

Abstract
Ralstonia metallidurans CH34, a soil bacterium resistant to a variety of metals, is known to reduce selenite to intracellular granules of elemental selenium (Se0). We have studied the kinetics of selenite (SeIV) and selenate (SeVI) accumulation and used X-ray absorption spectroscopy to identify the accumulated form of selenate, as well as possible chemical intermediates during the transformation of these two oxyanions. When introduced during the lag phase, the presence of selenite increased the duration of this phase, as previously observed. Selenite introduction was followed by a period of slow uptake, during which the bacteria contained Se0 and alkyl selenide in equivalent proportions. This suggests that two reactions with similar kinetics take place: an assimilatory pathway leading to alkyl selenide and a slow detoxification pathway leading to Se0. Subsequently, selenite uptake strongly increased (up to 340 mg Se per g of proteins) and Se0 was the predominant transformation product, suggesting an activation of selenite transport and reduction systems after several hours of contact. Exposure to selenate did not induce an increase in the lag phase duration, and the bacteria accumulated approximately 25-fold less Se than when exposed to selenite. SeIV was detected as a transient species in the first 12 h after selenate introduction, Se0 also occurred as a minor species, and the major accumulated form was alkyl selenide. Thus, in the present experimental conditions, selenate mostly follows an assimilatory pathway and the reduction pathway is not activated upon selenate exposure. These results show that R. metallidurans CH34 may be suitable for the remediation of selenite-, but not selenate-, contaminated environments.
Microorganisms play a major role in the biogeochemical cycle of selenium in the environment (12). Certain strains that are resistant to selenium oxyanions, and reduce selenite (SeIV) and/or selenate (SeVI) to the less available elemental selenium (Se0) (7), could be potentially used for the bioremediation of contaminated soils, sediments, industrial effluents, and agricultural drainage waters.
Ralstonia metallidurans CH34 is a soil bacterium characteristic of metal-contaminated biotopes. It is resistant to a variety of heavy metals and metalloids including CrVI, CoII, NiII, CuII, ZnII, AsV, CdII, HgII, and PbII. The genes for metal resistance are located in two large plasmids (pMOL28 and pMOL30). Their function and regulation are well understood for some of these elements (18). This bacterial strain is also resistant to selenite, and detoxification is realized by the incorporation of this oxyanion and its subsequent reduction to red Se0, as shown by X-ray absorption spectroscopy (24). This study also revealed that the Se0 granules were localized mainly in the cytoplasm. In contrast to previously cited metals and metalloids, the genes involved in selenite resistance have not yet been identified, and the exact mechanism of selenite bioreduction is still unknown. R. metallidurans CH34 can also resist up to 16 mM selenate (2). The capacity of R. metallidurans CH34 to accumulate selenate and the fate of this oxyanion following incorporation have never been investigated. We have now studied the kinetics of selenite and selenate accumulation and used X-ray absorption near-edge structure (XANES) spectroscopy to determine Se speciation in order to identify the chemical intermediates putatively appearing during reduction. For such a purpose, XANES is the method of choice since it is nondestructive and enables direct determination of the target element speciation, i.e., its oxidation state and sometimes its exact chemical form. The results obtained on speciation were combined with the total metal content of each sample in order to deduce the concentration of each metal species. Such quantitative information is particularly useful to estimate the relative importance of several chemical pathways in a particular system.
MATERIALS AND METHODS
Bacterial strain and growth media.
R. metallidurans CH34 provided by Max Mergeay (SCK/CEN, Mol, Belgium) was grown aerobically at 29°C in Tris salt mineral medium with 2% gluconate as a carbon source (18, 19).
One-contaminant (SeIV or SeVI) exposure.
A preculture was obtained by growing the cells until mid-exponential phase (absorbance at 600 nm [A600] of 1.5). Cells were then appropriately diluted to inoculate 300 ml Tris salt mineral medium at an initial A600 of 0.3. The cultures were monitored by recording the A600 as a function of time. A first series of experiments was run by adding selenite or selenate at a final concentration of 2 mM at time zero (A600 = 0.3). In a second set of experiments, the selenium oxyanion was added during the first half of the exponential phase (A600 = 1). Finally, a third series was run by adding the selenium oxyanion at the beginning of the stationary phase (A600 = 3). Sodium selenite and sodium selenate were prepared as a 1 M stock solution in ultrapure water and sterilized by filtration. Control cultures were grown under identical conditions in the absence of the selenium oxyanions. Anywhere from 5 to 15 ml, depending on the turbidity of the cell suspension, was sampled at various time intervals over 6 days and centrifuged, and the pellets were freeze-dried and stored for further use. Cell yield was determined by recording the A600 and assaying the protein content (bicinchoninic acid method with bovine serum albumin as standard).
Two-contaminant (SeIV and SeVI) exposure.
Cultures of R. metallidurans CH34 were inoculated to an absorbance at 600 nm of 3 (stationary phase) and exposed to two different mixtures of selenite and selenate (2 mM selenite and 2 mM selenate or 1 mM selenite and 10 mM selenate). Three cases were tested: (i) selenite and selenate were both added immediately after inoculation, (ii) selenite was added immediately after inoculation and selenate 3 h later, or (iii) selenate was introduced immediately after inoculation and selenite 3 h later. A control culture, under identical conditions, was grown in the presence of selenite alone added immediately after inoculation. The appearance of the red color, a sign of the reduction of selenite to Se0, was checked after 24 h of exposure.
Inductively coupled plasma-mass spectrometry (ICP-MS) analyses.
Bacteria and culture medium were separated by centrifugation at 6,000 × g for 10 min. Cell pellets were washed twice with ultrapure water at 4°C and resuspended in a minimum volume of ultrapure water. A fraction of the pellet was digested in a mixture of 1 M NaOH-20% sodium dodecyl sulfate. In order to fully solubilize elemental selenium, H2O2 was added to the digested sample until the characteristic red color disappeared. These samples were used to determine total Se accumulation.
Selenium concentrations were measured by ICP-MS using an X7 series quadrupole instrument (Thermo Electron Corporation, Cergy-Pontoise, France). Calibration curves were obtained by analysis of a range of SPEX certiPrep selenium standards (Metuchen, NJ). Selenium concentrations were determined with the isotopes 79 and 82, and yttrium was used as an internal standard (1 μg liter−1). For digested bacteria analyses, samples were acidified with ultrapure 65% nitric acid (Normatom quality grade; Prolabo, Fontenay sous Bois, France) and diluted in ultrapure water.
X-ray absorption spectroscopy.
Selenium K-edge X-ray absorption experiments were performed on beamline FAME (BM30B) of the European Synchrotron Radiation Facility. The Se model compounds (all in solid state unless otherwise noted) used for this study were as follows: hexagonal (gray) elemental selenium, sodium selenate (solid and in solution), sodium selenite (solid and in solution), selenium sulfide, selenium dioxide, dimethyl selenide in solution, selenomethionine, S-methyl seleno l-cysteine, seleno-dl-cystine, selenocystamine, selenodiglutathione (RSSeSR) in solution, selenourea, and selenoguanosine. Selenodiglutathione was prepared by mixing sodium selenite and glutathione with a molar ratio of 1:4 in a dilute HCl solution (pH 1.3) (10). A bacterial pellet of R. metallidurans CH34 exposed to selenite over 10 days, and which was shown to contain monoclinic (red) Se0 (24), was used as a reference for this compound. The other compounds were purchased from Sigma-Aldrich. Freeze-dried bacteria were ground in an agate mortar and diluted with glucose when necessary. The mixture was pressed into 5-mm-diameter pellets prior to determining XANES measurements.
The spectra were recorded at room temperature in fluorescence mode, using a 30-element solid-state Ge detector (Canberra), for the most diluted bacterial samples and in transmission mode, using a diode, for the more concentrated samples. The monochromator was a Si(220) double crystal. Two to four scans of 10 min were summed, depending on Se concentration. The position of the beam on the pellet was changed between each scan in order to limit radiation damage. Hexagonal Se (0) was recorded simultaneously, and the spectra were energy calibrated by setting the energy of the maximum of the white line for this reference spectrum at 12.6592 keV. XANES spectra were normalized using polynomial functions of degrees 1 and 3 for the pre-and postedge parts, respectively. Each set of spectra for a given kinetics experiment was treated by using principal component analysis (PCA) (17, 30). This approach allows the determination of the number of Se species present in a set of samples and to identify these species, using a library of reference spectra. The number of principal components was determined based on the eigenvalue of each component and on the quality of the reconstructed spectra by using one, two, three, or more components with the total normalized sum-squares residual (total NSS):
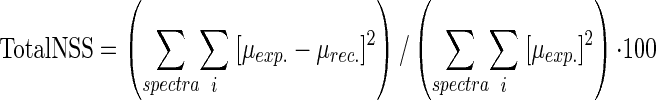 |
(1) |
where μexp. is the experimental normalized absorbance and μrec. is the reconstructed normalized absorbance. The principal components were identified by target transformation using the NSS criterion as follows:
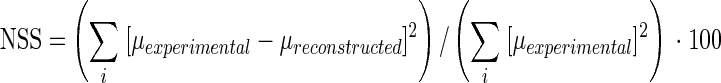 |
(2) |
The percentage of each species, in molar fraction of Se, was then determined by linear combination fitting of each spectrum with the spectra of the identified reference materials. The precision was estimated at ±5% of total Se. The percentages were then multiplied by total Se content, as determined by ICP-MS, in order to obtain the concentrations of each species (mg of Se per g of proteins).
RESULTS
The sensitivity of Se K-edge XANES spectroscopy for probing the oxidation state of Se is well established (20, 21). Figure 1 displays some of the reference spectra used in this study. The main peak of SeIV and SeVI is shifted by +4.5 eV and +7.5 eV, respectively, relative to that of Se0. The energy shift is much smaller for organoselenium compounds (+0.4 to +1.5 eV relative to that of Se0, depending on the type of compound). The position of the main peak is identical for compounds with similar Se environments, for instance, S-methyl selenocysteine and selenomethionine for alkyl selenide (RSeR), seleno-dl-cystine and selenocystamine for alkyl diselenide (RSeSeR), and selenourea and selenoguanosine for the Se-C double bond (not shown). The energy shift between two types of Se local structures, for instance, RSeSeR and RSSeSR, can be as small as 0.5 eV. The sensitivity of XANES is probably not sufficient to determine the distribution of several types of organic Se in a complex mixture, but it certainly enables the identification of the major organoselenium species.
FIG. 1.

XANES spectra for some reference compounds (in solid state unless otherwise specified). From top to bottom: sodium selenate, sodium selenite, selenourea, S-methyl seleno l-cysteine, seleno-dl-cystine, selenodiglutathione (in solution), red and gray elemental selenium. The position of the maximum of the white line is indicated in parentheses.
Selenite exposure.
In the first experiment, a culture of R. metallidurans CH34 was exposed to 2 mM selenite added at the beginning of growth (A600 = 0.3). As described previously (24), the presence of selenite induced an increase in the lag phase duration (approximately 48 h compared to 10 h in the absence of selenite). The accumulation of selenite was minimal during this phase (<40 mg Se per g of protein). However, at the end of the exponential phase, and during the stationary phase, selenium was strongly accumulated: at t = 144 h, Se accounted for one-third of the protein weight (Fig. 2).
FIG. 2.

Concentration of Se species in R. metallidurans CH34 exposed to selenite as determined by XANES linear combination fitting and ICP-MS analyses and time course of growth for the bacteria exposed to selenite (open circles). Error bars correspond to ±5% of total Se.
Selected XANES spectra for the bacteria at various exposure times are shown in Fig. 3. The position of the main peak for t = 0 h at 12.6637 keV and the presence of a shoulder on the left-hand part of the peak suggested that the bacteria contain selenite and a minor proportion of organoselenium. At higher exposure times (t = 1 h to 48 h), the spectra were identical and the maximum of the main peak was intermediate between RSeR and Se0 (12.6599 keV). The main peak was then slightly shifted to the left at t = 96 h (12.6596 keV) and matched the position of Se0 at t = 120 and 144 h (12.6592 keV). PCA showed that this set of spectra could be described by three components (eigenvalues of 80.0, 4.1, and 1.1). As expected, selenite and red Se0 were positively identified as principal components (NSS values of 4.7 10−3 and 2.9 10−4, respectively). Several organoselenium species including RSeR, RSeSeR, and RSSeSR were also correctly reconstructed (NSS values of 1.2 10−4 to 4.3 10−4). Among these five compounds retained, the most likely triplet of primary components should provide the best simulation of the whole set of bacterial spectra by linear combinations of these three spectra. Thus, all possible triplets were tested and selenite, red Se0, and RSeR provided the best results. Fits using RSeSeR, or RSSeSR instead of RSeR, were poorer, as shown by increases of the residual by 27%, 34%, and 47%, respectively. The fact that these species were correctly reconstructed by PCA is due to their intermediate position betweeen Se0 and RSeR (Fig. 1). The occurrence of selenocysteine (RSeH) was not tested since this compound reoxidized to selenocystine during the experiment. However, preliminary results obtained by high-performance liquid chromatography support the predominance of the RSeR form (L. Avoscan, R. Collins, G. Sarret, M. Carrière, J. Covès, and B. Gouget, Abstr. 227th ACS Natl. Meet., abstr. 016, p. U1107, 2004). In conclusion, alkyl selenide is believed to be the dominant organic form of Se in the bacteria exposed to selenite.
FIG. 3.

Selected Se K-edge XANES spectra for R. metallidurans CH34 at various incubation times after introduction of selenite into the culture medium at A600s (optical densities [O.D.]) of 0.3, 1, and 3 and the distribution of Se species determined by linear combination fitting.
Figure 2 shows the evolution in the concentration of Se species in the bacteria during growth. Immediately after the introduction of selenite, Se was distributed as 60% SeIV and 40% RSeR in the bacteria. This latter species is likely to be a reaction product of selenite rather than constitutive selenium contained in amino acids and/or proteins since control bacteria, not exposed to selenite, did not yield a detectable Se XANES signal. Subsequently, until the end of the lag phase (t = 1 h to 48 h), a mixture of RSeR and Se0 in equivalent proportions was observed. The concentration of RSeR was almost stable from 48 to 96 h (18 and 15 mg Se g−1 of proteins, respectively), whereas the Se0 concentration strongly increased (20 and 140 mg Se g−1 of proteins, respectively). At 120 and 144 h, Se0 was the only species detected. The amount of organoselenium species identified at 96 h might still be present in these two samples but was masked by the dominant Se0 form (estimated error of ±5% of total Se, i.e., 13 mg g−1 at 120 h and 17 mg g−1 at 144 h).
In parallel experiments, selenite was added at approximately mid-exponential phase (A600 = 1) and at the beginning of the stationary phase (A600 = 3). Similar evolutions of Se accumulation and speciation were observed: Se uptake was limited for several hours and then increased. RSeR and Se0 were observed during the slow uptake period, followed by Se0 only (Fig. 3). However, the production of Se0 was faster at higher A600 values. For instance, after 48-h exposure, the bacteria contained 99.4 mg g−1 Se0 compared to 19.8 mg g−1 Se0 when selenite was added at A600s of 1 and 0.3, respectively.
Selenate exposure.
In this experiment, the bacteria were exposed to 2 mM selenate at the beginning of the growth (A600 = 0.3) (Fig. 4). The highest accumulation also occurred during the exponential phase, but this oxyanion was much less accumulated than selenite (the maximum concentration is 14 compared to 340 mg of Se g−1 of proteins for selenite). In contrast with the selenite experiment, the presence of selenate did not increase the lag phase duration. During the exponential phase, bacteria in selenate-complemented media grew at rates comparable to those of bacteria grown in selenite-free media and maximal densities were very similar whether or not selenate was present in the culture medium (Fig. 4). The experiment was stopped after 72 h since the bacterial population started to decrease, probably due to the depletion of nutrients in the medium (the same decrease was observed for the control culture). This was indicated by the decrease in absorbance and verified by the numeration of cells forming colonies on Luria broth agar (data not shown).
FIG. 4.

Concentration of Se species in R. metallidurans CH34 exposed to selenate as determined by XANES linear combination fitting and ICP-MS analyses and time course of growth for the bacteria exposed to selenate (open circles) and for the control culture in the absence of added selenium oxyanion (filled circles). Error bars correspond to ±5% of total Se.
XANES spectra for the bacteria at various selenate exposure times are shown in Fig. 5. The PCA of this set of spectra showed that three Se species were present (eigenvalues of 73, 4.9, and 1.9): SeVI, SeIV, and organoselenium (NSS values of 3.7 10−3, 3.3 10−3, and 2.7 10−4 to 6.6 10−4, respectively). Se0 could not be considered a principal component (NSS value of 1.2 10−2). Using the same procedure as for the selenite experiment, we found that RSeR was the most likely organoselenium species. SeVI was detected in the bacterial pellets immediately after its introduction to the culture medium (t = 0 h) and at t = 12 h (Fig. 5). At t = 12 h, the bacteria contained SeIV, SeVI, and RSeR. The simulation of the spectrum for t = 24 h was significantly improved by adding Se0 to the simulation (NSS value decreased by 27%), although this species was not a principal component of the system. The occurrence of only 8% Se0 in one particular sample explains why it was not detected as principal component of the system. At longer exposure times, RSeR was the only species detected.
FIG. 5.

Selected Se K-edge XANES spectra for R. metallidurans CH34 at various incubation times after the introduction of selenate into the culture medium at A600s (optical densities [O.D.]) of 0.3 and 1 and the distribution of Se species determined by linear combination fitting.
In another experiment, selenate was added at mid-exponential phase (A600 = 1). The same evolution in the speciation of selenium was observed, although Se0 was more represented (up to 27% of total Se) (Fig. 5). The slight shift of the main peak for the spectrum at t = 6 h compared to at t = 168 h was indicative of this higher Se0 content (Fig. 5).
The composition of headspace gas was not investigated during the selenite and selenate experiments, but the formation of volatile methylated Se species is believed to be limited since >90% of the Se initially in solution could be accounted for upon ICP-MS analyses of the bacterial samples and solutions.
Two-contaminant (SeIV and SeVI) exposure.
As selenite is completely reduced to Se0, whereas selenate is not, although some selenite is produced, we have checked the hypothesis of the inhibition of the selenite reduction to Se0 by selenate. The bacteria were thus exposed to both selenite and selenate introduced at the same time or to one of these species first with the addition of the second one 3 h later. The day after exposure, the characteristic red color of Se0 was observed in the three experiments, both at equivalent selenite and selenate concentrations (2 mM) and with a 10-fold excess in selenate (1 mM selenite and 10 mM selenate) regardless of the order of introduction. Thus, the possible inhibition of selenite reduction by selenate could be ruled out.
DISCUSSION
The fast selenite uptake following several hours of slow uptake cannot be ascribed to the high metabolism of the cells during the exponential phase since the same profiles were observed when this oxyanion was added during the lag phase, during the exponential phase, or at the beginning of the stationary phase. This behavior might suggest the slow activation of some selenite transport system. To our knowledge, no specific selenite transporter has been characterized in microorganisms. In Escherichia coli, selenite can enter the cell through the sulfate transporter, but the repression of this carrier does not inhibit selenite uptake completely (28). In Rhodobacter sphaeroides, a polyol transporter is suggested as the transporting agent of selenite into the cytoplasm (4).
Could the organoselenium species observed during the period of slow uptake be an intermediate product of the formation of elemental selenium? For E. coli, selenite reduction can follow a nonenzymatic pathway involving glutathione, and organoselenium intermediates include RSSeSR and glutathioselenol (28). The fact that RSSeSR was not detected as transient species does not necessarily imply that this nonenzymatic reduction pathway does not exist in R. metallidurans CH34 (the occurrence of glutathioselenol was not tested since this compound was absent from our model compounds library). The nonenzymatic reduction of selenite is accompanied by the production of O2·− (14) and generates an oxidative stress. In the case of R. metallidurans CH34, the overexpression of an enzyme associated with oxidative stress, an iron-containing superoxide dismutase, has been recently observed in the presence of selenite (23). This might support the hypothesis that nonenzymatic reduction takes place in these bacteria. Alternatively, the RSeR species observed after selenite introduction might result from an assimilatory pathway. Such a pathway is thought to exist in bacteria since Se is a composite of some bacterial enzymes such as formate dehydrogenase (27). The most commonly (nonvolatile) alkyl selenide species found in microorganisms is selenomethionine (6). High-performance liquid chromatography analyses are under way to determine the exact nature of the RSeR species present in R. metallidurans CH34. To summarize, the presence of RSeR and Se0 species in equivalent amounts during the period of slow uptake suggests that selenite is accumulated through two competing pathways, an assimilatory pathway and a slow detoxification pathway, with both having similar kinetics.
During the period of fast selenite uptake, the reduction pathway becomes predominant. These contrasting behaviors between the period of slow and fast uptake mirror what has been previously observed for R. sphaeroides at low and high selenite exposure: this bacterium metabolized selenite into approximately 60% RSeR and 40% Se0 after exposure to 1.6 mg liter−1 of selenite and produced almost 100% Se0 after exposure to 160 mg liter−1 selenite (29). For some bacteria, selenite reduction is mediated by a single enzyme: a periplasmic nitrite reductase (16) in Thauera selenatis and a nitrite or a nitrate reductase (NR) in Enterobacter cloacae (9, 15). The proteome analysis of R. sphaeroides exposed to selenite did not reveal the overexpression of a single enzyme capable of reducing selenite but did confirm the presence of some chaperones, an elongation factor, and some enzymes associated with oxidative stress (4). Garbisu et al. (11) showed that selenite reduction by Bacillus subtilis was not affected by an excess of nitrate, nitrite, sulfate, or sulfite in the medium and suggested that selenite was reduced by an inducible detoxification system different from N- and S-related reductases. The kinetics of selenite accumulation and transformation by R. metallidurans CH34 suggest the induction of some selenite uptake and reduction systems whose nature remains unknown. The putative induction of selenite transport and reduction, which takes several hours, can be qualified as slow compared to the induction of the mer operon by Hg2+, which takes a few seconds (3).
The uptake of selenate by R. metallidurans CH34 was strongly limited, and the bacterial growth was not affected by this oxyanion. This behavior is consistent with the general idea that selenate is slowly transported inside the cells via the sulfate permease system (13). Selenite was detected during the first 12 h after selenate introduction. Several types of enzymes have been shown to reduce selenate to selenite. In T. selenatis, this is done by a specific selenate reductase (16, 26). In E. coli, reduction is catalyzed by a molybdenum enzyme distinct from a nitrate reductase (5). Evidence for the role of a molybdo-enzyme in selenate reduction was also shown for Enterobacter cloacae (31). In vitro studies showed that several NR, including some membrane-bound NR of E. coli (1) and some membrane-bound and periplasmic NR of R. sphaeroides, Paracoccus denitrificans, Paracoccus pantotrophus, and Ralstonia eutropha DSM 428 (25), were able to reduce selenate under anaerobic conditions. Selenate might also be reduced by the enzymes of the sulfate assimilation pathway. Such a process is known to occur in higher plants (22), but there is no direct evidence for the role of sulfate-reducing enzymes in bacterial selenate reduction. For the moment, we have no indication of the selenate-reducing agent in R. metallidurans CH34. The occurrence of selenite is followed by a mixture of Se0 and RSeR and then by RSeR only. The absence of Se0 at t = 72 h can be explained by the high RSeR content, which may mask Se0 (see the error bar in Fig. 4).
In summary, our results show that selenate is partly reduced to Se0 but that the main process is the transformation and accumulation of an RSeR-like organoselenium compound. A similar fate for selenate was observed in R. sphaeroides at both low and high selenate concentrations (29). de Souza et al. (8) found that selenate-treated Halomonas bacteria accumulate selenate and a minor selenomethionine-like species and suggested that selenate follows the sulfate assimilation pathway.
These results raise the question of why selenite and selenate follow different pathways, provided that selenate is first reduced to selenite. This study shows that reduction and assimilation pathways are taken by both oxyanions and that the former pathway seems to be activated upon selenite exposure only. The possible inhibition of the reduction of selenite to Se0 by selenate can be ruled out since the bacteria exposed to both oxyanions still produced the red color indicative of Se0. The nonactivation of the reduction pathway upon selenate exposure could be related to the much smaller uptake of selenate relative to selenite, supposing that this pathway is activated above a threshold concentration of selenite or any chemical agent derived from selenite. In this study, maximum measured selenite concentrations were comparable upon selenite and selenate exposure (around 3 mg Se per g of proteins) (Fig. 2 and 4), but these values are only snapshot images, rather than a direct monitoring of the selenite content.
In conclusion, this study showed that both selenite and selenate follow an assimilatory and a detoxification pathway in R. metallidurans CH34 and that transport and reduction are activated upon selenite exposure. The capacity of this bacterium to accumulate and reduce large amounts of selenite may qualify this strain as suitable for the bioremediation of selenite-contaminated soils, sediments, and waters. However, the same is not true for selenate, since the organoselenium species produced may represent some mobile and bioavailable forms of selenium. This study illustrates the potential of XANES spectroscopy combined with elemental analyses, which enable the quantification of Se species. This spectroscopic approach is complementary to analytical speciation techniques such as liquid or ionic chromatography or electrospray mass spectrometry (6), which are better suited to identify individual molecules.
Acknowledgments
We acknowledge J. L. Hazemann and O. Proux for their assistance during XANES measurements and the ESRF for the provision of beam time.
This research was supported by the CNRS and CEA through the Programme National Toxicologie Nucléaire Environnementale.
REFERENCES
- 1.Avazéri, C., R. Turner, J. Pommier, J. Weiner, G. Giordano, and A. Verméglio. 1997. Tellurite reductase activity of nitrate reductase is responsible for the basal resistance of Escherichia coli to tellurite. Microbiology 143:1181-1189. [DOI] [PubMed] [Google Scholar]
- 2.Avoscan, L., M. Carrière, F. Jehanneuf, R. Collins, F. Carrot, J. Covès, and B. Gouget. 2004. Ralstonia metallidurans CH34 resistance to selenium oxyanions: growth kinetics, bioaccumulation and reduction, p. 267-271. In J. A. Centeno, P. Collery, and G. Vernet (ed.), Metal ions in biology and medicine, vol. 8. John Libbey Eurotext, Montrouge, France.
- 3.Barkay, T., S. Miller, and A. Summers. 2003. Bacterial mercury resistance from atoms to ecosystems. FEMS Microbiol. Rev. 27:355-384. [DOI] [PubMed] [Google Scholar]
- 4.Bebien, M., J.-P. Chauvin, J.-M. Adriano, S. Grosse, and A. Verméglio. 2001. Effect of selenite on growth and protein synthesis in the phototrophic bacterium Rhodobacter sphaeroides. Appl. Environ. Microbiol. 67:4440-4447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bébien, M., J. Kirsch, V. Méjean, and A. Verméglio. 2002. Involvement of a putative molybdenum enzyme in the reduction of selenate by Escherichia coli. Microbiology 148:3865-3872. [DOI] [PubMed] [Google Scholar]
- 6.Chasteen, T., and R. Bentley. 2003. Biomethylation of selenium and tellurium: microorganisms and plants. Chem. Rev. 103:1-25. [DOI] [PubMed] [Google Scholar]
- 7.Combs, G. F., Jr., C. Garbisu, B. C. Yee, A. Yee, D. E. Carlson, N. R. Smith, A. C. Magyarosy, T. Leighton, and B. B. Buchanan. 1996. Bioavailability of selenium accumulated by selenite-reducing bacteria. Biol. Trace Elem. Res. 52:209-225. [DOI] [PubMed] [Google Scholar]
- 8.de Souza, M. P., A. Amini, M. A. Dojka, I. J. Pickering, S. C. Dawson, N. R. Pace, and N. Terry. 2001. Identification and characterization of bacteria in a selenium-contaminated hypersaline evaporation pond. Appl. Environ. Microbiol. 67:3785-3794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dungan, R. S., and W. T. Frankenberger. 1998. Reduction of selenite to elemental selenium by Enterobacter cloacae SLD1a-1. J. Environ. Qual. 27:1301-1306. [Google Scholar]
- 10.Ganther, H. 1968. Selenotrisulfides. Formation by the reaction of thiols with selenious acid. Biochemistry 7:2898-2905. [DOI] [PubMed] [Google Scholar]
- 11.Garbisu, C., S. Gonzalez, W. H. Yang, B. C. Yee, D. L. Carlson, A. Yee, N. R. Smith, R. Otero, B. B. Buchanan, and T. Leighton. 1995. Physiological mechanisms regulating the conversion of selenite to elemental selenium by Bacillus subtilis. Biofactors 5:29-37. [PubMed] [Google Scholar]
- 12.Haygarth, P. 1994. Global importance and global cycling of selenium, p. 1-27. In W. T. Frankenberger, Jr., and S. Benson (ed.), Selenium in the environment. Marcel Dekker, New York, N.Y.
- 13.Heider, J., and A. Böck. 1993. Selenium metabolism in micro-organisms. Adv. Microb. Physiol. 35:71-109. [DOI] [PubMed] [Google Scholar]
- 14.Kramer, G., and B. Ames. 1988. Mechanisms of mutagenicity and toxicity of sodium selenite (Na2SeO3) in Salmonella typhimurium. Mutat. Res. 201:169-180. [DOI] [PubMed] [Google Scholar]
- 15.Losi, M. E., and W. T. Frankenberger. 1998. Reduction of selenium oxyanions by Enterobacter cloacae strain SLDIa-I, p. 515-544. In W. T. Frankenberger and R. A. Engberg (ed.), Environmental chemistry of selenium, vol. 64. Marcel Dekker, New York, N.Y.
- 16.Macy, J. M. 1994. Biochemistry of selenium metabolism by Thauera selenatis gen. nov. sp. nov. and use of the organism for bioremediation of selenium oxyanions in San Joaquin Valley drainage water, p. 421-444. In W. T. Frankenberger, Jr., and S. Benson (ed.), Selenium in the environment. Marcel Dekker, New York, N.Y.
- 17.Manceau, A., M. A. Marcus, and N. Tamura. 2002. Quantitative speciation of heavy metals in soils and sediments by synchrotron X-ray techniques, p. 341-428. In P. Fenter, M. Rivers, N. Sturchio, and S. Sutton (ed.), Applications of synchrotron radiation in low-temperature geochemistry and environmental science, vol. 49. Reviews in mineralogy and geochemistry, Mineralogical Society of America, Washington, D.C.
- 18.Mergeay, M., S. Monchy, T. Vallaeys, V. Auquier, A. Benotmane, P. Bertin, S. Taghavi, J. Dunn, D. van der Lelie, and R. Wattiez. 2003. Ralstonia metallidurans, a bacterium specifically adapted to toxic metals: towards a catalogue of metal-responsive genes. FEMS Microbiol. Rev. 27:385-410. [DOI] [PubMed] [Google Scholar]
- 19.Mergeay, M., D. Nies, H. G. Schlegel, J. Gerits, P. Charles, and F. Van Gijsegem. 1985. Alcaligenes eutrophus CH34 is a facultative chemolithotroph with plasmid-bound resistance to heavy metals. J. Bacteriol. 162:328-334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pickering, I., G. George, V. Van Fleet-Stalder, T. Chasteen, and R. Prince. 1999. X-ray absorption spectroscopy of selenium-containing amino acids. J. Biol. Inorg. Chem. 4:791-794. [DOI] [PubMed] [Google Scholar]
- 21.Pickering, I. J., G. E. Brown, and T. K. Tokunaga. 1995. Quantitative speciation of selenium in soils using X-ray absorption spectroscopy. Environ. Sci. Technol. 29:2456-2459. [DOI] [PubMed] [Google Scholar]
- 22.Pilon-Smits, E. A. H., S. B. Hwang, C. M. Lytle, Y. L. Zhu, J. C. Tai, R. C. Bravo, Y. C. Chen, T. Leustek, and N. Terry. 1999. Overexpression of ATP sulfurylase in Indian mustard leads to increased selenate uptake, reduction, and tolerance. Plant Physiol. 119:123-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roux, M., and J. Covès. 2002. The iron-containing superoxide dismutase of Ralstonia metallidurans CH34. FEMS Microbiol. Lett. 210:129-133. [DOI] [PubMed] [Google Scholar]
- 24.Roux, M., G. Sarret, I. Pignot-Paintrand, M. Fontecave, and J. Covès. 2001. Mobilization of selenite by Ralstonia metallidurans CH34. Appl. Environ. Microbiol. 67:769-773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sabaty, M., C. Avazeri, D. Pignol, and A. Vermeglio. 2001. Characterization of the reduction of selenate and tellurite by nitrate reductases. Appl. Environ. Microbiol. 67:5122-5126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schröder, I., S. Rech, T. Krafft, and J. M. Macy. 1997. Purification and characterization of the selenate reductase from Thauera selenatis. J. Biol. Chem. 272:23765-23768. [DOI] [PubMed] [Google Scholar]
- 27.Stadtman, T. 1996. Selenocysteine. Annu. Rev. Biochem. 65:83-100. [DOI] [PubMed] [Google Scholar]
- 28.Turner, R. J., J. H. Weiner, and D. E. Taylor. 1998. Selenium metabolism in Escherichia coli. Biometals 11:223-227. [DOI] [PubMed] [Google Scholar]
- 29.Van Fleet-Stalder, V., T. G. Chasteen, I. J. Pickering, G. N. George, and R. C. Prince. 2000. Fate of selenate and selenite metabolized by Rhodobacter sphaeroides. Appl. Environ. Microbiol. 66:4849-4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wasserman, S. R., P. G. Allen, D. K. Shuh, J. J. Bucher, and N. M. Edelstein. 1999. EXAFS and principal component analysis: a new shell game. J. Synchrotron Rad. 6:284-286. [DOI] [PubMed] [Google Scholar]
- 31.Watts, C., H. Ridley, K. Condie, J. Leaver, D. Richardson, and C. Butler. 2003. Selenate reduction by Enterobacter cloacae SLD1a-1 is catalysed by a molybdenum-dependent membrane-bound enzyme that is distinct from the membrane-bound nitrate reductase. FEMS Microbiol. Lett. 228:273-279. [DOI] [PubMed] [Google Scholar]