

Abstract
Introduction.
Gastric cancer is a health disparity in the Alaska Native people. The incidence of Helicobacter pylori infection, a risk factor for non-cardia gastric adenocarcinoma, is also high. Gastric cancer is partially associated with the virulence of the infecting strain.
Aim.
To genotype the vacA s, m and i and cag pathogenicity island (cagPAI) genes in H. pylori from Alaskans and investigate associations with gastropathy.
Methodology.
We enrolled patients with gastritis, peptic ulcer disease (PUD) and intestinal metaplasia (IM) in 1998–2005 and patients with gastric cancer in 2011–2013. Gastric biopsies were collected and cultured and PCR was performed to detect the presence of the right and left ends of the cagPAI, the cagA, cagE, cagT and virD4 genes and to genotype the vacA s, m and i regions.
Results.
We recruited 263 people; 22 (8 %) had no/mild gastritis, 121 (46 %) had moderate gastritis, 40 (15%) had severe gastritis, 38 (14 %) had PUD, 30 (11 %) had IM and 12 (5 %) had gastric cancer. H. pylori isolates from 150 (57%) people had an intact cagPAI; those were associated with a more severe gastropathy (P≤0.02 for all comparisons). H. pylori isolates from 77 % of people had either the vacA s1/i1/m1 (40 %; 94/234) or s2/i2/m2 (37 %; 86/234) genotype. vacA s1/i1/m1 was associated with a more severe gastropathy (P≤0.03 for all comparisons).
Conclusions.
In this population with high rates of gastric cancer, we found that just over half of the H. pylori contained an intact cagPAI and 40 % had the vacA s1/i1/m1 genotype. Infection with these strains was associated with a more severe gastropathy.
Keywords: Helicobacter pylori, gastric cancer, cagPAI, vacA, Alaska
INTRODUCTION
Helicobacter pylori is a common human infection with over 70 % of people infected in some countries [1]. All H. pylori-infected persons have mild to severe gastric mucosal inflammation, but in some, infection leads to chronic active gastritis or peptic ulcer disease (PUD) [2–4]. Additionally, H. pylori-infected persons have at least a twofold increased gastric cancer risk when compared with uninfected persons [5]. Because of this, H. pylori is characterized as a group I carcinogen by the International Agency for Research on Cancer (World Health Organization) and a risk factor for non-cardia gastric adenocarcinoma [6–8]. Despite H. pylori’s role in disease aetiology development, it is estimated that infected persons have only a 10 to 20% lifetime risk of developing PUD and a 1 to 2% risk of developing gastric cancer [9, 10]. Acquiring these diseases depends upon the inflammatory response to chronic colonization, which is likely determined by a combination of factors, including the virulence of the infecting strain.
Two virulence markers are the cytotoxin-associated gene pathogenicity island (cagPAI) and the vacuolating cytotoxin gene A (vacA). Not all H. pylori contain a cagPAI and it is incomplete in some strains [11, 12]. cagPAI integrity is critical in the interaction between H. pylori and its host. The majority of cagPAI genes encode proteins that form type IV secretion system (T4SS) components; inactivation of some of these genes can result in a non-functional T4SS, causing strains to function similarly to those with a completely absent cagPAI [11–19]. CagA, coded for by the cagPAI cagA gene, is an oncoprotein that is inserted into host epithelial cells via the T4SS and is associated with increased gastric cancer risk [20–22]. CagA has several Glu-Pro-Ile-Tyr-Ala (EPIYA) motifs, which undergo tyrosine phosphorylation at the SHP-2 enzyme [23]. These tyrosine phosphorylation motifs (TPMs) are described as A-, B-, C-, or d- and are associated with gastric pathology with the d- motif having the highest affinity for SHP-2 and increased gastric cancer risk; multiple C- motifs may also increase cancer risk [24, 25].
VacA is a 95kD toxin that induces vacuolization in epithelial cells in vitro by loosening tight junctions and forming pores in cell membranes [26–28]. All H. pylori have a vacA gene, although there is vacuolating activity variation due to sequence heterogeneity at the 5′ end [signal (s), intermediate (i) and middle (m) regions] [29, 30]. Many groups have found a correlation between toxin activity and pathogenicity, with strains having vacA s1, i1 and m1 genotypes being more virulent and associated with increased gastric cancer risk [22, 29–37].
Gastric cancer morbidity and mortality is a significant health disparity in Alaska Native people. Data from 1999 to 2013 demonstrated an age-adjusted annual incidence rate of 22.6 (Alaska Native) vs 7.1 (US white) cases per 100 000 population [38]. In 2007–2011, the gastric cancer mortality rate in Alaska Native people was 11.6/100 000, nearly four times higher than the mortality rate in US whites (3.0/100,000) [38]. Alaska has high rates of H. pylori infection, antimicrobial-resistant H. pylori, H. pylori treatment failure and reinfection after successful H. pylori eradication [39–44], thus it is not practical to attempt to lower gastric cancer rates in Alaska Native people by treating all H. pylori-positive persons [45]. In addition, a massive eradication campaign could result in widespread antimicrobial resistance, which would be a concern not only for H. pylori but also for other organisms with high infection rates in Alaska, such as Streptococcus pneumoniae and Haemophilus influenzae. Another strategy is to identify factors that might increase a person’s risk of developing cancer. This could lead to programmes identifying persons at highest risk and offering H. pylori treatment or regular surveillance with esophagogastroduodenoscopy (EGD) for early gastric cancer detection [46].
A previous study has shown that a majority of H. pylori collected from Alaskans contain the cagA gene and nearly half have the vacA s1/m1 genotype [47]. As described earlier, CagA protein functionality is at least partially determined by the presence of the entire cagPAI as well as the CagA EPIYA motifs. The vacA i region may also play a role in H. pylori virulence. In this study, we genotyped the vacA s, i and m regions and identified the existence of an intact cagPAI from H. pylori isolated from Alaskans with a variety of gastric pathologies.
METHODS
Participants
The participants in this study were recruited via two mechanisms. Both studies were approved by the Centers for Disease Control and Prevention and the Alaska Area Indian Health Service Institutional Review Boards as well as review boards for the participating Tribal Health Organizations.
Twenty gastric cancer cases were prospectively recruited at the Alaska Native Medical Center (ANMC) as part of a 2011–2013 study investigating gastric cancer risk factors (Fig. 1). Twelve of those patients had a biopsy sample that grew H. pylori, so they were eligible for the present analysis. Persons ≥18 years old were eligible to participate if, after EGD, they had a newly diagnosed gastric cancer and received follow-up care at the ANMC. Participants initially received a brief description of the study from their clinical provider who contacted study staff if their patient indicated interest. Study staff met with the participant and collected informed consent.
Fig. 1.
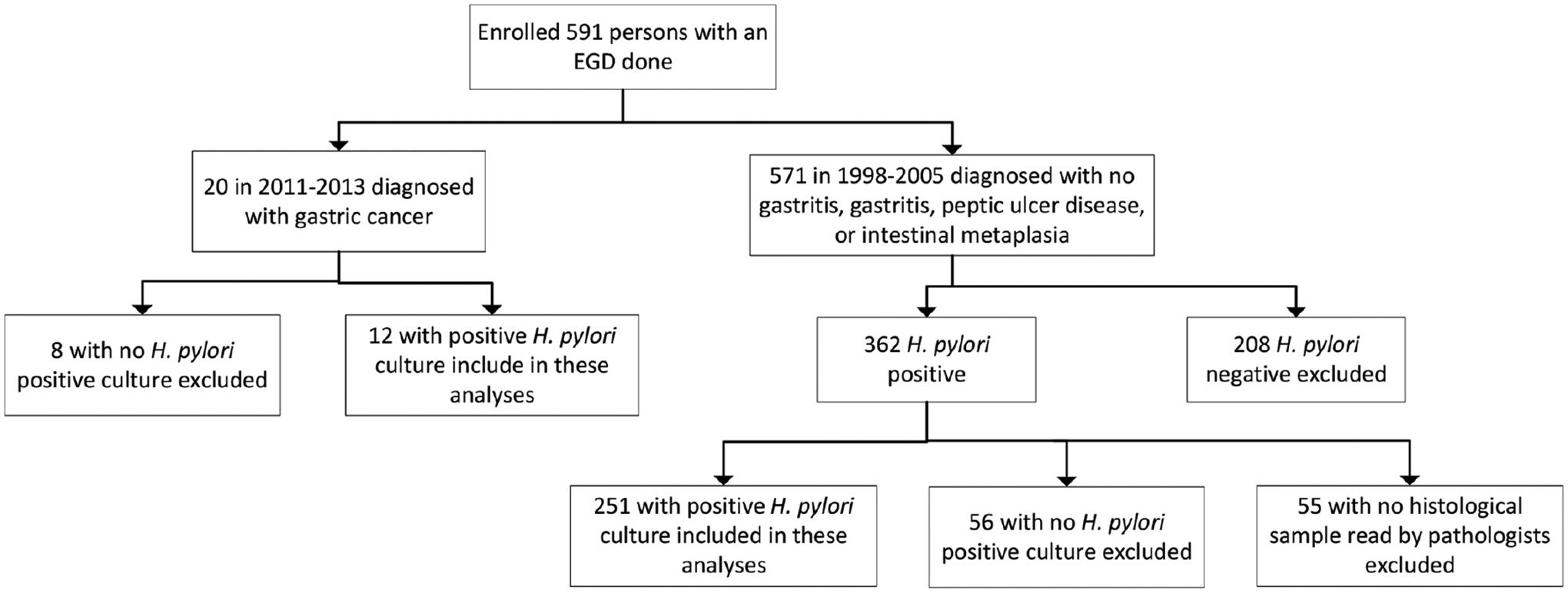
Flow diagram of enrolled participants.
Participants with gastritis, PUD and intestinal metaplasia (IM; n=251) were prospectively recruited as part of a previously described H. pylori reinfection study [44]. Briefly, persons ≥18 years old undergoing EGD from September 1998 through January 2005 were recruited at hospitals and clinics in four Alaskan communities (Fig. 1). Persons were eligible for enrolment into that study if they had a positive 13C-urea breath test (Meretek Diagnostics, Inc., Lafayette, CO, USA) at the time of their EGD. Persons were eligible for the present study if their H. pylori infection was also confirmed by culture and they had a histological sample read by study pathologists. Gastritis and IM diagnoses were determined by two study pathologists working independently of each other using standardized reporting criteria [48]; PUD was determined by endoscopy and chart review. For the purposes of this analysis, both acute and chronic gastritis were analysed together. Levels of acute gastritis were: none, mild (rare clustered neutrophils), moderate (neutrophils involved in several crypts) and severe (multiple extensive crypt lesions in nearly every field). Levels of chronic gastritis were: none, mild (lymphoplasmacytic infiltrates involving only the upper part of the mucosa), moderate (lamina propria involved at all levels with expiation by lymphoplasmacytic infiltrates) and severe (dense lymphoplasmacytic infiltrates at all levels with severe expansion or obliteration of epithelium). A sample was considered pathological if at least one pathologist considered it to have pathological changes. At the time of enrollment, study staff met with the participant and collected informed consent.
H. pylori genotyping
During the EGD, gastric biopsy specimens were collected, H. pylori was cultured, and DNA was extracted as described previously [47, 49]. PCR analysis was performed to detect the presence of the right and left ends of the cagPAI, the cagA, cagE, cagT and virD4 genes and to determine the genotype of the vacA s, i and m regions (Table 1). Due to potential sequence heterogeneity, two primer sets were used for some genes. In those cases, the first set listed in Table 1 was used, followed by testing with the second set on all extracts that were negative by the first. To determine the cagA EPIYA TPMs, positive cagA PCR products (CAGTF/CAGTR or A2530S/3000AS) were sequenced using the ABI 3130 genetic analyser.
Table 1.
PCR primer sequences for amplification of cag PAI and vacA genes
| Gene | Primer name | Sequence | Reference |
|---|---|---|---|
| cagA*,† | CAGTF | 5’ACCCTAGTCGGTAATGGG | [69] |
| CAGTR | 5’GCTTTAGCTTCTGAYACYGC | [69] | |
| A2530S | 5’GTTAARAATRGTGTRAAYGG | [70] | |
| 3000AS | 5’TTTAGCTTCTGATACCGC | [70] | |
| cagE † | cagEAF | 5’TTGAAAACTTCAAGGATAGGATAGAGC | [13] |
| cagEAR | 5’GCCTAGCGTAATATCACCATTACCC | [13] | |
| cagEBF | 5’AGTGATGCTTTGAGTCGCAAGTC | [71] | |
| cagEBR | 5’TGGGGCAATAGTGTGATGACG | [71] | |
| cagT † | cagTAF | 5’CCATGTTTATACGCCTGTGT | [54] |
| cagTAR | 5’CATCACCACACCCTTTTGAT | [54] | |
| cagTBF | 5’TCTAAAAAGATTACGCTCATAGGCG | [72] | |
| cagTBR | 5’CTTTGGCTTGCATGTTCAAGTTGCC | [72] | |
| virD4 † | virD4BF | 5’TTTCATAGGTTCTATGGCAAGCGGG | [73] |
| virD4BR | 5’TTAGCGCCATTCCTACCATAACC | [73] | |
| virD4AF | 5’TTTATGATGATAATCGATCGCC | [73] | |
| virD4AR | 5’GAACGCCTGCCCTGCGTAAGCG | [73] | |
| Right end cagPAI† | 3’F | 5’GGCTCAAGCTCGGAATGAT | |
| emptyR | 5’CTCTTTTTGTGCCTTTGATTGAA | ||
| Left end cagPAI† | emptyF | 5’CCAAATACATTTTGGCTAAATAAAC | |
| 5’R | 5’GCTTATCAGTCAAATTGTTTTTG | ||
| vacA s1a | SS1F | 5’GTCAGCATCACACCGCAAC | [29] |
| VAIR | 5’CTGCTTGAATGCGCCAAAC | [29] | |
| vacA s1b | SS3F | 5’AGCGCCATACCGCAAGAG | [29] |
| VAIR | 5’CTGCTTGAATGCGCCAAAC | [29] | |
| vacA s1c | S1cF | 5’CTYGCTTTAGTRGGGYTA | [74] |
| VAIR | 5’CTGCTTGAATGCGCCAAAC | [29] | |
| vacA s2 | SS2F | 5’GCTAACACGCCAAATGATCC | [29] |
| VAIR | 5’CTGCTTGAATGCGCCAAAC | [29] | |
| vacA m1a | VA3F | 5’GGTCAAAATGCGGTCATGG | [29] |
| VA3R | 5’CTAATGCCATTGGTACCTGTAGAAAC | [29] | |
| vacA m1b | VAMF | 5’CCCCAATGCAGTCATGGAT | [75] |
| VAMR | 5’GCTGTTAGTGCCTAAAGAAGCAT | [75] | |
| vacA m2 | VA4F | 5’GGAGCCCCAGGAAACATTG | [29] |
| VA4R | 5’TGTCATAACTAGCGCCTTGCAC | [29] | |
| vacA i1 | F1 | 5’GTTGGGATTGGGGGAATGCCG | [63] |
| c1R | 5’TTAATTTAACGCTGTTTGAAG | [63] | |
| vacA i2 | F1 | 5’GTTGGGATTGGGGGAATGCCG | [63] |
| c2R | 5’GATCAACGCTCTGATTTGA | [63] |
Also used to determine the EPIYA sequence for cagA-positive samples.
Sample is considered to have an intact cagPAI if all of these genes are present.
Statistical analysis
Persons were considered to have a particular H. pylori genotype if it was detected in ≥1 gastric biopsy specimen. We categorized the cagPAI as (1) intact, if the cagPAI left and right ends, cagA, cagE, cagT and virD4 genes were all detected; (2) partially deleted, if at least one, but not all, of the genes was detected; or (3) negative, if no cagPAI genes were detected. Analyses began with six disease categories ordered according to severity: no/mild gastritis, moderate gastritis, severe gastritis, PUD, IM, and gastric cancer. We tested for statistical significance for the prevalence of H. pylori genotypes across the entire clinical spectrum using a Mantel–Haenszel chi-square test with a modified ridit score [50]. The modified ridit score makes no assumption about the spacing between the categories of disease, only on the rank ordering. We used the likelihood ratio chi-square to test the prevalence of H. pylori genotypes between persons with gastritis, PUD, and IM/gastric cancer combined and between IM/gastric cancer combined and all others. Because gender differed between the two cohorts (1998–2005 and 2011–2013) and both gender and age differed between persons with different disease categories, we additionally ran age and gender adjusted P-values. The results remained similar and are not reported. Analyses were run within SAS software, version 9.4 (Cary, NC, USA). P-values were two-sided and exact when sample sizes necessitated and P<0.05 was considered significant.
RESULTS
Participants
Two hundred and sixty-three people were enrolled and had an H. pylori isolate available for genotyping (Table 2). Of these 263 participants, 255 (97 %) were Alaska Native, 6 (2 %) were Caucasian and 1 (0.4 %) each were African American and Asian American. Twenty-two out of 263 (8 %) participants had no/mild gastritis, 121/263 (46 %) had moderate gastritis, 40/263 (15 %) had severe gastritis, 38/263 (14 %) had peptic ulcer disease (PUD), 30/263 (11 %) had IM and 12/263 (5 %) had gastric cancer. An additional nine recruited gastric cancer patients are not included in these analyses because they either had no biopsy sample collected (n=6) or did not have H. pylori isolated from their biopsy (n=3). Older median age and male gender were associated with more severe gastric pathologies (P≤0.001 and ≤0.01, respectively, for all comparisons).
Table 2.
Study participant demographics; Alaska, 1998–2005 and 2011–2013
| Characteristic | Total (n=263) | No/mild gastritis (n=22) | Moderate gastritis (n=121) | Severe gastritis (n=40) | PUD* (n=38) | IM* (n=30) | GC* (n=12) | |||
|---|---|---|---|---|---|---|---|---|---|---|
| P-value clinical spectrum | P-value gastritis vs PUD vs IM/GC | P-value IM/GC vs others | ||||||||
| Age median (min, max) | 48 (18, 88) | 50 (24, 79) | 45 (18, 88) | 46 (24, 77) | 51 [21, 73] | 51 (18, 78) | 59 (38, 77) | 0.005 | 0.001 | 0.003 |
| Male gender | 115 (44 %) | 7 (32 %) | 42 (35 %) | 21 (53 %) | 19 (50 %) | 17 (57 %) | 9 (75 %) | 0.0005 | 0.01 | 0.01 |
Significant P-values are bolded.
PUD, peptic ulcer disease; IM, intestinal metaplasia; GC, gastric cancer.
cagPAI genotyping
H. pylori collected from 171 (65 %) people contained at least 1 cagPAI region. The presence of any of the six cagPAI regions was independently associated with a more severe gastric pathology regardless of how we grouped the pathologies (P≤0.04 for all but one comparison; Table 3). H. pylori collected from 150 (57 %) people had an intact cagPAI; those were also associated with a more severe gastric pathology (P≤0.02 for all comparisons). H. pylori collected from 92 (35 %) people had a completely deleted cagPAI; those were associated with less severe gastric pathologies (P≤0.02 for all comparisons).
Table 3.
cag pathogenicity island (cagPAI) genotyping of H. pylori strains isolated from Alaskans, 1998–2005 and 2011–2013
| cagPAI gene | Total (n=263) | No/mild gastritis (n=22) | Moderate gastritis (n=121) | Severe gastritis (n=40) | PUD* (n=38) | IM* (n=30) | GC* (n=12) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| P-value clinical spectrum | P-value gastritis vs PUD vs. IM/GC | P-value IM/GC vs others | |||||||||
| cagPAI left end | 159 (60 %) | 11 (50 %) | 70 (58 %) | 22 (55 %) | 23 (61 %) | 22 (73 %) | 11 (92 %) | 0.03 | 0.02 | 0.007 | |
| cagPAI right end | 156 (59 %) | 11 (50 %) | 67 (55 %) | 22 (55 %) | 23 (61 %) | 22 (73 %) | 11 (92 %) | 0.02 | 0.01 | 0.004 | |
| cagA | 158 (60 %) | 11 (50 %) | 67 (55 %) | 25 (63 %) | 23 (61 %) | 22 (73 %) | 10 (83 %) | 0.02 | 0.06 | 0.02 | |
| cagE | 162 (62 %) | 11 (50 %) | 71 (59 %) | 24 (60 %) | 22 (58 %) | 22 (73 %) | 12 (100 %) | 0.02 | 0.01 | 0.003 | |
| cagT | 158 (60 %) | 11 (50 %) | 68 (56 %) | 23 (58 %) | 22 (58 %) | 22 (73 %) | 12 (100 %) | 0.01 | 0.007 | 0.002 | |
| virD4 | 162 (62 %) | 11 (50 %) | 72 (60 %) | 24 (60 %) | 22 (58 %) | 22 (73 %) | 11 (92 %) | 0.04 | 0.04 | 0.01 | |
| Intact cagPAI† | 150 (57 %) | 11 (50 %) | 63 (52 %) | 22 (55 %) | 22 (58 %) | 22 (73 %) | 10 (83 %) | 0.02 | 0.02 | 0.006 | |
| Partially deleted cagPAI‡ | 21 (8 %) | 0 (0 %) | 15 (12 %) | 3 (8 %) | 1 (3 %) | 0 (0 %) | 2 (17 %) | 0.31 | 0.20 | 0.54 | |
| Completely deleted cagPAI§ | 92 (35 %) | 11 (50 %) | 43 (36 %) | 15 (38 %) | 15 (39 %) | 8 (27 %) | 0 (0 %) | 0.01 | 0.02 | 0.006 | |
| EPIYA | AB | 4 (2 %) | 0 (0 %) | 1 (1 %) | 1 (4 %) | 0 (0 %) | 2 (9 %) | 0 (0 %) | 0.45‖ | 0.69‖ | 0.77‖ |
| ABC | 133 (84 %) | 11 (100 %) | 56 (84 %) | 22 (88 %) | 19 (83 %) | 17 (77 %) | 8 (80 %) | ||||
| ABCC | 15 (9 %) | 0 (0 %) | 9 (13 %) | 1 (4 %) | 1 (4 %) | 3 (14 %) | 1 (10 %) | ||||
| ABD | 3 (2 %) | 0 (0 %) | 1 (1 %) | 0 (0 %) | 2 (9 %) | 0 (0 %) | 0 (0 %) | ||||
| ABCCC | 2 (1 %) | 0 (0 %) | 0 (0 %) | 0 (0 %) | 1 (4 %) | 0 (0 %) | 1 (10 %) | ||||
| ACC | 1 (1 %) | 0 (0 %) | 0 (0 %) | 1 (4 %) | 0 (0 %) | 0 (0 %) | 0 (0 %) | ||||
| B-TPM¶ | EPIYA | 74 | 5 (45 %) | 32 (48 %) | 13 (54 %) | 10 (43 %) | 9 (41 %) | 5 (50 %) | 0.77 | 0.84 | 0.64 |
| EPIYT | 58 | 3 (27 %) | 28 (42 %) | 5 (21 %) | 8 (35 %) | 11 (50 %) | 3 (30 %) | ||||
| Other# | 25 | 3 (27 %) | 7 (10 %) | 6 (25 %) | 5 (22 %) | 2 (9 %) | 2 (20 %) | ||||
Significant P-values are bolded.
PUD, peptic ulcer disease; IM, intestinal metaplasia; GC, gastric cancer.
Presence of all six cagPAI regions (cagPAI left end, cagPAI right end, cagA, cagE, cagT, virD4).
Presence of ≥1 but <6 cagPAI regions.
Absence of all cagPAI regions.
P-values for AB, ABC vs all others.
Tyrosine phosphorylation motif.
ESIYT (n=12), EDSIYT (n=6), EDPIYT (n=6) and ESIYA (n=1).
The cagA gene was present in H. pylori collected from 158 people. Of those, 133 (84 %) had a cagA gene with the ABC TPM (Table 3). When comparing cagA AB and ABC EPIYA TPMs with all others (ABCC, ABCCC, ACC, ABD), EPIYA TPMs were not associated with gastric pathologies. At the EPIYA-B TPM, the majority of the cagA genes had either an EPIYA (n=74; 47 %) or EPIYT sequence (n=58; 37 %). Other EPIYA-B TPM sequences identified were ESIYT (n=12; 8 %), EDSIYT (n=6; 4 %), EDPIYT (n=6; 4 %) and ESIYA (n=1; 1 %). The EPIYA-B TPM was not associated with gastric pathologies. There was no EPIYA heterogeneity at the EPIYA-A TPM and all but three people had an EPIYA sequence at the EPIYA-C TPM; the other EPIYA-C TPM sequences were one each of EPIYT, ESIYA and EPVYA.
vacA genotyping
Of the 263 people recruited, 250 (95 %) had H. pylori with the s region genotyped, 247 (94 %) with the i region genotyped, 257 (98 %) with the m region genotyped and 234 (89 %) with all 3 regions genotyped (Table 4). Of isolates with the s region genotyped, 140 (56 %) had an s1 subtype and 98 (39 %) an s2 subtype. Of isolates with the i region genotyped, 103 (42 %) had an i1 subtype and 127 (51 %) an i2 subtype. Of isolates with the m region genotyped, 112 (44 %) had an m1 subtype and 136 (53 %) an m2 subtype. Isolates containing multiple vacA genotypes (n=12, s region; n=17, i region; n=9, m region) are removed from further discussion and analysis. vacA subtypes s1, i1 and m1 were all independently associated with a more severe gastric pathology (P≤0.02 for all comparisons). The vacA s1/i1/m1 [94/234 40 %)] and s2/i2/m2 [86/234 (37 %)] genotypes accounted for 77 % of the H. pylori isolates. Of isolates with an s1/i1/m1 genotype, 93 (99 %) also had an intact cagPAI; of isolates with an s2/i2/m2 genotype, 77 (90 %) had a partially or completely deleted cagPAI. The vacA s1/i1/m1 genotype was associated with more severe gastric pathologies (P≤0.03 for all comparisons).
Table 4.
vacA genotyping of H. pylori strains isolated from Alaskans, 1998–2005 and 2011–2013
| vacA region genotype | Total (n=263) | No/mild gastritis (n=22) | Moderate gastritis (n=121) | Severe gastritis (n=40) | PUD* (n=38) | IM* (n=30) | GC* (n=12) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| P-value clinical spectrum | P-value gastritis vs PUD vs. IM/GC | P-value IM/GC vs others | |||||||||
| Signal (s) region (n=250) | s1 | 140 (56 %) | 11 (50 %) | 61 (52 %) | 20 (53 %) | 20 (57 %) | 17 (68 %) | 11 (92 %) | 0.02 | 0.006 | 0.002 |
| s2 | 98 (39 %) | 11 (50 %) | 50 (42 %) | 17 (45 %) | 14 (40 %) | 6 (24 %) | 0 (0 %) | ||||
| s1/s2† | 12 (5 %) | 0 (0 %) | 7 (6 %) | 1 (2 %) | 1 (3 %) | 2 (8 %) | 1 (8 %) | ||||
| Intermediate (i) region (n=247) | i1 | 103 (42 %) | 8 (36 %) | 41 (36 %) | 13 (35 %) | 16 (43 %) | 16 (64 %) | 9 (82 %) | 0.003 | 0.0004 | 0.0001 |
| i2 | 127 (51 %) | 14 (64 %) | 66 (57 %) | 18 (49 %) | 21 (57 %) | 7 (28 %) | 1 (9 %) | ||||
| i1/i2† | 17 (7 %) | 0 (0 %) | 8 (7 %) | 6 (16 %) | 0 (0 %) | 2 (8 %) | 1 (9 %) | ||||
| Mid (m) region (n=257) | m1 | 112 (44 %) | 9 (41 %) | 45 (38 %) | 16 (41%) | 16 (43 %) | 17 (57 %) | 9 (90 %) | 0.02 | 0.01 | 0.003 |
| m2 | 136 (53 %) | 13 (59 %) | 69 (58 %) | 21 (54 %) | 20 (54 %) | 13 (43 %) | 0 (0 %) | ||||
| m1/m2† | 9 (3 %) | 0 (0 %) | 5 (4 %) | 2 (5 %) | 1 (3 %) | 0 (0 %) | 1 (10 %) | ||||
| vacA genotype (n=234) | s1/i1/m1 | 94 (40 %) | 8 (36 %) | 38 (34 %) | 12 (34 %) | 15 (45 %) | 13 (57 %) | 8 (80 %) | 0.02 | 0.03 | 0.004 |
| s1/i2/m2 | 18 (8 %) | 2 (9 %) | 9 (8 %) | 4 (11 %) | 2 (6 %) | 1 (4 %) | 0 (0 %) | ||||
| s2/i2/m2 | 86 (37 %) | 11 (50 %) | 45 (41 %) | 11 (31 %) | 13 (39 %) | 6 (26 %) | 0 (0 %) | ||||
| Other‡ | 10 (4 %) | 1 (5 %) | 5 (5 %) | 2 (6 %) | 2 (6 %) | 0 (0 %) | 0 (0 %) | ||||
| mixed† | 26 (11 %) | 0 (0 %) | 14 (13 %) | 6 (17 %) | 1 (3 %) | 3 (13 %) | 2 (20 %) | ||||
Significant P-values are bolded.
PUD, peptic ulcer disease; IM, intestinal metaplasia; GC, gastric cancer.
Mixed infections removed from statistical analyses.
Other includes s1/i1/m2 (n=4) and s1/i2/m1 (n=6).
DISCUSSION
In 2011, we reported on H. pylori genotypes among Alaskans [47]. The current report enhances that data by including patients with IM and gastric cancer and by genotyping multiple cagPAI genes and three vacA regions in all isolates, including those described in the earlier report. Additionally, in that report, the physician performing the endoscopy graded the gastritis severity by visual observation of the gastric mucosa. Since then, we enlisted two pathologists to review the histological sections, allowing a more precise and objective estimate of gastritis presence and severity [48]. We found that the majority of H. pylori strains in the current study contained an intact cagPAI and either a vacA s1/i1/m1 or s2/i2/m2 genotype. The presence of any cagPAI region we investigated as well as a vacA s1, i1 or m1 subtype was associated with a more severe gastric pathology, as was the presence of an intact cagPAI and the vacA s1/i1/m1 genotype.
We detected at least one cagPAI gene in H. pylori isolates collected from nearly 2/3 of study participants; an intact cagPAI was present in just over half of the participants. Only 8 % of participants had a partially deleted cagPAI, thus the presence of any one cagPAI gene was suggestive of an intact cagPAI. The presence of H. pylori with an intact cagPAI varies geographically at least partially due to H. pylori having colonized humans for nearly 60 000 years as humans migrated around the planet [51]. In a large study of 877 isolates, Olbermann, et al. found the cagPAI intact in ≥95 % of strains associated with persons from East Asia, Asia, New Zealand, West Africa and South Africa, in 81 % of strains from persons from Northeast Africa, in 58 % of strains from persons from Europe, in 28 % of strains from persons from South American Indian populations, and in no strains from persons from some African populations [52]. In the current study, the presence of any cagPAI gene, as well as the presence of an intact cagPAI, was associated with a more severe gastric pathology. In our previous study investigating the presence of only the cagA portion of the cagPAI, the only association we discovered was one between the absence of the cagA gene and an endoscopic diagnosis of esophagitis [47]. With the exception of an ulcer diagnosis, which was evaluated using endoscopy, the clinical diagnoses described in this study were made by pathologists. We believe that is a more accurate method of diagnosing gastritis and IM and likely accounts for some of the difference in clinical associations between this study and our prior one. Furthermore, the data from this study support other reports that suggest the presence of an intact cagPAI confers an increase in the severity of a patient’s gastric pathology [13, 15, 16, 53, 54]. A possible explanation reported by Hanada, et al. is that the accumulation of DNA double-stranded breaks is significantly greater in cagPAI-positive compared with cagPAI-negative strains [55].
Although the number of gastric cancer patients recruited with H. pylori detected in their biopsy was small, we found the cagE and cagT genes in all of those isolates. This supports data from India where ≥95 and ≥90 %, respectively, of H. pylori-infected persons with gastric cancer were infected with H. pylori containing the cagE or cagT gene [53, 56]. Presence of the CagA oncoprotein in host cells and the host’s immunological response to infection contribute to a patient’s outcome from H. pylori. The cagT gene is required for a functional T4SS syringe and successful injection of CagA into the host cell [17, 57, 58]. The CagE protein is known to induce IL8 secretion and to mediate host-cell cytokine rearrangements in infected cells [15, 59]. Thus, the proteins encoded for by cagE and cagT are important in promoting inflammation and intestinal cell damage.
In this study, 151 participants were found to be infected with an H. pylori that was cagA-positive with some combination of EPIYA A-, B- and C- TPMs; these TPMs are associated with the western-type CagA [60]. Twenty-one participants were infected with strains with a single d- or multiple C- TPMs, both of which have been associated with increased virulence in some studies [24, 25]; however, that was not true in our study. Over half of our study participants were infected with H. pylori with a modified EPIYA-B TPM, with a little over a third of those having the EPIYT-B TPM. The EPIYT-B TPM has been associated with the induction of lower levels of cellular elongation and IL-8 secretion [61]. Additionally, a study of GenBank sequences found that, compared with gastritis alone, gastric cancer was significantly less associated with the EPIYT-B TPM compared with the EPIYA-B TPM [62]. Due to the small number of gastric cancer participants in our study, we were unable to perform a similar analysis.
Over 75 % of our study participants had an H. pylori with either the vacA s1/i1/m1 or s2/i2/m2 genotype. This is similar to our previous report of 83 % of participants having an isolate with the vacA s1/m1 or s2/m2 genotype [47]. In both studies, a slightly higher percentage of isolates had the genotype that produces more toxin (s1/m1 or s1/i1/m1). In our previous publication, the vacA s1/m1 genotype was associated with an increased risk of having an ulcer at enrollment or a history of PUD; in the current study, the vacA s1/i1/m1 genotype was also associated with a more severe gastric pathology. This is similar to results from our previous study in which we found an association between the vacA s1/m1 genotype and ulcer disease. Additionally, in this study the individual regions of s1, i1 and m1 were each associated with more severe disease. vacA i region data have not yet been reported from Alaska and our finding is not unique to this study. In the paper that first described the i region, Rhead, et al. reported that only infection with an i1 strain was independently associated with increased risk gastric cancer risk [63]. Other groups subsequently found similar associations, although it is not universally true, especially in isolates collected from persons living in East and Southeast Asia [64–68].
This study has three important limitations. First, there were only 12 persons diagnosed with gastric cancer from whose biopsy samples we were able to detect H. pylori. This limits our ability to detect associations between the organism’s genotype and gastric cancer specifically. However, IM is along the clinical spectrum leading to gastric cancer. When we include the 30 participants with that diagnosis along with the 12 with a cancer diagnosis, we are able to identify significant associations that seem to increase gastric cancer risk. Second, the gastric cancer patients were recruited during a different time period than the patients with the other disease categories. Due to the long-term (up to 2 years) nature of the 1998–2005 study [44] from which the majority (n=251) of participants in this analysis were originally recruited, persons with gastric cancer were ineligible for participation in that study. As it is important to understand what could be contributing to the high gastric cancer burden in the Alaska Native people, in this analysis we included persons recruited as part of the 2011–2013 study of gastric cancer. However, both of those studies were cross-sectional in nature, in that the collection of the samples from which H. pylori genotyping was performed was at the same time point that the disease status was determined. Due to the lack of a prospective study design and differential recruitment periods, time could be a confounder in our findings. Finally, the majority of participants in this study were Alaska Native, so the results may not be generalizable to other populations.
In conclusion, in this population with high rates of H. pylori infection and gastric cancer, we found that just over half of the H. pylori strains contained an intact cagPAI and 40 % had the high toxin-producing vacA s1/i1/m1 genotype. Infection with H. pylori containing an intact cagPAI or vacA s1/i1/m1 genotype was associated with a more severe gastric pathology. H. pylori infection and gastric cancer are also prevalent in indigenous populations in other Arctic countries, but there are few strain genotyping data from those countries. We would propose that similar studies to this one be conducted across the circumpolar north to give a more comprehensive picture of H. pylori infection among indigenous peoples of the Arctic.
Acknowledgements
We would like to thank Catherine Dentinger, Marilyn Getty, Jim Gove, Cindy Hamlin, Helen Peters and Susan Seidel for recruiting participants, Debra Parks and Richard Baum for data management, Alisa Reasonover for H. pylori culturing, and Julie Morris for performing most of the vacA genotyping. We would also like to thank staff of the gastroenterology departments at the ANMC, Yukon-Kuskokwim Delta Regional Hospital, Kanakanak Hospital, Norton Sound Hospital and the Anchorage Endoscopy Center for contributing patient data and biopsy specimens. Finally, we would like to thank staff at the oncology and surgical departments at the ANMC for participant referrals and the ANMC pathology department for reading the biopsy slides.
Funding information
This project was funded in part by Indian Health Service Native American Research Centers for Health grants U26IHS300410 and U2694 00005 as well as a grant from the Alaska Science and Technology Foundation. The funders did not have any role in this research, including in the study design, collection, analysis, and interpretation of the data, or in the writing of this report.
Abbreviations:
- cagPAI
cytotoxin-associated gene pathogenicity island
- EGD
esophagogastroduodenoscopy
- EPIYA
Glu-Pro-Ile-Tyr-Ala motifs
- GC
gastric cancer
- i
intermediate
- IM
intestinal metaplasia
- m
middle
- PUD
peptic ulcer disease
- s
signal
- TPM
tyrosine phosphorylation motif
- T4SS
type IV secretion system
- vacA
vacuolating cytotoxin gene A
Footnotes
Conflicts of interest
The authors declare that there are no conflicts of interest.
Ethical statement
Data were obtained from studies approved by the Centers for Disease Control and Prevention and the Alaska Area Indian Health Service Institutional Review Boards and review boards from participating Alaska Native Tribal Health Organizations. All participants provided written, informed consent to participate.
The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention.
References
- 1.Zamani M, Ebrahimtabar F, Zamani V, Miller WH, Alizadeh-Navaei R et al. Systematic review with meta-analysis: the world-wide prevalence of Helicobacter pylori infection. Aliment Pharmacol Ther 2018;47:868–876. [DOI] [PubMed] [Google Scholar]
- 2.Tytgat GN RE. Campylobacter pylori and its role in peptic ulcer disease. Gastroenterology Clin N Am 1990;19:183–196. [PubMed] [Google Scholar]
- 3.Rauws EAJ, Tytgat GNJ. Cure of duodenal ulcer associated with eradication of Helicobacter pylori. The Lancet 1990;335:1233–1235. [DOI] [PubMed] [Google Scholar]
- 4.The MBJ. Albert Lasker medical research award. Helicobacter pylori. The etiologic agent for peptic ulcer. JAMA 1995;1995:1064–1066. [DOI] [PubMed] [Google Scholar]
- 5.Huang JQ, Sridhar S, Chen Y, Hunt RH. Meta-Analysis of the relationship between Helicobacter pylori seropositivity and gastric cancer. Gastroenterology 1998;114:1169–1179. [DOI] [PubMed] [Google Scholar]
- 6.Bayerdörffer E, Rudolph B, Neubauer A, Thiede C, Lehn N et al. Regression of primary gastric lymphoma of mucosa-associated lymphoid tissue type after cure of Helicobacter pylori infection. The Lancet 1995;345:1591–1594. [DOI] [PubMed] [Google Scholar]
- 7.Nomura A, Stemmermann GN, Chyou PH, Kato I, Perez-Perez GI et al. Helicobacter pylori infection and gastric carci-noma among Japanese Americans in Hawaii. N Engl J Med 1991;325:1132–1136. [DOI] [PubMed] [Google Scholar]
- 8.IARC Working group on the evaluation of carcinogenic risks to humans. Biological Agents. A review ofhuman carcinogens; 2012. pp. 1–441. [PMC free article] [PubMed] [Google Scholar]
- 9.Kuipers EJ. Review article: exploring the link between Helicobacter pylori and gastric cancer. Aliment Pharmacol Ther 1999;13:3–11. [DOI] [PubMed] [Google Scholar]
- 10.Kuipers EJ, Thijs JC, Festen HP. The prevalence of Helicobacter pylori in peptic ulcer disease. Aliment Pharmacol Ther 1995;9:59–69. [PubMed] [Google Scholar]
- 11.Akopyants NS, Clifton SW, Kersulyte D, Crabtree JE, Youree BE et al. Analyses of the CAG pathogenicity island of Helicobacter pylori. Mol Microbiol 1998;28:37–53. [DOI] [PubMed] [Google Scholar]
- 12.Censini S, Lange C, Xiang Z, Crabtree JE, Ghiara P et al. Cag, a pathogenicity island of Helicobacter pylori, encodes type I-specific and disease-associated virulence factors. Proc Natl Acad Sci U S A 1996;93:14648–14653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sozzi M, Tomasini ML, Vindigni C, Zanussi S, Tedeschi R et al. Heterogeneity of CAG genotypes and clinical outcome of Helicobacter pylori infection. J Lab Clin Med 2005;146:262–270. [DOI] [PubMed] [Google Scholar]
- 14.Jenks PJ, Mégraud F, Labigne A. Clinical outcome after infection with Helicobacter pylori does not appear to be reliably predicted by the presence of any of the genes of the CAG pathogenicity island. Gut 1998;43:752–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maeda S, Yoshida H, Ikenoue T, Ogura K, Kanai F et al. Structure of CAG pathogenicity island in Japanese Helicobacter pylori isolates. Gut 1999;44:336–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nilsson C, Sillén A, Eriksson L, Strand M-L, Enroth H et al. Correlation between CAG pathogenicity island composition and Helicobacter pylori-associated gastroduodenal disease. Infect Immun 2003;71:6573–6581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fischer W, Püls J, Buhrdorf R, Gebert B, Odenbreit S et al. Systematic mutagenesis of the Helicobacter pylori CAG pathogenicity island: essential genes for CagA translocation in host cells and induction of interleukin-8. Mol Microbiol 2001;42:1337–1348. [DOI] [PubMed] [Google Scholar]
- 18.Waskito LA, Miftahussurur M, Lusida MI, Syam AF, Suzuki R et al. Distribution and clinical associations of integrating conjugative elements and CAG pathogenicity islands of Helicobacter pylori in Indonesia. Sci Rep 2018;8:6073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rohde M, Püls J, Buhrdorf R, Fischer W, Haas R et al. A novel sheathed surface organelle of the Helicobacter pylori CAG type IV secretion system. Mol Microbiol 2003;49:219–234. [DOI] [PubMed] [Google Scholar]
- 20.Blaser MJ, Perez-Perez GI, Kleanthous H et al. Infection with Helicobacter pylori strains possessing cagA is associated with an increased risk of developing adenocarcinoma of the stomach. Cancer Res 1995;55:2111–2115. [PubMed] [Google Scholar]
- 21.Parsonnet J, Friedman GD, Orentreich N, Vogelman H et al. Risk for gastric cancer in people with cagA positive or CagA negative Helicobacter pylori infection. Gut 1997;40:297–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.pormohammad A, Ghotaslou R, Leylabadlo HE, Nasiri MJ, Dabiri H et al. Risk of gastric cancer in association with Helicobacter pylori different virulence factors: a systematic review and meta-analysis. Microb Pathog 2018;118:214–219. [DOI] [PubMed] [Google Scholar]
- 23.Higashi H et al. Shp-2 tyrosine phosphatase as an intracellular target of Helicobacter pylori CagA protein. Science 2002;295:683–686. [DOI] [PubMed] [Google Scholar]
- 24.Argent RH, Kidd M, Owen RJ, Thomas RJ, Limb MC et al. Determinants and consequences of different levels of CagA phosphorylation for clinical isolates of Helicobacter pylori. Gastroenterology 2004;127:514–523. [DOI] [PubMed] [Google Scholar]
- 25.Hayashi T, Senda M, Suzuki N, Nishikawa H, Ben C et al. Differential mechanisms for SHP2 binding and activation are exploited by geographically distinct Helicobacter pylori CagA oncoproteins. Cell Rep 2017;20:2876–2890. [DOI] [PubMed] [Google Scholar]
- 26.Montecucco C, de Bernard M. Molecular and cellular mechanisms of action of the vacuolating cytotoxin (VacA) and neutrophil-activating protein (HP-NAP) virulence factors of Helicobacter pylori. Microbes and Infection 2003;5:715–721. [DOI] [PubMed] [Google Scholar]
- 27.Graham DY. Pathogenesis of increased sucrose permeability in H. pylori gastritis. Dig Dis Sci 2000;45:889. [DOI] [PubMed] [Google Scholar]
- 28.Papini E, Satin B, Norais N, de Bernard M, Telford JL et al. Selective increase of the permeability of polarized epithelial cell monolayers by Helicobacter pylori vacuolating toxin. J Clin Invest 1998;102:813–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Atherton JC, Cao P, Peek RM, Tummuru MK, Blaser MJ et al. Mosaicism in vacuolating cytotoxin alleles of Helicobacter pylori. Association of specific vacA types with cytotoxin production and peptic ulceration. J Biol Chem 1995;270:17771–17777. [DOI] [PubMed] [Google Scholar]
- 30.Ferreira RM, Machado JC, Letley D, Atherton JC, Pardo ML et al. A novel method for genotyping the Helicobacter pylori vacA intermediate region directly in gastric biopsy specimens. J Clin Microbiol 2012;50:3983–3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Atherton JC, Peek RM, Tham KT, Cover TL, Blaser MJ et al. Clinical and pathological importance of heterogeneity in vacA, the vacuolating cytotoxin gene of Helicobacter pylori. Gastroenterology 1997;112:92–99. [DOI] [PubMed] [Google Scholar]
- 32.Gunn MC, Stephens JC, Stewart JA, Rathbone BJ, West KP et al. The significance of cagA and vacA subtypes of Helicobacter pylori in the pathogenesis of inflammation and peptic ulceration. J Clin Pathol 1998;51:761–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang HJ, Kuo CH, Yeh AA, Chang PC, Wang WC et al. Vacuolating toxin production in clinical isolates of Helicobacter pylori with different vacA genotypes. J Infect Dis 1998;178:207–212. [DOI] [PubMed] [Google Scholar]
- 34.Basso D, Navaglia F, Brigato L, Piva MG, Toma A et al. Analysis of Helicobacter pylori vacA and cagA genotypes and serum antibody profile in benign and malignant gastroduodenal diseases. Gut 1998;43:182–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Navaglia F, Basso D, Piva MG, Brigato L, Stefani A et al. Helicobacter pylori cytotoxic genotype is associated with peptic ulcer and influences serology. Am J Gastroenterol 1998;93:227–230. [DOI] [PubMed] [Google Scholar]
- 36.Garza-González E, Bosques-Padilla FJ, Pérez-Pérez GI, Flores-Gutiérrez JP, Tijerina-Menchaca R et al. Association of gastric cancer, HLA-DQA1, and infection with Helicobacter pylori cagA+ and VacA+ in a Mexican population. J Gastroenterol 2004;39:1138–1142. [DOI] [PubMed] [Google Scholar]
- 37.Sugimoto M, Yamaoka Y. The association of vacA genotype and Helicobacter pylori-related disease in Latin American and African populations. Clin Microbiol Infect 2009;15:835–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carmack AM, Schade TL, Sallison I et al. Cancer in Alaska native people: 1969–2013 the 45-Year report 2015.
- 39.Parkinson AJ, Gold BD, Bulkow L, Wainwright RB, Swaminathan B et al. High prevalence of Helicobacter pylori in the Alaska native population and association with low serum ferritin levels in young adults. Clin Diagn Lab Immunol 2000;7:885–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miernyk KM, Bulkow LR, Gold BD, Bruce MG, Hurlburt DH et al. Prevalence of Helicobacter pylori among Alaskans: Factors associated with infection and comparison of urea breath test and anti-Helicobacter pylori IgG antibodies. Helicobacter 2018;23:e12482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mosites E, Bruden D, Morris J, Reasonover A, Rudolph K et al. Antimicrobial resistance among Helicobacter pylori isolates in Alaska, 2000–2016. J Glob Antimicrob Resist 2018;15:148–153. [DOI] [PubMed] [Google Scholar]
- 42.Bruce MG, Bruden D, McMahon BJ et al. The relationship between antimicrobial resistance and treatment outcome for Helicobacter pylori infections in native and non-native persons residing in Alaska. Helicobacter 2007;12:450–451. [Google Scholar]
- 43.McMahon BJ, Bruce MG, Hennessy TW, Bruden DL, Sacco F et al. Reinfection after successful eradication of Helicobacter pylori: a 2-year prospective study in Alaska natives. Aliment Pharmacol Ther 2006;23:1215–1223. [DOI] [PubMed] [Google Scholar]
- 44.Bruce MG, Bruden DL, Morris JM, Reasonover AL, Sacco F et al. Reinfection after successful eradication of Helicobacter pylori in three different populations in Alaska. Epidemiol Infect 2015;143:1236–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McMahon BJ, Bruce MG, Koch A, Goodman KJ, Tsukanov V et al. The diagnosis and treatment of Helicobacter pylori infection in Arctic regions with a high prevalence of infection: Expert Commentary. Epidemiol Infect 2016;144:225–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bruce MG, Miernyk K, Sacco F, Thomas T, McMahon B et al. Response to editorial. Helicobacter 2019;24:e12558. [DOI] [PubMed] [Google Scholar]
- 47.Miernyk K, Morris J, Bruden D, McMahon B, Hurlburt D et al. Characterization of Helicobacter pylori cagA and vacA Genotypes among Alaskans and Their Correlation with Clinical Disease. J Clin Microbiol 2011;49:3114–3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nolen LD, Bruden D, Miernyk K, McMahon BJ, Sacco F et al. H. pylori-associated pathologic findings among Alaska native patients. Int J Circumpolar Health 2018;77:1510715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McMahon BJ, Hennessy TW, Bensler JM, Bruden DL, Parkinson AJ et al. The Relationship among Previous Antimicrobial Use, Antimicrobial Resistance, and Treatment Outcomes for Helicobacter pylori Infections. Ann Intern Med 2003;139:463–469. [DOI] [PubMed] [Google Scholar]
- 50.Lehmann EL, D’Abrera HJM. Nonparametrics: Statistical Methods Based on Ranks. New York: Springer Science & Business Media; 2006. [Google Scholar]
- 51.Linz B, Balloux F, Moodley Y, Manica A, Liu H et al. An African origin for the intimate association between humans and Helicobacter pylori. Nature 2007;445:915–. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Olbermann P, Josenhans C, Moodley Y, Uhr M, Stamer C et al. A global overview of the genetic and functional diversity in the Helicobacter pylori CAG pathogenicity island. PLoS Genet 2010;6:e1001069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Khatoon J, Prasad KN, Prakash Rai R, Ghoshal UC, Krishnani N et al. Association of heterogenicity of Helicobacter pylori cag pathogenicity island with peptic ulcer diseases and gastric cancer. Br J Biomed Sci 2017;74:121–126. [DOI] [PubMed] [Google Scholar]
- 54.Ikenoue T, Maeda S, Ogura K, Akanuma M, Mitsuno Y et al. Determination of Helicobacter pylori virulence by simple gene analysis of the CAG pathogenicity island. Clinical and Vaccine Immunology 2001;8:181–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hanada K, Uchida T, Tsukamoto Y, Watada M, Yamaguchi N et al. Helicobacter pylori infection introduces DNA double-strand breaks in host cells. Infect Immun 2014;82:4182–4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ali M, Khan AA, Tiwari SK, Ahmed N, Rao LV et al. Association between cag-pathogenicity island in Helicobacter pylori isolates from peptic ulcer, gastric carcinoma, and non-ulcer dyspepsia subjects with histological changes. World J Gastroenterol 2005;11:6815–6822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Johnson EM, Gaddy JA, Voss BJ, Hennig EE, Cover TL et al. Genes required for assembly of pili associated with the Helicobacter pylori CAG type IV secretion system. Infect Immun 2014;82:3457–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Frick-Cheng AE, Pyburn TM, Voss BJ, McDonald WH, Ohi MD et al. Molecular and Structural Analysis of the Helicobacter pylori cag Type IV Secretion System Core Complex. mBio 2016;7:e02001–02015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kauser F, Hussain MA, Ahmed I, Srinivas S, Devi SM et al. Comparative genomics of Helicobacter pylori isolates recovered from ulcer disease patients in England. BMC Microbiol 2005;5:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Higashi H, Tsutsumi R, Fujita A, Yamazaki S, Asaka M et al. Biological activity of the Helicobacter pylori virulence factor CagA is determined by variation in the tyrosine phosphorylation sites. Proc Natl Acad Sci U S A 2002;99:14428–14433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Reyes-Leon A, Atherton JC, Argent RH, Puente JL, Torres J et al. Heterogeneity in the activity of Mexican Helicobacter pylori strains in gastric epithelial cells and its association with diversity in the cagA gene. Infect Immun 2007;75:3445–3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang X-S, Tegtmeyer N, Traube L, Jindal S, Perez-Perez G et al. A specific A/T polymorphism in Western tyrosine phosphorylation B-motifs regulates Helicobacter pylori CagA epithelial cell interactions. PLoS Pathog 2015;11:e1004621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rhead JL, Letley DP, Mohammadi M, Hussein N, Mohagheghi MA et al. A new Helicobacter pylori vacuolating cytotoxin determinant, the intermediate region, is associated with gastric cancer. Gastroenterology 2007;133:926–936. [DOI] [PubMed] [Google Scholar]
- 64.Basso D, Zambon C-F, Letley DP, Stranges A, Marchet A et al. Clinical relevance of Helicobacter pylori cagA and vacA gene polymorphisms. Gastroenterology 2008;135:91–99. [DOI] [PubMed] [Google Scholar]
- 65.Douraghi M, Talebkhan Y, Zeraati H, Ebrahimzadeh F, Nahvijoo A et al. Multiple gene status in Helicobacter pylori strains and risk of gastric cancer development. Digestion 2009;80:200–207. [DOI] [PubMed] [Google Scholar]
- 66.Ogiwara H, Sugimoto M, Ohno T, Vilaichone R-K, Mahachai V et al. Role of deletion located between the intermediate and middle regions of the Helicobacter pylori vacA gene in cases of gastroduodenal diseases. J Clin Microbiol 2009;47:3493–3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yordanov D, Boyanova L, Markovska R, Gergova G, Mitov I et al. Significance of Helicobacter pylori vacA intermediate region genotyping—a Bulgarian study. Diagn Microbiol Infect Dis 2012;74:253–257. [DOI] [PubMed] [Google Scholar]
- 68.Mottaghi B, Safaralizadeh R, Bonyadi M, Latifi-Navid S, Somi MH et al. Helicobacter pylori vacA I region polymorphism but not babA2 status associated to gastric cancer risk in northwestern Iran. Clin Exp Med 2016;16:57–63. [DOI] [PubMed] [Google Scholar]
- 69.Yamaoka Y, Osato MS, Sepulveda AR, Gutierrez O, Figura N et al. Molecular epidemiology of Helicobacter pylori: separation of H. pylori from East Asian and non-Asian countries. Epidemiol Infect 2000;124:91–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Panayotopoulou EG, Sgouras DN, Papadakos K, Kalliaropoulos A, Papatheodoridis G et al. Strategy to characterize the number and type of repeating EPIYA phosphorylation motifs in the carboxyl terminus of CagA protein in Helicobacter pylori clinical isolates. J Clin Microbiol 2007;45:488–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Antonio-Rincón F, López-Vidal Y, Castillo-Rojas G, Lazcano-Ponce EC, Ponce-de-León S et al. Pathogenicity island CAG, vacA and IS605 genotypes in Mexican strains of Helicobacter pylori associated with peptic ulcers. Ann Clin Microbiol Antimicrob 2011;10:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hsu P-I, Hwang I-ran, Cittelly D, Lai K-H, El-Zimaity HMT et al. Clinical presentation in relation to diversity within the Helicobacter pylori CAG pathogenicity island. Am J Gastroenterol 2002;97:2231–2238. [DOI] [PubMed] [Google Scholar]
- 73.Audibert C, Burucoa C, Janvier B, Fauchere JL et al. Implication of the structure of the Helicobacter pylori CAG pathogenicity island in induction of interleukin-8 secretion. Infect Immun 2001;69:1625–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yamaoka Y, Kodama T, Gutierrez O et al. Relationship between Helicobacter pylori iceA, cagA, and vacA status and clinical outcome: studies in four different countries. J Clin Microbiol 1999;37:2274–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mukhopadhyay AK, Kersulyte D, Jeong J-Y, Datta S, Ito Y et al. Distinctiveness of genotypes of Helicobacter pylori in Calcutta, India. J Bacteriol 2000;182:3219–3227. [DOI] [PMC free article] [PubMed] [Google Scholar]