

Significance
Uncontrolled cell division is a hallmark of cancer. Drugs that inhibit the cell cycle in tumor cells, such as palbociclib, have improved health outcomes in Estrogen receptor–positive, human epidermal growth factor 2 receptor–negative (ER+/HER2−) breast cancer patients. However, it is common for a small percentage of tumor cells to keep dividing in the presence of the drug, revealing a gap in our understanding of mechanisms that underlie resistance. Here, we show that flexibility, or plasticity, in the cell cycle allows some tumor cells to escape treatment by taking different cell cycle “paths.” Our work could lead to improved treatment strategies in ER+/HER2− breast cancer and points to cell cycle plasticity as a potential driver of therapeutic resistance in human tumors.
Keywords: ER+/HER2− breast cancer, cell cycle arrest, fractional resistance, single-cell imaging, single-cell proteomics
Abstract
The CDK4/6 inhibitor palbociclib blocks cell cycle progression in Estrogen receptor–positive, human epidermal growth factor 2 receptor–negative (ER+/HER2−) breast tumor cells. Despite the drug’s success in improving patient outcomes, a small percentage of tumor cells continues to divide in the presence of palbociclib—a phenomenon we refer to as fractional resistance. It is critical to understand the cellular mechanisms underlying fractional resistance because the precise percentage of resistant cells in patient tissue is a strong predictor of clinical outcomes. Here, we hypothesize that fractional resistance arises from cell-to-cell differences in core cell cycle regulators that allow a subset of cells to escape CDK4/6 inhibitor therapy. We used multiplex, single-cell imaging to identify fractionally resistant cells in both cultured and primary breast tumor samples resected from patients. Resistant cells showed premature accumulation of multiple G1 regulators including E2F1, retinoblastoma protein, and CDK2, as well as enhanced sensitivity to pharmacological inhibition of CDK2 activity. Using trajectory inference approaches, we show how plasticity among cell cycle regulators gives rise to alternate cell cycle “paths” that allow individual tumor cells to escape palbociclib treatment. Understanding drivers of cell cycle plasticity, and how to eliminate resistant cell cycle paths, could lead to improved cancer therapies targeting fractionally resistant cells to improve patient outcomes.
Estrogen receptor–positive, human epidermal growth factor 2 receptor–negative (ER+/HER2−) metastatic breast cancers show altered cell cycle behaviors that contribute to progression of the disease (1–3). Most ER+/HER2− tumors show elevated expression of the estrogen receptor (ER, ESR1) and its transcriptional target, cyclin D1 (CCND1) (4–6). Estrogen signaling up-regulates expression of cyclin D1, which works together with other cyclins (e.g., cyclin E) to activate cyclin-dependent kinases (CDKs) (7). Cyclin D1 forms complexes with CDK4 and CDK6 (8, 9), whereas cyclin E forms complexes with CDK2. Active cyclin-CDK complexes phosphorylate the retinoblastoma protein (RB) to its phosphorylated form (pRB) (10, 11). RB phosphorylation relieves repression of a large set of target genes controlled by the E2F family of transcription factors (e.g., E2F1) (12–14). Expression of E2F-regulated genes produces additional positive feedback mechanisms to initiate the S phase (13, 15). In general, ER+/HER2− breast tumors show enhanced signaling through cyclin-CDK signaling pathways that converge on phosphorylation of RB to initiate cell cycle entry and tumor cell proliferation (Fig. 1A).
Fig. 1.
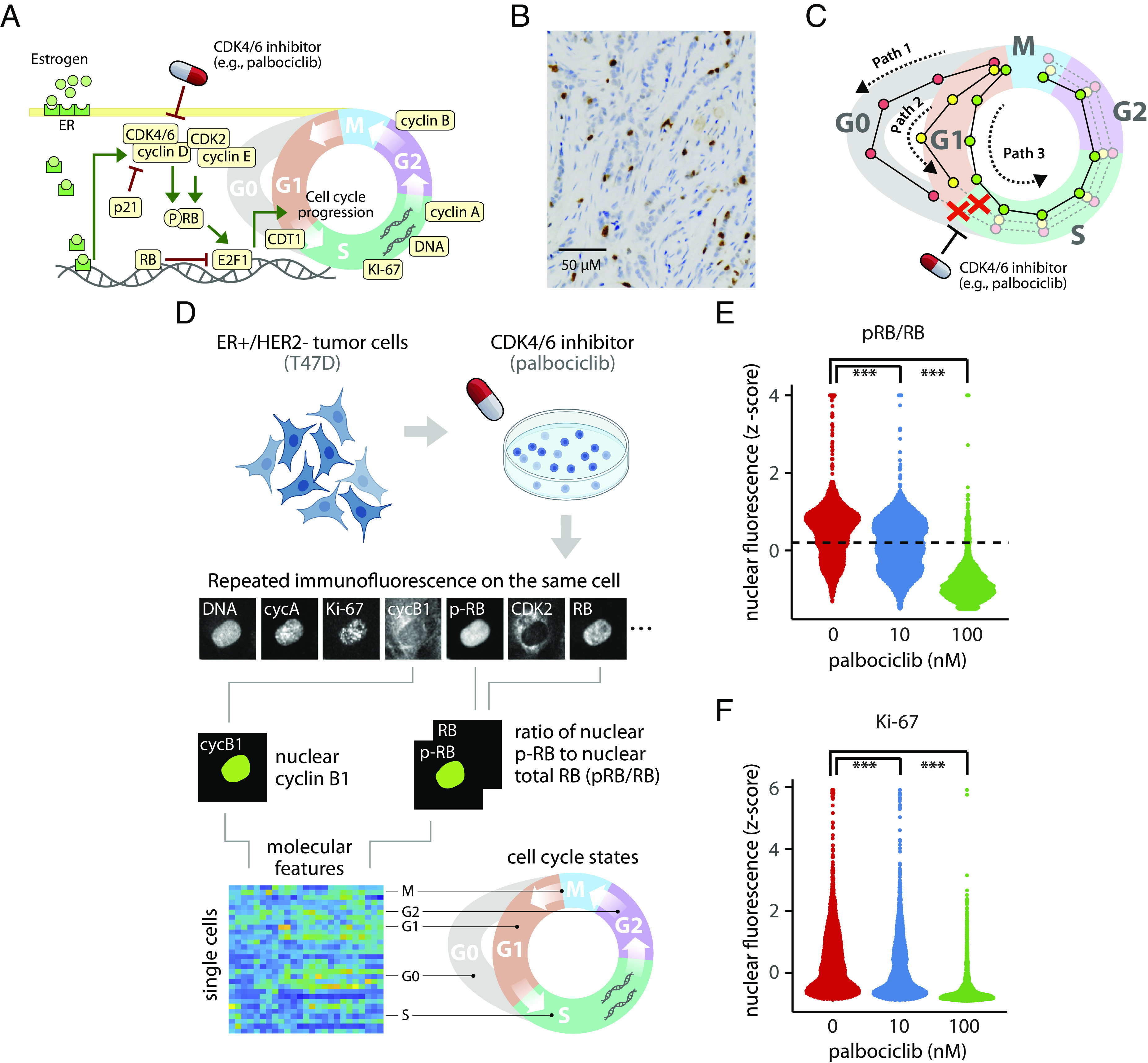
(A–C) Cell cycle regulation and fractional resistance to CDK4/6 inhibitors ER+/HER2− breast tumor cells. (A) Core cell cycle signaling network and points of drug activity in ER+/HER2− breast tumors. Cell cycle protein regulators shown in beige were measured in single tumor cells. (B) Ki-67 staining (brown) from an ER+/HER2− breast tumor with 14% Ki-67+ cells. (C) Hypothetical in which plasticity in cell cycle progression among individual cells could create different molecular “paths” with distinct sensitivities to CDK4/6 inhibitors. Red, yellow, and green paths represent distinct molecular trajectories for a single cell. In the proposed model of fractional resistance, CDK4/6 inhibitors do not block all paths, allowing some cells to complete the cell cycle in the presence of the drug. (D–F) Single-cell proteomic profiling reveals fractional resistance to palbociclib in ER+/HER2− breast tumor cells. (D) Workflow for 4i profiling of breast tumor cells. Asynchronous T47D cells were treated with increasing concentrations of palbociclib for 24 h. Cells were fixed and subjected to iterative indirect immunofluorescence imaging (4i) to quantify nuclear levels of pRB, RB, Ki-67, CDK2, CDK4, cyclin D1, cyclin E, Cdt1, E2F1, cyclin A, cyclin B1, p21, and DNA in each cell. The resulting data structure is a matrix containing 103,862 cells and imaging features representing the 14 cell cycle regulators. Because the cells are not synchronized, 4i captures a highly granular representation of proliferating and arrested cell cycle states. (E) Distribution of pRB/RB at 0, 10, or 100 nM palbociclib. The dotted line marks the statistically determined threshold for demarcating cells as proliferating despite palbociclib treatment. Cells above the dotted line were used for characterizing proliferating cells. (F) Distribution of nuclear Ki-67 levels at 0, 10, or 100 nM palbociclib. *** indicates a P-value < 0.001 using a two-sided Kolmogorov–Smirnov test between untreated and treated cells.
For over twenty years, the mainline treatments for ER+/HER2− breast tumors have been endocrine therapies such as tamoxifen, aromatase inhibitors, and fulvestrant. These “antiestrogen” agents partially block or antagonize estrogen receptor signaling to reduce cyclin D1 expression. More recently, several potent CDK4/6 inhibitors such as palbociclib, abemaciclib, or ribociclib are being given in combination with endocrine therapy to improve patient outcomes. These drugs have had a profound clinical impact. Data from multiple clinical trials show that combined antiestrogen and CDK4/6 inhibitor therapy nearly doubles progression-free survival, and many patients respond to CDK4/6 inhibitor therapy for several years (6, 16–18). However, many patients are initially resistant to the CDK4/6 inhibitors or acquire resistance within the first few months of treatment. Despite extensive investigation, the mechanisms of resistance to CDK4/6 inhibitors in ER+/HER2− breast cancers remain unclear (19, 20).
An important clue in understanding resistance to CDK4/6 inhibitors and endocrine therapy comes from Ki-67 staining, a common clinical diagnostic for breast tumors. Ki-67 is a nuclear protein that is expressed in actively proliferating cells but is absent in arrested cells (21). Ki-67 staining in paraffin-embedded tissues nearly always shows a subpopulation of proliferating cells (21, 22) (Fig. 1B). This percentage of proliferating cells is strongly predictive of clinical outcomes. After receiving treatment, for example, patients showing a low percentage (0 to 2%) of proliferating cells are considered “responsive” to therapy, whereas a higher percentage (3 to 15%) of Ki-67-positive cells predicts a poor response for ER+/HER2− patients (23). Recent work by Gaglia et al. shows that Ki-67 likely underestimates the number of proliferative cells in tumor tissues (24). These observations strongly suggest that a subpopulation of tumor cells continues to divide, even in responsive ER+/HER2− patients. We refer to this phenomenon in which a subpopulation of tumor cells continues to divide in the presence of a cell cycle–targeting drug as fractional resistance. Fractional resistance is conceptually similar to fractional killing, a term coined to describe the observation that each round of chemotherapy does not kill 100% of tumor cells (25). Previous work (26–33) showed that fractional killing is a consequence of nongenetic, cell-to-cell heterogeneity—that is, differences in the molecular makeup of individual cells that do arise from genetic mutations. Additional studies indicate that cell-to-cell heterogeneity plays a role in resistance in melanoma (28, 34, 35).
One source of cell-to-cell heterogeneity is cell cycle plasticity—differences in the cell cycle behavior driven by different combinations of cell cycle regulators. A recent meta-analysis of cancer cell lines by Kumarasamy and colleagues found that sensitivity to CDK4/6 inhibitors was associated with activation of RB, inhibition of CDK2 activity, and, in some cases, depletion of CDK4 and CDK6 (36). These results demonstrate how cell cycle plasticity across different cell lines leads to drug resistance, but it does not indicate what cell cycle plasticity may exist within a single type of tumor cell, potentially explaining its drug resistance. A growing body of evidence (36–42), including our own work (43–47), has shown that differences in cell cycle behavior occur at the level of individual cells. These differences include changes in the timing of key cell cycle events such as a shortened G1 duration (41, 43, 45, 48) as well as an altered ordering of events at the G1/S transition (49, 50). The cell cycle can also vary at the single-cell level in its pattern of cyclin expression (51), CDK activity (15, 37), and other molecular states (36, 42). These studies strongly suggest that individual cells can take distinct trajectories, or paths, through the cell cycle that are defined by a unique combination of molecular states over time (52, 53) (Fig. 1C).
Here, we show that ER+/HER2− tumor cells show subtle, cell-to-cell differences in core cell cycle regulators that allows a subset of tumor cells to escape CDK4/6 inhibitor therapy. We used multiplex, single-cell imaging to build a proteomic profile for individual tumor cells treated with palbociclib and identified fractionally resistant tumor cells both in a cell culture model of ER+/HER2− breast cancer as well as from live primary tumor cells resected from a patient. We found that resistant tumor cells harbored specific combinations of enriched and depleted cell cycle regulators including cyclin B1, Cdt1, CDK2, and p21, some of which were common to both tumor models. Using computational data integration and trajectory inference approaches to visualize resistant cells, our work shows how nongenetic plasticity in cell cycle regulators—at the single-cell level—creates alternate cell cycle paths that allow individual ER+/HER2− tumor cells to escape palbociclib treatment.
Results
Fractional Resistance in T47D Cells.
We first investigated fractional resistance in the T47D cell line, a well-established model of ER+/HER2− breast cancer (54, 55). Cells were allowed to proliferate freely or treated with either low (10 nM) or moderate (100 nM) doses of palbociclib for 24 h. We then profiled protein expression in 103,862 cells using iterative indirect immunofluorescence imaging, or “4i” (56) (Fig. 1D). For each cell, we quantified the abundance of 13 cell cycle regulators: pRB, RB, Ki-67, CDK2, CDK4, cyclin D1, cyclin E, Cdt1, E2F1, cyclin A, cyclin B1, p21, and integrated DNA (SI Appendix, Table S1). These core regulators cover a broad range of molecular mechanisms occurring throughout the cell cycle, including growth signaling (cyclins D and E and CDKs 2 and 4), the G1/S transition (RB, pRB, and E2F1), DNA replication (Cdt1, Ki-67, and DNA), and progression through G2 and M phases (cyclins A and B). Given that a freely dividing population of tumor cells is not synchronized to any particular cell cycle phase, 4i captures a diverse range of single-cell states across all phases of the entire cell cycle. To facilitate a direct comparison of cell cycle states across samples and treatment conditions, we performed principled downsampling of the data using kernel herding sketching (57). This approach identifies a limited subset of representative cells that preserves the original distribution of cell states. We selected an equal number of cells (n = 2,000) from each treatment condition (untreated, 10 nM, and 100 nM), resulting in a final downsampled dataset of 6,000 T47D cells (Materials and Methods).
To quantify the fraction of proliferating cells under each condition, we focused on the ratio of phosphorylated to total RB protein or pRB/RB. When quantified in individual cells, pRB/RB often shows a bimodal distribution: Low pRB/RB expression corresponds to a hypophosphorylated RB state and is characteristic of arrested cells. High pRB/RB expression represents the hyperphosphorylated form of the RB protein and comprises actively proliferating cells (58). As expected, palbociclib reduced the fraction of proliferating cells in a dose-dependent manner (Fig. 1E). Interestingly, however, we observed a small subset of cells that maintained high pRB/RB levels at both 10 nM (48.4%) and 100 nM (5.9%) palbociclib, indicating that some cells could evade drug treatment. Similarly, we observed a reduction in Ki-67 expression under increasing palbociclib concentration, but a fraction of cells maintained high Ki-67 expression in the presence of 10 nM and 100 nM palbociclib (Fig. 1F). We observed a similar level of fractional resistance in a biological replicate of T47D cells (SI Appendix, Fig. S1 A and B). To confirm the cells were truly capable of proliferating in the presence of palbociclib, and not merely finishing the previous cell cycle within the 24 h treatment time frame, we repeated the experiment in long-term culture, exposing cells to one week of continuous palbociclib treatment. Again, tumor cells showed fractional resistance as indicated by the continual presence of proliferating cells (SI Appendix, Fig. S1C). Taken together, these results reveal that a small subset of ER+/HER2− breast tumor cells continue to proliferate in the presence of palbociclib, implying that cell-to-cell variation may account for fractional resistance.
We next asked what intracellular features may be enriched in proliferating cells in the presence of palbociclib. We focused deliberately on differences in protein abundance—rather than epigenetic or transcriptomic differences—because palbociclib acts directly on the CDK4/6 proteins (59), and because the core cell cycle regulators are largely regulated through protein modification and degradation (60, 61). To identify only the proliferating cells, and exclude G0 cells, we set a threshold level of pRB/RB above which cells were confidently expected to be in the hyperphosphorylated state and therefore actively proliferating in either G1, S, G2, or M phase (37, 62, 63). We set a conservative cutoff (Materials and Methods) to capture this second peak of pRB/RB expression (dotted line in Fig. 1E). We then compared the single-cell profiles between these proliferating untreated and palbociclib-treated cells to identify differences in cell cycle regulators that may be responsible for fractional resistance. Shifts in the distributions of individual cell cycle proteins (Fig. 2A) were quantified using a two-sample t test between untreated cells and either the 10 nM or the 100 nM treatment condition, producing 95% CI for any observable differences in single-cell protein expression (Fig. 2B).
Fig. 2.
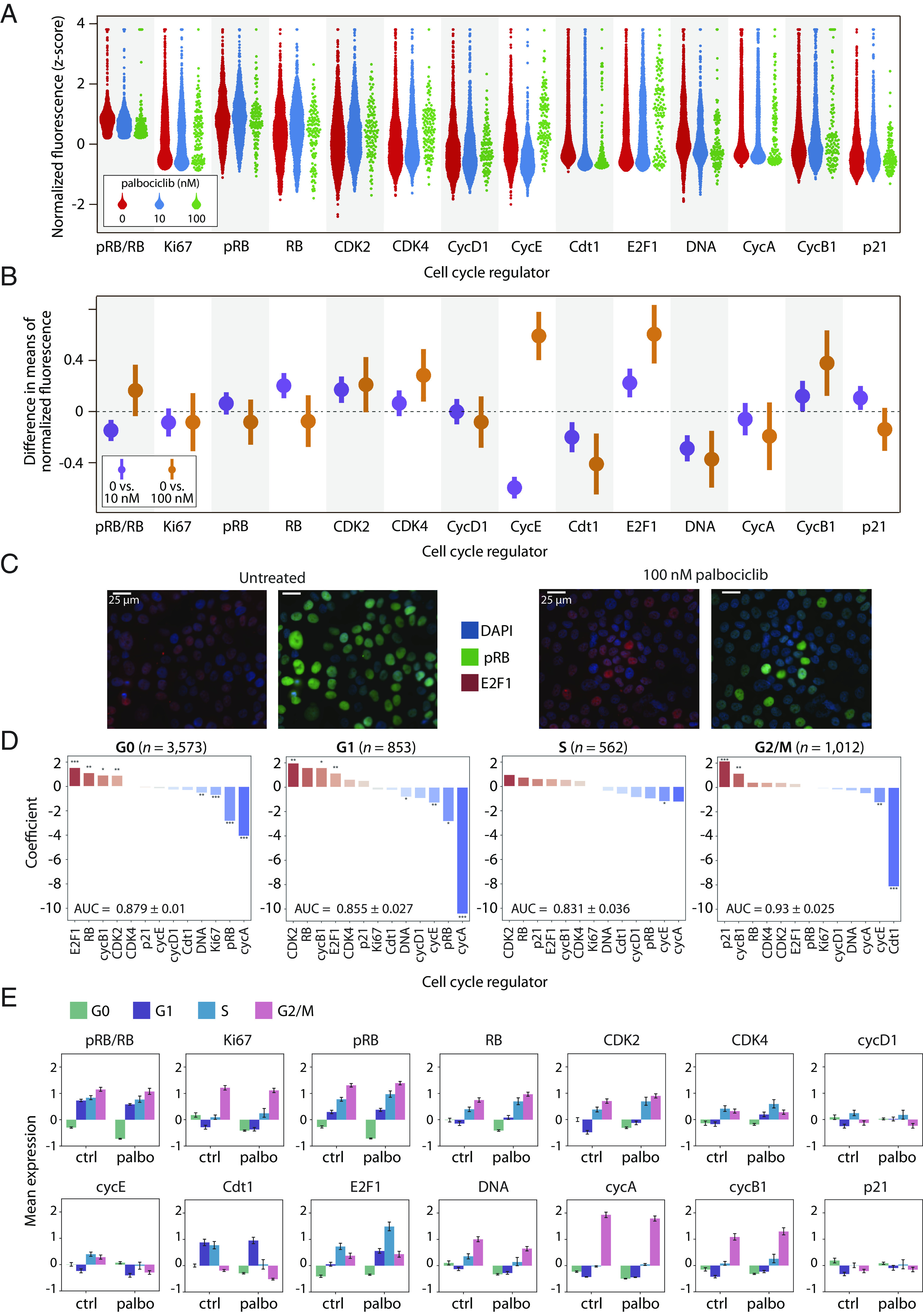
Shifts in expression of cell cycle proteins among fractionally resistant tumor cells. (A) Beginning with a downsampled dataset containing 2,000 cells from each condition (Materials and Methods), we identified the proliferating cells using the pRB/RB threshold defined in Fig. 1E. Proliferating single-cell distributions of cell cycle regulators at 0 nM, 10 nM, or 100 nM palbociclib, showing an expected reduction in the number of cells at higher drug concentrations. (B) 95% CI of proliferating cells for the differences in mean expression (normalized z-scores) between either untreated cells and 10 nM palbociclib (purple); or untreated and 100 nM palbociclib (orange). CI overlapping with the dashed line at 0 indicate a lack of statistical significance. CI are wider when comparing 0 vs. 100 nM due to lower sample sizes at the highest dose of palbociclib. (C) Representative images of the effect of palbociclib on cells quantified in panels A and B. We can visually observe the quantified depletion in pRB and upregulation of E2F1. (D) Logistic regression on all 6,000 cells predicting the odds that a given cell is either untreated (0 nM) or treated (10 nM or 100 nM) based on expression of its cell cycle regulators. Cell-to-cell increases in regulators shown in red (e.g., CDK2), or decreases in regulators shown in blue (e.g., cycA), increase the odds of association with treated (10 nM or 100 nM) vs. untreated cells. A separate regression was performed for each phase (G0, G1, S, and G2/M), where the last three are considered proliferating (high pRB/RB). This analysis was performed on all cells, including nonproliferating (G0) cells. Significance: *, P < 0.05; **, P < 0.01; ***, P < 0.001. (E) Z-score normalized expression levels of cell cycle features, stratified by cell cycle phase for all 6,000 cells. Bar height is the mean expression for untreated (ctrl) or palbociclib-treated (10 nM and 100 nM) cells. Error bars represent CI.
Under palbociclib treatment, many cell cycle regulators (e.g., pRB, Ki-67, cyclin A) showed similar distributions of expression compared to untreated cells. However, some regulators showed significant shifts in protein expression under 10 nM and/or 100 nM palbociclib treatment. For example, proliferating T47D cells showed elevated CDK2 levels under 10 nM and 100 nM palbociclib treatment [0 vs. 10 nM 95% CI (0.09, 0.26); 0 vs. 100 nM (0.014, 0.41)] and reduced expression of Cdt1 [0 vs. 10 nM (−0.30, −0.10,); 0 vs. 100 nM (−0.63, −0.19)] at both drug doses. Elevated expression of CDK2 activity is consistent with previous studies showing that ER+/HER2− tumor cells often become resistant to CDK4/6 inhibitors via increases in cyclin E/CDK2 activity (5, 16). However, we note that these studies are typically focused on genetic changes in cyclin E/CDK2 activity (e.g., mutations or copy number variation) and not cell-to-cell variability in protein expression. On the other hand, we uncovered reduced Cdt1 expression as a response to palbociclib treatment. Cdt1 encodes for a subunit of the prereplication complex necessary for DNA replication. Entry into the S phase with subnormal Cdt1 levels could potentially lead to replication stress (64). Finally, we noted that proliferating T47D cells showed a significant depletion of DNA content, likely reflecting a relative enrichment of G1-arrested cells with elevated pRB/RB levels.
We next asked how expression of cell cycle regulators shifted as cells progressed through individual cell cycle phases. To do this, we performed unsupervised clustering of the expression levels of DNA, cyclin A, and cyclin B1 to assign each cell to a specific cell cycle phase: G0, G1, S, or G2/M (Materials and Methods). We then trained a logistic regression model within each phase separately on the proteomic expression profiles from all 6,000 cells to classify whether a cell belonged to either the control (untreated) or treatment (10 nM and 100 nM palbociclib) group (Fig. 2D). When jointly analyzed with the average standardized expression of cell cycle regulators in cell cycle phases (Fig. 2E), this analysis reveals how changes in the normalized expression of cell cycle regulators—either positive (red bars in Fig. 2D) or negative changes (blue bars in Fig. 2D)—are associated with specific cell cycle phases under palbociclib treatment. The results were largely consistent with the previous t test analysis of proliferating cells. For example, we found that elevated CDK2 expression was a significant, positive predictor of palbociclib-treated cells in both G0 and G1 phases. However, this analysis also revealed several trends. For example, increases in three other core cell cycle regulators—E2F1, RB, and cyclin B1—were significantly associated with palbociclib-treated cells in G0 and/or G1 phases. E2F1 expression was among the strongest predictors of treatment in both G0 and G1 cells, providing further clarity for the overall E2F1 enrichment we observe under treatment in Fig. 2B. These results suggest that individual tumor cells with prematurely elevated E2F1 and CDK2 protein levels may be more likely to transition from G0 to G1 in the presence of palbociclib.
Overall, we found that fewer cell cycle regulators reached statistical significance in S or G2/M phases than in G0 or G1 phases, likely because of the smaller sample sizes for these subpopulations of cells, especially under palbociclib treatment where most cells were arrested in G0. Nevertheless, several overall trends were consistent across all phases in three biological replicates of T47D (Fig. 2 and SI Appendix, Figs. S2 and S3); specifically, these phase-specific analyses show that palbociclib either promotes or selects for proliferation of tumor cells with altered protein expression profiles characterized by increases in E2F1, CDK2, and RB and decreases in Cdt1.
Fractional Resistance in Primary ER+/HER2− Tumor Cells.
Although T47D is a well-established model of ER+/HER2− breast cancer, it may not reflect the physiology of primary tumor cells from an actual breast cancer patient. Thus, we developed an experimental strategy for studying fractional resistance in surgically resected primary human tumors (SI Appendix, Fig. S4 and Materials and Methods). Briefly, we obtained a specimen from a primary ER+/HER2− invasive lobular carcinoma which was delivered immediately from the operating room to the laboratory. After dissociation, cells were plated, treated with palbociclib or control media for 24 h, and subjected to 4i profiling. Because tissue samples contain complex mixtures of cell types, we used expression of the epithelial marker pan-cytokeratin (PanCK), as well as the estrogen and progesterone receptors (ER/PR) to computationally separate the tumor cells for downstream analysis. Out of a total population of 100,191 cells, we identified a subpopulation of 14,789 cells showing high expression of ER, PR, and PanCK (SI Appendix, Fig. S5). As above, we performed principled downsampling to a size of 6,000 cells (2,000 cells per condition) to match the T47D analysis.
We first looked for shifts in protein expression between untreated and palbociclib-treated primary tumor cells. As before, we focused initially only on the proliferating subpopulations (i.e., G1, S, and G2/M cells) using a conservative cutoff for pRB/RB levels to identify fractionally resistant tumor cells (SI Appendix, Fig. S6). Although we did not observe consistent changes in Cdt1 or CDK2 expression, primary tumor cells shared many of the same shifts in expression as T47D cells. For example, we observed a significant depletion of DNA content [0 vs. 100 nM 95% CI (−0.89, −0.21)] and additionally, Ki-67 expression [0 vs. 100 nM 95% CI (−0.95, −0.12)] (Fig. 3 A and B). As with T47D, the depletion of Ki-67 and DNA was likely due to the accumulation of G1 cells under treatment, which would be predicted to have lower DNA and Ki-67 content (65). We also observed a significant upregulation of E2F1 protein levels, and a second patient-derived tumor showed consistent increases in E2F1, CDK2, and RB expression (SI Appendix, Fig. S7), as previously observed in T47D.
Fig. 3.
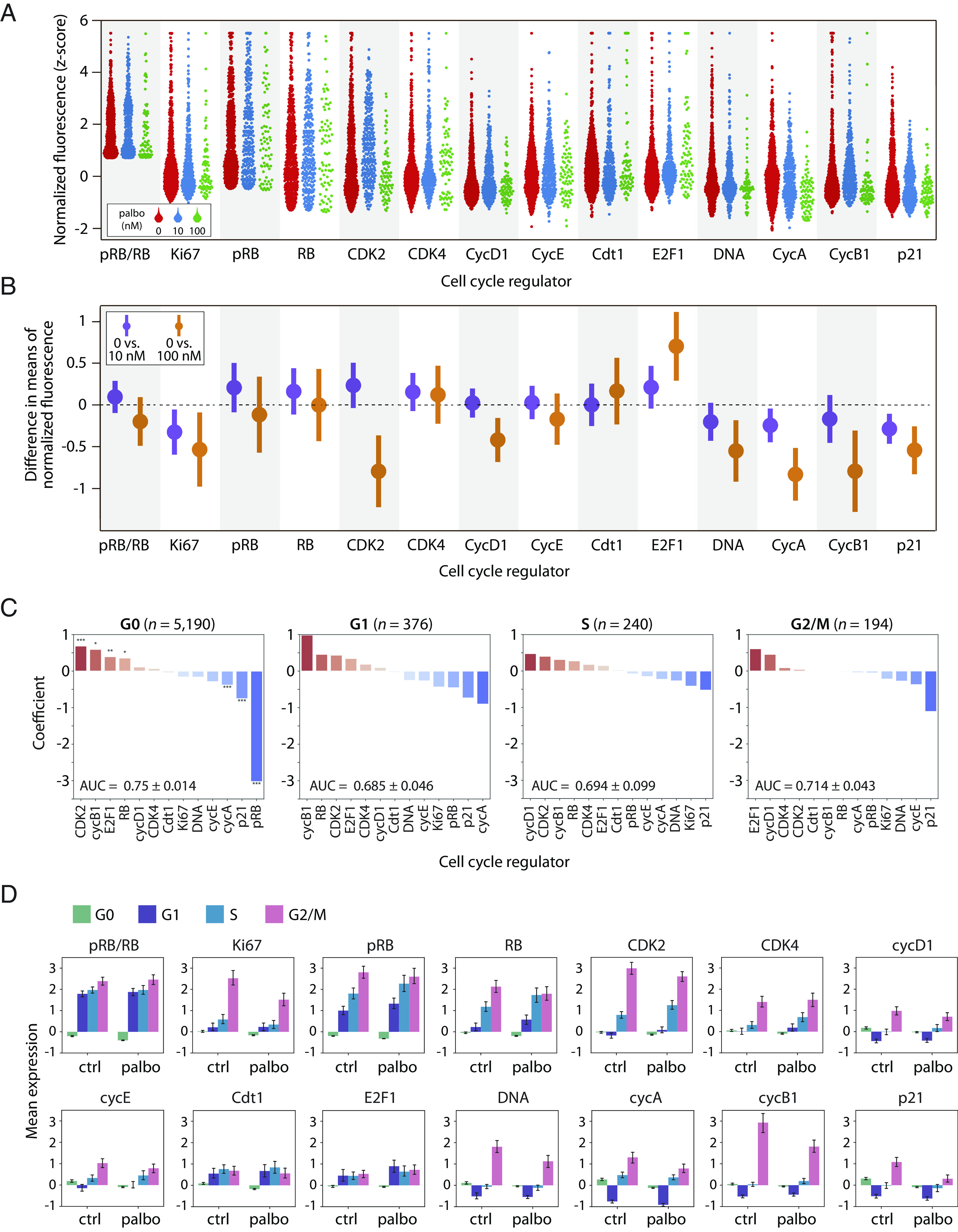
Palbociclib reveals shifts in expression of cell cycle proteins in primary tumor cells. (A) As with T47D, we identified proliferating primary tumor cells using the pRB/RB threshold defined in SI Appendix, Fig. S6. Proliferating single-cell distributions of cell cycle regulators at 0 nM, 10 nM, or 100 nM palbociclib, showing an expected reduction in the number of cells at higher palbociclib concentrations. (B) 95% CI of proliferating cells for the differences in mean expression (normalized z-scores) between either untreated cells and 10 nM palbociclib (purple) or untreated and 100 nM palbociclib (orange). CI overlapping with the dashed line at 0 indicate a lack of statistical significance. CI are wider when comparing 0 vs. 100 nM due to lower sample sizes at the highest dose of palbociclib. (C) Logistic regression on all 6,000 cells predicting the odds that a given cell is either untreated (0 nM) or treated (10 nM or 100 nM) based on expression of its cell cycle regulators. Cell-to-cell increases in regulators shown in red (e.g., cycB1), or decreases in regulators shown in blue (e.g., p21), increase the odds of association with treated (10 nM or 100 nM) vs. untreated cells. A separate regression was performed for each phase (G0, G1, S, and G2/M), where the last three are considered proliferating (i.e., high pRB/RB). This analysis was performed on all cells, including nonproliferating (G0) cells. Significance: *, P < 0.05; **, P < 0.01; ***, P < 0.001. (D) Z-score normalized expression levels of cell cycle features, stratified by cell cycle phase for all 6,000 cells. Bar height is the mean expression for untreated (ctrl) or palbociclib-treated (10 nM and 100 nM) cells. Error bars represent CI.
Although CDK2 showed an inconsistent pattern of expression characterized by apparent enrichment at 10 nM and depletion at 100 nM palbociclib treatment, this discrepancy was clarified by the logistic regression model, which identified elevated CDK2 expression as a top-ranking predictor of palbociclib treatment across all cell cycle phases (Fig. 3C and SI Appendix, Fig. S7C). By examining the average change in expression for each cell cycle regulator across the cell cycle phases (Fig. 3D), we observed a gradual accumulation of CDK2 as cells progressed from G1, through the S phase, peaking in G2/M, consistent with previously observed increases both in protein levels (53) and kinase activity (66, 67). Thus, accumulation of CDK2 protein in palbociclib-treated tumor cells suggests a potential axis of nongenetic resistance mediated by enhanced cyclin E/CDK2 activity. A more striking consistency with T47D cells was that three out of four cell cycle regulators—CDK2, E2F1, and RB—were again the highest-ranking and most significant positive factors associated with G0 arrest in primary tumors cells from two distinct patients with ER+/HER2− subtype (Fig. 3 and SI Appendix, Fig. S7). This result points to a potentially common mechanism of nongenetic drug resistance.
Interestingly, the cell cycle inhibitor, p21, which serves to block phosphorylation of RB by inhibiting cyclin-CDK complexes, was significantly downshifted under both 10 nM and 100 nM palbociclib treatment [Fig. 3B, 0 vs. 10 nM 95% CI (−0.44, −0.13), 0 vs. 100 nM (−0.79, −0.28)]. The logistic regression model also identified p21 as a strongly negative predictor of palbociclib treatment across all cell cycle phases (Fig. 3C), and p21 showed a consistent pattern of reduced expression across all phases (Fig. 3D). This result was observed in a replicate profiling of T47D cells (SI Appendix, Fig. S3) but not the second primary tumor (SI Appendix, Fig. S7). The palbociclib-induced reduction in p21 levels could potentially relax cell cycle arrest and allow cells to enter G1 with modest cyclin/CDK activity. This observation is consistent with enrichment of E2F1 under palbociclib treatment, which was significantly upshifted in primary human tumor cells.
Despite some discrepancies, both T47D and primary tumor cells showed consistent enrichment of CDK2, RB, and E2F1 in G0 and G1 phases, suggesting a potentially common mechanism of fractional resistance. The accumulation of such factors potentially allows a subpopulation of cells to prematurely enter G1 with elevated pRB/RB levels. These findings indicate that the tumor cell cycle is inherently plastic; individual cells can take different molecular paths through the cell cycle, some of which are resistant to CDK4/6 inhibitors.
Cell Cycle Paths in Resistant Cells.
We next sought to visualize resistant cell cycle paths taken by individual tumor cells. To this end, we first identified a shared latent space of T47D and primary tumor samples by performing data integration with tumor response assessment by nonlinear subspace alignment of cell lines and tumors (TRANSACT) (68) (Materials and Methods). We then applied potential of heat-diffusion for affinity-based trajectory embedding (PHATE) (69) on the joint latent space to generate a low-dimensional projection of the combined tumor cell cycle (52, 53). Fig. 4 shows the resulting saddle-shaped structure that captured the progression of four cell cycle phases and revealed significant variability in single-cell states. Each dot in the structure represents an individual cell, and dots nearer to each other have similar expression profiles of cell cycle regulators. By overlaying expression levels for individual cell cycle regulators (Fig. 4A), we observed well-established cell cycle events, including elevated pRB and Ki-67 in proliferating cells; a peak in Cdt1 expression in late G1; accumulation of E2F1 in the S phase; and sequential expression of cyclin A and cyclin B1. These temporal trends in core cell cycle regulators, which emerged without providing any input to the model, allowed us to estimate the general path of cells from G0 to G1, S, and G2/M phases (back curve in Fig. 4B). As expected, we found that palbociclib gradually depleted the number of cells in proliferative phases for both T47D (Fig. 4B) and primary tumor cells (Fig. 4C), potentially altering the path of proliferation through the cell cycle.
Fig. 4.
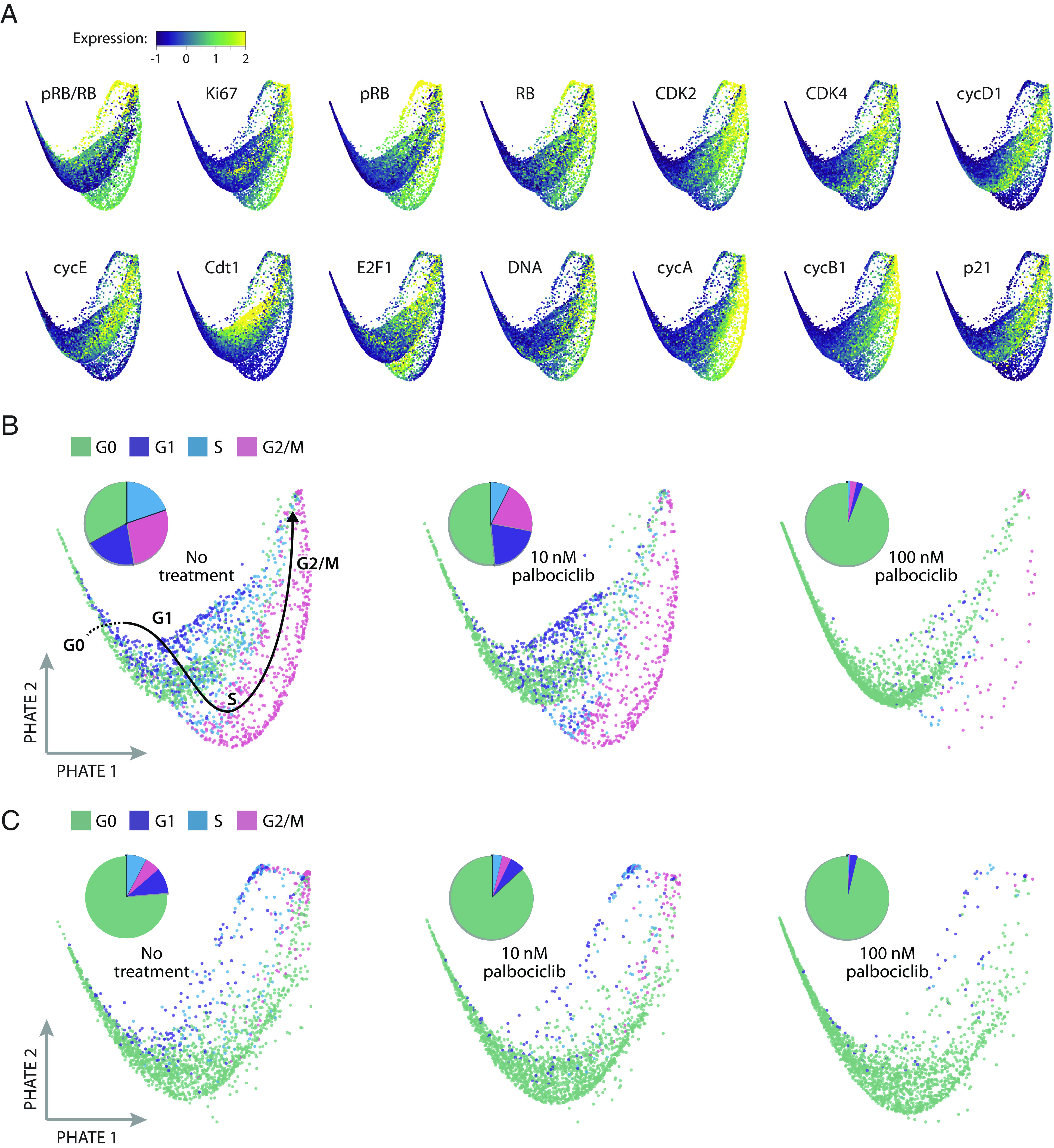
Visualization of single-cell states in ER+/HER2− breast tumors under palbociclib treatment. T47D and primary tumor samples were independently downsampled to yield a sample size of 6,000 cells each (2,000 cells from each of the three palbociclib treatment conditions). The datasets were then integrated into a joint latent space of 12,000 cells using TRANSACT. We then applied the nonlinear dimensionality method PHATE to produce a two-dimensional visualization of the cell cycle under each condition. Each dot is an individual cell. (A) Expression levels of each cell cycle regulator overlaid onto the PHATE embedding for both tumor models. (B) Visualization of cell cycle states for T47D cells, separated by treatment condition. Pie charts indicate the proportion of cells in each cell cycle phase. A hand-drawn estimate of cell cycle progression is shown on the first image based on the progression of overlaid features in panel A. (C) Visualization of cell cycle states for primary tumor cells, separated by treatment condition.
To obtain a more objective and quantitative calculation of the various paths cells take through the cell cycle under palbociclib treatment, we performed trajectory analysis using Slingshot (70) through the low-dimensional PHATE embedding. Without specifying the correct order of phases, this analysis determined the proper cell cycle ordering from G0, to G1, to S, and to G2/M phase in most of the trajectories (Materials and Methods). To allow direct comparisons between each path, we aligned the separate treatment trajectories into one shared axis for each data source using TrAGEDy (71). Fig. 5 shows a visualization of the different temporal expression trends of cell cycle regulators under each experimental condition. In both T47D and the primary tumor cells (Fig. 5 A and D), we observed a clear inward movement of the trajectories—away from proliferating subpopulations—as the palbociclib dose increased. Heatmaps in Fig. 5 B and E illustrate the scaled expression of cell cycle regulators ordered along the aligned pseudotime axis. As an alternative visualization, Fig. 5 C and F directly compare the aligned pseudotime traces for each cell cycle regulator across treatment conditions for T47D and primary tumor samples, respectively. To facilitate comparison among the drug doses, we defined a common transition point as the time at which 50% of the cells remained in G0 and 50% of the cells had progressed to a proliferative phase (vertical lines in panels B, C, E, and F of Fig. 5).
Fig. 5.

ER+/HER2− tumor cells take alternate cell cycle paths under palbociclib treatment. Trajectory inference was performed on cells from each treatment condition for T47D (A–C) and primary tumor (D–F) samples using Slingshot on the joint PHATE embedding. Each trajectory started in G0, progressed through the proliferative phases of the cell cycle, and ended in G2/M. A common pseudotime axis (B, C, E, and F) was determined by aligning trajectories across treatment conditions using TrAGEDy (Materials and Methods). (A and D) Trajectories for each treatment condition projected onto the joint two-dimensional PHATE embedding. (B and E) Heatmaps showing expression of cell cycle regulators along the pseudotime trajectories in panel A. The color strip above the heatmaps represents the cell cycle phase annotations for each cell in the pseudotime ordering. Vertical lines indicate the time at which approximately half of the cells have transitioned from G0 to a proliferative phase (G1, S, and G2/M). No line was detectable for primary tumor cells under 100 nM palbociclib treatment because too few cells entered proliferative phases. (C and F) Comparison of trajectories across treatment conditions for each cell cycle regulator. Vertical lines mark the same common transition point from G0 to proliferation for each drug dose. As in panels A and D, there was no calculable transition line for primary tumor cells under 100 nM palbociclib treatment.
As expected, the temporal trends of increasing pRB/RB and Ki-67 levels, representing escape from cell cycle arrest, were delayed (i.e., right-shifted) under palbociclib treatment in a dose-dependent fashion (Fig. 5 C and F). Despite these delays in cell cycle entry, however, many other cell cycle regulators nevertheless maintained synchronized trends. For example, in both tumor models, CDK2 and E2F1 showed temporally synchronized increases in expression among all treatment groups. Importantly, however, in palbociclib-treated cells, these factors accumulated for longer times and to greater extents before the population reached the G0/G1 transition point, when approximately half of the cells were determined to begin proliferation. More strikingly, other cell cycle regulators showed a reversal of temporal trends. For example, T47D cells treated with 100 nM of palbociclib showed early increases in CDK4, cyclin D1, and cyclin E compared to 10 nM and untreated cells. Conversely, Cdt1 expression was both reduced and delayed upon drug treatment (Fig. 5 A–C). In contrast to T47D, in the primary ER+/HER2− tumor (Fig. 5 D–F), expression of p21 was reduced among treated cells. Taken together, this analysis supports the observation that accumulation of distinct cell cycle-promoting factors, including E2F1 and CDK2, precedes—and potentially facilitates—escape from palbociclib-mediated arrest in T47D and primary tumor cells.
However, a key mechanistic question is whether increased expression of factors such as E2F1 and CDK2 are driving escape from palbociclib-mediated arrest, or merely accumulating in arrested cells or cells transitioning from G1 back to G0. To test this question, we hypothesized that palbociclib-resistant cells with elevated cyclin E/CDK2 expression should be especially sensitive to inhibition of Cyclin E/CDK2 activity, if cyclin E/CDK2 expression is driving escape. Indeed, we found that treatment of T47D cells with palbociclib increased their sensitivity to the CDK2 inhibitor CVT-313 in a dose-dependent fashion. In control cells, treatment with 2 µM CVT-313 resulted in no change in the percentage of pRB/RB positive cells, whereas in cells treated with palbociclib 50 nM, CVT-313 resulted in a 38.4 ± 2.4% reduction in pRB/RB positive cells from baseline, P-value = 0.021 (Fig. 6). This effect was augmented further at 5 µM CVT-313 where 50 nM palbociclib-treated cells had a 53.1 ± 3.3% reduction in pRB/RB+ cells with CVT treatment, 10 nM palbociclib cells had a 36.5 ± 10.9% reduction, and palbociclib untreated control cells had a 14.8 ± 10.8% reduction, P-value = 0.021 between control and palbociclib 50 nM treated cells (Fig. 6). These results—demonstrating a larger-than-additive effect of palbociclib and CDK2 inhibition—support a mechanism of elevated Cyclin E/CDK2 expression as a driver of escape from palbociclib-mediated arrest.
Fig. 6.

CyclinE/CDK2 may serve as a potential driver of escape from palbociclib-mediated arrest. T47D cells were cotreated with CDK4/6 inhibitor (control, 10 nM, and 50 nM palbociclib) and a range of CDK2 inhibitor concentrations (0, 0.01, 0.5, 1, 2, 5, 7.5, 10, and 20 µM CVT-313). (A) Proportion of pRB/RB positive cells following treatment with CDK2 inhibitor, CVT-313. (B) Fold changes in the proportion of pRB/RB positive cells under treatment with CVT-313. For each palbociclib condition, fold changes were computed by normalizing the proportion of pRB/RB positive cells to the average proportion of the first three doses of CVT-313 (0, 0.01, and 0.1 µM). Error bars represent the SD across four technical replicates. Dashed lines indicate (A) the proportion or (B) fold change of pRB/RB positive cells without treatment with the CDK2 inhibitor. * indicates a P-value < 0.05 using a two-sided Wilcoxon rank-sum test between control and 50 nM palbociclib-treated cells.
Discussion
In ER+/HER2− breast cancer, drugs that specifically inhibit cyclin-dependent kinases 4 and 6 (CDK4/6 inhibitors)—when given in combination with endocrine therapy—have dramatically improved oncologic outcomes and overall survival. Unfortunately, there is considerable heterogeneity in the clinical responses to CDK4/6 inhibitors, and most patients eventually develop drug resistance. The field has poured tremendous effort into genetic profiling studies with the hope of identifying molecular mechanisms that predict resistance to CDK4/6 inhibitors. Besides identifying a handful of genes associated with resistance, there is currently no biomarker in clinical use that can predict how an ER+/HER2− patient will respond to endocrine therapy and CDK4/6 inhibitors. This is not just a failure of precision medicine—it also reveals a serious gap in our understanding of the mechanisms that underlie drug resistance.
Here, we provide a framework for how drug resistance may arise in ER+/HER2− breast tumors. We show that cell-to-cell differences in core cell cycle regulators are associated with a subset of tumor cells that escape CDK4/6 inhibitor therapy. We refer to this phenomenon as fractional resistance—the incomplete arrest of tumor cells by a drug. By interrogating the multidimensional protein state of individual cells, we demonstrate the phenomenon of fractional resistance both in a well-established ER+/HER2− model, T47D, as well as two primary ER+/HER2− tumors resected from breast cancer patients. Through single-cell analysis, we found that tumor cells capable of proliferating in the presence of palbociclib showed unique combinations of enriched and depleted cell cycle regulators including E2F1, Cdt1, CDK2, and p21. Notably, resistant cells in both tumor models showed a common enrichment of the E2F1 transcription factor in G0 and G1 phases, suggesting that resistant cells may use a common molecular mechanism to overcome CDK4/6 inhibition. By tracing the trajectories of resistance in each tumor model, we visualize how plasticity in cell cycle regulators creates alternate cell cycle paths that allow some ER+/HER2− tumor cells to escape palbociclib treatment.
These findings explain several longstanding observations about ER+/HER2− breast cancer. For example, it has long been known that a correlation exists between the fraction of proliferating cells (i.e., Ki-67 staining) in a patient tumor and patient outcomes (23). Our work suggests that patients with more Ki-67-positive cells may have more plastic cell cycles that increase fractional resistance. Second, our work provides an alternative mechanism by which core cell cycle regulators can promote oncogenesis. Besides acquiring mutations in specific cell cycle genes, such as RB or cyclin E, we show that cell-to-cell variability in these same factors can promote drug resistance. Indeed, our observations of the enrichment of oncogenic protein factors may reflect the evolutionary pressure acting on tumor cells to select specific genetic alternations. Third, our work suggests a specific mechanism by which escaping tumor cells, through the accumulation of genetic events due to downregulation of the DNA licensing factor Cdt1, may be the seeds of genetically distinct tumor cells with more robust drug resistance. Supporting this idea, a recent study linked palbociclib-mediated arrest to downregulation of replisome components, defective origin licensing, and replication stress (72). Clinically, tumor mutational burden is associated with resistance to CDK4/6 inhibitors in patients with ER+/HER2− breast cancer (73), and deficient licensing due to downregulation of Cdt1 could be a key mechanism leading to accumulation of resistance promoting genetic events. Future work should investigate whether fractionally resistant cells are more prone to replication stress and genetic mutation.
Our work is consistent with the well-known additive effects of combining endocrine therapy and CDK4/6 inhibitors. If these drugs work together to reduce fractional resistance, then it may be profitable to consider additional combination therapies that further reduce the fractionally resistant subpopulation in ER+/HER2− breast tumors. Indeed, we found that the combination of palbociclib and tamoxifen, a drug that blocks estrogen receptor signaling, reduced fractional resistance in primary tumor cells (SI Appendix, Fig. S8). Although increasing doses of either drug are expected to reduce proliferation, high doses of palbociclib were unable to completely eliminate fractional resistance (SI Appendix, Fig. S9), suggesting the existence of a distinctly resistant subpopulation of cells. Further work is necessary to unambiguously distinguish between stochastic fluctuations in protein levels and a truly resistant subpopulation of cells. Clarifying this distinction could inform the development of biomarkers that predict patient response before treatment.
We identified the accumulation of cyclin E/CDK2 as a potential driver of escape from palbociclib-mediated arrest, which we demonstrated as a synergistic cotreatment strategy in vitro. Therefore, CDK4/6 inhibitors with activity against CDK2 may have greater efficacy by also arresting this fractionally resistant subpopulation (74). In fact, patients with ER+/HER2− breast cancer treated with abemaciclib or ribociclib, which have greater activity against CDK2 compared to the more CDK4/6 specific palbociclib, have improved responses (75–78). Thus, using this human tumor 4i model, we have uncovered a mechanism that explains clinical findings and supports the need for experimental systems using primary human tumors at the single-cell level to understand how tumors respond and resist therapies.
Evaluating therapeutic responses in terms of fractional resistance and cell cycle paths enhances our understanding of drug resistance mechanisms. Previous work has shown that cell cycle behaviors vary among tumors; here, we show that it also varies within the same tumor at a single-cell level. With the ability to fully profile the cell cycle behaviors in tumor cells—and distinguish among them—the field could make better predictions for when targeted therapy will work or how to develop new targeted treatments to arrest proliferating tumor cells. Future study will focus on understanding the range of sensitive and resistant cell cycle paths, and unique targetable drivers of resistant paths, both in ER+/HER2− tumors treated with CDK4/6 inhibitors, and in other human solid organ tumors. Identifying and characterizing tumor subpopulations with distinct sensitivities to targeted therapies could allow development of precision therapeutic regimens for individual patients based on specific tumor subpopulation drug sensitivities.
Materials and Methods
T47D ER+/HER2− breast cancer cells were obtained from the ATCC (catalog number HTB-133). We obtained tumor samples from two female patients with invasive breast carcinoma that were positive for expression of the estrogen receptor (ER+) and negative for HER2 (HER2−). Written informed consent was obtained under an operating room to laboratory protocol (47) approved by the Institutional Review Board at the University of North Carolina at Chapel Hill and included access to deidentified patient data which was obtained through an honest broker. Cells were treated with three concentrations of the CDK4/6 inhibitor palbociclib (0, 10, and 100 nM). Single-cell proteomic measurements for samples were obtained using iterative indirect immunofluorescent imaging (4i) by adapting the protocol previously described in ref. 56. Following image and data preprocessing, cell cycle phases were annotated using a three-component Gaussian Mixture Model (sklearn v0.24.1) to the log-transformed measurements of DNA content, cyclin A, and cyclin B1, as these features were shown previously to minimally represent the cell cycle (79). After representatively downsampling T47D and primary tumor samples to the same sample size using kernel herding sketching (57), we characterized fractionally resistant cells in response to palbociclib. To robustly represent and compare cell cycle trajectories under palbociclib treatment, we first identified a shared latent space, corresponding to biological processes that were present within both T47D and primary tumor samples, by performing data integration with TRANSACT (68) (transact-dr v1.0.1, kernel = radial basis function). High-dimensional single-cell profiles were subsequently visualized following nonlinear dimensionality reduction with PHATE (69) (phate v1.0.7; k = 150) on the integrated dataset of T47D and primary tumor samples. To identify and characterize trajectories through the cell cycle under palbociclib treatment, we performed trajectory inference using Slingshot (70) (slingshot v2.7.0). For the cotreatment experiment, cells were treated with a range of CDK2 inhibitor CVT-313 concentrations for 24 h. For more details on the methods outlined in this study and information on the data and code availability, please see SI Appendix, SI Materials and Methods.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
We thank the patients who donated tissue for this study. We also thank Jean Cook for her insightful discussions relating to the cell cycle. This work was supported by NIH F31HL156464 (T.M.Z.), NIH F31-HL156433 (J.S.R.), NIH 5T32-GM067553 (J.S.R.), NIH P50CA058223 (P.M.S.), NIH K08CA280388 (P.M.S.), R01-GM138834 (J.E.P.), NSF CAREER Award 1845796 (J.E.P.), and NSF Award 2242980 (J.E.P.). This work was also supported in part by P30CA016086 UNC Lineberger Comprehensive Cancer Center Core Support Grant and funding from the UNC School of Medicine for the Computational Medicine Pilot Award Program (P.M.S. and S.C.W.).
Author contributions
T.M.Z., S.C.W., M.R.K., P.M.S., and J.E.P. designed research; T.M.Z., S.C.W., J.S.R., H.M.D., A.N., N.L., A.A.W., K.M.K., and W.S. performed research; T.M.Z., S.C.W., J.S.R., A.A.W., and K.M.K. contributed new reagents/analytic tools; T.M.Z., S.C.W., J.S.R., and W.S. analyzed data; and T.M.Z., P.M.S., and J.E.P. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Contributor Information
Philip M. Spanheimer, Email: philip_spanheimer@med.unc.edu.
Jeremy E. Purvis, Email: jeremy_purvis@med.unc.edu.
Data, Materials, and Software Availability
Tabular data frames of preprocssed single-cell proteomic data for each data source have been deposited in Zenodo (80). Source code for image preprocessing, including cell segmentation, transformation, alignment, and quantification have been deposited in Github (81). Source code for computational analyses, including functions for preprocessing, sketching, integration, trajectory inference, and other computational analyses data have been deposited in Github (82).
Supporting Information
References
- 1.Foster J. S., Henley D. C., Ahamed S., Wimalasena J., Estrogens and cell-cycle regulation in breast cancer. Trends Endocrinol. Metab. 12, 320–327 (2001). [DOI] [PubMed] [Google Scholar]
- 2.Guarducci C., et al. , Cyclin E1 and Rb modulation as common events at time of resistance to palbociclib in hormone receptor-positive breast cancer. npj Breast Cancer 4, 38 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fassl A., Geng Y., Sicinski P., CDK4 and CDK6 kinases: From basic science to cancer therapy. Science 375, eabc1495 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tanioka M., et al. , Transcriptional CCND1 expression as a predictor of poor response to neoadjuvant chemotherapy with trastuzumab in HER2-positive/ER-positive breast cancer. Breast Cancer Res. Treat. 147, 513–525 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Herrera-Abreu M. T., et al. , Early adaptation and acquired resistance to CDK4/6 inhibition in estrogen receptor-positive breast cancer. Cancer Res. 76, 2301–2313 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DeMichele A., et al. , CDK 4/6 inhibitor palbociclib (PD0332991) in Rb+ advanced breast cancer: Phase II activity, safety, and predictive biomarker assessment. Clin. Cancer Res. 21, 995–1001 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Lundberg A. S., Weinberg R. A., Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Mol. Cell. Biol. 18, 753–761 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Malumbres M., Cyclin-dependent kinases. Genome Biol. 15, 122 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van den Heuvel S., Harlow E., Distinct roles for cyclin-dependent kinases in cell cycle control. Science 262, 2050–2054 (1993). [DOI] [PubMed] [Google Scholar]
- 10.Mittnacht S., Control of pRB phosphorylation. Curr. Opin. Genet. Dev. 8, 21–27 (1998). [DOI] [PubMed] [Google Scholar]
- 11.Morris L., Elizabeth Allen K., La Thangue N. B., Regulation of E2F transcription by cyclin E-Cdk2 kinase mediated through p300/CBP co-activators. Nat. Cell Biol. 2, 232–239 (2000). [DOI] [PubMed] [Google Scholar]
- 12.Knudsen E. S., Wang J. Y., Dual mechanisms for the inhibition of E2F binding to RB by cyclin-dependent kinase-mediated RB phosphorylation. Mol. Cell. Biol. 17, 5771–5783 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yao G., Lee T. J., Mori S., Nevins J. R., You L., A bistable Rb-E2F switch underlies the restriction point. Nat. Cell Biol. 10, 476–482 (2008). [DOI] [PubMed] [Google Scholar]
- 14.Black A. R., Azizkhan-Clifford J., Regulation of E2F: A family of transcription factors involved in proliferation control. Gene 237, 281–302 (1999). [DOI] [PubMed] [Google Scholar]
- 15.Cappell S. D., et al. , EMI1 switches from being a substrate to an inhibitor of APC/C(CDH1) to start the cell cycle. Nature 558, 313–317 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang Y., et al. , 442O—Cell cycle biomarker analysis from the PALOMA-1/TRIO18 palbociclib plus letrozole phase II study in ER-Positive/HER2-negative advanced breast cancer (ABC). Ann. Oncol. 25, iv146 (2014). [Google Scholar]
- 17.Finn R. S., et al. , The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): A randomised phase 2 study. Lancet Oncol. 16, 25–35 (2015). [DOI] [PubMed] [Google Scholar]
- 18.Finn R. S., et al. , Palbociclib and letrozole in advanced breast cancer. N. Engl. J. Med. 375, 1925–1936 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Schoninger S. F., Blain S. W., The ongoing search for biomarkers of CDK4/6 inhibitor responsiveness in breast cancer. Mol. Cancer Ther. 19, 3–12 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Álvarez-Fernández M., Malumbres M., Mechanisms of Sensitivity and resistance to CDK4/6 inhibition. Cancer Cell 37, 514–529 (2020). [DOI] [PubMed] [Google Scholar]
- 21.Yerushalmi R., Woods R., Ravdin P. M., Hayes M. M., Gelmon K. A., Ki67 in breast cancer: Prognostic and predictive potential. Lancet Oncol. 11, 174–183 (2010). [DOI] [PubMed] [Google Scholar]
- 22.Ellis M. J., et al. , Ki67 proliferation index as a tool for chemotherapy decisions during and after neoadjuvant aromatase inhibitor treatment of breast cancer: Results from the American College of Surgeons Oncology Group Z1031 Trial (Alliance). J. Clin. Oncol. 35, 1061–1069 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Inwald E. C., et al. , Ki-67 is a prognostic parameter in breast cancer patients: Results of a large population-based cohort of a cancer registry. Breast Cancer Res. Treat. 139, 539–552 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gaglia G., et al. , Temporal and spatial topography of cell proliferation in cancer. Nat. Cell. Biol. 24, 316–326 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berenbaum M. C., In vivo determination of the fractional kill of human tumor cells by chemotherapeutic agents. Cancer Chemother. Rep. 56, 563–571 (1972). [PubMed] [Google Scholar]
- 26.Cotter T. G., Glynn J. M., Echeverri F., Green D. R., The induction of apoptosis by chemotherapeutic agents occurs in all phases of the cell cycle. Anticancer Res. 12, 773–779 (1992). [PubMed] [Google Scholar]
- 27.Cohen A. A., et al. , Dynamic proteomics of individual cancer cells in response to a drug. Science 322, 1511–1516 (2008). [DOI] [PubMed] [Google Scholar]
- 28.Emert B. L., et al. , Variability within Rare Cell States Enables Multiple Paths towards Drug Resistance (Cold Spring Harbor Laboratory, 2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Spencer S. L., Gaudet S., Albeck J. G., Burke J. M., Sorger P. K., Non-genetic origins of cell-to-cell variability in TRAIL-induced apoptosis. Nature 459, 428–432 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schauer N. J., et al. , Selective USP7 inhibition elicits cancer cell killing through a p53-dependent mechanism. Sci. Rep. 10, 5324 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Palmer A. C., Chidley C., Sorger P. K., A curative combination cancer therapy achieves high fractional cell killing through low cross-resistance and drug additivity. eLife 8, e50036 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Flusberg D. A., Roux J., Spencer S. L., Sorger P. K., Cells surviving fractional killing by TRAIL exhibit transient but sustainable resistance and inflammatory phenotypes. Mol. Biol. Cell 24, 2186–2200 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roux J., et al. , Fractional killing arises from cell-to-cell variability in overcoming a caspase activity threshold. Mol. Syst. Biol. 11, 803 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang C., Tian C., Hoffman T. E., Jacobsen N. K., Spencer S. L., Rapidly Induced Drug Adaptation Mediates Escape from BRAF Inhibition in Single Melanoma Sells (Cold Spring Harbor Laboratory, 2020). [Google Scholar]
- 35.Torre E. A., et al. , Genetic screening for single-cell variability modulators driving therapy resistance. Nat. Genet. 53, 76–85 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kumarasamy V., Vail P., Nambiar R., Witkiewicz A. K., Knudsen E. S., Functional determinants of cell cycle plasticity and sensitivity to CDK4/6 inhibition. Cancer Res. 81, 1347–1360 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Spencer S. L., et al. , The proliferation-quiescence decision is controlled by a bifurcation in CDK2 activity at mitotic exit. Cell 155, 369–383 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Arora M., Moser J., Phadke H., Basha A. A., Spencer S. L., Endogenous replication stress in mother cells leads to quiescence of daughter cells. Cell Rep. 19, 1351–1364 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen J. Y., Lin J. R., Tsai F. C., Meyer T., Dosage of Dyrk1a shifts cells within a p21-cyclin D1 signaling map to control the decision to enter the cell cycle. Mol. Cell 52, 87–100 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Daigh L. H., Liu C., Chung M., Cimprich K. A., Meyer T., Stochastic endogenous replication stress causes ATR-triggered fluctuations in CDK2 activity that dynamically adjust global DNA synthesis rates. Cell Syst. 7, 17–27.e3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Araujo A. R., Gelens L., Sheriff R. S., Santos S. D., Positive feedback keeps duration of mitosis temporally insulated from upstream cell-cycle events. Mol. Cell 64, 362–375 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Knudsen E. S., et al. , CDK/cyclin dependencies define extreme cancer cell-cycle heterogeneity and collateral vulnerabilities. Cell Rep. 38, 110448 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chao H. X., et al. , Evidence that the human cell cycle is a series of uncoupled, memoryless phases. Mol. Syst. Biol. 15, e8604 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chao H. X., et al. , Orchestration of DNA damage checkpoint dynamics across the human cell cycle. Cell Syst 5, 445–459.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Coleman K. E., et al. , Sequential replication-coupled destruction at G1/S ensures genome stability. Genes Dev. 29, 1734–1746 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stallaert W., Kedziora K. M., Chao H. X., Purvis J. E., Bistable switches as integrators and actuators during cell cycle progression. FEBS Lett. 593, 2805–2816 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim H., et al. , Tamoxifen response at single cell resolution in estrogen receptor-positive primary human breast tumors. Clin. Cancer Res. 29, 4894–4907 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smith J. A., Martin L., Do cells cycle? Proc. Natl. Acad. Sci. U.S.A. 70, 1263–1267 (1973). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu C., et al. , Altered G1 signaling order and commitment point in cells proliferating without CDK4/6 activity. Nat. Commun. 11, 5305 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Grant G. D., Kedziora K. M., Limas J. C., Cook J. G., Purvis J. E., Accurate delineation of cell cycle phase transitions in living cells with PIP-FUCCI. Cell Cycle 17, 2496–2516 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gookin S., et al. , A map of protein dynamics during cell-cycle progression and cell-cycle exit. PLoS Biol. 15, e2003268 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stallaert W., et al. , The molecular architecture of cell cycle arrest. Mol. Syst. Biol. 18, e11087 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stallaert W., et al. , The structure of the human cell cycle. Cell Syst 13, 230–240.e3 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Holliday D. L., Speirs V., Choosing the right cell line for breast cancer research. Breast Cancer Res. 13, 215 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Neve R. M., et al. , A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 10, 515–527 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gut G., Herrmann M. D., Pelkmans L., Multiplexed protein maps link subcellular organization to cellular states. Science 361, eaar7042 (2018). [DOI] [PubMed] [Google Scholar]
- 57.Baskaran V. A., Ranek J., Shan S., Stanley N., Oliva J. B., "Distribution-based sketching of single-cell samples" in Proceedings of the 13th ACM International Conference on Bioinformatics, Computational Biology and Health Informatics (BCB '22) (Association for Computing Machinery, 2022), pp. 1–10. [Google Scholar]
- 58.Roufayel R., Mezher R., Storey K. B., The role of retinoblastoma protein in cell cycle regulation: An updated review. Curr. Mol. Med. 21, 620–629 (2021). [DOI] [PubMed] [Google Scholar]
- 59.Guiley K. Z., et al. , p27 allosterically activates cyclin-dependent kinase 4 and antagonizes palbociclib inhibition. Science 366, eaaw2106 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Otto T., Sicinski P., Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 17, 93–115 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Murray A. W., Recycling the cell cycle: Cyclins revisited. Cell 116, 221–234 (2004). [DOI] [PubMed] [Google Scholar]
- 62.Yang H. W., et al. , Stress-mediated exit to quiescence restricted by increasing persistence in CDK4/6 activation. eLife 9, e44571 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Moser J., Miller I., Carter D., Spencer S. L., Control of the restriction point by Rb and p21. Proc. Natl. Acad. Sci. U.S.A. 115, E8219–E8227 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pozo P. N., Cook J. G., Regulation and function of Cdt1: A key factor in cell proliferation and genome stability. Genes (Basel) 8, 2 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Miller I., et al. , Ki67 is a graded rather than a binary marker of proliferation versus quiescence. Cell Rep. 24, 1105–1112.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kim S., Leong A., Kim M., Yang H. W., CDK4/6 initiates Rb inactivation and CDK2 activity coordinates cell-cycle commitment and G1/S transition. Sci. Rep. 12, 16810 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tadesse S., et al. , Targeting CDK2 in cancer: Challenges and opportunities for therapy. Drug Discov. Today 25, 406–413 (2020). [DOI] [PubMed] [Google Scholar]
- 68.Mourragui S. M. C., et al. , Predicting patient response with models trained on cell lines and patient-derived xenografts by nonlinear transfer learning. Proc. Natl. Acad. Sci. U.S.A. 118, e2106682118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Moon K. R., et al. , Visualizing structure and transitions in high-dimensional biological data. Nat. Biotechnol. 37, 1482–1492 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Street K., et al. , Slingshot: Cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genomics 19, 477 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Laidlaw R. F., Briggs E. M., Matthews K. R., McCulloch R., Otto T. D., TrAGEDy: Trajectory alignment of gene expression dynamics. bioRxiv [Preprint] (2022). 10.1101/2022.12.21.521424 (Accessed 28 April 2023). [DOI]
- 72.Crozier L., et al. , CDK4/6 inhibitors induce replication stress to cause long-term cell cycle withdrawal. EMBO J. 41, e108599 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Davis A. A., et al. , Genomic complexity predicts resistance to endocrine therapy and CDK4/6 inhibition in hormone receptor-positive (HR+)/HER2-negative metastatic breast cancer. Clin. Cancer Res. 29, 1719–1729 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Freeman-Cook K., et al. , Expanding control of the tumor cell cycle with a CDK2/4/6 inhibitor. Cancer Cell 39, 1404–1421.e11 (2021). [DOI] [PubMed] [Google Scholar]
- 75.Hortobagyi G. N., et al. , Overall survival with ribociclib plus letrozole in advanced breast cancer. N. Engl. J. Med. 386, 942–950 (2022). [DOI] [PubMed] [Google Scholar]
- 76.Goetz M. P., et al. , MONARCH 3: Abemaciclib as initial therapy for advanced breast cancer. J. Clin. Oncol. 35, 3638–3646 (2017). [DOI] [PubMed] [Google Scholar]
- 77.Al-Ziftawi N. H., et al. , Cost-effectiveness and cost-utility of palbociclib versus ribociclib in women with stage IV breast cancer: A real-world data evaluation. Int. J. Environ. Res. Public Health 20 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kalinsky K., et al. , Randomized phase II trial of endocrine therapy with or without ribociclib after progression on cyclin-dependent kinase 4/6 inhibition in hormone receptor-positive, human epidermal growth factor receptor 2-negative metastatic breast cancer: MAINTAIN trial. J. Clin. Oncol. 41, 4004–4013 (2023). [DOI] [PubMed] [Google Scholar]
- 79.Ranek J. S., Stallaert W., Milner J., Stanley N., Purvis J. E., Feature selection for preserving biological trajectories in single-cell data. bioRxiv [Preprint] (2023). 10.1101/2023.05.09.540043 (Accessed 15 September 2023). [DOI] [PMC free article] [PubMed]
- 80.Zikry T. M., et al. , Single-cell datasets for cell cycle plasticity underlies fractional resistance to palbociclib in ER+/HER2- breast tumor cells. Zenodo. 10.5281/ZENODO.10063003. Accessed 12 May 2023. [DOI] [PMC free article] [PubMed]
- 81.Kedziora K. M., 4i_analysis. Github. https://github.com/fjorka/4i_analysis. Accessed 15 May 2022.
- 82.Zikry T. M., et al. , fractional_resistance: Characterizing fractional resistance in T47D and primary tumor samples. Github. https://github.com/purvislab/fractional_resistance. Accessed 16 May 2023.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
Tabular data frames of preprocssed single-cell proteomic data for each data source have been deposited in Zenodo (80). Source code for image preprocessing, including cell segmentation, transformation, alignment, and quantification have been deposited in Github (81). Source code for computational analyses, including functions for preprocessing, sketching, integration, trajectory inference, and other computational analyses data have been deposited in Github (82).