

Summary
Microglia (MG), the brain-resident macrophages, play major roles in health and disease via a diversity of cellular states. While embryonic MG display a large heterogeneity of cellular distribution and transcriptomic states, their functions remain poorly characterized. Here, we uncovered a role for MG in the maintenance of structural integrity at two fetal cortical boundaries. At these boundaries between structures that grow in distinct directions, embryonic MG accumulate, display a state resembling post-natal axon-tract-associated microglia (ATM) and prevent the progression of microcavities into large cavitary lesions, in part via a mechanism involving the ATM-factor Spp1. MG and Spp1 furthermore contribute to the rapid repair of lesions, collectively highlighting protective functions that preserve the fetal brain from physiological morphogenetic stress and injury. Our study thus highlights key major roles for embryonic MG and Spp1 in maintaining structural integrity during morphogenesis, with major implications for our understanding of MG functions and brain development.
Keywords: microglia, microglial state, development, cerebral cortex, repair, cavity, Spp1, osteopontin, amygdala, corpus callosum
Graphical abstract
Highlights
-
•
Embryonic ATM-like microglia accumulate at key cortical boundaries
-
•
Microglia prevent the formation of cavitary lesions due to morphogenetic stress
-
•
ATM-core factor Spp1 contributes to neuroprotective roles of microglia
-
•
Microglia and Spp1 contribute to the rapid repair of cavitary lesions
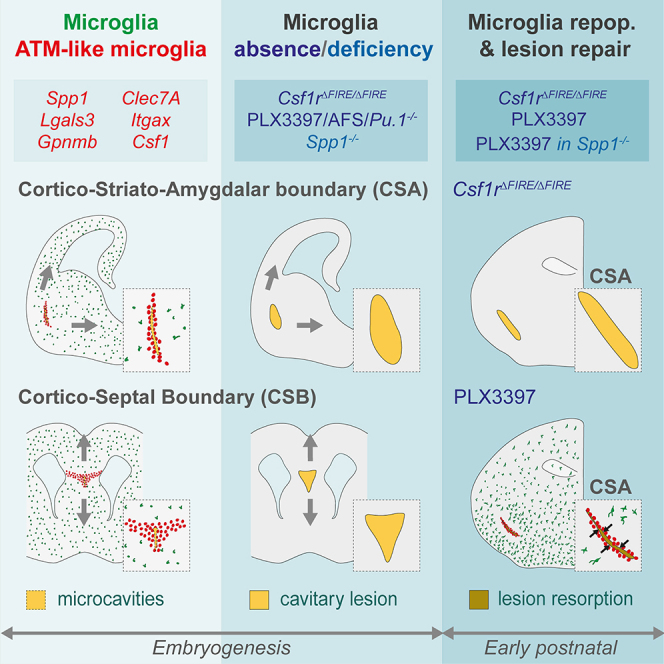
Microglia accumulate at fetal cortical boundaries where they preserve tissue integrity in the context of morphogenetic constraints. These immune cells prevent the formation of cavitary lesions and contribute to their rapid repair, highlighting protective functions during early brain development.
Introduction
Microglia (MG), the brain-resident macrophages, play key roles in the development and maintenance of brain circuits throughout life.1,2,3,4,5,6,7,8 These immune sentinels are involved in key steps of neural network assembly, for instance, by regulating neuronal numbers, synaptic development and refinement, maturation of myelin, synaptic transmission or neuronal excitability.1,2,3,4,5,6,7,8,9,10,11,12,13,14,15 In agreement with the diverse roles of MG, their dysfunction has been linked to almost all brain pathologies, ranging from developmental disorders to neurodegenerative diseases.1,2,3,4,5,6,7 Anatomical and recent single-cell (sc) transcriptomic studies have shown that MG exist in distinct cellular and transcriptomic states, particularly during development, aging, and neurodegeneration.16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34 This transcriptomic diversity, as shown in the white matter and cerebral cortex29,32 and in induced pluripotent cells,35 can be driven by the local environment, revealing a symbiotic relationship between MG and their local cerebral niches.33 Yet, how such heterogeneity relates to specific locations and functions of these multifaceted cells, especially during the dynamic period of prenatal development, remains poorly understood.
MG originate from yolk sac-derived macrophages that migrate and seed the brain primordium during early embryogenesis.36 This process starts from embryonic day 9 (E9) in mice, and gestational week 4 (GW4) in humans.2,37,38 MG subsequently colonize the parenchyma during the dynamic phases of cerebral development, characterized by a prolonged phase of neuronal generation, migration and wiring, as the brain grows, folds and changes of shape.39 In both human and mice, MG colonize the brain in a progressive, highly stereotyped manner2,4,38,40 characterized by an uneven distribution: MG accumulate at hotspots, avoid selective regions such as the developing cortical plate,41 and exhibit a diverse range of morphologies, cellular behaviors, and transcriptomic states.5,7,21,22,27,31,32 Although microglial heterogeneity is progressively lost as development proceeds, it can reemerge in the context of aging and disease.17,18,19,20,22,23,24,29,30,31,33 In physiological conditions, microglial heterogeneity spans both the prenatal and post-natal periods.2,4,21,22,40,42,43 In particular, specific populations of amoeboid MG present in the post-natal cortical white matter, including the corpus callosum (CC),21,22,42,43 display a specific transcriptomic state and have been variously termed axon-tract-associated microglia (ATM),22 proliferative-region associated microglia (PAM),2 youth-associated microglia (YAM),31 or CD11c-positive (+) MG.42,43 Post-natal ATM/PAM/YAM/CD11c+ MG, or ATM, are characterized by the expression of Spp1 (encoding osteopontin or OPN), Lgals3, Gpnmb, Clec7a, Itgax (encoding CD11c), Csf1, and Igf1, sharing transcriptomic features with disease-associated microglia (DAM), initially identified in mouse models of Alzheimer’s disease and observed in various neurodegenerative contexts.17,18,19,24,29,31 Functionally, post-natal ATM are involved in the engulfment of nascent glial cells, regulation of myelination, and promotion of neuronal survival, in part through Igf1 expression.21,22,42,43,44,45 While these studies highlight a link between post-natal microglial hotspots, specific transcriptomic states and functions, the roles of prenatal accumulations and their potential cellular heterogeneity remain largely to be deciphered.
Here, by combining transcriptomic analyses, imaging and genetically modified mouse models, we reveal that MG maintain the structural integrity at the fetal cortico-striato-amygdalar boundary (CSA) and cortico-septal boundary (CSB), where embryonic MG accumulate and display a specific state resembling post-natal ATM.21,22,42,43 These boundaries, which are vulnerable to developmental tensions linked to brain growth and morphogenesis, rely on MG to prevent the formation of large cavitary lesions, in part via a mechanism involving the pleiotropic ATM-factor Spp1/OPN. MG and Spp1 furthermore contribute to the rapid repair of large lesions, which highlights their protective functions in preserving the fetal brain from physiological morphogenetic tensions and injuries. Our study thus reveals that embryonic MG and the ATM-factor Spp1 play critical roles in maintaining the structural integrity of the developing brain during normal morphogenesis, with major implications for our understanding of MG functions and cerebral development.
Results
ATM-like MG accumulate at two embryonic cortical boundaries
While the developmental heterogeneity of MG has been established by anatomical2,4,38,40 and sc transcriptomic studies,21,22,27,32 whether microglial transient accumulations in the embryonic brain are composed of cells in specific states is yet to be determined.
As a first step toward exploring the heterogeneity of MG during embryogenesis, we took advantage of a longitudinal brain sc transcriptomic dataset generated by La Manno et al. between E9 and E18.30 By extracting and analyzing MG from this dataset, we identified 3 distinct clusters: cycling MG, non-cycling MG and embryonic MG that resemble post-natal ATM/PAM/YAM/CD11c+ MG30 (Figure 1A). Embryonic ATM-like cells are particularly present from E14 onward (Figures 1A–1C and S1A; Table S1) and share a core genetic signature with previously described post-natal white-matter ATM (Figures 1B and 1D)22 or PAM (Figures S1B–S1D)21: they notably express Spp1, which codes for OPN, Csf1, Igf1, Lgals3, which codes for Galectin3/Mac2 and Gpnmb (Figure S1A; Table S1).21,22 Consistently, embryonic ATM-like30 and PAM21 gene signatures are present mainly in the ATM cluster described by Hammond and colleagues22 (Figures S1D–S1F), highlighting similarities between these prenatal and post-natal transcriptomic signatures.
Figure 1.

ATM-like microglia accumulate at the embryonic CSA and CSB
(A) Single-cell transcriptomic uniform manifold approximation and projection (UMAP) plot of embryonic microglial cells (n = 1,711) extracted from the La Manno dataset,30 showing cycling MG (orange), non-cycling MG (purple), and embryonic ATM-like MG (green).
(B) Projection of the post-natal ATM signature (top-enriched differentially expressed genes [DEGs]) from the Hammond dataset22 onto (A).
(C) Proportion of cycling (orange), non-cycling (purple), and embryonic ATM-like MG (green) at different embryonic stages.
(D) Venn diagram showing the overlap between embryonic and post-natal ATM DEGs, respectively, identified from La Manno30 and Hammond22 datasets (fold change [FC] > 1.5, Bonferroni-adjusted p < 1e−10).
(E) Immunolabeling of brain sections from E14.5 or E15 Cx3cr1gfp/+ or CD11c-EYFP embryos showing co-expression of microglia and ATM markers at the CSA. CSA close ups are delineated by dotted lines. GFP-positive microglia in Cx3cr1gfp/+ brains fully colocalized with the microglial marker P2Y12 receptor, and IBA1 was used to label microglia in CD11c-EYFP brains (performed on brain sections of at least three mice from two different litters).
(F) Immunolabeling of coronal brain sections from E18.5 Cx3cr1gfp/+ or CD11c-EYFP embryos showing co-expression of microglia and ATM markers at the cortico-septal boundary (CSB), below the corpus callosum (CC). CSB close ups are delineated by dotted lines. GFP-positive microglia in Cx3cr1gfp/+ brains fully colocalized with the IBA1 marker, which was used to label all microglia in CD11c-EYFP brains (performed on brain sections of at least three mice from two different litters).
Graphs show means ± SEM. Scale bars: 500 μm in (E, upper left); 200 μm in (E, lower); 100 μm in (E, upper right) and (F); and 20 μm (E insets).
ATM, axon-tract-associated microglia; ATM-like, axon-tract-associated-like microglia; CC, corpus callosum; CSA, cortico-striato-amygdalar boundary; CSB, cortico-septal boundary; DEGs, differentially expressed genes; MG, microglia; Ncx, neocortex; Se, Septum; Str, striatum.
Figure S1.

Embryonic ATM-like cells resemble post-natal ATM, related to Figure 1
(A) Dot plot showing the relative expression levels of core ATM genes in embryonic ATM-like microglia (MG) compared with non-cycling or cycling embryonic MG in the La Manno scRNA-seq dataset.30 The color represents the normalized expression level across all cells within a cluster, while the dot size indicates the percentage of cells expressing each gene in that cluster.
(B) Projection of the post-natal PAM signature from the Li dataset21 onto the UMAP plot from Figure 1A showing embryonic ATM-like cells.
(C) Venn diagram highlighting the overlap between embryonic brain ATM differentially expressed genes (DEGs) identified in the La Manno dataset,30 post-natal ATM in the Hammond dataset,22 and post-natal PAM in the Li dataset.21
(D) Reproduced tSNE plot of the 76,149 cells characterized by scRNA-seq in the La Manno dataset.30
(E and F) Projections of the embryonic ATM signature defined in the La Manno dataset30 (E) and the PAM signature defined in the Li dataset21 (F), showing the overlap with the Hammond ATM cluster 4.22
(G) Low magnification of brain sections from embryonic mice at E15.0 and E18.5 showing that accumulations of SPP1-expressing microglia, identified as Cx3cr1gfp-positive or IBA1-positive parenchymal cells, are restricted to the CSA and CSB.
(H) Immunolabeling of embryonic E15.0 CD11c-EYFP brain section showing IBA1-positive microglia expressing YFP (n = 3) and ATM marker CLEC7A (n = 6) at the CSA.
(I) Comparison of ATM marker expression between microglia located in the neocortex and CSA in E15.0 CD11c-EYFP and Cx3cr1gfp-positive brains, showing examples of CSA and neocortical areas used for quantification; immunolabeling was performed on brain sections from at least 3 mice from 2 different litters.
(J) pHrodo assay conducted on ex vivo brain slices (at least n = 3 from 2 distinct litters) showing intense staining in CSA microglia at E14.5.
(K) Immunolabeling for CD68 showing intense staining inside CSA microglia at E14.5 in physiological conditions and comparison between mean CD68 coverage of SPP1-negative ramified microglia and SPP1-positive CSA ATM (nNcx = 28, nCSA = 29 cells in 4 mice from 2 distinct litters).
(L) Immunolabeling of human GW9 and GW14 human brain transverse sections showing co-expression of ATM markers with IBA1 at the CSA.
Graphs (ATM markers co-expression and CD68 coverage) show means ± SEM. Mann-Whitney U test were performed for statistical comparison, ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001. Scale bars: 200 μm in (G left) and (L right); 500 μm in (G, right); 150 μm in (I)–(K); 100 μm in (H); 1,000 μm in (L, upper left); and 1,500 μm in (L, lower left).
ATM, axon-tract-associated microglia; CC, corpus callosum; CSA, cortico-striato-amygdalar boundary; DEGs, differentially expressed genes; GW, gestational week; MG, microglia; Ncx, neocortex; PAM, proliferative-region associated microglia; Se, septum; Str, striatum.
To assess whether embryonic ATM-like cells may locate in specific regions, we first focused on mid-neurogenesis (E14.5), when microglial distribution starts to be noticeably uneven across the developing forebrain.40,41 Using the Cx3cr1gfp/+ mouse line, which labels all macrophages, we observed a significant accumulation of GFP+ MG at the boundary between the cerebral cortex, striatum , and amygdala ,40,41 which we called the CSA (Figures 1E and S1G–S1I). The bona fide microglial identity of GFP+ cells was confirmed by their location in the parenchyma (Figure 1E) and expression of the microglial marker P2Y12 receptor but not the perivascular macrophage marker LYVE1 (Figure 1E).2,23 These cells were most abundant at E15 and co-expressed ATM-specific markers at this time point (Figures1E and S1G–S1I). Using CD11c-eYFP mice and immunostainings we found that approximately 60% of CSA MG co-expressed proteins encoded by ATM “core” signature genes: CD11c, Mac2, Clec7A, Spp1, and GPNMB (Figures 1E and S1G–S1I). This contrasted with adjacent regions, where only few sparse cells expressed these markers (Figures 1E, S1G, and S1I), highlighting a local and dense CSA ATM-like accumulation between E14.5 and E16.5, with cells restricted to the ventrocaudal CSA at E18.5 (Figure S1G). In addition to their distinct transcriptomic signature, ATM-like cells exhibited an amoeboid morphology, phagocytic activity as assessed in ex vivo slices, and higher expression levels of the lysosomal marker CD68 than the neighboring MG (Figures S1J and S1K). Thus, like post-natal ATM,21,22 embryonic ATM-like MG seem highly phagocytic, reinforcing similarities between pre- and post-natal ATM states beyond their conserved transcriptomic signature. To investigate whether a similar accumulation occurs in humans, we labeled fetal brains from GW9 to GW14 and detected a conserved pattern of MG expressing ATM-core factors at the CSA throughout these stages (Figure S1L). These data show that, in both mice and humans, MG expressing an ATM-like signature accumulate at the embryonic CSA.
Since the ATM-like transcriptomic signature is detected throughout fetal life, we conducted a longitudinal study of the embryonic forebrain after E14.5 to investigate whether other accumulations were present at later prenatal stages. In addition to the CSA, we found only one large and dense accumulation of ATM-like cells at the midline between E16.5 and E18.5, at the CSB (Figures 1F and S1G). This hotspot was located below the CC, consistent with where post-natal ATM reside later in life21,22,42,43,46 and indicating a continuum of ATM accumulations that spans the pre- and post-natal periods. Using a conserved combination of core ATM markers (Spp1, Clec7A, GPNMB, and Mac2), we found that approximately 70% of MG accumulating at the CSB co-expressed ATM markers at E18.5 (Figure 1F).
These data show that MG expressing an ATM signature accumulate at the embryonic CSA and CSB, two boundaries between the neocortex and adjacent brain structures.
MG maintain structural integrity at ATM-dense cortical boundaries
Accumulation of ATM-like MG at the CSA and CSB occurs much earlier than the extensive generation of other glial cells or the development of myelin, both of which have been associated with post-natal ATM functions. To investigate the potential role of MG in the development of the CSA and CSB, we examined various established and novel models of macrophage and MG depletions by mainly targeting the colony-stimulating factor 1 receptor (CSF1R) signaling pathway, which is required for microglial survival.47,48 We first administered a CSF1R-blocking antibody (AFS98) to dams at E6.5 and E7.5, causing a transient depletion of microglial progenitors and a severe depletion of MG until E18.511,40,49 (Figure S2A), followed by a progressive microglial repopulation during the first post-natal week.11,40,49 We also achieved similar depletion/repopulation by feeding pregnant dams with PLX3397,50,51 a pharmacological inhibitor of CSF1R, between E6.5 and E15 (Figure S2B) and compared with Pu.1 mutant embryos that lack all myeloid cells, independently of the CSF1R signaling.52 As these models target both MG and other macrophage populations over a broad embryonic period, we also performed more transient depletions by treating pregnant dams with PLX3397 for only 3 days, between E12.5 and E15, overlapping the timing of microglial accumulation at the CSA (Figure S2C). Finally, and importantly, we examined Csf1r▵FIRE▵/FIRE mice that lack MG yet retain most other brain macrophages53,54 and enable to interrogate MG-specific roles in brain circuits.10,55 In all the mutants or depleted embryos, including the 3 days PLX3397 treatment and Csf1r▵FIRE▵/FIRE embryos, we found that the lack of MG induced a large “cavitary lesion” at the CSA starting at E14.5 (Figure 2A) and persisting until E18.5 (Figure 2A). These cavities, which formed where ATM-like MG normally accumulate (Figure 1E), lacked cell bodies and showed no contact with the ventricles, prompting us to examine them using electron microscopy (EM) (Figure S2D). As observed in PLX3397-treated embryos, the cavitary lesions did not contain cells or displayed a basal lamina (Figure S2D), but rather comprised sparse irregular membrane debris, resembling cysts, or pseudocysts that are reported in several human pathologies, including leukoencephalopathies.56 In contrast, we did not observe such cavitary lesions in models perturbing already known developmental functions of MG, such as Cx3cr1, Dap12/TyroBP, and complement receptor 3 (CR3) mutants, or in embryos exposed to a mild prenatal inflammation (maternal immune activation [MIA]) (Figure S2E). To further assess the impact of microglial depletion on the other cortical boundary where ATM-like cells accumulate, we examined the CSB in all the models of MG depletion (Figures 2A and S2A–S2C). Similar to what we observed at the CSA, the absence of MG at the CSB induced midline cavitary lesions, where embryonic ATM-like MG normally accumulate (Figure 2B).
Figure S2.

Models of macrophage depletion and functional alteration, related to Figure 2
(A–C) Confirmation of the depletion of microglia in brains of embryonic Cx3cr1gfp/+ mice at E14.5 and E18.5, where pregnant dams were subjected to (A) intraperitoneal injection of a CSF1R blocking antibody (AFS) at E6.5 and E7.5 (E14.5, ncontrols = 21, nAFS = 6; E18.5, ncontrols = 15, nAFS = 17); (B) feeding with PLX3397, a pharmacological inhibitor of the CSF1R pathway, from E6.5 to E15.0 (E14.5, ncontrols = 5, nPLX3397 = 5; E18.5, ncontrols = 12, nPLX3397 = 11); and (C) feeding with PLX3397 from E12.5 to E15.0 (E15.5, ncontrols = 5, nPLX3397-E12 = 9; E18.5, ncontrols = 12, nPLX3397-E12 = 10). Open and solid arrowheads indicate the accumulation of GFP-positive cells at the CSA and local CSA tissue lesion in the absence of GFP-positive cells, respectively.
(D) Transmission electron microscopy image of the CSA lesion in the brain of E14.5 embryos from PLX3397-treated dams, showing the presence of cell debris (solid arrowheads) but the absence of basal membrane (n = 3).
(E) Coronal sections through hemibrains of E14.5 embryos from wild-type mice; those exposed to mild maternal immune activation (MIA); and Cx3cr1−/−, Dap12/TyroBP−/−, and CR3−/− mutant embryos showing the absence of CSA lesions (open arrowheads) (at least n = 6 for each condition).
Scale bars: 500 μm in (A)–(C), and (E); 10 μm in (D, low magnification); and 2 μm in (D, high magnification).
CSA, cortico-striato-amygdalar boundary; Ncx, neocortex; Str, striatum.
Figure 2.

Microglia are required for tissue integrity at the embryonic CSA and CSB
(A) Hoechst staining of hemibrain coronal sections from E14.5 or E15.5 embryos, showing CSA integrity in controls (open arrowhead) and cavitary lesions in the absence of microglia (solid arrowheads) (ncontrols = 27, nPu1KO = 6, nAFS = 4, nPLX3397 = 8, nPLX3397-E12 = 11, nCSF1RFire = 7); and from E18.5 embryos, showing the localization of CSA cavitary lesions, bordering the neocortex (Ncx), striatum (Str), and amygdala (Am) (ncontrols = 35, nPu1KO = 5, nAFS = 7, nPLX3397 = 13, nPLX3397-E12 = 11, nCSF1RFire = 6).
(B) L1-immunolabeling stains the corpus callosum (CC) and fornix (Fx), highlighting CSB integrity in controls (open arrowheads) and cavitary lesions (solid arrowheads) in various models disrupting microglial colonization (ncontrols = 21, nPu1KO = 4, nAFS = 5, nPLX3397 = 9, nPLX3397-E12 = 11, nCSF1RFire = 5).
(C) Cavity reconstruction (yellow) after whole hemibrain clearing using iDISCO, highlighting the absence of cavities in controls and stereotypically located cavities in two depletion models (ncontrols = 4, nAFS = 3, nPLX3397 = 4) and enabling 3D quantification of lesion volumes (Imaris Software).
(D) Whole-head MRI scans of E15.5 or P0 mice, showing low-intensity signals confirming the formation of lesions at the CSA of PLX3397-treated embryos (yellow solid arrowheads) in contrast to controls (yellow open arrowheads) (ncontrols-E15 = 4, nPLX3397-E15 = 3, ncontrols-P0 = 3, nPLX3397-P0 = 3).
(E) Whole-head MRI scans of E15.5 or E18.5 mice, showing low-intensity signals confirming the formation of lesions at the CSA of Csf1rΔFIRE/ΔFIRE embryos (yellow solid arrowheads) in contrast to controls (yellow open arrowheads; ncontrols-E15 = 3, nCSF1RFire-E15 = 4, ncontrols-E18 = 4, nCSF1RFire-E18 = 4).
(F) Whole-head MRI scans of E18.5 mice, showing low-intensity signals confirming the formation of lesions at the CSB of Csf1rΔFIRE/ΔFIRE embryos (yellow solid arrowheads) in contrast to controls (yellow open arrowheads) (ncontrols-E18 = 4, nCSF1RFire-E18 = 4).
Graphs show means ± SEM. Mann-Whitney U tests were performed for statistical comparison, ∗∗p < 0.01. Scale bars: 200 μm in (A) and (B); 800 μm in (C); and 1 mm (D)–(F).
Am, amygdala; CC, corpus callosum; CSA, cortico-striato-amygdalar boundary; CSB, cortico-septal boundary; Fx, fornix; Ncx, neocortex; OB, olfactory bulb; Se, Septum; Str, striatum.
See also Figure S2 and Video S1. Lateral rotation of an AFS-treated E16.5 hemibrain cleared by iDISCO, related to Figure 2, Video S2. Lateral rotation of a PLX3397-treated E16.5 hemibrain cleared by iDISCO, related to Figure 2, Video S3. Rostrocaudal progression through an E16.5 control hemibrain cleared by iDISCO, related to Figure 2, Video S4. Rostrocaudal progression through a PLX3397-treated E16.5 hemibrain cleared by iDISCO, related to Figure 2.
To rule out the possibility that either CSA or CSB cavities were caused by tissue damage during sectioning, we analyzed whole brains using iDISCO clearing and magnetic resonance imaging (MRI).57,58 iDISCO clearing and 3D hemibrain reconstruction performed in both CSF1R depletion models revealed CSA lesions located in the caudal third of the telencephalic vesicles, at the border between the caudal insular cortex, claustrum, striatum, and amygdala (Figure 2C; Videos S1 and S2). Similarly, whole-head MRI scans of fixed PLX3397-treated and Csf1r▵FIRE▵/FIRE embryos, and newborns pups showed bright bilateral hyperintensities after contrast enhancement, unequivocally confirming the presence of CSA cavitary cystic lesions in the absence of MG (Figures 2D and 2E). Likewise, midline CSB cavitary lesions was observed with iDISCO transparization (Videos S3 and S4), and in MRI scans when the contrast at the midline enabled visualization (Figure 2F).
The hemibrain was imaged on a light-sheet microscope and reconstructed using the Imaris software. Green signal encompasses non-depleted CX3CR1-GFP+ cells, tissue autofluorescence delineates the hemibrain morphology and the cavitary lesion at the CSA was reconstructed and pseudocolored in yellow.
The hemibrain was imaged on a light-sheet microscope and reconstructed using the Imaris software. Green signal encompasses non-depleted CX3CR1-GFP+ cells, tissue autofluorescence delineates the hemibrain morphology and the cavitary lesion at the CSA was reconstructed and pseudocolored in yellow.
The hemibrain was imaged on a light-sheet microscope and reconstructed using the Imaris software, showing normal morphology at the CSA and CSB (open arrowheads). The movie progresses through light sheet photographs acquired along the coronal rostrocaudal axis of the sample at a 3 μm Z step size.
The hemibrain was imaged on a light-sheet microscope and reconstructed using the Imaris software, showing a cavitary lesion at the CSA (solid arrowheads).The movie progresses through light sheet photographs acquired along the coronal rostrocaudal axis of the sample at a 3 μm Z step size.
Overall, the embryonic absence of MG leads to a sequential loss of structural integrity at both the CSA and CSB, two sites where ATM-like MG normally accumulate, revealing an important physiological function of MG during prenatal development.
MG limit the formation of cavitary lesions due to morphogenetic stress
The involvement of MG in maintaining tissue cohesion at cortical boundaries raised the question of the specificities of these boundaries. The CSB, located at the border between the cerebral cortex and the septum, is subjected to morphogenetic constraints that increase as the cerebral cortex grows, and displays a normally occurring cavity called the cavum septum that resorbs over development.46,59 The CSA, similarly to the CSB, is located at the border between the cerebral cortex and structures growing at different rates and in distinct directions, making it a region of morphogenetic tensions and tissue reorganization.60,61,62 We therefore focused on the CSA to investigate whether such boundaries exhibit specific features and could be affected by morphogenetic constraints.
We first characterized the local CSA niche of ATM during normal physiological development at E14.5. Interestingly, the CSA was characterized by microcavities that were only sparsely populated by cells and that were bordered by MG expressing the ATM markers Mac2 and Spp1 (Figure 3A). EM confirmed the presence of microcavities surrounded by MG at the CSA (Figure 3B). These microcavities lacked a basal lamina, contained cell membrane fragments, and were systematically abutted by MG displaying an amoeboid morphology, which is a characteristic of CSA MG (Figure 3B). Our observations raised the possibility that such physiological microcavities might contribute to the local microglial recruitment and/or the induction of an ATM-like state. Furthermore, they suggested that in the absence of MG, microcavities might progress into larger lesions due to local morphogenetic constraints, with notable effects on fetal brain integrity.
Figure 3.

Microglia limit the progression of CSA microcavities into large lesions in response to morphogenetic constraints
(A) IBA1, Spp1, and Mac2 immunostaining findings indicate that ATM line microcavities (solid arrowheads), visible by Hoechst counterstaining at E14.5 (n = 6, from at least two distinct litters).
(B) Transmitted electron microscopy (EM) also reveals the presence of microcavities (solid arrowheads), lined with amoeboid microglia (green pseudo-color), and containing membrane fragments (open arrowheads) (n = 3, from at least two distinct litters).
(C and D) Coronal hemisections (C) from control and Emx1cre/+;RhoAfl/fl mice, the latter displaying a periventricular nodular heterotopia (PVNH) visible from E18.5 onward (dotted lines). Hoechst counterstaining shows the absence of CSA lesions in controls and mutants at E15.5 (open arrowheads), a striking CSA lesion (solid arrowheads) in 50% of the E18.5 mutants and in 100% of the mutants at P8 (ncontrols-E15 = 6, nheterotopia-E15 = 5, ncontrols-E18 = 8, nheterotopia-E18 = 10, ncontrols-P8 = 10, nheterotopia-P8 = 15). Quantification in (D) uses values that represent the scoring of lesion severity, scored from 0 to 2, as detailed in Table S2.
(E) Coronal hemisections of E15.5 brains from Brn4cre/+; Wnt3adta/+ embryos show an ablation of the thalamus (white asterisk) and a global change in brain shape but no marked impact on the CSA (ncontrols = 4, nthalamusdeleted = 5).
(F and G) Coronal hemisections (F) of E15. 5 brains from control and Brn4cre/+; Wnt3adta/+ mice exposed to PLX3397 between E12.5 and E15.5. While PLX3397-exposed controls consistently displayed lesions (open arrowheads), PLX3397-treated mutants exhibited smaller lesions or no lesions (solid arrowheads) despite effective local depletion as assessed by IBA1 staining (ncontrols = 4, nthalamusdeleted = 5, nPLX3397 = 8, nthalamusdeleted-PLX3397 = 5, from at least two distinct litters). Quantification in (G) uses values that represent the scoring of lesion severity, scored from 0 to 2, as detailed in Table S2.
Graphs show means ± SEM. Mann-Whitney U tests were performed for statistical comparison, ∗p < 0.05; ∗∗∗p < 0.001; ns, non significant (p > 0.05). Scale bars: 200 μm (A, left); 100 μm (A and F, high magnifications); 2.5 μm (B); and 500 μm (C, E, and F, low magnification).
Am, amygdala; CSA, cortico-striato-amygdalar boundary; PVNH, periventricular nodular heterotopia; Ncx, neocortex; Str, striatum; Th, thalamus.
See also Figure S3.
To experimentally test the hypothesis that CSA cavitary lesions form due to unrestrained stress linked to morphogenesis, we examined the embryonic brains of mice in which genetic mutations generate increased or decreased developmental constraints (Figures 3C–3G). First, we used a genetic model of conditional RhoA inactivation (Emx1cre/+;RhoAfl/fl), in which a large periventricular nodular heterotopia (PVNH) forms from E15.563 (Figures 3C and 3D), increasing constraints on the developing cortex and affecting its thickness and organization. At E18.5 and onward, when the heterotopia is large and cortical morphogenesis affected, we observed visible lesions at the CSA in 50% of mutant embryos, progressing into large lesions observed in all cases at P8 (Figures 3C and 3D). While the distribution of MG was not drastically altered at early stages, the growth of the heterotopia correlated with the recruitment of MG at the CSA at E18.5, even in embryos that did not show yet marked lesions at this time point (Figure S3). Our findings hence reveal that drastically increasing morphogenetic tensions can, even in the presence of MG, ultimately lead to CSA lesions, thereby revealing that this region is a site of morphogenetic vulnerability.
Figure S3.

Microglial recruitment is modulated by global changes in brain morphogenesis, related to Figure 3
(A) IBA1 immunolabeling in coronal sections shows the distribution of microglia in control and Emx1cre/+; RhoAfl/fl mutant brains at E18.5. Mutant mice display accumulations of microglia at the lesioned CSA and reduced numbers in surrounding areas, despite lack of overall difference in microglial numbers in the caudal CSA region or the rostral neocortex (n = 4 at least from 2 distinct litters for both controls and mutants).
(B) Quantifications of the IBA1+ cells in all the CSA region, rostral neocortex and within CSA subregions (1–3).
Graphs show means ± SEM. Mann-Whitney U tests were performed for statistical comparison, ∗p < 0.05. Scale bars, 200 μm.
Am, amygdala; CSA, cortico-striato-amygdalar boundary; Ncx, neocortex; Str, striatum.
Although MG were not able to compensate for such drastic developmental changes, they could nonetheless limit the progression of cavitary lesions at the CSA in the context of milder morphogenetic stress, as occurring in physiological conditions. To directly investigate this possibility, we tested whether alleviating morphogenetic constraints would limit lesion formation in the context of microglial absence. We took advantage of Brn4cre/+; Wnt3Adta/+ embryos, which lack a large brain structure, the thalamus (Th),64 and therefore experience reduced morphogenetic stress in the developing forebrain (Figures 3E–3G). As expected, the CSA was preserved in E15.5 mutants (Figure 3E). Remarkably, the treatment of Brn4cre/+; Wnt3Adta/+ dams with PLX3397 during a short time window between E12.5 and E15.5 revealed that mutant embryos with reduced morphogenetic stress showed fewer and smaller lesions when MG are depleted (Figures 3F and 3G). These observations thus show that the extent of CSA lesions is driven by morphogenetic constraints.
Taken together, our results indicate that the CSA is a site of developmental vulnerability, where MG accumulate in response to morphogenetic constraints. In physiological conditions, MG prevent the progression of cavitary lesions due to morphogenetic stress, thereby preserving the structural integrity of this fetal cortical boundary.
ATM-like MG are induced by morphogenetic stress and tissue lesions
Our results indicate that increasing or alleviating morphological tensions alters the recruitment of MG and tissue integrity at the CSA. Since ATM-like MG are normally detected at this boundary, we wondered whether changes associated with morphogenetic stress could contribute to the induction of the ATM-like state.
To address this question, we first examined whether core ATM markers were expressed at the CSA of both Emx1cre/+; RhoAfl/fl and Brn4cre/+; Wnt3Adta/+ embryos (Figures 4A–4D). We found that the very local and specific recruitment of MG at the CSA of Emx1cre/+; RhoAfl/fl embryos (Figure S3) was characterized by a marked increase in proportion of amoeboid cells expressing the ATM marker Spp1 at E18.5 (Figures 4A and 4B). Conversely, we observed a conserved recruitment of MG at the CSA in Brn4cre/+; Wnt3Adta/+ embryos, although with a reduced proportion of cells co-expressing the ATM-factor Spp1 (Figures 4C and 4D). Thus, drastic changes in morphogenetic constraints correlated with the recruitment of MG at the CSA and the local and specific induction of ATM-like features.
Figure 4.

ATM-like microglia are induced by morphogenetic constraints and tissue mechanical lesions
(A and B) Coronal E18.5 brain hemisections immunostained with IBA1 and Spp1 showing a marked recruitment of Spp1-expressing microglia at the CSA of Emx1cre/+;RhoAfl/fl mice, with approximately 75% of Spp1-positive CSA microglia in mutants, but 8% of CSA cells detected in controls at this stage (n = 4 at least for each condition, from a minimum of two distinct litters).
(C and D) Coronal E15.5 brain hemisections immunostained for IBA1 and Spp1 showing a significantly diminished percentage of CSA microglia expressing Spp1 in E15.5 Brn4cre/+; Wnt3adta/+ mutant mice compared to controls, despite a conserved number of accumulating cells (n = 4 at least for each condition, from a minimum of two distinct litters).
(E) Schematic representation of in utero lesion (IUL) procedure induced by mechanical poking of the neocortex using a glass capillary.
(F) Coronal sections through the E14.5 neocortex of control and IUL embryos collected 2.5 h after lesion induction, showing amoeboid Cx3cr1gfp-positive cells accumulating at the lesion site and the co-expression of Spp1 and Mac2 (solid arrowheads) in IUL embryos but dispersed Cx3cr1gfp-positive cells and no expression of ATM markers in controls (ncontrols = 9, nIUL = 8, from at least two distinct litters). Quantification of the percentage of Cx3cr1gfp-positive cells at the lesion site co-expressing ATM markers (nSPP1 = 4, nMAC2 = 3, nGPNMB = 4 from at least two distinct litters).
Graphs show means ± SEM. Mann-Whitney U tests were performed for statistical comparison, ∗p < 0.05. Scale bars: 200 μm in (A) and 100 μm in (C) and (F).
Am, amygdala; CSA, cortico-striato-amygdalar boundary; Ncx, neocortex; Str, striatum.
Since modifications in morphogenetic stress are linked to tissue damage (Figure 3), we further tested whether creating exogenous lesions was sufficient to recruit embryonic MG and/or induce ATM-like features. To this aim, we induced in utero lesions (IULs) in the neocortex of E14.5 Cx3cr1gfp/+ embryos by performing a stab-wound using a glass pipette (Figure 4E). We examined the brains 2.5 h after the injury to assess the initial response (Figures 4E and 4F). After 2.5 h, we already observed a marked accumulation of Cx3cr1gfp-positive cells at the lesion site, with approximately 30% of these cells also expressing the ATM markers Spp1 or Mac2, and 10% expressing GPNMB (Figure 4F). Thus, in utero mechanical stab-lesions led to rapid recruitment of Cx3cr1gfp-positive cells that expressed some ATM-core markers at the site of lesion (Figure 4F).
Overall, these results indicate that morphogenetic stress and induced tissue damage can drive the local recruitment of MG with ATM-like features, which in turn, could contribute to limit the progression of large cavitary lesions at the CSA.
ATM-factor Spp1 contributes to structural integrity at cortical boundaries
The role of MG in tissue cohesion at ATM-like hotspots raises the question of the underlying molecular mechanisms. As mentioned, we did not observe CSA cavitary lesions in genetic models perturbing known developmental functions of MG, such as Cx3cr1, Dap12/TyroBP, and CR3 mutants (Figure S2E), suggesting the involvement of alternative pathways. We focused on ATM-specific molecules known to modulate immune functions and contribute to adhesion and tissue remodeling, such as Spp1 and GPNMB.65,66 In particular, Spp1, which encodes OPN, has been linked to tissue remodeling, wound healing and repair processes, and its secreted form has recently been shown to promote microglial phagocytosis in the context of neuronal damage and in early stages of Alzheimer’s disease.67,68 We took advantage of available global knockouts, as these factors are highly and specifically expressed by MG in the prenatal brain at these developmental time points (Figure S4A). While mice lacking Gpnmb displayed normal brain structure (Figures S4B and S4C), approximately 50% of those lacking Spp1 showed cavitary lesions at the CSA at E14.5 (Figure 5A), and 70% had lesions at the CSB at E18.5 (Figure 5B). The cavities rapidly resorbed overtime, within 24 h, suggesting that while Spp1/OPN contributes to maintaining structural integrity in the developing brain (Figure S4D), additional factors or microglial properties appear to be implicated. Importantly, ATM factors, including Mac2 and GPNMB, were still expressed in Spp1 mutants at both the CSA and CSB (Figures 5C and 5D), indicating that Spp1 is not required for the induction of these ATM-core factors but rather contributes to microglial function in brain integrity at cortical boundaries.
Figure S4.

Spp1 inactivation transiently alters CSA and CSB integrity, related to Figure 5
(A) Dot plot reporting the average expression level of Spp1 and Gpnmb in various cell types of La Manno et al.30 scRNA-seq data across different embryonic time points (229,948 cells). Color represents the average expression level across all cells within a cluster, while the dot size indicates the percentage of cells expressed in that cluster.
(B) E14.5 coronal hemisection stained with Hoechst, showing the integrity of the CSA in control mice and Gpnmb−/− mutants (open arrowheads) (ncontrols = 18, nGpnmbKO = 13, from at least 2 distinct litters).
(C) L1 immunolabeling showing the integrity at the CSB in control mice and Gpnmb−/− mutants (open arrowheads) (ncontrols = 23, nGpnmbKO = 8, from at least two distinct litters).
(D) CSA and CSB lesions in Spp1−/− mutants are transient and have resorbed, respectively, by E18.5 (ncontrols = 9; nSpp1KO = 3, from at least 2 distinct litters) and P3 (ncontrols = 6; nSpp1KO = 4, from at least two distinct litters). Graphs show the percentages of brain with lesions, but individual dots represent brains with lesion (100) or no lesion (0), to illustrate sample numbers.
(E) UMAP visualization of all sorted cells (from wild-type [WT] and Spp1−/− E14.5 and E18.5 embryos) colored by annotated clusters (BroadCellType).
(F) UMAP of all sorted cells split and colored by WT (light gray) and Spp1−/− (dark gray) conditions.
(G) Dot plot of scaled average expression and percentage of top 5 differentially expressed genes (DEGs) by annotated macrophage clusters (RefinedCellType).
Scale bars, 200 μm.
ATM, axon-tract associated microglia; BAM, border associated macrophages; CC, corpus callosum; CSA, cortico-striato-amygdalar boundary; CSB, cortico-septal boundary; Fx, fornix; Ncx, neocortex; MG, microglia; PVM, perivascular macrophages; Se, septum; Str, striatum; WT, wild-type.
Figure 5.

ATM-core factor Spp1 contributes to tissue integrity at the CSA and CSB
(A) Coronal hemisections of E14.5 brains stained with Hoechst reveal CSA disruption in 50% of Spp1−/− mutants (solid arrowheads) compared to controls (open arrowheads) and in 100% of PLX3397-treated embryos (solid arrowheads) (ncontrols = 18, nSpp1KO = 21, nPLX3397 = 13). Quantification of CSA lesions across models.
(B) L1 immunolabeling enables axon visualization (open arrowheads) and midline lesions (solid arrowheads) in approximately 70% of Spp1−/− mutants compared with 100% in PLX3397-exposed embryos (ncontrols = 23, nSpp1KO = 20, nPLX3397 = 8). Quantification of CSB lesions across models.
(C and D) E14.5 (C) and E18.5 (D) coronal hemisections showing no differences in Mac2 and GPNMB co-expressing microglia at the CSA and CSB of controls (open arrowheads) and Spp1−/− embryos (ncontrols = 3, nSpp1KO = 3, at each stage from at least two distinct litters).
(E) UMAP visualization of single-cell RNA sequencing (scRNA-seq) data representing macrophage subsets extracted from wild-type (WT) and Spp1−/− E14.5 and E18.5 embryos colored by annotated clusters (RefinedCellType).
(F) Violin plot of normalized and scaled Spp1 expression across annotated clusters between wild-type (WT) (light gray) and Spp1−/− (KO)(dark gray) mice showing that Spp1 expression is largely restricted to WT ATM (cluster 5).
(G) Volcano plot of differentially expressed genes (DEGs) between WT and Spp1−/− conditions in ATM cells (false discovery rate[FDR]-adjusted p < 0.05 and avgerage_log2FC > 0.3). Genes downregulated in Spp1−/− embryos are displayed in orange, while the upregulated ones are shown in green, and some genes were manually annotated.
(H) Bar plots of top Metascape gene set enrichment of DEGs (G) in both WT or Spp1−/− conditions, highlighting upregulated (green) and downregulated pathways (orange) in Spp1−/− embryos versus controls, with pathways related to phagocytosis highlighted by a red arrowhead.
(I) Brain sections from E15 Cx3cr1gfp/+ mice showing specific fibronectin 1 (FN1) labeling within GFP- and Mac2-positive ATM microglia at the CSA (performed on brain sections of at least three mice from two distinct litters).
(J) High magnification confocal acquisition and 3D cell reconstructions (Imaris software) of immunolabeled sections from E14.5 embryonic brains showing Cx3cr1gfp-positive CSA microglia with FN1 signal inside cell bodies (performed on brain sections of at least three mice from two distinct litters).
(K) Comparison of the percentage of FN1 volume measured (Imaris software) inside individual CSA microglia shows a reduction in Spp1 mutants versus controls (ncontrols = 17, nSpp1KO = 14, from at least two distinct litters).
Graphs show percentages in (A) and (B) and means ± SEM for all others. Fisher’s exact test was performed to compare distributions of cases with lesions in controls, Spp1−/−, and PLX3397-exposed embryos (A and B), and Mann-Whitney U tests were performed for statistical comparison in all other graphs,∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ns, non significant (p > 0.5). Scale bars: 200 μm (A, B low magnification, and D); 100 μm (C and F); 5 μm (G, left); and 2 μm (3D reconstructed cells in F and G).
ATM, axon-tract-associated microglia; BAM, border associated macrophages; CC, corpus callosum; CSA, cortico-striato-amygdalar boundary; CSB, cortico-septal boundary; DEGs, differentially expressed genes; Fx, fornix; Intravasc mac, intravascular macrophages; Ncx, neocortex; PVM, perivascular macrophages; Se, Septum; Str, striatum; WT, wild type.
See also Figure S4 and Table S3. Genes defining the clusters identified by single-cell transcriptomic analyses on all the sorted cells from wild type and Spp1−/−, related to Figures 5 and S4, Table S4. Genes defining the clusters identified by single-cell transcriptomic analyses on the macrophages from wild type and Spp1−/−, related to Figure 5, Table S5. DEGs in ATM in wild type versus Spp1−/−, related to Figure 5, Table S6. Metascape analyses of ATM in wild type versus Spp1−/−, related to Figure 5.
To investigate how Spp1 inactivation may affect embryonic microglial functions, we performed sc transcriptomic analyses on macrophages from both wild-type (WT) and Spp1 mutants at E14.5 and E18.5 (Figures 5E–5H and S4E–S4G). Using MACS-sorting of CD45-positive cells from forebrains at these two time points, in which Spp1 inactivation impairs CSA and CSB integrity, we identified individual clusters corresponding to different cell types and states (Figure S4E; Table S3), that were all present in control and mutant conditions (Figure S4F). We next focused on macrophages (Figures 5E–5H; Table S4) and observed high Spp1 expression selectively in the ATM cluster of controls (Figure 5F), confirming our transcriptomic (Figure S4A) and immunostaining analyses (Figures 5C and 5D). We further assessed differentially expressed genes (DEGs) between ATM-like cells in controls and mutant embryos (Table S5) and found that the transcriptomic signature of ATM-like cells is altered in mutant embryos (Figures 5G and 5H). Both direct comparison (Figure 5G) and pathway analyses using Metascape (Figure 5H; Table S6) indicated that Spp1 inactivation impacts specific functional pathways in ATM-like cells: the expression of several ATM-core markers (such as Lgals3, ApoE, and Ftl1) was decreased and, among several altered pathways, phagocytosis-related pathways were downregulated, consistent with previously described effects of secreted Spp1 on MG during brain damage and Alzheimer’s disease.67,68
To further confirm whether Spp1 inactivation alters phagocytic properties in vivo, we first searched for substrates that ATM-like MG may engulf, focusing on the CSA (Figures 5I and 5J). In the context of tissue integrity, we examined components of the extracellular matrix (ECM) and observed an intense and punctuated staining of fibronectin 1 (FN1) in CSA MG using immunolabeling and 3D reconstructions (Figures 5I and 5J). Since Fn1 is not notably expressed by MG (Figure 5J), and FN1 punctuated staining was located in intracellular domains resembling vesicles (Figures 5I and 5J), our observations support that CSA MG engulf FN1 in physiological conditions. FN1 deposition and remodeling is a key factor in repair and wound healing,69,70,71 suggesting that its engulfment by MG might modify tissue properties. To test whether Spp1 inactivation alters this process, we measured the relative volume of FN1 within CSA MG in both controls and Spp1−/− and detected a significant reduction in mutant embryos (Figure 5K).
Our data thus show that Spp1, which regulates ATM-like microglial properties such as phagocytosis, is one of the factors contributing to maintain tissue integrity at cortical boundaries.
MG and Spp1 contribute to the rapid repair of cavitary lesions
Our results indicate that MG prevent lesion formation at sites of morphogenetic tensions in part through Spp1. Since MG are known to help repair exogenous damage in the adult brain and neonatal spinal cord,72 we further investigated whether MG and Spp1 could act by promoting tissue repair.
To address this issue, we examined whether MG contribute to the resorption of large lesions at the CSA and CSB. We first delineated the time window during which the permanent absence of MG in Csf1r▵FIRE▵/FIRE mice led to cavitary lesions at cortical boundaries. Consistently with the fact that Csf1r▵FIRE▵/FIRE mice did not show gross morphological alterations in adults,10,53 we found that while lesions were still observed in all Csf1r▵FIRE▵/FIRE pups at P7, they progressively resorbed during the second and third post-natal week, with no lesions detected by P28/P30 (Figures 6A, S5A, and S6A). While these results reveal the existence of potential compensatory mechanisms for lesion closure later in post-natal life, they highlight that MG are required to not only maintain structural integrity prenatally but also during the first post-natal week.
Figure 6.

Local microglia repopulation at the CSA drives the rapid repair of cavitary lesions
(A) Coronal sections showing the CSA region in controls and Csf1rΔFIRE/ΔFIRE pups, highlighting that cavitary lesions are systematically observed in P7 Csf1rΔFIRE/ΔFIRE pups (solid arrowheads), but absent in P7 controls or at P30 in both conditions (open arrowheads) (ncontrols-E18 = 5, nCSF1RFire-E18 = 6, ncontrols-P7 = 3, nCSF1RFire-P7 = 6, ncontrols-P30 = 3, nCSF1RFire-P30 = 5, from at least two distinct litters for each stage and condition). Graphs show percentages of brain with lesions, but individual dots represent brains with lesion (100) or no lesion (0) to illustrate variability.
(B and C) Coronal sections of brains prenatally exposed to PLX3397 showing the CSA region (solid arrowheads) at P0, P3, P7, and P20. Cavitary lesions progressively resorbed, with a significant proportion being resorbed at P3, and almost all achieved by P7, concurrently with the overall repopulation of Cx3cr1gfp-positive cells, which accumulated at the site of lesion closure (solid arrowhead) (ncontrols-P0 = 6; nPLX3397-P0 = 6; ncontrols-P3 = 6; nPLX3397-P3 = 5; ncontrols-P7 = 5; nPLX3397-P7 = 8; ncontrols-P20 = 7; nPLX3397-P20 = 7, from at least two distinct litters for each stage and condition). Graph in (B) is presented as in (A), while graph in (C) displays means ± SEM.
(D) Repopulating microglia numbers are comparable at the CSA of controls and PLX3397-exposed embryos at P3, even if microglia numbers remain lower in the surrounding area of resorbing brains (ncontrols-P3 = 6; nPLX3397-P3 = 5, from at least two distinct litters). Graph in (D) show means ± SEM.
Fisher’s exact test (A and B) and Mann-Whitney U test (C and D) were performed for statistical comparison, ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ns, non significant (p > 0.5). Scale bars: 300 μm in (B) and (C) and 200 μm in (A) and (D).
Am, amygdala; CSA, cortico-striato-amygdalar boundary; Ncx, neocortex; Str, striatum.
See also Figures S5 and S6.
Figure S5.

CSB lesion closure timeline is similar to the one at the CSA, related to Figure 6
(A) Coronal sections counterstained with Hoechst showing the CSB region in Csf1rΔFIRE/ΔFIRE mice. Cavitary lesions (solid arrowheads) were systematically observed in mutants up to P7, while they were absent in controls or resorbed in mutants at P30 (open arrowheads), similarly to what was observed at the CSA (ncontrols-E18.5 = 6, nCSF1RFire-E18.5 = 5, ncontrols-P7 = 3, nCSF1RFire-P7 = 6, ncontrols-P30 = 3, nCSF1RFire-P30 = 5).
(B) Coronal sections showing the CSB region (open arrowheads) in pups prenatally exposed to PLX3397 at P0, P3, P7, and P20. Cavitary lesions (solid arrowheads) progressively resorbed during the first post-natal week, with a similar timeline to the CSA (ncontrols-P0 = 6; nPLX3397-P0 = 3; ncontrols-P3 = 6; nPLX3397-P3 = 4; ncontrols-P7 = 5; nPLX3397-P7 = 10; ncontrols-P20 = 6; nPLX3397-P20 = 6).
Graphs show the percentages of brain with lesions, but individual dots represent brains with lesion (100) or no lesion (0) to illustrate variability. Fisher’s exact test was performed for statistical comparison, ∗p < 0.05; ∗∗p < 0.01; ns, non significant (p > 0.05). Scale bars, 200 μm.
CC, corpus callosum; Ncx, neocortex; Se, septum.
Figure S6.

Long-term impact on the CSA region after lesion closure, related to Figure 6
(A) P28 coronal hemisections of brains immunostained with the pan axonal neurofilament marker SMI-312 and myelin basic protein (MBP) and Hoechst reveal alterations in the CSA region in Csf1rΔFIRE/ΔFIRE mice. Axonal tracts of the amygdalar capsule, located between the neocortex and the basolateral nucleus of the amygdala (BLA), are disorganized in mutants (solid arrowheads) compared with controls (open arrowheads), highlighting long-term morphological consequences of microglial absence and early life CSA lesions, even after lesion closure (ncontrols = 4, nCSF1RFire = 4, from at least two distinct litters).
(B) P20 coronal hemisections of brains immunostained with the pan axonal neurofilament marker SMI-312 and myelin basic protein (MBP) (left) or with MBP and FOXP2 (right) reveal alterations in the CSA region of mice prenatally exposed to PLX3397 (solid arrowheads) compared with controls (open arrowheads). Axonal tracts of the amygdalar capsule and associated FOXP2-positive inhibitory interneurons are disorganized, highlighting long-term morphological consequences of early microglial absence and transient CSA lesions (ncontrols-SMI/MBP = 9; nPLX3397-SMI/MBP = 7; ncontrols-MBP/Foxp2 = 5; nPLX3397-MBP/Foxp2 = 5; from at least two distinct litters for each condition).
(C) Schematic representation of the experimental approach (left) used to record in P60 slices both excitatory post-synaptic currents (EPSCs) and inhibitory post-synaptic currents (IPSCs) from BLA pyramidal neurons in response to stimulation of the amygdalar capsule. Importantly, amygdalar capsule stimulation was designed to trigger EPSCs with an amplitude between −150 and −350 pA in both mutants and PLX3397-prenatally exposed mice, and IPSCs were subsequently measured by changing the holding potential in order to evaluate the inhibition/excitation ratio (I/E). EPSCs and IPSCs amplitude and I/E ratio (right) show an altered balance in PLX3397-exposed embryos, compared with controls at P60 (ncontrols = 18 cells from 5 animals and at least 2 distinct litters, nPLX3397 = 21 cells, from 5 animals and at least 2 distinct litters).
Scale bars, 500 μm (A and B, low magnification) and 100 μm (B, high magnification). Graphs show means ± SEM. Mann-Whitney U tests were performed for statistical comparison, ∗∗∗∗p < 0.0001.
Am, amygdala; ampl, amplitude; BLA, basolateral nucleus of the amygdala; CSA, cortico-striato-amygdalar boundary; EPSCs, excitatory post-synaptic currents; I/E, inhibition/excitation ratio; IPSCs, inhibitory post-synaptic currents; Ncx, neocortex.
To assess whether MG contribute to the repair of CSA and CSB lesions during the first post-natal week, we took advantage of the fact that MG start to repopulate the brain around birth after PLX3397-prenatal treatment. We found that in pups prenatally exposed to PLX3397, CSA lesions rapidly resorbed within the first post-natal days, concomitantly to the progressive microglial repopulation (Figures 6B and 6C): while all mice bore cavities at P0, 60% had already fully closed by P3 (Figure 6B). Remarkably, lesion closure was consistently associated with a dense microglial accumulation at the scar at P3, in spite of a reduced MG density in the surrounding region (Figures 6C and 6D), supporting a local role of MG at the CSA during resorption. At P7, over 70% of the mice had their CSA lesions closed and all CSA cavities were systematically resorbed by P20 (Figure 6B). We observed a similar timeline of resorption at the CSB (Figure S5B), showing that MG induced a rapid repair of lesions at both boundaries.
Despite lesion repair, long-lasting morphological defects persisted at the CSA (Figures S6A–S6C), underscoring the importance of preserving structural integrity during morphogenesis. Indeed, even in juvenile mice prenatally exposed to PLX3397 which display more transient lesions, we observed a disorganization of an axonal tract abutting the CSA, a capsule that conveys inputs to the Am, an essential brain structure which dysregulation is linked to neurodevelopmental disorders.73,74,75 Both axonal tracts and Foxp2-positive inhibitory interneurons, important to gate inputs to the Am,76,77,78 were disorganized (Figures S6A and S6B). Electrophysiological slice recordings in adult mice revealed that morphological deficits correlated with a consistent imbalance of inhibition/excitation (I/E) responses of Am pyramidal neurons following capsule stimulation (Figure S6C). Thus, even after the rapid repair of CSA lesions, this physiologically important region persistently displayed anatomical and functional wiring deficits up to adulthood.
To further explore the repair process and its potential dependence on Spp1, we focused on the CSA and examined microglial properties during the first post-natal week (Figures 7 and S7). We found that densely accumulating MG at the scar co-expressed ATM markers Spp1, GPNMB, and Mac2 at P3 (Figure 7A). These cells furthermore showed an amoeboid morphology and marked engulfment of FN1, which accumulates at the scar (Figure S7). In addition, we observed that Spp1 labeling was not only detected in and around microglial cells but also accumulating in the extracellular space at higher levels than in the adjacent neocortex or amygdala (Figure 7B). These results show that the repair process correlates with a local induction of ATM-like MG, as illustrated by the expression of core markers and FN1 engulfment, as well as the extracellular accumulation of Spp1. Like for the prevention of cavitary lesion formation, we found that the rapid repair process relied on Spp1 (Figures 7C and 7D). Indeed, by comparing at P7 WT and Spp1−/− pups exposed to PLX3397 prenatally, we observed a comparable microglial recruitment and Mac2 induction (Figure 7D), but a significant inhibition of lesion closure in mice lacking Spp1 (Figure 7C).
Figure 7.

ATM-factor Spp1 contributes to lesion repair
(A) Cx3cr1gfp-positive cells accumulating at the site of lesion closure co-express ATM markers Spp1, Mac2, and GPNMB, as shown and quantified at P3 (ncontrols = 4; nPLX3397 = 4 for each marker, from at least two distinct litters).
(B) Extracellular Spp1 signal, delineated by immunostaining and Hoechst labeling, accumulates at the resorbing CSA at P3. Graphs show the increased signal intensity at the CSA (dotted lines) compared with a mean between signal intensity measured in the surrounding neocortex (dotted lines) and amygdala (dotted lines) in PLX3397-exposed pups versus controls (ncontrols = 4; nPLX3397 = 4, from two distinct litters).
(C) In contrast to controls, Spp1−/− mutants, and PLX3397-exposed controls, PLX3397-exposed Spp1−/− mutants reproducibly displayed lesions visible by Hoechst staining (solid versus open arrowheads) (ncontrols = 7; nSpp1KO = 8; ncontrol-PLX3397 = 11; nSpp1KO-PLX3397 = 7). Values represent the scoring of lesion severity, scored from 0 to 2, as detailed in Table S2.
(D) While CSA IBA1-positive cells co-expressed Mac2 in resorbed PLX3397-treated controls at P7, they also accumulated around the lesions in PLX3397-treated Spp1 mutants, indicating that Spp1 inactivation did not prevent the expression of selected ATM markers (ncontrol-PLX3397 = 3; nSpp1KO-PLX3397 = 7).
Graphs show means ± SEM. Mann-Whitney U test was performed for statistical comparison, ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ns, non significant (p > 0.5). Scale bars: 200 μm in (A); 150 μm in (C) and (D); and 100 μm in (B).
Am, amygdala; CSA, cortico-striato-amygdalar boundary; Ncx, neocortex; Str, striatum.
See also Figure S7.
Figure S7.

Microglia engulf fibronectin 1 during CSA lesion repair, related to Figure 7
(A) Fibronectin 1 (FN1) signal after immunostaining accumulates at the P3 resorbing CSA in Cx3cr1gfp/+ pups prenatally exposed to PLX3397 in both repopulating microglia and in the tissue surrounding them, whereas little FN1 signal is detected beyond blood vessels in controls. Dotted boxes represent the areas in which the intensity of the FN1 signal in the extracellular space was measured in the CSA, neocortex (Ncx) and amygdala (Am). The quantification (right) shows the intensity of CSA FN1 signal, normalized relative to the mean of neocortex and amygdala signals in both controls and prenatally exposed PLX3397 pups (ncontrols = 4; nPLX3397 = 4, from at least two distinct litters).
(B) High magnification and 3D reconstruction of CSA microglia identified as Cx3cr1gfp-positive cells show a slight increase in the volume of FN1 inside microglia (ncontrols = 4 mice; nPLX3397 = 4 mice; from at least two distinct litters and at least 2 cells quantified and averaged per animal) and a significant increase in the number of microglia with FN1 inclusions in P3 resorbing brains compared with controls (ncontrols = 4; nPLX3397 = 5; mice from at least two distinct litters). Graphs show means ± SEM. Mann-Whitney U test was performed for statistical comparison, ∗p < 0.05; ns, non significant (p > 0.05). Scale bars: 100 μm in (A); 5 μm in (B, immunolabelings); and 2 μm in (3D reconstructions).
Am, amygdala; CSA, cortico-striato-amygdalar boundary; Ncx, neocortex.
Taken together, these data show that MG and Spp1 play crucial roles in preventing the progression of CSA microcavities into large lesions and in promoting their prompt closure, which is essential for preserving brain integrity during development. Our study reveals key functions of MG and Spp1 in maintaining structural integrity in response to morphogenetic stress and lesions, underscoring the importance of these immune cells in early brain development.
Discussion
MG have been associated with multiple functions, including axonal progression, cortical interneuron wiring, synaptic development and refinement, through a variety of receptors and signaling pathways including Cx3cr1/Cx3cl1, Trem2/Dap12, Complement, and purinergic P2Y12 receptors.2,11,12,13,14,37,79,80,81,82 Our study reveals additional essential roles of MG in preserving tissue integrity during the development of the cerebral cortex, before the emergence of potentially redundant or complementary glial cells, such as astrocytes.39,83 Indeed, because MG colonize the brain from early embryonic stages and astrocytes or oligodendrocytes are mostly produced later, MG constitute a unique and a large contingent of embryonic glial cells.2,37
In the absence of MG, lesions form at specific cortical boundaries that constitute sites of fragility and normally host accumulations of MG in a state resembling post-natal ATM.21,27,42,43,45 Our study uses a combination of global, transient and selective models of microglial depletion, to unambiguously establish a role for these immune cells in preventing the progression of microcavities into large cavitary lesions at cortical boundaries (Figures 2, 3, 6, S2, S3, and S5). By comparing permanent and transient depletions we showed that (1) the absence of MG leads to cavitary lesions during embryogenesis and up to P7 at least; (2) in the lasting absence of MG in Csf1rΔFIRE/ΔFIRE mice, lesions eventually repair after P7; (3) early post-natal repopulation of MG is sufficient to drive the rapid repair of lesions (Figures 2, 6, S2, and S5). The later closure of lesions in Csf1rΔFIRE/ΔFIRE mice highlights compensatory mechanisms potentially including fill-in of lesions due to brain growth or redundant functions of later developing cells, and is consistent with a lack of gross anatomical defects in adult Csf1rΔFIRE/ΔFIRE mice.10,53 Nonetheless, transient depletions and lesions had lasting impact on circuits, as assessed at the CSA (Figure S6), supporting that maintenance of structural integrity during morphogenesis is essential for proper brain development. Our study thus establishes that MG maintain CSA and CSB integrity and promote the rapid repair of lesions, revealing important physiological functions of MG at critical steps of early brain morphogenesis.
Both the CSA and CSB are characterized by dense accumulations of embryonic ATM-like MG, that display several common features with previously identified post-natal ATM.21,22,31,42,43 In particular, embryonic ATM-like cells and post-natal ATM/PAM share not only highly overlapping transcriptomic signatures (Figures 1 and S1), but also an amoeboid morphology and highly phagocytic features (Figure S1).21,22 As we were unable to efficiently locally deplete MG or manipulate transient cellular states, it is possible that the local accumulation of ATM-like cells at these sites might not be the only factor responsible for the observed phenotypes. Nonetheless, we discovered that transient microglial hotspots at the CSA and CSB contained dense accumulations of ATM-like cells that were remarkably similar to their post-natal counterparts, confirming that the ATM state spans the prenatal and post-natal periods.22,30
Interestingly, while the functions of post-natal ATM have largely been attributed to Igf1,42,43,44 our study reveals that Spp1/OPN, an atypical ECM-interacting factor previously linked to bone development, wound healing, inflammation,65,66,67 and microglial phagocytosis,67,68 contributes to tissue integrity and the rapid repair of large lesions in the developing brain (Figures 5, 7, S5, and S7). While inactivating Spp1 alone did not fully mimic the impact of microglial depletion, suggesting that other factors are likely involved, our findings support a contribution of the Spp1 core ATM factor to microglial functions in both tissue integrity and repair. At the mechanistic level, extracellular Spp1 accumulates at the repair site (Figure 7) and Spp1 inactivation impairs phagocytic pathways and the capacity of CSA MG to efficiently engulf FN1 (Figure 5). FN1 is an ECM component that accumulates after tissue damage or microlesions, and which remodeling is key for proper repair.69,70,71 Our results thus raise the possibility that Spp1 acts by regulating multiple processes, including its extracellular secretion and the promotion of phagocytosis, as shown by FN1 engulfment. While embryonic ATM likely engulf a variety of other cellular substrates that remain to be identified, our study suggests that Spp1 could, at least in part, act by regulating ECM remodeling, a microglial feature that has been previously involved in spinal cord repair72 and synapse regulation.84 Importantly, we also show that Spp1 and other ATM factors are very quickly induced in MG recruited to prenatal lesions (Figure 4), revealing a remarkable ability of these cells to switch states and exhibit plasticity in response to experimentally induced injury.
Core ATM factors, including Spp1/OPN, are involved in immune cell responses, ECM organization, wound healing, and regulation of inflammation and have been detected in a wide range of macrophages, during skin lesion, heart-repair after myocardial infarction or liver fibrosis.65,66,67,85,86,87 This observation indicates that at least some aspects of the “ATM program” involved in brain integrity and repair are likely not unique to MG but rather represent a basic feature shared across macrophages, which is important for the general maintenance of tissue homeostasis, particularly in response to exogenous damage or lesions. In MG, Spp1 and ATM factors are also expressed by damage-induced MG during stroke88 or neonatal spinal cord repair,72 as well as in DAM and TAM, which are respectively associated with neurodegeneration and tumors.18,20,23,27,89 Thus, the ATM program may overlap with a basic repair program in macrophages that is triggered by tissue disruption in different contexts of health and disease. Whether Spp1 is consistently involved and how it acts, which other factors contribute to ATM functions in tissue cohesion and repair, and whether other cell types could exert similar functions in the post-natal and adult brain constitute key questions to address in future studies.
The genuinely glial role of developing MG described in this study has not been reported in other species, such as zebrafish, which provides various genetic models to examine brain development in the absence of MG.90 While we identified dense ATM hotspots in both mouse and human, they have so far not been reported in zebrafish.90,91 These observations raise the intriguing possibility that the embryonic ATM-like state, and associated repair properties, might have been co-opted during evolution to ensure the proper morphogenesis of a growing cerebral cortex, which is a hallmark of mammals. In agreement, we found that increasing morphogenetic constraints in a model of cortical PVNH63 can lead to a selective breaking at the CSA boundary, strengthening the hypothesis that the CSA represents a site of morphogenetic fragility in mammals (Figures 3 and S3). In contrast, when morphogenetic constraints were alleviated in a genetic model of Th ablation, MG, and embryonic ATM were found to be less crucial in preventing lesions (Figure 3). However, in both models, we observed that the recruitment of ATM-like cells was influenced by morphogenetic constraints and the presence of lesions (Figure 4), underscoring the interaction between immune brain cells and their local niches, as seen in the aging white matter and post-natal cerebral cortex.29,32 There is thus a delicate balance between tissue fragility, embryonic ATM localization and microglial functions, which warrants further investigation into the pathways involved in ATM induction and their role in mice and across species. Overall, our findings demonstrate that the proper development of the cerebral cortex depends on the mitigation of tissue damage by brain-resident macrophages, allowing for the morphogenesis of complex structures, which is a hallmark of brain evolution.
In the context of human pathological brain wiring, lesions are typically associated with damage induced by abnormally “activated” MG in response to various triggers, such as hypoxia, inflammation, preterm birth, or congenital viral infections. For example, bilateral cysts in the temporal lobes, where the CSA is located, or midline cavities such as cavum septum pellucidum and cystic lesions, have been reported in several neurodevelopmental disorders.92,93 Our study demonstrates key roles of MG and Spp1 in maintaining tissue integrity, suggesting that such lesions may also result from a loss of physiological functions of MG during crucial stages of morphogenesis.
These findings have not only significant implications for our understanding of the fundamental mechanisms that govern brain morphogenesis and the state-specific functions of MG but also open avenues for exploring microglial contributions to brain pathology.
Limitations of the study
While our study identified a key role for MG and Spp1-dependent repair properties at cortical boundaries, it is important to acknowledge several limitations of our research. First, morphogenetic stress was indirectly assessed through genetic manipulations that distort brain morphogenesis. However, we believe that this caveat is compensated by leveraging two models that selectively increase and reduce morphogenetic constraints. Second, it was not possible to conduct local or state-specific manipulations of CSA and CSB MG. Despite several attempts to selectively manipulate this state, we were unable to efficiently and selectively eliminate this transient cell population, as reported for post-natal ATM,42,43 and consistent with our findings that this state is quickly induced in response to the local niche. Nonetheless, our results provide insights into the contribution of MG and their diversity to brain development, evolution, and pathology, highlighting their bona fide role in fetal cortical morphogenesis.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat anti-CD68 | Bio-Rad | Cat# MCA1957; RRID:AB_322219 |
| Rabbit anti-FN1 | Millipore | Cat# AB2033; RRID:AB_2105702 |
| Goat anti-FOXP2 | Santa Cruz Biotechnology | Cat# sc-21069; RRID:AB_2107124 |
| Chicken anti-GFP | Aves Labs | Cat# GFP-1020; RRID:AB_10000240 |
| Rabbit anti-IBA1 | FUJIFILM Wako Shibayagi | Cat# 019-19741; RRID:AB_839504 |
| Rabbit anti-IBA1 (human) | Abcam | Cat# ab178846; RRID:AB_2636859 |
| Chicken anti-IBA1 | Synaptic Systems | Cat# 234009; RRID:AB_2891282 |
| Rat anti-Lgals3 (MAC2) | CEDARLANE | Cat# CL8942AP; RRID:AB_10060357 |
| Rat anti-L1 | Millipore | Cat# MAB5272; RRID:AB_2133200 |
| Biotinylated rat anti-LYVE1 | Thermo Fisher Scientific | Cat# 13-0443-82; RRID:AB_1724157 |
| Rat anti-Myelin Basic Protein (MBP) | Millipore | Cat# MAB386; RRID:AB_94975 |
| Rat anti-mDectin-1 (CLEC7A) | InvivoGen | Cat# mabg-mdect; RRID:AB_2753143 |
| Mouse anti-neurofilament marker SMI-312 | BioLegend | Cat# 837904; RRID:AB_2566782 |
| Goat anti-mouse Osteoactivin (GPNMB) | R and D Systems | Cat# AF2330; RRID:AB_2112934 |
| Rabbit anti-P2Y12 receptor | AnaSpec; EGT Group | Cat# 55043A; RRID:AB_2298886 |
| Goat anti-SPP1 | R and D Systems | Cat# AF808; RRID:AB_2194992 |
| Donkey anti-chicken Alexa 10 Fluor® 488-conjugated | Jackson ImmunoResearch Labs | Cat# 703-545-155; RRID:AB_2340375 |
| Donkey anti-goat Alexa 10 Fluor® 488-conjugated | Jackson ImmunoResearch Labs | Cat# 705-545-147; RRID:AB_2336933 |
| Donkey anti-rat Alexa 10 Fluor® 488-conjugated | Jackson ImmunoResearch Labs | Cat# 712-545-150; RRID:AB_2340683 |
| Donkey anti-rabbit Alexa 10 Fluor® 488-conjugated | Jackson ImmunoResearch Labs | Cat# 711-545-152; RRID:AB_2313584 |
| Donkey anti-goat Cy3-conjugated | Jackson ImmunoResearch Labs | Cat# 705-165-147; RRID:AB_2307351 |
| Donkey anti-rabbit Cy3-conjugated | Jackson ImmunoResearch Labs | Cat# 711-165-152; RRID:AB_2307443 |
| Donkey anti-rat Cy3-conjugated | Jackson ImmunoResearch Labs | Cat# 712-165-150; RRID:AB_2340666 |
| Donkey anti-goat Alexa 10 Fluor® 647-conjugated | Jackson ImmunoResearch Labs | Cat# 705-605-147; RRID:AB_2340437 |
| Donkey anti-goat Cy5-conjugated | Jackson ImmunoResearch Labs | Cat# 705-175-147; RRID:AB_2340415 |
| Donkey anti-rat Cy5-conjugated | Jackson ImmunoResearch Labs | Cat# 712-175-150; RRID:AB_2340671 |
| Biological samples | ||
| Human fetal brain tissue | Gynaecology Department, Jeanne de Flandre Hospital, Lille, France | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Lipopolysaccharide | InvivoGen | Cat# tlrl-pelps; CAS: 93572-42-0 |
| Pexidartinib (PLX3397) | Plexxikon | CAS: 1029044-16-3 |
| Anti-CSF1R mAb (αCSF1R, clone AFS98) | Florent Ginhoux laboratory | N/A |
| Triton 100X | Eurobio | Cat# GAUTTR00-07 |
| Hoechst | Sigma Aldrich | Cat# 33342 |
| Paraformaldehyde | Sigma Aldrich | Cat# P6148 |
| Gelatin | VWR chemical | Cat# 24350.262 |
| Dibenzylether | Sigma Aldrich | Cat# 33630 |
| Dichloromethane | Sigma Aldrich | Cat# 270997 |
| Methanol | Sigma Aldrich | Cat# 34860 |
| Critical commercial assays | ||
| BD™ Ms Single Cell Sample Multiplexing Kit | BD Biosciences | Cat# 633793 |
| BD Rhapsody™ Whole Transcriptome Analysis (WTA) Amplification Kit | BD Biosciences | Cat# 633801 |
| BD Rhapsody™ Whole Transcriptome Analysis (WTA) Reagent Kit | BD Biosciences | Cat# 665915 |
| pHrodo™ BioParticles™ Conjugates for Phagocytosis and Phagocytosis Kit | Thermo Fisher Scientific | Cat# P35361 |
| Deposited data | ||
| scRNA-seq for embryonic microglia (C57BL/6) | La Manno et al.30 | http://mousebrain.org/development (“dev_all.loom”) |
| scRNA-seq for postnatal microglia (C57BL/6) | Hammond et al.22 | GSE: 121654 |
| scRNA-seq for postnatal microglia (C57BL/6) | Li et al.21 | GSE: 123025 |
| scRNA-seq for Spp1-/- and +/+ (C57BL/6) | This study | ArrayExpress accession E-MTAB-13581 https://www.ebi.ac.uk/biostudies/arrayexpress |
| Experimental models: Organisms/strains | ||
| Mouse: Cx3cr1gfp/+ | The Jackson Laboratory | RRID:IMSR_JAX:005582 |
| Mouse: Pu.1+/− | Back et al.52 | N/A |
| Mouse: Cd11c-eYFP | The Jackson Laboratory | RRID:IMSR_JAX:007567 |
| Mouse: Spp1-/- | The Jackson Laboratory | RRID:IMSR_JAX:004936 |
| Mouse: Csf1rΔFIRE/+ | Rojo et al.53 | RRID:IMSR_JAX:032783 |
| Mouse: Cd11b−/− | The Jackson Laboratory | RRID:IMSR_JAX:003991 |
| Mouse: Dap12/TyroBP−/− | Tomasello et al.94 | RRID:MGI:3818477 |
| Mouse: RhoAfl/fl | Jackson et al.95 | N/A |
| Mouse: Emx1cre/+; RhoAfl/+ | Cappello et al.63 | N/A |
| Mouse: WntA3dta/+; Brn4cre/+ | Deck et al.64 | N/A |
| Mouse: C57BL/6J | The Jackson Laboratory | Cat# 000664; RRID: IMSR_JAX:000664 |
| Software and algorithms | ||
| FIJI (ImageJ) 1.50 g | National Institute of Health | https://fiji.sc/; https://imagej.nih.gov/ij/index.html; RRID: SCR_003070 |
| LAS AF 4.0 | Leica Microsystems |
https://www.leica-microsystems.com/; RRID: SCR_013673 |
| GraphPad Prism 9.5 | GraphPad Software | RRID: SCR_000306 |
| Adobe Photoshop CS6 | Adobe Systems | RRID: SCR_014199 |
| Adobe Illustrator CS6 | Adobe Systems | RRID: SCR_010279 |
| R software 4.2.2 | GNU Project | https://www.r-project.org/; RRID:SCR_001905 |
| R package: Metascape 3.5.20230501 | Metascape Team | http://metascape.org/gp/index.html#/main/step1; RRID:SCR_016620 |
| R package: Seurat 4.3.0.1 | N/A | https://satijalab.org/seurat/get_started.html; RRID:SCR_016341 |
| R package: Tidyverse 2.0.0 | N/A |
https://CRAN.R-project.org/ package=tidyverse ; RRID:SCR_019186 |
| R package: Viridis 0.6.4 | N/A | https://cran.r-project.org/web/packages/viridis/vignettes/intro-to-viridis.html ; RRID:SCR_016696 |
| R package: Clustree 0.5.0 | N/A | https://CRAN.R-project.org/package=clustree ; RRID:SCR_016293 |
| R package: EnhancedVolcano 1.16.0 | N/A | https://bioconductor.org/packages/EnhancedVolcano/ ; RRID:SCR_018931 |
| R package: Paletteer 1.5.0 | N/A | https://CRAN.R-project.org/package=paletteer |
| R package: Ggplot2 3.4.3 | N/A | https://cran.r-project.org/web/packages/ggplot2/index.html ; RRID:SCR_014601 |
| R package: Sctransform 0.3.5 | N/A | https://github.com/satijalab/sctransform ; RRID:SCR_022146 |
| R package: GlmGamPoi 1.10.2 | N/A | https://bioconductor.org/packages/glmGamPoi/ |
| Bowtie 2 | N/A | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml ; RRID:SCR_016368 |
| Illumina HiSeq 4000 system | Ilumina | https://www.illumina.com/systems/sequencing-platforms/hiseq-3000-4000.html ; RRID:SCR_020127 |
| Rhapsody analysis pipeline | BD Biosciences | https://www.bdbiosciences.com |
| t-SNE | GitHub | https://github.com/jkrijthe/Rtsne |
| UMAP | GitHub | https://github.com/lmcinnes/umap |
| Imaris x64 software version 10.0 | Bitplane | RRID:SCR_007370 |
| Imspector Pro software | Miltenyi BioTec | https://www.miltenyibiotec.com |
| Gatan DigitalMicrograph software | Gatan | https://www.gatan.com |
| pClamp 10.3 software | Molecular devices | https://support.moleculardevices.com |
| Other | ||
| Leica TCS-SP8 confocal microscope | Leica | https://www.leica-microsystems.com/ |
| Leica TCS SP5 confocal microscope | Leica | https://www.leica-microsystems.com/ |
| Ultramicroscope II lightsheet microscope | Miltenyi BioTec | https://www.miltenyibiotec.com |
| Leica DMi8 fluorescence microscope | Leica | https://www.leica-microsystems.com/ |
| Philips Tecnai 12 Transmission Electron Microscope | Philips/FEI | https://nano.tau.ac.il |
| Leica CM 3050S cryostat | Leica | https://www.leica-microsystems.com/ |
| Olympus BX51WIF microscope | Olympus | https://www.olympus-lifescience.com |
| ssniff standard mouse chow | ssniff Spezialdiäten GmbH | https://www.ssniff.com |
Resource availability
Lead contact
Further requests and information concerning this study should be addressed to the lead contact, Sonia Garel (sonia.garel@bio.ens.psl.eu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
Single-cell RNA-seq dataset have been deposited into ArrayExpress with accession number E-MTAB-13581 and are publicly available as of the date of publication. Data generated in Tables S1, S3, and S4 is available for mining. Microscopy data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
Experimental model and study participant details
Mouse Lines
Cx3cr1gfp/+96 (RRID:IMSR_JAX:005582), Pu.1+/−,52 Cd11c-eYFP97 (RRID:IMSR_JAX:007567), Csf1rΔFIRE/+,53 Spp1-/-65 (RRID:IMSR_JAX:004936), CR3−/− (Cd11b−/−)98 (RRID:IMSR_JAX:003991), Dap12/TyroBP−/−94 (RRID:MGI:3818477), RhoAfl/fl ,95 Emx1cre/+;RhoAfl/+,63 WntA3dta and Brn4cre mice64 were maintained on a C57BL/6J background, except for Csf1rΔFIRE/+ mice that were kept on a mixed C57BL/6J CBA background. C57BL/6J wild-type mice or heterozygote littermates were used as controls for mutant mice, as they did not exhibit any phenotype, with the exception of repair analyses (Figure 7), in which we compared Spp1+/+ and Spp1-/-, in periventricular nodular heterotopia analyses (Figures 3 and 4), in which we compared Emx1cre/+;RhoAfl/fl with cre- littermates, and studies of the Csf1rΔFIRE/ΔFIRE mice in which we compared mutants with Csf1r+/+ littermates (Figures 2, 6, S5, and S6). We checked that the prenatal phenotype of Csf1rΔFIRE/ΔFIRE mice was similar on mixed C57BL/6J CBA and on C57BL/6J background (n=3 for E14.5 and E18.5, from two distinct litters), whereas the postnatal phenotype could not be assessed on C57BL/6J background because mutant mice developed hydrocephalus. The day of vaginal plug formation was considered E0.5. Most embryos were harvested in the morning, except E15.0 that were harvested on the afternoon of the 14th day of gestation. For all experiments, mice were age and sex matched and animals were housed in the animal facility of the IBENS, and handled in accordance with the regulations of the European Union and the local ethics committee.
Human fetuses
Fetal tissues were made available in accordance with French bylaws (Good Practice Concerning the Conservation, Transformation, and Transportation of Human Tissue to Be Used Therapeutically, published on December 29, 1998). The studies on human fetal tissue were approved by the French agency for biomedical research (Agence de la Biomédecine, Saint-Denis la Plaine, France, protocol n°: PFS16-002). Three human fetuses without known pathologies were obtained at gestational weeks (GW) 9, GW11 and GW14 from voluntarily terminated pregnancies upon obtaining written informed consent from the parents (Gynaecology Department, Jeanne de Flandre Hospital, Lille, France). Personal data (i.e. ethnicity, race, genetics, date of birth and other similar data) is not available in compliance with privacy regulations in France. Fetuses were fixed by immersion in 4% paraformaldehyde (PFA) at 4°C for 3 (GW9 fetus) or 5 days (GW11 and GW14 fetuses). The tissues were cryoprotected in 30% sucrose/PBS at 4°C overnight, embedded in Tissue-Tek OCT compound (Sakura Finetek, USA), frozen on dry ice and stored at -80°C until sectioning. Frozen samples were cut serially at 16 μm using a Leica CM 3050S cryostat (Leica Biosystems Nussloch GmbH, Germany). Sections were kept at -80°C.
Method details
Transcriptomic reanalysis
Single-cell RNA-seq data reanalysis
10X scRNA-seq microglia data from Hammond et al.22 and La Manno et al.30 were downloaded from the GSE121654 series and loom file http://mousebrain.org/development (“dev_all.loom”), respectively. The raw count matrix and metadata for microglia-annotated cells in La Manno et al.30 were extracted from the loom file using R v4.1.2. The data include a total of 1,711 cells (510 atm, 415 cycling microglia, 786 non-cycling microglia cells). Genes were filtered as described by the authors (expressed in at least 10 cells for La Manno et al.30 and at least 20 cells for Hammond et al.22). For the data set from Hammond et al.,22 additional metadata such as published t-distributed stochastic neighbor embedding (tSNE) coordinates and reported clusters were provided by the authors to select the 76,149 cells, including 2,517 atm cells.
The Seurat v4.1.199 scRNA-seq pipeline was used to produce uniform manifold approximation and projection (UMAP) and tSNE plots of the microglia data.22,30 Default parameters were used, unless stated otherwise. For,30 data were first normalized and scaled using SCTransform.100 PCA was performed and UMAP coordinates were computed using 5 principal components. Plots were created using Seurat v4.1.1, ggplot2 v3.3.6, scCustomize v1.1.1, and the viridis v0.6.2 palette.
Differential expression analysis
Differentially expressed genes (DEGs) of ATM-annotated cells from La Manno et al.30 were identified using the “FindAllMarkers” Seurat function (Wilcoxon signed-rank test, assay = “RNA”, min.pct=0.1, only.pos=TRUE). DEGs were further filtered (Fold Change > 1.5, Bonferroni adjusted p value < 1e-10). To construct Venn diagrams, the same threshold was applied to published DEGs of ATM22 and PAM.21
Gene set enrichment analysis
To determine gene set enrichment in the scRNA-seq dataset, the “AddModuleScore” Seurat function (ctrl.size = length of gene list, nbin=24) was used. Several gene signatures were tested for enrichment in the microglia scRNA-seq data from La Manno et al.30: (i) top enriched ATM DEGs from Hammond et al.,22 corresponding to the 9 reported markers (i.e., Spp1, Gpnmb, Igf1, Lgals3, Fapb5, Lpl, Lgals1, Ctsl, Anxa5), and (ii) top enriched PAM DEGs from Li et al.21 (i.e., Spp1, Clec7a, Gpnmb, Igf1, Lpl, Pld3, Ctsl, Ctsb, Slc23a2, Gpx3) (Table S1).
Transcriptomic analysis in controls and Spp1 mutants
scRNA-sequencing Whole Transcriptome using Rhapsody
For the Rhapsody experiment, the whole process was done by following manufacturer’s (BD Biosciences) protocol. Prior CD45+ cells were enriched using CD45 microbeads from Miltenyi and the Automacs using possel function. 115,256 cells were captured in a double run, 6 barcoded samples for each run, pooled using BD™ Ms Single Cell Sample Multiplexing Kit (Cat No: 633793). The sample was processed according to the BD Rhapsody™ Whole Transcriptome Analysis (WTA) Amplification Kit (Cat No: 633801) and the BD Rhapsody™ Whole Transcriptome Analysis (WTA) Reagent Kit (Cat No: 665915). The 2 librairies were then subjected to an indexed paired-end sequencing run of 2x151 cycles on an Illumina HiSeq 4000 system (Illumina, San Diego, CA, USA) with 20% PhiX spike in. A total of 5,830 million reads were sequenced for 115,256 cells.
Alignment and pre-processing of single-cell RNA sequencing data
Transcriptomics Fastq files were processed via the standard Rhapsody analysis pipeline (BD Biosciences) per the manufacturer’s recommendations. First, R1 and R2 reads are filtered for high-quality reads, dropping reads too short (less than 66 bases for R1 and 64 bases for R2) or have a base quality score of less than 20. R1 reads are annotated to identify cell label sequences and unique molecular identifiers (UMIs), and R2 reads are mapped to the respective reference sequences using Bowtie2.101 Finally, all passing R1 and R2 reads are combined and annotated to the respective molecules. For quality control of the reads, recursive substation error correction (RSEC) and distribution-based error correction (DBEC) were applied, which are manufacturer-developed algorithms correcting for PCR and sequencing errors. For determining putative cells (which will contain many more reads than noise cell labels), a filtering algorithm takes the number of DBEC-corrected reads into account, calculating the minimum second derivative along with the cumulative reads as the cut-off point. Finally, the expression matrix was obtained from the DBEC-adjusted molecule counts in a CSV format. A cell was determined as a singlet if the minimum read count of a single sample tag is above the threshold of 75%. A cell was classified as a multiplet if the cell exceeds the threshold for more than one sample tag. A cell that does not meet the threshold was labelled as undetermined. Both multiplets and undetermined cells were excluded from the analysis as described below.
Processing and analysis of single-cell RNA sequencing data
RSEC-adjusted molecule counts matrices from Seven Bridges BD Rhapsody alignment pipeline were loaded and analyzed using Seurat v4.3.0.1. Samples were demultiplexed using Sample Tag Call matrix from Seven Bridges BD Rhapsody alignment pipeline and metadata was manually assigned to each Tag. High quality cells were filtered and selected based on <25% of RNA from mitochondrial genes, >200 of expressed unique genes and >100 of UMI counts. Count matrix was normalized and scaled using gamma-poisson generalized linear model based on the 8,000 most variable genes using SCTransform v0.3.5102 and glmGamPoi v1.10.2102 packages. PCA, UMAP dimension reduction, nearest neighbors’ determination and Louvain clustering were performed using standard Seurat’s functions using standard parameters. The most stable resolution of clustering (SCT_snn_res.0.3) was chosen according to clustree v0.5.0 package guidelines103 Differential gene expression was performed with the “FindAllMarkers” Seurat function using Wilcoxon signed-rank test, FDR-adjusted p-value and standard parameters.
The first round of broad annotation was manually conducted based on the most differentially expressed genes across clusters (adjusted p-value < 0.05) and the top 5 DEGs were represented using “DotPlot” Seurat function. To refine the analysis on subsets of interest and get a more granular annotation, macrophages were manually selected and the whole process was performed again using the same parameters. The violin plot of gene expression was generated using “VlnPlot” function. Differential gene expression within ATM subset between wild-type and Spp1-/- conditions was conducted using “FindMarkers” Seurat function using Wilcoxon rank-sum test, FDR-adjusted p-value and standard parameters. DEGs were represented in a Volcano plot using EnhancedVolcano v1.16.0 filtered based on adjusted p-value < 0.05 and then input in Metascape (v3.5.20230501) web-based portal to get gene set functional enrichment terms using several databases. Bar plots were generated using ggplot2 v3.4.3 package from Metascape output zip files. All analyses were conducted on R v4.2.2.
Microglial Depletion
Pregnant C57BL/6J females were given anti-CSF1R mAb (αCSF1R, clone AFS98) or the rat IgG2a isotype control (clone R35-95; BD Biosciences) by intraperitoneal injection at E6.5 and E7.5, as described previously.40 Alternatively, pregnant mice were given the CSF1R inhibitor PLX3397 (Plexxikon) mixed into standard chow (Ssniff) from E6.5 or E12.5 until E15.5. The dose of PLX3397 was 290 mg/kg and respective controls received standard chow. The efficiency of depletion procedures was verified by immunohistochemistry at embryonic stages and in one newborn P0 per litter for postnatal litters.
Maternal Immune activation
Lipopolysaccharide in sterile PBS (0.12 μg/g mouse; InvivoGen and Sigma) was injected intraperitoneally into pregnant mice at E13.5. Sterile PBS was injected into control pregnant females by the same route and at the same timepoint, without detectable effects on embryonic phenotype.
Immunohistochemistry on sections
For immunohistochemistry, mouse embryonic brains were fixed in 4% PFA at 4°C for 2 h to overnight, depending on the developmental stage. For the analysis of postnatal brains, animals were perfused with 4% PFA, brains were dissected out, then post-fixed overnight at 4°C before cutting into sections in PBS. The preparation of human fetal brain tissue is described above. Immunohistochemistry was performed on free-floating 40-100 μm-thick vibratome-cut mouse brain sections or 25 μm-thick cryostat-cut human tissue sections. Slices were first incubated for 1 h at room temperature (RT) in 0.2% Triton X-100, 0.2% Gelatin in PBS (blocking solution), and then incubated at 4°C overnight in the same blocking solution with the following primary antibodies: rat anti-CD68 (1/500; Bio-Rad Cat# MCA1957, RRID:AB_322219), rabbit anti-FN1 (1/200; Millipore Cat# AB2033, RRID:AB_2105702), goat anti-FOXP2 (1/500; Santa Cruz Biotechnology Cat# sc-21069, RRID:AB_2107124), chicken anti-GFP (1/1000; Aves Labs Cat# GFP-1020, RRID:AB_10000240), rabbit anti-IBA1 (1/500; FUJIFILM Wako Shibayagi Cat# 019-19741, RRID:AB_839504), rabbit anti-IBA1 (human) (1/500; Abcam Cat# ab178846, RRID:AB_2636859), chicken anti-IBA1 (1/500; Synaptic Systems Cat# 234009, RRID:AB_2891282), rat anti-Lgals3 (MAC2) (1/1000; CEDARLANE Cat# CL8942AP, RRID:AB_10060357), rat anti-L1 (1/100; Millipore Cat# MAB5272, RRID:AB_2133200), biotinylated rat anti-LYVE1 (1/200; Thermo Fisher Scientific Cat# 13-0443-82, RRID:AB_1724157), rat anti-Myelin Basic Protein (MBP) (1/300; Millipore Cat# MAB386, RRID:AB_94975), rat anti-mDectin-1 (CLEC7A) (1/30; InvivoGen Cat# mabg-mdect, RRID:AB_2753143), mouse anti-neurofilament marker SMI-312 (1/300; BioLegend Cat# 837904, RRID:AB_2566782), goat anti-mouse Osteoactivin (GPNMB) (1/200; R and D Systems Cat# AF2330, RRID:AB_2112934), rabbit anti-P2Y12 receptor (1/500; AnaSpec; EGT Group Cat# 55043A, RRID:AB_2298886) and goat anti-SPP1 (1/400; R and D Systems Cat# AF808, RRID:AB_2194992). Sections were rinsed in PBS-0.1% TritonX-100 and incubated at 4°C from 2 h to overnight with the following secondary antibodies (1/400 in PBS, Jackson ImmunoResearch Labs): Alexa 10 Fluor® 488-conjugated donkey anti-chicken (Cat# 703-545-155, RRID:AB_2340375), Alexa 10 Fluor® 488-conjugated donkey anti-goat (Cat# 705-545-147, RRID:AB_2336933), Alexa 10 Fluor® 488-conjugated donkey anti-rat (Cat# 712-545-150, RRID:AB_2340683), Alexa 10 Fluor® 488-conjugated donkey rabbit (Cat# 711-545-152, RRID:AB_2313584), Cy3-conjugated donkey anti-goat (Cat# 705-165-147, RRID:AB_2307351), Cy3-conjugated donkey anti-rabbit (Cat# 711-165-152, RRID:AB_2307443), Cy3-conjugated donkey anti-rat (Cat# 712-165-150, RRID:AB_2340666), Alexa 10 Fluor® 647-conjugated donkey anti-goat (Cat# 705-605-147, RRID:AB_2340437), Cy5-conjugated donkey anti-goat (Cat# 705-175-147, RRID:AB_2340415), Cy5-conjugated donkey anti-goat (Jackson ImmunoResearch Labs Cat# 705-175-147, RRID:AB_2340415) and Cy5-conjugated donkey anti-rat (Cat# 712-175-150, RRID:AB_2340671). Hoechst (1/1000; Sigma) was used for fluorescent nuclear counterstaining.
Tissue Clearing
We used an adapted version of the previously published iDISCO+ clearing protocol.57,58 All incubation steps were performed at RT in a fume hood, on a tube rotator (SB3, Stuart) at 0.045 g, using a 15 mL centrifuge tube (Falcon) covered with aluminum foil to block light. E16.5 brain samples were first dehydrated by sequential 90 min incubation in a graded series (20%, 40%, 60%, 80%, and 100%) of methanol (MeOH, Sigma-Aldrich) diluted in H2O. This was followed by de-lipidation in dichloromethane (DCM; Sigma-Aldrich) for 30 min. Finally, samples were cleared overnight in dibenzylether (DBE; Sigma-Aldrich) and then stored in brown glass vials filled with DBE in the dark at RT.
Tissue preparation for transmission electron microscopy (TEM)
E14.5 mouse embryos were perfused with 4% PFA, 2.5% glutaraldehyde (EM grade) in PBS. Fixed embryos, wrapped in aluminum foil, were kept on ice for 30 min to allow glutaraldehyde impregnation. Brains were then dissected out of embryos and incubated at 4°C overnight in a buffer of 4% PFA in PBS. 100 μm vibratome sections were collected, washed 3 times in PBS and fixed in 2% osmium tetroxide in PBS for 2 h. Samples were colored en bloc with 1.5% aqueous uranyl acetate at 4°C for 1.5 h. Then, the samples were then dehydrated by sequential 10 min incubation in graded concentrations of ethanol (25%, 50%, 70%, 90% and three times 100%) and rinsed by incubation in anhydrous acetone 3 times for 10 min. Samples were infiltrated with graded concentrations of Araldite 502 resin (50%, 90%, 1 hour per step) and incubated for 2 h in freshly prepared pure resin. Samples were then mounted in a minimal amount of resin between two ACLAR 33C films, with polymerization performed at 60°C for 48 h. Before cutting, sample blocks were glued parallel to the flat surface of a cylinder bloc of resin used as a support. 70 nm ultrathin sections of the samples were obtained using an ultramicrotome (UC6, Leica). Ultrathin sections were collected on formvar-coated slot (2 x 1 mm) grids. Positive staining of grids was performed by 2 min incubation in UranyLess aqueous solution (Delta Microscopy) followed by lead citrate staining for 1 min.
MRI scans
After fixation, embryos were stored in a 1:250 mixture of 0.5 mmol gadoteric acid (Dotarem®, Guerbet) in PBS for at least 72 h. For imaging, the embryos were embedded in 2% low melting point agar gel (Sigma-Aldrich) and were placed in small Plexiglass containers. Acquisitions were performed on a 17.2 T (1H Larmor frequency = 730.2 MHz) Bruker Biospec preclinical scanner equipped with a 25 mm inner diameter quadrature birdcage volume coil (Rapid Biomedical). T1 weighted 3D images were acquired using a Fast-Low Angle Shot (FLASH) sequence with 40 micrometers isotropic resolution.
Induction of in utero cortical lesions
Pregnant female mice at E14.5 were anesthetized with isoflurane (3.5% for induction, 2% during the surgery) and given 0.1 mg/kg of buprenorphine injected subcutaneously for analgesia. The uterine horns were exposed after laparotomy. Cortical in utero lesions were induced unilaterally using a 100 μm diameter glass capillary, by poking through the cortical plate up to the lateral ventricle, as usually done for in utero electroporation or viral infection.104 Lesioned embryos were left to recover in the mother’s womb for 2.5 h after surgery, after which their brains were collected and fixed in 4% PFA at 4°C overnight.
Ex vivo phagocytic assay
Slice preparation was performed as previously described105 with the following modifications. Dissecting medium was prepared from minimum essential medium (MEM) (Gibco) with 20 mM TRIS powder pH7-9 (Sigma) and 45 mM D-glucose (Sigma). After telencephalic brains were cut into 250 μm vibratome coronal sections, slices of interest were stored in this medium on ice until use. For ex vivo phagocytosis, pH-rodo (Life technologies) was resuspended at 0.5 mg/mL in dissection medium containing 2.5 mM CaCl2, 2.5 mM MgSO4, 1 mM NaHCO3 and with 300 μL HEPES (Gibco) per 50 mL. After careful removal of MEM, 15 μL of pH-rodo solution was added to the top of each slice and incubated for 1 h at 37°C in 5% CO2. The reaction was stopped by adding cold pH-rodo suspension medium. After three washes, slices were fixed in 4% PFA in PBS for 45 min, followed by immunohistochemistry as described above.
Preparation of brain slices and electrophysiological recordings
Adult (∼P60) mice were deeply anesthetized with isoflurane and decapitated. The brain was quickly removed and placed in ice-cold sucrose-based ACSF, which contained (in mM): 86 NaCl, 2.5 KCl, 0.5 CaCl2, 7 MgCl2, 1.2 NaH2PO4, 25 NaHCO3, 25 glucose and 75 sucrose continuously bubbled with carbogen (95% O2/ 5% CO2). Acute coronal slices (300 μm-thick) containing the amygdala were prepared using a 7000 SMZ-2 Vibratome (Camden Instruments Ltd, UK) in ice-cold sucrose-based ACSF. Slices were transferred for 30 minutes in warm (34°C) ACSF which contained (in mM): 125 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 1.25 NaH2PO4, 25 NaHCO3 and 25 glucose, saturated with 95 % O2 - 5 % CO2., and then placed in a recording chamber and continuously perfused with ACSF bubbled with 95% O2/5% CO2 (3-4 mL/min; 30-34°C). Recorded neurons were visualized with an Olympus BX51WIF microscope (Olympus, France) equipped with a Qimaging RETIGA 2000R camera (Teledyne Photometrics, USA) run by Micro-Manager (Vale Lab, USCF, USA). Electrophysiological signals were recorded with a Multiclamp 700B amplifier (CV-7B headstage), a Digidata 1440A acquisition board and pClamp 10.3 software (Molecular Devices, USA). Electrophysiological signals were filtered at 2 kHz and sampled at 10 kHz. Data were analysed off-line (pClamp-10 software, Axon Instruments). Series resistances were compensated up to 65% maximum. Whole-cell patch-clamp recordings were performed in pyramidal neurons of the basolateral amygdala (BLA) using borosilicate glass pipettes (3–5 MΩ resistance) containing (in mM): 120 CsMeSO3; 10 HEPES; 4.6 MgCl2; 10 K2- creatine phosphate; 15 BAPTA; 4 Na2-ATP; 0.4 Na2-GTP; 1 QX314. Values were not corrected for the liquid junction potential. Glutamatergic currents (EPSCs) were measured at the reversal potential for GABA-A receptor mediated events (-70 mV) and GABA-A receptor mediated currents (IPSCs) were measured at the reversal potential of glutamatergic events (+5 mV) in external solution (ACSF) containing the NMDA receptor antagonist (50 μM D-AP5) (Figure S6C). Extracellular synaptic stimulation was obtained by applying extracellular voltage pulses (0.1 Hz, A-M systems USA) delivered using a second patch pipette filled with HEPES-buffered solution placed within the vicinity the amygdalar capsule (Figure S6C). The intensity of the stimulation has been adapted for each cell in order to evoke a comparable EPSC. The I/E ratio was then calculated by dividing IPSC and EPSC amplitudes measured in the same neuron by keeping the same position and intensity of the stimulation.
Image acquisition and analysis
Slice imaging
Images of immunohistochemistry on sections were acquired with a fluorescence binocular microscope (Leica MZ16 F), a fluorescence microscope (Leica DMi8) or a confocal microscope (Leica TCS SP5 and TSP8). Image analyses were performed with FIJI (ImageJ; RRID: SCR_003070), Imaris (Bitplane; RRID:SCR_007370) and Adobe Photoshop CS6 software (Adobe Sytems; RRID:SCR_014199).
iDISCO 3D Imaging and Processing
3D imaging was performed with an ultramicroscope II (LaVision BioTec) using ImspectorPro software (LaVision BioTec). The light sheet was generated by a laser (wavelength 488 Coherent Sapphire Laser, LaVision BioTec) and two cylindrical lenses. Samples were placed in an imaging reservoir made of 100% quartz (LaVision BioTec) filled with ethyl cinnamate and illuminated from the side by the laser light. Images were acquired with a PCO Edge SCMOS CCD camera (2.560×2.160-pixel size, LaVision BioTec). The step size between each image was fixed at 3 μm. For all samples, background fluorescence recorded from exposure to the 488 nm wavelength was acquired in order to reconstruct brain morphology and lesions. Images, 3D volume, and movies were generated using Imaris x64 software (version 10.0, Bitplane; RRID:SCR_007370). Stack images were first converted to an imaris file (.ims) using ImarisFileConverter. File size was next reduced to 8 bits. 3D reconstruction of the sample was performed using “volume rendering” (Imaris). The sample could be optically sliced at any angle using the “orthoslicer” or “obliqueslicer” tools to validate phenotypic alterations. Lesions due to microglial depletion were highlighted by creating a mask around the volume using the “surface” tool. To this end, low intensity pixels resulting from low background fluorescence (i.e. absence of cells in the lesions) in the 488nm exposure condition were selected in order to create the “surface” volume. The volume values of the created surfaces were automatically obtained from the Imaris software and compared between different conditions (Figure 2C). 3D pictures and movies were generated using the “snapshot” and “animation” tools. Movie legends were generated using FIJI ImageJ software.
3D Imaging and processing of microglia and Fibronectin 1 staining
3D reconstruction of microglia and Fibronectin 1 (FN1) staining (Figures 5J, 5K, and S7B) was performed using Imaris 10.0.0 (Bitplane; Zurich, Switzerland). For each animal, two to five CSA microglial cells were acquired as z-stacks on a confocal microscope (Leica TCS SP8) with a 63x objective and a 3x zoom factor, using an optimal z-step size of 0.33 μm and identical settings. Only microglial cells within the focal plane of FN1 staining were used for the analyses. Acquisitions were then smoothed and background was subtracted in FIJI ImageJ software (NIH) according to106 The resulting images were converted to an Imaris file (.ims) using ImarisFileConverter, then processed and analyzed in Imaris 10.0.0 (Bitplane; Zurich, Switzerland) as previously described.106 Briefly, 3D reconstruction of the sample was performed using the Surface rendering option in the Surpass view, with a thresholding method based on the Absolute Intensity of the signal. For 3D rendering of the microglial cell volume, threshold of IBA1 (for E14.5/E15 time point) or Cx3cr1-GFP (for P3 time point) signal was manually adjusted to surface the cell appropriately. For 3D rendering of the FN1 signal, threshold for reconstruction was set at 2 times the automatically detected threshold, to surface the high intensity accumulations. For quantification of FN1 signal within microglia, a channel was created by using the mask function, masking the FN1 signal within the microglia surface. 3D rendering of the microglia-masked FN1 signal was performed using the same threshold value that was set for rendering the total FN1 signal. The volume of each surface expressed in μm3 was recorded from the Graph tab. Then, the volume of the masked FN1 was expressed as a percentage of microglial volume (using the IBA1/Cx3cr1-GFP volume) for normalization. Unbiased quantification of all images was performed blind to treatment of animals. 3D pictures were generated using the Imaris “snapshot” tool.
Transmission electron microscopy (TEM) imaging
TEM was performed using a Philips Tecnai 12 Transmission Electron Microscope (Philips/FEI, Eindhoven, The Netherlands) at the electronic imaging department of the Imachem imaging platform (France BioImaging) at the Institut de Biologie de l’Ecole Normale Supérieure, Paris. The Gatan DigitalMicrograph software was used to acquire TEM images at various magnifications with a 4K CCD Orius 1000 camera (Gatan).
Quantification and statistical analysis
Co-labeling of IBA1+ or Cx3cr1-GFP+ macrophages with P2Y12 microglial marker and ATM markers was performed on 25x, zoom 1x confocal stacks (Leica DM6 FS) with 70 μm thickness and 2 μm z-step size and quantified specifically in cells accumulated along the CSA, adjacent cortical ramified microglia, as well as cells accumulated at the CSB and around in utero-induced lesions in the cortex using the Cell Counter tool in FIJI ImageJ software (Figures 1, 4F, and S1). Co-labeling of Cx3cr1-GFP+ macrophages with ATM markers was performed on 10x, zoom 1x confocal stacks (Leica DM6 FS) with 60 μm thickness and 2 μm z-step size and quantified specifically in cells accumulated along the CSA in control and PLX3397-treated pups at P3 using the Cell Counter tool in FIJI ImageJ software (Figure 7A).
Analysis of CD68 coverage in ATMs at E14.5 was performed on 40x, zoom 1x confocal stacks (Leica TCS SP8) with 22 μm thickness and 2 μm z-step size (Figure S1K). At least 5 amoeboid ATM-like microglia at the CSA (Spp1+) and at least 5 adjacent neocortex ramified microglia (Spp1-) were analysed per animal for a total of 4 Cx3cr1gfp/+ E14.5 embryos. For each cell, masks were created for GFP (cell outline) and CD68 (intracellular lysosomal signal) stainings and the area of each mask was measured in FIJI ImageJ software. CD68 coverage was calculated as the percentage of CD68 signal (CD68 mask) covering microglial area (GFP mask area).
Microglial distribution in the CSA region in E18.5 control and Emx1cre/+;RhoAfl/fl mutant mice was quantified using the Cell Counter tool in FIJI ImageJ on 10x, zoom 1x confocal stacks with a thickness of 60 μm and 3 μm z-stack confocal acquisitions (Leica DM6 FS). Quantification was performed in 3 distinct areas defined as follows: (1) the CSA, centered on the boundary and with a width of 100 μm, (2) the Surrounding area to the CSA with a width of 200 μm and (3) the Remote area, which encircles the Surrounding area (2) with a width of 250 μm (Figure S3). Microglial distribution in the CSA region in P3 control and PLX3397 mice was quantified using the Cell Counter tool in FIJI ImageJ on 10x, zoom 1x confocal stacks with a thickness of 60 μm and 2 μm z-stack confocal acquisitions (Leica DM6 FS) (Figure 6D). Quantification was performed in 2 distinct areas defined as follows: (1) the CSA, centered on the boundary and with a width of 100 μm, (2) the Surrounding area to the CSA with a width of 400 μm.
For assessing post-depletion lesion severity in Emx1cre/+;RhoAfl/fl mice, Brn4cre; Wnt3Adta mice (Figures 3C–3G), or PLX3397-exposed embryos and pups (Figure 7) we established a scoring system based on the lesion area measured via FIJI ImageJ software, as controls may display some microcavities (Figures 3A and 3B). For each brain, the area of the largest lesion observed on the slice was selected, and scores were assigned based on the absence of lesion (score 0), mild lesion (score 1) or severe lesions (score 2). Table S2 displays the detailed scoring of lesions. The mean scores for lesion severity across different experimental conditions were then compared statistically.
Quantification of extracellular staining of Spp1 and FN1 at the CSA in P3 control and PLX3397 was performed on 20x, zoom 1x confocal stacks (Leica TCS SP8) with 100 μm thickness and 3 μm z-step size (Figures 7 and S7). Mean signal intensity was measured in FIJI ImageJ software in 3 regions of interest (25x25 μm squares) that excluded microglial cell bodies (GFP+) for each brain: at the level of the CSA, as well as the adjacent the neocortex and BLA. For both Spp1 and FN1, a ratio was then calculated as the mean signal intensity at the CSA divided by the averaged mean intensity of the adjacent neocortex and BLA.
Data are presented as mean ± standard error of the mean (SEM), except when explicitly stated in the Figure legend. Non-parametric two-sided Mann-Whitney U-tests were used to compare two distributions in the co-labeling, CD68 coverage, lesion severity and signal intensity experiments. Fisher’s exact test was used to compare contingent presence or absence of CSA/CSB lesions in controls, Spp1-/-, and PLX3397-treated mice. All graphs and statistical analyses were generated using GraphPad Prism 8.0 software (GraphPad Software; RRID:SCR_002798). ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ns, non significant (p > 0.05).
For single cell transcriptomic analyses on wild-type and Spp1-/- mutants, all statistical analyses were performed using R (version 4.2.2).
Acknowledgments
We thank M. Keita for excellent technical assistance and E. Touzalin, A. Delecourt, and C. Le Moal for assistance with mouse colonies. We are grateful to Denis Jabaudon and Garel lab members for helpful discussions and critical review of the manuscript, to Ana-Maria Lennon-Duménil, Silvia Cappello, and Cord Brackebusch for the kind gifts of reagents and mouse models, and to Kristine Zengeler and John Lukens for discussions during the course of this work. We thank Lucy Robinson and Ilya Demchenko of Insight Editing London for scientific editing of the manuscript. We thank the IBENS Imaging Facility (France BioImaging, supported by ANR-10-INBS-04, ANR-10-LABX-54 MEMO LIFE, and ANR-11-IDEX-000-02 PSL∗ Research University, “Investments for the future”) and the midwives of the Gynecology Department, Jeanne de Flandre Hospital of Lille (Centre d’Orthogenie), for their kind assistance and support. The authors acknowledge the support of the Inserm Cross-Cutting Scientific Program (HuDeCA to P.G.). This work was supported by grants to S.G. from INSERM, CNRS, the ERC Consolidator NImO 616080, MicroSENSO ANR-19-CE16-0018, FSER, Fondation du Collège de France (Fonds St Michel), and Fondation pour la Recherche Médicale (FRM) (EQU202003010195). A.R.L. obtained a fellowship from Ecole des Neurosciences de Paris Ile-de-France network and an ATER position from the College de France; C.B. is supported by an AMX PhD fellowship; N.O. by the FRM (FRM, EQU202003010195); C.L. obtained a FRM PhD fellowship (FRM, ECO202006011600).
Author contributions
Conceptualization, A.R.L., C.B., M.S.T., L.L., and S.G.; formal analysis, A.R.L., A. Canzi, C.B., N.O., C.L., C.C., L.P., B.K., A.S., D.B., L.C, A. Candat, A.G., D.T., L.C., M.S.T., and L.L.; investigation, A.R.L., A. Canzi, C.B., N.O., C.L., C.C., L.P., B.K., A.S., F.Z., H.C., S.V., G.O., P.S., L. Cantini, L. Ciobanu, M.S.T., and L.L.; resources, D.M., A.F., C.A., F.W., J.-B.M., P.P., C.P., J.P., P.G., and F.G.; writing– original draft, A.R.L., M.S.T., L.L., and S.G.; all authors contributed to manuscript editing; visualization, A.R.L., A. Canzi, C.B., N.O., C.L., C.C., L.P., B.K., M.S.T., and L.L.; project administration, M.S.T., L.L., and S.G.; supervision, M.S.T., L.L., and S.G.; funding acquisition, S.G.
Declaration of interests
The authors declare no competing interests.
Published: February 2, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.cell.2024.01.012.
Supplemental information
This Table S1 (excel sheet) provides a comparison of overlap in differentially expressed genes and their mean raw count expression between embryonic ATM from La Manno et al.,30 ATM from Hammond et al.,22 and PAM from Li et al.,21 related to the Venn diagrams Figure1C and Figure1C.
This table (excel sheet) provides the list of genes specific of each cluster.
This table (excel sheet) provides the list of genes specific of each cluster.
This table (excel sheet) provides a listing of the DEGs in ATM cluster from wild-type and Spp1-/- mutants extracted from single cell analysis on the macrophage population.
This table provides the details of the metascape analyses of differential pathways, listing the genes corresponding to the altered pathways.
References
- 1.Colonna M., Butovsky O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017;35:441–468. doi: 10.1146/annurev-immunol-051116-052358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li Q., Barres B.A. Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 2018;18:225–242. doi: 10.1038/nri.2017.125. [DOI] [PubMed] [Google Scholar]
- 3.Hammond T.R., Robinton D., Stevens B. Microglia and the Brain: Complementary Partners in Development and Disease. Annu. Rev. Cell Dev. Biol. 2018;34:523–544. doi: 10.1146/annurev-cellbio-100616-060509. [DOI] [PubMed] [Google Scholar]
- 4.Thion M.S., Ginhoux F., Garel S. Microglia and early brain development: An intimate journey. Science. 2018;362:185–189. doi: 10.1126/science.aat0474. [DOI] [PubMed] [Google Scholar]
- 5.Sierra A., Paolicelli R.C., Kettenmann H. Cien Anos de Microglia: Milestones in a Century of Microglial Research. Trends Neurosci. 2019;42:778–792. doi: 10.1016/j.tins.2019.09.004. [DOI] [PubMed] [Google Scholar]
- 6.Prinz M., Jung S., Priller J. Microglia Biology: One Century of Evolving Concepts. Cell. 2019;179:292–311. doi: 10.1016/j.cell.2019.08.053. [DOI] [PubMed] [Google Scholar]
- 7.Prinz M., Masuda T., Wheeler M.A., Quintana F.J. Microglia and Central Nervous System-Associated Macrophages-From Origin to Disease Modulation. Annu. Rev. Immunol. 2021;39:251–277. doi: 10.1146/annurev-immunol-093019-110159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cserép C., Pósfai B., Dénes Á. Shaping Neuronal Fate: Functional Heterogeneity of Direct Microglia-Neuron Interactions. Neuron. 2021;109:222–240. doi: 10.1016/j.neuron.2020.11.007. [DOI] [PubMed] [Google Scholar]
- 9.Badimon A., Strasburger H.J., Ayata P., Chen X., Nair A., Ikegami A., Hwang P., Chan A.T., Graves S.M., Uweru J.O., et al. Negative feedback control of neuronal activity by microglia. Nature. 2020;586:417–423. doi: 10.1038/s41586-020-2777-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McNamara N.B., Munro D.A.D., Bestard-Cuche N., Uyeda A., Bogie J.F.J., Hoffmann A., Holloway R.K., Molina-Gonzalez I., Askew K.E., Mitchell S., et al. Microglia regulate central nervous system myelin growth and integrity. Nature. 2023;613:120–129. doi: 10.1038/s41586-022-05534-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thion M.S., Mosser C.A., Férézou I., Grisel P., Baptista S., Low D., Ginhoux F., Garel S., Audinat E. Biphasic Impact of Prenatal Inflammation and Macrophage Depletion on the Wiring of Neocortical Inhibitory Circuits. Cell Rep. 2019;28:1119–1126.e4. doi: 10.1016/j.celrep.2019.06.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scott-Hewitt N., Perrucci F., Morini R., Erreni M., Mahoney M., Witkowska A., Carey A., Faggiani E., Schuetz L.T., Mason S., et al. Local externalization of phosphatidylserine mediates developmental synaptic pruning by microglia. EMBO J. 2020;39 doi: 10.15252/embj.2020105380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Favuzzi E., Huang S., Saldi G.A., Binan L., Ibrahim L.A., Fernández-Otero M., Cao Y., Zeine A., Sefah A., Zheng K., et al. GABA-receptive microglia selectively sculpt developing inhibitory circuits. Cell. 2021;184 doi: 10.1016/j.cell.2021.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cserép C., Schwarcz A.D., Pósfai B., László Z.I., Kellermayer A., Környei Z., Kisfali M., Nyerges M., Lele Z., Katona I., Adam D. Microglial control of neuronal development via somatic purinergic junctions. Cell Rep. 2022;40 doi: 10.1016/j.celrep.2022.111369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gallo N.B., Berisha A., Van Aelst L. Microglia regulate chandelier cell axo-axonic synaptogenesis. Proc. Natl. Acad. Sci. USA. 2022;119 doi: 10.1073/pnas.2114476119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bisht K., Sharma K.P., Lecours C., Sánchez M.G., El Hajj H., Milior G., Olmos-Alonso A., Gómez-Nicola D., Luheshi G., Vallières L., et al. Dark microglia: A new phenotype predominantly associated with pathological states. Glia. 2016;64:826–839. doi: 10.1002/glia.22966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krasemann S., Madore C., Cialic R., Baufeld C., Calcagno N., El Fatimy R., Beckers L., O'Loughlin E., Xu Y., Fanek Z., et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity. 2017;47:566–581.e9. doi: 10.1016/j.immuni.2017.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Keren-Shaul H., Spinrad A., Weiner A., Matcovitch-Natan O., Dvir-Szternfeld R., Ulland T.K., David E., Baruch K., Lara-Astaiso D., Toth B., et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer's Disease. Cell. 2017;169:1276–1290.e17. doi: 10.1016/j.cell.2017.05.018. [DOI] [PubMed] [Google Scholar]
- 19.Deczkowska A., Keren-Shaul H., Weiner A., Colonna M., Schwartz M., Amit I. Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration. Cell. 2018;173:1073–1081. doi: 10.1016/j.cell.2018.05.003. [DOI] [PubMed] [Google Scholar]
- 20.Stratoulias V., Venero J.L., Tremblay M.E., Joseph B. Microglial subtypes: diversity within the microglial community. EMBO J. 2019;38 doi: 10.15252/embj.2019101997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Q., Cheng Z., Zhou L., Darmanis S., Neff N.F., Okamoto J., Gulati G., Bennett M.L., Sun L.O., Clarke L.E., et al. Developmental Heterogeneity of Microglia and Brain Myeloid Cells Revealed by Deep Single-Cell RNA Sequencing. Neuron. 2019;101:207–223.e10. doi: 10.1016/j.neuron.2018.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hammond T.R., Dufort C., Dissing-Olesen L., Giera S., Young A., Wysoker A., Walker A.J., Gergits F., Segel M., Nemesh J., et al. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity. 2019;50:253–271.e6. doi: 10.1016/j.immuni.2018.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Masuda T., Sankowski R., Staszewski O., Böttcher C., Amann L., Sagar S., Scheiwe C., Nessler S., Kunz P., van Loo G., et al. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature. 2019;566:388–392. doi: 10.1038/s41586-019-0924-x. [DOI] [PubMed] [Google Scholar]
- 24.Sala Frigerio C., Wolfs L., Fattorelli N., Thrupp N., Voytyuk I., Schmidt I., Mancuso R., Chen W.T., Woodbury M.E., Srivastava G., et al. The Major Risk Factors for Alzheimer's Disease: Age, Sex, and Genes Modulate the Microglia Response to Abeta Plaques. Cell Rep. 2019;27:1293–1306.e6. doi: 10.1016/j.celrep.2019.03.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kracht L., Borggrewe M., Eskandar S., Brouwer N., Chuva de Sousa Lopes S.M., Laman J.D., Scherjon S.A., Prins J.R., Kooistra S.M., Eggen B.J.L. Human fetal microglia acquire homeostatic immune-sensing properties early in development. Science. 2020;369:530–537. doi: 10.1126/science.aba5906. [DOI] [PubMed] [Google Scholar]
- 26.Bian Z., Gong Y., Huang T., Lee C.Z.W., Bian L., Bai Z., Shi H., Zeng Y., Liu C., He J., et al. Deciphering human macrophage development at single-cell resolution. Nature. 2020;582:571–576. doi: 10.1038/s41586-020-2316-7. [DOI] [PubMed] [Google Scholar]
- 27.Masuda T., Sankowski R., Staszewski O., Prinz M. Microglia Heterogeneity in the Single-Cell Era. Cell Rep. 2020;30:1271–1281. doi: 10.1016/j.celrep.2020.01.010. [DOI] [PubMed] [Google Scholar]
- 28.Marschallinger J., Iram T., Zardeneta M., Lee S.E., Lehallier B., Haney M.S., Pluvinage J.V., Mathur V., Hahn O., Morgens D.W., et al. Lipid-droplet-accumulating microglia represent a dysfunctional and proinflammatory state in the aging brain. Nat. Neurosci. 2020;23:194–208. doi: 10.1038/s41593-019-0566-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Safaiyan S., Besson-Girard S., Kaya T., Cantuti-Castelvetri L., Liu L., Ji H., Schifferer M., Gouna G., Usifo F., Kannaiyan N., et al. White matter aging drives microglial diversity. Neuron. 2021;109:1100–1117.e10. doi: 10.1016/j.neuron.2021.01.027. [DOI] [PubMed] [Google Scholar]
- 30.La Manno G., Siletti K., Furlan A., Gyllborg D., Vinsland E., Mossi Albiach A., Mattsson Langseth C., Khven I., Lederer A.R., Dratva L.M., et al. Molecular architecture of the developing mouse brain. Nature. 2021;596:92–96. doi: 10.1038/s41586-021-03775-x. [DOI] [PubMed] [Google Scholar]
- 31.Silvin A., Uderhardt S., Piot C., Da Mesquita S., Yang K., Geirsdottir L., Mulder K., Eyal D., Liu Z., Bridlance C., et al. Dual ontogeny of disease-associated microglia and disease inflammatory macrophages in aging and neurodegeneration. Immunity. 2022;55:1448–1465.e6. doi: 10.1016/j.immuni.2022.07.004. [DOI] [PubMed] [Google Scholar]
- 32.Stogsdill J.A., Kim K., Binan L., Farhi S.L., Levin J.Z., Arlotta P. Pyramidal neuron subtype diversity governs microglia states in the neocortex. Nature. 2022;608:750–756. doi: 10.1038/s41586-022-05056-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paolicelli R.C., Sierra A., Stevens B., Tremblay M.E., Aguzzi A., Ajami B., Amit I., Audinat E., Bechmann I., Bennett M., et al. Microglia states and nomenclature: A field at its crossroads. Neuron. 2022;110:3458–3483. doi: 10.1016/j.neuron.2022.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stratoulias V., Ruiz R., Kanatani S., Osman A.M., Keane L., Armengol J.A., Rodríguez-Moreno A., Murgoci A.N., García-Domínguez I., Alonso-Bellido I., et al. ARG1-expressing microglia show a distinct molecular signature and modulate post-natal development and function of the mouse brain. Nat. Neurosci. 2023;26:1008–1020. doi: 10.1038/s41593-023-01326-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dolan M.J., Therrien M., Jereb S., Kamath T., Gazestani V., Atkeson T., Marsh S.E., Goeva A., Lojek N.M., Murphy S., et al. Exposure of iPSC-derived human microglia to brain substrates enables the generation and manipulation of diverse transcriptional states in vitro. Nat. Immunol. 2023;24:1382–1390. doi: 10.1038/s41590-023-01558-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Park M.D., Silvin A., Ginhoux F., Merad M. Macrophages in health and disease. Cell. 2022;185:4259–4279. doi: 10.1016/j.cell.2022.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thion M.S., Garel S. Microglial ontogeny, diversity and neurodevelopmental functions. Curr. Opin. Genet. Dev. 2020;65:186–194. doi: 10.1016/j.gde.2020.06.013. [DOI] [PubMed] [Google Scholar]
- 38.Menassa D.A., Muntslag T.A.O., Martin-Estebané M., Barry-Carroll L., Chapman M.A., Adorjan I., Tyler T., Turnbull B., Rose-Zerilli M.J.J., Nicoll J.A.R., et al. The spatiotemporal dynamics of microglia across the human lifespan. Dev. Cell. 2022;57:2127–2139.e6. doi: 10.1016/j.devcel.2022.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cossart R., Garel S. Step by step: cells with multiple functions in cortical circuit assembly. Nat. Rev. Neurosci. 2022;23:395–410. doi: 10.1038/s41583-022-00585-6. [DOI] [PubMed] [Google Scholar]
- 40.Squarzoni P., Oller G., Hoeffel G., Pont-Lezica L., Rostaing P., Low D., Bessis A., Ginhoux F., Garel S. Microglia modulate wiring of the embryonic forebrain. Cell Rep. 2014;8:1271–1279. doi: 10.1016/j.celrep.2014.07.042. [DOI] [PubMed] [Google Scholar]
- 41.Thion M.S., Garel S. On place and time: microglia in embryonic and perinatal brain development. Curr. Opin. Neurobiol. 2017;47:121–130. doi: 10.1016/j.conb.2017.10.004. [DOI] [PubMed] [Google Scholar]
- 42.Wlodarczyk A., Holtman I.R., Krueger M., Yogev N., Bruttger J., Khorooshi R., Benmamar-Badel A., de Boer-Bergsma J.J., Martin N.A., Karram K., et al. A novel microglial subset plays a key role in myelinogenesis in developing brain. EMBO J. 2017;36:3292–3308. doi: 10.15252/embj.201696056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hagemeyer N., Hanft K.M., Akriditou M.A., Unger N., Park E.S., Stanley E.R., Staszewski O., Dimou L., Prinz M. Microglia contribute to normal myelinogenesis and to oligodendrocyte progenitor maintenance during adulthood. Acta Neuropathol. 2017;134:441–458. doi: 10.1007/s00401-017-1747-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ueno M., Fujita Y., Tanaka T., Nakamura Y., Kikuta J., Ishii M., Yamashita T. Layer V cortical neurons require microglial support for survival during post-natal development. Nat. Neurosci. 2013;16:543–551. doi: 10.1038/nn.3358. [DOI] [PubMed] [Google Scholar]
- 45.Nemes-Baran A.D., White D.R., DeSilva T.M. Fractalkine-Dependent Microglial Pruning of Viable Oligodendrocyte Progenitor Cells Regulates Myelination. Cell Rep. 2020;32 doi: 10.1016/j.celrep.2020.108047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hankin M.H., Schneider B.F., Silver J. Death of the subcallosal glial sling is correlated with formation of the cavum septi pellucidi. J. Comp. Neurol. 1988;272:191–202. doi: 10.1002/cne.902720204. [DOI] [PubMed] [Google Scholar]
- 47.Erblich B., Zhu L., Etgen A.M., Dobrenis K., Pollard J.W. Absence of colony stimulation factor-1 receptor results in loss of microglia, disrupted brain development and olfactory deficits. PLoS One. 2011;6 doi: 10.1371/journal.pone.0026317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pridans C., Raper A., Davis G.M., Alves J., Sauter K.A., Lefevre L., Regan T., Meek S., Sutherland L., Thomson A.J., et al. Pleiotropic Impacts of Macrophage and Microglial Deficiency on Development in Rats with Targeted Mutation of the Csf1r Locus. J. Immunol. 2018;201:2683–2699. doi: 10.4049/jimmunol.1701783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hoeffel G., Wang Y., Greter M., See P., Teo P., Malleret B., Leboeuf M., Low D., Oller G., Almeida F., et al. Adult Langerhans cells derive predominantly from embryonic fetal liver monocytes with a minor contribution of yolk sac-derived macrophages. J. Exp. Med. 2012;209:1167–1181. doi: 10.1084/jem.20120340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Elmore M.P., Najafi A.R., Koike M.A., Dagher N.N., Spangenberg E.E., Rice R.A., Kitazawa M., Matusow B., Nguyen H., West B.L., Green K.N. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron. 2014;82:380–397. doi: 10.1016/j.neuron.2014.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Green K.N., Crapser J.D., Hohsfield L.A. To Kill a Microglia: A Case for CSF1R Inhibitors. Trends Immunol. 2020;41:771–784. doi: 10.1016/j.it.2020.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Back J., Dierich A., Bronn C., Kastner P., Chan S. PU.1 determines the self-renewal capacity of erythroid progenitor cells. Blood. 2004;103:3615–3623. doi: 10.1182/blood-2003-11-4089. [DOI] [PubMed] [Google Scholar]
- 53.Rojo R., Raper A., Ozdemir D.D., Lefevre L., Grabert K., Wollscheid-Lengeling E., Bradford B., Caruso M., Gazova I., Sánchez A., et al. Deletion of a Csf1r enhancer selectively impacts CSF1R expression and development of tissue macrophage populations. Nat. Commun. 2019;10 doi: 10.1038/s41467-019-11053-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Munro D.A.D., Bradford B.M., Mariani S.A., Hampton D.W., Vink C.S., Chandran S., Hume D.A., Pridans C., Priller J. CNS macrophages differentially rely on an intronic Csf1r enhancer for their development. Development. 2020;147 doi: 10.1242/dev.194449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kiani Shabestari S., Morabito S., Danhash E.P., McQuade A., Sanchez J.R., Miyoshi E., Chadarevian J.P., Claes C., Coburn M.A., Hasselmann J., et al. Absence of microglia promotes diverse pathologies and early lethality in Alzheimer's disease mice. Cell Rep. 2022;39 doi: 10.1016/j.celrep.2022.110961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Esteban H., Blondiaux E., Audureau E., Sileo C., Moutard M.L., Gelot A., Jouannic J.M., Ducou le Pointe H., Garel C. Prenatal features of isolated subependymal pseudocysts associated with adverse pregnancy outcome. Ultrasound in Obstet. Gyne. 2015;46:678–687. doi: 10.1002/uog.14820. [DOI] [PubMed] [Google Scholar]
- 57.Belle M., Godefroy D., Dominici C., Heitz-Marchaland C., Zelina P., Hellal F., Bradke F., Chédotal A. A simple method for 3D analysis of immunolabeled axonal tracts in a transparent nervous system. Cell Rep. 2014;9:1191–1201. doi: 10.1016/j.celrep.2014.10.037. [DOI] [PubMed] [Google Scholar]
- 58.Renier N., Wu Z., Simon D.J., Yang J., Ariel P., Tessier-Lavigne M. iDISCO: a simple, rapid method to immunolabel large tissue samples for volume imaging. Cell. 2014;159:896–910. doi: 10.1016/j.cell.2014.10.010. [DOI] [PubMed] [Google Scholar]
- 59.Kaur C., Ling E.A. Transitory cystic cavities in the developing mammalian brain - normal or anomalous? J. Anat. 2017;230:197–202. doi: 10.1111/joa.12556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Saito K., Okamoto M., Watanabe Y., Noguchi N., Nagasaka A., Nishina Y., Shinoda T., Sakakibara A., Miyata T. Dorsal-to-Ventral Cortical Expansion Is Physically Primed by Ventral Streaming of Early Embryonic Preplate Neurons. Cell Rep. 2019;29:1555–1567.e5. doi: 10.1016/j.celrep.2019.09.075. [DOI] [PubMed] [Google Scholar]
- 61.Van Essen D.C. A 2020 view of tension-based cortical morphogenesis. Proc. Natl. Acad. Sci. USA. 2020;117:32868–32879. doi: 10.1073/pnas.2016830117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Das J.M., Dossani R.H. StatPearls. StatPearls Publishing; 2022. Cavum Septum Pellucidum. [PubMed] [Google Scholar]
- 63.Cappello S., Böhringer C.J., Bergami M., Conzelmann K.K., Ghanem A., Tomassy G.S., Arlotta P., Mainardi M., Allegra M., Caleo M., et al. A radial glia-specific role of RhoA in double cortex formation. Neuron. 2012;73:911–924. doi: 10.1016/j.neuron.2011.12.030. [DOI] [PubMed] [Google Scholar]
- 64.Deck M., Lokmane L., Chauvet S., Mailhes C., Keita M., Niquille M., Yoshida M., Yoshida Y., Lebrand C., Mann F., et al. Pathfinding of corticothalamic axons relies on a rendezvous with thalamic projections. Neuron. 2013;77:472–484. doi: 10.1016/j.neuron.2012.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liaw L., Birk D.E., Ballas C.B., Whitsitt J.S., Davidson J.M., Hogan B.L. Altered wound healing in mice lacking a functional osteopontin gene (spp1) J. Clin. Invest. 1998;101:1468–1478. doi: 10.1172/JCI1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rosmus D.D., Lange C., Ludwig F., Ajami B., Wieghofer P. The Role of Osteopontin in Microglia Biology: Current Concepts and Future Perspectives. Biomedicines. 2022;10 doi: 10.3390/biomedicines10040840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shen X., Qiu Y., Wight A.E., Kim H.J., Cantor H. Definition of a mouse microglial subset that regulates neuronal development and proinflammatory responses in the brain. Proc. Natl. Acad. Sci. USA. 2022;119 doi: 10.1073/pnas.2116241119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.De Schepper S., Ge J.Z., Crowley G., Ferreira L.S.S., Garceau D., Toomey C.E., Sokolova D., Rueda-Carrasco J., Shin S.H., Kim J.S., et al. Perivascular cells induce microglial phagocytic states and synaptic engulfment via SPP1 in mouse models of Alzheimer's disease. Nat. Neurosci. 2023;26:406–415. doi: 10.1038/s41593-023-01257-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bonnans C., Chou J., Werb Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014;15:786–801. doi: 10.1038/nrm3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Patten J., Wang K. Fibronectin in development and wound healing. Adv. Drug Deliv. Rev. 2021;170:353–368. doi: 10.1016/j.addr.2020.09.005. [DOI] [PubMed] [Google Scholar]
- 71.Moretti L., Stalfort J., Barker T.H., Abebayehu D. The interplay of fibroblasts, the extracellular matrix, and inflammation in scar formation. J. Biol. Chem. 2022;298 doi: 10.1016/j.jbc.2021.101530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li Y., He X., Kawaguchi R., Zhang Y., Wang Q., Monavarfeshani A., Yang Z., Chen B., Shi Z., Meng H., et al. Microglia-organized scar-free spinal cord repair in neonatal mice. Nature. 2020;587:613–618. doi: 10.1038/s41586-020-2795-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Meisner O.C., Nair A., Chang S.W.C. Amygdala connectivity and implications for social cognition and disorders. Handb. Clin. Neurol. 2022;187:381–403. doi: 10.1016/B978-0-12-823493-8.00017-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Varghese M., Keshav N., Jacot-Descombes S., Warda T., Wicinski B., Dickstein D.L., Harony-Nicolas H., De Rubeis S., Drapeau E., Buxbaum J.D., et al. Autism spectrum disorder: neuropathology and animal models. Acta Neuropathol. 2017;134:537–566. doi: 10.1007/s00401-017-1736-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Amaral D.G., Schumann C.M., Nordahl C.W. Neuroanatomy of autism. Trends Neurosci. 2008;31:137–145. doi: 10.1016/j.tins.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 76.Paré D., Quirk G.J., Ledoux J.E. New vistas on amygdala networks in conditioned fear. J. Neurophysiol. 2004;92:1–9. doi: 10.1152/jn.00153.2004. [DOI] [PubMed] [Google Scholar]
- 77.Tovote P., Fadok J.P., Lüthi A. Neuronal circuits for fear and anxiety. Nat. Rev. Neurosci. 2015;16:317–331. doi: 10.1038/nrn3945. [DOI] [PubMed] [Google Scholar]
- 78.Asede D., Doddapaneni D., Bolton M.M. Amygdala Intercalated Cells: Gate Keepers and Conveyors of Internal State to the Circuits of Emotion. J. Neurosci. 2022;42:9098–9109. doi: 10.1523/JNEUROSCI.1176-22.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Paolicelli R.C., Bolasco G., Pagani F., Maggi L., Scianni M., Panzanelli P., Giustetto M., Ferreira T.A., Guiducci E., Dumas L., et al. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333:1456–1458. doi: 10.1126/science.1202529. [DOI] [PubMed] [Google Scholar]
- 80.Schafer D.P., Lehrman E.K., Kautzman A.G., Koyama R., Mardinly A.R., Yamasaki R., Ransohoff R.M., Greenberg M.E., Barres B.A., Stevens B. Microglia sculpt post-natal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74:691–705. doi: 10.1016/j.neuron.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Miyamoto A., Wake H., Ishikawa A.W., Eto K., Shibata K., Murakoshi H., Koizumi S., Moorhouse A.J., Yoshimura Y., Nabekura J. Microglia contact induces synapse formation in developing somatosensory cortex. Nat. Commun. 2016;7 doi: 10.1038/ncomms12540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Weinhard L., di Bartolomei G., Bolasco G., Machado P., Schieber N.L., Neniskyte U., Exiga M., Vadisiute A., Raggioli A., Schertel A., et al. Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nat. Commun. 2018;9 doi: 10.1038/s41467-018-03566-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Allen N.J., Lyons D.A. Glia as architects of central nervous system formation and function. Science. 2018;362:181–185. doi: 10.1126/science.aat0473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nguyen P.T., Dorman L.C., Pan S., Vainchtein I.D., Han R.T., Nakao-Inoue H., Taloma S.E., Barron J.J., Molofsky A.B., Kheirbek M.A., Molofsky A.V. Microglial Remodeling of the Extracellular Matrix Promotes Synapse Plasticity. Cell. 2020;182:388–403.e15. doi: 10.1016/j.cell.2020.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Frangogiannis N.G. The inflammatory response in myocardial injury, repair, and remodelling. Nat. Rev. Cardiol. 2014;11:255–265. doi: 10.1038/nrcardio.2014.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pellicoro A., Ramachandran P., Iredale J.P., Fallowfield J.A. Liver fibrosis and repair: immune regulation of wound healing in a solid organ. Nat. Rev. Immunol. 2014;14:181–194. doi: 10.1038/nri3623. [DOI] [PubMed] [Google Scholar]
- 87.Vannella K.M., Wynn T.A. Mechanisms of Organ Injury and Repair by Macrophages. Annu. Rev. Physiol. 2017;79:593–617. doi: 10.1146/annurev-physiol-022516-034356. [DOI] [PubMed] [Google Scholar]
- 88.Shin Y.J., Kim H.L., Choi J.S., Choi J.Y., Cha J.H., Lee M.Y. Osteopontin: correlation with phagocytosis by brain macrophages in a rat model of stroke. Glia. 2011;59:413–423. doi: 10.1002/glia.21110. [DOI] [PubMed] [Google Scholar]
- 89.Rentsendorj A., Sheyn J., Fuchs D.T., Daley D., Salumbides B.C., Schubloom H.E., Hart N.J., Li S., Hayden E.Y., Teplow D.B., et al. A novel role for osteopontin in macrophage-mediated amyloid-beta clearance in Alzheimer's models. Brain Behav. Immun. 2018;67:163–180. doi: 10.1016/j.bbi.2017.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lyons D.A., Talbot W.S. Glial cell development and function in zebrafish. Cold Spring Harb. Perspect. Biol. 2014;7 doi: 10.1101/cshperspect.a020586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sharma K., Bisht K., Eyo U.B. A Comparative Biology of Microglia Across Species. Front. Cell Dev. Biol. 2021;9 doi: 10.3389/fcell.2021.652748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Teissier N., Fallet-Bianco C., Delezoide A.L., Laquerrière A., Marcorelles P., Khung-Savatovsky S., Nardelli J., Cipriani S., Csaba Z., Picone O., et al. Cytomegalovirus-induced brain malformations in fetuses. J. Neuropathol. Exp. Neurol. 2014;73:143–158. doi: 10.1097/NEN.0000000000000038. [DOI] [PubMed] [Google Scholar]
- 93.Nunes R.H., Pacheco F.T., da Rocha A.J. Magnetic resonance imaging of anterior temporal lobe cysts in children: discriminating special imaging features in a particular group of diseases. Neuroradiology. 2014;56:569–577. doi: 10.1007/s00234-014-1356-9. [DOI] [PubMed] [Google Scholar]
- 94.Tomasello E., Desmoulins P.O., Chemin K., Guia S., Cremer H., Ortaldo J., Love P., Kaiserlian D., Vivier E. Combined natural killer cell and dendritic cell functional deficiency in KARAP/DAP12 loss-of-function mutant mice. Immunity. 2000;13:355–364. doi: 10.1016/s1074-7613(00)00035-2. [DOI] [PubMed] [Google Scholar]
- 95.Jackson B., Peyrollier K., Pedersen E., Basse A., Karlsson R., Wang Z., Lefever T., Ochsenbein A.M., Schmidt G., Aktories K., et al. RhoA is dispensable for skin development, but crucial for contraction and directed migration of keratinocytes. Mol. Biol. Cell. 2011;22:593–605. doi: 10.1091/mbc.E09-10-0859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jung S., Aliberti J., Graemmel P., Sunshine M.J., Kreutzberg G.W., Sher A., Littman D.R. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol. Cell. Biol. 2000;20:4106–4114. doi: 10.1128/mcb.20.11.4106-4114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lindquist R.L., Shakhar G., Dudziak D., Wardemann H., Eisenreich T., Dustin M.L., Nussenzweig M.C. Visualizing dendritic cell networks in vivo. Nat. Immunol. 2004;5:1243–1250. doi: 10.1038/ni1139. [DOI] [PubMed] [Google Scholar]
- 98.van Spriel A.B., Leusen J.H.W., van Egmond M., Dijkman H.B.P.M., Assmann K.J.M., Mayadas T.N., van de Winkel J.G.J. Mac-1 (CD11b/CD18) is essential for Fc receptor-mediated neutrophil cytotoxicity and immunologic synapse formation. Blood. 2001;97:2478–2486. doi: 10.1182/blood.v97.8.2478. [DOI] [PubMed] [Google Scholar]
- 99.Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., 3rd, Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M., et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587.e29. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hafemeister C., Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019;20:296. doi: 10.1186/s13059-019-1874-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ahlmann-Eltze C., Huber W. glmGamPoi: fitting Gamma-Poisson generalized linear models on single cell count data. Bioinformatics. 2021;36:5701–5702. doi: 10.1093/bioinformatics/btaa1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zappia L., Oshlack A. Clustering trees: a visualization for evaluating clusterings at multiple resolutions. GigaScience. 2018;7 doi: 10.1093/gigascience/giy083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Genescu I., Aníbal-Martínez M., Kouskoff V., Chenouard N., Mailhes-Hamon C., Cartonnet H., Lokmane L., Rijli F.M., López-Bendito G., Gambino F., Garel S. Dynamic interplay between thalamic activity and Cajal-Retzius cells regulates the wiring of cortical layer 1. Cell Rep. 2022;39 doi: 10.1016/j.celrep.2022.110667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.de Frutos C.A., Bouvier G., Arai Y., Thion M.S., Lokmane L., Keita M., Garcia-Dominguez M., Charnay P., Hirata T., Riethmacher D., et al. Reallocation of Olfactory Cajal-Retzius Cells Shapes Neocortex Architecture. Neuron. 2016;92:435–448. doi: 10.1016/j.neuron.2016.09.020. [DOI] [PubMed] [Google Scholar]
- 106.Schafer D.P., Lehrman E.K., Heller C.T., Stevens B. An engulfment assay: a protocol to assess interactions between CNS phagocytes and neurons. JoVE. 2014 doi: 10.3791/51482. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The hemibrain was imaged on a light-sheet microscope and reconstructed using the Imaris software. Green signal encompasses non-depleted CX3CR1-GFP+ cells, tissue autofluorescence delineates the hemibrain morphology and the cavitary lesion at the CSA was reconstructed and pseudocolored in yellow.
The hemibrain was imaged on a light-sheet microscope and reconstructed using the Imaris software. Green signal encompasses non-depleted CX3CR1-GFP+ cells, tissue autofluorescence delineates the hemibrain morphology and the cavitary lesion at the CSA was reconstructed and pseudocolored in yellow.
The hemibrain was imaged on a light-sheet microscope and reconstructed using the Imaris software, showing normal morphology at the CSA and CSB (open arrowheads). The movie progresses through light sheet photographs acquired along the coronal rostrocaudal axis of the sample at a 3 μm Z step size.
The hemibrain was imaged on a light-sheet microscope and reconstructed using the Imaris software, showing a cavitary lesion at the CSA (solid arrowheads).The movie progresses through light sheet photographs acquired along the coronal rostrocaudal axis of the sample at a 3 μm Z step size.
This Table S1 (excel sheet) provides a comparison of overlap in differentially expressed genes and their mean raw count expression between embryonic ATM from La Manno et al.,30 ATM from Hammond et al.,22 and PAM from Li et al.,21 related to the Venn diagrams Figure1C and Figure1C.
This table (excel sheet) provides the list of genes specific of each cluster.
This table (excel sheet) provides the list of genes specific of each cluster.
This table (excel sheet) provides a listing of the DEGs in ATM cluster from wild-type and Spp1-/- mutants extracted from single cell analysis on the macrophage population.
This table provides the details of the metascape analyses of differential pathways, listing the genes corresponding to the altered pathways.
Data Availability Statement
-
•
Single-cell RNA-seq dataset have been deposited into ArrayExpress with accession number E-MTAB-13581 and are publicly available as of the date of publication. Data generated in Tables S1, S3, and S4 is available for mining. Microscopy data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.