editing. The safety of infusing up to three gene-edited neoTCR T cell products has also been demonstrated. In summary, the success of the first ACT trials was based on the presence of NeoAg-specific TILs and is now moving toward the genetic engineering of T cells with neoTCR. However, neoTCR identification remains challenging and the process must be tailored to clinical timelines.
In conclusion, currently used immunotherapies, including those based on ICI and adoptive cell transfer, have an important component associated with neoAg-specific responses, indicating the relevance of potentiating immunity against these antigenic targets.
Vaccines based on neoAgs
Upon identification of mutation-derived neoAgs in murine tumor models, immunization experiments using the mutation-containing peptides demonstrated not only the immunogenicity of these sequences but also their capacity to reject established tumors,111,112 suggesting the potential applicability of these antigens in vaccination strategies. There are several examples of neoAg-based vaccines carried out in murine models where the antitumor properties of these vaccines have been demonstrated.113–116 They include not only CD8 epitopes, initially considered as major rejection antigens, but also CD4 epitopes,117 which in addition to priming CD4 T cells with antitumor properties, may contribute to strengthen activation of CD8 responses.56
Given the specificity and potency of neoAg-based vaccines in the preclinical setting, this prompted the development of vaccination clinical trials in cancer patients, becoming vaccination the most common strategy based on neoAgs. To date, around 150 neoAg-based vaccine clinical trials have been initiated.118 Pioneer studies were carried out in melanoma patients, due to the high neoAg load present in these individuals. Carreno et al. used dendritic cells pulsed with CD8 epitope peptides to demonstrate induction of tumor – specific responses, where a broadening of antigenic breadth and clonal diversity was observed.119 Additional clinical trials have been reported (reviewed in Niemi et al.118), not only in melanoma,119–121 but also in glioblastoma,122,123 gastrointestinal cancer,124 lung cancer,125 pancreatic cancer126 and hepatocellular carcinoma,127 among others, indicating that vaccination is not only feasible in tumors with high TMB but also in those with a lower TMB. In general, the results of these studies have demonstrated safety in the vast majority of cases, and immunogenicity has also been described, resulting in the induction or boosting of CD8+ and CD4+ tumor-specific T cell responses. Indeed, in a pan-cancer clinical study administering neoAg peptide pools, similar safety and immunogenicity results were reported. Twenty out of 22 patients had no or mild adverse effects, 90% of them had measurable T cell activation and around 80% of peptides elicited immune responses.128 Although most clinical trials are based on the administration of neoAgs derived from ns-SNVs, there are examples of vaccines containing immunogenic frameshift peptide neoAgs. As in other trials, colon cancer patients with MMR-deficient tumors included in this trial did not experience severe adverse effects, and humoral and cellular immune responses were observed in all patients.129 Similarly, the Nous-209 “off-the-shelf” vaccine encoding frameshift peptides130 has progressed to clinical phases, with a demonstrated immunogenicity in all vaccinated patients.131
In addition to safety and immunogenicity, interesting clinical responses have been observed in melanoma cohorts. As described by Ott et al.,120 four out of six patients showed no recurrence for up to 25 months and two of the recurrent patients that were subsequently treated with anti-PD-1 presented complete tumor regressions. This was also described by Sahin et al.,121 whose metastatic melanoma cohort treated with a neoAg RNA-based vaccine showed two out of five patients with complete responses, and a third patient who was treated with anti-PD-1 therapy after vaccination also reached complete response. In a cohort of 29 patients with glioma vaccinated with the H3.3K27M shared neoAg, 39% demonstrated detectable T cell responses, associated with prolonged overall survival.132
Despite these results demonstrating the feasibility, safety and immunogenicity of neoAg-based vaccines, just a few patients presented clinically relevant responses when vaccines were administered as monotherapy. Since vaccines boost the first step of the cancer-immunity cycle and the generated response can enhance the expression of immune checkpoints as an escape mechanism, the combination of neoAg-based vaccines with ICI appears as a better therapeutic strategy. In fact, some clinical trials based on the administration of vaccines + anti-PD-1/PD-L1 have demonstrated clinical efficacy, exampled by the combination of the neoAg vaccine NEO PV 01 and nivolumab in melanoma, NSCLC and bladder cancer patients. In this clinical trial, considerable ORR rates were observed in the three cohorts (59%, 39% and 27%, respectively) and longer progression-free survival.133 These results have encouraged the development of vaccines in combination with ICI, which are currently being studied in different solid tumors with the inhibition of the PD-1/PD-L1 axis (NCT02287428, NCT03359239 and NCT04397003) or also in combination with anti-CTLA-4 monoclonal antibodies (NCT04117087, NCT03606967). Clinically relevant results have been achieved in tumors with a high TMB such as melanoma or lung cancer.119–121 This type of tumors also presents better clinical responses when receiving other therapies such as ICI, probably due to the presence of a high number of driver and clonal mutations that generate neoAgs and makes them more immunogenic. Nevertheless, as mentioned above, patients with tumors containing a lower TMB have also been enrolled in these trials. In line with combinatorial strategies, a recent work has been shared in the American Association for Cancer Research (AACR), where Khattak A. et al.134 presented their phase II clinical trial results of the combination of mRNA-4157/V940 vaccine with pembrolizumab in patients with advanced melanoma with a high risk of recurrence after surgery. Patients receiving this combination presented 78.6% of recurrence-free survival, while the control arm that received pembrolizumab alone achieved a 62.2%. These data indicate a 44% reduction in the risk of recurrence and death in patients receiving the combination. When considering the risk of recurrence according to TMB (dividing their cohort using the threshold of 10 mutations per megabase (mut/Mb)) a similar reduction in the risk of recurrence or death was observed, presumably due to the high overall TMB values in both groups. In the same line, Rojas et al. have reported results of their neoAg RNA vaccine in 16 patients with pancreatic cancer, administered in combination with atezolizumab (anti-PD-L1) and chemotherapy in the adjuvant setting. A significantly longer median recurrence-free survival was observed in those patients with vaccine-induced T cell expansion.135 In summary, all these results indicate the feasibility of neoAg vaccine preparation and administration to a variety of cancer patients, which in most cases are immunogenic and, when applied mainly in combination with ICI, yields promising clinical results. Notwithstanding, several challenges are ahead, and improvements in their design, generation and application are still needed.136
Non-classical tumor antigens
Other than classical tumor-associated antigens and neoAgs generated by mutations described above,137 novel tumor antigens can derive from additional alterations or changes in RNA maturation and translation or in protein processing (reviewed in Xie et al. and Nagel et al.19,138) Antigens deriving from transposable elements have also been described, but they are not discussed in this review.139
Tumor antigens derived from altered RNA processing or non-canonical translation
Several mechanisms related to RNA processing and RNA translation into proteins may suffer from aberrant events, originating new sequences (Figure 1b).
Antigens derived from altered splicing
Several cis-acting sequences and trans-acting factors are involved in the modification of newly transcribed RNA that eliminates large intronic sequences and joins neighboring exonic regions. This is a well-controlled and cell-type specific process known as splicing. Mutation or deregulation in splicing factors or genomic changes affecting cis-acting regions are abundant in tumors, resulting in elimination of exons (exon skipping) or intron retention, which is more prevalent in cancer.140–142 This altered splicing may result in novel cancer-specific proteins and tumor antigens, some of which are shared among patients.143,144 A well-known example of a tumor antigen derived from altered splicing is the EGFRvIII driver mutation mentioned above as a candidate for antigen-specific CAR-T therapies.145,146 In some cases, altered splicing results in transcripts with premature termination codons that are degraded by NMD. Thus, the antigens derived from these are poorly abundant (see below, altered RNA stability). However, some introns have coding potential and lack stop codons (exitrons). Retention of these introns results in transcripts that are invisible for the NMD machinery and high levels of tumor antigens.147 Also, resistant to NMD are transcripts with skipped exons that keep the reading frame. Proteins translated from these transcripts produce tumor antigens at the novel exon–exon junctions or neojunctions, which are more likely to be shared among patients than those generated from SNV mutations.148 In spite of this, additional work is required to ensure that neoAgs derived by altered splicing are indeed cancer-specific. Also, further experiments are required to evaluate the potential of drugs that affect splicing to increase the immunogenicity of cold tumors.149,150
Antigens derived from altered RNA modification and stability
As indicated above, NMD decreases the stability of transcripts with premature termination codons (PTCs). Interestingly, there is a pioneer round of translation prior to NMD, and the protein resulting from this translation is preferentially degraded and transported to MHC molecules.151 However, the bulk of transcripts with PTCs, including those caused by frameshifting, is degraded by NMD, and therefore, they cannot produce antigens. Therefore, pharmacological inhibition of NMD has been shown to produce larger amounts of tumor antigens.152
RNA stability is also controlled by polyadenylation. When this occurs at introns or non-canonical sites it can alter transcript stability but also nuclear export, cell localization and protein translation, leading to the production of truncated or novel proteins and tumor antigens.153 RNA modification can also affect different steps in RNA processing and translation. RNA editing can change specific nucleotides in the RNA sequence, leading to novel antigens, and it is also altered in several cancers.154 Most studied is adenosine-to-inosine (A-to-I) editing, which produces highly immunogenic neoepitopes.155 Further studies are required to demonstrate whether they are tumor specific.
Antigens derived from non-canonical translation
It has been established that only ~2% of the genome is transcribed and translated into canonical proteins.156,157 Recently, this has been revisited, as many regions previously classified as “non-coding” have been shown to contain short or non-canonical open reading frames (ncORFs).158 NcORFs can be translated to proteins shorter than 100 amino acids or microproteins or to proteins that do not follow the rules of canonical translation: they do not start with ATG or they are located upstream, downstream or overlapping with well-described coding sequences.159 Most of them have been ignored by the scientific community until very recently, as most are not conserved and many are not stable.160–162 Interestingly, ncORFs have been described in cancer-specific transcripts expressed in several cancer patients and classified as long non-coding RNAs.163,164 As these ncORFs are predicted to contain disordered and hydrophobic regions, it is expected that they are more visible to the proteasome and with stronger potential to generate epitopes.162,165 NcORFs may contribute to over 10% of the total immunopeptidome.166–168 This number could be underestimated as some MS spectra originally assigned to canonical proteins could indeed correspond to non-canonical ones.169 In addition, the total number of human ncORFs is yet unclear. While conservative estimates consider around 7,000, others find certain evidence for the existence for several hundred thousand.170
Antigens derived from aberrant mRNA translation
Aberrant translation deserves a special chapter within non-canonical translation. Hypoxia and amino acid starvation caused by tumor growth, coupled with inefficient angiogenesis and insufficient blood flow, activate the integrated stress response (ISR).171 ISR signaling alters canonical translation initiation, resulting in a stress-associated proteome devoted to activate survival in the hostile environment or induce apoptosis. Such stress-associated proteome could comprise novel cancer-specific tumor antigens. Among all deficient amino acids, tryptophan has a special effect. Under low tryptophan, the ribosome is stalled at tryptophan codons resulting in frameshifting and codon reassignment to phenylalanine, leading to the generation of novel proteins that are a source of tumor antigens.172–174 Interestingly, this is cancer-specific.
Tumor antigens derived from altered protein modification and processing
In addition to DNA- and RNA-related alterations, proteins may also suffer different modifications that can render them as antigenic (Figure 1c).
Antigens derived from altered protein modifications
After translation, many proteins are modified by post-translational modifications (PTMs), resulting in specific epitopes. One example among several is phosphorylation. Aberrant phosphorylation can generate novel epitopes or can increase the efficacy of MHC binding compared to that of the non-phosphorylated form.175 Some peptides have been described that are modified only in cancer cells that could lead, so far, to a limited number of tumor antigens.175–178
Antigens derived from altered protein processing
Protein splicing can also be considered as a special modification. Similar to RNA, protein splicing allows the deletion of “intein” sequences while neighboring “exteins” are fused. This has been well studied for FGF5, where the proteasome is the catalytic agent causing proteasome-catalyzed peptide splicing (PCPS).179–183 Epitopes from the junction region or splicetopes can bind HLA molecules and induce T cell responses, but it is yet unclear whether they are cancer-specific and how widespread is PCPS. While some estimates considered that 30% of the HLA ligandome could be originated from PCPS, more recent, conservative and cautious studies indicate that only around 2–6% could be explained as derived from spliced protein fragments, being unclear whether all originate from PCPS.184–186
Methods to detect non-classical antigens
In addition to the methods described above for the identification of neoAgs based on whole genome/exome and RNA sequencing, novel technologies have been established that quantify canonical and non-canonical translation or that look for the peptides bound to the MHC molecules by immunopeptidomics (Figure 1d).187 The finest method to evaluate translation is ribosome profiling, capable of detecting canonical and non-canonical translation but blind to further protein stability, processing or modification.188 Instead, mass spectrometry (MS) should be capable of identifying any protein sequence but requires refining to detect less abundant and shorter proteins and a robust database with reliable information about the complete proteome. As we are far from the latter, the best method nowadays involves proteogenomic approaches where genomic, transcriptomic and ribosome profiling data are combined with immunopeptidomics.189
Ribosome profiling
Ribosome profiling (RiboSeq) enables deep sequencing of ribosome-protected RNA fragments to determine regions of active translation.190 The precision of the technology is such that it provides a fingerprint of ribosome function: it visualizes the codon shifts at base-pair resolution, allowing the identification of the complete translated ORFs. Given its high precision and unbiased acquisition, RiboSeq is capable of identifying ORFs shorter than 8 or 15 amino acids in length, which would be fundamentally challenging with other methods.160,191,192 However, depending on the quality of the data and the pipeline followed for analysis, many false positives can be retrieved. High-quality RiboSeq datasets should have a codon periodicity (percentage of reads 28–30 nucleotides long that identify known ORFs) over 70%, a good library complexity (meaning that most reads are unique and do not result from PCR duplicates) and allow the identification of 9–10 thousand canonical proteins in humans.170 When less canonical proteins are detected, it is expected that also far less ncORFs will be identified. In a good RiboSeq library, 70–85% of the reads will be discarded and correspond to rRNA, tRNA, nuclear RNAs and even repetitive regions from retrotransposons or pseudogenes, which are difficult to resolve. The remaining 15–30% of the reads will be mapped as ribosome footprints and 80% of them will correspond to canonical ORFs.193 Thus, from a sequencing depth of 150 million reads, just around 5 million reads are left for ORF discovery.
Interestingly, this leaves enough space to identify ORFs that are not found in proteomic studies. The major reason for this is that, unlike with conventional Liquid Chromatography with tandem MS (LC-MS/MS), with RiboSeq data there is no penalty in the identification of short proteins, proteins enriched in lysines or arginines (generating peptides that are too small for MS after trypsin digestion) or proteins with low stability or low abundance, as sequencing methods have a PCR amplification step.160 More importantly, databases comprising ncORFs predicted by RiboSeq (with more or less confidence) can be used to interpret spectra obtained by LC-MS/MS.
Immunopeptidomics
As indicated above, identification of small non-canonical proteins that could serve as novel tumor antigens was inefficient following conventional tryptic or semi-tryptic (resulting from total or partial protein cleavage at the C-terminal side of a lysine or arginine) LC-MS/MS techniques.194–196 Protocols to enhance identification of non-canonical proteins included enrichment for short proteins with size exclusion chromatography or gel filtration and were not as successful as originally expected. This changed drastically with the advent of immunopeptidomics, where peptides are dissociated from MHC molecules prior to peptide sequencing and quantitation via MS.45,46,187 Information about peptide mass (MS1 spectra) and mass-to-charge (m/z) ratio (MS2 or fragment spectra) are matched to those from reference proteomes and custom databases using protein inference algorithms. The use of robust non-canonical custom databases is mandatory, as more than 70% of MS-detected peptides cannot be assigned with high confidence when they are only matched to reference proteomes.197 This already suggests that a relevant part of the immunopeptidome may be derived from non-canonical sequences.
Different from trypsin-based conventional strategies, in immunopeptidomics:
- The peptides are not trypsin products but the results of the natural degradation of the proteome by the proteasome or by cellular proteases, decreasing the bias over lysine or arginine residues. This also helps the identification of peptides derived from proteins with low stability or defective ribosome products, as the half-lives of HLA-binding motifs (hours) are in general longer than those of rapidly degraded proteins (minutes).198
- The peptides are bound to MHC molecules after degradation in the cell. Reagents used for MHC immunoaffinity purification may involve native conditions instead of stringent lysis buffers. Peptide enrichment is obtained after high-affinity binding to MHC antibodies.199
- The peptides that bind to class I and class II MHC molecules have specific characteristics that may serve as an additional quality control step to ensure proper peptide identification. These include peptide size, of nine amino acids for HLA-I and 12–25 for HLA-II.200,201 In addition, specific HLA molecules have distinct binding motifs, and the affinity of a given peptide to the HLA of the sample under study can be predicted with computational approaches such as MHCflurry or NetMHCpan.202,203
- The experimental HLA ligandome data define the landscape of peptides presented to T cells, excellent candidates for immunotherapy. This is a great advantage of immunopeptidomics over other approaches to identify novel tumor antigens.
In spite of the outstanding performance of updated immunopeptidomic protocols for antigen identification, novel pipelines should be developed to improve the confidence of individual candidates. Proteogenomic data for non-canonical proteins that lack RiboSeq validation has too high false-positive rates.204 Therefore, in addition to features related to specific HLAs (size, binding motives, etc.), the spectra and retention times can be validated with synthetic peptides, which can be not realistic for high-throughput screens, or predicted with specific machine-learning bioinformatic tools.205–207
The Human Proteome Organization (HUPO) establishes that novel proteins should be detected by at least two independent uniquely-mapping peptides longer than eight amino acids with publicly available, high-quality peptide-spectrum matches (PSMs).170,208 However, this may be intolerable due to the short length of MHC-bound peptides and some non-canonical proteins. Additional strategies should be considered to define true non-canonical proteins. Deep learning tools can help novel peptide and protein discovery by MS. Dubious identifications can be filtered with deepLC and Prosit, which enable the validation of peptide retention time and fragment ion intensity.205,207 Combination of these tools with excellent immunopeptidomic and RiboSeq data should increase the confidence of what has been described as “novel presented proteins,” which may lack a function but can be good antigens for immunotherapy.170
Concluding remarks
Tumor antigens play a pivotal role in cancer development, by affecting the TME and the efficacy of most immunotherapies. The relevance of these molecules has led to important efforts for their discovery and classification, characterizing their main properties. These studies have demonstrated that ideal tumor antigens should have the highest specificity for the tumor, preferably in a clonal manner (such as many driver mutations), lacking thymic expression that would generate central tolerance mechanisms (dissimilarity with self-proteins) while appearing at sufficient levels in tumor cells for efficient recognition by TCRs. This enforced recognition should be favored by promiscuous presentation by different HLA alleles and analogy with microbial epitopes, leading in the best scenario to the activation of polyclonal T cell responses, avoiding escape mechanisms mediated by antigen or HLA loss, frequently observed in tumors. Although some of the antigens identified so far do not fulfill all these properties, development of new combined methodologies is helping to broaden the antigen range, generating a new spectrum of targetable antigens useful for cancer immunotherapies.
Acknowledgments
PF is supported by Gobierno de Navarra (0011-1411-2021-000070 and 0011‐1383‐2022‐ 000014 projects), RTI2018-101759-B-I00, PDC2022-133961-100 and PID2021-128791OB-I00 MCIN/AEI/10.13039/501100011033/FEDER, UE, Scientific Foundation of the Spanish Association Against Cancer (AECC IDEAS20169FORT to P.F.) and by the Instituto de Salud Carlos III, which finances Centro de Investigación Biomédica en Red de Enfermedades Hepáticas y Digestivas (CIBEREhd) and Red Española de Terapias Avanzadas TERAV ISCIII (RICORS: (RD21/0017), financed by the EU (NextGenerationEU. Plan de Recuperación Transformación y Resiliencia). PS is funded by Instituto de Salud Carlos III (ISCIII) through the project “PI20/00260” and co-funded by the European Union and FEDER and by Gobierno de Navarra (grants GNS_54-2021, 0011-1411-2020-000011 and 0011-1411-2020-000010). S.H.-S. is funded by ISCIII (PI21/00292) and Gobierno de Navarra (Dpto. de Salud -045-2017/HEPATIL), co-financed with “Fondos FEDER” (“Una manera de hacer Europa”), and Gobierno de Navarra (Dpto. Industria – GN2020 PC196-197/SOLIDET- and Proyectos Estrategicos de I+D 2023-2026-0011-1411-2023-000072/PITAGORAS).
Funding Statement
The work was supported by the Gobierno de Navarra [045-2017/HEPATIL]; Gobierno de Navarra [0011-1411-2020-000011]; Gobierno de Navarra [GNS_54-2021]; Gobierno de Navarra [0011‐1383‐2022‐ 000014]; Gobierno de Navarra [0011-1411-2021-000070]; Gobierno de Navarra [0011-1411-2023-000072/PITAGORAS]; Gobierno de Navarra [GN2020 PC196-197/SOLIDET]; Gobierno de Navarra [0011-1411-2020-000010]; Instituto de Salud Carlos III [PI20/00260]; Instituto de Salud Carlos III [CIBERejd TERAv]; Instituto de Salud Carlos III [PI21/00292]; Ministerio de Ciencia e Innovación [PID2021-128791OBI00 MCIN/AEI/10.13039/501100011033]; Ministerio de Ciencia e Innovación [PDC2022-133961-100]; Ministerio de Ciencia e Innovación [RTI2018-101759-B-I00]; Foundation of the Spanish Association Against Cancer [AECC IDEAS20169FORT].
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- 1.Dunn GP, Old LJ, Schreiber RD.. The three Es of cancer immunoediting. Annu Rev Immunol. 2004;22(1):329–18. doi: 10.1146/annurev.immunol.22.012703.104803. [DOI] [PubMed] [Google Scholar]
- 2.Schumacher TN, Scheper W, Kvistborg P. Cancer Neoantigens. Annu Rev Immunol. 2019;37(1):173–200. doi: 10.1146/annurev-immunol-042617-053402. [DOI] [PubMed] [Google Scholar]
- 3.Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3(11):991–8. doi: 10.1038/NI1102-991. [DOI] [PubMed] [Google Scholar]
- 4.Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer. 2014;14(2):135–46. doi: 10.1038/nrc3670. [DOI] [PubMed] [Google Scholar]
- 5.Haen SP, Löffler MW, Rammensee HG, Brossart P. Towards new horizons: characterization, classification and implications of the tumour antigenic repertoire. Nat Rev Clin Oncol. 2020;17(10):595–610. doi: 10.1038/S41571-020-0387-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tashiro H, Brenner MK. Immunotherapy against cancer-related viruses. Cell Res. 2017;27(1):59–73. doi: 10.1038/CR.2016.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang W, Lee KW, Srivastava RM, Kuo F, Krishna C, Chowell D, Makarov V, Hoen D, Dalin MG, Wexler L. et al. Immunogenic neoantigens derived from gene fusions stimulate T cell responses. Nat Med. 2019;25(5):767–75. doi: 10.1038/S41591-019-0434-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xia H, McMichael J, Becker-Hapak M, Onyeador OC, Buchli R, McClain E, Pence P, Supabphol S, Richters MM, Basu A. et al. Computational prediction of MHC anchor locations guides neoantigen identification and prioritization. Sci Immunol. 2023;8(82). doi: 10.1126/SCIIMMUNOL.ABG2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Turajlic S, Litchfield K, Xu H, Rosenthal R, McGranahan N, Reading JL, Wong YNS, Rowan A, Kanu N, Al Bakir M. et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: a pan-cancer analysis. Lancet Oncol. 2017;18(8):1009–21. doi: 10.1016/S1470-2045(17)30516-8. [DOI] [PubMed] [Google Scholar]
- 10.van Belzen IAEM, Schönhuth A, Kemmeren P, Hehir-Kwa JY. Structural variant detection in cancer genomes: computational challenges and perspectives for precision oncology. Npj Precis Oncol. 2021;5(1). doi: 10.1038/S41698-021-00155-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang N, Lysenkov V, Orte K, Kairisto V, Aakko J, Khan S, Elo LL, Kosakovsky Pond SL. Tool evaluation for the detection of variably sized indels from next generation whole genome and targeted sequencing data. PLoS Comput Biol. 2022;18(2):e1009269. doi: 10.1371/JOURNAL.PCBI.1009269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Monaghan L, Longman D, Cáceres JF. Translation-coupled mRNA quality control mechanisms. EMBO J. 2023;42(19):e114378. doi: 10.15252/EMBJ.2023114378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deniger DC, Pasetto A, Robbins PF, Gartner JJ, Prickett TD, Paria BC, Malekzadeh P, Jia L, Yossef R, Langhan MM. et al. T-cell responses to TP53 “hotspot” mutations and unique neoantigens expressed by human ovarian cancers. Clin Cancer Res. 2018;24(22):5562–73. doi: 10.1158/1078-0432.CCR-18-0573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parkhurst MR, Robbins PF, Tran E, Prickett TD, Gartner JJ, Jia L, Ivey G, Li YF, El-Gamil M, Lalani A. et al. Unique Neoantigens Arise from Somatic Mutations in Patients with Gastrointestinal Cancers. Cancer Discov. 2019;9(8):1022–35. doi: 10.1158/2159-8290.CD-18-1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cohen CJ, Gartner JJ, Horovitz-Fried M, Shamalov K, Trebska-McGowan K, Bliskovsky VV, Parkhurst MR, Ankri C, Prickett TD, Crystal JS. et al. Isolation of neoantigen-specific T cells from tumor and peripheral lymphocytes. J Clin Invest. 2015;125(10):3981–91. doi: 10.1172/JCI82416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Buscail L, Bournet B, Cordelier P. Role of oncogenic KRAS in the diagnosis, prognosis and treatment of pancreatic cancer. Nat Rev Gastroenterol Hepatol. 2020;17(3):153–68. doi: 10.1038/S41575-019-0245-4. [DOI] [PubMed] [Google Scholar]
- 17.Tran E, Robbins PF, Lu Y-C, Prickett TD, Gartner JJ, Jia L, Pasetto A, Zheng Z, Ray S, Groh EM. et al. T-Cell transfer therapy targeting mutant KRAS in cancer. N Engl J Med. 2016;375(23):2255–62. doi: 10.1056/NEJMOA1609279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leko V, Rosenberg SA. Identifying and targeting human tumor antigens for T cell-based immunotherapy of solid tumors. Cancer Cell. 2020;38(4):454–72. doi: 10.1016/J.CCELL.2020.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xie N, Shen G, Gao W, Huang Z, Huang C, Fu L. Neoantigens: promising targets for cancer therapy. Signal Transduct Target Ther. 2023;8(1). doi: 10.1038/S41392-022-01270-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pearlman AH, Hwang MS, Konig MF, Hsiue EHC, Douglass J, DiNapoli SR, Mog BJ, Bettegowda C, Pardoll DM, Gabelli SB. et al. Targeting public neoantigens for cancer immunotherapy. Nat Cancer. 2021;2(5):487–97. doi: 10.1038/S43018-021-00210-Y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gurung HR, Heidersbach AJ, Darwish M, Chan PPF, Li J, Beresini M, Zill OA, Wallace A, Tong AJ, Hascall D. et al. Systematic discovery of neoepitope-HLA pairs for neoantigens shared among patients and tumor types. Nat Biotechnol. 2023. doi: 10.1038/S41587-023-01945-Y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu H-X, Wang Z-X, Zhao Q, Chen D-L, He M-M, Yang L-P, Wang Y-N, Jin Y, Ren C, Luo H-Y. et al. Tumor mutational and indel burden: a systematic pan-cancer evaluation as prognostic biomarkers. Ann Transl Med. 2019;7(22):640–640. doi: 10.21037/ATM.2019.10.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DI VL, Reijmers RM, Honders MW, Hagedoorn RS, De Jong RCM, Kester MGD, Van Der Steen DM, De Ru AH, Kweekel C, Bijen HM. et al. Mutated nucleophosmin 1 as immunotherapy target in acute myeloid leukemia. J Clin Invest. 2019;129(2):774–85. doi: 10.1172/JCI97482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Linnebacher M, Gebert J, Rudy W, Woerner S, Yuan YP, Bork P, von Knebel Doeberitz MK. Frameshift peptide-derived T-cell epitopes: a source of novel tumor-specific antigens. Int J Cancer. 2001;93(1):6–11. doi: 10.1002/IJC.1298. [DOI] [PubMed] [Google Scholar]
- 25.Huang J, El-Gamil M, Dudley ME, Li YF, Rosenberg SA, Robbins PF. T cells associated with tumor regression recognize frameshifted products of the CDKN2A tumor suppressor gene locus and a mutated HLA class I gene product. J Immunol. 2004;172(10):6057–64. doi: 10.4049/JIMMUNOL.172.10.6057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hansen UK, Ramskov S, Bjerregaard AM, Borch A, Andersen R, Draghi A, Donia M, Bentzen AK, Marquard AM, Szallasi Z. et al. Tumor-infiltrating T cells from clear cell renal cell carcinoma patients recognize neoepitopes derived from point and frameshift mutations. Front Immunol. 2020;11:11. doi: 10.3389/FIMMU.2020.00373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Y, Shi T, Song X, Liu B, Wei J. Gene fusion neoantigens: emerging targets for cancer immunotherapy. Cancer Lett. 2021;506:45–54. doi: 10.1016/j.canlet.2021.02.023. [DOI] [PubMed] [Google Scholar]
- 28.Comoli P, Basso S, Riva G, Barozzi P, Guido I, Gurrado A, Quartuccio G, Rubert L, Lagreca I, Vallerini D. et al. BCR-ABL-specific T-cell therapy in Ph+ ALL patients on tyrosine-kinase inhibitors. Blood. 2017;129(5):582–6. doi: 10.1182/BLOOD-2016-07-731091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wei Z, Zhou C, Zhang Z, Guan M, Zhang C, Liu Z, Liu Q. The landscape of tumor fusion neoantigens: a pan-cancer analysis. iScience. 2019;21:249–60. doi: 10.1016/J.ISCI.2019.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Biswas N, Chakrabarti S, Padul V, Jones LD, Ashili S. Designing neoantigen cancer vaccines, trials, and outcomes. Front Immunol. 2023;14. doi: 10.3389/FIMMU.2023.1105420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Okada M, Shimizu K, Fujii SI. Identification of neoantigens in cancer cells as targets for immunotherapy. Int J Mol Sci. 2022;23(5):2594. doi: 10.3390/IJMS23052594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koboldt DC, Chen K, Wylie T, Larson DE, McLellan MD, Mardis ER, Weinstock GM, Wilson RK, Ding L. VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics. 2009;25(17):2283–5. doi: 10.1093/BIOINFORMATICS/BTP373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Larson DE, Harris CC, Chen K, Koboldt DC, Abbott TE, Dooling DJ, Ley TJ, Mardis ER, Wilson RK, Ding L. SomaticSniper: identification of somatic point mutations in whole genome sequencing data. Bioinformatics. 2012;28(3):311–7. doi: 10.1093/BIOINFORMATICS/BTR665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang R, Van Etten JL, Dehm SM. Indel detection from DNA and RNA sequencing data with transIndel. Bmc Genom. 2018;19(1). doi: 10.1186/S12864-018-4671-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wajnberg G, Passetti F. Using high-throughput sequencing transcriptome data for INDEL detection: challenges for cancer drug discovery. Expert Opin Drug Discov. 2016;11(3):257–68. doi: 10.1517/17460441.2016.1143813. [DOI] [PubMed] [Google Scholar]
- 36.Sun Z, Bhagwate A, Prodduturi N, Yang P, Kocher JPA. Indel detection from RNA-seq data: tool evaluation and strategies for accurate detection of actionable mutations. Brief Bioinform. 2017;18(6):973–83. doi: 10.1093/BIB/BBW069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31(3):213–9. doi: 10.1038/NBT.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Richters MM, Xia H, Campbell KM, Gillanders WE, Griffith OL, Griffith M. Best practices for bioinformatic characterization of neoantigens for clinical utility. Genome Med. 2019;11(1). doi: 10.1186/S13073-019-0666-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hundal J, Kiwala S, McMichael J, Miller CA, Xia H, Wollam AT, Liu CJ, Zhao S, Feng YY, Graubert AP. et al. PVACtools: a computational toolkit to identify and visualize cancer neoantigens. Cancer Immunol Res. 2020;8(3):409–20. doi: 10.1158/2326-6066.CIR-19-0401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lybaert L, Lefever S, Fant B, Smits E, De Geest B, Breckpot K, Dirix L, Feldman SA, van Criekinge W, Thielemans K. et al. Challenges in neoantigen-directed therapeutics. Cancer Cell. 2023;41(1):15–40. doi: 10.1016/J.CCELL.2022.10.013. [DOI] [PubMed] [Google Scholar]
- 41.Wells DK, van Buuren MM, Dang KK, Hubbard-Lucey VM, Sheehan KCF, Campbell KM, Lamb A, Ward JP, Sidney J, Blazquez AB. et al. Key parameters of tumor epitope immunogenicity revealed through a consortium approach improve neoantigen prediction. Cell. 2020;183(3):818–34.e13. doi: 10.1016/j.cell.2020.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, Jamal-Hanjani M, Wilson GA, Birkbak NJ, Hiley CT. et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science (80-). 2016;351(6280):1463–9. doi: 10.1126/science.aaf1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cuevas MVR, Hardy MP, Larouche JD, Apavaloaei A, Kina E, Vincent K, Gendron P, Laverdure JP, Durette C, Thibault P. et al. BamQuery: a proteogenomic tool to explore the immunopeptidome and prioritize actionable tumor antigens. Genome Biol. 2023;24(1). doi: 10.1186/S13059-023-03029-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liepe J, Marino F, Sidney J, Jeko A, Bunting DE, Sette A, Kloetzel PM, Stumpf MPH, Heck AJR, Mishto M. A large fraction of HLA class I ligands are proteasome-generated spliced peptides. Sci. 2016;354(6310):354–8. doi: 10.1126/SCIENCE.AAF4384. [DOI] [PubMed] [Google Scholar]
- 45.Falk K, Rötzschke O, Stevanovié S, Jung G, Rammensee HG. Allele-specific motifs revealed by sequencing of self-peptides eluted from MHC molecules. Nature. 1991;351(6324):290–6. doi: 10.1038/351290A0. [DOI] [PubMed] [Google Scholar]
- 46.Slingluff CL, Cox AL, Stover JM, Moore MM, Hunt DF, Engelhard VH. Cytotoxic T-lymphocyte response to autologous human squamous cell cancer of the lung: epitope reconstitution with peptides extracted from HLA-Aw68. Cancer Res. 1994;54:2731–7. [PubMed] [Google Scholar]
- 47.Shapiro IE, Bassani-Sternberg M. The impact of immunopeptidomics: from basic research to clinical implementation. Semin Immunol. 2023;66:101727. doi: 10.1016/J.SMIM.2023.101727. [DOI] [PubMed] [Google Scholar]
- 48.Olsson N, Heberling ML, Zhang L, Jhunjhunwala S, Phung QT, Lin S, Anania VG, Lill JR, Elias JE. An integrated genomic, proteomic, and immunopeptidomic approach to discover treatment-induced neoantigens. Front Immunol. 2021;12:12. doi: 10.3389/FIMMU.2021.662443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li X, Pak HS, Huber F, Michaux J, Taillandier-Coindard M, Altimiras ER, Bassani-Sternberg M. A microfluidics-enabled automated workflow of sample preparation for MS-based immunopeptidomics. Cell Reports Methods. 2023;3(6):100479. doi: 10.1016/J.CRMETH.2023.100479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stutzmann C, Peng J, Wu Z, Savoie C, Sirois I, Thibault P, Wheeler AR, Caron E. Unlocking the potential of microfluidics in mass spectrometry-based immunopeptidomics for tumor antigen discovery. Cell Reports Methods. 2023;3(6):100511. doi: 10.1016/J.CRMETH.2023.100511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Robbins PF, Lu YC, El-Gamil M, Li YF, Gross C, Gartner J, Lin JC, Teer JK, Cliften P, Tycksen E. et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med. 2013;19(6):747–52. doi: 10.1038/NM.3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Danilova L, Anagnostou V, Caushi JX, Sidhom JW, Guo H, Chan HY, Suri P, Tam A, Zhang J, Asmar ME. et al. The mutation-associated neoantigen functional expansion of specific T cells (MANAFEST) assay: a sensitive platform for monitoring antitumor immunity. Cancer Immunol Res. 2018;6(8):888–99. doi: 10.1158/2326-6066.CIR-18-0129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bentzen AK, Marquard AM, Lyngaa R, Saini SK, Ramskov S, Donia M, Such L, Furness AJS, McGranahan N, Rosenthal R. et al. Large-scale detection of antigen-specific T cells using peptide-MHC-I multimers labeled with DNA barcodes. Nat Biotechnol. 2016;34(10):1037–45. doi: 10.1038/NBT.3662. [DOI] [PubMed] [Google Scholar]
- 54.Tran E, Robbins PF, Rosenberg SA. “Final common pathway” of human cancer immunotherapy: targeting random somatic mutations. Nat Immunol. 2017;18(3):255–62. doi: 10.1038/NI.3682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lozano-Rabella M, Garcia-Garijo A, Palomero J, Yuste-Estevanez A, Erhard F, Farriol-Duran R, Martín-Liberal J, Ochoa-de-Olza M, Matos I, Gartner JJ. et al. Exploring the immunogenicity of noncanonical HLA-I tumor ligands identified through proteogenomics. Clin Cancer Res. 2023;29(12):2250–65. doi: 10.1158/1078-0432.CCR-22-3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Repáraz D, Ruiz M, Llopiz D, Silva L, Vercher E, Aparicio B, Egea J, Tamayo-Uria I, Hervás-Stubbs S, García-Balduz J. et al. Neoantigens as potential vaccines in hepatocellular carcinoma. J Immunother Cancer. 2022;10(2):e003978. doi: 10.1136/JITC-2021-003978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Aparicio B, Repáraz D, Ruiz M, Llopiz D, Silva L, Theunissen P, Tamayo I, Smerdou C, Igea A, Santisteban M. et al. Identification of HLA class I-restricted immunogenic neoantigens in triple negative breast cancer. Front Immunol; 2022;13:985886. doi: 10.3389/fimmu.2022.985886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jardim DL, Goodman A, de Melo Gagliato D, Kurzrock R. The Challenges of Tumor Mutational Burden as an Immunotherapy Biomarker. Cancer Cell. 2021;39(2):154–73. doi: 10.1016/j.ccell.2020.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McGrail DJ, Pilié PG, Rashid NU, Voorwerk L, Slagter M, Kok M, Jonasch E, Khasraw M, Heimberger AB, Lim B. et al. High tumor mutation burden fails to predict immune checkpoint blockade response across all cancer types. Ann Oncol. 2021;32(5):661–72. doi: 10.1016/j.annonc.2021.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Montesion M, Murugesan K, Jin DX, Sharaf R, Sanchez N, Guria A, Minker M, Li G, Fisher V, Sokol ES. et al. Somatic HLA class I loss is a widespread mechanism of immune evasion which refines the use of tumor mutational burden as a biomarker of checkpoint inhibitor response. Cancer Discov. 2021;11(2):282–92. doi: 10.1158/2159-8290.CD-20-0672. [DOI] [PubMed] [Google Scholar]
- 61.Koopman LA, Corver WE, VanDer Slik AR, Giphart MJ, Fleuren GJ. Multiple genetic alterations cause frequent and heterogeneous human histocompatibility leukocyte antigen class I loss in cervical cancer. J Exp Med. 2000;191(6):961–75. doi: 10.1084/JEM.191.6.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McGranahan N, Rosenthal R, Hiley CT, Rowan AJ, Watkins TBK, Wilson GA, Birkbak NJ, Veeriah S, Van Loo P, Herrero J. et al. Allele-specific HLA loss and immune escape in lung cancer evolution. Cell. 2017;171(6):1259–71.e11. doi: 10.1016/J.CELL.2017.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shukla SA, Rooney MS, Rajasagi M, Tiao G, Dixon PM, Lawrence MS, Stevens J, Lane WJ, Dellagatta JL, Steelman S. et al. Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes. Nat Biotechnol. 2015;33(11):1152–8. doi: 10.1038/NBT.3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature. 2015;523(7559):231–5. doi: 10.1038/nature14404. [DOI] [PubMed] [Google Scholar]
- 65.Spranger S, Dai D, Horton B, Gajewski TF. Tumor-residing batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell. 2017;31(5):711–23.e4. doi: 10.1016/J.CCELL.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Luke JJ, Bao R, Sweis RF, Spranger S, Gajewski TF. WNT/β-catenin pathway activation correlates with immune exclusion across human cancers. Clin Cancer Res. 2019;25(10):3074–83. doi: 10.1158/1078-0432.CCR-18-1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ishino T, Kawashima S, Tanji E, Ueno T, Ueda Y, Ogasawara S, Sato K, Mano H, Ishihara S, Kato N. et al. Somatic mutations can induce a noninflamed tumour microenvironment via their original gene functions, despite deriving neoantigens. Br J Cancer. 2023;128(6):1166–75. doi: 10.1038/S41416-023-02165-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L. et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med. 2016;375(9):819–29. doi: 10.1056/NEJMOA1604958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, Grasso CS, Hugo W, Sandoval S, Torrejon DY. et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov. 2017;7(2):188–201. doi: 10.1158/2159-8290.CD-16-1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sucker A, Zhao F, Pieper N, Heeke C, Maltaner R, Stadtler N, Real B, Bielefeld N, Howe S, Weide B. et al. Acquired IFNγ resistance impairs anti-tumor immunity and gives rise to T-cell-resistant melanoma lesions. Nat Commun. 2017;8(1). doi: 10.1038/NCOMMS15440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, Xu C, McKenzie JA, Zhang C, Liang X. et al. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov. 2016;6(2):202–16. doi: 10.1158/2159-8290.CD-15-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kumagai S, Togashi Y, Sakai C, Kawazoe A, Kawazu M, Ueno T, Sato E, Kuwata T, Kinoshita T, Yamamoto M. et al. An oncogenic alteration creates a microenvironment that promotes tumor progression by conferring a metabolic advantage to regulatory T cells. Immunity. 2020;53(1):187–203.e8. doi: 10.1016/J.IMMUNI.2020.06.016. [DOI] [PubMed] [Google Scholar]
- 73.Peña CG, Nakada Y, Saatcioglu HD, Aloisio GM, Cuevas I, Zhang S, Miller DS, Lea JS, Wong KK, DeBerardinis RJ. et al. LKB1 loss promotes endometrial cancer progression via CCL2-dependent macrophage recruitment. J Clin Invest. 2015;125(11):4063–76. doi: 10.1172/JCI82152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Koyama S, Akbay EA, Li YY, Aref AR, Skoulidis F, Herter-Sprie GS, Buczkowski KA, Liu Y, Awad MM, Denning WL. et al. STK11/LKB1 deficiency promotes neutrophil recruitment and proinflammatory cytokine production to suppress T-cell activity in the lung tumor microenvironment. Cancer Res. 2016;76(5):999–1008. doi: 10.1158/0008-5472.CAN-15-1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Quigley D, Silwal-Pandit L, Dannenfelser R, Langerød A, Vollan HKM, Vaske C, Siegel JU, Troyanskaya O, Chin SF, Caldas C. et al. Lymphocyte invasion in IC10/Basal-like breast tumors is associated with wild-type TP53. Mol Cancer Res. 2015;13(3):493–501. doi: 10.1158/1541-7786.MCR-14-0387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Iannello A, Thompson TW, Ardolino M, Lowe SW, Raulet DH. p53-dependent chemokine production by senescent tumor cells supports NKG2D-dependent tumor elimination by natural killer cells. J Exp Med. 2013;210(10):2057–69. doi: 10.1084/JEM.20130783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC. et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462(7274):739–44. doi: 10.1038/NATURE08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kohanbash G, Carrera DA, Shrivastav S, Ahn BJ, Jahan N, Mazor T, Chheda ZS, Downey KM, Watchmaker PB, Beppler C. et al. Isocitrate dehydrogenase mutations suppress STAT1 and CD8+ T cell accumulation in gliomas. J Clin Invest. 2017;127(4):1425–37. doi: 10.1172/JCI90644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ilyas S, Yang JC. Landscape of tumor antigens in T cell immunotherapy. J Immunol. 2015;195(11):5117–22. doi: 10.4049/JIMMUNOL.1501657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vigneron N. Human tumor antigens and cancer immunotherapy. Biomed Res Int. 2015;2015:1–17. doi: 10.1155/2015/948501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science (80-). 1996;271(5256):1734–6. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- 82.Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A. 2002;99(19):12293–7. doi: 10.1073/pnas.192461099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sharma P, Siddiqui BA, Anandhan S, Yadav SS, Subudhi SK, Gao J, Goswami S, Allison JP. The next decade of immune checkpoint therapy. Cancer Discov. 2021;11(4):838–57. doi: 10.1158/2159-8290.CD-20-1680. [DOI] [PubMed] [Google Scholar]
- 84.Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS. et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371(23):2189–99. doi: 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, Sucker A, Hillen U, Foppen MHG, Goldinger SM. et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Sci. 2015;350(6257):207–11. doi: 10.1126/SCIENCE.AAD0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS. et al. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science (80-). 2015;348(6230):124–8. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Diaz LA, Shiu KK, Kim TW, Jensen BV, Jensen LH, Punt C, Smith D, Garcia-Carbonero R, Benavides M, Gibbs P. et al. Pembrolizumab versus chemotherapy for microsatellite instability-high or mismatch repair-deficient metastatic colorectal cancer (KEYNOTE-177): final analysis of a randomised, open-label, phase 3 study. Lancet Oncol. 2022;23(5):659–70. doi: 10.1016/S1470-2045(22)00197-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D. et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372(26):2509–20. doi: 10.1056/NEJMOA1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yarchoan M, Hopkins A, Jaffee EM. Tumor mutational burden and response rate to PD-1 inhibition. N Engl J Med. 2017;377(25):2500–1. doi: 10.1056/NEJMc1713444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zou XL, Li XB, Ke H, Zhang GY, Tang Q, Yuan J, Zhou CJ, Zhang JL, Zhang R, Chen WY. Prognostic value of neoantigen load in immune checkpoint inhibitor therapy for cancer. Front Immunol. 2021;12:12. doi: 10.3389/FIMMU.2021.689076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Florou V, Floudas CS, Maoz A, Naqash AR, Norton C, Tan AC, Sokol ES, Frampton G, Soares HP, Puri S. et al. Real-world pan-cancer landscape of frameshift mutations and their role in predicting responses to immune checkpoint inhibitors in cancers with low tumor mutational burden. J Immunother Cancer. 2023;11(8):e007440. doi: 10.1136/JITC-2023-007440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.McGrail DJ, Pilié PG, Rashid NU, Voorwerk L, Slagter M, Kok M, Jonasch E, Khasraw M, Heimberger AB, Lim B. et al. High tumor mutation burden fails to predict immune checkpoint blockade response across all cancer types. Ann Oncol Off J Eur Soc Med Oncol. 2021;32(5):661–72. doi: 10.1016/J.ANNONC.2021.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Puig-Saus C, Sennino B, Peng S, Wang CL, Pan Z, Yuen B, Purandare B, An D, Quach BB, Nguyen D. et al. Neoantigen-targeted CD8+ T cell responses with PD-1 blockade therapy. Nature. 2023;615(7953):697–704. doi: 10.1038/S41586-023-05787-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Holm JS, Funt SA, Borch A, Munk KK, Bjerregaard AM, Reading JL, Maher C, Regazzi A, Wong P, Al-Ahmadie H. et al. Neoantigen-specific CD8 T cell responses in the peripheral blood following PD-L1 blockade might predict therapy outcome in metastatic urothelial carcinoma. Nat Commun. 2022;13(1). doi: 10.1038/S41467-022-29342-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Goff SL, Dudley ME, Citrin DE, Somerville RP, Wunderlich JR, Danforth DN, Zlott DA, Yang JC, Sherry RM, Kammula US. et al. Randomized, prospective evaluation comparing intensity of lymphodepletion before adoptive transfer of tumor-infiltrating lymphocytes for patients with metastatic melanoma. J Clin Oncol. 2016;34(20):2389–97. doi: 10.1200/JCO.2016.66.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lu Y-C, Yao X, Li YF, El-Gamil M, Dudley ME, Yang JC, Almeida JR, Douek DC, Samuels Y, Rosenberg SA. et al. Mutated PPP1R3B is recognized by T cells used to treat a melanoma patient who experienced a durable complete tumor regression. J Immunol. 2013;190(12):6034–42. doi: 10.4049/JIMMUNOL.1202830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lu YC, Yao X, Crystal JS, Li YF, El-Gamil M, Gross C, Davis L, Dudley ME, Yang JC, Samuels Y. et al. Efficient identification of mutated cancer antigens recognized by T cells associated with durable tumor regressions. Clin Cancer Res. 2014;20(13):3401–10. doi: 10.1158/1078-0432.CCR-14-0433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Prickett TD, Crystal JS, Cohen CJ, Pasetto A, Parkhurst MR, Gartner JJ, Yao X, Wang R, Gros A, Li YF. et al. Durable complete response from metastatic melanoma after transfer of autologous T cells recognizing 10 mutated tumor antigens. Cancer Immunol Res. 2016;4(8):669–78. doi: 10.1158/2326-6066.CIR-15-0215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhou J, Dudley ME, Rosenberg SA, Robbins PF. Persistence of multiple tumor-specific T-cell clones is associated with complete tumor regression in a melanoma patient receiving adoptive cell transfer therapy. J Immunother (1991). 2005;28(1):53–62. doi: 10.1097/00002371-200501000-00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yannelli JR, Hyatt C, McConnell S, Hines K, Jacknin L, Parker L, Sanders M, Rosenberg SA. Growth of tumor-infiltrating lymphocytes from human solid cancers: summary of a 5-year experience. Int J Cancer. 1996;65(4):413–21. doi: 10.1002/(SICI)1097-0215(19960208)65:4<413:AID-IJC3>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 101.Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Sci. 2015;348(6230):62–8. doi: 10.1126/SCIENCE.AAA4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, Wunderlich JR, Somerville RP, Hogan K, Hinrichs CS. et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Sci. 2014;344(6184):641–5. doi: 10.1126/SCIENCE.1251102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zacharakis N, Chinnasamy H, Black M, Xu H, Lu YC, Zheng Z, Pasetto A, Langhan M, Shelton T, Prickett T. et al. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat Med. 2018;24(6):724–30. doi: 10.1038/S41591-018-0040-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kim SP, Vale NR, Zacharakis N, Krishna S, Yu Z, Gasmi B, Gartner JJ, Sindiri S, Malekzadeh P, Deniger DC. et al. Adoptive cellular therapy with autologous tumor-infiltrating lymphocytes and T-cell receptor-engineered T cells targeting common p53 neoantigens in human solid tumors. Cancer Immunol Res. 2022;10(8):932–46. doi: 10.1158/2326-6066.CIR-22-0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Feldman SA, Assadipour Y, Kriley I, Goff SL, Rosenberg SA. Adoptive cell therapy–tumor-infiltrating lymphocytes, T-Cell receptors, and chimeric antigen receptors. Semin Oncol. 2015;42(4):626–39. doi: 10.1053/J.SEMINONCOL.2015.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, Dudley ME, Feldman SA, Yang JC, Sherry RM. et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother (1991). 2013;36(2):133–51. doi: 10.1097/CJI.0b013e3182829903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Leidner R, Sanjuan Silva N, Huang H, Sprott D, Zheng C, Shih Y-P, Leung A, Payne R, Sutcliffe K, Cramer J. et al. Neoantigen T-Cell receptor gene therapy in pancreatic cancer. N Engl J Med. 2022;386(22):2112–9. doi: 10.1056/NEJMOA2119662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wang QJ, Yu Z, Griffith K, Hanada KI, Restifo NP, Yang JC. Identification of T-cell receptors targeting KRAS-Mutated human tumors. Cancer Immunol Res. 2016;4(3):204–14. doi: 10.1158/2326-6066.CIR-15-0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bear AS, Blanchard T, Cesare J, Ford MJ, Richman LP, Xu C, Baroja ML, McCuaig S, Costeas C, Gabunia K. et al. Biochemical and functional characterization of mutant KRAS epitopes validates this oncoprotein for immunological targeting. Nat Commun. 2021;12(1). doi: 10.1038/S41467-021-24562-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Cafri G, Yossef R, Pasetto A, Deniger DC, Lu YC, Parkhurst M, Gartner JJ, Jia L, Ray S, Ngo LT. et al. Memory T cells targeting oncogenic mutations detected in peripheral blood of epithelial cancer patients. Nat Commun. 2019;10(1). doi: 10.1038/S41467-019-08304-Z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Monach PA, SC M, Siegel CT, Schreiber H. A unique tumor antigen produced by a single amino acid substitution. Immunity. 1995;2(1):45–59. doi: 10.1016/1074-7613(95)90078-0. [DOI] [PubMed] [Google Scholar]
- 112.Matsutake T, Srivastava PK. The immunoprotective MHC II epitope of a chemically induced tumor harbors a unique mutation in a ribosomal protein. Proc Natl Acad Sci U S A. 2001;98(7):3992–7. doi: 10.1073/PNAS.071523398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Castle JC, Kreiter S, Diekmann J, Lower M, van de Roemer N, de Graaf J, Selmi A, Diken M, Boegel S, Paret C. et al. Exploiting the mutanome for tumor vaccination. Cancer Res. 2012;72(5):1081–91. doi: 10.1158/0008-5472.CAN-11-3722. [DOI] [PubMed] [Google Scholar]
- 114.Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, Ivanova Y, Hundal J, Arthur CD, Krebber WJ. et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014;515(7528):577–81. doi: 10.1038/NATURE13988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Duan F, Duitama J, Al Seesi S, Ayres CM, Corcelli SA, Pawashe AP, Blanchard T, McMahon D, Sidney J, Sette A. et al. Genomic and bioinformatic profiling of mutational neoepitopes reveals new rules to predict anticancer immunogenicity. J Exp Med. 2014;211(11):2231–48. doi: 10.1084/jem.20141308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, Franci C, Cheung TK, Fritsche J, Weinschenk T. et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature. 2014;515(7528):572–6. doi: 10.1038/nature14001. [DOI] [PubMed] [Google Scholar]
- 117.Kreiter S, Vormehr M, van de Roemer N, Diken M, Lower M, Diekmann J, Boegel S, Schrors B, Vascotto F, Castle JC. et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature. 2015;520(7549):692–6. doi: 10.1038/nature14426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Niemi JVL, Sokolov AV, Schiöth HB. Neoantigen vaccines; clinical trials, classes, indications, adjuvants and combinatorial treatments. Cancers Basel. 2022;14(20):5163. doi: 10.3390/cancers14205163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, Ly A, Lie W-R, Hildebrand WH, Mardis ER. et al. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Sci. 2015;348(6236):803–8. doi: 10.1126/science.aaa3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, Zhang W, Luoma A, Giobbie-Hurder A, Peter L. et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547(7662):217–21. doi: 10.1038/nature22991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Löwer M, Bukur V, Tadmor AD, Luxemburger U, Schrörs B. et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 2017;547(7662):222–6. doi: 10.1038/nature23003. [DOI] [PubMed] [Google Scholar]
- 122.Hilf N, Kuttruff-Coqui S, Frenzel K, Bukur V, Stevanović S, Gouttefangeas C, Platten M, Tabatabai G, Dutoit V, van der Burg SH. et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature. 2019;565(7738):240–5. doi: 10.1038/S41586-018-0810-Y. [DOI] [PubMed] [Google Scholar]
- 123.Keskin DB, Anandappa AJ, Sun J, Tirosh I, Mathewson ND, Li S, Oliveira G, Giobbie-Hurder A, Felt K, Gjini E. et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature. 2019;565(7738):234–9. doi: 10.1038/s41586-018-0792-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Cafri G, Gartner JJ, Zaks T, Hopson K, Levin N, Paria BC, Parkhurst MR, Yossef R, Lowery FJ, Jafferji MS. et al. mRNA vaccine-induced neoantigen-specific T cell immunity in patients with gastrointestinal cancer. J Clin Invest. 2020;130(11):5976–88. doi: 10.1172/JCI134915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ding Z, Li Q, Zhang R, Xie L, Shu Y, Gao S, Wang P, Su X, Qin Y, Wang Y. et al. Personalized neoantigen pulsed dendritic cell vaccine for advanced lung cancer. Signal Transduct Target Ther. 2021. 61;6(1):1–12. doi: 10.1038/s41392-020-00448-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Chen Z, Zhang S, Han N, Jiang J, Xu Y, Ma D, Lu L, Guo X, Qiu M, Huang Q. et al. A neoantigen-based peptide vaccine for patients with advanced pancreatic cancer refractory to standard treatment. Front Immunol. 2021;12. doi: 10.3389/FIMMU.2021.691605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Cai Z, Su X, Qiu L, Li Z, Li X, Dong X, Wei F, Zhou Y, Luo L, Chen G. et al. Personalized neoantigen vaccine prevents postoperative recurrence in hepatocellular carcinoma patients with vascular invasion. Mol Cancer. 2021;20(1):164. doi: 10.1186/S12943-021-01467-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Fang Y, Mo F, Shou J, Wang H, Luo K, Zhang S, Han N, Li H, Ye S, Zhou Z. et al. A pan-cancer clinical study of personalized neoantigen vaccine monotherapy in treating patients with various types of advanced solid tumors. Clin Cancer Res. 2020;26(17):4511–20. doi: 10.1158/1078-0432.CCR-19-2881. [DOI] [PubMed] [Google Scholar]
- 129.Kloor M, Reuschenbach M, Pauligk C, Karbach J, Rafiyan MR, Al-Batran SE, Tariverdian M, Jäger E, von Knebel Doeberitz M. A frameshift peptide neoantigen-based vaccine for mismatch repair-deficient cancers: a phase I/IIa clinical trial. Clin Cancer Res. 2020;26(17):4503–10. doi: 10.1158/1078-0432.CCR-19-3517. [DOI] [PubMed] [Google Scholar]
- 130.Leoni G, D’Alise AM, Cotugno G, Langone F, Garzia I, de Lucia M, Fichera I, Vitale R, Bignone V, Tucci FG. et al. A genetic vaccine encoding shared cancer neoantigens to treat tumors with microsatellite instability. Cancer Res. 2020;80(18):3972–82. doi: 10.1158/0008-5472.CAN-20-1072. [DOI] [PubMed] [Google Scholar]
- 131.D’Alise AM, Brasu N, De Intinis C, Leoni G, Russo V, Langone F, Baev D, Micarelli E, Petiti L, Picelli S. et al. Adenoviral-based vaccine promotes neoantigen-specific CD8+ T cell stemness and tumor rejection. Sci Transl Med. 2022;14(657). doi: 10.1126/scitranslmed.abo7604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Mueller S, Taitt JM, Villanueva-Meyer JE, Bonner ER, Nejo T, Lulla RR, Goldman S, Banerjee A, Chi SN, Whipple NS. et al. Mass cytometry detects H3.3K27M-specific vaccine responses in diffuse midline glioma. J Clin Invest. 2020;130(12):6325–37. doi: 10.1172/JCI140378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ott PA, Hu-Lieskovan S, Chmielowski B, Govindan R, Naing A, Bhardwaj N, Margolin K, Awad MM, Hellmann MD, Lin JJ. et al. A phase IB trial of personalized neoantigen therapy plus anti-PD-1 in patients with advanced melanoma, non-small cell lung cancer, or bladder cancer. Cell. 2020;183(2):347–62.e24. doi: 10.1016/j.cell.2020.08.053. [DOI] [PubMed] [Google Scholar]
- 134.Khattak A, Carlino M, Meniawy T, Ansstas G, Medina T, Taylor MH, Kim KB, McKean M, Long GV, Sullivan R. et al. A personalized cancer vaccine, mRNA-4157, combined with pembrolizumab versus pembrolizumab in patients with resected high-risk melanoma: efficacy and safety results from the randomized, open-label phase 2 mRNA-4157-P201/Keynote-942 trial. AACR Annu Meet. 2023:CT001. doi: 10.1158/1538-7445.AM2023-CT001 [DOI] [Google Scholar]
- 135.Rojas LA, Sethna Z, Soares KC, Olcese C, Pang N, Patterson E, Lihm J, Ceglia N, Guasp P, Chu A. et al. Personalized RNA neoantigen vaccines stimulate T cells in pancreatic cancer. Nature. 2023;618(7963):144–50. doi: 10.1038/S41586-023-06063-Y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Fritsch EF, Burkhardt UE, Hacohen N, Wu CJ. Personal neoantigen cancer vaccines: a road not fully paved. Cancer Immunol Res. 2020;8(12):1465–9. doi: 10.1158/2326-6066.CIR-20-0526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wu J, Chen W, Zhou Y, Chi Y, Hua X, Wu J, Gu X, Chen S, Zhou Z. Tsnadb v2.0: the updated version of tumor-specific neoantigen database. Genomics, Proteomics Bioinform. 2022. doi: 10.1016/J.GPB.2022.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Nagel R, Pataskar A, Champagne J, Agami R. Boosting antitumor immunity with an expanded neoepitope landscape. Cancer Res. 2022;82(20):3637–49. doi: 10.1158/0008-5472.CAN-22-1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Shah NM, Jang HJ, Liang Y, Maeng JH, Tzeng SC, Wu A, Basri NL, Qu X, Fan C, Li A. et al. Pan-cancer analysis identifies tumor-specific antigens derived from transposable elements. Nat Genet. 2023;55(4):631–9. doi: 10.1038/S41588-023-01349-3. [DOI] [PubMed] [Google Scholar]
- 140.Shiraishi Y, Kataoka K, Chiba K, Okada A, Kogure Y, Tanaka H, Ogawa S, Miyano S. A comprehensive characterization of cis-acting splicing-associated variants in human cancer. Genome Res. 2018;28(8):1111–25. doi: 10.1101/gr.231951.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Jung H, Lee D, Lee J, Park D, Kim YJ, Park WY, Hong D, Park PJ, Lee E. Intron retention is a widespread mechanism of tumor-suppressor inactivation. Nat Genet. 2015;47(11):1242–8. doi: 10.1038/NG.3414. [DOI] [PubMed] [Google Scholar]
- 142.Koh CM, Bezzi M, Low DHP, Ang WX, Teo SX, Gay FPH, Al-Haddawi M, Tan SY, Osato M, Sabò A. et al. MYC regulates the core pre-mRNA splicing machinery as an essential step in lymphomagenesis. Nature. 2015;523(7558):96–100. doi: 10.1038/NATURE14351. [DOI] [PubMed] [Google Scholar]
- 143.Hoyos LE, Abdel-Wahab O. Cancer-specific splicing changes and the potential for splicing-derived neoantigens. Cancer Cell. 2018;34(2):181–3. doi: 10.1016/J.CCELL.2018.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ehx G, Larouche JD, Durette C, Laverdure JP, Hesnard L, Vincent K, Hardy MP, Thériault C, Rulleau C, Lanoix J. et al. Atypical acute myeloid leukemia-specific transcripts generate shared and immunogenic MHC class-I-associated epitopes. Immunity. 2021;54(4):737–52.e10. doi: 10.1016/j.immuni.2021.03.001. [DOI] [PubMed] [Google Scholar]
- 145.O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, Martinez-Lage M, Brem S, Maloney E, Shen A. et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. 2017;9(399). doi: 10.1126/SCITRANSLMED.AAA0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Goff SL, Morgan RA, Yang JC, Sherry RM, Robbins PF, Restifo NP, Feldman SA, Lu YC, Lu L, Zheng Z. et al. Pilot trial of adoptive transfer of chimeric antigen receptor-transduced T cells targeting EGFRvIII in patients with glioblastoma. J Immunother (1991). 2019;42(4):126–35. doi: 10.1097/CJI.0000000000000260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wang TY, Liu Q, Ren Y, Alam SK, Wang L, Zhu Z, Hoeppner LH, Dehm SM, Cao Q, Yang R. A pan-cancer transcriptome analysis of exitron splicing identifies novel cancer driver genes and neoepitopes. Mol Cell. 2021;81(10):2246–60.e12. doi: 10.1016/J.MOLCEL.2021.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kahles A, Van LK, Toussaint NC, Hüser M, Stark SG, Sachsenberg T, Stegle O, Kohlbacher O, Sander C, Caesar-Johnson SJ. et al. Comprehensive analysis of alternative splicing across tumors from 8,705 patients. Cancer Cell. 2018;34(2):211–24.e6. doi: 10.1016/J.CCELL.2018.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Bowling EA, Wang JH, Gong F, Wu W, Neill NJ, Kim IS, Tyagi S, Orellana M, Kurley SJ, Dominguez-Vidaña R. et al. Spliceosome-targeted therapies trigger an antiviral immune response in triple-negative breast cancer. Cell. 2021;184(2):384–403.e21. doi: 10.1016/J.CELL.2020.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lu SX, De Neef E, Thomas JD, Sabio E, Rousseau B, Gigoux M, Knorr DA, Greenbaum B, Elhanati Y, Hogg SJ. et al. Pharmacologic modulation of RNA splicing enhances anti-tumor immunity. Cell. 2021;184(15):4032–47.e31. doi: 10.1016/J.CELL.2021.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Weinstein-Marom H, Hendel L, Laron EA, Sharabi-Nov A, Margalit A, Gross G. MHC-I presentation of peptides derived from intact protein products of the pioneer round of translation. FASEB J. 2019;33(10):11458–68. doi: 10.1096/FJ.201802717RRR. [DOI] [PubMed] [Google Scholar]
- 152.Becker JP, Helm D, Rettel M, Stein F, Hernandez-Sanchez A, Urban K, Gebert J, Kloor M, Neu-Yilik G, von Knebel Doeberitz M. et al. NMD inhibition by 5-azacytidine augments presentation of immunogenic frameshift-derived neoepitopes. iScience. 2021;24(4):102389. doi: 10.1016/J.ISCI.2021.102389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Singh I, Lee SH, Sperling AS, Samur MK, Tai YT, Fulciniti M, Munshi NC, Mayr C, Leslie CS. Widespread intronic polyadenylation diversifies immune cell transcriptomes. Nat Commun. 2018;9(1). doi: 10.1038/S41467-018-04112-Z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Avesson L, Barry G. The emerging role of RNA and DNA editing in cancer. Biochim Biophys Acta. 2014;1845(2):308–16. doi: 10.1016/J.BBCAN.2014.03.001. [DOI] [PubMed] [Google Scholar]
- 155.Zhang M, Fritsche J, Roszik J, Williams LJ, Peng X, Chiu Y, Tsou CC, Hoffgaard F, Goldfinger V, Schoor O. et al. RNA editing derived epitopes function as cancer antigens to elicit immune responses. Nat Commun. 2018;9(1). doi: 10.1038/S41467-018-06405-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Garitano-Trojaola A, Agirre X, Prósper F, Fortes P. Long non-coding RNAs in haematological malignancies. Int J Mol Sci. 2013;14(8):15386–422. doi: 10.3390/IJMS140815386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Unfried JP, Sangro P, Prats-Mari L, Sangro B, Fortes P. The landscape of lncRNAs in hepatocellular carcinoma: a translational perspective. Cancers Basel. 2021;13(11):2651. doi: 10.3390/CANCERS13112651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Unfried JP, Fortes P. SMIM30, a tiny protein with a big role in liver cancer. J Hepatol. 2020;73(5):1010–12. doi: 10.1016/J.JHEP.2020.07.015. [DOI] [PubMed] [Google Scholar]
- 159.Mudge JM, Ruiz-Orera J, Prensner JR, Brunet MA, Calvet F, Jungreis I, Gonzalez JM, Magrane M, Martinez TF, Schulz JF. et al. Standardized annotation of translated open reading frames. Nat Biotechnol. 2022;40(7):994–9. doi: 10.1038/S41587-022-01369-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Sandmann CL, Schulz JF, Ruiz-Orera J, Kirchner M, Ziehm M, Adami E, Marczenke M, Christ A, Liebe N, Greiner J. et al. Evolutionary origins and interactomes of human, young microproteins and small peptides translated from short open reading frames. Mol Cell. 2023;83(6):994–1011.e18. doi: 10.1016/J.MOLCEL.2023.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Broeils LA, Ruiz-Orera J, Snel B, Hubner N, van Heesch S. Evolution and implications of de novo genes in humans. Nat Ecol Evol. 2023;7(6):804–15. doi: 10.1038/S41559-023-02014-Y. [DOI] [PubMed] [Google Scholar]
- 162.Ruiz Cuevas MV, Hardy MP, Hollý J, É B, Durette C, Courcelles M, Lanoix J, Côté C, Staudt LM, Lemieux S. et al. Most non-canonical proteins uniquely populate the proteome or immunopeptidome. Cell Rep. 2021;34(10):108815. doi: 10.1016/J.CELREP.2021.108815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Laumont CM, Vincent K, Hesnard L, Audemard É, Bonneil É, Laverdure JP, Gendron P, Courcelles M, Hardy MP, Côté C. et al. Noncoding regions are the main source of targetable tumor-specific antigens. Sci Transl Med. 2018;10(470). doi: 10.1126/SCITRANSLMED.AAU5516. [DOI] [PubMed] [Google Scholar]
- 164.Chothani SP, Adami E, Widjaja AA, Langley SR, Viswanathan S, Pua CJ, Zhihao NT, Harmston N, D’Agostino G, Whiffin N. et al. A high-resolution map of human RNA translation. Mol Cell. 2022;82(15):2885–99.e8. doi: 10.1016/J.MOLCEL.2022.06.023. [DOI] [PubMed] [Google Scholar]
- 165.Kesner JS, Chen Z, Shi P, Aparicio AO, Murphy MR, Guo Y, Trehan A, Lipponen JE, Recinos Y, Myeku N. et al. Noncoding translation mitigation. Nature. 2023;617(7960):395–402. doi: 10.1038/S41586-023-05946-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Laumont CM, Daouda T, Laverdure JP, É B, Caron-Lizotte O, Hardy MP, Granados DP, Durette C, Lemieux S, Thibault P. et al. Global proteogenomic analysis of human MHC class I-associated peptides derived from non-canonical reading frames. Nat Commun. 2016;7(1). doi: 10.1038/NCOMMS10238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Chong C, Müller M, Pak HS, Harnett D, Huber F, Grun D, Leleu M, Auger A, Arnaud M, Stevenson BJ. et al. Integrated proteogenomic deep sequencing and analytics accurately identify non-canonical peptides in tumor immunopeptidomes. Nat Commun. 2020;11(1). doi: 10.1038/S41467-020-14968-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Erhard F, Dölken L, Schilling B, Schlosser A. Identification of the cryptic HLA-I immunopeptidome. Cancer Immunol Res. 2020;8(8):1018–26. doi: 10.1158/2326-6066.CIR-19-0886. [DOI] [PubMed] [Google Scholar]
- 169.Guilloy N, Brunet MA, Leblanc S, Jacques JF, Hardy MP, Ehx G, Lanoix J, Thibault P, Perreault C, Roucou X. OpenCustomDB: integration of unannotated open reading frames and genetic variants to generate more comprehensive customized protein databases. J Proteome Res. 2023;22(5):1492–500. doi: 10.1021/acs.jproteome.3c00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Prensner JR, Abelin JG, Kok LW, Clauser KR, Mudge JM, Ruiz-Orera J, Bassani-Sternberg M, Moritz RL, Deutsch EW, van Heesch S. What can ribo-seq, immunopeptidomics, and Proteomics tell us about the noncanonical proteome? Mol Cell Proteom: MCP. 2023;22(9):100631. doi: 10.1016/J.MCPRO.2023.100631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Marlin E, Viu-Idocin C, Arrasate M, Aragón T. The role and therapeutic potential of the integrated stress response in amyotrophic lateral sclerosis. Int J Mol Sci. 2022;23(14):7823. doi: 10.3390/IJMS23147823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Bartok O, Pataskar A, Nagel R, Laos M, Goldfarb E, Hayoun D, Levy R, Körner PR, Kreuger IZM, Champagne J. et al. Anti-tumour immunity induces aberrant peptide presentation in melanoma. Nature. 2021;590(7845):332–7. doi: 10.1038/S41586-020-03054-1. [DOI] [PubMed] [Google Scholar]
- 173.Champagne J, Pataskar A, Blommaert N, Nagel R, Wernaart D, Ramalho S, Kenski J, Bleijerveld OB, Zaal EA, Berkers CR. et al. Oncogene-dependent sloppiness in mRNA translation. Mol Cell. 2021;81(22):4709–21.e9. doi: 10.1016/J.MOLCEL.2021.09.002. [DOI] [PubMed] [Google Scholar]
- 174.Pataskar A, Champagne J, Nagel R, Kenski J, Laos M, Michaux J, Pak HS, Bleijerveld OB, Mordente K, Navarro JM. et al. Tryptophan depletion results in tryptophan-to-phenylalanine substitutants. Nature. 2022;603(7902):721–7. doi: 10.1038/S41586-022-04499-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Mohammed F, Cobbold M, Zarling AL, Salim M, Barrett-Wilt GA, Shabanowitz J, Hunt DF, Engelhard VH, Willcox BE. Phosphorylation-dependent interaction between antigenic peptides and MHC class I: a molecular basis for the presentation of transformed self. Nat Immunol. 2008;9(11):1236–43. doi: 10.1038/NI.1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Zarling AL, Polefrone JM, Evans AM, Mikesh LM, Shabanowitz J, Lewis ST, Engelhard VH, Hunt DF. Identification of class I MHC-associated phosphopeptides as targets for cancer immunotherapy. Proc Natl Acad Sci U S A. 2006;103(40):14889–94. doi: 10.1073/PNAS.0604045103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Meyer VS, Drews O, Günder M, Hennenlotter J, Rammensee HG, Stevanovic S. Identification of natural MHC class II presented phosphopeptides and tumor-derived MHC class I phospholigands. J Proteome Res. 2009;8(7):3666–74. doi: 10.1021/PR800937K. [DOI] [PubMed] [Google Scholar]
- 178.Petersen J, Wurzbacher SJ, Williamson NA, Ramarathinam SH, Reid HH, Nair AKN, Zhao AY, Nastovska R, Rudge G, Rossjohn J. et al. Phosphorylated self-peptides alter human leukocyte antigen class I-restricted antigen presentation and generate tumor-specific epitopes. Proc Natl Acad Sci U S A. 2009;106(8):2776–81. doi: 10.1073/PNAS.0812901106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Hanada KI, Yewdell JW, Yang JC. Immune recognition of a human renal cancer antigen through post-translational protein splicing. Nature. 2004;427(6971):252–6. doi: 10.1038/NATURE02240. [DOI] [PubMed] [Google Scholar]
- 180.Vigneron N, Stroobant V, Chapiro J, Ooms A, Degiovanni G, Morel S, Van Der Bruggen P, Boon T, Van Den Eynde BJ. An antigenic peptide produced by peptide splicing in the proteasome. Sci. 2004;304(5670):587–90. doi: 10.1126/SCIENCE.1095522. [DOI] [PubMed] [Google Scholar]
- 181.Groettrup M, Kirk CJ, Basler M. Proteasomes in immune cells: more than peptide producers? Nat Rev Immunol. 2010;10(1):73–8. doi: 10.1038/NRI2687. [DOI] [PubMed] [Google Scholar]
- 182.Michaux A, Larrieu P, Stroobant V, Fonteneau J-F, Jotereau F, Van den Eynde BJ, Moreau-Aubry A, Vigneron N. A spliced antigenic peptide comprising a single spliced amino acid is produced in the proteasome by reverse splicing of a longer peptide fragment followed by trimming. J Immunol Res. 2014;192(4):1962–71. doi: 10.4049/JIMMUNOL.1302032. [DOI] [PubMed] [Google Scholar]
- 183.Berkers CR, de Jong A, Schuurman KG, Linnemann C, Geenevasen JAJ, Schumacher TNM, Rodenko B, Ovaa H. Peptide splicing in the proteasome creates a novel type of antigen with an isopeptide linkage. J Immunol. 2015;195(9):4075–84. doi: 10.4049/JIMMUNOL.1402454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Mylonas R, Beer I, Iseli C, Chong C, Pak HS, Gfeller D, Coukos G, Xenarios I, Müller M, Bassani-Sternberg M. Estimating the contribution of proteasomal spliced peptides to the HLA-I ligandome. Molecular & Cellular Proteomics: MCP. 2018;17(12):2347–57. doi: 10.1074/MCP.RA118.000877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Kloetzel PM. Neo-splicetopes in tumor therapy: a lost case? Front Immunol. 2022;13. doi: 10.3389/FIMMU.2022.849863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Lichti CF, Vigneron N, Clauser KR, Van den Eynde BJ, Bassani-Sternberg M. Navigating critical challenges associated with immunopeptidomics-based detection of proteasomal spliced peptide candidates. Cancer Immunol Res. 2022;10(3):275–84. doi: 10.1158/2326-6066.CIR-21-0727. [DOI] [PubMed] [Google Scholar]
- 187.Chong C, Coukos G, Bassani-Sternberg M. Identification of tumor antigens with immunopeptidomics. Nat Biotechnol. 2022;40(2):175–88. doi: 10.1038/S41587-021-01038-8. [DOI] [PubMed] [Google Scholar]
- 188.Ingolia NT, Hussmann JA, Weissman JS. Ribosome profiling: global views of translation. Cold Spring Harb Perspect Biol. 2019;11(5):a032698. doi: 10.1101/CSHPERSPECT.A032698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Menschaert G, Van Criekinge W, Notelaers T, Koch A, Crappé J, Gevaert K, Van Damme P. Deep proteome coverage based on ribosome profiling aids mass spectrometry-based protein and peptide discovery and provides evidence of alternative translation products and near-cognate translation initiation events. Mol Cell Proteom: MCP. 2013;12(7):1780–90. doi: 10.1074/MCP.M113.027540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Ingolia NT, Ghaemmaghami S, Newman JRS, Weissman JS. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Sci. 2009;324(5924):218–23. doi: 10.1126/SCIENCE.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Tanaka M, Sotta N, Yamazumi Y, Yamashita Y, Miwa K, Murota K, Chiba Y, Hirai MY, Akiyama T, Onouchi H. et al. The minimum open reading frame, AUG-stop, induces boron-dependent ribosome stalling and mRNA degradation. Plant Cell. 2016;28(11):2830–49. doi: 10.1105/TPC.16.00481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Al-Turki TM, Griffith JD. Mammalian telomeric RNA (TERRA) can be translated to produce valine-arginine and glycine-leucine dipeptide repeat proteins. Proc Natl Acad Sci U S A. 2023;120(9). doi: 10.1073/PNAS.2221529120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Calviello L, Ohler U. Beyond read-counts: ribo-seq data analysis to understand the functions of the transcriptome. Trends Genet. 2017;33(10):728–44. doi: 10.1016/J.TIG.2017.08.003. [DOI] [PubMed] [Google Scholar]
- 194.Cao X, Khitun A, Na Z, Dumitrescu DG, Kubica M, Olatunji E, Slavoff SA. Comparative proteomic profiling of unannotated microproteins and alternative proteins in human cell lines. J Proteome Res. 2020;19(8):3418–26. doi: 10.1021/ACS.JPROTEOME.0C00254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Ma J, Diedrich JK, Jungreis I, Donaldson C, Vaughan J, Kellis M, Yates JR, Saghatelian A. Improved identification and analysis of small open reading frame encoded polypeptides. Anal Chem. 2016;88(7):3967–75. doi: 10.1021/ACS.ANALCHEM.6B00191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Ouspenskaia T, Law T, Clauser KR, Klaeger S, Sarkizova S, Aguet F, Li B, Christian E, Knisbacher BA, Le PM. et al. Unannotated proteins expand the MHC-I-restricted immunopeptidome in cancer. Nat Biotechnol. 2022;40(2):209–17. doi: 10.1038/S41587-021-01021-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Creech AL, Ting YS, Goulding SP, Sauld JFK, Barthelme D, Rooney MS, Addona TA, Abelin JG. The role of mass spectrometry and proteogenomics in the advancement of HLA epitope prediction. Proteomics. 2018;18(12). doi: 10.1002/PMIC.201700259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Bassani-Sternberg M, Pletscher-Frankild S, Jensen LJ, Mann M. Mass spectrometry of human leukocyte antigen class I peptidomes reveals strong effects of protein abundance and turnover on antigen presentation. Mol Cell Proteom: MCP. 2015;14(3):658–73. doi: 10.1074/MCP.M114.042812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Illing PT, Ramarathinam SH, Purcell AW. New insights and approaches for analyses of immunopeptidomes. Curr Opin Immunol. 2022;77:102216. doi: 10.1016/J.COI.2022.102216. [DOI] [PubMed] [Google Scholar]
- 200.MacLachlan BJ, Dolton G, Papakyriakou A, Greenshields-Watson A, Mason GH, Schauenburg A, Besneux M, Szomolay B, Elliott T, Sewell AK. et al. Human leukocyte antigen (HLA) class II peptide flanking residues tune the immunogenicity of a human tumor-derived epitope. J Biol Chem. 2019;294(52):20246–58. doi: 10.1074/JBC.RA119.009437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.van Hateren A, Elliott T. Visualising tapasin- and TAPBPR-assisted editing of major histocompatibility complex class-I immunopeptidomes. Curr Opin Immunol. 2023;83:102340. doi: 10.1016/J.COI.2023.102340. [DOI] [PubMed] [Google Scholar]
- 202.O’Donnell TJ, Rubinsteyn A, Laserson U. Mhcflurry 2.0: improved pan-allele prediction of MHC class I-presented peptides by incorporating antigen processing. Cell Syst. 2020;11(1):42–8.e7. doi: 10.1016/J.CELS.2020.06.010. [DOI] [PubMed] [Google Scholar]
- 203.Reynisson B, Alvarez B, Paul S, Peters B, Nielsen M. NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 2020;48(W1):W449–W54. doi: 10.1093/NAR/GKAA379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Rock KL, Reits E, Neefjes J. Present yourself! By MHC class I and MHC class II molecules. Trends Immunol. 2016;37(11):724–37. doi: 10.1016/J.IT.2016.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Gessulat S, Schmidt T, Zolg DP, Samaras P, Schnatbaum K, Zerweck J, Knaute T, Rechenberger J, Delanghe B, Huhmer A. et al. Prosit: proteome-wide prediction of peptide tandem mass spectra by deep learning. Nat Methods. 2019;16(6):509–18. doi: 10.1038/S41592-019-0426-7. [DOI] [PubMed] [Google Scholar]
- 206.Wilhelm M, Zolg DP, Graber M, Gessulat S, Schmidt T, Schnatbaum K, Schwencke-Westphal C, Seifert P, de Andrade Krätzig N, Zerweck J. et al. Deep learning boosts sensitivity of mass spectrometry-based immunopeptidomics. Nat Commun. 2021;12(1). doi: 10.1038/s41467-021-24263-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Bouwmeester R, Gabriels R, Hulstaert N, Martens L, Degroeve S. DeepLC can predict retention times for peptides that carry as-yet unseen modifications. Nat Methods. 2021;18(11):1363–9. doi: 10.1038/S41592-021-01301-5. [DOI] [PubMed] [Google Scholar]
- 208.Omenn GS. Reflections on the HUPO human proteome project, the flagship project of the human proteome organization, at 10 years. Mol Cell Proteom: MCP. 2021;20:100062. doi: 10.1016/J.MCPRO.2021.100062. [DOI] [PMC free article] [PubMed] [Google Scholar]