

Abstract
Recessive diseases arise when both copies of a gene are impacted by a damaging genetic variant. When a patient carries two potentially causal variants in a gene, accurate diagnosis requires determining that these variants occur on different copies of the chromosome (i.e., are in trans) rather than on the same copy (i.e., in cis). However, current approaches for determining phase, beyond parental testing, are limited in clinical settings. Here, we developed a strategy for inferring phase for rare variant pairs within genes, leveraging genotypes observed in the Genome Aggregation Database (gnomAD v2, n=125,748 exomes). Our approach estimates phase with 96% accuracy, both in trio data and in patients with Mendelian conditions and presumed causal compound heterozygous variants. We provide a public resource of phasing estimates for coding variants and counts per gene of rare variants in trans that can aid interpretation of rare co-occurring variants in the context of recessive disease.
Determination of phase has important implications in clinical genetics in the diagnosis of recessive diseases that result from disruption of both copies of a gene, either by homozygous variants or compound heterozygous variants. Compound heterozygous variants present a challenge because two variants observed within a gene can occur in trans or in cis, and only the former results in compound heterozygosity. Currently, phasing in clinical settings is performed using parental data, which is expensive and not always available. Thus, there is an important need for other approaches to determine phase of variants accurately, easily, and cheaply.
There are several approaches for directly inferring phase for variant pairs observed in an individual. Phase may be determined directly using data from sequencing reads. However, for typical short-read sequencing technologies, read-based phasing methods are generally only possible for variants in close proximity to each other1, although sophisticated algorithms can phase some variant pairs at slightly longer distances2–4. Long-read sequencing technologies that would allow for direct phasing are more expensive and have not yet been widely applied in clinical settings5,6, while laboratory-based molecular methods for determining phase of variant pairs are low-throughput and technically challenging7. Phase can be determined based on transmission of variants from parents to offspring, but this approach increases cost and parental DNA is often not feasible to obtain or available. Thus, these direct phasing approaches all present critical limitations for determining phase of variant pairs within an individual in a clinical setting.
In contrast, indirect approaches for phasing rely on statistical methods applied to population data by identifying shared haplotypes among individuals in a population8–11. However, these methods (reviewed in Tewhey et al.12 and Browning and Browning13) require large numbers of reference samples (typically n >105 individuals), are computationally intensive, and perform less well for rare variants. Furthermore, these approaches cannot be readily applied to exome sequencing data, which does not provide enough density of surrounding variants. Despite these limitations, these population-based approaches are attractive because they do not require sequencing of additional family members or application of expensive sequencing approaches.
We sought to address existing challenges of phasing in clinical settings, particularly for rare variants observed in exome sequencing data. We leverage the Genome Aggregation Database (gnomAD), which performed aggregation and joint genotyping of exome sequencing data from 125,748 individuals14. We use these haplotype patterns to generate a resource for phasing rare coding variants observed in an individual and identify factors that influence the accuracy of our approach. Additionally, to provide a contextualization of the background rate when observing biallelic rare variants in individuals with rare diseases, we provide statistics for how often variant pairs are observed in trans within gnomAD, stratified by allele frequency (AF) and mutational consequence. Finally, we disseminate these resources in a user-friendly fashion via the gnomAD browser for community use.
Results
Inference of phase in gnomAD
We sought to address the challenges of phasing variants observed in individuals in clinical settings by applying the principle that haplotypes are usually shared across individuals in a population (Fig. 1a). If two variants are in trans in many individuals in a population, then they are likely to be trans in any given individual’s genome and vice versa. The presence of a variant pair in trans in the population also indicates that the variant combination may be tolerated in trans. We reasoned that by generating phasing estimates from a large reference population, we could infer the phase of variants observed in an individual.
Fig. 1: Overview of phasing approach using Expectation-Maximization method in gnomAD.
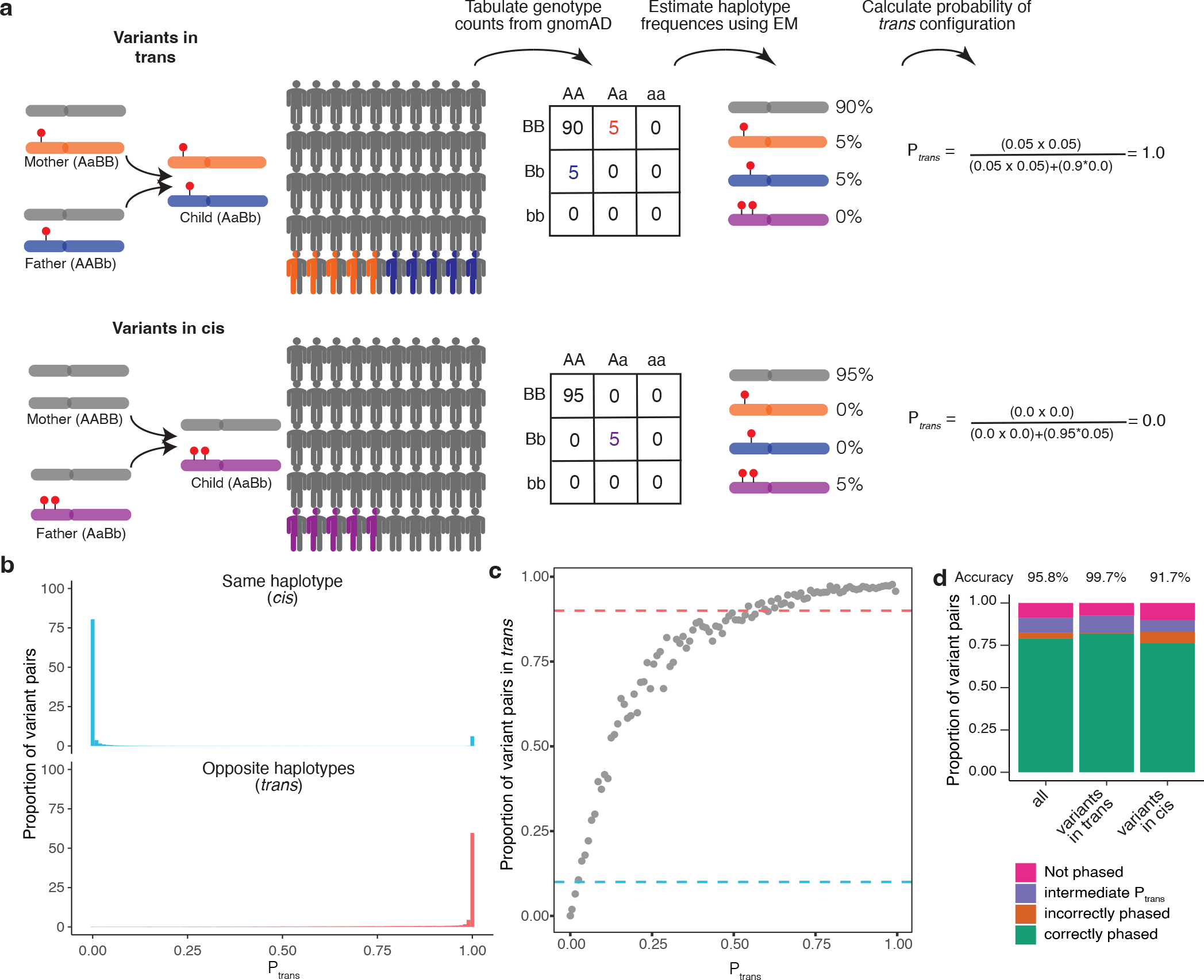
a, Schematic of phasing approach. b, Histogram of scores for variant pairs in cis (top, blue) and in trans (bottom, red). c, Proportion of variant pairs in each bin that are in trans. Each point represents variant pairs with bin size of 0.01. Blue dashed line at 10% indicates the threshold at which ≥ 90% of variant pairs in bin are on the same haplotype (). Red dashed line at 90% indicates the threshold at which ≥ 90% of variant pairs in bin are on opposite haplotypes (). Calculations are performed using variant pairs with population AF ≥ 1×10−4. d, Performance of for distinguishing variant pairs in cis and trans. Accuracy is calculated as the proportion of variant pairs correctly phased (green bars) divided by the proportion of variant pairs phased using (orange plus green bars). b-d, scores are population-specific.
To predict phase, we need to first estimate the haplotype frequencies in the population for a given pair of variants. To estimate haplotype frequencies, we used exome sequencing samples from gnomAD v2, a large sequencing aggregation database with 125,748 samples after rigorous quality control (Methods)14. There are several key advantages of using gnomAD as a reference dataset for calculating haplotype frequencies. First, samples in gnomAD undergo uniform processing and variant-calling, mitigating the impact of technical artifacts. Second, gnomAD provides sufficient sample size to estimate haplotype frequencies below 1×10−5. Lastly, gnomAD offers significant diversity, allowing results of our study to be applied beyond samples with European ancestry.
We focus on pairs of rare exonic variants occurring in the same gene, which are of the greatest interest in the context of Mendelian conditions. We required both variants to have a global minor AF in gnomAD exomes <5% and to be coding, flanking intronic (from position −1 to −3 in acceptor sites, and +1 to +8 in donor sites) or in the 5’/3’ UTRs. Across 19,877 genes, there were 5,320,037,963 unique variant pairs. 11,786,014 variant pairs are carried by the same individual at least once in gnomAD of which 105,322 are both singleton variants and observed in the same individual, where we are unable to make a phase prediction. We performed estimates based on all exome sequencing samples in gnomAD v2, as well as separate estimates within each genetic ancestry group (African/African American [AFR]: n=8128; Admixed American [AMR]: 17296; Ashkenazi Jewish [ASJ]: 5040; East Asian [EAS]: 9179; Finnish [FIN]: 10824; non-Finnish European [NFE]: 56885; Other: 3070; South Asian [SAS]: 15308).
For each pair of variants, we first generated pairwise genotype counts in gnomAD, with nine possible pairwise genotypes for each pair of variants (Fig. 1a). We then applied the Expectation-Maximization (EM) algorithm to each pair of variants to generate haplotype frequency estimates based on the observed pairwise genotype counts15. For a given pair of variants observed in an individual, the probability of two variants being in trans () is the probability of inheriting each of the haplotypes that contain only one of the two variant alleles.
Validation of phasing estimates using trio data
To measure the accuracy of our approach, we analyzed variants in a set of 4,992 trios that underwent exome sequencing and joint processing with gnomAD. In this trio structure, we could use parental transmission as a gold standard for phase and could compare with phase as predicted using the EM algorithm in gnomAD samples. We first estimated the genetic ancestry of each individual in the trios by projecting on the principal components of ancestry in the gnomAD v2 samples (Supplementary Fig. 1). Of the 4,992 children from the trios, 4,775 were assigned to one of seven genetic ancestry groups (AFR: 73; AMR: 358; ASJ: 62; EAS: 1252; FIN: 149; NFE: 2815; SAS: 46). We removed any samples in our trio dataset that did not fall into one of the seven aforementioned genetic ancestry groups. We used our approach leveraging gnomAD data to estimate phase for every pair of rare (global AF < 5% and population AF < 5%) coding and flanking intronic/UTR variants within genes observed in either of the parents in the trios. Across the 4,775 trio samples, we identified 339,857 unique variant pairs and 1,115,347 total variant pairs (mean 241.7 variant pairs per trio sample) (Supplementary Fig. 2a). On average, each trio sample had 64.4 variant pairs where both variants were missense, inframe insertions/deletions (indels), or predicted loss-of-function (pLoF), and 0.35 pLoF/pLoF variant pairs (Supplementary Fig. 2b–c). Nearly all of the variants identified in the trios were single nucleotide variants, with only 2.7% being short indels (functional consequences depicted in Supplementary Fig. 3a).
The majority (91.1%) of unique variant pairs in the trio samples were observed in gnomAD at least once and thus amenable to our phasing approach (Fig. 1d). By contrast, only 2.1% of variant pairs in these samples were within 10 bp of each other, the range in which we previously found read-back phasing of the physical read data to be most effective1 (Supplementary Fig. 3b). 8.2% of variant pairs were within 150 bp, the typical length of an Illumina exome sequencing read. Thus, our approach has a much higher ability to phase variants than physical read-back phasing data.
For each variant pair, we calculated the probability of being in trans () based on the haplotype frequencies estimated using the EM algorithm applied to gnomAD as described above. We found a bimodal distribution of scores: the majority of probabilities were either very high (> 0.99; suggesting a high likelihood of being in trans), or they were very low (< 0.01; suggesting a high likelihood of being in cis) (Fig. 1b, Supplementary Fig. 4a–g). Using trio phasing-by-transmission as a gold standard, we generated receiver-operator curves for distinguishing whether a variant pair is likely in trans and found high sensitivity and specificity (area under curve [AUC] ranging from 0.892 to 0.997 across the component genetic ancestry groups) (Supplementary Fig. 5a) and high precision and recall (Supplementary Fig. 5b).
We next defined thresholds for classifying variant pairs as being in cis versus trans (see Methods). To set these thresholds, we binned variant pairs observed in the trio data based on their scores calculated from gnomAD samples from the same genetic ancestry group. We used only variants on odd chromosomes (i.e., chromosomes 1, 3, 5, etc) to determine thresholds. For each bin, we calculated the proportion of trio variant pairs that were in cis or trans based on phasing-by-transmission. The threshold for variant pairs in trans was defined as the minimum such that ≥90% of variant pairs in that bin were in trans based on trio phasing-by-transmission, with a similar approach used for the threshold for variants in cis. This resulted in values of ≤ 0.02 and ≥ 0.55 as the threshold for variants in cis and trans, respectively (Fig. 1c).
We assessed how well our thresholds performed by measuring phasing accuracy using the phasing estimates generated by the EM algorithm applied to gnomAD against trio phasing-by-transmission. For measuring accuracy, we used only variant pairs observed on even chromosomes (i.e., chromosomes 2, 4, 6, etc). Of the 91.1% unique variant pairs that were amenable to phasing using the EM algorithm in gnomAD, only a minority (8.9%) of unique variant pairs had an intermediate score (i.e., ) and therefore an indeterminate phase (Fig. 1d). We calculated accuracy as the percentage of phaseable variant pairs (i.e., both variants present in gnomAD and score ≤ 0.02 or ≥ 0.55) that were correctly phased. Based on these thresholds, the overall phasing accuracy was 95.8%. The accuracy for unique variant pairs that are in cis based on trio data was 91.7%, and the accuracy for variant pairs in trans was 99.7%. Further exploration of the limitations of this approach, including how sample size impacts the number of variant pairs that can be phased, are detailed in the Supplementary Note and Supplementary Figure 6.
We calculated the overall percentage of variants correctly phased in a given individual (i.e., variants are counted more than once if seen multiple times in the trio data). 96.9% variant pairs per individual had both variants present in gnomAD and therefore were amenable to phasing, and 92.3% of variant pairs observed in a given individual were correctly phased using our approach. For rarer variant pairs (AF < 0.1%), 80.1% of variant pairs per individual were correctly phased. Together, these results suggest that our approach can generate highly accurate phasing estimates.
Accuracy of phasing across allele frequencies
Since rare variants are most likely to be of interest in clinical genetics, we assessed the accuracy of phasing at different AF bins. We found high accuracy (i.e., proportion correct classifications) ranging from 0.779 to 0.988 across pairs of AF bins (Fig. 2). Accuracy remained high across allele frequencies for variant pairs in trans. For variant pairs in cis based on trio phasing data, accuracy was high when both variants in the pair were more common (AF ≥ 0.001). However, accuracy was much lower for rare variants in cis (AF < 1×10−4), particularly when one variant in the pair is rare and the other is more common (Fig. 2c). Variant pairs where both variants are singletons (i.e., observed once in gnomAD) were phased fairly accurately for variants in trans based on the trio phasing data (accuracy of 0.993). Given the lack of information, we do not report the phasing estimates for singleton/singleton variant pairs in cis (see Supplementary Note).
Fig. 2: Phasing accuracy as a function of variant allele frequency (AF).

Phasing accuracy at different AF bins for all variant pairs (a), variant pairs in trans (b), and variant pairs in cis (c). Shading of squares and numbers in each square represent the phasing accuracy. Y-axis labels refer to the more frequent variant in each variant pair and X-axis labels refer to the rarer variant in each variant pair. Accuracy is the proportion of correct classifications (i.e., correct classifications / all classifications) and is calculated for all unique variant pairs seen in the trio data across all genetic ancestry groups using population-specific calculations.
Accuracy of phasing across genetic ancestry groups
In the above analyses, we used estimates calculated from samples in gnomAD with the same genetic ancestry group (“population”) in which the variant pair was seen in the trio data. We next asked if using all samples in gnomAD to calculate (“cosmopolitan”) would improve accuracy given larger sample sizes from which to calculate (Supplementary Fig. 7), with the caveat that using the full set of gnomAD samples would result in some genetic ancestry mismatching. We found that accuracy was generally similar when using population-specific ancestry estimates as compared to cosmopolitan estimates (Fig. 3a–b). However, for AFR and EAS, accuracy was slightly lower when using cosmopolitan estimates as compared to population-specific estimates specifically for variants in trans in these populations. For example, the phasing accuracy for variants in trans in the AFR ancestry group was 0.995 when using AFR-specific estimates, but dropped to 0.952 when using cosmopolitan estimates. These results suggest that cosmopolitan estimates allow a greater proportion of variants to be phased with generally similar accuracy as population-specific estimates, though we do identify certain scenarios where more caution is required.
Fig. 3: Phasing accuracy using population-specific versus cosmopolitan estimates.
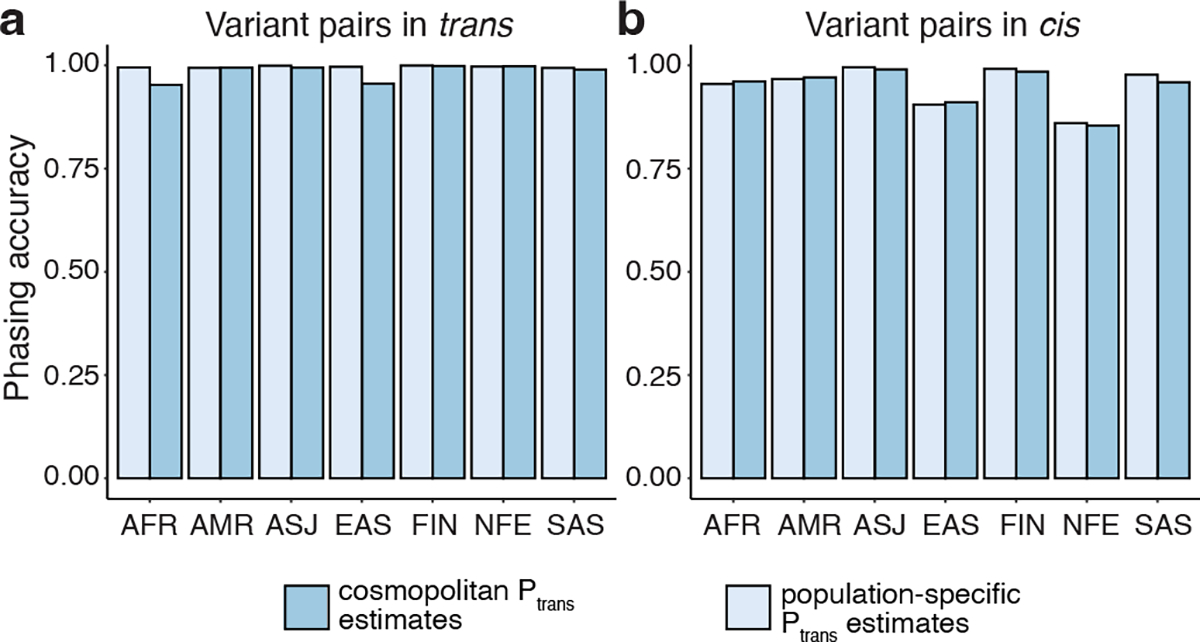
Population-specific estimates are shown in light blue and cosmopolitan estimates are shown in medium blue. Accuracies are shown separately for variants in trans (a, left) and variants in cis (b, right).
Effect of distance and mutation rate on phasing accuracy
Recombination events, which disrupt the haplotype configuration of variant pairs, should influence phasing accuracy. To explore the impact of recombination, we plotted the accuracy of our phasing estimates as a function of physical distance between variant pairs. For variant pairs in trans, phasing accuracy was maintained across physical distances. However, for variant pairs in cis, accuracy rapidly decreased with longer physical distances (Fig. 4a). Since physical distance is only a proxy for recombination frequency, we also performed this analysis using interpolated genetic distances (Fig. 4b). We found again that variants in trans had preserved phasing accuracy across genetic distances, while variants in cis had phasing estimates that decreased substantially with genetic distance, particularly at distances greater than 0.1 centiMorgan. We also tested the effect of recombination using a set of multinucleotide variants, which are variant pairs in cis and very close in physical distance (see Supplementary Note).
Fig. 4: Phasing accuracy as a function of distance between variant pairs.
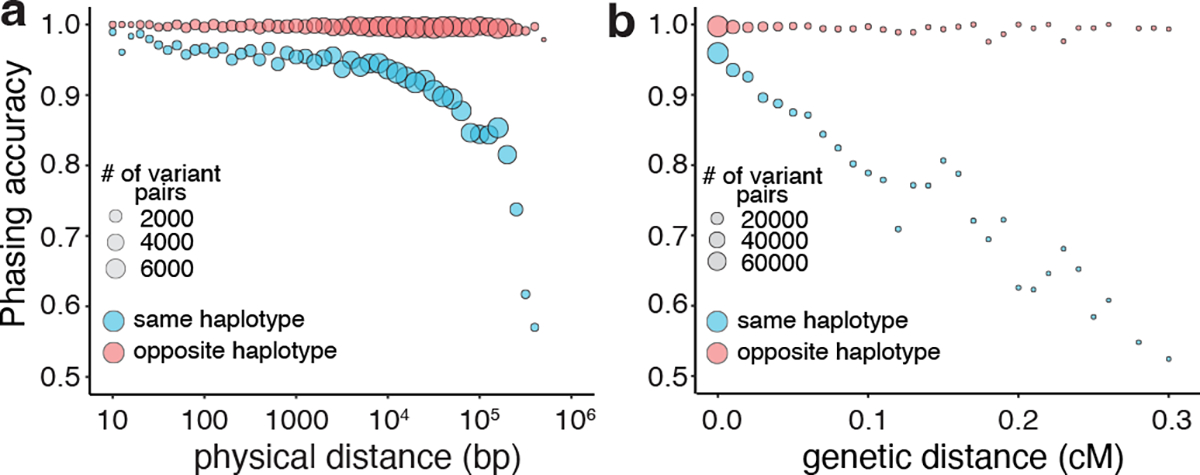
a, Phasing accuracy (y-axis) as a function of physical distance (in base pairs on log10 scale) between variants (x-axis). Blue represents variants on the same haplotype (in cis), and red represents variants on opposite haplotypes (in trans). b, Same as a, except the x-axis shows genetic distance (in centiMorgans). Accuracies for a and b are calculated based on unique variant pairs observed across all genetic ancestry groups using population-specific estimates.
Recurrent germline mutations can also result in inaccurate phasing. Rates of recurrent mutations are dependent on mutation type (e.g., transition versus transversion) and epigenetic marks (particularly CpG methylation), among other factors16–20. Notably, transitions have higher mutation rates than transversions18,21 and CpG transitions have the highest mutation rates, which increase with higher levels of methylation at the CpG14. To better understand the impact of mutation rates on phasing accuracy, we classified each single nucleotide variant in the trio data as a transversion, non-CpG transition, or CpG transition, with further subclassifications of these as having low, medium, or high DNA methylation as before14. We then calculated phasing accuracy as a function of combinations of mutation types using the trio data. We found similar accuracy for transversions and transitions (~0.97) (Supplementary Fig. 8a). However, mutation rates had a strong impact on accuracy for variant pairs in cis but not those in trans (Supplementary Fig. 8b–c). For variant pairs in cis, the phasing accuracies were lower at medium and high methylation CpG sites (0.82–0.89) than they were for low methylation sites (0.96). These results are consistent with recurrent mutations contributing to inaccurate phasing estimates, particularly for variant pairs in cis.
Accuracy in a cohort of patients with Mendelian disorders
To demonstrate our approach in a clinically relevant scenario, we turned to a set of 627 patients from the Broad Institute Center for Mendelian Genetics (CMG)22. All patients had a confident or strong candidate genetic diagnosis of a Mendelian condition based on carrying two rare variants in a recessive disease gene consistent with the patient’s phenotype. For 293 of the 627 patients, both variants were present in gnomAD and thus amenable to phasing (Supplementary Table 1). For these 293 variant pairs, we used population-specific estimates when available (n=215), and cosmopolitan estimates for the remaining 78 variant pairs. Our phasing approach predicted 281 (95.9%) variant pairs to be in trans, seven variant pairs (2.4%) to be in cis, and five (1.7%) as indeterminate ( or singleton/singleton variant in the same individual). Had only cosmopolitan estimates been used, one of the 281 in trans predictions would have been predicted in cis and one indeterminate. Of the seven variant pairs predicted to be in cis, six were from patients with proband-only sequencing. For these patients, the responsible clinician was contacted to ensure phenotype overlap with the disease gene and to pursue parental Sanger sequencing for confirmatory phasing by transmission or long read sequencing, where possible. The remaining variant pair predicted to be in cis originated from a patient with parental data confirming trans phase and thus our inferred phase to be incorrect (Supplementary Table 1). Overall, the results suggest that our phasing approach is highly accurate in clinical scenarios in patients with suspected Mendelian conditions and can be applied to a large fraction (~50% in our cohort) of candidate diagnoses.
Bi-allelic predicted damaging variants
We tabulated for each gene the number of individuals in gnomAD who carry two rare heterozygous variants, stratified by the predicted phase using cutoffs (i.e., in trans, unphased [intermediate ], and in cis), AF, and the predicted functional consequence of the least damaging variant in the pair. For comparison, we tabulated individuals with homozygous variants in the same manner. We classified predicted functional consequences as pLoF, missense with deleteriousness scored by REVEL23 in line with recent ClinGen recommendations24, or synonymous.
Overall, the number of individuals with rare, compound heterozygous (in trans), predicted damaging variants was low (median 0 individuals per gene with compound heterozygous loss-of-function variants at ≤ 1% AF, range 0–9) and only occurred in a small number of genes (Fig. 5 and Supplementary Fig. 9). 28 genes carried compound heterozygous pLoF variants (in 56 individuals) and an additional four genes carried compound heterozygous variants with at least a strong REVEL missense predicted consequence (in six individuals) at ≤ 1% AF cutoff. The vast majority of these genes have not, to date, been associated with disease (Fig. 5b). Manual curation of the pLoF variants resulted in seven high confidence “human knock-out” genes (ARHGEF37, CCDC66, FAM81B, FYB2, GNLY, RBKS, and SDSL). These genes are not associated with Mendelian disease nor are they known to be essential (see Methods). In the remaining 21 of the 28 genes with compound heterozygous pLoF variants, true loss-of-function was found to be uncertain or unlikely following manual curation, due, for example, to the variant falling in the last exon of the gene, in a weakly conserved exon, or in a minority of isoforms (Supplementary Table 2).
Fig. 5: Counts of genes with variants in trans in gnomAD.
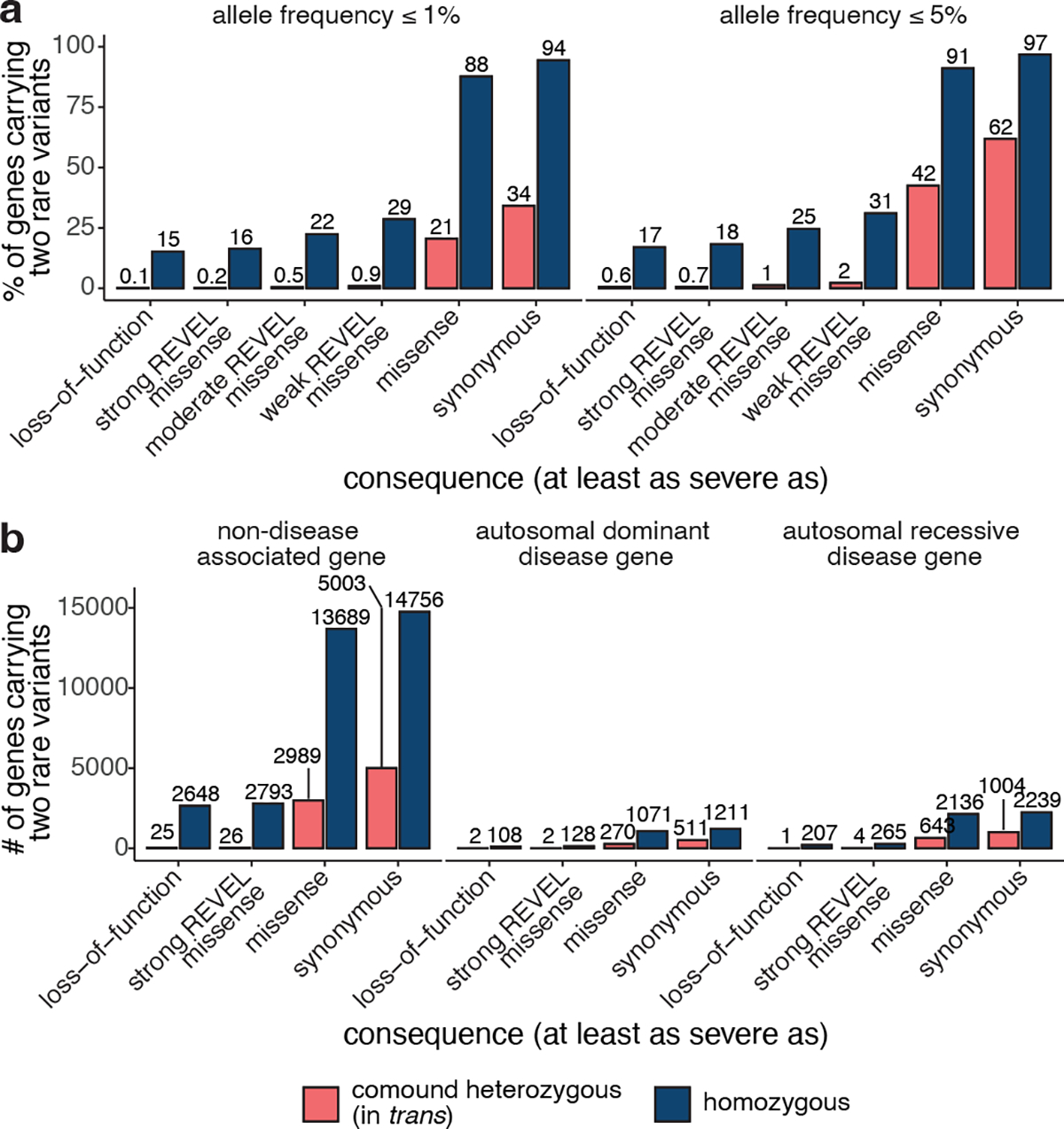
a, Proportion of genes with one or more individuals in gnomAD carrying predicted compound heterozygous (in trans) variants or a homozygous variant at ≤ 1% and ≤ 5% AF stratified by predicted functional consequence. b, Number of genes with ≥ 1 individual in gnomAD carrying compound heterozygous (in trans) or homozygous predicted damaging variants at ≤ 1% AF, stratified by predicted functional consequence and Mendelian disease-association in the Online Mendelian Inheritance in Man database. In total, 28 genes (25 non-disease, 2 autosomal dominant, and 1 autosomal recessive) carried predicted compound heterozygous loss-of-function variants at ≤ 1% AF, only seven of which were high confidence “human knock-out” events following manual curation. For predicted compound heterozygous variants, both variants in the variant pair must be annotated with a consequence at least as severe as the consequence listed (i.e., a compound heterozygous loss-of-function variant would be counted under the pLoF category but also included with a less deleterious variant under the other categories). All homozygous pLoF variants previously underwent manual curation as part of Karczewski et al14.
Generation of public resource
To make our resource widely usable to clinicians and researchers, we have calculated and released pairwise genotype counts and phasing estimates for each pair of rare coding variants occurring in the same gene for gnomAD. We have included all variant pairs within a gene where both variants have global minor AF in gnomAD exomes < 5%, and are either coding, flanking intronic (from position −1 to −3 in acceptor sites, and +1 to +8 in donor sites) or in the 5’/3’ UTRs. We have integrated these data into the gnomAD browser so that users can easily look up a pair of variants to obtain the genotype counts, haplotype frequency estimates, estimates, and likely co-occurrence pattern (Extended Data Fig. 1a). These results are shown for each individual genetic ancestry group and across all genetic ancestries in gnomAD v2. In addition, the data are available as a downloadable table for all variant pairs that were seen in at least one individual.
Furthermore, we have incorporated counts tables detailing the number of individuals carrying two rare variants stratified by AF, and functional consequence. The first table counts individuals carrying two rare heterozygous variants by predicted phase (in trans, unphased, and in cis) and the second table counts individuals carrying homozygous variants (Extended Data Fig. 1b). We envision that these data will aid the medical genetics community in interpreting the clinical significance of co-occurring variants in the context of recessive conditions. The data for all genes are also available as a downloadable table within gnomAD v2.
Discussion
In this work, we leveraged a large exome sequencing cohort to estimate haplotype frequencies for pairs of rare variants within genes and show that these haplotype frequency estimates can be utilized to predict phase of pairs of variants. We achieve high accuracy across a range of allele frequencies and across genetic ancestries and demonstrate that our approach is able to distinguish variants that are likely compound heterozygous in a clinical setting. We freely disseminate our results in an easy-to-use browser for the community.
Our work focuses on the challenging, yet common, scenario of determining phase for rare variants identified in exome sequencing of rare disease patients. While this scenario is common in medical genetics, other phasing approaches such as phasing-by-transmission or population-based phasing are challenging to apply. Our approach of using estimated haplotype frequencies from gnomAD to predict phase of variant pairs was generally accurate across a range of AFs (even for singleton variants) and across genetic ancestries. Most notably, 96.9% of rare (AF < 5%) variant pairs in a given individual had both variants present in gnomAD and therefore were amenable to phasing using our approach, which is much higher than the proportion amenable to phasing using physical read data. Overall, 92.3% of variant pairs observed per individual were correctly phased using our approach. We did find that our approach was less accurate for rare variant pairs in cis (see Supplementary Note). We also found that using “cosmopolitan” phasing estimates that leverage more samples in gnomAD generally had similar accuracy to using population-specific estimates, except for individuals of EAS and AFR genetic ancestry (see Supplementary Note). Thus, our approach can be applied to nearly all rare variant pairs and can generate accurate phasing estimates for variants of medical importance in rare recessive genetic diseases.
We utilized the EM algorithm to phase pairs of variants instead of more sophisticated population-based phasing approaches for several reasons8–11. First, exome and targeted gene panel sequencing data are sparse, precluding the use of common non-coding variants as a “scaffold” for population-based phasing approaches. Recent work performed population-based phasing of rare variants from exome sequencing data by combining exome data with SNP genotyping arrays11,25. However, SNP genotyping data are not usually generated in conjunction with a clinical sequencing test and were not readily available for much of gnomAD. Second, rare variants, which are of the greatest interest in Mendelian diseases, are challenging to phase using population-based approaches given the small numbers of shared haplotypes from which to derive phasing estimates in the population. Recent methods have shown accurate phasing of rare variants using genome sequencing data10,11,26, but rely on a large genome reference panel. As the numbers of whole genome sequencing samples increases in future releases of gnomAD, this may represent a tractable and more accurate approach for phasing of rare variants. Exome sequencing and targeted gene sequencing remain commonly used in clinical settings, and thus we anticipate that our approach and the resources we have generated will remain useful. Third, we found that application of the EM algorithm to pairs of variants was more intuitive to illustrate how phasing estimates were derived from genotype data, allowing users to more easily assess the reliability of phasing estimates. Together, the EM algorithm provided us with the unique ability to phase pairs of rare variants in exome data in an intuitive fashion.
We found that there are only a small number of “human knock-out” genes affected by predicted compound heterozygous (in trans) loss-of-function variants, and that this number is substantially lower than is observed for homozygous loss-of-function variants. These compound heterozygous “human knock-out” events occurred in genes that are not known to be essential, an expected finding given that gnomAD is largely depleted of individuals with severe and early-onset conditions. When analyzing the 23,672 individuals that carry two rare (AF ≤ 1%) pLoF variants, we predict that in 20,421 (86%) of those individuals the variant pair is in cis and only ~0.2% confidently predicted to be in trans. This observation emphasizes that when a pair of rare pLoF variants is observed in the same gene in an individual from a general population sample, it is vastly more likely that these variants are carried on the same haplotype than that the individual is a genuine “knock-out” due to compound heterozygosity. We note, however, that our ability to identify rare variant pairs in trans in gnomAD v2 individuals is limited by the fact the same dataset was used for training (see Supplementary Note). We have released counts of co-occurring variant pairs within genes as observed in gnomAD, which will aid with interpretation of the clinical significance of co-occurring variant pairs.
There are several other important limitations to our work. First, to limit computational burden, we only report phasing estimates for rare coding and flanking intronic/UTR variant pairs within genes. These are the variant pairs of most interest to the medical genetics community, though we acknowledge that phase of deeper intronic variation will become important as more genome sequencing is performed. Second, future studies would benefit from even larger sample sizes, especially for genetic ancestry groups not well represented in our present study. Finally, we have only tested our phasing accuracy in a clinical setting in a retrospective manner and future prospective studies will be needed to confirm the clinical utility of our approach.
Methods
Ethical compliance and informed consent statement
Our collaborators obtained informed consent for all participants in the Broad Institute Center for Mendelian Genetics (CMG), and individual-level data, including genomics and clinical data, were de-identified and coded prior to our analyses in this work. We have complied with all relevant ethical regulations. The Broad Institute of MIT and Harvard, and Mass General Brigham IRB approved this work
gnomAD characteristics and data processing
In this work, we used exome sequencing data from the gnomAD v2.1 dataset (n = 125,748 individuals, GRCh37). These data were uniformly processed, underwent joint variant calling, and rigorous quality control, as described in Karczewski et al.14. Briefly, we aggregated ~200k exome and ~20k genome sequencing samples, primarily from case-control studies of common adult-onset conditions, and applied a BWA-Picard-GATK pipeline27. Using Hail (https://github.com/hail-is/hail), we then removed samples that (1) failed population- and platform-specific quality control, (2) had second-degree or closer relations in the dataset, (3) did not have appropriate consent for release, and (4) had known severe, early-onset conditions. For variant quality control, we trained a random forest on site-level and genotype-level metrics (e.g., the quality by depth, QD), and demonstrated that it achieved both high precision and recall for both common and rare variants.
We subsetted the final cleaned gnomAD dataset for variants with global AF in gnomAD exomes < 5% that were either coding, flanking intronic (from position −1 to −3 in acceptor sites, and +1 to +8 in donor sites) or in the 5’/3’ UTRs. In total, this encompasses 5,320,037,963 unique variant pairs across 19,877 genes when removing singleton/singleton variant pairs observed in the same individual. We specifically extracted 20,921,100 pairs of variants, most of which were observed at least once in the same individual to create a more manageable downloadable file.
We performed analysis in this manuscript using Hail version 0.2.10528, and analysis code is available at https://github.com/broadinstitute/gnomad_chets.
Haplotype estimates
Consider two variants, A and B. A and B represent the major alleles, and a and b represent the respective minor alleles. There are thus 9 pairwise genotypes for A and B: AABB, AaBB, aaBB, AABb, AaBb, aaBb, AAbb, Aabb, and aabb. Of these pairwise genotypes, only the phase for the double heterozygote (AaBb) is unknown. From these 9 possible genotypes, there are four possible haplotype configurations: AB, Ab, aB, and ab.
For each pair of variants, we applied the expectation-maximization (EM) algorithm15 to estimate haplotype frequencies from genotype counts. We set the initial conditions of the EM algorithm by partitioning the doubly heterozygous (AaBb) genotype counts equally between the AB|ab and Ab|aB haplotype configurations. We ran the EM algorithm until convergence or until the absolute value of the difference between consecutive maximum likelihood function values was less than 1×10−7. We calculated haplotype frequencies based on genotypes present within the same genetic ancestry group (“population-specific”) or using all samples from gnomAD (“cosmopolitan”). We performed these analyses of haplotype frequency estimates using Hail.
We then calculate as the likelihood that any given pair of doubly heterozygous variants (AaBb) in a patient is compound heterozygous (Ab|aB). can be calculated simply from the haplotype frequency estimates (AB, Ab, aB, and ab):
Thus, simply represents the probability that the patient is compound heterozygous by inheriting both the Ab and aB haplotypes.
Determination of cutoffs
To determine cutoffs for classifying variants as occurring in cis or trans, we binned variant pairs on odd chromosomes (chromosome 1, 3, 5, etc) in increments of 0.01. For each bin, we calculated the proportion of variant pairs in that bin that are in trans based on phasing by trio data. We determined the threshold for variants in trans as the minimum such that 90% of variants in the bin are in trans based on trio data. We determined the threshold for variants in cis as the maximum such that 90% of variants in the bin are in cis based on trio data. For these calculations, we used only variants where both variants had a population AF ≥ 1×10−4. We used trio samples across all genetic ancestry groups and population-specific values for determination of the cutoffs.
Trio validation data
For validation of our phasing approach, we utilized 4,992 parent-child trios that were jointly processed and variant-called with gnomAD. Having access to parental genotypes allows us to perform phase-by-transmission and accurately determine whether two co-occurring variants in the same gene are in cis or in trans.
First, we estimated genetic ancestry of each individual in the trios by using ancestry inference estimates from the full gnomAD dataset, as previously described14. Briefly, we selected bi-allelic variants that passed all hard filters, had allele frequencies in a joint exome and genome callset > 0.001, and high joint call rates (> 0.99). The variants were then LD-pruned (r2 = 0.1) and used in a principal component analysis (PCA). We previously used samples with known genetic ancestry to train a random forest on the first 20 principal components (PCs) and assigned samples to a genetic ancestry group based on having a random forest probability > 0.9. For the trios in this cohort, we projected their PCs for genetic ancestry onto the same gnomAD v2 samples to infer the genetic ancestry used here (Supplementary Fig. 1). Of these 4,922 trios, 4,775 of the children from the trios were assigned to one of the seven genetic ancestry groups in this study based on PCA and were used in this study.
We then phased the trio data using the Hail phase_by_transmission (https://hail.is/docs/0.2/experimental/index.html#hail.experimental.phase_by_transmission) function, which uses Mendelian transmission of alleles to infer haplotypes in trios for all sites that are not heterozygous in all members of the trio. Assigning haplotypes in trios based on parental genotype has traditionally been the gold standard, has switch error rates below 0.1%, and importantly errors aren’t dependent on the allele frequency of the variants phased29. To maximize our confidence in the genotypes and phasing, we filtered genotypes to include only those with genotype quality (GQ) > 20, depth > 10 and allele balance > 0.2 for heterozygous genotypes prior to phasing. Sex chromosomes were excluded. In total, there were 339,857 unique variant pairs and 1,115,347 total variant pairs.
We compared trio phasing-by-transmission with phasing using our approach on even chromosomes (e.g., chromosomes 2, 4, 6, etc). 3,836 of the 4,775 trio samples were in the full release of gnomAD and were removed from gnomAD for trio validation. This resulted in a set of 121,912 gnomAD samples from which we derived haplotype estimates. We then performed phasing using the EM algorithm and calculated as above.
Based on the estimates, we classified trio variant pairs into 1) unable to phase using our approach (either variant not seen in gnomAD, or singleton/singleton variant pairs observed in the same individual in gnomAD), 2) indeterminate phase (those with intermediate ), 3) incorrectly phased, or 4) correctly phased. We calculated accuracy as the number of variant pairs correctly phased divided by the number of pairs correctly and incorrectly phased.
CpG analysis
We divided single nucleotide variants seen in the trio data into transitions and transversions. Transitions were further subdivided into those that are CpG mutations (5’-CpG-3’ mutating to 5’-TpG-3’) and those that are not. For each CpG transition, we calculated the mean DNA methylation values across 37 tissues in the Roadmap Epigenomics Project30 and then stratified CpG transitions into 3 levels: low (missing or < 0.2), medium (0.2–0.6), and high (> 0.6) methylation, as detailed in Karczewski et al14. We calculated phasing accuracy as the number of variant pairs correctly phased divided by the number of pairs correctly and incorrectly phased. We calculated phasing accuracy for all pairwise combinations of transversions, non-CpG transitions, low methylation CpG transitions, medium methylation CpG transitions, and high methylation CpG transitions. We included all single nucleotide variants in the analysis and utilized population-specific EM estimates.
Calculating accuracy as a function of genetic distance
To estimate the genetic distance between pairs of genetic variants, we interpolated genetic distances between variant pairs using a genetic map from HapMap231 (https://github.com/joepickrell/1000-genomes-genetic-maps). We utilized a HapMap2 genetic map representing average over recombination rates in the CEU, YRI, and ASN populations. We then ran interpolate_maps.py (downloaded from https://github.com/joepickrell/1000-genomes-genetic-maps/blob/master/scripts/interpolate_maps.py) for all variant pairs in the phasing trio data. We used population-specific estimates and calculated phasing accuracy as the number of variant pairs correctly phased divided by the number of pairs correctly and incorrectly phased.
MNV analysis
We obtained multi-nucleotide variant pairs for which read-back phasing had previously been calculated1. We included all multi-nucleotide variant pairs where each constituent variant was analyzed in our study. We utilized cosmopolitan estimates and calculated phasing accuracy as the number of variant pairs correctly phased divided by the number of pairs correctly and incorrectly phased.
Rare disease patient analysis
We selected 627 patients from the Broad Institute Center for Mendelian Genetics (CMG)22 with a confident or strong candidate genetic diagnosis of a Mendelian condition. Each patient carried two presumed bi-allelic variants in an autosomal recessive disease gene consistent with the patient’s phenotype. For 293 of the patients, both variants were present in gnomAD and thus were amenable to our phasing approach. For 168 of the 293 patients, trio-sequencing (i.e., sequencing of the proband and the two unaffected biological parents) had been performed. For these 168 individuals with parental DNA sequencing, we were able to confirm phasing of the two variants via phase-by-transmission.
Determining counts of individuals with two rare, damaging variants
We annotated variants by the worst consequence on the canonical transcript by the Ensembl Variant Effect Predictor (VEP)32. We applied LOFTEE14 to annotate LoF variants and counted only high confidence LoF variants as “pLoF”. We used REVEL23 in line with recent ClinGen recommendations24: we counted REVEL scores ≥ 0.932 as “strong_revel_missense”, ≥ 0.773 as “moderate_to_strong_revel_missense”, and ≥ 0.644 as “weak_to_strong_revel_missense”.
We predicted phase (cis or trans) based on the thresholds for all variant pairs. All singleton/singleton variant pairs (AC = 1) and variant pairs with an indeterminate values () were annotated as unphased.
We selected five AF thresholds for analysis and filtered variant pairs based on the highest global AF and, where available, the “popmax” AF of each variant in gnomAD (i.e., the highest AF information for the non-bottlenecked population [AFR, AMR, EAS, NFE, SAS]): 0.5%, 1%, 1.5%, 2%, and 5%. We also filtered out all variant pairs containing a variant with an AF > 5% in a bottlenecked population.
We performed gene-wise calculations of the number of individuals carrying a variant pair (irrespective of phase) and the number predicted to be in trans, unphased (indeterminate), and the number predicted to be in cis. We performed gene-wise calculations separately by AF threshold and functional consequences (26 consequence groups). If individuals carried multiple variant pairs in the same gene with different phase predictions, we counted the individual in only one phase group, prioritizing in trans over unphased and unphased over in cis. These gene-wise counts are displayed in the “Variant Co-occurrence” gnomAD browser feature. For individuals carrying multiple variant pairs in the same gene with different phase predictions, we also performed separate calculations allowing these individuals to be counted in multiple phase groups (data available to download).
Essential gene lists
We queried the following essential gene lists for the presence of the true “human knock-out” genes identified in this study:
2,454 genes essential in mice from Georgi et al. 201333
553 pan-cancer core fitness genes from Behan et al., 201934
360 core essential genes from genomic perturbation screens from Hart et al. 201435
684 genes essential in culture by CRISPR screening from Hart et al. 201736
1,075 genes annotated by the ADaM analysis of a large collection of gene dependency profiles (CRISPR-Cas9 screens) across human cancer cell lines from Vinceti et al. 202137
Extended Data
Extended Data Fig. 1: Publicly available browser for sharing phasing data.
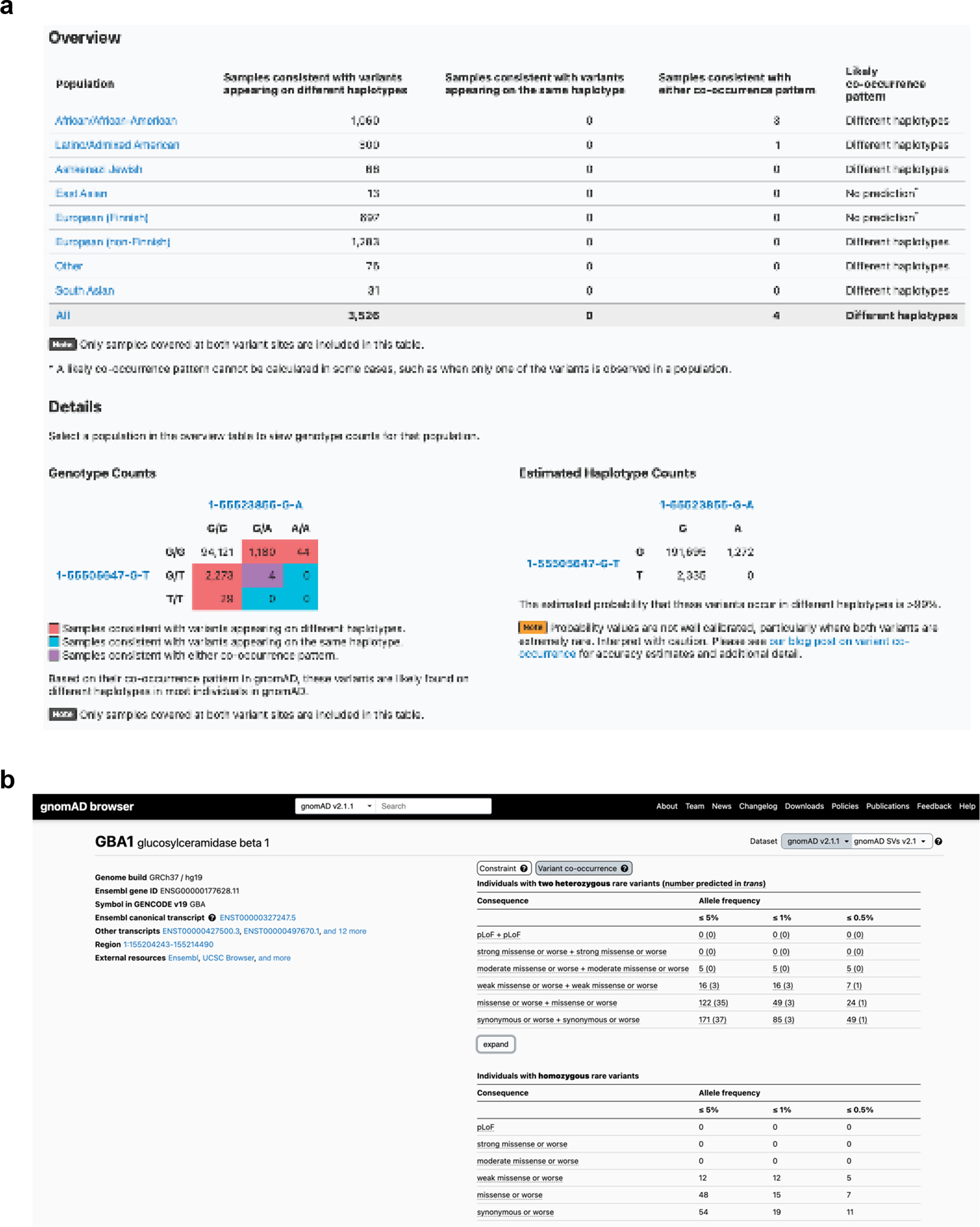
a, Sample gnomAD browser output for two variants (GRCh37 1–55505647-G-T and 1–55523855-G-A) in the gene PCSK9. On the top, a table subdivided by genetic ancestry group displays how many individuals in gnomAD v2 from that genetic ancestry are consistent with the two variants occurring on different haplotypes (trans), and how many individuals are consistent with their occurring on the same haplotype (cis). Below that, there is a 3×3 table that contains the 9 possible combinations of genotypes for the two variants of interest. The number of individuals in gnomAD v2 that fall in each of these combinations are shown and are colored by whether they are consistent with variants falling on different haplotypes (red) or the same haplotype (blue), or whether they are indeterminate (purple). The estimated haplotype counts for the four possible haplotypes for the two variants as calculated by the EM algorithm is displayed on the bottom right. The probability of being in trans for this particular pair of variants is >99%. b, Variant co-occurrence tables on the gene landing page. For each gene (GBA1 shown), the top table lists the number of individuals carrying pairs of rare heterozygous variants by inferred phase, allele frequency (AF), and predicted functional consequence. The number of individuals with homozygous variants are tabulated in the same manner and presented as a comparison below. AF thresholds of ≤ 5%, ≤ 1%, and ≤ 0.5% are displayed across six predicted functional consequences (combinations of pLoF, various evidence strengths of predicted pathogenicity for missense variants, and synonymous variants). Both variants in the variant pair must be annotated with a consequence at least as severe as the consequence listed (that is, pLoF + strong missense also includes pLoF + pLoF).
Supplementary Material
Acknowledgements
We would like to thank all members of the gnomAD team for helpful comments and suggestions, and to particularly recognize the members of the gnomAD methods and browser teams who worked hard over many years to provide cleaned datasets, easy-to-use browsers, and visualizations. This work was supported by NHGRI U24HG011450 (H.L.R., M.J.D.), UM1HG008900 (D.G.M., H.L.R.), and U01HG011755 (A.O.D.L., H.L.R.).
Genome Aggregation Database Consortium
Maria Abreu14, Carlos A. Aguilar Salinas15, Tariq Ahmad16, Christine M. Albert17,18, Jessica Alföldi2,3, Diego Ardissino19, Irina M. Armean2,3,20, Gil Atzmon21,22, Eric Banks23, John Barnard24, Samantha M. Baxter2, Laurent Beaugerie25, Emelia J. Benjamin26,27,28, David Benjamin23, Louis Bergelson23, Michael Boehnke29, Lori L. Bonnycastle30, Erwin P. Bottinger31, Donald W. Bowden32,33,34, Matthew J. Bown35,36, Steven Brant37, Sarah E. Calvo2,10, Hannia Campos38,39, John C. Chambers40,41,42, Juliana C. Chan43, Katherine R. Chao2, Sinéad Chapman2,3,5, Daniel Chasman17,44, Siwei Chen2,3, Rex L. Chisholm45, Judy Cho31, Rajiv Chowdhury46, Mina K. Chung47, Wendy K. Chung48,49,50, Kristian Cibulskis23, Bruce Cohen44,51, Ryan L. Collins2,10,52, Kristen M. Connolly53, Adolfo Correa54, Miguel Covarrubias23, Beryl Cummings2,52, Dana Dabelea55, Mark J. Daly2,3,11, John Danesh46, Dawood Darbar56, Joshua Denny57, Stacey Donnelly2, Ravindranath Duggirala58, Josée Dupuis59,60, Patrick T. Ellinor2,61, Roberto Elosua62,63,64, James Emery23, Eleina England2, Jeanette Erdmann65,66,67, Tõnu Esko2,68, Emily Evangelista2, Yossi Farjoun23, Diane Fatkin69,70,71, Steven Ferriera72, Jose Florez44,73,74, Laurent C. Francioli2,3, Andre Franke75, Martti Färkkilä76, Stacey Gabriel77, Kiran Garimella23, Laura D. Gauthier23, Jeff Gentry23, Gad Getz44,77,78, David C. Glahn79,80, Benjamin Glaser81, Stephen J. Glatt82, David Goldstein83,84, Clicerio Gonzalez85, Julia K. Goodrich2,3, Leif Groop86,87, Sanna Gudmundsson2,3,4, Namrata Gupta2,72, Andrea Haessly23, Christopher Haiman88, Ira Hall89, Craig Hanis90, Matthew Harms91,92, Mikko Hiltunen93, Matti M. Holi94, Christina M. Hultman95,96, Chaim Jalas97, Thibault Jeandet23, Mikko Kallela98, Diane Kaplan23, Jaakko Kaprio87, Konrad J. Karczewski2,3,6,10, Sekar Kathiresan10,44,99, Eimear Kenny96,100, Bong-Jo Kim101, Young Jin Kim101, George Kirov102, Zan Koenig2, Jaspal Kooner41,103,104, Seppo Koskinen105, Harlan M. Krumholz106, Subra Kugathasan107, Soo Heon Kwak108, Markku Laakso109,110, Nicole Lake111, Trevyn Langsford23, Kristen M. Laricchia2,3, Terho Lehtimäki112, Monkol Lek111, Emily Lipscomb2, Christopher Llanwarne23, Ruth J.F. Loos31,113, Steven A. Lubitz2,61, Teresa Tusie Luna114,115, Ronald C.W. Ma43,116,117, Daniel G. MacArthur2,3,12,13, Gregory M. Marcus118, Jaume Marrugat64,119, Alicia R. Martin2, Kari M. Mattila112, Steven McCarroll5,120, Mark I. McCarthy121,122,123, Jacob McCauley124,125, Dermot McGovern126, Ruth McPherson127, James B. Meigs2,44,128, Olle Melander129, Andres Metspalu130, Deborah Meyers131, Eric V. Minikel2, Braxton D. Mitchell132, Vamsi K. Mootha2,133, Ruchi Munshi23, Aliya Naheed134, Saman Nazarian135,136, Benjamin M. Neale2,3,5,6, Peter M. Nilsson137, Sam Novod23, Anne H. O’Donnell-Luria2,3,4,10, Michael C. O’Donovan102, Yukinori Okada138,139,140, Dost Ongur44,51, Lorena Orozco141, Michael J. Owen102, Colin Palmer142, Nicholette D. Palmer143, Aarno Palotie3,5,87, Kyong Soo Park108,144, Carlos Pato145, Nikelle Petrillo23, William Phu2,4, Timothy Poterba2,3,5, Ann E. Pulver146, Dan Rader135,147, Nazneen Rahman148, Heidi L. Rehm2,3,10, Alex Reiner149,150, Anne M. Remes151, Dan Rhodes2, Stephen Rich152,153, John D. Rioux154,155, Samuli Ripatti87,156,157, David Roazen23, Dan M. Roden158,159, Jerome I. Rotter160, Valentin Ruano-Rubio23, Nareh Sahakian23, Danish Saleheen161,162,163, Veikko Salomaa164, Andrea Saltzman2, Nilesh J. Samani35,36, Kaitlin E. Samocha2,3,10, Jeremiah Scharf2,5,10, Molly Schleicher2, Heribert Schunkert165,166, Sebastian Schönherr167, Eleanor Seaby2, Cotton Seed3,5, Svati H. Shah168, Megan Shand23, Moore B. Shoemaker169, Tai Shyong170,171, Edwin K. Silverman172,173, Moriel Singer-Berk2, Pamela Sklar174,175,176, J. Gustav Smith157,177,178, Jonathan T. Smith23, Hilkka Soininen179, Harry Sokol180,181,182, Matthew Solomonson2,3, Rachel G. Son2, Jose Soto23, Tim Spector183, Christine Stevens2,3,5, Nathan Stitziel89,184, Patrick F. Sullivan95,185, Jaana Suvisaari164, E. Shyong Tai186,187,188, Michael E. Talkowski2,5,10, Yekaterina Tarasova2, Kent D. Taylor160, Yik Ying Teo186,189,190, Grace Tiao2,3, Kathleen Tibbetts23, Charlotte Tolonen23, Ming Tsuang191,192, Tiinamaija Tuomi87,193,194, Dan Turner195, Teresa Tusie-Luna196,197, Erkki Vartiainen198, Marquis Vawter199, Christopher Vittal2,3, Gordon Wade23, Arcturus Wang2,3,5, Qingbo Wang2,138, James S. Ware2,200,201, Hugh Watkins202, Nicholas A. Watts2,3, Rinse K. Weersma203, Ben Weisburd23, Maija Wessman87,204, Nicola Whiffin2,205,206, Michael W. Wilson2,3, James G. Wilson207, Ramnik J. Xavier208,209, Mary T. Yohannes2
14University of Miami Miller School of Medicine, Gastroenterology, Miami, USA
15Unidad de Investigacion de Enfermedades Metabolicas, Instituto Nacional de Ciencias Medicas y Nutricion, Mexico City, Mexico
16Peninsula College of Medicine and Dentistry, Exeter, UK
17Division of Preventive Medicine, Brigham and Women’s Hospital, Boston, MA, USA
18Division of Cardiovascular Medicine, Brigham and Women’s Hospital and Harvard Medical School, Boston, MA, USA
19Department of Cardiology University Hospital, Parma, Italy
20European Molecular Biology Laboratory, European Bioinformatics Institute, Wellcome Genome Campus, Hinxton, Cambridge, UK
21Department of Biology Faculty of Natural Sciences, University of Haifa, Haifa, Israel
22Departments of Medicine and Genetics, Albert Einstein College of Medicine, Bronx, NY, USA
23Data Science Platform, Broad Institute of MIT and Harvard, Cambridge, MA, USA
24Department of Quantitative Health Sciences, Lerner Research Institute Cleveland Clinic, Cleveland, OH, USA
25Sorbonne Université, APHP, Gastroenterology Department Saint Antoine Hospital, Paris, France
26NHLBI and Boston University’s Framingham Heart Study, Framingham, MA, USA
27Department of Medicine, Boston University Chobanian and Avedisian School of Medicine, Boston, MA, USAD
28Department of Epidemiology, Boston University School of Public Health, Boston, MA, USA
29Department of Biostatistics and Center for Statistical Genetics, University of Michigan, Ann Arbor, MI, USA
30National Human Genome Research Institute, National Institutes of Health Bethesda, MD, USA
31The Charles Bronfman Institute for Personalized Medicine, Icahn School of Medicine at Mount Sinai, New York, NY, USA
32Department of Biochemistry, Wake Forest School of Medicine, Winston-Salem, NC, USA
33Center for Genomics and Personalized Medicine Research, Wake Forest School of Medicine, Winston-Salem, NC, USA
34Center for Diabetes Research, Wake Forest School of Medicine, Winston-Salem, NC, USA
35Department of Cardiovascular Sciences, University of Leicester, Leicester, UK
36NIHR Leicester Biomedical Research Centre, Glenfield Hospital, Leicester, UK
37John Hopkins Bloomberg School of Public Health, Baltimore, MD, USA
38Harvard School of Public Health, Boston, MA, USA
39Central American Population Center, San Pedro, Costa Rica
40Department of Epidemiology and Biostatistics, Imperial College London, London, UK
41Department of Cardiology, Ealing Hospital, NHS Trust, Southall, UK
42Imperial College, Healthcare NHS Trust Imperial College London, London, UK
43Department of Medicine and Therapeutics, The Chinese University of Hong Kong, Hong Kong, China
44Department of Medicine, Harvard Medical School, Boston, MA, USA
45Northwestern University Feinberg School of Medicine, Chicago, IL, USA
46University of Cambridge, Cambridge, England
47Departments of Cardiovascular, Medicine Cellular and Molecular Medicine Molecular Cardiology, Quantitative Health Sciences, Cleveland Clinic, Cleveland, OH, USA
48Department of Pediatrics, Columbia University Irving Medical Center, New York, NY, USA
49Herbert Irving Comprehensive Cancer Center, Columbia University Medical Center, New York, NY, USA
50Department of Medicine, Columbia University Medical Center, New York, NY, USA
51McLean Hospital, Belmont, MA, USA
52Division of Medical Sciences, Harvard Medical School, Boston, MA, USA
53Genomics Platform, Broad Institute of MIT and Harvard, Cambridge, MA, USA
54Department of Medicine, University of Mississippi Medical Center, Jackson, MI, USA
55Department of Epidemiology Colorado School of Public Health Aurora, CO, USA
56Department of Medicine and Pharmacology, University of Illinois at Chicago, Chicago, IL, USA
57Vanderbilt University Medical Center, Nashville, TN, USA
58Department of Genetics, Texas Biomedical Research Institute, San Antonio, TX, USA
59Department of Biostatistics, Boston University School of Public Health, Boston, MA, USA
60National Heart Lung and Blood Institute’s Framingham Heart Study, Framingham, MA, USA
61Cardiac Arrhythmia Service and Cardiovascular Research Center, Massachusetts General Hospital, Boston, MA, USA
62Cardiovascular Epidemiology and Genetics Hospital del Mar Medical Research Institute, (IMIM) Barcelona Catalonia, Spain
63CIBER CV Barcelona, Catalonia, Spain
64Departament of Medicine, Medical School University of Vic-Central, University of Catalonia, Vic Catalonia, Spain
65Institute for Cardiogenetics, University of Lübeck, Lübeck, Germany
66German Research Centre for Cardiovascular Research, Hamburg/Lübeck/Kiel, Lübeck, Germany
67University Heart Center Lübeck, Lübeck, Germany
68Estonian Genome Center, Institute of Genomics University of Tartu, Tartu, Estonia
69Victor Chang Cardiac Research Institute, Darlinghurst, NSW, Australia
70Faculty of Medicine, UNSW Sydney, Kensington, NSW, Australia
71Cardiology Department, St Vincent’s Hospital, Darlinghurst, NSW, Australia
72Broad Genomics, Broad Institute of MIT and Harvard, Cambridge, MA, USA
73Diabetes Unit and Center for Genomic Medicine, Massachusetts General Hospital, Boston, MA, USA
74Programs in Metabolism and Medical & Population Genetics, Broad Institute of MIT and Harvard, Cambridge, MA, USA
75Institute of Clinical Molecular Biology, (IKMB) Christian-Albrechts-University of Kiel, Kiel, Germany
76Helsinki University and Helsinki University Hospital Clinic of Gastroenterology, Helsinki, Finland
77Bioinformatics Program MGH Cancer Center and Department of Pathology, Boston, MA, USA
78Cancer Genome Computational Analysis, Broad Institute of MIT and Harvard, Cambridge, MA, USA
79Department of Psychiatry and Behavioral Sciences, Boston Children’s Hospital and Harvard Medical School, Boston, MA, USA
80Harvard Medical School Teaching Hospital, Boston, MA, USA
81Department of Endocrinology and Metabolism, Hadassah Medical Center and Faculty of Medicine, Hebrew University of Jerusalem, Israel
82Department of Psychiatry and Behavioral Sciences, SUNY Upstate Medical University, Syracuse, NY, USA
83Institute for Genomic Medicine, Columbia University Medical Center Hammer Health Sciences, New York, NY, USA
84Department of Genetics & Development Columbia University Medical Center, Hammer Health Sciences, New York, NY, USA
85Centro de Investigacion en Salud Poblacional, Instituto Nacional de Salud Publica, Mexico
86Lund University Sweden, Sweden
87Institute for Molecular Medicine Finland, (FIMM) HiLIFE University of Helsinki, Helsinki, Finland
88Lund University Diabetes Centre, Malmö, Skåne County, Sweden
89Washington School of Medicine, St Louis, MI, USA
90Human Genetics Center University of Texas Health Science Center at Houston, Houston, TX, USA
91Department of Neurology Columbia University, New York City, NY, USA
92Institute of Genomic Medicine, Columbia University, New York City, NY, USA
93Institute of Biomedicine, University of Eastern Finland, Kuopio, Finland
94Department of Psychiatry, Helsinki University Central Hospital Lapinlahdentie, Helsinki, Finland
95Department of Medical Epidemiology and Biostatistics, Karolinska Institutet, Stockholm, Sweden
96Icahn School of Medicine at Mount Sinai, New York, NY, USA
97Bonei Olam, Center for Rare Jewish Genetic Diseases, Brooklyn, NY, USA
98Department of Neurology, Helsinki University, Central Hospital, Helsinki, Finland
99Cardiovascular Disease Initiative and Program in Medical and Population Genetics, Broad Institute of MIT and Harvard, Cambridge, MA, USA
100Charles Bronfman Institute for Personalized Medicine, New York, NY, USA
101Division of Genome Science, Department of Precision Medicine, National Institute of Health, Republic of Korea
102MRC Centre for Neuropsychiatric Genetics & Genomics, Cardiff University School of Medicine, Cardiff, Wales
103Imperial College, Healthcare NHS Trust, London, UK
104National Heart and Lung Institute Cardiovascular Sciences, Hammersmith Campus, Imperial College London, London, UK
105Department of Health THL-National Institute for Health and Welfare, Helsinki, Finland
106Section of Cardiovascular Medicine, Department of Internal Medicine, Yale School of Medicine, Center for Outcomes Research and Evaluation Yale-New Haven Hospital, New Haven, CT, USA
107Division of Pediatric Gastroenterology, Emory University School of Medicine, Atlanta, GA, USA
108Department of Internal Medicine, Seoul National University Hospital, Seoul, Republic of Korea
109The University of Eastern Finland, Institute of Clinical Medicine, Kuopio, Finland
110Kuopio University Hospital, Kuopio, Finland
111Department of Genetics, Yale School of Medicine, New Haven, CT, USA
112Department of Clinical Chemistry Fimlab Laboratories and Finnish Cardiovascular Research Center-Tampere Faculty of Medicine and Health Technology, Tampere University, Finland
113The Mindich Child Health and Development, Institute Icahn School of Medicine at Mount Sinai, New York, NY, USA
114National Autonomous University of Mexico, Mexico City, Mexico
115Salvador Zubirán National Institute of Health Sciences and Nutrition, Mexico City, Mexico
116Li Ka Shing Institute of Health Sciences, The Chinese University of Hong Kong, Hong Kong, China
117Hong Kong Institute of Diabetes and Obesity, The Chinese University of Hong Kong, Hong Kong, China
118University of California San Francisco Parnassus Campus, San Francisco, CA, USA
119Cardiovascular Research REGICOR Group, Hospital del Mar Medical Research Institute, (IMIM) Barcelona, Catalonia, Spain
120Department of Genetics, Harvard Medical School, Boston, MA, USA
121Oxford Centre for Diabetes, Endocrinology and Metabolism, University of Oxford, Churchill Hospital Old Road Headington, Oxford, OX, LJ, UK
122Welcome Centre for Human Genetics, University of Oxford, Oxford, OX, BN, UK
123Oxford NIHR Biomedical Research Centre, Oxford University Hospitals, NHS Foundation Trust, John Radcliffe Hospital, Oxford, OX, DU, UK
124John P. Hussman Institute for Human Genomics, Leonard M. Miller School of Medicine, University of Miami, Miami, FL, USA
125The Dr. John T. Macdonald Foundation Department of Human Genetics, Leonard M. Miller School of Medicine, University of Miami, Miami, FL, USA
126F. Widjaja Foundation Inflammatory Bowel and Immunobiology Research Institute Cedars-Sinai Medical Center, Los Angeles, CA, USA
127Atherogenomics Laboratory University of Ottawa, Heart Institute, Ottawa, Canada
128Division of General Internal Medicine, Massachusetts General Hospital, Boston, MA, USA
129Department of Clinical Sciences University, Hospital Malmo Clinical Research Center, Lund University, Malmö, Sweden
130Estonian Genome Center, Institute of Genomics, University of Tartu, Tartu, Estonia
131University of Arizona Health Science, Tuscon, AZ, USA
132Department of Medicine, University of Maryland School of Medicine, Baltimore, MD, USA
133Howard Hughes Medical Institute and Department of Molecular Biology, Massachusetts General Hospital, Boston, MA, USA
134International Centre for Diarrhoeal Disease Research, Bangladesh
135Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, USA
136Johns Hopkins Bloomberg School of Public Health, Baltimore, MD, USA
137Lund University, Dept. Clinical Sciences, Skåne University Hospital, Malmö, Sweden
138Department of Statistical Genetics, Osaka University Graduate School of Medicine, Suita, Japan
139Laboratory of Statistical Immunology, Immunology Frontier Research Center (WPI-IFReC), Osaka University, Suita, Japan
140Integrated Frontier Research for Medical Science Division, Institute for Open and Transdisciplinary Research Initiatives, Osaka University, Suita, Japan
141Instituto Nacional de Medicina Genómica, (INMEGEN) Mexico City, Mexico
142Medical Research Institute, Ninewells Hospital and Medical School University of Dundee, Dundee, UK
143Wake Forest School of Medicine, Winston-Salem, NC, USA
144Department of Molecular Medicine and Biopharmaceutical Sciences, Graduate School of Convergence Science and Technology, Seoul National University, Seoul, Republic of Korea
145Department of Psychiatry Keck School of Medicine at the University of Southern California, Los Angeles, CA, USA
146Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore, MD, USA
147Children’s Hospital of Philadelphia, Philadelphia, PA, USA
148Division of Genetics and Epidemiology, Institute of Cancer Research, London, SM, NG
149University of Washington, Seattle, WA, USA
150Fred Hutchinson Cancer Research Center, Seattle, WA, USA
151Medical Research Center, Oulu University Hospital, Oulu Finland and Research Unit of Clinical Neuroscience Neurology University of Oulu, Oulu, Finland
152Center for Public Health Genomics, University of Virginia, Charlottesville, VA, USA
153Department of Public Health Sciences, University of Virginia, Charlottesville, VA, USA
154Research Center Montreal Heart Institute, Montreal, Quebec, Canada
155Department of Medicine, Faculty of Medicine Université de Montréal, Québec, Canada
156Department of Public Health Faculty of Medicine, University of Helsinki, Helsinki, Finland
157Broad Institute of MIT and Harvard, Cambridge, MA, USA
158Department of Biomedical Informatics Vanderbilt, University Medical Center, Nashville, TN, USA
159Department of Medicine, Vanderbilt University Medical Center, Nashville, TN, USA
160The Institute for Translational Genomics and Population Sciences, Department of Pediatrics, The Lundquist Institute for Biomedical Innovation at Harbor-UCLA Medical Center, Torrance, CA, USA
161Department of Biostatistics and Epidemiology, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, USA
162Department of Medicine, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA, USA
163Center for Non-Communicable Diseases, Karachi, Pakistan
164National Institute for Health and Welfare, Helsinki, Finland
165Deutsches Herzzentrum, München, Germany
166Technische Universität München, Germany
167Institute of Genetic Epidemiology, Department of Genetics and Pharmacology, Medical University of Innsbruck, 6020 Innsbruck, Austria
168Duke Molecular Physiology Institute, Durham, NC
169Division of Cardiovascular Medicine, Nashville VA Medical Center, Vanderbilt University School of Medicine, Nashville, TN, USA
170Division of Endocrinology, National University Hospital, Singapore
171NUS Saw Swee Hock School of Public Health, Singapore
172Channing Division of Network Medicine, Brigham and Women’s Hospital, Boston, MA, USA
173Harvard Medical School, Boston, MA, USA
174Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York, NY, USA
175Department of Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai, New York, NY, USA
176Institute for Genomics and Multiscale Biology, Icahn School of Medicine at Mount Sinai, New York, NY, USA
177The Wallenberg Laboratory/Department of Molecular and Clinical Medicine, Institute of Medicine, Gothenburg University and the Department of Cardiology, Sahlgrenska University Hospital, Gothenburg, Sweden
178Department of Cardiology, Wallenberg Center for Molecular Medicine and Lund University Diabetes Center, Clinical Sciences, Lund University and Skåne University Hospital, Lund, Sweden
179Institute of Clinical Medicine Neurology, University of Eastern Finad, Kuopio, Finland
180Sorbonne Université, INSERM, Centre de Recherche Saint-Antoine, CRSA, AP-HP, Saint Antoine Hospital, Gastroenterology department, F-75012 Paris, France
181INRA, UMR1319 Micalis & AgroParisTech, Jouy en Josas, France
182Paris Center for Microbiome Medicine, (PaCeMM) FHU, Paris, France
183Department of Twin Research and Genetic Epidemiology King’s College London, London, UK
184The McDonnell Genome Institute at Washington University, Seattle, WA, USA
185Departments of Genetics and Psychiatry, University of North Carolina, Chapel Hill, NC, USA
186Saw Swee Hock School of Public Health National University of Singapore, National University Health System, Singapore
187Department of Medicine, Yong Loo Lin School of Medicine National University of Singapore, Singapore
188Duke-NUS Graduate Medical School, Singapore
189Life Sciences Institute, National University of Singapore, Singapore
190Department of Statistics and Applied Probability, National University of Singapore, Singapore
191Center for Behavioral Genomics, Department of Psychiatry, University of California, San Diego, CA, USA
192Institute of Genomic Medicine, University of California San Diego, San Diego, CA, USA
193Endocrinology, Abdominal Center, Helsinki University Hospital, Helsinki, Finland
194Institute of Genetics, Folkhalsan Research Center, Helsinki, Finland
196Juliet Keidan Institute of Pediatric Gastroenterology Shaare Zedek Medical Center, The Hebrew University of Jerusalem, Jerusalem, Israel
196Instituto de Investigaciones Biomédicas, UNAM, Mexico City, Mexico
197Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán, Mexico City, Mexico
198Department of Public Health Faculty of Medicine University of Helsinki, Helsinki, Finland
199Department of Psychiatry and Human Behavior, University of California Irvine, Irvine, CA, USA
200National Heart & Lung Institute & MRC London Institute of Medical Sciences, Imperial College, London, UK
201Royal Brompton & Harefield Hospitals, Guy’s and St. Thomas’ NHS Foundation Trust, London, UK
202Radcliffe Department of Medicine, University of Oxford, Oxford, UK
203Department of Gastroenterology and Hepatology, University of Groningen and University Medical Center Groningen, Groningen, Netherlands
204Folkhälsan Institute of Genetics, Folkhälsan Research Center, Helsinki, Finland
205National Heart & Lung Institute and MRC London Institute of Medical Sciences, Imperial College London, London, UK
206Cardiovascular Research Centre, Royal Brompton & Harefield Hospitals NHS Trust, London, UK
207Department of Physiology and Biophysics, University of Mississippi Medical Center, Jackson, MS, USA
208Program in Infectious Disease and Microbiome, Broad Institute of MIT and Harvard, Cambridge, MA, USA
209Center for Computational and Integrative Biology, Massachusetts General Hospital, Boston, MA, USA
Footnotes
Competing interests
L.C.F. is currently an employee of, and owns stock in, Vertex Pharmaceuticals. B.M.N. is a member of the scientific advisory board at Deep Genomics and Neumora, Inc. (f/k/a RBNC Therapeutics). H.L.R. has received support from Illumina and Microsoft to support rare disease gene discovery and diagnosis. M.J.D. is a founder of Maze Therapeutics and Neumora Therapeutics, Inc. (f/k/a RBNC Therapeutics). A.O.D.L. has consulted for Tome Biosciences and Ono Pharma USA Inc, and is member of the scientific advisory board for Congenica Inc and the Simons Foundation SPARK for Autism study. K.J.K. is a consultant for Tome Biosciences and Vor Biosciences, and a member of the Scientific Advisory Board of Nurture Genomics. D.G.M. is a paid advisor to GlaxoSmithKline, Insitro, Variant Bio and Overtone Therapeutics, and has received research support from AbbVie, Astellas, Biogen, BioMarin, Eisai, Google, Merck, Microsoft, Pfizer, and Sanofi-Genzyme. K.E.S. has received support from Microsoft for work related to rare disease diagnostics. The remaining authors declare no competing interests.
Code Availability
The code used to estimate estimates for variant pairs and to determine the number of individuals carrying rare, compound heterozygous variants can be found at:https://github.com/broadinstitute/gnomad_chets
The code has also been uploaded to Zenodo (https://doi.org/10.5281/zenodo.10034663).
Data Availability
The gnomAD v2 dataset can be accessed at https://gnomad.broadinstitute.org. We made use of prior quality control processing of these and related data. In addition, we downloaded HapMap2 genetic maps from https://github.com/joepickrell/1000-genomes-genetic-maps.
We provide both web-based look up tools and downloads for the data generated here. A look-up tool to find the likely co-occurrence pattern between two rare (global allele frequency in gnomAD exomes < 5%) coding, flanking intronic (from position −1 to −3 in acceptor sites, and +1 to +8 in donor sites) or 5’/3’ UTR variants can be found at:https://gnomad.broadinstitute.org/variant-cooccurrence
Additionally, we display the per-gene counts tables that detail the number of individuals with two rare variants, stratified by AF and functional consequence, on each gene’s main page. One table details counts of individuals with two heterozygous variants and includes predicted phase, while the second details individuals with homozygous variants. Both can be found by clicking on the “Variant Co-occurrence” tab on each gene’s main page.
All variant co-occurrence tables can be downloaded from:https://gnomad.broadinstitute.org/downloads#v2-variant-cooccurrence
References
- 1.Wang Q et al. Landscape of multi-nucleotide variants in 125,748 human exomes and 15,708 genomes. Nat. Commun. 11, 2539 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bansal V, Halpern AL, Axelrod N & Bafna V An MCMC algorithm for haplotype assembly from whole-genome sequence data. Genome Res. 18, 1336–1346 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Patterson M et al. WhatsHap: Weighted Haplotype Assembly for Future-Generation Sequencing Reads. J. Comput. Biol. 22, 498–509 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Hager P, Mewes H-W, Rohlfs M, Klein C & Jeske T SmartPhase: Accurate and fast phasing of heterozygous variant pairs for genetic diagnosis of rare diseases. PLoS Comput. Biol. 16, e1007613 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maestri S et al. A Long-Read Sequencing Approach for Direct Haplotype Phasing in Clinical Settings. Int. J. Mol. Sci. 21, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mantere T, Kersten S & Hoischen A Long-Read Sequencing Emerging in Medical Genetics. Front. Genet. 10, 426 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Snyder MW, Adey A, Kitzman JO & Shendure J Haplotype-resolved genome sequencing: experimental methods and applications. Nat. Rev. Genet. 16, 344–358 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Li N & Stephens M Modeling linkage disequilibrium and identifying recombination hotspots using single-nucleotide polymorphism data. Genetics 165, 2213–2233 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Loh P-R et al. Reference-based phasing using the Haplotype Reference Consortium panel. Nat. Genet. 48, 1443–1448 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Browning BL, Tian X, Zhou Y & Browning SR Fast two-stage phasing of large-scale sequence data. Am. J. Hum. Genet. 108, 1880–1890 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hofmeister RJ, Ribeiro DM, Rubinacci S & Delaneau O Accurate rare variant phasing of whole-genome and whole-exome sequencing data in the UK Biobank. Nat. Genet. 55, 1243–1249 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tewhey R, Bansal V, Torkamani A, Topol EJ & Schork NJ The importance of phase information for human genomics. Nat. Rev. Genet. 12, 215–223 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Browning SR & Browning BL Haplotype phasing: existing methods and new developments. Nat. Rev. Genet. 12, 703–714 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karczewski KJ et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Excoffier L & Slatkin M Maximum-likelihood estimation of molecular haplotype frequencies in a diploid population. Mol. Biol. Evol. 12, 921–927 (1995). [DOI] [PubMed] [Google Scholar]
- 16.Hodgkinson A & Eyre-Walker A Variation in the mutation rate across mammalian genomes. Nature Reviews Genetics vol. 12 756–766 Preprint at 10.1038/nrg3098 (2011). [DOI] [PubMed] [Google Scholar]
- 17.Ségurel L, Wyman MJ & Przeworski M Determinants of mutation rate variation in the human germline. Annu. Rev. Genomics Hum. Genet. 15, 47–70 (2014). [DOI] [PubMed] [Google Scholar]
- 18.Rahbari R et al. Timing, rates and spectra of human germline mutation. Nat. Genet. 48, 126–133 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lek M et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carlson J et al. Extremely rare variants reveal patterns of germline mutation rate heterogeneity in humans. Nat. Commun. 9, 3753 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lynch M Rate, molecular spectrum, and consequences of human mutation. Proc. Natl. Acad. Sci. U. S. A. 107, 961–968 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baxter SM et al. Centers for Mendelian Genomics: A decade of facilitating gene discovery. Genet. Med. 24, 784–797 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ioannidis NM et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 99, 877–885 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pejaver V et al. Calibration of computational tools for missense variant pathogenicity classification and ClinGen recommendations for PP3/BP4 criteria. Am. J. Hum. Genet. 109, 2163–2177 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lassen FH et al. Exome-wide evidence of compound heterozygous effects across common phenotypes in the UK Biobank. medRxiv 2023.06.29.23291992 (2023) doi: 10.1101/2023.06.29.23291992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sharp K, Kretzschmar W, Delaneau O & Marchini J Phasing for medical sequencing using rare variants and large haplotype reference panels. Bioinformatics 32, 1974–1980 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods-only references
- 27.Van der Auwera GA et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinformatics 43, 11.10.1–11.10.33 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hail Team. Hail 0.2.105-acd89e80c345. GitHub; https://github.com/hail-is/hail/commit/acd89e80c345. [Google Scholar]
- 29.Choi Y, Chan AP, Kirkness E, Telenti A & Schork NJ Comparison of phasing strategies for whole human genomes. PLoS Genet. 14, e1007308 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roadmap Epigenomics Consortium et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.International HapMap Consortium et al. A second generation human haplotype map of over 3.1 million SNPs. Nature 449, 851–861 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McLaren W et al. The Ensembl Variant Effect Predictor. Genome Biol. 17, 122 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Georgi B, Voight BF & Bućan M From mouse to human: evolutionary genomics analysis of human orthologs of essential genes. PLoS Genet. 9, e1003484 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Behan FM et al. Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature 568, 511–516 (2019). [DOI] [PubMed] [Google Scholar]
- 35.Hart T, Brown KR, Sircoulomb F, Rottapel R & Moffat J Measuring error rates in genomic perturbation screens: gold standards for human functional genomics. Mol. Syst. Biol. 10, 733 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hart T et al. Evaluation and Design of Genome-Wide CRISPR/SpCas9 Knockout Screens. G3 7, 2719–2727 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vinceti A et al. CoRe: a robustly benchmarked R package for identifying core-fitness genes in genome-wide pooled CRISPR-Cas9 screens. BMC Genomics 22, 828 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The gnomAD v2 dataset can be accessed at https://gnomad.broadinstitute.org. We made use of prior quality control processing of these and related data. In addition, we downloaded HapMap2 genetic maps from https://github.com/joepickrell/1000-genomes-genetic-maps.
We provide both web-based look up tools and downloads for the data generated here. A look-up tool to find the likely co-occurrence pattern between two rare (global allele frequency in gnomAD exomes < 5%) coding, flanking intronic (from position −1 to −3 in acceptor sites, and +1 to +8 in donor sites) or 5’/3’ UTR variants can be found at:https://gnomad.broadinstitute.org/variant-cooccurrence
Additionally, we display the per-gene counts tables that detail the number of individuals with two rare variants, stratified by AF and functional consequence, on each gene’s main page. One table details counts of individuals with two heterozygous variants and includes predicted phase, while the second details individuals with homozygous variants. Both can be found by clicking on the “Variant Co-occurrence” tab on each gene’s main page.
All variant co-occurrence tables can be downloaded from:https://gnomad.broadinstitute.org/downloads#v2-variant-cooccurrence