

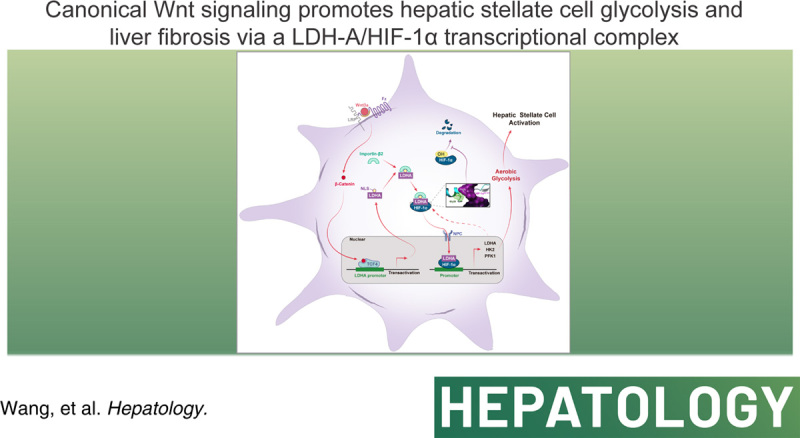
Abstract
Background and Aims:
Aerobic glycolysis reprogramming occurs during HSC activation, but how it is initiated and sustained remains unknown. We investigated the mechanisms by which canonical Wnt signaling regulated HSC glycolysis and the therapeutic implication for liver fibrosis.
Approach and Results:
Glycolysis was examined in HSC-LX2 cells upon manipulation of Wnt/β-catenin signaling. Nuclear translocation of lactate dehydrogenase A (LDH-A) and its interaction with hypoxia-inducible factor-1α (HIF-1α) were investigated using molecular simulation and site-directed mutation assays. The pharmacological relevance of molecular discoveries was intensified in primary cultures, rodent models, and human samples. HSC glycolysis was enhanced by Wnt3a but reduced by β-catenin inhibitor or small interfering RNA (siRNA). Wnt3a-induced rapid transactivation and high expression of LDH-A dependent on TCF4. Wnt/β-catenin signaling also stimulated LDH-A nuclear translocation through importin β2 interplay with a noncanonical nuclear location signal of LDH-A. Mechanically, LDH-A bound to HIF-1α and enhanced its stability by obstructing hydroxylation-mediated proteasome degradation, leading to increased transactivation of glycolytic genes. The Gly28 residue of LDH-A was identified to be responsible for the formation of the LDH-A/HIF-1α transcription complex and stabilization of HIF-1α. Furthermore, LDH-A-mediated glycolysis was required for HSC activation in the presence of Wnt3a. Results in vivo showed that HSC activation and liver fibrosis were alleviated by HSC-specific knockdown of LDH-A in mice. β-catenin inhibitor XAV-939 mitigated HSC activation and liver fibrosis, which were abrogated by HSC-specific LDH-A overexpression in mice with fibrosis.
Conclusions:
Inhibition of HSC glycolysis by targeting Wnt/β-catenin signaling and LDH-A had therapeutic promise for liver fibrosis.
INTRODUCTION
Liver fibrosis is a pathological process characterized by chronic liver injury due to diverse aetiologies concomitant with the accumulation of extracellular matrix. Without effective treatment, it may progress to cirrhosis, HCC or liver failure. HSCs located in the space of Disse are the primary pro-fibrogenic cells in the liver.1 Upon activation, the quiescent HSCs transdifferentiate into proliferative, migratory, and contractile myofibroblasts. These cells produce massive extracellular matrix components forming scar tissue and disrupting the liver’s sinusoidal structure.2 Inhibition of the pro-fibrogenic phenotype of activated HSCs is an important strategy for liver fibrosis therapy.3
Recent understanding reveals that rapid metabolism reprogramming occurs during HSC activation. The proliferation and growth of HSCs are largely dependent on aerobic glycolysis, a phenomenon similar to the Warburg effect in cancer cells.4 Glycolysis is a multistep process initiated by the transportation of glucose into cells followed by conversion to pyruvate. This process is successively catalyzed by the rate-limiting enzymes hexokinase 2 (HK2), phosphofructokinase 1 (PFK1), and pyruvate kinase type M2.5 The final conversion of pyruvate to lactate is a crucial step catalyzed by lactate dehydrogenase A (LDH-A), whose high expression or activity allows for rapid glycolysis flux.6 Studies have demonstrated that glycolytic activity was significantly increased to promote HSC activation, in which the hedgehog signaling through hypoxia-inducible factor-1α (HIF-1α) upregulated several glycolytic enzymes under normoxic conditions.7 This metabolic switch may be due to the much shorter path of glycolysis than oxidative phosphorylation, leading to faster generation of ATP and metabolites and thereby meeting the high demands of rapid cell proliferation. These metabolic perturbations provide a high possibility of selective inhibition of HSC fibrogenic transdifferentiation by means of interference with metabolism.
Although the molecular mechanisms underlying HSC aerobic glycolysis reprogramming remain to be defined, it has been accepted that this metabolic signature is commonly induced by signal transduction pathways.8 The Wnt pathway has recently emerged to play a pivotal role in liver pathophysiology.9 Wnt proteins interact with various receptors and co-receptors at the cell membrane and activate an intracellular signaling network to induce diverse biological responses.10 β-catenin-mediated cascade is termed canonical Wnt signaling. In this case, binding of Wnt proteins to the Frizzled receptor and LRP5/6 co-receptor results in the stabilization of β-catenin, which then translocates into the nucleus and interacts with the T-cell factor (TCF)/lymphoid enhancer-binding factor transcription factors to activate the transcription of Wnt-targeted genes.10 Here, we investigated how the Wnt signaling regulated HSC glycolytic metabolic reprogramming implicated in the therapy of liver fibrosis.
METHODS
Cell culture, treatment, and transfection
Immortalized human HSC-LX2 cell line (Procell Life Science & Technology, Wuhan, China) and HEK-293T cells (Cell Bank of Chinese Academy of Sciences, Shanghai, China) were characterized by human short tandem repeat markers. Mouse primary HSCs were isolated based on the well-established procedures.11 Cells were cultured in DMEM (Keygen Biotech, Nanjing, China) with 10% fetal bovine serum (Royacel Biotechnology, Lanzhou, China) and 1% antibiotics and grown in a 5% CO2 humidified atmosphere at 37°C. Cells at 60% confluence were transfected with small interfering RNA (siRNAs) or plasmids using Lipofectamine RNAiMAX Transfection Reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s recommendations. Information of reagents, chemicals, siRNAs, and plasmids are shown in Supplemental Tables S1-S3, http://links.lww.com/HEP/H940.
RNA sequencing
Total RNA was extracted from LX2 cells treated with Wnt3a using TRIzol reagent. RNA sample quality control, cDNA library preparation, RNA sequencing, and bioinformatics analysis were performed by Personal Biotechnology (Shanghai, China). Deseq2 was used to estimate the differentially expressed genes between samples. These genes were plotted with the mean values centered using the heat map package, followed by gene ontology and Kyoto Encyclopedia of Genes and Genomes pathway enrichment analyses using the MSigDB gene sets.
Dual-luciferase reporter assay
The promoter sequences of HK2, PFK1, and LDH-A were cloned into pGL4.10 vectors to construct luciferase reporter plasmids. In some experiments, the predicted TCF4 binding sequences at LDH-A promoter were mutated individually and cloned into LDH-A luciferase reporter plasmids. LX2 cells were treated with different reagents and/or co-transfected with siRNA or plasmids and with the LDH-A luciferase reporter plasmids. Transfection efficiency was normalized by co-transfection with renilla luciferase reporter plasmids. Luciferase activity was measured using the Dual-luciferase reporter gene assay Kit (Yeasen, Shanghai, China).
Co-immunoprecipitation
LDH-A interaction with importin β2 or HIF-1α in Wnt3a-treated LX2 cells, HIF-1 interaction with ARNT or p300 in the nucleus of LX2 cells transfected with LDH-A Gly28 mutant plasmids, and the interactions between hyaluronic acid-labeled HIF-1α plasmids and Flag-tagged N-terminal LDH-A, Flag-tagged C-terminal LDH-A, or Flag-tagged full-length LDH-A plasmids in HEK-293T cells were probed using co-immunoprecipitation (Co-IP) according to the methods we reported.12
EMSA
EMSA was performed using the Lightshift Chemiluminescent EMSA Kit (Thermo Scientific, USA), as we described.13 Nuclear extracts were prepared from Wnt3a-treated LX2 cells and subjected to polyacrylamide gel electrophoresis. A biotin-labeled, double-stranded oligonucleotide containing the consensus-binding sequence for HIF-1α from HK2 or PFK1 promoter was used to evaluate the binding activity. A double-stranded oligonucleotide with the same sequence but without biotin labeling was used as a competitor (in 200-fold excess) to determine the specificity.
Animal experiments
Animal experimental procedures were approved by the Institutional and Local Committee on the Care and Use of Animals of Nanjing University of Chinese Medicine (Approval No. 201912A003). All animals received humane care according to the National Institutes of Health (USA) guidelines. We complied with the ARRIVE reporting guidelines.14 Male Institute of Cancer Research mice (20–25 g body weight, 6-week old; Nanjing Qinglongshan Animal, Nanjing, China) were housed in vented cages under standardized conditions at 20°±2°C room temperature, 40%±5% relative humidity and a 12 h light/dark cycle. The sample size was determined by the Online Animal Sample Size Calculator provided by the Centre for Comparative Medicine Research, the University of Hong Kong. Liver fibrosis in mice was induced by i.p. injection with a mixture of carbon tetrachloride (CCl4) and olive oil [1:9 (v/v)] at a dose of 5 mL/kg for 4 weeks (3 times/week). Vitamin A-liposome-LDH-A short hairpin RNA (shRNA) and vitamin A-liposome-LDH-A overexpression plasmids were prepared according to the well-established methods,15 which have been successfully used in our studies.16 Vitamin A-liposome-LDH-A shRNA and vitamin A-liposome-LDH-A overexpression plasmids were administrated to mice through tail vein to achieve HSC-specific knockdown or overexpression of LDH-A according to the established methods.17 Briefly, in study 1, 40 mice were randomly divided into 4 groups (n=10 per cage). Liver fibrosis was induced in the mice of groups 2–4 for 4 weeks, and the mice of group 1 were given olive oil (i.p.) as negative control. At the beginning of week 1 and 3, the mice of group 4 were administrated with vitamin A-liposome-LDH-A shRNA twice, and the mice of group 3 were administrated with vitamin A-liposome-control shRNA twice. In study 2, 50 mice were randomly divided into 5 groups (n = 10 per cage). Liver fibrosis was induced in the mice of groups 2–5 for 4 weeks, and the mice of group 1 were given olive oil (i.p.) as negative control. After 4 weeks, the mice of groups 3–5 were orally given β-catenin inhibitor XAV (20 mg/kg/day) during weeks 5–8, and the mice of group 2 were orally given vehicle as negative control. At the beginning of week 5 and 7, the mice of group 5 were administrated with vitamin A-liposome-LDH-A overexpression plasmids twice, and the mice of group 4 were administrated with vitamin A-liposome-vector as a negative control twice. Notably, the mice with established fibrosis were still given CCl4 (i.p., 2 mL/kg, 1 time per week) during XAV treatment, so as to eliminate the possible impacts of natural regression of fibrosis on the results. At the end of both studies, 5 mice in each group were killed for the isolation of primary HSCs, and the remaining 5 mice were killed for blood collection and harvest of livers. Mice inhaled carbon dioxide for anesthesia, and no criteria were set for exclusion during the analysis.
Statistical analysis
Data are presented as mean ± SD and were analyzed using GraphPad Prism 7.0 (San Diego, CA, USA). For the normally distributed data, a significant difference was determined using Student t-test for comparing 2 groups and 1-way ANOVA with post hoc Tukey’s test for comparing multiple groups under the condition that F achieved p<0.05 and there was no significant variance inhomogeneity. For the non-normally distributed data, a significant difference was determined using the Mann-Whitney U test for comparing 2 groups and the Kruskal-Wallis H test with the post hoc Steel Dwass test for comparing multiple groups. Statistical significance was defined as p<0.05.
Other materials and methods are described in the Supplemental Materials and Methods, http://links.lww.com/HEP/H941 and Tables S4-S5, http://links.lww.com/HEP/H940.
RESULTS
Wnt/β-catenin signaling promotes aerobic glycolysis in HSCs
Initially, we examined the effects of Wnt on HSC glucose metabolism. We used recombinant Wnt3a to activate the signaling and employed β-catenin specific inhibitor XAV or targeted siRNA to test whether β-catenin was involved. Wnt3a increased glucose uptake and consumption, which was abolished by XAV or β-catenin siRNA in HSCs (Supplemental Figure S1A, B, http://links.lww.com/HEP/H942). Wnt3a-treated HSCs had elevated capacity of lactate production, which was diminished by XAV or β-catenin knockdown (Supplemental Figure S1C, http://links.lww.com/HEP/H942). To confirm the influence of Wnt on glycolysis and oxidative phosphorylation, oxygen consumption rate (OCR) and extracellular acidification rate were measured. Wnt3a markedly increased glycolytic rate and glycolytic capacity in HSCs, which were abrogated by XAV or β-catenin siRNA (Supplemental Figure S1D, http://links.lww.com/HEP/H942). Interestingly, Wnt3a suppressed oxidative phosphorylation as reflected by the alterations in basal and maximal OCR, but β-catenin inhibition or knockdown restored OCR at both basal and maximal levels (Supplemental Figure S1E, http://links.lww.com/HEP/H942). These results overtly demonstrated that Wnt/β-catenin signaling promoted HSC aerobic glycolysis.
TCF4-dependent transactivation of LDH-A contributes to canonical Wnt enhancement of aerobic glycolysis in HSCs
We explored the mechanisms by which Wnt/β-catenin signaling facilitated HSC aerobic glycolysis. RNA-sequencing analyses showed significant activation of Wnt signaling and upregulation of a range of glycolytic genes in Wnt3a-treated HSCs (Figure 1A). Consistently, HK2, PFK1, and LDH-A, the pivotal genes controlling glycolytic metabolism, had the most remarkable increases in mRNA expression upon Wnt3a stimulation (Supplemental Figure S2, http://links.lww.com/HEP/H942), suggesting that these molecules could be critically involved in the current context. β-catenin inhibitor or siRNA interference abolished Wnt3a-induced elevation in the mRNA and protein levels of HK2, PFK1 and LDH-A (Figure 1B, Supplemental Figure S3, http://links.lww.com/HEP/H942). Consistently, 13C metabolic flux analyses confirmed that Wnt3a increased glycolytic flux and weakened the tricarboxylic acid cycle, which was abolished by XAV or β-catenin siRNA (Figure 1C). We next probed the regulatory patterns of glycolytic genes by canonical Wnt. Time-course analyses by PCR revealed that LDH-A mRNA expression reached peak levels after 15 h stimulation with Wnt3a and thereafter changed little, whereas the mRNA levels of HK2 and PFK1 were steadily upregulated during the whole course (Figure 1D). This raised a hypothesis that there could be an acute regulation of LDH-A by canonical Wnt and an indirect regulation for HK2 and PFK1. To confirm this, luciferase activity analyses of LDH-A promoter activity were performed, demonstrating that Wnt3a increased the transcription of LDH-A, but XAV or β-catenin siRNA abrogated the Wnt3a effect in HSCs (Figure 1E). Furthermore, a complex with TCF/lymphoid enhancer-binding factor family transcription factors is required for β-catenin regulation of target gene transcription.10 To determine which TCF was involved, HSCs were transfected with siRNAs targeting TCF1, TCF3, or TCF4, respectively. Only knockdown of TCF4 eliminated Wnt3a-induced increases in the promoter activity and expression of LDH-A (Figure 1F-H). These data suggested LDH-A as a direct target gene of the β-catenin/TCF4 complex (Figure 1I). Next, we determined the exact binding site at the LDH-A promoter for β-catenin/TCF4. We predicted 3 most likely sequences and then mutated them individually and found that only mutation of sequence 2 abolished Wnt3a-enhanced LDH-A promoter activity (Figure 1J). This sequence was thus responsible for the transactivation of LDH-A induced by β-catenin/TCF4 complex. Moreover, LDH-A was sometimes transcriptionally upregulated by c-Myc18,19 or SIX120 in glycolytic cells. Here, Wnt3a did not affect the nuclear translocation of c-Myc and SIX1 (Supplemental Figure S4A, http://links.lww.com/HEP/H942), and neither c-Myc specific inhibitor nor SIX1 siRNA affected Wnt3a-induced LDH-A mRNA expression (Supplemental Figure S4B, http://links.lww.com/HEP/H942), excluding the involvement of c-Myc and SIX1 in current system. Overall, these discoveries demonstrated that canonical Wnt enhanced HSC aerobic glycolysis associated with β-catenin/TCF4-mediated transactivation of LDH-A.
FIGURE 1.

TCF4-dependent transactivation of LDH-A contributes to canonical Wnt enhancement of aerobic glycolysis in HSCs. (A) RNA sequencing analysis of LX2 cells treated with Wnt3a (100 ng/mL) for 24 h. Significantly altered genes were demonstrated by heat map, followed by Kyoto Encyclopedia of Genes and Genomes and gene ontology enrichment analyses. (B) Real-time PCR analysis of mRNA expression of glycolytic genes in LX2 cells treated with Wnt3a (100 ng/mL), and/or β-catenin inhibitor XAV (10 μM), or transfected with β-catenin siRNA for 24 h. *p<0.05 versus Ctrl, # p<0.05 versus Wnt3a. (C) 13C-labeled glucose metabolic flux analysis using LC-HRMS in LX2 cells treated with Wnt3a (100 ng/mL) and/or XAV (10 μM), or transfected with β-catenin siRNA for 24 h. Significantly changed metabolites in glycolysis and tricarboxylic acid cycle were quantified. *p<0.05 versus Ctrl, # p<0.05 versus Wnt3a. (D) Real-time PCR analysis of mRNA expression of glycolytic genes in LX2 cells treated with Wnt3a (100 ng/mL) for indicated time durations. *p<0.05 versus 0 h. (E) Dual-luciferase reporter assay for LDH-A promoter activity in LX2 cells treated with Wnt3a (100 ng/mL), and/or XAV (10 μM), or transfected with β-catenin siRNA for 24 h. *p<0.05 versus Ctrl, # p<0.05 versus Wnt3a. (F–H) LX2 cells were treated with Wnt3a (100 ng/mL), and/or transfected with siRNAs targeting TCF1, TCF2, and TCF3 for 24 h. LDH-A promoter activity was evaluated by dual-luciferase reporter assay (F), LDH-A mRNA expression was detected by real-time PCR (G), LDH-A protein expression was examined by western blotting assay with quantification (H). *p<0.05 versus Ctrl, # p<0.05 versus Wnt3a. (I) Illustration of the regulatory mechanism. (J) Three possible sites at LDH-A promoter for TCF4 binding to were predicted using the JASPAR database and their mutant luciferase reporter plasmids were constructed, respectively. LX2 cells were treated with Wnt3a (100 ng/mL) and/or transfected with TCF4 siRNA or the mutant plasmids for 24 h. LDH-A promoter activity was evaluated by dual-luciferase reporter assays. *p<0.05 versus Ctrl, # p<0.05 versus Wnt3a. Abbreviations: LDH-A, lactate dehydrogenase A; siRNA, small interfering RNA.
Canonical Wnt signaling specifically stimulates importin β2-dependent nuclear translocation of LDH-A in HSCs
The distinct expression pattern of LDH-A during Wnt3a stimulation led us to ask whether LDH-A had certain moonlighting functions in HSCs. Classical nuclear location signal (NLS) mapper analysis revealed the presence of a noncanonical nuclear location signal in LDH-A protein structure (Figure 2A, Supplemental Figure S5A, http://links.lww.com/HEP/H942). Intriguingly, Wnt3a increased the nuclear abundance of LDH-A time-dependently in HSCs (Supplemental Figure S5B, http://links.lww.com/HEP/H942), but LDH-A nuclear translocation was halted by β-catenin inhibition or knockdown as evidenced by Western blotting (Figure 2B) and immunofluorescence assays (Figure 2C). To test whether LDH-A nuclear translocation was specific for Wnt activation, HSCs were treated with TGF-β and PDGF-BB, which can potently activate HSCs, or β-catenin agonist LiCl. The 2 cytokines had little effects on LDH-A protein abundance and nuclear distribution, but LiCl, similar to Wnt3a, considerably upregulated LDH-A proteins and facilitated its nuclear translocation (Supplemental Figure S5C, http://links.lww.com/HEP/H942). To address why this happened, we examined protein nuclear-shuttling machinery. Neither TGF-β nor PDGF-BB changed the expression of importin β1 and importin β2, whereas Wnt3a specifically upregulated importin β2 (Figure 2D). Importin β inhibitor importazole abrogated Wnt3a-induced LDH-A nuclear translocation (Figure 2E). Co-IP showed that LDH-A had physical interaction with importin β2 in Wnt3a-treated HSCs (Figure 2F). Furthermore, the GFP fusion vector of LDH-A NLS sequence was able to translocate into the nucleus upon Wnt3a stimulation (Supplemental Figure S5D, http://links.lww.com/HEP/H942). Single mutation of NLS reduced the nuclear abundance of LDH-A, and double mutation more effectively prevented LDH-A nuclear translocation (Figure 2G,H). Taken together, these data demonstrated that canonical Wnt specifically stimulated LDH-A nuclear translocation in an importin β2-dependent manner in HSCs (Figure 2I).
FIGURE 2.
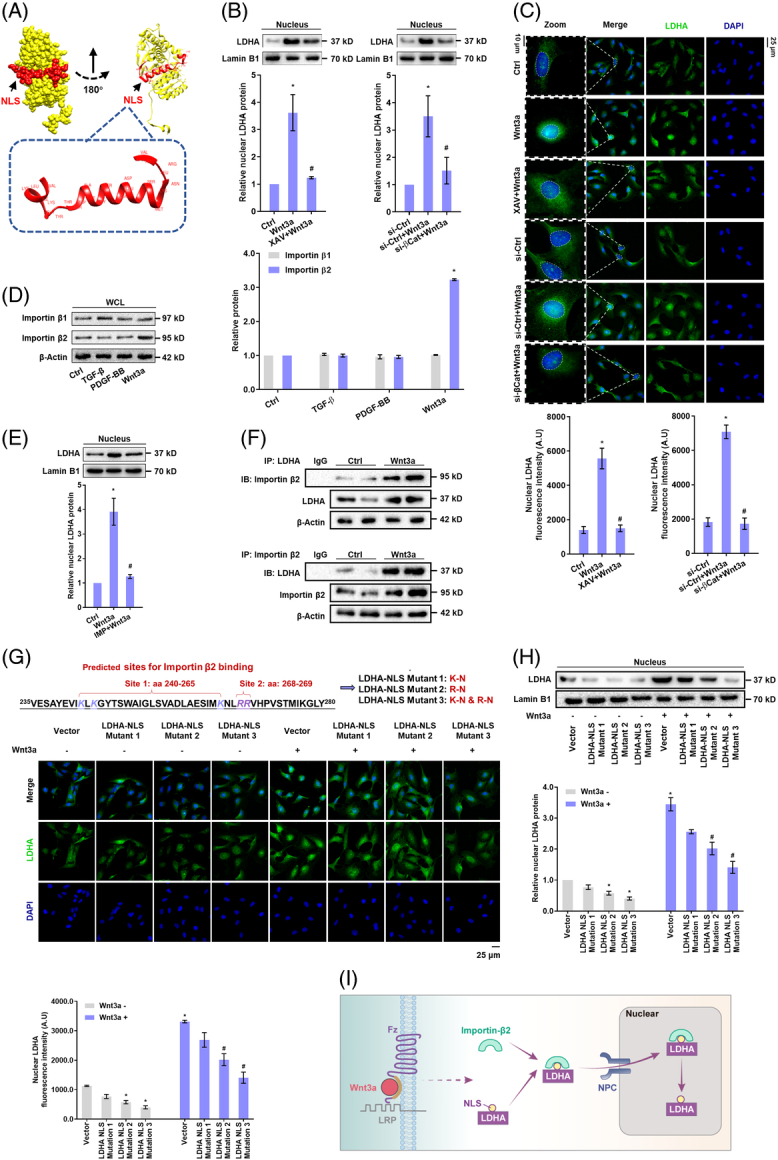
Canonical Wnt signaling specifically stimulates importin β2-dependent nuclear translocation of LDH-A in HSCs. (A) Structure information of predicted NLS of LDH-A using the classical nuclear localization signal mapper. (B) Western blotting analysis of the nuclear abundance of LDH-A with quantification in LX2 cells treated with Wnt3a (100 ng/mL) and/or XAV (10 μM) or transfected with β-catenin siRNA for 24 h. *p<0.05 versus Ctrl, # p<0.05 versus Wnt3a. (C) Immunofluorescence analysis of LDH-A nuclear localization with quantification in LX2 cells treated with Wnt3a (100 ng/mL), and/or XAV (10 μM), or transfected with β-catenin siRNA for 24 h. The nucleus was stained with DAPI. *p<0.05 versus Ctrl, # p<0.05 versus Wnt3a. (D) Western blotting analysis of protein expression of importin β1 and importin β2 with quantification in LX2 cells treated with TGF-β (20 ng/mL), PDGF-BB (20 ng/mL), or Wnt3a (100 ng/mL) for 24 h. *p<0.05 versus Ctrl. (E) Western blotting analysis of nuclear abundance of LDH-A with quantification in LX2 cells treated with Wnt3a (100 ng/mL) and/or importin β inhibitor importazole (IMP, 10 μM) for 24 h. *p<0.05 versus Ctrl, # p<0.05 versus Wnt3a. (F) Co-IP analysis of the interaction between LDH-A and importin β2 in LX2 cells treated with Wnt3a (100 ng/mL) for 24 h. (G) Two possible sites at LDH-A NLS for importin β2 binding were predicted using classical nuclear localization signal mapper and subjected to single or double mutations, and thereby 3 LDH-A NLS mutant plasmids were constructed. Immunofluorescence analysis of LDH-A nuclear localization with quantification in LX2 cells treated with Wnt3a (100 ng/mL) and/or transfected with LDH-A NLS mutant plasmids for 24 h. Nucleus was stained with DAPI. *p<0.05 versus Vector, # p<0.05 versus Vector+Wnt3a. (H) Western blotting analysis of the nuclear abundance of LDH-A with quantification in LX2 cells treated with Wnt3a (100 ng/mL) and/or transfected with LDH-A NLS mutant plasmids for 24 h. *p<0.05 versus Vector, # p<0.05 versus Wnt3a+Vector. (I) Illustration of the regulatory mechanism. Abbreviations: Co-IP, co-immunoprecipitation; LDH-A, lactate dehydrogenase A; NLS, nuclear location signal; siRNA, small interfering RNA.
Direct interaction between LDH-A and HIF-1α stabilizes HIF-1α protein and promotes transactivation of glycolytic genes in HSCs
We next investigated the functions of nuclear LDH-A. LDH-A inhibitor FX-11 or siRNA remarkably abrogated Wnt3a-increased promoter activity of HK2 and PFK1 in HSCs (Supplemental Figure S6A, http://links.lww.com/HEP/H942), suggesting that LDH-A was involved in transactivation of glycolytic enzymes. However, bioinformatics analysis compared the protein sequences of LDH-A and HIF-1α (3 isoforms), showing low similarities between them and excluding the possibility of LDH-A as a transcription factor like HIF-1α (Supplemental Figure S6B, http://links.lww.com/HEP/H942). Interestingly, Wnt3a did not alter HIF-1α mRNA levels (Figure 3A) but remarkably increased its total protein levels and nuclear abundance (Figure 3B,C). Electrophoretic mobility shift assay showed that HIF-1α had more combination with the DNA sequences of HK2 and PFK1 in Wnt3a-treated HSCs (Figure 3D). Importantly, immunofluorescence analyses demonstrated a remarkable co-localization of LDH-A and HIF-1α in the HSC nucleus upon Wnt3a treatment (Figure 3E). Indeed, Co-IP and GST pull-down assays revealed that LDH-A had increased physical connection with HIF-1α following Wnt3a stimulation (Figure 3F,G). Then, we assessed the consequences of this direct interaction. After blockade of protein synthesis by cycloheximide, HIF-1α degradation was accelerated in HSCs with siRNA-mediated depletion of LDH-A but was retarded by ectopic expression of LDH-A (Figure 3H). To address whether the faster reduction of HIF-1α protein in LDH-A–deficient HSCs was associated with its proteasome-dependent degradation, we used the proteasome inhibitor MG132. It remarkably boosted Wnt3a-induced upregulation of HIF-1α protein, which was abrogated by FX-11 or LDH-A siRNA (Figure 3I). The central event in HIF-1α proteasome degradation is the hydroxylation of Pro402 and Pro564 residues by prolyl hydroxylase 2 (PHD2).21 We hence monitored HIF-1α hydroxylation in HSCs pretreated with MG132. FX-11 or LDH-A siRNA restored hydroxyl-HIF-1α levels in the presence or absence of Wnt3a (Figure 3J), indicating that LDH-A maintained HIF-1α stabilization by obstructing hydroxylation-mediated proteasome degradation (Figure 3K).
FIGURE 3.

Direct interaction between LDH-A and HIF-1α stabilizes HIF-1α protein and promotes transactivation of glycolytic genes in HSCs. (A-C) LX2 cells were treated with Wnt3a (100 ng/mL) for 24 h. Real-time PCR analysis of HIF-1α mRNA expression (A); Western blotting analysis of HIF-1α protein abundance in WCL (B) and in nuclear lysates (C) with quantification. *p<0.05 versus Ctrl. (D) EMSA for determining HIF-1α binding to DNA sequences of HK2 and PFK1 in LX2 cells treated with Wnt3a (100 ng/mL) for 24 h. (E) Immunofluorescence analysis for nuclear co-localization of LDH-A and HIF-1α in LX2 cells treated with Wnt3a (100 ng/mL) for 24 h. The nucleus was stained with DAPI. (F) Co-IP analysis of the interaction between LDH-A and HIF-1α in LX2 cells treated with Wnt3a (100 ng/mL) for 24 h. (G) GST pull-down analysis of interaction between LDH-A and HIF-1α in HEK-293T cells transfected with GST-HIF-1α and His-LDH-A, followed by treatment with Wnt3a (100 ng/mL) for 24 h. (H) Western blotting analysis of HIF-1α protein degradation with quantification in LX2 cells transfected with LDH-A siRNA or LDH-A OE plasmid for 24 h, followed by treatment with protein synthesis inhibitor cycloheximide (10 μM) for indicated time durations. *p<0.05 versus Ctrl. (I) Western blotting analysis of HIF-1α protein abundance with quantification in LX2 cells treated with Wnt3a (100 ng/mL), and/or proteasome inhibitor MG132 (10 μM), or LDH-A inhibitor FX-11 (10 μM), or transfected with LDH-A siRNA for 24 h. *p<0.05 versus Ctrl, # p<0.05 versus Wnt3a, & p<0.05 versus Wnt3a+MG132. (J) Western blotting analysis of hydroxyl-HIF-1α protein abundance with quantification in LX2 cells treated with MG132 (10 μM), and/or Wnt3a (100 ng/mL), or FX-11 (10 μM), or transfected with LDH-A siRNA for 24 h. *p<0.05 versus Ctrl, # p<0.05 versus Wnt3a. (K) Illustration of the regulatory mechanism. Abbreviations: Co-IP, co-immunoprecipitation; HIF-1α, hypoxia-inducible factor-1α; HK2, hexokinase 2; LDH-A, lactate dehydrogenase A; OE, overexpression; PFK1, phosphofructokinase 1; siRNA, small interfering RNA.
Subsequently, we accessed the effect of LDH-A/HIF-1α interaction on HSC glycolysis. LDH-A inhibitor FX-11 or targeted siRNA markedly impaired Wnt3a-facilitated HIF-1α nuclear accumulation (Supplemental Figure S6C, http://links.lww.com/HEP/H942), implicating an indispensable role for LDH-A. Furthermore, upregulation of HIF-1α by deferoxamine increased the transcription of HK2 and PFK1 in HSCs with or without LDH-A ectopic expression (Supplemental Figure S6D, http://links.lww.com/HEP/H942), indicating a critical role for HIF-1α here. Knockdown of LDH-A reduced the transcription of HK2 and PFK1 in HSCs with or without deferoxamine-induced HIF-1α upregulation (Supplemental Figure S6E, http://links.lww.com/HEP/H942), suggesting the importance of LDH-A in the current context. We further confirmed the importance of LDH-A nuclear translocation. Double mutation of LDH-A NLS significantly reduced Wnt3a-increased pyruvate and lactate levels (Supplemental Figure S6F, http://links.lww.com/HEP/H942), indicating that the enzymatic activity of LDH-A was also affected by its nuclear translocation. Moreover, Wnt3a-stimulated nuclear accumulation of HIF-1α and transcription of HK2 and PFK1 were eliminated by LDH-A NLS double mutation (Supplemental Figure S6G-J, http://links.lww.com/HEP/H942). In aggregate, these discoveries disclosed that a direct combination of LDH-A to HIF-1α increased the stability of HIF-1α and enhanced its transactivation of glycolytic genes in HSCs.
LDH-A uses its Gly28 residue to interact with HIF-1α, which is required for HIF-1α stabilization and transactivation of glycolytic genes in HSCs
We speculated that the sites at HIF-1α for interaction with PHD2 could be obstructed by LDH-A. We thus further investigated the precise interaction of the LDH-A/HIF-1α complex. Co-IP showed that the N-terminal domain of LDH-A was involved in its physical connection with HIF-1α (Figure 4A). Given that the peptide segment Leu557-Leu574 in the oxygen-dependent degradation domain of HIF-1α is mainly responsible for its direct interaction with PHD2,22 we performed molecular docking and dynamics simulations showing that the segment Leu557-Leu574 of HIF-1α could stably bind to LDH-A (Figure 4B, Supplemental Figure S7A,B, http://links.lww.com/HEP/H942), in which the Pro564 residue was completely sheltered by LDH-A. We next carried out virtual saturation mutations to mutate the interaction interface residues of LDH-A to all essential amino acids individually followed by analysis of mutation energy, indicating that the Gly26 and Gly28 residues could be responsible for LDH-A binding to HIF-1α segment (Supplemental Figure S7C, http://links.lww.com/HEP/H942) and the Gly28 was much closer to the fragment (Supplemental Figure S7D, http://links.lww.com/HEP/H942). To strengthen the results, we subsequently performed virtual alanine scanning by mutating the interaction interface residues of LDH-A to alanine individually, suggesting that the Gly28-to-alanine mutation had the highest mutation energy indicative of considerably decreased binding ability of LDH-A to HIF-1α (Figure 4C). This mutation also generated steric clashes with the Asp569 and Asp570 residues of the HIF-1α segment (Figure 4D). All these results highlighted the critical importance of the Gly28 residue of LDH-A in the current context. Thereafter, we performed functional analyses using Gly28 mutant LDH-A plasmids in HSCs. HIF-1α degradation was increased by ectopic expression of mutant LDH-A (Figure 4E). HSCs with ectopic expression of mutant LDH-A had markedly less nuclear accumulation of HIF-1α (Figure 4F,G). Measurements of glycolysis showed that LDH-A Gly28 mutation reduced the transcription of HK2 and PFK1 (Supplemental Figure S7E,F, http://links.lww.com/HEP/H942), and resultantly decreased lactate production in HSCs (Supplemental Figure S7G, http://links.lww.com/HEP/H942). In addition, LDH-A here did not affect HIF-1α interaction with ARNT or p300 in the HSC nucleus (Supplemental Figure S7H, http://links.lww.com/HEP/H942). Collectively, these results unveiled that LDH-A required its Gly28 residue to directly interact with HIF-1α, which was vital for HIF-1α stabilization and transactivation of glycolytic genes in HSCs.
FIGURE 4.

LDH-A uses its Gly28 residue to interact with HIF-1α, which is required for HIF-1α stabilization and transactivation of glycolytic genes in HSCs. (A) Co-IP analysis of interaction between HIF-1α and LDH-A in HEK-293T cells transfected with HA-labeled HIF-1α plasmid and Flag-tagged N-terminal (LDH-A-N), C-terminal (LDH-A-C) and full length (LDH-A-FL) of plasmids of LDH-A. (B) Molecular docking and dynamics simulation of HIF-1α/LDH-A complex using the ZDOCK modular of Discovery Studios 2019. The peptide segment Leu557-Leu574 of HIF-1α was docked to LDH-A protein structure, and the direct interaction residues were illustrated. (C) Virtual alanine scanning was performed by mutating all the amino acid residues of LDH-A to alanine individually, and mutation energy was calculated to indicate the binding ability between LDH-A and HIF-1α. (D) Spatial conformation illustration of the interaction between the Gly28-to-alanine mutant LDH-A and the segment Leu557-Leu574 of HIF-1α, showing the presence of steric clashes between the Ala28 residue of LDH-A and the Asp569 and Asp570 residues of HIF-1α segment. (E) Western blotting analysis of HIF-1α protein degradation with quantification in LX2 cells transfected with LDH-A wide type plasmid or LDH-A Gly28 mutant plasmid for 24 h, followed by treatment with cycloheximide (10 μM) for indicated time durations. *p<0.05 versus LDH-A wide type plasmid. (F, G) LX2 cells were transfected with LDH-A wide type plasmid or LDH-A Gly28 mutant plasmid for 24 h. Western blotting analysis of the nuclear abundance of HIF-1α with quantification (F); Immunofluorescence analysis of HIF-1α nuclear translocation with quantification, and nucleus was stained with DAPI (G). *p<0.05 versus LDH-A WT plasmid. Abbreviations: Co-IP, co-immunoprecipitation; HA, hyaluronic acid; HIF-1α, hypoxia-inducible factor-1α; LDH-A, lactate dehydrogenase A.
LDH-A-mediated aerobic glycolysis is required for HSC activation and liver fibrosis
We subsequently tested the role of LDH-A in Wnt-driven HSC activation. LDH-A inhibitor FX-11 or targeted siRNA abolished Wnt3a-enhanced glucose uptake and consumption and lactate production in HSCs (Supplemental Figure S8A-C, http://links.lww.com/HEP/H942). Extracellular acidification rate was dropped and OCR was restored in HSCs treated with FX-11 or transfected with LDH-A siRNA (Figure 5A,B). The transcription and expression of HK2 and PFK1 were diminished by inhibition or knockdown of LDH-A in Wnt3a-stimulated HSCs (Supplemental Figure S8D,E, http://links.lww.com/HEP/H942). These observations demonstrated LDH-A as a critical mediator for canonical Wnt to sustain the glycolytic phenotype of HSCs. We then monitored HSC activation. HSC viability enhanced by Wnt3a was lowered by glycolysis inhibitor 2-DG, LDH-A inhibitor FX-11, or siRNA-mediated LDH-A depletion but was reinforced by ectopic expression of LDH-A (Supplemental Figure S8F, http://links.lww.com/HEP/H942). The mRNA and protein levels of α-smooth muscle actin (α-SMA) and collagen 1α1 (COL1α1), 2 well-established HSC activation markers, were decreased by 2-DG, FX-11, or LDH-A siRNA, but were further increased by LDH-A overexpression in Wnt3a-treated HSCs (Figure 5C,D). Besides, blockade of glycolysis or LDH-A abolished Wnt3a-induced HSC contraction, but LDH-A overexpression intensified Wnt3a-enhanced HSC contraction (Supplemental Figure S8G, http://links.lww.com/HEP/H942). In parallel, we evaluated the effects of rotenone, a mitochondrial electron transport chain complex I inhibitor, on HSC activation. Wnt3a-increased viability, activation maker expression, and contraction were not apparently affected by rotenone (Figure 5C,D, Supplemental Figure S8F,G, http://links.lww.com/HEP/H942). To test the contribution of LDH-A/HIF-1α complex to HSC activation, Gly28 mutant LDH-A plasmids were transfected into HSCs, which decreased the mRNA expression of α-SMA and COL1α1 (Figure 5E). Furthermore, we validated the correlation between β-catenin and LDH-A during primary HSC activation. The fresh primary HSCs from healthy mice were spontaneously activated as evidenced by increased expression of α-SMA, concomitant with upregulation of β-catenin expression during culture for different days (Figure 5F,G). The mRNA and protein levels of HK2, PFK1, and LDH-A were all increased during HSC activation (Figure 5H,I). Besides, the nuclear LDH-A and intracellular lactate levels were increasingly upregulated during this process (Figure 5J,K). Overall, these discoveries revealed that LDH-A-mediated aerobic glycolysis was necessary for canonical Wnt-induced HSC activation.
FIGURE 5.
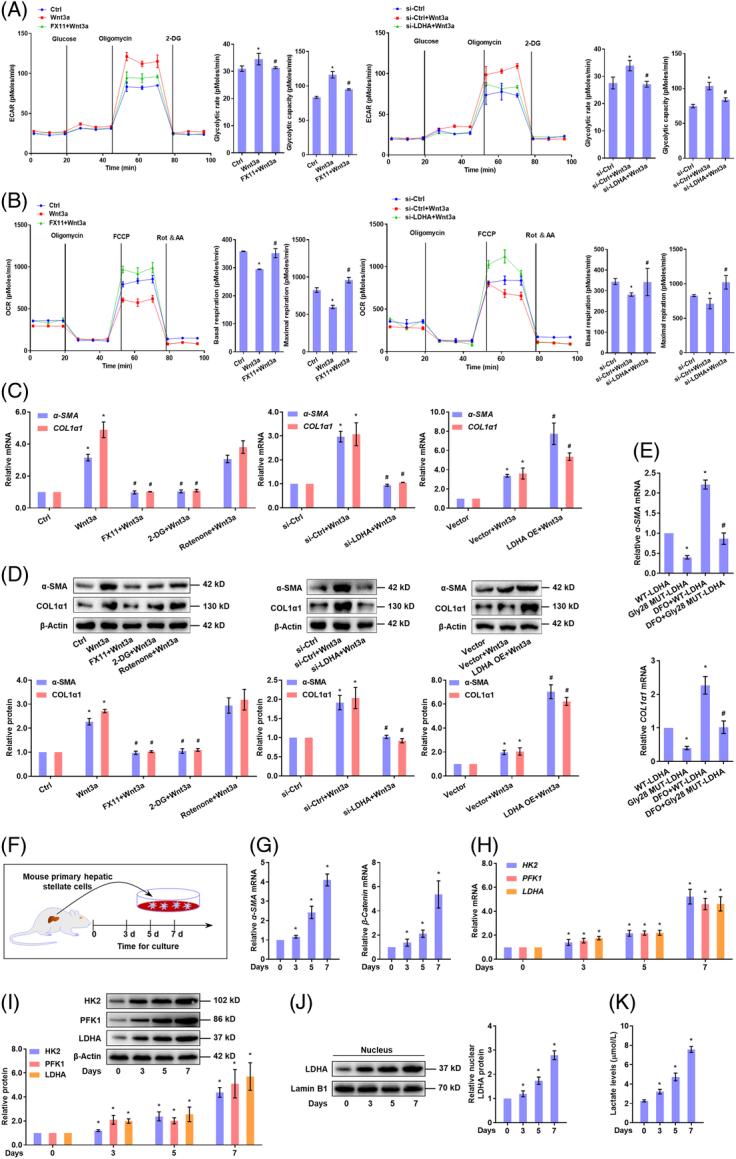
LDH-A-mediated aerobic glycolysis is required for canonical Wnt-induced HSC activation. (A, B) LX2 cells were treated with Wnt3a (100 ng/mL), and/or FX-11 (10 μM), or transfected with LDH-A siRNA for 24 h. Measurement of ECAR with the calculation of glycolytic rate and glycolytic capacity (A); Measurement of OCR with the calculation of basal respiratory and maximal respiratory capacities (B). *p<0.05 versus Ctrl, # p<0.05 versus Wnt3a. (C, D) LX2 cells were treated with Wnt3a (100 ng/mL), and/or FX-11(10 μM) or glycolysis inhibitor 2-DG (10 μM) or mitochondrial electron transport inhibitor rotenone (10 μM), or transfected with LDH-A siRNA or LDH-A OE plasmid for 24 h. Real-time PCR analysis of mRNA expression of α-SMA and COL1α1 (C); western blotting analysis of protein expression of α-SMA and COL1α1 with quantification (D). *p<0.05 versus Ctrl, # p<0.05 versus Wnt3a. (E) Real-time PCR analysis of mRNA expression of α-SMA and COL1α1 in LX2 cells transfected with Gly28 mutant LDH-A plasmid and/or treated with HIF-1α activator DFO (10 μM) for 24 h. *p<0.05 versus WT-LDH-A, # p<0.05 versus DFO+WT-LDH-A. (F–K) Freshly isolated mouse primary HSCs were cultured for indicated time durations. Real-time PCR analysis of mRNA expression of α-SMA and β-catenin (G), and glycolytic genes HK2, PFK1, and LDH-A (H) Western blotting analysis of protein abundance of glycolytic enzymes HK2, PFK1, and LDH-A in WCL (I), and nuclear abundance of LDH-A with quantification (J) Measurement of intracellular lactate levels (K). *p<0.05 versus 0 day. Abbreviations: α-SMA, α-smooth muscle actin; COL1α1, collagen 1α1; DFO, deferoxamine; ECAR, extracellular acidification rate; HK2, hexokinase 2; LDH-A, lactate dehydrogenase A; OCR, oxygen consumption rate; OE, overexpression; PFK1, phosphofructokinase 1; siRNA, small interfering RNA; WCL, whole cell lysates.
To establish the in vivo relevance, mice were intravenously administrated vitamin A-liposome-LDH-A shRNA, accompanied by CCl4-induced liver fibrosis (Figure 6A), which caused selective knockdown of LDH-A in primary HSCs (Supplemental Figure S9A, http://links.lww.com/HEP/H942). Using this model, we observed that HSC-specific deficiency of LDH-A mitigated hepatic injury and collagen deposition and reduced hepatic hydroxyproline contents in mouse fibrotic liver (Figure 6B,C), concomitant with a decrease in α-SMA-positive HSCs (Supplemental Figure S9B, http://links.lww.com/HEP/H942). Serum biochemical parameters were also decreased in the mice with fibrosis with HSC-specific LDH-A depletion (Supplemental Figure S9C, http://links.lww.com/HEP/H942). Consistent with the findings in culture, LDH-A co-localization with HIF-1α or importin β2 was observed in mouse HSCs, evidenced by immunohistofluorescence analysis (Figure 6D). Next, primary HSCs were isolated for examining fibrotic and glycolytic molecules. LDH-A depletion decreased the mRNA and protein levels of α-SMA and COL1α1 in mouse primary HSCs (Supplemental Figure S9D,E, http://links.lww.com/HEP/H942). The nuclear abundance of LDH-A and HIF-1α was diminished in the primary HSCs from LDH-A–deficient mice with fibrosis (Figure 6E). The expression of HK2 and PFK1 was also downregulated by LDH-A deficiency in the primary HSCs (Figure 6F,G), concomitant with reduced intracellular levels of lactate and ATP (Figure 6H,I). In addition, 13C metabolic flux analyses were performed with primary HSCs, showing that glycolytic flux was remarkably enhanced in fibrotic HSCs, which was reduced by LDH-A deficiency (Supplemental Figure S10, http://links.lww.com/HEP/H942). Altogether, these data highlighted a key role of LDH-A in HSC activation and liver fibrosis.
FIGURE 6.
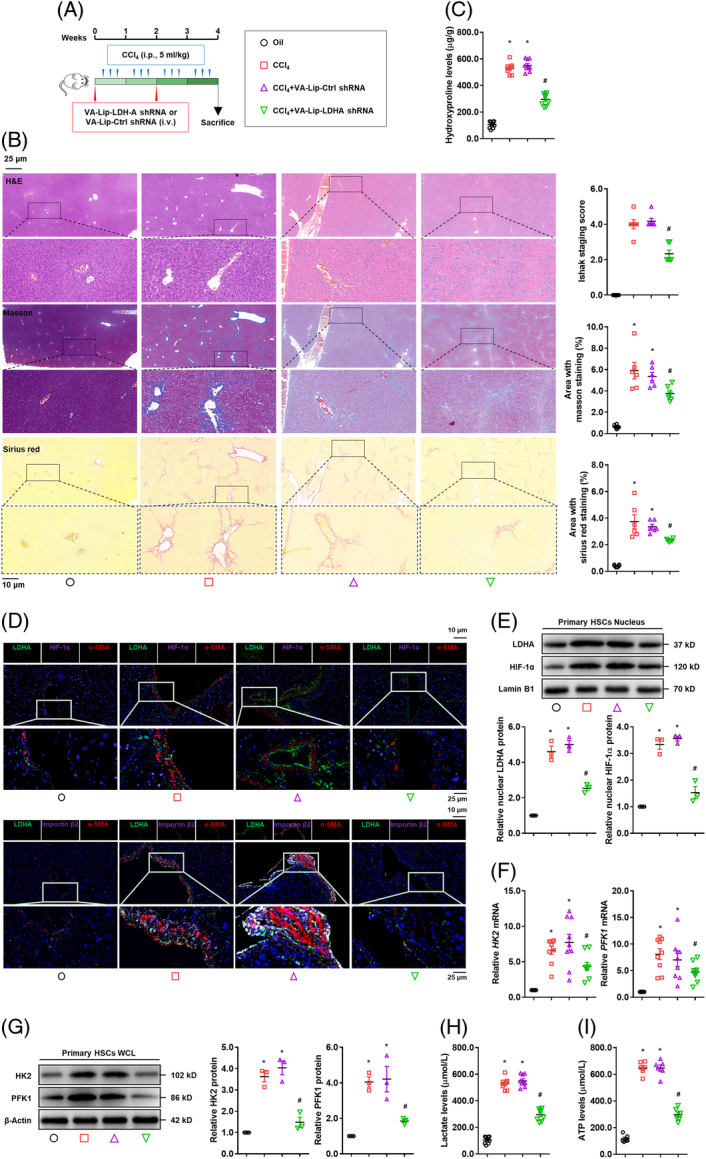
Hepatic-specific deficiency of LDH-A mitigates liver fibrosis and inhibits HSC glycolysis. Mice were intravenously administrated vitamin A-liposome-LDH-A shRNA accompanied by CCl4-induced liver fibrosis (n = 10). (A) Scheme of the experiments. (B) Examinations of liver histopathology and collagens using HE staining, Masson staining, and Sirius red staining with Ishak stage scoring and quantification of positive staining area. (C) Measurement of hepatic hydroxyproline contents. (D) Immunofluorescence analysis of expression and localization of LDH-A, HIF-1α, importin β2, and α-SMA in liver tissues. The nucleus was stained with DAPI. (E–I) Mouse-primary HSCs were isolated for molecular examinations. Western blotting analysis of nuclear abundance of LDH-A and HIF-1α with quantification (E); real-time PCR analysis of mRNA expression of HK2 and PFK1 (F); Western blotting analysis of protein expression of HK2 and PFK1 with quantification (G); measurement of intracellular lactate levels (H), and intracellular ATP levels (I). For this figure, *p<0.05 versus oil, # p<0.05 versus CCl4+VA-Lip-Ctrl shRNA. Abbreviations: α-SMA, α-smooth muscle actin; HIF-1α, hypoxia-inducible factor-1α; HK2, hexokinase 2; LDH-A, lactate dehydrogenase A; PFK1, phosphofructokinase 1; shRNA, short hairpin RNA.
Pharmacological inhibition of β-catenin reduces HSC glycolysis and mitigates liver fibrosis through blocking LDH-A/HIF-1α complex in mice
To further intensify the pharmacological implication of current findings, mice with CCl4-induced liver fibrosis were orally treated with β-catenin inhibitor XAV with HSC-targeted LDH-A overexpression (Figure 7A). XAV improved liver histology and collagen deposition and reduced hepatic hydroxyproline contents accompanied by a reduction in α-SMA-positive HSCs in mice with fibrosis, but HSC-specific overexpression of LDH-A considerably abrogated these effects (Figure 7B,C, Supplemental Figure S11A, http://links.lww.com/HEP/H942). Similar alterations were recaptured in serum levels of hepatic injury and fibrotic markers (Supplemental Figure S11B, http://links.lww.com/HEP/H942). LDH-A co-localization with HIF-1α or importin β2 in fibrotic HSCs was inhibited by XAV, which was abolished by LDH-A overexpression (Figure 7D). Furthermore, the expression of α-SMA and COL1α1 was considerably decreased in the primary HSCs from mice with fibrosis treated with XAV, which were rescued by HSC-specific overexpression of LDH-A (Supplemental Figure S11C,D, http://links.lww.com/HEP/H942). In addition, the nuclear abundance of LDH-A and HIF-1α and the expression of HK2 and PFK1 were all downregulated in the primary HSCs from XAV-treated mice with fibrosis, whereas their downregulation was reversed by HSC-specific LDH-A overexpression (Figure 7E-G). Similar changes were observed in the intracellular levels of lactate and ATP in primary HSCs (Figure 7H,I). These observations disclosed that LDH-A was a critical mediator for the pharmacological inhibition of β-catenin to reduce liver fibrosis in vivo.
FIGURE 7.
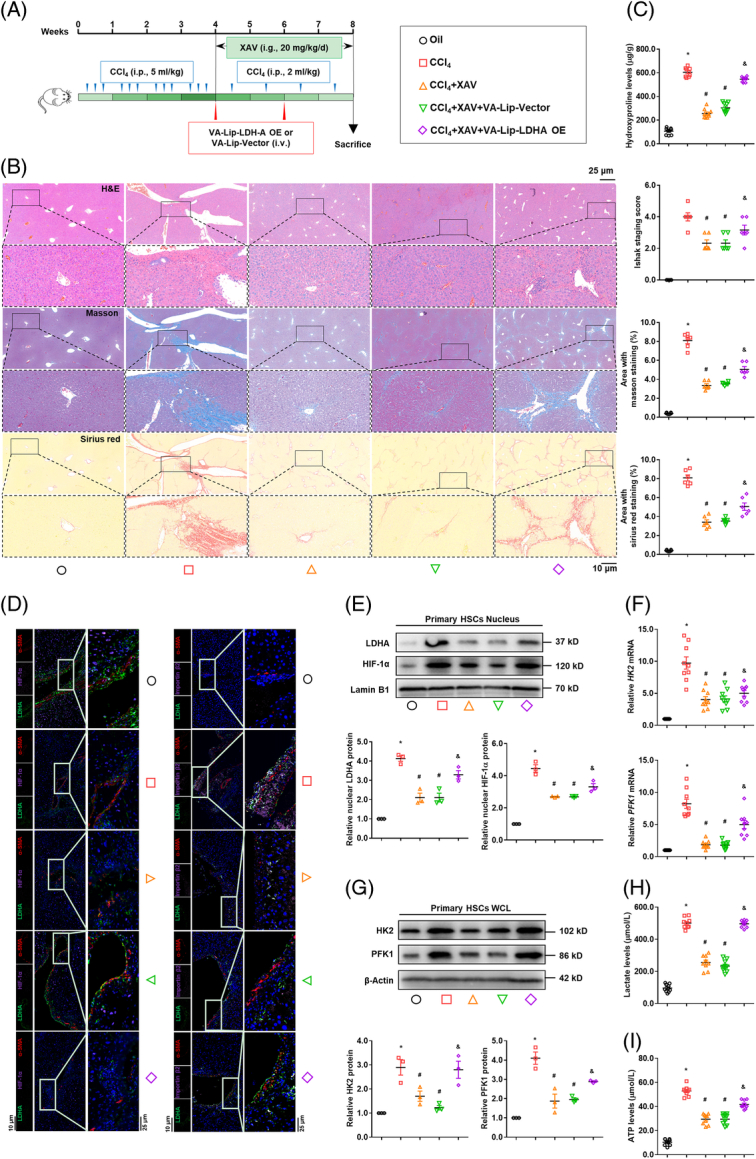
Pharmacological inhibition of β-catenin attenuates liver fibrosis and reduces HSC glycolysis through blocking LDH-A/HIF-1α complex in mice. Mice with CCl4-induced liver fibrosis were orally treated with β-catenin inhibitor XAV (20 mg/kg/day) accompanied by i.v. administration with vitamin A-liposome-LDH-A overexpression plasmid (n=10). (A) Scheme of the experiments. (B) Examinations of liver histopathology and collagens using HE staining, Masson staining, and Sirius red staining with Ishak stage scoring and quantification of positive staining area. (C) Measurement of hepatic hydroxyproline contents. (D) Immunofluorescence analysis of expression and localization of LDH-A, HIF-1α, importin β2, and α-SMA in liver tissues. The nucleus was stained with DAPI. (E-I) Mouse primary HSCs were isolated for molecular examinations. Western blotting analysis of the nuclear abundance of LDH-A and HIF-1α with quantification (E); Real-time PCR analysis of mRNA expression of HK2 and PFK1 (F); Western blotting analysis of the protein expression of HK2 and PFK1 with quantification (G); Measurement of intracellular lactate levels (H); and intracellular ATP levels (I). For this figure, *p<0.05 versus Oil, # p<0.05 versus CCl4, & p<0.05 versus CCl4 +XAV+ VA-Lip-Vector. Abbreviations: α-SMA, α-smooth muscle actin; HIF-1α, hypoxia-inducible factor-1α; HK2, hexokinase 2; LDH-A, lactate dehydrogenase A; PFK1, phosphofructokinase 1.
We finally examined hepatic biopsy samples of patients with fibrosis. The hepatic lactate levels in cirrhotic liver (Metavir F4) were significantly higher than that in mild fibrotic liver (Metavir F1) (Figure 8A). β-catenin mRNA was markedly increased in cirrhotic liver and was positively correlated with the considerable upregulation of mRNA of HK2, PFK1, and LDH-A (Figure 8B). Dual immunofluorescence analysis revealed an increasing amount of α-SMA-positive HSCs with these glycolytic molecules in the human cirrhotic liver (Figure 8C). These results collectively validated the clinical relevance of current findings.
FIGURE 8.

β-Catenin positively correlates with LDH-A and HSC glycolysis in the human fibrotic liver. Hepatic biopsy samples from patients with mild fibrosis (Metavir F1, n=8) and cirrhosis (Metavir F4, n=11) were used for examinations. (A) Measurement of hepatic lactate levels. *p<0.05 versus Metavir F1. (B) Real-time PCR analysis of mRNA expression of β-catenin and glycolytic genes. *p<0.05 versus Metavir F1. The correlations between β-catenin and HK2, PFK1, or LDH-A in the cirrhotic liver were analyzed using Pearson correlation test, and the correlation coefficients (r) and p values were calculated. (C) Immunofluorescence analysis of expression and localization of α-SMA and HK2, PFK1, or LDH-A in liver tissues. The nucleus was stained with DAPI. (D) Scheme of the molecular mechanisms underlying how HSC glycolysis reprogramming is driven and maintained through a novel positive feedback regulatory pathway embedded in glycolysis requiring a transcriptional loop of LDH-A/HIF-1α. Abbreviations: α-SMA, α-smooth muscle actin; HK2, hexokinase 2; LDH-A, lactate dehydrogenase A; PFK1, phosphofructokinase 1.
DISCUSSION
Wnt can interplay with metabolic cascades,23 but little is known about its role in HSC glucose metabolism. We speculated that Wnt was a prime candidate for directing HSC aerobic glycolysis. This speculation was supported by the high glycolytic phenotype of HSCs induced by Wnt3a. We also unveiled the critical involvement of β-catenin in the current setting and substantiated the positive correlation between aberrant β-catenin activation and increased aerobic glycolysis in mouse primary HSC activation and human fibrotic liver. We confirmed the concept that the same pro-fibrogenic pathways that potentiate HSC activation must also favor metabolic pathways that support this process.
We identified that HK2, PFK1, and LDH-A have different transcription patterns in Wnt3a-treated HSCs, suggesting an acute regulation of LDH-A and a relatively long regulatory loop of the other 2 molecules. Indeed, LDH-A was characterized to be a direct target gene of the β-catenin/TCF4 complex. This novel finding explicated the previous ambiguous concept that β-catenin activation was associated with LDH-A expression under some pathophysiological circumstances.24–26 Intriguingly, we disclosed that LDH-A specifically translocated into HSC nucleus upon Wnt activation. Nuclear pores are tunnels that allow small proteins (usually below 40 kDa) to diffuse in while forcing large molecules to employ an active transport system to pass. The initial step of this facilitative system is the recognition of NLS on nuclear cargos by carrier proteins such as importins to form a transport complex.27 However, the classical NLS is neither necessary nor sufficient in and of themselves in all cases to effectively transport a protein into the nucleus.28 Here, LDH-A has a permissible molecular weight, and Wnt upregulation of LDH-A protein might generate a concentration gradient between cytoplasm and nucleus, presumably urging LDH-A nuclear shuttling. Furthermore, importin β2 was found to be required for this process, an observation consistent with the notion that importin β2-binding motifs have a greater diversity than classical NLS motifs that bind to a complex of importin α and importin β1.29 Further evidence confirmed that the predicted non-classical NLS was indispensable for Wnt3a-driven LDH-A nuclear entry in HSCs, indicating an active and finely regulated mechanism in this process.
HIF-1α is a master transcriptional regulator of glycolysis through transactivating many glycolytic genes.22 Here, LDH-A is directly bound to and stabilized HIF-1α protein, followed by transactivation of HK2 and PFK1 in HSCs. We inferred that the presence of NLS in LDH-A robustly assisted the nuclear transport of the LDH-A/HIF-1α complex and that the nuclear LDH-A served as a transcription co-activator of HIF-1α. We unveiled a novel moonlighting function of LDH-A for the regulation of HIF-1α biology implicated in aerobic glycolysis. Interestingly, the nuclear LDH-A here played a transcription regulatory role, which was different from Liu’s observation that LDH-A gained an alternative enzymatic activity to produce α-hydroxybutyrate in the nucleus of cervical cancer cells.30 Based on these results, we explained the different transcription patterns between HK2, PFK1, and LDH-A as follows. (1) The accumulated LDH-A formed a complex with HIF-1α, entering the nucleus and transactivating HK2 and PFK1, and resultantly sustaining the increase in their mRNA levels. (2) LDH-A is also transcriptionally upreguated by HIF-1α.31,32 There could be a positive feedback loop between LDH-A and HIF-1α. The LDH-A/HIF-1α complex continuously transactivated LDH-A and thus maintained the high transcript levels of LDH-A during Wnt3a stimulation.
Molecular simulation indicated that LDH-A used its N-terminal Gly28 residue for the physical connection, which was supported by the Co-IP evidence that HIF-1α bound to the N-terminal domain of LDH-A. Site-directed mutation analyses underscored this residue as a requisite for LDH-A/HIF-1α regulation of HSC aerobic glycolysis. In addition, the HIF-1α protein contains several functional domains, among which the oxygen-dependent degradation (ODD) domain is critical for HIF-1α stability and controlled by an oxygen-sensitive prolyl hydroxylase that can hydroxylate the Pro402 and Pro564 residues in the ODD domain under normoxic conditions.21 The hydroxylated HIF-1α then interacts with VHL and enables itself to be ubiquitinated, triggering proteasome degradation.33 Here, our molecular simulation revealed that LDH-A is bound to the ODD domain of HIF-1α, contributing to HIF-1α stabilization. This was substantiated by the experiments using proteasome inhibitors and examining hydroxylated HIF-1α, suggesting that LDH-A masked the ODD domain, acting as a molecular barrier to prevent HIF-1α from ubiquitination. These results confirmed the recognition that the ODD domain is also necessary for an oxygen-independent degradation pathway.34 Our discovered mechanism was different from that found in breast cancer cells, where direct interaction with PHD2 was required for lncRNA HISLA to stabilize HIF-1α.35 Interestingly, β-catenin directly interacted with the N-terminal domain of HIF-1α in hypoxia colorectal cancer cells, inhibiting the transcriptional activity of TCF4.36 Here in Wnt3a-activated HSCs, the β-catenin/HIF-1α interaction might not occur because TCF4 still maintained its transcriptional function. Besides, the β-catenin/TCF4 complex induced the expression of VHL, a ubiquitin ligase facilitating hydroxylated HIF-1α degradation, and hence suppressed HIF-1α activity in colorectal cancer cells.37 This might not be true in the current scenario because LDH-A obstructed the hydroxylation sites of HIF-1α, preventing VHL-mediated ubiquitination, despite the possible upregulation of VHL induced by β-catenin/TCF4 complex. Here, the nonenzymatic moonlighting function of LDH-A regulated the oxygen-independent posttranslational modification and transcription capacity of HIF-1α implicated in HSC glycolytic metabolism.
In summary, canonical Wnt signaling promoted HSC activation through enhanced aerobic glycolysis, requiring LDH-A to use its nonenzymatic function to directly interact with HIF-1α and stabilize it, followed by nuclear shuttling and transactivation of glycolytic genes (Figure 8E). HSCs-targeted inhibition of β-catenin and/or LDH-A may produce potent therapeutic efficacy against liver fibrosis.
Supplementary Material
Acknowledgments
DATA AVAILABILITY STATEMENT
RNA sequencing data that support the findings of this study are available from the corresponding author upon reasonable request.
AUTHOR CONTRIBUTIONS
Feixia Wang, Shizhong Zheng and Feng Zhang conceived the study. Feixia Wang, Li Chen, Desong Kong, Siwei Xia, Baoyu Liang, Yang Li, and Ya Zhou conducted experiments and analyzed the data. Xiaojin Zhang performed computational molecular simulation. Zili Zhang and Jiangjuan Shao provided assistance in some experiments. Feixia Wang and Feng Zhang wrote and revised the manuscript.
FUNDING INFORMATION
This work was financially supported by the National Natural Science Foundation of China (82173874, 82073914), the Qing Lan Project of Jiangsu Higher Institutions (Young and Middle-Aged Academic Leader), and the Open Project of Chinese Materia Medica First-Class Discipline of Nanjing University of Chinese Medicine (2020YLKX022, 2020YLKX023).
CONFLICTS OF INTEREST
The authors have no conflicts to report.
Footnotes
Abbreviations: α-SMA, α-smooth muscle actin; CCl4, carbon tetrachloride; Co-IP, co-immunoprecipitation; COL1α1, collagen 1α1; DFO, deferoxamine; ECAR, extracellular acidification rate; HA, hyaluronic acid; HIF-1α, hypoxia-inducible factor-1α; HK2, hexokinase 2; LDH-A, lactate dehydrogenase A; NLS, nuclear location signal; OCR, oxygen consumption rate; ODD, oxygen-dependent degradation; OE, overexpression; PFK1, phosphofructokinase 1; PHD2, prolyl hydroxylase 2; shRNA, short hairpin RNA; siRNA, small interfering RNA; TCF, T-cell factor; WCL, whole cell lysates.
Supplemental Digital Content is available for this article. Direct URL citations are provided in the HTML and PDF versions of this article on the journal’s website, www.hepjournal.com.
Contributor Information
Feixia Wang, Email: feixiawang526@163.com.
Li Chen, Email: nucmcl@163.com.
Desong Kong, Email: kongds0675@126.com.
Xiaojin Zhang, Email: zhilictfw@163.com.
Siwei Xia, Email: 1473751931@qq.com.
Baoyu Liang, Email: 312347814@qq.com.
Yang Li, Email: 1536766094@qq.com.
Ya Zhou, Email: 2738614096@qq.com.
Zili Zhang, Email: 543843183@qq.com.
Jiangjuan Shao, Email: 1084645255@qq.com.
Shizhong Zheng, Email: nytws@njucm.edu.cn.
Feng Zhang, Email: zhangfeng2013@njucm.edu.cn.
REFERENCES
- 1. Lackner C, Tiniakos D. Fibrosis and alcohol-related liver disease. J Hepatol. 2019;70:294–304. [DOI] [PubMed] [Google Scholar]
- 2. Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. 2017;14:397–411. [DOI] [PubMed] [Google Scholar]
- 3. Higashi T, Friedman SL, Hoshida Y. Hepatic stellate cells as key target in liver fibrosis. Adv Drug Deliv Rev. 2017;121:27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hou W, Syn WK. Role of Metabolism in Hepatic Stellate Cell Activation and Fibrogenesis. Front Cell Dev Biol. 2018;6:150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. 2011;27:441–464. [DOI] [PubMed] [Google Scholar]
- 6. Feng Y, Xiong Y, Qiao T, Li X, Jia L, Han Y. Lactate dehydrogenase A: A key player in carcinogenesis and potential target in cancer therapy. Cancer Med. 2018;7:6124–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen Y, Choi SS, Michelotti GA, Chan IS, Swiderska-Syn M, Karaca GF, et al. Hedgehog controls hepatic stellate cell fate by regulating metabolism. Gastroenterology. 2012;143:1319–29 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Talekar M, Boreddy SR, Singh A, Amiji M. Tumor aerobic glycolysis: new insights into therapeutic strategies with targeted delivery. Expert Opin Biol Ther. 2014;14:1145–1159. [DOI] [PubMed] [Google Scholar]
- 9. Russell JO, Monga SP. Wnt/beta-Catenin Signaling in Liver Development, Homeostasis, and Pathobiology. Annu Rev Pathol. 2018;13:351–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Perugorria MJ, Olaizola P, Labiano I, Esparza-Baquer A, Marzioni M, Marin JJG, et al. Wnt-beta-catenin signalling in liver development, health and disease. Nat Rev Gastroenterol Hepatol. 2019;16:121–36. [DOI] [PubMed] [Google Scholar]
- 11. Mederacke I, Dapito DH, Affo S, Uchinami H, Schwabe RF. High-yield and high-purity isolation of hepatic stellate cells from normal and fibrotic mouse livers. Nat Protoc. 2015;10:305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang F, Hao M, Jin H, Yao Z, Lian N, Wu L, et al. Canonical hedgehog signalling regulates hepatic stellate cell-mediated angiogenesis in liver fibrosis. Br J Pharmacol. 2017;174:409–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lian N, Jiang Y, Zhang F, Jin H, Lu C, Wu X, et al. Curcumin regulates cell fate and metabolism by inhibiting hedgehog signaling in hepatic stellate cells. Lab Invest. 2015;95:790–803. [DOI] [PubMed] [Google Scholar]
- 14. Percie du Sert N, Hurst V, Ahluwalia A, Alam S, Avey MT, Baker M, et al. The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. Br J Pharmacol. 2020;177:3617–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sato Y, Murase K, Kato J, Kobune M, Sato T, Kawano Y, et al. Resolution of liver cirrhosis using vitamin A-coupled liposomes to deliver siRNA against a collagen-specific chaperone. Nat Biotechnol. 2008;26:431–442. [DOI] [PubMed] [Google Scholar]
- 16. Wang F, Li Z, Chen L, Yang T, Liang B, Zhang Z, et al. Inhibition of ASCT2 induces hepatic stellate cell senescence with modified proinflammatory secretome through an IL-1alpha/NF-kappaB feedback pathway to inhibit liver fibrosis. Acta Pharm Sin B. 2022;12:3618–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wu X, Ma Y, Shao F, Tan Y, Tan T, Gu L, et al. CUG-binding protein 1 regulates HSC activation and liver fibrogenesis. Nat Commun. 2016;7:13498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shim H, Dolde C, Lewis BC, Wu CS, Dang G, Jungmann RA, et al. c-Myc transactivation of LDH-A: implications for tumor metabolism and growth. Proc Natl Acad Sci U S A. 1997;94:6658–6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lewis BC, Prescott JE, Campbell SE, Shim H, Orlowski RZ, Dang CV. Tumor induction by the c-Myc target genes rcl and lactate dehydrogenase A. Cancer Res. 2000;60:6178–6183. [PubMed] [Google Scholar]
- 20. Li L, Liang Y, Kang L, Liu Y, Gao S, Chen S, et al. Transcriptional Regulation of the Warburg Effect in Cancer by SIX1. Cancer Cell. 2018;33:368–85 e7. [DOI] [PubMed] [Google Scholar]
- 21. Bruick RK. Oxygen sensing in the hypoxic response pathway: regulation of the hypoxia-inducible transcription factor. Genes Dev. 2003;17:2614–2623. [DOI] [PubMed] [Google Scholar]
- 22. Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148:399–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sethi JK, Vidal-Puig A. Wnt signalling and the control of cellular metabolism. Biochem J. 2010;427:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vallee A, Lecarpentier Y, Guillevin R, Vallee JN. Aerobic Glycolysis Hypothesis Through WNT/Beta-Catenin Pathway in Exudative Age-Related Macular Degeneration. J Mol Neurosci. 2017;62:368–79. [DOI] [PubMed] [Google Scholar]
- 25. Chafey P, Finzi L, Boisgard R, Cauzac M, Clary G, Broussard C, et al. Proteomic analysis of beta-catenin activation in mouse liver by DIGE analysis identifies glucose metabolism as a new target of the Wnt pathway. Proteomics. 2009;9:3889–3900. [DOI] [PubMed] [Google Scholar]
- 26. Vallee A, Vallee JN. Warburg effect hypothesis in autism Spectrum disorders. Mol Brain. 2018;11:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stewart M. Molecular mechanism of the nuclear protein import cycle. Nat Rev Mol Cell Biol. 2007;8:195–208. [DOI] [PubMed] [Google Scholar]
- 28. Lange A, Mills RE, Lange CJ, Stewart M, Devine SE, Corbett AH. Classical nuclear localization signals: definition, function, and interaction with importin alpha. J Biol Chem. 2007;282:5101–5105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen A, Akhshi TK, Lavoie BD, Wilde A. Importin beta2 Mediates the Spatio-temporal Regulation of Anillin through a Noncanonical Nuclear Localization Signal. J Biol Chem. 2015;290:13500–13509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liu Y, Guo JZ, Liu Y, Wang K, Ding W, Wang H, Liu X, et al. Nuclear lactate dehydrogenase A senses ROS to produce alpha-hydroxybutyrate for HPV-induced cervical tumor growth. Nat Commun. 2018;9:4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Firth JD, Ebert BL, Ratcliffe PJ. Hypoxic regulation of lactate dehydrogenase A. Interaction between hypoxia-inducible factor 1 and cAMP response elements. J Biol Chem. 1995;270:21021–21027. [DOI] [PubMed] [Google Scholar]
- 32. Ebert BL, Bunn HF. Regulation of transcription by hypoxia requires a multiprotein complex that includes hypoxia-inducible factor 1, an adjacent transcription factor, and p300/CREB binding protein. Mol Cell Biol. 1998;18:4089–4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lisy K, Peet DJ. Turn me on: regulating HIF transcriptional activity. Cell Death Differ. 2008;15:642–649. [DOI] [PubMed] [Google Scholar]
- 34. Kong X, Alvarez-Castelao B, Lin Z, Castano JG, Caro J. Constitutive/hypoxic degradation of HIF-alpha proteins by the proteasome is independent of von Hippel Lindau protein ubiquitylation and the transactivation activity of the protein. J Biol Chem. 2007;282:15498–15505. [DOI] [PubMed] [Google Scholar]
- 35. Chen F, Chen J, Yang L, Liu J, Zhang X, Zhang Y, et al. Extracellular vesicle-packaged HIF-1alpha-stabilizing lncRNA from tumour-associated macrophages regulates aerobic glycolysis of breast cancer cells. Nat Cell Biol. 2019;21:498–510. [DOI] [PubMed] [Google Scholar]
- 36. Kaidi A, Williams AC, Paraskeva C. Interaction between beta-catenin and HIF-1 promotes cellular adaptation to hypoxia. Nat Cell Biol. 2007;9:210–217. [DOI] [PubMed] [Google Scholar]
- 37. Giles RH, Lolkema MP, Snijckers CM, Belderbos M, van der Groep P, Mans DA, et al. Interplay between VHL/HIF1alpha and Wnt/beta-catenin pathways during colorectal tumorigenesis. Oncogene. 2006;25:3065–3070. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
RNA sequencing data that support the findings of this study are available from the corresponding author upon reasonable request.