

Abstract
Effector CD8+ T cell (TE) proliferation and cytokine production depends on enhanced glucose metabolism. However, circulating T cells continuously adapt to glucose fluctuations caused by diet and inter-organ metabolite exchange. Here we show that transient glucose restriction (TGR) in activated CD8+ TE metabolically primes effector functions and enhances tumor clearance in mice. Tumor-specific TGR CD8+ TE co-cultured with tumor spheroids in replete conditions display enhanced effector molecule expression, and adoptive transfer of these cells in a murine lymphoma model leads to greater numbers of immunologically functional circulating donor cells and complete tumor clearance. Mechanistically, TGR TE undergo metabolic remodelling that upon glucose re-exposure supports enhanced glucose uptake, increased carbon allocation to the pentose phosphate pathway (PPP), and a cellular redox shift toward a more reduced state, all indicators of a more anabolic program to support their enhanced functionality. Thus, metabolic conditioning could be utilized to promote efficiency of T cell products for adoptive cellular therapy.
Introduction
During T cell activation, metabolism is altered to increase glycolytic flux crucial to support TE function 1–4. When glucose is limiting, TE rewire central carbon metabolism to sustain survival, which is accompanied by reduced mTOR signaling 5, engagement of the cellular energy sensor AMPK 6, reduced proliferation, and elongation of mitochondria associated with higher mitochondrial spare respiratory capacity (SRC) in a CD28-dependent manner 7. These mitochondrial changes in T cells are crucial for long-term protection against tumor recurrence 7–12, and augmented mitochondrial fitness can contribute to enhanced capacity to clear tumors 8,12. In addition, glucose is crucial for optimal effector function in vivo and in vitro, and metabolic limitations contribute to dysfunction of T cells in the tumor microenvironment (TME) 13. Short-term glucose depletion reversibly limits IFN-γ production in T cells 4, while, long-term glucose depletion is detrimental to T cell function and can lead to epigenetic modifications that cannot be reversed by glucose re-exposure 14. Also, transient dampening of glycolysis during T cell activation is beneficial for the generation of long-lived memory T (TM) cells, which leads to greater immune protection in vivo 9,10,15. Like the effects of dampening glycolysis during activation, selecting cells with reduced mitochondrial membrane potential 10, or transient interference with glutamine metabolism during activation, benefits anti-tumor CD8+ T cell responses 16.
Despite these observations, how T cells adapt to nutrients as they encounter substrate fluctuations remains unclear. To address the changes TE undergo during temporary substrate limitation, we utilized transient glucose restriction (TGR) during which we monitored metabolic and functional changes. By re-exposing substrate-depleted T cells to glucose, we found that glucose carbon allocation was altered in cells after TGR. TGR led to greater anabolic engagement after glucose re-exposure, and was associated with better anti-tumor function in vitro and in vivo. Our results suggest that TGR treatment of fully activated CD8+ TE could benefit the in vivo functional capacity of T cells for adoptive cellular therapy in cancer.
Results
TGR enhances CD8+ TE cytokine production upon glucose re-exposure
To model the ability of TGR TE to mount anti-tumor responses in the TME we generated spheroids of the murine melanoma cell line B16 expressing ovalbumin (B16-F10–0VA). Spheroids were established in T cell culture media for 7 days. OT-I splenocytes were activated with OVA-derived SIINFEKL peptide for 2 days followed by 24h expansion in IL-2. Cultures were then split into media containing either 10mM (control, black) or 1mM glucose (TGR, orange) for 20h (Fig. 1a). The control or TGR TE were added to B16-F10-OVA spheroids in fresh media containing 10mM glucose and IL-2. 24h after co-culture the TGR TE cells produced significantly more IFN-γ (Fig. 1b,c) both by level of IFN-γ production per cell (MFI, Fig. 1c left panel) and by percentage of IFN-γ positive cells (Fig. 1c right panel). In addition, the TGR TE produced more granzyme B (GZMB) upon encountering OVA-positive tumor spheroids by MFI and percentage of GZMBhi expressing cells (Fig. 1b,d). Further addition of glucose did not increase effector molecule expression in either of the cell populations.
Fig. 1. TGR enhances CD8+ TE effector molecule expression upon glucose re-exposure.

(a) To generate OVA-specific CD8+ TE, OT-I splenocytes were activated with SIINFEKL peptide and IL-2 (100 U/mL), expanded for 72 hours total, followed by 20 hours of exposure to 10mM (control, black) or 1mM (TGR, orange) glucose in cultures set to 1 million per ml. B16-F10-OVA cells were established as multicellular spheroids for 8 days. (b) 100.000 OT-I TE cells generated as in (a) were combined with spheroid cultures in refreshed media containing 10mM glucose and IL-2, co-cultured for 20 hours, treated with brefeldin-A and assessed for intracellular cytokine expression induced by tumor-expressed antigen. Representative scatter plots are shown. (c-d) Quantification of IFN-γ and GZMB per cell (MFI, left) and number of IFN-γ+ or GZMBhi cells (right). Points represent averaged data from 4 independent co-cultures. data shown for 3 biological replicates. Statistical significance was calculated by 2-tailed Student’s t test. * p<0.05. (e-k) WT CD8+ T cells isolated from spleens of C7Bl/6 mice were activated with anti-CD3 (5 mg/mL), anti-CD28 (0.5 mg/mL), IL-2 (100 U/mL), expanded for a total of 72 hours, to 10mM (control, black) or 1mM (TGR, orange) glucose in cultures set to 1 million per ml. (e-f) Cells were restimulated using PMA/ionomycin with brefeldin-A and assessed for intracellular levels of (e) GZMB and (f) IFN-γ. Data show 3 biological replicates and are representative of 3 independent experiments. Statistical significance was calculated by 2-tailed Student’s t test. * p<0.05, ** p<0.01. (g-h) WT CD8+ T were isolated, activated and expanded as in (e-f), and exposed to 2-deoxy-glucose (2-DG, 0.5mM) for 20h. 2-DG was removed, cell were restimulated as above and assessed for intracellular (g) IFN-γ expression and (h) number of IFN-γ+ cells. Data show 3 biological replicates and statistical significance was calculated by 2-tailed Student’s t test. * p<0.05. (i-k) WT CD8+ T cells activated and starved as in (e,f) or exposed to 100u IL-15 for 20h. Expression of (i) CD25, (j) CD98 and (k) p-4E-BP1Thr37/46 were assessed. Data show n=4 biological replicates. Statistical significance was calculated by 2-tailed Student’s t test. * p<0.05; ** p<0.01; *** p<0.001. All error bars show SEM.
In the spheroid co-cultures, both control and TGR TE experienced a comparable nutrient microenvironment with similar antigen exposure, suggesting that pre-treatment with TGR allows CD8+ TE to produce more effector molecules. To assess whether the enhanced effects were cell-intrinsic, or caused by altered resistance to immunomodulatory signals from the tumor spheroids, we activated WT CD8+ T cells with anti-CD3/28 in IL-2 for 48h, followed by expansion for 24h in IL-2. Like in Fig. 1a, the cultures were then exposed to either 10mM glucose (black) or TGR (orange) for 20h. Control or TGR TE were re-stimulated with PMA-ionomycin in fresh media containing 10mM glucose and IL-2. TGR-TE increased expression of granzyme B (Fig. 1e) and augmented IFN-γ production (Fig. 1f). Similarly, 2-deoxyglucose (2-DG)-mediated inhibition of glucose metabolism followed by 2-DG washout and re-stimulation with PMA-ionomycin in the presence of glucose augmented IFN-γ production (Fig. 1g, h). Thus, T cells transiently exposed to limiting glucose responded more vigorously to antigen re-stimulation, both in the context of tumor spheroids, or non-TCR-dependent stimulation by PMA/ionomycin, when exogenous glucose was replenished.
Given that TM cells, including TM-like cells generated with IL-15 in vitro, respond more vigorously to antigen 13 we assessed whether TGR induced a TM-like phenotype in TE during the 20h culture period. TRG TE do not resemble memory-like T cells, suggested by their comparatively higher expression of the effector cell-surface markers CD25 (Fig. 1i) and CD98 (Fig. 1j), as well as the mTORC1 target p-4E-BP1 (Fig. 1k), when compared to TE cells exposed to IL-15 for 20h.
TGR CD8+ TE have depleted sugar metabolism, but sustain mitochondrial activity
To assess metabolic changes, WT CD8+ T cells were activated with anti-CD3/28 for 48h, expanded for 24h in IL-2, then exposed to 20h of 10mM glucose or TGR as before. Polar metabolites were extracted, followed by untargeted metabolomic analysis, which identified glucose and the aerobic glycolysis product lactate to be significantly lower in the TGR TE (Fig. 2a). Kegg analysis showed that the significantly deceased metabolites in the TGR TE were restricted to sugar metabolism intermediates and intermediates of the PPP (Fig. 2b). These observations suggested that the cells adapted to reduced glucose by limiting glucose-dependent anabolic processes, while maintaining allocation to metabolic pathways needed for survival. To assess whether the reduction in metabolite pool sizes was associated with differential glucose carbon allocation during TGR we incubated cells for 20h with uniformly-labeled (U13C) glucose. Extracellular glucose was incorporated in a glucose concentration-dependent manner, confirming the reduction in pool sizes of central glycolytic intermediates as presented in Fig. 2a, b. We measured reduced glucose carbon allocation to glycolytic intermediates over the 20h of TGR (Fig. 2c, open bars), however, neither TCA intermediate pool sizes (Fig. 2d, open and closed bars), nor the percent allocation of 13C label for 2 out of 3 TCA intermediates (Fig. 2e) were affected. Thus, glucose carbon allocation to the TCA cycle was maintained, even with limiting extracellular glucose and depletion of glycolytic intermediates. The preferential maintenance of glucose-derived TCA intermediates during TGR appeared sufficient as no compensatory increase in glutamine uptake from the media (Extended Data Fig. 1a) or glutamine carbon allocation to TCA intermediates (Extended Data Fig. 1b, open bars) were observed. Given the increased functional ability of TGR TE in spheroid co-cultures, we also tested whether lactate, a metabolite present at high concentrations in the TME could be used as an alternative carbon source during TGR. Both control and TGR TE incorporated lactate-derived carbons in the TCA intermediate citrate to a similar degree, indicating that TGR TE do not switch to lactate as an alternative substrate (Extended Data Fig. 1c).
Fig. 2. TGR TE have depleted sugar metabolism, but sustain TCA cycle metabolites.

WT CD8+ T cells were isolated, activated and expanded as in (Fig. 1e–k), followed by 20h exposure to 10mM, 1mM glucose. in cultures set to 1 million per ml. (a) Polar metabolites were extracted followed by untargeted metabolomic analysis. Volcano plot compares relative metabolite pools between control and TGR cells, orange circles represent the first and last metabolites of aerobic glycolysis, significantly reduced in TGR TE. (b) Table showing Kegg-pathway analysis of the significantly reduced metabolites in TGR TE identified in (a). (c-e) WT TE were generated as above, but during the final 20 hours, normal (U-12C) glucose was exchanged for heavy labeled (U-13C) glucose in the annotated concentrations. Polar metabolites were extracted and isotopologue distribution assessed by targeted mass spectrometry. (c-d) Relative contribution to glycolytic intermediates and TCA intermediates respectively. Statistical analysis was carried out on n=3 biological replicates, representative of 3 independent experiments, using a 2way ANOVA. Statistical significance is indicated for the 13C portion (open bars). *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001; ns=not significant. No statistical significance was observed in the 12C portion (filled bars), with the exception of succinate, which significantly increased in 1mM compared to 10mM (p<0.01). (e) Percent 13C contribution to TCA intermediates. Statistical analysis was carried out on 3 biological replicates, representative of 3 independent experiments, using an Ordinary one-way ANOVA. *p<0.05; ns=not significant. (f) WT CD8+ T cells isolated from spleens of C7Bl/6 mice were activated with anti-CD3 (5 mg/mL), anti-CD28 (0.5 mg/mL), IL-2 (100 U/mL), expanded for a total of 72 hours, and exposed to 10mM (black) 3mM (grey) or 1mM (orange) glucose in cultures set to 1 million per ml. (g-h) 2 ×105 T cells were plated in triplicate in assay medium containing the indicated concentration of glucose for analysis in a Seahorse extracellular flux analyzer. (g) Basal oxygen consumption rate (OCR) and (h) extracellular acidification rate (ECAR) were measured 3 times over a span of 20 minutes. Data represent average of n=3 independent biological replicates. Statistical significance was calculated using a 2-tailed Student’s t test. * p< 0.05; ** p<0.01, data representative of >3 independent experiments. All error bars show SEM.
To further assess global metabolic activity of TE during TGR, we measured the effects of glucose starvation using extracellular flux analysis. We activated CD8+ T cells in IL-2 followed by a 20h exposure to a glucose titration (Fig. 2f). We measured basal metabolism in unbuffered XF media with a titration of glucose matching the overnight cultures. Upon lowering glucose concentrations, we observed a shift in the relative metabolic state of the TGR cells, as assessed by the oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) as a proxy for oxidative phosphorylation and lactate secretion from the cells, respectively. Basal OCR levels in TGR TE were increased when glucose was limiting (Fig. 2g), whereas ECAR levels were reduced (Fig. 2h), as predicted by the reduced lactate pool (Fig. 2a) and glucose allocation to lactate (Fig. 2c). The relative shift to more mitochondrial activity (increased OCR, decreased ECAR) was consistent with the sustained allocation of glucose carbon to the TCA cycle (Fig. 2d, e). Taken together these data suggest that during TGR, CD8+ TE alter the allocation of acquired glucose carbons, dialing down the loss of carbons through the generation of the metabolic waste product lactate, and maintaining allocation to the TCA cycle, likely to increase energy efficiency and remain energetically viable.
To assess whether these metabolic alterations were mediated by global transcriptional changes we compared the single cell transcriptomes from control or TGR TE (generated as in Fig. 2f) and found limited differences (Extended Data Fig. 2a). While there were significant changes in individual gene expression patterns between clusters (Extended Data Fig. 2b), Kegg pathway analysis did not detect central carbon pathway alterations, but flagged OXPHOS related gene expression as significantly regulated between clusters 0 and 1 (Extended Data Fig. 2c). There was a slight shift in the proportion of TGR TE toward cluster 1, in agreement with the enhanced OCR (Fig. 2h). Although OXPHOS genes were altered, they related to ETC complexes, and while the OXPHOS module score between clusters did vary, it was not significantly driven by the control or TGR condition (Extended Data Fig. 2d). Therefore, we postulated that the altered metabolic state and glucose carbon allocation was likely not mediated by global transcriptional remodeling of TE in response to TGR. To further highlight this, we compared expression of key enzymes that are involved in OXPHOS, glycolysis, and the PPP generated from the single cell data, which showed no significant differential expression between the two TE cultures (Extended Data Fig. 2e).
CD8+ TE restore energetic balance during TGR
Since glucose is an important carbon source to generate energy in activated T cells 17, we assessed the activation state of AMP-kinase, an indicator of energy stress 18. TGR TE did not show enhanced AMPK phosphorylation after 20h, but rather a reduction in phosphorylation after TGR (Fig. 3a), suggesting that cellular energy homeostasis was not negatively affected. Also, despite lower available carbon substrate, we did not detect significant differences in ATP/AMP ratios in TGR TE (Fig. 3b), suggesting that the amount of energy in TGR T cells was not rate-limiting, which was also consistent with the lack of compensation by glutamine metabolism (Extended Data Fig. 1). Given that previous studies highlighted increased AMPK activity in T cells upon glucose depletion 5,6, we assessed the extracellular metabolite concentrations in the media in 2h intervals over the 20h window. While the control TE had half the original concentration of glucose in the media after 20h, the TGR cultures depleted extracellular glucose after 12h (Fig. 3c). The glutamine concentration in the media was comparable between the two cultures (Fig. 3d), agreeing with the stable glutamine allocation in both cell types (Extended Data Fig. 1). Glucose consumption was mirrored by lactate production, which increased between cell types over the first 8 hours, but as glucose was depleted in the TGR cultures, lactate production plateaued (Fig. 3e).
Fig. 3. TGR CD8+ TE are metabolically active and energetically balanced.

(a-f) WT CD8+ T cells isolated from spleens of C7Bl/6 mice were activated with anti-CD3 (5 mg/mL), anti-CD28 (0.5 mg/mL), IL-2 (100 U/mL), expanded for a total of 72 hours, and exposed to 10mM (black) or 1mM (orange) glucose in cultures set to 1 million per ml. (a) Immunoblot analysis of protein extracts from equal cell numbers probed for phosphorylated AMPK at Thr172 (p-AMPkThr172) and total AMPK. Tubulin was used as a loading control. Data are representative of 3 independent experiments, and 4 biological replicates. (b) Polar metabolites were extracted, levels of ATP (a.u.) and AMP (a.u.) were measured by liquid-chromatography mass spectrometry and ratios (a.u.) were calculated for 3 independent biological replicates. No significant differences were observed using a 2-tailed Student’s t test. Representative of 2 independent experiments. (c-e) Media samples and (f) protein isolates were collected every 2h over the 20h exposure to limiting glucose concentrations. Changes in media (c) glucose, (d) glutamine, and (e) lactate concentration were measured over the 20h timecourse using a Cedex Bio Analyzer. Statistical significance was calculated from 3 biological replicates using 2way ANOVA. * p<0.05; *** p<0.001; ****p<0.0001. (f) Immunoblot analysis of protein extracts from equal cell numbers probed for phosphorylated acetyl-coA carboxylase at Ser79 (p-ACC1Thr79), total ACC1, phosphorylated AMPK at Thr172 (p-AMPkThr172), total AMPK, phosphorylated ribosomal protein S6 at Ser235/236 (p-S6Ser235/236), total S6, phosphorylated 4E-binding protein 1 at Thr37/46 (p-4E-BP1Thr37/46), total 4E-BP1, and Glut1. Tubulin was used as a loading control. Optical density was measured and changes in phosphorylation are shown relative to total protein and as direct measurements. Representative of 3 biological independent samples. Biological replicate data is shown in Extended Data Fig. 3. All error bars show SEM.
To visualize both the acute and long-term effects of glucose depletion, we measured AMPK and mTORCI signaling pathways by western blot over the 20h culture. In response to metabolic stress, activated AMPK phosphorylates acetyl-coA carboxylase 1 (ACC1) on serine 79 6. Neither p-ACC nor p-AMPK was increased at 20 hours after TGR when compared to their levels earlier in the timecourse (Fig. 3f, Extended Data Fig. 3). AMPK phosphorylation did not precede the p-ACC signal in this setting (Fig. 3f, Extended Data Fig. 3). mTORCI signaling, visible as S6 phosphorylation, decreased after glucose depletion in the cultures. Although S6 phosphorylation was decreased at 20h in TGR TE, we observed no phosphorylation differences of the direct mTORCI target 4E-BP1, which regulates CAP-dependent translation 22. Despite being depleted of glucose, the TGR TE increased over time glucose transporter GLUT1, a transporter induced by TCR signals that is important for T cell activation 19,20. Our scRNA sequencing data did not identify Glut1 transcripts as differentially regulated between control and TGR TE cultures, suggesting that Glut1 expression was post-transcriptionally regulated by glucose depletion in TGR cultures, perhaps as a stress response to be able to better compete for substrate. Taken together these data suggest that glucose depletion during TGR induced transient AMPK signaling, which is resolved after 20h in culture. Our data showing reduced AMPK signaling, maintained ATP/AMP ratios, and upregulated Glut1, indicate that TGR TE cells restore energetic balance after the 20h culture period.
TGR TE have decreased reducing equivalents, but do not accumulate ROS
To further assess TE health, we measured the relative subcellular redox state in TGR TE compared to control cells by exogenous expression of a targeted redox sensitive green fluorescent protein mutant (roGFPcyto, cytosolic roGFP; roGFPmito, mitochondrial roGFP (Fig. 4a,b) 23. TGR TE shifted to a more oxidized state in the cytosolic and mitochondrial compartments (Fig. 4b), along with a decreased GSH/GSSG ratio (Fig. 4c), although the NADH/NAD+ ratio was not significantly altered (Fig. 4d). Many factors can influence the redox state of a subcellular compartment including the availability of reducing equivalents (NAD(P)H) or reactive oxygen species (ROS) 24–26. Although the TGR TE showed a more oxidized state, and presented higher basal mitochondrial activity (Fig. 2h), we did not detect an increase in cellular or mitochondrial ROS (Fig. 4e,f). The altered redox state might be explained by the altered glucose carbon allocation (Fig. 2b), the utilization of reducing equivalents to sustain complex I of the ETC (Fig. 2h), or the activity of ROS scavengers such as glutathione (GSH), which was also more oxidized (GSSG) in the TGR TE, preventing the accumulation of ROS (Fig. 4c). Given the important role of GSH in T cell function 27, we measured the de novo GSH synthesis from U-13C-glutamine during the 20h culture period using m+5 labeled GSH. There was no significant difference in de novo GSH synthesis from glutamine between cultures (Fig. 4g), although there was an accumulation of m+10 GSSG (Fig. 4h). This suggests that although GSH synthesis from glutamine is not perturbed by TGR, the recycling of GSSG to GSH might be the reason for the altered GSH/GSSG ratio (Fig. 4c).
Fig. 4. TGR TE have reversible redox balance without accumulating cellular ROS.

(a-b) WT CD8+ T cells were activated with anti-CD3 (5 mg/mL), anti-CD28 (0.5 mg/mL), and IL-2 (100 U/mL) for 36h, transduced with retrovirally encoded roGFP or mitochondrially-targeted roGFP (MOI=1), expanded for 36 hours and exposed to 10mM (black) or 1mM (orange) glucose for 20h in cultures set to 1 million per ml. (a) Micrographs show cytosolic roGFP and mitochondrial roGFP localization assessed with confocal microscopy (scale bar 10μm). (b). Flowcytometric quantification of signal ratios, taking the derived parameter per cell (488nm-excited (reduced roGFP) / 405nm-excited (oxidized roGFP). WT-GFP was used as a control. (c-d) Polar metabolites were extracted from cells as generated in Fig. 2f, analyzed using a QTOF mass spectrometer. Redox-couple ratios were computed in extracts from cells grown in 10mM (black) or 1mM (orange) glucose. (c) ratio of reduced glutathione (GSH) / oxidized glutathione (GSSG). (d) ratio of reduced nicotinamide adenine dinucleotide (NADH) / oxidized dinucleotide (NAD+). (a-d) Data shown for n=3 biological replicates, representative of 3 independent experiments. (e) Reactive oxygen species (ROS) were assessed by incubation of control TE (black) and TGR TE (orange) in cellular ROS dye (CellROS, left panel) or mitochondrial ROS dye (MitoSOX, right panel). Staining levels were measured by flow cytometry and compared to unstained cells (grey). Histograms represent 3 biological replicates. (f) Quantification of the histograms shown in (e) showing n=3 biological replicates. (g-h) WT CD8+ T cells isolated from spleens of C7Bl/6 mice were activated with anti-CD3 (5 mg/mL), anti-CD28 (0.5 mg/mL), IL-2 (100 U/mL), expanded for a total of 72 hours, and exposed to 10mM (black) or 1mM (orange) glucose and U-13C-glutamine in cultures set to 1 million per ml.. Cells were washed in cold PBS and polar metabolites were extracted. Samples were measured by LC-MS and peak areas for (g) the m+5 isotopologue of GSH and (h) the m+10 isotopologue of GSSG were extracted. Data of n=3 biological replicates. (b-h) Significance was calculated using 2-tailed Student’s t tests, ns; non-signifcant, * p<0.05; ** p<0.01; *** p<0.01. All error bars show SEM.
TGR CD8+ TE have enhanced anabolic metabolism upon glucose re-exposure
We assessed the effects of TGR on glucose metabolism when TE were re-exposed to glucose. It was evident that stimulated TGR TE were better equipped to produce effector molecules in higher glucose concentrations (Fig. 1). We asked whether glucose metabolism after glucose re-exposure was altered after TGR, perhaps mediating the augmented function. We activated, expanded, and glucose-restricted WT CD8+ T cells as in Fig. 3a, and then performed extracellular flux analysis as in Fig. 3b. After baseline measurements of both OCR and ECAR in the context of a glucose titration, we exposed cells to 10mM glucose. TGR TE increased glycolytic reserve (ECAR after oligomycin injection) (Fig. 5a), and decreased OCR (Fig. 5b, orange lines). These data showed that glucose re-exposure rapidly reversed the higher mitochondrial activity of TGR TE (Fig. 2h), with a slight increase in the generation of glucose-derived lactate (ECAR) over the 30-minute window after glucose addition. The slight increase in ECAR and decrease in OCR after glucose addition was not evident in TE exposed to control glucose concentrations from the beginning of the assay (black lines).
Fig. 5. TGR CD8+ TE have enhanced anabolic metabolism upon glucose re-exposure.

TE were generated as in (Fig. 2f). (a) Basal ECAR and (b) basal OCR (percentage of baseline) with 10mM glucose (GLC), Oligomycin (Oligo), and 2-deoxyglucose (2-DG) treatment. Data shows average of n=3 biological replicates. (c) roGFP+ control (black) or TGR (orange) TE generated as in (Fig. 3a–b) were pulsed with 10mM glucose. roGFP ratios were measured continuously for 5-mins. Data depicts average normalized ratio in 1-second bins from n=3 biological replicates. (d-e) Table depicts overlapping Kegg-Pathway hits with increased total pool size and 13C incorporation in TGR-treated TE following 5-minute glucose re-exposure. (e) control (black) or TGR (orange) TE were pulsed with 10mM U-13C-glucose for 30, 60, and 300 seconds. 20h cultures were started at 1×106 (control) and 1.5×106 (TGR) cells per/ml to generate similar end concentrations for glucose pulse experiments. Data are averages from n=3 biological replicates. Statistical significance was calculated using 2way-ANOVA, shown for U-13C allocation of time-matched pairs. * p<0.05, ** p<0.01; *** p<0.001; **** p<0.0001. (f) Reduced nicotinamide-adenine-dinucleotide-phosphate (NADP) and oxidized nicotinamide-adenine-dinucleotide-phosphate (NADPH). (g) Ratio of reduced glutathione (GSH)/oxidized glutathione (GSSG). (h) Ratio of reduced NADH/oxidized NAD. (f-h) Significance was calculated using 2-tailed Student’s t tests on n=3 biological replicates. ** p<0.01; * p<0.05; ns=not significant. (i) Percent 13C labeled ATP derived from 6h 13C-glucose re-exposure. Significance was calculated using 2-tailed Student’s t tests on n=3 biological replicates. * p<0.05. (j) TGR TE were pretreated with solvent or 6-aminonicotinamide (6-AN, 250 μM) for 30 minutes, followed by re-stimulation using PMA/ionomycin + 6-AN, 10mM glucose and brefeldin-A, and assessed for (j) intracellular GZMB and (k) IFN-γ. Data shows n=3 biological replicates, significance was calculated by 2-tailed Student’s t-test. * p<0.05. (l) TE pretreated with control or AMPK-inhibitor (CompC, 10 μM) for 30 minutes were pulsed with U-13C-glucose for 5 minutes followed by polar metabolite extraction. Incorporation of 13C label in Pentose-phosphate was measured by LC-MS. Data show on n=3 biological replicates, significance was calculated by 2way-ANOVA. Significance is shown for relative 13C incorporation (open bars). **** p<0.001. All data panels are representative of 3 independent experiments, all error bars show SEM.
Since we observed a reduction in reducing equivalents upon glucose depletion (Fig. 4c) and a shift to an oxidized redox state (Fig. 4a,b), we asked if glucose re-addition would impact cellular redox. We observed a rapid reversal toward a reduced redox state in TGR TE following glucose re-exposure (Fig. 5c). This glucose-mediated induction of a reduced cytosolic redox state could be a result of altered glucose carbon allocation, leading to the regeneration of reducing equivalents 25. To evaluate whether TGR TE allocate glucose carbon differently upon glucose re-exposure, we pulsed control or TGR TE with 10mM U-12C-glucose or U-13C-glucose for 5 minutes followed by an untargeted analysis of the polar metabolome using XCMS 28, or X13CMS 28,29 software, respectively. By overlaying Kegg pathway hits derived from data of significantly increased total metabolite pools (Extended Data Fig.4a) and 13C-isotope incorporation (Extended Data Fig. 4b), we found anabolic pathways, including the PPP and pyrimidine metabolism, to be enriched in refed TGR TE (Fig. 5d and Extended Data Fig. 4c). To test assimilation of glucose carbons into these anabolic intermediates, we pulsed control TE and TGR TE with 10mM U-13C-glucose followed by isotope tracing into glycolytic and PPP intermediates. By extracting metabolites at 30, 60, and 300 seconds after U-13C-glucose addition we could establish enhanced accumulation of glucose-derived carbons in TGR TE in intracellular glucose, and assimilation into glycolytic intermediates glucose-6P, and triose-P, but not lactate, at these early timepoints (Fig. 5e, orange open bars). The TGR TE shunted glucose carbons into the NAPDH-regenerating oxidative PPP as shown by labeled pentose-P, and the non-oxidative PPP, by labelled sedoheptulose-P and erythrose-P (Fig. 5e, open orange bars). The enhanced carbon allocation to the PPP in TGR TE was also evident in the enhanced NADPH regeneration (Fig. 5f, left panel) and drop in NADP pools (Fig. 5f, right panel), which could in part be responsible for, and in agreement with, the reversal of the cytosolic redox state (Fig. 5c). The observed redox state reversal was also evident in the increase of GSH/GSSG ratio (Fig. 5g), but not in the NADH/NAD+ ratio which changed neither during TGR (Fig. 4d), nor after glucose re-exposure (Fig. 5h).
Given the allocation to the PPP shortly after glucose re-exposure (Fig. 5e, orange open bars), we measured glucose carbon allocation in PPP-generated nucleotides. 6h after glucose re-exposure, TGR cells synthesized significantly more AMP, and UMP (Extended Data Fig. 5a, orange open bars), and the TGR TE also made more ATP with glucose-derived carbons (Fig. 5i, orange open bars). In agreement with enhanced glucose anabolism, 6h after refeeding we there was an increase in glucose allocation to the amino acid serine (Extended Data Fig. 5b), important for T cell proliferation 30. Although glucose carbon allocation into the PPP could be driven by the altered redox state, the normalized redox balance in TGR TE 5 minutes after glucose re-exposure (Fig. 5g) remained stable after 6h, as suggested by a comparable GSH/GSSG ratio at this time point (Extended Data Fig. 5c), suggesting that the anabolic regulation might be impacted by other factors than solely redox balance.
To assess the functional relevance of the PPP in the TGR-mediated enhanced effector function (Fig. 1), we treated control and TGR cultures with the PPP inhibitor 6-aminonicotinamide (6-AN), followed by PMA/ionomycin restimulation. 6-AN treatment significantly blocked GZMB expression (Fig. 5j) and blunted IFN-γ expression (Fig. 5k), highlighting an important functional role for the enhanced PPP allocation. To determine the role of AMPK signaling in glucose-carbon allocation to the PPP after TGR (Fig. 3a, f), we inhibited AMPK with Compound C during glucose re-exposure. Glucose-carbon allocation to the PPP was similar between TGR TE and untreated controls (Fig. 5l). Taken together these data suggest that TGR induces metabolic reprogramming leading to sustained anabolic metabolism upon activation and glucose re-exposure, further underwritten by significantly reduced lactate pools even 6h after glucose re-exposure (Extended Data Fig. 5d).
TGR enhances tumor-specific CD8+ TE function in vivo
Previous studies have highlighted the importance of enhanced mitochondrial metabolism 7–12, SRC 7,8 and dampened glucose metabolism during activation for the prolonged in vivo function of anti-tumor CD8+ TE 9,10,31. TGR TE presented with sustained mitochondrial metabolism (Fig. 2), enhanced SRC 7, altered redox state without augmented ROS (Fig. 4), and enhanced capacity to engage anabolism upon glucose re-exposure (Fig. 5). To ask whether TGR could condition ex-vivo generated antigen-specific CD8+ TE cells for enhanced efficacy of adoptive cellular therapy, we performed adoptive T cell transfers of TGR and control TE into mice bearing EL4-OVA lymphoma tumors on their flanks (Fig. 6a). By injecting suboptimal numbers of T cells (3×106 per mouse) after the tumors had already established (day 8 post-injection, 8mm diameter average, Fig. 6b) we achieved a model by which the control TE were able to control ~50% of the tumors with responses ranging from tumor clearance (black lines reverting to non-measureable tumors), stable disease control (no further increase or reduction in tumor size), to 4/9 mice showing limited tumor control followed by progressive disease, ultimately leading to mice having to be sacrificed (Fig. 6b, black lines). Strikingly, injection of 3 ×106 TGR TE per mouse cleared tumors in 10/10 mice (Fig. 6b, orange lines). These data demonstrated that TGR treatment augmented in vivo protective immunity against tumors. We bled the tumor bearing mice 8 days after T cell transfer (16 days post-tumor inoculation) and observed significantly higher numbers of circulating donor CD8+ T cells in all mice receiving TGR TE (Fig. 6c), despite the loss of tumor mass in all of the TGR TE recipient mice (Fig. 6b, orange lines). In addition, circulating TGR TE donor cells showed higher percentages of IFN-γ producers when re-stimulated ex vivo 8 days post-transplantation (Fig. 6d flow panels, quantified in Fig. 6e left panel), although there was no significant difference between subgroups in MFI of the IFN-γ expression (Fig. 6e right panel). When mice were bled 20 days after T cell transfer (28 days post-tumor inoculation), the augmented numbers of TGR TE donor cells were persistent (Fig. 6f). Together these data suggested the better persistence and enhanced function of anti-tumor TGR TE provided greater protection against tumor recurrence.
Fig. 6. TGR enhances tumor-specific CD8+ TE function in vivo.

(a) Ova-specific CD8+ T cells (OT-I) from CD45.2 female mice were activated by SIINFEKL peptide and IL-2 (100 U/mL), expanded for 72h, then exposed to 20h to 10mM (Control, black) or 1mM (TGR, orange) glucose. CD45.1 female recipient mice were injected with 1×106 E.G7-OVA lymphoma cells, tumors were established for 8 days, and 3 ×106 OT-I+ TE were injected per tumor bearing mouse. (b) Average tumor diameter of control (black n=9) or TGR (orange n=10) TE recipient animals. (c-e) Blood was collected 16 days post tumor injection and the percent and function of donor (CD45.2) CD8+ T cells was assessed by (c) congenic markers and (d-e) IFN-γ expression after re-stimulation with PMA/ionomycin + brefeldin-A. (f) Blood was collected day 28 post tumor injection to assess percent of donor (CD45.2) CD8+ T cells. (c-f) Statistical significance was calculated on n=9 and n=10 animals by 2-tailed Student’s t-test. * p<0.05; *** p<0.001. (g) TE cells were generated as described in (a). CD45.1 female recipient mice were injected subcutaneously with 1×106 B16-OVA melanoma cells and tumors established for 5 days. 5 ×106 control (black) or TGR (orange) OT-I+ TE were injected intravenously per tumor bearing mouse. Mice receiving no TE were used as control. 3, 6 and 9 days after tumor inoculation, mice received 200 μg anti-PD-1 antibody or lgG2a isotype control in 100 μl PBS intraperitoneally. (h-i) Lines depict average tumor diameter of grouped mice. Statistical significance was calculated by multiple 2-tailed Student’s t tests comparing control TE and TGR TE over the timecourse on n=5 animals. * p<0.05; ** p<0.01. (j-k) 21 days after tumor inoculation mice were humanely euthanized and the tumor mass was excised. Single cell suspensions were analyzed by flow cytometry. The number of donor (CD45.2) CD8+ T cells per mg tissue (j) and the percent of donor (CD45.2) CD8+ T cells (k) was quantified. Statistical significance was calculated by 2-tailed Student’s t test. * p<0.05; ** p<0.01. Tumor size measurements were performed by blinded researchers. All error bars show SEM.
To extend our findings we tested the added benefit of TGR TE in a murine melanoma model, in which we administered anti-PD-L1 checkpoint-therapy to mice bearing B16-F10-OVA flank tumors. T cell infusions of TGR TE or control TE were combined with 3 intraperitoneal injections with anti-PD-L1 or isotype control antibodies (Fig. 6g). We observed increased circulating donor-derived T cells in the TGR conditions as compared to controls 16 days post T cell infusion (Extended Data Fig. 6a). While adoptive transfer of donor-derived control or TGR TE led to better tumor control compared to isotype only treated mice, TGR TE (Fig. 6h orange squares) significantly slowed tumor progression compared to the control TE (Fig. 6h black squares). These findings agreed with our previous in vivo experiments using EL4-OVA tumors both in terms of increased circulating numbers (Fig. 6c,f) and increased tumor control (Fig. 6b) in the TGR TE condition.
Anti-PD-L1 therapy significantly improved tumor control in the control TE recipient mice (Fig. 6i, black squares), but anti-PD-L1 therapy did not further improve the enhanced tumor control mediated by TGR TE (Fig. 6i, orange squares). The increase in numbers of donor-derived TGR TE cells was not only evident in the circulation (Extended Data Fig. 6a), but also in the tumors 16 days after T cell infusion (Fig. 6j,k left groups). Although TGR enhanced donor-derived T cells in circulation, independent of PD-L1 treatment (Extended Data Fig. 6a), the TGR-mediated enhanced number of donor-derived T cells inside the tumors was no longer evident when combined with anti-PD-L1 treatment (Fig. 6j,k right groups). Note that the percentage of donor-derived CD8+ T cells is lower in the case of anti-PD-L1 treatment (Fig. 6k), reflecting the enhanced tumor-infiltration of host CD8+ T cells after anti-PD-L1 treatment (Extended Data Fig. 6b). Taken together, these data suggest that 1) TGR leads to an augmented number of circulating donor-derived T cells increasing the likelihood of anti-tumor immunity. 2) TGR leads to an enhanced ability to infiltrate or survive inside B16-F10-OVA tumors. 3) This TGR-mediated increased ability to infiltrate or survive in the tumor was not further enhanced by anti-PD-L1 treatment.
Discussion
Previous studies have highlighted the benefit of blunting glycolysis during T cell activation on the development of long-lived functional memory T cells 9,10,31. Aerobic glycolysis is a hallmark of fast-dividing cells 32,33, and dampening this pathway can be associated with slower proliferation 34. Rather than limiting glucose during activation, we depleted glucose after the cells engaged multiple rounds of division and were fully activated. Although this was associated with reduced proliferation over 20h of glucose-restriction, expansion in glucose-replete media before TGR allowed us to generate large numbers of activated CD8+ TE. This approach might be valuable in the generation of T cell products for adoptive cellular therapy, especially given the large numbers of cells infused into patients 35.
After acute glucose starvation in vitro, CD8+ T cells become energetically stressed, as shown by activation of the energy sensor AMPK 5,6, which is also associated with dampened S6 phosphorylation and Glut1 upregulation (Fig. 3f). However, after TGR CD8+ TE had lower p-AMPK compared to control cells, and a non-significant, but reproducible increase in the ATP/AMP ratio (Fig. 3e), suggesting that TGR TE have an improved energetic balance, and increased Glut1 expression, which may underlie their ability to robustly respond to glucose re-exposure. Although AMPK inhibition did not alter glucose carbon allocation upon refeeding, how the transient induction of AMPK activity and gradual upregulation of GLUT1 contributes to this phenotype is to be determined. Decreased ACC1 phosphorylation and GLUT1 upregulation coincide with the time that extracellular glucose is depleted, and could indicate that this is the metabolic switch leading to the adaptation observed at the 20h time point.
The sustained energy homeostasis in TGR TE could in part be explained by their enhanced mitochondrial metabolism. Although we observed decreased metabolite pools in pathways associated with sugar metabolism, including amino- and nucleotide-sugar metabolism, and glycolysis, TCA metabolites did not decrease to the same extent. As the relative incorporation of glucose and glutamine carbons remained stable, we inferred that the T cells specifically allocated remaining glucose carbons away from anabolic pathways to sustain mitochondrial ATP production. Preferential glucose carbon allocation into the TCA cycle allows the full oxidation and generation of up to 10-fold more ATP per glucose molecule when compared to aerobic glycolysis. Another explanation for the sustained ATP pools is that TGR TE do not have sufficient substrate for proliferation and cytokine production and therefore have a reduced ATP demand to fuel TE functions.
Along with sustained ATP pools, the ability to handle oxidative stress is important for cellular health. While mitochondrial ROS is crucial for T cell activation 27,36, ROS accumulation can cause damage, leading to functional perturbation or programmed cell death 37. Although TGR led to the relative oxidation of both the cytosolic and mitochondrial compartments, TGR TE did not deplete the ROS scavenging molecule glutathione, in part supported by sustained glutathione synthesis from glutamine (Fig. 4g, h). This in combination with sustained reducing equivalents could be the metabolic rewiring that TGR TE utilize to maintain basal metabolic activity in the cell, and then benefit from when carbons become available upon glucose re-exposure. A previous study highlighted that the accumulation of the redox co-factor NAD+ allowed increased anti-tumor function of pro-inflammatory CD4+ T cells 38. A longer window of TGR could lead to depletion of these factors and lead to dysfunction that is no longer responsive to glucose re-exposure 14.
The differences in glucose carbon allocation after TGR showed that depletion of a dominant biofuel alters allocation of the same biofuel when re-encountered (Fig. 5e). Whether altered carbon allocation is a mediator of increased anti-tumor functions after glutamine depletion has not yet been addressed 16,39. Redox state changes, as measured by roGFP and ratios of redox pairs, could account for this altered carbon allocation, as there is a depletion of redox cofactors no longer allowing reducing reactions to take place. Whether this is the cause or effect of altered glucose metabolism induced by TGR remains undetermined. The enhanced glucose carbon allocation into the PPP upon glucose re-addition could be supported by the accumulated NADP+ in the TGR TE (Fig. 5f). The concomitant increase in NADPH and decrease in NADP+ upon glucose re-exposure could result from the allocation of glucose carbons to the PPP, as measured by increased levels of pentose-P derived from newly added glucose (Fig. 5e, open bars). This is also corroborated by the observed normalization of the GSH/GSSG ratios after glucose re-exposure.
How the enhanced carbon allocation to the oxidative and non-oxidative PPP augments effector function remains unclear. While PPP inhibition with 6-AN dampened TGR TE cytokine production, PPP engagement is unlikely the sole contributor to enhanced function in vivo. Glucose 6-phosphate dehydrogenase (G6PD) inhibition abrogates pro-inflammatory cytokine production in CD8+ T cells, in part by reduced IFN-γ transcription 40. While this study links the PPP to cytokine production upon activation, other studies have shown that the flux through glycolysis is important for cytokine production. The dampened ability of TE cells to produce IFN-γ in the absence of glucose 14, or when exposed to galactose 4, which enhances PPP flux and mitochondrial metabolism, suggests that hyperactivation of the PPP alone cannot compensate for a lack of glycolytic flux. A lack of increased IFN-γ transcription suggests a role for either NADPH or the biosynthetic contribution of the PPP in the augmented function of the TGR cells, but will require further investigation.
OT-I transgenic TGR TE co-cultured with OVA-expressing B16 spheroids displayed increased anti-tumor immune function, as measured by effector molecule expression. The TE were added to complete media containing IL-2, so the extracellular environment was identical in the cocultures. By using transgenic OT-I CD8+ TE, we normalized the TCR signal the cells received from the tumor cells. Regardless, effector cytokine production was augmented in the TE exposed to TGR for 20h preceding the coculture, suggesting differences in cell-intrinsic metabolic signals associated with enhanced anti-tumor function. A recent study demonstrated that CD8+ T cells activated in vivo shed fewer carbons as lactate and had greater glucose carbon allocation to anabolic pathways and the TCA cycle compared to T cells activated in vitro 41, suggesting that our TGR protocol may prepare cells for a more ‘in vivo-like’ environment. Results showing that glucose only accounts for 10% of the biomass in actively proliferating lymphocytes 17 could therefore suggest that in vitro conditions lead to T cells being wasteful, no longer optimally utilizing the substrates that are plentiful in media.
Our data suggest that human T cell expansion protocols, such as those used for tumor-infiltrating lymphocyte (TIL) or CAR T cell therapy 42, could be amended to include a transient glucose starvation before transplantation. We reproducibly observed significantly more circulating donor cells after 8- and 20-days post-transplantation in mice, suggesting that TGR confers survival and/or proliferative advantages to the cells after infusion. In addition to the increase in circulating donor-derived tumor-specific T cells, we also detected higher percentages of IFN-γ expressing cells that produced more GZMB upon ex vivo re-stimulation. The enhanced tumor clearance reflected the observation that not only ex vivo reactivation, but also in vivo function of these cells was significantly enhanced, leading to 100% tumor clearance in the EL4-OVA bearing mice receiving TGR-treated cells. Data from the B16-F10-OVA melanoma model suggested that this difference in tumor control is perhaps not solely due to enhanced anti-tumor function and in vivo persistence, but also increased tumor infiltration. Although we did not observe further therapeutic benefit by combining anti-PD-L1 therapy with TGR TE adoptive transfer, the TGR TE showed enhanced persistence in these mice as well. Future studies could determine which parameter is the driving force behind the anti-tumor activity and in vivo persistence observed after treating TE with 20h TGR before infusion, but it is likely to be a combination of factors. Previous studies in murine tumor models identified that anti-PD-L1 therapy-induced increased respiration, and SRC, supporting enhanced tumor infiltration and CD8-mediated tumor control 12,43,44. Comparably, TGR increases respiration (Fig. 2h), SRC 7, and tumor infiltration (Fig. 6j,k) perhaps explaining the lack of further therapeutic benefit of the anti-PD-L1 therapy on TGR TE-infused mice.
We suspect that there is a limited time during which T cells can respond vigorously to added glucose. In a recent study we showed that prolonged glucose-restriction leads to epigenetic remodeling through loss of acetyl-CoA pools 14. Whereas the cells undergoing transient starvation reacted vigorously to newly added glucose, when the depletion of extracellular glucose was prolonged for days, cells failed to augment effector cytokine production upon re-addition of glucose. These data showed that there is a window of opportunity to reinvigorate CD8+ TE upon transient glucose deprivation. We found that transient glucose-depletion after activation leads to metabolic reprogramming of CD8+ TE, enhancing their longevity and functional capacity in vivo. Our study highlights that dynamic changes in nutrient allocation are associated with functional outcomes in primary cells.
Further information and requests for resources and reagents should be directed to and will be made available upon reasonable request by the corresponding author, Erika L. Pearce (pearce@ie-freiburg.mpg.de).
Methods
Mouse lines
C57BL/6J (RRID: IMSR_JAX:000664), major histocompatibility complex (MHC) class I-restricted OVA specific TCR OT-I transgenic mice (RRID: IMSR_JAX:003831), CD45.1+ C57NL/6J (B6.SJL-Ptprca Pepcb/BoyJ; Jax, 002014) and Thy1.1 congenic (Jax strain number 000406) mouse strains were purchased from The Jackson Laboratory and were maintained at the Max Planck institute for Immunobiology and cared for according to the Institutional Animal Use and Care Guidelines. Tumor studies were approved by the animal care committee of the Regierungspraesidium Freiburg. Mice were bred under specific pathogen free standards, and transferred to open top cages for experimental procedures. For tumor experiments 9-11-week-old female mice were randomized to match age across experimental groups. Temperature and humidity of the holding rooms was monitored daily and Tumor bearing mice were weighed every 2 days. Mouse body condition score was monitored over the experimental period, assuring the mice maintained expected weight gain, did not show signs of distress, tumors did not ulcerate and tumor size did not interfere with feeding or drinking of the animals and did not exceed 20mm diameter. No association between age or weight with response was observed or expected.
Primary T cell cultures
CD8+ T cells were obtained from spleens and lymph nodes isolated from 8-12-week-old C57Bl/6 mice using the EasySep CD8 T cell isolation kit (Stem Cell technologies catalogue # 19753) according to the manufacturers protocol. Within experiments mice were age and sex matched. Sample size is indicated in the figure legends. Isolated T cells were activated using plate bound anti-CD3 (5 μg/ml) (InVivoMab anti-mouse CD3, BioXCell catalogue #BE0002); and soluble anti-CD28 (0.5 μg/ml) (InVivoMab anti-mouse CD28, BioXcell catalogue # BE0015), in 1640 media (invitrogen) supplemented with 10% fetal calf serum (Gibco), 4mM L-glutamine, 1% penicillin/streptomycin, 100 U/ml hrIL-2 (Peprotech), 55 μM beta-mercaptoethanol, in a humidified incubator at 37°C, atmospheric oxygen supplemented with 5% CO2. For OT-I cultures, single cell suspensions of splenocytes were incubated for 48 hr in IL-2 containing media as above in the presence of SIINFEKL peptide. 48 hours after activation, media was replaced with expansion media containing 11mM glucose and 100U/ml IL-2. After 24 hours of expansion, cells were exposed to experimental media for 20 hours, containing a dilution series of glucose as indicated in figure legends (control U12C or stable isotope U13C to assess carbon allocation). Cells exposed to limiting glucose allocation (1mM) were plated at 1.5 × 106/ml to achieve equal cell number / ml after 20 hours for analysis of acute carbon allocation over time. For acute carbon allocation, 5 × 106 T cells/mouse/glucose concentration/time point were added to 5ml Eppendorf Safe-Lock tubes, and incubated in a 37°C aluminum bead bath. 10mM glucose (control U12C for pool size measurements or stable isotope U13C to assess glucose carbon allocation) was added to the cells, mixed and incubated for 30, 60, or 300 seconds followed by transfer to a watery ice bath to instantly cool the cultures. For stable isotope tracing, cells were incubated in differential U-13C-glucose, U-13C-glutamine or U-13C-lactate for 6 or 20 hours as indicated in the figure legends. Pharmacological inhibition of glycolysis in TE cells was achieved by treatment with low dose 0.5mM 2-deoxy-glucose (Sigma, L07338.14) or solvent (media) control for 20h, followed by drug washout and restimulation with PMA/ionomycin, as indicated in the figure legend. To block the PPP during glucose reexposure, cells were pretreated with 250μM 6-aminonicotinamide (Sigma, A68203) or solvent (DMSO) control for 30 minutes, followed by PMA/ionomycin restimulation as indicated in the figure legends. For AMPK pathway assessment TGR TE cells were pretreated with 10μM Compound C (Merck, 171260) or solvent (DMSO) control for 30 minutes, followed by a 5 minute pulse with U13C glucose as indicated in the figure legend.
Metabolite extraction
Cells were collected by centrifugation at 4°C, washed in ice cold PBS, and polar metabolites were extracted twice using 500μl ice cold extraction buffer (50:30:20, methanol:acetonitrile:water) (extraction buffer was pre-cooled on dry ice for 30 mins before extraction). Samples were centrifuged at maximum speed for 10 minutes to remove protein debris, and extracts were dried using an EZ2-Elite evaporator (Genevac). Metabolites were resuspended in 50ul extraction buffer and and stored at −80°C until acquisition.
Metabolite measurement by LC-MS
LC-MS was carried out using an Agilent 1290 Infinity II UHPLC inline with a Bruker Impact II QTOF operating in negative ion mode. Scan range was from 20 to 1000 Da. Mass calibration was performed at the beginning of each run. LC separation was on a Phenomenex Luna propylamine column (50 × 2 mm, 3 um particles) using a solvent gradient of 100% buffer B (5 mM ammonium carbonate in 90% acetonitrile) to 90% buffer A (10 mM NH4 in water). Flow rate was from 1000 to 750 uL/min. Autosampler temperature was 5 degrees and injection volume was 2uL.
Data analysis of metabolite measurements
For targeted analysis, metabolites were quantified using AssayR 45, and identified by matching accurate mass and retention time to standards. For untargeted analysis, samples were compared using XCMS 28, or X13CMS 29 (isotope-labeled samples), and significantly different metabolite identities (p-value cutoff <0.05) were defined by accurate mass using the Human Metabolomics Database (HMDB). For pathway analysis, metabolites assigned a Kegg ID in the HMDB were searched using the Kegg Mapper-Search Pathway tool. Kegg Pathways were ranked according to the number of metabolite hits in that pathway (from highest to lowest). Metabolites in the highest ranked pathways were further validated by targeted analysis.
Extracellular flux analysis (Seahorse assays)
Extracellular acidification rate (ECAR) and oxygen consumption rate (OCR) were measured using a Seahorse XFe96 bioanalyzer (Agilent). 2 ×105 T cells per well (minimum 3 wells per sample) were spun into previously poly-D-lysine coated seahorse 96 well plates and preincubated CO2-free 37°C incubator for a minimum of 45 min. Basal OCR and ECAR were measured in cells incubated in unbuffered 1640 media supplemented with 1 mM pyruvate and 4 mM L-glutamine adjusted to pH 7.4 (for detailed protocols see ref 46) in the presence of differential glucose concentrations, followed by the addition of 10 mM glucose, 1 μM oligomycin, and 50 mM 2-deoxyglucose (2-DG) (all Sigma) as indicated.
Cell lines
The murine EG.7 lymphoblast cell line expressing OVA (EL4-OVA; E.G7-OVA), was purchased from ATCC (ATCC Cat# CRL-2113, RRID: CVCL_3505), cultured for three passages before being implanted in the flank of naive mice. The murine melanoma cell line B16-F10-OVA was a kind gift of Dr. Dietmar Zehn. Further authentication of the cell lines was not performed. Cells were maintained in 1640 media (E.G7-OVA) or DMEM media (B16-F10-OVA) supplemented with 10% fetal calf serum, 4 mM L-glutamine, 1% penicillin/streptomycin, 55 μM beta-mercaptoethanol, in a humidified incubator at 37°C, atmospheric oxygen supplemented with 5% CO2. Spheroids were allowed to establish for 8 days on ultra-low attaching plates. 96 well U-bottom plates were coated with a solution of 12mg/ml poly-2-hydroxyethyl methacrylate (poly-HEMA (sigma)) dissolved in 95% ethanol. 150 μl of sterile poly-HEMA per well was allowed to evaporate in a sterile biosafety cabinet, dried plates were washed with PBS and stored at 4°C before use. Sub-confluent cultures of B16-F10-OVA were dissociated into single cell suspensions by incubation with accutase for 5 minutes at 37°C and resuspension in growth media. Cells were resuspended at 50.000 cells/ml and plated at 200 μl per well. Cells were concentrated at the bottom of the non-attaching wells by low speed centrifugation at 100 × g for 10 mins. Effector T cells were added after the spheroid media was removed by aspiration, and co-cultures were performed in T cell expansion medium as described above.
Virus generation and transduction
Cyto-roGFP and matrix-roGFP were a gift from Paul Schumacker (Addgene plasmid # 49435 and # 49437). roGFP coding sequences were transferred to an MSCV backbone (MIGR1, addgene plasmid #27490, digested with Bglll and Sall to remove the IRES-GFP cassette) and packaged into ecotropic virus using 293T cells as previously described 47. CD8+ T cells were isolated and activated as above and after 36 hours, roGFP expressing MSCV-γ retrovirus containing supernatant was added to the cultures in the presence of polybrene (8μg/ml) followed by a 90-minute spin at 30°C at 1000 × G to enhance transduction efficiency. To maintain sensitivity to small changes in redox, cells were infected at an MOI of 1, to achieve low level roGFP expression.
Flow cytometry
Fluorochrome-conjugate monoclonal antibodies were purchased from eBioscience, BDBioscience, or Life Technologies. Staining was performed in 1% FBS/PBS for 30 min on ice, dead cells were excluded with the LIVE/DEAD Fixable Blue Dead Cell Stain Kit (Thermo scientific). Donor-derived CD8+ T cells from blood were quantified after bed blood cell lysis, by direct staining for CD90.1 and CD90.2 congenic markers. For intracellular cytokine staining cells were reactivated with phorbol 12-myristate 13-acetate (PMA, 50ng/ml, Sigma) and ionomycin (500ng/ml, Sigma), in the presence of Brefeldin A (0.1%, Biolegend) for 5 hours prior to fixation using Cytofix Cytoperm (BDBioscience), except for the spheroid cocultures, there Brefeldin was added directly into the coculture (without extra restimulation) 5 hours before staining. For analysis of redox status cells in cell expressing roGFP, the excitation with a 488 nm laser and a 405 nm laser (detection using a was used to quantify reduced and oxidized signal, respectively. FlowJo software was used to generate the derived parameter of the two fluorescent signals. For dynamic assessment of changes in redox state signals were collected for 5 minutes after adding 10mM glucose on an LSRII flow cytometer while the samples were kept at 37°C. The data was plotted as dynamic data after splitting signals into 1 second bins over the full 5-minute period, deriving the mean of the two signals per bin. The gating strategy is shown in the Supplementary Information.
Western blotting
For western blot analysis cells were washed with ice cold PBS and lysed in lysis buffer (20 mM Tris-HCl, [pH 7.5], 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 μg/mL leupeptin (Cell Signaling Technologies) supplemented with 1 mM PMSF. Samples were frozen and thawed 3 times followed by centrifugation at 20000g for 10 min at +4° C. Cleared protein lysate was denatured with LDS loading buffer for 10 min at 70° C, and loaded on precast 4% to 12% bis-tris protein gels (Life Technologies). Proteins were transferred onto nitrocellulose membranes using the iBLOT 2 system (Life Technologies) following the manufacturer’s protocols. Membranes were blocked with 5% w/v milk and 0.1% Tween-20 in TBS and incubated with the appropriate antibodies in 5% w/v BSA in TBS with 0.1% Tween-20 overnight at 4°C (primary antibodies were at 1:2000 unless otherwise stated below). The following antibodies were used: anti-phospho-ACC1Ser79 (cell signaling catalogue #11818); anti-ACC1 (cell signaling catalogue #3676); anti-phospho-AMPKThr172 (Cell Signaling catalogue #2535); anti-AMPK (Cell Signaling catalogue #5831), anti-phospho-S6Ser235/236 (cell signaling catalogue #2211); anti-S6 (cell signaling catalogue #2317); anti-phospho-4E-BP1Thr37/45 (cell signaling catalogue #2855, at 1:4000); anti-4E-BP1 (cell signaling catalogue #9644, at 1:4000); anti-Glut1 (cell signaling catalogue #12939); and anti-tubulin (Sigma catalogue #T6199, at 1:5000). All primary antibody incubations were followed by incubation with secondary HRP-conjugated antibody (Pierce) in 5% milk and 0.1% Tween-20 in TBS (at 1:20000) and visualized on radiosensitive film (Amersham) using chemiluminescent substrate (Supersignal west-pico, Pierce). Relative optical density was measured using Image J software, and plotted as relative phospho/total protein amount, and as phospho signal optical density.
Single cell RNA sequencing
Single cell RNA sequencing was performed using a 10X Genomics Chromium Controller. OT-I TE were generated as before and exposed to control or TGR for 20 hours. Single cells were processed with GemCode Single Cell Platform using GemCode Gel Beads, Chip and Library Kits (v2) following the manufacturer’s protocol. An estimated 5,000 cells were added to each channel with an average of 3,000 cells recovered. Libraries were sequenced on HiSeq 3000 (Illumina). Samples were demultiplexed and aligned using Cell Ranger 2.2 (10X genomics) then processed and analyzed in R using Seurat 48 and Uniform Manifold Approximation and Projection (UMAP) as a dimensionality reduction approach 49. Genes from specific metabolic pathways were retrieved from Kegg 50 and expression scores were calculated using Seurat. Differentially expressed genes, with greater than a 1.2 fold change and an adjusted p value of less than 0.1, were analyzed for pathway enrichment using STRING 51.
In vivo tumor experiments
Congenically marked female C57Bl/6 mice (Thy1.1) were shaved, and injected subcutaneously into the right flank of mice with 1 ×106 E.G7-OVA in 100μl PBS. After 8 days, donor cells from 3 female mice were mixed before injection and 3 ×106 OT-I+ TE were injected intravenously per tumor bearing mouse, during which the investigator injecting the mice was blinded to the identity of the donor TE. 3 × 106 OT-I TE, Right before injection, the size of tumors was measured and equally distributed over both groups (average size was 7mm diameter at D8 in all mice). The grouping of the donor cells was blinded to the investigator injecting the cells, and tumor volumes were measured with calipers every 2 days after T cell infusion, by 2 investigators, one of which was blinded to the identity of the donor cells, to ensure reproducible, unbiased measurements. A small quantity of blood was obtained by facial vein laceration at indicated time points to quantify congenically marked CD8+ T cells by flow cytometry. Congenically marked female C57BL/6 mice (CD45.1) were shaved, and injected subcutaneously into the right flank of mice with 1×106 B16-OVA cells in 100 μl PBS. Tumor growth was monitored every 2 days by measuring the diameter of the tumor mass. After 5 days, 5×106 OT-I TE that had previously been activated in vitro with SIINFELK peptide, were transferred i.v. into tumor bearing mice. Control groups did not receive TE injection. The grouping of the donor cells was blinded to the investigator injecting the cells. On day 3, 6 and 9 after tumor inoculation, mice were treated with an intraperitoneal injection of 200 μg anti-PD-1 antibody or IgG2a isotype control in 100 μl PBS. On day 21 after tumor inoculation, mice were humanely euthanized and the tumor mass was excised. Isolated tumors were homogenized to obtain single cell suspensions and analyzed by flow cytometry
Statistics
Statistical analysis was performed using prism 8 software (Graph pad) and results are represented as mean ± SEM, unless otherwise indicated. Comparisons for two groups were calculated using unpaired two-tailed Student’s t tests, comparisons of more than two groups were calculated using an Ordinary one-way ANOVA with Bonferroni’s multiple comparison tests or 2way ANOVA with Bonferroni’s multiple comparison tests. We observed normal distribution and no difference in variance between groups in individual comparisons. Selection of sample size was based on extensive experience with metabolic and in vivo tumor immunology assays. Also see Reporting Summary.
Data Availability
The data that support the findings of this study are available from the corresponding author upon request. Single cell RNA sequencing data has been deposited in the Gene Ontology Omnibus under accession number GSE152018.
Extended Data
Extended Data Fig. 1. TGR CD8+ TE do not sustain mitochondrial metabolism by incorporating more glutamine-derived carbons into the TCA cycle.

WT CD8+ T cells isolated from spleens of C7BI/6 mice were activated with anti-CD3 (5 mg/mL), anti-CD28 (0.5 mg/mL), IL-2 (100 U/mL), expanded for a total of 72 hours, and exposed to 10mM, 3mM, or 1mM glucose as indicated in cultures set to 1 million per ml. (a) Polar metabolites were extracted from 500μl media supernatant from 20h cultures of 10mM and 1mM cells. Bar graphs represent glutamine concentration in unused culture media for n=3 biological replicates. Significance was calculated using 2-tailed Student’s t tests, no significant changes (ns) were observed. (b) TE were generated as above, but during the final 20h all groups were cultured in 4mM heavy labeled (U-13C) glutamine (100% U-13C glutamine). Polar metabolites were extracted and isotopologue distribution assessed by targeted mass spectrometry. TCA intermediates are plotted as percent label from newly metabolized U-13C (open bars) or remaining U-12C (black bars) glutamine carbons. Data are from n=3 biological replicates, representative of 2 independent experiments. Significance was calculated using 2-tailed Student’s t tests, no significant changes were observed. (c) TE were generated as above, but during the final 6h 2mM U-13C-lactate was added to the culture. Polar metabolites were extracted and isotopologue distribution assessed by targeted mass spectrometry. Significance was calculated using 2-tailed Student’s t tests comparing the predominant m+2 isotopologue group in both conditions. Data shown for n=3 biological replicates. Significance was calculated using 2-tailed Student’s t tests, No significant changes were observed. All error bars show SEM.
Extended Data Fig. 2. Limited transcriptional reprogramming during TGR.
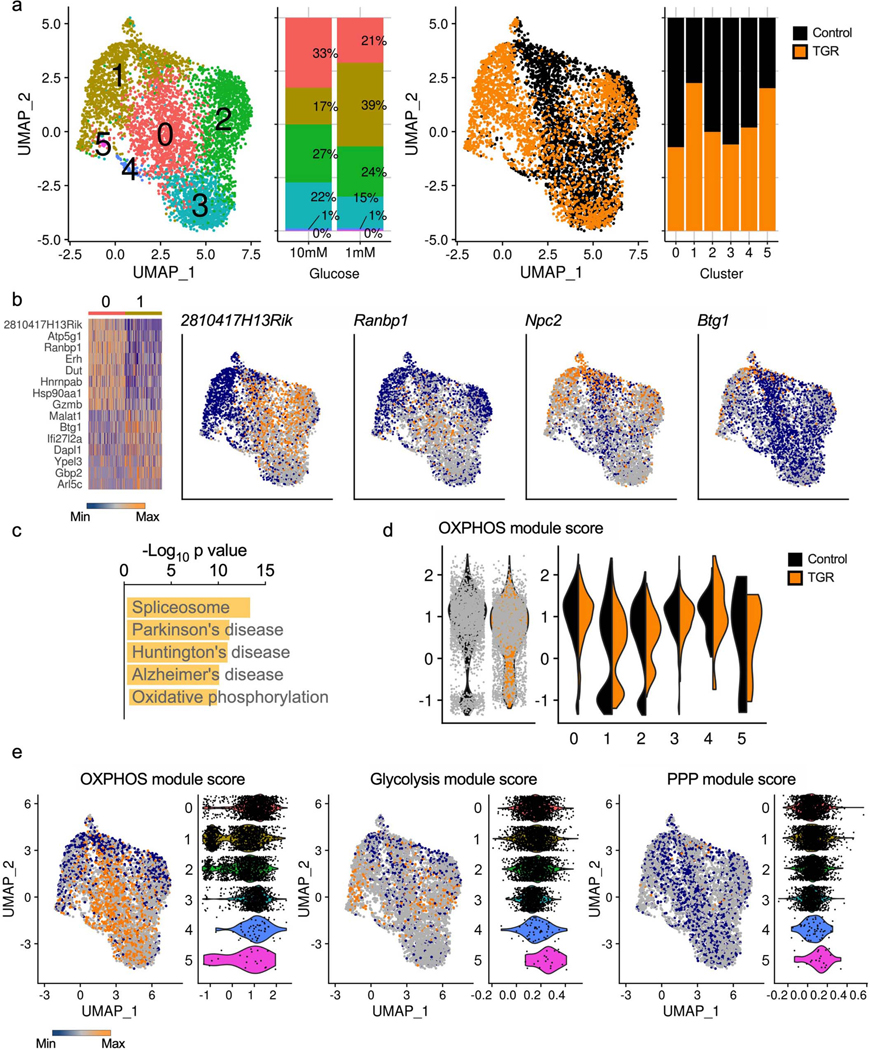
WT CD8+ T cells isolated from spleens of C7Bl/6 mice were activated with anti-CD3 (5 mg/mL), anti-CD28 (0.5 mg/mL), IL-2 (100 U/mL), expanded for a total of 72 hours, and exposed to 10mM (black) or 1mM (orange) glucose in cultures set to 1 million per ml. RNA isolated from single cells was sequenced using the 10X genomics platform and analyzed to explore transcriptional changes in each population of cells. (a) A Uniform Manifold Approximation and Projection (UMAP) of the overlapped cells from each treatment (control, black; TGR, orange), clustered on the basis of transcriptional similarity is shown highlighting clusters and treatment distribution. Bar graphs depicting cluster distribution for each condition are also shown. (b) Clusters 0 and 1, where least overlap between treatments was observed, were contrasted to look for differentially expressed genes (> 1.2-fold change and < 0.1 adjusted p-value); a heatmap of top up and down regulated genes is shown, along with UMAPs of example genes. (c) The top 5 most enriched pathways in differentially regulated genes between cluster 0 and 1. (d) Module scores based on average expression levels of gene programs were calculated for 18 differentially expressed OXPHOS genes (Atp5g1, Atp5g3, Cox5a, Cox6a1, Cyc1, Ndufa11, Ndufa12, Ndufa4, Ndufa8, Ndufabl, Ndufb7, Ndufb8, Ndufcl, Ndufc2, Ndufs6, Sdhb, Uqcr10, Uqcr11). Violin plots depict the global OXPHOS module score per condition (left) or per cluster and condition (right). (e) Module scores based on average expression levels of gene programs were also calculated for enzyme-coding genes involved in glycolysis (mmu00010) or the pentose phosphate pathway (PPP, mmu00030) based on Kegg annotation. UMAP and Violin plots illustrate the OXPHOS, glycolysis and PPP modules, with the violin plots divided per cluster.
Extended Data Fig. 3. Signaling timecourse during TGR.

Protein isolates were prepared as described in (figure 3f), taking samples every 2h over the 20h exposure to limiting glucose concentration. Immunoblot analysis of protein extracts from equal cell numbers probed for phosphorylated acetyl-coA carboxylase at Ser79 (p-ACC1Thr79), total ACC1, phosphorylated AMPK at Thr172 (p-AMPkThr172), total AMPK, phosphorylated ribosomal protein S6 at Ser235/236 (p-S6Ser235/236), total S6, phosphorylated 4E-binding protein 1 at Thr37/46 (p-4E-BP1Thr37/46), total 4E-BP1, and Glut1. Tubulin was used as a loading control. Optical density was measured and changes in phosphorylation are shown relative to total protein and as direct measurements. Representative of 3 biological independent samples. Biological replicate data is shown in Fig. 3f.
Extended Data Fig. 4. TGR TE have altered glucose reallocation upon re-exposure to drive anabolic metabolism.

WT CD8+ T cells isolated from spleens of C7Bl/6 mice were activated with anti-CD3 (5 mg/mL), anti-CD28 (0.5 mg/mL), IL-2 (100 U/mL), expanded for a total of 72 hours, and exposed to 10mM (control) or 1mM (TGR) glucose during the final 5 minutes cells were re-exposed to normal (U-12C) glucose or heavy labeled (U-13C) glucose. 20h cultures were started at 1×106 (control) and 1.5×106 (TGR) cells per/ml to generate similar end concentrations for glucose pulse experiments. (a) Table showing the results of a Kegg-pathway analysis of the significantly increased and decreased metabolite pools in TGR TE after normal (U-12C) glucose re-feeding when compared to re-fed control TE. (b) Table shows the results of a Kegg pathway analysis of the metabolites that exhibited increased glucose-derived U-13C assimilation (as determined by X13CMS software) after re-feeding TGR TE with heavy labeled (U-13C) glucose compared to re-fed control TE. Data are from 3 biological replicates, representative of 3 independent experiments. Kegg Pathways were ranked according to the number of metabolite hits in that pathway (from highest to lowest). Metabolites in the highest ranked pathways were further validated by targeted analysis. (c) Polar metabolites were extracted and untargeted metabolomic analysis was performed using XCMS on cells re-exposed to normal (U-12C) glucose. Volcano plot depicts relative metabolite pools compared between control and TGR cells. Orange circle represents a pentose phosphate pathway metabolite Pentose-phosphate, significantly increased in glucose re-fed TGR TE. Statistical significance was calculated using a Welch’s t-test, comparing relative intensities of each isotopologue in labeled samples of control TE versus those of TGR TE.
Extended Data Fig. 5. 6h U-13C-glucose re-exposure following TGR.

WT CD8+ T cells isolated from spleens of C7Bl/6 mice were activated with anti-CD3 (5 mg/mL), anti-CD28 (0.5 mg/mL), IL-2 (100 U/mL), expanded for a total of 72 hours, and exposed to 10mM (black) or 1mM (orange) glucose. Cells were re-exposed to 10mM U-13C-glucose for 6h prior to polar metabolite extraction and analysis by LC-MS. 20h cultures were started at 1×106 (control) and 1.5×106 (TGR) cells per/ml to generate similar end concentrations for glucose pulse experiments. (a) Percent 13C label incorporation into nucleotide monophosphates was analyzed. Data are from n=3 biological replicates. Significance was calculated using 2-tailed Student’s t tests. * p<0.05. (b) Relative 13C label incorporation into serine was analyzed. Data are from n=3 biological replicates. Significance was calculated using 2way ANOVA. Significance is indicated for the 13C portion (open bars). *** p<0.001. The 12C portion (filled bars) was also significant. ** p<0.01. (c) The GSH/GSSG ratio was calculated from summed isotopologues for each metabolite. Data are from n=3 biological replicates. Significance was calculated using 2-tailed Student’s t tests, no significant difference was found. (d) Relative 13C label incorporation into lactate was analyzed. Data are from n=3 biological replicates. Significance was calculated using 2way ANOVA. Significance is indicated for the 13C portion (open bars), which was not significant. The 12C portion (filled bars) was significant. *** p<0.001. All error bars show SEM.
Extended Data Fig. 6. TGR enhances CD8+ tumour-specific antitumour function in vivo.

As depicted in Fig. 6f, CD45.1 female recipient mice were injected subcutaneously with 1×106 B16-OVA melanoma cells and tumors established for 5 days. 5 ×106 control (black) or TGR (orange) OT-I+ TE were injected intravenously per tumor bearing mouse. Mice receiving no TE were used as control. 3, 6 and 9 days after tumor inoculation, mice received 200 μg anti-PD-1 antibody or IgG2a isotype control in 100 μl PBS intraperitoneally. (a) 21 days after tumor inoculation mice were humanely euthanized, blood was collected, red cells were lysed, and the white blood cell fraction stained for congenic markers. Data are presented as % of donor-derived (CD45.2+) CD8+ T cells as a fraction to total circulating CD8+ T cells and each dot represents an individual mouse (n=5). Statistical significance was calculated by 2-tailed Student’s t test. * p<0.05; ** p<0.01. (b) 21 days after tumor inoculation mice were humanely euthanized and the tumor mass was excised. Single cell suspensions were analyzed by flow cytometry. The number of host (CD45.1) CD8+ T cells per mg tissue was quantified and each dot represents an individual mouse (n=5). Statistical significance was calculated by 2-tailed Student’s t test. ns not significant. All error bars show SEM.
Supplementary Material
Acknowledgements
We thank members of the Pearce laboratories for support and helpful discussions and Andrea Quintana, and John Sutherland mouse colony management. This work was funded by the NIH (CA181125 to E.L.P. and AI110481 to E.J.P.), and the Max Planck Society. R.Z. was supported by the DFG (SFB1160, B09, TRR167, B06) and ERC (GVHD Cure n° 681012).
Footnotes
Competing Interests Statement
E.L.P. is an SAB member of ImmunoMet Therapeutics, and E.L.P and E.J.P. are Founders of Rheos Medicines. The other authors declare no competing interests.
References
- 1.Michalek RD et al. in J Immunol Vol. 186 3299–3303 (American Association of Immunologists, 2011).21317389 [Google Scholar]
- 2.Maciver NJ et al. in J. Leukoc. Biol Vol. 84 949–957 (Society for Leukocyte Biology, 2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chang C-H et al. in Cell Vol. 162 1229–1241 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chang C-H et al. in Cell Vol. 153 1239–1251 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blagih J. et al. The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity 42, 41–54, doi: 10.1016/j.immuni.2014.12.030 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Rolf J. et al. AMPKalpha1: a glucose sensor that controls CD8 T-cell memory. Eur J Immunol 43, 889–896, doi: 10.1002/eji.201243008 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klein Geltink RI et al. Mitochondrial Priming by Cd28. Cell 171, 385–397 e311, doi: 10.1016/j.cell.2017.08.018 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buck MD et al. Mitochondrial Dynamics Controls T Cell Fate through Metabolic Programming. Cell 166, 63–76, doi: 10.1016/j.cell.2016.05.035 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sukumar M. et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Invest 123, 4479–4488, doi: 10.1172/JCI69589 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sukumar M. et al. in Cell Metabolism Vol. 23 63–76 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scharping NE et al. in Immunity Vol. 45 374–388 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chowdhury PS, Chamoto K, Kumar A. & Honjo T. PPAR-Induced Fatty Acid Oxidation in T Cells Increases the Number of Tumor-Reactive CD8. Cancer Immunol Res 6, 1375–1387, doi: 10.1158/2326-6066.CIR-18-0095 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Buck MD, Sowell RT, Kaech SM & Pearce EL in Cell Vol. 169 570–586 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qiu J. et al. Acetate Promotes T Cell Effector Function during Glucose Restriction. Cell Rep 27, 2063–2074.e2065, doi: 10.1016/j.celrep.2019.04.022 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Crompton JG, Sukumar M. & Restifo NP Targeting Akt in cell transfer immunotherapy for cancer. Oncoimmunology 5, e1014776, doi: 10.1080/2162402X.2015.1014776 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nabe S. et al. Reinforce the antitumor activity of CD8. Cancer Sci 109, 3737–3750, doi: 10.1111/cas.13827 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hosios AM et al. in Dev. Cell Vol. 36 540–549 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garcia D. & Shaw RJ AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol Cell 66, 789–800, doi: 10.1016/j.molcel.2017.05.032 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jacobs SR et al. in J Immunol Vol. 180 4476–4486 (American Association of Immunologists, 2008).18354169 [Google Scholar]
- 20.Macintyre AN et al. in Cell Metabolism Vol. 20 61–72 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Siska PJ et al. in J Immunol Vol. 197 2532–2540 (American Association of Immunologists, 2016).27511728 [Google Scholar]
- 22.Saxton RA & Sabatini DM in Cell Vol. 168 960–976 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Waypa GB et al. Hypoxia triggers subcellular compartmental redox signaling in vascular smooth muscle cells. Circ Res 106, 526–535, doi: 10.1161/CIRCRESAHA.109.206334 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schieber M. & Chandel NS ROS function in redox signaling and oxidative stress. Curr Biol 24, R453–462, doi: 10.1016/j.cub.2014.03.034 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hosios AM & Vander Heiden MG The redox requirements of proliferating mammalian cells. J Biol Chem 293, 7490–7498, doi: 10.1074/jbc.TM117.000239 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kong H. & Chandel NS Regulation of redox balance in cancer and T cells. J Biol Chem 293, 7499–7507, doi: 10.1074/jbc.TM117.000257 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mak TW et al. in Immunity Vol. 46 675–689 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Smith CA, Want EJ, O’Maille G, Abagyan R. & Siuzdak G. XCMS: processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal Chem 78, 779–787, doi: 10.1021/ac051437y (2006). [DOI] [PubMed] [Google Scholar]
- 29.Huang X. et al. X13CMS: global tracking of isotopic labels in untargeted metabolomics. Anal Chem 86, 1632–1639, doi: 10.1021/ac403384n (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ma EH et al. in Cell Metab. Vol. 25 345–357 (2017). [DOI] [PubMed] [Google Scholar]
- 31.Crompton JG et al. Akt inhibition enhances expansion of potent tumor-specific lymphocytes with memory cell characteristics. Cancer Res 75, 296–305, doi: 10.1158/0008-5472.CAN-14-2277 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vander Heiden MG, Cantley LC & Thompson CB Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033, doi: 10.1126/science.1160809 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Warburg O. in The Journal of Cancer Research Vol. 9 148–163 (American Association for Cancer Research Journals, 1925). [Google Scholar]
- 34.Liberti MV & Locasale JW in Trends Biochem. Sci Vol. 41 211–218 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao J, Song Y. & Liu D. Clinical trials of dual-target CAR T cells, donor-derived CAR T cells, and universal CAR T cells for acute lymphoid leukemia. J Hematol Oncol 12, 17, doi: 10.1186/s13045-019-0705-x (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sena LA et al. in Immunity Vol. 38 225–236 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nathan C. & Cunningham-Bussel A. Beyond oxidative stress: an immunologist’s guide to reactive oxygen species. Nat Rev Immunol 13, 349–361, doi: 10.1038/nri3423 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chatterjee S. et al. CD38-NAD +Axis Regulates Immunotherapeutic Anti-Tumor T Cell Response. Cell Metab 27, 85–100.e108, doi: 10.1016/j.cmet.2017.10.006 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Johnson MO et al. Distinct Regulation of Th17 and Th1 Cell Differentiation by Glutaminase-Dependent Metabolism. Cell 175, 1780–1795.e1719, doi: 10.1016/j.cell.2018.10.001 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ghergurovich JM et al. A small molecule G6PD inhibitor reveals immune dependence on pentose phosphate pathway. Nat Chem Biol, doi: 10.1038/s41589-020-0533-x (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ma EH et al. Metabolic Profiling Using Stable Isotope Tracing Reveals Distinct Patterns of Glucose Utilization by Physiologically Activated CD8. Immunity 51, 856–870.e855, doi: 10.1016/j.immuni.2019.09.003 (2019). [DOI] [PubMed] [Google Scholar]
- 42.Yang JC & Rosenberg SA Adoptive T-Cell Therapy for Cancer. Adv Immunol 130, 279–294, doi: 10.1016/bs.ai.2015.12.006 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chamoto K. et al. Mitochondrial activation chemicals synergize with surface receptor PD-1 blockade for T cell-dependent antitumor activity. Proc Natl Acad Sci U S A 114, E761–E770, doi: 10.1073/pnas.1620433114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kumar A, Chamoto K, Chowdhury PS & Honjo T. Tumors attenuating the mitochondrial activity in T cells escape from PD-1 blockade therapy. Elife 9, doi: 10.7554/eLife.52330 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods-only References
- 45.Wills J, Edwards-Hicks J. & Finch AJ AssayR: A Simple Mass Spectrometry Software Tool for Targeted Metabolic and Stable Isotope Tracer Analyses. Anal Chem 89, 9616–9619, doi: 10.1021/acs.analchem.7b02401 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van der Windt GJW, Chang CH & Pearce EL Measuring Bioenergetics in T Cells Using a Seahorse Extracellular Flux Analyzer. Curr Protoc Immunol 113, 3.16B.11–13.16B.14, doi: 10.1002/0471142735.im0316bs113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pearce EL et al. in Nature Vol. 460 103–107 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Butler A, Hoffman P, Smibert P, Papalexi E. & Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36, 411–420, doi: 10.1038/nbt.4096 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Diaz-Papkovich A, Anderson-Trocme L, Ben-Eghan C. & Gravel S. UMAP reveals cryptic population structure and phenotype heterogeneity in large genomic cohorts. PLoS Genet 15, e1008432, doi: 10.1371/journal.pgen.1008432 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Aoki-Kinoshita KF & Kanehisa M. Gene annotation and pathway mapping in KEGG. Methods Mol Biol 396, 71–91, doi: 10.1007/978-1-59745-515-2_6 (2007). [DOI] [PubMed] [Google Scholar]
- 51.Szklarczyk D. et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 47, D607–D613, doi: 10.1093/nar/gky1131 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon request. Single cell RNA sequencing data has been deposited in the Gene Ontology Omnibus under accession number GSE152018.