

Summary
The discovery of novel structural classes of antibiotics is urgently needed to address the ongoing antibiotic resistance crisis1–9. Deep learning approaches have aided in exploring chemical spaces1,10–15; yet, these models are typically black box in nature and do not provide chemical insights. Here, we reasoned that the chemical substructures associated with antibiotic activity learned by neural network models can be identified and used to predict structural classes of antibiotics. We tested this hypothesis by developing an explainable, substructure-based approach for the efficient, deep learning-guided exploration of chemical spaces. We determined the antibiotic activities and human cell cytotoxicity profiles of 39,312 compounds and applied ensembles of graph neural networks to predict antibiotic activity and cytotoxicity for 12,076,365 compounds. Using explainable graph algorithms, we identified substructure-based rationales for compounds with high predicted antibiotic activity and low predicted cytotoxicity. We empirically tested 283 compounds and found that compounds exhibiting antibiotic activity against Staphylococcus aureus were enriched in putative structural classes arising from rationales. Of these structural classes of compounds, one is selective against methicillin-resistant S. aureus (MRSA) and vancomycin-resistant enterococci, evades substantial resistance, and reduces bacterial titers in mouse models of MRSA skin and systemic thigh infection. Our approach enables the deep learning-guided discovery of structural classes of antibiotics and demonstrates that machine learning models in drug discovery can be explainable, providing insights into the chemical substructures that underlie selective antibiotic activity.
Introduction
The ongoing antibiotic resistance crisis threatens to render current antibiotics ineffective and increase morbidity from bacterial infections. This crisis has been exacerbated by a lack of new antibiotics, without which global deaths due to resistant infections are projected to reach 10 million per year by 2050.16 Antibiotic candidates have been discovered in the past decade through various approaches based on natural product mining2,3, high-throughput screening4, evolution and phylogeny analyses5,6, structure-guided and rational design7,8, and in silico screens using machine learning1,12–14. Nevertheless, developing effective approaches to antibiotic discovery that better leverage the large structural diversity of chemical space remains a challenge, and novel approaches to antibiotic discovery are urgently needed.
We recently developed a deep learning approach to antibiotic discovery and showed that it identifies potential antibiotics from large chemical libraries, resulting in the discovery of halicin1 and abaucin14 from the Drug Repurposing Hub17 (comprising ~6,000 molecules) and other antibacterial compounds from ~107 million molecules in the ZINC15 library18. This approach relies on Chemprop, a platform for graph neural networks10,11, which are typically black-box models19, or models that are not readily interpreted or explained. By definition, interpreting or explaining such models reveals the patterns of decision-making steps the models perform to arrive at their predictions (interpretability), or renders such predictions human-understandable (explainability)20. Here, we aimed to vastly expand graph neural network models for antibiotic discovery by training on large datasets measuring antibiotic activity and human cell cytotoxicity, and we hypothesized that model predictions could be explained on the level of chemical substructures using graph search algorithms (Fig. 1a). As antibiotic classes are typically defined based on shared substructures, we reasoned that substructure identification may, by better explaining model predictions, allow for the efficient exploration of chemical spaces and facilitate the discovery of novel structural classes, in lieu of lone compounds.
Fig. 1. Ensembles of deep learning models for predicting antibiotic activity and human cell cytotoxicity.
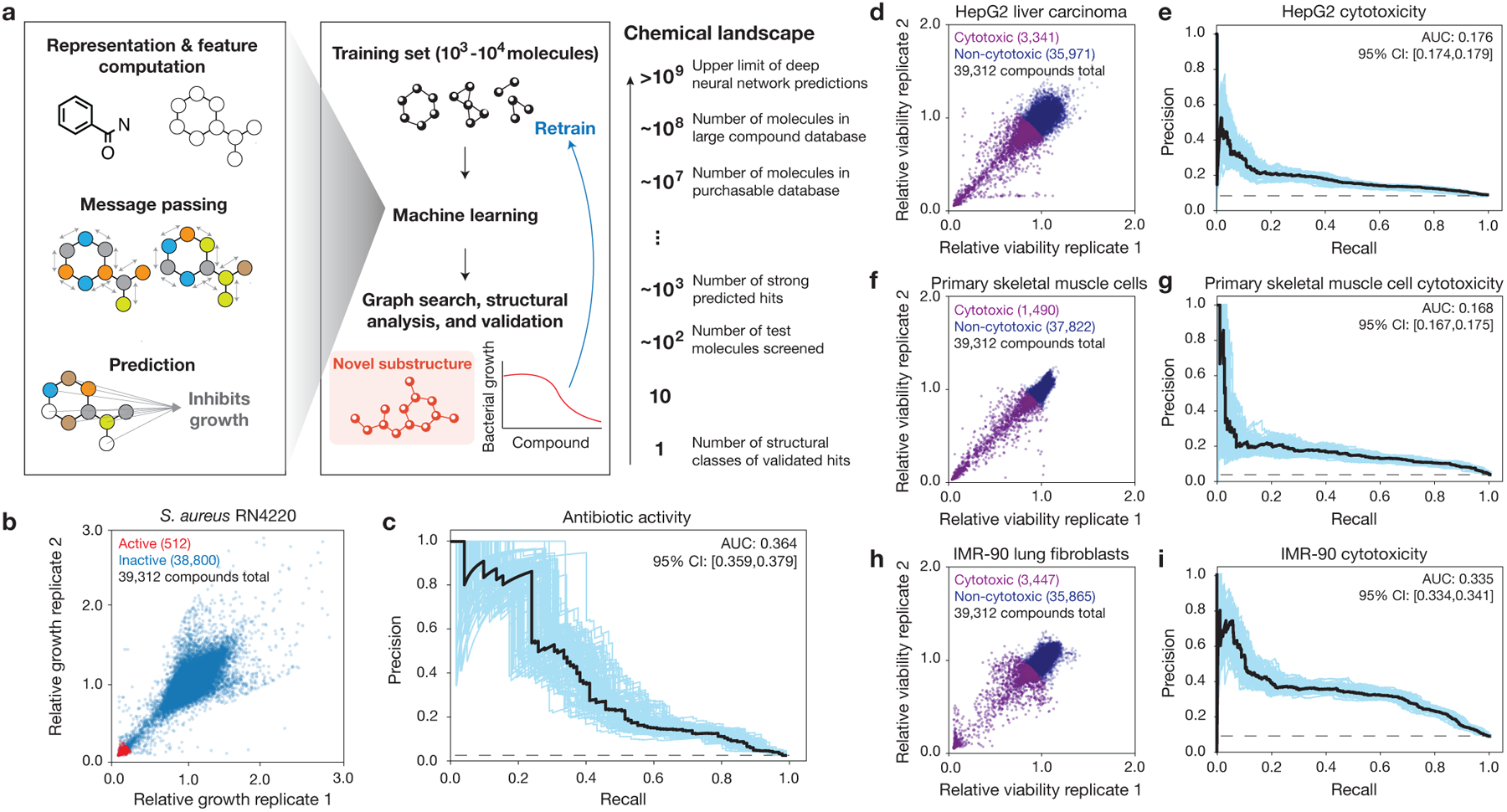
a, Schematic of the approach. Graph neural networks predict the chemical properties of >109 molecules in silico, in contrast to expensive and time-consuming experimental screening of large chemical libraries. Here, the growth inhibition activities of 39,312 chemically diverse compounds are used to train the model, the model is applied to virtual chemical databases comprising 12,076,365 molecules that can be readily procured, and compounds with high prediction scores (“hits”) are analyzed according to structural class, procured, and tested. This approach can be iterated, and the model can be retrained to generate new predictions.
b, S. aureus RN4220 growth inhibition data for a screen of 39,312 compounds at a final concentration of 50 μM. Data are from two biological replicates. Active compounds are those for which the mean relative growth is <0.2.
c, Precision-recall curves for an ensemble of 10 Chemprop models, augmented with RDKit features, trained and tested on the data in (b). The black dashed line represents the baseline fraction of active compounds in the dataset (1.3%). Blue curves and the 95% confidence interval (CI) indicate variation from bootstrapping. AUC, area under the curve.
d, f, h, HepG2 (d), HSkMC (f), and IMR-90 (h) viability data for screens of 39,312 compounds at a final concentration of 10 μM. Data are from two biological replicates for each cell type. Cytotoxic compounds are those for which the mean relative viability is <0.9.
e, g, i, Precision-recall curves for an ensemble of 10 Chemprop models, augmented with RDKit features, trained and tested on the data in (d,f,h). Black dashed lines represent the baseline fractions of cytotoxic compounds in the datasets (e, 8.5%; g, 3.8%; i, 8.8%). Blue curves and the 95% confidence interval (CI) indicate variation from bootstrapping.
Models for antibiotic activity
In this study, we focus on discovering structural classes of antibiotics that are effective against Staphylococcus aureus, a Gram-positive pathogen resistant to many first-line antibiotics and a major cause of difficult-to-treat nosocomial and bloodstream infections21. We first screened an original set of 39,312 compounds containing most known antibiotics, natural products, and structurally diverse molecules, with molecular weights between 40 Da and 4,200 Da, for growth inhibitory activity against a methicillin-susceptible strain, S. aureus RN4220 (Fig. 1b, Extended Data Fig. 1, and Supplementary Data 1). These compounds were screened for overnight growth inhibitory activity in nutrient-rich media at a final concentration of 50 μM, and their effects were binarized as active or inactive using an 80% normalized growth inhibition cut-off, resulting in a total of 512 active compounds (1.3% of all compounds).
Using Chemprop, we trained ensembles of graph neural networks on our screening data to make binary classification predictions of whether or not a new compound will inhibit bacterial growth based on its chemical structure. Each graph neural network operates by performing convolution steps that depend on the atoms and bonds of each input chemical structure, which is viewed as a mathematical graph with vertices (atoms) and edges (bonds; Fig. 1a)10,11. After successive convolution steps which pool together information from neighboring atoms and bonds, each model generates a final prediction score between 0 and 1, representing its estimate of the probability that the molecule is active. To provide additional data that may improve model performance, each model was supplied a list of RDKit-computed molecular features for each input (e.g., the number of hydrogen donors and acceptors and partition coefficient estimates; see Supplementary Data 1). The prediction scores from multiple models within an ensemble were then averaged to improve robustness. Each model was trained and validated, then tested, on the same 80%−20% splits of the training dataset. For an ensemble of ten models applied to the withheld test data, the area under the precision-recall curve (AUPRC) was 0.364, indicating good performance while accounting for the imbalance of active compounds in the training data (Fig. 1c). We observed decreased performance, as measured by the AUPRC for the test set, of alternative models including an ensemble of ten Chemprop models without RDKit features and the best-performing random forest classifier model based on Morgan fingerprints as the molecular representation (Extended Data Fig. 2). While the statistical significance of these differences in performance varied (Supplementary Table 1), these findings indicate that Chemprop models with RDKit-computed molecular features produce promising predictions of antibiotic activity and can outperform simpler or shallower (i.e., random forest) deep learning models.
Models for human cell cytotoxicity
To better identify compounds that are selective against S. aureus, we developed orthogonal models that predict cytotoxicity in human cells. We first counter-screened our training set of 39,312 compounds for cytotoxicity in human liver carcinoma cells (HepG2), human primary skeletal muscle cells (HSkMCs), and human lung fibroblast cells (IMR-90). HepG2 cells are commonly used to study hepatotoxicity and general cytotoxicity, while HSkMCs and IMR-90 cells may better model in vivo toxicity than do immortal cell lines. Cellular viability was measured after 2–3 days of treatment with each compound at 10 μM, a concentration appropriate to, and widely used for, human cell cultures15. Compound activities were then binarized using a stringent 90% cell viability cut-off, resulting in a total of 3,341 (8.5%), 1,490 (3.8%), and 3,447 (8.8%) compounds classified as cytotoxic for HepG2 cells, HSkMCs, and IMR-90 cells, respectively, and of the 512 active antibacterial compounds, 306 were non-cytotoxic for all three cell types (Fig. 1d,f,h and Supplementary Data 1). As above, these data were used to train binary classification models that predict the probability of whether or not a new compound is cytotoxic to HepG2 cells, HSkMCs, or IMR-90 cells based on the compound’s chemical structure. For ensembles of 10 Chemprop models trained and validated, then tested, on the same 80%−20% splits of the data, the AUPRC values for the HepG2, HSkMC, and IMR-90 models were 0.176, 0.168, and 0.335, respectively (Fig. 1e,g,i). This indicated positive, but less predictive, performance than our models for antibiotic activity, a result which may arise due to our more stringent criteria for declaring compounds as non-cytotoxic. The cytotoxicity models were most predictive for IMR-90 cells, which may arise from having more cytotoxic compounds—and more learning examples—against this cell type in the screening data. Similar to our findings for antibiotic activity, for cytotoxicity of all cell types we found decreased AUPRCs using alternative models, including an ensemble of ten Chemprop models without RDKit features and the best-performing random forest classifier models using Morgan fingerprints (Extended Data Fig. 3), with varying statistical significance of these differences in performance (Supplementary Table 1). Further benchmarking using two Tox21 datasets22 and a human metabolites database23, as well as experimental testing of 190 compounds, support that these models can productively filter out cytotoxic compounds (Supplementary Note 1 and Methods).
Filtering and visualizing chemical space
Satisfied with the performance of our models, we retrained ensembles of 20 Chemprop models with the entirety of each of the training datasets, resulting in four ensembles predicting antibiotic activity, HepG2 cytotoxicity, HSkMC cytotoxicity, and IMR-90 cytotoxicity. We applied the ensembles to predict the antibiotic activities and cytotoxicity profiles of 12,076,365 compounds, comprising 11,277,225 compounds from the Mcule purchasable database24—in which most compounds can be readily purchased without recourse to in-house chemical synthesis—in addition to 799,140 compounds from a Broad Institute database (Fig. 2a–e and Supplementary Data 2). We filtered chemical compounds of interest based on the predicted antibiotic activities and cytotoxicity, retaining at first only the 3,004 compounds with antibiotic prediction scores >0.4 from the Mcule purchasable database and, due to better access to compounds in this database, the 7,306 compounds with antibiotic prediction scores >0.2 from the Broad Institute database (Fig. 2a,b). We then retained only those compounds with HepG2, HSkMC, and IMR-90 cytotoxicity prediction scores <0.2, a stringent filter resulting in 3,646 compounds—1,210 compounds from the Mcule purchasable database and 2,436 compounds from the Broad Institute database—or 0.03% of all compounds assessed (Fig. 2a,c-e).
Fig. 2. Filtering and visualizing chemical space.
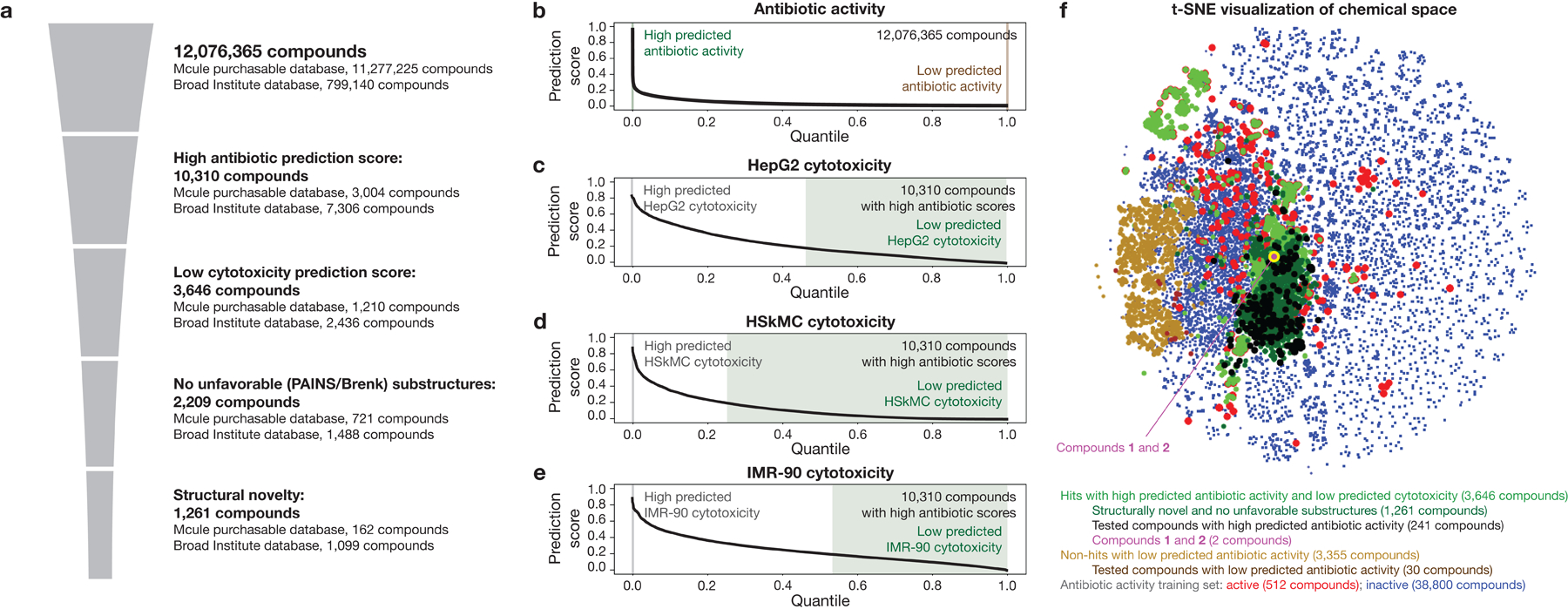
a, In silico filtering procedure. Trained graph neural networks are applied to make predictions of antibiotic activity for 12,076,365 compounds from the Mcule purchasable database and a Broad Institute database. Compounds with high (>0.4 for the Mcule database, and >0.2 for the Broad Institute database) prediction scores for antibiotic activity are retained, and similar graph neural networks are applied to predict the cytotoxicity of these compounds for HepG2 cells, HSkMCs, and IMR-90 cells. Compounds with low (<0.2) cytotoxicity prediction scores for all cell types are retained, then computationally tested for the presence of promiscuously reactive or unfavorable chemical substructures (PAINS and Brenk substructures). Finally, the remaining compounds are filtered for structural novelty, as defined by a Tanimoto similarity score of <0.5 with respect to any active compound in the training dataset and lack of a quinolone bicyclic core or β-lactam ring.
b, Rank-ordered antibiotic activity prediction scores of all 12,076,365 compounds for which antibiotic activity was predicted.
c-e, Rank-ordered HepG2 (c), HSkMC (d), and IMR-90 (e) cytotoxicity prediction scores of 10,310 compounds with high antibiotic activity prediction scores.
f, t-Distributed neighbor embedding (t-SNE) plot of compounds with high and low antibiotic prediction scores, in addition to compounds in the training set. The plot shows the chemical similarity or dissimilarity of various compounds, and active compounds in the training set (red dots) are seen to largely separate compounds with high prediction scores (green, black, and purple dots) from compounds with low prediction scores (brown dots).
In contrast to compounds passing the aforementioned filters (“hits”), we consolidated 3,355 compounds with low (<10−6) antibiotic prediction scores (“non-hits”). These prediction score cutoffs were chosen to generate computationally tractable groups of ~103 compounds, but the following results are general across different prediction score cutoffs (Extended Data Fig. 4). We visualized the chemical space using t-distributed stochastic neighbor embedding25 (t-SNE) applied to Morgan fingerprints as the molecular representation. This revealed that hits were structurally dissimilar to non-hits, and the training set, which includes compounds from diverse classes of known antibiotics, largely separates non-hits from hits (Fig. 2f). Intriguingly, as indicated by t-SNE and our subsequent substructure-based analyses (Fig. 3), multiple hits were structurally dissimilar to active compounds in the training set, suggesting that our models generalize to unseen chemical spaces.
Fig. 3. Graph-based rationales reveal scaffolds for prospective antibiotic classes.

a, Illustration of the Monte Carlo tree search method resulting in chemical structure rationales (graph substructures) with high predicted antibiotic activity.
b, A rationale (red) determined using a Monte Carlo tree search for cefmenoxime, an example hit compound. Here, the rationale overlaps with the cephalosporin core and results, by itself, in an antibiotic prediction score of 0.149. For comparison, the cephalosporin core is shown in black.
c, Rank-ordered Tanimoto similarity scores of all hits with respect to active compounds in the training set. A threshold of 0.5 was used to threshold predicted hits that are structurally distinct from active compounds in the training set.
d, Rank-ordered numbers of hits with rationales in rationale groups with conserved scaffolds, for 186 hits with rationales found in 1,261 structurally novel hits containing no unfavorable substructures. Here, 16 hits with rationales were associated with five scaffolds, G1-G5.
e, Rank-ordered antibiotic activity prediction scores of 253 compounds with high (>0.2) antibiotic prediction scores and 30 compounds with low (<0.1) antibiotic prediction scores procured for empirical testing. True positives are colored in purple, and true negatives are colored in brown.
f, Chemical structures of compounds 1 and 2, two structurally novel hits associated with rationale group G2 that possess no unfavorable substructures and were found to inhibit the growth of S. aureus RN4220. The rationales (red) are identical for both compounds, resulting in an antibiotic prediction score of 0.144.
g, S. aureus MIC and human cell IC50 values of compounds 1 and 2, shown on a log scale. Bars show the means of two biological replicates (points) and are colored by the bacterial strain, human cell type, or media condition tested. Asterisks indicate values larger than 128 μg/mL.
Rationales predict antibiotic classes
As graph neural networks make predictions based on the information contained in the atoms and bonds of each molecule, we hypothesized that compounds with high antibiotic prediction scores contain substructures (“rationales”) that largely determine their scores. Identifying such rationales would provide guarantees of model explainability for the hits of interest: namely, any hit’s high antibiotic prediction score would be directly attributable to its rationale, such that the rationale—when viewed as a molecular input to Chemprop in its own right—possesses a high antibiotic prediction score. The ability to classify such rationales would render Chemprop’s predictions more human-understandable and enable subsequent machine learning-guided substructure analyses.
Given our trained Chemprop models, we computed such rationales by employing graph-based search algorithms. These graph search algorithms allowed us to determine, in the context of a single molecule, the smallest rationale with a prespecified threshold number of atoms identified to have positive predictive value (Fig. 3a, Extended Data Fig. 5, and Methods). We aimed to determine rationales containing at least eight atoms and exhibiting high antibiotic prediction scores >0.1 using Monte Carlo tree searches, which have been used to inform deep learning models including AlphaGo26. Monte Carlo tree searches comprise of selecting an initial substructure, iteratively pruning the substructure, and selecting for deletions resulting in high prediction scores when the subgraphs are passed as inputs into Chemprop (Fig. 3a, Extended Data Fig. 5, and Methods). This graph search outputs a rationale explaining a threshold amount (at least 0.1) of the compound’s prediction score if it converges; otherwise, no rationale is found, and the hit of interest is not explainable in this way. While other approaches centered on maximal common substructure (MCS) identification have been used to study the chemical motifs shared among groups of compounds in high-throughput screens and cheminformatics analyses27, we found that MCS-based approaches did not necessarily yield substructures that were diagnostic of high predicted antibiotic activity when applied to deep learning model predictions (Supplementary Note 2 and Extended Data Fig. 6).
We first validated that the calculation of rationales could recapitulate the discovery of structural classes of antibiotics not found in the training data using leave-one-out analyses with quinolones and β-lactams, two structural classes highly enriched in the training data. We trained ensembles of Chemprop models similarly to our final models for antibiotic activity, but with all 31 or 505 compounds containing the quinolone bicyclic core or β-lactam ring, respectively, withheld from the training. When the corresponding trained models were applied to the withheld test sets and the prediction score threshold was set to 0.2, active quinolone and β-lactam compounds were predicted to have antibacterial activity, with modest true positive rates of 0.294 and 0.060, respectively; additionally, for a subset of these compounds, the models produced rationales that contain the relevant core rings (Supplementary Data 2). These analyses underscore our approach’s ability to identify new antibiotic scaffolds, including those not previously seen by the model during training, based on the arrangements of molecular atoms and bonds in chemical structures. Importantly, similar results cannot be accomplished using traditional quantitative structure-activity relationship (QSAR) analyses, which assume knowledge of an active scaffold a priori and aim to design chemical analogs containing the scaffold.28
Applying this rationale analysis to the filtered hits emerging from our full model, we computed rationales for 380 of the 3,646 hits (10.4%). As expected, many rationales coincided with known fragments of structural classes, including the quinolone bicyclic core and the cephalosporin and β-lactam rings (Fig. 3b, Extended Data Fig. 6, and Supplementary Data 2). Intriguingly, we also found rationales that were not associated with any known antibiotic classes. We therefore aimed to better filter structurally novel hits of interest and investigate their corresponding rationales.
Novel, filtered substructures
Building on the emergence of known antibiotic classes from our analyses and the ability of graph-based rationales to predict substructures diagnostic of high antibiotic prediction scores (Fig. 3a,b and Extended Data Fig. 5), we sought to identify structurally novel antibiotic classes predicted by our models. In order to consider chemical structures with favorable medicinal chemistry properties, we removed all hits containing PAINS and Brenk alerts29,30, which refer to substructures that may be promiscuously reactive, mutagenic, or pharmacokinetically unfavorable. This narrowed down the 3,646 predicted hits to 2,209 hits (Fig. 2a). Next, we focused on procuring compounds dissimilar to those in the training set. We computed the maximal Tanimoto similarity of each hit to any active compound in the training set and shortlisted hits with maximal similarity scores ≤0.5 as a rudimentary cut-off (Fig. 3c), as well as those not containing a β-lactam ring or a quinolone bicyclic core. This yielded a final set of 1,261 hits, of which 162 were from the Mcule purchasable database and 1,099 were from the Broad Institute database (Fig. 2a). For this more focused set of hits, our rationale calculations revealed that 186 hits (14.8%) possessed rationales (Supplementary Data 2).
In order to leverage these rationales for clear predictions of structural classes, we reasoned that studying the chemical scaffolds shared across rationales would highlight the most salient predictions of structural classes. This is especially useful for down-sampling, as typical rationales possess large numbers (>17) of atoms and differ from each other by minor modifications. We computationally identified chemical scaffolds with at least 12 atoms that were conserved across rationales (see Methods for details). With this approach, we found that 16 of the 186 hits with rationales (8.6%) could be grouped using five distinct scaffolds, G1-G5 (Fig. 3d), with each group containing at least two hits with associated rationales. Intriguingly, three of the five scaffolds were chlorine-containing, suggesting that our models view the presence of a chlorine atom in these chemical contexts as an important factor influencing antibiotic activity.
Due to the tractable number of hits remaining from our filtering steps and analyses, we directly tested our model predictions by procuring nine hits associated with the rationales in groups G1-G5. As a positive control, we procured 12 cephalosporin- and quinolone-like hits, which shared common substructures with cephalosporin- and quinolone-containing rationales (Extended Data Fig. 6). For comparison, we also procured 45 hits (out of the filtered 1,261 hits) with computed rationales that were not associated with G1-G5, 187 hits (out of the filtered 1,261 hits) with no computed rationale, and 30 structurally dissimilar compounds with low (<0.1) prediction scores. This approach resulted in a set of 283 compounds (Fig. 3e and Supplementary Data 2), which we experimentally tested.
A structural class of antibiotics from rationales
Testing for growth inhibition, we found that four out of the nine procured hits (44%) associated with groups G1-G5 exhibited activity against S. aureus, with minimal inhibitory concentrations (MICs) ≤32 μg/mL (Fig. 3f,g, Supplementary Table 2, and Extended Data Fig. 7). Intriguingly, none of the 45 procured hits with rationales not associated with G1-G5, and 17 of the 187 procured hits with no rationale (9.1%), exhibited activity (Fig. 3e and Supplementary Table 2). The working true discovery rates associated with all tested structurally novel hits with rationales (7.4%) and across all tested structurally novel hits (8.7%) were higher than the fraction of active compounds in our training set (1.3%), suggesting the utility of our approach when generalizing to diverse chemical spaces. These values suggest that compound testing efforts can be as productive as testing one-off hits when they focus on the structural classes predicted by deep learning models. Additionally, as expected, all 12 cephalosporin and quinolone-like hits inhibited growth and exhibited antibiotic cross-resistance in methicillin-resistant S. aureus (MRSA, strain USA300), confirming their likely mechanisms of action (Supplementary Table 2). Consistent with a low false omission rate for the model, none of the 30 procured compounds with low prediction scores inhibited the growth of S. aureus (Fig. 3e).
Of the four hits found to be active against S. aureus associated with G1-G5, no compound had previously been studied against the pathogens considered here (Supplementary Note 3), and together, these hits are associated with three rationale groups—G1, G2, and G5 (Fig. 3d and Extended Data Fig. 7). Of note, G2 was associated with two validated (active) hits (compounds 1 and 2; Fig. 3f), indicating that this rationale group may represent an active structural class, and compounds 1 and 2 simultaneously satisfy the Lipinski’s rule of five31 and the Ghose criteria32 for druglikeness, suggesting favorable oral bioavailability and druglike properties for further development (Supplementary Table 3). Additional properties, including O’Shea and Moser’s physicochemical observations for antibiotics33 (Supplementary Table 3), may further narrow down chemical space and inform subsequent development, especially when considering candidates from larger libraries such as ZINC15 (ref. 18) and specific routes of administration. While we have not filtered our hits based on these or other physicochemical properties, we note that the validated hits were smaller and less polar than typical Gram-positive antibiotics (Supplementary Table 3).
Performing additional growth inhibition experiments, we found that compounds 1 and 2, as well as nearly all of the other structurally novel validated hits, were also active against MRSA USA300 with MICs comparable to their methicillin-susceptible analogues (Fig. 3g and Supplementary Table 2). Counter-screening all structurally novel validated hits for cytotoxicity against HepG2 cells, HSkMCs, and IMR-90 cells, we found that 20 out of the 21 structurally novel, validated hits were non-cytotoxic at a concentration of 10 μM. Compounds 1 and 2 exhibited half-maximal inhibitory concentration (IC50) values ≥128 μg/mL for all cell types, indicating robust selectivity against S. aureus (Fig. 3g and Supplementary Table 2). In contrast, the therapeutic windows of all the other structurally novel validated hits, including the two other validated hits associated with G1 and G5, were less than those of compounds 1 and 2 (Fig. 3g and Supplementary Table 2).
As a final empirical filter, we measured the S. aureus MICs of the validated hits associated with G1-G5 in media supplemented with 10% fetal bovine serum as a control for binding of the compounds to serum proteins (Fig. 3g). We found that the MICs of compounds 1 and 2 increased 4- to 8-fold to 16 μg/mL, but remained substantively (≥8-fold) less than their human cell IC50 values; in contrast, the MICs of the other two compounds increased to ≥64 μg/mL in serum (Extended Data Fig. 7). Together with their favorable MIC values in serum-free media (≥64-fold less than their human cell IC50 values), these observations suggested that compounds 1 and 2 were the most selective of all the validated hits and merited further study.
Mechanism of action and resistance
Compounds 1 and 2 share an N-[2-(2-chlorophenoxy)ethyl]aniline core, which was predicted to be diagnostic of antibiotic activity based on our Monte Carlo tree search-based rationales (Fig. 3f). The common substructure suggests that the compounds may share a similar mechanism of action, which we studied using traditional microbiological assays. Time-kill experiments for log-phase S. aureus RN4220 and B. subtilis 168 showed that treatment with both compounds at supra-MIC concentrations led to decreases in colony forming units (CFU)/mL compared to non-treatment after four hours, which was typically similar to, but less bactericidal, than vancomycin treatment (Fig. 4a). Moreover, MRSA USA300 exhibits at least 16-fold increased MICs relative to the methicillin-susceptible strain for ampicillin, ciprofloxacin, and tetracycline but exhibits only two-fold increased MICs for compounds 1 and 2 (Extended Data Fig. 8), suggesting that these compounds may not share similar mechanisms of action with β-lactams, fluoroquinolones, and tetracyclines. These compounds were specific against Gram-positive bacteria, as they did not inhibit the growth of Escherichia coli, Acinetobacter baumannii, or Pseudomonas aeruginosa, with the exception of permeable or efflux-impaired E. coli (lptD4213 and ΔtolC832), for which both compounds exhibited MICs of 2 μg/mL (Supplementary Tables 2 and 4).
Fig. 4. Resistance and mechanism of action of a structural class.
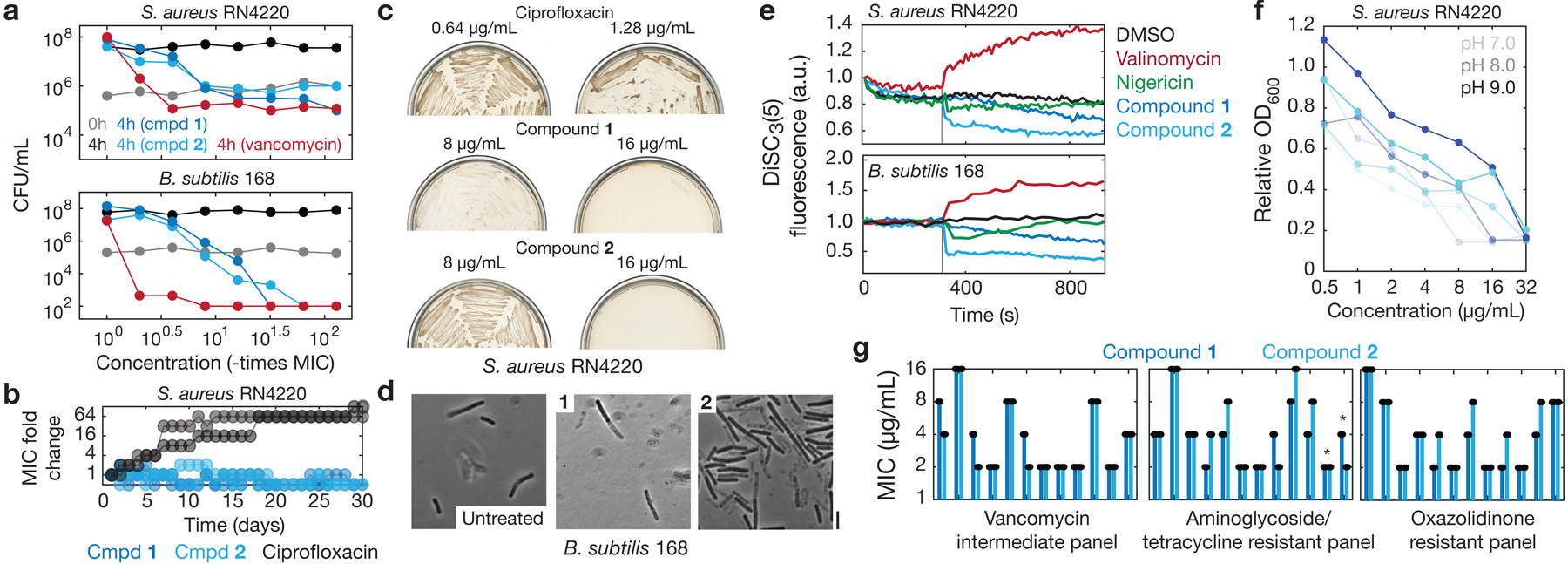
a, Time-kill measurements for log-phase S. aureus RN4220 and B. subtilis 168 treated with compounds 1 and 2, vancomycin, or untreated. Data are from two biological replicates, and points indicate mean values. Where applicable, CFU/mL values less than 102 were truncated to a value of 102 to reflect the lower limit of quantification.
b, MIC fold changes in serial passaging experiments, in which S. aureus RN4220 was passaged in liquid LB every 24 h for 30 days. Two biological replicates (individual curves) are shown for each compound, and fold change is on a log scale.
c, Growth of suppressor mutants in evolution experiments, in which S. aureus RN4220 was plated at 109 CFU on LB agar plates containing compound, incubated for 5 days, then streaked on fresh compound-containing LB agar plates. Each image represents two biological replicates.
d, Phase contrast images of log-phase B. subtilis 168 cells treated with compounds 1 and 2 (16 μg/mL) for 3 h. Scale bar, 3 μm. Results shown represent three biological replicates.
e, DiSC3(5) fluorescence in log-phase S. aureus RN4220 and B. subtilis 168 during treatment with DMSO (1%), valinomycin and nigericin (~1 mg/mL), and compounds 1 and 2 (32 μg/mL). Cells were treated at time 300 s (vertical lines). Results shown represent three biological replicates.
g, OD600 measurements from S. aureus RN4220 cultures incubated overnight with compounds 1 and 2 across different media pH levels. Each growth curve shows one biological replicate, and results shown represent two biological replicates.
h, MIC values of compounds 1 and 2 against CDC MRSA and VRE isolates, shown on a log scale. Bars show the means of two biological replicates (points). Asterisks denote bars corresponding to VRE isolates. All other bars correspond to MRSA isolates.
We therefore further investigated the mechanisms of action of these compounds through the evolution of resistant mutants. We serially passaged S. aureus RN4220 treated with each of compounds 1 and 2 in liquid culture, and found that MICs remained essentially unchanged after 30 days (Fig. 4b). In contrast, cultures exhibited ≥64-fold increased MICs to ciprofloxacin after 30 days (Fig. 4b). Additionally, in suppressor mutant generation experiments, we plated S. aureus RN4220 at high inocula on solid media in the presence of supra-MIC levels of compounds 1 and 2, and found that colonies grew at 4× but not 8× MIC after 5 days (Fig. 4c), suggestive of low-level resistance (frequency of resistance at 4× MIC, ~10−8). For comparison, suppressor mutants grew in ciprofloxacin at concentrations corresponding to 4× and 8× MIC (Fig. 4c; frequency of resistance at 4× and 8× MIC, ~10−6 and ~10−7, respectively). In order to study these cells further, we subcultured cells from the endpoints of both experiments and selected individual colonies in biological duplicate for sequencing. Whole-genome sequencing of these colonies indicated that the main mutations to arise were inconsistent between colonies and largely in genes involved in osmoregulation and virulence pathways, as opposed to mutations arising consistently across different colonies (as in DNA topoisomerase for ciprofloxacin; see Supplementary Data 3). Taken together, these findings suggest that compounds 1 and 2 can evade substantial resistance.
In order to investigate the phenotypic effects of compounds 1 and 2 further, we combined microscopic observation with cellular physiology measurements. As we have previously done for other classes of antibiotics34–37, we first performed single-cell imaging; here, we focused on B. subtilis, whose rod-like shape exhibits more salient morphological changes than does S. aureus. Single-cell imaging revealed that cells treated with compound 1 or 2 lysed (Fig. 4d), consistent with the bactericidal activity of these compounds (Fig. 4a) and suggestive of a cell envelope-targeting mechanism of action. To study this suggestion further, we used a dye sensitive to the membrane proton motive force (PMF), DiSC3(5), in bulk culture experiments. In S. aureus and B. subtilis, the PMF is generated by two components, the membrane potential, ΔΨ, and the pH gradient, ΔpH, across the membrane, and bacterial cultures treated with DiSC3(5) display increases (decreases) in fluorescence when ΔΨ (ΔpH) is disrupted38. We found that treatment with both compounds 1 and 2 resulted in fluorescence quenching of DiSC3(5) in S. aureus and B. subtilis, indicating that both compounds disrupt ΔpH (Fig. 4e). Furthermore, we found that the growth inhibitory effects of both compounds were antagonized by higher media pH levels, which result in increases in ΔpH (ref. 1; Fig. 4f). Together, these findings establish dissipation of ΔpH as a primary mechanism of action of compounds 1 and 2. Notably, while halicin has been shown to exhibit a similar mechanism of action1 and bacterial membrane-sensitive mechanisms of action have often been de-prioritized in antibiotic drug discovery due, in part, to potential lack of selectivity39, compounds 1 and 2 selectively target Gram-positive bacteria over Gram-negative bacteria and human cells. Additional studies measuring DiSC3(5) in S. aureus cells and leveraging Chemprop to predict PMF alterations suggest, intriguingly, that the mechanism of action of compounds 1 and 2 might be accurately predicted from chemical structure (Methods and Supplementary Data 4).
Given that compounds 1 and 2 exhibit a structural scaffold distinct from those of known antibiotics and dissipate ΔpH, we further expected that these compounds would be active against diverse antibiotic-resistant pathogens. We found that both compounds were active (MIC ≤16 μg/mL) against 40 CDC isolates of different bacterial species containing various resistance factors, including vancomycin, aminoglycoside/tetracycline (AG/TC), and oxazolidinone resistance (Fig. 4g and Supplementary Table 4). Across these isolates, the median MICs for compounds 1 and 2 were 4 and 3 μg/mL, respectively, and both compounds exhibited MIC ranges of 2 to 16 μg/mL. Of note, both compounds were active against vancomycin-resistant enterococci (VRE), a serious antimicrobial resistance threat40 (Fig. 4g and Supplementary Table 4). Moreover, time-kill experiments indicate that both compounds were effective against B. subtilis persisters, resulting in the eradication of a log-phase culture after treatment with kanamycin (Extended Data Fig. 8). These findings suggest that compounds 1 and 2 can overcome common resistance determinants and antibiotic tolerance in Gram-positive bacteria.
Toxicology, chemical properties, and in vivo efficacy
Given the favorable in vitro selectivity of compounds 1 and 2 (Fig. 3g), we investigated whether these compounds may be useful for the treatment of Gram-positive pathogens in clinical contexts. We first investigated their toxicological and chemical properties, including hemolysis, metal ion binding, genotoxicity, and chemical stability. Hemolysis is a severe toxic liability; metal iron binding may suggest compound reactivity, an undesirable property; genotoxicity often arises from alkylating agents; and chemical stability is predictive of compound availability in solution. We found that compounds 1 and 2 are non-hemolytic, do not chelate iron, are not genotoxic, are chemically stable in solutions of various pH, and are non-toxic when applied topically (1%) to ex vivo human skin and injected intraperitoneally (80 mg/kg) in mice (Extended Data Fig. 9 and Methods).
We next investigated the efficacy of compound 1 in the treatment of MRSA when administered topically and systemically to mice. We tested topical administration in a neutropenic mouse superficial skin infection1,6,14 model using an aminoglycoside and tetracycline-resistant clinical isolate of MRSA. Treatment with compound 1 decreased mean bacterial load by ~1.2 logs relative to vehicle (Fig. 5a), demonstrating efficacy similar to that of complestatin and corbomycin, two Gram-positive antibiotics recently discovered through phylogeny and evolution analyses6. We further tested systemic administration of compound 1 in a mouse neutropenic thigh infection model41 using an oxazolidinone-resistant clinical isolate of MRSA. Treatment with compound 1 at 80 mg/kg significantly decreased mean bacterial load by ~1.2 logs relative to vehicle treatment (Fig. 5b). The efficacy of compound 1 in a thigh infection model indicates that compounds 1 and 2, and structural analogs thereof, represent a promising chemical series for development as novel antibiotic candidates. Indeed, structure-activity relationship analyses indicate that the structure-activity space of our rationale of interest is not flat, supporting the suggestion that compounds 1 and 2 hold promise for further optimization (Supplementary Note 4 and Extended Data Fig. 10).
Fig. 5. In vivo efficacy.
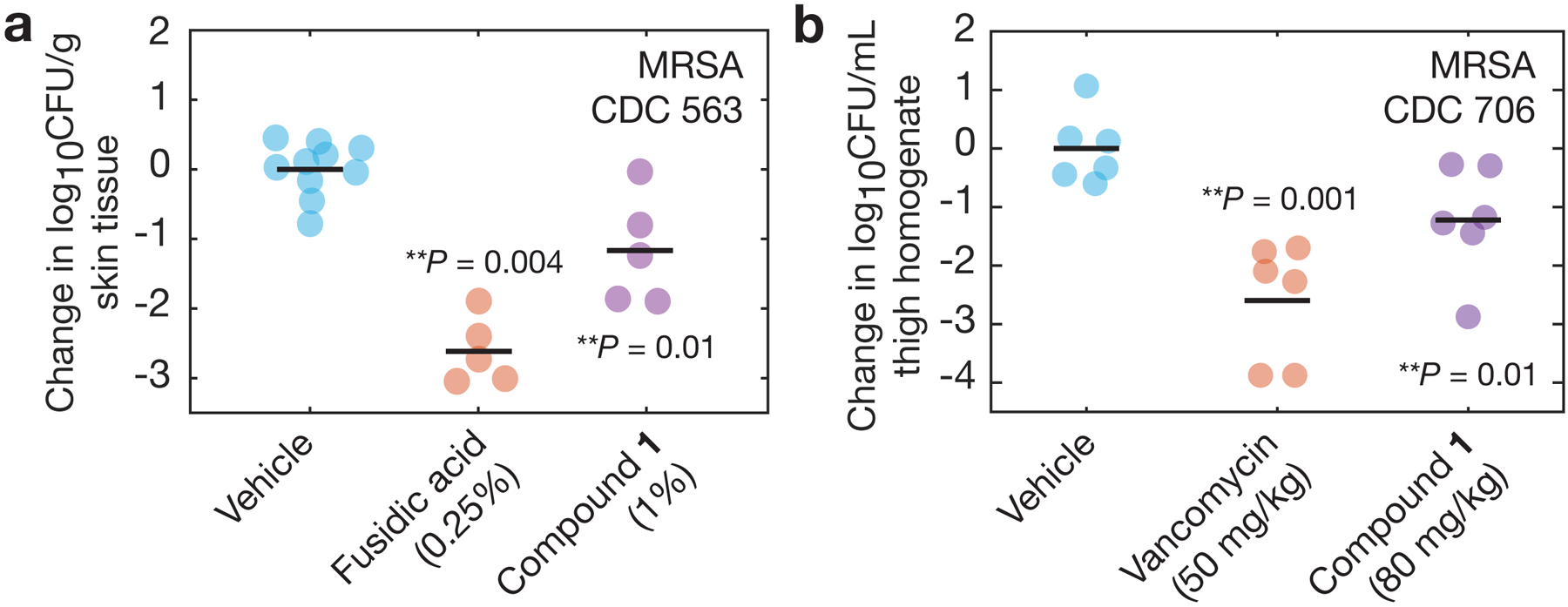
a, b, In vivo study of a neutropenic mouse wound infection model using MRSA CDC 563 (a) and a neutropenic mouse thigh infection model using MRSA CDC 706 (b), as described in Methods. In a, treatment was administered topically beginning 1 h post-infection and at 4, 8, 12, 20, and 24 h post-infection. n = 5 mice were used in each group, and the fusidic acid and compound 1 treatment arms were tested against vehicle treatment on separate occasions; points for both vehicle groups are overlaid. In b, treatment was administered single-dose intraperitoneally at 1 h post-infection, and n = 6 mice were used in each treatment group. Horizontal lines indicate mean log10 CFU/g values. One-sided, two-sample permutation test compared to vehicle treatment: **p ≤ 10−2.
Discussion
The need to discover novel structural classes of antibiotics is pressing given the antibiotic resistance crisis. This challenge has manifested in the 38-year interval between the introduction of the fluoroquinolone class of antibiotics in 1962 and the next new structural class, the oxazolidinones, in 2000.42 In the present study, we identified putative structural classes of antibiotics using graph-based explanations of deep learning model predictions of antibiotic activity and cytotoxicity in a space of 12,076,365 compounds. Our approach revealed multiple compounds with antibiotic activity against S. aureus. Of these, we found that one structural class exhibits high selectivity, overcomes resistance, possesses favorable toxicological and chemical properties, and is effective in both the topical and systemic treatment of MRSA in mouse infection models. Mechanistic and structure-activity relationship analyses additionally suggest that this structural class can be further optimized for higher selectivity against Gram-positive pathogens and increased permeability against Gram-negative pathogens.
This work demonstrates a deep learning approach to discovering structural classes of antibiotics, one which systematically builds on predictions of lone compound hits and allows for the efficient, substructure-based exploration of vast chemical spaces. In addition to down-sampling chemical space, a useful feature of our approach is the ability to automate the identification of unprecedented structural motifs, particularly in the context of deep learning models. This capability provides a source of chemical novelty that can suggest chemical spaces to explore and productively augment current discovery pipelines, for instance, by generating chemical fragments of interest for de novo design efforts. Importantly, this capability cannot be accomplished using alternative approaches, such as traditional QSAR analyses, that build on known scaffolds and do not identify novel scaffolds based on generalizing the patterns of molecular atoms and bonds in chemical structures28. We anticipate that a better understanding of graph-based rationale predictions could aid the discovery and design of additional, much-needed classes of antibiotics—for instance, those active against Gram-negative bacteria—as well as drug classes that target other biological processes and diseases, including anti-viral and anti-cancer drugs.
An alluring implication of the present study is that deep learning models in drug discovery can be made explainable. Indeed, a fundamental limitation of the black-box models that are commonly used in machine learning has been that such models typically do not provide information into the underlying decision-making processes20. Yet, model explainability may lead to generalizable insights that could better inform the use and development of next-generation approaches to exploring chemical spaces. Our study demonstrates that graph neural networks can be better understood and explained using graph-based searches for chemical substructure rationales that recapitulate model predictions. This provides meaningful chemical insights into what was learned by a particular model or ensemble of models. We anticipate that future work will build on this and similar approaches43,44 to further analyze and understand the predictions generated by deep learning models, for instance by using methods centered on perturbing model inputs45 for additional tests of explainability, as well as perturbing neural network structure for interpretability.
The approach presented here—which includes in silico predictions of compound cytotoxicity and stringent medicinal chemistry filtering steps that might inform work in other areas of drug discovery—could be further refined to consider more detailed representations of chemical space and factors important to antibiotic activity, such as protein binding in serum. By iterating the tasks of data generation, model retraining, and substructure identification, more complete representations of chemical space may be constructed, and promising predictions may be better identified and triaged. The discovery of structural classes using explainable deep learning could facilitate the process of identifying and optimizing potential leads by focusing on key scaffolds of interest, with which we may begin to efficiently explore novel chemical spaces and gain specific insights into the chemical substructures that underlie biological activity.
Methods
Deep learning model.
The deep learning approach used in this work builds on that applied in ref. 1. For each compound, RDKit was used to generate a graph-based molecular representation from the compound’s simplified molecular-input line-entry system (SMILES) string. A feature vector for each atom and bond in the compound was generated based on the following computable features: atom features include the atomic number, number of bonds for each atom, formal charge, chirality, number of bonded hydrogen atoms, hybridization, aromaticity, and atomic mass; bond features include the bond type (single, double, tripe, or aromatic), conjugation, ring membership, and stereochemistry. The model then implements the bond-based message-passing convolutional neural network described in refs. 1 and 11, which builds on the atom-based message-passing approach developed in ref. 10. Here, each message (a real number) associated with a bond is updated by summing the messages from neighboring bonds, concatenating the current bond’s message with the sum, and then applying a single neural network layer with a nonlinear activation function. After a fixed number of message-passing steps, the messages across the molecule are summed to produce a final message representing the molecule. This message is passed through a feed-forward neural network that outputs a prediction of the compound’s activity. For models predicting antibiotic activity, the final output is a real number between 0 (does not inhibit bacterial growth) and 1 (inhibits bacterial growth), describing the probability that the compound inhibits growth of S. aureus RN4220. For models predicting cytotoxicity, the final output is a real number between 0 (is not cytotoxic) and 1 (is cytotoxic), describing the probability that the compound is cytotoxic to HepG2 cells, HSkMCs, or IMR-90 cells. For models predicting proton motive force-altering activity, the final output is a real number between 0 (does not alter the proton motive force) and 1 (alters the proton motive force), describing the probability that the compound either increases or decreases DiSC3(5) fluorescence in S. aureus RN4220.
Model optimization.
Building on ref. 1, three model optimizations were employed to improve model performance. First, 200 additional molecule-level features computed with RDKit, as summarized in Supplementary Data 1, were added to the graph-based representation of each compound. This step was performed in order to provide additional information about global properties of each compound, which the local message-passing approach may not encapsulate. Second, we used hyperparameter optimization in order to select best-performing hyperparameters for each antibiotics model. For all Chemprop models with RDKit features predicting antibiotic activity, a limited grid search was used to find hyperparameters resulting in good performance; the parameter search ranges used are indicated in Supplementary Table 5. The same hyperparameters were used for the Chemprop models without RDKit features and without further optimization. For random forest classifiers based on Morgan fingerprints (radius = 2 and number of bits = 2,048), we used an exhaustive grid search in the preselected region of hyperparameter space indicated in Supplementary Table 5. We note here that, in contrast to our Chemprop embedding (which produces vectors of dimension NF, where N is the number of atoms in a molecule and F is the number of features), the Morgan fingerprint representation encodes only a count of F substructures and produces vectors of dimension F; for this reason, Morgan fingerprints are better suited as inputs to random forest models and the t-SNE analyses described below. For all Chemprop models predicting cytotoxicity, a more limited grid search suggested that the same hyperparameters as those for Chemprop models predicting antibiotic activity were suitable, and no further optimization was performed. For all models, the final hyperparameters used are tabulated in Supplementary Table 5. Finally, we used ensembling to increase the robustness of the model predictions. For each Chemprop model, 20 models were trained on a different random split of the training data. For benchmarking, the highest-scoring 10 models, according to the AUPRC on the withheld test set, were used in the ensemble. For predictions, all 20 models were used in the ensemble. We note here that training for all final models was performed using data from the full screening dataset of 39,312 compounds; requirements for structural novelty were enforced after making predictions (as described below), as opposed to removing known structural motifs from model training.
Model evaluation.
Screening data for 39,312 compounds were acquired experimentally, as described below. To evaluate model performance using the AUPRC, the training dataset was partitioned, such that 80% of the compounds (~31,647 compounds) were reserved for training and validation and 20% of the compounds (~7,911 compounds) were withheld for testing and calculation of PRCs. Active compounds in each group were distributed similarly as in the overall dataset (1.3% for antibiotic activity, 8.5% for HepG2 cytotoxicity, 3.8% for HSkMC cytotoxicity, and 8.8% for IMR-90 cytotoxicity). For each Chemprop model, training was performed for 30 epochs using random 80%−10%−10% training-validation-testing splits of the training subset, with each model being assigned a different random seed. All models were then pooled together to complete an ensemble. The ensemble of models was then applied to the withheld testing subset, and prediction scores of the ensemble were taken as the average of the prediction scores of all models in the ensemble. Random forest classifiers were trained using the software package scikit-learn. Bootstrapping with 100 subsamples, where each subsample had size equal to the test set, was used to calculate 95% AUPRC confidence intervals and variations of PRCs. The area under the receiver operating characteristic curve (AUROC) values shown in Supplementary Table 1 were calculated using the sklearn package in Python, and exact p-values for DeLong’s test of the statistical significance of the difference in AUROC values46 were calculated using a Python implementation47.
After selection of the best-performing type of model based on our benchmarks (for each predicted output property, an ensemble of Chemprop models with RDKit features), 20 models were retrained on the entire training dataset and applied to make predictions on a total of 12,076,365 compounds. While previous work has used a similar model for E. coli to predict the antibiotic activity of 107 million molecules in the ZINC15 database18, here we were interested in assessing compounds that could be readily procured, without recourse to in-house or specialized chemical synthesis. We therefore applied the final models to the entire Mcule purchasable database of 11,277,249 compounds (ver. June 2020)24, combined with an in-house database of 799,140 compounds from the Broad Institute. Prediction score thresholds for hits and non-hits were chosen to generate computationally tractable groups of ~103 compounds, but we note that the ability of our final models of antibiotic activity to discriminate between hits and non-hits is generally similar across different prediction score cutoffs (Extended Data Fig. 4).
Given the lower AUPRC values of all our models predicting cytotoxicity, as compared to our models predicting antibiotic activity, we aimed to further validate the performance of our cytotoxicity models. The final, trained cytotoxicity models were further benchmarked on two Tox21 datasets22 and a human metabolites database23, as described in Supplementary Note 1 and Supplementary Tables 7 and 8. Here, 7,151 compounds independently screened for cytotoxicity against HepG2 cells and 5,726 compounds screened for mitochondria toxicity from the Tox21 dataset were evaluated, and we found AUPRC values of ~0.3 for both datasets and all three Chemprop models (HepG2, HSkMC, and IMR-90). Consistent with the expected model performance, evaluating 3,126 human metabolites that are putatively non-cytotoxic resulted in false-positive rates of ~1% to ~10%, with lower false positive rates associated with higher cytotoxicity prediction score thresholds (Supplementary Note 1). Additionally, we procured and tested 100 structurally dissimilar compounds that were predicted to be cytotoxic by all Chemprop models (prediction score >0.4 across all models) and 90 compounds that were predicted to be non-cytotoxic (prediction score <0.05 across all models). Assessing these compounds tested the models’ generalizability, as the Tanimoto similarity values were <0.5 with respect to all cytotoxic compounds for any cell type in the training set (Supplementary Data 1). We found that 24 and 8 compounds, respectively, were cytotoxic to all three cell types (reducing cell viability by ≥10%), suggesting a working true positive rate of 0.75. Taken together, these findings support the suggestion that our models can be productively used to filter out cytotoxic compounds, thereby augmenting our antibiotic discovery efforts.
t-SNE and visualization.
For t-SNE analyses, we used sklearn.manifold’s TSNE() function in conjunction with Morgan fingerprint representations of all compounds (radius = 2 and number of bits = 2048) to visualize compounds in two dimensions. Following previous work1,14, the Jaccard distance, which is another name for Tanimoto distance for binary variables, was used as the distance metric; the Tanimoto distance is defined as Tanimoto distance = 1 – Tanimoto similarity, and the Tanimoto similarity between two fingerprints is given by the quotient of the number of 1-bits in the intersection of both fingerprints divided by the number of 1-bits found in their union. All calculations of Tanimoto similarity used in this work are based on Morgan fingerprint representations of all compounds (radius = 2 and number of bits = 2,048). The choice of the Jaccard metric for the t-SNE plot implies that the distance between points reflects the Tanimoto similarity of the corresponding compounds, with greater t-SNE distance indicating lower Tanimoto similarity1. We note here that the Tanimoto similarity depends on the global chemical structures of both inputs, and thus, does not necessarily quantify hits with common substructures or rationales. A perplexity parameter of 30 was found to produce clear visualizations and used for all plots. The initialization of embedding used was PCA.
Monte Carlo tree search for substructure rationales.
We employed graph neural network-based rationale explanations to determine, for each molecule with high predicted antibiotic activity, the smallest subgraph resulting in the molecule being classified as active (Fig. 3, Extended Data Figs. 5 and 6, and Supplementary Data 2). Formally, a rationale should satisfy three properties. First, its maximum size must be no more than a set number of atoms. Second, it must be a connected subgraph. Third, its predicted property must be greater than an activity threshold. We used Chemprop’s built-in “interpret” function to produce rationales yielding a minimal prediction score of 0.1. Given any input molecule with high prediction score, the rationale search proceeds by running a Monte Carlo tree search (MCTS; described below). An initial substructure size of 8 atoms was chosen to produce reasonably-sized outputs, a batch size of 500 parallel runs were used, and at each node, 10 rollout steps were performed wherein the rationale was expanded to distinct nodes. The expanded rationale was then scored with the same trained Chemprop models used to make the initial hit prediction. For searches in which no rationale producing a prediction score above 0.1 could be obtained after 10 minutes of search using all available CPUs on a Google Cloud c2-standard-60 instance, no rationales were deemed to have been computed for the hit of interest.
Finding the rationale of a molecule is a discrete optimization problem, which can be solved by the MCTS algorithm. The root of the search tree is the original active molecule and each state in the search tree is a subgraph derived from a sequence of bond or ring deletions. To ensure that each state is chemically valid and remains connected, we only allow deletion of one peripheral bond or ring from each state. A bond or ring is called peripheral if a molecule remains connected after deleting it.
During the search process, each state S in the search tree stores the following statistics:
N(S) is the number of times state S has been visited during the search process, and is a quantity used for exploration-exploitation tradeoff in the MCTS algorithm.
W(S) is the total long-term reward, which indicates how likely state S will eventually lead to a valid rationale.
R(S) is the predicted activity score of S, viewed as a subgroup and input to Chemprop in its own right, which indicates the immediate reward by choosing this state.
Guided by these statistics, the MCTS algorithm searches for rationales through an iterative process. Each iteration consists of two phases:
- Forward pass: The MCTS algorithm selects a path from the root (the starting compound) to a leaf state, Sleaf (a candidate rationale). At each intermediate state S, a deletion action is selected based on the mean action value:
where the parameter cs controls the trade-off between the long-term reward, W(s), and immediate reward, R(s). This parameter is set according to the well-known PUCT (predictor upper confidence bound applied to trees) equation48.
Backward pass: The state statistics are updated for each visited state in the selected path: N(S) ⟵ N(S) + 1; W(S) ⟵ W(S) + R(Sleaf).
Based on the backward pass update, W(S) represents the sum of the predicted activity of all valid rationales (leaf nodes) derived from state S. Different from the immediate reward R(S), W(S) measures long-term reward because it focuses on the predicted activity of the leaf nodes. The intuition is that the immediate reward is useful for filtering poor choices: states are unlikely to contain a rationale if R(S) is low. Among states with similar R(S) values, W(S) aids in selecting those with higher long-term reward. To better illustrate the MCTS algorithm, we provide an example in Extended Data Fig. 5 using compound 1: Extended Data Fig. 5a illustrates the MCTS forward pass, and Extended Data Fig. 5b shows a complete search path from the root to a rationale.
As described in the main text, we reasoned that further exploring the scaffolds of the rationales would better inform the chemical motifs underlying structural classes. The focus on scaffolds that are conserved across rationales is important, as we found that rationales were often large (>17 atoms), could contain most of the hit structures of interest, and may differ from hits and other structurally similar rationales by a small (<3) number of atoms. These observations imply that a direct matching of rationales will often result in groups of large rationales that may not be as productive or informative for structural class-based discovery efforts. Accordingly, here we have calculated the scaffold conserved between two randomly chosen rationales using RDKit’s FindMCS() function (as described in detail below) and assigned any remaining rationale to this scaffold if the scaffold contained at least 12 atoms—a threshold chosen to exclude small and generic substructures. We then repeated this process for at least 103 iterations, in order to sample the combinatorial space of all scaffolds defined by the rationales. Independent runs of this sampling procedure resulted in samples with similar scaffolds. All rationales and scaffolds presented in this work are provided as SMILES arbitrary target specification (SMARTS) strings in Supplementary Data 2.
Leave-one-out analyses.
Compounds in the training set were checked for the presence of the quinolone bicyclic core or β-lactam ring using RDKit’s FindMCS() function as below, with respect to the molecules described by two SMILES: “C1=CC=C2C(=C1)C(=O)C=CN2” (quinolone) or “C1CNC1=O” (β-lactam). Compounds (active or inactive) whose MCSs shared ≥11 (quinolone) or ≥4 (β-lactam) atoms with the respective substructures were withheld. The remaining training sets were checked visually to confirm the absence of any quinolone or β-lactam structure, respectively. Given the similarity in size of the remaining training sets to the full training set, we used the same Chemprop model hyperparameters as with the final model (Supplementary Table 5) and trained ensembles of 20 Chemprop models with RDKit features to make binary classification predictions of antibiotic activity. The models were then applied to make predictions of the antibiotic activities of the respective withheld quinolone and β-lactam compounds (Supplementary Data 2).
Maximal common substructure identification and analyses.
The importance of maximal common substructures and their identification have been acknowledged in prior studies27,49. As mentioned in the main text, we found that MCS-based approaches did not necessarily yield substructures that were diagnostic of high predicted antibiotic activity when applied to deep learning model predictions (Supplementary Note 2, Supplementary Table 9, and Extended Data Fig. 6). Indeed, Supplementary Note 2 shows that MCSs shared between hits can have antibiotic prediction scores <0.005, demonstrating that MCSs have low predictive capability as compared to rationales. In Supplementary Note 2, we were interested in quickly identifying maximal common substructures (MCSs) enriched in sets of compounds. Methods for addressing this problem remain limited: the mismatch tolerant matching mode of the fmcsR package49 allows for integer atom or bond mismatches that often effectively lower the atom threshold for MCS matches, while typical molecular fingerprinting methods rely on the deconstruction of a chemical structure into rigid substructures. We therefore employed a simple method. Given an integer N0 and a list, N, of compounds, we first chose, at random, two compounds n1 and n2 from N. Using RDKit’s FindMCS() function with the options of bondCompare set to rdFMCS.BondCompare.CompareOrderExact (bonds are equivalent if and only if they have the same bond type) and completeRingsOnly set to True (if an atom is part of the MCS and the atom is in a ring of the entire molecule, then that atom is also in a ring of the MCS), we computed the MCS, M, shared by n1 and n2. If the number of atoms of M was less than N0, then M was discarded and the combination of n1 and n2 not chosen again; otherwise, N was transversed, and whether or not each compound n ∈ N (n ≠ n1, n2) properly contained M was determined using the HasSubstructMatch() function in RDKit. If n properly contained M, then n was eliminated from N and said to be associated with M; otherwise, n remained in N. This process was repeated for a predetermined number of iterations or until a prespecified fraction of all compounds remained, which were not associated with any M. In the best case that all elements of N are associated with any MCS between any two members of N, this method requires |N|−1 MCS or substructure matching computations; in the worst case that no elements of N are associated with any suitable MCS, this method requires |N|(|N|−1)(|N|−2) MCS or substructure matching computations. We implemented this method in a Python notebook, available as described below in Code availability.
We applied the foregoing method on hits and non-hits with varying atom number thresholds and the number of iterations set to 5,000, which resulted in the identification of MCSs A1-A12, B1-B12, C1-C12, and D1-D12 (Extended Data Fig. 6). We note here that increasing the number of iterations did not substantially change the MCSs identified. MCSs A1-A12, B1-B12, C1-C12, and D1-D12 are provided as SMARTS strings in Supplementary Data 2.
The MCS prediction scores shown in Extended Data Fig. 6 were calculated by calculating Chemprop model predictions for the SMARTS strings computed above, viewed as inputs in their own right. For a small subset of MCSs, the corresponding SMARTS strings were invalid inputs due to ambiguity in the bond type (single or double) of specific bonds. In these cases, the bond type was manually chosen either as single or double bonds to create valid SMILES strings, which were then inputted into the Chemprop models to generate MCS prediction scores.
Computational hit analyses.
The PAINS and Brenk alerts29,30 refer to chemical substructures that may be promiscuous or toxic. PAINS and Brenk substructures were calculated for each compound passing antibiotic activity prediction score and cytotoxicity prediction score thresholds (Fig. 2) using RDKit’s FilterCatalogParams.FilterCatalogs.PAINS and FilterCatalogParams.FilterCatalogs.BRENK classifications, respectively. We calculated Tanimoto similarity scores of each remaining compound with respect to all active compounds in the training set using the FingerprintSimilarity() function in RDKit, in conjunction with Morgan fingerprint representations of all compounds (radius = 2 and number of bits = 2048), as mentioned above. Compounds were then checked for the presence of the β-lactam ring or the quinolone bicyclic core using RDKit’s FindMCS() function as above, with respect to the molecules described by two SMILES: “C1CNC1=O” (β-lactam) or “C1=CC=C2C(=C1)C(=O)C=CN2” (quinolone). Compounds whose MCSs shared ≥4 (β-lactam) or 11 atoms (quinolone) with the respective substructures were discarded. The medicinal chemistry property predictions shown in Supplementary Table 3 were performed using SwissADME50. Of note, Lipinski’s rule of five31, which is often used as a guideline for oral bioavailability but also viewed as a guideline for druglikeness, demands that a compound possesses (1) number of H-bond donors ≤5; (2) number of H-bond acceptors ≤10; (3) molecular weight ≤500 Da; and (4) an octanol-water partition coefficient (log P) ≤5. The Ghose criteria32 for druglikeness demand that a compound possesses (1) molecular refractivity ≥40 and ≤130; (2) number of atoms ≥20 and ≤70; (3) an octanol-water partition coefficient (log P) ≥−0.4 and ≤5.6; and (4) a molecular weight ≥160 and ≤480.
Chemical compound sourcing.
In order to systematically source compounds for testing, we developed a custom Python script which queries the PubChem database for vendors of each compound, according to its SMILES string. Of note, while the Mcule purchasable database contains compounds that are readily purchasable, compounds may not be purchasable from Mcule. The query results were tabulated for all compounds, and we shortlisted a subset of compounds which were available in high purity (>90%) and could be purchased from common vendors. Compounds were then sourced from multiple suppliers, including ChemBridge (San Diego, CA), Vitas-M (Hong Kong, China), and Enamine (Kyiv, Ukraine); catalogue details for each procured compound are provided in Supplementary Data 2.
Bacterial strains.
A list of all common bacterial strains used in this study is provided in Supplementary Table 6. Main strains include Staphylococcus aureus RN4220, FPR3757 (MRSA USA300; ATCC BAA-1556), Bacillus subtilis 168 (ATCC 23857), Escherichia coli BW25113, Acinetobacter baumannii ATCC 17978, and Pseudomonas aeruginosa PAO1. The resistance phenotype of S. aureus FPR3757 was verified by comparing growth inhibition against S. aureus RN4220 on 2 and 4 μg/mL oxacillin salt-containing Mueller Hinton agar (Becton Dickinson 225250; oxacillin, MilliporeSigma 28221). Additional bacterial isolates, as shown in Supplementary Table 4, were obtained from the Centers for Disease Prevention’s AR Isolate Bank (Atlanta, Georgia).
Bacterial culture and growth.
All cells were grown in liquid LB medium (Becton Dickinson 244620). LB media containing 1.5% Difco agar (Becton Dickinson 244520) was used to grow individual colonies. Cells were grown from single colonies aerobically at 37°C in 14 mL Falcon tubes using 2 mL working volumes without antibiotic selection. Cell cultures were incubated in a light-insulated, humidity-controlled incubation chamber with shaking at 300 rpm.
Antibiotics.
Unless otherwise stated, stock solutions and serial dilutions of all antibiotics were freshly prepared in dimethyl sulfoxide (DMSO; MilliporeSigma D5879) before each experiment. Stock solutions and serial dilutions of kanamycin, ampicillin, fosfomycin, vancomycin, and teicoplanin were prepared with ultrapure Milli-Q water. Stock solutions of ciprofloxacin and tetracycline were prepared by dissolving in weak acid (0.1 M HCl), then diluted in ultrapure Milli-Q water.
Compound screening and antibiotic activity training data generation.
The compound library used in this work builds on the one used to screen for growth inhibition in E. coli in previous work from our lab51. Compounds were sourced and dissolved in DMSO to generate working stocks of 5 mM concentration. Stock solutions were maintained at −20°C for long-term storage. S. aureus RN4220 was grown overnight in LB media as described above, then diluted 1:10,000 in fresh LB and plated into either (1) 96-well flat-bottom clear plates (Corning 9018) using 100 μL final working volumes or (2) 384-well clear plates (Corning 3702) using 50 μL final working volumes. Compounds were added to a final concentration of 50 μM and automatically mixed to facilitate homogeneous distribution, and plates were incubated at 37°C without shaking overnight (16 to 24 h) in sealed plastic bags. The optical density (OD600) was then read using a SpectraMax M3 plate reader and SoftMax Pro software (version 7.1, Molecular Devices, San Jose, CA) to quantify cell growth. Plate data were normalized by the interquartile mean of each plate to calculate relative growth. All screens were performed in biological replicate. After screening all 39,312 compounds in this way, a subset of 51 randomly chosen active compounds were rescreened for secondary validation according to the same procedures described above. The replicate results for all 51 active compounds were consistent with the results of the main screen. Furthermore, we note here that the Pearson’s correlation coefficient between relative growth values of replicates in the screen, respectively, was R = 0.8 (p < 10−14), demonstrating good reproducibility between replicates (Fig. 1b).
Cytotoxicity screening and testing.
Cytotoxicity in human cells was assayed using a resazurin (alamarBlue) assay. HepG2 cells were obtained from ATCC (ATCC HB-8065), passaged <10 times, and grown to log phase in high-glucose Dulbecco’s Modified Eagle Medium (DMEM; Corning 10–013-CV) supplemented with 10% fetal bovine serum (FBS; ThermoFisher 16140071) and 1% penicillin-streptomycin (ThermoFisher 15070063). HSkMCs were obtained from ATCC (ATCC PCS-950–010), passaged <5 times, and grown to log phase in mesenchymal stem cell basal medium for adipose, umbilical and bone marrow-derived MSCs (ATCC PCS-500–030) supplemented with ATCC’s primary skeletal muscle growth kit (ATCC PCS-950–040) and 1% penicillin-streptomycin. IMR-90 cells were obtained from ATCC (ATCC CCL-186), passaged <10 times, and grown to log phase in Eagle’s Minimum Essential Medium (EMEM; ATCC 30–2003) supplemented with 10% FBS and 1% penicillin-streptomycin. Cells were tested for mycoplasma contamination by the supplier, and the HepG2 and IMR-90 cell lines were authenticated by the supplier using short tandem repeat profiling. For IMR-90 cytotoxicity, data for a subset of 2,335 compounds, corresponding to the Pharmacon and natural products library used to screen for growth inhibition in E. coli in previous work from our lab1, have previously been generated by us for cells treated with 0.5% DMSO15; as the experimental conditions of the screen are similar to those considered here, these data were used and expanded upon for the current IMR-90 dataset in lieu of screening the same subset of compounds again. For all other compounds or cell types, cells were plated into either (1) 96-well clear flat-bottom black tissue-culture-treated plates (Corning 3603) at a density of 104 cells/well using 100 μL working volumes or (2) 384-well clear flat-bottom black tissue-culture-treated plates (Corning 3764) at a density of 5,000 cells/well using 30 to 50 μL working volumes, then incubated at 37°C with 5% CO2. Twenty-four h after plating, test compounds were added to a final concentration of 10 μM (final DMSO concentration of 0.5%) and automatically mixed to facilitate homogeneous distribution of compounds. Cells were re-incubated for either 2 days (HepG2 and HSkMCs) or 3 days (IMR-90), with the incubation period chosen to reflect the relative timescales of cell doubling for each cell type, after which resazurin (MilliporeSigma R7017) was added to each well to a final concentration of 0.15 mM. After an additional 4 to 24 h of incubation, the fluorescence excitation/emission at 550/590 nm was read using a SpectraMax M3 plate reader or an EnVision plate reader and EnVision Workstation software (version 1.14.3049.1193, PerkinElmer, Waltham, MA). Plate data were normalized by the interquartile mean of each plate to calculate relative cell viability (Fig. 1d,f,h). All screens were performed in biological replicate. We note here that the Pearson’s correlation coefficients between relative cell viability values of replicates in the screens, respectively, were R = 0.9 (HepG2), R = 0.96 (HSkMC) and R = 0.81 (IMR-90; p < 10−14 for all cell types), demonstrating good reproducibility between replicates (Fig. 1d,f,h). For testing cytotoxicity model predictions, 190 compounds were procured from commercial vendors and assayed in the same manner for each cell type, with the exception that relative viability values were normalized by the mean of two DMSO (final concentration, 0.5%) controls.
MIC and bacterial growth inhibition assays.
We used the microbroth dilution method for determining MICs in this study, including the values shown in Fig. 3g. A 1:10,000 dilution of overnight cell culture in fresh LB was plated into 96-well flat-bottom clear plates using 99 μL working volumes. One μL of a serial dilution of compound in DMSO was added to each well, with two-fold serial dilutions across wells. Plates were sealed with breathable membranes (MilliporeSigma Z763624) and incubated at 37°C with shaking at 900 rpm. The MIC was determined as the concentration of compound resulting in inhibited growth of the culture (OD600 < 0.2) after overnight (16 to 24 h) incubation. Where applicable, FBS was added to fresh LB to a final concentration of 10% before addition of bacterial inocula and compounds. All MIC experiments were replicated at least in biological duplicate, and optical density was read using a SpectraMax M3 plate reader.
Cytotoxicity IC50 assays.
Cells were cultured as described above in Cytotoxicity screening and seeded at a density of ~2×104 cells/well into 96-well clear flat-bottom black tissue-culture-treated plates. For each compound, 1 μL of two-fold serial dilutions in DMSO was added to 99 μL of medium containing cells. Addition of 1 μL DMSO to 99 μL of medium containing cells was used as a negative control, and doxorubicin (Cayman Chemical Company 15007) was used as a positive control. To facilitate comparison across cell types, plates for all cell types were incubated for ~2 days. IC50 values were calculated as the minimal concentration used for which the fluorescence intensity values were decreased by at least 50% from those of negative controls (DMSO), with baseline values being those of blank wells containing medium with resazurin only. The effects of vehicle (1% DMSO) were found to be minimal (<10% decrease) on cell viability, as determined by comparing values from negative controls to those of untreated wells containing cells only. Experiments were performed at least in biological replicate on two independent occasions.
Bacterial time-kill assays and CFU measurements.
Cells were diluted 1:10,000 or 1:100 from an overnight culture into fresh LB and plated into 96-well flat-bottom clear plates using 99 μL working volumes. Plates were then sealed with breathable membranes, and cells were grown to early exponential phase, OD600 ~ 0.01 or 0.1—corresponding to ~106 or ~107 CFU/mL—in a 37°C incubator with shaking at 900 rpm. Unless otherwise indicated, 1 μL of compound in two-fold serial dilutions in DMSO was then added to each well to the final concentrations indicated, and bacterial cell cultures were sealed and re-incubated at 37°C with shaking at 900 rpm. At the indicated times, cells were removed from incubation, serially diluted in room-temperature LB, and spotted on LB agar. We performed serial dilutions of cells in LB instead of other media, like PBS, in order to better control for osmolarity and nutrient shifts (as we have previously done34,35). Petri dishes containing plated cells on LB agar were allowed to dry at room temperature before stationary incubation at 37°C overnight (16 to 24 h). CFUs were determined by manual counting, and all measurements are based on counts containing at least six colonies.
Serial passaging experiments.
S. aureus RN4220 was diluted 1:10,000 from an overnight culture in fresh LB and plated into 96-well flat-bottom clear plates using 99 μL working volumes. One μL of a serial dilution of compound in DMSO was added to each well, with two-fold serial dilutions across wells. Cells were incubated at 37°C with shaking at 900 rpm. After 24 h, plates were read using a SpectraMax M3 plate reader, and cells that grew (OD600 > 0.3) in the presence of the highest concentration of compound were diluted into fresh LB at the optical density equivalent of 1:10,000 of an overnight culture. Cells were then plated using 99 μL working volumes into 96-well flat-bottom clear plates. One μL of a serial dilution of compound in DMSO was again added to each well, with two-fold serial dilutions across wells, and this process was repeated every 24 h over 30 days. Stock serial dilutions in DMSO of all compounds used for passaging were prepared at day 0 and stored at −20°C. For all compounds tested, 64 or 128× baseline MIC was the highest concentration used. After 30 days, cells that grew in the presence of the highest concentration of compound were streaked on blank LB agar plates to isolate individual colonies. Individual colonies picked from LB agar plates were grown in blank LB overnight, serial dilutions of all tested compounds were prepared fresh, and the MIC values were determined again. MIC values were compared to those determined using overnight cultures of non-passaged S. aureus RN4220 cells, in order to confirm MIC changes where applicable. As a negative control, cells were serially passaged in 1% DMSO as described above, and without selection, for 30 days, and all MICs were confirmed to be identical to those of the ancestral strain in two biological replicates.
Suppressor mutant generation experiments.
S. aureus RN4220 was picked from single colonies and grown overnight in fresh LB. For each replicate in each tested condition, 1 mL of overnight culture (~109 CFU) was aliquoted and centrifuged at 3700 × g for 5 min. The cell pellet was resuspended to a final volume of 50 μL in fresh LB, then pipetted onto the surface of LB agar plates containing the indicated concentrations of compounds. Cells were then spread using a bent, sterile inoculating loop, and plates were dried and inverted before stationary incubation at 37°C for 5 days. At the end of 5 days, plates were removed from incubation, and colonies that grew on each plate were picked and streaked on fresh compound-containing LB agar plates (up to 6 colonies streaked per plate). These plates were then incubated overnight in a stationary incubator at 37°C, and bacterial growth was assessed by eye.
Genomic sequencing.
For serial passaging experiments, passaged cells were streaked onto blank LB agar as described above. Following MIC determination and validation, cells from the same liquid culture were struck again on blank LB agar and incubated overnight. Single colonies were picked and grown in 2 mL blank LB overnight at 37°C with shaking at 300 rpm. One mL of cell culture was then aliquoted and pelleted by centrifugation at 3700 × g for 5 min. The supernatant was discarded, and cell pellets were frozen and kept at −80°C until sequencing. For suppressor mutant generation experiments, plates with bacterial growth after the last overnight incubation step were taken, and bacterial cells were sampled from each streak and used to inoculate 2 mL of fresh LB. Liquid cultures were then incubated overnight at 37°C with shaking at 300 rpm, and cell pellets were prepared as described above for serially passaged cells.
On the day of sequencing, gDNA was extracted after pre-treating cells with lysostaphin (MilliporeSigma SAE0091) for 30 min, using a Qiagen DNeasy Blood and Tissue Kit (Qiagen 69504) according to the manufacturer’s instructions. Illumina (San Diego, CA) DNA library preparations were used following the manufacturer’s instructions. gDNA extraction and sequencing were performed at the Microbial Genome Sequencing Center (Pittsburgh, PA).
Sequencing analysis.
Sequencing results were analyzed by aligning each read set to the finished RN4220 genome (GCF_018732165.1) using the BWA-MEM algorithm. Pilon52 was used to call variants for each read set. Variants with low mapping quality (<10) were filtered from the final results (Supplementary Data 3).
Phase-contrast microscopy.
As in previous work34–36, microscopy experiments were performed with cells sandwiched between agarose pads and glass slides unless otherwise stated. B. subtilis 168 was grown from a 1:100 dilution of an overnight culture in 14-mL Falcon tubes to early exponential phase (OD600 ~ 0.1), and cells were treated with the indicated compounds for the indicated durations at 37°C with shaking at 300 rpm. Cells were concentrated by centrifugation at 7000 × g for 5 min and resuspended in a smaller volume of supernatant. We placed 2 μL of the resuspended bacterial culture between 3”×1”×1” microscope slides (Fisher Scientific 125444) and 1 mm thick agarose (1.5%) pads made from growth media (agarose: MilliporeSigma A2576). Cells were imaged immediately afterward at room temperature using a Zeiss Axioscope A1 upright microscope equipped with a Zeiss Axiocam 503 camera and a Zeiss 100× NA 1.3 Plan-neofluar objective (Zeiss, Jena, Germany). Images were recorded using Zen Lite Blue (version 2.3, Zeiss) software. All microscopy experiments were replicated at least in biological duplicate.
DiSC3(5) fluorescence.
S. aureus RN4220 and B. subtilis 168 were picked from individual colonies and grown in liquid LB overnight at 37°C with shaking at 300 rpm. Cells were then diluted 1:100 from the overnight cultures into liquid LB and grown to mid-log phase, OD600 ~ 0.5, at 37°C with shaking at 300 rpm. DiSC3(5) (Invitrogen D306) was dissolved in DMSO and added to liquid cultures at a final concentration of 1 μM. After additional incubation in the presence of DiSC3(5) for 1 to 2 h, cells were plated in 200 μL working volumes in black, opaque flat-bottom 96-well plates, after which fluorescence was measured every 10 to 30 s at an excitation/emission of 622/670 nm using a SpectraMax M3 plate reader. Cells were then treated with DMSO (1%) as a negative control, valinomycin (MilliporeSigma V0627) and nigericin (MilliporeSigma N7143) at a final concentration of 1 mM as positive controls, and compounds 1 and 2 at a final concentration of 32 μg/mL. Fluorescence was measured immediately following treatment according to the same specifications as above.
pH-dependent growth inhibition.
S. aureus RN4220 was picked from individual colonies and grown in liquid LB overnight at 37°C with shaking at 300 rpm. Cells were then diluted 1:10,000 into liquid LB titrated to pH 8.0 and 9.0 using ammonium hydroxide (MilliporeSigma 09859), and MIC values were determined as detailed above in MIC and bacterial growth inhibition assays.
Membrane-specific activity model development.
Bacterial membrane-sensitive mechanisms of action, such as that of compounds 1 and 2, have often been de-prioritized in antibiotic drug discovery due, in part, to potential lack of selectivity39. In order to study the generality of this mechanism of action, we further quantified and trained Chemprop models to predict membrane-specific activity. Additional screens of membrane disruption for a subsample of 475 active antibacterial compounds emerging from our initial screen (Fig. 1b), used to treat exponentially-growing S. aureus cells at a final concentration of 50 μM, indicate that 35 compounds (7.3%) induce alterations in the proton motive force, as measured by relative changes of ≥30% in DiSC3(5) fluorescence (Supplementary Data 4). In brief, this subset of 475 active compounds, comprising all compounds for which additional compound stock was available, was procured at 10 mM for stock solutions in DMSO. S. aureus RN4220 was picked from individual colonies and grown in liquid LB overnight at 37°C with shaking at 300 rpm. Cells were then diluted 1:100 from the overnight cultures into liquid LB and grown to mid-log phase, OD600 ~ 0.8 to 1.0, at 37°C with shaking at 300 rpm. As above, DiSC3(5) was dissolved in DMSO and added to liquid cultures at a final concentration of 1 μM. After additional incubation in the presence of DiSC3(5) for 1 h, cells were plated in 20 μL working volumes in black, clear- and flat-bottom 384-well plates, after which each of the 475 procured compounds were immediately added to a final concentration of 50 μM. After a 5 min incubation at room temperature, fluorescence was measured at an excitation/emission of 625/660–720 nm using a GloMax Discover microplate reader and GloMax Discover software (version 4.0.0, Promega, Madison, WI). Relative DiSC3(5) fluorescence was calculated by normalizing with respect to values for vehicle (DMSO) treatment, and experiments were performed in biological duplicate (Supplementary Data 4).
Compounds increasing or decreasing DiSC3(5) fluorescence by 30% relative to DMSO control were declared as active (35 compounds). This suggests that alteration of the proton motive force is not necessarily a widespread mechanism of action of antibacterial compounds. Building on these data, we trained Chemprop models to predict the probability that any given compound induces alterations in the proton motive force. The 35 compounds declared active, together with the inactive tested compounds and all inactive antibacterial compounds (which were assumed to not alter proton motive force), were used to train an ensemble of 20 Chemprop models. Model hyperparameters were determined using Bayesian hyperparameter optimization (Chemprop’s “hyperopt” function) with ten iterations (Supplementary Table 5). The trained models were then applied to make binary classification predictions on the Broad Institute database of 799,140 compounds. We identified 5,759 compounds (0.72% of the Broad Institute database) with activity prediction scores greater than the prediction scores of compounds 1 and 2 (0.040 and 0.043, respectively); these compounds were then shortlisted and filtered to ensure that the Tanimoto similarity with respect to the 35 active training set compounds was <0.5, with no other filters applied. Fifteen readily available filtered compounds were procured from the Broad Institute and tested as above to determine proton motive force-altering activity (Supplementary Data 4). Defining active compounds as above, we found that these models have an encouraging working positive predicted value of 0.4, supporting the notion that the membrane-specific mechanism of action of compounds 1 and 2 might be accurately predicted from chemical structure (Supplementary Data 4). We anticipate that these and additional models based on bacterial cytological profiling will guide further in silico screens of membrane-targeting compounds.
Hemolysis measurements.
Following previous work53, for the hemolysis experiments shown in Extended Data Fig. 9, whole human blood containing EDTA (Innovative Resarch IWB1K2E) was centrifuged at 120 × g at 4°C for 5 min and resuspended in Dulbecco’s PBS (DPBS; VWR 02–0119-0500). These washing steps were repeated until the supernatant was clear (at least 10 times). Red blood cells were then resuspended in DPBS to a density of 5 × 108 cells/mL, and 100 μL of cells was plated into each well of a 96-well round-bottom clear plate (Corning 3788). Compounds were added to the indicated final concentrations, and DMSO was used as a vehicle. Samples were incubated for 1 h at 37°C without shaking, after which plates were centrifuged at 1500 × g at room temperature for 5 min to pellet cells. 60 μL of the supernatant from each sample was then transferred to a 96-well flat-bottom clear plate, and the optical density was read at 405 nm using a SpectraMax M3 plate reader to quantify the amount of soluble hemoglobin. Fractional hemolysis was determined by linearly interpolating absorbance values with respect to a positive control (saturation with 10% Triton X-100) and a negative control (1% DMSO vehicle). We found that treatment with compounds 1 and 2 did not induce substantial hemolysis up to a final concentration of 128 μg/mL, the highest tested (64× MIC; Extended Data Fig. 9).
Iron chelation measurements.
In Extended Data Fig. 9, iron chelation was assayed based on the ferrous iron chelating assay kit from ZenBio (AOX-15) with modifications. Briefly, FeSO4 stock solutions were prepared by adding 1.8 mL of ultrapure Milli-Q water to 5 mg FeSO4. Ferrozine stock solution was prepared by adding 400 μL of ultrapure Milli-Q water to 5 mg ferrozine. Both stock solutions were diluted 100-fold in water, and 99 μL of working FeSO4 solution was plated into each well of a 96-well flat-bottom clear plate. One μL of test compound in DMSO or EDTA (MilliporeSigma E7889) was added into each well to the final concentrations indicated and mixed via pipette. After 10 min incubation at room temperature, 100 μL of working ferrozine solution was added to each well, and the plate was incubated again at room temperature for 10 min. The absorbance at 562 nm was then read using a SpectraMax M3 plate reader. Fractional ferrous iron chelating activity was determined by linearly interpolating absorbance values with respect to untreated and EDTA-treated (128 μg/mL final concentration) controls. We found that treatment with compounds 1 and 2 did not result in substantial iron chelation up to a final concentration of 128 μg/mL (Extended Data Fig. 9).
Bacterial Ames assay for genotoxicity.
For the mutagenesis experiments shown in Extended Data Fig. 9, a 5041 Modifed Ames ISO from Environmental Bio-Detection Products, Inc. was used following the manufacturer’s instructions. Briefly, Salmonella typhimurium TA100 was grown overnight (16–18 h) at 37°C with shaking at 300 rpm and treated with the provided exposure media and compound samples at the final concentrations indicated. Treatment with the provided sodium azide, a mutagen, was used as a positive control. Cells were added to the provided reversion solution, and each sample was aliquoted into 48 wells of 96-well plates. Plates were incubated at 37°C for 3 days, after which the number of revertant (yellow-colored) wells corresponding to each sample was counted by eye. Additionally, we verified that each test compound did not inhibit the growth of S. typhimurium TA100. An overnight bacterial culture was diluted 1:10,000 in LB medium and plated using 99 μL working volumes into the wells of a 96-well flat-bottom clear plate. One μL of two-fold dilutions of each test compound in DMSO, starting from a final concentration of 500 μM, was added across wells, and plates were sealed and incubated overnight at 37°C to determine bacterial growth. In contrast to treatment with 5 μg/mL sodium azide, a potent mutagen, treatment with compounds 1 and 2 up to a final concentration of 128 μg/mL did not induce substantial reversion of bacterial cultures (Extended Data Fig. 9).
Chemical stability measurements.
To assess the chemical stability of compound 1 in various solutions, we injected the compound into acidic (pH 5.0), neutral (pH 7.0), and basic (pH 10.0) media. Acetate buffer (0.1 M, pH 5.0), PBS (pH 7.1), and glycine buffer (0.08 M, pH 10.0) were prepared as aqueous solutions using ultrapure Milli-Q water. Ten μL of a 500 μM stock solution of compound 1 in DMSO was then added to 990 μL of buffer in 1.5 mL centrifuge tubes (final compound concentration, 5 μM), vortexed, and incubated at 37°C with shaking at 300 rpm and protected from light for 0, 45, or 120 min. Samples were then flash-frozen on dry ice and kept at −80°C until processing at the Harvard Center for Mass Spectrometry using LC-MS, as described in Liquid chromatography-mass spectrometry. We found that compound 1 was stable across the three buffers used at 0, 45, and 120 min after compound addition, with no substantial decrease in the concentration of free compound across all timepoints measured (Extended Data Fig. 9).
Liquid chromatography-mass spectrometry.
All reagents used were LC-MS-grade. For sample preparation, 100 μL of each sample was mixed with 100 μL of water containing 10 μM of compound 2 as an internal standard. Next, 800 μL of methanol was added, and samples were stored overnight at −20°C. Samples were centrifuged for 10 min at max speed at 4°C, and the supernatants were transferred to microcentrifuge tubes and dried under N2 flow. Dried samples were resuspended in 100 μL of acetonitrile:water (1:1 w/w) and centrifuged for 10 min at max speed at 4°C. The supernatants were then transferred to microinserts. A standard curve was prepared using seven 1/3 dilution series of a 100 μM solution of compound 1 in water. One hundred μL of each standard was prepared similarly to samples, and the lower limit of quantification was determined to be 150 nM.
All samples were run on an Agilent Triple Quadrupole. The column used was Phenomenex Kinetex EVO C18, 2.6 μm, 100 Å, 150 × 2.1 mm. The source used was AJS ESI negative. MS parameters were as follows: gas 350°C at 9 L/min, nebulizer 30 psi, sheath 350°C at 10 L/min, nozzle at 1300 V, capillary at 2200 V. The mobile phases were A: water and 0.1% NH4OH and B: acetonitrile, 0.03% NH4OH. The following gradient was used: 5 min at 0% B, then to 50% B at 5 min, then to 100% B at 7.01 min, followed by 0% B at 12.01 min. The column was then equilibrated at 0% B for 5 min. The flow rate was 0.2 mL/min, the column was maintained at 35°C, and 5 μL of each sample was injected.
Ex vivo human skin toxicity.
WoundSkin 11 mm models were procured from Genoskin (Salem, MA) from a 46-year-old Hispanic female donor. Upon arrival, 1 mL of the provided ex vivo culture medium was added to each well containing WoundSkin sample and samples were incubated at 37°C with 5% CO2 for 1 h. Compound 1 was prepared as a stock solution in DMSO, then formulated using 50% polyethylene glycol 300 (PEG300, MilliporeSigma 202371) and 50% water for injection as solvent. Thirty μL of a 1% formulation of compound 1 was administered topically by pipetting directly onto each of six WoundSkin models. As controls, 30 μL of a corresponding formulation of DMSO was administered topically by pipetting directly onto each of six WoundSkin models. All models were incubated at 37°C with 5% CO2 for 24 h and assessed for typical signs of toxicity, including tissue death, skin discoloration, and irritation. Consistent with the predictions of our cytotoxicity models and its characterized selectivity profile, we found that compound 1 was non-toxic when applied topically (1%) to ex vivo human skin (Extended Data Fig. 9).
In vivo mouse toxicity.
Studies were performed at the Wyss Institute at Harvard in accordance with protocol IS00000852–6, approved by the Harvard Medical School Institutional Animal Care and Use Committee and the Committee on Microbiological Safety. Female C57BL/6J mice, 6–8 weeks old, 22 ± 2 g, received from The Jackson Laboratory, were quarantined at least 2 days prior to use. Compound 1 was prepared as a stock solution in DMSO, then formulated using PEG300 and water for injection as solvent so that the final formulation was 10%:45%:45% DMSO stock of compound 1:PEG300:water for injection (w/w). The formulation was injected intraperitoneally to a final concentration of 80 mg/kg, and mice were observed for at least 24 h for typical signs of toxicity, including impaired movement, lethality, and irritation. We found that compound 1 was well-tolerated after intraperitoneal injection in all mice, with results representative of three mice (n = 3) injected with compound 1.
Mouse topical wound infection model.
Studies were performed at the Wyss Institute at Harvard in accordance with protocol IS00000852–6, approved by the Harvard Medical School Institutional Animal Care and Use Committee and the Committee on Microbiological Safety. Female C57BL/6J mice, 6–8 weeks old, 22 ± 2 g, received from The Jackson Laboratory, were quarantined at least 2 days prior to use. Animals were housed in a facility maintained at 20–26°C ambient temperature, 40–65% relative humidity, and a 12:12 light-dark cycle. Enrichment devices were included in the animal environments as required and changed bi-weekly. As illustrated in Extended Data Fig. 9, mice were rendered neutropenic by a 0.2 mL intraperitoneal injection of cyclophosphamide (Cytoxan) at 150 mg/kg (Day −4) and at 100 mg/kg (Day −1) pre-infection. Each mouse was anesthetized and kept sedated during the initial procedure under isoflurane vapors (3%). For each mouse, the fur on the back dorsal surface was shaved, then sterilized with alcohol. An area of the shaved skin was abraded using a sterile gauze pad. Following this procedure, the skin became visibly damaged and was characterized by reddening and glistening, but no bleeding. The skin was then wiped with an alcohol swab and allowed to dry completely. The resulting surface area for infection and treatment was ~1.5 cm2. The S. aureus AR Bank # 0563 isolate was struck onto LB agar plates from a freezer stock and incubated at 37°C overnight. Overnight cultures were grown from single colonies in LB to 109 CFU/mL (OD600 ~ 1), then diluted in LB to achieve the indicated inoculum concentration. The diluted overnight culture was serially diluted in PBS and plated onto LB agar to determine input CFU. Five μL of the diluted culture, corresponding to an inoculum of ~105 CFU, was placed on the skin to initiate the bacterial infection. Treatment was initiated at 1 h post-infection, then continued at 4, 8, 12, 20, and 24 h post-infection. Compound 1 (1% final concentration) was prepared as a stock solution in DMSO, then formulated using PEG300 and water for injection as solvent so that the final formulation was 10%:45%:45% DMSO stock of compound 1:PEG300:water for injection (w/w). A 1% formulation of compound 1 was chosen for our preliminary experiments, as higher concentrations of compound 1 were found to result in cloudy suspensions, suggestive of limits to compound solubility. Fusidic acid (0.25% final concentration) was used as a positive control, and appropriate vehicle treatments of DMSO:PEG300:water for injection (10%:45%:45%) were included. For each treatment, ~40 μL of formulation was applied topically on the infected skin at the indicated times. At ~25 hrs post-infection (~1 h following the last topical treatment), all mice were euthanized by CO2 asphyxiation, and wounds were wiped with an alcohol pad, excised, weighed, rinsed in sterile saline, and homogenized together with 3 mL of sterile PBS using a Polytron PT10–35 with a 12 mm aggregate. Homogenized wounds were serially diluted and plated onto LB agar to determine bacterial titers (CFU/g tissue), and each data point represents the mean of two technical replicates for plating and CFU enumeration.
Mouse systemic thigh infection model.
Studies were performed at the Wyss Institute at Harvard in accordance with protocol IS00000852–6, approved by the Harvard Medical School Institutional Animal Care and Use Committee and the Committee on Microbiological Safety. Female C57BL/6J mice, 6–8 weeks old, 18 ± 2 g, received from Charles River, were quarantined at least 2 days prior to use and kept under the housing conditions described above. As illustrated in Extended Data Fig. 9, mice were rendered neutropenic by a 0.2 mL intraperitoneal injection of cyclophosphamide (Cytoxan) at 150 mg/kg (Day −4) and at 100 mg/kg (Day −1) pre-infection. S. aureus AR Bank # 0706 was cultured overnight on tryptic soy agar plates at 37°C. Isolated colonies were suspended in PBS to achieve an OD600 of 0.1, then further diluted 1:1000 in tryptic soy broth to prepare the infecting inoculum of ~0.5 × 107 CFU/mL. Under anesthesia and sedation, mice were intramuscularly injected with 50 μL of the infecting inoculum into the right thigh. One hour post-infection, mice received a single intraperitoneal injection of compound 1 (80 mg/kg in 10% DMSO, 45% PEG300, 45% water; 200 μL, 6 mice), vancomycin (50 mg/kg in endotoxin-free water; 200 μL, 6 mice), or vehicle control (10% DMSO, 45% PEG300, 45% water; 200 μL, 6 mice). At ~25 hrs post-infection (~24 h after treatment), mice were euthanized by CO2 asphyxiation, and thighs were aseptically removed and homogenized in 2 mL of ice-cold sterile PBS using a Polytron PT10–35 with a 12 mm aggregate. For each sample, 200 μL of homogenized thigh were serially diluted and plated onto LB and MRSA CHROMagar to determine bacterial titers (CFU/mL thigh homogenate), and each data point represents one technical replicate for plating and CFU enumeration.
Structure-activity relationship analyses.
The analogues of compounds 1 and 2 procured for the structure-activity relationship analyses shown in Supplementary Note 4 and Extended Data Fig. 10 were chosen based on the following criteria: (1) the compound of interest contains the rationale shown in Extended Data Fig. 10; (2) the antibiotic prediction score for the compound of interest was at least 0.15; and (3) the compound of interest did not contain any PAINS or Brenk substructures, which may confound interpretation of structure-activity relationship results. This resulted in a list of 17 additional commercially available compounds (Supplementary Data 2), which we procured from multiple suppliers including ChemBridge, Vitas-M, and Specs. The compounds were dissolved in DMSO to prepare stock solutions and, where applicable, MIC and IC50 values were determined as described above in MIC and bacterial growth inhibition assays and Cytotoxicity IC50 assays.
Statistics and reproducibility.
No statistical method was used to predetermine sample size for all mouse experiments in this study, but our sample sizes are similar to those reported in previous publications (refs. 1–4, 6–8, 14). We were not blinded to allocation during experiments and outcome assessment, and data collection and analysis were not performed blind to the conditions of the experiments. For mouse experiments, no significant bias was observed across initial groups. No data were excluded from the analyses in this study. One-sided, two-sample permutation tests for differences in mean value54 were performed using MATLAB (Mathworks, Natick, MA) in Fig. 5a,b to test the hypothesis that log10 CFU/g or log10 CFU/mL titers were different from vehicle values for mouse model experiments. Exact permutation tests, in which all possible combinations were considered, were used for all comparisons.
Extended Data
Extended Data Fig. 1. Molecular weight distribution of the 39,312 compounds screened.
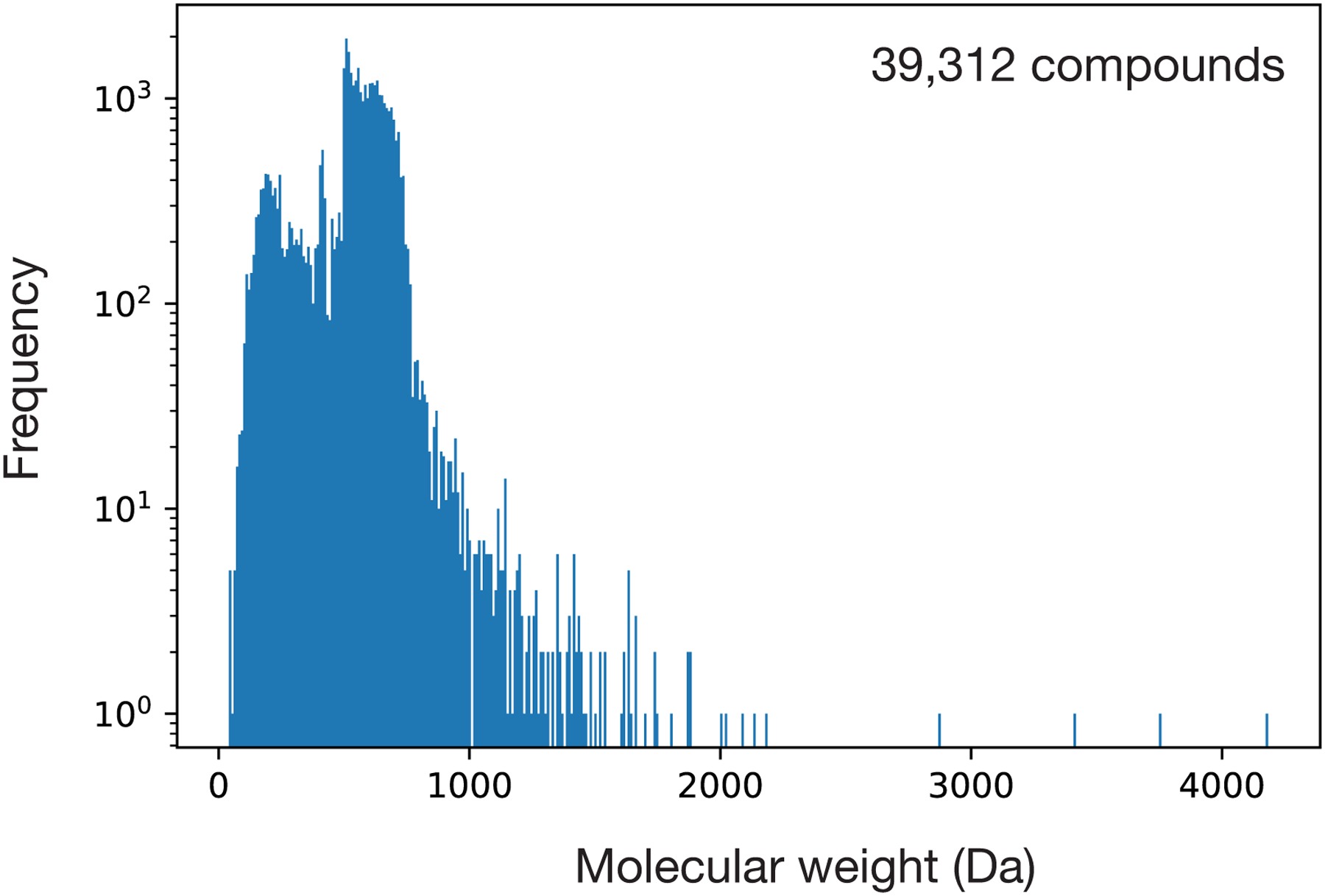
Data are from an original set of 39,312 compounds containing most known antibiotics, natural products, and structurally diverse molecules, with molecular weights between 40 Da and 4,200 Da. Frequency is shown on a log scale.
Extended Data Fig. 2. Comparison of deep learning models for predicting antibiotic activity.
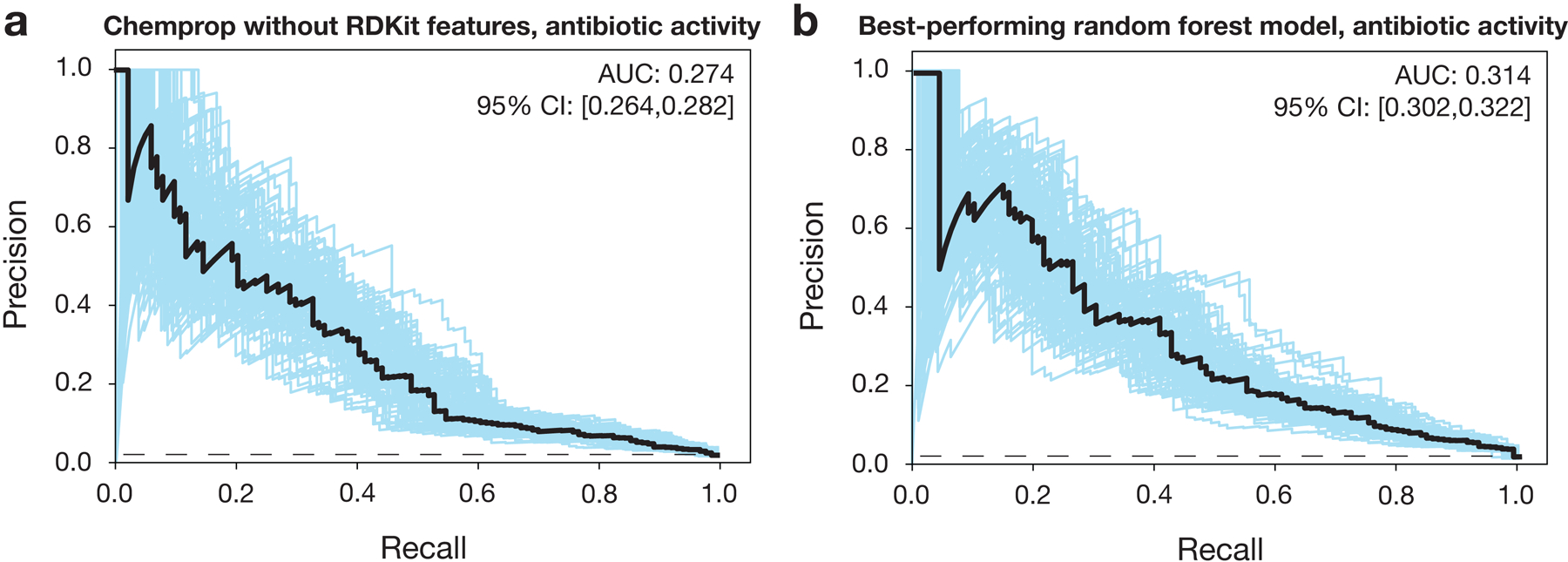
a, b, Precision-recall curves for predictions of antibiotic activity, for an ensemble of 10 Chemprop models without RDKit features (a) and the best-performing random forest classifier model based on Morgan fingerprints (b), trained and tested using data from a screen of 39,312 molecules (Fig. 1 of the main text). The black dashed line represents the baseline fraction of active compounds in the training set (1.3%). Blue curves and the 95% confidence interval indicate the variation generated by bootstrapping. AUC, area under the curve.
Extended Data Fig. 3. Comparison of deep learning models for predicting human cell cytotoxicity.
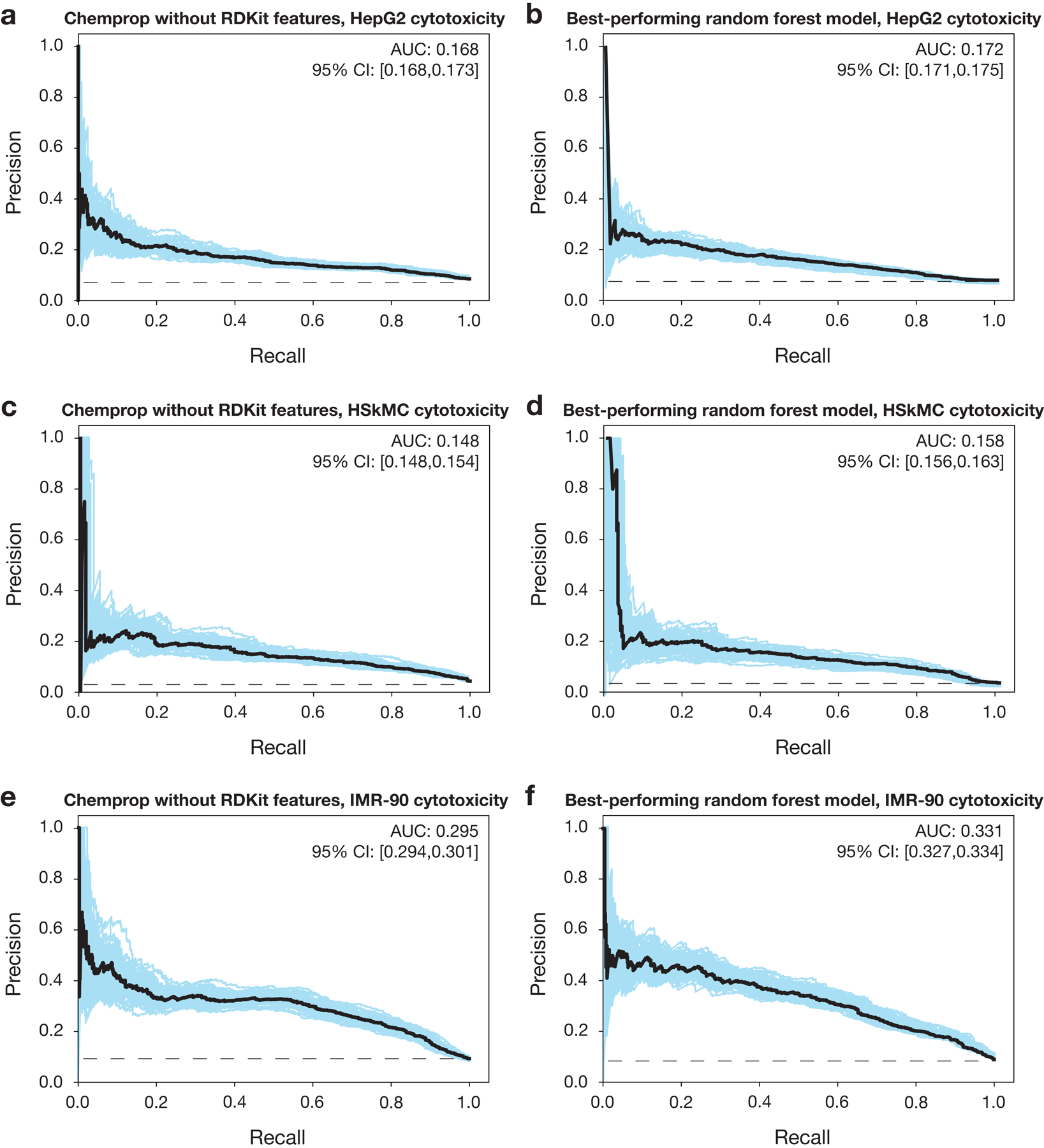
a, b, Precision-recall curves for predictions of HepG2 cytotoxicity, for an ensemble of 10 Chemprop models without RDKit features (a) and the best-performing random forest classifier model based on Morgan fingerprints (b), trained and tested using data from a screen of 39,312 molecules (Fig. 1 of the main text). The black dashed line represents the baseline fraction of active compounds in the training set (8.5%). Blue curves and the 95% confidence interval indicate the variation generated by bootstrapping. AUC, area under the curve.
c, d, Precision-recall curves for predictions of HSkMC cytotoxicity, for an ensemble of 10 Chemprop models without RDKit features (c) and the best-performing random forest classifier model based on Morgan fingerprints (d), trained and tested using data from a screen of 39,312 molecules (Fig. 1 of the main text). The black dashed line represents the baseline fraction of active compounds in the training set (3.8%). Blue curves and the 95% confidence interval indicate the variation generated by bootstrapping.
e, f, Precision-recall curves for predictions of IMR-90 cytotoxicity, for an ensemble of 10 Chemprop models without RDKit features (e) and the best-performing random forest classifier model based on Morgan fingerprints (f), trained and tested using data from a screen of 39,312 molecules (Fig. 1 of the main text). The black dashed line represents the baseline fraction of active compounds in the training set (8.8%). Blue curves and the 95% confidence interval indicate the variation generated by bootstrapping.
Extended Data Fig. 4. Visualizing chemical space across different prediction score thresholds.
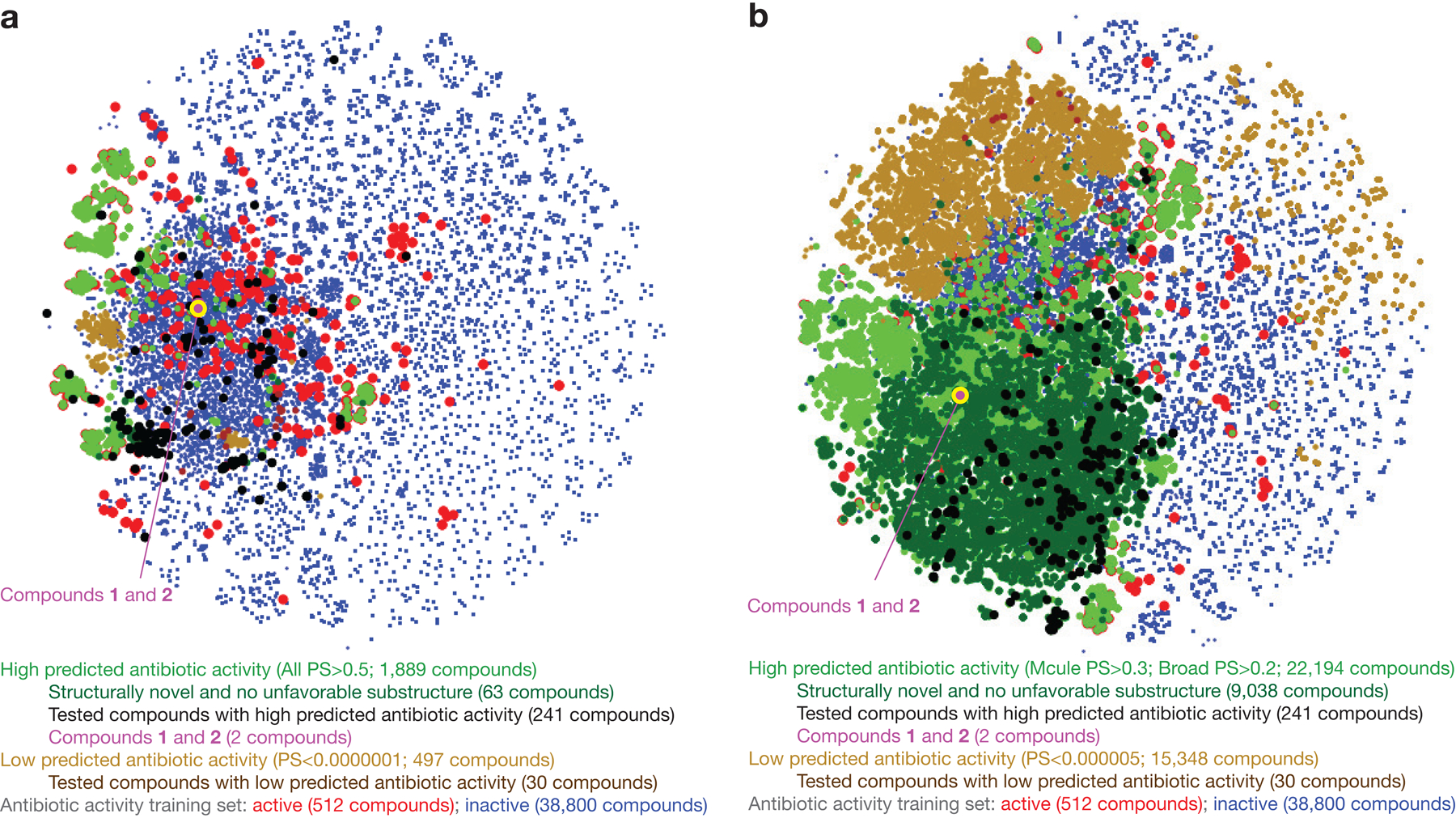
a, b, t-Distributed neighbor embedding (t-SNE) plot of compounds with high and low antibiotic prediction scores, in addition to compounds in the training set, for different prediction score thresholds. The plot shows the chemical similarity or dissimilarity of various compounds, and active compounds in the training set (red dots) are seen to largely separate compounds with high prediction scores (green, black, and purple dots) from compounds with low prediction scores (brown dots).
Extended Data Fig. 5. Examples of rationale calculations using Monte-Carlo tree search.
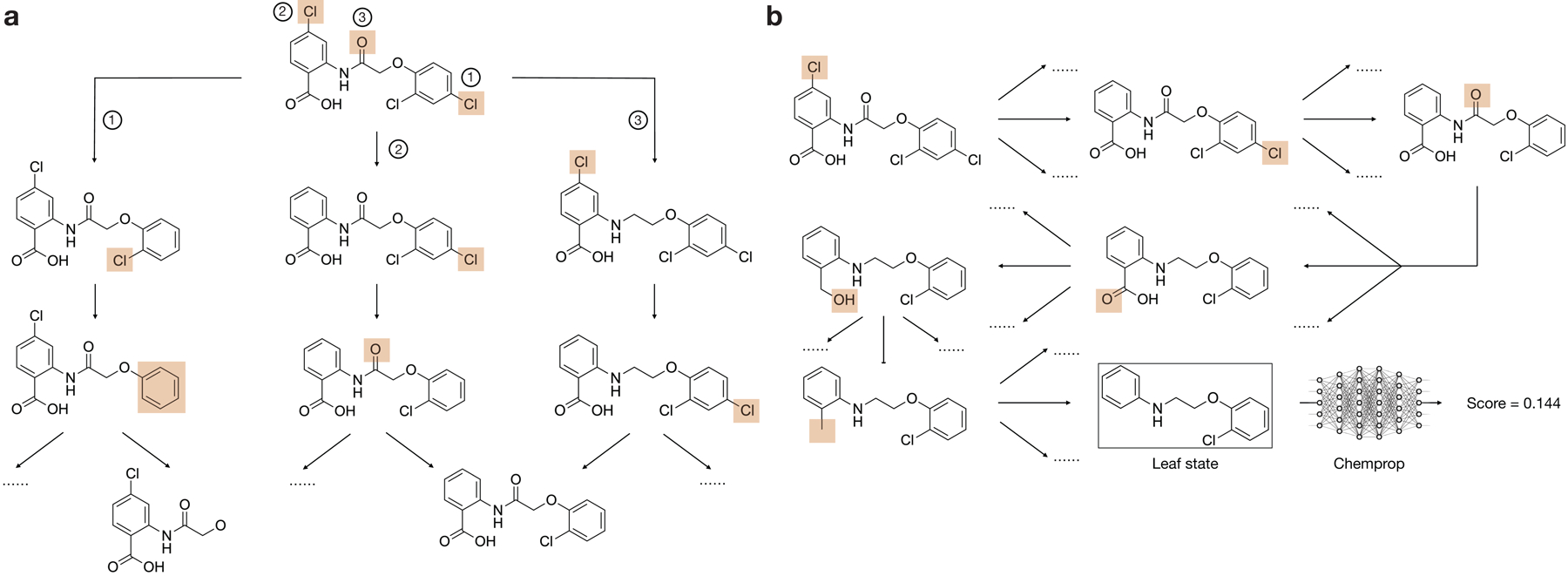
a, Illustration of the MCTS forward pass using compound 1. The figure shows three possible search paths from the root (compound 1) by deleting peripheral bonds or rings (highlighted in red). Due to space limitations, only three steps from the root are shown.
b, Illustration of a complete search path from the root (compound 1) to a leaf node (the rationale). Chemprop is used to predict the activity of each leaf node, and these predictions are used to make updates to the statistics of each intermediate node in the backward pass.
Extended Data Fig. 6. Maximal common substructure identification reveals known antibiotic classes, but are less predictive than Chemprop rationales across all hits.
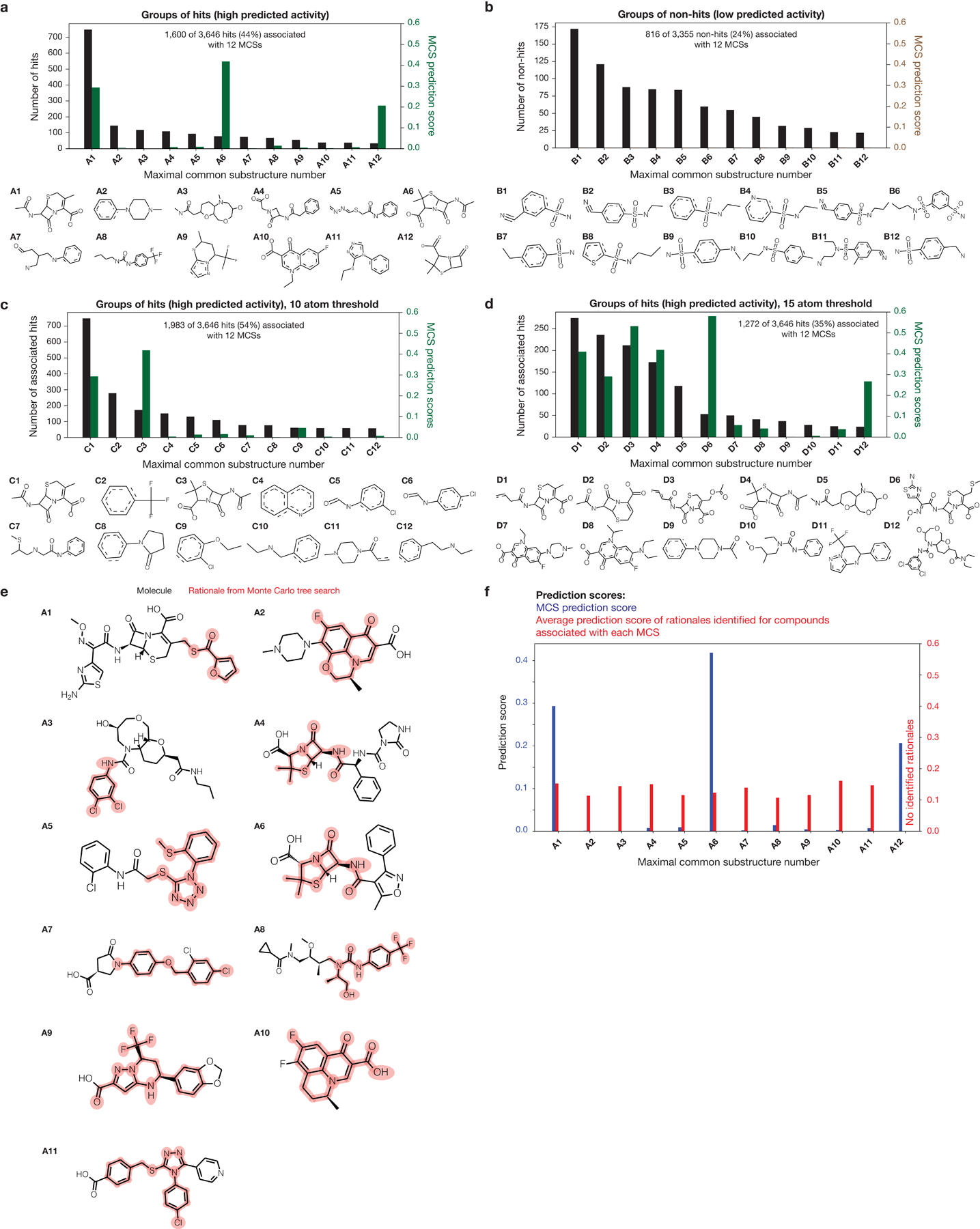
a, b, Rank-ordered numbers of hits (a) and non-hits (b) associated with maximal common substructures (MCSs) identified by a grouping method. Here, any hit associated with any of the MCSs shown shares a minimum of 12 atoms with the MCS. Dashed lines in MCSs indicate either single or double bonds. Each green or brown bar shows the prediction score of each MCS viewed as a molecule in its own right. Where bars are thin, the corresponding MCS prediction scores are approximately zero (including all brown bars in (b)).
c, d, Similar to (a), but here, any hit associated with any of the MCSs shown shares a minimum of 10 (c) or 15 (d) atoms with the MCS.
e, Illustration of the rationales (red) determined using a Monte Carlo tree search for example hits (black) associated with MCSs A1-A12. No hit associated with MCS A12 possessed a rationale.
f, MCS prediction scores (blue bars) and the average prediction scores of all rationales of all hits associated with MCSs A1-A12 (red bars). Where blue bars are thin, the corresponding MCS prediction scores are approximately zero. No hit associated with MCS A12 possessed a rationale.
Extended Data Fig. 7. Closest active training set compounds to, and selectivities of, four validated hits associated with rationale groups G1-G5.
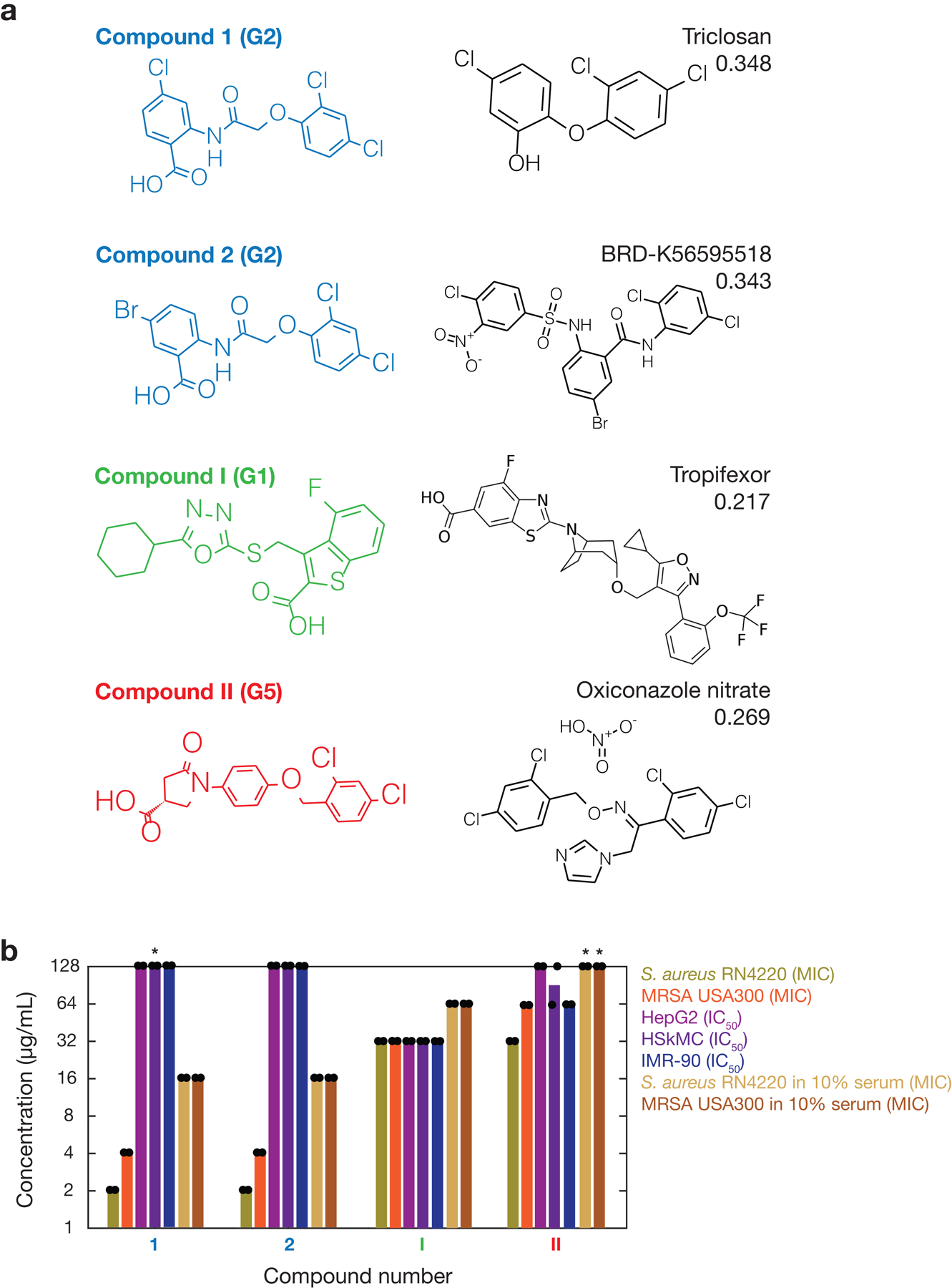
a, Closest active compounds (right), as measured by Tanimoto similarity, are from the training set of 39,312 compounds. Compounds are colored according to associated rationale groups (as indicated in parentheses), and the identifier and Tanimoto similarity score of each closest active compound are displayed.
b, S. aureus MIC and human cell IC50 values of the four compounds in (a), shown on a log scale. Bars show the means of two biological replicates (points) and are colored by the bacterial strain, human cell type, or media condition tested. Asterisks indicate values larger than 128 μg/mL.
Extended Data Fig. 8. Comparison of MICs of different compounds against methicillin-susceptible and methicillin-resistant S. aureus, and eradication of kanamycin persisters by treatment with compounds 1 and 2.
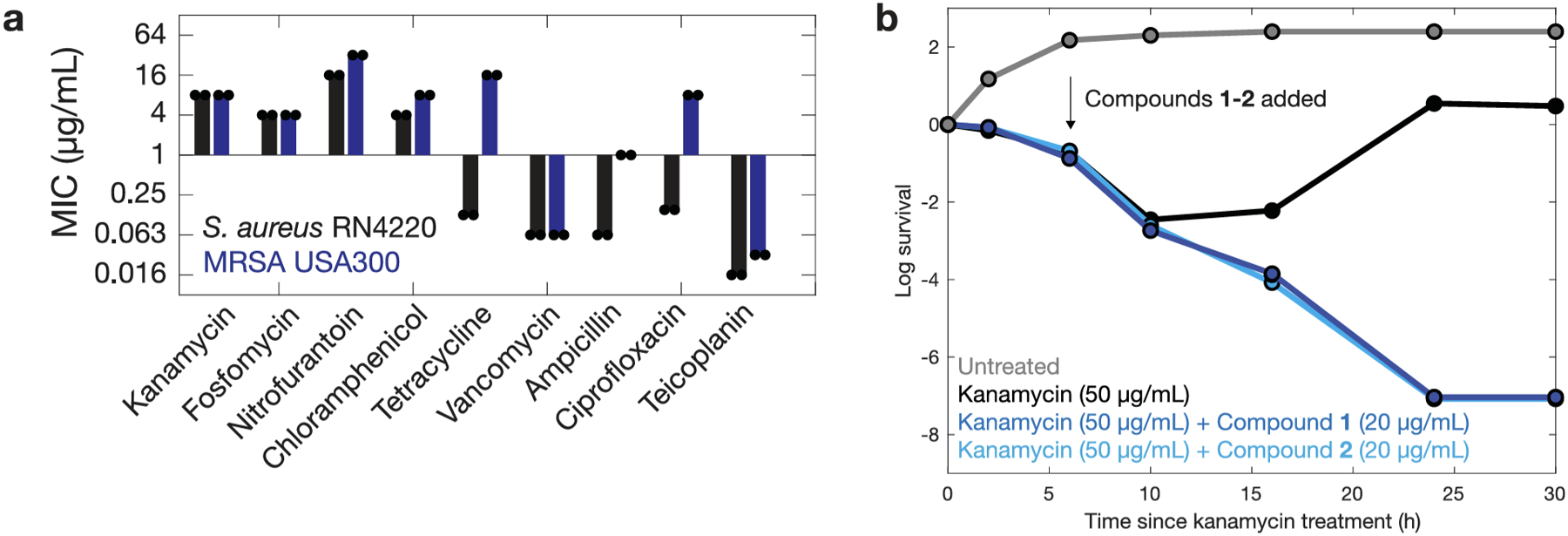
a, MICs of various antibiotics against S. aureus RN4220 (black) and S. aureus USA300 (blue) on a log scale. Bars show the mean of two biological replicates (individual points).
b, Survival curves of B. subtilis 168 after combination treatment with kanamycin and compounds 1 and 2, respectively, as determined by plating and CFU counting. Initial CFU values are ~107. Each point is representative of the mean of two biological replicates. Cultures treated with kanamycin in addition to compounds 1 and 2 were eradicated after 24 h (CFU/mL = 0), and these values were truncated to a log survival value of −7 on this plot.
Extended Data Fig. 9. Toxicity, chemical properties, and in vivo efficacy of compounds 1 and 2.
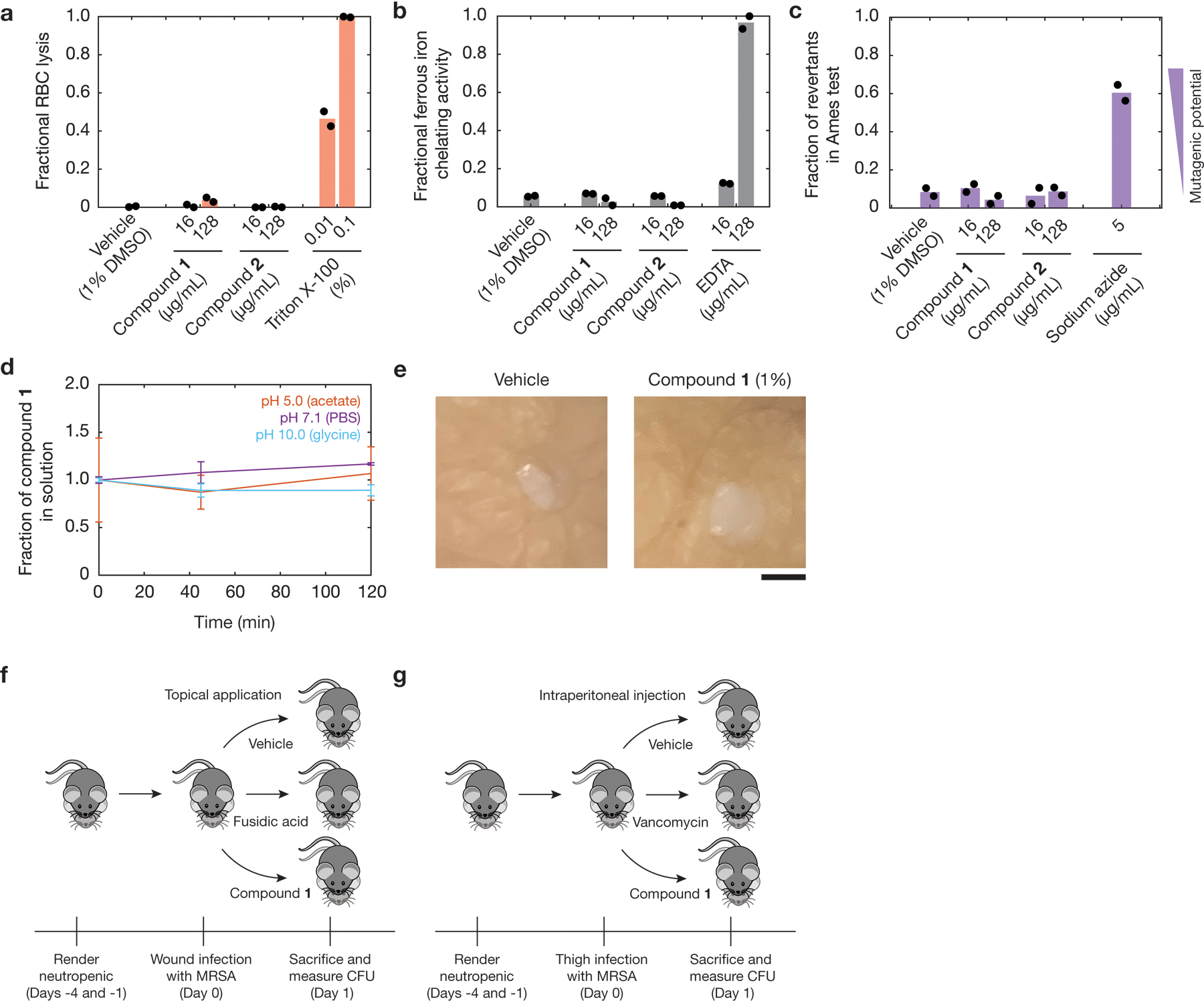
a, Fractional hemolysis measurements of human red blood cells (RBCs) treated with compounds 1 and 2 at the indicated final concentrations. Vehicle (1% DMSO) was used as a negative control, and Triton X-100, a detergent, was used as a positive control. Black points indicate values from two biological replicates, and red bars indicate average values.
b, Ferrous iron chelation measurements of compounds 1 and 2. Vehicle (1% DMSO) was used as a negative control, and ethylenediaminetetraacetic acid (EDTA), an iron chelator, was used as a positive control. Black points indicate values from two biological replicates, and gray bars indicate average values.
c, Ames test mutagenesis measurements of the fractions of revertant S. typhimurium TA100 cultures treated with compounds 1 and 2 at the indicated final concentrations. Vehicle (1% DMSO) was used as a negative control, and 5 μg/mL sodium azide was used as a positive control. Black points indicate values from two biological replicates, and purple bars indicate average values. Higher fractions of revertant cultures indicate higher mutagenic potential (inset).
d, Chemical stability of compound 1 in various buffers as a function of incubation time at 37°C. Values are normalized to the mean measurement at time zero, and each point is representative of the mean of two biological replicates. Error bars indicate the full range of values arising from two biological replicates.
e, Photographs of WoundSkin models 24 h after topical treatment with compound 1 (1%) or DMSO vehicle. Images are representative of six biological replicates in each treatment group. Scale bar, 2 mm.
f, Illustration of the in vivo study of a neutropenic mouse wound infection model using MRSA CDC 563 shown in Fig. 5a of the main text.
g, Illustration of the in vivo study of a neutropenic mouse thigh infection model using MRSA CDC 706 shown in Fig. 5b of the main text.
Extended Data Fig. 10. Exploration of a structural class through structure-activity relationships.
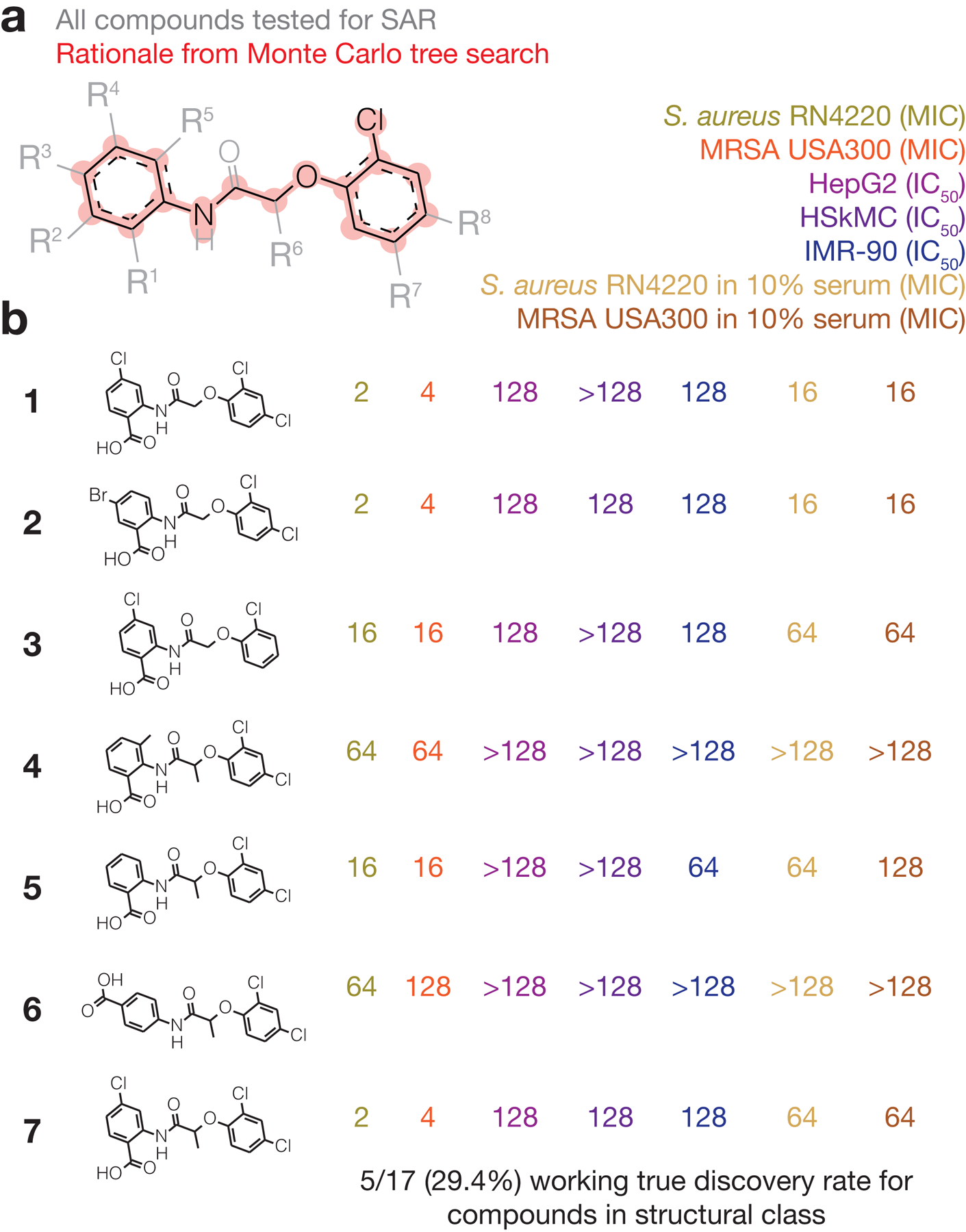
a, The rationale of compounds 1 and 2, overlaid with chemical modifications (R1-R8) that encompass all compounds used to test SAR (Supplementary Data 2). SAR, structure-activity relationships.
b, Analogues of compounds 1 and 2 found to have varying degrees of activity against S. aureus. Corresponding MIC and IC50 values are representative of two biological replicates.
Supplementary Material
Acknowledgements:
We thank the editor and all the reviewers for important comments and suggestions on previous versions of this manuscript. We thank the past and present members of the Collins lab for helpful discussions, members of the Broad Institute Center for the Development of Therapeutics (CDoT) for helpful feedback, the Microbial Genome Sequencing Center (Pittsburgh, PA) for assistance with sequencing, the Harvard Center for Mass Spectrometry for assistance with LC-MS experiments, Sandy Gould and Ritu Singh at the Broad Institute for medicinal chemistry feedback, Anita Vrcic and Taline Dawson at the Broad Institute for assistance with compound management, Amanda Graveline at the Wyss Institute for assistance with mouse experiments, and Zemer Gitai at Princeton University for Escherichia coli strains RFM795 and JW5503-KanS. F.W. was supported by the James S. McDonnell Foundation and the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award number K25AI168451. A.K. was supported by the Swiss National Science Foundation under grant number SNSF_ 203071. A.M.E. and A.L.M. were supported by federal funds from the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under grant number U19AI110818 to the Broad Institute. J.M.S. was supported by the Banting Fellowships Program (393360). L.D.R. was supported by the Volkswagen Foundation. J.J.C. was supported by the Defense Threat Reduction Agency (grant number HDTRA12210032), the National Institutes of Health (grant number R01-AI146194), and the Broad Institute of MIT and Harvard. This work is part of the Antibiotics-AI Project, which is directed by J.J.C. and supported by the Audacious Project, Flu Lab, LLC, the Sea Grape Foundation, Rosamund Zander and Hansjorg Wyss for the Wyss Foundation, and an anonymous donor.
Footnotes
Competing interests: J.J.C. is an academic co-founder and Scientific Advisory Board chair of EnBiotix, an antibiotic drug discovery company, and Phare Bio, a non-profit venture focused on antibiotic drug development. J.J.C. is also an academic co-founder and board member of Cellarity and the founding Scientific Advisory Board chair of Integrated Biosciences. J.M.S. is scientific co-founder and scientific director of Phare Bio. F.W. is a co-founder of Integrated Biosciences. S.O. and A.L. contributed to this work as employees of Integrated Biosciences, and S.O. may have an equity interest in Integrated Biosciences. F.W. and J.J.C. have filed a patent based on the results of this work. The remaining authors declare no competing interests.
Ethics statement: The human skin biopsy experiment shown in Extended Fig. 9 involved skin tissue obtained with inform consent of human donors by Genoskin, in compliance with all applicable regulations and approved and authorized by the French Ministry of Research and Higher Education. All tissue donors support the use of human skin tissue for experiments and research purposes, in compliance with the Declaration of Helsinki.
Supplementary Information is available for this paper.
Code availability: Chemprop is available at https://github.com/chemprop/chemprop. The Chemprop checkpoints for the final antibiotic activity, cytotoxicity, and proton motive force-alteration models, along with a code platform for performing and adapting the analyses developed in this work, are available at https://github.com/felixjwong/antibioticsai and https://zenodo.org/records/1009587955.
Data availability:
Data generated from chemical screens, machine learning models, and whole-genome sequencing experiments are available as Supplementary Data 1–4. Source Data are available for Figs. 4 and 5 and Extended Data Figs. 8 and 9. Data from whole-genome sequencing reads have been deposited on BioProject under accession number PRJNA1026995. A copy of model predictions for the Mcule purchasable database (ver. 200601) and the Broad Institute database used in this work is available at https://github.com/felixjwong/antibioticsai.
References
- 1.Stokes JM et al. A deep learning approach to antibiotic discovery. Cell 180, 688–702 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Imai Y et al. A new antibiotic selectively kills Gram-negative pathogens. Nature 576, 459–464 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ling LL et al. A new antibiotic kills pathogens without detectable resistance. Nature 517, 455–459 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martin JK II et al. A dual-mechanism antibiotic kills Gram-negative bacteria and avoids drug resistance. Cell 181, 1–15 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lewis K Platforms for antibiotic discovery. Nat. Rev. Drug Dis 12, 371–387 (2013). [DOI] [PubMed] [Google Scholar]
- 6.Culp EJ et al. Evolution-guided discovery of antibiotics that inhibit peptidoglycan remodelling. Nature 578, 582–587 (2020). [DOI] [PubMed] [Google Scholar]
- 7.Mitcheltree MJ et al. A synthetic antibiotic class overcoming bacterial multidrug resistance. Nature 599, 507–512 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Durand-Reville TF et al. Rational design of a new antibiotic class for drug-resistant infections. Nature 597, 698–702 (2021). [DOI] [PubMed] [Google Scholar]
- 9.Silver LL Challenges of antibacterial discovery. Clin. Microbiol. Rev 24, 71–109 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gilmer J et al. Neural message passing for quantum chemistry. In Proceedings of the 34th International Conference on Machine Learning (2017). [Google Scholar]
- 11.Yang K et al. Analyzing learned molecular representations for property prediction. J. Chem. Inf. Model 59, 3370–3388 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wong F et al. Leveraging artificial intelligence in the fight against infectious diseases. Science 381, 164–170 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Melo MCR, Maasch JRMA, and de la Fuente-Nunez C Accelerating antibiotic discovery through artificial intelligence. Commun. Biol 4, 1050 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu G et al. Deep learning-guided discovery of an antibiotic targeting Acinetobacter baumannii. Nat. Chem. Biol (2023). [DOI] [PubMed] [Google Scholar]
- 15.Wong F et al. Discovering small-molecule senolytics with deep neural networks. Nat. Aging 3, 734–750 (2023). [DOI] [PubMed] [Google Scholar]
- 16.The Review on Antimicrobial Resistance. Antimicrobial resistance: tackling a crisis for the health and wealth of nations. (2014) [Google Scholar]
- 17.Corsello SM et al. The Drug Repurposing Hub: a next-generation drug library and information resource. Nat. Med 23, 405–408 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sterling T and Irwin JJ ZINC 15 – ligand discovery for everyone. J. Chem. Inf. Model 55, 2324–2337 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Camacho DM et al. Next-generation machine learning for biological networks. Cell 173, 1581–1592 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Rudin C Stop explaining black box machine learning models for high stakes decisions and use interpretable models instead. Nat. Mach. Intell 1, 206–215 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee AS et al. Methicillin-resistant Staphylococcus aureus. Nat. Rev. Dis. Primers 4, 18033 (2018). [DOI] [PubMed] [Google Scholar]
- 22.Toxicology in the 21st century. Accessed 20 October 2022 at https://tripod.nih.gov/tox/.
- 23.The Human Metabolome Database. Accessed 20 October 2022 at https://hmdb.ca/metabolites.
- 24.M-cule purchaseable database (in-stock), ver. 200601. Accessed 27 June 2020 at https://mcule.com/database/.
- 25.Van der Maaten L and Hinton G Visualizing data using t-SNE. J. Mach. Learn. Res 9, 2579–2605 (2008). [Google Scholar]
- 26.Silver D et al. Mastering the game of Go without human knowledge. Nature 550, 354–359 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Cao Y, Jiang T, and Girke T A maximum common substructure-based algorithm for searching and predicting drug-like compounds. Bioinformatics 24, i366–i374 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Muratov EN et al. QSAR without borders. Chem. Soc. Rev 49, 3525–3564 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baell JB, and Holloway GA New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem 53, 2719–2740 (2010). [DOI] [PubMed] [Google Scholar]
- 30.Brenk R et al. Lessons learnt from assembling screening libraries for drug discovery for neglected diseases. ChemMedChem 3, 435–444 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lipinski CA, Lombardo F, Dominy BW, and Feeney PJ Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug. Dis. Rev 23, 3–25 (1997). [DOI] [PubMed] [Google Scholar]
- 32.Ghose AK, Viswanadhan VN, and Wendoloski JJ A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem 1, 55–68 (1999). [DOI] [PubMed] [Google Scholar]
- 33.O’Shea R and Moser HE Physicochemical properties of antibacterial compounds: implications for drug discovery. J. Med. Chem 51, 2871–2878 (2008). [DOI] [PubMed] [Google Scholar]
- 34.Wong F et al. Reactive metabolic byproducts contribute to antibiotic lethality under anaerobic conditions. Mol. Cell 82, 3499–3512 (2022) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wong F et al. Cytoplasmic condensation induced by membrane damage is associated with antibiotic lethality. Nat. Commun 12, 2321 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wong F et al. Understanding beta-lactam-induced lysis at the single-cell level. Front. Microbiol 12, 712007 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wong F et al. Mechanics and dynamics of bacterial cell lysis. Biophys. J 116, 2378–2389 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Farha MA, Verschoor CP, Bowdish D, and Brown ED Collapsing the proton motive force to identify synergistic combinations against Staphylococcus aureus. Chem. Biol 20, 1168–1178 (2013). [DOI] [PubMed] [Google Scholar]
- 39.Hurdle JG Targeting bacterial membrane function: an underexploited mechanism for treating persistent infections. Nat. Rev. Microbiol 9, 62–75 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Centers for Disease Control and Prevention. Antibiotic Resistance Threats in the United States, 2019. Accessed 20 September 2021 at https://www.cdc.gov/drugresistance/pdf/threats-report/2019-ar-threats-report-508.pdf.
- 41.Lewis K The science of antibiotic discovery. Cell 181, 29–45 (2020). [DOI] [PubMed] [Google Scholar]
- 42.Walsh C Where will new antibiotics come from? Nat. Rev. Microbiol 1, 65–70 (2003). [DOI] [PubMed] [Google Scholar]
- 43.Ying R, Bourgeois D, You J, Zitnik M, and Leskovic J GNNExplainer: Generating explanations for graph neural networks. Adv. Neural. Inf. Process. Syst 32, 9240–9251 (2019). [PMC free article] [PubMed] [Google Scholar]
- 44.Jiménez-Luna J, Grisoni F, and Schneider G Drug discovery with explainable artificial intelligence. Nat. Mach. Intell 2, 573–584 (2020). [Google Scholar]
- 45.Yuan H, Yu H, Gui S, and Ji S Explainability in graph neural networks: a taxonomic survey. IEEE Trans. Pattern Anal. Mach. Intell 45, 5782–5799 (2023). [DOI] [PubMed] [Google Scholar]
- 46.DeLong ER, DeLong DM, Clarke-Pearson DL Comparing the areas under two or more correlated receiver operating characteristic curves: a nonparametric approach. Biometrics 44, 837–845 (1988). [PubMed] [Google Scholar]
- 47.Kazeev N The fast version of DeLong’s method for computing the covariance of unadjusted AUC. Accessed 21 July 2023 at https://github.com/yandexdataschool/roc_comparison.
- 48.Rosin CD Multi-armed bandits with episode context. Ann. Math. Artif. Intell 61, 203–230 (2011). [Google Scholar]
- 49.Wang Y, Backman TWH, Horan K, and Girke T fmcsR: mismatch tolerant maximum common substructure searching in R. Bioinformatics 29, 2792–2794 (2013). [DOI] [PubMed] [Google Scholar]
- 50.Daina A, Michielin O, and Zoete V SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep 7, 42717 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wong F et al. Benchmarking AlphaFold‐enabled molecular docking predictions for antibiotic discovery. Mol. Syst. Biol 18, e11081 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Walker BJ et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9, e112963 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Greco I et al. Correlation between hemolytic activity, cytotoxicity and systemic in vivo toxicity of synthetic antimicrobial peptides. Sci. Rep 6, 13206 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Krol LR Permutation Test. Accessed 22 July 2023 at https://github.com/lrkrol/permutationTest.
- 55.Wong F et al. Supporting code for: Discovery of a structural class of antibiotics with explainable deep learning (2023). 10.5281/zenodo.10095879. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data generated from chemical screens, machine learning models, and whole-genome sequencing experiments are available as Supplementary Data 1–4. Source Data are available for Figs. 4 and 5 and Extended Data Figs. 8 and 9. Data from whole-genome sequencing reads have been deposited on BioProject under accession number PRJNA1026995. A copy of model predictions for the Mcule purchasable database (ver. 200601) and the Broad Institute database used in this work is available at https://github.com/felixjwong/antibioticsai.