

Abstract
INTRODUCTION
Neurogranin (Ng) is considered a biomarker for synaptic dysfunction in Alzheimer's disease (AD). In contrast, the inflammasome complex has been shown to exacerbate AD pathology.
METHODS
We investigated the protein expression, morphological differences of Ng, and correlated Ng to hyperphosphorylated tau in the post mortem brains of 17 AD cases and 17 age‐ and sex‐matched controls. In addition, we correlated the Ng expression with two different epitopes of apoptosis‐associated speck‐like protein containing a caspase recruitment domain (ASC).
RESULTS
We show a reduction of Ng immunopositive neurons and morphological differences in AD compared to controls. Ng immunostaining was negatively correlated with neurofibrillary tangles, humanized anti‐ASC (IC100) positive neurons and anti‐ASC positive microglia, in AD.
DISCUSSION
The finding of a negative correlation between Ng and ASC speck protein expression in post mortem brains of AD suggests that the activation of inflammasome/ASC speck pathway may play an important role in synaptic degeneration in AD.
Highlights
We show the role that neurogranin plays on post‐synaptic signaling in specific hippocampal regions.
We demonstrate that there could be clinical implications of using neurogranin as a biomarker for dementia.
We describe the loss of plasticity and neuronal scaffolding proteins in the present of AD pathology.
We show the response of neuroinflammation when tau proteins phosphorylate in hippocampal neurons.
We show that there is a potential therapeutic target for the inflammasome, and future studies may show that IC100, a humanized monoclonal antibody directed against ASC, may slow the progression of neurodegeneration.
Keywords: Alzheimer's disease, axon, inflammasome, microglia, neurogranin
1. INTRODUCTION
Neuropathological changes in Alzheimer's disease (AD) include the accumulation of dysfunctional proteins in the form of extracellular amyloid beta (Aβ), intracellular neurofibrillary tangles (NFTs) with hyperphosphorylated tau (p‐tau), and synaptic loss. 1 , 2 , 3
Anatomically, in the hippocampus, the cornu ammonis (CA) 1 region of the hippocampus is affected by NFTs early in the disease. As the neurodegeneration progresses, pathological changes are more prevalent in the CA2, CA3, and dentate gyrus (DG) regions, then spread to the cerebral cortex. 4 Along with the progression of NFTs in the neurons, dendritic spines density and synaptic connections are dramatically reduced. 5 , 6 As the synaptic proteins spill out of the damaged neurons in the early stages of AD, some synaptic proteins such as neurogranin (Ng) have been found to accumulate in the cerebrospinal fluid (CSF). 7
Ng is a post‐synaptic protein involved in long‐term potentiation and synaptic plasticity, which plays an important role in learning and memory. 8 Recently, clinical studies have shown that the level of Ng in the CSF is elevated and associated with longitudinal cognitive decline in patients with mild cognitive impairment (MCI) and AD. 9 , 10 , 11 , 12 , 13 Moreover, it has been reported that peptide‐to‐total full‐length Ng ratios in brain tissue are increased in AD, indicating synaptic damage by loss of intact Ng. 14 At the genetic level, Ng encoding NRGN gene expression is associated with Aβ and tau pathology. 15 , 16 Taken together, it is suggested that Ng plays an important role in the pathophysiological process of AD.
The microtubule‐associated proteins (MAPs) of type II (MAP2) stabilize the dendrites and the axons of neurons. Studies show that p‐tau is mislocated to dendritic spines leading to microtubule destabilization and synaptic dysfunction. 17 , 18 , 19 , 20 In addition, synaptic loss is reported to be associated with microglia‐mediated neuroinflammation in AD. 21 , 22
Recently, we have reported that in intermediate AD there are increases in inflammasome proteins compared to age‐matched controls. 23 The inflammasome is an inflammatory complex comprised of caspase‐1, apoptosis‐associated speck‐like protein containing a caspase activation and recruitment domain (ASC), and sensor proteins, either NOD‐like receptor1 (NLRP1) present in neurons or NLRP3 primarily expressed in microglia. 23 The studies show that inflammasome/ASC speck protein‐mediated aggregation of tau. 24 Recently, it has been shown that inflammasome activation via NLRP3 sensor in microglia leads to synaptic loss and impairment of excitatory neurons using in vitro cell culture studies of sepsis, 25 but the role of the inflammasome pathway in synaptic loss seen in AD has not been well studied. A study focused on the relationship between synaptic proteins and inflammasome/ASC expression will provide significant insight into the mechanism underlying synaptic degeneration in AD.
Clinically, cases with mild to moderate cognitive impairment correlate with intermediate AD post mortem neuropathological changes. Intermediate neuropathological changes are not normally associated with high levels of neuronal loss and gliosis but have Aβ plaques and tauopathy seen in the CA1‐CA3 hippocampal regions; 1 therefore, intermediate post mortem AD pathology offers an ideal disease state to study the protein distribution of Ng in pyramidal cells of the hippocampus and ASC expression.
In this study, we performed an immunohistochemical analysis of Ng, MAP2, tau, Aβ, and ASC in the hippocampus of AD brains with intermediate AD pathology and age‐matched controls. We then compared the region‐specific hippocampal immunostaining of Ng between the AD and control brains. Next, we examined the relationship between immunostaining of Ng and tau and Aβ pathology in the hippocampus. Finally, we correlated the quantitative Ng assessment with immunostaining using mouse anti‐ASC directed against the caspase activation and recruitment domain (CARD) which has been shown to be expressed in microglia, and IC100, a humanized monoclonal antibody directed against the PYRIN domain (PYD) of ASC which is expressed in neurons. 26
RESEARCH IN CONTEXT
Systematic review: The authors reviewed the literature using traditional (e.g., PubMed) sources, meeting abstracts, and presentations. While the pathophysiology of neurodegeneration has been well documented other aspects of Alzheimer's disease (AD) neuropathology need to be investigated. There have been several recent publications describing the clinical implications of using neurogranin as a biomarker for dementia. These relevant citations are appropriately cited.
Interpretation: Our findings led to an integrated hypothesis describing the loss of plasticity and neuronal scaffolding proteins in the presence of AD pathology.
Future directions: The article proposes a framework for the generation of new hypotheses and the conduct of additional studies. Examples include further understanding: (1) the role that neurogranin plays on post‐synaptic signaling, (2) the role of that neuroinflammation has on the brain when tau proteins phosphorylate, and (3) the potential that 1C100, humanized monoclonal antibody raised against ASC, may slow the progression of neurodegeneration.
Here we find morphological differences in Ng expression in the hippocampus from post mortem brains with AD and control. In addition, we show that Ng negatively correlated with p‐tau (Ser203/Thr205; AT‐8) and two different epitopes ASC in AD brains.
2. METHODS
2.1. Post mortem human brains
Research study ethics was obtained from the Human Subjects Research Office, University of Miami, Miami, Florida (Institutional Review Board ethics number, 19920348 [CR00012340]).
2.1.1. Tissue preparation
Case details, tissue preparation, the regions of interest (ROIs), and neuropathological analyses have already been described in Vontell et al. 23 and are summarized in Data S1 in supporting information.
2.2. Immunohistochemistry
Standard immunohistochemistry procedures have been described previously in Vontell et al. 23 and Gober et al. 27 and are summarized in Data S1.
2.3. Immunofluorescence labeling
To identify the cellular location of the Ng protein in our tissue immunofluorescence (IF) double labeling was performed using immunofluorescence protocols described in Vontell et al. 23 and in Data S1.
2.4. NFTs, neuritic plaques, Ng, MAP2, and synaptophysin assessments
In this study, scoring analysis of the Ng, MAP2, Tau (AT‐8 and GT‐38), and synaptophysin and the ROIs were determined by the cellular architecture as described in Vontell et al. 23 and Mai et al. 28 and in Data S1.
2.5. Data analysis
The Student t test was used to compare the variables between two groups (AD and control) in all experiments. . Two‐tailed Pearson's analysis was used to test the correlation of Ng loss and NFT numbers (AT‐8 and GT‐38), ASC accumulation, and synaptophysin as described in Vontell et al. 23 for each of the ROIs. All statistical analyses and generation of plots were performed using GraphPad Prism 9.0 (GraphPad Software). Data were presented as mean ± SD for the t test and the correlation coefficient Pearson's analyses; significance was assumed at P < 0.05.
3. RESULTS
3.1. Donors and AD pathology
Experiments in this study were designed to determine the density and distribution of the post‐synaptic marker Ng in relation to the expression of MAP2, Aβ, synaptophysin, tau, and ASC in post mortem human brains with and without intermediate AD neuropathological changes.
3.2. MAP2‐positive neurons decrease in the CA regions of the hippocampus in AD brains
NFTs accumulate in the entorhinal cortex, the subiculum, and in CA1 of the hippocampal region, 1 , 2 in the control cases, demonstrating age‐related neuropathological changes. Although the rate of p‐tau accumulation and the degree of destabilization of MAP2 occur independently, 29 it is thought that as NFTs increase, MAP2 destabilizes, the neurons lose plasticity, and neuronal atrophy will follow. 18 Here we assessed the change in the number of MAP2‐positive neurons in the hippocampus and entorhinal cortex. There was a significant decrease in the number of MAP2‐positive neurons seen in the intermediate AD cases compared to controls in the entorhinal cortex, and in the CA hippocampal sectors. However, there was no significant change in the subiculum or the DG between intermediate AD cases and controls (Table 1).
TABLE 1.
MAP2 immunopositive neurons.
| Region | Control | Intermediate AD | P value | Significance |
|---|---|---|---|---|
| Entorhinal cortex | 299 ± 81 | 161 ± 84 | 0.013 | * |
| Subiculum | 253 ± 57 | 199 ± 28 | 0.3 | ns |
| CA1 | 196 ± 20 | 123 ± 19 | 0.02 | * |
| CA2 | 260 ± 32 | 146 ± 21 | 0.009 | ** |
| CA3 | 278 ± 32 | 140 ± 20 | 0.005 | ** |
| Dentate gyrus | 238 ± 23 | 219 ± 63 | 0.41 | ns |
| Ng immunopositive neurons | ||||
| Region | Control | Intermediate AD | P value | Significance |
|---|---|---|---|---|
| Entorhinal cortex | 225 ± 20 | 149 ± 22 | 0.016 | * |
| Subiculum | 269 ± 63 | 207 ± 54 | 0.08 | * |
| CA1 | 149 ± 36 | 112 ± 42 | 0.02 | * |
| CA2 | 223 ± 60 | 104 ± 45 | <0.0001 | *** |
| CA3 | 296 ± 17 | 104 ± 12 | <0.0001 | *** |
| Dentate gyrus | 233 ± 49 | 218 ± 61 | 0.43 | ns |
Note. t test for each ROI, mean ± SD, P value, and significance. Counts are expressed as MAP2‐ or Ng‐positive neurons in 1 mm2 area in AD cases and in controls.
Abbreviations: AD, Alzheimer's disease; CA, cornu ammonis; MAP2, microtubule‐associated proteins type II; Ng, neurogranin; ns, not significant; ROI, region of interest; SD, standard deviation.
P < 0.05.
P < 0.01.
P < 0.001.
3.3. Ng neuronal counts differ between intermediate AD and controls
Pathologically dysregulated MAP2 could lead to a reduction in neuronal outgrowth, synaptic plasticity, and dendritic structure, 18 but how MAP2 relates to the distribution and location of Ng and other neuronal supporting proteins has not been well characterized in the hippocampal formation in AD cases. We investigated each of the ROIs for Ng distribution patterns and compared the morphology to that of MAP2 immunostaining. There was no significant change in the number of Ng positive neurons in the intermediate AD cases in the DG. In regions where NFTs increase in the hippocampus in intermediate staging of AD pathology, there was a significant decrease in Ng in the intermediate AD group. (Data are summarized in Table 1.)
3.4. MAP2 and Ng cell morphology differ between intermediate AD and controls
In the control cases, extracellular neurofibrillary threads are seen in the CA2 hippocampal region (Figure 1A). In addition, the anti‐MAP2 and anti‐Ng stains identified the neurons and the prominent apical dendrites in the CA2 hippocampal region (Figure 1B and C). However, in intermediate AD cases, the neurons in the CA2 hippocampal region showed more prominent NFTs and neurofibrillary threads (Figure 1E). Moreover, the MAP2 and the Ng expression that we identified were more concentrated adjacent to the cell body while the neuronal apical dendrite was not detected (Figure 1F and G).
FIGURE 1.
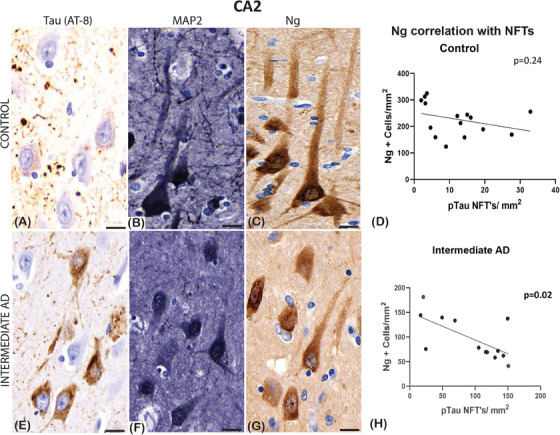
Examples of immunoreactivity of mouse‐anti‐tau (AT‐8), mouse‐anti‐microtubule‐associated protein 2 (MAP2), and mouse‐anti‐neurogranin (Ng) in normal control and in a case with intermediate Alzheimer's disease (AD) in a section from the hippocampal region cornu ammonis (CA2). The control (A) shows sparse apical primary dendritic staining of tau in the pyramidal neurons; in the AD case there are moderate (E) amounts of staining spreading into the apical dendrite. The MAP2 staining was seen in both groups, but the processes are well defined in the control (B) compared to cases of intermediate AD (F). Ng protein expression differs between the groups in pyramidal neurons, as this appears denser and more concentrated around the nucleus of pyramidal neurons in the intermediate AD cases (G), whereas in the control cases the staining throughout the apical dendritic processes (C). No significant Pearson's correlation, in the controls, was seen between the number of Ng2 positive neurons and the number of neurofibrillary tangles (NFTs) defined with phosphorylated tau (pTau; D), whereas a significant negative correlation between Ng and NFTs was seen in the intermediate AD cases (H). Scale bars = 10 μm (A, B, C, E, F, G)
In the CA3 hippocampal region of control brains, there are a few neurofibrillary threads and NFTs (Figure 2A) and multiple dendritic processes highlighted with MAP2 immunostaining in the control cases (Figure 2B). Anti‐Ng stained the neuronal cell bodies and there was a dense extracellular expression seen within extracellular matrix the of the CA3 region in the control cases (Figure 2C).
FIGURE 2.
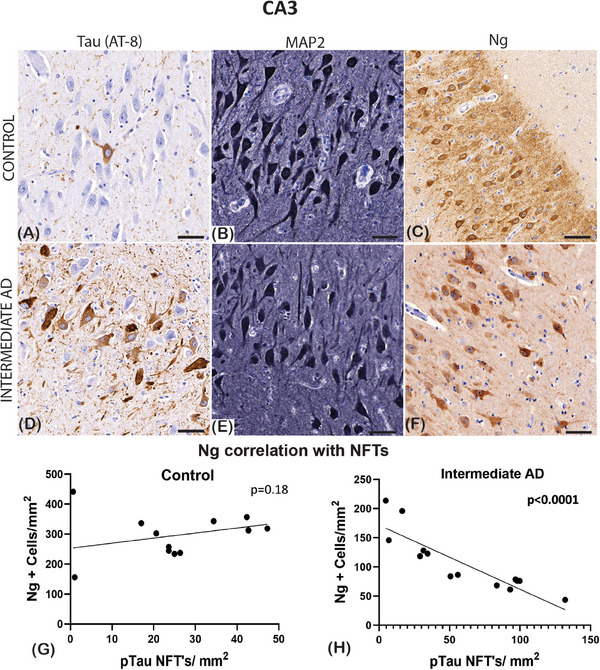
Photomicrographs of the immunostaining seen in the cornu ammonis (CA3) hippocampal region. Mouse‐anti‐phosphorylated tau (pTau; AT‐8), mouse‐anti‐microtubule‐associated protein 2 (MAP2), and mouse‐anti‐neurogranin (Ng) is identified in controls and intermediate Alzheimer's disease (AD). In (A), control cases, there is a sparse density staining of the neurofibrillary tangles (NFTs) in the pyramidal neurons, in the intermediate AD case (D) there is a frequent amount of staining that spreads the dendritic processes. The MAP2 staining was seen in both the control (B) and intermediate AD cases (E). Extracellular and intracellular Ng2 expression is seen the controls (C), but in the intermediate AD group, the expression is seen in the pyramidal neuronal bodies (F). Pearson's correlation analysis did not reveal any significant relationship between the number of Ng2‐positive neurons and the number of NFTs defined with pTau in the control group (G), whereas there was a significant negative correlation between Ng and NFTs in the intermediate AD cases (H). Scale bars = 30 μm (A, B, C, E, F, G)
In the CA3 hippocampal region of the intermediate AD cases, NFTs were more numerous (Figure 2D). Similarly, there was a reduction in the MAP2‐positive neuronal population in the CA3 region of the intermediate AD cases (Figure 2E). The anti‐Ng staining demonstrated a loss of extracellular expression in the CA3 region in the intermediate AD cases (Figure 2F).
The Ng‐positive neurons in the intermediate AD cases appeared more circular and lacked linear dendritic processes. To quantify this morphological change, we used a measure of circularity to decipher the changes in morphology seen between intermediate AD and control cases. In the regions where neurodegeneration begins to occur with age, such as the entorhinal cortex, the subiculum, and the CA1 hippocampal region there was no change between controls and intermediate AD in the circularity measures using the Ng expression (P > 0.05). However, as illustrated in Figure 1, the apical dendrite was not as prominent in the CA2 hippocampal region in the intermediate AD cases. Moreover, the circularity analysis showed a significant increase in Ng‐positive neurons to display a more circular morphology (P < 0.001) in the intermediate AD cases. In the CA3 hippocampal region of the intermediate AD cases, there is a loss of dendritic processes as seen with the MAP2 immunostaining, and a significant change in the neurons to have a perinuclear concentrated expression of Ng (P < 0.05; Table 2).
TABLE 2.
Ng circularity measure.
| Region | Control | Intermediate AD | P value | Significance |
|---|---|---|---|---|
| Entorhinal cortex | 0.47 ± 0.03 | 0.53 ± 0.02 | 0.07 | ns |
| Subiculum | 0.49 ± 0.03 | 0.52 ± 0.02 | 0.88 | ns |
| CA1 | 0.67 ± 0.05 | 0.64 ± 0.03 | 0.6 | ns |
| CA2 | 0.37 ± 0.03 | 0.67 ± 0.04 | 0.0007 | *** |
| CA3 | 0.40 ± 0.05 | 0.54 ± 0.02 | 0.013 | * |
| Dentate gyrus | 0.77 ± 0.04 | 0.75 ± 0.05 | 0.95 | ns |
Note. t test for each ROI, mean ± SD, P value, and significance.
Abbreviations: AD, Alzheimer's disease; CA, cornu ammonis; Ng, neurogranin; ns, not significant; ROI, region of interest; SD, standard deviation.
P < 0.05.
P < 0.001.
3.5. Presynaptic vesicle loss is seen in specific hippocampal regions
The presynaptic vesicle protein, synaptophysin, showed a significant reduction in the subiculum, CA3, and DG, but not in the CA1, CA2, or entorhinal cortex in the intermediate AD cases. Of interest, there was no correlation seen between synaptophysin reduction and Ng expression (see Data S2 in supporting information).
3.6. Ng (+) cell count negatively correlates with p‐tau (AT‐8) NFT counts
Because there was a significant decrease in MAP2, synaptophysin, and Ng proteins that are important to neuronal stability and plasticity, we determined the relationship between the number of NFTs (AT‐8) described in Vontell et al. 23 and the expression of these proteins. To establish whether NFTs correlates with Ng loss, we carried out a correlation analysis of Ng neuronal counts and NFTs across several hippocampal regions in brains with intermediate AD and controls. Interestingly, in the intermediate AD groups, our results indicated a significant negative correlation between the increase of NFTs and the number of Ng cells in the entorhinal cortex (r = −0.63; P = 0.05; Figure 3B), the CA1 (r = −0.56; P = 0.03; Figure 3F), the CA2 (r = −0.43; P = 0.015; Figure 1H) and the CA3 (r = −0.77; P < 0.0001; Figure 2H), the dentate gyrus (r = −0.59; P = 0.046; Figure 3H), but not in the subiculum (r = 0.16; P = 0.25; Figure 3D).
FIGURE 3.
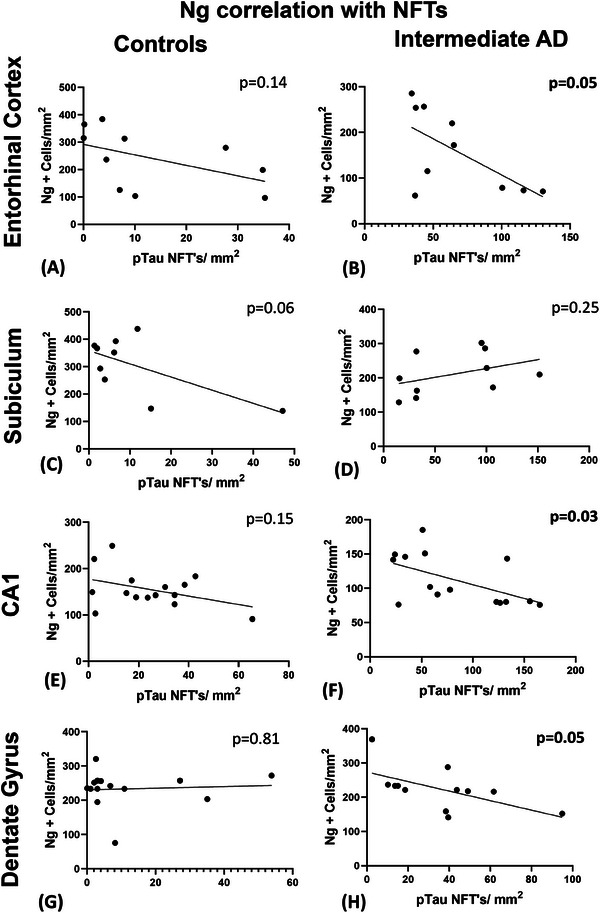
Pearson's correlation analysis of Ng and NFTs (AT‐8) in the CA1 hippocampal region and surrounding structures. The correlation analysis did not reveal any significant relationship between the number of Ng‐positive neurons and the number of NFTs defined with pTau in the control group in the entorhinal cortex (A), the subiculum (C), the CA1 (E), and the dentate gyrus (G). The intermediate AD cases showed a significant negative correlation between Ng and NFTs in the entorhinal cortex (B) and the CA1 hippocampal region (F), and in the subiculum (D) but not in the dentate gyrus (H). AD, Alzheimer's disease; CA, cornu ammonis; NFTs, neurofibrillary tangles; Ng, neurogranin; pTau, hyperphosphorylated tau
In the control group, there was no correlation observed between the Ng‐positive cells and the number of NFTs identified in the entorhinal cortex (r = −0.5; P = 0.14; Figure 3A), the subiculum (r = −0.64; P = 0.06; Figure 3C) and in the CA1 hippocampal region (r = −0.15; P = 0.15; Figure 3E), in the CA2 (r = −0.32; P = 0.24; Figure 1D), the CA3 (r = −0.34; P = 0.18; Figure 2G), and in the DG (r = 0.07; P = 0.81; Figure 3G). Together, the results of this correlation analysis demonstrated that as p‐tau NFT density increases the Ng cell counts decrease in specific regions of the brain (see Table 3).
TABLE 3.
Correlation: Ng+ cell counts and p‐tau (AT‐8) NFTs.
| Control | |||
|---|---|---|---|
| Region | r | P value | Significance |
| Entorhinal cortex | –0.5 | 0.14 | ns |
| Subiculum | –0.64 | 0.06 | ns |
| CA1 | –0.15 | 0.15 | ns |
| CA2 | –0.32 | 0.24 | ns |
| CA3 | –0.34 | 0.18 | ns |
| Dentate gyrus | 0.07 | 0.81 | ns |
| Intermediate AD | |||
| Entorhinal cortex | –0.63 | 0.05 | * |
| Subiculum | 0.16 | 0.25 | ns |
| CA1 | –0.56 | 0.03 | * |
| CA2 | –0.43 | 0.015 | * |
| CA3 | –0.77 | <0.0001 | *** |
| Dentate gyrus | –0.59 | 0.046 | * |
Note. Pearson's correlation coefficient for each ROI, the r statistic, P value, and significance.
Abbreviations: CA, cornu ammonis; NFTs, neurofibrillary tangles; Ng, neurogranin; ns, not significant; p‐tau, phosphorylated tau; ROI region of interest.
P < 0.05.
P < 0.001.
3.7. Conformation‐selective tau immunostaining is increased in cases of intermediate AD
Antibodies that detect p‐tau (i.e., AT‐8 or PHF1) are standard for neuropathological analysis. GT‐38 detects pathological aggregates which will include neurons showing insoluble paired helical filaments. 30 , 31 Our results show that GT‐38 NFTs increased in the intermediate AD cases in all the ROIs. Interestingly, there was no significant correlation found between Ng loss and GT‐38–positive NFTs in any of the ROIs in the intermediate AD group or in the controls.
Our data support what others have described, as more strands of tau aggregates are seen in the neuropil of neurons when stained with standard p‐tau antibodies, than neurons showing double helical conformation changes using anti‐GT‐38 in stages of Braak 0 to III. 32 Data are summarized in Data S3 in supporting information.
3.8. Neurogranin loss does not correlate with Aβ clusters
We carried out a correlation analysis of Ng (+) cell counts and the number of Aβ clusters described in Vontell et al. 23 across hippocampal regions in brains with intermediate AD and controls. Interestingly, our results indicated no significant correlation between the number of Ng+ neurons and Aβ clusters in any of the ROIs in both the intermediate AD cases and in the control group. Data summarized in Data S4 in supporting information.
3.9. Neurogranin loss correlates with increases in human IC100 antibody detection in the CA1 and CA2 hippocampal regions
The correlation between tau positivity and Ng loss demonstrated that there are key synaptic changes that occur as AD advances from low AD to intermediate AD pathology. Next, we correlated the neuronal positive ASC (IC100) counts to the density changes of Ng. In the controls, there was no significant correlation seen between the number of Ng‐positive cells and the number of IC100 labeled neurons in all three of the hippocampal CA regions (CA1, r = −0.25; P = 0.17, Figure 4A and C; CA2; r = −0.03; P = 0.9, Figure 4E; CA3 r = −0.03; P = 0.90, Data S5 in supporting information). However, in the intermediate AD group, there was a negative correlation found in the CA1 (r = −0.60; P = 0.004; Figure 4B and D), the CA2 (r = −0.5; P = 0.02; Figure 4F), but not the CA3 hippocampal region (r = −0.03; P = 0.28; Data S5; summarized in Table 4).
FIGURE 4.
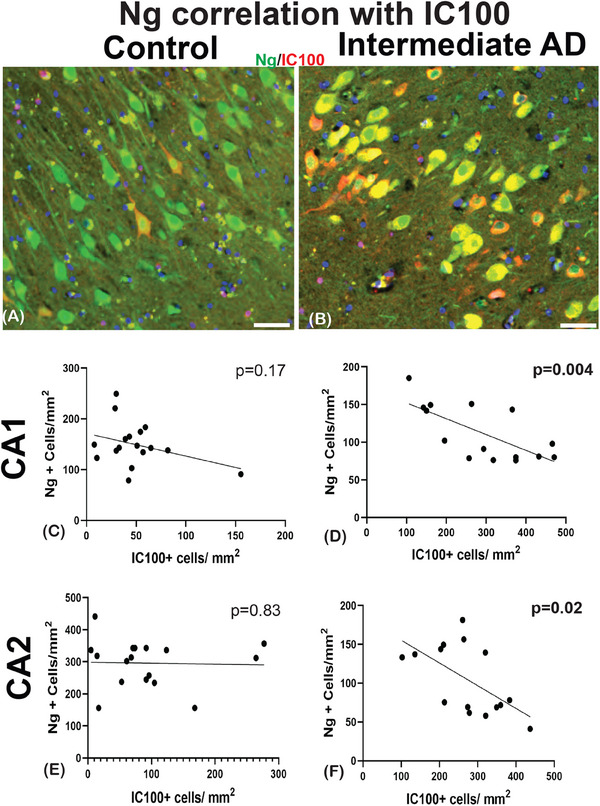
Pearson's correlation analysis of Ng and humanized anti‐ASC (IC100) in the hippocampal subfields. An example of double‐labeling immunostaining of Ng (green) and 1C100 (red) of controls (A) in intermediate AD (B). In the controls, the correlation analysis did not reveal any significant relationship between the number of Ng positive neurons and the number of IC100 ‐positive neurons (C, and E). The intermediate AD cases showed a significant negative correlation between Ng and IC100 in the CA1 (D), and the CA2 (F) hippocampal regions. AD, Alzheimer's disease; ASC, apoptosis‐associated speck‐like protein containing a caspase recruitment domain; CA, cornu ammonis; Ng, neurogranin. Scale bars = 30 μm (A) and (B)
TABLE 4.
Correlation: Ng+ cell counts and IC100.
| Control | |||
|---|---|---|---|
| Region | r | P value | Significance |
| CA1 | –0.25 | 0.17 | ns |
| CA2 | –0.03 | 0.9 | ns |
| CA3 | –0.03 | 0.9 | ns |
| Intermediate AD | |||
| CA1 | –0.6 | 0.004 | ** |
| CA2 | –0.59 | 0.02 | * |
| CA3 | –0.3 | 0.28 | ns |
Note. Pearson's correlation coefficient for each ROI, the r statistic, P value. and significance.
Abbreviations: AD, Alzheimer's disease; CA, cornu ammonis; Ng, neurogranin; ns, not significant; ROI, region of interest.
P < 0.05.
P < 0.01.
Next, we correlate the Ng counts with the number of ASC‐positive microglia to investigate if ASC in microglia increases with loss of synaptic proteins. The controls showed no significant correlation between the number of Ng‐positive cells and the number of ASC‐labeled microglia in the CA1 region (r = −0.54; P = 0.11, Figure 5A and C), and the CA2 region (r = −0.05; P = 0.88, Figure 5E) and in the CA3 hippocampal region (r = −0.60; P = 0.071, Data S5). In the cases of intermediate AD, there was a negative correlation in the CA1 (r = −0.14; P = 0.03; Figure 5B and D), in the CA2 (r = −0.69; P = 0.008; Figure 5F), and in the CA3 hippocampal region (r = −0.6; P = 0.038; Table 5 and Data S5).
FIGURE 5.
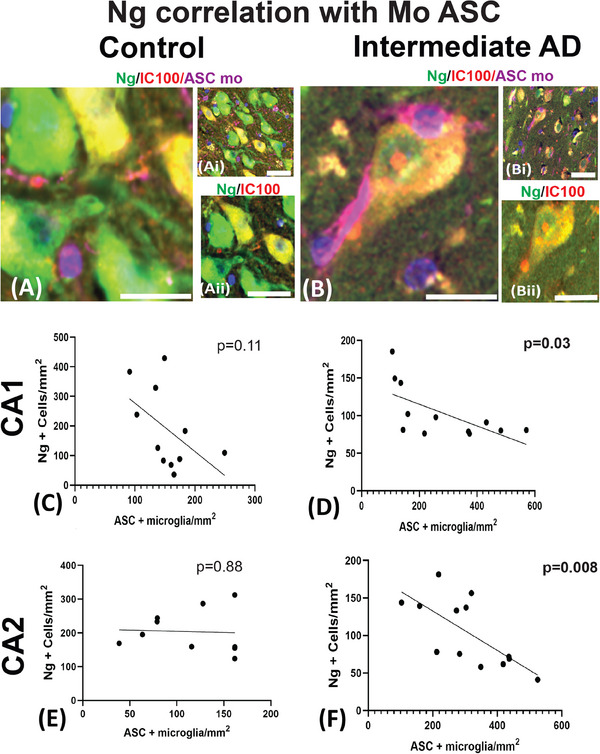
Pearson's correlation analysis of Ng and mouse anti‐ASC proteins seen in the hippocampal subfields. A photomicrograph of Ng (green), 1C100 (red), and mouse anti‐ASC (purple) are seen in control (A, Ai, and Aii) and the intermediate AD cases (B, Bi, and Bii). In the controls, the correlation analysis did not reveal any significant relationship between the number of Ng‐positive neurons and the number of ASC‐positive microglia in CA1 and CA2 (C and E). The intermediate AD cases showed a significant negative correlation between Ng counts and ASC positive microglia in the CA1 (C) and in the CA2 (F) hippocampal regions. AD, Alzheimer's disease; ASC, apoptosis‐associated speck‐like protein containing a caspase recruitment domain; CA, cornu ammonis; Mo, mouse; Ng, neurogranin (Ng). Scale bars = 20 μm (A), (Ai), (Aii), and (B), (Bi), (Bii)
TABLE 5.
Correlation: Ng+ cell counts and mouse ASC.
| Region | r | P value | Significance |
|---|---|---|---|
| CA1 | –0.54 | 0.11 | ns |
| CA2 | –0.05 | 0.88 | ns |
| CA3 | –0.6 | 0.07 | ns |
| CA1 | –0.14 | 0.03 | * |
| CA2 | –0.69 | 0.008 | ** |
| CA3 | –0.6 | 0.038 | * |
Note. Pearson's correlation coefficient for each ROI, the r statistic, P value, and significance.
Abbreviations: ASC, apoptosis‐associated speck‐like protein containing a caspase recruitment domain; CA, cornu ammonis; Ng, neurogranin; ns, not significant.
P < 0.05.
P < 0.01.
4. DISCUSSION
In this study, we have identified the morphological expression of MAP2 and Ng in neurons of the hippocampus in cases of intermediate AD and in controls. This study contributes unique data that show that the expression of the post‐synaptic marker Ng can be seen in the different subfields of the hippocampal region in post mortem cases of AD. In addition, we show that Ng expression was altered in AD cases compared to controls. The effect that post‐synaptic connections are lost with NFT accumulation, is important to fully comprehend the pathophysiological changes that occur in AD.
Our results demonstrate that during the intermediate stage of AD, the number of MAP2‐positive neurons decreases, except in the subiculum and DG. In addition to a decrease in the MAP2 positivity, the apical dendrite was not as defined in the CA2. However, in the CA3 hippocampal region, a region that has fewer NFTs, the MAP2 population significantly decreased, yet‐ the axonal and the dendritic processes were well defined, which suggests that MAP2 may compensate for or expand the neuronal processes during the intermediate stage of AD pathology.
Along with the changes in the microtubule protein MAP2, the post‐synaptic protein Ng significantly decreased in the CA hippocampal regions and in entorhinal cortex and subiculum, but not in the DG. Recently, Saunders et al. demonstrated reductions in Ng, in the hippocampus, and frontal, temporal, and visual cortices; also they demonstrated increases in CSF Ng 33 in AD cases. Here, we demonstrate that there is a region‐specific reduction in Ng in the hippocampus subfields, which supports the concept that synaptic loss in the hippocampus is a part of the neurodegenerative process, leading to clinical presentation of memory and learning deficits in AD.
Besides determining counts, we sought to investigate the histological characteristics of Ng seen in the hippocampal region. Similar to the MAP2, we found that Ng in the CA2 region was not present in the apical dendrite. Using a test of circularity, we found that the neurons with Ng expression in the CA2 and CA3 regions of AD cases had increased circularity compared to the controls. This finding indicates that the loss of synaptic structure underlies the impaired synaptic function and loss of plasticity, which accounts for the cognitive impairment associated with intermediate AD pathology via the loss of neuronal structure. 34
Tau is expressed in six isoforms and has two N‐terminal domains and four microtubule‐binding repeat domains composed of (3R or 4R). In AD pathology, tau phosphorylates and forms insoluble paired helical filaments that contain both 3R and 4R tau. 32 Aggregation of p‐tau is noted in NFTs and can be detected using antibodies raised against AT8 and PHF1. In age‐related changes, NFTs stained with AT‐8 or PHF1 are sparsely detected in the hippocampus of the donors who had little or no cognitive deficits. 32 In more progressive dementias (e.g., AD [Braak III–VI]) tauopathies are more widespread in the brain and NFTs take on different morphologies.
The increases in NFTs seen in the CA2 and CA3 hippocampal regions of the AD cases define the intermediate staging of AD pathology. The negative correlation between Ng and NFTs using the AT‐8 antibody was only significant in the intermediate AD cases. Our study supports that loss of Ng correlates with AD progression and supports the use of Ng as a biomarker of AD progression and possibly cognitive decline seen in patients with AD.
We found a significant increase in the number of neurons showing conformational NFTs using anti‐GT‐38 antibody, in the AD cases. Although the number of NFTs that were detected with anti‐GT‐38 did not correlate with the loss of Ng expression, it may indicate that the morphological changes of the GT‐38–positive neurons represent a more toxic or degenerative state in the intermediate AD cases. 35 Gibbons et al. describe similar NFT staining using GT‐38 and p‐tau antibody PHF1, but neuropil accumulation was not seen with GT‐38. 32 Future studies that compare the somatodendrite densities of GT‐38 NFTs and other isoforms of tau may indicate the pathological state of tau and shed light on the progression of AD pathology and other tauopathies (e.g., frontal temporal lobe dementia).
In this study, neuronal ASC expression identified by IC100 demonstrated that the reduction of Ng in neurons is in concert with the increases of neuronal ASC expression in the CA1 and CA2 hippocampal subfields. This finding suggests that as neurons become more unstable with the progression of hyperphosphorylation of tau proteins, inflammatory processes are recruiting ASC molecules in a temporal progression of AD pathology. Our data emphasize that the synapse may be more vulnerable when the inflammatory machinery is activated. These data support the importance of targeting ASC specks during the progression of AD pathology.
In addition to a neuronal expression of ASC, we found that there is a negative correlation with Ng expression across all three hippocampal subfields (i.e., CA1–CA3) using an ASC antibody identified in microglia. During the progression of AD pathology, tau aggregates intracellularly and Aβ accumulates in the parenchyma, which boosts the toxicity of microglia. 36 , 37 , 38 Hence it seems possible that as AD pathology progresses from CA1 to the other hippocampal subfields ASC would oligomerize in the microglia.
In conclusion, this is the first study to show that ASC expression is increased as Ng expression is decreased in the subfields of the hippocampus of the post mortem brains with AD pathology. It is not known if the inhibition of the inflammasome sensor protein would prevent synaptic loss but there seems to be evidence to support that when Ng expression is maintained in the apical dendrites, as seen in controls, ASC oligomerization did not occur in the neurons, even in the presence of neighboring NFTs. Taken together these data demonstrate that the loss of synaptic plasticity is in concert with the formation of the inflammasome complex in both neurons and microglia.
This post mortem study is limited as it only offers one a snapshot of one time period after death; however, other investigations show that the inflammasome/ASC pathway may affect synaptic function when activated in microglia, 25 thus supporting that the inflammasome may open a novel avenue for drug development in AD. Future studies that include a more diverse population are important to assess the relationship between synaptic degeneration and inflammasome/ASC pathway in AD.
CONFLICT OF INTEREST STATEMENT
KB and HZ have served on scientific advisory boards and/or as consultants for Abbvie, Acumen, Alector, Alzinova, ALZPath, Annexon, Apellis, Artery Therapeutics, AZTherapies, Cognito Therapeutics, CogRx, Denali, Eisai, Nervgen, Novo Nordisk, Optoceutics, Passage Bio, Pinteon Therapeutics, Prothena, Red Abbey Labs, reMYND, Roche, Samumed, Siemens Healthineers, Triplet Therapeutics, and Wave, have given lectures in symposia sponsored by Cellectricon, Fujirebio, Alzecure, Biogen, and Roche, and are co‐founders of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program (outside submitted work).
JPdRV, HMB, RWK, and WDD are co‐founders and managing members of InflamaCORE, LLC and have licensed patents on inflammasome proteins as biomarkers of injury and disease as well as on targeting inflammasome proteins for therapeutic purposes. JPdRV, HMB, RWK, and WDD are Scientific Advisory Board Members of ZyVersa Therapeutics. The other authors declare no conflicts of interest. Author disclosures are available in the supporting information.
CONSENT STATEMENT
All human subjects and or their families provided informed consent under the guidance of the Human Subjects Research Office, University of Miami, Miami, Florida (IRB ethics number, 19920348 [CR00012340]) Brain Endowment Bank.
Supporting information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
The authors would like to thank the donors and their families for allowing us to be the custodians of this precious brain collection. The authors thank the Brain Endowment Bank and the National Institutes of Health (NIH) for the use of the post mortem human tissue samples. This research was funded by an 1Florida ADRC for the Alzheimer's Science Training to Advance Research Success (AlzSTARS;RV), GR019010 and Team Science Award, University of Miami (BG003848; RV, JPdRV) and an RF1 grant from the NIH/NINDS/NIA (1RF1NS125578‐01) to WDD and JPdRV. HZ is a Wallenberg Scholar supported by grants from the Swedish Research Council (#2022‐01018 and #2019‐02397), the European Union's Horizon Europe research and innovation programme under grant agreement No 101053962, and Swedish State Support for Clinical Research (#ALFGBG‐71320).
Vontell RT, Gober R, Dallmeier J, et al. Association of region‐specific hippocampal reduction of neurogranin with inflammasome proteins in post mortem brains of Alzheimer's disease. Alzheimer's Dement. 2024;10:e12444. 10.1002/trc2.12444
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding author.
REFERENCES
- 1. Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease‐associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006;112(4):389‐404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Montine TJ, Phelps CH, Beach TG, et al. National Institute on Aging‐Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol. 2012;123(1):1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298(5594):789‐791. [DOI] [PubMed] [Google Scholar]
- 4. Lace G, Savva GM, Forster G, et al. Hippocampal tau pathology is related to neuroanatomical connections: an ageing population‐based study. Brain. 2009;132(Pt 5):1324‐1334. [DOI] [PubMed] [Google Scholar]
- 5. Subramanian J, Savage JC, Tremblay M‐È. Synaptic loss in Alzheimer's disease: mechanistic insights provided by two‐photon in vivo imaging of transgenic mouse models. Front Cellular Neurosci. 2020;14:592607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353‐356. [DOI] [PubMed] [Google Scholar]
- 7. Duits FH, Brinkmalm G, Teunissen CE, et al. Synaptic proteins in CSF as potential novel biomarkers for prognosis in prodromal Alzheimer's disease. Alzheimer's Research & Therapy. 2018;10(1):5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Xiang Y, Xin J, Le W, Yang Y. Neurogranin: a potential biomarker of neurological and mental diseases. Front Aging Neurosci. 2020;12:584743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liu W, Lin H, He X, et al. Neurogranin as a cognitive biomarker in cerebrospinal fluid and blood exosomes for Alzheimer's disease and mild cognitive impairment. Transl Psychiatry. 2020;10(1):125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kvartsberg H, Duits FH, Ingelsson M, et al. Cerebrospinal fluid levels of the synaptic protein neurogranin correlates with cognitive decline in prodromal Alzheimer's disease. Alzheimers Dement. 2015;11(10):1180‐1190. [DOI] [PubMed] [Google Scholar]
- 11. De Vos A, Jacobs D, Struyfs H, et al. C‐terminal neurogranin is increased in cerebrospinal fluid but unchanged in plasma in Alzheimer's disease. Alzheimers Dement. 2015;11(12):1461‐1469. [DOI] [PubMed] [Google Scholar]
- 12. Hellwig K, Kvartsberg H, Portelius E, et al. Neurogranin and YKL‐40: independent markers of synaptic degeneration and neuroinflammation in Alzheimer's disease. Alzheimers Res Ther. 2015;7:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Headley A, De Leon‐Benedetti A, Dong C, et al. Neurogranin as a predictor of memory and executive function decline in MCI patients. Neurology. 2018;90(10):e887‐e895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kvartsberg H, Lashley T, Murray CE, et al. The intact postsynaptic protein neurogranin is reduced in brain tissue from patients with familial and sporadic Alzheimer's disease. Acta Neuropathol. 2019;137(1):89‐102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sun X, Wang Q, Blennow K, et al. Association of neurogranin gene expression with Alzheimer's disease pathology in the perirhinal cortex. Alzheimers Dement (N Y). 2021;7(1):e12162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sun X, Dong C, Levin B, et al. APOE ε4 carriers may undergo synaptic damage conferring risk of Alzheimer's disease. Alzheimers Dement. 2016;12(11):1159‐1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dehmelt L, Halpain S. The MAP2/Tau family of microtubule‐associated proteins. Genome Biol. 2004;6(1):204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. DeGiosio RA, Grubisha MJ, MacDonald ML, McKinney BC, Camacho CJ, Sweet RA. More than a marker: potential pathogenic functions of MAP2. Frontiers in Molecular Neuroscience. 2022;15:974890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Guo T, Noble W, Hanger DP. Roles of tau protein in health and disease. Acta Neuropathol. 2017;133(5):665‐704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol Med. 2016;8(6):595‐608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rajendran L, Paolicelli RC. Microglia‐mediated synapse loss in Alzheimer's disease. J Neurosci. 2018;38(12):2911‐2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Heneka MT, Carson MJ, El Khoury J, et al. Neuroinflammation in Alzheimer's disease. Lancet Neurol. 2015;14(4):388‐405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vontell RT, de Rivero Vaccari JP, Sun X, et al. Identification of inflammasome signaling proteins in neurons and microglia in early and intermediate stages of Alzheimer's disease. Brain Pathol. 2022;33(4):e13142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hulse J, Bhaskar K. Crosstalk between the NLRP3 inflammasome/ASC speck and amyloid protein aggregates drives disease progression in Alzheimer's and Parkinson's disease. Front Mol Neurosci. 2022;15:805169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Moraes CA, Hottz ED, Dos Santos Ornellas D, et al. Microglial NLRP3 inflammasome induces excitatory synaptic loss through il‐1β‐enriched microvesicle release: implications for sepsis‐associated encephalopathy. Mol Neurobiol. 2023;60(2):481‐494. [DOI] [PubMed] [Google Scholar]
- 26. de Rivero Vaccari JP, Mim C, Hadad R, Cyr B, Stefansdottir TA, Keane RW. Mechanism of action of IC 100, a humanized IgG4 monoclonal antibody targeting apoptosis‐associated speck‐like protein containing a caspase recruitment domain (ASC). Transl Res. 2023;251:27‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gober R, Ardalan M, Shiadeh SMJ, et al. Microglia activation in postmortem brains with schizophrenia demonstrates distinct morphological changes between brain regions. Brain Pathol. 2022;32(1):e13003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. JrK Mai, Assheuer J, Paxinos G. Atlas of the human brain. 2nd ed. Elsevier Academic Press; 2004. [Google Scholar]
- 29. Xie C, Miyasaka T, Yoshimura S, et al. The homologous carboxyl‐terminal domains of microtubule‐associated protein 2 and TAU induce neuronal dysfunction and have differential fates in the evolution of neurofibrillary tangles. PLoS One. 2014;9(2):e89796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Callahan LM, Vaules WA, Coleman PD. Progressive reduction of synaptophysin message in single neurons in Alzheimer disease. Journal of Neuropathology & Experimental Neurology. 2002;61(5):384‐395. [DOI] [PubMed] [Google Scholar]
- 31. Hernández F, Ferrer I, Pérez M, Zabala JC, del Rio JA, Avila J. Tau Aggregation. Neuroscience. 2023;518:64‐69. [DOI] [PubMed] [Google Scholar]
- 32. Gibbons GS, Kim S‐J, Robinson JL, et al. Detection of Alzheimer's disease (AD) specific tau pathology with conformation‐selective anti‐tau monoclonal antibody in co‐morbid frontotemporal lobar degeneration‐tau (FTLD‐tau). Acta Neuropathologica Communications. 2019;7(1):34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Saunders T, Gunn C, Blennow K, et al. Neurogranin in Alzheimer's disease and ageing: a human post‐mortem study. Neurobiol Dis. 2023;177:105991. [DOI] [PubMed] [Google Scholar]
- 34. He M, Sun L, Cao W, et al. Association between plasma exosome neurogranin and brain structure in patients with Alzheimer's disease: a protocol study. BMJ Open. 2020;10(8):e036990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. He Z, McBride JD, Xu H, et al. Transmission of tauopathy strains is independent of their isoform composition. Nat Commun. 2020;11(1):7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Venegas C, Kumar S, Franklin BS, et al. Microglia‐derived ASC specks cross‐seed amyloid‐beta in Alzheimer's disease. Nature. 2017;552(7685):355‐361. [DOI] [PubMed] [Google Scholar]
- 37. Friker LL, Scheiblich H, Hochheiser IV, et al. β‐amyloid clustering around ASC fibrils boosts its toxicity in microglia. Cell Rep. 2020;30(11):3743‐3754.e3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Konstantoulea K, Guerreiro P, Ramakers M, et al. Heterotypic Amyloid β interactions facilitate amyloid assembly and modify amyloid structure. Embo j. 2022;41(2):e108591. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author.