

Abstract
Lithium ortho-thiophosphate (Li3PS4) has emerged as a promising candidate for solid-state electrolyte batteries, thanks to its highly conductive phases, cheap components, and large electrochemical stability range. Nonetheless, the microscopic mechanisms of Li-ion transport in Li3PS4 are far from being fully understood, the role of PS4 dynamics in charge transport still being controversial. In this work, we build machine learning potentials targeting state-of-the-art DFT references (PBEsol, r2SCAN, and PBE0) to tackle this problem in all known phases of Li3PS4 (α, β, and γ), for large system sizes and time scales. We discuss the physical origin of the observed superionic behavior of Li3PS4: the activation of PS4 flipping drives a structural transition to a highly conductive phase, characterized by an increase in Li-site availability and by a drastic reduction in the activation energy of Li-ion diffusion. We also rule out any paddle-wheel effects of PS4 tetrahedra in the superionic phases—previously claimed to enhance Li-ion diffusion—due to the orders-of-magnitude difference between the rate of PS4 flips and Li-ion hops at all temperatures below melting. We finally elucidate the role of interionic dynamical correlations in charge transport, by highlighting the failure of the Nernst–Einstein approximation to estimate the electrical conductivity. Our results show a strong dependence on the target DFT reference, with PBE0 yielding the best quantitative agreement with experimental measurements not only for the electronic band gap but also for the electrical conductivity of β- and α-Li3PS4.
I. Introduction
The growing demand for portable electronic products and electric vehicles has stimulated the creation of energy storage systems that offers better safety and higher energy density than current Li-ion battery systems.1 While commercial Li-ion batteries use organic liquid electrolytes and additives to achieve a high working voltage,2,3 these materials pose safety concerns due to their flammability and susceptibility to thermal runaway.4,5 To address these issues, researchers are developing all-solid-state batteries (ASSBs) with inorganic solid electrolytes (SEs) to provide a sustainable solution for energy storage, exploiting their expected longer lifespan and improved energy efficiency.6,7 Many families of SEs have been considered and studied during these years.8−10 Sulfides are recognized as uniquely promising materials due to their remarkable mechanical stability and room-temperature ionic conductivity.11−16 In particular, the family of lithium thiophosphates (LPS), with its archetypal Li3PS4 compound, is widely recognized as one of the most promising families of sulfide electrolytes, and it has been the subject of many experimental and computational studies.1,17−27
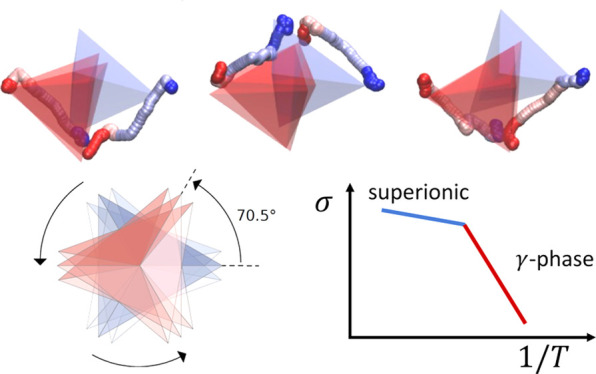
Li3PS4 has three main polymorphs: α-Li3PS4 (with space group Cmcm(28)), β-Li3PS4 (Pnma), and γ-Li3PS4 (Pmn21, Figure 1). Whereas the γ polymorph is the most stable at room temperature, it also exhibits low room-temperature ionic conductivity (≈ 3 × 10–7 S cm–1, see ref (20)). The system transforms into the metastable β-polymorph at 573 K and then into the α-polymorph at 746 K.20 Despite their great relevance, in the past years computational studies have been limited by the use of empirical potentials23 ,24 and ab initio molecular dynamics (AIMD),22 based on density functional theory (DFT) with generalized gradient approximation (GGA).29,30 The former can provide useful mechanistic insights but fail to correctly predict the activation energies of the conductive phases23 and are inherently limited in their accuracy and transferability. Quantum mechanical approaches, on the other hand, are more accurate, but they are burdened by a higher computational cost, which hinders their applicability to realistic systems. For example, recent studies based on AIMD-PBE simulations attributed the superionic conductivity of glassy 75Li2S–25P2S5 and that of bulk β-Li3PS4 to the presence of fast cation–polyanion correlations—the so-called paddlewheel effect.31,32 While providing evidence of this effect, the simulations carried out in these works are clearly limited in the simulation times and system sizes that they can achieve, potentially leading to unphysical outcomes.
Figure 1.

Sketch of the α, β, and γ phases of Li3PS4, showing the difference of the relative alignment of the PS4 tetrahedra along reference (010) crystallographic planes (here numbered between 1 and 6 for clarity). The γ structure has all tetrahedra aligned along the [100] direction for all of this set of planes, while the β structure has the tetrahedra aligned along both the [100] direction and the [1̅00] across each plane. Finally, the α phase has a staggered ordering: the tetrahedra are aligned along [100]/[1̅00] for the planes that are numbered with even/odd numbers.
In the past decade, the advent of machine learning has allowed the construction of interatomic potentials possessing quantum mechanical accuracy at a cost that is only marginally higher than that of classical force fields.33−45 Machine learning potentials (MLP) rely on the construction of physically motivated representations to predict a given target property. In particular, representations of atomic configurations should preserve key physical symmetries: global translational and rotational invariance, as well as invariance with respect to the permutation of atoms of the same chemical species.46 Among the numerous potential representations, the Smooth Overlap of Atomic Positions (SOAP)47 used in combination with appropriate regression schemes has facilitated the development of ML potentials for simulating a variety of materials properties via extensive finite-temperature thermodynamic sampling.25,26,48−58 Notable examples of the use of MLPs to study the ionic conductivity in solid-state electrolytes are the Gaussian Approximation Potential (GAP) for lithium thiophosphate developed by Staacke et al.26 and the Deep Neural Network (DNN) for Li10GeP2S12-type compounds developed by Huang and co-workers.59 Both of these studies were able to characterize the diffusion properties of their respective target compounds and overcome some known limitations of AIMD, namely, the small size and the short simulation times that are accessible by this type of modeling. Despite these important breakthroughs, two main aspects are still missing to provide a comprehensive study of transport properties in this class of materials. First, the accuracy of the aforementioned potentials is limited to the GGA level of theory due to the choice of the reference DFT functional (PBE and PBEsol) for the calculation of the training set structures. While this is a standard choice for performing first-principles calculations in solids, the relatively small number of reference single-point calculations (usually a few thousands) that are needed to reach the desired target ML accuracy enables the use of more accurate references, like meta-GGA and hybrid functionals.60,61 To our knowledge, no systematic study comparing different DFT references exists to date for this class of materials. Second, these studies neglect the contribution of interionic correlations to the electrical conductivity and its relation with polyanion rotations.
In this work, we train three MLPs to investigate the physical mechanisms of charge transport in Li3PS4 and their effect on the electrical conductivity of its stable polymorphs. Each potential is trained over data sets computed at a different level of theory: GGA, metaGGA, and hybrid functionals. In particular, we use the Perdew–Burke–Ernzerhof functional revisited for solids (PBEsol),29,62 the regularized version of the strongly constrained and appropriately normed (r2SCAN) functional,63 and the PBE0 functional.64 We explore the temperature dependence of the ionic conductivity of Li3PS4 showing that different functionals predict different critical temperatures for the onset of the conductive regime, which is roughly associated with the onset of a structural phase transition. We also elucidate the importance of including the effects of the interionic correlation in the conductivity, by computing it with the full Green–Kubo (GK) theory of linear response,65,66 instead of the Nernst–Einstein approximation commonly employed in the literature. Overall, we find that the PBE0 functional gives the best quantitative agreement with existing experimental measurements of the ionic conductivity of β-Li3PS4. Furthermore, we relate the onset of the superionic phase of the Li3PS4 compound with the PS4 flipping dynamics and find that discrete P–S flips induce a structural phase transition from the nonconductive γ to a mixture of the β and α structures, that cannot be fully resolved at the size and time scale of these simulations. This structural change determines a drastic decrease of the slope of the Arrhenius curve and thus a significant reduction of the activation energy of Li-ion diffusion (by a factor of 6 compared to the γ phase). Finally, we detect a second transition to a disordered phase with freely rotating polyanions at even higher temperatures that we attribute to melting of the PS3–4 sublattice. Both the transition to the conductive phase and the Li3PS4 melting appear as peaks of the heat capacity and are thus associated with separate first-order phase transitions of this material.
II. Methods
II.A. Training Set Construction and Validation of the ML Models
We constructed the training set for the ML models in an iterative fashion. A starting data set of structures is generated by running NVT Car–Parrinello Molecular Dynamics67 with the PBEsol functional62 as provided by the Quantum ESPRESSO package,68−71 for a set of selected temperatures (250, 500, and 1000 K) and volumes. This initial data set is then computed via more converged single-point DFT calculations with the PBEsol functional. Further details can be found in the Supporting Information.
As a next step, we fit a preliminary MLP on this data set and run finite-temperature Molecular Dynamics (MD) with i-PI72 over the entire temperature range of interest (between 200 and 1000 K). Among the uncorrelated structures generated in the resulting trajectories, only a subset consisting of the most diverse ones according to the Farthest-Point Sampling (FPS) method73 is selected and recomputed using DFT. This active-learning loop, consisting of the regression of an MLP, MD simulations, and recalculation of a set of structures by DFT, allows us to extend the data set until the model is deemed sufficiently accurate and robust. In order to generate data sets for the ML-SCAN and ML-PBE0 models, we select via FPS a subset of snapshots out of the whole PBEsol data set and we use a two-level machine learning (2LML) scheme74 to train accurate potentials from a minimal number of expensive r2SCAN or PBE0 calculations. The 2LML is a specific case of the general multilevel machine learning scheme and consists of training a ML model on the large PBEsol data set, then computing energy and forces at the r2SCAN (PBE0) level, and finally training a new ML potential on the difference between the ML-PBEsol predicted energies and forces and the true r2SCAN (PBE0) references.74,75 The final data sets consist of 2400 structures for the ML-PBEsol model, 740 structures for the ML-r2SCAN model, and 790 structures for the ML-PBE0 model. Within these data sets, a subset of 100 randomly selected structures is used as a test set for the ML-PBEsol model, while a subset of 40 structures is used for both the ML-r2SCAN and the ML-PBEsol model. PBEsol calculations are performed with Quantum ESPRESSO, while r2SCAN and PBE0 calculations are performed with VASP.76−78 The training of all of the models is performed targeting the cohesive energies to avoid offset issues induced by different pseudopotentials. Figure 2 shows the parity plots for the forces of the three models over their respective test sets. Table 1 contains the root-mean-square-errors (RMSEs) for all models, showing that our model can achieve errors similar (or better) than those obtained in other similar works.25,26,59 The learning curves for each of these models are reported in the Supporting Information (Section I). The Supporting Information also reports results from kernel principal component analysis79 (Section II) and the newly introduced local prediction rigidity(80) (Section III) to check the distribution of the environments in our data set and along the MD trajectories, and to verify that our training set can reliably represent the complex local environments that occur during PS4 flips. Dynamical properties, like the mean square displacement and atomic diffusivity of Li ions, also appear to be properly reproduced by our ML potentials, as we have directly tested via MD simulations of the α phase at high temperature, obtained with PBEsol ab initio potential and with its corresponding ML model, as reported in Section IV of the Supporting Information.
Figure 2.
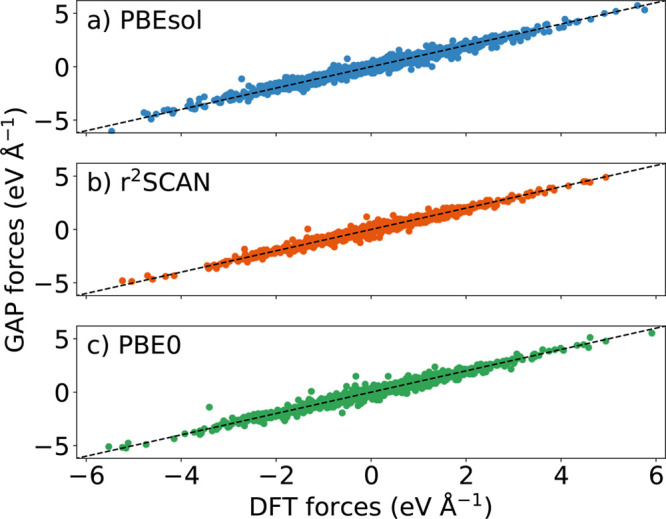
Parity plot of the atomic forces for each model: (a) PBEsol; (b) r2SCAN; and (c) PBE0.
Table 1. Root-Mean-Square-Error (RMSE) of the Energy (Second Column) and Forces (Third Column) for All of the Modelsa.
| model | RMSE [meV/atom] | RMSE (%RMSE) [meV/Å] |
|---|---|---|
| PBEsol | 7.0 | 120 (14.8%) |
| r2SCAN | 6.5 | 141 (15.8%) |
| PBE0 | 6.5 | 165 (17.2%) |
In the third column, the number in parentheses indicates the %RMSE relative to the standard deviation of forces within the test set.
II.B. Collective Variables for Li3PS4
The three main polymorphs of Li3PS4 are differentiated by the relative orientation of PS4 tetrahedra. In order to distinguish them and identify phase transitions in MD simulations, we construct two collective variables (CVs), based on the alignment, along the [100] direction, of the tetrahedra of the (010) planes (Figure 1). To this aim, we first compute the polar angle θSP, spanned by the vector rSP ≡ rS – rP that connects, for any PS4 group, a given S atom with the central P atom, with respect to the x-axis shown in Figure 1:
| 1 |
One can then define an order parameter that measures the average alignment of PS4 tetrahedra within each (010) plane, as follows:
| 2 |
where C = 1, ..., 6 labels the (010) planes as shown in Figure 1, NCP = 16 is the number of P atoms in each plane, and ⟨s, p⟩ represents the S atoms that are first nearest neighbors of P atom p. In other words, the outer sum runs over P atoms that belong to a given crystallographic plane, while the inner sum runs on the four S atoms that belong to the tetrahedron centered around atom p. Whenever one tetrahedron is perfectly aligned along ±x, the cosine of one P–S angle is close to ±1, while the remaining three have a value of cos(109.5°) ≈ −0.3338 or cos(70.5°) ≈ +0.3338 for +x and −x, respectively, due to tetrahedral symmetry. We thus raise the cosine to the fifth power in eq 2, so that the result of the inner sum will be approximately equal to ±1. While any odd power greater that 1 would serve this scope, the fifth power gives the best results on preliminary tests. [Note: This observation stems from the symmetry of the tetrahedra, ∑⟨s,p⟩cos(θsp) = 0.]
Since the final aim is to define a global measure of the relative alignment across planes, so as to capture the staggered ordering of the α structure, we construct two intermediate order parameters seven and sodd, by averaging sC for even and odd values of C, and a total of NC = 6 planes. We finally construct the following pair of collective variables:
| 3 |
| 4 |
In Section III.B we demonstrate that s1 completely distinguishes the three polymorphs, as it takes on values close to +1 when the structure is similar to the perfect γ, −1 when it is similar to α, and 0 when the structure is close to β or fully disordered. s2 instead measures a global alignment of the entire structure and does not resolve the β and α structure, as they both contain mixed PS4 orientations in equal proportions. Still, this CV is meaningful when combined with s1 as it carries information about the relative number of tetrahedra that are aligned along the positive and negative directions of the x-axis. For instance, it can distinguish between two perfect γ structures that are mirror-symmetric with respect to a (100) plane.
II.C. Green–Kubo Theory
The Green–Kubo (GK) theory of linear response65,66 provides a rigorous and elegant framework to compute transport coefficients of extended systems in terms of the stationary time series of suitable fluxes evaluated at thermal equilibrium with MD. For an isotropic system of N interacting particles, the GK expression for the electrical conductivity reads:
| 5 |
where kB is the Boltzmann constant, T the temperature, and Γt indicates the time evolution of a point in phase space from the initial condition Γ0, over which the average ⟨·⟩ is performed. Jq is the charge flux, that can be easily computed from MD, knowing the velocities of the atoms, vi, and their charges, qi:
| 6 |
Here, the sum runs over all the atoms, e is the electron charge, and the qi are equal to the nominal oxidation number of the atoms:81 in the absence of electronic conductivity due to conduction electrons or polaronic states, the overall electrical conductivity coincides with that obtained from eq 6 using integer, time-independent ionic charges.82
A commonly used approximation of eq 5 is the Nernst–Einstein (NE) equation:
| 7 |
where DLi and NLi represent the diffusion coefficient and the total number of the lithium atoms respectively. Equation 7 is widely used in practice to estimate the ionic conductivity, due to the high statistical accuracy with which atomic diffusion coefficients can be computed from numerical simulations.83 Nevertheless, its application to solid-state-electrolytes (SSEs) is burdened by systematic errors:84 in fact, the large interatomic dynamical correlations, both between carriers (Li+) and the solid matrix (PS3–4) and among the carriers themselves, which is typical in systems with a high carrier concentration like SSEs, is completely neglected by eq 7. The discrepancy between σ and σNE can be quantified by the Haven ratio:84−86
| 8 |
We redirect the reader to Section III.D for a thorough comparison between σ and σNE in the different polymorphs of Li3PS4.
From a methodological standpoint, eq 5 can be expressed in an equivalent formulation, called the Helfand–Einstein (HE) formula, which exhibits better statistical behavior:87
| 9 |
The Li diffusivity appearing in eq 7 is obtained from the asymptotic slope of the mean square displacement of the Li ions:
| 10 |
In this case, care has to be taken to compute DLi in the reference frame where the solid matrix is fixed to avoid nonphysical contribution to the calculations of the electrical conductivity. These spurious effects arise for simulations run in the barycenter reference frame, where the position of the center of mass of the entire system is fixed. In practice, the difference between DLi computed in these two reference frames vanishes when the box size is increased.88
Due to its very general formulation, the GK expression of eq 5 can be used to investigate, with minimal variations, other characteristic properties of Li3PS4. In Section III.C we will characterize the rotational properties of the PS4 polyanions at high temperature by computing a rotational diffusion coefficient as follows:
| 11 |
In this equation j runs over every P–S bond of each PS4 tetrahedron and ωj(t) represents the time series of its angular velocity:
| 12 |
where rjP,S (t) and vjP,S (t) are the positions and the velocities of the P and the S atom belonging the jth bond.
II.D. ML-MD Computational Details
We use the MLPs constructed in Section II.A to investigate the physics of Li3PS4 via constant-temperature MD simulations using a combination of i-PI,72,89lammps,90,91 and librascal.92 In order to simulate the phase transitions and the charge transport in Li3PS4, we perform MD simulations in the NpT ensemble of a quasi-cubic 768-atom cell in all stable α, β, and γ phases with a constant isotropic pressure of p = 0 atm for a set of temperatures between 200 and 1000 K. The system’s center of mass is kept fixed during the simulations. A generalized Langevin equation (GLE) thermostat93,94 is used to equilibrate the cell volume, while a stochastic velocity rescaling (SVR) thermostat95 is used to thermalize the velocity distribution of the atoms without affecting significantly the dynamical properties. The characteristic times of the barostat, the SVR thermostat, and the MD time step are set to 1 ps, 10 fs, and 2 fs, respectively. We run these simulations long enough to ensure statistical convergence of the ionic conductivity (see Section II.C). Specifically, we run the weakly conductive simulations of the γ phase for ∼6 ns, the β phase for ∼4 ns, and the α phase for ∼2 ns. As discussed in Section III.B, this setup ensures that the system can sample configurations within the range of stability of each phase without explicitly requiring a quantitative prediction of the temperature-dependent phase diagram of Li3PS4. A validation of the setup via a heat-quench simulation in the NST ensemble is found in the Supporting Information (Section VII).
III. Results
In this section, we compare the results obtained with the PBEsol, PBE0, and r2SCAN functional and the corresponding ML models, as described in Section II. In Section III.A, we compute the electronic band structure of the β polymorph using DFT and show that the band gap predicted by the PBE0 functional is in good agreement with experiments, while PBEsol and r2SCAN considerably underestimate it. In the following sections, we investigate the finite-temperature predictions of the ML models. First, in Section III.B, we analyze the MD simulations of the Li3PS4 polymorphs using the collective variable introduced in Section II.A and discuss the onset of a phase transition from the γ structure to a structure with a mixed α and β arrangement by increasing the temperature. In Section III.C, we investigate the rotational dynamics of PS4 tetrahedra and relate the occurrence of phase transitions with the thermal activation of correlated polyanion flips. Furthermore, we detect the presence of a high-temperature liquid phase characterized by freely rotating polyanions. In Section III.D, we compute the ionic conductivity from MD NpT simulations using both the NE and HE expressions introduced in Section II.C and the Haven ratio of Li3PS4. Finally, in Section III.E, we discuss the role of spatial correlations and their effect on the calculated ionic conductivity.
III.A. Electronic Band Structure
The Generalized Gradient Approximation (GGA) functionals offer a good compromise between accuracy and computational efficiency, making them a practical choice for a broad range of materials and systems. It is known, however, that they often fall short when it comes to accurately characterizing critical electronic properties, such as the electronic band gap, which is frequently underestimated in GGA, and the density of states.98−100 In order to solve this problem, different functionals have been developed, such as meta-GGA60 and hybrid functionals,61 that offer more accurate approximations of the exchange-correlation functional and are able to better describe long-range electron–electron interactions. These new functionals have enabled more accurate predictions of electronic properties in a variety of different materials101−106 and notably solid-state electrolytes.107−109 In Figure 3 we compare the band structure and the density of states (DOS) for the β phase computed with the PBEsol (GGA), r2SCAN (meta-GGA), and PBE0 (hybrid) functionals. [Note: The results for the γ phase can be found in the SI.] Since in the β phase the Wyckoff sites of the Li atoms have partial occupations, we perform the calculation using one (B3C1108) of the known configurations with minimum energy, since the electronic bands and the gap are only weakly dependent on this choice.108 Furthermore, in Table 2 we compare the band gaps predicted by the different functionals in the γ and β phases and recent experimental measurements. We note that both PBEsol and r2SCAN considerably underestimate the electronic band gap, while PBE0 shows a remarkably good agreement, thus further motivating the use of this functional as a reference for the training of a dedicated ML model. [Note: We remark that the reported experimental value from ref (97) is obtained as the electrochemical window and, as such, represents an upper limit of the band gap.]
Figure 3.
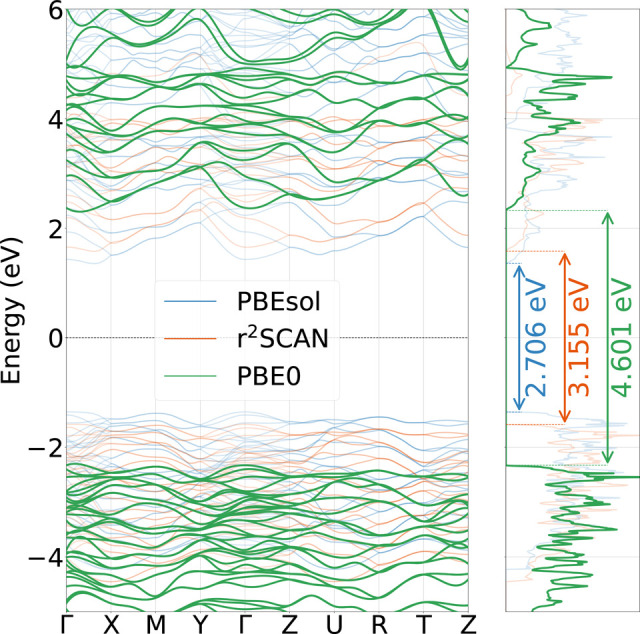
Electronic bands and density of states of Li3PS4 for the β-phase.
Table 2. Energy Band Gap with Different Models for the γ and β Phasesa.
The PBE0 values are in best agreement with the reported experimental value from ref (97).
III.B. Structural Phase Transitions
We used the pair of CVs s1 and s2, introduced in Section II.B, to investigate the presence of structural phase transitions appearing in the MD trajectories. As anticipated, s1 characterizes the mutual orientation of adjacent (010) planes (i.e., even and odd numbers in Figure 1) along the [100] direction. Consequently, s1 = −1 for the α phase, where the PS4 orientation of adjacent planes is antiparallel, s1 = 0 for the β phase (each plane has no net orientation of PS4 units), and s1 = +1 for the γ phase, where adjacent planes share the same orientation of PS4 tetrahedra along the positive x axis. Conversely, s2 measures the global orientation of PS4 tetrahedra and vanishes for both the α and β phases, while it is equal to 1 for the γ phase. Figure 4 displays, with red dots, the evolution of the CVs across a set of MD simulations run with the ML-PBE0 model at different temperatures, T, and initialized in the γ phase. The green markers of three different shades represent reference points sampled from MD simulations in the α, β, and γ phases below Tc = 750 K, and are used as a guide to interpret the T-dependent results. For T < Tc, the red dots are all concentrated in a region around (s1, s2) = (1, 1), which is typical of the pure γ phase, the small deviations being due to the thermal motion of the atoms only. As T is raised above ≈Tc, a structural transition occurs, and the CVs approach a region between the α and β phases. Although the total time scale and size of the simulations are not sufficient to allow for a complete transition of the γ phase to a specific polymorph, but only to intermediate configurations, our MD simulations capture the microscopic driving mechanism, i.e., the onset of concerted reorientations of PS4 tetrahedra.
Figure 4.
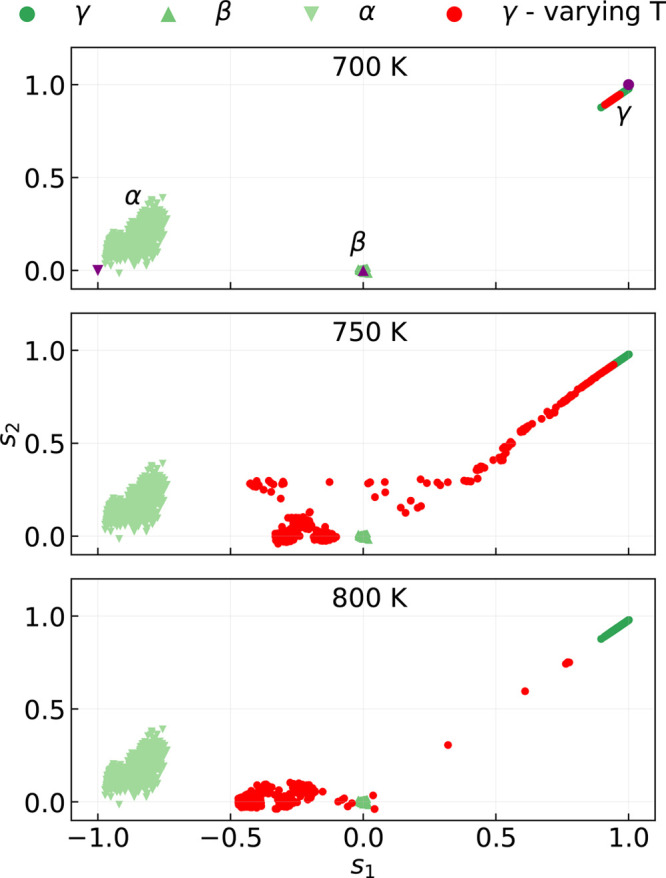
Evolution of the collective variables of γ-Li3PS4 (red points) sampled over a set of MD trajectories generated with the ML-PBE0 model. Green markers of three different shades represent a sample of reference points extracted from all MD simulations in the α, β, and γ phase below Tc = 750 K where no phase transitions are observed. The purple markers in the top panel indicate the CVs for the ideal crystalline structures.
Figure 5 shows a typical example of this phenomenon, occurring during an ≈15 ps segment of a MD trajectory: the starting (ending) configuration is depicted in red (blue). The color fades from red to blue (with an RWB scheme) in a continuous manner as the transition occurs. The trajectories of the S atoms lying at the vertices of the PS4 tetrahedra clearly indicate that the reorientation of the entire row occurs coherently and not as a collection of individual, decorrelated flips. Figure 5 also shows that the transition is purely orientational and does not occur through the hopping of S anions between adjacent PS4 groups. Additional information and a comparison of this mechanism with a heat-quench simulation showing a similar behavior can be found in the Supporting Information (Sections VII and VIII).
Figure 5.
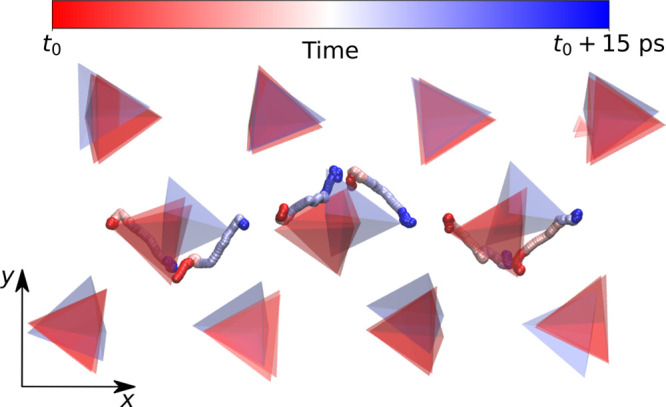
Transition corresponding to the reorientation of one line of PS4 tetrahedra. Tetrahedra colored red and blue correspond to snapshots of the solid matrix taken over the transition for one trajectory at 750 K starting from the γ structure and run with the ML-PBE0 model. The trajectory of two vertices of each tetrahedron is displayed with lines, that are colored with an RWB scheme and with a smoothening window of 2.5 ps. The red end of these lines corresponds to t0, while the blue end corresponds to t1 = t0 + 15 ps.
III.C. PS4 Rotational Dynamics and Heat Capacity
Further insights into the rotational reorientation of PS4 planes and their relation with structural transitions can be obtained by a direct investigation of the rotational dynamics of PS4 tetrahedra and specifically those that form [100] rows. Figure 6 shows the dynamics of the polar angles θSP, as defined in eq 1, for a set of four tetrahedra (row panels) that belong to the same [100] row in NpT simulations run with the ML-PBE0 model. [Note: The ML-PBEsol and the ML-r2SCAN models give the same qualitative behavior of the ML-PBE0 model. Notably, all of them display the same phase transitions, albeit at different temperatures (see also Figure 7).] We also compare three trajectories initialized in the γ phase and equilibrated at T = 725 K, i.e., just below Tc = 750 K (left column); at T = Tc (central column); and above melting, at T = 900 K (right column). The four lines in the plots correspond to the dynamics of the four bonds that constitute each PS4 tetrahedron.
Figure 6.
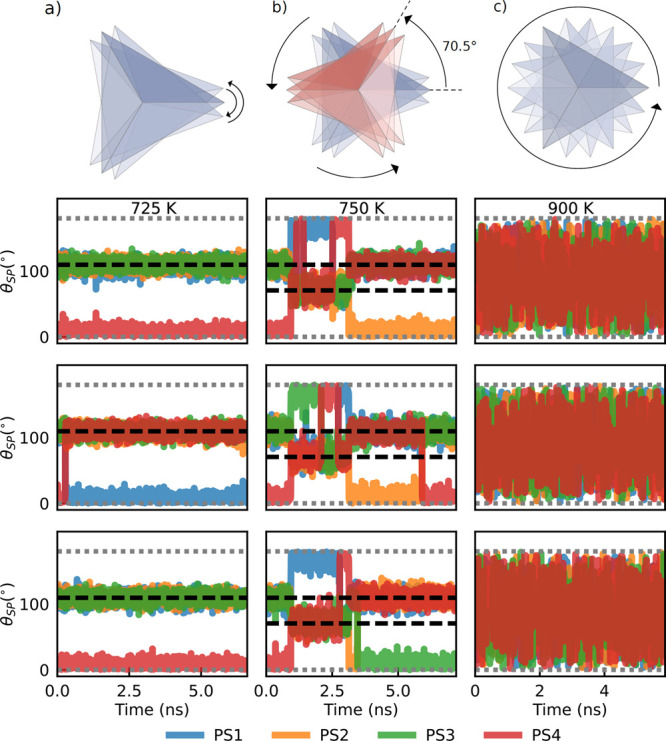
Sketch of the rotational dynamics of the PS4 groups: low temperature (panel a), where only small librations with respect to the initial configuration occur, at intermediate temperature (panel b) where PS4 flips determine the structural transition observed in Figure 4 and at high temperature (panel c) where the system is melted. The lower plots show the time evolution of the polar angle θSP as defined in eq 1 (angle with respect to the x-axis) for a set of three distinct tetrahedra, forming a [100] row. Each panel represents the dynamics of the four PS bonds forming each tetrahedron. Rows correspond to different tetrahedra, while columns correspond to different NpT trajectories at T = 725, 750, and 900 K. Horizontal dashed black lines indicate the position of the ideal tetrahedral angles at 70.5° and 109.5°, while gray dotted lines mark the extremes of the domain of θSP (i.e., 0° and 180°).
Figure 7.
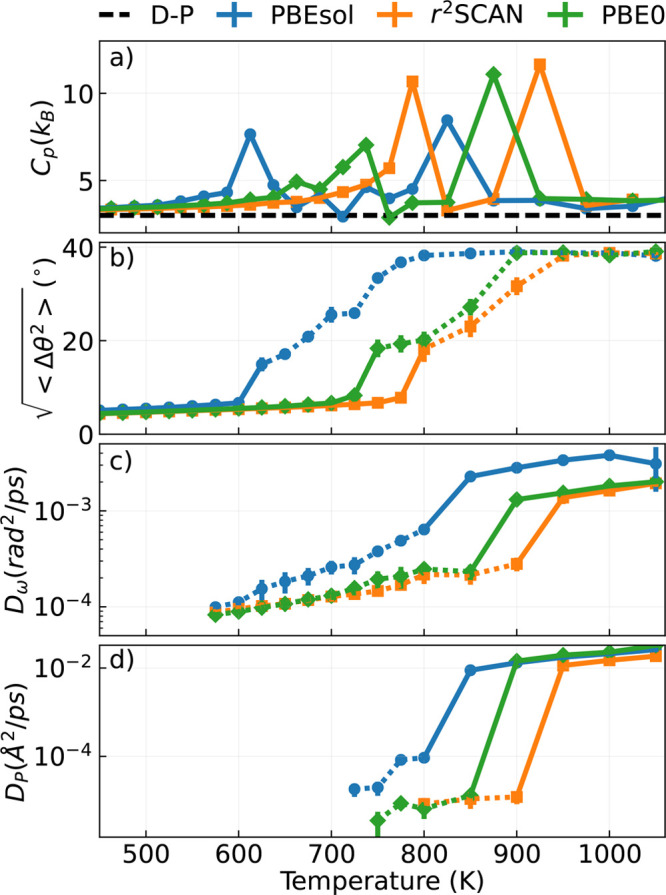
(a) Heat
capacity (Cp), (b) fluctuations
of the P–S polar angle averaged over all
bonds of every PS4 tetrahedron in the simulation box (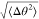 ), (c) PS4 rotational
diffusion
coefficient (Dω), and (d) the linear
diffusion coefficient of the P atoms (DP) as a function of the simulated temperature. Solid/dashed lines
represent each quantity in the temperature regime, where it is well/ill-defined.
The dashed black line of panel (a) represents the Dulong–Petit
limit, 3kB.
), (c) PS4 rotational
diffusion
coefficient (Dω), and (d) the linear
diffusion coefficient of the P atoms (DP) as a function of the simulated temperature. Solid/dashed lines
represent each quantity in the temperature regime, where it is well/ill-defined.
The dashed black line of panel (a) represents the Dulong–Petit
limit, 3kB.
At 725 K, only small angular fluctuations occur with no reorientation of the tetrahedra. The average angles of the P–S bonds define the mean orientation of each tetrahedron: as one of the P–S bonds always has an average value that is close to 0, the orientation is along the positive x-axis (+x) during the entire simulation time, which is typical of the γ phase. Instead, the angles of the other three bonds oscillate around θ0 = 109.5° (black dashed line), corresponding to the P–S bond angles in a perfect tetrahedral geometry. The jump observed in the central panel of the left column corresponds to a rotation of the tetrahedron, that does not involve a reorientation toward the negative x-axis.
Instead, at the transition temperature (Tc = 750 K), simultaneous flips of one P–S bond from 0 to 70.5° and another P–S bond from 109° to 180° correspond to the reorientations of the tetrahedra from +x to −x, consistently with the mechanism shown Figure 5. Notably, these flips occur at the same instants of time for every tetrahedron in a row (see, e.g., the central column of Figure 6 between 1 and 2.5 ns), confirming that they are highly correlated across [100] rows. This effect is the basis of the phase transition observed in Figure 4 as it modifies the relative orientation of tetrahedra across (010) crystallographic planes. It is crucial to note, in this respect, that the spatial correlation of these flips at the transition extends up to the edge of the simulation box, potentially leading to finite-size effects. Specifically, we expect this transition to manifest in larger boxes by nucleation of ordered clusters with opposite orientation as a result of the formation of defects with sudden changes of the PS4 orientation. We also note from panels (b) of Figure 6 that the time lag between subsequent flips is of the order of 1 ns. As we will see in Section III.D, the presence of this long time scale will be important to elucidate the mechanism of charge transport in this material.
At 900 K, the dynamics of the tetrahedra changes dramatically, and a second phase transition occurs. In particular, all PS bonds across every tetrahedron span the entire range of angles between 0° and 180°. This suggests that unlike the phases observed at lower temperatures, the tetrahedra are freely rotating in the simulation box. A direct inspection of the simulations indicates that this behavior is accompanied by a melting of the system into a mixture of Li+ cations and PS3–4 anions (see also the P–P and P–S radial distribution functions shown in the Supporting Information, Figure S9).
The onset of these two phase transitions, from the nonconductive γ phase to the superionic β-α hybrid phase and the melting of Li3PS4, can be quantitatively investigated for all ML models by computing the temperature dependence of a set of relevant quantities. Panel (a) of Figure 7 shows, for each of the ML models, the isobaric heat capacity Cp(T) computed from the finite-difference derivative, with respect to T, of the mean enthalpy collected in the NpT simulations. Panel (b) shows the temperature dependence of the mean squared fluctuation of the polar angle θSP as defined in eq 1 and further averaged over every P–S bond. Panel (c) shows the PS4 rotational diffusion coefficient Dω, defined in eq 11, and panel (d) the linear diffusion coefficient of the P atoms, DP.
Cp(T) displays two distinct peaks, characteristic of the phase transitions observed in Figure 6, while the associated critical temperatures depend on the specific functional. We can characterize more clearly the position of these peaks by analyzing their relation with the microscopic quantities shown in panels (b), (c), and (d). Since the first transition is associated with discrete PS4 flips, the increase of Cp is accompanied by a sudden change of slope of the angular fluctuations. Conversely, the transition to the molten phase occurs with a dramatic increase of both Dω and DP by one and 2 orders of magnitude, respectively. In other words, the action of thermal fluctuations at this high temperature destroys the periodic arrangement of P atoms, while the tetrahedra are still intact and can freely rotate at a rate given by Dω. Notably, the transition temperature to the molten salt as predicted by the ML-PBE0 model is in agreement with a previous experimental measurement of the binary phase diagram of β-Li3PS4–Li4GeS4 solid solutions obtained through differential thermal analysis.110 More specifically, the transition point upon heating is reported to be 600 °C when the concentration of β-Li3PS4 is equal to 98% (P-rich regime), which is compatible with our PBE0 estimate.
We stress that the angular deviations of panel (b) can be defined only with respect to a local equilibrium for each P–S bond and are thus meaningful only in the low-T phase. Conversely, the diffusion coefficients of panels (c) and (d) are physically meaningful when the simulations sample sufficiently many configurations with displaced P atoms and rotated PS4 anions. They are thus not well-defined if the MD simulations are not fully ergodic.111 In Figure 7, we display each of the quantities with solid lines in the regions where they are well-defined; otherwise, they are shown with dotted lines. The phase transitions investigated so far have strong implications on electrical conduction, as we describe in the following section.
III.D. Ionic Conductivity and Haven Ratio
The ionic conductivity, σ, is a crucial property to identify promising solid-state electrolytes. As discussed in Section II.C, the GK theory of linear response in its HE formulation gives us an efficient and statistically robust method to obtain an estimate of σ from equilibrium MD simulations at any target temperature.
The upper panels of Figure 8 show the temperature dependence of the ionic conductivity at zero pressure for a set of NpT simulations that start from the ideal α, β, and γ polymorph. Imperfect ergodicity, and the constraints on cell shape, make simulations dependent on the initial conditions. Even though simulations can only be considered converged within the stability range of each phase and target functional, results outside this range still report useful information about their behavior when metastable. Thus, results are shown for every target functional and for all temperatures where the ionic conductivity is nonzero within the errorbars.
Figure 8.

Temperature dependence of σ and Haven ratio. Panels (a), (b), and (c) show a comparison between the ionic conductivities predicted by the ML models as a function of the inverse temperature, computed via the Green–Kubo relation. Panels (d), (e), and (f) show the behavior of the Haven ratio, HR = σNE/σ, as a function of the inverse temperature. Error bars obtained from standard block analysis over eight blocks are displayed. Vertical dashed lines indicate the experimental stability boundaries for the three phases.
All of the ML models predict the γ(α) phase to be the least (most) conductive, while the β phase has intermediate values of σ over a wide temperature window (up to 600–800 K depending on the model). This result is in agreement with previous computational studies26,112 and experimental measurements of the ionic conductivity20,28 on the known crystalline phases of Li3PS4. The negative slope of the profiles of σ with respect to the inverse temperature is typical of Arrhenius plots. In fact, since lithium-ion diffusion occurs via thermal activation, one can mathematically relate the Nernst–Einstein conductivity σNE with the activation energy of the Li-hopping process. In particular, higher negative slopes correspond to higher activation energies. The γ (α) phase is thus not only the least (most) conductive phase but also the one that has the largest (smallest) activation barrier for lithium diffusion.
The high-temperature ends of the conductivity profiles of Figure 8 (for T < 670 K for ML-PBEsol, T < 880 K for ML-r2SCAN, and T < 770 K for ML-PBE0) give us additional insights. While the curves related to the three phases clearly span different conductivity ranges and show different slopes at low temperatures, this difference is not noticeable any more at high temperatures, and the conductivity and the activation energies all approach the values of the α phase, with a characteristic kink that is most visible for simulations initialized in the γ phase. The critical temperature at which this kink occurs depends on the reference functional. This is a clear effect of the structural transition studied in Section III.B and further investigated in Section III.C by an analysis of the PS4 rotational dynamics. In other words, the PS4 flips that induce the transition from the γ polymorph to the partly ordered β–α structure are responsible for the changes of the ionic conductivity due to the larger availability of hopping sites for Li-ions,20 as well as a reduction of the Li-hopping activation barrier. This conclusion is also highlighted by simulations of the β and α phases at low temperatures, that show large σ although PS4 flips are suppressed and librations are weak. To quantify the reduction of the activation energy due to the structural transition, in Table 3, we report the activation energies that are fit to the Nernst–Einstein conductivities below and above the transition temperatures of each model (see Section X of the Supporting Information for additional details). The effect of the transition is remarkable, as we observe a reduction of the activation energies of up to a factor of 6 depending on the reference DFT functional. Furthermore, the activation energies are very small above the transition, with values ranging between 0.25 and 0.32 eV. These values are very close to the values observed for the α phase and smaller than those for the β phase (see Table 4), indicating superionic behavior. In contrast, we note that the transition to the molten salt that we observed for T > 800 K in Section III.C has practically no effect on the conductivity profiles.
Table 3. Activation Energies for Li-Ion Diffusion for the Simulations Initialized in the γ Phase for Temperatures below the Phase Transition Temperature (T > Tc) Observed in Section III.C and above Tca.
| model | EA(T < Tc) (eV) | EA(T > Tc) (eV) | ratio |
|---|---|---|---|
| ML-PBEsol | 0.93 ± 0.07 | 0.249 ± 0.004 | 3.4 ± 0.3 |
| ML-r2SCAN | 1.64 ± 0.05 | 0.32 ± 0.01 | 5.1 ± 0.2 |
| ML-PBE0 | 1.43 ± 0.06 | 0.269 ± 0.007 | 5.3 ± 0.3 |
See the SI for details on the computation of the activation energies. The last column is the ratio between the EA(T < Tc) and EA(T > Tc) and quantifies the reduction of the Li-diffusion activation energy due to the phase transition.
Table 4. Comparison between the Predicted Activation Energies and Conductivities (Computed at 500 K and Extrapolated at Room Temperature; See SI for Details) of the β and α Phases with Both Experimental References28,96 and Previous ab initio MD Studies22,114,115.
| EβA (eV) | EαA (eV) | σβ298K (S cm–1) | σβ500K (S cm–1) | σα298K (S cm–1) | |
|---|---|---|---|---|---|
| PBEsol | 0.38 | 0.17 | 7.7 × 10–4 | 2.8 × 10–1 | 1.9 × 10–1 |
| r2SCAN | 0.57 | 0.21 | 2.1 × 10–5 | 5.1 × 10–2 | 9.2 × 10–2 |
| PBE0 | 0.62 | 0.19 | 8.7 × 10–6 | 3.9 × 10–2 | 1.6 × 10–1 |
| exp. | 0.47 [96], 0.36 [28] | 0.22 [28] | 8.9 × 10–7 [96,113] | 3.0 × 10–2 [20] | |
| AIMD-PBE | 0.40 [22] | 0.18 [114] | 8.0 × 10–2 [114] |
This analysis, combined with the results of Sections III.B and III.C, also allows us to rule out any paddle-wheel effect, whereby PS4 motion is time-correlated with Li-hopping and increases Li-ion diffusion. In fact, due to the different rates of PS4 flipping (≈ one every nanosecond) and Li-ion hopping (≈ one every picosecond) even at large temperature right below melting, the two mechanisms cannot be related. This point is strengthened in Figure S12 of the Supporting Information, showing the fast and linear increase of the mean square displacement of lithium ions in the simulation at 750 K of Figure 5 over a 30 ps time window. Instead, the interaction between Li-ion diffusion and PS4 libration, which share the same time scale, may be important, as we have directly inspected in Section III.E by analyzing the local contributions to σ stemming from the interaction between Li and S ions.
The lower panels of Figure 8 show the temperature dependence of the Haven ratio, HR, computed by using eq 8. As anticipated in Section II.C, this coefficient quantifies the discrepancy between σ and σNE. HR < 1 for almost every temperature and only for the γ phase it approaches one at low temperatures, where the system is weakly conductive. This indicates the presence of interatomic correlations in the ionic conductivity, both between carriers (Li+) and the solid matrix (PS4), that cannot be captured by the NE approach (see eq 7), as the latter only estimates the conductivity based on the self-diffusion of the lithium ions. This effect is most pronounced in the α phase, where HR ≈ 0.4 below melting, meaning that the NE estimate underestimates the GK conductivity by more than a factor of 2. At high temperatures, the Haven ratio slightly increases, indicating that the material becomes disordered. This might be a result of the phase transformations of the solid matrix, which we expect weaken the interionic correlations. Still, the Haven ratio never exceeds 0.8, even at 1000K for any of the ML models studied.
In Table 4 we quantitatively compare the activation energies and the ionic conductivities predicted by our ML models with recent experimental measurements28,96 and with computational studies based on AIMD at the PBE level.22,114,115 The first two columns of Table 4 compare the activation energies in the β and α phases, while the remaining three report the estimates of the ionic conductivity of the β phase at 298 K and 500 K and the ionic conductivity of the α phase at 298 K. [Note: This value is extrapolated from a fit of the temperature profiles of the β structure (see Figure 8) for each ML model; see SI for details on the computation of the activation energies.] The ML-PBEsol model predicts activation energies in agreement with previous AIMD for both the β and the α structure. In the case of the β phase, ML-r2SCAN and ML-PBE0 predict an activation energy that is greater than the one computed with the ML-PBEsol model, but overall the values are close to the experimental results. For the α phase, the activation energies are particularly close to the experiment. These last results are remarkably good in particular when comparing them with the prediction of the classical empirical potential recently introduced by Forrester et al.23 For this model, the activation energy for stoichiometric α-Li3PS4 is much larger (0.40 eV) than the experimental result and similar to the value of the β phase. The empirical potential also predicts the α phase to be slightly more conductive than the β phase for all temperatures.
In conclusion, our analysis shows that the ML potentials are the only possible solution to accurately predict the properties of Li3PS4, given the unreliability of empirical potentials and the prohibitive cost of ab initio simulations, in particular at the PBE0 level.
III.E. Spatial Correlations
In this last section, we investigate the role of local correlations in determining the full ionic conductivity by computing the spatial dependence of the integral of the partial cross-correlation functions, ILiA, between lithium atoms and other atomic species A = Li, P, and S. However, before we start this analysis, it is necessary to make a few technical considerations. While the total conductivity does not depend on the frame of reference, due to the charge neutrality of the simulation cell, the value of any partial correlation does depend on it, as we show in the SI. For instance, in the reference frame of the matrix, all the Green–Kubo integrals of the partial correlation between Li and the other species (P and S) vanish. In contrast, in the reference frame of the center of mass of the entire system, the solid matrix recoils due to the diffusion of the center of mass of Li atoms (see SI). To carry out our analysis, we choose the reference frame where the center of mass of the PS solid matrix remains stationary, e.g., ∑i∈P,Svi = 0 at every instant. This choice is motivated by the fact that this reference frame is in principle the same in which the lithium diffusivity, entering the NE relation, eq 7, should be computed. Moreover, this is the most natural choice for a battery, since in this reference frame the solid matrix of the battery is not moving.
To study the spatial dependence of the integral of the correlation functions we perform a Gaussian kernel density estimation (KDE)116 of the correlation functions:
| 13 |
| 14 |
| 15 |
where i runs over all Li atoms and j runs over all the atoms of type A; wij(t, r) is a Gaussian weight with width ς, which we set to 0.33 Å; and w̅ij(r) is the time average of wij(r) over the whole trajectory. The connection between eq 13 and the ionic conductivity of eq 5 is readily established. In fact, within the KDE,
| 16 |
where gLiA(r) is the radial distribution function between the Li and the species A. Therefore, the integral over r of ILiLi(r) plays the role of the correction to the NE relation that is needed to recover the full GK conductivity:
| 17 |
Figure 9 shows ILiA(r) for different values of r. The most relevant correlations come from the Li–Li and Li–S interactions, while the correlations with the P are compatible with zero for any r. As expected, for large r values, all of the correlations become compatible with zero. The oscillations in the correlations functions follow, as expected, the peaks in the radial distribution function gLiA(r) (lower panel) and show that only the first shell of Li atoms and the first two of S atoms are important for the conductivity. The correlation of the first shell of Li is strongly positive in agreement with ref (117), where it was shown that the hopping of Li atom in one direction facilitates the movement of the other Li in the same direction.
Figure 9.
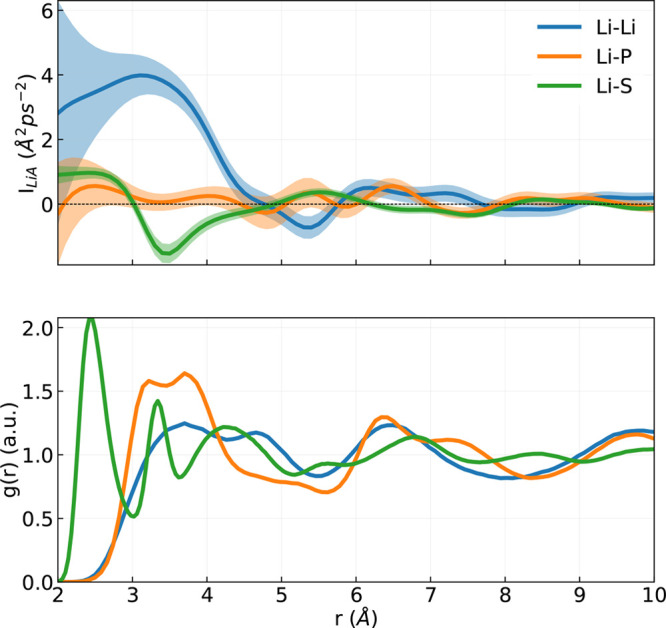
(Upper panel) Integrals of the cross correlation functions as defined in eq 13, for different pairs of atomic species and as a function of r. All of the velocities are computed in the system of reference of the center of mass of the solid matrix of the battery. The shaded area indicates the uncertainty on the mean value, obtained from block analysis on 10 blocks. (Lower panel) Radial distribution function between the Li and all the other atomic species. The data reported in the figure are obtained from 190 ps of a simulation of the α-phase at 650 K.
IV. Conclusions
In this work, we have presented a computational study of lithium ortho-thiophosphate via multiple machine learning models, targeting DFT references of increasing accuracy and elucidating the critical role of PS4 flips and phase transitions in determining the observed superionic behavior of Li3PS4. We find that all the ML models predict two distinct phase transitions. First, we observe a transition from the γ to a partly ordered phase with both β and α alignments that cannot be fully resolved at the time and length scale of the MD simulations. While this limitation does not allow us to provide a prediction of the full phase diagram of Li3PS4, we identify the presence of collective PS4 flips as the driving mechanism. Second, we observe the melting of the system into its constituent Li+ cations and PS3–4 polyanions at elevated temperatures (>800 K). Both of these transitions are associated with drastic changes in the rotational dynamics of the PS4 groups as a function of the temperature and appear as distinct peaks of the heat capacity.
We also compute the ionic conductivity of Li3PS4 in all its stable polymorphs and elucidate the importance of including the effects of interatomic correlations by computing it with the full Green–Kubo theory of linear response and with the Nernst–Einstein approximation. We find that the interionic correlations account for considerable deviations between the NE and the GK estimates, as quantified by a Haven ratio that is smaller than one in every polymorph at all temperatures, except for the weakly conductive γ structure at low temperatures. Notably, the Haven ratio reaches values of 0.4 in the highly conductive α-phase, suggesting that a pure NE approach can result in the underestimation of the conductivity by more than a factor of 2. From a spatially resolved analysis performed in the reference frame of the solid matrix, we find that most of these interionic correlations come from the first shell of Li–Li neighbors, thus indicating that a concerted Li-ion hopping is a key aspect of charge transport in this material, in agreement with ref (117).
Finally, we investigate how the observed phase transitions of Li3PS4 affect the ionic conductivity. We find that the occurrence of correlated PS4 flips results in a dramatic decrease of the activation energy (up to a factor of 6) when the system transitions from the γ to a mixed β–α phase. Furthermore, we show that subsequent PS4 flips occur at the time scale of nanoseconds, that is much larger than the typical time laps between two subsequent lithium ion hoppings, in agreement with ref (117). We thus conclude that the sudden change in the PES of the lithium ions that is due to the rearrangement of the PS4 tetrahedra is the physical mechanism for the observed superionic behavior of Li3PS4. Crucially, this mechanism is fundamentally different from the one proposed in ref (31). There, a characteristic paddlewheel effect was observed in AIMD-PBE simulations at elevated temperatures and invoked to explain the neutron diffraction measurements showing polyanion reorientational disorder. The AIMD simulations were however limited in size and thus showed much larger finite-size effects than the ones we observe in this work, including an artificial stabilization of the solid phase up to temperatures (1200 K) that are much larger than the nominal melting point of Li3PS4. We also stress that the paddlewheel effect itself was observed in AIMD simulations at these very high temperatures, thus likely making it an artifact of the small simulation box that was used there. The ML models that we present in this work overcome these limitations and offer a more natural interpretation of the experimental results. This finding also suggests additional directions of research in the quest for a promising solid electrolyte and potentially a way to design new target compounds. In particular, we expect that tentative modifications of Li3PS4 to stabilize its superionic phases at room temperature, by, e.g., atomic substitution and amorphization, should be accompanied by a reduction of the polyanion rotational free energy barrier, that limits the spatial extension of the PS4 fluctuations. Further developments of this work thus imply a detailed thermodynamic study of the phase transitions observed here and a comparison of multiple different SE compounds with the aim of suggesting a target compound for experimental synthesis.
While providing these useful mechanistic insights, our ML models show remarkable agreement with experiment in the prediction of a number of independent quantities. Specifically, the PBE0 functional provides the best agreement on the prediction of the electronic band gap, while the associated ML-PBE0 model reaches overall the best accuracy on the prediction of lithium activation energies in the β and α phase, the ionic conductivities at 298 and 500 K, and the melting temperature. In particular, our results proved to be much more accurate than empirical potentials,23 that are often used to overcome the high computational cost of AIMD. These results and the observed dependence of the finite-temperature predictions of the ML models on the DFT reference indicate the necessity of using more accurate functionals for the description of transport properties in solid electrolytes, than state-of-the-art GGA functionals. Machine learning becomes then a necessary step in modeling this class of materials, as ab initio studies with the PBE0 functional are far beyond reach because of their very high computational cost.
In conclusion, we have shown how the use of machine learning potentials for a prototypical solid electrolyte captures the mechanisms of the transition to its superionic phase and the quantitative values of the ionic conductivity, while also allowing us to investigate the role of interionic correlations. This work thus opens up a new frontier in the exploration of superionic materials, as it allows their large-scale simulations at hybrid-DFT accuracy for hundreds of nanoseconds. This will offer crucial insights into the fundamental properties of solid electrolytes as well as guidance for the experimental realization of new candidate compounds.
Acknowledgments
We thank M. L. Kellner for a critical review of an early version of this manuscript and S. Lombardi for technical help in improving the figures. L.G., D.T., and M.C. acknowledge funding from the Swiss National Science Foundation (SNSF) under the Sinergia Project CRSII5_202296 and support from the MARVEL National Centre of Competence in Research (NCCR) for computational resources. F.G. and M.C. acknowledge funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme Grant No. 101001890-FIAMMA. F.G. also acknowledges funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie Action IF-EF-ST, Grant Agreement No. 101018557-TRANQUIL. This work was supported by grants from the Swiss National Supercomputing Centre (CSCS) under the Projects s1092 and s1219 and by the Platform for Advanced Scientific Computing (PASC) project “Machine learning for materials and molecules: toward the exascale”.
Data Availability Statement
The Li3PS4 data sets and the ML-potentials generated in this work, as well as the input files of the simulations, the notebooks, and the scripts employed are available on the Materials Cloud Platform125 at 10.24435/materialscloud:g2-fp.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.chemmater.3c02726.
Section I contains the parameters of the ML models and the learning curves;33,36,74,92,118 Section II shows the results from kernel principal component analysis;79,119 Section III contains the analysis of the local prediction rigidity;80 Section IV shows the comparison between the mean square displacement from an ab initio simulation and a simulation with the ML-PBEsol model; Section V contains the details of the DFT calculations;62,67−71,76−78,120−123 Section VI shows the band structure of the γ phase; Section VII contains the analysis of a heat-quench trajectory; Section VIII shows the behavior of the order parameter during a collective plane flipping; Section IX contains the study of the g(r) during melting; Section X shows results for the Nernst–Einstein approximation to the ionic conductivity; and Section XI contains theoretical insights84,87,124 on the frame dependence of the breakdown of the electrical conductivity into species-dependent terms (PDF)
Author Contributions
† (L.G. and D.T.) These authors equally contributed to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Kwade A.; Haselrieder W.; Leithoff R.; Modlinger A.; Dietrich F.; Droeder K. Current status and challenges for automotive battery production technologies. Nature Energy 2018, 3, 290. 10.1038/s41560-018-0130-3. [DOI] [Google Scholar]
- Aurbach D. Review of selected electrode–solution interactions which determine the performance of Li and Li ion batteries. J. Power Sources 2000, 89, 206. 10.1016/S0378-7753(00)00431-6. [DOI] [Google Scholar]
- Etacheri V.; Marom R.; Elazari R.; Salitra G.; Aurbach D. Challenges in the development of advanced Li-ion batteries: a review. Energy Environ. Sci. 2011, 4, 3243. 10.1039/c1ee01598b. [DOI] [Google Scholar]
- Arbizzani C.; Gabrielli G.; Mastragostino M. Thermal stability and flammability of electrolytes for lithium-ion batteries. J. Power Sources 2011, 196, 4801. 10.1016/j.jpowsour.2011.01.068. [DOI] [Google Scholar]
- Murmann P.; Schmitz R.; Nowak S.; Ignatiev N.; Sartori P.; Cekic-Laskovic I.; Winter M. Electrochemical performance and thermal stability studies of two lithium sulfonyl methide salts in lithium-ion battery electrolytes. J. Electrochem. Soc. 2015, 162, A1738. 10.1149/2.0261509jes. [DOI] [Google Scholar]
- Janek J.; Zeier W. G. A solid future for battery development. Nature Energy 2016, 1, 16141. 10.1038/nenergy.2016.141. [DOI] [Google Scholar]
- Ahniyaz A.; de Meatza I.; Kvasha A.; Garcia-Calvo O.; Ahmed I.; Sgroi M. F.; Giuliano M.; Dotoli M.; Dumitrescu M.-A.; Jahn M.; Zhang N. Progress in solid-state high voltage lithium-ion battery electrolytes. Advances in Applied Energy 2021, 4, 100070. 10.1016/j.adapen.2021.100070. [DOI] [Google Scholar]
- Bachman J. C.; Muy S.; Grimaud A.; Chang H.-H.; Pour N.; Lux S. F.; Paschos O.; Maglia F.; Lupart S.; Lamp P.; et al. Inorganic solid-state electrolytes for lithium batteries: mechanisms and properties governing ion conduction. Chem. Rev. 2016, 116, 140. 10.1021/acs.chemrev.5b00563. [DOI] [PubMed] [Google Scholar]
- Famprikis T.; Canepa P.; Dawson J. A.; Islam M. S.; Masquelier C. Fundamentals of inorganic solid-state electrolytes for batteries. Nature materials 2019, 18, 1278. 10.1038/s41563-019-0431-3. [DOI] [PubMed] [Google Scholar]
- Castelli I. E.; Zorko M.; Østergaard T. M.; Martins P. F. B. D.; Lopes P. P.; Antonopoulos B. K.; Maglia F.; Markovic N. M.; Strmcnik D.; Rossmeisl J. The role of an interface in stabilizing reaction intermediates for hydrogen evolution in aprotic electrolytes. Chemical Science 2020, 11, 3914. 10.1039/C9SC05768D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seino Y.; Ota T.; Takada K.; Hayashi A.; Tatsumisago M. A sulphide lithium super ion conductor is superior to liquid ion conductors for use in rechargeable batteries. Energy Environ. Sci. 2014, 7, 627. 10.1039/C3EE41655K. [DOI] [Google Scholar]
- 12 Kato Y.; Hori S.; Saito T.; Suzuki K.; Hirayama M.; Mitsui A.; Yonemura M.; Iba H.; Kanno R. High-power all-solid-state batteries using sulfide superionic conductors. Nature Energy 2016, 1, 16030. 10.1038/nenergy.2016.30. [DOI] [Google Scholar]
- Sakuda A.; Hayashi A.; Tatsumisago M. Recent progress on interface formation in all-solid-state batteries. Current Opinion in Electrochemistry 2017, 6, 108. 10.1016/j.coelec.2017.10.008. [DOI] [Google Scholar]
- Takada K. Progress in solid electrolytes toward realizing solid-state lithium batteries. J. Power Sources 2018, 394, 74. 10.1016/j.jpowsour.2018.05.003. [DOI] [Google Scholar]
- Hakari T.; Deguchi M.; Mitsuhara K.; Ohta T.; Saito K.; Orikasa Y.; Uchimoto Y.; Kowada Y.; Hayashi A.; Tatsumisago M. Structural and electronic-state changes of a sulfide solid electrolyte during the Li deinsertion–insertion processes. Chem. Mater. 2017, 29, 4768. 10.1021/acs.chemmater.7b00551. [DOI] [Google Scholar]
- Materzanini G.; Kahle L.; Marcolongo A.; Marzari N. High Li-ion conductivity in tetragonal LGPO: A comparative first-principles study against known LISICON and LGPS phases. Physical Review Materials 2021, 5, 035408 10.1103/PhysRevMaterials.5.035408. [DOI] [Google Scholar]
- Pasta M.; Armstrong D.; Brown Z. L.; Bu J.; Castell M. R.; Chen P.; Cocks A.; Corr S. A.; Cussen E. J.; Darnbrough E.; Deshpande V.; Doerrer C.; Dyer M. S.; El-Shinawi H.; Fleck N.; Grant P.; Gregory G. L.; Grovenor C.; Hardwick L. J.; Irvine J. T. S.; Lee H. J.; Li G.; Liberti E.; McClelland I.; Monroe C.; Nellist P. D.; Shearing P. R.; Shoko E.; Song W.; Jolly D. S.; Thomas C. I.; Turrell S. J.; Vestli M.; Williams C. K.; Zhou Y.; Bruce P. G. 2020 roadmap on solid-state batteries. Journal of Physics: Energy 2020, 2, 032008 10.1088/2515-7655/ab95f4. [DOI] [Google Scholar]
- Li C.; Wang Z.-y.; He Z.-j.; Li Y.-j.; Mao J.; Dai K.-h.; Yan C.; Zheng J.-c. An advance review of solid-state battery: Challenges, progress and prospects. Sustainable Materials and Technologies 2021, 29, e00297 10.1016/j.susmat.2021.e00297. [DOI] [Google Scholar]
- Kudu Ö. U.; Famprikis T.; Cretu S.; Porcheron B.; Salager E.; Demortiere A.; Courty M.; Viallet V.; Mercier T. L.; Fleutot B.; Braida M.-D.; Masquelier C. Structural details in Li3PS4: Variety in thiophosphate building blocks and correlation to ion transport. Energy Storage Materials 2022, 44, 168. 10.1016/j.ensm.2021.10.021. [DOI] [Google Scholar]
- Homma K.; Yonemura M.; Kobayashi T.; Nagao M.; Hirayama M.; Kanno R. Crystal structure and phase transitions of the lithium ionic conductor Li3PS4. Solid State Ionics 2011, 182, 53. 10.1016/j.ssi.2010.10.001. [DOI] [Google Scholar]
- Kimura T.; Inaoka T.; Izawa R.; Nakano T.; Hotehama C.; Sakuda A.; Tatsumisago M.; Hayashi A. Stabilizing high-temperature α-Li3PS4 by rapidly heating the glass. J. Am. Chem. Soc. 2023, 145, 14466. 10.1021/jacs.3c03827. [DOI] [PubMed] [Google Scholar]
- de Klerk N. J.; van der Maas E.; Wagemaker M. Analysis of diffusion in solid-state electrolytes through md simulations, improvement of the Li-ion conductivity in β-Li3PS4 as an example. ACS applied energy materials 2018, 1, 3230. 10.1021/acsaem.8b00457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrester F. N.; Quirk J. A.; Famprikis T.; Dawson J. A. Disentangling cation and anion dynamics in Li3PS4 solid electrolytes. Chem. Mater. 2022, 34, 10561. 10.1021/acs.chemmater.2c02637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariga S.; Ohkubo T.; Urata S.; Imamura Y.; Taniguchi T. A new universal force-field for the Li2S–P2S5 system. Phys. Chem. Chem. Phys. 2022, 24, 2567. 10.1039/D1CP05393K. [DOI] [PubMed] [Google Scholar]
- Staacke C. G.; Heenen H. H.; Scheurer C.; Csányi G.; Reuter K.; Margraf J. T. On the role of long-range electrostatics in machine-learned interatomic potentials for complex battery materials. ACS Applied Energy Materials 2021, 4, 12562. 10.1021/acsaem.1c02363. [DOI] [Google Scholar]
- Staacke C. G.; Huss T.; Margraf J. T.; Reuter K.; Scheurer C. Tackling structural complexity in Li2S-P2S5 solid-state electrolytes using machine learning potentials. Nanomaterials 2022, 12, 2950. 10.3390/nano12172950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H.; Wang Q.; Urban A.; Artrith N. Artificial Intelligence-Aided Mapping of the Structure–Composition–Conductivity Relationships of Glass–Ceramic Lithium Thiophosphate Electrolytes. Chem. Mater. 2022, 34, 6702. 10.1021/acs.chemmater.2c00267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaup K.; Zhou L.; Huq A.; Nazar L. F. Impact of the Li substructure on the diffusion pathways in alpha and beta Li3PS4: an in situ high temperature neutron diffraction study. Journal of Materials Chemistry A 2020, 8, 12446. 10.1039/D0TA02805C. [DOI] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized gradient approximation made simple. Physical review letters 1996, 77, 3865. 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Burke K.; Wang Y. Generalized gradient approximation for the exchange-correlation hole of a many-electron system. Phys. Rev. B 1996, 54, 16533. 10.1103/PhysRevB.54.16533. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Li H.; Kaup K.; Zhou L.; Roy P.-N.; Nazar L. F. Targeting Superionic Conductivity by Turning on Anion Rotation at Room Temperature in Fast Ion Conductors. Matter 2020, 2, 1667. 10.1016/j.matt.2020.04.027. [DOI] [Google Scholar]
- Smith J. G.; Siegel D. J. Low-temperature paddlewheel effect in glassy solid electrolytes. Nat. Commun. 2020, 11, 1483. 10.1038/s41467-020-15245-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deringer V. L.; Bartók A. P.; Bernstein N.; Wilkins D. M.; Ceriotti M.; Csányi G. Gaussian process regression for materials and molecules. Chem. Rev. 2021, 121, 10073. 10.1021/acs.chemrev.1c00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Zhang L.; Han J.; E W. DeePMD-kit: A deep learning package for many-body potential energy representation and molecular dynamics. Comput. Phys. Commun. 2018, 228, 178. 10.1016/j.cpc.2018.03.016. [DOI] [Google Scholar]
- Zeng J.; Zhang D.; Lu D.; Mo P.; Li Z.; Chen Y.; Rynik M.; Huang L.; Li Z.; Shi S.; Wang Y.; Ye H.; Tuo P.; Yang J.; Ding Y.; Li Y.; Tisi D.; Zeng Q.; Bao H.; Xia Y.; Huang J.; Muraoka K.; Wang Y.; Chang J.; Yuan F.; Bore S. L.; Cai C.; Lin Y.; Wang B.; Xu J.; Zhu J.-X.; Luo C.; Zhang Y.; Goodall R. E. A.; Liang W.; Singh A. K.; Yao S.; Zhang J.; Wentzcovitch R.; Han J.; Liu J.; Jia W.; York D. M.; E W.; Car R.; Zhang L.; Wang H. DeePMD-kit v2: A software package for deep potential models. J. Chem. Phys. 2023, 159, 054801 10.1063/5.0155600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartók A. P.; Payne M. C.; Kondor R.; Csányi G. Gaussian Approximation Potentials: The Accuracy of Quantum Mechanics, without the Electrons. Phys. Rev. Lett. 2010, 104, 136403. 10.1103/PhysRevLett.104.136403. [DOI] [PubMed] [Google Scholar]
- Behler J.; Parrinello M. Generalized Neural-Network Representation of High-Dimensional Potential-Energy Surfaces. Phys. Rev. Lett. 2007, 98, 146401. 10.1103/PhysRevLett.98.146401. [DOI] [PubMed] [Google Scholar]
- Smith J. S.; Isayev O.; Roitberg A. E. ANI-1: an extensible neural network potential with DFT accuracy at force field computational cost. Chemical Science 2017, 8, 3192. 10.1039/C6SC05720A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schütt K. T.; Hessmann S. S. P.; Gebauer N. W. A.; Lederer J.; Gastegger M. SchNetPack 2.0: A neural network toolbox for atomistic machine learning. J. Chem. Phys. 2023, 158, 144801. 10.1063/5.0138367. [DOI] [PubMed] [Google Scholar]
- Rupp M.; Tkatchenko A.; Müller K.-R.; von Lilienfeld O. A. Fast and Accurate Modeling of Molecular Atomization Energies with Machine Learning. Phys. Rev. Lett. 2012, 108, 058301 10.1103/PhysRevLett.108.058301. [DOI] [PubMed] [Google Scholar]
- Butler K. T.; Davies D. W.; Cartwright H.; Isayev O.; Walsh A. Machine learning for molecular and materials science. Nature 2018, 559, 547. 10.1038/s41586-018-0337-2. [DOI] [PubMed] [Google Scholar]
- Ko T. W.; Finkler J. A.; Goedecker S.; Behler J. A fourth-generation high-dimensional neural network potential with accurate electrostatics including non-local charge transfer. Nat. Commun. 2021, 12, 398. 10.1038/s41467-020-20427-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unke O. T.; Meuwly M. Physnet: A neural network for predicting energies, forces, dipole moments, and partial charges. J. Chem. Theory Comput. 2019, 15, 3678. 10.1021/acs.jctc.9b00181. [DOI] [PubMed] [Google Scholar]
- Batzner S.; Musaelian A.; Sun L.; Geiger M.; Mailoa J. P.; Kornbluth M.; Molinari N.; Smidt T. E.; Kozinsky B. E(3)-equivariant graph neural networks for data-efficient and accurate interatomic potentials. Nat. Commun. 2022, 13, 2453. 10.1038/s41467-022-29939-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Z.; Zeng Z.; Zhang C.; Wang Y.; Song K.; Dong H.; Chen Y.; Ala-Nissila T. Neuroevolution machine learning potentials: Combining high accuracy and low cost in atomistic simulations and application to heat transport. Phys. Rev. B 2021, 104, 104309. 10.1103/PhysRevB.104.104309. [DOI] [Google Scholar]
- Willatt M. J.; Musil F.; Ceriotti M. Atom-density representations for machine learning. J. Chem. Phys. 2019, 150, 154110. 10.1063/1.5090481. [DOI] [PubMed] [Google Scholar]
- Bartók A. P.; Kondor R.; Csányi G. On representing chemical environments. Phys. Rev. B 2013, 87, 184115. 10.1103/PhysRevB.87.184115. [DOI] [Google Scholar]
- Imbalzano G.; Ceriotti M. Modeling the Ga/As binary system across temperatures and compositions from first principles. Phys. Rev. Materials 2021, 5, 063804 10.1103/PhysRevMaterials.5.063804. [DOI] [Google Scholar]
- Lopanitsyna N.; Ben Mahmoud C.; Ceriotti M. Finite-temperature materials modeling from the quantum nuclei to the hot electron regime. Phys. Rev. Mater. 2021, 5, 043802 10.1103/PhysRevMaterials.5.043802. [DOI] [Google Scholar]
- Gigli L.; Veit M.; Kotiuga M.; Pizzi G.; Marzari N.; Ceriotti M. Thermodynamics and dielectric response of BaTiO3 by data-driven modeling. npj Computational Materials 2022, 8, 209. 10.1038/s41524-022-00845-0. [DOI] [Google Scholar]
- Gigli L.; Goscinski A.; Ceriotti M.; Tribello G. A.. Modeling the ferroelectric phase transition in barium titanate with DFT accuracy and converged sampling. arXiv, 2023, 2310.12579 [cond-mat], accessed: 2023-12-23.
- Deringer V. L.; Csányi G. Machine learning based interatomic potential for amorphous carbon. Phys. Rev. B 2017, 95, 094203 10.1103/PhysRevB.95.094203. [DOI] [Google Scholar]
- Bartók A. P.; Kermode J.; Bernstein N.; Csányi G. Machine learning a general-purpose interatomic potential for silicon. Phys. Rev. X 2018, 8, 041048 10.1103/PhysRevX.8.041048. [DOI] [Google Scholar]
- Maresca F.; Dragoni D.; Csányi G.; Marzari N.; Curtin W. A. Screw dislocation structure and mobility in body centered cubic Fe predicted by a Gaussian Approximation Potential. npj Computational Materials 2018, 4, 69. 10.1038/s41524-018-0125-4. [DOI] [Google Scholar]
- Sivaraman G.; Krishnamoorthy A. N.; Baur M.; Holm C.; Stan M.; Csányi G.; Benmore C.; Vázquez-Mayagoitia Á. Machine-learned interatomic potentials by active learning: amorphous and liquid hafnium dioxide. npj Computational Materials 2020, 6, 104. 10.1038/s41524-020-00367-7. [DOI] [Google Scholar]
- Deringer V. L.; Caro M. A.; Csányi G. A general-purpose machine-learning force field for bulk and nanostructured phosphorus. Nat. Commun. 2020, 11, 5461. 10.1038/s41467-020-19168-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deringer V. L.; Caro M. A.; Csányi G. Machine learning interatomic potentials as emerging tools for materials science. Adv. Mater. 2019, 31, 1902765. 10.1002/adma.201902765. [DOI] [PubMed] [Google Scholar]
- Rosenbrock C. W.; Gubaev K.; Shapeev A. V.; Pártay L. B.; Bernstein N.; Csányi G.; Hart G. L. W. Machine-learned interatomic potentials for alloys and alloy phase diagrams. npj Computational Materials 2021, 7, 24. 10.1038/s41524-020-00477-2. [DOI] [Google Scholar]
- Huang J.; Zhang L.; Wang H.; Zhao J.; Cheng J.; E W. Deep potential generation scheme and simulation protocol for the Li10GeP2S12-type superionic conductors. J. Chem. Phys. 2021, 154, 094703 10.1063/5.0041849. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Schmidt K. Jacob’s ladder of density functional approximations for the exchange-correlation energy. AIP Conf. Proc. 2001, 577, 1. 10.1063/1.1390175. [DOI] [Google Scholar]
- Becke A. D. A new mixing of Hartree–Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372. 10.1063/1.464304. [DOI] [Google Scholar]
- Perdew J. P.; Ruzsinszky A.; Csonka G. I.; Vydrov O. A.; Scuseria G. E.; Constantin L. A.; Zhou X.; Burke K. Restoring the density-gradient expansion for exchange in solids and surfaces. Phys. Rev. Lett. 2008, 100, 136406. 10.1103/PhysRevLett.100.136406. [DOI] [PubMed] [Google Scholar]
- Furness J. W.; Kaplan A. D.; Ning J.; Perdew J. P.; Sun J. Accurate and numerically efficient r2SCAN meta-generalized gradient approximation. J. Phys. Chem. Lett. 2020, 11, 8208. 10.1021/acs.jpclett.0c02405. [DOI] [PubMed] [Google Scholar]
- Adamo C.; Barone V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158. 10.1063/1.478522. [DOI] [Google Scholar]
- Green M. S. Markoff random processes and the statistical mechanics of time-dependent phenomena, II. Irreversible processes in fluids. J. Chem. Phys. 1954, 22, 398. 10.1063/1.1740082. [DOI] [Google Scholar]
- Kubo R. Statistical-mechanical theory of irreversible processes. I. General Theory and Simple Applications to Magnetic and Conduction Problems. J. Phys. Soc. Jpn. 1957, 12, 570. 10.1143/JPSJ.12.570. [DOI] [Google Scholar]
- Car R.; Parrinello M. Unified Approach for Molecular Dynamics and Density-Functional Theory. Phys. Rev. Lett. 1985, 55, 2471. 10.1103/PhysRevLett.55.2471. [DOI] [PubMed] [Google Scholar]
- Giannozzi P.; Baroni S.; Bonini N.; Calandra M.; Car R.; Cavazzoni C.; Ceresoli D.; Chiarotti G. L.; Cococcioni M.; Dabo I.; Dal Corso A.; de Gironcoli S.; Fabris S.; Fratesi G.; Gebauer R.; Gerstmann U.; Gougoussis C.; Kokalj A.; Lazzeri M.; Martin-Samos L.; Marzari N.; Mauri F.; Mazzarello R.; Paolini S.; Pasquarello A.; Paulatto L.; Sbraccia C.; Scandolo S.; Sclauzero G.; Seitsonen A. P.; Smogunov A.; Umari P.; Wentzcovitch R. M. QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J. Phys. Cond. Matt. 2009, 21, 395502. 10.1088/0953-8984/21/39/395502. [DOI] [PubMed] [Google Scholar]
- Giannozzi P.; Andreussi O.; Brumme T.; Bunau O.; Buongiorno Nardelli M.; Calandra M.; Car R.; Cavazzoni C.; Ceresoli D.; Cococcioni M.; Colonna N.; Carnimeo I.; Dal Corso A.; de Gironcoli S.; Delugas P.; DiStasio R. A.; Ferretti A.; Floris A.; Fratesi G.; Fugallo G.; Gebauer R.; Gerstmann U.; Giustino F.; Gorni T.; Jia J.; Kawamura M.; Ko H.-Y.; Kokalj A.; Küçükbenli E.; Lazzeri M.; Marsili M.; Marzari N.; Mauri F.; Nguyen N. L.; Nguyen H.-V.; Otero-de-la-Roza A.; Paulatto L.; Poncé S.; Rocca D.; Sabatini R.; Santra B.; Schlipf M.; Seitsonen A. P.; Smogunov A.; Timrov I.; Thonhauser T.; Umari P.; Vast N.; Wu X.; Baroni S. Advanced capabilities for materials modelling with Quantum ESPRESSO. J. Phys.: Condens. Matter 2017, 29, 465901. 10.1088/1361-648X/aa8f79. [DOI] [PubMed] [Google Scholar]
- Giannozzi P.; Baseggio O.; Bonfà P.; Brunato D.; Car R.; Carnimeo I.; Cavazzoni C.; de Gironcoli S.; Delugas P.; Ferrari Ruffino F.; Ferretti A.; Marzari N.; Timrov I.; Urru A.; Baroni S. Quantum ESPRESSO toward the exascale. J. Chem. Phys. 2020, 152, 154105. 10.1063/5.0005082. [DOI] [PubMed] [Google Scholar]
- Carnimeo I.; Affinito F.; Baroni S.; Baseggio O.; Bellentani L.; Bertossa R.; Delugas P. D.; Ruffino F. F.; Orlandini S.; Spiga F.; Giannozzi P. Quantum espresso: One further step toward the exascale. J. Chem. Theory Comput. 2023, 19, 6992. 10.1021/acs.jctc.3c00249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapil V.; Rossi M.; Marsalek O.; Petraglia R.; Litman Y.; Spura T.; Cheng B.; Cuzzocrea A.; Meißner R. H.; Wilkins D. M.; Helfrecht B. A.; Juda P.; Bienvenue S. P.; Fang W.; Kessler J.; Poltavsky I.; Vandenbrande S.; Wieme J.; Corminboeuf C.; Kühne T. D.; Manolopoulos D. E.; Markland T. E.; Richardson J. O.; Tkatchenko A.; Tribello G. A.; Van Speybroeck V.; Ceriotti M. i-PI 2.0: A universal force engine for advanced molecular simulations. Comput. Phys. Commun. 2019, 236, 214. 10.1016/j.cpc.2018.09.020. [DOI] [Google Scholar]
- Imbalzano G.; Anelli A.; Giofré D.; Klees S.; Behler J.; Ceriotti M. Automatic selection of atomic fingerprints and reference configurations for machine-learning potentials. J. Chem. Phys. 2018, 148, 241730. 10.1063/1.5024611. [DOI] [PubMed] [Google Scholar]
- Zaspel P.; Huang B.; Harbrecht H.; von Lilienfeld O. A. Boosting quantum machine learning models with a multilevel combination technique: Pople diagrams revisited. J. Chem. Theory Comput. 2019, 15, 1546. 10.1021/acs.jctc.8b00832. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan R.; Dral P. O.; Rupp M.; von Lilienfeld O. A. Big data meets quantum chemistry approximations: The δ-machine learning approach. J. Chem. Theory Comput. 2015, 11, 2087. 10.1021/acs.jctc.5b00099. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Hafner J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558. 10.1103/PhysRevB.47.558. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Furthmüller J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15. 10.1016/0927-0256(96)00008-0. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Furthmüller J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. 10.1103/PhysRevB.54.11169. [DOI] [PubMed] [Google Scholar]
- Schölkopf B.; Smola A.; Müller K.-R.. Kernel principal component analysis. In International conference on artificial neural networks; Springer: 1997; pp 583–588. [Google Scholar]
- Chong S.; Grasselli F.; Ben Mahmoud C.; Morrow J. D.; Deringer V. L.; Ceriotti M. Robustness of local predictions in atomistic machine learning models. J. Chem. Theory Comput. 2023, 19, 8020. 10.1021/acs.jctc.3c00704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasselli F.; Baroni S. Topological quantization and gauge invariance of charge transport in liquid insulators. Nat. Phys. 2019, 15, 967. 10.1038/s41567-019-0562-0. [DOI] [Google Scholar]
- Pegolo P.; Grasselli F.; Baroni S. Oxidation states, Thouless’ pumps, and nontrivial ionic transport in nonstoichiometric electrolytes. Physical Review X 2020, 10, 041031 10.1103/PhysRevX.10.041031. [DOI] [Google Scholar]
- Allen M. P.; Tildesley D. J.: Computer simulation of liquids; Oxford University Press: 1990. [Google Scholar]
- Marcolongo A.; Marzari N. Ionic correlations and failure of Nernst-Einstein relation in solid-state electrolytes. Phys. Rev. Mater. 2017, 1, 025402 10.1103/PhysRevMaterials.1.025402. [DOI] [Google Scholar]
- Murch G. The haven ratio in fast ionic conductors. Solid State Ionics 1982, 7, 177. 10.1016/0167-2738(82)90050-9. [DOI] [Google Scholar]
- Kuhn A.; Duppel V.; Lotsch B. V. Tetragonal Li10GeP2S12 and Li7GePS8 – exploring the Li ion dynamics in LGPS Li electrolytes. Energy Environ. Sci. 2013, 6, 3548. 10.1039/c3ee41728j. [DOI] [Google Scholar]
- Grasselli F.; Baroni S. Invariance principles in the theory and computation of transport coefficients. European Physical Journal B 2021, 94, 160. 10.1140/epjb/s10051-021-00152-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasselli F. Investigating finite-size effects in molecular dynamics simulations of ion diffusion, heat transport, and thermal motion in superionic materials. J. Chem. Phys. 2022, 156, 134705 10.1063/5.0087382. [DOI] [PubMed] [Google Scholar]
- Ceriotti M.; More J.; Manolopoulos D. E. i-PI: A Python interface for ab initio path integral molecular dynamics simulations. Comput. Phys. Commun. 2014, 185, 1019. 10.1016/j.cpc.2013.10.027. [DOI] [Google Scholar]
- Plimpton S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1. 10.1006/jcph.1995.1039. [DOI] [Google Scholar]
- Thompson A. P.; Aktulga H. M.; Berger R.; Bolintineanu D. S.; Brown W. M.; Crozier P. S.; in ‘t Veld P. J.; Kohlmeyer A.; Moore S. G.; Nguyen T. D.; Shan R.; Stevens M. J.; Tranchida J.; Trott C.; Plimpton S. J. LAMMPS - a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales. Comput. Phys. Commun. 2022, 271, 108171. 10.1016/j.cpc.2021.108171. [DOI] [Google Scholar]
- Musil F.; Veit M.; Goscinski A.; Fraux G.; Willatt M. J.; Stricker M.; Junge T.; Ceriotti M. Efficient implementation of atom-density representations. J. Chem. Phys. 2021, 154, 114109. 10.1063/5.0044689. [DOI] [PubMed] [Google Scholar]
- Ceriotti M.; Bussi G.; Parrinello M. Langevin equation with colored noise for constant-temperature molecular dynamics simulations. Phys. Rev. Lett. 2009, 102, 020601 10.1103/PhysRevLett.102.020601. [DOI] [PubMed] [Google Scholar]
- Ceriotti M.; Bussi G.; Parrinello M. Colored-noise thermostats à la carte. J. Chem. Theory Comput. 2010, 6, 1170. 10.1021/ct900563s. [DOI] [Google Scholar]
- Bussi G.; Donadio D.; Parrinello M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- Liu Z.; Fu W.; Payzant E. A.; Yu X.; Wu Z.; Dudney N. J.; Kiggans J.; Hong K.; Rondinone A. J.; Liang C. Anomalous high ionic conductivity of nanoporous β-Li3PS4. J. Am. Chem. Soc. 2013, 135, 975. 10.1021/ja3110895. [DOI] [PubMed] [Google Scholar]
- Rangasamy E.; Li J.; Sahu G.; Dudney N.; Liang C. Pushing the theoretical limit of Li-CFx batteries: A tale of bifunctional electrolyte. J. Am. Chem. Soc. 2014, 136, 6874. 10.1021/ja5026358. [DOI] [PubMed] [Google Scholar]
- Stroppa A.; Kresse G. The shortcomings of semi-local and hybrid functionals: what we can learn from surface science studies. New J. Phys. 2008, 10, 063020 10.1088/1367-2630/10/6/063020. [DOI] [Google Scholar]
- Csonka G. I.; Perdew J. P.; Ruzsinszky A.; Philipsen P. H. T.; Lebègue S.; Paier J.; Vydrov O. A.; Ángyán J. G. Assessing the performance of recent density functionals for bulk solids. Phys. Rev. B 2009, 79, 155107. 10.1103/PhysRevB.79.155107. [DOI] [Google Scholar]
- Hermet J.; Adamo C.; Cortona P.. Towards a greater accuracy in dft calculations: From gga to hybrid functionals. In Quantum Simulations of Materials and Biological Systems; Zeng J., Zhang R.-Q., Treutlein H. R., Eds.; Springer Netherlands: Dordrecht, 2012; pp 3–15. [Google Scholar]
- Mardirossian N.; Head-Gordon M. Thirty years of density functional theory in computational chemistry: an overview and extensive assessment of 200 density functionals. Mol. Phys. 2017, 115, 2315. 10.1080/00268976.2017.1333644. [DOI] [Google Scholar]
- Yang J.; Falletta S.; Pasquarello A. Range-separated hybrid functionals for accurate prediction of band gaps of extended systems. npj Computational Materials 2023, 9, 108. 10.1038/s41524-023-01064-x. [DOI] [Google Scholar]
- Zhang L.; Wang H.; Car R.; E W. Phase diagram of a deep potential water model. Phys. Rev. Lett. 2021, 126, 236001. 10.1103/PhysRevLett.126.236001. [DOI] [PubMed] [Google Scholar]
- Schmidt J.; Wang H.-C.; Cerqueira T. F. T.; Botti S.; Marques M. A. L. A dataset of 175k stable and metastable materials calculated with the PBEsol and SCAN functionals. Sci. Data 2022, 9, 64. 10.1038/s41597-022-01177-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tisi D.; Zhang L.; Bertossa R.; Wang H.; Car R.; Baroni S. Heat transport in liquid water from first-principles and deep neural network simulations. Phys. Rev. B 2021, 104, 224202. 10.1103/PhysRevB.104.224202. [DOI] [Google Scholar]
- Malosso C.; Zhang L.; Car R.; Baroni S.; Tisi D. Viscosity in water from first-principles and deep-neural-network simulations. npj Computational Materials 2022, 8, 139. 10.1038/s41524-022-00830-7. [DOI] [Google Scholar]
- Marana N. L.; Sgroi M. F.; Maschio L.; Ferrari A. M.; D’Amore M.; Casassa S. Computational characterization of β-Li3PS4 solid electrolyte: From bulk and surfaces to nanocrystals. Nanomaterials 2022, 12, 2795. 10.3390/nano12162795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim M.-S.; Jhi S.-H. First-principles study of lithium-ion diffusion in β-Li3PS4 for solid-state electrolytes. Curr. Appl. Phys. 2018, 18, 541. 10.1016/j.cap.2018.03.002. [DOI] [Google Scholar]
- Pegolo P.; Baroni S.; Grasselli F. Temperature- and vacancy-concentration-dependence of heat transport in Li3ClO from multi-method numerical simulations. npj Comput. Mater. 2022, 8, 24. 10.1038/s41524-021-00693-4. [DOI] [Google Scholar]
- Hori S.; Kato M.; Suzuki K.; Hirayama M.; Kato Y.; Kanno R. Phase Diagram of the Li4GeS4–Li3PS4 Quasi-Binary System Containing the Superionic Conductor Li10GeP2S12. J. Am. Ceram. Soc. 2015, 98, 3352. 10.1111/jace.13694. [DOI] [Google Scholar]
- Poletayev A. D.; Dawson J. A.; Islam M. S.; Lindenberg A. M. Defect-driven anomalous transport in fast-ion conducting solid electrolytes. Nat. Mater. 2022, 21, 1066. 10.1038/s41563-022-01316-z. [DOI] [PubMed] [Google Scholar]
- Yang J.; Tse J. S. First-principles molecular simulations of Li diffusion in solid electrolytes Li3PS4. Comput. Mater. Sci. 2015, 107, 134. 10.1016/j.commatsci.2015.05.022. [DOI] [Google Scholar]
- Self E. C.; Hood Z. D.; Brahmbhatt T.; Delnick F. M.; Meyer H. M.; Yang G.; Rupp J. L. M.; Nanda J. Solvent-Mediated Synthesis of Amorphous Li3PS4/Polyethylene Oxide Composite Solid Electrolytes with High Li+ Conductivity. Chem. Mater. 2020, 32, 8789. 10.1021/acs.chemmater.0c01990. [DOI] [Google Scholar]
- Kim J.-S.; Jung W. D.; Choi S.; Son J.-W.; Kim B.-K.; Lee J.-H.; Kim H. Thermally induced S-sublattice transition of Li3PS4 for fast lithium-ion conduction. J. Phys. Chem. Lett. 2018, 9, 5592. 10.1021/acs.jpclett.8b01989. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Wu Q.; Cui Y.; Chen Y.; Shi S.; Wang R.-Z.; Yan H. Elastic Properties, Defect Thermodynamics, Electrochemical Window, Phase Stability, and Li+ Mobility of Li3PS4: Insights from First-Principles Calculations. ACS Appl. Mater. Interfaces 2016, 8, 25229. 10.1021/acsami.6b06754. [DOI] [PubMed] [Google Scholar]
- Hill P. D. Kernel estimation of a distribution function. Communications in Statistics - Theory and Methods 1985, 14, 605. 10.1080/03610928508828937. [DOI] [Google Scholar]
- He X.; Zhu Y.; Mo Y. Origin of fast ion diffusion in super-ionic conductors. Nat. Commun. 2017, 8, 15893. 10.1038/ncomms15893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goscinski A.; Principe V.; Fraux G.; Kliavinek S.; Helfrecht B.; Loche P.; Ceriotti M.; Cersonsky R. scikit-matter: A suite of generalisable machine learning methods born out of chemistry and materials science. Open Research Europe 2023, 3, 81. 10.12688/openreseurope.15789.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeni C.; Anelli A.; Glielmo A.; Rossi K. Exploring the robust extrapolation of high-dimensional machine learning potentials. Phys. Rev. B 2022, 105, 165141. 10.1103/PhysRevB.105.165141. [DOI] [Google Scholar]
- Blöchl P. E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953. 10.1103/PhysRevB.50.17953. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Joubert D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758. 10.1103/PhysRevB.59.1758. [DOI] [Google Scholar]
- Nosé S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511. 10.1063/1.447334. [DOI] [Google Scholar]
- Rappe A. M.; Rabe K. M.; Kaxiras E.; Joannopoulos J. D. Optimized pseudopotentials. Phys. Rev. B 1990, 41, 1227. 10.1103/PhysRevB.41.1227. [DOI] [PubMed] [Google Scholar]
- Marcolongo A.; Umari P.; Baroni S. Microscopic theory and quantum simulation of atomic heat transport. Nat. Phys. 2016, 12, 80. 10.1038/nphys3509. [DOI] [Google Scholar]
- Talirz L.; Kumbhar S.; Passaro E.; Yakutovich A. V.; Granata V.; Gargiulo F.; Borelli M.; Uhrin M.; Huber S. P.; Zoupanos S.; et al. Materials Cloud, a platform for open computational science. Scientific data 2020, 7, 299. 10.1038/s41597-020-00637-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The Li3PS4 data sets and the ML-potentials generated in this work, as well as the input files of the simulations, the notebooks, and the scripts employed are available on the Materials Cloud Platform125 at 10.24435/materialscloud:g2-fp.