

Summary
Our ability to determine the clinical impact of variants in 3′ untranslated regions (UTRs) of genes remains poor. We provide a thorough analysis of 3′ UTR variants from several datasets. Variants in putative regulatory elements, including RNA-binding protein motifs, eCLIP peaks, and microRNA sites, are up to 16 times more likely than variants not in these elements to have gene expression and phenotype associations. Variants in regulatory motifs result in allele-specific protein binding in cell lines and allele-specific gene expression differences in population studies. In addition, variants in shared regions of alternatively polyadenylated isoforms and those proximal to polyA sites are more likely to affect gene expression and phenotype. Finally, pathogenic 3′ UTR variants in ClinVar are up to 20 times more likely than benign variants to fall in a regulatory site. We incorporated these findings into RegVar, a software tool that interprets regulatory elements and annotations for any 3′ UTR variant and predicts whether the variant is likely to affect gene expression or phenotype. This tool will help prioritize variants for experimental studies and identify pathogenic variants in individuals.
Keywords: genetic variants, 3ʹ UTR, RNA-binding proteins, polyadenylation, miRNA, variant interpretation, regulatory motifs
Graphical abstract
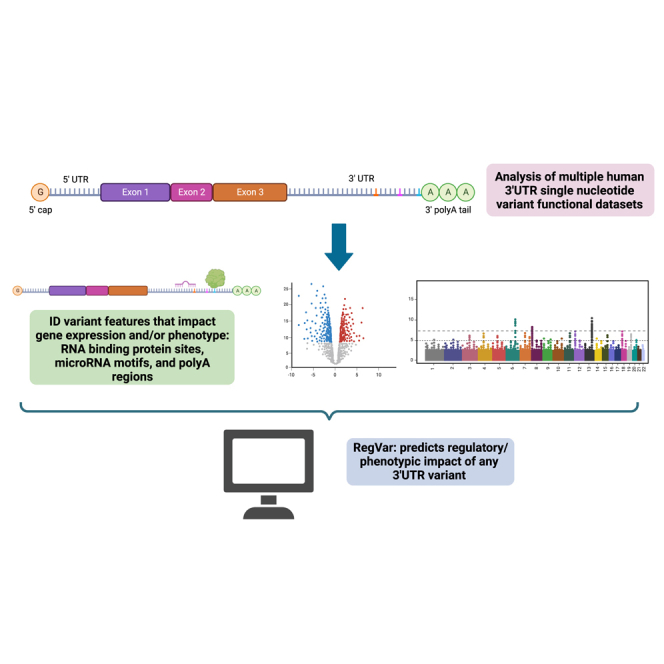
Genetic variants in noncoding regions such as 3′ UTRs are associated with disease and phenotypes, but their functional interpretation remains challenging. We have identified specific regulatory elements in 3′ UTRs that are enriched for variants that impact gene expression or phenotype and have developed a tool specifically designed to interpret 3′ UTR variants.
Introduction
Despite the ubiquity of exome sequencing in modern human genetics, our ability to determine the clinical impact of genetic variants remains limited. The average person has 20,000 variants in their exome, most of which are rare in the population.1 The effect of rare variants in coding regions can be predicted by their impact on protein amino acid composition.2,3,4 However, many variants in the ClinVar variant database are noncoding variants of uncertain significance.5 The impact of these variants is difficult to predict due to our incomplete knowledge of the function of noncoding regions.
The 3′ untranslated region (3′ UTR) comprises the bulk of noncoding sequences present in exomes and is important for regulation of messenger RNA (mRNA) processing, stability, translation, and localization. Sequence-specific RNA-binding proteins (RBPs) interact with cognate RNA motifs at specific 3′ UTR positions.6 Such RBPs often recruit effector proteins to the mRNA that can alter transcript stability, translational efficiency, and intracellular mRNA localization.7 Altered transcript stability and translation impact protein abundance, while altered transcript localization can impact protein function.7 Recognition of polyadenylation signals (PASs) within 3′UTRs directs cleavage and polyadenylation of the mRNA transcript. Many 3′UTRs contain more than one functional PAS, and alternative polyadenylation (APA) yields transcripts of different lengths containing different sets of RBP and microRNA (miRNA) target sites.8 miRNAs are small noncoding RNAs that bind to 3′ UTRs when complexed with proteins in miRNPs.9 Binding is mediated primarily by complementarity between the 3′ UTR target and nucleotides 2–7 or 2–8 of the miRNA called the seed sequence. miRNA binding decreases transcript stability or represses translation.10
RBP motifs in the 3′ UTR can be predicted from in vitro binding studies using RNA Bind-n-Seq (RBNS), RNACompete, or other assays or can be identified in cells via enhanced crosslinking and immunoprecipitation (eCLIP).11,12 miRNA sites can be predicted in silico from base complementarity and sequence conservation or can be identified in cells using crosslinking and sequencing.13,14 Variants in the 3′ UTR, especially those that disrupt RBP interactions, miRNA binding, or cleavage and polyadenylation, are likely to impact mRNA function and may be deleterious, potentially contributing to disease.
Several metrics have been developed to predict the pathogenicity of noncoding variants.15,16,17,18,19,20 However, these metrics don’t take into account the unique regulatory features of 3′ UTRs, such as APA, RBP interactions, or miRNA binding. More general methods that may implicate 3′ UTR variants include expression quantitative trait loci (eQTLs), which link variants to gene-expression changes, and genome wide association studies (GWASs), which link variants to phenotypes.21,22,23,24 However, these methods don’t suggest mechanisms, detect association rather than causality, and can only interrogate common variants. Experimental methods, such as massively parallel 3′ UTR reporter assays, can address molecular functions of variants, with the caveats that variants are assayed in artificial genomic contexts and are over expressed.25,26 Saturation genome editing can generate possible genomic variants in clinically important regions, with clonal cell lines used to assess phenotypes.27 Though effective in identifying pathogenic variants, this method is laborious and expensive.
Exome and genome sequencing are becoming less expensive and more rapid, but data interpretation remains the limiting step. For many individuals, exome or genome sequencing results in a diagnosis that alters clinical management and can be lifesaving.28,29 More general and accessible methods of variant characterization could therefore be highly impactful. We propose that 3′ UTR variants that disrupt (or create) specific types of regulatory elements are more likely to alter function and contribute to disease, aiding in the identification and interpretation of pathogenic 3′ UTR variants.
Here, we show that single-nucleotide variants (SNVs) in locations that overlap RBP motifs, eCLIP peaks, and miRNA sites are both more evolutionarily conserved and more likely than other variants to be associated with phenotypic or gene-expression changes. Many of these variants are causal, as they are enriched for allele-specific RBP binding in cell lines and allele-specific gene-expression differences in population studies. eQTL variants in potential regulatory elements are more likely to be GWAS hits, and pathogenic 3′ UTR variants in ClinVar are more likely to fall in a regulatory element than benign variants. We also show that variants in certain 3′ UTR regions, e.g., proximal to polyA sites, are more likely to be linked to gene expression changes or phenotypes. Finally, we provide a high-throughput R package, RegVar, which assesses regulatory elements and annotations associated with any 3′ UTR variant(s) of interest and predicts whether the variant is likely to affect gene expression or phenotype. We expect the program will help prioritize variants for experimental studies and identify thousands of pathogenic variants.
Material and methods
eQTL processing
eQTL variant call files fine-mapped using the deterministic approximation of posteriors method (DAP-G) were downloaded from the GTex project (https://storage.cloud.google.com/adult-gtex/bulk-qtl/v8/fine-mapping-cis-eqtl/GTEx_v8_finemapping_DAPG.tar) and intersected with terminal 3′ UTRs using BEDTools, as defined by the region from the GENCODE stop codon to the most distal polyA Database peak.24,30,31,32 Because many variants in 3′ UTRs (and elsewhere) are in linkage disequilibrium, it can be difficult to discern causal variants. Fine-mapping is a statistical method to distinguish the effects of variants in linkage disequilibrium blocks.33 Fine-mapping eQTLs or GWAS hits results in a posterior inclusion probability (PIP) for each variant that represents the likelihood that each is causal of expression differences or phenotypes.34 For each variant-tissue combination, only the tissue with the highest PIP was used, except for the transcript expression analysis (see below).
GWAS processing
Sum of single effects (SuSiE) fine-mapped UK Biobank GWAS variant call files were downloaded from the Finucane lab (https://www.dropbox.com/s/cdsdgwxkxkcq8cn/UKBB_94traits_release1.1.tar.gz?dl=0) and intersected with 3′ UTRs, as above.35 All variant-phenotype combinations were considered in analysis.
Identifying variants in putative 3′ UTR regulatory elements
3′ UTRs were defined as above. To identify variants in putative RBP motifs, reference/alternative alleles and their surrounding genomic sequence were processed with RBPamp.36 Variants overlapping an RBNS motif with an affinity of >0.33 of the ideal motif were considered to be in RBP motifs. Alternate alleles overlapping a motif with an affinity of 0.66 or more compared to the ideal motif were considered preserving, and alternate alleles that caused the motif affinity to drop below 0.33 compared to the ideal were considered disrupting. To identify variants in eCLIP peaks, variants were intersected with eCLIP coordinates downloaded from the Encyclopedia of DNA Elements (ENCODE) (https://www.encodeproject.org/search/?type=Experiment&assay_title=eCLIP&files.file_type=tsv).12 For Figure 3A, we considered RBPs with at least fifty variants in peaks to allow sufficient power to detect PIP differences. RBPamp eCLIP-Proximal (ReP) sites were defined as motifs matching the highest affinity RBPamp motif in the vicinity of each of the eCLIP peaks. Variants in possible conserved family miRNA sites and their seed types and site conservation were defined by TargetScan (https://www.targetscan.org/cgi-bin/targetscan/data_download.vert80.cgi).13
Figure 3.

Certain RBP motifs and eCLIP peaks are enriched among eQTLs and alter expression
(A) Proportion causal (with 95% confidence interval) for variants in eCLIP peaks for different RBPs, for all RBPs with ≥50 variants in eCLIP peaks. The proportion causal for variants not in any eCLIP peak was 0.05 (dashed line). ∗p < 0.01.
(B) Mean PIP for eQTLs in eCLIP peaks but not RBP motifs (y axis) versus mean PIP for eQTLs in RBP motifs but not eCLIP peaks, for all RBPs in both datasets. Shown is the regression line with Pearson correlation coefficient.
(C) Distribution of RBPs among ReP sites at different minimum eQTL PIP cutoffs. Shown are RBPs representing at least 0.1% of all ReP sites.
(D) Variant NES (from GTEx) on gene expression for high-confidence (PIP > 0.9) eQTLs not in RBP motifs or eCLIP peaks (gray), and for high-confidence eQTLs predicted to disrupt (red) or preserve (blue) HNRNPK motifs. ∗p < 0.01.
Annotation of 3′ UTR variants
PolyA signals/sites and 3′ UTR isoforms were identified from aggregate 3′-seq data generated from multiple tissues and cell lines (http://cbio.mskcc.org/leslielab/ApA/atlas/#).37 Calling of polyA sites and removal of peaks from genomic polyA priming was performed on 3′-seq data as described previously.38 We considered variants proximal to polyA sites if they fell within 50 nucleotides of a 3′-seq peak. The relationship between distance to polyA site and PIP becomes nonsignificant in regression models for variants more than 50 nucleotides away. Shared regions of APA isoforms were regions proximal to the first polyA site. Partially shared regions were between the first and penultimate polyA site, whereas unique regions were distal to the penultimate polyA site.
Conservation
Variants were merged with conservation information from PhastCons 100-way scores to identify conserved (PhastCons > 0.5) or non-conserved (PhastCons < 0.5) variants (https://bioconductor.org/packages/release/data/annotation/html/phastCons100way.UCSC.hg38.html).39 For miRNA sites, the TargetScan conservation designation was used.13
Statistical analysis
The proportion of causal variants in an element (the fraction with a PIP of greater than 0.25) was calculated using pairwise proportion z tests. The proportions causal for variants in elements was compared to that for variants not in elements using a Fisher’s exact test. When comparing PIPs, a paired Wilcoxon rank-sum test was used. For odds ratios, a Fisher’s exact test was used to determine confidence intervals and significance. For the generalized linear model, we used a binomial distribution to model a binary score (GWAS or eQTL PIP greater or less than/equal to 0.5). Goodness of fit was assessed via Hosmer-Lemeshow test. All p values were corrected using the false discovery rate method.
eQTL expression analysis
The relative expression of transcripts with the alternate versus reference allele was determined by the transcript normalized effect size (NES) from GTex.24 The NES is the slope of the eQTL regression line comparing expression of transcripts with the alternative and reference alleles; more positive NES values indicate higher gene expression in individuals with the alternative allele and vice versa.24 Each PIP-tissue combination was considered for every eQTL.
Binding Estimation of Allele-specific Protein-RNA (BEAPR) analysis
All variants with heterozygous genotypes in eCLIP peaks in K562 and HepG2 cells, as well as their predicted eCLIP allele specificity as defined by BEAPR analysis, were kindly provided by the Xiao lab (personal communication).40 Only 3′ UTR variants were considered, and variants in RBP motifs (with reference allele affinity >0.05 of alternate allele) matching the eCLIP RBPs (defined as above) were compared to variants only in eCLIP peaks but not in motifs.
Expression control
To control APA results for gene expression, HepG2 and K562 RNA sequencing data was downloaded from ENCODE (https://www.encodeproject.org/rnaget-report/?type=RNAExpression).12 The fraction of causal eQTL variants was compared between genes with various numbers of canonical APA isoforms but nonsignificant differences in mean transcripts per million (TPM) expression.
ClinVar analysis
Variant call files were downloaded from ClinVar (https://ftp.ncbi.nlm.nih.gov/pub/clinvar/vcf_GRCh38/clinvar.vcf.gz) and intersected with 3′ UTR coordinates/annotations and putative regulatory elements as above.5 Only variants with known clinical impact (pathogenic or benign) were considered.
CADD scores
Raw as well as scaled Combined Annotation Dependent Depletion (CADD) scores for all gnomAD variants were downloaded from the CADD website (https://krishna.gs.washington.edu/download/CADD/v1.6/GRCh38/gnomad.genomes.r3.0.snv.tsv.gz) and intersected with eQTL variants.15,41 The raw CADD scores of variants in putative regulatory elements were compared to those for controls.
RegVar
The development version of the RegVar R package is available for download on GitHub at https://github.com/RomoL2/RegVar. The RegVar tool characterizes user-provided 3′ UTR variants by their regulatory features as described. A variant is predicted to be an eQTL or GWAS hit if its log-odds are greater than 0.01 (eQTL) or 0.0075 (GWAS) using our logistic regression model (see statistical analysis). These thresholds maximize sensitivity and specificity.
Gene annotation
Disease-associated genes were defined as the union of Blekhman 2008 and Berg 2013 and downloaded from GitHub (https://github.com/QingboWang/gene_lists).42,43
TIA1 cytotoxic granule associated RNA binding protein (TIA1)-knockdown gene-expression analysis
GC-corrected RNA sequencing data from before and after TIA1 shRNA knock down in K562 and HepG2 cells was downloaded from ENCODE (https://www.encodeproject.org/experiments/ENCSR694LKY/and https://www.encodeproject.org/experiments/ENCSR057GCF/).12 Genes with TIA1 eCLIP peaks or ReP sites were identified via RegVar and then intersected with the TIA1 knockdown dataset.
In vitro allele parallel reporter assay
Four pairs of 101 nucleotide 3ʹ UTR fragments consisting of reference or variant alleles +/− 50 bases of flanking sequence were ordered in oPools Oligo Pool format (IDT) and cloned into a modified version of the pmirGLO (Promega) downstream of a GFP open reading frame via BsaI sites introduced by site-directed mutagenesis (Agilent). The plasmid library was transfected into human embryonic kidney (HEK) 293 cells using Lipofectamine 3000 (Life Technologies) and total RNA was collected 48 h later using the RNeasy mini kit (Qiagen). Plasmid RNA was reverse transcribed with a gene-specific primer (5′GCATTCTAGTTGTGGTTTGTCCA3′) and Superscript IV (Life Technologies). Libraries were amplified, and two replicates of each sample were uniquely dual indexed using custom primers. RNA and plasmid DNA input read counts for each fragment were obtained using Illumina Miseq. For each variant, the alt: ref variant activity is the odds ratio calculated as (RNAalt/DNAalt)/(RNAref/DNAref). These data are published in aggregate in Findlay et al.44
Results
Identification of causal 3′ UTR variants
We developed an analysis pipeline to identify features that might differentiate 3′ UTR variants that impact gene expression or phenotype (Figure 1). We used three sources of 3′ UTR variants: fine-mapped eQTLs identified by GTEx (82,903 variants), fine-mapped GWAS hits from the UK Biobank (174,065 variants), and variants with heterozygous genotypes in eCLIP peaks (2,856 variants).22,24,40 Fine-mapping is a statistical method that yields a PIP for each eQTL or GWAS hit representing the likelihood that each is causal for the observed association.33,34
Figure 1.

Variant processing pipeline schematic
We compared the probability that eQTLs and GWAS hits affect gene expression (eQTLs) or phenotype (GWAS hits) for variants in eCLIP peaks, RBP motifs, and various 3′ UTR annotation categories. We compared the likelihood of overlapping an RBP motif for variants with and without allele-specific eCLIP binding. SNV, single nucletoide variant; eCLIP, enhanced crosslinking and immunoprecipitation; RBNS, RNA Bind-n-Seq; PIP, posterior inclusion probability; RBP, RNA binding protein.
Variants were first annotated by APA isoform location and then intersected with putative regulatory elements. The specific categories of elements studied included RBP-binding motifs from in vitro studies, eCLIP peaks, ReP sites, and miRNA sites (see material and methods).11,12,13,37 Here, we defined ReP sites for RBPs with both in vitro and in vivo binding data as the highest-affinity motif for the RBP in the vicinity of each of its eCLIP peaks.11,12 To determine whether variants in specific regulatory element or annotation categories preferentially impact gene expression or phenotype, we compared the PIP for variants located in these elements versus controls. Allele-specific eCLIP binding events were used to assess whether variants in motifs altered RBP binding.40
3′ UTR variants in putative regulatory elements are associated with altered gene expression
We hypothesized that variants in RBP motifs and/or eCLIP peaks often impact transcript expression by altering binding of regulatory RBPs. Overall, most eQTL variants have low fine-mapped PIP values (Figure S1). However, we found that high-PIP eQTLs are slightly more likely than non-eQTLs to be located in an in vitro-derived RBP motif, over four times more likely to be in an eCLIP peak, and over nine times more likely to be in a ReP site (Figure 2A). These observations support our hypothesis that each of these classes of elements is enriched for variants that alter expression. Variants located in eCLIP peaks and ReP sites also have significantly higher PhastCons 100-way conservation scores than controls even after matching for gene-expression level, providing further evidence that these classes are enriched for regulatory function (Figure 2B).45
Figure 2.

3′ UTR variants in RBP motifs, eCLIP peaks, and miRNA sites are associated with gene-expression changes
(A) Odds of an eQTL variant being in predicted RBP sites versus control variants (PIP < 0.001) as minimum PIP increases; odds ratio is shown with 95% confidence intervals.
(B) Comparison of PhastCons score distributions for eQTL variants in ReP sites (green), eCLIP peaks (purple), RBP motifs (yellow), or outside of known regulatory elements (black).
(C) Proportion causal (PIP > 0.25) with 95% confidence intervals for eQTL variants not in RBP motifs or eCLIP peaks compared to variants in RBP motifs or eCLIP peaks.
(D) Odds of an eQTL variant being in predicted miRNA site versus control variants (PIP < 0.001) as minimum PIP increases, as in (A).
(E) Proportion causal with 95% confidence intervals for variants not in miRNA sites compared to variants in miRNA sites of different seed types.
(F) Variant NES (from GTEx) on gene expression for high-confidence (PIP > 0.9) eQTLs disrupting miR motifs (green), or not in RBP/miR motifs or eCLIP peaks (gray).
(G) Proportion causal with 95% confidence intervals (left) for variants not in RBP motifs or eCLIP peaks compared to variants in ReP sites or eCLIP peaks, matched for raw CADD score (right).
(H) Odds of variants with heterozygous genotypes in HepG2 and K562 cells being in an RBP motif for decreasing allele-specific eCLIP binding p values, with 95% confidence intervals.
For all panels, ∗p < 0.01.
We considered the proportion of eQTL variants with PIP >0.25 (i.e., variants at least 25% likely to alter expression) as a summary statistic, which we call “proportion causal.” Although the precise PIP cutoff used is somewhat arbitrary, repeating our key analyses using different PIP thresholds yielded similar results (Figure S2). We found that eQTLs in RBP motifs and those in eCLIP peaks had a higher proportion causal than variants outside of these elements, and that variants in RBP motifs were more likely to be causal if conserved (Figure 2C). Unfortunately, the number of eQTL variants in ReP sites was too low to perform a similar analysis.
We hypothesized that variants in miRNA target sites typically disrupt miRNA binding, resulting in increased mRNA levels. Indeed, high-PIP eQTLs are over twice as likely to fall in conserved miRNA sites than non-eQTL variants (Figure 2D). miRNAs with greater complementarity to target transcripts (longer seed matches: 8mer > 7mer-m8 > 7mer-a1) exert stronger regulatory effects.13 We found that eQTLs in conserved 8mer sites of miRNAs in broadly conserved families have three times higher proportion causal than controls (Figure 2E).
3′ UTR variants in putative regulatory elements likely cause expression changes
To determine whether variants in miRNA sites increase gene expression as expected, we compared the NES of causal (PIP > 0.25) eQTLs that disrupt miRNA motifs to those outside of predicted regulatory elements. The NES measures the magnitude and direction in which eQTLs change gene expression.24 Variants predicted to disrupt conserved miRNA motifs predominantly have positive NES (p < 0.001, Wilcoxon rank-sum test), suggesting a direct relationship between disrupted miRNA binding and increased expression (Figure 2F).
If variants in RBP motifs and eCLIP peaks commonly alter gene expression, we would expect some to be pathogenic. The CADD score is a metric that discriminates between benign and pathogenic variants based on their evolutionary deleteriousness. We found that variants in eCLIP peaks and RBP motifs have significantly higher CADD scores (Figure S3). As CADD score does not incorporate RBP motif or eCLIP information, higher scores reflect other features of RBP motifs, such as conservation and base composition. We found that variants in eCLIP peaks, conserved miRNA sites, or ReP sites are more likely to cause gene-expression changes than controls even when comparing sets with matched CADD scores (Figure 2G). Thus, considering eCLIP, miRNA, and ReP information can substantially add to the information in CADD scores for identification of functional variants.
To test the idea that expression differences associated with eQTLs in RBP motifs result from differential binding of RBPs, we analyzed allele-specific eCLIP binding.40 Variants with heterozygous genotypes that exhibit allele-specific eCLIP enrichment are up to four times more likely to be located in a motif for the corresponding RBP than variants that do not (Figure 2H). This observation supports that the gene-expression changes associated with variants in eCLIP peaks and RBP motifs noted above commonly result from changes in RBP binding.
Enrichment for RBP motifs and eCLIP peaks among eQTLs is driven by a subset of RBPs
We next examined which RBPs are responsible for increased PIPs among eQTLs in eCLIP peaks. RBPs with high-PIP eQTLs in eCLIP peaks included those known to bind the 3′ UTR to alter mRNA stability, such as polyA binding protein cytoplasmic 4 (PABPC4) and La ribonucleoprotein 4 (LARP4).46,47 In contrast, RBPs with fewer high-PIP eQTLs in eCLIP peaks included transcription factors or repressors, such as BCL2 associated transcription factor 1 (BCLAF1), as well as primarily nuclear proteins, such as heterogeneous nuclear ribonucleoprotein L (HNRNPL), KH-type splicing regulatory protein (KHSRP), and KH domain containing RNA binding (QKI) (Figure 3A). Mean PIP values were positively correlated across RBPs for variants in RBP motifs and those in eCLIP peaks (Figure 3B), suggesting that these two subsets of variants function similarly, with some RBPs impacting expression more often than others.
To further explore which RBPs may most commonly impact expression, we examined ReP sites among high-PIP eQTLs and found that several motifs, including those for heterogenous nuclear ribonucleoprotein K (HNRNPK), are highly represented (Figure 3C). HNRNPK is a multifunctional RBP involved in both transcriptional and post-transcriptional mRNA processing that binds 3′ UTRs at C-rich motifs to alter stability of target mRNAs.48,49,50 We found that high-confidence eQTLs that disrupt HNRNPK motifs are associated with higher transcript expression than those that preserve motifs or are located outside of HNRNPK motifs (Figure 3D). This observation suggests that most HNRNPK binding tends to destabilize mRNAs in tissues assessed for eQTLs.
3′ UTR variants in putative regulatory elements likely result in phenotypic changes
Recent studies have demonstrated limited overlap between GWAS hits and eQTLs and found that many genes with eQTLs are under weak selective constraint and are likely less functionally important than genes with GWAS hits.51 Therefore, it was of interest to explore the extent to which eQTL variants that change gene expression by disruption of RBP motifs have phenotypes. To assess the effect of 3′ UTR variants on phenotypes, we analyzed GWAS data generated by the UK Biobank and fine-mapped by the Finucane lab.22 Overall, GWAS variants have lower PIPs than eQTL variants after fine-mapping. Otherwise, the distribution of GWAS and eQTL variants along the 3′ UTR was similar and uniform (Figure S1).
GWAS hits in eCLIP peaks had much higher conservation scores than those outside of eCLIP peaks, even those in an RBP motif (Figure 4A). To determine whether variants in regulatory elements result in phenotype changes, we compared PIPs for variants in RBP motifs, eCLIP peaks, and controls. GWAS hits in these elements have higher PIPs compared to variants outside of these sites (Figure 4B). (Too few ReP sites overlapped to permit similar analyses of this class.) Conservation has a larger impact on GWAS variant PIP than on eQTL PIP (Figure 4B), likely because variants that affect phenotype are under stronger selection than those that merely affect gene expression, consistent with recent studies comparing GWAS hits and eQTL variants.51 Also potentially contributing to observed differences in PIP is the fact that most GWAS traits are categorical whereas eQTLs are continuous.
Figure 4.

3′ UTR variants in RBP motifs, eCLIP peaks, and miRNA sites are associated with phenotypes
(A) Comparison of PhastCons score distributions for GWAS variants in eCLIP peaks, RBP motifs, or no known regulatory elements.
(B) Proportion causal (proportion of GWAS hits with PIP > 0.25) with 95% confidence intervals for variants not in RBP motifs or eCLIP peaks compared to variants in RBP motifs or eCLIP peaks.
(C) As in (B), but for variants in a single motif or CLIP peak compared to variants in more than one motif or peak in genes matched by gene expression.
(D) Odds of a GWAS variant being in predicted miRNA site versus control variants (PIP < 0.001) as minimum PIP increases; shown is odds ratio with 95% confidence intervals.
(E) Proportion causal with 95% confidence intervals for variants not in miRNA sites compared to variants in miRNA sites with increasing predicted seed strength.
(F) Proportion causal with 95% confidence intervals (left) for variants not in RBP motifs or eCLIP peaks compared to variants in ReP sites or eCLIP peaks, matched for raw CADD score (right).
For all panels, ∗p < 0.01.
Recent research has suggested that variants in “RBP hubs”—locations where multiple RBPs bind–have lower allele frequencies compared to variants in single eCLIP peaks. However, these studies did not control for the bias in eCLIP data toward genes with higher expression, a property that is associated with higher conservation.52 Regardless, we found that variants located in multiple overlapping eCLIP peaks or RBP motifs are slightly more likely to be causal of phenotypes than those in single peaks/motifs, even after controlling for gene expression (Figure 4C), supporting that such variants are enriched for function. Binding of multiple RBPs to a region may increase the likelihood that the variant alters binding of at least one protein or may enrich for sites that have multiple or important functions.
Variants in miRNA sites are also enriched for association with GWAS phenotypes. As PIP increases, GWAS variants are up to sixteen times more likely to be located in conserved miRNA target sites. However, GWAS variants are actually mildly depleted from non-conserved miRNA sites, suggesting that such sites rarely impact phenotype (Figure 4D). Similar to eQTLs, GWAS hits in miRNA sites have higher PIPs, especially those in 8mer seeds, which have up to 15-fold higher proportion of causal variants than variants outside of regulatory elements (Figure 4E). We did not see a significant association between the number of distinct miRNA family targets overlapping a variant and the proportion of high-PIP GWAS variants, but statistical power was limited (Figure S4). Concerns have been raised regarding the accuracy of computational methods for predicting miRNA sites and the ability of these predicted sites to impact phenotype.53 Our results suggest that computationally predicted miRNA sites, especially conserved targets for conserved miRNA families, are strongly enriched for causal variants affecting both gene expression and phenotype.
As observed for eQTLs, we found that GWAS hits in eCLIP peaks and RBP motifs have significantly higher CADD scores (Figure S3). Variants in eCLIP peaks, conserved miRNA sites, or ReP sites are more likely to be causal, even when CADD scores are matched, again supporting the argument for supplementing CADD scoring with regulatory information to improve discrimination of pathogenic variants (Figure 4F).
Effects of 3′ UTR variants on expression and phenotype depend on APA isoforms
APA can impact mRNA localization, translation, and/or steady-state transcript expression.8 We found that the proportion causal was 2-fold higher for eQTLs proximal to polyA sites (within 50 nt) than for eQTLs further from the PAS, for major isoform categories (Figure 5A). eQTL variants within 50 nt of a polyA site tend to occur in more proximal APA isoforms compared to other eQTLs (Figure S5A). eQTLs falling in this region where core PAS motifs are located likely impact expression via changing the location or efficiency of cleavage and polyadenylation.8
Figure 5.

Variants in single or common 3′ UTRs more often impact gene expression and phenotype
(A) Proportion causal (proportion of eQTL variants with PIP greater than 0.25) with 95% confidence intervals for variants in various 3′ UTRs and proximal (<50 nucleotides) or distal to polyA sites.
(B) Proportion causal with 95% confidence intervals for eQTL variants in genes with various numbers of canonical APA isoforms (total genes include 5,335 APA and 3,360 single isoform).
(C) Proportion causal for GWAS variants in various 3′ UTRs, as in (A).
(D) Proportion causal for GWAS variants in genes with various numbers of APA isoforms, as in (B).
For all panels, ∗p < 0.01 compared to leftmost group, and error bars represent 95% confidence intervals.
The eQTL enrichment in single 3′ UTR isoform genes, especially proximal to PAS, is not unexpected. A variant in a regulatory element in a single 3′ UTR gene, especially a PAS-proximal variant, may be more likely to affect expression than a similar variant in an APA gene because single-isoform UTRs tend to be shorter and have fewer regulatory elements, whereas APA genes tend to have more redundant regulatory elements and PAS.8 Thus, a variant in a single-isoform gene is more likely to disrupt a unique regulatory element or PAS, whereas a variant in an APA gene is more likely to disrupt a redundant regulatory element or PAS. Another possibility that is supported by prior studies is that single-UTR genes are more likely to be regulated by expression changes from RBP and miRNA binding, whereas multi-UTR genes are more likely to be regulated by isoform abundance changes from APA.37 Therefore eQTLs, which are variants that impact expression, are more likely to be identified in single-UTR genes, as these genes are more likely than APA genes to be regulated by expression changes.
We further examined which subsets of RBPs may be responsible for the higher proportion of causal eQTLs proximal to PAS. As expected, we found motifs for RBPs known to bind near PAS such as LARP4 are enriched among PAS-proximal eQTLs, and motifs for RBPs known to bind distal to PAS, such as UPF1 RNA helicase and ATPase (UPF1), are depleted Figure S6(Figure S5B).
We also observed that the PIPs of eQTLs are higher in genes with single 3′ UTR isoforms and in common regions of 3′ UTRs from APA genes than in “partially shared” (alternative) UTR regions of the same genes (Figure 5A). The likely explanation is that presence in all transcripts from a gene versus only some transcripts increases the magnitude of the impact on gene expression for common types of variants. Considering the relationship between eQTL PIP and the number of APA isoforms, we found that variants in genes with fewer APA isoforms have a higher proportion of causal variants, likely for similar reasons (Figure 5B). These findings persisted after controlling for 3′ UTR length, distance to the stop codon, and gene expression, and the number of eQTL variants per gene did not vary substantially with APA isoform number Figure S5 (Figure S6). Unlike eQTLs, GWAS hits did not have higher PIPs near polyA sites; however, we do see a trend toward higher PIP for GWAS variants in common regions or in 3′ UTRs with a single PAS (Figure 5C). Variants in genes with fewer APA isoforms also have higher GWAS PIPs, as seen for eQTLs (Figure 5D).
Regulatory features help to identify pathogenic 3′ UTR variants
Here, we have shown that conserved variants in RBP and miRNA sites within common 3′ UTR regions of genes with fewer APA isoforms are more likely to impact gene expression and phenotype. These features can be incorporated into a generalized linear model to predict whether a variant is an eQTL or GWAS hit based on its regulatory features (Figure S7). The model coefficients for each feature represent the increase in the odds of a variant being a causal GWAS hit or causal eQTL (with PIP > 0.5) if the variant overlaps the feature. As expected, conservation score has the largest predictive value for GWAS hits, whereas proximity to PAS is most predictive for eQTLs (Figure 6A). Similar to recent studies, we found limited overlap between eQTLs and GWAS hits.51 This limited overlap may be due to differences in discovery methods and gene features, as discussed by Mostafavi et al.51 In addition, many genes are expected to tolerate differences in expression without impacting phenotype; thus, most eQTLs are not expected to be GWAS hits. For our variants overall, only 17% of GWAS hits were supported by eQTL evidence. We hypothesized that incorporation of regulatory information can improve overlap between GWAS and eQTLs. Indeed, we found that 35% of 6,764 eQTLs in eCLIP peaks and all 140 eQTLs in conserved miRNA sites were GWAS variants (PIP > 0.001, Figure 6B). Additionally, whereas only 39% of eQTLs are GWAS variants, nearly 50% of variants in regulatory elements are GWAS variants (data not shown). Thus, regulatory elements are enriched for both eQTL and GWAS variants and explain more phenotypic differences than eQTLs alone, likely because some regulatory elements affect aspects of mRNA function (translation, localization, etc.) that are not reflected in gene-expression measurements. Incorporation of regulatory features significantly improves the overlap between GWAS and eQTL variants compared to prior analyses.51
Figure 6.

Characterization of 3′ UTR variants into their annotations and regulatory elements helps prioritize variants for functional analysis and disease classification
(A) Exponential of logistic regression model coefficients with 95% confidence intervals. The model independent variable is binary (PIP greater or less than/equal to 0.5), and ∗p < 0.05.
(B) Intersection of eQTL variants in different putative regulatory elements with GWAS hits; dots are weighted by intersect size and error bars are 95% confidence intervals.
(C) Odds of a ClinVar 3′ UTR variant in RBP motifs or eCLIP peaks being pathogenic versus variants not in a predicted regulatory element; shown is odds ratio with 95% confidence intervals.
(D) RegVar workflow and example output for two ClinVar 3′ UTR variants of uncertain clinical significance (genomic coordinates are in hg38).
(E) Odds of a pathogenic ClinVar 3′ UTR variant being predicted by RegVar to be an eQTL versus all 3′ UTR gnomAD variants, with 95% confidence intervals.
(F) Odds of a pathogenic ClinVar 3′ UTR variant being in a predicted regulatory element (miRNA site, RBP motif, or eCLIP peak) versus all 3′ UTR gnomAD variants, with 95% confidence intervals.
(G) Odds of a gnomAD 3′ UTR variant predicted by RegVar to be an eQTL to be in a disease-associated gene versus 3′ UTR gnomAD variants not predicted to be eQTLs, with 95% confidence intervals.
(H) Difference in reference versus alternative allele reporter RNA expression in cell lines for select ClinVar variants. Confidence intervals are standard deviation of two technical replicates.
For all panels except (A), ∗p < 0.01.
We show that regulatory analysis of noncoding variants using several orthogonal methods aids in identification of causal eQTLs and GWAS hits, many of which are expected to be pathogenic. Of conserved 3′ UTR variants with known clinical significance in the ClinVar database, variants in RBP motifs are 3 times more likely and variants in eCLIP peaks are over 20 times more likely than variants not in regulatory elements to be pathogenic (Figure 6C). (Too few ClinVar variants were located in conserved miRNA sites to permit analysis.) Conservation had little if any impact on these findings as the degree of conservation between these ClinVar variant subsets was similar (Figure S8).
Our results indicate that regulatory analysis of 3′ UTR variants can aid in prioritization of variants for functional analysis and detection of pathogenic variants in individuals. To this end, we developed a program, RegVar, that characterizes 3′ UTR variants by their annotations, conservation, and predicted regulatory elements (Figure 6D). This tool will enable prioritization of 3′ UTR variants for functional analysis, potentially contributing to improved diagnosis and treatment. Many variants in ClinVar are located in UTRs, and most of these variants are of uncertain significance.5 Prioritization of specific variants for experimental analysis will be beneficial to the thousands of people who have no genetic diagnosis despite whole-exome or whole-genome sequencing.
Variants causing disease typically have low allele frequency in the general population as a result of natural selection, whereas the GWAS and eQTL analyses have statistical power only to detect variants of relatively high frequency. RegVar enables functional analysis of both common and rare variants due to its descriptive and predictive abilities. Any single 3′ UTR variant or variant call format file can be supplied to RegVar, and RegVar will describe potential functional impacts of each variant based on sequence (RBP motifs, miRNA seeds), functional datasets (GWAS, eCLIP, and eQTLs), and clinical annotations (ClinVar). In addition, RegVar will predict whether the variant is likely to be an eQTL or GWAS variant based on our regression model (Figure 6A).
We provide several analyses to demonstrate the ability of RegVar to discriminate functional and/or pathogenic variants from variant datasets. RegVar identified significant enrichment for predicted eQTL variants among known pathogenic variants in ClinVar compared to gnomAD variants, which are expected to be benign (Figure 6E). In addition, pathogenic ClinVar variants are over 20 times more likely than gnomAD variants to fall in a regulatory element identified by RegVar (Figure 6F). Finally, for the subset of gnomAD variants that fall in 3'seq gene annotations, variants that are predicted to be eQTL or GWAS variants are more likely to occur in disease-associated genes than those that are not predicted to impact expression or phenotype (Figure 6G).
We selected several ClinVar variants that we characterized by RegVar to further illustrate its potential (ClinVar variants c.∗1622A>T [ClinVar: RCV000319698.5] [GenBank: NM_007375.4(TARDBP)]; c.∗1289A>T [ClinVar: VCV000874917.4] [GenBank: NM_001033044.4(GLUL)]; c.∗36A>G [ClinVar: VCV000879046.4] [GenBank: NM_005445.4(SMC3)]; and c.∗64T>C [ClinVar: VCV000883795.5] [GenBank: NM_001065.4(TNFRSF1A)].
All of these variants are predicted by RegVar to overlap multiple regulatory sites. To test the variants for their ability to directly modulate transcript levels, a parallel reporter assay was conducted via transfection of allelic pairs of fragments for each variant in HEK293 cells (see material and methods). RNA sequencing demonstrated allele-biased expression for each variant (Figure 6H).
Variant RCV000319698.5 is a variant of uncertain significance for amyotrophic lateral sclerosis type 10 in the TAR DNA binding protein (TARDBP) gene. This disease is known to occur in an autosomal-dominant fashion due to haploinsufficiency.54 The variant is in a ReP site for heterogeneous nuclear ribonucleoprotein C (HNRNPC), and is predicted to disrupt motifs for far upstream element binding protein 3 (FUBP3), heterogeneous nuclear ribonucleoprotein C like 1 (HNRNPCL1), heterogeneous nuclear ribonucleoprotein D like (HNRNPDL), RNA binding motif protein 15B (RBM15B), TIA1, and tRNA selenocysteine 1 associated protein 1 (TRNAU1AP). It is also in seeds for miR-4282 and miR-5096 and in a common region of a gene with only two isoforms. The variant is predicted by RegVar to be an eQTL. VCV000874917.4 is a variant of uncertain significance for congenital brain dysgenesis due to glutamine synthetase deficiency in the gene glutamate-ammonia ligase (GLUL). It is in a TIA1 ReP site; in motifs for FUBP3, HNRNPCL1, HNRNPDL, RBM15B, TIA, TRNAU1AP, and ELAV like RNA binding protein 4 (ELAVL4); and in a miR-5692 seed. It is in a common region of an APA gene and is less than 50 nucleotides away from the nearest PAS.
VCV000879046.4 is a variant of uncertain significance for Cornelia de Lange syndrome 3 in the structural maintenance of chromosome 3 (SMC3) gene. This is an autosomal dominant disease caused by haploinsufficiency, and SMC3 is intolerant to loss-of-function mutations.55 The variant falls in a pumilio RNA binding family member 2 (PUM2) ReP site, motif, and eCLIP peak. PUM2 is known to destabilize RNA.56 There is higher expression of the alternative allele compared to the reference allele in cells, as expected for a variant that disrupts binding of a destabilizing RBP. This variant is predicted by RegVar to be both an eQTL and a GWAS variant, is highly conserved, is in the common region of a gene with two isoforms, and is less than 50 nucleotides to a PAS, all supporting its functionality.
VCV000883795.5 is a variant of uncertain significance for tumor necrosis factor (TNF) receptor-associated periodic fever syndrome in the TNF receptor superfamily member 1A (TNFRSF1A) gene. This is an autosomal dominant disease, with haploinsufficiency as a mechanism. There is higher expression of the alternative allele in cells, and the variant disrupts a TIA1 site. To confirm that TIA1 is expected to destabilize target RNAs, we analyzed RNA sequencing data from the ENCODE database before and after TIA1 knock down in HepG2 and K562 cells. As expected for a destabilizing RBP, we found genes with TIA1 eCLIP peaks exhibited increased expression after TIA1 knock down in both cell lines (Figure S9A). In addition, genes that exhibited differential expression after TIA1 knock down were significantly enriched for TIA1 ReP motifs (Figure S9B). TNFRSF1A itself exhibits a 1.3-fold increase in expression after TIA1 knockdown.
These variants should be investigated as potentially pathogenic, as in vitro data support their regulatory activity, and RegVar suggests several possible mechanisms of action for each. These examples highlight the power of RegVar to quickly analyze any 3′ UTR variant and provide possible mechanisms of pathogenicity, unlike more time-intensive strategies, such as cellular assays.25 In addition, as RegVar provides analysis of variants based on population datasets and functional in vitro studies of variants in their natural context, including eCLIP and RBNS, it avoids false positives and negatives generated by these other methods due to their reliance on overexpressed variants in artificial genomic contexts.
Discussion
Characterization of noncoding variants is required to expand the impact of exome and genome sequencing on the clinical sphere. Here, we show that 3′ UTR variants in eCLIP peaks, RBP motifs, miRNA seed sites, and common APA isoform regions proximal to polyA sites are associated (often strongly) with gene expression changes, phenotypes, and pathology. We provide a tool, RegVar, to help researchers and clinicians prioritize noncoding variants for functional analysis based on their location in regulatory elements. RegVar can process many variants in parallel and can be readily integrated into bioinformatic pipelines for systematic variant annotation. We anticipate that this program will be used to interpret the over 10,000 ClinVar variants of uncertain significance in putative 3′ UTR regulatory elements.
New models are being developed to predict whether variants have cis-regulatory effects on gene expression or phenotype.57,58,59,60 These models incorporate many variant annotations, but have not incorporated RBP motif, miRNA target, or eCLIP peak data. Our general linearized model suggests that up to 10% more high-confidence GWAS hits or eQTL variants can be explained with incorporation of these features. Our findings will improve discrimination of pathogenic variants, as we show that 3′ UTR variants with the same CADD score are more likely to affect gene expression or phenotype if they fall in these regulatory elements. In addition, our tool, RegVar, fills an important unmet need in noncoding variant interpretation, providing variant effect prediction.
Recent findings show that eQTLs are mostly found in less constrained genes with simple regulatory architecture, compared to GWAS hits, which are more likely to be found in functionally important genes.51 This could suggest that predicting variants that impact gene expression has limited clinical utility. However, we found that eQTL variants in regulatory elements are more likely to be GWAS hits, indicating that including regulatory features into eQTL models will help distinguish phenotypically important eQTLs.
Most individuals with undiagnosed rare diseases have exome rather than genome sequencing performed despite the increased power of genome sequencing.61 This is largely due to limitations on interpretation of noncoding variants.62 Our findings argue that clinical sequencing should extend further into the 3′ UTR to improve pathogenic variant detection. This could be done by extending exome capture slightly without significantly increasing the cost of sequencing. Recently published guidelines for interpretation of noncoding variants require querying multiple databases.63 We propose noncoding variants should be systematically assessed using RegVar and reported with sequencing results. RegVar will decrease the workload for noncoding variant interpretation by incorporating multiple datasets into one user-friendly tool.
Despite the advances our study makes into interpretation of noncoding variants, there remain some limitations. Our findings are based on GWAS and eQTL variants, which are more common in the population than Mendelian disease-causing variants. In addition, GWAS hits and eQTL variants are mostly of low PIP after fine-mapping, suggesting that most variants in these datasets are not truly causal. However, we found that pathogenic ClinVar variants, like eQTLs and GWAS hits, are more likely to be in RBP motifs and eCLIP peaks, suggesting our results are generalizable to rare, pathogenic variants. We anticipate that our work will result in targeted experimental studies of individual variants that will aid in disease diagnosis.
Our data provide a thorough analysis of 3′ UTR variants from several computational and population-wide datasets. We found variants that exhibit allele-specific binding in cells are more likely to be in predicted motifs, suggesting these computational methods predict in vivo regulation. We limited our study to the 3′ UTR because this is an important regulatory region ignored by current methods of variant effect prediction; however, RBPs, and to a more limited extent miRNAs, also bind other noncoding regions such as introns or 5′ UTRs. Our results may extend to these areas as well, and these will be important future areas of study as variants in these regions also remain difficult to interpret.
Web resources
GitHub, RegVar R package, https://github.com/RomoL2/RegVar
Acknowledgments
We thank members of the Burge laboratory for their helpful discussions and comments on the manuscript, especially Michael McGurk and Hannah Jacobs for their insights. In addition, we thank Sarah Stenton and Anne O’Donnell Luria for their helpful comments on the manuscript. Graphical abstract was created with BioRender.com. L.R. was supported by a Fred Lovejoy Resident Research and Education Fund award. This work was funded by grants from the NIH (GM085319 and HG002439 to C.B.B.).
Author contributions
L.R. designed the study with input from C.B.B. and S.D.F.; L.R. performed analysis with contributions from S.D.F.; L.R. wrote the RegVar package; L.R. wrote the draft manuscript; and L.R. and C.B.B. finalized the manuscript with input from S.D.F.
Declaration of interests
The authors declare no competing interests.
Published: January 17, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ajhg.2023.12.017.
Contributor Information
Lindsay Romo, Email: lindsay.romo@childrens.harvard.edu.
Christopher B. Burge, Email: cburge@mit.edu.
Supplemental information
References
- 1.Dewey F.E., Murray M.F., Overton J.D., Habegger L., Leader J.B., Fetterolf S.N., O’Dushlaine C., Van Hout C.V., Staples J., Gonzaga-Jauregui C., et al. Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the DiscovEHR study. Science. 2016;354:aaf6814-1–aaf6814-10. doi: 10.1126/science.aaf6814. [DOI] [PubMed] [Google Scholar]
- 2.Ng P.C., Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Karczewski K.J., Francioli L.C., Tiao G., Cummings B.B., Alföldi J., Wang Q., Collins R.L., Laricchia K.M., Ganna A., Birnbaum D.P., et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–443. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pérez-Palma E., Gramm M., Nürnberg P., May P., Lal D. Simple ClinVar: an interactive web server to explore and retrieve gene and disease variants aggregated in ClinVar database. Nucleic Acids Res. 2019;47:W99–W105. doi: 10.1093/nar/gkz411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gebauer F., Schwarzl T., Valcárcel J., Hentze M.W. RNA-binding proteins in human genetic disease. Nat. Rev. Genet. 2021;22:185–198. doi: 10.1038/s41576-020-00302-y. [DOI] [PubMed] [Google Scholar]
- 7.Mayya V.K., Duchaine T.F. Ciphers and executioners: How 3 0 -untranslated regions determine the fate of messenger RNAs. Front. Genet. 2019;10:1–18. doi: 10.3389/fgene.2019.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tian B., Manley J.L. Alternative polyadenylation of mRNA precursors. Nat. Rev. Mol. Cell Biol. 2017;18:18–30. doi: 10.1038/nrm.2016.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiang P., Coller H. Functional Interactions Between microRNAs and RNA Binding Proteins. MicroRNA. 2012;1:70–79. doi: 10.2174/2211536611201010070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gebert L.F.R., MacRae I.J. Regulation of microRNA function in animals. Nat. Rev. Mol. Cell Biol. 2019;20:21–37. doi: 10.1038/s41580-018-0045-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lambert N., Robertson A., Jangi M., McGeary S., Sharp P.A., Burge C.B. RNA Bind-n-Seq: Quantitative Assessment of the Sequence and Structural Binding Specificity of RNA Binding Proteins. Mol. Cell. 2014;54:887–900. doi: 10.1016/j.molcel.2014.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Van Nostrand E.L., Pratt G.A., Shishkin A.A., Gelboin-Burkhart C., Fang M.Y., Sundararaman B., Blue S.M., Nguyen T.B., Surka C., Elkins K., et al. Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP) Nat. Methods. 2016;13:508–514. doi: 10.1038/nmeth.3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Agarwal V., Bell G.W., Nam J.W., Bartel D.P. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015;4:e05005. doi: 10.7554/eLife.05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fields C.J., Li L., Hiers N.M., Li T., Sheng P., Huda T., Shan J., Gay L., Gu T., Bian J., et al. Sequencing of Argonaute-bound microRNA/mRNA hybrids reveals regulation of the unfolded protein response by microRNA-320a. PLoS Genet. 2021;17:e1009934. doi: 10.1371/journal.pgen.1009934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rentzsch P., Witten D., Cooper G.M., Shendure J., Kircher M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47:D886–D894. doi: 10.1093/nar/gky1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.ENCODE Project Consortium. Moore J.E., Purcaro M.J., Pratt H.E., Epstein C.B., Shoresh N., Adrian J., Kawli T., Davis C.A., Dobin A., et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature. 2020;583:699–710. doi: 10.1038/s41586-020-2493-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ionita-Laza I., Mccallum K., Xu B., Buxbaum J.D. A spectral approach integrating functional genomic annotations for coding and noncoding variants. Nat. Genet. 2016;48:214–220. doi: 10.1038/ng.3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Quang D., Chen Y., Xie X. DANN: A deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics. 2015;31:761–763. doi: 10.1093/bioinformatics/btu703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hassan M.S., Shaalan A.A., Dessouky M.I., Abdelnaiem A.E., ElHefnawi M. A review study: Computational techniques for expecting the impact of non-synonymous single nucleotide variants in human diseases. Gene. 2019;680:20–33. doi: 10.1016/j.gene.2018.09.028. [DOI] [PubMed] [Google Scholar]
- 20.Frazer J., Notin P., Dias M., Gomez A., Min J.K., Brock K., Gal Y., Marks D.S. Disease variant prediction with deep generative models of evolutionary data. Nature. 2021;599:91–95. doi: 10.1038/s41586-021-04043-8. [DOI] [PubMed] [Google Scholar]
- 21.Mohammadi P., Castel S.E., Brown A.A., Lappalainen T. Quantifying the regulatory effect size of cis-acting genetic variation using allelic fold change. Genome Res. 2017;27:1872–1884. doi: 10.1101/gr.216747.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bycroft C., Freeman C., Petkova D., Band G., Elliott L.T., Sharp K., Motyer A., Vukcevic D., Delaneau O., O’Connell J., et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562:203–209. doi: 10.1038/s41586-018-0579-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wen X., Pique-Regi R., Luca F., Walter K., Min J.L., Huang J., Crooks L., Memari Y., McCarthy S., Perry J.R.B., et al. The UK10K project identifies rare variants in health and disease. Nature. 2019;10:1–29. doi: 10.1038/nature14962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.GTEx Consortium. Laboratory Data Analysis &Coordinating Center LDACC—Analysis Working Group. Statistical Methods groups—Analysis Working Group. Enhancing GTEx eGTEx groups. NIH Common Fund. Jo B., Mohammadi P., Park Y.S., Parsana P., et al. Biospecimen Collection Source Site—NDRI Genetic effects on gene expression across human tissues. Nature. 2017;550:204–213. doi: 10.1038/nature24277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Griesemer D., Xue J.R., Reilly S.K., Ulirsch J.C., Kukreja K., Davis J.R., Kanai M., Yang D.K., Butts J.C., Guney M.H., et al. Genome-Wide Functional Screen of 3’UTR Variants Uncovers Causal Variants for Human Disease and Evolution. Cell. 2021;184:5247–5260.e19. doi: 10.1016/j.cell.2021.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klein J.C., Keith A., Rice S.J., Shepherd C., Agarwal V., Loughlin J., Shendure J. Functional testing of thousands of osteoarthritis-associated variants for regulatory activity. Nat. Commun. 2019;10:2434. doi: 10.1038/s41467-019-10439-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Findlay G.M., Daza R.M., Martin B., Zhang M.D., Leith A.P., Gasperini M., Janizek J.D., Huang X., Starita L.M., Shendure J. Accurate classification of BRCA1 variants with saturation genome editing. Nature. 2018;562:217–222. doi: 10.1038/s41586-018-0461-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meng L., Pammi M., Saronwala A., Magoulas P., Ghazi A.R., Vetrini F., Zhang J., He W., Dharmadhikari A.V., Qu C., et al. Use of exome sequencing for infants in intensive care units ascertainment of severe single-gene disorders and effect on medical management. JAMA Pediatr. 2017;171:e173438. doi: 10.1001/jamapediatrics.2017.3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.NICUSeq Study Group. Krantz I.D., Medne L., Weatherly J.M., Wild K.T., Biswas S., Devkota B., Hartman T., Brunelli L., Fishler K.P., et al. Effect of Whole-Genome Sequencing on the Clinical Management of Acutely Ill Infants with Suspected Genetic Disease: A Randomized Clinical Trial. JAMA Pediatr. 2021;175:1218–1226. doi: 10.1001/jamapediatrics.2021.3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wen X., Pique-Regi R., Luca F. Integrating molecular QTL data into genome-wide genetic association analysis: Probabilistic assessment of enrichment and colocalization. PLoS Genet. 2017;13:e1006646. doi: 10.1371/journal.pgen.1006646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang R., Nambiar R., Zheng D., Tian B. PolyA-DB 3 catalogs cleavage and polyadenylation sites identified by deep sequencing in multiple genomes. Nucleic Acids Res. 2018;46:D315–D319. doi: 10.1093/nar/gkx1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Quinlan A.R., Hall I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schaid D.J., Chen W., Larson N.B. From genome-wide associations to candidate causal variants by statistical fine-mapping. Nat. Rev. Genet. 2018;19:491–504. doi: 10.1038/s41576-018-0016-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wen X., Luca F., Pique-Regi R. Cross-Population Joint Analysis of eQTLs: Fine Mapping and Functional Annotation. PLoS Genet. 2015;11:e1005176. doi: 10.1371/journal.pgen.1005176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang G., Sarkar A., Carbonetto P., Stephens M. A simple new approach to variable selection in regression, with application to genetic fine mapping. J. R. Stat. Soc. Series B Stat. Methodol. 2020;82:1273–1300. doi: 10.1111/rssb.12388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jens M., Mcgurk M., Bundschuh R., Burge C.B. RBPamp: Quantitative Modeling of Protein-RNA Interactions in vitro Predicts in vivo Binding. bioRxiv. 2022 Preprint at. [Google Scholar]
- 37.Lianoglou S., Garg V., Yang J.L., Leslie C.S., Mayr C. Ubiquitously transcribed genes use alternative polyadenylation to achieve tissue-specific expression. Genes Dev. 2013;27:2380–2396. doi: 10.1101/gad.229328.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Romo L., Ashar-Patel A., Pfister E., Aronin N. Alterations in mRNA 3′UTR isoform abundance accompany gene expression changes in human Huntington’s disease brains. Cell Rep. 2017;20:3057–3070. doi: 10.1016/j.celrep.2017.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Siepel A., Bejerano G., Pedersen J.S., Hinrichs A.S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L.W., Richards S., et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang E.W., Bahn J.H., Hsiao E.Y.H., Tan B.X., Sun Y., Fu T., Zhou B., Van Nostrand E.L., Pratt G.A., Freese P., et al. Allele-specific binding of RNA-binding proteins reveals functional genetic variants in the RNA. Nat. Commun. 2019;10:1338. doi: 10.1038/s41467-019-09292-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kircher M., Witten D.M., Jain P., O’roak B.J., Cooper G.M., Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014;46:310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blekhman R., Man O., Herrmann L., Boyko A.R., Indap A., Kosiol C., Bustamante C.D., Teshima K.M., Przeworski M. Natural Selection on Genes that Underlie Human Disease Susceptibility. Curr. Biol. 2008;18:883–889. doi: 10.1016/j.cub.2008.04.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Berg J.S., Adams M., Nassar N., Bizon C., Lee K., Schmitt C.P., Wilhelmsen K.C., Evans J.P. An informatics approach to analyzing the incidentalome. Genet. Med. 2013;15:36–44. doi: 10.1038/gim.2012.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Findlay S.D., Romo L., Burge C.B. Quantifying negative selection in human 3’ UTRs uncovers constrained targets of RNA-binding proteins. Nat. Commun. 2024;15:85. doi: 10.1038/s41467-023-44456-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Felsenstein J., Churchill G.A. A Hidden Markov Model approach to variation among sites in rate of evolution. Mol. Biol. Evol. 1996;13:93–104. doi: 10.1093/oxfordjournals.molbev.a025575. [DOI] [PubMed] [Google Scholar]
- 46.Yang R., Gaidamakov S.A., Xie J., Lee J., Martino L., Kozlov G., Crawford A.K., Russo A.N., Conte M.R., Gehring K., Maraia R.J. La-Related Protein 4 Binds Poly(A), Interacts with the Poly(A)-Binding Protein MLLE Domain via a Variant PAM2w Motif, and Can Promote mRNA Stability. Mol. Cell Biol. 2011;31:542–556. doi: 10.1128/MCB.01162-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang H., Duckett C.S., Lindsten T. iPABP, an inducible poly(A)-binding protein detected in activated human T cells. Mol. Cell Biol. 1995;15:6770–6776. doi: 10.1128/mcb.15.12.6770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dominguez D., Freese P., Alexis M.S., Su A., Hochman M., Palden T., Bazile C., Lambert N.J., Van Nostrand E.L., Pratt G.A., et al. Sequence, Structure, and Context Preferences of Human RNA Binding Proteins. Mol. Cell. 2018;70:854–867.e9. doi: 10.1016/j.molcel.2018.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Natarajan K., Sundaramoorthy A., Shanmugam N. HnRNPK and lysine specific histone demethylase-1 regulates IP-10 mRNA stability in monocytes. Eur. J. Pharmacol. 2022;920 doi: 10.1016/j.ejphar.2021.174683. [DOI] [PubMed] [Google Scholar]
- 50.Liu X.H., Ma J., Feng J.X., Feng Y., Zhang Y.F., Liu L.X. Regulation and related mechanism of GSN mRNA level by hnRNPK in lung adenocarcinoma cells. Biol. Chem. 2019;400:951–963. doi: 10.1515/hsz-2018-0417. [DOI] [PubMed] [Google Scholar]
- 51.Mostafavi H., Spence J.P., Naqvi S., Pritchard J.K. Limited overlap of eQTLs and GWAS hits due to systematic differences in discovery. bioRxiv. 2022 Preprint at. [Google Scholar]
- 52.Zhang J., Liu J., Lee D., Feng J.J., Lochovsky L., Lou S., Rutenberg-Schoenberg M., Gerstein M. RADAR: annotation and prioritization of variants in the post-transcriptional regulome of RNA-binding proteins. Genome Biol. 2020;21:151. doi: 10.1186/s13059-020-01979-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Seitz H. Issues in current microRNA target identification methods. RNA Biol. 2017;14:831–834. doi: 10.1080/15476286.2017.1320469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Barmada S.J., Finkbeiner S. Pathogenic TARDBP mutations in amyotrophic lateral sclerosis and frontotemporal dementia: Disease-associated pathways. Rev. Neurosci. 2010;21:251–272. doi: 10.1515/revneuro.2010.21.4.251. [DOI] [PubMed] [Google Scholar]
- 55.Kruszka P., Berger S.I., Casa V., Dekker M.R., Gaesser J., Weiss K., Martinez A.F., Murdock D.R., Louie R.J., Prijoles E.J., et al. Cohesin complex-associated holoprosencephaly. Brain. 2019;142:2631–2643. doi: 10.1093/brain/awz210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wolfe M.B., Schagat T.L., Paulsen M.T., Magnuson B., Ljungman M., Park D., Zhang C., Campbell Z.T., Goldstrohm A.C., Freddolino P.L. Principles of mRNA control by human PUM proteins elucidated from multimodal experiments and integrative data analysis. RNA. 2020;26:1680–1703. doi: 10.1261/rna.077362.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang Q.S., Kelley D.R., Ulirsch J., Kanai M., Sadhuka S., Cui R., Albors C., Cheng N., Okada Y., et al. Biobank Japan Project Leveraging supervised learning for functionally informed fine-mapping of cis-eQTLs identifies an additional 20,913 putative causal eQTLs. Nat. Commun. 2021;12:3394. doi: 10.1038/s41467-021-23134-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Agarwal V., Shendure J. Predicting mRNA Abundance Directly from Genomic Sequence Using Deep Convolutional Neural Networks. Cell Rep. 2020;31 doi: 10.1016/j.celrep.2020.107663. [DOI] [PubMed] [Google Scholar]
- 59.Zhou J., Troyanskaya O.G. Predicting effects of noncoding variants with deep learning-based sequence model. Nat. Methods. 2015;12:931–934. doi: 10.1038/nmeth.3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhou J., Theesfeld C.L., Yao K., Chen K.M., Wong A.K., Troyanskaya O.G. Deep learning sequence-based ab initio prediction of variant effects on expression and disease risk. Nat. Genet. 2018;50:1171–1179. doi: 10.1038/s41588-018-0160-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Belkadi A., Bolze A., Itan Y., Cobat A., Vincent Q.B., Antipenko A., Shang L., Boisson B., Casanova J.L., Abel L. Whole-genome sequencing is more powerful than whole-exome sequencing for detecting exome variants. Proc. Natl. Acad. Sci. USA. 2015;112:5473–5478. doi: 10.1073/pnas.1418631112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Petersen B.S., Fredrich B., Hoeppner M.P., Ellinghaus D., Franke A. Opportunities and challenges of whole-genome and -exome sequencing. BMC Genet. 2017;18:14. doi: 10.1186/s12863-017-0479-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ellingford J.M., Ahn J.W., Bagnall R.D., Baralle D., Barton S., Campbell C., Downes K., Ellard S., Duff-Farrier C., FitzPatrick D.R., et al. Recommendations for clinical interpretation of variants found in non-coding regions of the genome. Genome Med. 2022;14:73. doi: 10.1186/s13073-022-01073-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.