

Abstract
The APOE4 allele is the strongest genetic risk factor for late-onset Alzheimer’s disease (AD). The contribution of microglial APOE4 to AD pathogenesis is unknown, although APOE has the most enriched gene expression in neurodegenerative microglia (MGnD). Here, we show in mice and humans a negative role of microglial APOE4 in the induction of MGnD response to neurodegeneration. Deletion of microglial APOE4 restores the MGnD phenotype associated with neuroprotection in P301S tau transgenic mice and decreases pathology in APP/PS1 mice. MGnD-astrocyte cross-talk associated with β-amyloid (Aβ) plaque encapsulation and clearance are mediated via LGALS3 signaling following microglial APOE4 deletion. In the brain of AD donors carrying the APOE4 allele, we found a sex-dependent reciprocal induction of AD risk factors associated with suppression of MGnD genes in females, including LGALS3, compared to individuals homozygous for the APOE3 allele. Mechanistically, APOE4-mediated induction of ITGB8-transforming growth factor-β (TGFβ) signaling impairs the MGnD response via upregulation of microglial homeostatic checkpoints, including Inpp5d, in mice. Deletion of Inpp5d in microglia restores MGnD-astrocyte cross-talk and facilitates plaque clearance in APP/PS1 mice. We identify the microglial APOE4-ITGB8-TGFβ pathway as a negative regulator of microglial response to AD pathology, and restoring the MGnD phenotype via blocking ITGB8-TGFβ signaling provides a promising therapeutic intervention for AD.
Genome-wide association meta-analysis studies (GWASs)1, 2 and interactome investigation3 identified microglia gene expression patterns to be strongly correlated with late onset Alzheimer’s disease (AD). We identified that neurodegenerative microglia (MGnD)4, also referred to as disease associated microglia5, are regulated by the reciprocal suppression of transforming growth factor β (TGFβ) and induction of apolipoprotein E (APOE) signaling in different neurodegenerative models including AD4. The APOE4 allele is a major genetic risk factor for late-onset AD and was previously shown to accelerate AD progression in humans and in mouse models6, 7, 8, 9, 10, 11. In the central nervous system, APOE is upregulated in MGnD4, 5. A recent report showed that deletion of APOE4 in astrocytes markedly reduced neurodegeneration in a mouse model of tauopathy12. Several studies investigated the molecular signature of microglia in mice globally expressing APOE410, 11; however, the cell-autonomous role of APOE4 expressed by microglia in the regulation of AD pathology has not been addressed.
Results
APOE4 impairs microglial response to neurodegeneration
To study the role of APOE variants in regulating the microglial signature, we sorted microglia from APOE3-knock-in (APOE3-KI) and APOE4-KI naïve mice at 4 months of age. Bulk RNA-sequencing (RNA-seq) analysis of isolated microglia identified significant upregulation of homeostatic genes in APOE4-expressing microglia compared to in APOE3-expressing microglia at 4 months of age, including Tgfbr2, Inpp5d, Spi1 and Smad3 (Fig. 1a,b and Supplementary Table 1). Epigenetic analysis of genomic DNA bound to lysine 9 acetylated histone 3 (H3K9ac) by anti-H3K9ac chromatin immunoprecipitation sequencing (ChIP-seq) analysis of isolated microglia revealed that APOE4-expressing microglia displayed reduced chromatin acetylation compared to APOE3-expressing microglia (Extended Data Fig. 1a and Supplementary Table 1). Furthermore, genomic regions associated with a homeostatic microglia signature, such as Inpp5d, Havcr2 and Smad3, showed enrichment for H3K9ac in APOE4-expressing microglia compared to in APOE3-expressing microglia (Extended Data Fig. 1b and Supplementary Table 1). To study the effect of APOE variants on the microglial response to acute neurodegeneration, we injected apoptotic neurons into the brains of APOE3-KI and APOE4-KI mice and sorted phagocytic and non-phagocytic microglia from the injection sites (Fig. 1c). Fluorescence-activated cell sorting (FACS) analysis of the injection site using the microglia specific marker FCRLS4, 13, 14 demonstrated impaired phagocytosis of dead neurons by FCRLS+CD11b+ APOE4-expressing microglia compared with APOE3-expressing microglia (Fig. 1d,e). APOE3-expressing phagocytic microglia showed induction of an MGnD profile compared to non-phagocytic microglia isolated from the same mice (Fig. 1f,g and Supplementary Table 1). APOE4-expressing phagocytic microglia, however, failed to induce an MGnD response of key genes, including Clec7a, Itgax, Lilr4b, Lpl, Lgals3, and Spp1 (Fig. 1g, Extended Data Fig. 1c–e, and Supplementary Table 1). Moreover, APOE4-expressing microglia failed to upregulate the expression of genes associated with phagocytosis, antigen presentation, interferon-γ (IFNγ) signaling and autophagosome maturation in response to phagocytosis of apoptotic neurons (Fig. 1h and Supplementary Table 1). Ingenuity pathway analysis (IPA) comparing phagocytic to non-phagocytic APOE3- and APOE4-expressing microglia revealed a dysfunctional response of APOE4-expressing microglia to phagocytosis of dead neurons and antigen presentation (Extended Data Fig. 1f and Supplementary Table 1). Immunostaining of microglia in the injection site showed accumulation of LAMP1+ lysosomes in IBA1+ APOE4-expressing microglia compared to in APOE3-expressing microglia (Fig. 1i,j). Furthermore, APOE4-expressing microglia demonstrated an impaired response to acute neurodegeneration and exhibited reduced phagocytic IBA1+ cell numbers in the injection site compared to APOE3-expressing microglia (Fig. 1i and Extended Data Fig. 1g,h). The reduction in LGALS3 immunoreactivity, a key MGnD marker4, 14, was also observed in APOE4 mice compared to APOE3 mice in response to apoptotic neurons (Fig. 1k,l). To address the cell-autonomous role of microglial APOE variants, we crossed Cx3cr1CreERT2 mice15 with APOE3-KIfl/fl (APOE3-conditonal knockout (cKO)) and APOE4-KIfl/fl (APOE4-cKO) mice, which were recently described and used to study the role of APOE variants in astrocytes in tauopathy12. APOE3-KIfl/fl and APOE4-KIfl/fl mice expressing Cx3cr1CreERT2/WT and Cx3cr1WT/WT genes were treated with tamoxifen at the age of 1.5 months to conditionally delete APOE variants in microglia. Of note, using Cx3cr1CreERT2 mice to delete microglia genes may also affect border associated macrophages (BAMs)16, albeit to a lesser extent as BAMs were shown to be continuously replenished from skull bone marrow unlike microglia17. FACS analysis of cells isolated from the injection site showed increased recruitment of FCRLS+CD11b+ microglia in APOE4-cKO mice compared to APOE4-KI mice (Fig. 1m,n). Deletion of microglial APOE4 restored the expression of key MGnD genes and downregulation of homeostatic genes in response to phagocytosis of apoptotic neurons (Fig. 1o, Extended Data Fig. 1i, and Supplementary Table 1). Furthermore, LGALS3 immunoreactivity was increased in the injection site of APOE4-cKO mice compared to APOE4-KI mice (Fig. 1p,q). NicheNetR analysis of differentially regulated ligands in phagocytic microglia and their autocrine interactions with microglial receptors identified LGALS3 as one of the top ligands that is highly affected by APOE4 deletion (Extended Data Fig. 1i and Supplementary Table 1).
Fig. 1. APOE4 impairs the microglial response to acute neurodegeneration.
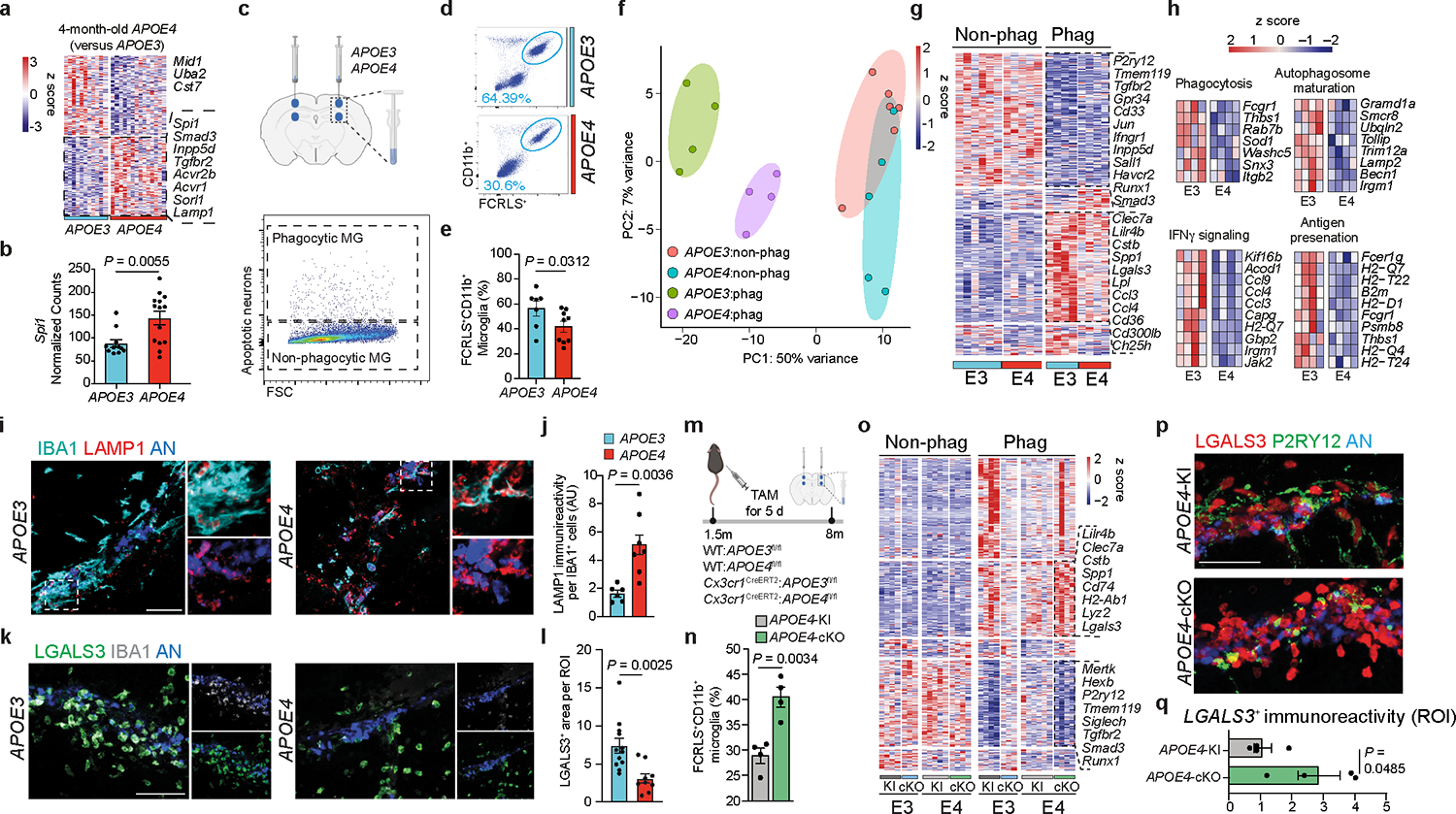
a, Heat map of differentially expressed genes from APOE4-KI vs. APOE3-KI microglia at 4 months of age (n = 11–14 mice per group, P < 0.05). b, Spi1 normalized counts (n = 11–14 mice per group). c, Top, schematics of apoptotic neurons injection to the cortex and hippocampus of 8-months-old APOE3-KI and APOE4-KI mice; Bottom, sorting strategy 16 h after injection of phagocytic and non-phagocytic microglia (MG) for labeled apoptotic neurons; Figure created with Biorender.com; FSC, forward scatter. d, Gating strategy for FCRLS+CD11b+ microglia from the apoptotic neuron injection site in APOE3-KI and APOE4-KI mice. e, Bar plot showing the percentage of FCRLS+CD11b+ cells (n = 7 APOE3-KI mice, n = 9 APOE4-KI mice). f, Principal component analysis (PCA) of each group; APOE3:non-phag, APOE4:non-phag; APOE3:phag, APOE4:phag. g, Heat map of phagocytic and non-phagocytic microglia from APOE3-KI (E3) and APOE4-KI (E4) mice. Differentially expressed genes were identified using DESeq2 analysis with a likelihood ratio test (LRT; n = 4–6 mice per group, P < 0.05). h, Gene ontology analysis of differentially expressed genes for phagocytosis, autophagosome maturation, IFNγ signaling and antigen presentation (P < 0.05). i, Confocal microscopy images of IBA1, LAMP1 and apoptotic neurons (AN) at the injection sites. j, Quantification of LAMP1 immunoreactivity per IBA1+ cell (n = 6 APOE3-KI mice, n = 7 APOE4-KI mice); AU, arbitrary units. k, Confocal microscopy images of IBA1, LGALS3 and apoptotic neurons at the injection sites. l, Quantification of LGALS3+ area per region of interest (ROI) at the injection site (n = 12 ROIs from the APOE3-KI group, n = 9 ROIs from the APOE4-KI group). m, Schematics of tamoxifen (TAM) administration at 1.5 months of age and the injection of apoptotic neuron to cortex and hippocampus of 8-months-old APOE3-KI, APOE4-KI, APOE3-cKO and APOE4-cKO mice. Figure created with Biorender.com. n, Percentage of FCRLS+CD11b+ microglia in the injection site in APOE4-KI and APOE4-cKO mice (n = 4 mice per group). o, Heat map of non-phagocytic and phagocytic microglia isolated from APOE3-KI, APOE3-cKO, APOE4-KI, and APOE4-cKO injected with apoptotic neurons. Differentially expressed genes were identified using DESeq2 analysis with an LRT (n = 3–6 mice per group, P < 0.05). p, Confocal images of P2RY12, LGALS3, and apoptotic neurons from APOE4-KI and APOE4-cKO mice. q, Quantification of LGALS3+ area per ROI at injection sites (n = 4 mice per group). Data were analyzed by two-tailed Student’s t-test. Scale bars, 50 μm. Data are presented as mean ± s.e.m.
APOE4 impairs microglial response via PU.1
Spi1, also known as PU.1, binds SMAD3 to establish a homeostatic gene-regulatory-landscape in microglia18. Importantly, low expression of PU.1 was suggested by a recent GWAS to be protective against AD19. To address whether high expression of Spi1 in APOE4-expressing microglia (Fig. 1a) maintains a homeostatic signature, we conditionally deleted microglial Spi1 in 2-month-old mice using an MGnD paradigm4 (Fig. 2a). Deletion of Spi1 in microglia enhanced expression of MGnD genes including Clec7a and Lgals3, whereas homeostatic genes were downregulated, including Tgfb1, Cd33 and Inpp5d (Fig. 2b and Supplementary Table 2). Moreover, by comparing Spi1 deletion and APOE4 deletion in phagocytic microglia (Fig. 1o), we identified common MGnD and homeostatic genes induced and downregulated, respectively, in both conditions (Fig. 2c). To validate the enhanced MGnD response to amyloid pathology, we crossed APP/PS1:Tmem119CreERT2 to Spi1fl/WT mice to evaluate plaque pathology at 4 months of age after tamoxifen administration at 1.5 months of age (Fig. 2d,e). Microglial Spi1 deletion in APP/PS1 mice resulted in reduction in plaque load and enhanced CLEC7A+ MGnD around plaques (Fig. 2d–g). RNA-seq analysis of microglia isolated from APP/PS1:APOE4-KI mice treated with PU.1 pharmacological inhibitors showed an induction of the MGnD signature and suppression of homeostatic genes (Fig. 2h). Immunohistochemistry showed enhanced CLEC7A+ MGnD response and increased astrocytic activation, as determined by Serpina3n expression around Aβ-plaques (Fig. 2i–m and Supplementary Table 2). These findings indicate that APOE4 predisposes microglia towards a homeostatic signature that interferes with microglial responses to acute neurodegeneration, and targeting microglial APOE4 or PU.1 can restore MGnD induction.
Fig. 2. APOE4 impairs the microglial response to neurodegeneration via PU.1.
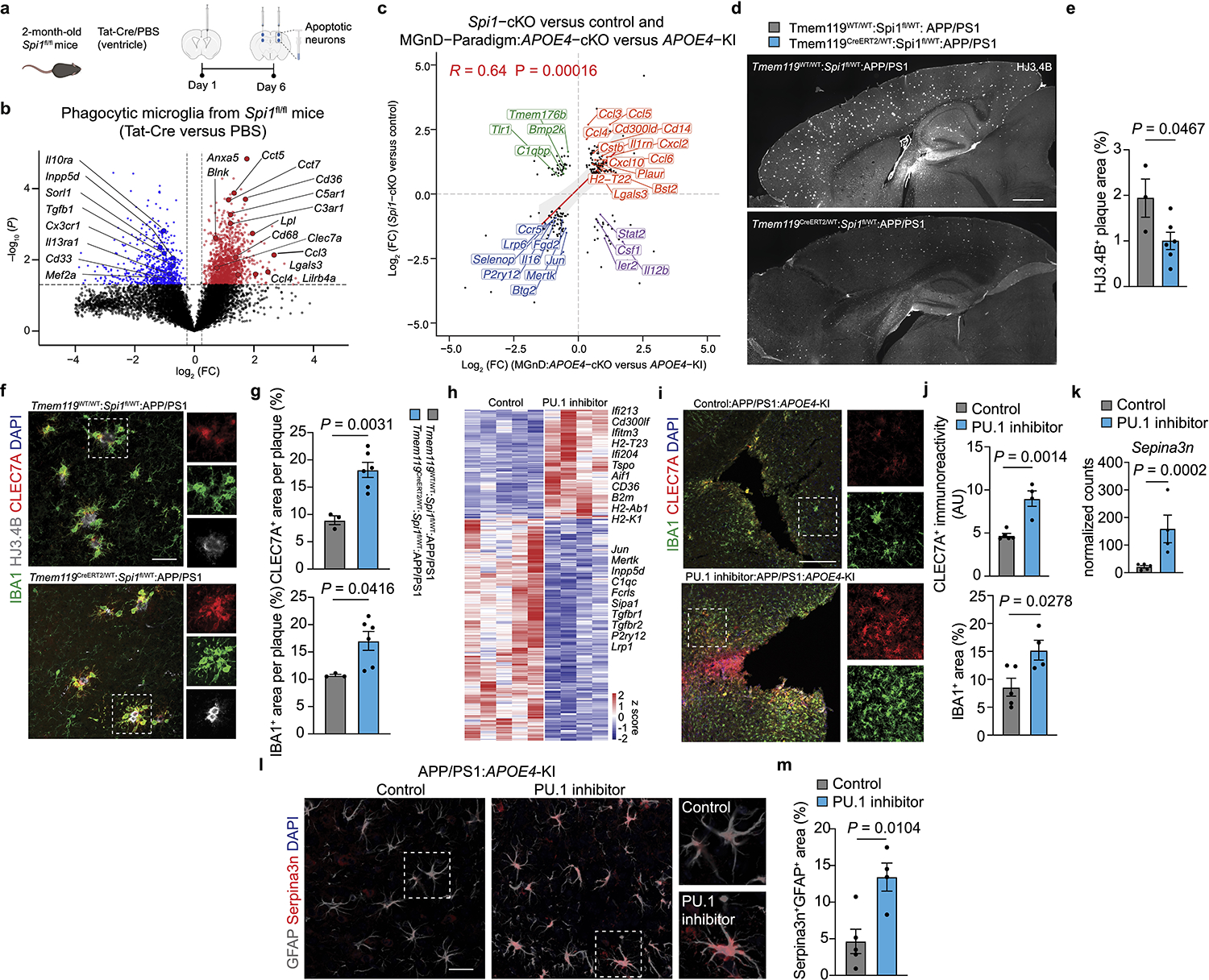
a, Schematics of Tat-Cre or PBS injection to the brains of Spi1fl/fl mice followed by injection of apoptotic neurons. Figure created with Biorender.com. b, Volcano plot showing differentially expressed genes in phagocytic microglia isolated from Spi1fl/fl mice treated with Tat-Cre or PBS. Differentially expressed genes were identified using DESeq2 analysis with an LRT (n = 5 mice per group, P < 0.05). c, Scatter plot comparing the differentially expressed genes of microglia from Spi1-cKO and APOE4-cKO mice. Differentially expressed genes were identified using DESeq2 analysis with an LRT (P < 0.05, log2 (fold change) (log2 (FC) of >0.25 or <–0.25). d, Representative images of brain sections from Tmem119WT/WT:Spi1fl/WT:APP/PS1 and Tmem119CreERT2/WT:Spi1fl/WT:APP/PS1 mice stained for HJ3.4B; scale bar, 1mm. e, Quantification of HJ3.4B+ area per ROI (n = 3 Tmem119WT/WT:Spi1fl/WT:APP/PS1 mice, n = 6 Tmem119CreERT2/WT:Spi1fl/WT:APP/PS1 mice). f, Confocal images of CLEC7A, HJ3.4B and IBA1 in Tmem119WT/WT:Spi1fl/WT:APP/PS1 and Tmem119CreERT2/WT:Spi1fl/WT:APP/PS1 mice; scale bar, 50 μm. g, Quantification of CLEC7A+ and IBA1+ area per plaque (n = 3 Tmem119WT/WT:Spi1fl/WT:APP/PS1 mice, n = 6 Tmem119CreERT2/WT:Spi1fl/WT:APP/PS1 mice). h, Heat map of differentially expressed genes from microglia isolated from PU.1 inhibitor versus vehicle control-injected APP/PS1:APOE4-KI mice. Differentially expressed genes were identified using DESeq2 analysis with an LRT (n = 4–5 mice per group, P < 0.05). i, Confocal images of IBA1 and CLEC7A in PU.1 inhibitor- and control-injected APP/PS1:APOE4-KI mice; scale bar, 200 μm. j, Quantification of CLEC7A immunoreactivity and IBA1+ area per ROI (n = 5 mice from control group, n = 4 mice from PU.1 inhibitor group). k, Normalized counts of Serpina3n in astrocytes isolated from PU.1 inhibitor- and vehicle control-injected APP/PS1:APOE4-KI mice (n = 5 mice from the control group, n = 4 mice from the PU.1 inhibitor group). l, Confocal images of GFAP and Serpina3n in PU.1 inhibitor- and vehicle control-injected APP/PS1:APOE4-KI mice; scale bar, 50 μm. m, Quantification of Serpina3n+GFAP+ area per ROI (n = 5 mice from the control group, n = 4 mice from the PU.1 inhibitor group). Data were analyzed by two-tailed Student’s t-test and are presented as mean ± s.e.m.
Deletion of microglial APOE4 restores protective MGnD
Neurodegeneration is a pathological hallmark of AD pathology20, which is exacerbated in a mouse model of tauopathy expressing APOE412, 21. To dissect the impact of microglial APOE variants on tau-induced neurodegeneration, Cx3cr1CreERT2/WT:APOE3-KIfl/fl and Cx3cr1CreERT2/WT:APOE4-KIfl/fl mice were crossed to P301S mice, which develop tau pathology and neurodegeneration between 6 to 9.5 months of age22. These mice were treated with tamoxifen at the age of 1.5 months and evaluated at the age of 9 months (Fig. 3a). Quantitative reverse transcription PCR (qPCR) analysis confirmed the deletion of the human APOE gene in microglia expressing Cx3cr1CreERT2 (Fig. 3b), with no significant difference in APOE immunoreactivity comparing P301S:APOE3-KI and P301S:APOE4-KI (Extended Data Fig. 2a,b). Microglia sorted from the brains of P301S:APOE3-KI mice at 9 months of age demonstrated upregulation of key MGnD genes, including Clec7a, Lgals3 and Itgax, and downregulation of TGFβ-signaling molecules, including Smad3 and Tgfb1, compared to microglia isolated from WT:APOE3-KI mice (Fig. 3c and Supplementary Table 3). By contrast, microglia from P301S:APOE4-KI mice showed reduced MGnD and an increased TGFβ-signaling signature compared to microglia from age-matched P301S:APOE3-KI mice (Fig. 3c and Supplementary Table 3). Immunohistochemistry confirmed decreased CLEC7A immunoreactivity in cortical microglia of P301S:APOE4-KI mice, which exhibited increased tau hyperphosphorylation compared to age-matched P301S:APOE3-KI mice (Fig. 3d,e). In previous studies, brain regions of P301S:APOE4-KI mice demonstrate marked neurodegeneration compared to P301S:APOE3-KI mice at 9.5 months of age, although the MGnD phenotype was similar between the two groups12, 21. We found that microglial APOE4 deletion was sufficient to restore CLEC7A immunoreactivity and the MGnD signature in P301S mice, which was associated with reduced tau hyperphosphorylation (Fig. 3d–f and Supplementary Table 3). To determine the effect of microglial deletion of APOE variants on neurodegeneration, we quantified neurons stained with Cresyl Violet in cortical layer 5, as previously described in tau mice23. P301S:APOE4-KI mice showed increased neuronal loss in cortical layer 5 compared to P301S:APOE3-KI mice, whereas neuronal loss was significantly reduced in P301S mice with a deletion in microglial APOE4 compared to age-matched P301S:APOE4-KI mice (Fig. 3g,h). These results suggest that microglial expression of APOE4 is important for the MGnD response to tau-mediated neurodegeneration in the cortex and its deletion in microglia delays neuronal loss.
Fig. 3. Deletion of microglial APOE4 restores MGnD response to chronic neurodegeneration and promotes neuroprotection.
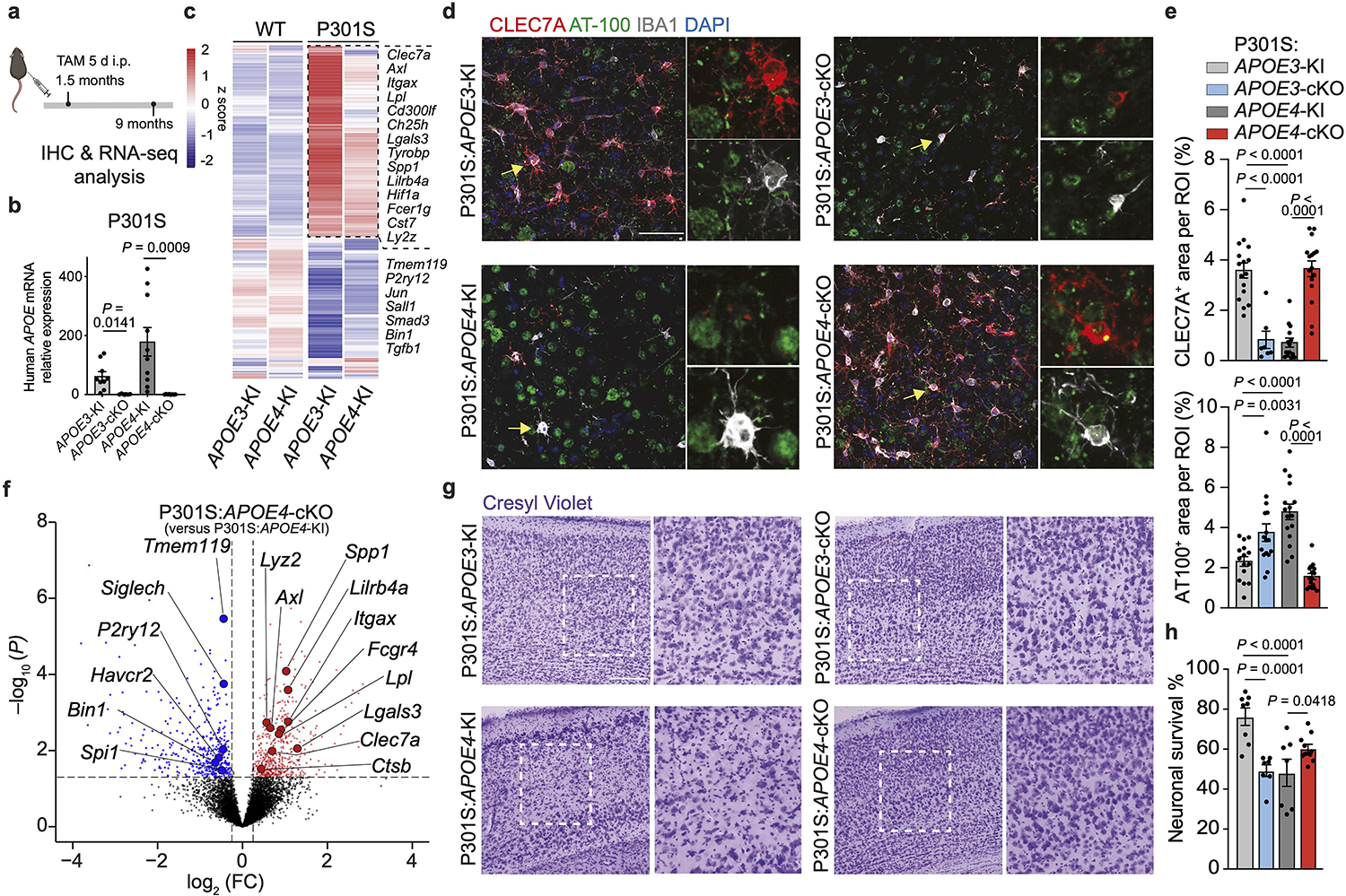
a, Schematics of tamoxifen administration at 1.5 months of age and analysis of P301S mice at 9 months of age; Figure created with Biorender.com; i.p., intraperitoneal. b, RT-qPCR validation of human APOE expression in sorted microglia (n = 9 P301S:APOE3-KI mice, n = 6 P301S:APOE3-cKO mice, n = 10 P301S:APOE4-KI mice, n = 8 P301S:APOE4-cKO mice). c, Differentially expressed genes of aggregated samples for WT and tau (P301S) mice with APOE variants. Differentially expressed genes were identified using DESeq2 analysis with an LRT (n = 3–11 mice per group, P < 0.01). d, Confocal images of CLEC7A, phospho-tau (AT-100) and IBA1. Arrowheads indicate CLEC7A+ microglia associated with phospho-tau in the cortex in P301S:APOE3-KI, P301S:APOE3-cKO, P301S:APOE4-KI, and P301S:APOE4-cKO mice; Scale bar, 50 μm. e, Quantification of CLEC7A+ area and AT-100+ area in the cortex (n = 16 ROIs from P301S:APOE3-KI mice, n = 7 ROIs from P301S:APOE3-cKO mice, n = 14 ROIs from P301S:APOE4-KI mice, n = 16 ROIs from P301S:APOE4-cKO mice). f, Volcano plots of differentially expressed genes of P301S:APOE4-cKO versus P301S:APOE4-KI mice. Differentially expressed genes were identified using DESeq2 analysis with an LRT (n = 3–8 mice per group, P < 0.05). g, Representative images of Cresyl Violet staining of P301S mice carrying different APOE variants. Dashed squares indicate area of interest; Scale bar, 200 μm. h, Quantification of cortical neurons in WT and P301S mice carrying different APOE variants (n = 8 P301S:APOE3-KI mice, n = 7 P301S:APOE3-cKO mice, n = 7 P301S:APOE4-KI mice, n = 10 P301S:APOE4-cKO mice). Data were analyzed by one-way analysis of variance (ANOVA) and are presented as mean ± s.e.m.
Deletion of microglial APOE4 mitigates AD pathology in mice
To investigate the cell-intrinsic regulation of APOE4 in microglia, we used APP/PS1 mice, which develop plaque pathology as early as 2 months of age24 crossed to Cx3cr1CreERT2/WT:APOE3-KIfl/fl and Cx3cr1CreERT2/WT:APOE4-KIfl/fl mice, referred as APP/PS1:APOE3-cKO and APP/PS1:APOE4-cKO, respectively. These mice were treated with tamoxifen at the age of 1.5 months and evaluated at the age of 4 months to validate the deletion of human APOE in microglia (Fig. 4a,b). RNA-seq analysis of sorted microglia showed restoration of the MGnD signature in APP/PS1:APOE4-cKO mice compared to APP/PS1:APOE4-KI mice (Fig. 4c and Supplementary Table 4). To characterize microglial response in APP/PS1 mice, we performed single-cell RNA-seq (scRNA-seq) of brain cells isolated from APP/PS1:APOE4-KI and APP/PS1:APOE4-cKO mice. Unsupervised clustering identified six clusters showing microglia-like expression profiles (Extended Data Fig. 3a–c and Supplementary Table 4). To distinguish peripheral recruited myeloid cells and BAMs from microglia, we reclustered all microglia-like cells and identified Ms4a7+ and Mrc1+ macrophages25, 26 and Lyve1+ BAMs27, 28 (Extended Data Fig. 3d–f and Supplementary Table 4) and identified three major subtypes, which we labeled as (1) M0 (homeostatic microglia), which express Tmem119, P2ry12 and Gpr34, (2) MGnD, which express Clec7a, Lgals3 and Spp1, and (3) IFN microglia, which are enriched for Stat1, Irf7, and Ilfit1 (Extended Data Fig. 3f and Supplementary Table 4). An increased proportion of Clec7ahi/Lgals3hi MGnD was detected following microglial deletion of APOE4 in APP/PS1 mice (Fig. 4d,e, Extended Data Fig. 3g,h, and Supplementary Table 4). Importantly, Clec7ahiLgals3hi microglia exhibited an enhanced MGnD signature compared to Clec7ahiLgals3lo microglia in APP/PS1:APOE4-cKO mice (Fig. 4f and Supplementary Table 4). The proportion of CLEC7A+ and LGALS3+ microglia associated with Aβ plaques was significantly reduced in APP/PS1:APOE4-KI mice, despite the increased plaque pathology in APP/PS1 mice expressing APOE4 compared to APOE3 (Fig. 4g–j). Deletion of microglial APOE4 in APP/PS1 mice resulted in increased recruitment of CLEC7A+ and LGALS3+ MGnD to Aβ plaques (Fig. 4g–j). The restoration of an MGnD phenotype in APP/PS1:APOE4-cKO was accompanied by a significant reduction in HJ3.4B+ Aβ plaque pathology (Fig. 4k,l) and LAMP1+ dystrophic neurites (Fig. 4m,n). Notably, the immunoreactivity of LAMP1+ dystrophic neurites was enhanced in microglia with an APOE3 deletion (Fig. 4n). To study whether APOE4-KI microglia display an altered metabolic profile, we performed lipidome analysis of sorted microglia from mice harboring APOE variants on APP/PS1 and wild-type (WT) backgrounds (Extended Data Fig. 4a). We detected increased levels of certain lipid species in APOE4-KI microglia compared to that observed in APOE3-KI microglia, which were more pronounced on the APP/PS1 background (Extended Data Fig. 4a and Supplementary Table 4). Immunohistochemistry confirmed the increased accumulation of Plin2+ lipid droplets in microglia in APP/PS1:APOE4-KI mice, which was reduced following the conditional deletion of APOE4 in microglia (Extended Data Fig. 4b,c). These results suggest a beneficial role of MGnD in limiting AD pathology, which is disrupted by APOE4 and the deletion of APOE4 in microglia is sufficient to restore MGnD response to phagocytic stress and neurodegeneration.
Fig. 4. Targeting microglial APOE4 restricts Aβ pathology in APP/PS1 mice.
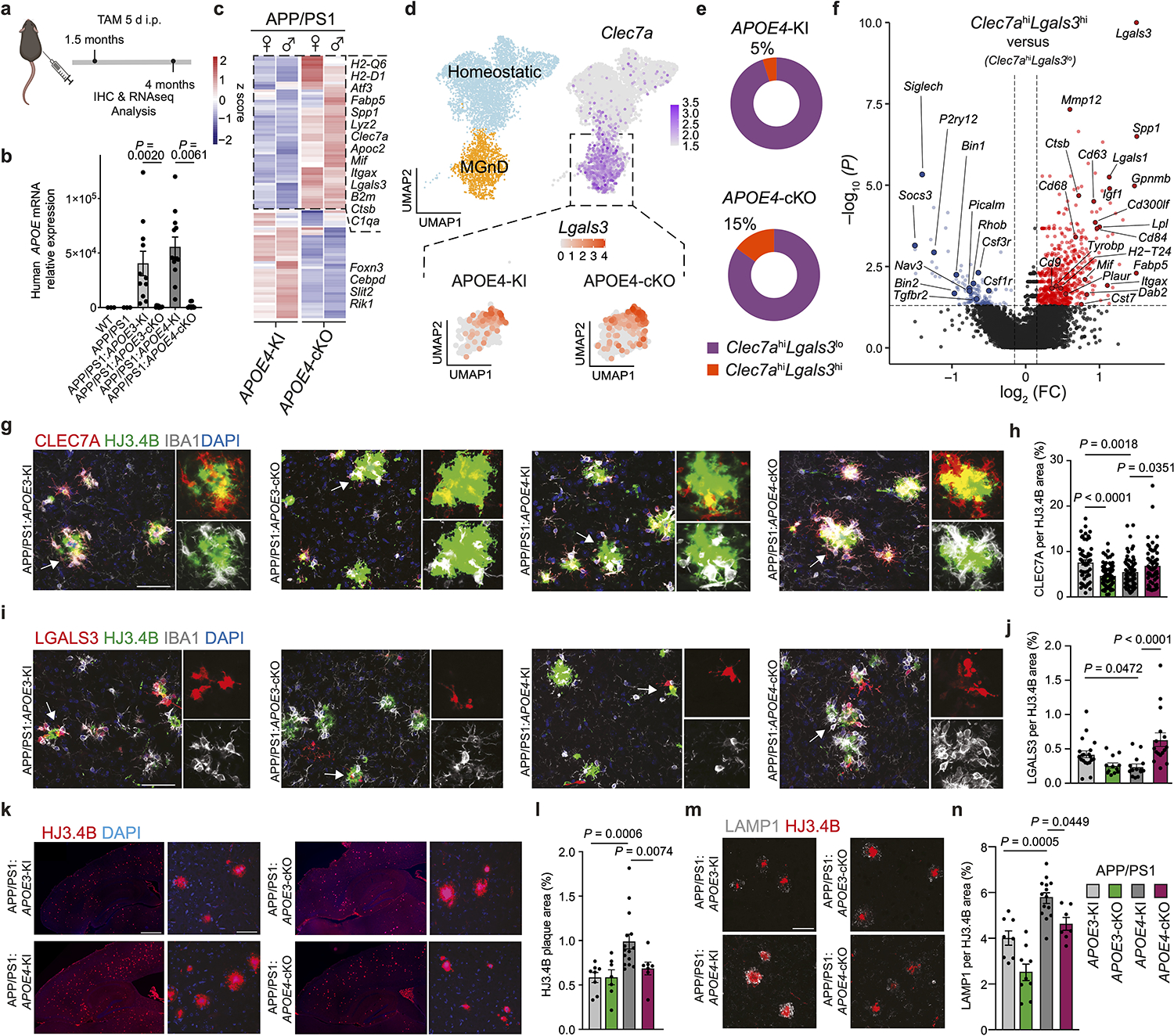
a, Schematics of tamoxifen administration at 1.5 months of age and analysis of APP/PS1 mice at 4 months of age; Figure created with Biorender.com. b, RT-qPCR validation of human APOE expression in sorted microglia (n = 3 WT mice, n = 3 APP/PS1 mice, n = 11 APP/PS1:APOE3-KI mice, n = 11 APP/PS1:APOE3-cKO mice, n = 12 APP/PS1:APOE4-KI mice, n = 8 APP/PS1:APOE4-cKO mice). c, Heat map showing the top 100 differentially expressed genes in microglia isolated from APP/PS1:APOE4-KI versus APP/PS1:APOE4-cKO mice. Differentially expressed genes were identified using DESeq2 analysis with an LRT (n = 5–9 mice per group, P < 0.01). d, Uniform manifold approximation and projection (UMAP) plot of scRNA-seq analysis of microglia from both APP/PS1:APOE4-KI and APP/PS1:APOE4-cKO mice indicating subclusters of homeostatic microglia and MGnD. Dashed box indicates Clec7a+ MGnD co-expressing Lgals3 (n = 2 mice per group). e, Donut charts showing the percentage of Clec7ahiLgals3hi and Clec7ahiLgals3lo microglia. f, Volcano plot comparing Clec7ahiLgals3hi and Clec7ahiLgals3lo microglia. Differentially expressed genes were identified using FindMarkers Seurat function (P < 0.05). g, Confocal images of CLEC7A, IBA1 and HJ3.4B (plaques) in APP/PS1:APOE3-KI, APP/PS1:APOE3-cKO, APP/PS1:APOE4-KI, and APP/PS1:APOE4-cKO mice. Arrowheads indicate magnified ROI; scale bar, 50 μm. h, Quantification of CLEC7A immunoreactivity per plaque. (n = 55 plaques (APP/PS1:APOE3-KI), n = 73 plaques (APP/PS1:APOE3-cKO) n = 62 plaques (APP/PS1:APOE4-KI), n = 62 plaques (APP/PS1:APOE4-cKO)). i, Confocal images of LGALS3, IBA1 and HJ3.4B; scale bar, 50 μm. j, Quantification of LGALS3 immunoreactivity. (n = 20 plaques (APP/PS1:APOE3-KI), n = 12 plaques (APP/PS1:APOE3-cKO), n = 14 plaques (APP/PS1:APOE4-KI), n = 16 plaques (APP/PS1:APOE4-cKO)). k, Representative images of brain sections from all APOE genotypes stained for HJ3.4B; scale bar, 500 μm (left) and 50 μm (right). l, Quantification of cortical HJ3.4B+ plaque number per ROI (n = 8 APP/PS1:APOE3-KI mice, n = 7 APP/PS1:APOE3-cKO mice, n = 14 APP/PS1:APOE4-KI mice, n = 7 APP/PS1:APOE4-cKO mice. m, Confocal images of LAMP1 and HJ3.4B; scale bar, 50 μm. n, Quantification of LAMP1+ area in cortex (n = 8 APP/PS1:APOE3-KI mice, n = 7 APP/PS1:APOE3-cKO mice, n = 14 APP/PS1:APOE4-KI mice, n = 7 APP/PS1:APOE4-cKO mice). Data were analyzed by one-way ANOVA and are presented as mean ± s.e.m.
MGnD activates astrocytic plaque encapsulation via LGALS3
The impact of microglial APOE4 on astrocyte phenotype and function is unknown. scRNA-seq analysis of brain cells isolated from APP/PS1:APOE4-KI vs. APP/PS1:APOE4-cKO mice identified two astrocyte clusters (3 and 5). Cluster 3 was enriched for reactive astrocyte genes, including Gfap, Vim, Cd9 and Serpina3n, compared to Cluster 529 (Fig. 5a, Extended Data Fig. 5a,b, and Supplementary Table 5). Microglial APOE4 deletion further induced expression of Apoe, Ttr, Cd9, Vim, Gfap, and Serpina3n in Cluster 3 (Fig. 5b). Furthermore, microglial APOE4 deletion increased the ratio of GfaphiApoehi, GfaphiSerpina3nhi, GfaphiVimhi, and GfaphiCd9hi astrocytes (Fig. 5c and Extended Data Fig. 5c). IPA of astrocytes from APP/PS1:APOE4-cKO mice showed upregulation of metabolic pathways, including cholesterol biosynthesis, response to oxidative stress, HIF1α and IGF-1 signaling (Fig. 5d). Immunohistochemistry showed decreased GFAP immunoreactivity around Aβ plaques in APP/PS1:APOE4-KI mice compared to that observed in APP/PS1:APOE3-KI mice, whereas APP/PS1:APOE4-cKO mice restored GFAP immunoreactivity (Fig. 5e,f). Immunostaining for human APOE protein and GFAP validated the deletion of APOE in IBA1+ microglia of APP/PS1:APOE3-cKO and APP/PS1:APOE4-cKO mice, despite the detection of APOE immunoreactivity in plaques and astrocytes (Fig. 5e). Moreover, APOE immunoreactivity in GFAP+ astrocytes was also restored in APP/PS1:APOE4-cKO mice (Fig. 5e,g), confirming results from the scRNA-seq analysis (Fig. 5b). Furthermore, expression of Serpina3n in GFAP+ astrocytes was reduced in APP/PS1:APOE3-cKO and APP/PS1:APOE4-KI mice compared to in APP/PS1:APOE3-KI mice (Fig. 5h,i), whereas deletion of APOE4 in microglia increased Serpina3n expression in GFAP+ astrocytes encapsulating Aβ plaques (Fig. 5h). To address direct microglia-astrocyte cross-talk affected by APOE3 versus APOE4 alleles expressed in microglia, we transplanted phagocytic MGnD from APOE3-KI, APOE4-KI and APOE4-cKO donor mice injected with apoptotic neurons to WT naive mice (Fig. 5j and Supplementary Table 5). Astrocytes were isolated from the injection sites 16 h later and were analyzed by bulk RNA-seq. We found reduced expression of Serpina3n and Gfap in mice injected with APOE4-MGnD compared to APOE3-MGnD (Fig. 5k). Importantly, microglial APOE4 deletion restored their cross-talk with astrocytes (Fig. 5l and Supplementary Table 5). IPA identified IGF-1 as the most activated upstream regulator, whereas TGFβ1 was the top suppressed upstream regulator in astrocytes in response to APOE4-cKO MGnD (Fig. 5m). To determine whether microglia-astrocyte cross-talk is affected by microglial APOE4 in the presence of amyloid pathology, we analyzed ligand-receptor interactions in APP/PS1:APOE4-cKO and APP/PS1:APOE4-KI mice. Cross-talk analysis identified LGALS3 as the top microglial ligand interacting with astrocyte receptors after deletion of APOE4 in microglia (Fig. 6a and Supplementary Table 5). Of note, Lgals3 expression was absent in astrocytes (Supplementary Table 4). To determine whether LGALS3 can induce astrocyte activation, we treated APP/PS1 mice with 2 different doses of recombinant LGALS3 protein (rLGALS3) or with PBS, injected to the hippocampus and cortex in APP/PS1 mice at the age of 4 months. RNA-seq analysis of astrocytes 3 d after rLGALS3 injection showed upregulation of Serpina3n and Vim29 in a dose-depended manner (Fig. 6b,c). rLGALS3 suppressed the astrocytic expression of Itgb8 and Itgav (subunits of integrin αvβ8)30 (Fig. 6b,c and Supplementary Table 5), which plays a critical role in TGFβ activation and microglia homeostatic maturation31. Immunohistochemistry of APP/PS1 mice 14 d after treatment with 100ng of rLGALS3 demonstrated increased astrocyte association with Aβ plaques and reduced plaque load compared to APP/PS1 mice treated with PBS (Fig. 6d,e). To address the role of microglial APOE4 in the regulation of astrocytes in response acute and chronic neurodegeneration4, we analyzed astrocytes sorted from mice described in Fig. 1m–o using bulk RNA-seq and from P301S:APOE4-cKO mice described in Fig. 3, respectively. These astrocytes showed marked overlap with Cluster 3 astrocytes from APP/PS1:APOE4-cKO mice, including reciprocal upregulation of Gfap, Serpina3n, Ttr, and Vim (Fig. 6f) and downregulation of Itgav and Tgfb1 (Fig. 6g, and Supplementary Table 5). NichNetR receptor-ligand analysis of microglia-astrocyte cross-talk in response to apoptotic neurons (Fig. 1m) identified LGALS3 as the top ligand (Fig. 6h). Importantly, LGALS3-mediated interactions with astrocytes were reduced in APOE4-expressing microglia phagocytosing apoptotic neurons, compared to APOE3 microglia, whereas microglial APOE4 deletion restored these interactions (Fig. 6h–j and Supplementary Table 5). Importantly, the injection of rLGALS3 into APP/PS1:APOE4-KI mice restored CLEC7A and GFAP induction and reduced plaque load (Fig. 6k,l). These findings support the critical role of MGnD expressing LGALS3 in promoting astrocytes activation and their recruitment to plaques.
Fig. 5. Targeting microglial APOE4 promotes astrocyte activation and their recruitment towards plaque in APP/PS1 mice.
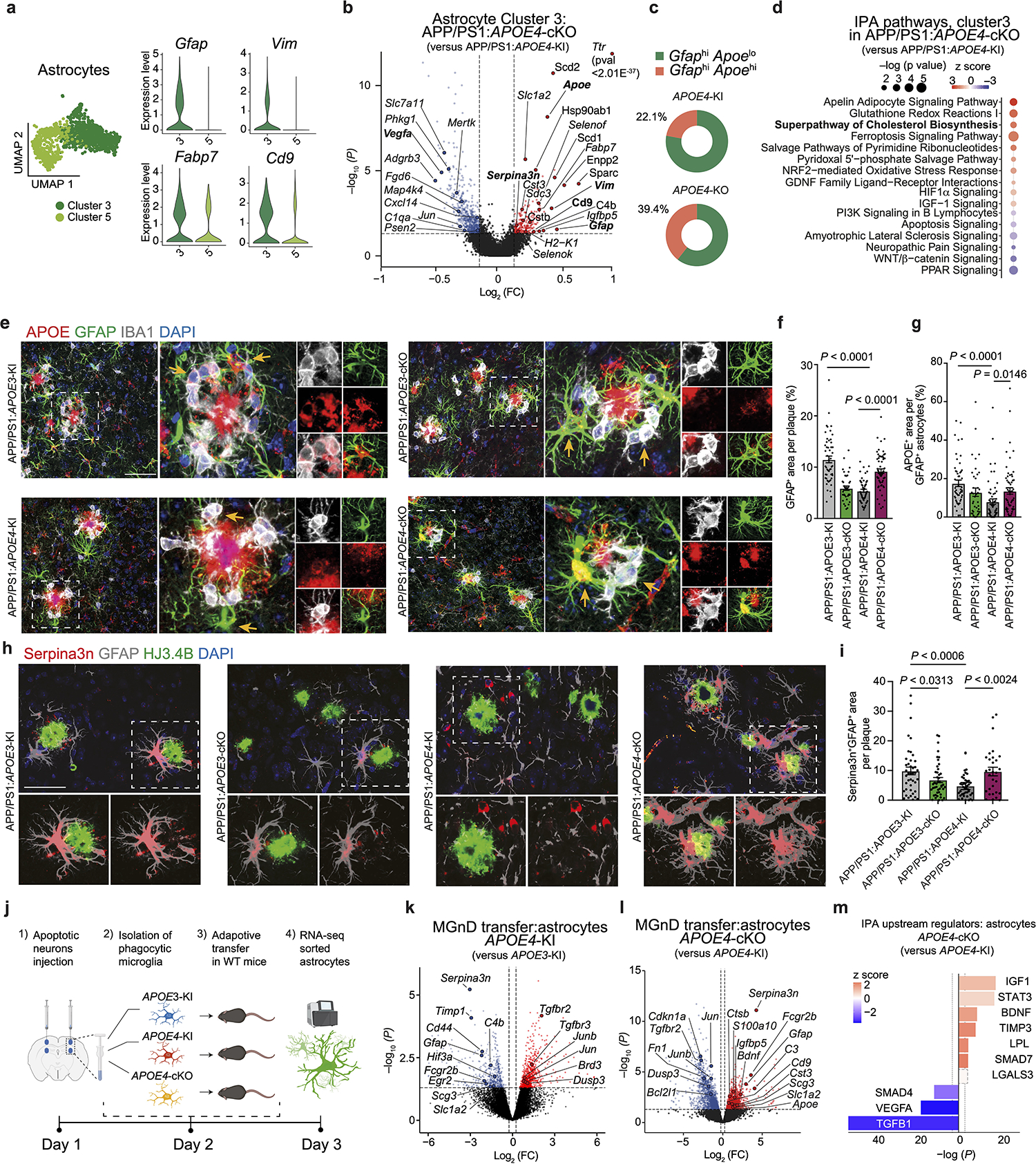
a, UMAP of scRNA-seq analysis of astrocytes showing clusters 3 and 5. Violin plots of key activation genes (Gfap, Vim and Cd9 representing clusters 3 and 5) are shown on the right. b, Volcano plot of cluster 3. c, Donut charts showing the percentage of GfaphiApoelo and GfaphiApoehi astrocyte clusters. d, Top upregulated canonical pathways in astrocytes from cluster 3 identified using IPA. e, Confocal images of GFAP, IBA1, and human APOE in the cortex of 4-month-old mice. Arrows indicate human APOE immunoreactivity or its loss in GFAP+ and IBA1+ cells. f,g, Quantification of GFAP+ area (f) and APOE+ immunoreactivity in GFAP+ cells associated with plaques (g; n = 51 plaques from APP/PS1:APOE3-KI mice, n = 39 plaques from APP/PS1:APOE3-cKO mice, n = 59 plaques from APP/PS1:APOE4-KI mice, n = 61 plaques from APP/PS1:APOE4-cKO mice). h, Confocal images of Serpina3n, GFAP, and HJ3.4B. i, Quantification of Sepina3n+ immunoreactivity in GFAP+ cells (n = 42 plaques from APP/PS1:APOE3-KI mice, n = 44 plaques from APP/PS1:APOE3-cKO mice, n = 41 plaques from APP/PS1:APOE4-KI mice, n = 30 plaques from APP/PS1:APOE4-cKO mice). j, Schematics of experimental design of adoptive transfer of phagocytic microglia showing the isolation of MGnD from APOE3-KI, APOE4-KI and APOE4-cKO mice and injection to 2 months-old WT recipient mice, followed by isolation of astrocytes from recipient mice 16h later. Figure created with Biorender.com. k, Volcano plot showing differentially expressed genes in astrocytes isolated from WT recipient mice injected with MGnD sorted from APOE4-KI or APOE3-KI mice (n = 3 mice per group, P < 0.05). l, Volcano plot showing differentially expressed genes of astrocytes isolated from WT recipient mice injected with MGnD cells sorted from APOE4-cKO mice or APOE4-KI mice (n = 3 mice per group, P < 0.05). m, IPA of top-affected upstream regulators in WT astrocytes isolated from recipient mice following the injection of APOE4-cKO MGnD versus APOE4-KI MGnD (n = 3 mice per group, P < 0.05). Upstream regulators with a P value of < 0.05 were selected. Data were analyzed by one-way ANOVA and are presented as mean ± s.e.m.; scale bars, 50 μm.
Fig. 6. Deletion of microglial APOE4 promotes astrocyte activation and their recruitment to plaque via LGALS3 in APP/PS1 and P301S mice.
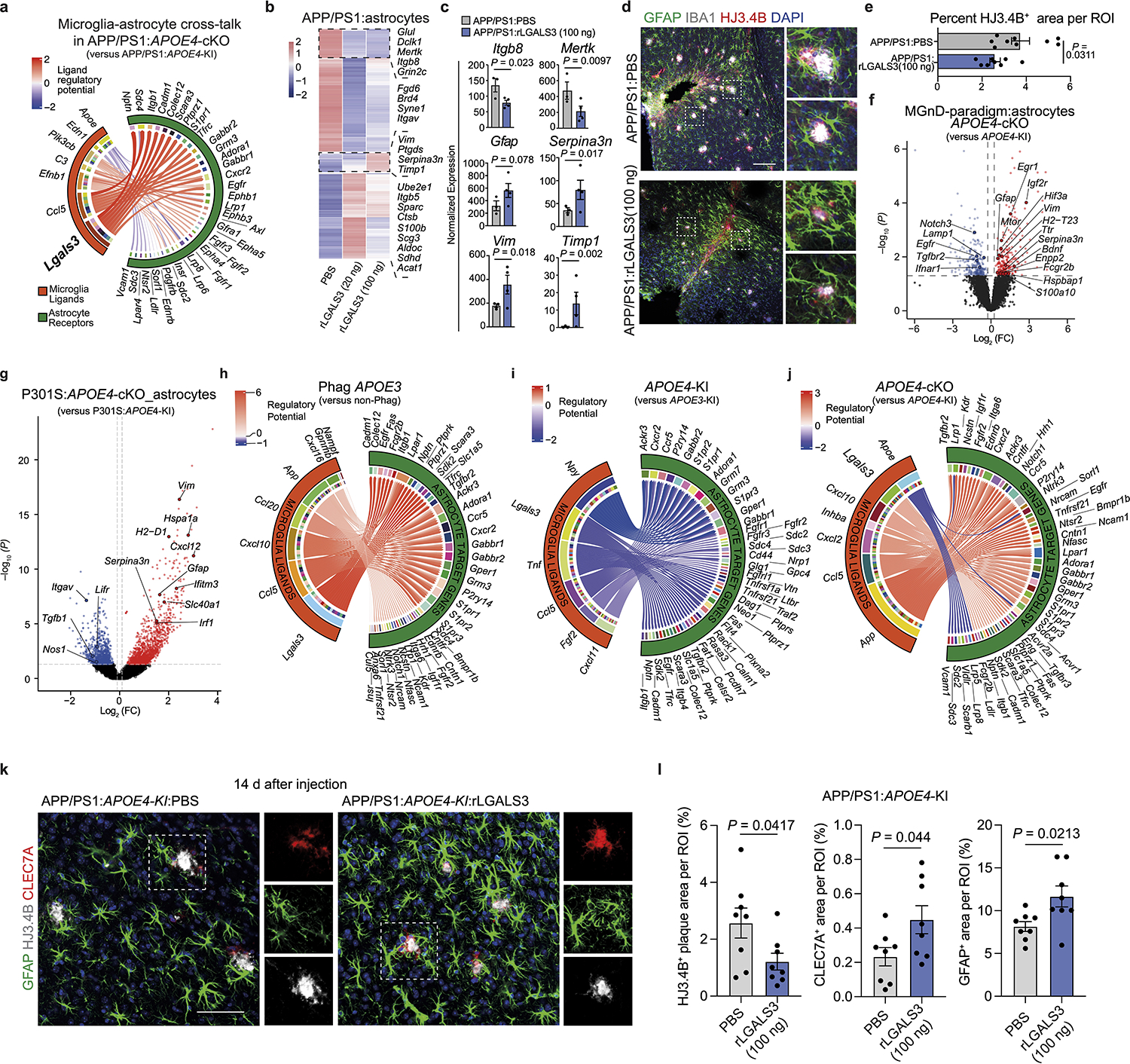
a, Circos plot illustrating prioritized cross-talk between microglia ligands and astrocyte receptors. b, Heat map of the top 200 differentially expressed genes in astrocytes isolated from APP/PS1 mice injected with PBS, 20 ng and 100 ng of rLGALS3. Differentially expressed genes were identified using DESeq2 analysis with an LRT (n = 3–4 mice per group, P < 0.05). c, Normalized counts of key astrocytic genes isolated from APP/PS1 mice treated with PBS and 100ng rLGALS3 (n = 3 PBS-treated mice, n = 4 rLGALS3-treated mice). d, Confocal images of IBA1, GFAP, and HJ3.4B in APP/PS1 mice treated with PBS and 100ng of rLGALS3; scale bar, 200 μm. e, Quantification of plaque load at the injection site (n = 9 ROIs from PBS-treated mice, n = 8 ROIs from rLGALS3-treated mice). f, Volcano plot showing differentially expressed genes in astrocytes isolated from apoptotic neuron-injected 8-month-old APOE4-cKO mice compared to those isolated from APOE4-KI mice. Differentially expressed genes were identified using DESeq2 analysis with an LRT (n = 4 mice per group, P < 0.05). g, Volcano plot showing differentially expressed genes in astrocytes isolated from 9-month-old P301S:APOE4-cKO mice compared to those isolated from P301S:APOE4-KI mice. Differentially expressed genes were identified using DESeq2 analysis with an LRT (n = 4–7 mice per group, P < 0.05). h–j, Circos plots illustrating prioritized ligand-receptor interactions between microglia and astrocytes, comparing phagocytic APOE3-KI versus non-phagocytic APOE3-KI conditions (h), phagocytic APOE4-KI versus APOE3-KI (i) and phagocytic APOE4-cKO versus APOE4-KI (j). Regulatory potential demonstrates directionality of comparison. k, Confocal images of GFAP, HJ3.4B and CLEC7A in APP/PS1:APOE4-KI mice treated with PBS or 100ng of rLGALS3; scale bar, 50 μm. l, Quantification of HJ3.4B, CLEC7A and GFAP (n = 8 ROIs from five mice per group). Data were analyzed by two-tailed Student’s t-test and are presented as mean ± s.e.m.
Impaired MGnD and astrocyte activation in individuals carrying the APOE4 allele
To study the sex differences related to APOE4 in individuals with AD6, we used RNA-seq analysis of whole human brains. We found a significant reduction in the expression levels of MGnD genes including CLEC7A, AXL, LYZ, CD300LG, HLA-DQB2 and an induction in expression of the upstream regulator of TGFβ signaling ITGB8 in the brains of male individuals with AD heterozygous for the APOE3 and APOE4 alleles (Fig. 7a and Supplementary Tables 6 and 7). Moreover, in line with a reduction of astrocyte activation in APP/PS1:APOE4-KI mice (Fig. 5), we found reduced expression of S100A, GFAP, VIM and LGALS3 in the brains of female individuals with AD heterozygous for the APOE3 and APOE4 alleles (Fig. 7b,c and Supplementary Tables 6 and 7). Importantly, among the top genes upregulated in the brains of females with AD heterozygous for the APOE3 and APOE4 alleles were several AD risk factors including ABCA7, IFNAR1, APP, BIN1, CD33 and HAVCR2 (Fig. 7b,d and Supplementary Table 6), the latter of which encodes the checkpoint molecule TIM3, one of the most promising new therapeutic approaches in cancer32, that was recently described in a GWAS as an AD risk gene2. In addition, the expression of SMAD3, a downstream adaptor of TGFβ signaling33, was upregulated in the brains of females with AD heterozygous for the APOE3 and APOE4 alleles (Fig. 7b,d and Supplementary Tables 6 and 7). KEGG analysis showed enrichment of pathways associated with neurodegenerative diseases, reactive oxygen species, phagosome formation and proteolysis in females with AD that are homozygous for the APOE3 allele (Fig. 7e). Immunohistochemistry of cortical tissue from females with AD that are heterozygous for the APOE3 and APOE4 alleles confirmed the reduction in LGALS3 immunoreactivity and its colocalization with IBA1+ microglia associated with Aβ plaques (Fig. 7f,g and Supplementary Table 7). Analysis of publicly available human brain scRNA-seq data34 showed increased expression of TGFβ-signaling genes and downstream AD risk factors, including INPP5D, in females that are heterozygous for the APOE3 and APOE4 alleles. Notably, the expression of MGnD genes, such as APOE, SPP1, HLA-DQB1 and LGALS3, were reduced in females that are heterozygous for the APOE3 and APOE4 alleles (Extended Data Fig. 6a). Importantly, we found a similar observation in an independent cohort described by Zhou et al.35 showing increased expression of SPI1, TGFB1 and GRN in microglia from the brains of individuals with AD that are heterozygous for the APOE3 and APOE4 alleles (Fig. 7h and Extended Data Fig. 6b,c). Moreover, RNAscope analysis confirmed the increased expression of the homeostatic checkpoint gene INPP5D in IBA1+ microglia in individuals with AD that are heterozygous for the APOE3 and APOE4 alleles compared to those that are homozygous for the APOE3 allele (Extended Data Fig. 6d,e). These findings in humans are in line with our previous results demonstrating enriched Spi1 expression in APOE4-KI mice (Fig. 1a,b) and restoration of the MGnD signature following genetic deletion of Spi1 (Fig. 2a,b and Supplementary Table 2). Immunohistochemistry confirmed SMAD3 activation and phosphorylated SMAD3 (pSMAD3) immunoreactivity in IBA1+ microglia in females with AD that are heterozygous for the APOE3 and APOE4 alleles (Fig. 7i,j and Supplementary Table 7). We also confirmed that GFAP immunoreactivity associated with Aβ plaques was reduced in females that are heterozygous for the APOE3 and APOE4 alleles compared those that are homozygous for the APOE3 allele (Fig. 7k,l and Supplementary Table 7). Analysis of public single-nucleus RNA-seq data35 of astrocytes from the brains of individuals with AD that are homozygous for the APOE3 allele and those that are heterozygous for the APOE3 and APOE4 alleles confirmed that the percentage of GFAPhi and SERPINA3+ astrocytes is increased in donors that are homozygous for the APOE3 allele compared to in those that are heterozygous for the APOE3 and APOE4 alleles (Fig. 7m and Extended Data Fig. 6f). Furthermore, the expression of astrocyte activation genes, including CST3, HSP90AB1 and ALDOC, was reduced, while TGFβ1-signaling-related genes, including TGFBR3, TGFB2 and ITGB8, were significantly increased in expression in individuals that are heterozygous for the APOE3 and APOE4 alleles (Fig. 7n). RNAscope analysis confirmed the induction of ITGB8 in GFAP+ astrocytes in males with AD that are heterozygous for the APOE3 and APOE4 alleles compared to those that are homozygous for the APOE3 allele (Extended Data Fig. 6g–i). Furthermore, RNAscope showed reduced Itgb8 expression in astrocytes from APP/PS1:APOE4-cKO mice compared to in those from APP/PS1:APOE4-KI mice (Extended Data Fig. 6j,k). Together, these findings show increased TGFβ signaling in APOE4 carriers, which may predispose the development of AD via the inhibition of MGnD and astrocytes response to neurodegeneration.
Fig. 7. Impaired induction of the MGnD signature and astrocytes activation in individuals with AD that carry the APOE4 allele.
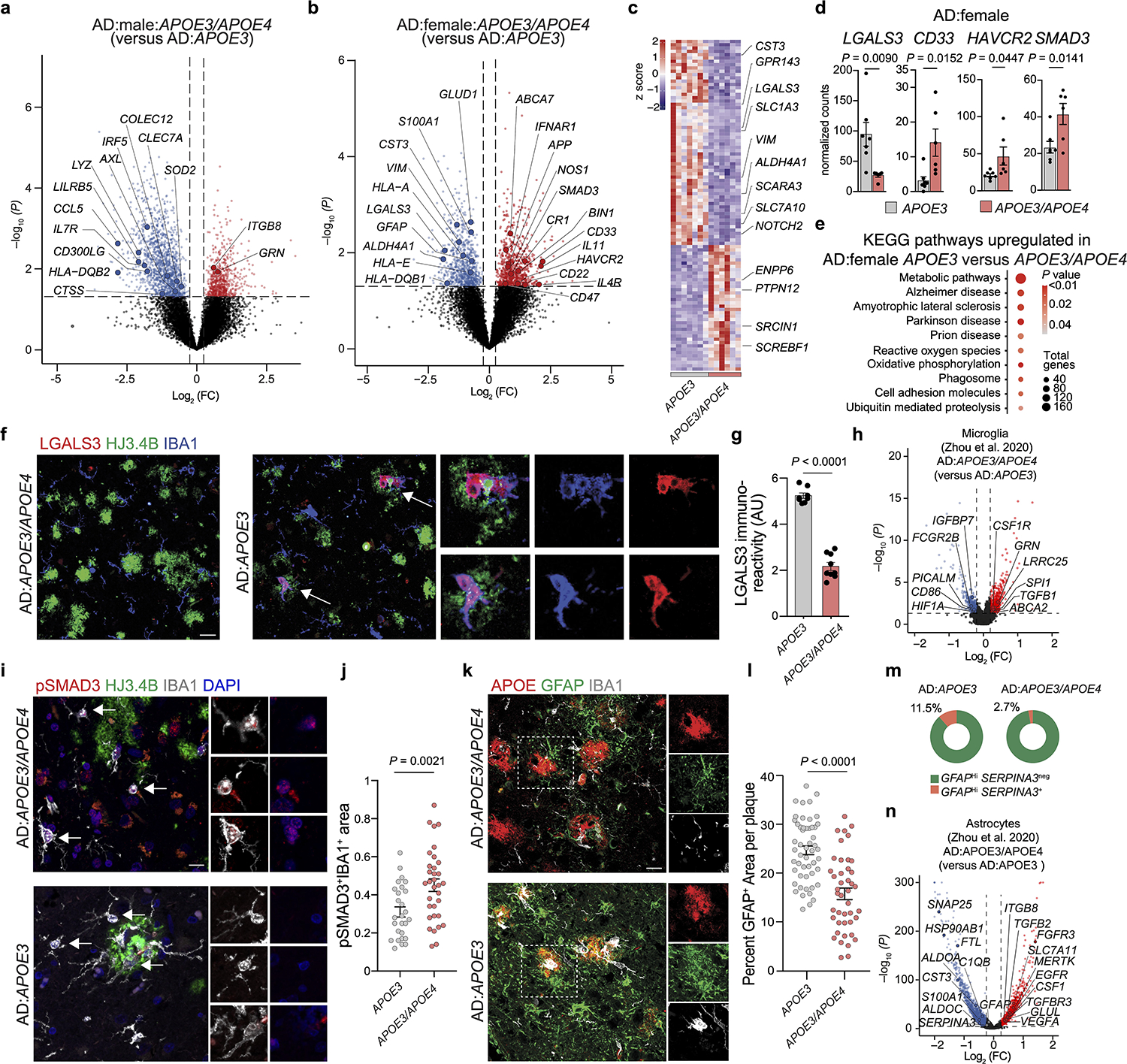
a,b, Volcano plot of bulk RNA-seq analysis of total brain tissue isolated from males (a) and females (b) showing selected differentially expressed genes in individuals that are heterozygous for the APOE3 and APOE4 alleles compared to those that are homozygous for the APOE3 allele (male n = 5–7 donors, female n = 6–7 donors, P < 0.05). c, Top 100 differentially expressed genes from female AD:heterozygous APOE3/APOE4 carriers compared to AD:homozygous APOE3 carriers (P < 0.05, n = 6–7 donors per group). d, Normalized counts of key affected genes (n = 7 AD:homozygous APOE3 carriers, n = 6 AD:heterozygous APOE3/APOE4 carriers). e, Top-affected KEGG pathways in female AD:homozygous APOE3 carriers compared to AD:heterozygous APOE3/APOE4 carriers. f, Confocal microscopy images of brain sections from females with AD that are homozygous for the APOE3 allele or heterozygous for the APOE3 and APOE4 alleles stained for LGALS3, HJ3.4B-plaque and IBA1. g, Quantification of LGALS3+ immunoreactivity (n = 8 AD:homozygous APOE3 carriers, n = 9 AD:heterozygous APOE3/APOE4 carriers). h, Volcano plot of microglial differentially expressed genes in AD:heterozygous APOE3/APOE4 carriers compared to AD:homozygous APOE3 carriers analyzed from dataset by Zhou et al.35 (n = 6 AD:homozygous APOE3 carriers, n = 4 AD:heterozygous APOE3/APOE4 carriers, P < 0.05). i, Confocal images of brain sections from female individuals with AD homozygous for the APOE3 allele or heterozygous for the APOE3 and APOE4 alleles stained for pSMAD3, IBA1, and HJ3.4B. j, Quantification of pSMAD3 immunoreactivity in IBA1+ cells (n = 27 cells from AD:homozygous APOE3 carriers, n = 33 cells from AD:heterozygous APOE3/APOE4 carriers ). k, Confocal microscopy images of GFAP in the brains of individuals with AD that are homozygous for the APOE3 allele or heterozygous for the APOE3 and APOE4 alleles. l, Quantification of GFAP immunoreactivity per plaque (n = 52 plaques from AD:homozygous APOE3 carriers, n = 43 plaques from AD:heterozygous APOE3/APOE4 carrier). m, Donut plots representing analysis from dataset by Zhou et al.35, showing the percentage of GFAPhiSERPINA3+ and GFAPhiSERPINA3− astrocyte clusters in AD:heterozygous APOE3/APOE4 carriers and AD:homozygous APOE3 carriers. n, Volcano plot of astrocytic differentially expressed genes of AD:heterozygous APOE3/APOE4 carriers compared to AD:homozygous APOE3 carriers analyzed from the dataset by Zhou et al.35 (n = 6 AD:homozygous APOE3 carriers, n = 4 AD:heterozygous APOE3/APOE4 carriers, P < 0.05). Arrows indicate ROIs for f and i. Data were analyzed by two-tailed Student’s t-test and are presented as mean ± s.e.m.; scale bars, 50 μm.
Targeting ITGB8 enhances MGnD response
To gain molecular insights into the role of APOE4-mediated induction of TGFβ signaling in AD brains, we genetically and pharmacologically inhibited ITGB8, which is critical for the activation of latent TGFβ136, in WT and APP/PS1 mice. ITGB8–TdTomato (Itgb8-tdT) reporter mice showed ITGB8 expression in cortical astrocytes37, mature oligodendrocytes, and oligodendrocyte precursors cells, but not in microglia, neurons, or endothelial cells (Extended Data Fig. 7), as previously reported38. Genetic deletion of Itgb8 specifically in the cortex in Emx1Cre:Itgb8fl/fl (Itgb8-cKO) mice showed a complete absence of homeostatic TMEM119+ microglia, while LGALS3 and CLEC7A were highly expressed in cortical microglia (Fig. 8a and Extended Data Fig. 8a–c). This mouse model allows for the investigation of the effect of Itgb8 deletion in the cortex and hippocampus, while the rest of the brain is not affected. We also observed increased GFAP immunoreactivity colocalized with CLEC7A+ MGnD in the cortex of Itgb8-cKO mice (Fig. 8b–d). RNA-seq analysis of cortical microglia revealed an induction of MGnD-related genes (Apoe, Cd300ld, Cd74 and Axl), while the expression of homeostatic genes (Tmem119, Siglech, Mertk and Havcr2) were suppressed (Fig. 8e and Supplementary Table 8). Of note, cortical microglia in Itgb8-cKO mice did not express peripheral monocyte lineage gene Ms4a339 (Supplementary Table 8). Functional characterization showed induced phagosome formation, antigen presentation, chemokine signaling, and IFNγ signaling in cortical microglia of Itgb8-cKO mice (Extended Data Fig. 8d). Additional characterization of microglia using the acute response to neurodegeneration via cranial injection of apoptotic neurons4 showed upregulation of antigen presentation- and IFNγ signaling-related genes in Itgb8-cKO mice compared to in control mice (Extended Data Fig. 8e–g and Supplementary Table 8). Furthermore, deletion of Itgb8 promoted microglial Aβ phagocytosis in response to acute cranial injection of Aβ (Extended Data Fig. 8h,i). Moreover, we found reduced SMAD3 phosphorylation and increased APOE immunoreactivity in Itgb8-cKO mice (Fig. 8g–i). Using Cx3cr1Cre:Smad2/Smad3fl/fl mice, we confirmed that microglial Smad2/Smad3 deletion results in the suppression of Tmem119 and enhanced expression of MGnD molecules, including Cd68 and Apoe, associated with astrocyte activation (Extended Data Fig. 9a–d). Gene expression profiles of cortical microglia from Emx1Cre:Itgb8fl/fl mice were correlated with Tgfbr2-cKO microglia (Fig. 8j) and previously published microglia transcriptomes from Nrros-KO40 and Smad2/Smad3-cKO mice (Extended Data Fig. 9e–h). Importantly, AD risk factor genes, including Bin1, Inpp5d and Havcr2, were reduced in expression in all datasets (Fig. 8j, Extended Data Fig. 9i–l, and Supplementary Table 8). The reproducibility in microglial gene expression profiles indicates that regional deletion of Itgb8 suppresses TGFβ signaling, leading to an induction of the MGnD phenotype.
Fig. 8. Blocking ITGB8-TGFβ signaling enhances MGnD response and reduces AD pathology in APP/PS1 mice.
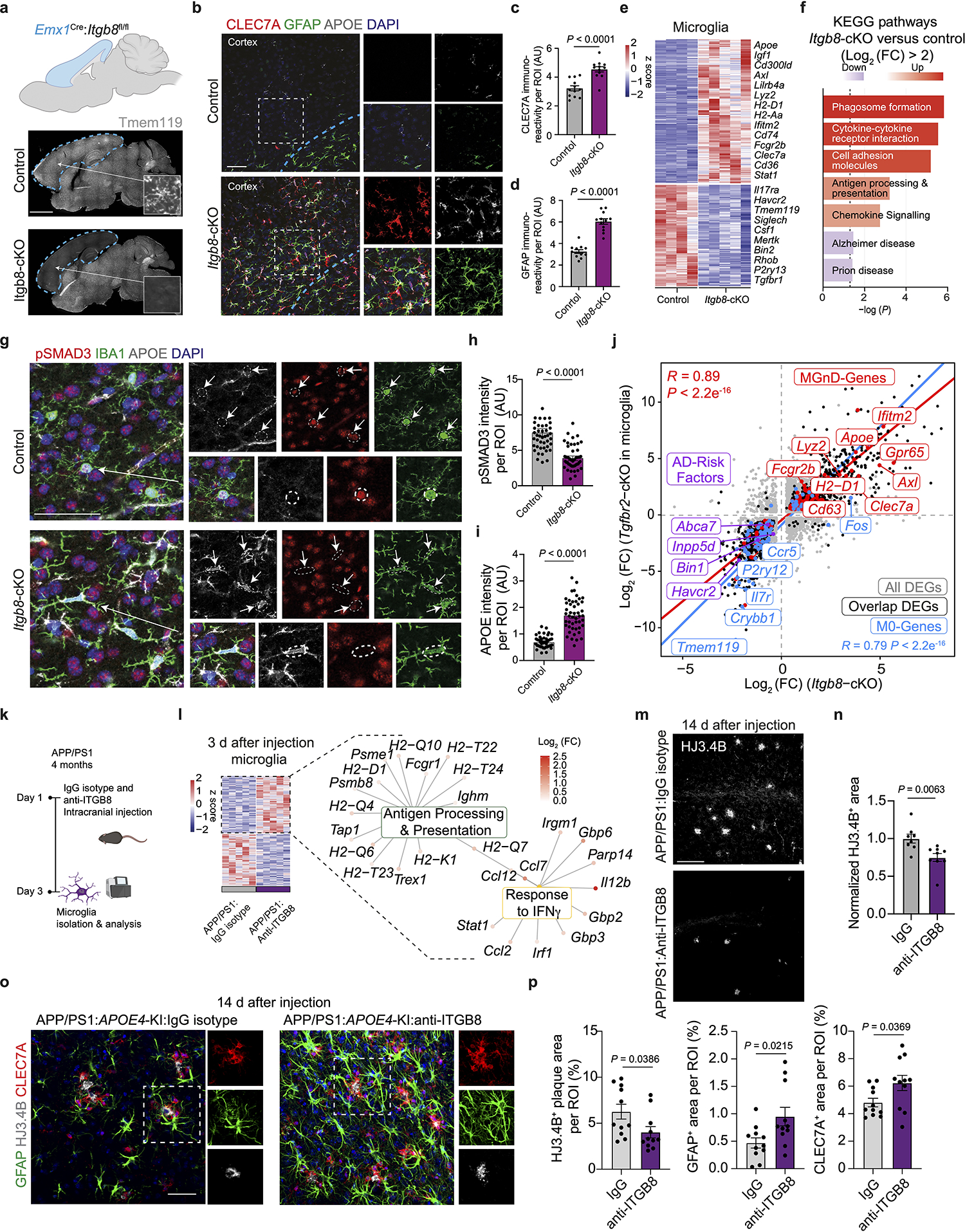
a, Representative images of TMEM119 immunoreactivity in sagittal brains of Itgb8-cKO mice and control littermates. Arrows indicate magnified ROIs. b, Images of CLEC7A, GFAP, and APOE in Itgb8-cKO and control mice. c,d, Quantification of CLEC7A+(c) and GFAP+(d) immunoreactivity in the cortex (n = 12 ROIs per group). e, Heat map of differentially expressed genes for microglia isolated from Itgb8-cKO versus control mice (n = 4–5 mice per group, P < 0.05). f, Top KEGG pathways in Itgb8-cKO microglia versus control microglia. g, Images of pSMAD3, APOE, and IBA1 in Itgb8-cKO and control mice. h, Quantification of pSmad3 immunoreactivity in IBA1+ cells (n = 45 cells in the control group, n = 47 cells in the Itgb8-cKO group). i, Quantification of Apoe immunoreactivity in IBA1+ cells (n = 45 cells in the control group, n = 47 in the Itgb8-cKO group). j, Scatter plot comparing differentially expressed genes in microglia from Itgb8-cKO and Tgfbr2-cKO mice, described by Lund et al.61 (for Lund et al., n = 3 mice per group; for Itgb8-cKO, n = 4–5 mice per group; P < 0.05, Log2(fold change) of >0.25 or <–0.25). k, Schematics of the administration of neutralizing antibody to ITGB8 or IgG isotype control into the brains of APP/PS1 mice and analysis 3 d later. l, Heat map of microglia isolated from APP/PS1 mice treated with neutralizing antibody to ITGB8 or IgG isotype control and top Gene ontology pathways affected (n = 5 mice per group, P < 0.05). m, Images of HJ3.4B+ plaques at the injection site 14 days after the treatment of APP/PS1 mice with neutralizing antibody to ITGB8 or IgG isotype control. n, Quantification of HJ3.4B+ plaques at the injection sites (n = 8 IgG-treated mice, n = 9 anti-ITGB8-treated mice). o, Images of GFAP+, CLEC7A+, and HJ3.4B+ plaques at the injection site 14 days after the treatment of APP/PS1:APOE4 KI mice with neutralizing antibody to ITGB8 or IgG isotype control. p, Quantification of HJ3.4B+ plaques area and GFAP+ and CLEC7A+ area per ROI at the injection sites (n = 11 ROIs from 6–7 mice per group). Data were analyzed by two-tailed Student’s t-test and are shown as mean ± s.e.m.; scale bars, 2mm for a; 100 μm for b and m; 50 μm for g and o.
Microglial Inpp5d deletion ameliorates pathology in AD mice
The induction of MGnD genes in microglia deleted from Itgb8, Tgfbr2, Nrros or Smad2/Smad3 (Extended Data Fig. 9e–h) is associated with the suppression of AD risk factor genes, including Bin1, Havcr2 and Inpp5d1, 2 (Fig. 8j, Extended Data Fig. 9i–l, and Supplementary Table 8). In addition, APOE4-expressing microglia showed increased Inpp5d expression levels (Fig. 1a, Extended Data Fig. 6d,e) and enriched Inpp5d locus histone acetylation (Extended Data Fig. 1b). To address the impact of microglial-specific Inpp5d expression on AD pathology and MGnD response, we crossed Cx3cr1CreERT2/WT:Inpp5dfl/fl mice with APP/PS1 mice. Microglial deletion of Inpp5d induced the MGnD response in APP/PS1 mice (Extended Data Fig. 10a,b and Supplementary Table 8) associated with reduced Aβ plaque load, as determined by Thioflavin-S and HJ3.4B staining (Extended Data Fig. 10c,d,h,j). Moreover, deletion of Inpp5d in microglia showed increased CLEC7A and LGALS3 immunoreactivity in association with Aβ-plaques (Extended Data Fig. 10e–i). Importantly, Inpp5d deletion in microglia was sufficient to reduce LAMP1+ dystrophic neurites in APP/PS1 mice (Extended Data Fig. 10k–l). Furthermore, the increased MGnD response in association with Aβ plaques was accompanied by increased GFAP+ immunoreactivity following the deletion of microglial Inpp5d (Extended Data Fig. 10m,n). Similarly, recent studies showed that microglial deletion of Inpp5d protects against plaque-induced neuronal dystrophy in transgenic AD mice41, 42. These results support the role of INPP5D as a microglial homeostatic checkpoint, and its deletion is beneficial in mitigating AD pathology via the induction of MGnD response.
Blocking ITGB8 signaling mitigates AD pathology in AD mice
To test whether enhanced microglia activation and phagocytosis can reduce pathological hallmarks of AD in adult APP/PS1 mice, we used a monoclonal neutralizing antibody to ITGB8 (ADWA-11)43. Three days after injection in 4-month-old APP/PS1 mice, microglia increased antigen-presentation and IFNγ signaling (Fig. 8k,l). Immunohistochemistry confirmed increased major histocompatibility complex class II+ (MHC II+) immunoreactivity around the injection site (Extended Data Fig. 8j,k). Moreover, 14 d after injection, we found a significant decrease in Aβ plaque size (Fig. 8m,n) in mice treated with anti-ITGB8 compared to control-treated mice. In APP/PS1:APOE4-KI mice, treatment with monoclonal neutralizing antibody to ITGB8 restored the expression of CLEC7A and GFAP associated with reduced plaque pathology (Fig. 8o,p). Overall, these results strongly support a critical role of ITGB8-TGFβ signaling in the regulation of an MGnD response in AD pathology. Thus, pharmacological targeting of ITGB8-TGFβ signaling in AD can promote MGnD response and astrocyte activation and provide a novel approach for therapeutic modulation of innate immunity in AD and dementia.
Discussion
Several mechanisms have been proposed by which APOE4 increases the risk of developing AD, and most proposed mechanisms involve models harboring global APOE4 expression or astrocyte specific deletion10, 11, 12, 44. Here we show a cell-autonomous negative regulation of microglial APOE4 in the acquisition of MGnD phenotype in response to neurodegeneration. Induction of an MGnD response was restored in mice with tau and amyloid pathology in which microglial APOE4 was deleted, resulting in improved neuronal survival, reduction in plaque pathology and increased microglial and astrocytic association with Aβ plaques. Deletion of microglial APOE3 reduced CLEC7A expression to levels comparable to APOE4-KI in P301S mice. We recently showed impaired MGnD responses in APOE4-KI glaucoma mice, similarly to Apoe−/− mice14. Thus, microglial expression of APOE3 is critical for the induction of MGnD response to neurodegeneration, and its deletion accelerates neuronal loss, whereas deletion of microglial APOE4 favors neuroprotection in tau mice. In addition to the classical role of APOE in cholesterol transport, APOE4 acquires a novel function in the nucleus, where it translocates and directly binds DNA to affect the transcription of genes associated with AD45, 46. Furthermore, microglial APOE4 shows increased nuclear localization in AD brains45. These results suggest that nuclear localization of APOE4 may directly repress the transcription of MGnD genes, a function absent in APOE3-expressing microglia, which is alleviated following the deletion of APOE4. Thus, APOE may have a dual role in controlling MGnD responses based on its subcellular localization, which may be altered in APOE4-expressing microglia, accounting for the different impact on neurodegeneration detected in tau mice following the deletion of microglial APOE3 compared to APOE4. Consistent with these findings, we found that the expression of the transcription factor PU.1 was upregulated in APOE4-KI microglia, and its deletion in microglia resulted in a robust induction of MGnD genes, including Lgals3. In the brains of individuals with AD that carry the APOE4 allele, we found a sex-dependent impairment of MGnD signature, with a pronounced reduction in astrocyte activation observed in females. Several AD risk factor genes, including CD33, BIN1, ABCA7, CR1, INPP5D and HAVCR22, 47, were enriched in the brains of females with AD that carry the APOE4 allele, associated with downregulation of key MGnD genes4, 13, 14. These results support previous reports demonstrating that higher PU.1 expression in humans is likely to result in earlier AD onset19, 48. In brains from females with AD that carry the APOE4 allele, PU.1 expression was associated with increased SMAD3 expression levels, which would reinforce the microglial homeostatic signature as a downstream molecule of TGFβ signaling13, 18, 49. Moreover, microglial of pSMAD3 immunoreactivity as a marker of TGFβ signaling was increased in the brains of females with AD that carry the APOE4 allele, indicative of a homeostatic microglial signature. A TGFβ-enriched milieu in the brains of individuals with AD that carry the APOE4 allele suppressed astrocyte activation, consistent with previous reports showing astrocyte suppression in response to TGFβ ligand50, and deletion of Tgfbr231 or Smad2/Smad3 activates astrocytes. We also found that deletion of microglial APOE4 in APP/PS1 mice promotes astrocyte activation and encapsulation of Aβ plaques. Moreover, reactive astrocytes play a beneficial role in limiting AD pathology, and their depletion in AD mice results in increased plaque load, synaptic dysfunction, and memory loss51. These findings in mice and in humans suggest that APOE4 predisposes to AD pathology in part via the induction of TGFβ-dependent microglia homeostatic regulators13 that impair microglia-astrocyte cross-talk in response to neurodegeneration. In line with these findings, Liu et al. demonstrated that APOE4 reduces activated microglia signature in response to AD pathology in human brains and in human induced pluripotent stem cell-derived microglia with the APOE4 allele. We identified LGALS3 as a critical MGnD molecule that is restored in microglia following the deletion of APOE4 and is associated with increased astrocytic activation and reduced plaque pathology. Although LGALS3 was reported to promote Aβ oligomerization and toxicity, these studies were performed either using global Lgals3-KO mice or Aβ monomers precultured with LGALS3 prior to the injection to the brain52, 53. Thus, these data should be interpreted with caution, especially regarding the role of microglial-derived LGALS3 in AD pathology. Importantly, we found LGALS3 reduction among the top 100 differentially regulated genes in the brain of female AD donors with the APOE4 allele. Interestingly, APOE4 was shown to be associated with a decreased risk of glaucoma54, 55, 56. We recently demonstrated that APOE4 impairs the response of neurodegenerative retinal microglia via LGALS3 signaling associated with neuroprotection in glaucoma14. Thus, APOE4 converges to impair MGnD response to neurodegeneration, which plays a protective role in glaucoma14 and is detrimental in AD. Although TGFβ signaling was suggested to play both beneficial and detrimental roles in AD57, its overproduction in astrocytes promoted cerebrovascular fibrosis and amyloidosis58. Moreover, TGFβ1 was implicated in vascular dementia to promote abnormal vascular remodeling and was proposed as a therapeutic target for AD59. We detected upregulation of ITGB8, which plays a critical role in the activation of latent TGFβ136 in males with AD that carry the APOE4 allele. In the brains of females with AD that carry the APOE4 allele, we identified the induction of SMAD3 and INPP5D signaling associated with downregulation of MGnD genes. Mechanistically, the genetic deletion of Itgb8 or Inpp5d was sufficient to restore MGnD response and astrocyte activation and reduce plaque load in AD mice. Furthermore, pharmacological blockade of ITGB8-TGFβ signaling enhanced the response of MGnD associated with increased plaque clearance in mice with AD. Importantly, ITGB8 blockade resulted in an increased IFNγ response, which we recently showed to play a critical role in the acquisition of beneficial pre-MGnD in AD mice60. These data support the beneficial role of MGnD in restricting AD pathology and identify ITGB8-TGFβ axis as a therapeutic intervention for AD.
Methods
Human brain tissue and neuropathology
Frozen brain tissue blocks from the superior parietal gyrus for RNA-seq were obtained from the Netherlands Brain Bank (NBB). Donors gave informed consent to donate their brains to NBB. Post-mortem interval was limited to 10 h to ensure tissue quality. Paraffin-embedded brain tissue sections were obtained from NBB and Massachusetts Alzheimer’s Disease Research Center (MADRC). The study was approved by the Ethics Committees of the Brigham Women’s Hospital and compliant with the World Medical Association Declaration of Helsinki on Ethical principles for medical research involving human subjects. For tissue selection, neuropathological staging of the brains was performed by NBB and Massachusetts Alzheimer’s Disease Research Center. Details about the brain samples were reported in Supplementary Table 7.
Mice
B6.129P2(Cg)-Cx3cr1tm2.1(cre/ERT2)Litt/WganJ mice (Cx3cr1CreERT2, stock #021160)15, C57BL/6-Tmem119em1(cre/ERT2)Gfng/J (Tmem119-CREERT2, stock #031820)62, B6.129S6-Inpp5dtm1Wgk/J mice (Inpp5dflox, stock #028255)63, Smad2tm1.1Epb/J mice (Smad2flox, stock #022074)64 and Smad3tm1Zuk mice (Smad3flox, MGI: 3822465)65 were purchased from the Jackson Laboratory. Cx3cr1CreERT2 mice were crossed with human APOE knock-in mice, APOE3fl/fl and APOE4fl/fl mice, respectively66. These transgenic mice were then triple crossed with APP/PS124 or P301S22 mice. B6.Cg-Tg(Thy1-APPSw,Thy1-PSEN1*L166P)21Jckr (APP/PS1)24 mice were kindly provided by Dr. Mathias Jucker (University of Tubingen). B6;C3-Tg(Prnp-MAPT*P301S)PS19Vle/J (P301S, Jax stock #008169)22 mice have been backcrossed to C57BL/6 mice for more than ten generations. B6.129P2-Apoetm2(APOE*3)Mae N8 (APOE3-KI, MGI #4838571)67 and B6.129P2-Apoetm3(APOE*4)Mae N8 (APOE4-KI, MGI # 4838572)68 were obtained from Taconic. The Spi1fl/fl mice69 were generously provided by Daniel Tenen and Junyan Zhang from Beth Israel Deaconess Medical Center. Emx1Cremice (B6.129S2-Emx1tm1(cre)Krj/J, JAX stock 005628) and Itgb8fl/fl mice70 were kindly provided by Dr. Thomas Arnold from University of California, San Francisco. The Itgb8-tdT mice were provided by Dr. Helen Paidassi37 (Université Claude Bernard Lyon 1). Cx3cr1CreERT2 mice were crossed with Inpp5dfl/fl mice both on WT and APP/PS1 background. Cx3cr1Cre (stock Tg(Cx3cr1-cre)MW126Gsat/Mmucd, MMRRC_036395-UCD) mice71 were obtained from the Mouse Resource and Research Center (MMRRC) at University of California Davis, and were crossed with Smad2flox and Smad3flox mice. All the experimental procedures using animals were approved by the Institutional Animal Care and Use Committee at Brigham and Women’s Hospital, Harvard Medical School and University of California, San Francisco.
Tamoxifen treatment
At six weeks of age, mice were intraperitoneally (ip) injected with 75 mg/kg of tamoxifen (Sigma Aldrich, T5648–5G) dissolved in corn oil per day for five consecutive days.
Microglia isolation
Mice were euthanized in a CO2 chamber and transcardially perfused with ice-cold Hanks’ Balanced Salt Solution (HBSS, Thermo Fisher, 14175103). The whole brain was removed from the skull and separated into sections with a sagittal brain matrix for further processing. The left hemisphere was used for sorting and the right hemisphere was used for immunohistochemistry. The left hemisphere was homogenized to form a single cell suspension, then resuspended and centrifuged in a 37%/70% Percoll Plus (GE Healthcare, 17–5445-02) gradient in HBSS at 800 g, 23°C, for 25 min with an acceleration of 3 and a deceleration of 1. Mononuclear cells were taken from the interface layer. The cells were stained with rat allophycocyanin (APC)-conjugated anti-mouse FCRLS (1:1000, clone 4G11, Butovsky Lab)13, PE-Cy7-conjugated anti-mouse CD11b (1:300, eBioscience, 50–154-54), and PerCP/Cy5.5-conjugated anti-mouse Ly-6C (1:300, Biolegend, 128012). After staining, Ly-6C−CD11b+FCRLS+ cells were washed and sorted using BD FACSAria™ II (BD Bioscience). FACS data were collected by BD FACS Diva Software. The analysis was done using FlowJo™ 10. For mice with stereotaxic injections of apoptotic neuron, a 3 × 6mm2 coronal slice around the injection site was taken. Microglia were enriched using the same method mentioned above. The apoptotic neurons were labeled with Alexa Fluor™ 405 dye. During sorting, phagocytic microglia (apoptotic neuron-Alexa Fluor 405+Ly-6C−CD11b+FCRLS+) and non-phagocytic microglia (apoptotic neuron-Alexa Fluor 405−Ly-6C−CD11b+FCRLS+) were collected as previously described4.
Primary neuron culture
Primary Neurons were obtained from mouse embryos at embryonic day 18 (E18). Cerebral hemispheres were isolated. The meninges were then removed from the hemispheres under a dissection microscope. The cerebral hemispheres were digested in a 10 U/mL papain solution for 15 min at 37°C, and then triturated in a trituration solution of 19 mL Neurobasal media (Gibco, 21103–049), 133 μL BSA (Sigma, A-9576), 0.5 mL Pen/Strep/Glucose/Pyruvate, and 200 μl DNase (Sigma, D-5025) with fire-polished glass pipettes until a single cell suspension was created. The suspension was filtered through a 40-μm cell strainer, spun down at 400 g for 5 min, and resuspended in 1mL HBSS. The cell number was counted using a trypan blue stain (Gibco™, 15–250-061) and a cellometer (Nexcelom). Cells were plated on a 6 well, poly-D-lysine coated plate with approximately 1 million cells per well in a minimum volume of 2 mL growth media made of Neurobasal, 1:50 B-27 supplement (Gibco 17504–044), 1:200 Pen/Strep (Gibco, 15140–122), 1:400 Glutamax (Gibco, 35050–061), and 1:50 fetal bovine serum (FBS, Gibco, 10–438-026). After 24 h of incubation at 37°C, cell viability and contamination were checked under the microscope. Half of the growth media was replaced with fresh growth media containing 5 μM Ara-C (Sigma-Aldrich, C1768–100MG) without FBS. After five days, half of the media was removed and replaced with new growth media without FBS.
Induction of apoptotic neurons
Primary neurons (d7-d10) were typically cultured one week after the initiation of culture. Neurons were removed from the surface of the plate by multiple washing with PBS. The neurons were then incubated under UV light (302 nm) at an intensity of 6315 W for 20 min to induce apoptosis. After this step, the neurons were kept on ice. The cells were collected, spun down via centrifugation, and resuspended in 1 mL PBS. Next, they were stained with the labeling dye (Alexa Fluor™ 405 NHS Ester, Invitrogen, A3000) for 15 min at 37°C, protected from light. Neurons were then washed, spun, and resuspended. The number of apoptotic cells was determined using a trypan blue stain and a cellometer. Neurons were resuspended at a density of 25,000 dead cells per μL PBS.
RT-qPCR
Total RNA from mouse tissue was extracted using RNeasy Plus Micro Kit (Qiagen, 74034) according to the manufacturer’s protocol. Total RNA from human tissue was extracted using mirVana miRNA isolation kit (Invitrogen, AM1560) according to the manufacturer’s protocol. For quantitative reverse transcription polymerase chain reaction (RT-qPCR), RNA was used after the reverse transcription reaction (high-capacity cDNA Reverse Transcription Kit; Applied Biosystems, 436884). Total mRNA amplifications were performed with commercially available FAM-labeled Taqman probe for human APOE (Thermo Fisher Scientific, Hs00171168_m1) and mouse Gapdh (Thermo Fisher Scientific, Mm9999915_g1). A real-time PCR reaction was performed using QuantStudio™ 7 (Applied Biosystems). All RT-qPCRs were performed in duplicate, and the level of mRNAs were presented as relative expression normalized to Gapdh as mean ± s.e.m.
Stereotaxic injection
Mice were anesthetized via intraperitoneal injection of ketamine (100 mg per kg (body weight)) and xylazine (10 mg per kg (body weight)). To induce MGnD paradigm, 2 μl of the apoptotic neuron mixture was injected into the hippocampus (medial/lateral (ML): ±1.5 mm, anterior/posterior (AP): –2 mm, dorsal/ventral (DV): –2 mm) and cortex (ML: ±1.5 mm, AP: –2 mm, DV: –1 mm) bilaterally using a stereotactic injection apparatus. Animals were processed 16 hours after injection. To delete Spi1 from microglia, 6 to 8-week-old Spi1fl/fl mice were stereotaxically injected with TAT-Cre (MilliporeSigma, SCR508) into ventricles (ML: ±1 mm, AP: –1 mm, DV: –2.5 mm; 2 μl per injection site, 10 μg μl–1, 40 μg per brain). To induce MGnD paradigm, 5 days after the i.c.v. injection of TAT-Cre, Alexa Flour™ 405 dye labeled apoptotic neurons, premixed with TAT-Cre, were injected into the cortex and hippocampus as described above. Phagocytic and non-phagocytic microglia were sorted from the brain regions narrow down to the injection sites, followed by SmartSeq2 analysis. To study the role of PU.1, we used PU.1 inhibitor (DB1876, MCE, HY-135797A) to inhibit PU.1. PU.1 inhibitor was dissolved in 10% DMSO (Invitrogen, D12345) to get the concentration as 2.08 mg ml–1 (4 mM) according to the manufacture’s instruction. DMSO (10%) was used as control. The PU.1 inhibitor or control was injected into the cortex and hippocampus as described above. After 7 days, microglia and astrocytes were isolated from the brain regions restricted to the injection sites, followed by IHC and Smart-seq2 analysis. To study the role of MGnD carrying different APOE alleles in the regulation of astrocytes response, the apoptotic neurons were injected into the cortex and hippocampus of APOE3-KI, APOE4-KI, and APOE4-cKO mice to induce MGnD paradigm as described above. After 16 hours, phagocytic microglia were sorted out of the brains, then injected into the cortex and hippocampus of WT mice (ML: ±1.5 mm, AP: –2 mm, DV: –1 and –2 mm; 2 μl per injection site; 3000 microglia per injection site). After 16 hours, astrocytes were isolated from the brain regions restricted to the injection sites, followed by Smart-seq2 analysis. To explore the effect of LGALS3 on AD pathology, recombinant mouse LGALS3 protein (R&D systems, 9039-GAB-050) were diluted in sterile PBS to 10 ng μl–1 or 50 ng μl–1. The rLGALS3 or PBS were injected into the cortex and hippocampus (ML: ±1.5 mm, AP: –2 mm, DV: –1 and –2 mm; 2 μl per injection site). After 3 days and 14 days, astrocytes were isolated from the brain regions restricted to the injection sites, followed by immunohistochemistry (IHC) and Smart-seq2 analysis. To study the role of Itgb8, anti-ITGB8 antibody (ADWA11, kindly provided by Dr. Dean Sheppard from University of California, San Francisco) was used to block mouse Itgb8. Mouse IgG1 was used as isotype control (BioXCell, Catalog# BP0083). Both ADWA11 and IgG1 were diluted to 0.5 μg μl–1 in sterile PBS. Anti-ITGB8 antibody or isotype control was injected into the cortex and hippocampus (ML: ±1.5 mm, AP: –2 mm, DV: –1 and –2 mm; 2 μl per injection site). After 14 days, microglia were isolated from the brain regions restricted to the injection sites, followed by IHC and Smart-seq2 analysis. Fluorescence-labeled amyloid-β peptide (1–42, Anaspec, AS-60480–01) were reconstituted by NH4OH, then diluted in sterile PBS to 1 μg μL–1. The Aβ was injected into the cortex and hippocampus (ML: ±1.5 mm, AP: –2 mm, DV: –1 and –2 mm; 2 μl per injection site). After 6 hours, phagocytic and non-phagocytic microglia were isolated from the brain regions restricted to the injection sites, followed by FACS analysis.
scRNA-seq
Brain tissue isolated from mice following perfusion was performed as previously described72. Briefly, tissue was finely minced and incubated in 10 mL of enzyme digestion solution consisting of 75 μL Papain suspension (Worthington, LS003126) diluted in enzyme stock solution (ESS) and equilibrated to 37°C. 200 mL ESS medium consisted of 20 mL 10× EBSS (Sigma-Aldrich, E7510), 2.4 mL 30% D(+)-glucose (Sigma-Aldrich, G8769), 5.2 mL 1M NaHCO3 (VWR, AAJ62495-AP), 200 μL 500 mM EDTA (Thermo Fisher Scientific, 15575020), and 168.2 mL ddH2O, filter-sterilized through a 0.22-μm filter. Samples were shaken at 80 rpm for 40 min at 37°C. Enzymatic digestion was stopped with 1 mL of 10× hi ovomucoid inhibitor solution and 20 μL 0.4% DNase (Worthington, LS002007) diluted in 10 mL inhibitor stock solution (ISS). 10 mL 10× hi ovomucoid inhibitor stock solution contained 300 mg BSA (Sigma-Aldrich, A8806), 300 mg ovomucoid trypsin inhibitor (Worthington, LS003086) diluted in 10 mL 1× PBS and filter sterilized using at 0.22-μm filter. ISS medium contained 50 mL 10× EBSS (Sigma-Aldrich, E7510), 6 mL 30% D(+)-glucose (Sigma-Aldrich, G8769), 13 mL 1M NaHCO3 (VWR, AAJ62495-AP) diluted in 170.4 mL ddH2O and filter-sterilized through a 0.22-μm filter. Tissue was mechanically dissociated using a 5 mL serological pipette and filtered through at a 70-μm cell strainer (Thermo Fisher Scientific, 22363548) into a fresh 50 mL conical tube. Tissue was centrifuged at 500 g for 5 min and resuspended in 10 mL of 30% Percoll solution (2.7 mL Percoll Plus (GE Healthcare Biosciences, 17–5445-01), 1 mL 10× PBS, 6.3 mL ddH2O). The samples were centrifuged at 800 g, 23°C, for 25 min with an acceleration of 4 and a deceleration of 3. Samples were loaded onto a 10X Genomics Chromium platform for GEM and cDNA generation carrying cell- and transcript-specific barcodes and sequencing libraries constructed using the Chromium Single Cell 3′ Library & Gel Bead Kit v3. Libraries were sequenced on the Illumina. NovaSeq S1 was used targeting a depth of 100,000 reads per cell. Gene counts were obtained by aligning reads to the mm10 genome (refdata-gex-GCRm38–2020-A) using CellRanger software (v.4.0.0) (10x Genomics). Single-cell clustering and differential expression analyses were carried out using Seurat (v.4.0.6) (https://satijalab.org/Seurat/index.html)73. Cells with more than 200 sequencing reads and less than 20 percent mitochondrial transcripts were selected, and genes with more than two reads across all samples entered downstream analyses. Expression counts were normalized by the “LogNormalize” method and scaled for mitochondrial read count using linear regression as implemented in Seurat’s “Regress Out” function. Variable genes were identified using the “vst” selection method. The data was then centered and scaled, analyzed by principal component analysis (PCA), and dimensionally reduced to the top 30 principal components. The cells were clustered on PCA space using gene expression data as implemented in FindNeighbors and FindClusters commands in Seurat-v4. The method returned 28 cell clusters which were then visualized on a UMAP created by the top 30 principal components. The differentially expressed genes in each cluster were output by FindAllMarkers. The clusters were identified based on cell-type-specific key signature genes. 26 clusters were confidently assigned with one cluster showing no confident cell-type signature. For identified astrocytic cells (clusters 3 & 5), the differentially expressed genes in cluster 3 and cluster 5 comparison were identified by using the FindMarkers Function. Donut plots for astrocytes were created using a cutoff of > 2 for Gfap and Apoe > 3, Vim > 1, Serpina3n > 0.9, Cd9 > 0.9. For identified microglial cells (clusters 1, 4, 9, 15, 19 & 26), re-clustering of the cells was performed to extract any lost variations from the original clustering of all cells. The data were centered and scaled, analyzed by principal component analysis (PCA), and dimensionally reduced to the top 26 principal components. The method returned 11 cell clusters which were then visualized on a UMAP created by the top 26 principal components. The differentially expressed genes in homeostatic and MGnD sample comparisons were identified by cell-type-specific key signature genes using the FindMarkers Function. Clec7a+/Lgals3+ cells were identified using high cutoffs using the Ridgeplot function for each gene. Gene expression was visualized using FeaturePlot, DittoHeatmap, and VlnPlot functions from Seurat-v4. Processed data from Zhou et al.35 and Olah et al.34 was downloaded from the AD Knowledge Portal and processed using workflow described above with some adjustments. For Zhou et al.35, cells with 5% mitochondrial content or more were removed. Cell types were annotated using the scType74. For Olah et al. only cells with less than 10 percent mitochondrial transcripts were selected. (For detailed steps, see ‘Code availability’).
Bulk RNA-seq
Smart-Seq2 libraries were prepared by the Broad Technology Labs and sequenced by the Broad Genomics Platform. cDNA libraries were generated from sorted cells using the Smart-seq2 protocol75. RNA sequencing was performed using Illumina NextSeq500 using a High Output v2 kit to generate 2 × 25 bp reads. Count files (fastq) were downloaded and aligned using Salmon (v1.7) to mm10 genome (GCA_000001635.2) for mouse data and GRCh38 (GCA_000001405.15) for human data and checked for sequencing quality using Multiqc (v1.11). Potential technical outliers were removed for further analysis. All analysis was carried out using DESeq2 (v.1.34.00)76. Biological outliers were determined using PCA plotting and heatmap visualization and removed for final analysis. Low abundance genes below a mean count of 5 reads per sample were filtered out. Comparisons were run using LRT, and the cutoff for significant genes was either P < 0.01 or P < 0.05. Heat maps were visualized using the pheatmap package (v.1.0.12), volcano plots were generated using the EnhancedVolcano package (v1.12.0), and violin plots were generated using the function geom_violin from the ggplot2 package (v3.3.5) (For detailed steps see ‘Code availability’).
Astrocytes sorting
Astrocytes were isolated following perfusion using enzymatic digestion as described in the isolation of mouse brain cells for scRNA-seq. Cells were stained for 30 minutes in the dark on ice. The following antibodies were used for negative selection: PE anti-CD45R/B220 (BD, 553089, 1:100), PE anti-Ter119 (Biolegend, 116207, 1:100), PE anti-Olig4 (R&D Systems, FAB1326P, 1:100), PE anti-CD105 (eBioscience, 12–1051-82, 1:100), PE anti-CD140a (eBioscience, 12–1401-81, 1:100), PE anti-Ly6G (Biolegend, 127608, 1:100), PE-Cy7 anti-CD11b (eBioscience, 50–154-54, 1:300), BV421™ anti-CD45 (Biolegend, 103133, 1:100) and Alexa Fluor® 700 anti-O1 (R&D Systems, FAB1327N, 1:100). APC anti-ACSA2 (Miltenyi Biotec, 130–117-535, 1:100) was used for positive selection of sorted astrocytes.
Cross-Dataset comparison
Bulk RNA-seq data from Lund et al.61 were cross-compared with our Itgb8-cKO data in WT mice. Differentially expressed genes (P < 0.05, Log2 Fold Change (FC) > (0.25)) were selected from both studies and differentially expressed genes were plotted on a scatter plot. MGnD-related genes were determined by Krasemann et al. 20174. Linear regression was calculated using geom_smooth() for MGnD genes and M0 genes (For detailed steps, see ‘Code availability’).
Ingenuity pathway analysis
Pathway analysis was performed using gene ontology (GO) enrichment analysis (http://geneontology.org). Differentially expressed genes were used to detect the pathways associated with biological processes. Differentially expressed genes with corresponding fold changes and adjusted p values were applied to gene set enrichment analysis (GSEA, https://www.gsea-msigdb.org/gsea/index.jsp)77,78 and Ingenuity pathway analysis (IPA, https://digitalinsights.qiagen.com/products-overview/discovery-insights-portfolio/analysis-and-visualization/qiagen-ipa/). In IPA, canonical pathways and biological functions were tested for generating biological networks as described previously13.
Cell-Cell Crosstalk
Microglia/Astrocyte cross-talk in Fig. 6 was determined using the Receptor-Ligand database CellTalkDB (Supplementary Table 5). Receptors in astrocytes were selected from DESeq2 normalized counts, and only receptors with expression > 10 were chosen. Ligands in Microglia were selected based on differential expression between conditions (P < 0.05). The regulatory potential for each ligand was calculated by multiplying the number of receptor interactions and its Log2FC. The Log2FC of ligands from Fig. 6i,j was calculated from Supplementary Table 1 (Fig. 1o). Log2FC of ligands from Fig. 6h was calculated from Supplementary Table 1 (Fig. 1g). The expression of astrocyte receptors from Fig. 6h–j was determined from Supplementary Table 5 (Fig. 6f) with normalized counts >10. Crosstalk in Fig. 6a was determined using the NichenetR bulk RNA-seq workflow with adjustments. (https://github.com/saeyslab/nichenetr)79. Prioritized ligands were selected from differentially expressed genes calculated from Supplementary Table 4 (Fig. 4c, P < 0.05). The expression of astrocyte receptors was determined from Supplementary Table 5 (Fig. 5b) with normalized counts > 10. The geneset of interest (geneset_oi) was selected from differentially expressed genes in astrocytes of cluster 3 (For detailed steps, see ‘Code availability’). The lr_network_top_df_large was exported as excel. To determine the regulatory potential, the Log2(fold change) of ligands was multiplied by the number of interactions, subsequently divided by 10 (for visualization purposes), and then the NichenetR-defined receptor-ligand weight was added (See Supplementary Table 5 for details). All circos plots were visualized using circlize package (v.0.4.14) (https://jokergoo.github.io/circlize_book/book/)80.
Anti-H3K9ac ChIP-seq
The anti-H3K9ac ChIP was performed using iDeal ChIP-seq kit for Histones (Diagenode, C01010059) following the manufacturer’s instruction with mild modifications. Briefly, sorted microglia were crosslinked with 1% formaldehyde at 20°C for 8 min then quenched with 0.125 M Glycine. The nuclei were isolated and then sonicated using a Bioruptor® Plus (Diagenode, B01020001) for 20 cycles (30 seconds “ON”, 30 seconds “OFF”, high power). Chromatin were then pooled from 3 individuals with same genotype for ChIP. Two μl of anti-H3K9ac antibody (Millipore, 07–352) was added per IP reaction. The DNA libraries were prepared using NEBNext® Ultra™ II DNA Library Prep Kit for Illumina® (NEB, E7103S), following the manufacturer’s instructions. The DNA libraries were analyzed using Qubit 4 Fluorometer (Invitrogen, Q33238) and 2100 Bioanalyzer DNA system (Agilent G2939BA). The pooled libraries were then sent to Azenta Life Sciences for paired-end sequencing (2×150 bp) on Illumina HiSeq platform.
ChIP-seq data analysis
Raw fastq files were first checked for quality using Multiqc sequence analysis. Cutadapt (v.4.0) was used to cut adaptors (-a AGATCGGAAGAGCACACGTCTGAACTCCAGTC -A AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT), and reads were aligned to the mouse genome (mm10) using bowtie2 (2.3.4.3)81. Subsequently, SAM files were sorted and filtered using Sambamba (v.0.8.2)82. We discarded unmapped and duplicate fragments. Sorted BAM files were indexed using samtools (v.1.15.1)83. BAM files were then converted to BigWig files for visualization using deeptools (v.3.5.0)84. Normalization for all BigWig files were carried out against effective genome size (2652783500 for mice H3K9ac ChIP-seq). Peak analysis was carried out with Macs2 (v.2.2.7)85 using stringent parameters (-f BAMPE --mfold 5 50 -p 0.001). A master-peak file was created for extracting counts from BAM files. The threshold of peak pile was set to >10 and the cutoff for fold change was set at > 2 or < 0.5 to determine significantly different peaks (Supplementary Table 1) (For more details, see ‘Code availability’). Peaks were visualized using IGV (v.2.11.4), exported as .png, and edited in Adobe Illustrator.
Lipidomics
Analyses of polar and non-polar lipids were conducted using a liquid chromatography/mass spectrometry (LC-MS) system comprised of a Shimadzu Nexera X2 U-HPLC (Shimadzu Corp.) coupled to an Exactive™ Plus orbitrap mass spectrometer (Thermo Fisher Scientific). 30K sorted microglia were prepared in 50 μl isopropanol. After centrifugation, 10 μL supernatant was injected directly onto a 100 × 2.1 mm, 1.7 μm ACQUITY BEH C8 column (Waters). The column was eluted isocratically with 80% mobile phase A (95:5:0.1 vol/vol/vol 10mM ammonium acetate/methanol/formic acid) for 1 minute followed by a linear gradient to 80% mobile-phase B (99.9:0.1 vol/vol methanol/formic acid) over 2 minutes, a linear gradient to 100% mobile phase B over 7 minutes, then 3 minutes at 100% mobile-phase B. Mass spectrometry (MS) analyses were carried out using electrospray ionization in the positive ion mode using full scan analysis over 200–1100 m/z at 70,000 resolution and 3 Hz data acquisition rate. Other MS settings were: sheath gas 50, in source CID 5 eV, sweep gas 5, spray voltage 3 kV, capillary temperature 300°C, S-lens RF 60, heater temperature 300°C, microscans 1, automatic gain control target 1e6, and maximum ion time 100 ms. Raw data were processed using TraceFinder software (Thermo Fisher Scientific) for targeted peak integration and manual review of a subset of identified lipids and using Progenesis QI (Nonlinear Dynamics) for peak detection and integration of both lipids of known identity and unknowns. Lipid identities were determined based on a comparison to reference extracts and are denoted by the total number of carbons in the lipid acyl chain(s) and total number of double bonds in the lipid acyl chain(s). All analysis was carried out using R (v.4.1.1). A total of 199 metabolites were reduced to 170 by removing any metabolites with missing data in one or more of the samples. Data were normalized using the z-score method. The heatmap of the 170 metabolites was plotted using the pheatmap package (v.1.0.12).
Immunohistochemistry
Mice were transcardially perfused with cold HBSS before tissue excision and fixation. Tissues that were not adequately perfused were not further analyzed to eliminate autofluorescence associated with blood contamination. Two different tissue preparation protocols (paraffin-embedded for human brain tissue or microtome free-floating sections) were applied, as previously described23. Briefly, sections were blocked in PBS with 20% horse serum (Thermo Fisher Scientific, NC9909742) and 0.3% Triton X-100 (Sigma) for 1 hour at room temperature. Primary antibodies were incubated overnight at 4oC in PBS containing 2% horse serum and 0.3% Triton X-100. The following primary antibodies were mouse anti-Aβ (1:300, BioLegend, 803001); chicken anti-GFAP (1:400, Abcam, 4674); mouse anti-Phospho-Tau (1:50, Thermo Fisher Scientific, AT-100, MN1060); goat anti-IBA1 (1:100, Abcam 5076); rabbit anti-IBA1 (1:200, Wako, 019–19741); mouse anti-HJ3.4B86 (1:600, Holtzman Lab); rat anti-LAMP1 (1:100, DSHB, 1D4B); rat anti-LGALS3 (1:200, Cedarlane Labs, CL8942AP); goat anti-LGALS3 (1:100, R&D, 9039-GAB-050); rat anti-CLEC7A (1:100, Invivogen, Clone: R1–8g7); rabbit anti-APOE (1:400, Cell Signaling Technology, 13366S); rabbit anti-pSMAD3 (1:100, Abcam, ab52903); guinea pig anti-Plin2 (1:200, Fitzgerald industries international, 20R-AP002); goat anti-Serpina3n (1:200, R&D systems, AF4709) and anti-MHC II (1:200, BioLegend, I-A/I-E, 107601). Secondary antibodies included: Cy2/Cy3/Cy5-conjugated donkey anti-mouse/goat/rabbit/rat/chicken/guinea pig antibodies (1:200; all from Jackson Immunoresearch). Sections were imaged on a Zeiss LSM710 confocal using 20x or 40x objective. Two negative controls were routinely used in immunostaining procedures, staining with isotype control antibody followed by the secondary antibody, or staining with the secondary antibody alone. The tissues were applied to slides, mounted with ProLong™ Gold Antifade Mountant with DAPI (Thermo Scientific, P36931) or with VECTASHIELD® Antifade Mounting Medium (Vector Laboratories, H-1000), and sealed with coverslips. The mouse brains of Itgb8-tdT and Smad2/3-cKO line were harvested following transcardial perfusion with 20 mL cold PBS and 20 mL cold 4% formaldehyde. Tissue was fixed in 4% formaldehyde overnight at 4°C, followed by overnight incubation in 30% sucrose. Samples were embedded (Tissue-Plus™ O.C.T. Compound, Fisher Scientific, 23–730-571) and cryosectioned at 20 μm. Sections were mounted onto glass, blocked with PBS containing 1–2% BSA, 5% donkey serum and 0.5% TritonX-100. Primary and secondary antibodies were diluted in PBS containing 1% BSA and 0.25%–0.5% TritonX-100. The primary antibodies were goat anti-Sox9 (1:300, R&D systems, AF3075); rabbit anti-Olig2 (1:300, Millipore, AF2418); mouse anti-NeuN (1:300, Millipore, MAB377); rabbit anti-GFAP (1:300, DAKO, Z0334); rat anti-GFAP (1:300, Invitrogen 13–0300); goat anti-Pdgfra (1:300, R&D Systems, AF1062); Goat anti-IBA1 (1:300, Novus, NB100–1028); Rat anti-Cd68 (1:300, Bio-Rad, MCA1957); Rabbit anti-Apoe (1:300, Abcam, ab183596). Secondary antibodies included: Cy2/Cy3/Cy5-conjugated donkey anti-mouse/goat/rabbit antibodies (1:200; Jackson Immunoresearch).
Thioflavin-S staining and quantification
Free-floating brain sections with a section thickness of 30 μm were incubated in filtered 1% aqueous Thioflavin-S (Sigma T1892) for 8 minutes at room temperature. The sections were then sequentially washed in 80%, 95% ethanol and distilled water. Entire cortical images were taken with Leica Microsystems DMi8 Microscope using Tile scan. The positive area of Thioflavin-S was quantified using the automatic thresholding method in Fiji with “Otsu”. Seven animals were analyzed per group.
Cresyl Violet staining and neuron counting
Free-floating brain sections with a section thickness of 30 μm were stained with Cresyl Violet (Sigma) to estimate neuronal survival. Staining was performed to visualize neurons. Sample slices were incubated in 100% ethanol for 6 min and defatted in xylene for 15 min, followed by another 10 min in 100% ethanol. After rinsing in distilled water, the slides were stained with 0.5% Cresyl Violet acetate for 15 min and again rinsed in distilled water. The sections were then placed in differentiation buffer (0.2% acetic acid in 95% ethanol) for 2 min, dehydrated with ethanol and xylene, and mounted with Depex medium. Pyramidal neurons were counted in each brain from serial sections located 30 μm apart and analyzed using One-way ANOVA with Fisher’s LSD post hoc test. The % neuronal survival is quantified relative to the number of the pyramidal neurons in the age-matched APOE3-KI mice.
RNAscope
RNAscope® ISH technology was applied on human formalin-fixed, paraffin-embedded (FFPE) sections and mouse 4% PFA-fixed (fixed-frozen) sections using RNAscope® Multiplex Fluorescent Reagent kit (v2, ACD, 323100), ITGB8 human probe (Hs-ITGB8-XMfa, ACD, 515881), Itgb8 mouse probe (Mm-Itgb8, ACD, 407931), INPP5D human probe (Hs-INPP5D, ACD, 465051), negative control probe DapB (ACD, 321831), and positive control probe (Hs-PPIB, ACD, 313901) according to the manufacturer instructions. The sections were further blocked with 5% BSA, 5% Normal Donkey serum, 0.3% Triton in PBS for 1 hour, followed by primary antibody overnight at 4°C (anti-GFAP, 1:400, Abcam, 4674; anti-IBA1, 1:200, Wako, 019–19741; anti-HJ3.4B86, 1:600, Holtzman Lab). The secondary antibodies (AlexaFluor 488 Donkey anti-mouse, 1:300, AlexaFluor 546 Donkey anti-goat, 1:300, AlexaFluor 546 Donkey anti-rabbit, 1:300, AlexaFluor 647 Donkey anti-mouse, 1:300) were applied for 2 hours. After PBS wash, the sections were mounted with Fluoromount-G™ mounting medium (Thermo Fisher, 00–4958-02) with DAPI and sealed with coverslips.
Image visualization
Images were cropped, merged, and optimized using Fiji, Photoshop CS6 13.0 (Adobe) and were arranged using Adobe Illustrator CS5 15.1.
Image analysis
To assess the amount of MGnD and neuritic dystrophy, randomly selected 40x magnification images were taken close to the injection sites of APOE3-KI, APOE4-KI, and APOE4-cKO mice. The positive area of LGALS3 and LAMP1 of APOE3-KI and APOE4-KI mice were quantified using the automatic thresholding method in Fiji with “Otsu”. The positive area of LAMP1 was further normalized to the positive area of each IBA1+ cell. 5–7 animals were analyzed per experimental group. The integrated density of the LGALS3 of APOE4-KI and APOE4-cKO mice was measured using Fiji. Four animals were analyzed per group. To quantify the HJ3.4B+ plaque load in Tmem119CreERT2/WT:Spi1fl/WT:APP/PS1 and Tmem119WT/WT:Spi1fl/WT:APP/PS1 mice, 10x magnification images were taken from the cortex. The positive area of HJ3.4B was quantified using the automatic thresholding method “RenyiEntropy” in Fiji. The positive areas of CLEC7A and IBA1 were quantified using the automatic thresholding method “Moments” in Fiji. 3–6 mice were analyzed per experimental group. To assess the amount of CLEC7A, IBA1 and Serpina3N, 10x and 40x magnification images were taken from the cortex of PU.1 inhibitor or control injected APP/PS1:APOE4-KI mice. The integrated density of the CLEC7A was measured using Fiji. The automatic thresholding methods in Fiji were used to quantify the positive areas (“Moments” for IBA1, “Triangle/Triangle” for Serpina3N/Gfap). To assess the amount of CLEC7A and phospho-Tau (AT-100), 40x magnification images were taken from the cortex of P301S:APOE3-KI, P301S:APOE3-cKO, P301S:APOE4-KI, and P301S:APOE4-cKO mice. The positive areas of CLEC7A and AT-100 were quantified using the automatic thresholding method in Fiji with “Triangle”. 7–16 ROIs from 4 animals were analyzed per experimental group. To assess the amount of APOE, 40x magnification images were taken from the cortex of P301S:APOE4-KI and P301S:APOE4-cKO mice. The positive area of APOE was quantified using the automatic thresholding method in Fiji with “Triangle”. Five mice per group were analyzed per experimental group. To measure the area of CLEC7A, LGALS3, GFAP, APOE, Serpina3n and Plin2 in the plaque area, 20x or 40x magnification images were taken from the cortex of APP/PS1:APOE3-KI, APP/PS1:APOE3-cKO, APP/PS1:APOE4-KI, and APP/PS1:APOE4-cKO mice. The images were exported to Fiji and individual plaques were selected and cropped for analysis. The positive area of CLEC7A, LGALS3, and GFAP per plaque were quantified using automatic thresholding methods in Fiji (“RenyiEntropy” for CLEC7A, “Otsu” for LGALS3 and GFAP). The positive area of CLEC7A, LGALS3, and GFAP was further normalized to plaque size in each image. 55–73 plaques for CLEC7A, 12–20 plaques for LGALS3, and 39–61 plaques for GFAP were analyzed per experimental group. For quantifying the percentage of APOE+ or Serpina3n+ area in GFAP+ cells, 39–61 plaque regions for APOE and 30–44 plaque regions for Serpina3n per experimental group were analyzed. The automatic thresholding methods in Fiji were used to quantify the positive areas (“Otsu/Otsu” for APOE/GFAP; “RenyiEntropy/Triangle” for Serpina3n/GFAP). The overlay regions were calculated using customized macros in Fiji. To calculate the percentage of Plin2 in IBA1+ cells, The area of the IBA1+ cells and Plin2+ were selected by freehand selection and measured in Fiji. 8–24 cells per group were analyzed. To quantify the HJ3.4B+ plaque load and LAMP1 area, 10x magnification images were taken from the cortex of APP/PS1:APOE3-KI, APP/PS1:APOE3-cKO, APP/PS1:APOE4-KI, and APP/PS1:APOE4-cKO mice. The positive area of HJ3.4B and LAMP1 were quantified using automatic thresholding methods in Fiji with “Otsu” and “Triangle” separately. The average positive area was calculated for each animal. The number of plaques were quantified using customized macros in Fiji. 3–6 images per animal were taken, and 7–14 mice were quantified per group. To quantify the LGALS3, GFAP and pSMAD3 in human brain, 40x magnification images were taken from the cortical section of human AD subjects carrying APOE ε3/3 and APOE ε3/4 alleles. The integrated density of LGALS3 was measured using Fiji. 8–9 samples were analyzed per group. For GFAP quantification, individual plaques were selected and cropped for analysis in Fiji. The positive area of GFAP per plaque was quantified using the automatic thresholding method “Otsu” in Fiji. The positive area of LGALS3 was further normalized to plaque size in each image. 43–52 plaques per group were analyzed. For pSMAD3 quantification, individual IBA1+ cells were selected and cropped for analysis in Fiji. The positive area of pSMAD3 per IBA1+ cell was quantified using the automatic thresholding method “Otsu” in Fiji. The positive area of pSMAD3 was further normalized to the IBA1+ cell area, and 27–33 cells per group were analyzed. To quantify the fluorescence of INPP5D in IBA1+ cells and ITGB8 in GFAP+ cells, 2–3 images per sample were collected in the gray matter. The automatic thresholding methods in Fiji were used to quantify the positive areas (“RenyiEntropy/Triangle” for INPP5D/IBA1; “Otsu/Triangle” for ITGB8/GFAP). The overlay regions were calculated using customized macros in Fiji. 7–8 samples per sex per group were analyzed for INPP5D, and for ITGB8, 7–8 male samples per group were analyzed. To quantify the fluorescence of Itgb8 in GFAP+ cells, 3–4 images per sample were collected in the cortex of APP/PS1:APOE4-KI and APP/PS1:APOE4-cKO mice. Itgb8+ positive areas in GFAP+ astrocytes were quantified using automatic thresholding methods in Fiji with “RenyiEntropy/Triangle” for Itgb8/GFAP. The overlay regions were calculated using customized macros in Fiji. 3–5 mice per group were analyzed. To measure the immunoreactivity of CLEC7A and GFAP, 20x magnification images were taken from the cortex of control and Itgb8-cKO mice. The integrated density was measured in Fiji. Twelve ROIs per group were analyzed. To quantify the area of plaques in the rLGALS3-injected APP/PS1 brains, 20x magnification images were taken from the cortical section close to the injection site. The images were exported to Fiji for analysis. The positive area of HJ3.4B was quantified using the automatic thresholding method “Intermodes” in Fiji. 8–9 ROIs from 5 mice were analyzed per experimental group. To quantify the area of plaques, CLEC7A and GFAP in the rLGALS3-injected APP/PS1:APOE4-KI brains, 10x and 40x magnification images were taken from the cortical section close to the injection site. The positive areas were quantified using the automatic thresholding method with “Moments” for GFAP and “RenyiEntropy” for HJ3.4B and CLEC7A in Fiji. Eight ROIs from 5 mice were analyzed per experimental group. To quantify the plaque load and MHC II area in the anti-ITGB8-antibody-injected APP/PS1 brain, 20x and 40x magnification images were taken from the cortical section close to the injection site. The images were exported to Fiji, and the positive area of HJ3.4B or MHC II was quantified using the automatic thresholding method “Otsu” in Fiji. 4–7 mice per group were analyzed for MHC II. 8–9 mice per group were analyzed for HJ3.4B (two independent experiments combined, normalized to control group). To quantify the area of plaques, CLEC7A and GFAP in the anti-ITGB8-antibody-injected APP/PS1:APOE4-KI brain, 20x and 40x magnification images were taken from the cortical section close to the injection site. The positive areas were quantified using the automatic thresholding method with “Otsu” for CLEC7A, “Triangle” for HJ3.4B, and “RenyiEntropy” for GFAP in Fiji. Eleven ROIs from 6–7 mice per group were analyzed. To measure the load of CLEC7A, IBA1, LGALS3, LAMP1, GFAP in the plaque area, 10x or 20x magnification images were taken from the cortex of APP/PS1 and APP/PS1:Inpp5d-cKO mice. The integrated density was measured using Fiji. 115–218 plaque regions for CLEC7A and IBA1, 126–138 plaques for LAMP1, 141–186 plaques for LGALS3, and 127–154 plaques for GFAP per experimental group were analyzed. To quantify the HJ3.4B+ plaque load in the cortex, 10x magnification images were taken from the cortex of APP/PS1 and APP/PS1:Inpp5d-cKO mice. The positive area of HJ3.4B was quantified using the automatic thresholding method “Otsu” in Fiji. Seven mice (3 ROIs per mouse) were analyzed per experimental group.
Statistical analysis
Sample sizes for experiments were chosen based on prior publications using APP/PS1, P301S mice, and human analysis, which defined the expectations for the effect size and variance. Statistical analyses were performed using GraphPad Prism statistical software. All comparisons groups were assessed for normal distribution. We used Student’s t-test for comparisons with only two groups and one-way ANOVA with Fisher’s LSD post hoc test for multiple groups.
Extended Data
Extended Data Fig. 1 |. APOE4 impairs microglial response to acute neurodegeneration.
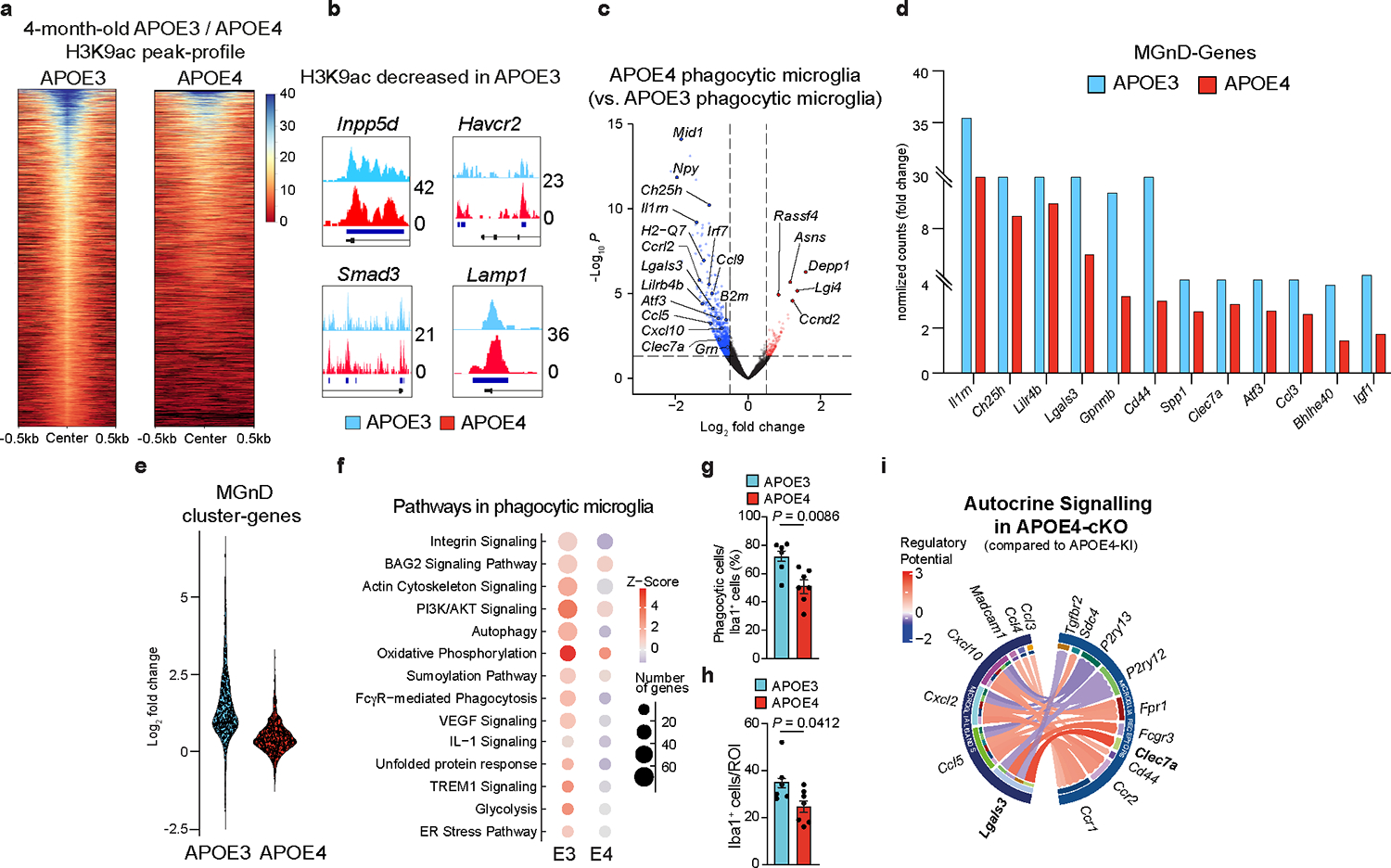
a, H3K9ac peak-plot heat map of microglia from 4-month-old APOE3-KI or APOE4-KI mice (n = 3 mice/group). b, H3K9ac peaks of key genes. c, Volcano plot of DEGs of phagocytic microglia isolated from APOE4-KI vs. APOE3-KI mice. DEGs were identified using DESeq2 analysis with an LRT (n = 4–6 mice/group, P < 0.05). d, Fold change induction of key MGnD genes expressed in phagocytic microglia relative to non-phagocytic microglia, isolated from APOE3-KI and APOE4-KI mice. DEGs were identified using DESeq2 analysis with an LRT and key MGnD genes were selected from Krasemann et al.4. e, Violin plot of top-300 induced MGnD genes in phagocytic microglia isolated from APOE3-KI and APOE4-KI mice (Fig. 1g). f, Top-affected canonical pathways in phagocytic microglia compared to non-phagocytic microglia from APOE3-KI and APOE4-KI mice (P < 0.05). g, Quantification of phagocytic Iba1+ cell numbers from AN-injected APOE3-KI and APOE4-KI mice (n = 6 APOE3-KI mice, n = 7 APOE4-KI mice). h, Quantification of Iba1+ cells from AN-injected APOE3-KI and APOE4-KI mice (n = 6 APOE3-KI mice, n = 7 APOE4-KI mice). i, Circos plot illustrating ligand- receptor interactions in APOE4-cKO microglia compared to APOE4-KI microglia. Two-tailed Student’s t-test. Data were presented as mean ± s.e.m.
Extended Data Fig. 2 |. No differences of APOE expression between P301S:APOE3-KI and P301S:APOE4-KI mice.
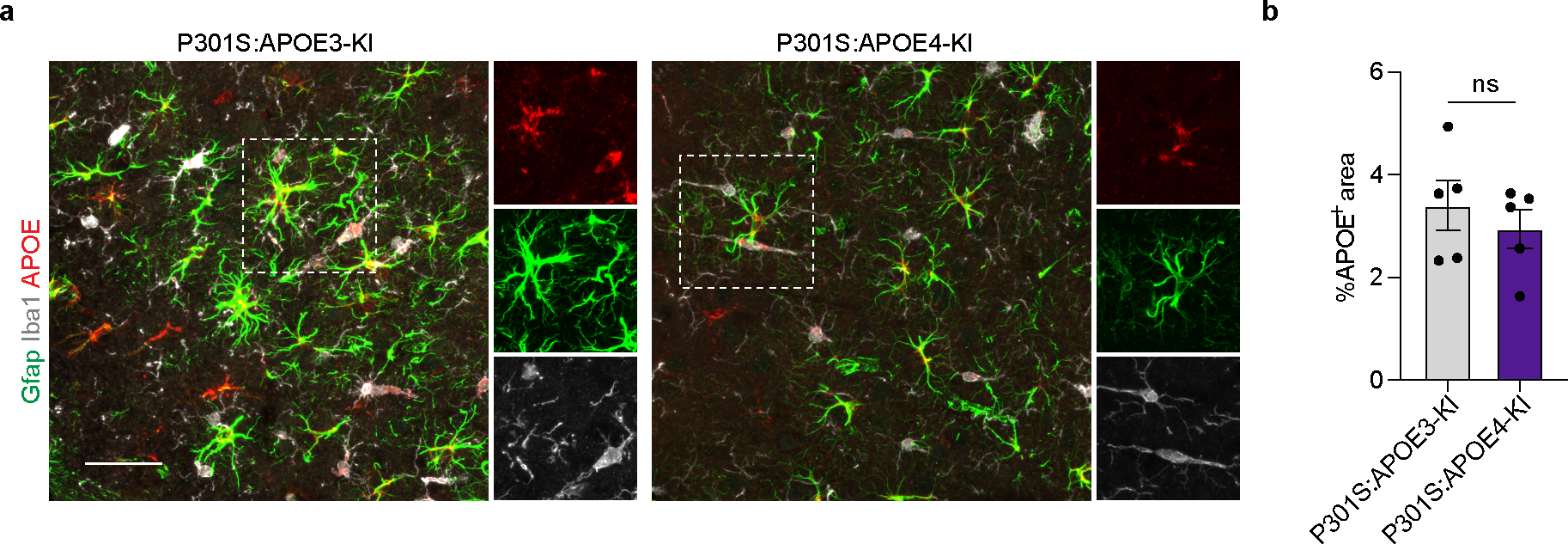
a, Confocal images of GFAP, APOE and Iba1 in P301S:APOE3-KI and P301S:APOE4-KI mice. Scale bar: 50 μm. b, Quantification of APOE in the cortex (n = 5 mice/group). Two-tailed Student’s t-test. Data presented as mean ± s.e.m.
Extended Data Fig. 3 |. Targeting microglial APOE4 restores neurodegenerative microglia in APP/PS1 mice.
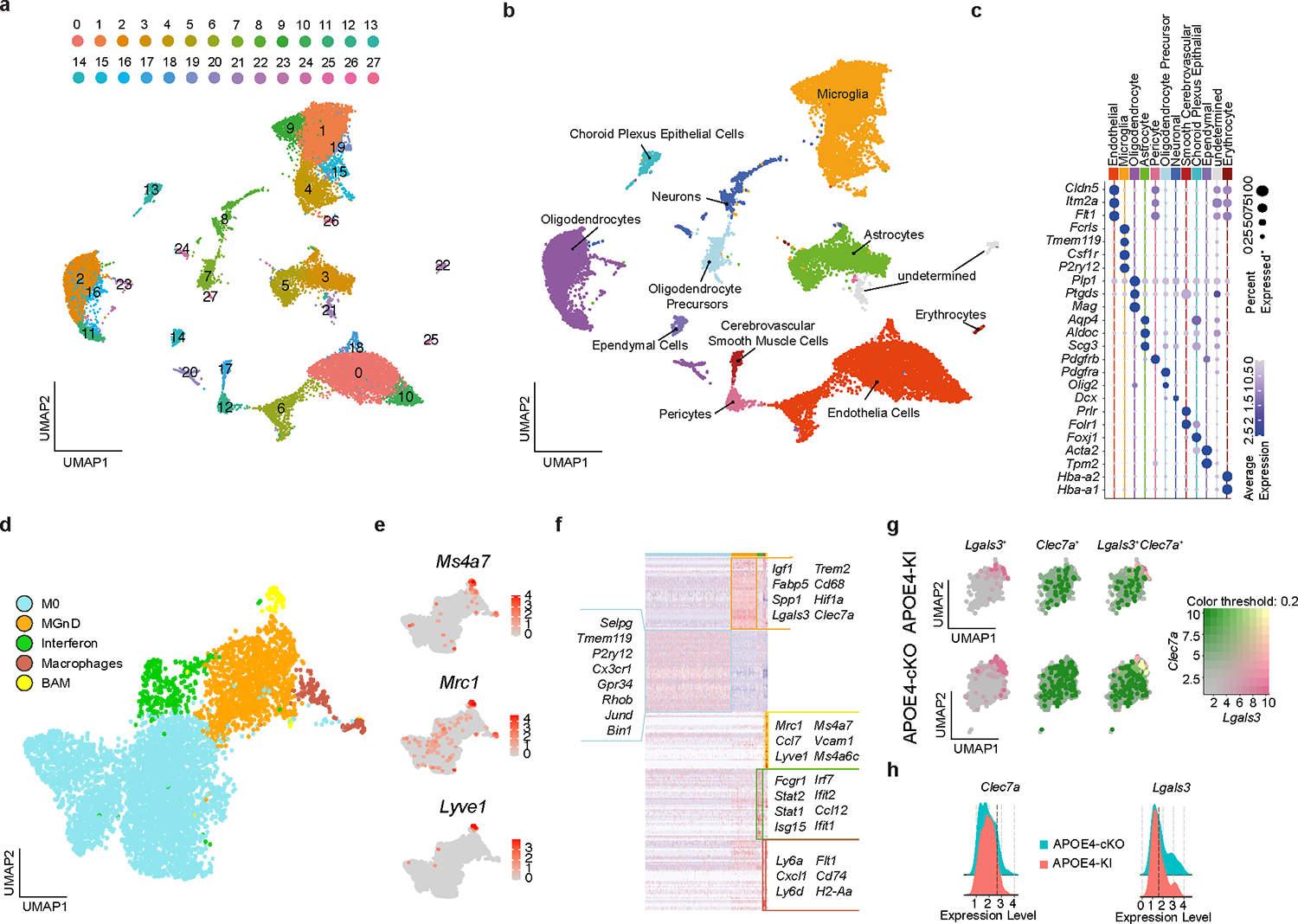
a, UMAP plot of unsupervised seurat clusters from scRNAseq analysis of brain cells isolated from APP/PS1:APOE4-KI and APP/PS1:APOE4-cKO mice (n = 2). b, UMAP plot of labelled cell types.c, Cell-specific genes used to identify cell types from seurat clusters. d, UMAP plot of microglia/myeloid cells re-clustered. e, Feature plot showing expression of border-associated macrophages genes. f, Heat map of DEGs representing the microglia/myeloid cluster. DEGs were identified using FindAllMarker Seurat function (P < 0.05). g, Feature plot showing co-expression of Lgals3 and Clec7a microglia. h, Ridgeplot of Clec7a and Lgals3 in APOE4-cKO and APOE4-KI in MGnD microglia showing gene expression cut-off for Clec7aHi (2.65) and Lgals3Hi (1.68) microglia.
Extended Data Fig. 4 |. Deletion of microglial APOE4 alleviates lipid dysregulation in APP/PS1 mice.
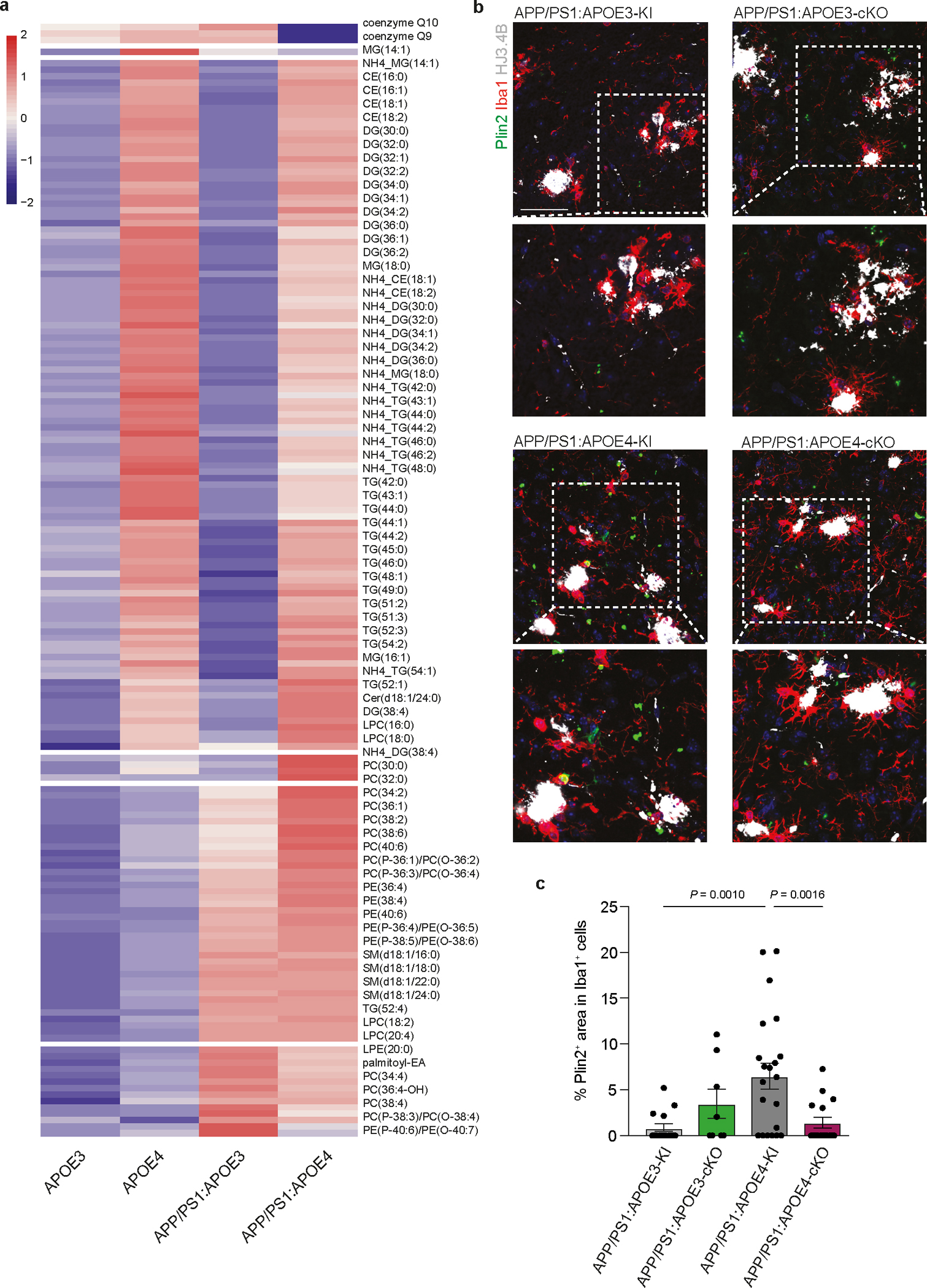
a, Heat map of lipids significantly altered by genotype in microglia isolated from APOE3-KI, APOE4-KI, APP/PS1:APOE3-KI, and APP/PS1:APOE4-KI. DEGs were identified using DESeq2 analysis with an LRT (n = 2 mice, P < 0.05). b, Confocal images of Plin2, Iba1 and HJ3.4B in APP/PS1:APOE3-KI, APP/PS1:APOE3-cKO, APP/PS1:APOE4-KI, and APP/PS1:APOE4-cKO mice. Scale bar: 50 μm. c, Quantification of Plin2+ area in Iba1+ cells (n = 15 cells from APP/PS1:APOE3-KI mice, n = 8 cells from APP/PS1:APOE3-cKO mice, n = 24 cells from APP/PS1:APOE4-KI mice, n = 16 cells from APP/PS1:APOE4-cKO mice). One-way ANOVA. Data presented as mean ± s.e.m.
Extended Data Fig. 5 |. Deletion of microglial APOE4 promotes astrocyte activation in APP/PS1 mice.
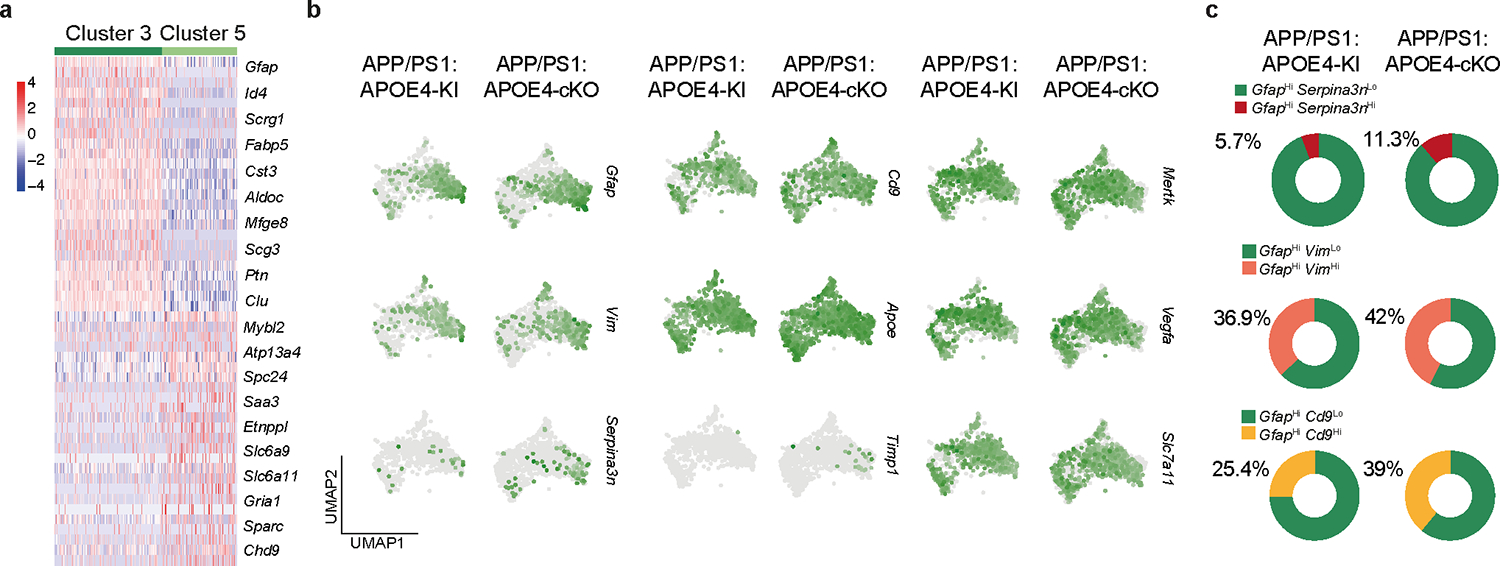
a, Heat map showing DEGs of clusters 3 and 5 (astrocyte clusters). DEGs were identified using FindMarkers Seurat function (P < 0.05, n = 2). b, UMAP projection of disease-associated astrocyte genes29 in APP/PS1:APOE4-KI and APP/PS1:APOE4-cKO mice. c, Donut charts showing percentage of GfapHi:Serpina3nLo, GfapHi:Serpina3nHi, GfapHi:VimLo, GfapHi:VimHi, GfapHi:Cd9Lo, and GfapHi:Cd9Hi microglia between APP/PS1:APOE4-KI and APP/PS1:APOE4-cKO mice.
Extended Data Fig. 6 |. Impaired induction of MGnD signature and astrocyte activation in APOE ε4 AD brains.
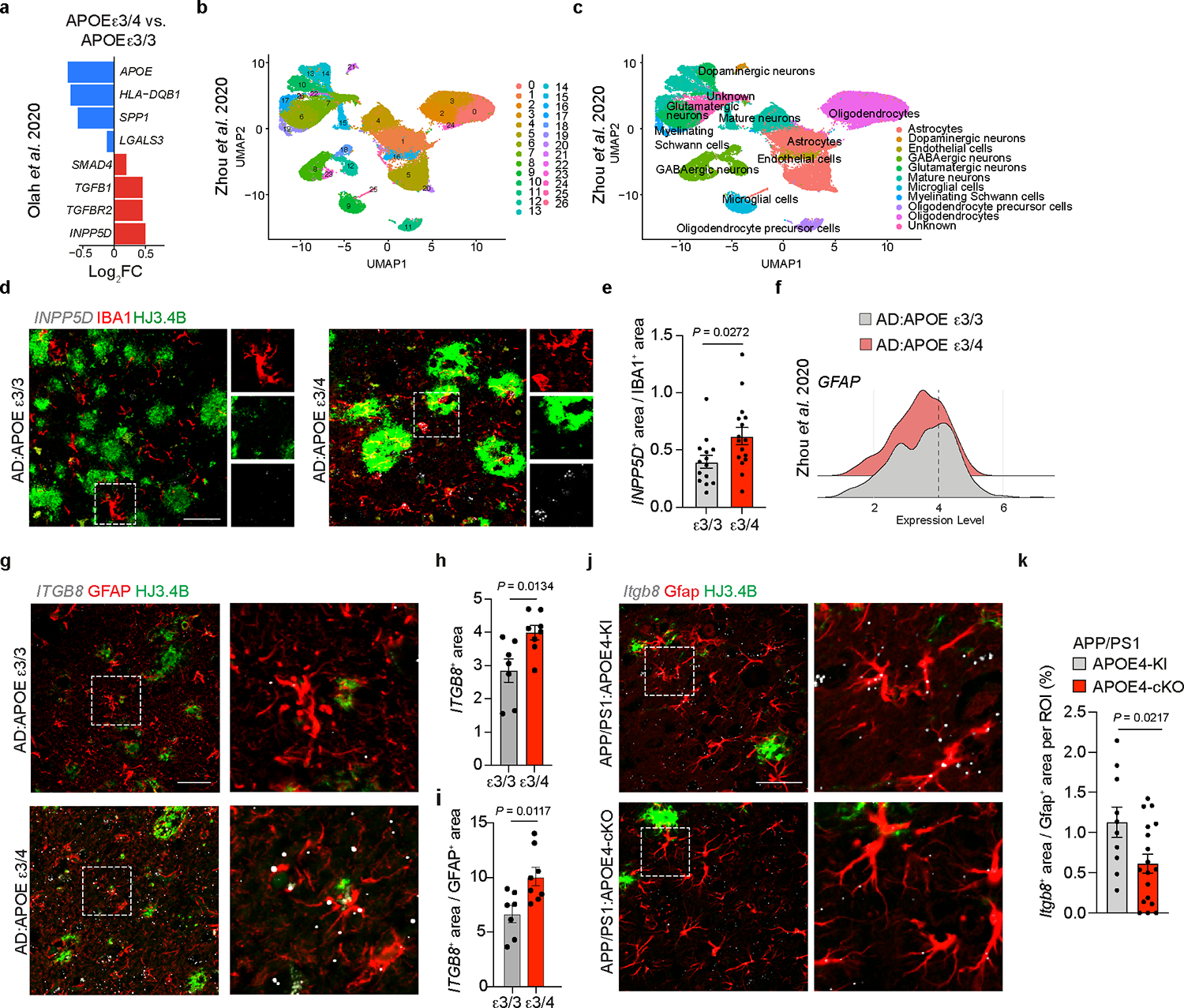
a, DEGs of female APOE ε3/3 carriers (n = 4) vs. APOE ε3/4 (n = 3) carriers analyzed from Olah et al.34. DEGs were identified using FindMarkers Seurat function (P < 0.01). b, UMAP plot of brain scRNAseq showing Seurat clusters of AD:APOE ε3/3 carriers and AD:APOE ε3/4 carriers analyzed from dataset by Zhou et al.35. c, UMAP plot indicating cell type assignment to clusters analyzed from dataset by Zhou et al.35. d, Confocal images of IBA1 and HJ3.4B immunoreactivity and detection of INPP5D gene expression using RNAscope in AD:APOE ε3/4 carriers compared to AD:APOE ε3/3 carriers. e, Quantification of INPP5D fluorescence in IBA1+ microglia in AD:APOE ε3/4 carriers compared to AD:APOE ε3/3 carriers (n = 14 AD:APOE ε3/3 carriers, n = 16 AD:APOE ε3/4 carriers). f, Ridgeplot of GFAPHi vs. GFAPLo astrocytes analyzed from dataset by Zhou et al.35. g, Confocal images of GFAP and HJ3.4B immunoreactivity and detection of ITGB8 gene expression using RNAscope in AD:APOE ε3/4 males compared to AD:APOE ε3/3 males. h,i, Quantification of ITGB8 fluorescence in ROI (h) and in GFAP+ astrocytes (i) in AD:APOE ε3/4 males compared to AD:APOE ε3/3 males (n = 7 AD:APOE ε3/3 carriers, n = 8 AD:APOE ε3/4 carriers). j, Confocal images of Gfap and HJ3.4B immunoreactivity and detection of Itgb8 mRNA expression using RNAscope in APP/PS1:APOE4-cKO mice compared to APP/PS1:APOE4-KI mice. k, Quantification of Itgb8 fluorescence in Gfap+ astrocytes (n = 10 ROIs from 3 APP/PS1:APOE4-KI mice, 18 ROIs from 5 APP/PS1:APOE4-cKO mice). Two-tailed unpaired Student’s t-test. Scale bar: 50 μm. Data were shown as mean ± s.e.m.
Extended Data Fig. 7 |. Expression of Itgb8 in the adult mouse brain.
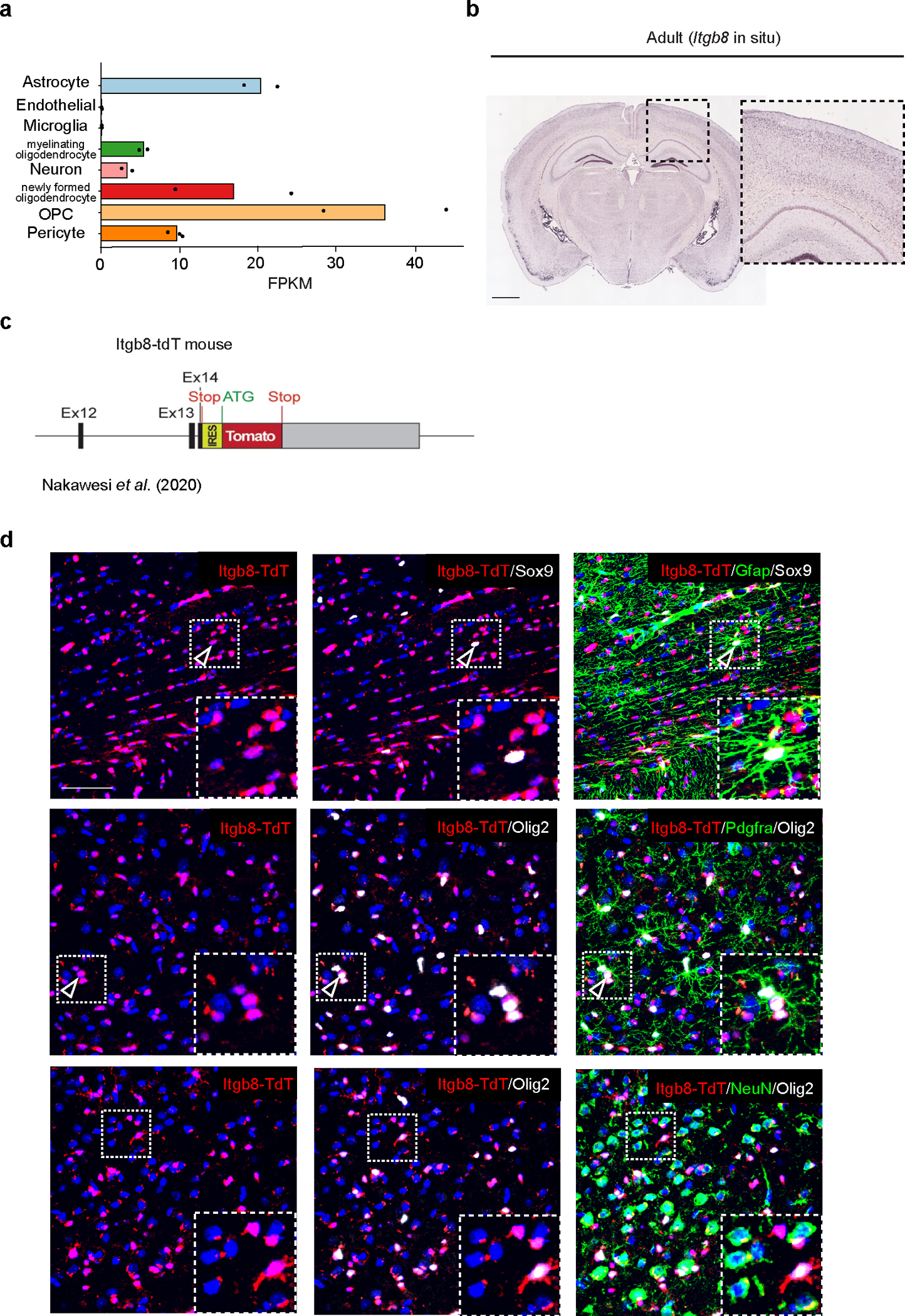
a, Dot plot showing expression level of Itgb8 in multiple CNS cell types (adopted from Zhang et al.38). b, Representative in situ image of Itgb8 in the cortex of adult mouse brain (Allen Brain Atlas: https://mouse.brain-map.org). n = 2 (the Allen Brain Atlas tested the Itgb8 probe on two brains; one sectioned sagitally and another sectioned coronally). Scale bar: 1 mm. c, Schematics of Itgb8−tdT transgenic mouse strain37. d, Representative images of Itgb8-TdT, Sox9, Gfap (top); Itgb8-TdT, Pdgfra, Olig2 (middle); and Itgb8-TdT, NeuN, Olig2 (bottom). Arrowheads indicate magnified ROIs (n = 6). Data are representative of 2 independent experiments. Scale bar: 50 μm.
Extended Data Fig. 8 |. Blocking Itgb8 signaling enhances MGnD response and reduces AD pathology in APP/PS1 mice.
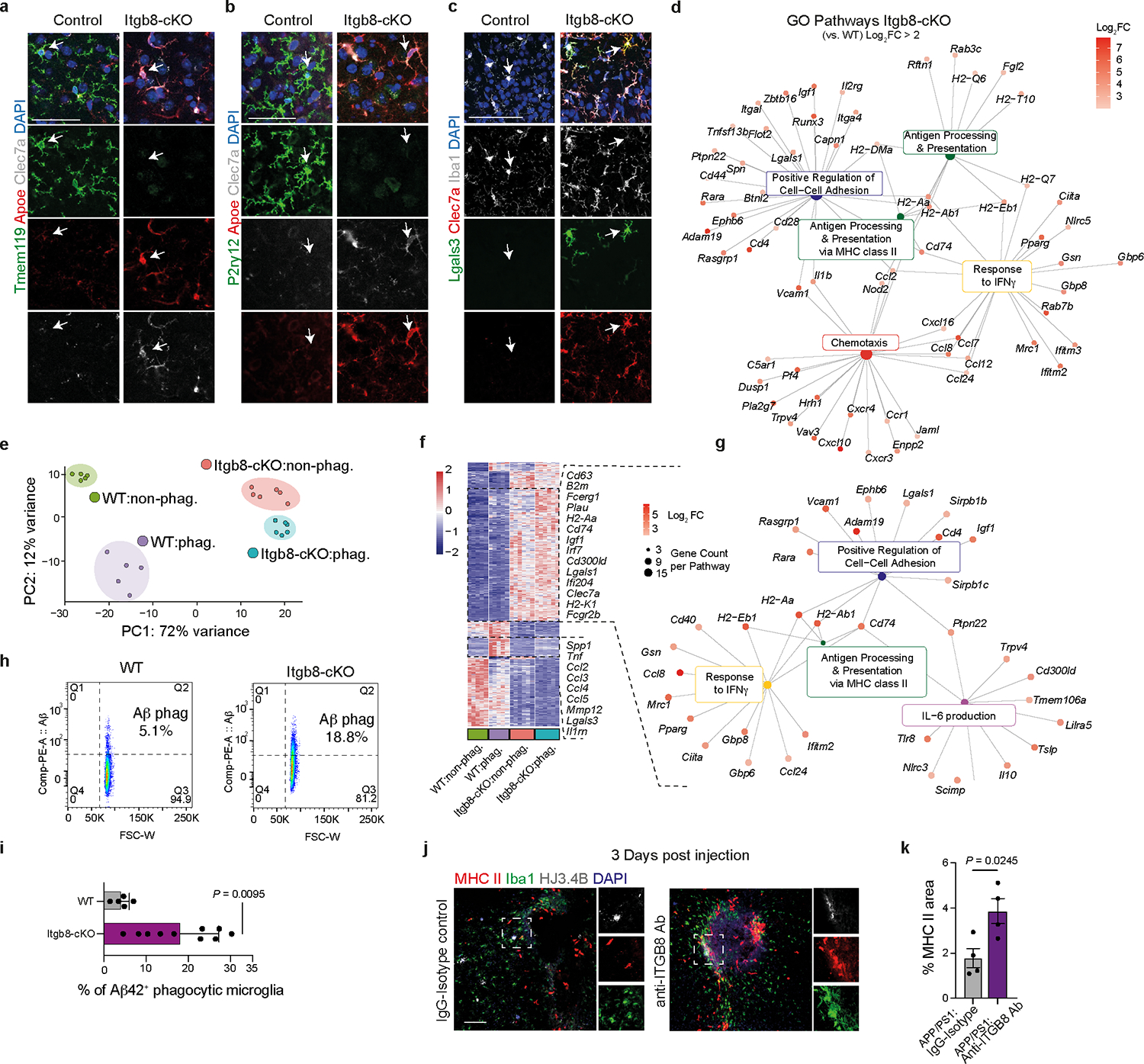
a–c, Confocal images of Tmem119, Apoe, and Clec7a staining (a); P2ry12, Apoe, and Clec7a staining (b); Lgals3, Iba1, and Clec7a staining (c) in the cortex of Itgb8-cKO mice and littermate controls. Arrows indicate microglia. Data are representative of 2 independent experiments. Scale bar: 50 μm. d, Gene ontology network of increased DEGs in microglia from Itgb8-cKO mice compared to WT littermates. DEGs were identified using DESeq2 analysis with an LRT and gene ontology pathways selected with P < 0.05. e, PCA of each genotype. f, Heat map of DEGs of phagocytic- and non-phagocytic microglia isolated from WT and Itgb8-cKO mice. DEGs were identified using DESeq2 analysis with an LRT (n = 5–6 mice/group, P < 0.05). g, Gene ontology network of top pathways in phagocytic- and non-phagocytic microglia from Itgb8-cKO mice compared with WT mice. DEGs were identified using DESeq2 analysis with an LRT and gene ontology pathways selected with P < 0.05. h, FACS plot of Aβ phagocytosis in WT and Itgb8-cKO mice. Percentage calculated as Aβ42-Alexa Fluor 555+ out of Fcrls+CD11b+Ly6C− microglia. i, Quantification of percentage of Aβ−42 phagocytic microglia in WT and Itgb8-cKO mice (n = 5 WT mice, n = 10 Itgb8-cKO mice). j, Confocal images of MHC II, Iba1, and HJ3.4B in APP/PS1 mice injected with anti-ITGB8 neutralizing antibody and IgG isotype control. Scale bar: 100 μm. k, Quantification of MHC II+ immunoreactivity at the injection site (n = 4 mice/group). Two-tailed Student’s t-test. Data were presented as mean ± s.e.m.
Extended Data Fig. 9 |. Microglial deletion of Smad2/3 induces MGnD phenotype.
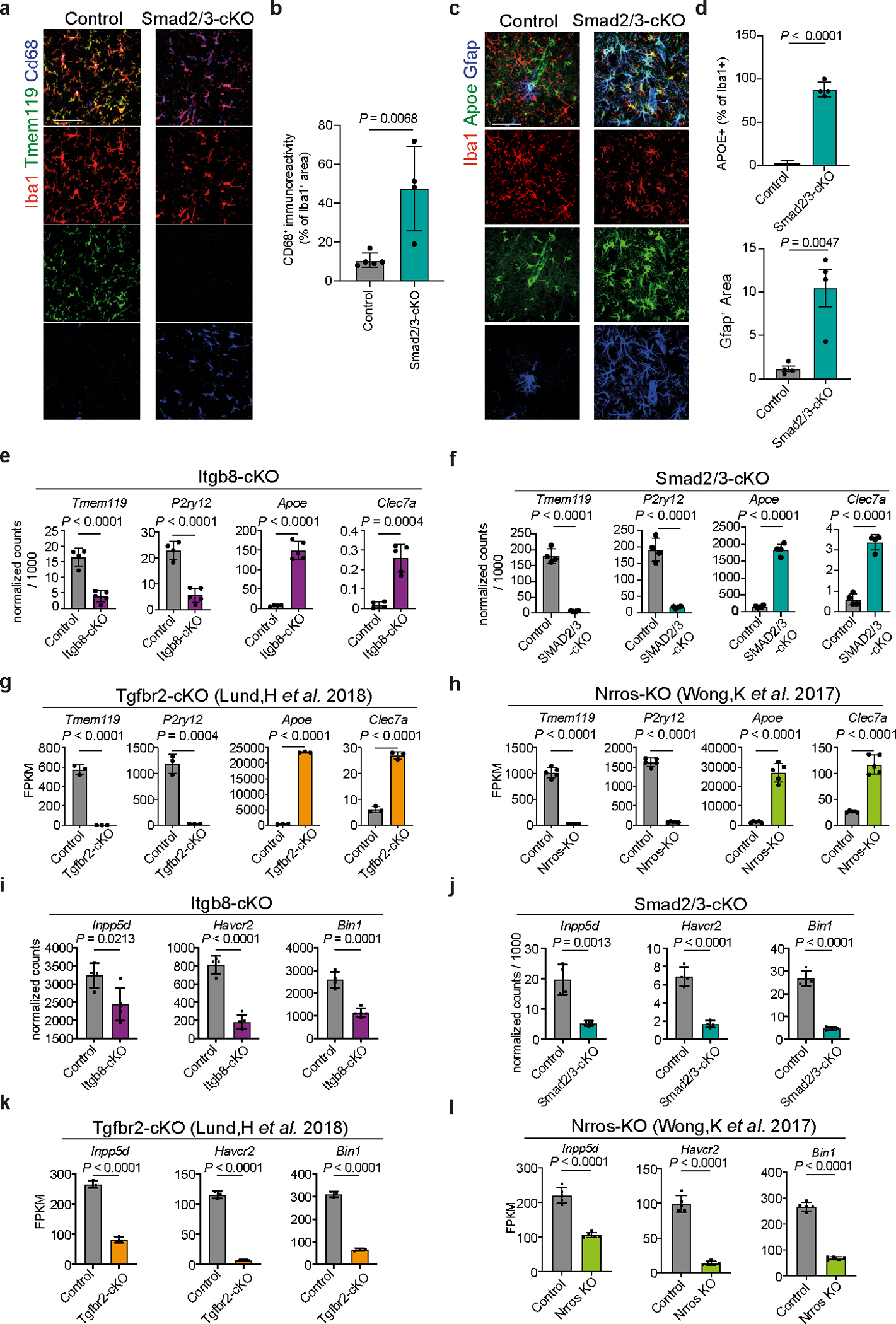
a, Confocal images showing Iba1, Tmem119 and Cd68 staining in control and Smad2/3-cKO mice. Scale bar: 50 μm. b, Quantification of Cd68+ immunoreactivity (n = 5 WT mice, n = 4 Smad2/3-cKO mice). c, Confocal images of Iba1, Apoe and Gfap in control and Smad2/3-cKO mice. Scale bar: 50 μm. d, Quantification of Apoe+ and Gfap+ immunoreactivity (n = 4 mice/group).e–h, Expression of key homeostatic and MGnD genes in microglia from Itgb8-cKO (n = 4–5 mice/group) (e), Smad2/3-cKO (n = 4 mice/group) (f), Tgfbr2-cKO (n = 3 mice/group) (g) and Nrros-KO (n = 5 mice/group) (h) relative to non-transgenic control mice. i–l, Expression levels of key AD-risk factor genes (Inpp5d, Havcr2 and Bin1) in microglia from Itgb8-cKO in microglia from Itgb8-cKO (n = 4–5 mice/ group) (i), Smad2/3-cKO (n = 4 mice/group) (j), Tgfbr2-cKO (n = 3 mice/group) (k) and Nrros-KO (n = 5 mice/group) (l), relative to non-transgenic control mice. Two-tailed Student’s t-test. Data were shown as mean ± s.e.m.
Extended Data Fig. 10 |. Microglial deletion of the homeostatic checkpoint Inpp5d facilitates plaque clearance via the induction of MGnD response.
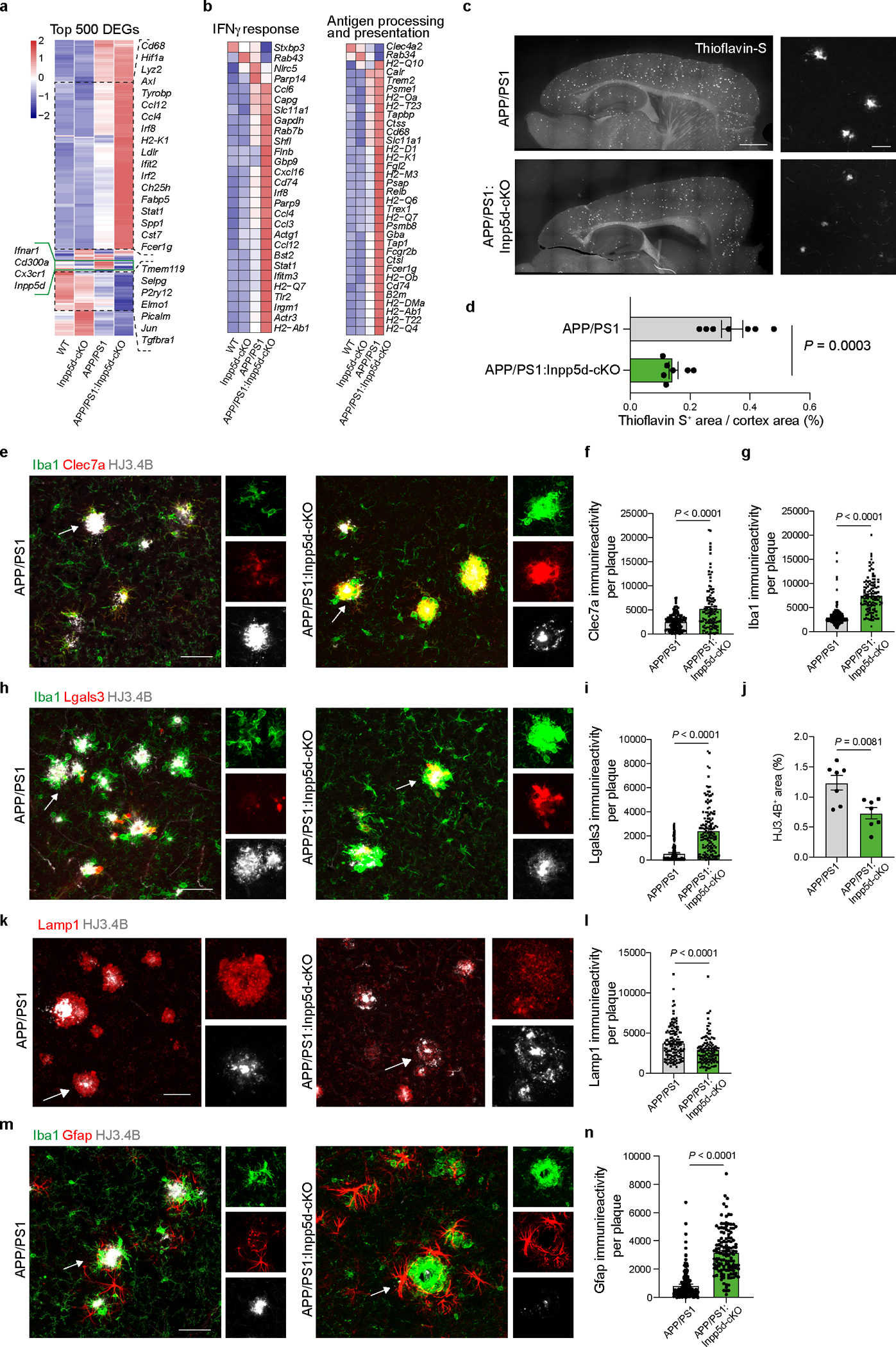
a, Heat map of top-500 DEGs in WT:control vs. WT:Inpp5d-cKO vs. APP/ PS1:control vs. APP/PS1:Inpp5d-cKO microglia. DEGs were identified using DESeq2 analysis with an LRT (n = 2–5 mice/group, P < 0.05). b, Gene ontology analysis of DEGs for IFNγ response, antigen processing and presentation in microglia. DEGs were identified using DESeq2 analysis with an LRT and gene ontology pathways selected with P < 0.05. c, Representative images of Thioflavin-S staining. Scale bar: 1 mm (left), 50 μm (right). d, Quantification of Thioflavin-S staining in APP/PS1 (n = 7) and APP/PS1:Inpp5d-cKO (n = 7) mice. e, Confocal images of Iba1, Clec7a and HJ3.4B staining. Scale bar: 50 μm. f,g, Quantification of Clec7a (f) and Iba1 (g) immunoreactivity in association with Aβ plaques (n = 218 plaques from APP/PS1 mice, n = 115 plaques from APP/PS1:Inpp5d-cKO mice). h, Confocal images of Iba1, Lgals3 and HJ3.4B staining. Scale bar: 50 μm. i,j, Quantification of Lgals3 (i) and HJ3.4B (j) immunoreactivity in association with Aβ plaques (n = 141 plaques from APP/PS1 mice, n = 186 plaques from APP/PS1:Inpp5d-cKO mice for Lgals3, n = 7 mice for HJ3.4B). k, Confocal images of Lamp1 and HJ3.4B staining. Scale bar: 50 μm. l, Quantification of Lamp1 immunoreactivity in association with Aβ plaques (n = 126 plaques from APP/PS1 mice, n = 138 plaques from APP/PS1:Inpp5d-cKO mice). m, Confocal images of Iba1, Gfap and HJ3.4B staining. Scale bar: 50 μm. n, Quantification of Gfap immunoreactivity in association with Aβ plaques (n = 154 plaques from APP/PS1 mice, n = 127 plaques from APP/PS1:Inpp5d-cKO mice). Two-tailed Student’s t-test. Data were presented as mean ± s.e.m.
Supplementary Material
Acknowledgements
We thank the NeuroTechnology Studio at Brigham and Women’s Hospital for providing Zeiss LSM 710 confocal microscope and Leica DMi8 microscope access and Dr. Lai Ding consultation on data acquisition and data analysis; Broad Institute of MIT and Harvard for Smartseq2 RNA-seq and single-cell RNA-seq; Dr. Mathias Jucker for providing APP/PS1 mice; Daniel Tenen and Junyan Zhang for providing Spi1-flox mice; Dr. Dean Sheppard for providing anti-ITGB8 antibody. This study was supported by the Cure Alzheimer’s Fund (to O.B. and D.M.H.); BrightFocus Foundation 2020A016806 (O.B.); NIH-NIA R01AG051812 (O.B.), R01AG054672 (O.B.), R01AG075509 (O.B.), R21AG076982 (O.B.), R01AG080992 (O.B.); NIH-NEI R01EY027921 (O.B.); NIH-NINDS R01NS088137 (O.B.), R21NS104609 (O.B.), R21NS101673 (O.B.); NIH-NIGMS R01GM132668 (O.B.); Nancy Davis Foundation innovative Award (O.B.). RF1NS090934 (D.M.H). Alzheimer’s association research fellowship AARF-21-846786 (N.R.); National Multiple Sclerosis Society FG-2108-38372 (W.B.); Department of Defense MS210257 (W.B.). NIH-NEI K12EY016335 (M.A.M.), K08 EY030160 (M.A.M.), Research to Prevent Blindness Career Development Award (M.A.M.), Glaucoma Research Foundation Catalyst for a Cure Initiative to Prevent and Cure Neurodegeneration (M.A.M.), Alcon Research Institute Young Investigator Award (M.A.M.).
Footnotes
Competing Interests: O.B., Z.Y. and N.R. are inventors of a patent licensed by Brigham and Women’s Hospital for use of LGALS3 for treatment of neurodegenerative diseases. O.B., Z.Y., N.R., X.Z., T.D.A. and D.S. are inventors for a patent licensed by Brigham and Women’s Hospital and University of California, San Francisco, for use of an ITGB8 blocker for treatment of neurodegenerative diseases. O.B. has a collaboration with GSK and Regulus Therapeutics, receives research funding from Sanofi and GSK and receives honoraria for lectures and consultancy from UCB, Camp4, Ono Pharma USA and General Biophysics. D.M.H. is an inventor on a patent licensed by Washington University to C2N Diagnostics on the therapeutic use of antibodies to tau and on a patent licensed to NextCure on antibodies to APOE. D.M.H. cofounded and is on the scientific advisory board of C2N Diagnostics. D.M.H. is on the scientific advisory board of Denali, Genentech and Cajal Neuroscience and consults for Alector. All other authors have no competing interests.
Code availability: Customized code is available at: https://github.com/The-Butovsky-Lab/APOE4-impairs-the-microglial-response-in-Alzheimer-disease-by-inducing-TGFB-mediated-checkpoint
Data availability
Our Smartseq2 RNA-seq, single-cell RNA-seq, ChIP-seq data that support the findings of this study have been deposited into Gene Expression Omnibus (GEO) under SuperSeries GSE239603. The raw data from Olah et al. are available through Synapse (https://www.synapse.org/#!Synapse:syn21438358). The raw data from Zhou et al. are available through Synapse (https://doi.org/10.7303/syn21125841). The raw data from Zhang et al. are available under accession code GSE52564. The raw data from Lund et al. are available through accession code GSE111385. The raw data from Wong et al. are available through accession code GSE84148.
References
- 1.Jansen IE et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet 51, 404–413 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wightman DP et al. A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer’s disease. Nat Genet 53, 1276–1282 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nott A et al. Brain cell type-specific enhancer-promoter interactome maps and disease-risk association. Science 366, 1134–1139 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krasemann S et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 47, 566–581 e569 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Keren-Shaul H et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 169, 1276–1290 e1217 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Farrer LA et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 278, 1349–1356 (1997). [PubMed] [Google Scholar]
- 7.Corder EH et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–923 (1993). [DOI] [PubMed] [Google Scholar]
- 8.Strittmatter WJ et al. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A 90, 1977–1981 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Raber J et al. Isoform-specific effects of human apolipoprotein E on brain function revealed in ApoE knockout mice: increased susceptibility of females. Proc Natl Acad Sci U S A 95, 10914–10919 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Youmans KL et al. APOE4-specific changes in Abeta accumulation in a new transgenic mouse model of Alzheimer disease. J Biol Chem 287, 41774–41786 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu DS et al. APOE4 enhances age-dependent decline in cognitive function by down-regulating an NMDA receptor pathway in EFAD-Tg mice. Mol Neurodegener 10, 7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang C et al. Selective removal of astrocytic APOE4 strongly protects against tau-mediated neurodegeneration and decreases synaptic phagocytosis by microglia. Neuron 109, 1657–1674 e1657 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Butovsky O et al. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat Neurosci 17, 131–143 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Margeta MA et al. Apolipoprotein E4 impairs the response of neurodegenerative retinal microglia and prevents neuronal loss in glaucoma. Immunity (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parkhurst CN et al. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155, 1596–1609 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goldmann T et al. Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat Immunol 17, 797–805 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cugurra A et al. Skull and vertebral bone marrow are myeloid cell reservoirs for the meninges and CNS parenchyma. Science 373 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gosselin D et al. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell 159, 1327–1340 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang KL et al. A common haplotype lowers PU.1 expression in myeloid cells and delays onset of Alzheimer’s disease. Nat Neurosci 20, 1052–1061 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Serrano-Pozo A, Frosch MP, Masliah E & Hyman BT Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med 1, a006189 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shi Y et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549, 523–527 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yoshiyama Y et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53, 337–351 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Rosenzweig N et al. PD-1/PD-L1 checkpoint blockade harnesses monocyte-derived macrophages to combat cognitive impairment in a tauopathy mouse model. Nat Commun 10, 465 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Radde R et al. Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep 7, 940–946 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bennett FC et al. A Combination of Ontogeny and CNS Environment Establishes Microglial Identity. Neuron 98, 1170–1183 e1178 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Van Hove H et al. A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat Neurosci 22, 1021–1035 (2019). [DOI] [PubMed] [Google Scholar]
- 27.Mrdjen D et al. High-Dimensional Single-Cell Mapping of Central Nervous System Immune Cells Reveals Distinct Myeloid Subsets in Health, Aging, and Disease. Immunity 48, 380–395 e386 (2018). [DOI] [PubMed] [Google Scholar]
- 28.Jordao MJC et al. Single-cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science 363 (2019). [DOI] [PubMed] [Google Scholar]
- 29.Habib N et al. Disease-associated astrocytes in Alzheimer’s disease and aging. Nat Neurosci 23, 701–706 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Munger JS & Sheppard D Cross talk among TGF-beta signaling pathways, integrins, and the extracellular matrix. Cold Spring Harb Perspect Biol 3, a005017 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arnold TD et al. Impaired alphaVbeta8 and TGFbeta signaling lead to microglial dysmaturation and neuromotor dysfunction. J Exp Med 216, 900–915 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wolf Y, Anderson AC & Kuchroo VK TIM3 comes of age as an inhibitory receptor. Nat Rev Immunol 20, 173–185 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakao A et al. TGF-beta receptor-mediated signalling through Smad2, Smad3 and Smad4. EMBO J 16, 5353–5362 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Olah M et al. Single cell RNA sequencing of human microglia uncovers a subset associated with Alzheimer’s disease. Nat Commun 11, 6129 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou Y et al. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat Med 26, 131–142 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Travis MA et al. Loss of integrin alpha(v)beta8 on dendritic cells causes autoimmunity and colitis in mice. Nature 449, 361–365 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nakawesi J et al. alphavbeta8 integrin-expression by BATF3-dependent dendritic cells facilitates early IgA responses to Rotavirus. Mucosal Immunol 14, 53–67 (2021). [DOI] [PubMed] [Google Scholar]
- 38.Zhang Y et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34, 11929–11947 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu Z et al. Fate Mapping via Ms4a3-Expression History Traces Monocyte-Derived Cells. Cell 178, 1509–1525 e1519 (2019). [DOI] [PubMed] [Google Scholar]
- 40.Wong K et al. Mice deficient in NRROS show abnormal microglial development and neurological disorders. Nat Immunol 18, 633–641 (2017). [DOI] [PubMed] [Google Scholar]
- 41.Samuels JD et al. The Alzheimer’s disease risk factor INPP5D restricts neuroprotective microglial responses in amyloid beta-mediated pathology. Alzheimers Dement (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Iguchi A et al. INPP5D modulates TREM2 loss-of-function phenotypes in a beta-amyloidosis mouse model. iScience 26, 106375 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dodagatta-Marri E et al. Integrin alphavbeta8 on T cells suppresses anti-tumor immunity in multiple models and is a promising target for tumor immunotherapy. Cell Rep 36, 109309 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mahan TE et al. Selective reduction of astrocyte apoE3 and apoE4 strongly reduces Abeta accumulation and plaque-related pathology in a mouse model of amyloidosis. Mol Neurodegener 17, 13 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rohn TT & Moore ZD Nuclear Localization of Apolipoprotein E4: A New Trick for an Old Protein. Int J Neurol Neurother 4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Theendakara V et al. Direct Transcriptional Effects of Apolipoprotein E. J Neurosci 36, 685–700 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hollingworth P et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet 43, 429–435 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pimenova AA et al. Alzheimer’s-associated PU.1 expression levels regulate microglial inflammatory response. Neurobiol Dis 148, 105217 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gosselin D et al. An environment-dependent transcriptional network specifies human microglia identity. Science 356 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liddelow SA et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Katsouri L et al. Ablation of reactive astrocytes exacerbates disease pathology in a model of Alzheimer’s disease. Glia 68, 1017–1030 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Boza-Serrano A et al. Galectin-3, a novel endogenous TREM2 ligand, detrimentally regulates inflammatory response in Alzheimer’s disease. Acta Neuropathol 138, 251–273 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tao CC et al. Galectin-3 promotes Abeta oligomerization and Abeta toxicity in a mouse model of Alzheimer’s disease. Cell Death Differ 27, 192–209 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mabuchi F et al. The apolipoprotein E gene polymorphism is associated with open angle glaucoma in the Japanese population. Mol Vis 11, 609–612 (2005). [PubMed] [Google Scholar]
- 55.Lam CY et al. Association of apolipoprotein E polymorphisms with normal tension glaucoma in a Chinese population. J Glaucoma 15, 218–222 (2006). [DOI] [PubMed] [Google Scholar]
- 56.Margeta MA et al. Association of APOE With Primary Open-Angle Glaucoma Suggests a Protective Effect for APOE epsilon4. Invest Ophthalmol Vis Sci 61, 3 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tesseur I et al. Deficiency in neuronal TGF-beta signaling promotes neurodegeneration and Alzheimer’s pathology. J Clin Invest 116, 3060–3069 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wyss-Coray T et al. Amyloidogenic role of cytokine TGF-beta1 in transgenic mice and in Alzheimer’s disease. Nature 389, 603–606 (1997). [DOI] [PubMed] [Google Scholar]
- 59.Kandasamy M et al. TGF-beta Signaling: A Therapeutic Target to Reinstate Regenerative Plasticity in Vascular Dementia? Aging Dis 11, 828–850 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yin Z et al. Identification of a protective microglial state mediated by miR-155 and interferon-gamma signaling in a mouse model of Alzheimer’s disease. Nat Neurosci 26, 1196–1207 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lund H et al. Fatal demyelinating disease is induced by monocyte-derived macrophages in the absence of TGF-β signaling. Nat Immunol 19, 1–7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kaiser T & Feng G Tmem119-EGFP and Tmem119-CreERT2 Transgenic Mice for Labeling and Manipulating Microglia. eNeuro 6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang JW et al. Influence of SHIP on the NK repertoire and allogeneic bone marrow transplantation. Science 295, 2094–2097 (2002). [DOI] [PubMed] [Google Scholar]
- 64.Ju W et al. Deletion of Smad2 in mouse liver reveals novel functions in hepatocyte growth and differentiation. Mol Cell Biol 26, 654–667 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li Q et al. Redundant roles of SMAD2 and SMAD3 in ovarian granulosa cells in vivo. Mol Cell Biol 28, 7001–7011 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Huynh TV et al. Lack of hepatic apoE does not influence early Abeta deposition: observations from a new APOE knock-in model. Mol Neurodegener 14, 37 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sullivan PM et al. Targeted replacement of the mouse apolipoprotein E gene with the common human APOE3 allele enhances diet-induced hypercholesterolemia and atherosclerosis. J Biol Chem 272, 17972–17980 (1997). [DOI] [PubMed] [Google Scholar]
- 68.Knouff C et al. Apo E structure determines VLDL clearance and atherosclerosis risk in mice. J Clin Invest 103, 1579–1586 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Iwasaki H et al. Distinctive and indispensable roles of PU.1 in maintenance of hematopoietic stem cells and their differentiation. Blood 106, 1590–1600 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Proctor JM, Zang K, Wang D, Wang R & Reichardt LF Vascular development of the brain requires beta8 integrin expression in the neuroepithelium. J Neurosci 25, 9940–9948 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhao XF et al. Targeting Microglia Using Cx3cr1-Cre Lines: Revisiting the Specificity. eNeuro 6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sanmarco LM et al. Gut-licensed IFNgamma(+) NK cells drive LAMP1(+)TRAIL(+) anti-inflammatory astrocytes. Nature 590, 473–479 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hao Y et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 e3529 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ianevski A, Giri AK & Aittokallio T Fully-automated and ultra-fast cell-type identification using specific marker combinations from single-cell transcriptomic data. Nat Commun 13, 1246 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Picelli S et al. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat Methods 10, 1096–1098 (2013). [DOI] [PubMed] [Google Scholar]
- 76.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mootha VK et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34, 267–273 (2003). [DOI] [PubMed] [Google Scholar]
- 78.Subramanian A et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Browaeys R, Saelens W & Saeys Y NicheNet: modeling intercellular communication by linking ligands to target genes. Nat Methods 17, 159–162 (2020). [DOI] [PubMed] [Google Scholar]
- 80.Gu Z, Gu L, Eils R, Schlesner M & Brors B circlize Implements and enhances circular visualization in R. Bioinformatics 30, 2811–2812 (2014). [DOI] [PubMed] [Google Scholar]
- 81.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tarasov A, Vilella AJ, Cuppen E, Nijman IJ & Prins P Sambamba: fast processing of NGS alignment formats. Bioinformatics 31, 2032–2034 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li H et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ramirez F, Dundar F, Diehl S, Gruning BA & Manke T deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res 42, W187–191 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhang Y et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bero AW et al. Bidirectional relationship between functional connectivity and amyloid-beta deposition in mouse brain. J Neurosci 32, 4334–4340 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Our Smartseq2 RNA-seq, single-cell RNA-seq, ChIP-seq data that support the findings of this study have been deposited into Gene Expression Omnibus (GEO) under SuperSeries GSE239603. The raw data from Olah et al. are available through Synapse (https://www.synapse.org/#!Synapse:syn21438358). The raw data from Zhou et al. are available through Synapse (https://doi.org/10.7303/syn21125841). The raw data from Zhang et al. are available under accession code GSE52564. The raw data from Lund et al. are available through accession code GSE111385. The raw data from Wong et al. are available through accession code GSE84148.