

Abstract
N 6-methyladenosine (m6A) in eukaryotes is the most common and widespread internal modification in mRNA. The modification regulates mRNA stability, translation efficiency, and splicing, thereby fine-tuning gene regulation. In plants, m6A is dynamic and critical for various growth stages, embryonic development, morphogenesis, flowering, stress response, crop yield, and biomass. Although recent high-throughput sequencing approaches have enabled the rapid identification of m6A modification sites, the site-specific mechanism of this modification remains unclear in trees. In this review, we discuss the functional significance of m6A in trees under different stress conditions and discuss recent advancements in the quantification of m6A. Quantitative and functional insights into the dynamic aspect of m6A modification could assist researchers in engineering tree crops for better productivity and resistance to various stress conditions.
Introduction
RNA modifications that naturally occur in eukaryotic messenger RNA (mRNA), long non-coding RNA (lncRNA), ribosomal RNA (rRNA), transfer RNA (tRNA), small nuclear RNA (snRNA), small nucleolar RNA (snoRNA), circular RNA (circRNA), etc. have been shown to be an important biological mechanism for regulating RNA activity [1–4]. To date, more than 170 RNA modifications constitute the ‘epitranscriptome’ repertoire [5, 6]. Among the various RNA modifications, the functions of N6-methyladenosine (m6A), 5-methylcytosine (m5C), N1-methyladenosine (m1A), pseudouridine (Ψ), and 2′-O-methylation (Nm) are better documented than other modifications [2, 7]. m6A was first discovered in eukaryotic mRNAs and accounts for over 80% of all RNA methylation [8]. Compared with m5C, m1A, Ψ, and Nm, m6A is the most common modification in plants, and its function has been better elucidated in several plants, including Arabidopsis, maize, wheat, oats, and rice [9, 10].
The m6A modification varies among different plant tissues [2, 9, 11], and serves as a key switch for translation efficiency, nuclear retention, splicing, and RNA decay, through the recruitment of m6A-binding proteins [12, 13]. Moreover, the regulation of m6A is crucial for plant development under various stress conditions, including embryonic development, leaf initiation, shoot stem cell fate, trichome morphogenesis, flower transition, and root development [14–17]. Although m6A is evolutionarily conserved and plays important roles in cellular and biological processes [18, 19], the functional role of m6A modification in trees is less clear. Though the physiological functions of m6A in Arabidopsis have been well reviewed [2, 20, 21], reviews focusing on the function of m6A in trees are limited. In this review we summarize the biological functions of m6A in trees in response to biotic and abiotic stresses. In addition, we also summarize recent technologies capable of both detection and quantification of m6A.
How m6A modifications are created and removed in trees
The m6A pathway involves three core RNA-binding proteins (RBPs), namely m6A writers (methyltransferases), m6A readers, and m6A erasers (demethylases), that collectively make m6A dynamic and reversible. These proteins are referred to as WERs (writers, erasers, and readers). Depending on downstream genes being targeted, m6A WERs can have different functions or the same function under different conditions [15, 22]. Although m6A WERs have been identified in both Arabidopsis and trees, the majority of these discoveries are restricted to Arabidopsis. This limitation restricts our comprehensive understanding of m6A modifications in trees.
Trees are characterized by their larger and more complex genomes and are perennial species exposed to different environmental conditions throughout their lifespan. Therefore, trees might exhibit different m6A modification patterns compared with Arabidopsis, thereby presenting a unique challenge in deciphering the intricacies of m6A modifications. However, the differences in modification pattern and its stoichiometry between trees and Arabidopsis, and the functions of WERs in trees remain unclear despite the identification of several WERs in tree species (Fig. 1). Furthermore, the economic and ecological importance of trees underscores the importance of elucidating m6A modifications in tree species. m6A modifications can significantly influence tree growth, development, and stress responses [19, 23, 24]. Consequently, understanding m6A in tree species is of paramount importance. Such an understanding will not only enrich our understanding of m6A modifications in trees but also provide insight into how this modification contributes to tree adaptation and resilience in different ecosystems. This perspective emphasizes the need to overcome the limitations of using Arabidopsis and highlights the potential implications for our understanding of m6A modifications in tree species. The following subsections focus on the functional significance of m6A WERs in tree crops.
Figure 1.

Schematic overview of the m6A writers, erasers, and readers so far identified and their known biological functions in trees. Created with BioRender.com.
m6A writers
The m6A writer is a methyltransferase protein that adds a methyl group to adenosines. These m6A writer complexes were first identified in HeLa cells [25–27]. Later, orthologs of animal m6A writers, including MTA (an ortholog of METTL3, a SAM-binding protein) [28], MTB (an orthologue of METLL14, an mRNA target-binding protein) [29], and FIP37 (an orthologue of WTAP) [30], were identified in Arabidopsis. Besides this, Arabidopsis also contains the E3 ubiquitin-protein ligase Hakai (HAKAI), which is involved in the regulation of m6A writer components [29]. In rice, an alternative m6A pathway, mediated through ENHANCED DOWNY MILDEW 2 protein (EDM2), has been identified [31].
Recent studies reveal that mutants of m6A writer components, such as MTA, MTB, VIRILIZER (VIR), and HAKAI, show salt-sensitive phenotypes in an m6A-dependent manner. VIR-mediated m6A in mRNA was found to be correlated with mRNA stability by altering the length of 3′-UTR transcripts through alternative polyadenylation [32]. To date, only two studies have reported the function of m6A writers in trees. PtrMTA in poplar [33] and MdMTA in apple trees [24] have been identified as critical for m6A modification. The overexpression of PtrMTA increased drought tolerance by promoting trichome and root development in poplar [33]. MdMTA is important for drought tolerance [34], and this might prove to be helpful in the search for potential genes for developing stress-tolerant apple cultivars.
m6A readers
The m6A reader proteins, which recognize m6A marks on target sites and regulate biological functions, were first discovered in animals [35, 36]. The YTH protein family functions as a group of specific reader proteins [37]. Depending on the type of YTH protein, m6A can affect mRNA metabolism in different ways, such as modulating stability, promoting translation, or affecting splicing [27].
In contrast to the extensive studies performed on YTH domain-containing proteins (YTHDs) in animals, only three evolutionarily conserved proteins of the C-terminal region (ECT) family have recently been functionally characterized as YTHD homologs in Arabidopsis [15, 16]. The m6A reader proteins mainly include ECT2, ECT3, ECT4, and CPSF30 [38]. YTH proteins are abundant in GL-3 (Royal Gala) apple, Malus hupehensis (Chinese crab apple) [39], cucumber [40], and rice [41]. In Arabidopsis, ECT2 controls proteasome activity, trichome morphology, and developmental timing and morphogenesis by fine-tuning mRNA stabilization. Moreover, ECT2, ECT3, and ECT4 relocalize to cytoplasmic foci during osmotic stress, whereas only ECT2 relocalizes to cytoplasmic stress granules during heat stress [16, 42, 43]. In addition, ECT1 and ECT2 also interact with the stress response protein CALCINEURIN B-LIKE-INTERACTING PROTEIN KINASE1 (CIPK1) to facilitate the transmission of calcium signals into the nucleus during various external stimuli [44].
In apple, m6A reader MhYTP2 overexpression enhances apple powdery mildew resistance by regulating the mRNA stability of MdMLO19 and MdMLO19-X1 and improving the translation efficiency of glutamate dehydrogenase 1-like MdGDH1L [45]. In addition, MhYTP2 also improves water use efficiency by increasing the photosynthetic rate and water uptake by roots and decreasing stomatal opening through increased abscisic acid concentrations and activated ethylene signaling [46]. However, the overexpression of MhYTP1 or MhYTP2 in GL-3 (Royal Gala) apple led to higher sensitivity to infection caused by Diplocarpon mali (Marssonina apple blotch), salinity, and heat stress, and enhanced resistance to water-logging, chilling, drought, and nutritional deficiency [39]. Furthermore, the overexpression of MhYTP1 and MhYTP2 was found to promote leaf senescence in Arabidopsis and GL-3 (Royal Gala) apple, and fruit ripening in tomato [47]. In poplar 84K (Populus alba × Populus glandulosa), a fast-growing poplar hybrid, the expression pattern of m6A pathway genes was tissue-specific and there was differential expression in leaves, xylem, phloem, and roots [48].
m6A erasers
m6A erasers, also known as demethylases, are responsible for removing the methyl groups from the methylated adenosine [49]. The first identified RNA demethylase, Fat mass and obesity-associated protein (FTO), catalyzes the demethylation of m6A in human mRNA [50]. Introducing human FTO to rice and potato increased crop yield and biomass by 50%. FTO stimulates root meristem, cell proliferation, and tiller bud formation and promotes photosynthetic efficiency and drought tolerance by mediating substantial m6A demethylation [51]. ALKBH (α-ketoglutarate-dependent dioxygenase homolog) proteins are another class of erasers that remove RNA methylation marks [52, 53]. Under normal and salt stress conditions, the m6A levels in alkbh10b mutants in Arabidopsis were higher than those of the wild type [54]. In addition to stress response, ALKBH proteins also perform various other functions. Depletion of m6A demethylase suppresses vegetative growth and floral transition by affecting the stability of the target mRNA [14]. ALKBH10B affects Arabidopsis floral transition and modifies the abscisic acid response during seed germination [14, 55]. ALKBH9B regulates m6A abundance on viral RNAs and is required for the vascular movement of Alfalfa mosaic virus in Arabidopsis [56]. Although many eraser proteins targeting specific methylation marks have been identified in model plants such as Arabidopsis, only three studies have reported m6A eraser proteins in trees under drought stress and for leaf color variations: HrALKBH10 in sea buckthorn [57], PtrALBH8B in poplar [58], and CfALKBH5 in Catalpa fargesii [59]. The level of activity of most ALKBH family members in trees has yet to be determined.
Identification of RNA-binding sites for m6A writers, erasers, and readers
WERs associated with RNAs and their modifications play crucial roles in various cellular processes. Therefore, identification of binding sites for RBPs is important in order to understand how m6A modifications are generated and removed. To address this issue, TRIBE (Targets of RNA-binding proteins identified by editing) was introduced, which expresses RBP fused to the catalytic domain of the RNA-editing enzyme ADAR (ADARcd) in vivo to identify RBP binding sites [60]. ADARcd tags target RNA transcripts by converting A to I near the RBP binding sites. However, the editing efficiency is low, and the editing sequence is biased [61]. Thus, HyperTRIBE with the hyperactive mutation E488Q was incorporated into ADARcd to increase editing efficiency and reduce the sequence bias [62, 63]. Using HyperTRIBE, the targets of ECT2 and ECT3 were identified [64]. HyperADARcd in plants showed better performance than other RNA editing enzymes and identifies RBPs and their targets in a simple manner [65]. These methods can be used to identify m6A-associated targets of m6A writers, readers, and erasers in trees in future.
Functions of m6A modification in trees under stress conditions and normal development
In a cellular context, different types of mRNA may have different amounts of m6A. For example, in Arabidopsis there are ~5000 mRNA transcripts that contain 0.5–0.7 m6A peaks per 1000 nucleotides or 0.7–1.0 m6A peaks per actively expressed transcript. m6A has a significant influence on nuclear processes such as splicing and epigenetic activity and it plays an important role in promoting cytoplasmic mRNA degradation, affecting the corresponding cellular processes and pathways [66]. Since mRNA metabolism regulates nuclear export, alternative splicing, translation, degradation etc., m6A has a profound impact on all mRNA-associated processes [67, 68]. Thus, m6A influences almost every stage of the mRNA life cycle [28, 69].
As in the case of mammals, m6A plays a crucial role in plant development and maintains circadian and seasonal rhythms in plants [14, 70, 71]. In the coding regions, m6A affects translational dynamics [72, 73], and in the 5′-UTRs it promotes cap-independent translation [74]. However, unlike in mammals, m6A in plants is enriched around the stop codon and within the 3′-UTRs [69]. Though m6A has been discovered in almost all plant species, more attention has been paid to the model plant Arabidopsis [71, 75] and crop plants such as maize [76], wheat [77], and rice [51, 78]. Only a few studies have investigated m6A in trees (Table 1), such as apple [79], poplar [33], tea [80], citrus [81], sea buckthorn [57], and moso bamboo [19, 82]. In the following subsections, we will discuss the functional significance of m6A in tree mRNA under stress conditions.
Table 1.
Physiological functions of N6-methyladenosine (m6A) in trees.
| Tree species | Function and possible involvement in the stress response | Reference |
|---|---|---|
| Apple (Malus domestica) | MdMTA, the m6A methyltransferase complex, is responsible for m6A development in mRNA under drought conditions. Transcribing mRNAs are involved in oxidative stress and lignin deposition. The m6A modification in mRNA promotes mRNA stability and translational efficiency of the genes in response to drought and oxidative stress. | [24] |
| Apple | Under drought stress, m6A modification changes gene expression in drought-responsive genes such as HEAT SHOCK PROTEIN 60 (HSP60), JASMONATE-ZIM-DOMAIN PROTEIN 3 (JAZ3), Scarecrow-Like 1 (SCL1), and ETHYLENE RESPONSE FACTOR1 (ERF1). | [79] |
| Apple | Overexpression of the m6A reader MhYTP2 increases mRNA m6A and regulates the mRNA stability of MdMLO19 and the translational efficiency of antioxidant genes, resulting in resistance to powdery mildew in apples. | [45] |
| Chinese crab apple (Malus hupehensis) | MhYTP1 and MhYTP2 were induced by methyl jasmonate and salicylic acid, and their overexpression made apple trees more susceptible to Marssonina apple blotch, heat stress, and high salinity, and more resistant to oxygen and nutrient deficiency. The promoter regions contain many stress-related cis-elements. | [39] |
| Poplar (Populus trichocarpa) | Overexpressed PtrMTA (methyltransferase) participates in m6A formation, improves drought tolerance, increases trichome density, and results in a better root system. | [33] |
| Poplar | m6A sites were mainly enriched in the coding regions and the 3′-UTRs and associated with drought-induced genes. PtrMTA transcripts showed a positive correlation with the protein content. Cellulose- and lignin-related genes in response to drought stress were associated with m6A ratio, and m6A interacted with poly(A) tail length (PAL) and polyadenylation. | [58] |
| Cotton (Gossypium hirsutum) | m6A is dynamic under salt stress. Genes containing m6A are differentially expressed in response to salt stress. m6A reader protein regulates salt tolerance. | [83] |
| Sea buckthorn (Hippophae rhamnoides) | The m6A modification genes are associated with metabolic biosynthesis. Three m6A demethylases, HrALKBH10B, HrALKBH10C, and HrALKBH10D, were significantly upregulated under drought stress. m6A demethylase genes decrease m6A methylation during drought stress. | [57] |
| Tea (Camellia sinensis) | m6A regulatory genes are driven by whole-genome duplication and segmental duplication events. m6A duplicated regulatory gene pairs evolved by purifying selection. Sequence variation of the regulatory genes contributes to m6A functional diversification. Regulatory genes are differentially expressed under stress conditions and in tea-withering stages. RNA methylation and DNA methylation develop negative feedback through interaction with other regulatory genes. | [80] |
| Citrus (Citrus grandis) | Comprehensive analysis of m6A regulatory genes reveals different expression patterns during different growth stages. The genes are divided into writers, erasers, and readers and are distributed across nine chromosomes. The domain structures are diverse among the m6A enzymes. | [81] |
| Moso bamboo (Phyllostachys edulis) | m6A sites enriched at different growth stages during rapid growth were higher in 2-m shoots than in 4-m shoots of the 18th internode, enriched mainly in the 3′-UTRs, and maintained the stability of the transcripts associated with rapid growth. | [19] |
| Moso bamboo | A novel method was developed to detect m6A, and m6A sites may also drive the translation of circularized transcripts expressed under GA3 treatments. | [82] |
| Moso bamboo | Decreased m6A level in the 3′-UTR region and increased m6A level in the coding region (as observed in genes such as PedMKK3 and PedMTA) promoted the lateral root growth and development, increased expression of m6A writers, and increased expression of exon junction complexes such as MAGO, Y14, and EIF4A-III. | [23] |
| Maiyuanjinqiu derived from Catalpa fargesii | m6A in the 3′-UTRs showed a negative correlation with mRNA abundance, m6A pathways were related to photosynthesis and stress response, and increased m6A in yellow-green leaves decreased CfALKBH5 gene expression and was associated with a chlorotic phenotype. | [59] |
m6A response to biotic stress in trees
In apples, the overexpression of MhYTP2 increased m6A in mRNA and enhanced the translational efficiency of antioxidant genes, in the process improving resistance to powdery mildew [45]. MhYTP2-induced m6A modifications in the exon regions destabilized associated mRNAs, whereas m6A modifications in untranslated regions were positively correlated with mRNA abundance and improved powdery mildew resistance in apple trees through the rapid degradation of bound mRNAs of MdMLO19 and MdMLO19-X1 and the translation efficiency of the glutamate dehydrogenase-1-like gene, MdGDH1L [45]. In another study, in GL-3 (Royal Gala) apples, the overexpression of MhYTP1 and MhYTP2 (m6A readers) resulted in increased susceptibility to Marssonina apple blotch. Moreover, several cis-acting elements related to biotic and abiotic stresses were identified in the promoter regions of MhYTP1 and MhYTP2. Therefore, these two genes showed inducibility in response to various stress treatments and are actively involved in various stress responses [39]. Furthermore, it will be useful to investigate whether m6A has longer-term effects in apple species than in annual plants. While m6A in cereal crops may be temporary and present only during specific seasons, m6A in tree mRNA could potentially promote long-term stress responses. However, further evidence is required to support this hypothesis.
m6A response to drought stress in trees
In response to drought stress in apple trees, the presence of m6A has been shown to affect both mRNA stability and the translational efficiency of drought- and oxidative stress-related genes [24]. In addition, MdMTA is involved in oxidative stress and lignin deposition under drought conditions [24]. A recent study conducted on apple seedlings revealed that m6A modulates drought response genes, including HEAT SHOCK PROTEIN 60 (HSP60), JASMONATE-ZIM-DOMAIN PROTEIN 3 (JAZ3), Scarecrow-Like 1 (SCL1), and ETHYLENE RESPONSE FACTOR1 (ERF1), which are related to drought tolerance [79]. In sea buckthorn, drought stress significantly induces the expression of three m6A demethylases (HrALKBH10B, HrALKBH10C, and HrALKBH10D), suggesting that decreased m6A methylation plays important roles in drought tolerance in sea buckthorn [57]. In poplar, Poplar methyltransferase (PtrMTA), m6A erasers (ALBH8B), and m6A readers (ECT2 and ECT8) were upregulated under drought stress. The m6A ratio was associated with cellulose- and lignin-related genes in response to drought stress and reduced transcription and translation levels, suggesting that m6A represses wood formation under drought stress [58]. In another study, the overexpression of PtrMTA increased the root system, lignin deposition, and the scavenging of reactive oxygen species, leading to enhanced drought tolerance in poplar [33].
m6A response to salt stress in trees
In cotton, m6A deposition was positively correlated with gene expression under salt stress [83]. m6A prevents RNA cleavage in stress-responsive transcripts and stabilizes mRNAs under salt and osmotic stresses [84]. Furthermore, m6A and RNA-associated secondary structure in Arabidopsis increased mRNA stability and ultimately protein levels during salt stress [85]. Rice stress tolerance requires the m6A-YTH system [86]. GhECT6, a YTH domain gene in cotton, plays an important role in salt tolerance [83]. In citrus, the cis-acting elements of 26 m6A regulatory genes encoding the regulatory proteins are related to the abscisic acid (ABA)-responsive element (ABRE) [81]. In tea, downregulation was observed in all m6A writer genes under drought treatment [80].
m6A response to leaf color variations
Analysis of m6A composition in mRNA derived from leaves with different colors revealed varying correlation with gene expression in Maiyuanjinqiu and C. fargesii [59]. Maiyuanjinqiu is a new natural variety with yellow-green leaves derived from C. fargesii, the Chinese bean tree with green leaves. Increased m6A and decreased CfALKBH5 gene expression (an m6A eraser) were positively correlated with yellow-green leaves, suggesting a chlorotic phenotype. In contrast, m6A levels were significantly lower in the seedlings of C. fargesii (green leaves) than in those of Maiyuanjinqiu (yellow-green leaves), further supporting a positive correlation between increased m6A, decreased CfALKBH5 gene expression, and the occurrence of a chlorotic phenotype. This implies that m6A levels may serve as a crucial epitranscriptomic marker for identifying leaf color variations in trees. Furthermore, m6A enrichment in the 3′-UTR was negatively correlated with global gene expression, and m6A-containing mRNAs were related to photosynthesis, pigment biosynthesis and metabolism, oxidation–reduction, and stress response [59]. This underscores the potential impact of m6A on broader physiological pathways related to leaf color variations in trees.
m6A response in growth and development
In poplar, the overexpression of PtrMTA, which colocalized with PtrFIP37 in the nucleus, led to increased trichome density and a more developed root system compared with the wild type. Furthermore, the m6A levels in the roots were higher than those of the wild type, indicating that m6A formation affects the development of trichomes and the root system, thereby enhancing drought tolerance [33]. In moso bamboo, which grows rapidly (up to 114.5 cm per day), m6A regulates the rapid cell division and elongation of each internode [87]. The m6A levels were higher in 2-m shoots than in 4-m shoots of the 18th internode, suggesting that m6A was slowly demethylated during rapid growth. m6A was also found to maintain the stability of mRNAs of the genes related to lignin biosynthesis during rapid growth [19]. Additionally, m6A modifications drive the regulation of circularized transcripts in moso bamboo seedlings treated with gibberellic acid (GA3) [82]. A recent study by the same group revealed the importance of m6A in moso bamboo root development [23]. This study observed that reducing m6A levels in the 3′-UTR regions while increasing them in the coding region, as seen in genes like PedMKK3 and PedMTA due to the RNA methylation inhibitor (DZnepA), led to increased lateral root growth. This coincided with elevated gene expression, an increased full-length ratio, enhanced proximal poly(A) site utilization, and a shorter poly(A) tail length. Notably, common motifs in this process were AAACA and AAACT [23]. These findings underscore the indispensable role of m6A in moso bamboo development. A summary of all recent studies describing the functional role of WREs in tree crops is listed in Table 1.
Emerging technologies to identify and quantify nucleotide-specific m6A
Many technological advances have helped to detect RNA modifications and uncover new functions and regulators involved in RNA modifications [88]. In the early stages, chromatographic methods, including high-performance liquid chromatography–mass spectrometry (HPLC–MS), thin-layer chromatography (TLC), and gas chromatography (GC), have been used to identify methylated nucleotides. HPLC–MS, dot-blot, and high-performance liquid chromatography coupled to triple-quadrupole mass spectrometry (LC–MS/MS) [89–91] are reliable and provide quantitative information on RNA modifications, making them suitable for analyzing a wide range of biological samples [92]. However, precise identification of RNA modification sites is essential for understanding the interaction between RNA functions and regulatory mechanisms.
To date, there are numerous methods for profiling m6A modifications, including antibody-based immunoprecipitation, digestion-based detection, m6A sensing-reverse transcription-based detection, ligation-based detection, gene editing-based detection, metabolic labeling, and direct RNA-based detection (Fig. 2). The advantages and disadvantages of each method for m6A detection are listed in Table 2. Due to the word limit, only recent methods such as digestion-based detection, deamination of unmethylated adenosines, cryo-electron microscopy (cryo-EM), and nanopore direct RNA sequencing (DRS) are briefly described in the following sections, and the other methods are briefly described in Supplementary Data Appendix S1.
Figure 2.

Different methods for m6A detection and profiling. Created with BioRender.com.
Table 2.
Working principle, advantages, and disadvantages of methods for the identification, quantification and localization of m6A.
| Detection methods for m 6 A profiling | Working principle | Advantages | Disadvantages |
|---|---|---|---|
| Antibody-based detection | Use of antibody enrichment followed by RNA sequencing | Identification of m6A from total RNA or mRNA | Library preparation, poor reproducibility, and no site-specific identification |
| Photo-crosslinking-assisted sequencing (PA-m6A-seq) | Incorporating photoactivatable nucleosides and then crosslinking mRNA at protein binding sites using light followed by m6A sequencing | Accurate identification of m6A sites and the best method for cultured cells | Requirement for cell pre-treatment, inconsistent quantification, and errors in transition sites and in exact positions |
| Reverse transcription-based method | Combination of reverse transcriptase and polymerases followed by RNA sequencing | Lower error rate in m6A identification, accurate site recognition, and no use of antibodies | Unquantifiable blocking effect, false-positive results, unable to detect in 5′-UTRs, and database limitations |
| Digestion-based detection | Digests into single nucleotides with RNase T1 and RNase A, followed by LC–MS, LC–MS /MS, TLC, SCARLET or MAZTER-seq and m6A- REF-seq | Precise quantification of m6A from nucleotides, nucleosides, and bases at low levels, identification at ACA sites and selectivity, sensitivity, and simplicity | Unable to detect exact positions and low abundance of m6A, loss of sequence context, requirement for expensive chemicals and many enzymes, and time-consuming processes |
| Ligation-based detection | Ligation of RNA templates using DNA probes ligated with T3 DNA ligase and amplification of ligated nucleotides | Accurate detection and quantification at one nucleotide resolution and higher specificity even in low abundance RNA in real biological samples | No genome-wide identification and accurate quantification, false-positive results, time-consuming processes, and requirement for PCR amplification |
| Gene editing-based detection | Conversion of C to U at targeted RNA sites using a base editing approach by fusing APOBEC1 (cytidine deaminase enzyme for RNA editing) to the m6A-binding YTH domain | Identification of global m6A in cells and single cells at low levels of RNA, isoform-specific m6A patterns, and m6A RNA-binding proteins, and antibody-free technique | False-positive results, requiring multiple molecular techniques (e.g. cloning and transfection), expensive chemicals and many enzymes, and time-consuming processes |
| Metabolic labeling-based detection (m6A-label-seq) | Feeding cells with m6A methyltransferase to generate and transfer methyl groups to specific mRNA adenosine to convert m6A to a6A by native m6A methylation enzymes | Detection of m6A at single-base resolution, detection of clustered m6A sites, and localization of nascent m6A in the nucleus | Requirement for several molecular techniques, expensive chemicals and many enzymes, and time-consuming processes |
| Direct RNA-based detection | Direct RNA sequencing (native/full-length RNA molecules in their native context) with Oxford Nanopore sequencing | Direct detection and quantification of m6A on native/full-length RNA molecules at single-base resolution, detection of novel m6A sites and no requirement for reverse transcription, amplification, antibodies, digestion, ligation, library preparation etc. | High error rates, database limitations in data analysis, unable to distinguish m6A from other modifications, and the requirement for computationally intensive tools |
| Cryo-EM | Imaging modified nucleotides within cellular conditions using cryo-EM with high resolution and 3D representation | Direct visualization of m6A nucleotides in macromolecular structures such as the ribosome, accurate structure-based rRNA and deep mapping | Ribosomes are prepared from a large quantity of cells, collecting cryo-EM data requires very expensive instruments and microscopes, and solving the structure takes a longer time |
| Deamination of unmethylated adenosines (similar to bisulfite sequencing) | Deamination of unmethylated adenosines to inosines followed by RNA sequencing | Transcriptome-wide absolute quantification of m6A at single-base high resolution, unbiased, and convenient | Requirement for multistep library preparation, short-read sequencing, inability to detect m6A interactions with other modifications, and need for improvement for standard applications of m6A |
Digestion-based detection
Nucleotides with or without m6A modification have different chemical and physical properties. Therefore, a liquid chromatography–mass spectrometry (LC–MS)-based approach has become the standard method for the quantification of RNA modifications by single nucleotide digestion and ultraviolet detection [93, 94]. RNase T1 and RNase A are commonly used to enzymatically degrade nucleic acids into single nucleotides. LC–MS compares single nucleotides with standard nucleotides to quantify m6A. The method features excellent selectivity, sensitivity, and simplicity. However, this method cannot determine the position of m6A in RNA molecules. Furthermore, enzymatic digestion of RNA into single nucleotides loses the sequence context [95, 96].
To address this issue, a strategy of targeted RNA fragmentation using specific enzymes along with LC–MS/MS analysis, similar to the methods utilized in proteomics, was implemented [97, 98]. However, the low abundance of modified mRNA made single-nucleotide resolution detection challenging. Another method for the accurate and quantitative identification of mRNA is site-specific cleavage and radiolabeling, followed by ligation-assisted extraction and TLC (SCARLET). SCARLET has the capability to detect low levels of m6A modification at specific sites and is more sensitive compared with methylated RNA immunoprecipitation sequencing (MeRIP-Seq)–qPCR. However, SCARLET also requires expensive chemicals and time-consuming processes, including many enzyme transformations and separations [99].
The ACA sequence-specific RNA endoribonuclease MazF, which was discovered in 2017, is sensitive to m6A and cleaves RNA only at the ACA sequence motif [100]. This characteristic allows the detection of m6A demethylase and methyltransferase activities. Normally, a reduction in MazF cleavage efficiency can indicate the presence of m6A residues within the ACA motif. To map m6A at single-nucleotide resolution with systematic quantitative profiling, MAZTER-seq (RNA digestion via m6A sensitive RNase) and m6A-REF-seq (m6A-sensitive RNA-endoribonuclease-facilitated sequencing) have also been developed using MazF [101, 102]. However, this method does not identify other known motifs as the enzyme only targets ACA sites. Thus, the universality of this method is limited, making it challenging to quantify m6A in all DRACH motifs, despite the availability of appropriate bioinformatics tools and resources for MAZTER-seq and m6A-REF-seq.
Direct RNA-based detection
The direct RNA-based detection method provides significant advantage for quantitative m6A modification as it does not require reverse transcription, amplification, antibodies, digestion, ligation, etc. [103]. Nanopore DRS, a promising technology, can detect various RNA modifications in full-length RNA molecules [104]. During DRS, modified nucleotides and unmodified nucleotides emit different signal intensities when RNA molecules pass through the nanopore. This allows m6A identification using either comparative or supervised approaches. Nanopore m6A detection can be classified into two categories: the electrical signal and the basecalling error. Over the past 3 years, several algorithms and software packages (Tombo, EpiNano, DiffErr, ELIGOS, DRUMMER, MINES, XPore, Nanom6A, nanoDoc, Yanocomp, etc.) have been developed for the analysis of direct RNA nanopore sequencing [105–107]. Tombo, ELIGOS, DRUMMER, Yanocomp, xPore, nanoDoc, and Nanocompore use a comparative approach to distinguish m6A modification by comparing with control samples without m6A modifications [such as knockout or knock-down of m6A writer enzyme or in vitro transcribed (IVT) RNAs] [108]. On the other hand, MINES and Nanom6A use a supervised approach that utilizes training data or experimental protocols to identify m6A modification and overcome the limitations of the comparative approach. However, the supervised approach is limited to a specific nucleotide content. The advantages and disadvantages and the basic classification and GitHub repository of these packages are available in Supplementary Data Table S1.
EpiNano predicts m6A from DRS datasets by increasing mismatches and decreasing basecalling. However, it does not distinguish between m6A and other modifications such as m1A [109, 110]. MINES is another tool that provides the qualitative profiles of m6A sites, but it can only recognize m6A sites in four specific contexts: GGACA, GGACT, GGACC, and AGACT [111]. On the other hand, the Nanom6A pipeline utilizes an Extreme Gradient Boosting (XGBoost) model based on ion current signals from DRS to identify and quantify a transcriptome-wide m6A at single-base resolution with high accuracy [112]. This method also presents the m6A quantification of an individual transcript in different samples. In particular, in poplar transcripts [112] this method identified thousands of m6A sites enriched near the stop codon and 3′-UTR regions, as well as alternative polyadenylation sites. This suggests that Nanom6A is capable of conducting a transcriptome-wide analysis of m6A modifications and helps to understand the role of m6A in stabilizing highly expressed transcripts in genes associated with wood formation in tree species [112]. DiffErr (differr_nanopore_DRS) detects transcriptome-wide m6A using basecalling error [71]. Nanocompore, a robust and flexible analysis system with several unique features, was introduced to identify m6A with positional accuracy without the need for a training set. This method modifies the current signal and uses a model-free approach to compare m6A with different samples at single-molecule high resolution [106]. Recently, m6Anet, a neural-network-based method, has been reported for the quantification of m6A from DRS. However, m6A detection is still challenging due to the high error rates of DRS, which complicates the analysis of nanopore sequencing data. Comparison of 10 different tools used for identification of m6A from DRS data reveals that integrating analyses from multiple tools significantly enhances the effectiveness [113]. Further studies will aim to improve the ability for m6A detection based on nanopore DRS and algorithms in future.
Cryo-electron microscopy
Recent advancements in single-particle cryo-EM have enabled direct visualization of modified nucleotides in macromolecular structures like the ribosome [114]. A recent study using a 2.38-Å resolution cryo-EM reconstruction of the tomato ribosome was able to assign density for rRNA modifications [114]. The analysis of these ribosomes revealed 89 rRNA modifications, including Ψ, Nm (Fig. 3A), and base modifications such as m1acp3Ψ, N4-acetylcytidine (ac4C), N7-methylguanosine (m7G), and m6A (Fig. 3B). Although the method is not considered quantitative, this method provides nucleotide-specific detection and also could be used to study the function of RNA modification in the local and global structure of the RNAs they localize.
Figure 3.
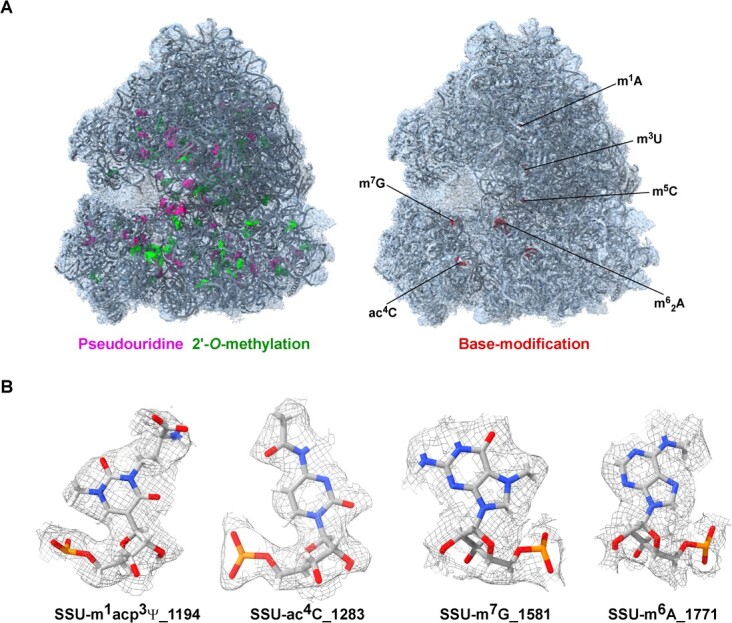
Detection and visualization of rRNA modification in plants. A Distribution of pseudouridine (Ψ) and 2′-O-methylation (Nm) in the tomato ribosome. The cryo-EM model and map are presented for the entire ribosome. B Cryo-EM density map of selected rRNA modification in plant ribosomes. The cryo-EM data ware derived from Cottilli et al. [114], deposited in PDB (PDB ID-7QIZ and EMD-14004).
Deamination of unmethylated adenosines
Bisulfite sequencing is widely regarded as the most reliable method for measuring DNA methylation at individual nucleotide resolution [115]. NOseq (amplicon sequencing evaluation method for RNA m6A sites after chemical deamination), similar to bisulfite sequencing, was the first method developed to detect m6A in RNA by chemical deamination (A to I). However, this method also introduces the G-to-X effect (deamination of guanosine into xanthosine) [116], which complicates transcriptome-wide studies. Recently, GLORI (glyoxal and nitrite-mediated deamination of unmethylated adenosines) has been developed for the absolute quantification of m6A at a single-base high resolution. GLORI consists of three steps: preventing deamination by protecting guanosine with glyoxal, deaminating all unmethylated adenosines except m6A to inosines using nitrite, and deprotecting guanosine under alkaline conditions (Fig. 4). The reverse transcription of inosines to guanosines allows comprehensive detection and stoichiometric quantification of m6A in mRNA fragments. GLORI has detected >175 000 m6A sites in HEK293T cells at a single-base resolution [117, 118]. Another method, evolved transfer RNA adenosine deaminase (TadA)-assisted m6A sequencing (eTAM-seq), is an enzyme-assisted sequencing method, which detects and quantifies site-specific m6A by inducing deamination of global adenosines to inosines using a hyperactive TadA variant. eTAM-seq facilitates transcriptome-wide m6A profiling without deep-sequencing by preserving RNA integrity from limited input samples [119]. Both GLORI and eTAM-seq are unbiased and convenient methods for the absolute quantification of m6A, with the potential to become a gold standard for m6A profiling.
Figure 4.
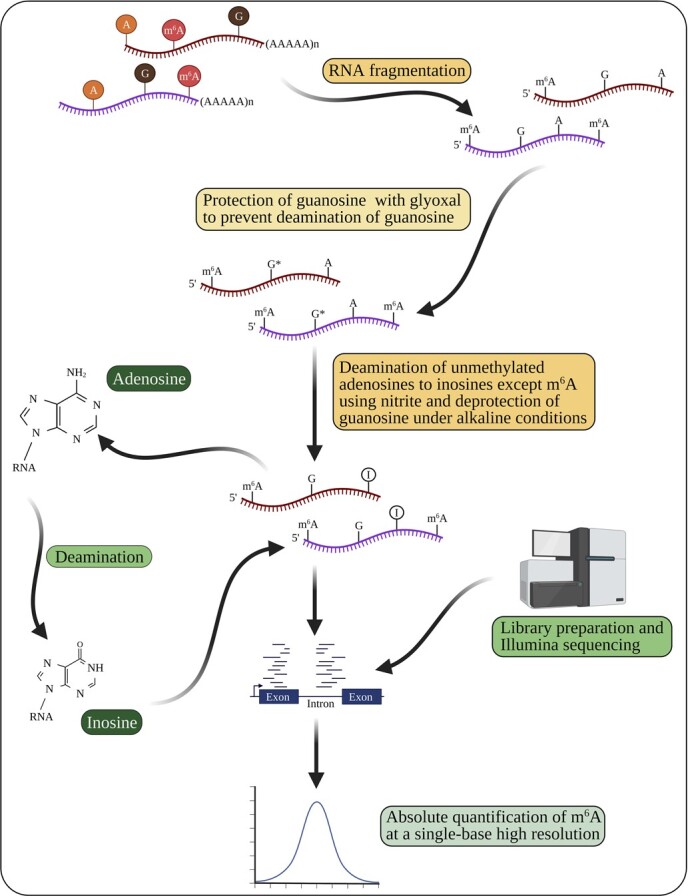
Deamination of unmethylated adenosines to inosines for a transcriptome-wide absolute quantification of m6A using GLORI (glyoxal- and nitrite-mediated deamination of unmethylated adenosines). This method consists of three steps: guanosine protection, adenosine deamination, and guanosine deprotection, followed by sequencing and absolute quantification of m6A. The schematic representation is based on Liu et al. [117] and Jones et al. [118]. Created with BioRender.com.
Databases for RNA modifications
Numerous RNA databases have also been developed to deposit RNA modifications. These RNA databases facilitate various tasks, such as RNA methylation site detection, motif discovery, differential RNA methylation analysis, and functional analysis. Currently, the MODOMICS database comprises a comprehensive list of RNA modifications discovered in all types of RNA molecules across all life kingdoms [120, 121]. Several other databases, such as RNAmod, also exist to catalog the RNA modification in model plants such as Arabidopsis (Supplementary Data Table S2). RNAmod is an interactive, web-based database designed for the functional annotation of mRNA modifications [122]. The RNAMDB database serves as a focal point for 109 known naturally occurring RNA modifications and continues to expand each year with new additions [123]. The m6Avar database contains 1 678 126 variants associated with nine types of RNA modifications, including m6A [124]. RNAWRE [125] and M6A2Target [126] are two dedicated databases for WERs, while RMBase is a comprehensive database for RBPs [127]. REPIC [128] and m6A-Atlas [129] are two dedicated databases for m6A and its site-specific interactions. DirectRMDB is the first database based on Oxford Nanopore Technologies, covering 16 types of modifications across 25 species [130]. However, there is currently no database that catalogs RNA modifications in tree species. Therefore, it is crucial to develop databases that provide information on mRNA modifications in tree species.
Differences and similarities between Arabidopsis and trees with respect to m6A
Due to the contrast between the life cycles of Arabidopsis, a short-lived and small model plant, and tree species, long-lived and larger plants, differences in m6A patterns and stoichiometry are likely to exist. However, the precise functions of m6A in trees remain unexplored. This raises a crucial question: how does m6A modification differ between Arabidopsis and tree species in terms of functions, frequency of occurrence, and other relevant aspects? Thus, a comparative analysis of m6A modification between tree species and Arabidopsis would provide valuable insights into their similarities and differences. Therefore, through the analysis of m6A-related genes, their promoters, and their amino acids in trees and Arabidopsis, this review observed the differences and similarities in m6A WERs between these two species. A concise overview of the gene structure of m6A WERs in trees and Arabidopsis is listed in Supplementary Data Table S3.
Specificity of m6A in tree species
Distinct repertoire of m6A WERs. Tree species possess a unique repertoire of m6A WERs compared with Arabidopsis, playing crucial roles in regulating m6A levels and patterns. For instance, in poplar the number of genes involved in the m6A pathway is significantly higher, comprising 61 m6A genes, including 14 m6A erasers, 14 m6A writers, and 33 m6A readers, in contrast to Arabidopsis, which has 28 m6A genes [48].
Diverse functional roles. m6A modifications in tree species are involved in a broad spectrum of biological processes (Fig. 1). While some functions may overlap with Arabidopsis, tree species exhibit distinct m6A-mediated regulatory mechanisms tailored to their unique life cycles and stress conditions [2, 45]. For instance, in Arabidopsis the m6A writer VIR and the m6A eraser ALKBH10B regulate salt tolerance, promoting seedling growth and seed germination [32, 54, 55]. In contrast, the m6A writer MTA plays a role in drought tolerance in apple and poplar [24, 33], while the m6A reader YTP2 regulates powdery mildew resistance in apple [45].
Distinct target genes. Although the primary function of WERs is to add methyl groups to adenosine residues, the specific target genes and biological consequences of m6A could differ between Arabidopsis and trees [2]. For instance, in apple, MhYTP2 targets MdMLO19 (known to influence apple powdery mildew susceptibility) and antioxidant genes, reflecting the specific roles in tree species [45].
Higher ratio of m6A. Due to the higher abundance and activity of WERs in tree species [2, 48], the m6A ratio appears to be higher in tree species compared with Arabidopsis, suggesting that the m6A ratio may be linked to changes in the mRNA stability and translation efficiency of specific genes and may exert long-term effects in trees, aligning with their perennial life cycle and distinct developmental stages.
Dynamic regulation of m6A. Despite their evolutionary conservation across plant taxa, global and individual m6A levels display dynamic regulation in response to various stresses, exhibiting distinct patterns between tree species and Arabidopsis [2]. For instance, global m6A levels increase under salt stress in Arabidopsis [32], while they decrease under drought stress in sea buckthorn [57]. Interestingly, global m6A levels remain unchanged in apple under drought stress [24, 79], underscoring the dynamic and stress- or species-specific nature of m6A modifications. This dynamic regulation is likely mediated by altered expression of m6A WERs.
Transcript-specific m6A. While m6A modifications can occur on various transcripts, the extent and specific locations of m6A marks vary between tree species and Arabidopsis, depending on their unique life cycles and stress conditions. For instance, salt stress in Arabidopsis triggers increased m6A deposition in the 5′- and 3′-UTRs but not in the coding regions [32]. Conversely, increased m6A levels in the coding region of moso bamboo promote lateral root growth [23]. This suggests that m6A modifications are not randomly distributed across transcripts, but specific RRACH motifs are selectively targeted for m6A modification, despite every transcript comprising several RRACH motifs [2].
Differential gene architecture. Although m6A is evolutionarily conserved across plant taxa, the total number of cis-acting and trans-acting elements in m6A-related genes differs between Arabidopsis and trees. Moreover, gene structures, including the numbers of exons and introns, 5′-UTR and 3′-UTR lengths, numbers of domains and motifs, number of amino acids, cellular localization, signaling, and the number of transcripts, vary between Arabidopsis and trees (Supplementary Data Table S3). These differences suggest that the regulation of m6A expression varies between these two groups of plants.
Similarities between Arabidopsis and trees in m6A
Despite their distinct m6A patterns and stoichiometry, Arabidopsis and trees share some fundamental similarities in m6A mechanism.
Conserved core m6A machinery. The core m6A machinery, including WERs, is conserved between tree species and Arabidopsis, suggesting that the fundamental mechanism of recognizing adenosine residues and catalyzing m6A is likely conserved between Arabidospis and trees. This similarity also applies to m6A-mediated gene regulation, which influences gene expression, mRNA stability, translation efficiency, and splicing patterns [2, 23, 24, 45].
Shared m6A biological functions. Certain m6A biological functions, such as increased trichome density and enhanced root development, are shared between tree species (e.g. poplar and bamboo) and Arabidopsis [15, 23, 33].
m6A detection technologies. The methods are currently universal across various organisms, including trees, Arabidopsis, humans, and animals [48] (Table 2). There are no specialized detection techniques specifically developed for trees.
Challenges in studying m6A modifications in tree species
Arabidopsis has been extensively studied to elucidate the mechanisms of m6A modification. However, m6A modification in tree species presents unique features and challenges compared with Arabidopsis. Further, while Arabidopsis is a valuable model plant, there is a need for a model plant specifically for forestry and horticultural research, such as poplar, to emphasize the importance of m6A in trees [131–133]. Below we present the challenges involved in gaining a better understanding of m6A modifications in tree species and suggest possible ways to overcome existing limitations.
Comparative studies and functional roles. Understanding the differences and similarities in the m6A mechanism between tree species and Arabidopsis can help us to identify the conserved and divergent features of the m6A pathway in trees as there are no comparative studies between trees and Arabidopsis. Characterizing tree-specific m6A WERs, as well as determining the functional roles of m6A in different tree species, are priorities.
Functional characterization of m6A pathway genes. Functionally characterizing m6A pathway genes in tree species, including their tissue-specific expression patterns and roles in plant development and stress responses, is vital for a comprehensive understanding of m6A-mediated gene regulation in trees.
Manipulating m6A levels. Developing efficient methods to manipulate m6A levels in tree species is essential for understanding the functional implications of m6A modifications and for developing m6A-based biotechnological tools for tree improvement. This includes exploring the potential of m6A-based gene editing tools to develop mutant trees with desirable traits, such as improved resistance to pests and diseases or increased tolerance to abiotic stresses.
Molecular mechanisms and interplay. Elucidating the molecular mechanisms governing the recognition of the RRACH motif by m6A writers in plants is a pivotal step in understanding m6A modification. Investigating the crosstalk between m6A and other epigenetic modifications, such as DNA methylation, offers valuable insights into the interplay of different regulatory mechanisms. In addition, exploring the role of m6A in the regulation of alternative splicing in trees could provide insights into how m6A regulates gene expression and tree development.
Computational tools and resources. Existing computational tools and resources are mostly used for humans and Arabidopsis (Supplementary Data Tables S1 andS2). Refining the tools to analyze m6A data in tree species can help generate complex m6A datasets for tree species and identify m6A sites, predict m6A targets, and provide insights into the functional implications of m6A modifications.
By addressing these challenges, we can underscore the importance of m6A in tree species, given their economic and ecological significance, and pave the way for a more comprehensive understanding of m6A roles in tree biology.
Conclusions
m6A has emerged as a powerful gene regulator of all eukaryotes studied to date [134, 135]. It regulates mRNA stability, translation efficiency, splicing, and RNA binding proteins, particularly under different stress conditions [136–138]. However, a major limitation still exists in the need to identify m6A differential methylation in a genome-wide manner. Recent technologies advancements, such as chemical digestion coupled with next-generation sequencing (NGS), DRS using Oxford Nanopore Technologies [103], and single-cell deamination adjacent to RNA modification targets (DART-seq) (scDART-seq) [139], have significantly contributed to m6A research [140–142]. In addition, various software tools, including EpiNano and Nanom6A, have been developed to improve m6A quantification at single-molecule resolution. These technologies assist in deciphering m6A sites accurately and quantitatively, especially in cases where the modification is presented at low levels or at multiple isoform-specific sites.
Despite advancements in m6A detection, progress in investigating m6A in trees is extremely slow. Unlike cereal crops, where m6A is transient, m6A modification has long-term consequences in trees. Consequently, trees might be a new avenue for studying the long-term implications of m6A modification [19, 23, 24]. m6A WERs play important roles in executing specific modifications, influencing growth and development and enhancing stress tolerance. For instance, PtrMTA in poplar [33] and MdMTA in apple trees [24] deposit m6A in mRNA and promote mRNA stability and stress tolerance. In contrast, CfALKBH5 in C. fargesii removes m6A in mRNA and regulates leaf color variations [59]. Furthermore, m6A regulates lignin biosynthesis by maintaining mRNA stability during the rapid growth of trees. However, key differences, such as signaling pathways and protein interactions of m6A, exist between tree species and Arabidopsis. The molecular mechanisms involved in this process remain unclear, and it is uncertain whether other m6A members are involved in m6A modification and are conserved across different species. Therefore, the identification and characterization of m6A regulatory proteins, as well as the corresponding genes, along with comparative studies between tree species and Arabidopsis, may contribute to a better understanding of m6A modifications in trees.
Acknowledgements
The authors wish to thank the anonymous reviewers for their valuable time and insightful comments, which greatly improved the quality of the manuscript. We apologize to those whose original work(s) could not be included in this review owing to space limitations.
Preparation of this review was supported by a grant from the National Key Research & Development Program of China (2021YFD2200503-01), a grant from the National Natural Science Foundation of China (32071848), a grant from the Natural Science Foundation of Jiangsu Province (BK20231289), the Jiangxi ‘Shuangqian’ Program (S2019DQKJ2030), the Natural Science Foundation for Distinguished Young Scholars of Nanjing Forestry University (JC2019004), the Project for Groundbreaking Achievements of Nanjing Forestry University (202211), and a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions. The authors are also grateful for the Young Foreign Talent Program (QN2022014012L) and the support of Metasequoia Faculty Research Start-up Funding (163100028) at the Bamboo Research Institute, Nanjing Forestry University, for the first author, M.R. K.S.R. is supported by the Dean of Faculty Fellowship, the Koshland Prize, and a Sir Charles Clore Postdoctoral Fellowship from the Weizmann Institute.
Author contributions
M.R., K.S.R., Q.W., and S.M. planned, designed and wrote the review. M.R., K.S.R., Q.W., and M.Z. outlined and edited the review. M.R., S.M., and K.S.R. drew the images. M.R. and Q.W. made the tables. M.R., K.S.R., M.Z., A.S., Z.A., S.R., and Q.W. edited and revised the review.
Data availability
No additional data were generated or are associated with this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary data
Supplementary data is available at Horticulture Research online.
Supplementary Material
Contributor Information
Muthusamy Ramakrishnan, State Key Laboratory of Tree Genetics and Breeding, Co-Innovation Center for Sustainable Forestry in Southern China, Bamboo Research Institute, Key Laboratory of National Forestry and Grassland Administration on Subtropical Forest Biodiversity Conservation, School of Life Sciences, Nanjing Forestry University, Nanjing 210037, Jiangsu, China.
K Shanmugha Rajan, Department of Chemical and Structural Biology, Weizmann Institute of Science, 7610001 Rehovot, Israel.
Sileesh Mullasseri, Department of Zoology, St. Albert’s College (Autonomous), Kochi 682018, Kerala, India.
Zishan Ahmad, State Key Laboratory of Tree Genetics and Breeding, Co-Innovation Center for Sustainable Forestry in Southern China, Bamboo Research Institute, Key Laboratory of National Forestry and Grassland Administration on Subtropical Forest Biodiversity Conservation, School of Life Sciences, Nanjing Forestry University, Nanjing 210037, Jiangsu, China.
Mingbing Zhou, State Key Laboratory of Subtropical Silviculture, Bamboo Industry Institute, Zhejiang A&F University, Lin’an, Hangzhou 311300, Zhejiang, China; Zhejiang Provincial Collaborative Innovation Center for Bamboo Resources and High-Efficiency Utilization, Zhejiang A&F University, Lin’an, Hangzhou 311300, Zhejiang, China.
Anket Sharma, State Key Laboratory of Subtropical Silviculture, Bamboo Industry Institute, Zhejiang A&F University, Lin’an, Hangzhou 311300, Zhejiang, China.
Subbiah Ramasamy, Cardiac Metabolic Disease Laboratory, Department of Biochemistry, School of Biological Sciences, Madurai Kamaraj University, Madurai 625 021, Tamilnadu, India.
Qiang Wei, State Key Laboratory of Tree Genetics and Breeding, Co-Innovation Center for Sustainable Forestry in Southern China, Bamboo Research Institute, Key Laboratory of National Forestry and Grassland Administration on Subtropical Forest Biodiversity Conservation, School of Life Sciences, Nanjing Forestry University, Nanjing 210037, Jiangsu, China.
References
- 1. Liang Z, Riaz A, Chachar S. et al. Epigenetic modifications of mRNA and DNA in plants. Mol Plant. 2020;13:14–30 [DOI] [PubMed] [Google Scholar]
- 2. Hu J, Cai J, Xu T. et al. Epitranscriptomic mRNA modifications governing plant stress responses: underlying mechanism and potential application. Plant Biotechnol J. 2022;20:2245–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shi H, Wei J, He C. Where, when, and how: context-dependent functions of RNA methylation writers, readers, and erasers. Mol Cell. 2019;74:640–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yang X, Hu X, Liu J. et al. N 6 -methyladenine modification in noncoding RNAs and its function in cancer. Biomark Res. 2020;8:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ontiveros RJ, Stoute J, Liu KF. The chemical diversity of RNA modifications. Biochem J. 2019;476:1227–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wiener D, Schwartz S. The epitranscriptome beyond m6A. Nat Rev Genet. 2021;22:119–31 [DOI] [PubMed] [Google Scholar]
- 7. Wilkinson E, Cui YH, He YY. Context-dependent roles of RNA modifications in stress responses and diseases. Int J Mol Sci. 2021;22:1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Desrosiers R, Friderici K, Rottman F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc Natl Acad Sci USA. 1974;71:3971–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shen L, Liang Z, Wong CE. et al. Messenger RNA modifications in plants. Trends Plant Sci. 2019;24:328–41 [DOI] [PubMed] [Google Scholar]
- 10. Wang L, Zhuang H, Fan W. et al. m6A RNA methylation impairs gene expression variability and reproductive thermotolerance in Arabidopsis. Genome Biol. 2022;23:244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bhat SS, Bielewicz D, Gulanicz T. et al. mRNA adenosine methylase (MTA) deposits m6A on pri-miRNAs to modulate miRNA biogenesis in Arabidopsis thaliana. Proc Natl Acad Sci USA. 2020;117:21785–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kadumuri RV, Janga SC. Epitranscriptomic code and its alterations in human disease. Trends Mol Med. 2018;24:886–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xu Y, Song M, Hong Z. et al. The N6-methyladenosine METTL3 regulates tumorigenesis and glycolysis by mediating m6A methylation of the tumor suppressor LATS1 in breast cancer. J Exp Clin Cancer Res. 2023;42:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Duan HC, Wei LH, Zhang C. et al. ALKBH10B is an RNA N6-methyladenosine demethylase affecting Arabidopsis floral transition. Plant Cell. 2017;29:2995–3011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Arribas-Hernández L, Bressendorff S, Hansen MH. et al. An m6A-YTH module controls developmental timing and morphogenesis in Arabidopsis. Plant Cell. 2018;30:952–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Scutenaire J, Deragon J-M, Jean V. et al. The YTH domain protein ECT2 is an m6A reader required for normal trichome branching in Arabidopsis. Plant Cell. 2018;30:986–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ramakrishnan M, Rajan KS, Mullasseri S. et al. The plant epitranscriptome: revisiting pseudouridine and 2′-O-methyl RNA modifications. Plant Biotechnol J. 2022;20:1241–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ye T, Wang J, Zhao H. et al. Role of N6-methyladenosine in the pathogenesis, diagnosis and treatment of pancreatic cancer (review). Int J Oncol. 2023;62:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li T, Wang H, Zhang Y. et al. Comprehensive profiling of epigenetic modifications in fast-growing Moso bamboo shoots. Plant Physiol. 2022;191:1017–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bhat SS, Bielewicz D, Jarmolowski A. et al. N 6-methyladenosine (m6A): revisiting the old with focus on new, an Arabidopsis thaliana centered review. Genes (Basel). 2018;9:596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Reichel M, Köster T, Staiger D. Marking RNA: m6A writers, readers, and functions in Arabidopsis. J Mol Cell Biol. 2019;11:899–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Martínez-Pérez M, Aparicio F, López-Gresa MP. et al. Arabidopsis m6A demethylase activity modulates viral infection of a plant virus and the m6A abundance in its genomic RNAs. Proc Natl Acad Sci USA. 2017;114:10755–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liufu Y, Xi F, Wu L. et al. Inhibition of DNA and RNA methylation disturbs root development of moso bamboo. Tree Physiol. 2023;43:1653–74 [DOI] [PubMed] [Google Scholar]
- 24. Hou N, Li C, He J. et al. MdMTA-mediated m6A modification enhances drought tolerance by promoting mRNA stability and translation efficiency of genes involved in lignin deposition and oxidative stress. New Phytol. 2022;234:1294–314 [DOI] [PubMed] [Google Scholar]
- 25. Zhang W, Qian Y, Jia G. The detection and functions of RNA modification m6A based on m6A writers and erasers. J Biol Chem. 2021;297:100973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bokar JA, Rath-Shambaugh ME, Ludwiczak R. et al. Characterization and partial purification of mRNA N6-adenosine methyltransferase from HeLa cell nuclei. Internal mRNA methylation requires a multisubunit complex. J Biol Chem. 1994;269:17697–704 [PubMed] [Google Scholar]
- 27. Knuckles P, Bühler M. Adenosine methylation as a molecular imprint defining the fate of RNA. FEBS Lett. 2018;592:2845–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhong S, Li H, Bodi Z. et al. MTA is an Arabidopsis messenger RNA adenosine methylase and interacts with a homolog of a sex-specific splicing factor. Plant Cell. 2008;20:1278–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ruzicka K, Zhang M, Campilho A. et al. Identification of factors required for m6A mRNA methylation in Arabidopsis reveals a role for the conserved E3 ubiquitin ligase HAKAI. New Phytol. 2017;215:157–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shen L, Liang Z, Gu X. et al. N 6-methyladenosine RNA modification regulates shoot stem cell fate in Arabidopsis. Dev Cell. 2016;38:186–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ma K, Han J, Zhang Z. et al. OsEDM2L mediates m6A of EAT1 transcript for proper alternative splicing and polyadenylation regulating rice tapetal degradation. J Integr Plant Biol. 2021;63:1982–94 [DOI] [PubMed] [Google Scholar]
- 32. Hu J, Cai J, Park SJ. et al. N 6-methyladenosine mRNA methylation is important for salt stress tolerance in Arabidopsis. Plant J. 2021;106:1759–75 [DOI] [PubMed] [Google Scholar]
- 33. Lu L, Zhang Y, He Q. et al. MTA, an RNA m6A methyltransferase, enhances drought tolerance by regulating the development of trichomes and roots in poplar. Int J Mol Sci. 2020;21:2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hu J, Manduzio S, Kang H. Epitranscriptomic RNA methylation in plant development and abiotic stress responses. Front Plant Sci. 2019;10:500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dominissini D, Nachtergaele S, Moshitch-Moshkovitz S. et al. The dynamic N1-methyladenosine methylome in eukaryotic messenger RNA. Nature. 2016;530:441–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Arguello AE, DeLiberto AN, Kleiner RE. RNA chemical proteomics reveals the N6-methyladenosine (m6A)-regulated protein–RNA interactome. J Am Chem Soc. 2017;139:17249–52 [DOI] [PubMed] [Google Scholar]
- 37. Dai XY, Shi L, Li Z. et al. Main N6-methyladenosine readers: YTH family proteins in cancers. Front. Oncol. 2021;11:635329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yue H, Nie X, Yan Z. et al. N 6-methyladenosine regulatory machinery in plants: composition, function and evolution. Plant Biotechnol J. 2019;17:1194–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang N, Guo T, Sun X. et al. Functions of two Malus hupehensis (Pamp.) Rehd. YTPs (MhYTP1 and MhYTP2) in biotic- and abiotic-stress responses. Plant Sci. 2017;261:18–27 [DOI] [PubMed] [Google Scholar]
- 40. Zhou Y, Hu L, Jiang L. et al. Genome-wide identification and expression analysis of YTH domain-containing RNA-binding protein family in cucumber (Cucumis sativus). Genes Genomics. 2018;40:579–89 [DOI] [PubMed] [Google Scholar]
- 41. Zhou C, Wang C, Liu H. et al. Identification and analysis of adenine N6-methylation sites in the rice genome. Nature Plants. 2018;4:554–63 [DOI] [PubMed] [Google Scholar]
- 42. Wei LH, Song P, Wang Y. et al. The m6A reader ECT2 controls trichome morphology by affecting mRNA stability in Arabidopsis. Plant Cell. 2018;30:968–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wu J, Peled-Zehavi H, Galili G. The m6A reader ECT2 post-transcriptionally regulates proteasome activity in Arabidopsis. New Phytol. 2020;228:151–62 [DOI] [PubMed] [Google Scholar]
- 44. Ok SH, Jeong HJ, Bae JM. et al. Novel CIPK1-associated proteins in Arabidopsis contain an evolutionarily conserved C-terminal region that mediates nuclear localization. Plant Physiol. 2005;139:138–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Guo T, Liu C, Meng F. et al. The m6A reader MhYTP2 regulates MdMLO19 mRNA stability and antioxidant genes translation efficiency conferring powdery mildew resistance in apple. Plant Biotechnol J. 2022;20:511–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Liu C, Guo T, Wang N. et al. Overexpression of MhYTP2 enhances apple water-use efficiency by activating ABA and ethylene signaling. Environ Exp Bot. 2019;157:260–8 [Google Scholar]
- 47. Wang N, Guo T, Wang P. et al. Functional analysis of apple MhYTP1 and MhYTP2 genes in leaf senescence and fruit ripening. Sci Hortic. 2017;221:23–32 [Google Scholar]
- 48. Sun X, Wu W, Yang Y. et al. Genome-wide identification of m6A writers, erasers and readers in poplar 84K. Genes (Basel). 2022;13:1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang W, Huang Q, Liao Z. et al. ALKBH5 prevents hepatocellular carcinoma progression by post-transcriptional inhibition of PAQR4 in an m6A dependent manner. Exp. Hematol Oncol. 2023;12:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jia G, Fu Y, Zhao X. et al. N 6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7:885–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yu Q, Liu S, Yu L. et al. RNA demethylation increases the yield and biomass of rice and potato plants in field trials. Nat Biotechnol. 2021;39:1581–8 [DOI] [PubMed] [Google Scholar]
- 52. Alemu EA, He C, Klungland A. ALKBHs-facilitated RNA modifications and de-modifications. DNA Repair (Amst). 2016;44:87–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Scarrow M, Chen N, Sun G. Insights into the N6-methyladenosine mechanism and its functionality: progress and questions. Crit Rev Biotechnol. 2020;40:639–52 [DOI] [PubMed] [Google Scholar]
- 54. Shoaib Y, Hu J, Manduzio S. et al. Alpha-ketoglutarate-dependent dioxygenase homolog 10B, an N6-methyladenosine mRNA demethylase, plays a role in salt stress and abscisic acid responses in Arabidopsis thaliana. Physiol Plant. 2021;173:1078–89 [DOI] [PubMed] [Google Scholar]
- 55. Tang J, Yang J, Duan H. et al. ALKBH10B, an mRNA m6A demethylase, modulates ABA response during seed germination in Arabidopsis. Front. Plant Sci. 2021;12:712713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Martínez-Pérez M, Gómez-Mena C, Alvarado-Marchena L. et al. The m6A RNA demethylase ALKBH9B plays a critical role for vascular movement of alfalfa mosaic virus in Arabidopsis. Front Microbiol. 2021;12:745576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhang G, Lv Z, Diao S. et al. Unique features of the m6A methylome and its response to drought stress in sea buckthorn (Hippophae rhamnoides Linn.). RNA Biol. 2021;18:794–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gao Y, Liu X, Jin Y. et al. Drought induces epitranscriptome and proteome changes in stem-differentiating xylem of Populus trichocarpa. Plant Physiol. 2022;190:459–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhang Y, Wang J, Ma W. et al. Transcriptome-wide m6A methylation in natural yellow leaf of Catalpa fargesii. Front. Plant Sci. 2023;14:1167789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yin S, Chen Y, Chen Y. et al. Genome-wide profiling of rice double-stranded RNA-binding protein 1-associated RNAs by targeted RNA editing. Plant Physiol. 2023;192:805–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. McMahon AC, Rahman R, Jin H. et al. TRIBE: hijacking an RNA-editing enzyme to identify cell-specific targets of RNA-binding proteins. Cell. 2016;165:742–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Rahman R, Xu W, Jin H. et al. Identification of RNA-binding protein targets with HyperTRIBE. Nat Protoc. 2018;13:1829–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Xu W, Rahman R, Rosbash M. Mechanistic implications of enhanced editing by a HyperTRIBE RNA-binding protein. RNA. 2018;24:173–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Arribas-Hernández L, Rennie S, Schon M. et al. The YTHDF proteins ECT2 and ECT3 bind largely overlapping target sets and influence target mRNA abundance, not alternative polyadenylation. elife. 2021;10:e72377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zhou G, Niu R, Zhou Y. et al. Proximity editing to identify RNAs in phase-separated RNA binding protein condensates. Cell Discov. 2021;7:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Murakami S, Jaffrey SR. Hidden codes in mRNA: control of gene expression by m6A. Mol Cell. 2022;82:2236–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wang S, Lv W, Li T. et al. Dynamic regulation and functions of mRNA m6A modification. Cancer Cell Int. 2022;22:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Huang Q, Mo J, Liao Z. et al. The RNA m6A writer WTAP in diseases: structure, roles, and mechanisms. Cell Death Dis. 2022;13:852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Luo G-Z, MacQueen A, Zheng G. et al. Unique features of the m6A methylome in Arabidopsis thaliana. Nat Commun. 2014;5:5630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Arribas-Hernández L, Brodersen P. Occurrence and functions of m6A and other covalent modifications in plant mRNA. Plant Physiol. 2019;182:79–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Parker MT, Knop K, Sherwood AV. et al. Nanopore direct RNA sequencing maps the complexity of Arabidopsis mRNA processing and m6A modification. Elife. 2020;9:e49658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Choi J, Ieong KW, Demirci H. et al. N 6-methyladenosine in mRNA disrupts tRNA selection and translation-elongation dynamics. Nat Struct Mol Biol. 2016;23:110–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zhao BS, Roundtree IA, He C. Post-transcriptional gene regulation by mRNA modifications. Nat Rev Mol Cell Biol. 2017;18:31–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zhou J, Wan J, Gao X. et al. Dynamic m6A mRNA methylation directs translational control of heat shock response. Nature. 2015;526:591–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Xu T, Wu X, Wong CE. et al. FIONA1-mediated m6A modification regulates the floral transition in Arabidopsis. Adv Sci. 2022;9:e2103628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Liang Z, Zhang L, Chen H. et al. m6A-maize: weakly supervised prediction of m6A-carrying transcripts and m6A-affecting mutations in maize (Zea mays). Methods. 2022;203:226–32 [DOI] [PubMed] [Google Scholar]
- 77. Huang T, He W-J, Li C. et al. Transcriptome-wide analyses of RNA m6A methylation in hexaploid wheat reveal its roles in mRNA translation regulation. Front. Plant Sci. 2022;13:917335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Cheng Q, Wang P, Wu G. et al. Coordination of m6A mRNA methylation and gene transcriptome in rice response to cadmium stress. Rice. 2021;14:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Mao X, Hou N, Liu Z. et al. Profiling of N6-methyladenosine (m6A) modification landscape in response to drought stress in apple (Malus prunifolia (Willd.) Borkh). Plants. 2022;11:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Zhu C, Zhang S, Zhou C. et al. Genome-wide investigation of N6-methyladenosine regulatory genes and their roles in tea (Camellia sinensis) leaves during withering process. Front. Plant Sci. 2021;12:702303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Tian Y, Zeng J, Fan R. Comprehensive analysis of N6-methyladenosine regulatory genes from Citrus grandis and expression profilings in the fruits of “Huajuhong” (C. grandis “Tomentosa”) during various development stages. Horticulturae. 2022;8:462 [Google Scholar]
- 82. Wang Y, Wang H, Xi F. et al. Profiling of circular RNA N6-methyladenosine in moso bamboo (Phyllostachys edulis) using nanopore-based direct RNA sequencing. J Integr Plant Biol. 2020;62:1823–38 [DOI] [PubMed] [Google Scholar]
- 83. Wang W, Li W, Cheng Z. et al. Transcriptome-wide N6-methyladenosine profiling of cotton root provides insights for salt stress tolerance. Environ Exp Bot. 2022;194:104729 [Google Scholar]
- 84. Anderson SJ, Kramer MC, Gosai SJ. et al. N 6-methyladenosine inhibits local ribonucleolytic cleavage to stabilize mRNAs in Arabidopsis. Cell Rep. 2018;25:1146–1157.e3 [DOI] [PubMed] [Google Scholar]
- 85. Kramer MC, Janssen KA, Palos K. et al. N 6-methyladenosine and RNA secondary structure affect transcript stability and protein abundance during systemic salt stress in Arabidopsis. Plant. Direct. 2020;4:e00239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Ma W, Cui S, Lu Z. et al. YTH domain proteins play an essential role in rice growth and stress response. Plants (Basel). 2022;11:2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Chen M, Guo L, Ramakrishnan M. et al. Rapid growth of Moso bamboo (Phyllostachys edulis): cellular roadmaps, transcriptome dynamics, and environmental factors. Plant Cell. 2022;34:3577–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Janssen KA, Xie Y, Kramer MC. et al. Data-independent acquisition for the detection of mononucleoside RNA modifications by mass spectrometry. J Am Soc Mass Spectrom. 2022;33:885–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Tuncel G, Kalkan R. Importance of m N6-methyladenosine (m6A) RNA modification in cancer. Med Oncol. 2019;36:36. [DOI] [PubMed] [Google Scholar]
- 90. Zhang Y, Lu L, Li X. Detection technologies for RNA modifications. Exp Mol Med. 2022;54:1601–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Richter F, Plehn JE, Bessler L. et al. RNA marker modifications reveal the necessity for rigorous preparation protocols to avoid artifacts in epitranscriptomic analysis. Nucleic Acids Res. 2022;50:4201–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Peer E, Rechavi G, Dominissini D. Epitranscriptomics: regulation of mRNA metabolism through modifications. Curr Opin Chem Biol. 2017;41:93–8 [DOI] [PubMed] [Google Scholar]
- 93. Thüring K, Schmid K, Keller P. et al. LC-MS analysis of methylated RNA. Methods Mol Biol. 2017;1562:3–18 [DOI] [PubMed] [Google Scholar]
- 94. Zheng H-x, Zhang X-s, Sui N. Advances in the profiling of N6-methyladenosine (m6A) modifications. Biotechnol Adv. 2020;45:107656 [DOI] [PubMed] [Google Scholar]
- 95. Helm M, Motorin Y. Detecting RNA modifications in the epitranscriptome: predict and validate. Nat Rev Genet. 2017;18:275–91 [DOI] [PubMed] [Google Scholar]
- 96. Sarkar A, Gasperi W, Begley U. et al. Detecting the epitranscriptome. Wiley Interdiscip Rev. RNA. 2021;12:e1663 [DOI] [PubMed] [Google Scholar]
- 97. Jora M, Lobue PA, Ross RL. et al. Detection of ribonucleoside modifications by liquid chromatography coupled with mass spectrometry. Biochim Biophys Acta Gene Regul Mech. 2019;1862:280–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Limbach PA, Paulines MJ. Going global: the new era of mapping modifications in RNA. Wiley Interdiscip Rev RNA. 2017;8:e1367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Liu N, Parisien M, Dai Q. et al. Probing N6-methyladenosine RNA modification status at single nucleotide resolution in mRNA and long noncoding RNA. RNA. 2013;19:1848–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Imanishi M, Tsuji S, Suda A. et al. Detection of N6-methyladenosine based on the methyl-sensitivity of MazF RNA endonuclease. Chem Commun (Camb). 2017;53:12930–3 [DOI] [PubMed] [Google Scholar]
- 101. Garcia-Campos MA, Edelheit S, Toth U. et al. Deciphering the “m6A code” via antibody-independent quantitative profiling. Cell. 2019;178:731–747.e16 [DOI] [PubMed] [Google Scholar]
- 102. Zhang Z, Chen LQ, Zhao YL. et al. Single-base mapping of m6A by an antibody-independent method. Sci Adv. 2019;5:eaax0250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Stephenson W, Razaghi R, Busan S. et al. Direct detection of RNA modifications and structure using single-molecule nanopore sequencing. Cell Genom. 2022;2:100097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Begik O, Lucas MC, Pryszcz LP. et al. Quantitative profiling of pseudouridylation dynamics in native RNAs with nanopore sequencing. Nat Biotechnol. 2021;39:1278–91 [DOI] [PubMed] [Google Scholar]
- 105. Furlan M, Delgado-Tejedor A, Mulroney L. et al. Computational methods for RNA modification detection from nanopore direct RNA sequencing data. RNA Biol. 2021;18:31–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Leger A, Amaral PP, Pandolfini L. et al. RNA modifications detection by comparative nanopore direct RNA sequencing. Nat Commun. 2021;12:7198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Wan YK, Hendra C, Pratanwanich PN. et al. Beyond sequencing: machine learning algorithms extract biology hidden in Nanopore signal data. Trends Genet. 2022;38:246–57 [DOI] [PubMed] [Google Scholar]
- 108. Hendra C, Pratanwanich PN, Wan YK. et al. Detection of m6A from direct RNA sequencing using a multiple instance learning framework. Nat Methods. 2022;19:1590–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Liu H, Begik O, Lucas MC. et al. Accurate detection of m6A RNA modifications in native RNA sequences. Nat Commun. 2019;10:4079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Liu H, Begik O, Novoa EM. EpiNano: detection of m6A RNA modifications using Oxford Nanopore direct RNA sequencing. Methods Mol Biol. 2021;2298:31–52 [DOI] [PubMed] [Google Scholar]
- 111. Lorenz DA, Sathe S, Einstein JM. et al. Direct RNA sequencing enables m6A detection in endogenous transcript isoforms at base-specific resolution. RNA. 2020;26:19–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Gao Y, Liu X, Wu B. et al. Quantitative profiling of N6-methyladenosine at single-base resolution in stem-differentiating xylem of Populus trichocarpa using nanopore direct RNA sequencing. Genome Biol. 2021;22:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Zhong Z-D, Xie Y-Y, Chen H-X. et al. Systematic comparison of tools used for m6A mapping from nanopore direct RNA sequencing. Nat Commun. 2023;14:1906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Cottilli P, Itoh Y, Nobe Y. et al. Cryo-EM structure and rRNA modification sites of a plant ribosome. Plant Commun. 2022;3:100342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Li Y, Tollefsbol TO . DNA methylation detection: bisulfite genomic sequencing analysis. Methods Mol Biol. 2011;791:11–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Werner S, Galliot A, Pichot F. et al. NOseq: amplicon sequencing evaluation method for RNA m6A sites after chemical deamination. Nucleic Acids Res. 2021;49:e23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Liu C, Sun H, Yi Y. et al. Absolute quantification of single-base m6A methylation in the mammalian transcriptome using GLORI. Nat Biotechnol. 2022;41:355–66 [DOI] [PubMed] [Google Scholar]
- 118. Jones JD, Eyler DE, Koutmou KS. Mapping mRNA modifications for functional studies. Nat Biotechnol. 2022;41:324–5 [DOI] [PubMed] [Google Scholar]
- 119. Xiao Y-L, Liu S, Ge R. et al. Transcriptome-wide profiling and quantification of N6-methyladenosine by enzyme-assisted adenosine deamination. Nat Biotechnol. 2023;41:993–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Ma J, Zhang L, Chen S. et al. A brief review of RNA modification related database resources. Methods. 2022;203:342–53 [DOI] [PubMed] [Google Scholar]
- 121. Boccaletto P, Stefaniak F, Ray A. et al. MODOMICS: a database of RNA modification pathways 2021 update. Nucleic Acids Res. 2022;50:D231–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Liu Q, Gregory RI. RNAmod: an integrated system for the annotation of mRNA modifications. Nucleic Acids Res. 2019;47:W548–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Cantara WA, Crain PF, Rozenski J. et al. The RNA modification database, RNAMDB: 2011 update. Nucleic Acids Res. 2011;39:D195–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Luo X, Li H, Liang J. et al. RMVar: an updated database of functional variants involved in RNA modifications. Nucleic Acids Res. 2021;49:D1405–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Nie F, Feng P, Song X. et al. RNAWRE: a resource of writers, readers and erasers of RNA modifications. Database (Oxford). 2020;2020:baaa049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Deng S, Zhang H, Zhu K. et al. M6A2Target: a comprehensive database for targets of m6A writers, erasers and readers. Brief Bioinform. 2021;22:bbaa055. [DOI] [PubMed] [Google Scholar]
- 127. Xuan J-J, Sun W-J, Lin P-H. et al. RMBase v2.0: deciphering the map of RNA modifications from epitranscriptome sequencing data. Nucleic Acids Res. 2018;46:D327–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Liu S, Zhu A, He C. et al. REPIC: a database for exploring the N6-methyladenosine methylome. Genome Biol. 2020;21:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Tang Y, Chen K, Song B. et al. m6A-atlas: a comprehensive knowledgebase for unraveling the N6-methyladenosine (m6A) epitranscriptome. Nucleic Acids Res. 2021;49:D134–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Zhang Y, Jiang J, Ma J. et al. DirectRMDB: a database of post-transcriptional RNA modifications unveiled from direct RNA sequencing technology. Nucleic Acids Res. 2023;51:D106–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Douglas CJ. Populus as a model tree in comparative and evolutionary genomics of angiosperm trees. In: Groover A, Cronk Q, eds. Plant Genetics and Genomics: Crops and Models, Vol. 21. Cham, Switzerland, Springer International Publishing, 2017,61–84 [Google Scholar]
- 132. Jansson S, Douglas CJ. Populus: a model system for plant biology. Annu Rev Plant Biol. 2007;58:435–58 [DOI] [PubMed] [Google Scholar]
- 133. Taylor G. Populus: Arabidopsis for forestry. Do we need a model tree? Ann Bot. 2002;90:681–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Uzonyi A, Dierks D, Nir R. et al. Exclusion of m6A from splice-site proximal regions by the exon junction complex dictates m6A topologies and mRNA stability. Mol Cell. 2023;83:237–251.e7 [DOI] [PubMed] [Google Scholar]
- 135. Kan RL, Chen J, Sallam T. Crosstalk between epitranscriptomic and epigenetic mechanisms in gene regulation. Trends Genet. 2022;38:182–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Zhou L, Gao G, Tang R. et al. m6A-mediated regulation of crop development and stress responses. Plant Biotechnol J. 2022;20:1447–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Jiménez-Ramírez IA, Pijeira-Fernández G, Moreno-Cálix DM. et al. Same modification, different location: the mythical role of N6-adenine methylation in plant genomes. Planta. 2022;256:9. [DOI] [PubMed] [Google Scholar]
- 138. Yue J, Wei Y, Zhao M. The reversible methylation of m6A is involved in plant virus infection. Biology (Basel). 2022;11:271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Yao H, Yang Y, Yang YG. scDART-seq: mapping m6A at the single-cell level. Mol Cell. 2022;82:713–5 [DOI] [PubMed] [Google Scholar]
- 140. Motorin Y, Helm M. RNA nucleotide methylation: 2021 update. Wiley Interdiscip Rev RNA. 2021;13:e1691 [DOI] [PubMed] [Google Scholar]
- 141. Moshitch-Moshkovitz S, Dominissini D, Rechavi G. The epitranscriptome toolbox. Cell. 2022;185:764–76 [DOI] [PubMed] [Google Scholar]
- 142. Krusnauskas R, Stakaitis R, Steponaitis G. et al. Identification and comparison of m6A modifications in glioblastoma non-coding RNAs with MeRIP-seq and Nanopore dRNA-seq. Epigenetics. 2023;18:2163365. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No additional data were generated or are associated with this article.