

SUMMARY
RNA binding proteins (RBPs) provide precise control of neuronal RNA fate. Here we report a mechanism that the stimuli-induced neuronal translation is mediated by phosphorylation of a YTHDF1-binding protein FMRP. Mechanistically, YTHDF1 can condense with ribosomal proteins to promote the translation of its RNA targets. FMRP regulates this process by sequestering YTHDF1 away from the ribosome; upon neuronal stimulation FMRP becomes phosphorylated and releases YTHDF1 for translation upregulation. We show that a new, selective small molecule inhibitor of YTHDF1 can reverse fragile X syndrome (FXS) developmental defects associated with FMRP deficiency in an organoid model. Our study thus reveals FMRP and its phosphorylation as an important regulator of the activity-dependent translation during neuronal development and stimulation, and identifies YTHDF1 as a potential therapeutic target for FXS in which developmental defects caused by FMRP depletion could be reversed through YTHDF1 inhibition.
eTOC Blurb
YTHDF1, a main m6A reader protein exerts stimulation-induced translation modulation function. Zou et al. show that FMRP binds to YTHDF1 and inhibits its condensation with ribosomal proteins. FMRP serine 499 phosphorylation reverses this effect. YTHDF1 inhibition rebalances the hyperactive translation in FMRP-deficient FXS organoid model and rescues the developmental defects.
Graphical Abstract
INTRODUCTION
Translation activation of local synaptic mRNAs is critical to learning and memory1–3. In addition to the global translation activation, specific mRNAs are known to be activated upon neuronal stimulation to produce proteins that alter synaptic transmission to facilitate learning and memory4. For example, Calcium/calmodulin-dependent protein kinase II (CaMKII) is rapidly synthesized in the dendrites of neurons in the hippocampal CA1 region for long-term potentiation (LTP)5. By contrast, elevated Arc/Arg3.1 translation is required for metabotropic glutamate receptor (mGluR)-dependent long-term depression (LTD)6. Despite extensive studies on how phosphorylation of ribosomal proteins and translation factors enables timely response to exogenous stimuli7, those mechanisms do not explain the selectivity in activating mRNA translation for specific neuronal activities. Thus, there exists a pressing need to better understand how the selective translation activation of mRNAs in neuronal activities can be achieved8.
Selective binding by RNA binding proteins (RBPs) provide a mechanism to functionally segregate cellular mRNA into different pools. We have previously shown that YTHDF1 regulates depolarization-induced protein synthesis by promoting the translation of N6-methyladenosine (m6A)-modified transcripts9. In this context, the selective activation of mRNA translation can be achieved by the installation of m6A marks on specific mRNAs, followed by their recognition by RBPs. YTHDF1 preferentially binds m6A-modified transcripts10,11 to promote their translation12,13. In mouse hippocampi, YTHDF1 RNA targets are enriched in transcripts encoding proteins regulating synaptic transmission and LTP. YTHDF1-mediated translation of its target transcripts is not significant at resting state but is activated upon stimulation such as neuronal depolarization with potassium chloride (KCl)9,14,15. While this YTHDF1 activation mechanism is critical to learning and memory, the underlying regulatory mechanism has yet to be elucidated.
Post-translational modifications (PTMs) on proteins constitute a mechanism for timely responding to cellular stimuli. Changes in protein PTMs often require less time than DNA transcription and RNA translation to achieve substantial alterations of protein activities16. Here, by studying the post-translational modifications on YTHDF1-bound fractions, we found that FMRP is rapidly phosphorylated at the primary site serine 499 (S499) within ten minutes post-stimulation. This phosphorylation event inhibits the interaction between FMRP and YTHDF1, consequently inducing YTHDF1 condensation. Furthermore, we showed that YTHDF1 mainly condenses with ribosomal proteins to promote mRNA translation. Our findings provide a mechanism for how phase separation, RBP phosphorylation, and mRNA modification work together to dictate precise mRNA translation activation during neuronal activities.
We applied our findings to the context of fragile X syndrome (FXS), a condition caused by FMRP deficiency. FXS is the most common known cause of inherited intellectual disability and is caused by loss of FMRP protein during brain development17. FMRP is primarily thought to be a suppressor of mRNA translation. The hippocampal slices from Fmr1-KO mice incorporate 15–20% more [35S]methionine into nascent peptides than wild-type controls18. Consistent with the translational repression role, FMRP was also reported to be associated with stalled polyribosomes19. However, studies of FMRP targets also suggested that FMRP may enhance the translation of its target mRNA in certain neurons, because the association of FMRP-target mRNA with ribosome was reduced in FMRP-deficient neurons20. Another study conducted in Drosophila oocytes indicated that FMRP maintains translation of a subset of long mRNAs21. These conflicting roles of FMRP on translation may suggest the involvement of multiple pathways in FMRP-deficient systems; some of these pathways may play more determinant roles in FXS pathophysiology.
Our efforts led to the identification of a selective small molecule inhibitor of YTHDF1. YTHDF1 inhibition in mouse cortical neurons efficiently restores normal protein synthesis by dissolving YTHDF1 granules. Furthermore, application of YTHDF1 inhibitor reverses the developmental phenotypes in an FXS forebrain organoid model, and we showed that it could restore normal translation of key genes for FXS. This success confirms the FMRP-YTHDF1 axis in translation control and provides a potential therapeutic strategy to rebalance neuronal translation for FXS therapy.
RESULTS
FMRP inhibits neuronal translation mediated through YTHDF1
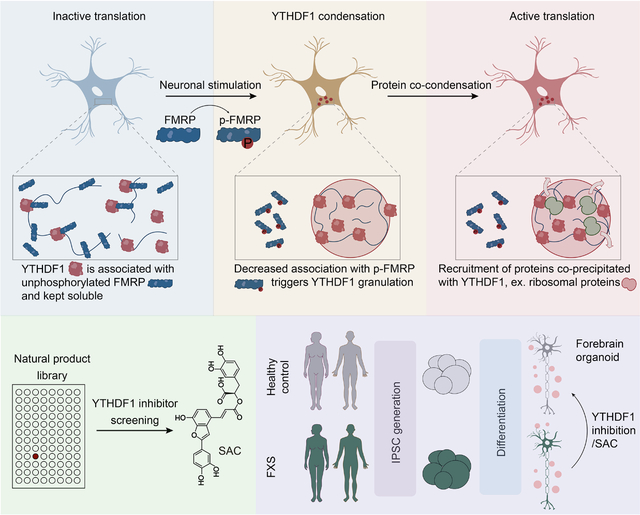
YTHDF1 does not promote mRNA translation in neurons without stimulation9. Instead, YTHDF1 is activated within two hours post KCl depolarization treatment, and then returns to an inactivated state after six hours9. This fast and reversible activation suggests a regulatory mechanism at the post-translational level, which is frequently implicated in neuronal processes22. Therefore, we analyzed the global post-translational modification (PTM) changes on cellular proteins upon neuronal depolarization; we examined serine phosphorylation (pS), ubiquitylation (Ub), O-linked β-N-acetylglucosamine (O-GlcNAc), and tyrosine phosphorylation (pY) by western blotting (Figure S1A). Only the global pS level displayed a temporal pattern change resembling YTHDF1 activation (Figure S1A), indicating a potential connection between serine phosphorylation and YTHDF1 activation.
We investigated whether YTHDF1 could be phosphorylated and if such phosphorylation could activate its functions in promoting translation. We isolated protein fractions bound by YTHDF1 following KCl depolarization in mouse neurons and noticed that the fractions with a molecular weight approximately at 80 kDa exhibited the most significant increase in serine phosphorylation (Figure 1A). This indicated that a YTHDF1-interacting protein was phosphorylated upon neuronal depolarization. We did not detect any noticeable YTHDF1 phosphorylation in the mouse brain upon fear conditioning or electroconvulsive treatments, nor did we observe such phosphorylation in cultured mouse neurons subjected to KCl depolarization (Figure S1B–C).
Figure 1. FMRP phosphorylation activates YTHDF1 upon neuronal stimulation.
(A) Western blot analysis of phosphorylated serine in YTHDF1 co-immunoprecipitated fractions.
(B) List of top 11 proteins identified from fractions co-immunoprecipitated with YTHDF1 with WT mice brain tissue. Proteins were listed by gene names and ordered by total peptide intensity for each protein.
(C) Western blot analysis of YTHDF1-FMRP interaction in cultured mouse neurons depolarized by 50 mM KCl for 10 minutes (KCl) or not (Control).
(D) Reporter assays of YTHDF1-mediated translation in HEK293T cells (n = 3). Proteins negatively regulating m6A-mediated translation were depleted individually.
(E) Western blots and dot plots showing the level of p-FMRP(S499) (p-FMRP) and p-Chk1(S345) (p-Chk1) in neuron lysates collected at different time points post KCl depolarization (Mock, 10 min, 2h and 6h).
(F) Quantification of protein synthesis rate measured by L-Homopropargylglycine (HPG) incorporation in mouse neurons (n = 6 for each condition).
(G) Translation efficiency of YTHDF1-mediated translation in cultured mouse neurons measured by reporter assays.
(H) Proximity ligation assays in cultured mouse neurons depolarized by 50 mM KCl (KCl) or not (Mock) against YTHDF1 and RPS6.
(I) Proximity ligation assays in cultured mouse neurons depolarized by 50 mM KCl (KCl) or not (Mock) against YTHDF1 and FMRP.
(J) Bar plots showing the effects of FMRP phosphor-null mutant (S499A) on the distribution of YTHDF1 targets in ribosomal fractions.
Data are mean ± s.e.m. (D), (E), (F), (G) and (J), P values were determined using a two-tailed t test with Welch’s correction. (H) and (I), P values were determined using a Wilcoxon ranked sum test. NS means not significant (P values > 0.05). Reported P values are for the individual comparisons. Box-plot elements: centre line, median; box limits, upper and lower quartiles; whiskers, 1–99%; error bars, 95% confidence interval of mean
In search for this YTHDF1-binding protein, we performed mass spectrometry analysis of proteins co-immunoprecipitated with YTHDF1 in mouse brain tissue (Figure S1D–E). Among the top proteins identified, fragile X mental retardation protein (FMRP) stood out as the only one previously known to regulate translation and has the matching molecular weight (~ 80 kDa) (Figure 1B). FMRP is a translation repressor in the brain19 known to be associated with m6A-methylated transcripts23 and YTHDF124. We further confirmed this interaction between YTHDF1 and FMRP in cultured mouse neurons (Figure 1C).
Consistent with its association with YTHDF1, our previous m6A-quantitative trait loci (QTL) mapping also revealed FMRP as one of the top hits that may suppress the translation of m6A-methylated mRNAs, including YBX3, IGF2BP2, FXR2, DDX3X, FMR1 and TIA125. We selected HEK293T cells to study YTHDF1 activation as YTHDF1 is known to have minimal effects on mRNA translation in the absence of stimulation in this cell line26. To test whether they regulate YTHDF1-mediated translation, we employed a YTHDF1-tethered firefly luciferase (F-Luc) reporter system, in which the N terminus of YTHDF1 was tethered to the 3’-UTR of the reporter RNA to mimic binding to its native targets (Figure S1F)12. Intriguingly, translation upregulation was only observed when we depleted FMRP (Figure 1D and Figure S1G). FMR1 knockdown alone does not affect the translation efficiency of untethered reporter mRNAs (Figure S1H). Therefore, these observations suggested that FMRP may bind to (Figure 1C) and sequester YTHDF1 from its translation function (Figure 1D).
Serine 499 (S499) in FMRP is known to be a critical phosphorylation site27,28. And we indeed observed increased FMRP phosphorylation as early as ten minutes post-stimulation in neurons (Figure 1E). The phosphorylation level of FMRP reduced to basal level after two to six hours, mirroring the temporal pattern of YTHDF1 activation. These changes are likely attributed to the mGluR activity, which is closely related to FXS29,30. This notion is supported by our findings demonstrating that treatment with a small molecule antagonist NPS 239031 or an agonist DHPG of mGluR32 bidirectionally changed the FMRP phosphorylation level, respectively. In contrast, treatment with TTX, a sodium channel blocker, did not produce a similar outcome (Figure S1I). As a negative control, Checkpoint kinase 1 (Chk1) phosphorylation, which responds to DNA damage33, remained unchanged throughout the process (Figure 1E). These results show that FMRP is phosphorylated upon neuronal stimulation and suggest that phosphorylated FMRP may play a role in releasing YTHDF1 for translation promotion.
Phosphorylation of FMRP regulates translation promotion by YTHDF1
To test whether FMRP phosphorylation is involved in YTHDF1 activation during neuronal depolarization, we isolated cortical neurons from three different mouse lines: Fmr1+/+ (WT), Fmr1−/− (Fmr1 KO), or FMRP(S499A) (see details in STAR Methods), a phosphor-S499 null mutant mice (Figure S1J). We used the isolated primary neurons as experimental models and assayed overall protein synthesis rate following KCl depolarization.
The basal protein synthesis rate of Fmr1 KO neurons is higher than that of WT and S499A neurons, which is consistent with the reported role of FMRP as a translation repressor17,19,34. Importantly, while KCl depolarization induced a burst of protein synthesis in WT neurons, neither Fmr1 KO nor S499A neurons displayed a similar response (Figure 1F). These results strongly indicate that FMRP S499 phosphorylation is key to the stimulation-induced protein synthesis. Consistently, translation of the YTHDF1 (N terminus)-tethered firefly luciferase reporter was elevated in Fmr1 KO neurons compared with WT neurons (Figure 1G). Depolarization also only induced translation upregulation of this reporter RNA in the wild-type neurons (Figure 1G).
Agreeing with the observed elevated translation rates, YTHDF1 was in closer spatial proximity to ribosome component protein RPS6 in WT neurons post-depolarization when compared to same neurons without stimulation (Figure 1H). However, the proximity between YTHDF1 and RPS6 did not change after depolarization in both the S499A mutant and Fmr1 KO neurons (Figure 1H). Specifically, the YTHDF1-RPS6 proximity is always high in Fmr1 KO neurons while is always low in S499A mutant neurons (Figure 1H). The opposite trend was observed for the YTHDF1-FMRP proximity (Figure 1I). We further validated the effect of FMRP phosphorylation on the translation of YTHDF1 target transcripts by examining the distribution of two YTHDF1 target mRNA transcripts, LRPAP1 and eEF1G35, in different polysome fractions. We observed that overexpression of phosphorylation-null FMRP S499A caused a decrease of those mRNAs in ribosomal fractions (Figure 1J). Our results therefore suggest an inhibitory role of unphosphorylated FMRP on YTHDF1 in mouse neurons, with FMRP phosphorylation during neuronal depolarization abolishes this inhibition.
FMRP phosphorylation modulates YTHDF1 condensation
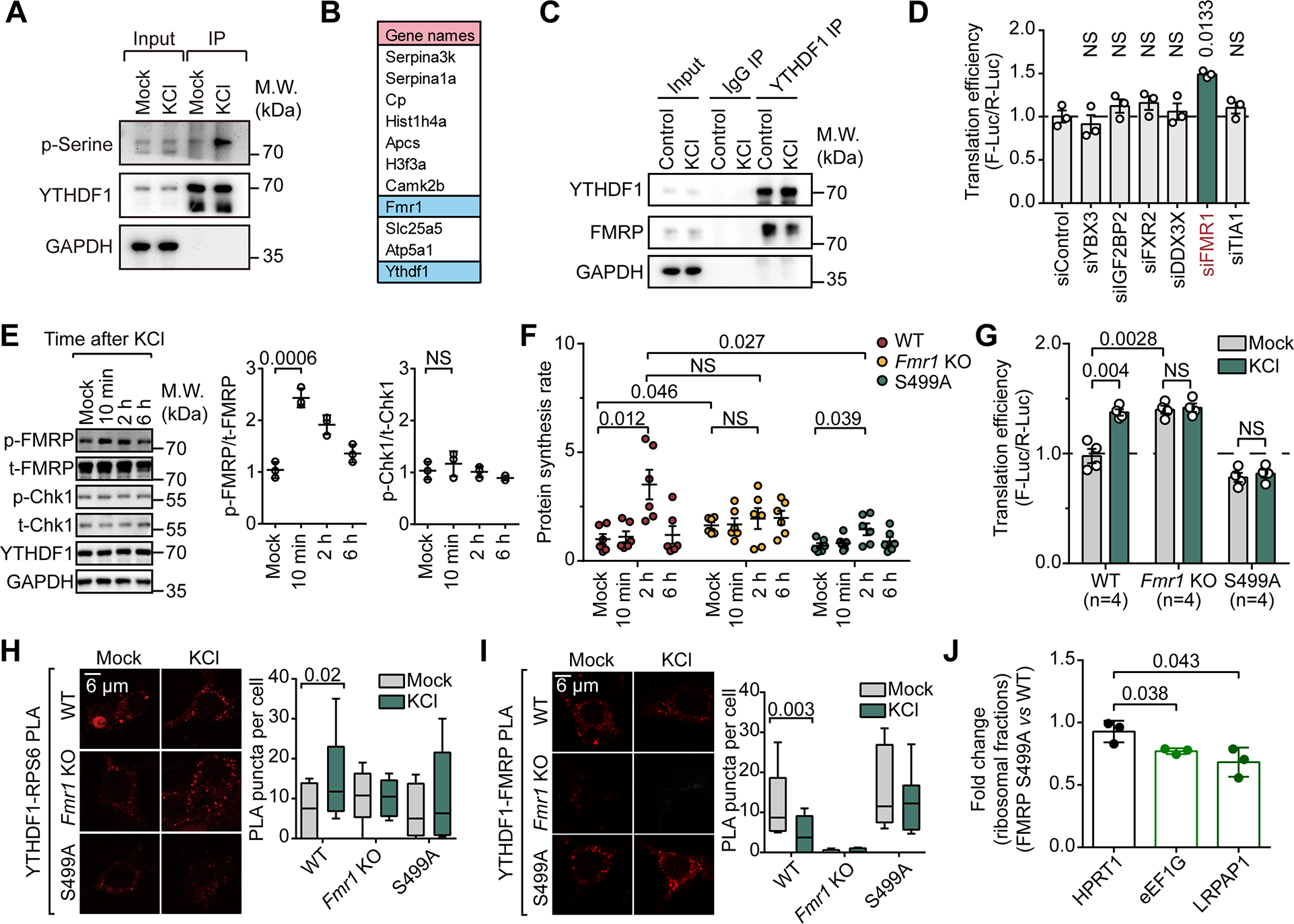
One of the key functions of FMRP S499 phosphorylation is to regulate its condensation behavior36,37. Notably, YTHDF1 and FMRP are both known to contain intrinsically-disordered domains and are prone to form condensates. YTHDF1 contains prion-like domains (PLDs) and thus can fold into distinct structures, which has been implicated in synaptic plasticity formation38. To this end, we monitored the condensation behavior of YTHDF1 in neurons. We observed that YTHDF1 puncta were formed within 10 minutes post-depolarization in WT neurons (Figure 2A). In contrast, the formation of visible YTHDF1 puncta in response to KCl depolarization was not observed in either Fmr1 KO nor S499A neurons (Figure 2A).
Figure 2. Unphosphorylated FMRP inhibits YTHDF1 condensation, phosphorylated FMRP does not.
(A) Representative images of cultured mouse neurons labeled using antibodies against YTHDF1. Left, representative images from three independent experiments; right, quantification of YTHDF1 puncta per cell.
(B) Line plots showing the condensation of YTHDF1 incubated with HeLa cell lysates. YTHDF1 condensation was quantified by turbidity measurement at optical density at 600 nm (OD600) in three independent experiments.
(C) Representative images of co-condensing of YTHDF1 with unphosphorylated or phosphorylated FMRP (n = 6). Intensity distribution on a line (as labeled in the figures) was quantified.
Data are mean ± s.e.m. P values were determined using a two-tailed unpaired t test with Welch’s correction. NS means not significant (P values > 0.05). Reported P values are for the individual comparisons. Box-plot elements: centre line, median; box limits, upper and lower quartiles; whiskers, 1–99%; error bars, 95% confidence interval of mean
We constructed an in vitro assay, where HeLa cells were either pre-treated with an FMRP kinase inhibitor (Figure S2A, CX-4945 inhibits FMRP phosphorylation at S49939) to eliminate FMRP phosphorylation, or with siRNA to deplete FMRP (Figure S2B). We monitored YTHDF1 condensation in the absence of the pretreated cell extracts by measuring optical density at 600 nm40. Our observations revealed that FMRP depletion promoted YTHDF1 condensation (Figure 2B, left panel) while the absence of FMRP phosphorylation impaired YTHDF1 condensation (Figure 2B, right panel). Furthermore, we isolated insoluble fractions within cellular extracts41 and confirmed that the knockdown of FMRP led to increased YTHDF1 and RPS6 in these insoluble fractions (Figure S2C). These findings collectively suggest that unphosphorylated FMRP inhibits YTHDF1 condensation.
To elucidate the direct interactions between YTHDF1 and FMRP, we constructed an in vitro condensate formation system using only recombinant FMRP and YTHDF1. Compared to YTHDF1 alone (Figure 2C, lane “YTHDF1”), the addition of unphosphorylated FMRP into the mixture notably inhibited YTHDF1 condensation (Figure 2C). YTHDF1 molecules coalesced to form visible condensates under the microscope in the absence of phosphorylated FMRP (Figure 2C and Figure S2D). To study the nature of these protein condensates, we induced YTHDF1 condensation by adding crowding reagent PEG800042. We found that unphosphorylated FMRP colocalizes with YTHDF1 while p-FMRP and YTHDF1 exhibited mutual exclusivity within the condensates (Figure 2C). This suggests a transition in its interaction with YTHDF1 when FMRP is phosphorylated.
Additionally, co-immunoprecipitation (co-IP) assay revealed a stronger association between YTHDF1 and FMRP (S499A) compared to FMRP (WT) (Figure S2E). This tighter association is further supported by the increased PLA signals in cells treated with CX-4945 (Figure S2F, left panel). Taken together, these findings support the conclusion that unphosphorylated FMRP binds YTHDF1 more strongly than phosphorylated FMRP, and this binding represses YTHDF1 condensation, likely with the ribosome translation initiation complex. Indeed, we noticed a significant decrease in spatial proximity between YTHDF1 and a small ribosome unit RPS6 when FMRP phosphorylation was inhibited with CX-4945 (Figure S2F, right panel). Therefore, unphosphorylated FMRP prevents YTHDF1 condensation, and phosphorylation of FMRP releases this inhibition. This dynamic regulation of YTHDF1 condensation in neurons during stimulation could be linked to mRNA translation.
YTHDF1 condenses with the ribosome to promote translation
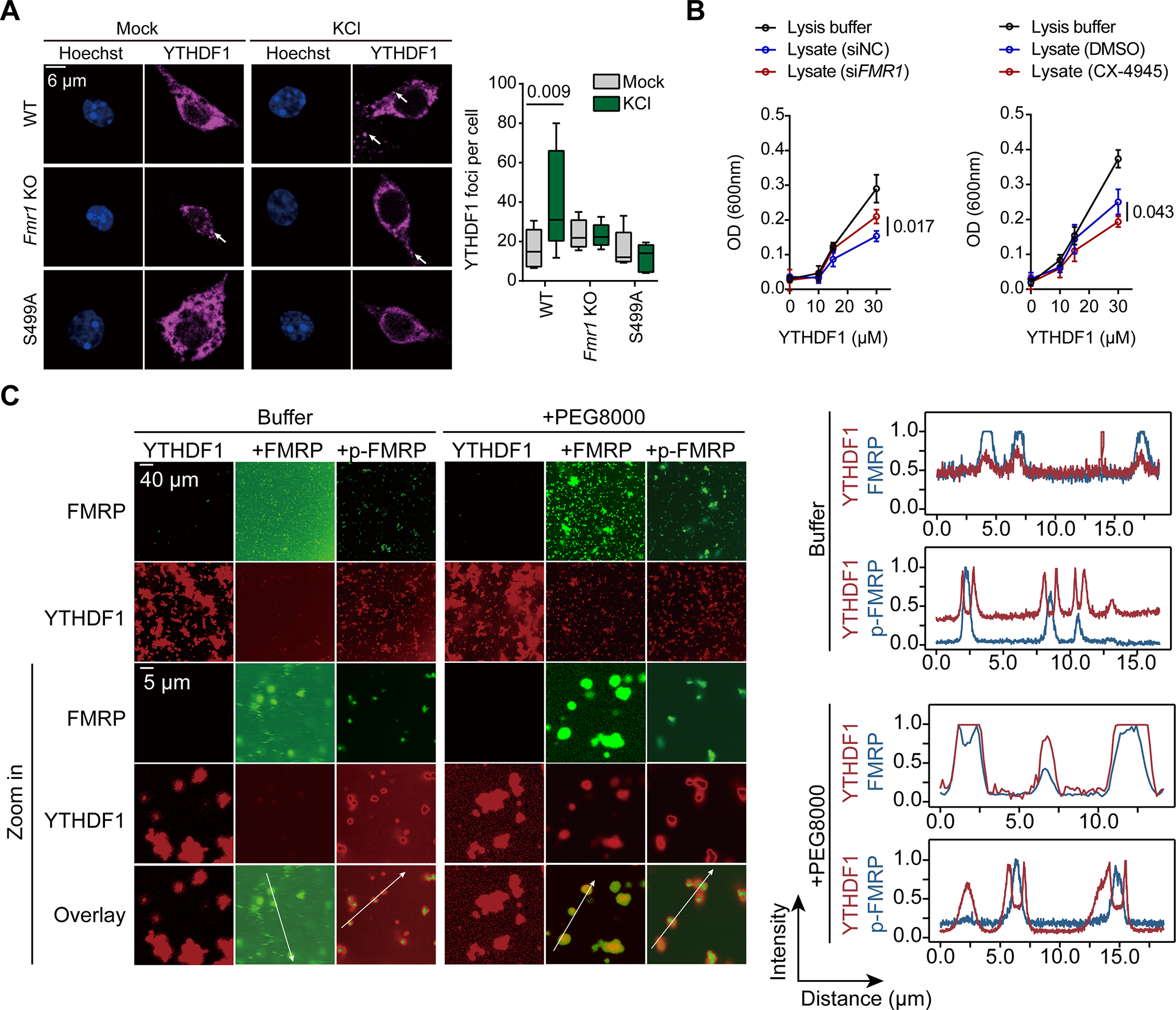
To study the functional implications of YTHDF1 condensation, we aimed to precipitate YTHDF1-containing condensates to reveal condensate-dependent interactions between YTHDF1 and other cellular biomolecules. According reported by Maharana et al., the condensation of proteins containing PLDs in cultured cells can be triggered by RNase microinjection43, such as YTHDF1. Other reports also found that excessive RNAs could dissolve phase-separated granules 44.
To validate this approach, we treated neuron or HEK293T lysates with RNase to induce formation of condensates, which were subsequently observed either under a microscope (Figure 3A) or with turbidity measurements (Figure S3A). These condensates are confirmed to be liquid-liquid phase separated granules as both 1,6-hexanediol (1,6-HD) and a high concentration of sodium chloride (NaCl) could partially dissolve them (Figure S3A).
Figure 3. YTHDF1 condenses with the ribosome to promote mRNA translation.
(A) Representative images of mouse neuron lysates in controls samples, samples treated by RNase, or samples treated by RNase and NaCl (n = 3).
(B) Schematic of using RNase to precipitate prion-like proteins.
(C) Protein partners of YTHDF1 revealed by co-immunoprecipitation with RNase treatment.
(D) Western blot analysis of YTHDF1 protein partners with RNase treatment.
(E) Illustration of the prion-like features of YTHDF1 and FMRP calculated by PLAAC.
(F) Illustration of YTHDF1 mutants with deletion of different domains.
(G) Granule formation propensity of YTHDF1 mutants.
(H) Relative translation efficiency measured by reporter assay showing effects of YTHDF1 mutants on mRNA translation (n = 3 for each condition).
(I) In vitro translation assay with rabbit reticulocyte lysate (RRL) and YTHDF1 (n = 3 each condition). Representative fluorescence images were shown to demonstrate formations of YTHDF1 granules and that 0.2% (w/v) 1,6-hexanediol dissolved them.
(J) Bar plots showing the translation efficiency of YTHDF1-tethered reporter mRNA normalized to untethered control.
Data are mean ± s.e.m. P values were determined using a two-tailed t test with Welch’s correction. NS means not significant (P values > 0.05). Reported P values are for the individual comparisons.
Subsequently, we isolated the RNase-induced condensates from HEK293T lysates using differential centrifugation (Figure S3B) and confirmed that YTHDF1 responded to RNase treatment and was enriched within the RNP granules (RG) (Figure S3C). Remarkably, ribosome component RPS6 was also significantly enriched in the RG phase after RNase treatment (Figure S3C). Thus, RNase treatment triggers precipitation of RNP granules containing YTHDF1, and thus could be utilized to study its protein interactions.
We designed a workflow to study condensation-dependent protein-protein interactions. Cells were lysed with hypotonic treatment to avoid the use of detergents and subjected to RNase treatment to induce granule formation (Figure 3B). Immunoprecipitation was then performed to study the interaction between a specific protein and other biomolecules. The co-purified proteins are subjected to further analysis using liquid chromatography-tandem mass spectrometry (LC-MS/MS). We applied this approach to investigate protein-protein interactions involving protein condensates formed by YTHDF1. Specifically, we focused on proteins co-purified with YTHDF1 under RNase treatment, as protein found within YTHDF1 condensates should be enriched in the portion precipitated by RNase treatment. Consistent with their decreased association upon KCl-induced depolarization (Figure 1I), a significant decrease in the binding of FMRP to YTHDF1 was found upon RNase treatment (Figure 3C, highlighted in green). Thus, FMRP could be depleted from YTHDF1 condensate. This observation was further validated by western blotting (Figure 3D) and is consistent with the notion that FMRP protein lacks clear prion-like domains (Figure 3E).
Notably, our analysis of the protein partners that depend on YTHDF1 condensation revealed that this repertoire primarily consists of ribosomal proteins (RPs), and is enriched in proteins involved in translation regulation (Figure 3C and Figure S3D). This finding provides a potential explanation for the dependency of the YTHDF1-promoted mRNA translation on condensate formation. YTHDF1 could form an active translation condensate with ribosome to promote the translation of its mRNA targets.
To establish a causal relationship between enhanced YTHDF1 condensation and mRNA translation efficiency, we constructed a panel of YTHDF1 mutants with deletion of individual or multiple domains based on prion-like score predicted by PLAAC (Prion-Like Amino Acid Composition, http://plaac.wi.mit.edu/) (Figure 3F). We first tested their condensing tendency in vitro. Interestingly, two mutants with individual deletions of amino acid 53–106 (Δ53–106), or PLD1 and 3 (ΔPLD1,3) showed the most significantly impaired condensation abilities (Figure 3G). They also exhibited lower translation promotion ability compared to wild-type YTHDF1 in a cell-based reporter assay to examine the functional outcomes (Figure 3H). We found that YTHDF1 mutants with impaired condensing ability are less capable of promoting translation of the reporter mRNA it binds (Figure 3G–H). This suggests a direct link between YTHDF1 condensation and its capacity to enhance mRNA translation.
Furthermore, we examined the interactions between endogenous FMRP and the YTHDF1 mutants. Deletion of any of the three portions of the PLD1-linker-PLD2 minimum structure (amino acid 41–155) of YTHDF1 impaired the binding of YTHDF1 to FMRP (Figure S3E). Therefore, different regions of YTHDF1 are used for FMRP binding (amino acid 41–155) and condensation formation (amino acid 41–52 and 286–395).
To further support the connection between YTHDF1 condensation and its capacity to enhance mRNA translation, we employed an in vitro translation system in rabbit reticulocyte lysates (RRLs). In this system, we can inhibit YTHDF1 condensation by adding 1,6-hexanediol (Figure 3I, left panel). Following the 1,6-hexanediol treatment, YTHDF1 protein condensation was diminished (Figure 3I). Moreover, the same treatment also abolished the elevated translation efficiency of the YTHDF1 (N terminus)-tethered firefly luciferase reporter mRNA (Figure 3J). Consistent with these findings, the distribution of YTHDF1 in the ribosomal fractions (40S, 60S and 80S) within cell lysates decreased upon salt or 1,6-hexanediol treatment (Figure S3F). We therefore conclude that the condensing propensity of YTHDF1 is important to its translation promotion function.
Collectively, these observations support a working model that the binding of unphosphorylated FMRP to YTHDF1 prevents its interaction with ribosomal components, thus inhibiting YTHDF1-mediated translation. When FMRP is phosphorylated, YTHDF1 is released to form condensates with the ribosome and to promote mRNA translation.
YTHDF1 may induce aberrant translation activation in fragile X syndrome
We conceived that FMRP deficiency may lead to activation of the YTHDF1-mediated translation in fragile X syndrome (FXS) systems, and this pathway could be physiologically relevant in FXS pathophysiology. Aberrantly active translation was implicated in FXS models19; and restoring normal translation has been explored as a strategy to ameliorate FXS-related phenotypes45,46. Our discoveries regarding FMRP-regulated mRNA translation activation through YTHDF1 could provide a potential explanation for the hyperactive translation in FXS when FMRP is depleted (Figure 4A). Thus, we decided to develop and test small molecule inhibitors that inhibit YTHDF1 binding to m6A and evaluate their potential for restoring normal translation in fragile X syndrome (Figure 4A).
Figure 4. SAC is a selective substrate-competitive inhibitor of YTHDF1.
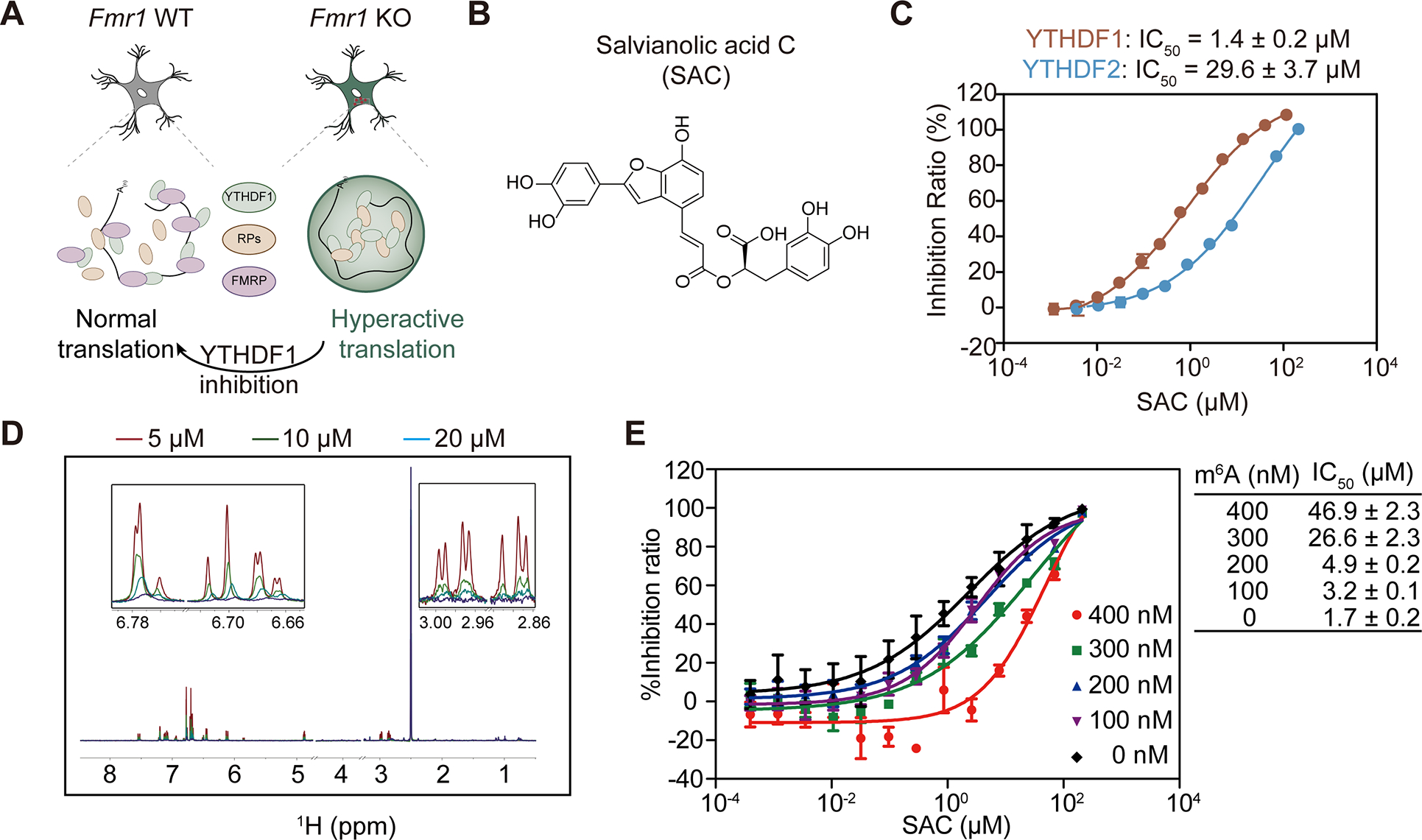
(A) Illustration of YTHDF1 inhibition in FMRP-deficient neurons to treat FXS.
(B) Chemical structure of a YTHDF1 inhibitor salvianolic acid C (SAC).
(C) IC50 value of SAC against YTHDF1 and YTHDF2 measured by AlphaScreen experiments. The selectivity of SAC between YTHDF1 and YTHDF2 is around 20:1.
(D) NMR CPMG binding assay between SAC and YTHDF1. The measurements were conducted under the conditions of 0 μM (red), 5 μM (green), 10 μM (blue) and 20 μM (purple) YTHDF1 protein, respectively.
(E) Competitive binding analysis between SAC and m6A-containing RNA oligos. The activities of SAC were summarized in the table, in which m6A-containing RNA oligos were shown as m6A. (Data are expressed as the mean ± se).
Identification of SAC as a selective inhibitor of YTHDF1
We started with an in-house small molecule library consisting of 320 natural products. We performed a fluorescence polarization (FP)-based high-throughput screen (HTS) to identify compounds capable of disrupting the binding of YTHDF1 to m6A-modified RNA. We validated that addition of unlabeled m6A-containing RNA efficiently disrupted this interaction at an IC50 of 0.71 μM (Figure S4A). The Z’ value of the FP based screening was 0.86 (Figure S4B), suggesting that the FP based method was stable and was suitable for high throughput screening.
During the screening, Salvianolic acid C (SAC) was found to be a potential hit (Figure 4B). To confirm its inhibitory effect, a half-maximal inhibitory concentration (IC50) value of 1.4 ± 0.2 μM was obtained using an in vitro binding assay (Figure 4C). We further confirmed the direct binding between SAC and YTHDF1 through nuclear magnetic resonance (NMR) and Carr-Purcell-Meiboom-Gill (CPMG) experiments (Figure 4D). The dissociation constant (KD) of SAC against YTHDF1 was determined to be 6.3 μM and 5.3 μM through individual isothermal titration calorimetry (ITC) assay and microscale thermophoresis (MST), respectively (Figure S4C–D). Moreover, AlphaScreen-based substrate competition experiment showed that the IC50 value of SAC against YTHDF1 increased from 1.7 ± 0.2 μM to 46.9 ± 2.3 μM with the addition of unlabeled m6A-containing RNA from 0 nM to 400 nM (Figure 4E). This suggests that SAC exhibits a competitive inhibition mode against YTHDF1, while an allosteric inhibition that affects substrate binding is also possible.
Because the YTH domains of YTHDF1 and YTHDF2 share similar sequences, we investigated the selectivity of SAC against YTHDF1 versus YTHDF2. By comparing IC50 values of the SAC inhibition on individual YTHDF protein interaction with m6A-containing RNA obtained from AlphaScreen assay (1.4 ± 0.2 μM for YTHDF1 and 29.6 ± 3.7 μM for YTHDF2), we can conclude that SAC possesses a decent selectivity against YTHDF1 over YTHDF2 (Figure 4C). To further demonstrate this selectivity, PRR5L mRNA, which is known to be a specific target of YTHDF2 but not YTHDF147, was only found to be destabilized with 50 μM or higher concentrations of SAC (Figure S4E). In contrast, translation efficiencies of YTHDF1 target RNA eEF1G and LRPAP1 started to decrease with SAC treatment at 6.25 μM (Figure S4F). Thus, SAC is a specific inhibitor against YTHDF1 and exhibits 20-fold selectivity over YTHDF2. We used 20 μM for most subsequent studies to maximize YTHDF1 inhibition while ensuring that YTHDF2 is not affected. This selectivity of SAC against YTHDF1 over YTHDF2 allows us to apply SAC as a tool compound and study biological consequences specifically caused by YTHDF1 inhibition.
SAC dissolves YTHDF1 condensates and counteracts hyperactive YTHDF1 in neurons
We next applied SAC to cultured neuronal cells to study the functional outcomes. Consistent with its ability to block YTHDF1 binding to m6A, SAC caused the disassembly of YTHDF1 oligomers at concentrations starting at 10 μM, while YTHDF2 oligomers remained unaffected at SAC concentrations up to 20 μM (Figure 5A). YTHDF1 and YTHDF2 differ in their N-terminal intrinsically disordered domain sequence (Figure S5A). This difference was reflected by their differences in a Thioflavin T (ThT) assay to study amyloid fibril formations48. Freshly-purified prion-like domains of YTHDF1 exhibited faster kinetics and higher tendency to form fibrils when compared with that of YTHDF2 within 100-hour monitoring (Figure S5B). Thus, SAC is demonstrated to be capable of selectively inhibit YTHDF1 condensation in neurons.
Figure 5. SAC inhibits YTHDF1 by suppressing YTHDF1 condensation.
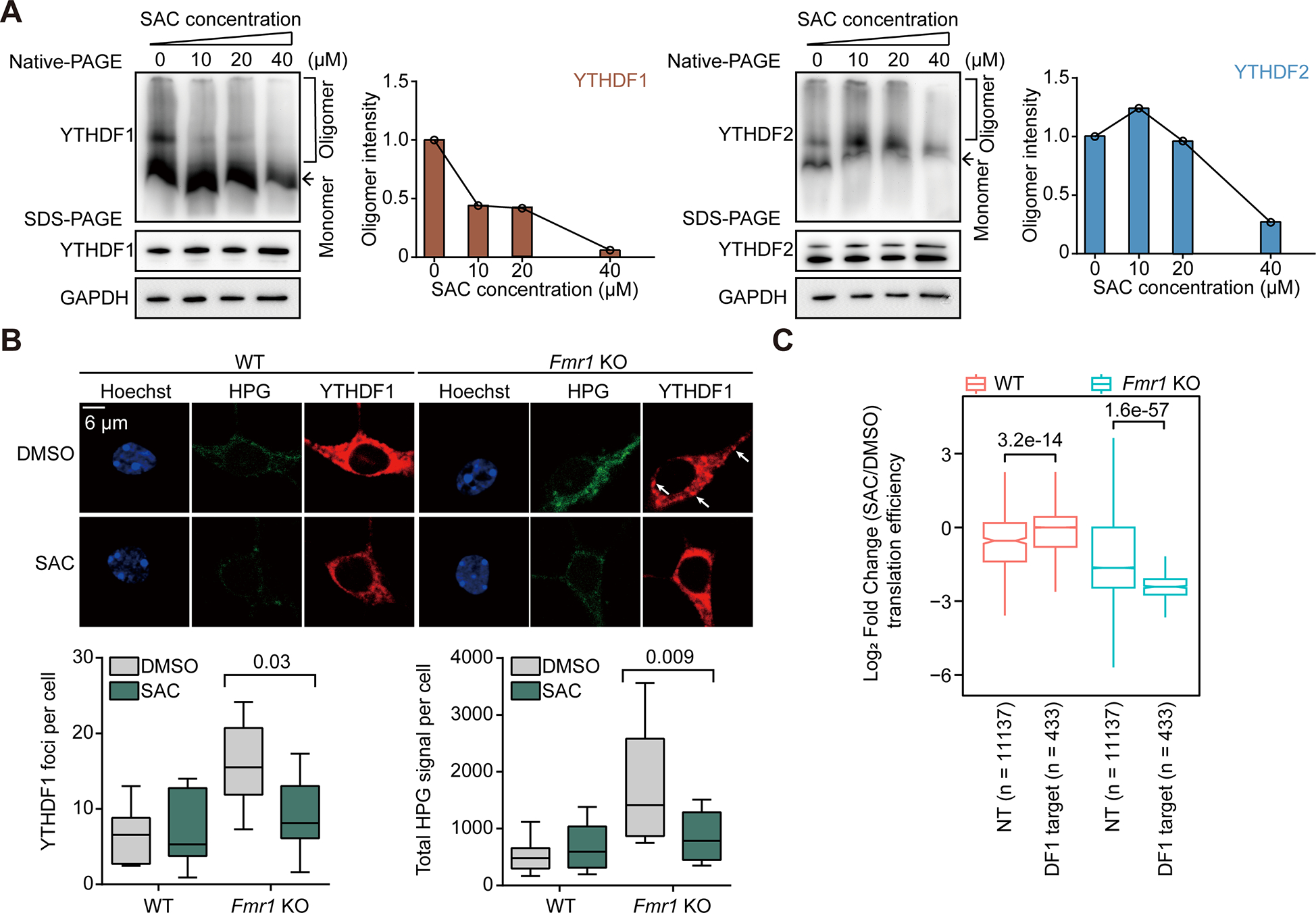
(A) Analysis of YTHDF1 (left) or YTHDF2 (right) protein oligomers in cultured mouse neurons treated with SAC. YTHDF protein oligomers were visualized by Native-PAGE and loading controls were performed with denaturing SDS-PAGE.
(B) SAC treatment rescues aberrant YTHDF1 condensation and protein synthesis in Fmr1 KO neurons. Protein synthesis rates were measured by HPG incorporation in mouse neurons (n = 6 for each condition).
(C) Box plots showing the log2-transformed fold changes of the translation efficiency of YTHDF1 target transcripts (DF1 target) or non-targets (NT) in WT or Fmr1 KO mouse neurons upon SAC.
(B) and (C), P values were determined using a Wilcoxon ranked sum test. NS means not significant (P values > 0.05). Reported P values are for the individual comparisons. Box-plot elements: centre line, median; box limits, upper and lower quartiles; whiskers, 1–99%; error bars, 95% confidence interval of mean
In Fmr1 KO neurons, SAC treatment restored normal protein synthesis rate and YTHDF1 granulation (Figure 5B) to levels comparable to those in WT neurons. To assess the translation efficiency of the entire transcriptome, we utilized riboLace, a method in which actively translating ribosomes stalled by puromycin are immunoprecipitated. This method therefore requires much less starting material than traditional ribo-seq49. We validated that our datasets captured ribosome-protected fragments (RPFs) with correct P-site periodicity (Figure S5C). Notably, SAC treatment caused a more significant impact on translation in Fmr1 KO neurons (Figure S5D).
Specifically, SAC treatment reversed the hyper-active translation of YTHDF1 target transcripts only in Fmr1 KO neurons (Figure 5C). We also showed that the genes with decreased translation efficiency after SAC treatment are enriched in FXS-related functional terms including axonogenesis and positive regulation of cell projection organization (Figure S5E, right panel), while the hyper-translated genes are less relevant based on their annotated functions (Figure S5E, left panel). These data suggest that YTHDF1 is more active in Fmr1 KO neurons, with more YTHDF1 targets hypo-translated (145) compared to hyper-translated (71) after SAC-induced inhibition (Figure S5D). Altogether, our results demonstrate that SAC not only inhibits RNA-binding of YTHDF1, but also disrupts YTHDF1 condensation, leading to the attenuated translation of YTHDF1 target RNAs.
SAC reverses neurodevelopmental deficits in FXS forebrain organoids
We next studied SAC activity on the tissue level with a mouse forebrain organoid system. One recent work studying FXS forebrain organoids indicated that loss of FMRP in human forebrain organoids could lead to various abnormalities, including reduced proliferation of neural progenitor cells, dysregulated neural differentiation, increased synapse formation, neuronal hyperexcitability, and a deficit in the production of GABAergic neurons (Figure 6A)50.
Figure 6. SAC partially rescues FXS pathology in a human forebrain organoid model.
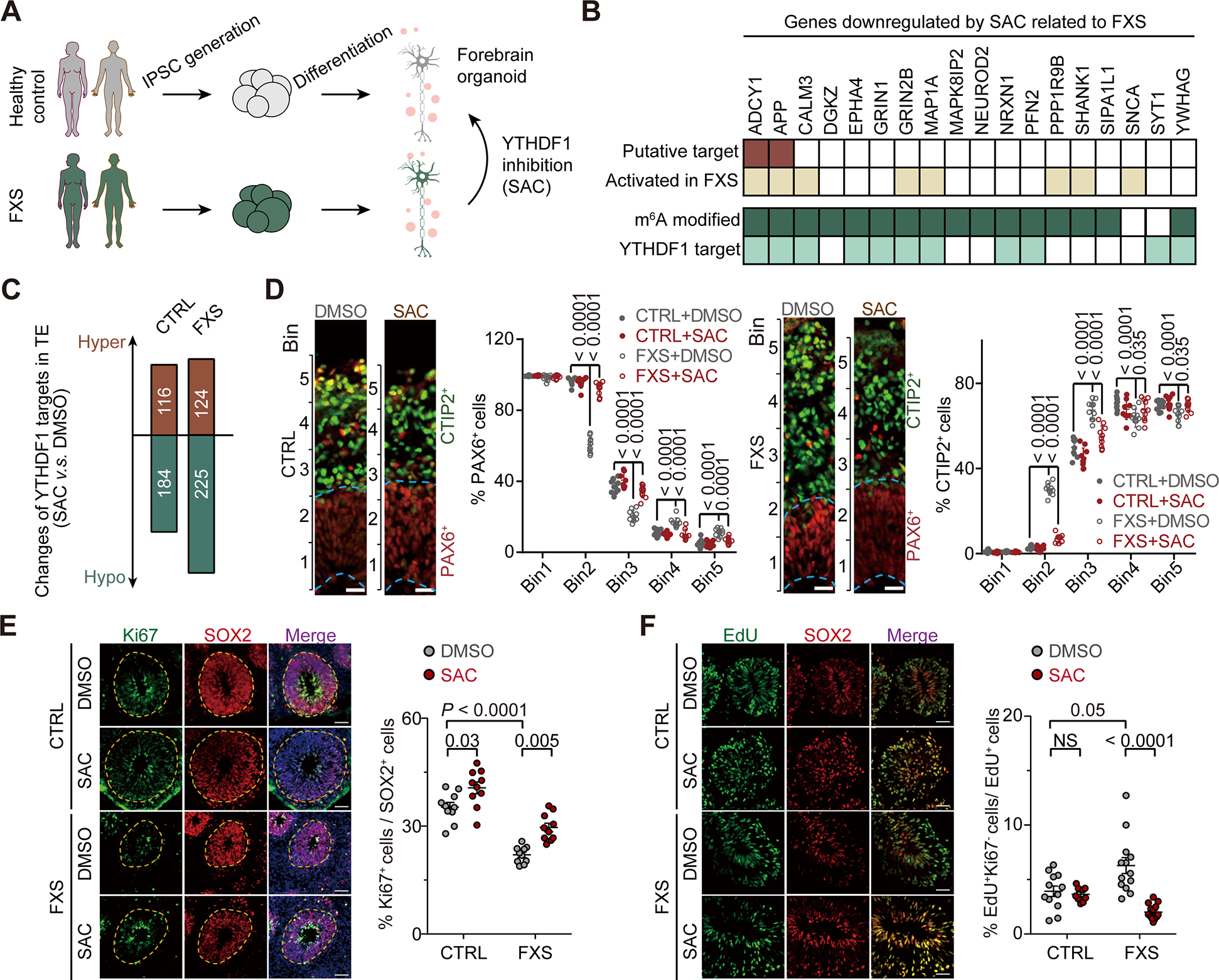
(A) Illustration of human forebrain organoid model derived from healthy control and FXS patients.
(B) Heat map showing selected downregulated genes by SAC treatment identified in the FXS organoid model. Eighteen genes known to be related to FXS were selected out of 30 downregulated genes enriched in the biological process: regulation of trans-synaptic signaling.
(C) Bar plot showing the numbers of YTHDF1 targets with altered translation in control and FXS organoids upon SAC treatment.
(D) YTHDF1 inhibition rescued deficit of altered neuronal differentiation in FXS forebrain organoids. Shown are sample images and quantification of PAX6+ (left) or CTIP2+ (right) cells in either control or FXS forebrain organoids under various treatment conditions at day 56. PAX6 staining and CTIP2 staining were shown in red and green, individually. Data are presented as mean ± s.d. (n = 10 sections from at least from 3 organoids each condition. Scale bars: 50 μm.
(E) Left: representative images showing the inhibition of YTHDF1 rescues the reduced NPC proliferation in FXS organoids. Right: Quantifications of NPC proliferation.
(F) Representative images showing the inhibition of YTHDF1 delays hastened cell cycle exit of cells in FXS organoids. Right: Quantifications of NPC cell cycle exit.
Data are mean ± s.e.m. P values were determined using a two-tailed t test with Welch’s correction. NS means not significant (P values > 0.05). Reported P values are for the individual comparisons.
We generated a translation profile of our human-derived forebrain organoid model, employing the same riboLace approach (Figure S6A)51. The treatment with SAC primarily led to changes in RPFs, rather than the input mRNA abundance in organoids, indicating that YTHDF1 inhibition alters translation efficiency (Figure S6B). Specifically, we found that the genes exhibiting decreased translation efficiency after SAC treatment are enriched in pathways regulating synaptic transmission and plasticity, which account for 13% of hypo-translated genes (Figure S6C). Amongst the twenty-nine hypo-translated mRNAs involved in regulating synaptic pathways, eighteen of them have been reported to be involved in FXS (Figure 6B). Most of these transcripts are modified by m6A and bound by YTHDF1 (Figure 6B). Notably, two of these genes, ADCY1 and APP are already identified to be therapeutic targets in FXS52,53, supporting our hypothesis that SAC-mediated YTHDF1 inhibition could be a strategy guiding drug development to treat FXS. Similar to our observations in neurons, we showed that SAC treatment led to more hypo-translated genes in FMRP depleted FXS organoid (FXS, 225) than control (CTRL, 184) (Figure 6C).
Next, we assessed the effects of SAC on the neuronal defects in FXS organoids. To delineate the effect of SAC on the neuronal differentiation defect caused by the loss of FMRP, we determined the distribution of specific cell types (PAX6+ or CTIP2+ cells) among equally sized bins spanning the neuroepithelium of forebrain organoids, as previously reported54. In FXS forebrain organoids compared to control organoids, the number of PAX6+ neural progenitor cells (NPCs) was significantly reduced in the ventricular zone (VZ)-like layer (lower bins) while increased in the cortical plate layer (higher bins) (Figure 6D). This indicated that the alteration in PAX6+ NPC differentiation in FXS organoids was reversed after treatment with SAC. Moreover, the proportion of CTIP2+ cortical plate neurons drastically increased in Bin 2 and 3 in DMSO-treated FXS forebrain organoids compared to DMSO-treated control organoids, illustrating the dysregulated neuronal differentiation and layer specification by FMRP loss (Figure 6D). When FXS organoids were treated with SAC, the dysregulated CTIP2+ neuronal differentiation and layer organization were rescued to a level comparable to that of control forebrain organoids (Figure 6D). Importantly, SAC treatment in control forebrain organoids did not alter CTIP2+ neuronal differentiation and layer organization (Figure 6D). Consistently, SAC treatment downregulates the translation of key targets such as ADCY1 and APP only in FXS organoids (Figure 6B).
We observed a partial rescue of the proliferation deficit in NPCs in FXS forebrain organoids following SAC treatment. This rescue was demonstrated by quantifying cells with proliferation marker Ki67 and NPC marker SOX255,56 using immunofluorescence (Figure 6E). While a slight increase in the proportion of Ki67+ NPCs in control forebrain organoids by SAC treatment was observed, this effect is not as significant as in FXS organoids (Figure 6E, P = 0.005 for FXS and P = 0.03 for control group).
Furthermore, we examined NPC cell cycle kinetics and found that bath application of SAC significantly reverted the phenotype of aberrantly accelerated cell cycle exit in FXS organoids compared to the control (Figure 6F, cell cycle exit was assessed by measuring the proportion of Ki67− and EdU+ cells among total EdU+ cells within 24 hours’ labeling with EdU labeling). Consistent with the notion YTHDF1 is inactive in control organoids, SAC treatment did not cause a change of cell cycle exit in control forebrain organoids (Figure 6F). Moreover, we found that excessive synapse formations57 confirmed in our FXS forebrain organoids could be rescued to a level comparable to controls by both shRNA-enabled YTHDF1 knockdown or SAC application (Figure S6D).
In summary, our findings highlight the significance of dysregulated YTHDF1 condensation and its translation function in contributing to disease phenotypes in FXS in the absence of FMRP. The small molecule YTHDF1 inhibitor, SAC, could rescue the defects of decreased proliferation of neural progenitor cells, hastened cell cycle exit and altered neural differentiation in FXS organoids by inhibiting the condensing behavior of YTHDF1. Moreover, these rescue effects are specific to FMRP-deficient systems, demonstrating the importance of activated YTHDF1 on FXS pathogenesis. These results further suggest YTHDF1 as a potential target for treating FXS, opening up possibilities for future therapy development (Figure 6A). Our study also suggests that targeting dysregulated RNP granule equilibrium components may guide drug target discovery in other human diseases such as amyotrophic lateral sclerosis (ALS) and Alzheimer’s disease (AD).
DISCUSSION
Proteins with prion-like features play important roles in local synaptic mRNA translation, which contributes to synaptic plasticity58. Here we report the first example of an activity-dependent phosphorylation of the FMRP/YTHDF1 system as a previously unrecognized functional module. Neurons utilize this mechanism to modulate translation in response to stimuli. This discovery presents an example of a post-translational modification (PTM) as a switch for segregation of RBPs into different functional RNP granules.
Absence of FMRP results in fragile X syndrome, which majorly leads to the subsequent aberrant translation. Rebalancing this aberrant translation has been thought to be a viable strategy to treat FXS45. We developed a selective non-covalent YTHDF1 inhibitor, salvianolic acid C (SAC), and characterized its functions in neurons and in an FXS forebrain organoid model. YTHDF1 inhibition rescued the defects of neural progenitor cells and an FXS organoid model, further supporting the involvement of YTHDF1 activation in the FMRP-deficient system.
FMRP was reported to be a repressor of global protein synthesis by inhibiting translation elongation19,46,59,60. However, FMRP only binds to 4% of mRNAs in the brain61 and it was also suggested to facilitate translation of its target mRNAs21. Our findings showing that FMRP inhibits YTHDF1-mediated translation revealed its role as a general translation regulator beyond its own target mRNAs. Considering the prevalence of m6A in mammalian mRNAs, our findings significantly expanded the RNA pool regulated by FMRP that includes YTHDF1 target mRNAs. This provides an explanation for the importance of FMRP to mRNA translation and its role as a translation repressor in the brain.
Our study not only suggests YTHDF1 as a potential drug target in treating FXS, but it may also inspire further research on other loss-of-function neuronal diseases or defects related to cytosol RNA processing and translation. Loss-of-function gene perturbations are usually challenging for therapy development because of the difficulty to supply protein agonists. Targeting other RNP granule components to compensate for protein loss could open up various rescue possibilities. With numeral dysfunctions of LLPS events reported in neuronal disorders such as amyotrophic lateral sclerosis (ALS) and Alzheimer’s disease (AD), targeting the dysregulated RNP granules could be a generally applicable strategy towards further understanding of their molecular mechanisms and for developing potential future therapies.
Limitations of the study
Our approach involving the use of RNase to precipitate protein condensates could be artificial and discoveries made should be validated by orthogonal experiments. Phosphorylation of FMRP was known to regulate its own phase separation. Thus, it may also impact the RNA binding abilities of FMRP itself, which may also contribute to FXS pathology. The pathogenesis of FXS in the absence of FMRP is a multifactored process and our proposed FMRP-YTHDF1 axis is most likely one of the many contributing pathways, although our FXS organoid model studies suggested a key role of YTHDF1 in FXS. Please also note that the current YTHDF1 inhibitor may not cross the blood-brain-barrier, and thus new inhibitors will need to be developed to further validate effects of YTHDF1 inhibition for FXS treatment in vivo in the future.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests may be directed to and will be fulfilled by the lead contact corresponding author Chuan He (chuanhe@uchicago.edu).
Materials availability
All unique reagents generated in this study will be made available upon reasonable request without restrictions.
Data and code availability
Sequencing data have been deposited into the Gene Expression Omnibus (GEO) under the accession number GSE214882 for RNA sequencings.
This work does not generate original codes.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact (Chuan He, chuanhe@uchicago.edu) upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Cell culture
HeLa or HEK293T cells were kept in DMEM (ThermoFisher Scientific) + 10% FBS (ThermoFisher Scientific). The Fmr1 KO mouse line was obtained from The Jackson Laboratory. The S499A mutant line was generated by homologous recombination. The expression of FMRP in S499A line is similar to wildtype, however, no phosphorylated FMRP could be detected in S499A line. The characterization of the new mutant line is under preparation for another manuscript (Jin, unpublished data). For the isolation of primary neurons from P1–3 wildtype, Fmr1 knockout or FMRP S499A mutant mouse, Papain dissociation system from Worthington Biochemical Corporation (LK003160) was used. Briefly, the whole cerebral hemispheres were dissected out, slightly minced and dissociated in papain dissociation solution containing 20 units/ml papain and 0.005% DNase with constant agitation on a rocker platform for 30 minutes at 37 °C. Then, the dissociated tissue mixture was triturated with 10 ml pipette. The undissociated tissue was allowed to settle down to the bottom of the tube. Only the cloudy cell suspension was centrifuged at 300 g for 5 minutes at room temperature. The pelleted cells were resuspended in reconstituted albumin-ovomucoid inhibitor solution. The discontinuous density gradient was made with albumin-inhibitor solution. The cell suspensions were carefully layered on top, then centrifuged at 70 g for 6 minutes at room temperature. The dissociated cells pelleted at the bottom of the tube, and the membrane fragments remained at the interface. The supernatant was discarded and pelleted cells were immediately resuspended in the medium for cell culture or frozen in Bambanker cell freezing media (bulldog bio BBO1) for storage freezing at the rate of −1 °C/minute. The cryopreserved neurons are then thawed on PDL (Gibco A3890401)-coated surface and kept in neurobasal medium (Gibco) with B-27 and N-2 supplement. DIV9 neurons are used for individual experiments.
METHOD DETAILS
Protein mass spectrometry
Mouse brain tissue was dissected for identification of YTHDF1 protein partners. Briefly, about 400 μl pellets were resuspended and lysed in 1,200 μl RIPA lysis buffer (Thermo Scientific, 89900) supplemented with Halt™ Protease and Phosphatase Inhibitor Cocktail (Thermo Scientific, 78442). Immunoprecipitation was performed with anti-YTHDF1 antibody (Abcam, 17722) conjugated to protein A magnetic beads (Invitrogen, 10001D) for 18 hours at 4°C. Beads were washed extensively with RIPA lysis buffer at room temperature for 5 times.
The beads were washed with PBS buffer and suspended in 200 μl of 1 M urea/TEAB (Triethylammonium bicarbonate) buffer. Proteins were then reduced by 10 mM dithiothreitol (DTT) at 37°C for 30 minutes and blocked using 20mM iodoacetamide (IAA) at 35°C for 30 minutes in dark. After trypsin digestion, samples were subjected to reductive dimethylation. Briefly, 8 μl of 4% HCHO, or D13CDO were added to two replicates, respectively and 8μL of 0.6M NaBH3CN was added to each sample on ice and incubated at 29°C for 1 h. The reactions were quenched by 32 μl of 1% ammonia solution and 16 μl formic acid. Two replicates were then combined, desalted, dried, and subjected to LC-MS/MS analysis using Orbitrap Eclipse™ Tribrid™ Mass Spectrometer. Protein searching and quantification analysis were performed using MaxQuant.
Small molecule treatment for cultured neurons
To avoid contacts with the air, all the small molecules were first dissolved in DMSO at a higher concentration and them diluted to full neuron culture medium at 2× the concentration. For precise control of drug incubation time, half of the pre-existing medium was replaced with medium with 2× drug concentrations. The final concentrations of individual small molecules were determined based on their reported IC50 values against their physiological targets (NPS 2390: 82 nM, DHPG, 7 μM, and TTX, 2 nM). For NPS 2390, DHPG and TTX treatments, cells were harvested and lysed after 5 minutes.
In vitro protein co-condensation
Recombinant protein fluorophore conjugation was performed as follows: purified protein in physiological buffer (20mM Tris-HCl (pH 7.4), 150mM NaCl) was treated by 5 mM TCEP at room temperature for 10 minutes. Then the reactive dye (Alexa Fluor 488 C5 Maleimide (ThermoFisher Scientific, A10254) or Alexa Fluore 647 C2 Maleimide (ThermoFisher Scientific, A20347)) was added into the reaction at 20× the protein concentrations. The reaction was conducted at room temperature for two hours with shaking. The residual reactive dye was isolated from the proteins via desalting with size-exclusion resin (Bio-Rad, 7576221) three times.
Fluorescence-labeled purified recombinant proteins were diluted to 1 μM and incubated in physiological buffer (20mM Tris-HCl (pH 7.4), 150mM NaCl). Droplet formation was initiated by the addition of TEV protease at a final concentration of 0.03 mg/ml and incubation at 30°C for 30 minutes. In some experiments, 10% PEG8000 was added to induce protein condensation. After 30 minutes of incubation, phase-separated droplets were imaged by Leica SP8 confocal microscopy.
In vitro protein phosphorylation with CK2
The phosphorylation of recombinant FMRP with CK2 was performed with commercial CK2 assay (NEB, P6010S) at 30 °C for one hour. The resulting phosphorylated FMRP was verified via western blotting.
Tethering assay
Tethering assay in HeLa and HEK293T cell lines were performed similarly. About 50 ng reporter plasmid (pmirGlo-Ptight-2BoxB-2MS2) and 250 ng of each effecter plasmid (Flag-MS2, Flag-λ, Flag-YTHDF1N-MS2, Flag-YTHDF1N-λ, or the combination indicated) were used to transfect HeLa or HEK293T in each well of six-well plate at 60%–80% confluency. Six hours later, transfection mixture was replaced with fresh media. Four hours later, each well was trypsin-digested and re-seeded into 96-well plate (1:30) and 12-well plate (1:3) to allow expression of reporter and effector plasmids. Eighteen hours after reseeding, cells in the 96-well plate were analyzed by Dual-Glo Luciferase Assay Systems (E2920, Promega). Firefly luciferase (F-Luc) activity was normalized by renilla luciferase (R-Luc) to evaluate protein production from the reporter. At the same time, samples in the 12-well plate were processed to extract total RNA (DNase I digested) by TRIzol™ reagent (Invitrogen, 15596026), followed by RT-qPCR quantification. The amount of F-Luc mRNA was also normalized by that of R-Luc mRNA. Translation efficiency (TE) of F-Luc mRNA was calculated as the ratio of normalized F-Luc activity (protein level) to normalized F-Luc mRNA level.
Tethering assay in neurons was performed by pre-coating 12-well plates with poly-D-lysine/laminin and seed neurons to coated plates, on day 6 post passaging, cells were transfected with AD1 Primary Cell 4D-nucleofector Y kit (Lonza, V4YP-1A24) with 2 μg reporter plasmid (pmirGlo-Ptight-2BoxB-2MS2) and 16 μg of each effecter plasmid (HA-MS2 or HA-YTHDF1N-MS2) to each well. Fourty-eight hours after nucleofection, neuronal cells are depolarized and collected at different timepoints, luciferase activity was probed with a procedure similar to HeLa and HEK293T cells.
Tethering assay in rabbit reticulocyte lysates was performed by applying in vitro transcribed and capped mRNA encoding mApple-YTHDF1N-MCP or mApple-MCP to the lysates (Promega, L4960) for effector protein translation. After one hour, applying in vitro transcribed and capped mRNA encoding F-Luc-2MS2 and R-Luc were added to the lysates for reporter translation. Luciferase signals were probed after 30 minutes. In vitro transcriptions were performed with a T7 system (Invitrogen, AM1345). 1,6-hexanediol was added to 0.2% (w/v) to dissociate YTHDF1 granules.
Translation efficiency was determined via dividing normalized protein levels by normalized RNA levels. The N terminus of YTHDF1 (amino acid 1–395) was used for all YTHDF1-tethered reporters.
Polysome profiling
We followed the procedure reported previously62. Briefly, cycloheximide (CHX) was added to the media at 100 μg/ml for 7 minutes before lysis and the cell lysate was cleared and digested with Turbo DNase (Invitrogen, AM2238). The cleared lysate was subjected to ultracentrifugation at 28,000 rpm for 3 hours at 4°C and separated on a 5% - 50% sucrose gradient. 1,6-hexanediol was added to 2% (w/v) to dissociate YTHDF1 granules.
Cell fractionation
For RNase treated cell fractionation, one 90% confluent 15-cm dish of WT HEK293T cells was washed with PBS at room temperature once, then harvested with a cell scraper in 2 ml PBS. Cell pellet was resuspended with 3 volumes of lysis buffer (150 mM KCl, 10 mM HEPES pH 7.6, 2 mM EDTA, 0.5% NP-40, 0.5 mM DTT, 1:100 protease inhibitor cocktail, 20 U/ml RNase inhibitor) and incubated on a rotor at 4°C for 15 minutes. Cell pellet was cleared by centrifuging at 15,000 g, 15 minutes, 4°C. Input samples were saved, and lysates were then separated into two equal volumes, one was subjected to 10 μl RNase A/T1 mix (ThermoFisher Scientific, EN0551), 10 μl lysis buffer was added to the control sample and both samples were incubated at 37°C for 10 minutes. Then the samples were spun at 4°C, 15,000 g for 15 minutes. Save supernatant as (Sol) fraction. Resuspend pellet from in 100 μl laemmli buffer (Bio-rad, 1610747) and heat at 90°C for 5 minutes as (RG) fraction. Load sample for SDS-PAGE followed by western blotting.
Protein co-immunoprecipitation (co-IP)
Cells, or frozen pellets were lysed in ice-cold RIPA buffer (ThermoFisher Scientific, 89900) with end-to-end rotation for 30 minutes. Lysates were cleared with centrifugation at 4 °C, 21,000 g for 15 minutes. Conjugated protein A/G beads (Invitrogen, 10015D) were applied to the lysate for overnight immunoprecipitation. The beads were separated from the supernatant on a magnet during washing steps with ice-cold RIPA buffer. Final elution was performed by resuspension in 1x laemmli buffer (Bio-rad, 1610747) and heating at 90 °C for 5 minutes.
RNase-assisted co-IP
For protein co-IP after RNase treatment, one 15-cm dish of confluent WT HeLa cells was washed once with DPBS. After washing, the cells were harvested in 0.6 ml ice-cold buffer L (50 mM Tris-HCl pH 7.6, 50 mM NaCl, 5 mM MgCl2, 0.1% NP-40, 1 mM DTT, 1× Halt™ protease & phosphatase inhibitor (ThermoFisher Scientific, 78440), 10 U/ml RNase inhibitor (ThermoFisher Scientific, AM2696)) on ice. All subsequent steps were conducted at 25°C. Cell suspensions were given 30 strokes (1 ml, in 30 s) in a Dounce homogenizer, followed by centrifugation at 2,000 g for 2 minutes. The supernatant (Cyt, cytosolic fraction) was collected without disrupting nuclei pellet (Nuc). Cytosol fraction was splitted into two equivalent fractions, 20 μl RNase A/T1 mix (or buffer L) was added to each fraction and incubated at room temperature for 20 minutes while rotating. The two fractions were split to two for IgG and YTHDF1 IP (2 μg each, rabbit IgG isotype control, Abcam ab37415; YTHDF1 antibody, Abcam ab220162) and incubated at room temperature for one hour. Then 10 μl protein A beads (Invitrogen, 1001D) were added to each fraction and tubes were incubated at room temperature for another one hour. Beads were separated on a magnetic device and washed extensively with 1 ml NT2 wash buffer (50 mM HEPES, pH 7.6, 200 mM NaCl, 2 mM EDTA, 0.05% NP-40, 0.5 mM DTT, 200 U/ml SUPERase·In (Invitrogen, AM2694)) at room temperature for 5 times. IP sample was eluted with 80 μl 1x laemmli buffer (Bio-rad, 1610747) at 90 °C for 10 minutes. The eluents were submitted to TMT labeling and MS-Spec identification.
Fluorescence microscopy
For imaging of nascent protein, neuronal cells were depolarized with 50 mM KCl for 10 minutes prior to incubation of L-homopropargylglycine (HPG) for 30 minutes. Then cells were fixed and clicked with Alexa Fluor 488 dye according to manufacturer’s instruction of Click-iT™ HPG Alexa Fluor™ 488 Protein Synthesis Assay Kit (ThermoFisher Scientific, C10428). Fluorescence intensity across different samples were quantified with Cellprofiler 3.0 with a custom workflow, total protein synthesis rate was obtained by multiplying average intensity in each cell by the area of each cell. For imaging of protein condensates, the mixtures were prepared in vitro on a 384-well plate with coverslip bottom (Cellvis, P384–1.5H-N). Image acquirement and processing were conducted similarly.
For proximity ligation assays (PLA), we followed the manufacturer’s instructions for Duolink® Proximity Ligation Assay (MilliporeSigma). PLA puncta were identified and quantified with Cellprofiler 3.0 with a custom workflow adapted from example: Speckle Counting.
For immunolabeling, neurons were fixed with 4% paraformaldehyde (PFA) in DPBS at 37°C for five minutes, permeabilized with MeOH at −20°C for eight minutes, dried at room temperature for ten minutes, then washed with DPBS at room temperature for three times. Chambers are blocked in blocking buffer (DPBS, 0.5% BSA, 0.05% Triton X-100, 1:100 SUPERase·In™ (Invitrogen, AM2694)) for one hour at room temperature and YTHDF1 antibody (Abcam, ab220162) was 1:100 diluted in blocking solution and incubate at room temperature for one hour. Chambers were washed with 0.05% Triton X-100 in DPBS for 3 times, then 1:100 diluted goat anti rabbit IgG-AF568 conjugate (ThermoFisher Scientific, A-11011) in blocking solution was added to each well and chambers were incubated at room temperature for one hour. Then chambers were washed with 0.05% Triton X-100 in DPBS for three times and fixed with 4% PFA in DPBS for thirty minutes at room temperature and washed for three times with DPBS. Nuclei was counterstained with 2 μg/ml Hoechst 33342 (Abcam, ab145597) in DPBS at room temperature for 20 minutes, wash with DPBS for 3 times. Chambers were stored at 4°C before proceeding to imaging.
All samples were imaged on a Leica SP8 laser scanning confocal microscope at the University of Chicago light facility.
Turbidity measurement
Purified YTHDF1 protein was dialyzed to lysis buffer (25 mM Tris-HCl (pH 7.4), 150 mM KCl, 2.5% Glycerol and 0.5 mM DTT). WT HeLa cells treated with DMSO or 2 μM CX-4945 (MedChemExpress, HY-50955) were lysed with lysis buffer on ice and cleared by centrifugation. Different ratios of cell lysates and purified YTHDF1 were mixed and incubated at 37°C for 10 minutes before analysis of optical density at 600 nm (OD600) on a Nanodrop instrument. Turbidity measurements for cell lysates were performed similarly.
RiboLace
Cultured neurons or organoids were treated with 100 μg/ml cycloheximide (MilliporeSigma, C1988) for seven minutes. Cells were spun down and washed once with ice-cold DPBS. Cell pellets were flash-frozen with liquid nitrogen and stored at −80 °C. Total RNA was extracted from cell pellets with standard TRIzol™ protocol. Library constructions were performed with SMARTer Stranded Total RNA-Seq Kit v2 (TaKaRa, 634417) following the manufacturer’s protocols. Ribosome protected fragments (RPFs) were extracted from the cell lysates following the riboLace protocol (IMMAGINA, RL001). RPFs were end-repaired and subjected to library contruction with NEB small RNA kit (NEB, E7560S). Libraries were pooled and sequenced on a NovaSeq6000 sequencer with paired-end mode 50bp setting.
Quantitative RT-PCR (RT-qPCR)
To quantify expression levels of transcripts, total RNA was reverse transcribed by using PrimeScript™ RT Master Mix (Takara, RR0361) with oligo dT primer and random 6 mers as primer. The cDNA was then subjected to real-time PCR (LightCycler 96 sytem, Roche) by using FastStart Essential DNA Green Master (Roche, 06402712001) with gene specific primers. Relative changes in expression were calculated using the ΔΔCt method.
Recombinant protein purification
Standard molecular cloning strategies were used to generate N-terminally 6×His tagged prion-like domains of YTHDF1 (residues 69–363), YTHDF2 (residues 69–384) and YTHDF3 (residues 71–390). Recombinant proteins were expressed in E. coli BL21 (DE3) cells grown to OD600 of 0.6 in LB medium. The expression was induced with 0.6 mM IPTG at 16°C for 20 hours and cells were harvested via centrifugation.
For purification of prion-like domain of YTHDF proteins, cell pellet was resuspended in a lysis buffer containing 25 mM Tris-HCl (pH 7.5), 200 mM NaCl, 20 mM imidazole, 10 mM β-mercaptoethanol (β-ME), and protease inhibitors (Ethylenediaminetetraacetic acid-free protease inhibitor cocktail tablet, MilliporeSigma) and disrupted by sonication for 3 minutes. Cell lysates were clarified via centrifugation at 36,000 g for 50 minutes and supernatant was applied to Ni2+-NTA resin and washed with lysis buffer, and bound proteins were eluted with lysis buffer supplemented with 300 mM imidazole. The eluted protein was analyzed by SDS-PAGE and concentrated by centrifugal filtration (Amicon Ultra-15). Final concentrated protein was aliquoted, flash frozen, and stored at −80 °C for future use.
For purification of FMRP and YTHDF1 (WT and mutants), cell pellet was resuspended in a lysis buffer containing 25 mM Tris-HCl (pH 7.5), 500 mM NaCl, 20 mM imidazole, 10 mM β-mercaptoethanol (β-ME), and protease inhibitors (Ethylenediaminetetraacetic acid-free protease inhibitor cocktail tablet, MilliporeSigma) and disrupted by sonication for 3 minutes. Cell lysates were clarified via centrifugation at 36000 g for 50 minutes and supernatant was applied to MBP resin and washed with lysis buffer, and bound proteins were eluted by on-column digestion by TEV enzyme (with His tag). The purified proteins were applied to Ni2+-NTA resin and washed with lysis buffer, and bound proteins were eluted with lysis buffer supplemented with 300 mM imidazole. The eluted protein was analyzed by SDS-PAGE and concentrated by centrifugal filtration (Amicon Ultra-15). Final concentrated protein was aliquoted, flash frozen, and stored at −80°C for future use.
Fluorescence polarization (FP)
For YTHDF1 inhibitor screening, the compounds were first diluted to 80 μM in the high throughput screening (HTS) experiment. Then the compounds were incubated with 1.25 μM YTHDF1 (aa 361–559) at room temperature for 30 minutes in the assay buffer (20 mM Hepes (pH 7.4), 50 mM NaCl, 0.01% (v/v) tween 20, 5% (v/v) glycerol). And 40 nM 5’-FAM labeled m6A-containing mRNA (5’-FAM-UUCUUCUGUGG (m6A) CUGUG-3’) was next added and incubated with the mixture at 4 °C for 1 hour before the measurements performed on Envision Readers (PerkinElmer). The fluorescently-labeled m6A-containing mRNA was used to adjust the gain factor. The same amount of DMSO and unlabeled m6A-containing mRNA were used as the negative and positive control, respectively.
The same amount of DMSO was used as negative control (no compound control, NCC). According to the results of unlabeled m6A-containg mRNA (m6A) competing experiments, 10 μM unlabeled m6A-containg mRNA, which has the same sequence of 5’-FAM labeled m6A-containg mRNA, was used as the positive control. To evaluate the stability of the screening method, we utilized the negative control (NCC) and the solution of 5’-FAM labeled m6A-containg mRNA (Gain), which has approximate value of positive control, to calculate the Z’ using the equation (3).
Where factor, = standard deviation of Gain, = standard deviation of NCC, = mean value of Gain, = mean value of NCC.
AlphaScreen
SAC was first diluted from 210 μM to a sequence of concentrations as indicated and incubated with 100 nM His-tagged YTHDF1 (aa 361–559) or His-tagged YTHDF2 (aa 380–579) at room temperature for 30 minutes in the assay buffer (20 mM Hepes (pH 7.4), 150 mM NaCl, 1 mg/ml BSA, 0.01% (v/v) TritonX-100). Then, 10 nM biotinylated m6A-containing mRNA (5’-biotin-UUCUUCUGUGG (m6A) CUGUG-3’) was added, followed by adding streptavidin donor beads and anti-His acceptor beads in the dark conditions. The samples were kept away from light and incubated at 4 °C for 1 hour. The Alpha signal was detected on Envision Readers (PerkinElmer). The same amount of DMSO was used as the negative control, and the unlabeled m6A-containing mRNA (5’-UUCUUCUGUGG (m6A) CUGUG-3’) was used as the positive control.
In the substrate competition experiment, unlabeled m6A-containing mRNA was first diluted to 400 nM, 300 nM, 200 nM and 100 nM and incubated with 200 nM His-tagged YTHDF1 (aa 361–559) in the assay buffer at 4 °C for 10 minutes. SAC was next diluted from 210 mM and incubated with samples in the assay buffer at room temperature for another 30 minutes. And 20 nM biotinylated m6A-containing mRNA was then added to samples. The following steps are as same as aforementioned.
NMR
SAC was dissolved in 5% deuterated DMSO to the concentration of 200 μM. YTHDF1 (aa 361–559) was diluted to 20 μM, 10 μM and 5 μM in phosphate buffer (20 mM NaH2PO4, 20 mM Na2HPO4, 150 mM NaCl, pH 7.4, D2O). NMR CPMG experiment was conducted at 25 °C using Bruker Avance III spectrometer (600 MHz proton frequency) with a cryogenically cooled probe (Bruker biospin, Germany). Through the pulse sequence (RD−90°−(τ−180°−τ) n−ACQ), the solvent-suppressed 1D 1H CPMG was obtained. And a 54.78 dB pulse in 4 s duration of the recycle delay (RD) was applied to eliminate water resonance in the presaturation procedure. Then, the 90° pulse length was modulated to 11.82 μs approximately. And at last, a total of 4 dummy scans and 64 free induction decays (FIDs) were collected into 64000 acquisition points, covering a spectral width of 12 kHz (20 ppm) with the acquisition time (ACQ) of 2.73 s.
Isothermal Titration Calorimetry (ITC)
Freshly purified YTHDF1 (aa 361–559) was first dialyzed at 4 °C overnight and diluted to 50 mM with dialysis buffer (20 mM Hepes (pH 7.4) and 200 mM NaCl) before loaded into the sample cell. SAC was then dissolved and diluted to 1 mM with dialysis buffer and loaded into the syringe. After one 0.4 μL injection, through titrating SAC into YTHDF1 (aa 361–559) by nineteen 2 μL injections at 180 s intervals, the isothermal titration calorimetry (ITC) experiments were performed on the Microcal ITC 200 isothermal titration calorimeter instrument (GE Healthcare) at 25°C. In order to exclude the thermal effect of background dilution, 1 mM SAA titrating into dialysis buffer was also performed as the control. The experimental data was analyzed using Microcal ORIGIN (v7.0) software (Microcal Software).
Microscale thermophoresis (MST)
Microscale Thermophoresis (MST) experiments was performed on Monolith NT. Automated instrument (NanoTemper Technologies) via the label-free method. SAC was diluted from 250 μM in a three quarters manner. Then diluted compounds were mixed with 4 μM purified YTHDF1 protein at the proportion of 1:1. Samples were incubated at room temperature for 20 minutes in MST buffer (20 mM Hepes, pH 7.4, 200 mM NaCl, 0.1mM Pluronic® F-127) and centrifuged at 13000 rpm for 10 minutes at 4 °C before the measurement. Then the samples were loaded into Monolith NT. Automated LabelFree Premium Capillary Chips (NanoTemper Technologies) and the measurements were started. MO. Affinity Analysis Software v2.3 (NanoTemper Technologies) was used to analyze the data and get the KD value of SAC.
Human forebrain-specific organoid cultures
The human iPSC lines were reprogrammed from skin biopsy samples of three FXS male patients and three age-matched healthy males at Emory University. All iPSC lines were cultured on irradiated MEFs in human iPSC medium consisting of D-MEM/F12 (Invitrogen), 20% Knockout Serum Replacement (KSR, Invitrogen), 1× Glutamax (Invitrogen), 1× MEM Non-essential Amino Acids (Invitrogen), 100 μM β-Mercaptoethanol (Invitrogen), and 10 ng/ml human basic FGF (PeproTech). Forebrain-specific organoids were generated using established protocols as previously described56. Briefly, human iPSC colonies were detached from the MEF feeder layer with 1 mg/ml collagenase treatment for 1 hour and suspended in embryonic body (EB) medium, consisting of FGF-2-free iPSC medium supplemented with 2 μM Dorsomorphin and 2 μM A-83 in non-treated polystyrene plates for 4 days with a daily medium change. On days 5–6, half of the medium was replaced with induction medium consisting of DMEM/F12, 1× N2 Supplement (Invitrogen), 10 μg/ml Heparin (MilliporeSigma), 1× Penicillin/Streptomycin, 1× Non-essential Amino Acids, 1× Glutamax, 4 ng/ml WNT-3A (R&D Systems), 1 μM CHIR99021 (Tocris), and 1 μM SB-431542 (Tocris). On day 7, organoids were embedded in Matrigel (BD Biosciences) and continued to grow in induction medium for 6 more days. On day 14, embedded organoids were mechanically dissociated from Matrigel by pipetting up and down onto the plate with a 5 ml pipette tip. Typically, 10–20 organoids were transferred to each well of a 12-well spinning bioreactor (SpinΩ) containing differentiation medium, consisting of DMEM/F12, 1× N2 and B27 Supplements (Invitrogen), 1× Penicillin/Streptomycin, 100 μM β-Mercaptoenthanol (Invitrogen), 1× MEM NEAA, 2.5 μg/ml Insulin (MilliporeSigma). All media was changed every other day. Control or FXS forebrain organoids were treated with a newly developed DC-Y22 (SAC) at 20 μM from day 49 to 56, fixed at day 56, and stained for immunocytochemistry.
Synapse formation assay
Control or FXS forebrain organoids were treated with SAC at 20 μM from day 65 day and, fixed at day 80, and stained with Synapsin I (Synaptic System) and MAP2 (abcam). Non-specific scramble or shRNA specifically against YTHDF1 (MilliporeSigma; CCCTACCTGTCCAGCTATT) lentivirus was infected at 2 multiplicity of infection (MOI), cultured for 4 days, fixed at 80 day, and stained with Synapsin I and MAP2 antibodies.
Immunocytochemistry
For immunocytochemistry, forebrain organoids were processed at day 56 as previously described56. Briefly, whole organoids were fixed in 4% Paraformaldehyde in Phosphate Buffered Saline (PBS) for 30–60 minutes at room temperature. Organoids were washed 3 times with PBS, incubated in 30% sucrose solution overnight, embedded in tissue freezing medium (General Data) and sectioned with a cryostat (Leica). For immunostaining, freezing medium was washed with PBS before permeabilization with 0.2% Triton-X in PBS for 1 hour. Tissues were then blocked with blocking medium consisting of 10% donkey serum in PBS with 0.1% Tween-20 (PBST) for 30 minutes. The following primary antibodies were used: anti-Ki67 (mouse; 1:500; BD-Pharmingen), anti-SOX2 (goat; 1:500; R&D Systems), anti-PAX6 (rabbit; 1:500; Thermo fisher), anti-CTIP2 (rat; 1:400; Abcam), Synapsin I (Rabbit; 1:200; Synaptic System) and MAP2 (chicken; 1:1000; abcam). Primary antibodies diluted in blocking solution were applied to the sections overnight at 4°C. After washing with PBST, secondary antibodies diluted in blocking solution were applied to the sections for 1 hour at room temperature, washed with PBST and stained with DAPI. For EdU labeling, forebrain organoids were pulsed labeled with EdU (10 μM) for 24 hours at day 56, followed by fixation and immunostaining. All images were captured by Nikon Eclipse Ti-E microscope. Quantitative analyses were conducted on randomly selected cortical structures in a blind fashion. The numbers of cells positive for each marker were measured using ImageJ software (NIH). To quantify the distribution of specific cell types in the neuroepithelium, the entire span of the neuroepithelium was divided into five equal portions (bins) and measured the distributions of specific cell types by calculating the percentage of each marker in the total DAPI+ cells in each bin.
RiboLace analysis
Adaptors for input and ribosome-protected fragments (RPFs) reads were trimmed with cutadapt (version 1.14)63 with the minimum length set to 1 (--minimum-length 1 and --pair-filter any), respectively. The trimmed reads were aligned to the hg38 or mm10 genome64 with STAR (version 2.5.2b)65 with the following parameters (--quantMode TranscriptomeSAM --alignEndsType Local). Then the output SAM files were converted to BAM files with samtools (version 1.6)66. Posterior_mean_estimate counts (PMC) values for each transcript were calculated with RSEM (version 1.2.28)67. Translation efficiency was determined by normalizing PMC of RPFs to input.
QUANTIFICATION AND STATISTICAL ANALYSIS
P values were determined using two-tailed t-test or Mann-Whitney test indicated in the figure legends. NS means not significant (P > 0.05). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Error bars represent mean ± s.d.. For box plots, the center line represents the median, the box limits show the upper and lower quartiles, whiskers represent 1.5 x the interquartile range unless otherwise specified.
Reference sequence and annotation data
The human and mice genome sequence (hg19, hg38, and mm10), the annotated gene models and the annotations of rRNAs were obtained from the UCSC genome browser database.
Supplementary Material
Table S3 Summarized read counts for high-throughput sequencing in this study, related to Figure 5–6, S5–S6.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-YTHDF1 antibody | Abcam | ab220162, RRID:AB_2892231 |
| Rabbit monoclonal anti-YTHDF2 antibody | Abcam | ab220163, RRID:AB_2868573 |
| Rabbit polyclonal anti-FMRP antibody | Abcam | ab17722, RRID:AB_2278530 |
| Rabbit polyclonal anti-FMRP(Ser499) antibody | PhosphoSolutions | P1125-499, RRID:AB_2492094 |
| Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary antibody, Alexa Fluor 568 | ThermoFisher Scientific | A-11011, RRID:AB_143157 |
| Rabbit monoclonal anti-GAPDH antibody | Cell Signaling | 5174, RRID:AB_10622025 |
| Rabbit monoclonal anti-RPS6 antibody | Cell Signaling | 2217, RRID:AB_331355 |
| Anti-rabbit IgG, HRP-linked antibody | Cell Signaling | 7074, RRID:AB_2099233 |
| Anti-mouse IgG, HRP-linked antibody | Cell Signaling | 7076, RRID:AB_330924 |
| Rabbit polyclonal anti-phosphoserine antibody | Abcam | Ab9332, RRID:AB_307184 |
| Mouse monoclonal anti-Ki67 antibody | BD Biosciences | 556003, RRID:AB_396287 |
| Goat polyclonal anti-SOX2 antibody | R&D systems | AF2018, RRID:AB_355110 |
| Rabbit polyclonal anti-PAX6 antibody | ThermoFisher Scientific | 42-6600, RRID:AB_2533534 |
| Rat monoclonal anti-CTIP2 antibody | Abcam | ab18465, RRID:AB_2064130 |
| Guinea pig anti-Synapsin I antibody | Synaptic Systems | 106104, RRID:AB_2721082 |
| Mouse monoclonal anti-O-linked N-acetylglucosamine (O-GlcNAc) antibody | ThermoFisher Scientific | MA1-072, RRID:AB_326364 |
| Rabbit monoclonal anti-ubiquitin antibody | Cell Signaling | 20326, RRID:AB_3064918 |
| Mouse monoclonal anti-phosphotyrosine antibody | MilliporeSigma | 05-321, RRID:AB_2891016 |
| Rabbit monoclonal anti-DDDDK tag | Abcam | ab205606, RRID:AB_2916341 |
| Rabbit polyclonal anti-eEF1G antibody | Abcam | ab72368, RRID:AB_1268721 |
| Rabbit monoclonal anti-LRPAP1 antibody | Abcam | ab76500, RRID:AB_1523899 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| DMEM | Gibco | 11995 |
| DMEM-F12 | Gibco | 11320-033 |
| FBS | Gibco | 10438-026 |
| Pen/Strep | Gibco | 15140 |
| Puromycin Dihydrochloride | Gibco | A1113802 |
| Lipofectamine 3000 Transfection Reagent | Invitrogen | L3000015 |
| Lipofectamine 2000 Transfection Reagent | Invitrogen | 11668030 |
| N-2 supplement | Gibco | 17502048 |
| B-27 serum free supplement | Gibco | 17504-044 |
| Neurobasal medium | Gibco | 21103049 |
| GlutaMAX-I | Gibco | 35050061 |
| SUPERNase·In. | ThermoFisher Scientific | AM2694 |
| Halt™ Protease and Phosphatase Inhibitor Cocktail, EDTA-free (100X) | ThermoFisher Scientific | 78441 |
| KCl (2 M), RNase-free | Invitrogen | AM9640G |
| CX-4945 | Abcam | ab141350 |
| Okadaic acid | Abcam | ab120385 |
| Hoechst 33342 | ThermoFisher Scientific | 62249 |
| RNase T1 | ThermoFisher Scientific | EN0541 |
| RNase A/T1 Mix | ThermoFisher Scientific | EN0551 |
| cOmplete™, Mini, EDTA-free Protease Inhibitor Cocktail | Roche | 11836170001 |
| TRIzol® reagent | Invitrogen | 15596018 |
| Critical Commercial Assays | ||
| Dynabeads® mRNA DIRECT™ kit | ThermoFisher Scientific | 61006 |
| TruSeq Stranded mRNA Library Prep Kit | Illumina | 20020594 |
| NEBNext® Multiplex Small RNA Library Prep Kit for Illumina® | NEB | E7560S |
| Click-iT™ HPG Alexa Fluor™ 488 Protein Synthesis Assay Kit | Invitrogen | C10428 |
| Dual-Glo® Luciferase Assay System | Promega | E2920 |
| P3 Primary Cell 4D-Nucleofector X Kit L | Lonza | V4XP-3024 |
| mMESSAGE mMACHINE™ T7 Transcription Kit | Invitrogen | AM1344 |
| Duolink® In Situ Detection Reagents Green | Millipore Sigma | DUO92014 |
| Duolink® In Situ PLA® Probe Anti-Mouse PLUS | Millipore Sigma | DUO92001 |
| Duolink® In Situ PLA® Probe Anti-Rabbit MINUS | Millipore Sigma | DUO92005 |
| Deposited Data | ||
| Raw data files for RNA sequencing | This paper | GSE214882 |
| Experimental Models: Cell Lines | ||
| WT HEK293T cells | Laboratory of Dr. Tao Pan | N/A |
| WT HeLa cells | ATCC | CCL-2™ |
| Experimental Models: Organisms/Strains | ||
| Human forebrain-specific organoid cultures | Laboratory of Dr. Peng Jin | https://pubmed.ncbi.nlm.nih.gov/34413513/ |
| Oligonucleotides | ||
| RNA oligo for AlphaScreen experiments: 5’-biotin-UUCUUCUGUGG (m6A) CUGUG-3’ |
This paper | N/A |
| Unlabeled control RNA oligo for AlphaScreen experiments: 5’-UUCUUCUGUGG (m6A) CUGUG-3’ |
This paper | N/A |
| Primers for qRT-PCR (see Table S1) | This paper | N/A |
| Software and Algorithms | ||
| FlowJo | Treestar | https://www.flowjo.com/ |
| Fiji | ImageJ | https://fiji.sc/ |
| Prism 7 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| R v3.3.0 /Bioconductor v3.3 | R core team | https://www.bioconductor.org/ |
| STAR | Dobin et al., 2012 | https://github.com/alexdobin/STAR |
| Trimmomatic | Bolger et al., 2014 | https://github.com/usadellab/Trimmomatic |
| SAMtools | Li et al., 2009 | http://www.htslib.org/ |
| CellProfiler | McQuin et al., 2018 | https://cellprofiler.org/ |
| Cutadapt | Martin et al., 2011 | https://github.com/marcelm/cutadapt |
| HISAT2 | Kim et al., 2015 | N/A |
| Other | ||
| See the list of siRNAs, qPCR primers, plasmids, used in this study in Table S1. Related to Figures 1–3; S2–4 | This paper | N/A |
| See the list of raw data for all the figure panels shown in this study in Table S2. Related to Figures 1–6; S1–6 | This paper | N/A |
| See the list of summarized read counts for high-throughput sequencings in this study in Table S3. Related to Figures 1,5,6; S1,6 | This paper | N/A |
HIGHLIGHTS.
FMRP sequesters YTHDF1 and inhibits its condensation with ribosomes
Phosphorylation of FMRP releases YTHDF1 to promote mRNA translation
FMRP deficiency leads to upregulated translation of YTHDF1 target transcripts
Small molecule inhibitor of YTHDF1 alleviates FXS pathology in an organoid model
ACKNOWLEDGEMENTS
This work was supported by NIH HG008935 (C.H.). NS111602, MH116441 and HG008935 to P.J., SAC inhibitor discovery was supported by the National Key Research and Development Program of China (2021ZD0203900 and 2022YFC3400500 to C.L.) National Natural Science Foundation of China (92253303 and 81821005 to C.L. and 21820102008 to H.J., the project of National Multidisciplinary Innovation Team of Traditional Chinese Medicine supported by National Administration of Traditional Chinese Medicine to C.L.. C.H. is an investigator of the Howard Hughes Medical Institute. We thank Ms. Lindsay Scarpitta and Genomics Facility of the University of Chicago for their generous help with high-throughput sequencing. We thank Ms. Linda Zhang for helping with SAC characterization in cell lines. We thank the Integrated Light Microscopy Core at the University of Chicago for providing imaging tools and Dr. April Pawluk from Life Science Editors for editing.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
Footnotes
DECLARATION OF INTERESTS
C.H. is a scientific founder, a member of the scientific advisory board, and an equity holder of Aferna Green, Inc. and AccuaDX Inc., and a scientific co-founder and equity holder of Accent Therapeutics, Inc. P.J., C.H. and Z.Z. are inventors of a patent WO 2023/064490 A1. The other authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
SUPPLEMENTAL INFORMATION
Supplemental Information including six figures and three tables can be found with this article online.
REFERENCES
- 1.Kandel ER (2001). The molecular biology of memory storage: a dialogue between genes and synapses. Science 294, 1030–1038. 10.1126/science.1067020. [DOI] [PubMed] [Google Scholar]
- 2.Huber KM, Kayser MS, and Bear MF (2000). Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science 288, 1254–1257. 10.1126/science.288.5469.1254. [DOI] [PubMed] [Google Scholar]
- 3.Martin KC, Casadio A, Zhu H, Yaping E, Rose JC, Chen M, Bailey CH, and Kandel ER (1997). Synapse-specific, long-term facilitation of aplysia sensory to motor synapses: a function for local protein synthesis in memory storage. Cell 91, 927–938. 10.1016/s0092-8674(00)80484-5. [DOI] [PubMed] [Google Scholar]
- 4.Kang H, and Schuman EM (1996). A requirement for local protein synthesis in neurotrophin-induced hippocampal synaptic plasticity. Science 273, 1402–1406. 10.1126/science.273.5280.1402. [DOI] [PubMed] [Google Scholar]
- 5.Ouyang Y, Kantor D, Harris KM, Schuman EM, and Kennedy MB (1997). Visualization of the distribution of autophosphorylated calcium/calmodulin-dependent protein kinase II after tetanic stimulation in the CA1 area of the hippocampus. J Neurosci 17, 5416–5427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Park S, Park JM, Kim S, Kim JA, Shepherd JD, Smith-Hicks CL, Chowdhury S, Kaufmann W, Kuhl D, Ryazanov AG, et al. (2008). Elongation factor 2 and fragile X mental retardation protein control the dynamic translation of Arc/Arg3.1 essential for mGluR-LTD. Neuron 59, 70–83. 10.1016/j.neuron.2008.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sonenberg N, and Hinnebusch AG (2009). Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell 136, 731–745. 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kapur M, Monaghan CE, and Ackerman SL (2017). Regulation of mRNA Translation in Neurons-A Matter of Life and Death. Neuron 96, 616–637. 10.1016/j.neuron.2017.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shi H, Zhang X, Weng YL, Lu Z, Liu Y, Lu Z, Li J, Hao P, Zhang Y, Zhang F, et al. (2018). m(6)A facilitates hippocampus-dependent learning and memory through YTHDF1. Nature 563, 249–253. 10.1038/s41586-018-0666-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Z, Theler D, Kaminska KH, Hiller M, de la Grange P, Pudimat R, Rafalska I, Heinrich B, Bujnicki JM, Allain FH, and Stamm S (2010). The YTH domain is a novel RNA binding domain. J Biol Chem 285, 14701–14710. 10.1074/jbc.M110.104711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu C, Wang X, Liu K, Roundtree IA, Tempel W, Li Y, Lu Z, He C, and Min J (2014). Structural basis for selective binding of m6A RNA by the YTHDC1 YTH domain. Nat Chem Biol 10, 927–929. 10.1038/nchembio.1654. [DOI] [PubMed] [Google Scholar]
- 12.Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, Weng X, Chen K, Shi H, and He C (2015). N(6)-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell 161, 1388–1399. 10.1016/j.cell.2015.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zou Z, Sepich-Poore C, Zhou X, Wei J, and He C (2023). The mechanism underlying redundant functions of the YTHDF proteins. Genome Biol 24, 17. 10.1186/s13059-023-02862-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weiler IJ, and Greenough WT (1993). Metabotropic glutamate receptors trigger postsynaptic protein synthesis. Proc Natl Acad Sci U S A 90, 7168–7171. 10.1073/pnas.90.15.7168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rienecker KDA, Poston RG, and Saha RN (2020). Merits and Limitations of Studying Neuronal Depolarization-Dependent Processes Using Elevated External Potassium. ASN Neuro 12, 1759091420974807. 10.1177/1759091420974807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Walsh CT, Garneau-Tsodikova S, and Gatto GJ Jr. (2005). Protein posttranslational modifications: the chemistry of proteome diversifications. Angew Chem Int Ed Engl 44, 7342–7372. 10.1002/anie.200501023. [DOI] [PubMed] [Google Scholar]
- 17.Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, Reiner O, Richards S, Victoria MF, Zhang FP, and et al. (1991). Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65, 905–914. 10.1016/0092-8674(91)90397-h. [DOI] [PubMed] [Google Scholar]
- 18.Dolen G, Osterweil E, Rao BS, Smith GB, Auerbach BD, Chattarji S, and Bear MF (2007). Correction of fragile X syndrome in mice. Neuron 56, 955–962. 10.1016/j.neuron.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, et al. (2011). FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146, 247–261. 10.1016/j.cell.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sawicka K, Hale CR, Park CY, Fak JJ, Gresack JE, Van Driesche SJ, Kang JJ, Darnell JC, and Darnell RB (2019). FMRP has a cell-type-specific role in CA1 pyramidal neurons to regulate autism-related transcripts and circadian memory. Elife 8. 10.7554/eLife.46919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Greenblatt EJ, and Spradling AC (2018). Fragile X mental retardation 1 gene enhances the translation of large autism-related proteins. Science 361, 709–712. 10.1126/science.aas9963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kandel ER (2012). The molecular biology of memory: cAMP, PKA, CRE, CREB-1, CREB-2, and CPEB. Mol Brain 5, 14. 10.1186/1756-6606-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Edupuganti RR, Geiger S, Lindeboom RGH, Shi H, Hsu PJ, Lu Z, Wang SY, Baltissen MPA, Jansen P, Rossa M, et al. (2017). N(6)-methyladenosine (m(6)A) recruits and repels proteins to regulate mRNA homeostasis. Nat Struct Mol Biol 24, 870–878. 10.1038/nsmb.3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Worpenberg L, Paolantoni C, Longhi S, Mulorz MM, Lence T, Wessels HH, Dassi E, Aiello G, Sutandy FXR, Scheibe M, et al. (2021). Ythdf is a N6-methyladenosine reader that modulates Fmr1 target mRNA selection and restricts axonal growth in Drosophila. EMBO J 40, e104975. 10.15252/embj.2020104975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Z, Luo K, Zou Z, Qiu M, Tian J, Sieh L, Shi H, Zou Y, Wang G, Morrison J, et al. (2020). Genetic analyses support the contribution of mRNA N(6)-methyladenosine (m(6)A) modification to human disease heritability. Nat Genet 52, 939–949. 10.1038/s41588-020-0644-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O, Pestova TV, Qian SB, and Jaffrey SR (2015). 5’ UTR m(6)A Promotes Cap-Independent Translation. Cell 163, 999–1010. 10.1016/j.cell.2015.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ceman S, O’Donnell WT, Reed M, Patton S, Pohl J, and Warren ST (2003). Phosphorylation influences the translation state of FMRP-associated polyribosomes. Hum Mol Genet 12, 3295–3305. 10.1093/hmg/ddg350. [DOI] [PubMed] [Google Scholar]
- 28.Mazroui R, Huot ME, Tremblay S, Boilard N, Labelle Y, and Khandjian EW (2003). Fragile X Mental Retardation protein determinants required for its association with polyribosomal mRNPs. Hum Mol Genet 12, 3087–3096. 10.1093/hmg/ddg335. [DOI] [PubMed] [Google Scholar]
- 29.Dolen G, and Bear MF (2008). Role for metabotropic glutamate receptor 5 (mGluR5) in the pathogenesis of fragile X syndrome. J Physiol 586, 1503–1508. 10.1113/jphysiol.2008.150722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gross C, Berry-Kravis EM, and Bassell GJ (2012). Therapeutic strategies in fragile X syndrome: dysregulated mGluR signaling and beyond. Neuropsychopharmacology 37, 178–195. 10.1038/npp.2011.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu YM, Jia Z, Janus C, Henderson JT, Gerlai R, Wojtowicz JM, and Roder JC (1997). Mice lacking metabotropic glutamate receptor 5 show impaired learning and reduced CA1 long-term potentiation (LTP) but normal CA3 LTP. J Neurosci 17, 5196–5205. 10.1523/JNEUROSCI.17-13-05196.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wisniewski K, and Car H (2002). (S)-3,5-DHPG: a review. CNS Drug Rev 8, 101–116. 10.1111/j.1527-3458.2002.tb00218.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F, Bradley A, et al. (2000). Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev 14, 1448–1459. [PMC free article] [PubMed] [Google Scholar]
- 34.Zalfa F, Giorgi M, Primerano B, Moro A, Di Penta A, Reis S, Oostra B, and Bagni C (2003). The fragile X syndrome protein FMRP associates with BC1 RNA and regulates the translation of specific mRNAs at synapses. Cell 112, 317–327. 10.1016/s0092-8674(03)00079-5. [DOI] [PubMed] [Google Scholar]
- 35.Li A, Chen YS, Ping XL, Yang X, Xiao W, Yang Y, Sun HY, Zhu Q, Baidya P, Wang X, et al. (2017). Cytoplasmic m(6)A reader YTHDF3 promotes mRNA translation. Cell Res 27, 444–447. 10.1038/cr.2017.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim TH, Tsang B, Vernon RM, Sonenberg N, Kay LE, and Forman-Kay JD (2019). Phospho-dependent phase separation of FMRP and CAPRIN1 recapitulates regulation of translation and deadenylation. Science 365, 825–829. 10.1126/science.aax4240. [DOI] [PubMed] [Google Scholar]
- 37.Tsang B, Arsenault J, Vernon RM, Lin H, Sonenberg N, Wang LY, Bah A, and Forman-Kay JD (2019). Phosphoregulated FMRP phase separation models activity-dependent translation through bidirectional control of mRNA granule formation. Proc Natl Acad Sci U S A 116, 4218–4227. 10.1073/pnas.1814385116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tompa P, and Friedrich P (1998). Prion proteins as memory molecules: An hypothesis. Neuroscience 86, 1037–1043. [DOI] [PubMed] [Google Scholar]
- 39.Bartley CM, O’Keefe RA, Blice-Baum A, Mihailescu MR, Gong X, Miyares L, Karaca E, and Bordey A (2016). Mammalian FMRP S499 Is Phosphorylated by CK2 and Promotes Secondary Phosphorylation of FMRP. eNeuro 3. 10.1523/ENEURO.0092-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saurabh S, Chong TN, Bayas C, Dahlberg PD, Cartwright HN, Moerner WE, and Shapiro L (2022). ATP-responsive biomolecular condensates tune bacterial kinase signaling. Sci Adv 8, eabm6570. 10.1126/sciadv.abm6570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Namkoong S, Ho A, Woo YM, Kwak H, and Lee JH (2018). Systematic Characterization of Stress-Induced RNA Granulation. Mol Cell 70, 175–187 e178. 10.1016/j.molcel.2018.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Phillip Y, Sherman E, Haran G, and Schreiber G (2009). Common crowding agents have only a small effect on protein-protein interactions. Biophys J 97, 875–885. 10.1016/j.bpj.2009.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maharana S, Wang J, Papadopoulos DK, Richter D, Pozniakovsky A, Poser I, Bickle M, Rizk S, Guillen-Boixet J, Franzmann TM, et al. (2018). RNA buffers the phase separation behavior of prion-like RNA binding proteins. Science 360, 918–921. 10.1126/science.aar7366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hondele M, Sachdev R, Heinrich S, Wang J, Vallotton P, Fontoura BMA, and Weis K (2019). DEAD-box ATPases are global regulators of phase-separated organelles. Nature 573, 144–148. 10.1038/s41586-019-1502-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bear MF, Huber KM, and Warren ST (2004). The mGluR theory of fragile X mental retardation. Trends Neurosci 27, 370–377. 10.1016/j.tins.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 46.McCamphill PK, Stoppel LJ, Senter RK, Lewis MC, Heynen AJ, Stoppel DC, Sridhar V, Collins KA, Shi X, Pan JQ, et al. (2020). Selective inhibition of glycogen synthase kinase 3alpha corrects pathophysiology in a mouse model of fragile X syndrome. Sci Transl Med 12. 10.1126/scitranslmed.aam8572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu J, Eckert MA, Harada BT, Liu SM, Lu Z, Yu K, Tienda SM, Chryplewicz A, Zhu AC, Yang Y, et al. (2018). m(6)A mRNA methylation regulates AKT activity to promote the proliferation and tumorigenicity of endometrial cancer. Nat Cell Biol 20, 1074–1083. 10.1038/s41556-018-0174-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hudson SA, Ecroyd H, Kee TW, and Carver JA (2009). The thioflavin T fluorescence assay for amyloid fibril detection can be biased by the presence of exogenous compounds. FEBS J 276, 5960–5972. 10.1111/j.1742-4658.2009.07307.x. [DOI] [PubMed] [Google Scholar]
- 49.Clamer M, Tebaldi T, Lauria F, Bernabo P, Gomez-Biagi RF, Marchioretto M, Kandala DT, Minati L, Perenthaler E, Gubert D, et al. (2018). Active Ribosome Profiling with RiboLace. Cell Rep 25, 1097–1108 e1095. 10.1016/j.celrep.2018.09.084. [DOI] [PubMed] [Google Scholar]
- 50.Kang Y, Zhou Y, Li Y, Han Y, Xu J, Niu W, Li Z, Liu S, Feng H, Huang W, et al. (2021). A human forebrain organoid model of fragile X syndrome exhibits altered neurogenesis and highlights new treatment strategies. Nat Neurosci 24, 1377–1391. 10.1038/s41593-021-00913-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang C, Wang M, Li Y, and Zhang Y (2022). Profiling and functional characterization of maternal mRNA translation during mouse maternal-to-zygotic transition. Sci Adv 8, eabj3967. 10.1126/sciadv.abj3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sethna F, Feng W, Ding Q, Robison AJ, Feng Y, and Wang H (2017). Enhanced expression of ADCY1 underlies aberrant neuronal signalling and behaviour in a syndromic autism model. Nat Commun 8, 14359. 10.1038/ncomms14359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Westmark CJ (2019). Fragile X and APP: a Decade in Review, a Vision for the Future. Mol Neurobiol 56, 3904–3921. 10.1007/s12035-018-1344-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Qian X, Su Y, Adam CD, Deutschmann AU, Pather SR, Goldberg EM, Su K, Li S, Lu L, Jacob F, et al. (2020). Sliced Human Cortical Organoids for Modeling Distinct Cortical Layer Formation. Cell Stem Cell 26, 766–781 e769. 10.1016/j.stem.2020.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, Hurles ME, Homfray T, Penninger JM, Jackson AP, and Knoblich JA (2013). Cerebral organoids model human brain development and microcephaly. Nature 501, 373–379. 10.1038/nature12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Qian X, Nguyen HN, Song MM, Hadiono C, Ogden SC, Hammack C, Yao B, Hamersky GR, Jacob F, Zhong C, et al. (2016). Brain-Region-Specific Organoids Using Mini-bioreactors for Modeling ZIKV Exposure. Cell 165, 1238–1254. 10.1016/j.cell.2016.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pfeiffer BE, and Huber KM (2009). The state of synapses in fragile X syndrome. Neuroscientist 15, 549–567. 10.1177/1073858409333075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Si K, Lindquist S, and Kandel ER (2003). A neuronal isoform of the aplysia CPEB has prion-like properties. Cell 115, 879–891. 10.1016/s0092-8674(03)01020-1. [DOI] [PubMed] [Google Scholar]
- 59.Jacquemont S, Pacini L, Jonch AE, Cencelli G, Rozenberg I, He Y, D’Andrea L, Pedini G, Eldeeb M, Willemsen R, et al. (2018). Protein synthesis levels are increased in a subset of individuals with fragile X syndrome. Hum Mol Genet 27, 2039–2051. 10.1093/hmg/ddy099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Utami KH, Yusof N, Kwa JE, Peteri UK, Castren ML, and Pouladi MA (2020). Elevated de novo protein synthesis in FMRP-deficient human neurons and its correction by metformin treatment. Mol Autism 11, 41. 10.1186/s13229-020-00350-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ashley CT Jr., Wilkinson KD, Reines D, and Warren ST (1993). FMR1 protein: conserved RNP family domains and selective RNA binding. Science 262, 563–566. 10.1126/science.7692601. [DOI] [PubMed] [Google Scholar]
- 62.Shi H, Wang X, Lu Z, Zhao BS, Ma H, Hsu PJ, Liu C, and He C (2017). YTHDF3 facilitates translation and decay of N(6)-methyladenosine-modified RNA. Cell Res 27, 315–328. 10.1038/cr.2017.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. 2011 17, 3. 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 64.Schneider VA, Graves-Lindsay T, Howe K, Bouk N, Chen HC, Kitts PA, Murphy TD, Pruitt KD, Thibaud-Nissen F, Albracht D, et al. (2017). Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res 27, 849–864. 10.1101/gr.213611.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323. 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S3 Summarized read counts for high-throughput sequencing in this study, related to Figure 5–6, S5–S6.
Data Availability Statement
Sequencing data have been deposited into the Gene Expression Omnibus (GEO) under the accession number GSE214882 for RNA sequencings.
This work does not generate original codes.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact (Chuan He, chuanhe@uchicago.edu) upon request.