

Abstract
Distinct morphologies of the mitochondrial network support divergent metabolic and regulatory processes that determine cell function and fate1–3. The mechanochemical GTPase optic atrophy 1 (OPA1) influences the architecture of cristae and catalyses the fusion of the mitochondrial inner membrane4,5. Despite its fundamental importance, the molecular mechanisms by which OPA1 modulates mitochondrial morphology are unclear. Here, using a combination of cellular and structural analyses, we illuminate the molecular mechanisms that are key to OPA1-dependent membrane remodelling and fusion. Human OPA1 embeds itself into cardiolipin-containing membranes through a lipid-binding paddle domain. A conserved loop within the paddle domain inserts deeply into the bilayer, further stabilizing the interactions with cardiolipin-enriched membranes. OPA1 dimerization through the paddle domain promotes the helical assembly of a flexible OPA1 lattice on the membrane, which drives mitochondrial fusion in cells. Moreover, the membrane-bending OPA1 oligomer undergoes conformational changes that pull the membrane-inserting loop out of the outer leaflet and contribute to the mechanics of membrane remodelling. Our findings provide a structural framework for understanding how human OPA1 shapes mitochondrial morphology and show us how human disease mutations compromise OPA1 functions.
Eukaryotic cells sequester specialized biochemical reactions into discrete membranous and non-membranous compartments. For membrane-delimited organelles, fusion and fission reactions driven by protein and lipid catalysts spatially organize the cell and its energy gradients. Fusion and fission reactions are also necessary for differentiation and communication within and between cells6–8. These principles are evident in organelles such as mitochondria, which are organized into dynamically interconnected tubules. The morphology of the mitochondrial network emerges from the balance of membrane fusion and fission reactions and therefore governs energy homeostasis and many additional functions9–12. A key protein in the modulation of mitochondrial shape and connectivity is OPA1—a mechano-chemical GTPase13,14. OPA1 catalyses the fusion of the mitochondrial inner membrane (IM) and shapes the dynamic structure of cristae4,5. These activities influence oxidative phosphorylation (OXPHOS), apoptosis, reactive oxygen species production and mitochondrial DNA maintenance4,5,15,16. Moreover, OPA1 is essential for the formation and function of differentiated cells, especially skeletal and cardiac muscle17, blood vessels18 and neurons19. OPA1 deficiency leads to mitochondrial dysfunction and cardiomyopathy20, metabolic stroke21, autosomal dominant optic atrophy13,14, Behr syndrome22 and other inherited conditions, including diverse neuropathies23.
In humans, eight isoforms of the OPA1 precursor are expressed and directed to the mitochondrial intermembrane space through a mitochondrial-targeting sequence (MTS)24. Inside the mitochondria, the MTS is cleaved away to produce the long form (L-OPA1), which is N-terminally anchored to the IM. Regulated proteolysis can generate the short form (S-OPA1), which lacks the transmembrane (TM) domain, and this proteolytic cleavage is essential for IM fusion25,26. Both the long and short isoforms, which assemble and co-assemble into oligomers to remodel membranes together, are necessary for the balanced organization of the mitochondrial network26. Thus, OPA1 assemblies in cells comprise heterogeneous proteins with variance arising from different splicing patterns, proteolysis patterns and post-translational modifications26–28. OPA1 also interacts with several other mitochondrial proteins, including the core MICOS component MIC60 and the rhomboid protease PARL, that further modulate its membrane remodelling properties29,30.
The activity of OPA1 is uniquely modulated by membrane binding. Previous studies showed that OPA1 preferentially binds to membranes containing cardiolipin (CL) and that CL enhances GTP hydrolysis activity27. This protein–lipid interaction is necessary for membrane fusion and crista shape, therefore highlighting the critical role of CL in mitochondrial morphology. Despite advances in structural and mechanistic studies of OPA1 and its homologues31–34, the CL-dependent membrane assembly and remodelling mechanism remain unclear. Here we sought to understand how conformational changes in OPA1 are transduced to its paddle domain (PD) and how the PD both perturbs bilayer structure and modulates the OPA1’s membrane-remodelling activities. To better define the subunit arrangement, membrane-binding sites and the molecular mechanism of OPA1-mediated membrane remodelling, we reconstituted human S-OPA1 on CL-enriched membranes to determine the structures of distinct S-OPA1 conformations that vary in curvature and membrane perturbations. We validated these structural models by comparing structure-based and disease-associated mutations with biochemical analyses in vitro and by measuring the activity of OPA1 variants in living cells.
To visualize how OPA1 molecules were arranged in the filaments, and how their architecture relates to membrane remodelling, we elucidated the structures of the human S-OPA1 assembled on the membrane using a combination of electron cryo-microscopy (cryo-EM) and helical reconstruction, cross-linking mass spectrometry (CX-MS) and molecular modelling (Fig. 1, Extended Data Figs. 1–4 and Supplementary Fig. 1). Cryo-EM 2D class averages revealed that the α-helical stalk domains are arranged as a tight helical bundle, whereas the GTPase domains appeared as a diffuse density, indicating substantial conformational heterogeneity (Extended Data Fig. 2b). We found that all of the segments showed similar subunit arrangements in three-dimensional (3D) reconstructions with the membrane-bound polymers displaying a wide range of inner leaflet diameters (from 7 nm to 22 nm) (Extended Data Figs. 2 and 3). 3D helical reconstruction of the most abundant diameter yielded a three-start helical map. Subsequent focused 3D classification and refinement resulted in a more homogeneous subset of particles that enabled the determination of the S-OPA1 structure to an overall resolution of 4.8 Å, enabling molecular modelling (Fig. 1 and Extended Data Figs. 2 and 3). This reconstruction has an approximately 49 nm outer diameter and a luminal inner leaflet diameter of around 19.3 nm (Extended Data Fig. 2c–f). Moreover, our analysis of a minor population in the dataset revealed a second state of the membrane-bound S-OPA1 filament in which the inner lumen distance was constricted to about 7.6 nm (Extended Data Fig. 2g–j). A similar workflow resulted in a 6.8 Å reconstruction of S-OPA1 in a second conformation, in which the density map shows secondary structure features, particularly for the bundle signalling element (BSE), stalk, membrane-associated PDs and some of the GTPase domain (Extended Data Figs. 2 and 3). Both states are narrower than the previously reported reconstructions of OPA1 and fungal orthologue MGM1 helical assemblies32,35. These results indicate that OPA1 lattices are flexible and can adopt multiple low-energy conformations under our solution conditions and when bound to CL-rich membranes (Extended Data Figs. 2 and 3).
Fig. 1 |. Model of the human S-OPA1 bound to membranes.
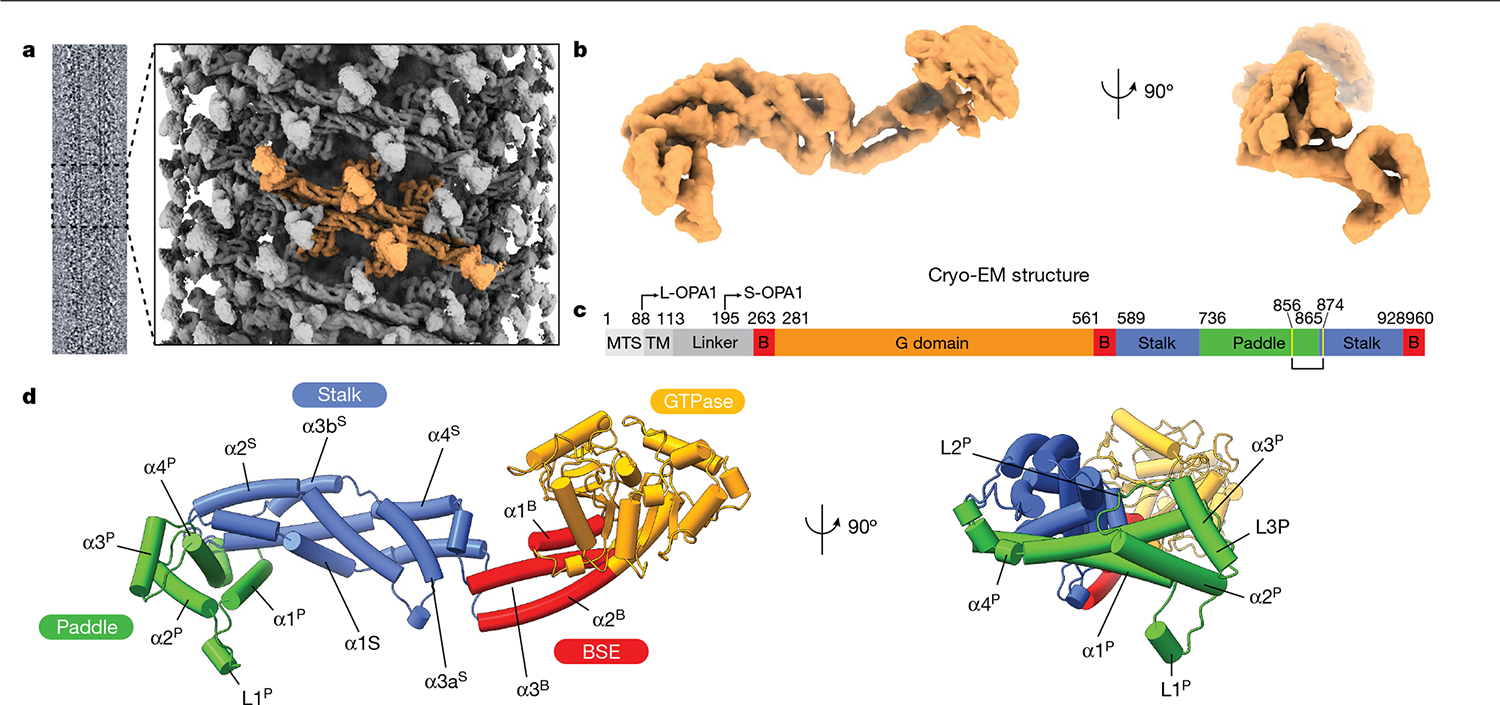
Lipid-binding triggers the formation of higher-order OPA1 assemblies. a, Cryo-EM image of the S-OPA1–membrane assembly and a side view of the cryo-EM density map of the membrane-bound S-OPA1 assembled in the presence of GMPPCP. Four S-OPA1 subunits forming a tetrameric assembly are coloured in orange. b, Side and back views of the molecular structure of the isolated S-OPA1 subunit. c, Schematic of the domain organization of full-length human OPA1. TM, transmembrane domain; B, BSE. L- and S-OPA1 describe the transmembrane-anchored long-form and proteolytically processed short form. Cysteine residues that form a disulfide bond are highlighted in yellow. d, Corresponding views of the structural model of the membrane-bound S-OPA1. Domains are coloured individually here and in all of the subsequent figures as in c. The GTPase, BSE and stalk domains are a conserved hallmark of the dynamin-related protein family. The GTPase domain consists of a central β-sheet surrounded by α-helices. The BSE was built of an extended three-helical bundle (α1B–α3B) that displays an amphipathic character, whereby the hydrophobic residues pack into a core and charged residues are accessible to the solvent. At the N terminus, the GTPase domain is connected to the stalk domain by the α1B of the BSE. The stalk domain of OPA1 consists of four antiparallel α-helices forming an extended bundle (α1S–α4S). The conformational changes in these domains are transmitted to the lipid-binding PD to reinforce its ability to remodel membranes.
The structural organization of the OPA1 lattice was further validated by CX-MS data, which provide an important context for modelling while illuminating the conformational variability of the polymer. In total, we identified 127 cross-links that were specific to monomeric OPA1 (Extended Data Fig. 4). Another 13 crosslinks were gained in the polymeric structure, and these corroborated the interactions observed between neighbouring S-OPA1 molecules in the membrane-bound conformation (Extended Data Fig. 4c). The BSE and stalk were the most extensively cross-linked domains throughout the polymer, suggesting that membrane binding and associated repositioning of the stalk domains leads to stable interactions between subunits (Extended Data Fig. 4d).
S-OPA1 has a modular architecture comprising an amino-terminal GTPase domain followed by a BSE domain, a stalk domain and a lipid-binding PD (Fig. 1). Although the stalk domain exhibits a similar structure to the MGM1 protein, the elongated α3S of the MGM1 stalk is kinked further in the OPA1 structure and broken into two helices connected with a short linker (Fig. 1d and Extended Data Fig. 5). Moreover, MGM1 contains a long flexible loop (MGM1 residues 598–625) between α1S and α2S of the stalk at the C terminus that strongly interacts with the α2P and α3P of the PD and may constrain the movement of the PD helices during membrane remodelling. In OPA1, this loop is replaced by a short five-residue linker (residues 641–645) that forms minimal interactions with the PD (Fig. 1d and Extended Data Fig. 5). Despite its extensive hydrophobic core, the flexible regions between the stalk helices, and the loose interactions between the stalk and PD of OPA1, may provide a higher degree of structural freedom to adopt alternative conformations in different contexts. These results are also supported by the low sequence conservation between the two proteins (Extended Data Fig. 5d).
The mechanism by which dynamin-related proteins interacts with membranes is highly variable and remains poorly characterized. Our structure reveals that the OPA1 PD, which is inserted between stalk helices α3S and α4S, extends perpendicularly from the stalk domain, representing to our knowledge a previously undescribed architecture (Fig. 1). The PD adopts a distinct hammer shape with dimensions of around 60 Å × 40 Å × 28 Å and is composed of four α-helices (α1P–α4P) and three flexible loops (L1P–L3P) (Fig. 1). A single disulfide bond (Cys856–Cys874) that is located between the α4P core PD helix and α4S stalk helix stabilizes the interactions between the proximal region of PD and stalk helices. The elongated architecture of the OPA1 PD is unique within the dynamin-related protein family and distinguished from fungal protein MGM1 by an additional conserved loop (L1P) between α-helices α1P and α2P and a short α3P helix35,36 (Fig. 1, Extended Data Fig. 5 and Supplementary Fig. 2).
Notably, our structure revealed at least two regions within the PD—the proximal end of α1P and α4P helices plus the L1P loop—that are tightly associated with CL-containing bilayers (Fig. 2a–c). Notably, the L1P loop, which we refer to hereafter as the membrane-inserting loop (MIL), was embedded in the lipid bilayer (Fig. 2a–c). The cryo-EM map suggests that the α1P and α4P helices facilitate the docking of S-OPA1 molecules on the lipid bilayer, while the MIL anchors S-OPA1 into the remodelled membrane (Fig. 2a–c). To investigate the function of the PD overall and the unique features of the MIL, we first engineered an S-OPA1 construct in which the PD (Ile735 to Asp869) is replaced by an eight-residue glycine–serine linker (GGS–GGS–GG) (Extended Data Fig. 1). We next tested this construct, which we refer to as ΔPD, in a membrane-remodelling assay to measure oligomerization and liposome deformation activity using negative-stain transmission EM (TEM). As expected, the ΔPD construct lacked lipid-binding activity and could not remodel CL-containing liposomes (Extended Data Fig. 6). Furthermore, several residues in the docking region, particularly Lys738, Arg857 and Arg858, were required for S-OPA1 to bind to membranes, whereas other non-interface residues in the vicinity of the membrane, Phe860, Tyr861 and His866, had no apparent contribution to lipid binding by S-OPA1 (Extended Data Figs. 6). By contrast, a polyalanine mutation (residues 770 to 782) targeting the MIL along with single-residue mutations within the MIL, including W771A, K772E, R774E and R781E, completely abolished the membrane-binding and membrane-remodelling activity of S-OPA1 (Fig. 2d–g, Extended Data Fig. 6 and Supplementary Fig. 1). Notably, an alanine mutation to Trp775 did not affect the membrane-binding activity of S-OPA1, but impaired its ability to remodel membranes (Extended Data Fig. 6). This suggests that this residue lowers the energy barrier required for local membrane bending. Moreover, co-sedimentation experiments confirmed that the mutant proteins bind less strongly to CL-enriched liposomes compared with the wild type (WT) (Extended Data Fig. 7 and Supplementary Fig. 1). Mutational studies of the conserved MGM1 membrane interface residues Arg748 and Lys749, which are similarly positioned to the OPA1 MIL, resulted in comparable assembly defects and hindered the membrane-remodelling activity of S-MGM135.
Fig. 2 |. Cryo-EM reconstruction of membrane-bound S-OPA1 polymer showing protein–lipid interactions.
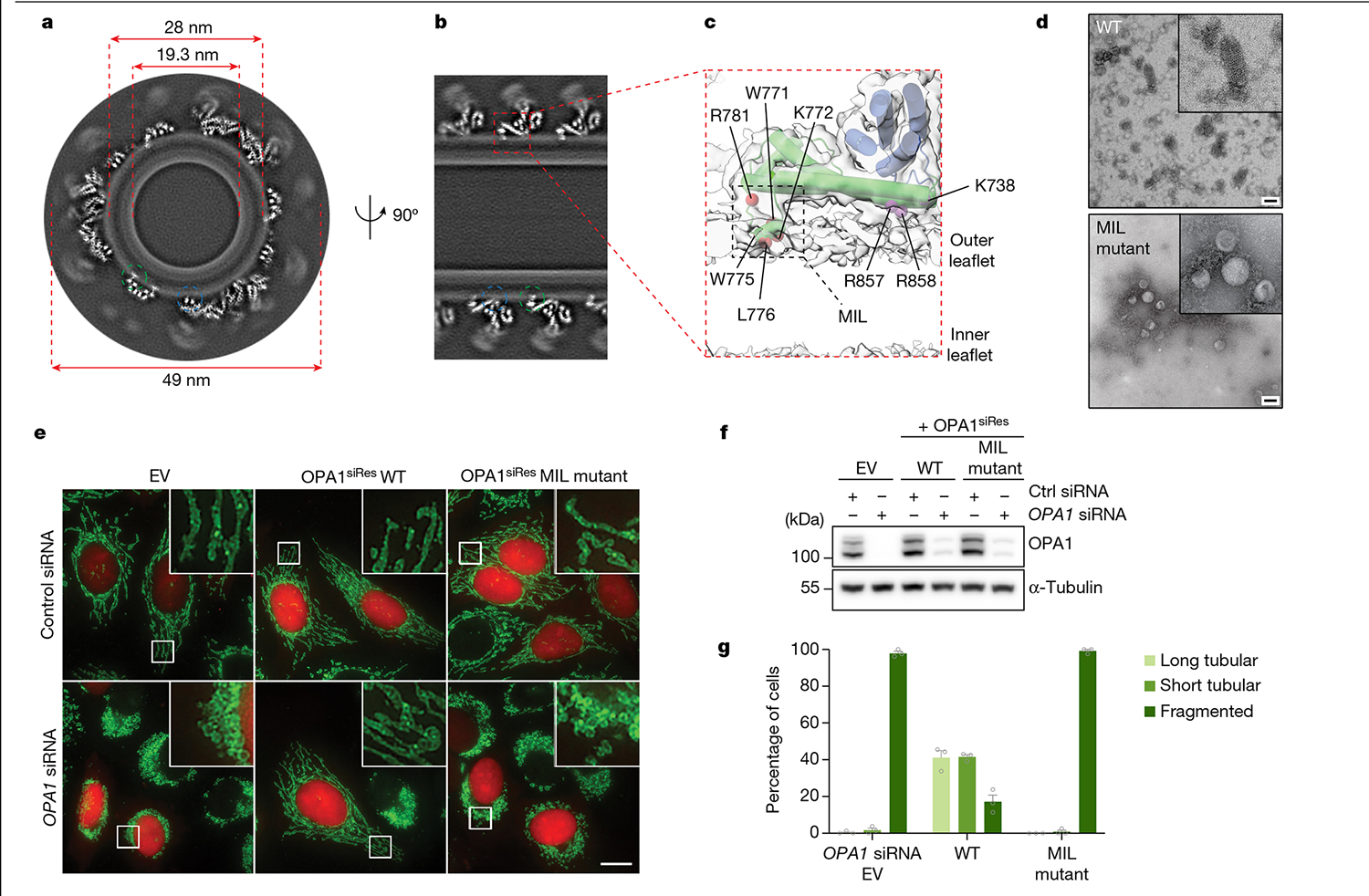
a, A grayscale top-down view of the central slices of the reconstruction. b, Side view of the central slices of the cryo-EM density map. Dashed circles indicate the MIL (green) and membrane-docking region (blue) of S-OPA1. c, Magnified view showing the membrane-docking and MIL regions of S-OPA1 inserted in the membrane. The area indicated by a box shows the position of the MIL. d, Negative-stain TEM analyses of the WT and polyalanine MIL mutant in the presence of CL-enriched liposomes. Inset: magnified views of selected liposomes. The experiment was performed in technical triplicate. Scale bars, 100 nm. e, Fluorescence microscopy images of the WT and MIL mutant in mitochondrial morphology analyses. HeLa cells were transfected with empty vector (EV) or the indicated short interfering RNA (siRNA)-resistant (siRes) OPA1 constructs and then processed for RNA interference using the indicated siRNAs. The mitochondrial network was visualized by staining for the outer-membrane protein TOMM22 (green), and co-transfected mCherry-NLS (red) to identify transfected cells. The experiment was performed in biological triplicate. Scale bar, 20 μm. f, The expression and stability of the siRNA-resistant OPA1 proteins were assessed by immunoblot analysis of lysates derived from transfected HeLa cells. n = 3 independent experiments. Mitochondrial phenotypes observed in HeLa cells transiently expressing human OPA1 variants as described in e. g, Quantification of microscopy images is reported as the average percentage of three biological replicates for three morphological phenotypes: WT (n = 238 cells), EV (n = 238 cells) and MIL mutant (n = 238 cells). The morphological phenotypes were categorized as long tubular morphology, showing elongated and connected phenotype; fragmented phenotype, showing the mitochondrial network being completely broken apart into small network pieces; and short tubular/intermediate phenotype, showing a few elongated pieces and not as fragmented. Data are the mean ± s.e.m. percentage of cells across three experimental replicates.
To assess whether our structure-based insights predict OPA1 loss-of-function phenotypes in living cells, we performed mutagenesis studies of the residues located in the PD and visualized the mitochondrial network morphology (Supplementary Figs. 1, 3 and 4 and Supplementary Data 1). Charge-reversal mutations of the α1P(K738E) and α4P(R858E) induced mitochondrial fragmentation and inhibited fusion (Extended Data Fig. 8 and Supplementary Fig. 1). On the other hand, the expression of the non-interface H866A mutant did not alter the mitochondrial network morphology (Supplementary Fig. 4e–g and Supplementary Data 2). While the polyalanine MIL mutant completely impaired the ability of OPA1 to elongate mitochondria (Fig. 2, Supplementary Fig. 5 and Supplementary Data 3), notable changes were observed with mutations to the specific residues within the MIL, including W775A, L776A and R781E, but not W771A (Extended Data Fig. 8). Moreover, we investigated the role of the OPA1 MIL in the development and stabilization of cristae. Cells expressing the MIL mutant displayed reduced numbers of cristae and lamellae compared with the WT cells. Ultrastructurally, expression of the MIL mutant disrupted tubular crista structure and resulted in more aberrant (short, swollen, disordered) morphologies (Extended Data Fig. 9). Examining the activity of the PD in living cells suggests that OPA1–membrane interactions are stabilized by the combined effects of charged and hydrophobic residues in this region. Consistent with these contacts and previously observed interactions32, mutations of the other conserved membrane interface residues (K797E, K800E, R824E) almost completely abrogated the membrane-binding and membrane-remodelling activity of S-OPA1 in vitro and, in the case of mutant R824E, hindered membrane fusion in living cells (Extended Data Figs. 6 and 8). Together, these cellular and structural observations of the OPA1–membrane interface reveal how OPA1 uses a pattern of charged and hydrophobic residues in this region to recognize and remodel CL-rich mitochondrial membranes in mammalian cells. Although the involvement of charged and hydrophobic residues in lipid association is similar to other dynamin-related proteins35,37–41, our models showed that the OPA1–membrane interface has a distinct residue composition.
The criss-cross association of the stalk domains through various interfaces are key determinants of higher-order assembly for dynamin-related proteins42–44, including human OPA1 (Supplementary Fig. 6). Unexpectedly, the human S-OPA1 structure revealed an intersubunit interface between the membrane-interacting PDs, which we refer to as interface 7 (Fig. 3a). We also identified two intersubunit crosslinks (Lys800–Lys800 and Lys797–Lys797) between the PDs, which are only satisfied in the polymeric assembly, confirming the interactions between neighbouring PDs (Extended Data Fig. 4c). The two-fold symmetry interface is formed by the α3P helix of the two opposing protomers and spans a buried solvent-accessible surface area of approximately 300 Å2 (Fig. 3a). The two protomers interact with each other through two sets of intermolecular salt bridges between the highly conserved Asp812 and Lys819 residues (Fig. 3a). A third critical residue, Thr816, makes additional contacts in the binding site and stabilizes the interactions between OPA1 molecules in the lattice as well as membranes (Fig. 3a).
Fig. 3 |. Interface 7 of the OPA1 assembly is essential for the regulation of mitochondrial morphology.
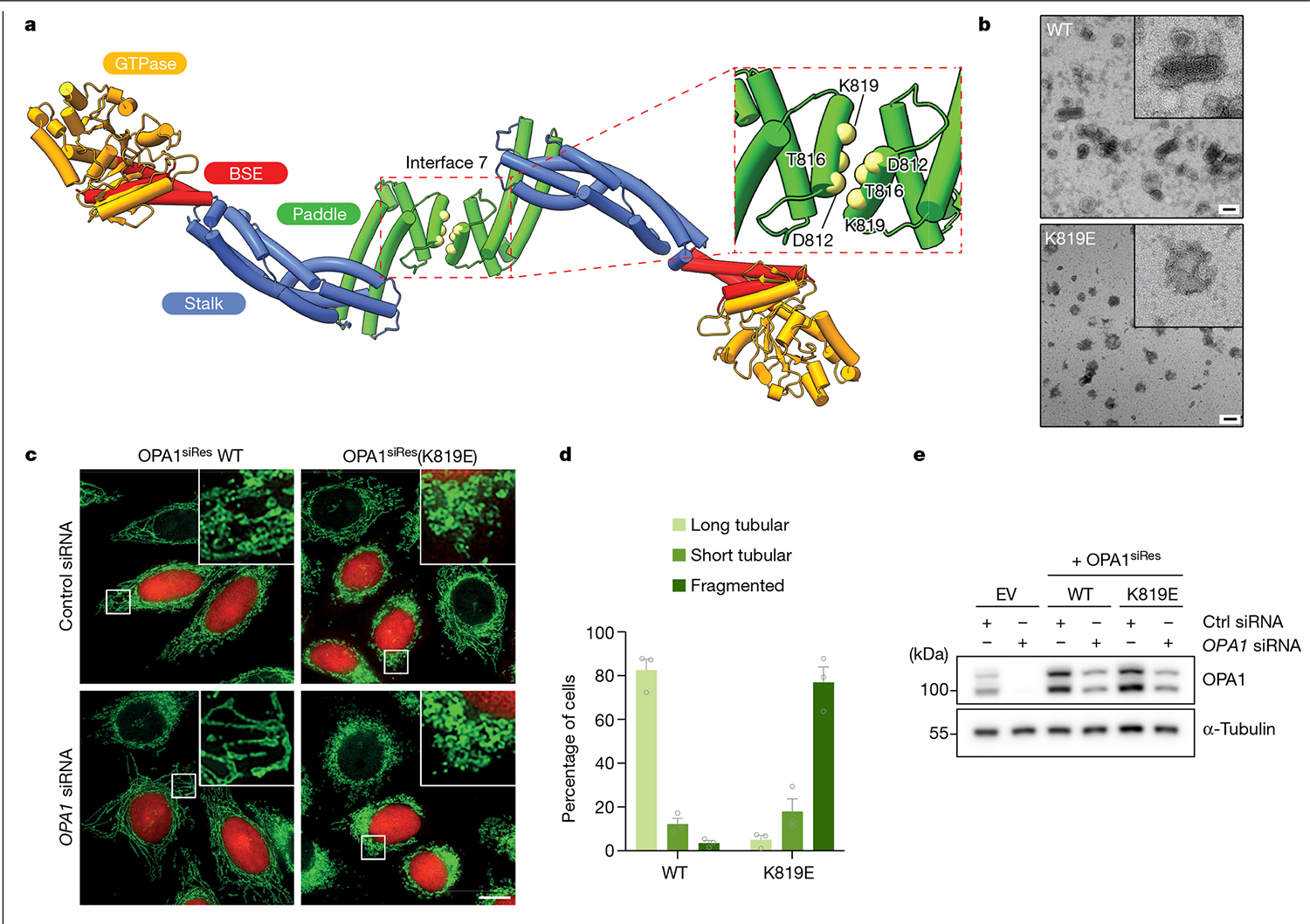
a, Top view of the structural model of the membrane-bound S-OPA1, revealing the dimerization of PDs. Top right, magnified view of the area indicated by the dashed box showing interface 7 between two PDs. α3P helix residues involved in interface 7 formation are shown as solid spheres (yellow) and numbered. b, Negative-stain TEM analyses of WT OPA1 and the OPA1(K819E) mutant in the presence of CL-enriched liposomes. Negative-stain electron micrographs show impaired liposome binding and remodelling activity of the OPA1(K819E) mutant compared with the WT. Inset, magnified views of selected liposomes. Scale bars, 100 nm. c, Fluorescence microscopy images of WT and interface-7-mutant OPA1(K819E) in mitochondrial morphology analyses. HeLa cells were transfected with the indicated siRNA-resistant OPA1 constructs and then subjected to RNA interference using the indicated siRNAs. The mitochondrial network was visualized by staining for the outer membrane protein TOMM22 (green), and co-transfected mCherry-NLS (red) was used to identify transfected cells. The experiment was performed in biological triplicate. Scale bar, 20 μm. d, Quantification of microscopy images of OPA1 WT and the K819E mutant. Mitochondrial phenotypes observed in HeLa cells transiently expressing human OPA1 variants as described in c. The average percentage of cells belonging to each of three morphological classifications across three experimental replicates. n = 238 cells expressing WT OPA1 over three experimental replicates and n = 283 cells expressing OPA1(K819E) over three experimental replicates. Data are the mean ± s.e.m. percentage of cells across three experimental replicates. e, Expression and stability of the siRNA-resistant OPA1 proteins were assessed by immunoblot analysis of lysates derived from HeLa cells transfected with either empty vector or the indicated siRNA-resistant OPA1 constructs before treatment with the indicated siRNAs. n = 3 independent experiments.
To investigate the functional importance of interface 7, we examined the membrane-remodelling activity of an OPA1(K819E) charge-reversal mutant in vitro and in living cells by visualizing the morphology of the mitochondrial network. We found that the K819E mutation completely abrogates membrane remodelling and results in severely fragmented mitochondrial morphology (Fig. 3b–e, Supplementary Figs. 1, 4e–g and 5 and Supplementary Data 2 and 3). Furthermore, the cristae of K819E-expressing cells exhibited a strongly aberrant architecture phenotype with reduced numbers of cristae per mitochondrion (Extended Data Fig. 9). Thus, the essential PD residue Lys819 at interface 7 promotes membrane remodelling to regulate both fusion and crista morphology in mammalian cells. When we generated a second construct lacking the entire α3P helix and the repulsive electrostatic interactions of the K819E mutant, we found that S-OPA1 Δα3P could bind to precurved membrane nanotubes but did not remodel spherical liposomes into cylinders in vitro (Extended Data Fig. 6). These findings demonstrate that the formation of interface 7 provides some of the free energy that is required for membrane bending during OPA1-mediated membrane fusion and crista shape generation.
In our cryo-EM analysis, we define the membrane-bound filaments, with an inner diameter of around 19.3 nm, as the membrane-proximal conformation of S-OPA1, where the PD is docked on the lipid bilayer and the MIL is deeply embedded in the membrane. Notably, the more constricted (around 7.6 nm) second conformation of the S-OPA1 polymer revealed that the entire S-OPA1 protomer, including the PD, was largely removed from the bilayer. We therefore describe this second state as the membrane-distal conformation of the helical filament (Fig. 4 and Extended Data Figs. 2g–j). Although the general domain arrangement in this second conformation is similar to the first conformation, the superimposition of the two states demonstrates informative differences, as indicated by the 2.3 Å root mean squared deviation over 698 Cα atoms (Fig. 4). First, the stalk domains are kinked further towards the membrane compared to the first structure (Fig. 4). Second, the BSEs undergo an opening exhibiting an unbent conformation that is widely separated from the stalk (Fig. 4). Third, a comparison of the membrane-distal and the membrane-proximal GTPase domain structures reveals that the GTPase domain moves about 20 Å towards neighbouring GTPase domains in the assembled polymer (Fig. 4). Notably, the GTPase domains facing one another do not form a dimer in the polymeric assembly (Figs. 1 and 4). The orientation of the GTPase domains in the membrane-distal conformation probably represents an intermediate state between membrane-stimulated oligomerization and nucleotide-dependent dimerization of the GTPase domain during membrane remodelling. These structural changes also corroborate the CX-MS analysis (Fig. 4b). Consistent with the large conformational change of the GTPase domain, a cross-link between the GTPase domain residue Lys372 and stalk residue Lys699 is only satisfied in the membrane-distal conformation, while three other crosslinks (Lys387–Lys568, Lys568–Lys705, Lys691–Lys800) were specific to the membrane-proximal state of S-OPA1 (Figs. 4a,b).
Fig. 4 |. Conformational changes in membrane-bound S-OPA1 result in the dissociation of the membrane-embedded fusion loop from the outer leaflet.
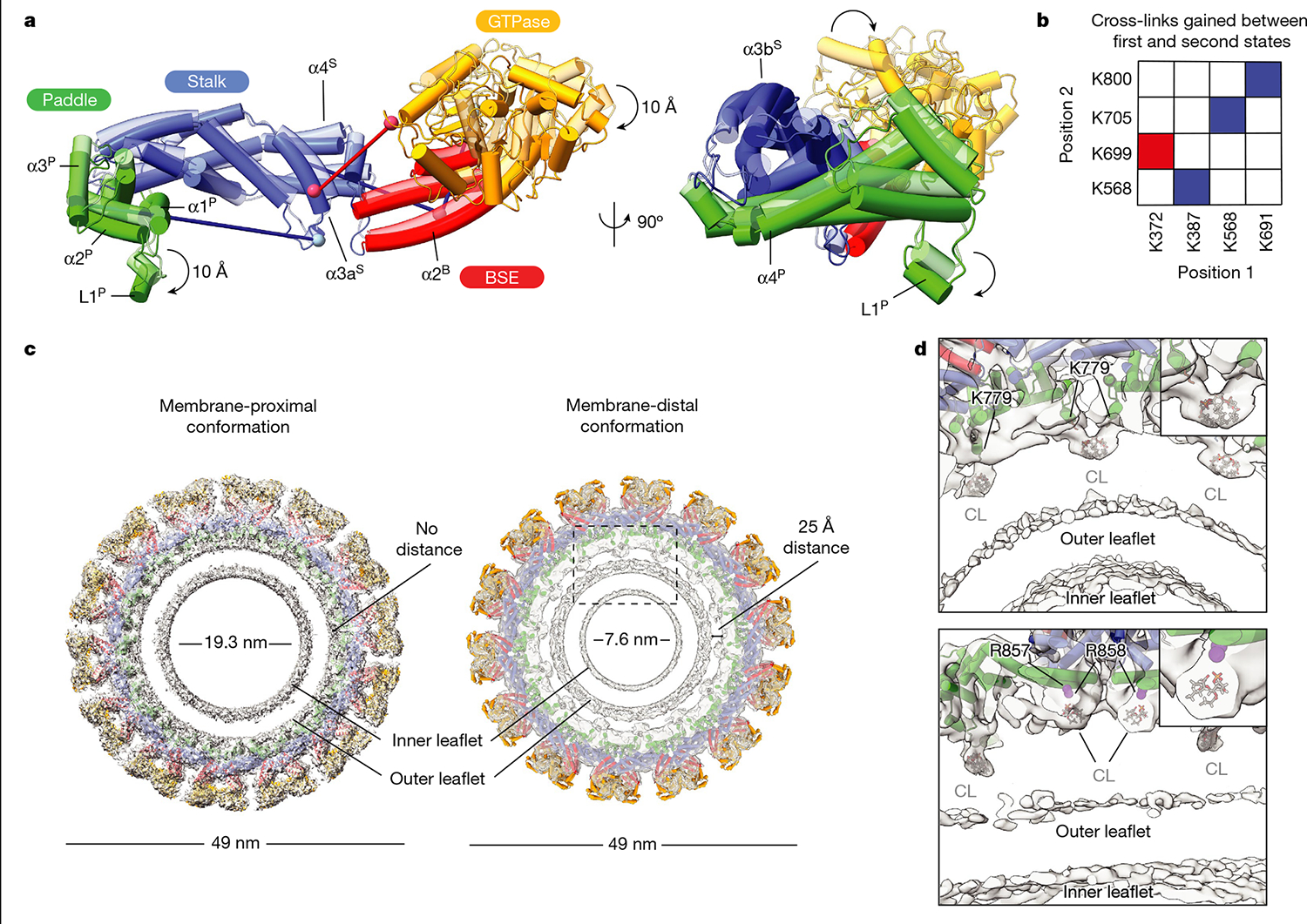
a, A comparison of membrane-proximal (transparent) and membrane-distal conformations (opaque) of S-OPA1 shows an approximately 20 Å shift in the GTPase domain towards a neighbouring GTPase domain density and an approximately 10 Å rotation of the PD L1P MIL towards the membrane. Additional conformational changes are also apparent for BSE and stalk domains. b, Cross-links that are only satisfied in membrane-proximal (blue) and membrane-distal (red) conformations of membrane-bound S-OPA1. Identified cross-link pairs are mapped on the S-OPA1 subunits and described as positions 1 and 2. c, Structural comparison of the membrane-proximal and membrane-distal conformations of the S-OPA1 polymer. The cryo-EM structures have the models placed in the density. In membrane-distal conformation, the PD is moved away from the outer leaflet creating a gap of about 25 Å between the membrane-facing surface of the PD and the lipid bilayer. The area indicated by a box is a magnified in d. d, Magnified views of the PD–membrane interface in membrane-distal conformation. The top view of the central slices indicates the positions of lipid-binding sites. Bound lipids at the protein–membrane contact sites are identified as CL, and the acyl tails are truncated according to the map density. CL molecules flanking PDs are shown in grey. The PD residues associated with lipids include two arginine residues (Arg857, Arg858) and one lysine residue (Lys779) and are numbered accordingly.
While the major interfaces are similar, the most notable change between the two conformations is the movement of the PD relative to the membrane bilayer. The entire PD moves up and away from the membrane bilayer in the membrane-distal conformation, whereas the MIL is pulled out from the outer leaflet, losing contact with the bilayer surface (Fig. 4c). These conformational changes drive further constriction of lipid nanotubes (Fig. 4c). Moreover, we observed alterations in leaflet mass intensity profiles when we quantified the scattering from each leaflet as observed by cryo-EM (Supplementary Fig. 7). Specifically, in the membrane-proximal conformation, the lipid tails of the outer, convex leaflet displayed less intense scattering than the inner, concave leaflet. This pattern was reversed in the membrane-distal conformation, with the tails of the convex and protein-associated leaflet scattering more intensely.
Following up on this observation, we identified multiple lipid headgroup-like densities that appeared to be partially extracted from the bilayer. These densities appeared engaged with the membrane-facing and positively charged surfaces of the PD (Fig. 4 and Supplementary Fig. 8). We hypothesize that, if these features correspond with partially extracted lipids, this probably generates packing defects in the outer leaflet that may influence the probability of IM fusion. Of the lipids present in the membrane mixture, the characteristic negative charge and unique headgroup of CL suggested to us that this lipid may be the extracted molecule because it has an excluded volume comparable with the features observed in the cryo-EM map (Fig. 4).
To further examine the changes in membrane integrity, we incubated liposomes mimicking the lipid composition of mitochondrial IM and containing fluorophores with S-OPA1. Fluorescence spectroscopy of liposomes containing the dye Nile Red dye or 7-nitrobenz-2-oxa-1,3-diazol-4-yl (NBD) acyl chain-labelled fluorescent phosphatidylcholine (NBD-PC)—which are strongly fluorescent molecules in hydrophobic environments but almost nonfluorescent in polar solvents—revealed that S-OPA1 reduces the fluorescence signal of Nile Red- and NBD-PC-containing liposomes by 50% and 30%, respectively (Extended Data Fig. 10). The reduction in fluorescence suggests that membrane binding by S-OPA1 exposes these molecules to the aqueous solvent by creating packing defects, consistent with the appearance of the membrane in our cryo-EM maps. We also used molecular dynamics simulations to analyse the protein-independent properties of CL-enriched lipid mixtures and showed that membranes remained stable in these simulations (Supplementary Fig. 9).
These headgroup-like densities are not randomly oriented but, rather, form stable interactions with the conserved membrane-proximal PD residues, Lys779, Arg857 and Arg858 identified previously (Fig. 4). The first site of lipid densities is coordinated by the membrane-docking interface residues Arg857 and Arg858, which are essential for membrane remodelling and regulation of mitochondrial morphology (Fig. 4 and Extended Data Figs. 6 and 8). A second site for well-defined electron densities for lipids was identified between opposing PDs, where the lipid headgroup is stabilized by the Lys779 residues located within the MIL (Fig. 4). As the MIL comes out of the outer leaflet, the Lys779 residue shifts around 10 Å towards the membrane and ends up in proximity with the membrane surface forming close contacts with negatively charged CL molecules (Fig. 4). To investigate how the Lys779 residue interacts with membrane lipids and introduces fusogenic bilayer perturbations, we introduced the K779E mutation into OPA1 and characterized its activity in vitro and in living cells (Extended Data Figs. 6–8). Compared with cells with WT OPA1, the OPA1(K779E) mutant displayed a reduced ability to elongate mitochondria (Extended Data Fig. 8). Thus, it is reasonable to conclude that specific protein–lipid interactions between conserved membrane interface residues and CL are required for OPA1 activity.
OPA1 isoforms are ubiquitously expressed in all human tissues, but the highest levels are detected in the retina, brain, heart and testis13. Mutations of the OPA1 gene cause 60–80% of the cases of autosomal dominant optic atrophy and an expanding spectrum of neurodegenerative disorders, including spastic paraplegia45, spinocerebellar ataxia22, syndromic parkinsonism23 and dementia46. We mapped the location of pathology-associated variants onto the structure of the membrane-bound human S-OPA1. Given that the PD forms the most extensive interface with the membranes, we focused on single-residue missense mutations in this region that may attenuate the membrane-remodelling activity of OPA1 (Extended Data Figs. 8). Among these, we identified three conserved disease alleles (Arg781, R824 and Arg857) that interact with membranes, and sought to functionally characterize their role in membrane remodelling. Notably, we found that charge-reversal mutations of these PD residues resulted in significant reductions in S-OPA1-mediated membrane binding and remodelling in vitro (Extended Data Figs. 6 and 7). We next analysed their impact on mitochondrial morphology in mammalian cells. As predicted, mutations of Arg781 and Arg824 to glutamic acid impaired OPA1’s activity to regulate mitochondrial morphology, indicating that heritable mutations close to the membrane interface of OPA1 lead to human diseases associated with mitochondrial dysfunction (Extended Data Fig. 8).
Discussion
Here we determined the structures of two functional states of membrane-bound human OPA1 using cryo-EM and CX-MS. These structures inspired cell-based mutagenesis experiments to probe the mechanisms of membrane remodelling and fusion that govern mitochondrial morphology and function. From these efforts, we show how OPA1 shapes lipid membranes and how these activities impact mitochondrial morphology through specific protein–protein and protein–lipid interactions. The structural flexibility of OPA1 domains suggests how OPA1 proteoforms can adapt to different conformations and recognize negatively curved surfaces, as has been shown for fungal MGM135. Our findings on positive-curvature surfaces uncover unique insights into how a large GTPase perturbs lipid leaflets through insertion and removal of the PD to regulate mitochondrial morphology. Our findings also provide molecular explanations for the pathologies associated with OPA1 mutations.
We highlight the essential role of the PD in membrane assembly and deformation. Within the PD, OPA1 contains a unique MIL between α-helices α1P and α2P, which can act as a deep-seated membrane anchor. Residues within the MIL form intimate interactions with lipids. Moreover, the presence of lipid-like densities associated with S-OPA1 in our reconstructions supports the idea that OPA1 can induce membrane-packing defects to promote membrane fusion. Our results suggest that the functional importance of PD interactions is to overcome the large energy barrier required for local membrane bending, ultimately leading to mitochondrial IM fusion in living cells. Eight OPA1 isoforms, and the L- and S-proteoforms, contain the α3P helix of the PD interface, so this feature is key to understanding the oligomeric OPA1 machine comprised of heterogeneous oligomers in vivo24,25.
We also investigated whether the narrower, membrane-distal conformation of the S-OPA1 lattice could be attributed to a powerstroke-like constriction mechanism47. In dynamin-family GTPases, a powerstroke refers to GTP-induced dimerization of trans-rung GTPase domains, plus the large-scale motions of the BSE-GTPase domains after GTP gamma phosphate hydrolysis and dimer dissociation47,48. The swing of the BSE is associated with a sliding of adjacent turns of the supermolecular helical lattice, powering constriction of the entire collar on the target membrane47,48. For S-OPA1, the membrane-proximal and membrane-distal conformations were both determined in the presence of GMPPCP. In both structures, the positions of the BSE and GTPase domains relative to the stalks are nearly identical—and are comparable with GTPγS-bound MGM1 structure35. By contrast, the PD position, relative to the stalks and to the membrane surface, is substantially altered between the membrane-proximal versus distal conformations—suggesting a new mechanism of membrane constriction unrelated to the canonical powerstroke. As the outer diameters of the filament are similar in both conformations, we postulate that the movement of the PDs relative to the stalk domains pushes the membrane layer radially inwards to generate high-curvature tubes and remove the MIL from the outer leaflet.
Previous studies of mitochondrial fusion showed that L-OPA1 and S-OPA1 synergistically catalyse membrane fusion33. Although the structural arrangements of L- and S-OPA1 that are required for efficient fusion within the cell are still not fully understood, the results of these studies indicate that L-OPA1 and S-OPA1 form higher-order assemblies to coordinate the fusion of mitochondrial membranes33. Our results suggest a mechanism for OPA1-mediated mitochondrial IM fusion in which S-OPA1 and its PD has a major role in driving IM fusion (Fig. 5). We propose that IM-anchored L-OPA1 molecules initiate membrane remodelling and recruit soluble S-OPA1 to rapidly polymerize into a flexible cylindrical scaffold encaging the mitochondrial IM. The energy provided by the dimerization of PDs within the outer leaflet, in particular, shapes the IM into a moderate-curvature tubule. Subsequently, concerted conformational changes in the angles between the stalk and the PD lead to PD extraction from the lipid bilayer and perturb outer leaflet lipid packing to produce additional curvature stress, which sum together to generate an unstable membrane that is prone to fusion. Further tubulation of membranes brings the two bilayers into close apposition and drives the fusion of high-curvature IM tubules into a single membrane (Fig. 5).
Fig. 5 |. Proposed mechanism of OPA1-mediated mitochondrial IM fusion.
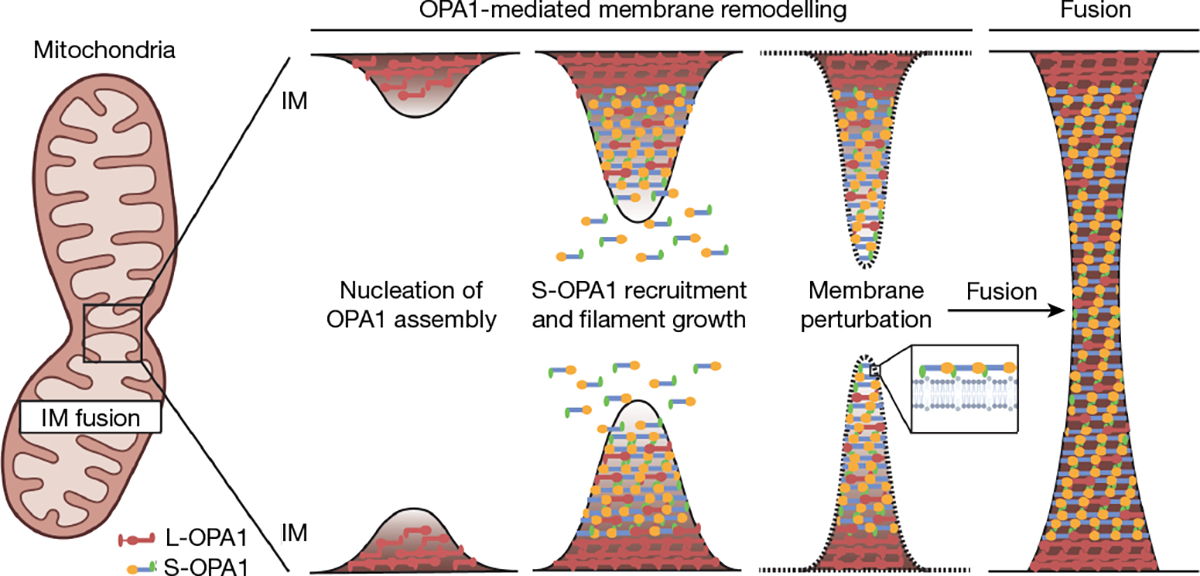
Human L-OPA1 molecules embedded on the surface of the mitochondrial IM may nucleate helical polymerization of MIL-embedded S-OPA1 to drive IM tubulation. Subsequent conformational changes within the OPA1 lattice extract the PD, further constricting the IM and introducing lipid-packing defects to lower the energy barrier to fusion. The diagram was created using BioRender.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41586-023-06441-6.
Methods
Data reporting
No statistical methods were used to predetermine the sample size. The experiments were not randomized, and the investigators were not blinded to allocation during the experiments and outcome assessment.
Protein expression and purification
The gene encoding full-length human OPA1 isoform 1 was obtained from Addgene (26047). The DNA sequence corresponding to the short form of human OPA1 (residues 196–960) was subcloned into the pCA528 vector using the Gibson cloning method. The resulting fused ORF consists of an N-terminal 10×His tag followed by a SUMO tag and residues 196–960 of OPA1. The vector was provided by the laboratory of W. I. Sundquist. OPA1 mutants were generated using a modified QuickChange PCR-based mutagenesis protocol, and all constructs were confirmed by Sanger sequencing. To improve expression and stability, the first 56 residues (Δ196–252) were omitted from the short form of OPA1 (S-OPA1). All WT or mutant OPA1 genes were transformed separately into Escherichia coli BL21 (DE3)-RIPL cells. A single colony was inoculated into lysogeny broth (LB) medium supplemented with 50 μg ml−1 kanamycin and 25 μg ml−1 chloramphenicol and grown overnight at 37 °C on an orbital shaker. A 10 ml overnight culture was used to inoculate 0.75 l of ZYP-5052 auto-induction medium49, and the cells were grown at 37 °C to an optical density at 600 nm (OD600) of 0.6 in baffled flasks. Subsequently, the temperature was lowered to 18 °C, and the culture was grown for another 16 h. Cells were collected by centrifugation at 4,000g for 20 min, and the bacterial pellets were stored at −80 °C. Thawed cells were resuspended in buffer A (50 mM HEPES-NaOH, pH 7.5, 500 mM NaCl, 20 mM imidazole, 5 mM MgCl2, 5 mM CHAPS (Millipore), 5 mM 2-mercaptoethanol, 10% (v/v) glycerol) supplemented with 0.5% (v/v) Triton X-100, 0.5 mg DNase I, 1× EDTA-free complete protease inhibitor cocktail (Roche) and lysozyme. Cells were then lysed with a probe sonicator and centrifuged at 35,000g for 45 min at 4 °C to remove cell debris. The supernatant was filtered using a 0.45 μm membrane (Millipore), loaded onto Ni-NTA Agarose beads (Qiagen) pre-equilibrated with lysis buffer and rotated at 4 °C for 1 h. The beads were collected on column support, and washed with around 10 column volumes of buffer A and buffer B (50 mM HEPES-NaOH, pH 7.5, 1 M NaCl, 20 mM imidazole, 5 mM MgCl2, 5 mM CHAPS, 5 mM 2-mercaptoethanol, 10% (v/v) glycerol), followed by an imidazole wash in sequential steps using increasing concentrations of imidazole (80 mM and 120 mM) in buffer A. The protein was eluted with buffer C (50 mM HEPES-NaOH, pH 7.5, 500 mM NaCl, 500 mM imidazole, 5 mM MgCl2, 5 mM CHAPS, 5 mM 2-mercaptoethanol, 10% (v/v) glycerol) and incubated with 6×His-tagged Ulp1 protease while dialysing overnight at 4 °C against buffer A. The dialysed sample was then applied again to Ni-NTA beads pre-equilibrated with dialysis buffer to remove the uncleaved protein, free purification tag and the His-tagged Ulp1 protease. The cleaved protein was further purified by size-exclusion chromatography on the Superdex-200 16/60 column (GE Healthcare) equilibrated with buffer D (50 mM HEPES-NaOH, pH 7.5, 500 mM NaCl, 5 mM MgCl2, 5 mM CHAPS, 5 mM 2-mercaptoethanol, 10% (v/v) glycerol). Peak fractions were pooled, concentrated to 2 mg ml−1 using a 50 kDa MWCO concentrator (Vivaspin20), flash-frozen as single-use aliquots in liquid nitrogen and stored at −80 °C. The protein concentration was determined by its absorbance at 280 nm, and protein purity was analysed by SDS–PAGE.
Mass photometry experiments
Mass photometry measurements were recorded on a OneMP mass photometer using glass coverslips. Before mass photometry measurements, coverslips were cleaned by sequential rinsing with H2O, isopropanol and H2O, followed by drying with a clean stream of nitrogen. The sample buffer (20 mM HEPES-NaOH, pH 7.5, 140 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1 mM DTT, 1% (v/v) glycerol) was filtered through a 0.22 μm membrane (Millipore), and the protein samples were diluted using the filtered stock buffer. A clean coverslip and gasket were assembled and placed onto the mass photometer. Focal calibration was determined using stock buffer and contrast-to-mass calibration was performed using purified bovine serum albumin (BSA) and apoferritin proteins. The calibration experiment was analysed using DiscoverMP 2.2 software (Refeyn), and the resulting contrast distribution was Gaussian-fitted using the maximum-likelihood procedure. Data acquisition was performed using AcquireMP (v.2.2; Refeyn). For each acquisition, 1 μl of 600 nM freshly purified OPA1 protein sample was applied to a 15 μl stock buffer on a coverslip, resulting in a final concentration of ~40 nM. After autofocus stabilization, mass photometry videos were acquired for 60 s with a frame rate of 994 Hz. Individual particle binding events and their interferometric scattering contrast were then detected and quantified from the analysis of each individual video and converted to mass using the contrast-to-mass calibration. The resulting mass distribution of individual particle binding events yields the solution distribution of oligomeric species. The accuracy of measured mass is assessed by the difference between the measured data and the linear fit of calibration proteins. Measurements were repeated at least three times.
Preparation of lipid vesicles and lipid nanotubes
The following lipids were purchased from Avanti Polar Lipids: 1-palmitoyl-2-oleoyl-glycero-3-phosphocholine (POPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE), l-α-lysophosphatidylinositol (Soy Lyso PI), 1′,3′-bis[1,2-dioleoyl-sn-glycero-3-phospho]-glycerol (CL (18:1)4), d-galactosyl-β-1,1′ N-nervonoyl-d-erythro-sphingosine (C24:1 galactosyl(β) ceramide, GalCer). Stock lipid solutions were resuspended in chloroform, methanol and water mixture (20:9:1, (v/v/v)). The lipid composition of vesicles (POPC:POPE:PI:CL; 45:22:8:25) was chosen to represent the lipid composition of the mitochondrial IM50 and prepared according to an established protocol51. The indicated ratio of lipid stock solutions was mixed in glass vials (Thermo Fisher Scientific), dried under a steady stream of nitrogen, and resuspended in chloroform to obtain a homogenous mixture. Thin lipid films were prepared by evaporating the chloroform under a gentle stream of nitrogen while rotating the vials. The residual chloroform was further removed by vacuum desiccation for 2 to 4 h. The resulting lipid film was rehydrated in liposome buffer (20 mM HEPES-NaOH, pH 7.5 and 150 mM NaCl) with vortexing and extruded through polycarbonate membranes with 50 nm pore diameter to yield unilamellar liposomes. Samples were flash-frozen as single-use aliquots in liquid nitrogen and stored at −80 °C. Lipid nanotubes composed of GalCer:CL (90:10) were prepared using the same protocol, with slight modifications. After rehydration in liposome buffer, the mixture was incubated for 15 min at room temperature with intermittent mixing. The resulting suspension of lipid nanotubes was vortexed briefly and sonicated using a bath sonicator at 50 °C for 5 min. After cooling to room temperature, the lipid nanotubes were immediately used in reconstitution assays.
Preparation of OPA1 reconstitution and membrane remodelling reactions
Before performing reconstitution assays, frozen aliquots of OPA1 were thawed and processed for size-exclusion chromatography on the Superose 6 10/300 GL column (GE Healthcare) equilibrated with reaction buffer (20 mM HEPES-NaOH, pH 7.5, 130 mM NaCl, 10 mM KCl, 2 mM MgCl2, 2 mM DTT, 2% (v/v) glycerol). For cryo-EM experiments, equal volumes of (50 μl) of ~12 μM purified protein and 2 mg ml−1 lipid nanotubes rehydrated in liposome buffer (20 mM HEPES-NaOH, pH 7.5, and 150 mM NaCl) were gently mixed and incubated with 500 μM β,γ-methyleneguanosine 5′-triphosphate sodium salt (GMPPCP) at room temperature for 4 h. The reconstitution of membrane-bound filaments was observed using negative-stain TEM, and the reaction was directly used for cryo-EM sample preparation. For negative-stain TEM analysis, 25 μl of purified WT or mutant OPA1 at a final concentration of 1.5 to 5 μM was added to an equal volume of lipid vesicles or nanotubes diluted to a final concentration of 0.5 mg ml−1 to allow direct visualization of OPA1 assemblies during membrane-remodelling reactions. The proteoliposome solutions were incubated in the presence or absence of 500 μM GMPPCP for 4 h at room temperature and immediately processed for EM sample preparation.
Liposome co-sedimentation assays
Sedimentation assays were performed as described previously35,52. In brief, aliquots of purified S-OPA1 were buffer-exchanged to the liposome buffer (20 mM HEPES-NaOH, pH 7.5, and 150 mM NaCl) with a micro-spin desalting column (Thermo Scientific Zeba) at 4 °C according to the manufacturer’s instructions. Aliquots of 25 μl of WT or mutant OPA1 at a final concentration of 2.4 to 3 μM were added to an equal volume of lipid vesicles (POPC:POPE:PI:CL, 45:22:8:25) diluted to a final concentration of 1.0 mg ml−1 and incubated for 30 min at room temperature. Subsequently, the protein–lipid mixture was centrifuged at 55,000 rpm for 30 min at 20 °C using a TLA-120.2 rotor (Beckman Coulter) and the supernatant and pellet fractions were analysed by SDS–PAGE. The experiments were performed in triplicates and the protein bands were quantified using ImageJ53.
Negative-stain EM
Grids of purified OPA1 for negative-stain EM were prepared according to an established protocol54. In brief, 4 μl of the sample at a concentration of around 0.03 mg ml−1 was applied to a glow-discharged 400 mesh copper grid coated with a thin layer of continuous carbon film (Ted Pella), blotted with filter paper and then stained with freshly prepared 0.75% (w/v) uranyl formate. Samples were visualized at room temperature using the Tecnai T12 microscope (FEI) equipped with a 4k × 4k CCD camera (UltraScan 4000, Gatan) and operated at a voltage of 120 kV. Images were recorded at a nominal magnification of ×52,000 at the sample level with a calibrated pixel size of 2.21 Å. A total of 51 and 52 micrographs were collected for S-OPA1 in the presence or absence of 500 μM GMPPCP, respectively. Contrast transfer function (CTF) parameters were determined using CTFFIND455. Particles were manually picked from the micrographs, extracted with a box size of 160 × 160 pixels and sorted into two-dimensional (2D) classes using the Relion software package56. The good 2D class averages generated from manually picked particles were then used as templates for semi-automated particle picking. A total of 17,981 and 82,892 particle projections of S-OPA1 monomer in the presence or absence of non-hydrolysable analogue GMPPCP were semi-automatically picked from the micrographs, respectively, and sorted through subsequent rounds of reference-free 2D classification in Relion. For analysing WT and mutant OPA1 reconstitution on membranes, 4 μl of purified OPA1-membrane mixture was applied onto glow-discharged carbon-coated grids, blotted with filter paper and stained with uranyl formate as described above. Negatively-stained grids were imaged at a nominal magnification of ×30,000, corresponding to a calibrated pixel size of 3.75 Å on the sample. Negative-stain TEM imaging of reconstitution assays in the presence of fluorescently labelled liposomes was performed using the Tecnai T12 Spirit LaB6 filament TEM (FEI) equipped with an AMT 2k × 2k side-mounted CCD camera and operated at a voltage of 100 kV. The micrographs were collected at a nominal magnification of ×98,000 at the sample level with a calibrated pixel size of 6.47 Å per pixel.
Cryo-EM sample preparation and data acquisition
An aliquot of 6 μl of the membrane reconstitution reaction was applied to glow-discharged R1.2/1.3 200 Cu mesh grids (Quantifoil) and incubated for an additional 30 s. Grids were blotted for 4 s under blot force of 0, and plunge-frozen in liquid ethane cooled by liquid nitrogen using the Vitrobot Mark IV (FEI) operated at 10 °C and 100% humidity. Cryo-EM datasets were acquired on the Titan Krios transmission electron microscope (FEI) operated at 300 kV equipped with a K3 direct electron detector (Gatan) and a GIF Quantum energy filter (Gatan) with a slit width of 20 eV. Automated data collection was performed using the SerialEM software57. Videos were recorded at a nominal magnification of ×105,000 in super-resolution mode with a super-resolution pixel size of 0.4165 Å px−1 at a dose rate of ~8 e− px−1 s−1. A total of 12,237 micrographs were acquired in three different sessions with a defocus range of approximately −0.5 to −1.2 μm. Most data were collected as dose-fractionated stacks of 120 frames at a per frame dose rate of ~0.69 e− Å−2 and recorded for 2 s corresponding to a total accumulated dose of ~82 e− Å−2. A subset of the dataset was recorded at a per-frame dose rate of ~0.62 electrons, corresponding to a total accumulated dose of ~74 e− Å−2. Image acquisition parameters are summarized in Extended Data Table 1.
Image analysis and 3D reconstruction
All dose-fractionated super-resolution image stacks were motion-corrected, 2× binned by Fourier cropping (1.666 Å px−1) and summed to a single micrograph with dose filtering using MotionCor258. CTF parameters for each motion-corrected sum without dose weighting were determined using CTFFIND455. Subsequent helical image processing steps for all datasets were performed with the dose-weighted micrographs in Relion (v.3.0)56,59. Filaments were manually picked, and overlapping segments were extracted using a box size of 700 Å and an inter-box distance of about 10% (72 Å) of the box length. A total of 623,000 segments was Fourier binned twice and then processed for multiple rounds of reference-free 2D classification to identify homogeneous subsets with a largely uniform diameter. The remaining 472,097 segments were re-extracted by resetting the refined offsets to zero and used for 3D classification. A featureless cylinder-shaped electron density map was generated as an initial alignment reference for 3D classification using the SPIDER software package60. All segments were processed for 3D classification and 3D auto-refine with helical priors but without imposing helical symmetry. A wide range of different helical parameters was tested and imposing an initial helical rise of 24 Å and a helical twist of 26.01° led to the 3D reconstruction of a right-handed 3-start helical polymer (with C1 point group symmetry) in which secondary structure features were clearly resolved. To harness the symmetry between the three separate helical strands of molecules, additional helical symmetry was imposed on the polymers by using a helical rise of 24/3 Å and a twist of (360° + 26.01°)/3 (ref. 59). After determining the helical symmetry as a 1-start helix (rise of 8 Å and a twist of 128.67°), a soft mask was applied to the central 30% of the helix along the z axis (helical_z_percentage = 0.3), and all segment images were 3D-auto-refined imposing helical priors and helical symmetry. Moreover, the previously imposed helical symmetry was further expanded to improve the density of the membrane-associated domains by using the symmetry expansion procedure (relion_particle_symmetry_expand) in Relion to generate 4,248,873 symmetry-expanded particles. The output volume was used as the reference model for subsequent 3D skip align classification with local optimization of the helical rise and twist. After 3D classification with a local mask on the BSE-stalk and PDs, the best-aligned classes comprising 497,314 (~12%) particles were processed for 3D refinement and CTF refinement, followed by another round of 3D refinement, resulting in a 5.1 Å density map. To further improve the density map, an additional round of 3D classification without alignment was performed, and a subset of 139,018 (~3.3%) particles was selected for refinement, which yielded a ~4.8 Å reconstruction of the membrane-bound OPA1 filament. The helical parameters converged to a rise of 7.73 Å and a twist of 128.634° per subunit.
For the second conformation of OPA1 on more constricted membranes, 3D skip align classification was performed without a soft mask around the protein density to unveil the variations in the diameter of the membrane nanotube. Analysis of the nine density maps obtained revealed human OPA1 in various conformations. The class displaying the highest membrane constriction, which contained a subset of 258,806 particles (~6%), was refined to a resolution of 7.4 Å. To reduce heterogeneity among these particles, a second round of 3D skip align classification with a soft mask was performed, and a single stable class of 96,152 particles (~2.3%) was selected for further 3D refinement. These particles were then processed for per-particle CTF refinement before another round of refinement, which led to a density map reconstruction at 6.8 Å resolution. The helical parameters were refined to rise = 8.04 Å and twist = 128.619°.
Model building and refinement
All model building was performed in Coot61. The crystal structure of nucleotide-free S-MGM1 (Protein Data Bank (PDB): 6QL4) was used as an initial model for the filament. The model of the S-MGM1 monomer was first manually fitted into the membrane-proximal cryo-EM density map in UCSF Chimera62 and rigid-body-refined using phenix.real_space_refine tool of the PHENIX software package63. In both maps, a clear density for the GTPase domain was observed, but the resolution of this region was insufficient for model building. As the resolution of the GTPase domain was insufficient for chain tracing in several areas, we unambiguously fitted the available crystal structure of the minimal GTPase domain of OPA1 (PDB: 6JTG) into the density map and adjusted residues manually. The density of the BSE, stalk and PDs was of sufficient quality for de novo model building. As the map showed that OPA1 is mostly helical, poly-alanine helices for BSE, stalk and PDs were first built manually and then connected by manually extending the chain at the N and C termini of each helix in Coot. Subsequent amino acid assignments were made on the basis of clearly defined densities, sequence and secondary structure predictions. The model was then refined during model building using real-space refinement in Coot with torsion, planar peptide and Ramachandran restraints applied. To obtain an initial model for an OPA1 monomer, a monomeric density of OPA1 from the cryo-EM map was extracted using the zone tool in the UCSF chimera. A starting PDB model, which was modelled ab initio using coot, was refined against this density using an all-atom refinement protocol in Rosetta (v.3.12)64. The lowest-energy, best-fit-to-density and highest-scoring models were assessed manually and advanced to the next stage of modelling the higher-order assembly. Once a near-complete initial model of a single OPA1 subunit was built, the structure was then processed for iterative rounds of refinement against the membrane-proximal cryo-EM density map with global minimization, local grid search, secondary structure and geometry restraints to improve the stereochemistry as well as the model-map correlation coefficient using phenix.real_space_refine (v.1.19). For the membrane-distal conformation of OPA1, the refined membrane-proximal model was used as a template for model building and fitted into the cryo-EM density map as a rigid body. The model was manually corrected, and mismatching regions were rebuilt to fit into the membrane-distal cryo-EM map in Coot and then refined in phenix. real_space_refine using the protocol described above. Subsequently, a filament model that included 34 subunits of OPA1 was generated for both conformations in real space using UCSF Chimera. After further iterations of manual adjustments in Coot, the subunits were refined against their respective cryo-EM maps with noncrystallographic symmetry constraints in phenix.real_space_refine until the models corresponded with the features of the map.
Validation and structure analysis
The quality of the final models was validated by MolProbity implementation in PHENIX65. For cross-validations, the final model was refined against the half maps generated by 3D auto-refine in Relion. Independent FSC curves for map-to-model correlations were calculated between the resulting models and the two half maps as well as between the resulting model and the combined map within PHENIX. The refinement and validation statistics for membrane-proximal and membrane-distal models are shown in Extended Data Table 1.
Electrostatic surface potentials for pre-constricted and constricted structures were calculated in the APBS program implemented in PyMOL using an AMBER force field and are displayed at ±5 kT per e− (ref. 66). Subunit interfaces within the OPA1 filaments and solvent-accessible and buried surface areas were calculated from the models using the PDBePISA server67. Analysis of the fold conservation of the OPA1 PD was performed by a 3D structural database search using the Dali server68. All structural illustrations were prepared using UCSF Chimera62, UCSF ChimeraX69 and PyMOL (The PyMOL Molecular Graphics System, v.2.0 Schrödinger).
Chemical cross-linking and MS
Two batches of membrane-bound OPA1 polymer (60 μg) were prepared as described and cross-linked with 2 mM DSS-H12 and DSG-H6 (Creative Molecules), respectively, for 30 min at 30 °C. Reactions were quenched (10 mM ammonium bicarbonate, 10 min, room temperature) and processed for MS as described previously70. Cross-linked products were enriched by size-exclusion chromatography (Superdex Peptide, GE Healthcare Life Sciences) as described previously71, and fractions eluting between 0.9 and 1.4 ml were dried and resuspended in 0.1% formic acid for MS analysis. The fractions starting at 0.9 ml and 1.3 ml were combined before evaporation to make four total SEC fractions.
MS experiments were performed using liquid chromatography using the NanoAcquity UPLC (Waters) system coupled to an Orbitrap QE HF-X MS (Thermo Fisher Scientific). A flow rate of 300 nl min−1 was used, where mobile phase A was 0.2% (v/v) aqueous formic acid, and mobile phase B was 0.2% (v/v) formic acid in acetonitrile. Analytical columns were pulled-and-packed and prepared in-house, with an internal diameter of 100 μm packed with NanoLCMS solutions 1.9 μm C18 stationary phase to an approximate length of 25 cm. Peptides were directly injected into the analytical column using a gradient (2% to 45% B, followed by a high-B wash) of 90 min. The mass spectrometer was operated in a data-dependent manner using higher-energy collision dissociation fragmentation for MS/MS spectra generation detected in the orbitrap.
For data analysis, the .raw data files were checked using Preview (Protein Metrics) to verify calibration and quality before further analysis. Data were then processed using Byonic (Protein Metrics; v.3.6.0) to identify peptides and infer proteins. Proteolysis was assumed to be fully specific to trypsin/LysC with up to two missed cleavages and allowed for DSS cross-links on K. Precursor and fragment mass accuracies were held within 12 ppm. Cross-link assignments were inspected and assessed using Byologic (Protein Metrics) to determine the assignment quality. The identified cross-links were filtered to include unique cross-links with a Byonic score bigger than 300 (ref. 72). In total, we identified 112 unique DSS cross-links and 89 unique DSG cross-links. Filtered cross-links were mapped to the primary protein structure using xiNET73 and to protein models using the Xlink Analyzer program implemented in Chimera74.
Molecular dynamics simulations
The bilayer systems were produced by using a membrane patch with the dimensions of 104 Å × 104 Å along the x and y axes and a length of 148 Å along the membrane normal z. The lipid compositions of Galcer:CL:POPC; 85:10:5 and POPC:CL; 90:10 were used for the atomistic simulations of lipids. The simulations were run for 1.1 μs and data analyses were performed using POPC as a probe of membrane fluidity, of which the order parameters can be properly calculated and compared across systems. All systems were built with CHARMM-GUI75 and parametrized with the CHARMM36m forcefield. The simulations were run with Gromacs (v.2021.5)76 using the standard settings from CHARMM-GUI’s membrane builder, and trajectories were analysed with VMD77 and the MEMBPLUGIN.
Fluorescent microscopy imaging of liposomes
To actively visualize the co-localization of WT S-OPA1 onto CL-enriched liposomes, a phospholipid labelled on the head group with red fluorescent Texas Red dye (1,2-dihexadecanoyl-sn-glycero-3-ph osphoethanolamine, triethylammonium salt (Texas Red-DHPE)) was purchased from Thermo Fisher Scientific (T1395MP). The liposome buffer and liposomes were prepared as described above. The lipid composition for the Texas-Red-containing liposomes was POPC:POPE:PI:CL:Texas Red, 45:21.6:8:25:0.4. Moreover, supportive lipid bilayers (SLBs) were prepared to support and stabilize the Texas Red liposomes within the fluid cell. The following lipids were purchased from Avanti Polar Lipids: 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), 1,2-dioleoyl-sn-glycero-3 phosphoethanolamine (DOPE), 1,2-dioleoyl-sn-glycero-3-phospho-(1′-myo-inositol) (PI), 1′,3′-bis[1,2-dioleoyl-sn-glycero-3-phospho]-glycerol (CL (18:1)4), and the lipid composition of DOPE:DOPC:PI:CL, 26:45:4:25 was used for the SLB preparation. The resulting suspension of vesicles was added onto the mica surface to form SLBs within the fluid cell and kept at 4 °C for up to 1 week. To visualize and track S-OPA1, a thiol-reactive Alexa Fluor 488 C5 Maleimide fluorescent marker (Thermo Fisher Scientific, A10254) was conjugated to thiol groups of the protein sample. Previously purified protein was thawed and dialysed against HP500 buffer (25 mM HEPES, 25 mM PIPES, 500 mM NaCl, pH 7.1) overnight at 4 °C. Subsequently, Alexa Fluor 488 was added dropwise to a final concentration of approximately 0.1 mg ml−1 and agitated for 5 min before the sample was loaded onto Superdex 200 gel filtration column for size-exclusion. A fluid cell encasing the SLB, Texas Red vesicles and S-OPA1 protein was created to visualize the protein–liposome interactions in solution. In brief, a thin cover slip (Millipore Sigma, CLS2975246) washed with 70% ethanol and Optic adhesive (VWR, MSPP-NOA8801) was used to attach a thin slice of mica (Sigma-Aldrich, AFM-71855–15-10) and cured with a 365 nm wavelength of light for 10 min. A piece of Scotch tape was then used to thin the mica and ensure an even surface. A plastic 4 ml screw cap lid with no centre (Millipore Sigma, 27120-U) was adhered to the mica using the same optic adhesive and cured for 5 min using 365 nm wavelength light. The previously prepared SLB was diluted to 0.2 mM and placed onto the mica surface of the fluid cell for 1 h at room temperature. Subsequently, the SLB was washed three times with liposome buffer by gently pipetting up and down three times and replacing it with fresh buffer every time. After washing, the Texas Red liposomes (0.08 mg ml−1), GMPPCP (500 μM) and Alexa Fluor 488-conjugated S-OPA1 protein (0.15 mg ml−1) were added to the fluid cell containing SLB (0.1 mM) and analysed immediately using fluorescent microscopy imaging. The samples were imaged using the Nikon A1R confocal laser-scanning microscope with a ×100 oil objective and a 488 laser line to detect S-OPA1 and a 561 laser line to detect Texas Red liposomes. All images were taken using perfect focus, the same gain and the same z height, and edited in ImageJ to merge.
Quantification of packing defects in lipid membranes
To determine the lipid-bilayer-packing defects caused by S-OPA1-mediated membrane remodelling, we acquired the following fluorescent molecules, which are fluorescent only in hydrophobic environments: Nile Red (NR) (Thermo Fisher Scientific, N1142) and 1-oleoyl-2-[12-[(7-nitro-2–1,3-benzoxadiazol-4-yl) amino]dodecanoyl]-sn-glycero-3-phosphocholine (NBD-PC, 18:1–12:0) (Avanti Polar Lipids). The liposome buffer and liposomes were prepared as described above. The lipid composition of POPC:POPE:PI:CL:NR, 45:21.75:8:25:0.25 was used for the preparation of the NR containing liposomes, while POPC:POPE:PI:CL:NBD-PC, 44:22:8:25:1 was chosen for the NBD-PC liposomes. Using the CLARIOstar microplate reader, we measured the fluorescence signal of NR at an excitation wavelength of 540 nm and an emission wavelength of 630 nm and NBD-PC at an excitation wavelength of 463 nm and an emission wavelength of 536 nm. To determine the S-OPA1-mediated changes in fluorescence signal, freshly purified S-OPA1 at 0.1 mg ml−1 concentration was incubated with 0.1 mg ml−1 of liposomes containing either NR or NBD-PC in the presence of 500 μM GMPPCP for 4 h before measuring the fluorescence in an end-point assay at the respective excitation and emission wavelengths. The following controls were also performed: (1) liposome buffer alone; (2) liposomes containing either NR or NBD-PC alone; (3) S-OPA1 alone; and (4) BSA with liposomes containing either NR or NBD-PC. All assays were performed using a 384-well solid black polystyrene assay plate (Corning) at room temperature and total reaction volumes were 20 μl. The reactions were run in five independent replicates consisting of four technical replicates. The fluorescence measurements were analysed using t-tests and Grubbs tests (α = 0.2) to determine the statistical significance and outliers (if any), respectively.
Cell culture
HeLa cells were provided by M. Ryan, and cell identity was confirmed by the human cell line authentication service from Eurofins (1 March 2022). Authentication was confirmed by PCR-single-locus-technology. Cells were tested negative for Mycoplasma. Cells were grown in DMEM supplemented with 10% FCS and 50 μg ml−1 uridine at 37 °C and 5% CO2.
Plasmid construction
The transfection control pcDNA3.1-mCherry-NLS was generated by Gibson cloning of mCherry (provided by D. Mick) into pcDNA3.1(−) (provided by B. Schrul) using the primers listed in Supplementary Table 1. The siRNA-resistant OPA1 construct was generated using a two-step mutagenesis PCR reaction. To render the construct siRNA-resistant, a total of ten mutations was introduced that left the amino acid sequence unaltered (CCGGACCTTAGTGAATATAAA to CCAGATTTGTCCGAGTACAAG). The resulting sequence was subcloned into the pCAGEN vector from Addgene (11160) using the Gibson-cloning method. Mutations were introduced by site-directed mutagenesis PCR using the primer pairs listed in Supplementary Table 1. The correct insertion of all mutations was confirmed by Sanger sequencing (Eurofins).
Plasmid transfection and RNA interference
HeLa cells were seeded in a six-well plate and transfected with 200 ng pcDNA3.1-mCherry-NLS, 700 ng pcDNA3.1(−), and either 100 ng pCAGEN or 100 ng of siRNA-resistant pCAGEN-OPA1 using Lipofectamine 3000 (Invitrogen, L3000015) according to the manufacturer’s instructions. After 24 h, the medium was changed, and the cells were transfected with 25 pmol of either control siRNA (Qiagen, 1027281) or OPA1 siRNA (Qiagen, SI03019429) using the RNAiMAX siRNA transfection kit (Invitrogen 13778150) according to the manufacturer’s instructions. Cells were incubated for 24 h and were then seeded onto poly-l-lysine precoated coverslips for microscopy or in six-well plates for immunoblot analysis and were incubated for another 48 h. Experiments were performed 72 h after siRNA transfection.
Fluorescence microscopy imaging of cell-based assays
Cells on coverslips were washed once with warm PBS and were then fixed for 20 min at room temperature with 4% PFA. After three PBS wash steps, the cells were permeabilized with PBS containing 0.1% Triton X-100 for 10 min at room temperature and were blocked in PBS with 2% BSA. Immunolabelling was performed in PBS with 2% BSA using a 1:500 dilution of a recombinant anti-TOMM22 antibodies (Abcam, ab179826) as the primary antibody for 1 h at room temperature. After ten wash steps, fluorescence staining was performed using a 1:500 dilution of a donkey anti-rabbit antibody coupled to Alexa488 (Invitrogen, A21206) in PBS with 2% BSA. Coverslips were washed ten times in PBS and another ten times in water and were then mounted on specimen slides using Fluoromount-G mounting medium (Invitrogen, 00–4958-02). Images of moderately mCherry–NLS expressing cells were taken on the Leica Dmi8 fluorescent microscope with a ×63/1.40 NA objective and a Leica DFC3000 G CCD camera. z-Stacks covering the whole sample were recorded to ensure that the entire mitochondrial network was covered. Maximum-intensity projections of the central three planes were created in ImageJ53. The contrast was adjusted linearly to correct for variations in TOMM22 staining.
For the OPA1 quantification experiments, transfected cells were treated as above and were stained with a 1:500 dilution of rabbit anti-OPA1 antibody (Cell Signaling, 67589) and mouse anti-TOMM22 (Abcam, ab57523), followed by staining with a 1:500 dilution of a donkey anti-rabbit antibody coupled to Alexa488 (Invitrogen, A21206) and a goat anti-mouse antibody coupled to Alexa647 (Invitrogen, A32728) with a 1:400 addition of Phalloidin-Alexa405 (Invitrogen, A30104). All pictures were recorded with the same settings and no adjustments were made. Regions of interest were defined and analysed using the LasX Software v.1.4.4 (Leica). For the statistical analysis, datasets were log-transformed to achieve a Gaussian distribution and three independent experiments were tested for significance using nested one-way ANOVA in GraphPad Prism (GraphPad).
Immunoblot analysis
Cells from a six-well plate were collected using trypsin. After washing with DMEM containing 10% FCS and PBS, the cell pellet was resuspended in lysis buffer (20 mM HEPES, pH 7.2, 150 mM NaCl, 0.5% Triton X-100 and 1× EDTA-free complete protease inhibitor cocktail (Roche)). The samples were incubated on ice for 30 min and then centrifuged at 4 °C for 5 min at 10,000g. The protein concentration of the resulting supernatant was determined by a Bradford assay (RotiQuant, Roth K015.1) using a Tecan Spark 10M microplate reader. The final protein concentration was adjusted to 2.5 μg μl−1 with lysis buffer. The samples were mixed with Laemmli buffer and were incubated for 10 min at 65 °C. A total of 20 μg of each sample was loaded onto a 4–12% Bis-Tris gel (Invitrogen, NW04125BOX). Proteins were separated in MOPS running buffer (50 mM MOPS, 50 mM Tris, 1 mM EDTA, 0.1% (w/v) SDS) at 100 V and were then blotted in transfer buffer (25 mM Tris, 192 mM glycine and 10% ethanol) on an ethanol-activated PVDF membrane using a wet-transfer blotting system. The membrane was blocked in TBS-T (20 mM Tris pH 7.5, 150 mM NaCl and 0.1% Tween-20) containing 5% (w/v) milk powder. The membrane was then cut horizontally at 70 kDa, and the slices were stained for OPA1 (>70 kDa, BD Biosciences, 612607) and α-tubulin (<70 kDa, Abcam, ab7291) for 1 h at room temperature or overnight at 4 °C. After washing, the membranes were incubated with a goat anti-mouse IgG HRP-coupled secondary antibody (Millipore, AP127P). After washing with TBS-T, the detection was carried out using a chemiluminescent substrate (Thermo Fisher Scientific, 34578) and a CCD camera system (Amersham ImageQuant 680). All antibodies were diluted 1:500 except for the HRP-coupled secondary antibody, which was diluted 1:2,500.
Quantification from fluorescence microscopy
Cells from three independent experiments were manually scored and sorted by apparent phenotype into three categories: long tubular, short tubular/intermediate and fragmented. Another criterion, network distance from the nucleus, was also considered for quantification. The long tubular phenotype was categorized as the mitochondrial network spanning long distances from the nucleus and/or all or some of the cell has longer pieces of mitochondrial network regardless of apparent breaks in the network itself. The short tubular/intermediate phenotype was characterized by the cell showing not as fragmented (pieces not as circular) as the fragmented phenotype and or the cells containing a few (1 or 2) more elongated pieces. Moreover, the network does not span as long of a distance away from the nucleus when compared to the long tubular phenotype. Finally, the fragmented phenotype consisted of the mitochondrial network being completely broken apart into small circular network pieces, parts of the network were gone entirely or clustered around the nucleus. Each cell was assessed on the basis of an apparent mitochondrial network morphology and classified into one of the three categories. Furthermore, morphology analysis was performed by the same person to preserve consistency and eliminate bias.
MitoSegNet Image Processing
Images taken from three independent experiments were processed into MitoSegNet78 for automated confirmation of manual sorting. Using the pretrained model provided by the creators of MitoSegNet, the data were first confirmed to be a good fit with the pretrained images by comparing the results of the overall number of objects from MitoSegNet. ImageJ automated processing and manual counting resulted in nearly 100% accuracy across all three tests. Individual cells were separated into their own .tiff file and MitoS was used for the segmentation of each cell. After confirming the quality of the segmentation, MitoA was used to analyse the comparison of mitochondrial fragments, specifically looking at the average branch length, average eccentricity and average major axis length of the empty vector, WT, MIL-mutant and K819E, and Microsoft Excel was used to plot the graphical data.
Transmission electron microscopy analysis of ultrathin sections
Transfected Hela cells were sorted for medium mCherry expression on the FACSAria III (BD) system. Then, approximately 10,000 cells were seeded onto poly-l-lysine-coated 1.4 mm sapphire discs (Leica) and incubated for 24 h at 37 °C and 5% CO2. Subsequently, the cells were vitrified in a high-pressure freezing system (EM PACT2; Leica) and embedded in Lowcryl (Polysciences). All of the samples were processed in an automatic freeze-substitution apparatus (AFS2; Leica) and transferred into the precooled (−130 °C) freeze-substitution chamber of the AFS2. The temperature was increased from −130 to −90 °C over a 2 h period. Cryo-substitution was performed in anhydrous acetone and 2% water. The temperature was increased linearly from −90 °C to −70 °C over 20 h, and from −70 °C to −60 °C over 20 h. Increasing concentrations of Lowicryl (50%, 75% and 100%) were added stepwise to the samples at 1 h intervals followed by a 5 h incubation in 100% Lowicryl. Polymerization was carried out under ultraviolet light for 24 h followed by a slow increase of temperature over 15 h to 20 °C. Ultrathin (70 nm) sections were cut using an ultramicrotome (EM UC7; Leica), collected on Pioloform-coated copper grids, stained with uranyl acetate and lead citrate, and analysed with a Tecnai G2 Biotwin electron microscope (Thermo Fisher Scientific). The TEM images were acquired using the Olympus iTEM 5.0 image software (build 1243).
Sequence alignment of OPA1 proteins
Sequence conservation analysis was calculated using Clustal Omega79, and alignments were formatted using ESPript80. In brief, a multiple-sequence alignment of human OPA1 isoform 1 to its mammalian homologues and orthologues was generated using the WT OPA1 protein sequences obtained from UniProt. The representation of surface residue conservation of the OPA1 polymer was calculated using the ConSurf server81 supplied with the multiple-sequence alignment that was generated previously.
Extended Data
Extended Data Fig. 1 |. Purification and functional characterization of the human S-OPA1 construct.
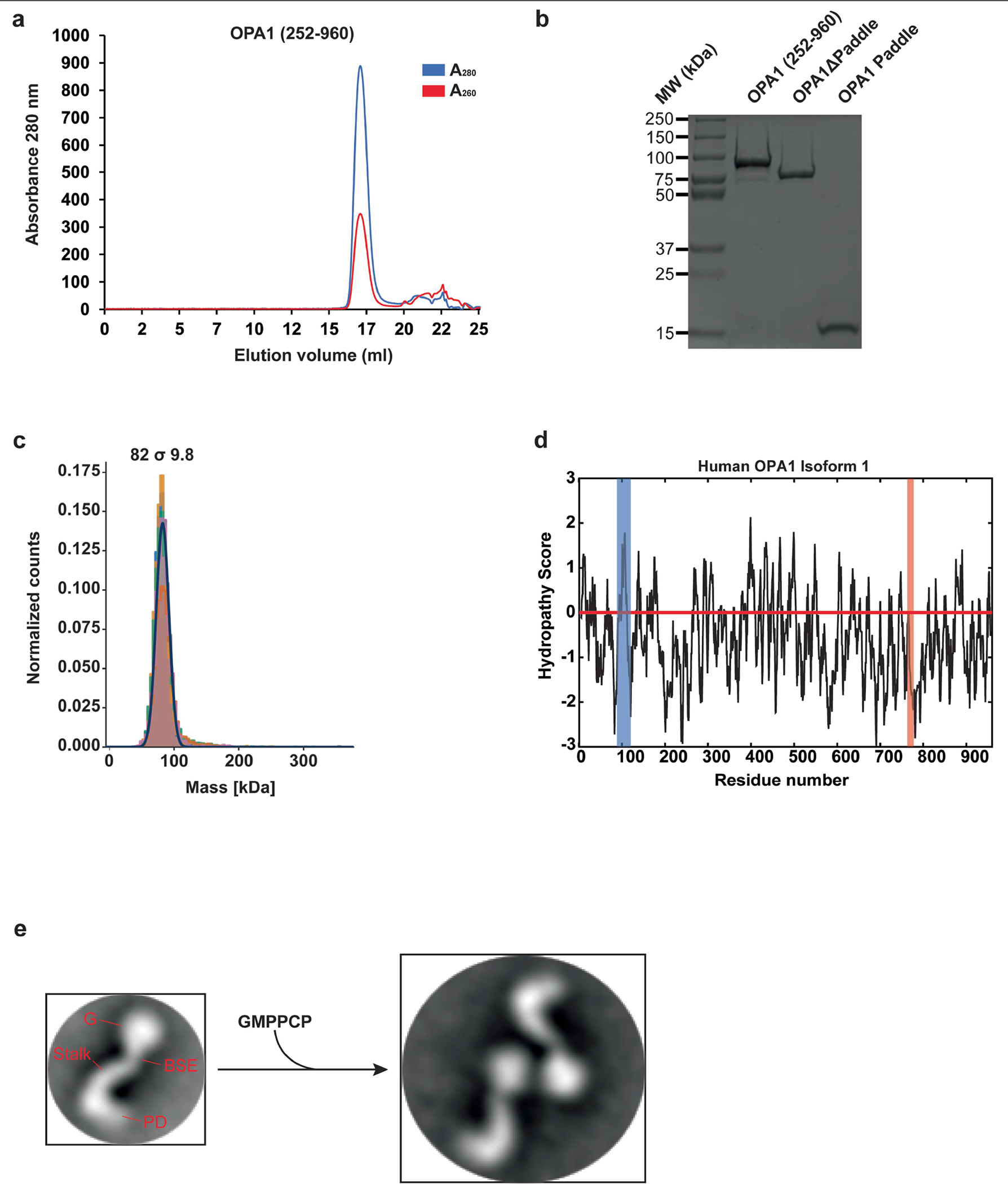
Using a recombinant Escherichia coli expression system, we expressed and purified human S-OPA1 (residues 252–960) in the presence of detergents by Ni2+-affinity and gel filtration chromatography. a, A representative size exclusion chromatogram of the human S-OPA1. b, Purified S-OPA1 constructs on an SDS-PAGE gel stained with Coomassie blue. Performed in technical triplicate. c, Mass photometry profile of nucleotide-free human S-OPA1 reveals an apparent molecular mass of 82 σ 9.8 kDa, which corresponds to a monomeric state. d, Kyte and Doolittle hydropathy plot of full-length human OPA1 isoform 1. Hydrophobic regions corresponding to the transmembrane (TM) region and fusion loop are highlighted in blue and red, respectively. The red line indicates the zero baseline on the hydropathy scale. e, To assess the proper folding and nucleotide-dependent dimerization of recombinant S-OPA1, we incubated the sample with the non-hydrolysable analogue GMPPCP, Mg2+, and K+. Then, using negative-stain transmission electron microscopy (TEM), we observed that S-OPA1 forms dimers via GTPase domain interactions. Representative negative-stain EM 2D class averages of human S-OPA1 from the negative-stain data collected on Tecnai T12 microscope equipped with CCD camera showing that particles have well-defined shapes with modular domain architecture. Monomeric human S-OPA1 forms G domain dimers in the presence of non-hydrolysable GTP analogue GMPPCP.
Extended Data Fig. 2 |. Reconstitution of human S-OPA1 assemblies and analysis by cryo-EM.
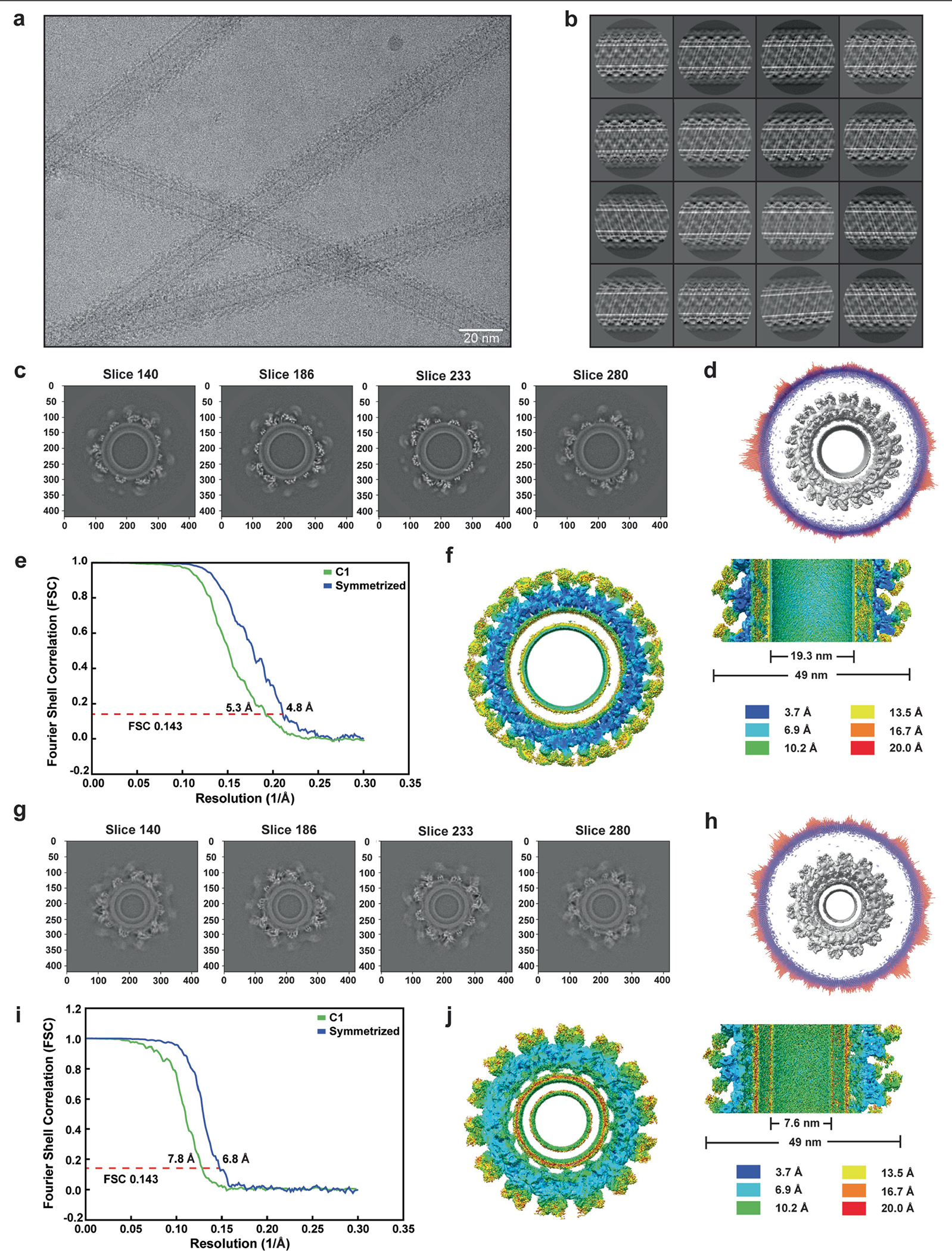
a, Representative motion-corrected electron micrograph of membrane nanotube-bound S-OPA1 filaments. b, Gallery of 2D class averages calculated from the cryo-EM data showing the S-OPA1 monomers assembled on the surface of cardiolipin-enriched lipid nanotubes. c, g, Slices through the unsharpened density map at distinct levels are shown in the top view. d, h, Angular distribution of the membrane-bound S-OPA1 filament for all particle images included in the calculation of the final 3D reconstruction of the membrane-proximal (d) and the membrane-distal conformations (h). e, i, Fourier shell coefficient (FSC) curves (threshold of 0.143) between two independently refined half maps before and after post-processing. f, j, Final 3D electron density maps of both structures are coloured according to local resolution and are shown in horizontal and vertical slices through cryo-EM densities. Local resolution was calculated by ResMap.
Extended Data Fig. 3 |. Flowchart for cryo-EM data processing of membrane-bound S-OPA1 assembly.
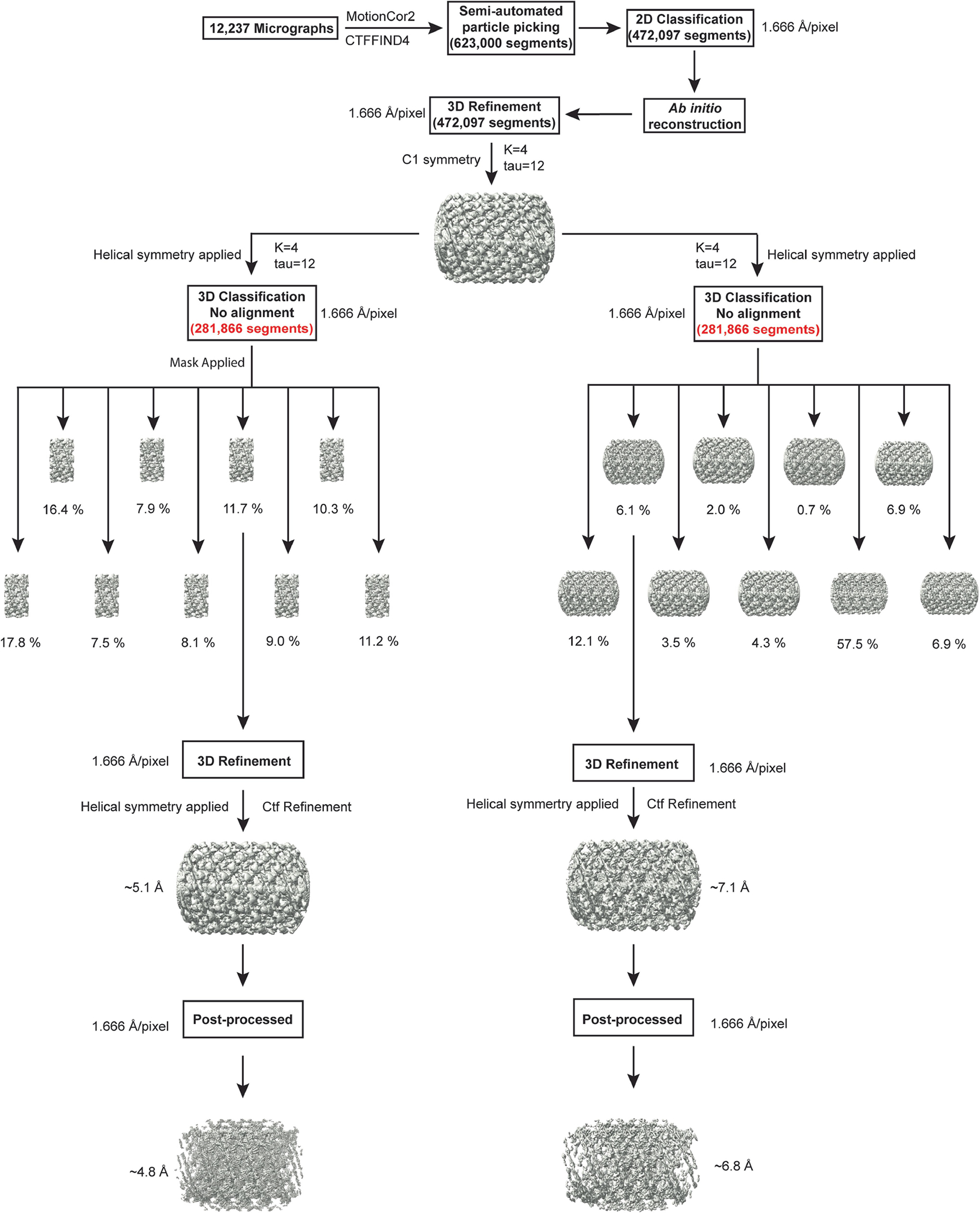
Details can be found in the image analysis and 3D reconstruction section of the Methods.
Extended Data Fig. 4 |. Crosslinks between all OPA1 subunits in membrane-bound conformation.
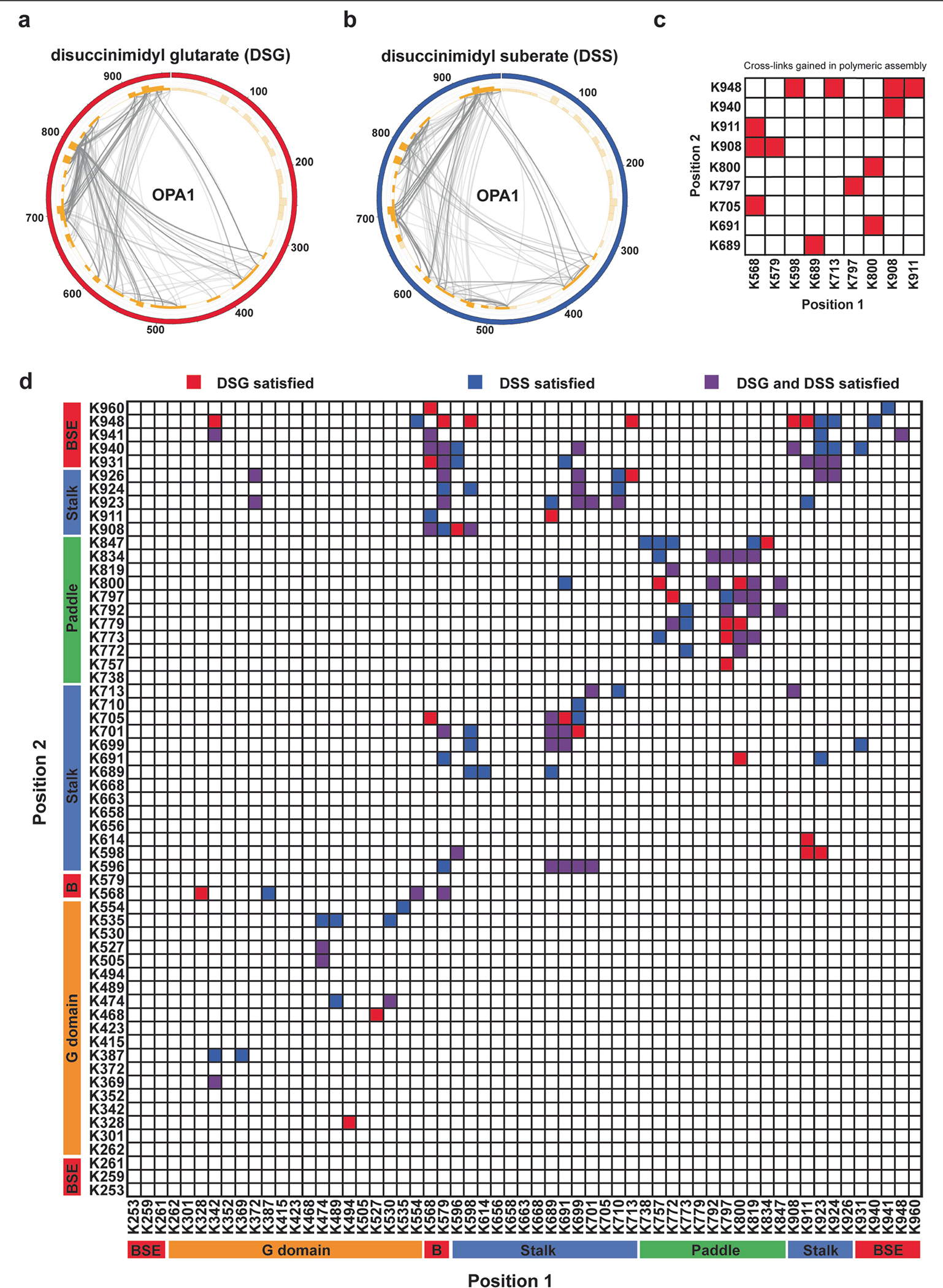
a, b, Chemical cross-linking and mass spectrometry reveals protein-protein interaction (PPI) maps of the human S-OPA1 polymers. Circular plot showing the distribution of DSG (a) and DSS (b) chemical crosslinks mapped to human OPA1 represented as coloured circles with amino acid positions labelled. Orange bars indicate the positions of lysine residues, and crosslinks connecting the lysine residues are represented by black lines between the corresponding amino acid pairs. c, Crosslinks that are gained (red) in the S-OPA1 polymer upon membrane binding. Identified inter-molecular crosslinks are mapped on the S-OPA1 subunits. CX-MS reveals a cluster of contacts between S-OPA1 paddle domains, indicating strong interactions in membrane-bound conformation. d, Crosslinks between all OPA1 domains. Domains are arranged based on sequence (coloured blocks). The DSG (red) and DSS (blue) crosslinks that satisfy the distance restraint (30 Å) are mapped onto the membrane-bound cryo-EM structure of human S-OPA1. Crosslinks identified in both DSG and DSS datasets are shown in purple.
Extended Data Fig. 5 |. Human S-OPA1 map quality, model building, and structural comparison with the yeast Mgm1.
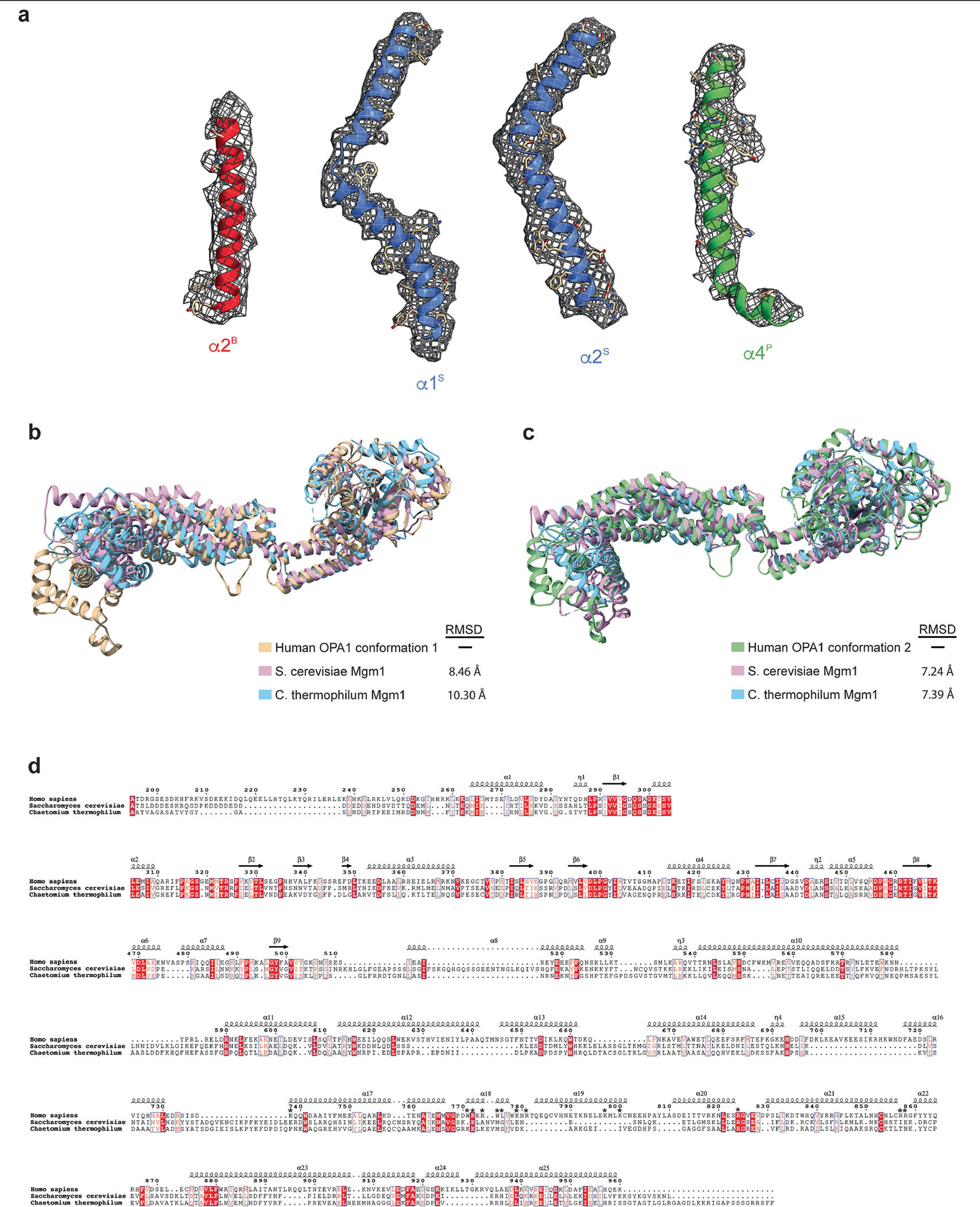
a, Isolated S-OPA1 monomer EM density from post-processed maps showing the quality of the map, build and fit. A single view of the cryo-EM density is depicted as a semi-transparent mesh and superimposed upon the model. Examples of model fit within B-factor sharpened cryo-EM density for the alpha-helices of S-OPA1 BSE (red), stalk (blue), paddle (green) are shown in the context of the atomic model with side chains are shown as sticks and the backbone as ribbons. b, A comparative analysis of the human S-OPA1 to yeast structures deposited in the PDB using topology independent comparison server CLICK revealed that the S-OPA1 structure displays a similar topology for the GTPase, BSE, and stalk domains; however, the paddle domain adopts a novel architecture with the addition of α3P helix, which facilitates the formation of interface 7. Structural comparison of membrane-proximal conformation of human S-OPA1 (beige) to S. cerevisiae (Pink, PDB ID: 6JSJ) and C. thermophilum (Light blue, PDB ID: 6QL4) Mgm1 crystal structures. Overall, the membrane-proximal conformation of human S-OPA1 and S. cerevisiae Mgm1 align with an r.m.s.d. of 8.46 Å, and human S-OPA1 and C. thermophilum Mgm1 align with an r.m.s.d. of 10.30 Å over all Cα atoms. c, Superimposition of membrane-distal conformation of human S-OPA1 (beige), S. cerevisiae (Pink), and C. thermophilum (Light blue) Mgm1 structures. Overall, the membrane-distal conformation of human S-OPA1 superimposes with S. cerevisiae Mgm1 and C. thermophilum Mgm1 with a rmsd of ~7.3 Å over all Cα atoms. d, Multiple sequence alignment of human S-OPA1, S. cerevisiae S-Mgm1, and C. thermophilum S-Mgm1. There is ~21% sequence conservation between human and yeast proteins. Most of the residues involved in binding to membranes are not conserved (highlighted with an asterisk).
Extended Data Fig. 6 |. Oligomerization and liposome deformation activity of wild-type S-OPA1 and mutants visualized with EM.

To gain further insights into the molecular mechanism of OPA1-mediated membrane remodelling, we reconstituted the human S-OPA1 polymer assembly in the presence of GMPPCP and CL-enriched membranes. Negative-stain electron micrographs showing the liposome binding and remodelling activity of wild-type and mutant S-OPA1 on cardiolipin-enriched liposomes and membrane nanotubes. S-OPA1 forms well-ordered filaments that wrapped around membrane tubules. Wild-type and mutant proteins were incubated with liposomes and membrane nanotubes for four hours at room temperature prior to grid preparation. Mutations to positively charged and hydrophobic residues within the membrane docking region, membrane-inserting loop (MIL), membrane-facing surface, and interface 7 resulted in severe defects in membrane binding and remodelling activity of human S-OPA1. Inset, close-up views of selected liposomes and membrane nanotubes. Images were taken in technical triplicate. Scale bars, 100 nm.
Extended Data Fig. 7 |. Liposome co-sedimentation experiments.
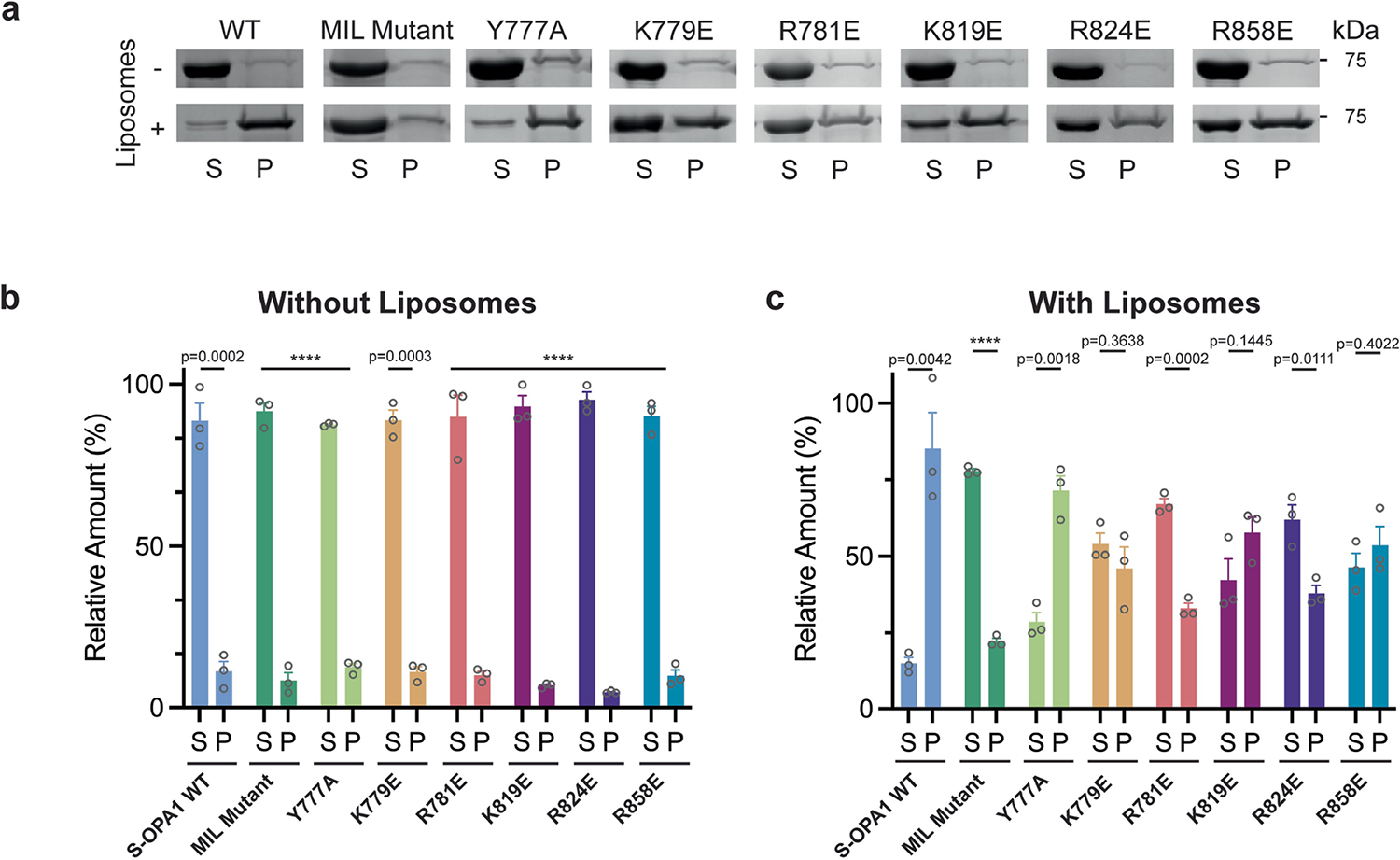
a, Representative SDS-PAGE gels showing the sedimentation of S-OPA1 WT and mutants in the presence and absence of CL-enriched liposomes. The samples were derived from the same experiment and analysed using multiple gels in parallel. P, pellet; S, supernatant; WT, wild type; MIL, membrane-inserting loop. b, c, Gel quantification depicting the relative amount of S-OPA1 (%) for pellet and supernatant fractions without liposomes (b) and with liposomes (c) in the co-sedimentation assays. The bar graphs represent the quantification of the Coomassie-stained protein bands from three biological replicates and expressed as mean ± s.e.m. Two-tailed unpaired t-tests were performed on each S-OPA1 variant comparing pellet verse supernatant. P < 0.0001 (****); P = 0.0002(***) or P = 0.0003(***); P = 0.001 (**); P = 0.01 (*); ns, not significant.
Extended Data Fig. 8 |. Mitochondrial morphology analyses for the WT, fusion loop, and disease mutants of OPA1.
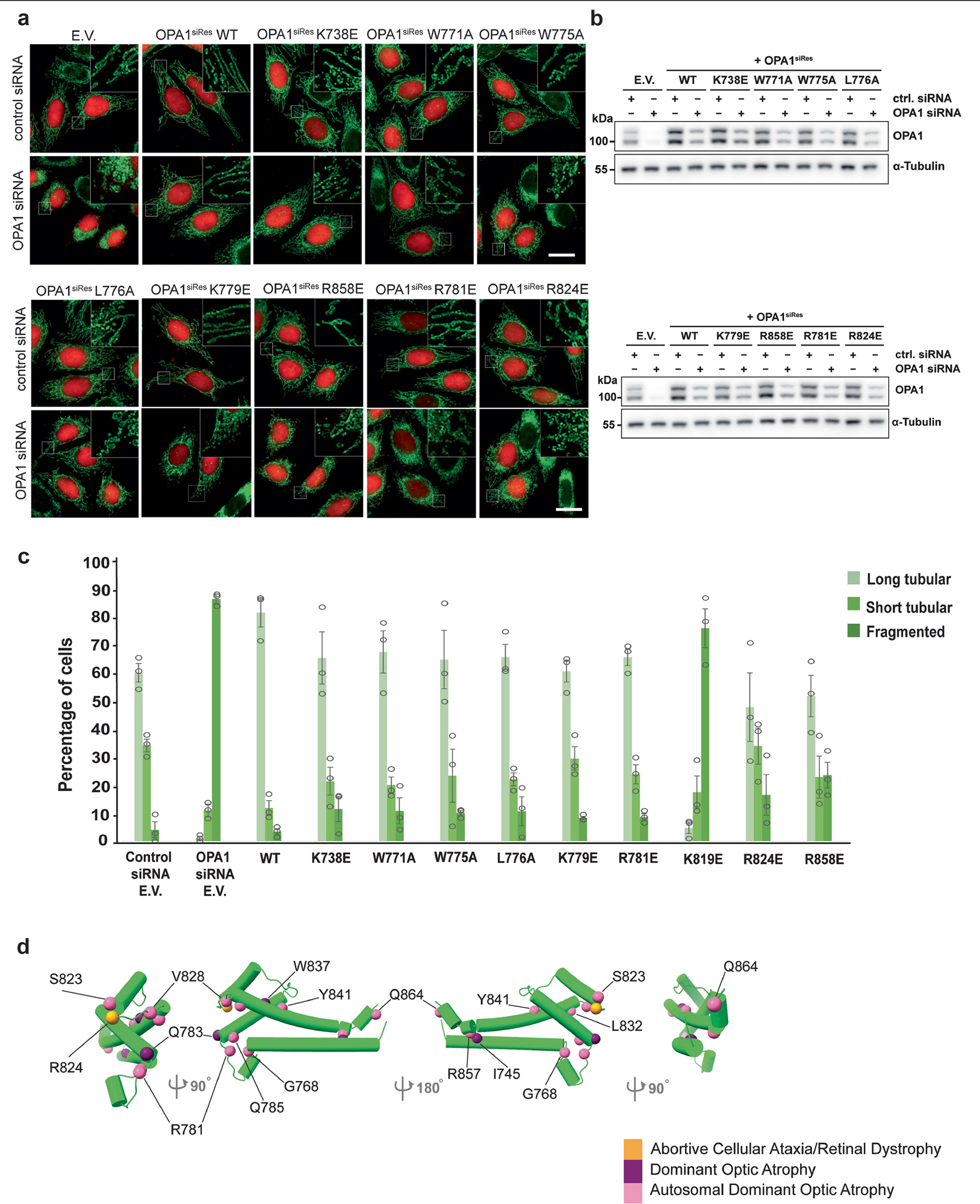
a, Fluorescence microscopy images of wild-type (WT), membrane docking (K738E, R858E), membrane-inserting loop (W771A, W775A, L776A, K779E), and disease (R781E, R824E) mutants in mitochondrial morphology analyses. HeLa cells were transfected with empty vector (E.V.) or the indicated siRNA-resistant OPA1 constructs for 24h and then subjected to RNA interference using the indicated siRNAs for 72h. The mitochondrial network was visualized by staining for the outer membrane protein TOMM22 (green), and co-transfected mCherry-NLS (red) was used to identify transfected cells. Performed in biological triplicate. Scale bar, 20 μm. b, Expression and stability of the siRNA-resistant OPA1 proteins were assessed by immunoblot analysis of Triton X-100 extracts derived from HeLa cells transfected with either empty vector (E.V.) or the indicated siRNA-resistant OPA1 constructs 24h before treatment with the indicated siRNAs for 72h. 20 μg per lane (n = 3 independent experiments). c, Quantification of microscopy images of wild-type, empty vector (E.V.), and mutants. Mitochondrial phenotypes observed in HeLa cells transiently expressing human OPA1 variants as described in a. Cells expressing the control siRNA E.V. (n = 280), OPA1 siRNA E.V. (n = 269 cells), wild-type OPA1 (n = 238 cells), OPA1 K738E (n = 235 cells), OPA1 W771A (n = 281 cells), OPA1 W775A (n = 264 cells), OPA1 W776A (n = 255 cells), OPA1 K779E (n = 282 cells), OPA1 R858E (n = 245 cells), OPA1 R781E (n = 262 cells), and OPA1 R824E (n = 283 cells) over three experimental replicates. Data points represent the average percentage of cells across three experimental replicates. Error bars indicate s.e.m. d, Human OPA1 mutations associated with optic atrophy, cerebellar ataxia, and other diseases mapped onto the paddle domain of OPA1 as solid spheres and numbered. Back and side views of the molecular structure of human S-OPA1 LBD (coloured in green).
Extended Data Fig. 9 |. OPA1 Mutations disrupt cristae architecture and overall mitochondrial morphology as compared to the WT.
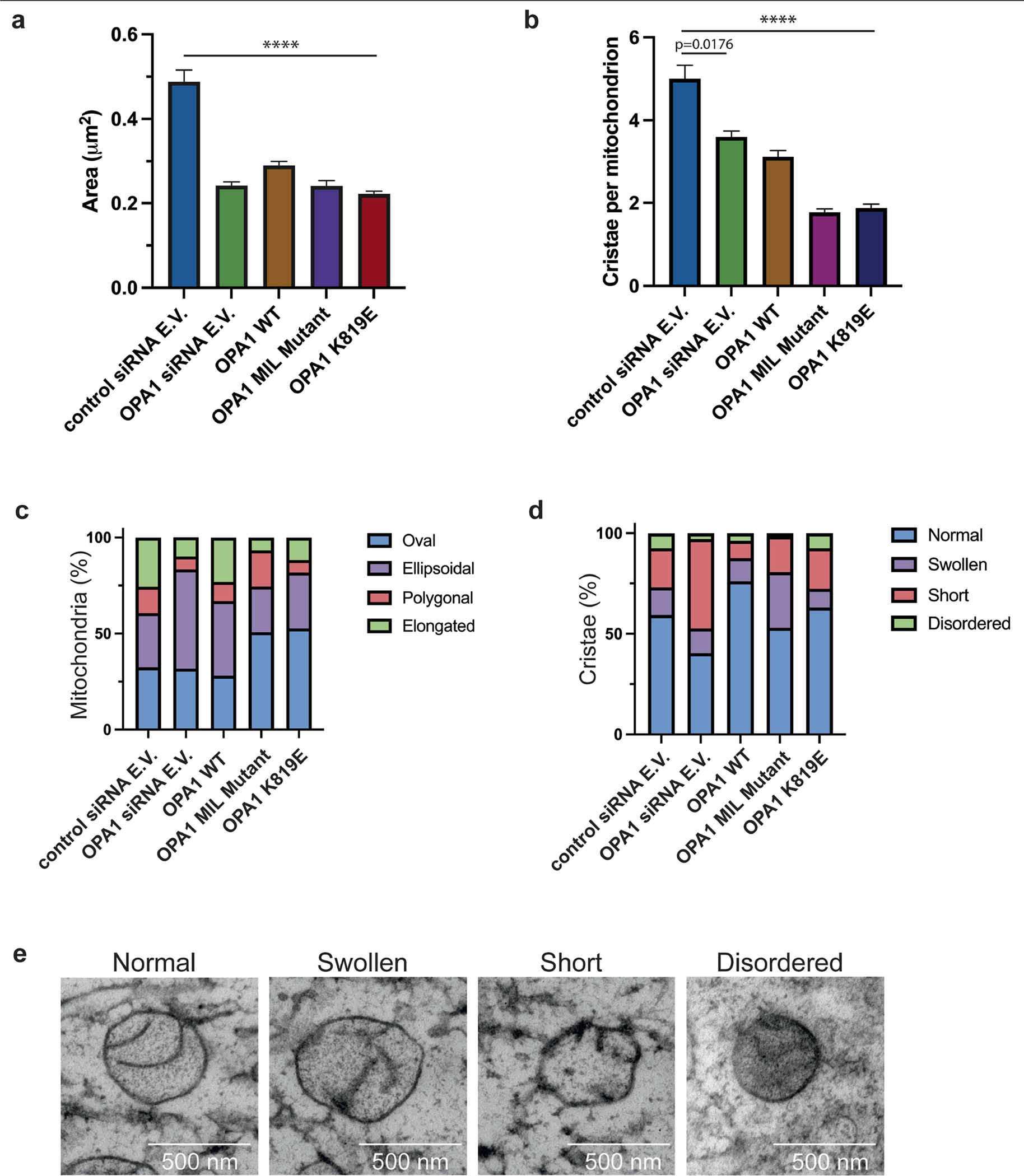
a, Mitochondrial surface area (μm2) was calculated from ultrathin sections of cells transfected with control siRNA empty vector (E.V.) (n = 312), OPA1 siRNA E.V. (n = 444), OPA1 WT (n = 543), OPA1 membrane-inserting loop (MIL) mutant (n = 523), and OPA1 K819E (n = 516). The standard error of the mean is reported with the mean of each dataset (n = 3 technical triplicates). Due to the variation in numbers for each dataset, two-tailed Mann-Whitney statistical test was utilized to calculate the statistical significance between control siRNA E.V. to each other sample. P < 0.0001 (****). b, Quantification of the number of cristae per mitochondrion. The mean number of cristae was calculated and reported with the standard error of the mean for each sample (n = 3 technical triplicates). The control siRNA E.V. and other samples were subjected to a two-tailed Mann-Whitney test to determine the statistical significance. P = 0.0176 (*); P < 0.0001 (****). c, Quantification of the mitochondrial morphology in WT and mutant OPA1 cells. The mitochondrial shape was assigned to four different classes, oval, ellipsoidal, polygonal, or elongated, and the relative distribution of each shape was reported as a bar graph. The percentage of each shape is reported for each sample. d, Quantification of cristae morphology in WT and mutant OPA1 cells. A bar graph representing the distribution of four different cristae morphology (normal, swollen, short, or disordered) observed in respective samples as percentages. e, Representative TEM images of the mitochondrion showing different types of cristae morphology assigned to four classes: normal, swollen, short, and disordered. Images were taken in technical triplicate. Scale bar, 500 nm.
Extended Data Fig. 10 |. S-OPA1 membrane binding and remodelling activities cause lipid bilayer deformations.
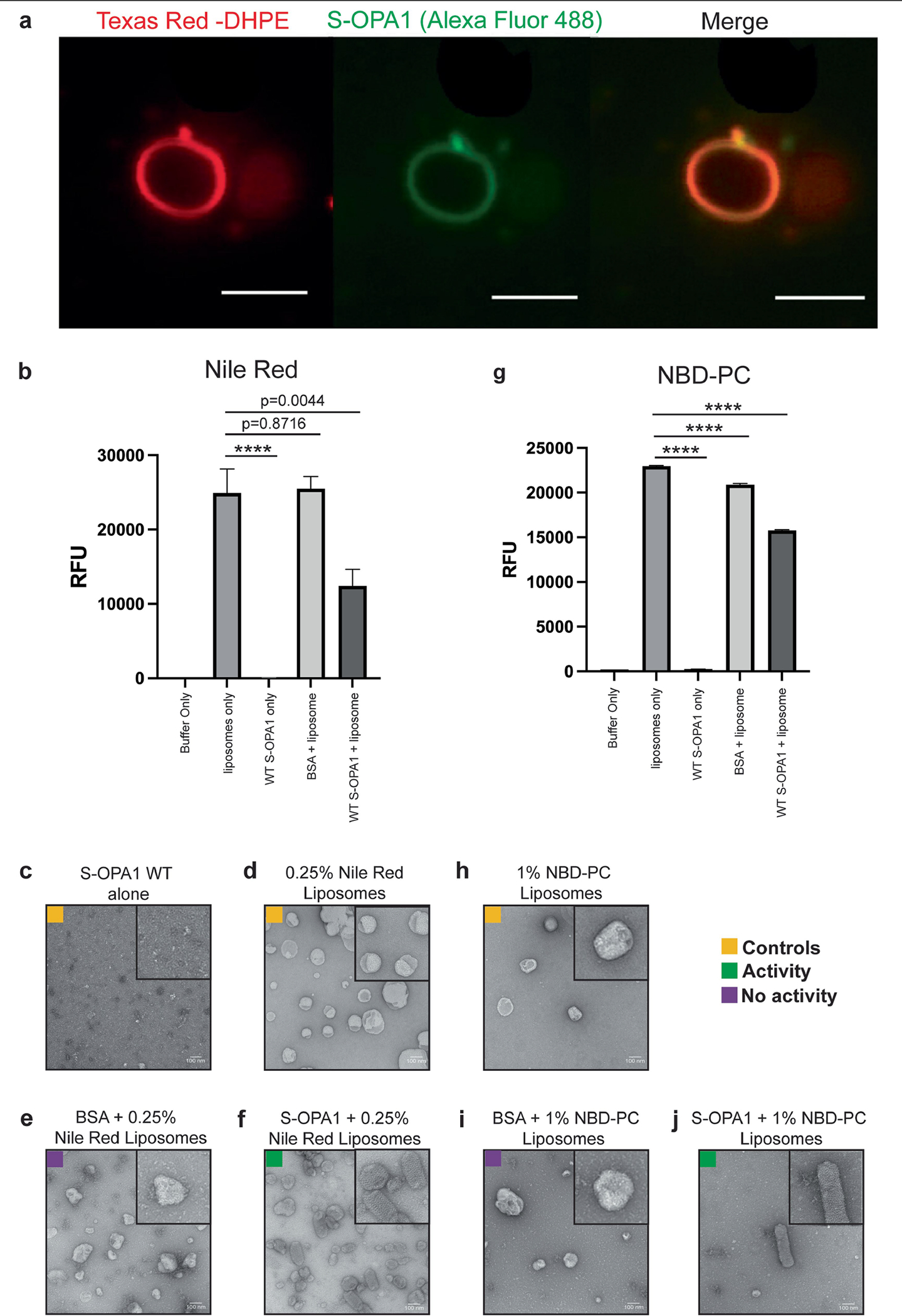
a, Representative fluorescent microscopy images show the co-localization of Alexa Fluor 488 labelled WT S-OPA1 onto the Texas Red-DHPE-containing liposomes after approximately 30min. Experiments performed in technical triplicate. Scale bar, 0.5 μm. b, Membrane deformation assays with WT S-OPA1 and liposomes containing 0.25% Nile Red; n = 5 biologically independent experiments and expressed as mean, error bars as ± s.e.m. Statistical analysis was performed using an unpaired two-tailed Student t-test (P = <0.0001, ****), and the Grubbs test removed one outlier from each dataset. c-f, Negative stain TEM analysis of membrane reconstitution assays with c, S-OPA1 alone; d, liposomes with 0.25% Nile Red alone; e, BSA with liposomes containing 0.25% Nile Red; and f, WT S-OPA1 with liposomes containing 0.25% Nile Red. g, Membrane deformation assay with WT S-OPA1 and liposomes containing 1% NBD-PC; n = 5 biologically independent experiments and expressed as mean error bars as ± s.e.m. An unpaired two-tailed Student t-test was performed on the NBD-PC dataset (P = <0.0001, ****). h, liposomes with 1% NBD-PC alone; i, BSA with liposomes containing 1% NBD-PC; and j, WT S-OPA1 with liposomes containing 1% NBD-PC. Inset, close-up view of selected liposomes from negative-stain TEM images. Scale bar, 100 nm. It has been proposed that the outer leaflet perturbations of CL-containing large unilamellar vesicles (LUV) result in vesicle fusion82. Atomic force microscopy (AFM) and fluorescence microscopy, moreover, have revealed that the incubation of yeast S-Mgm1 with labelled liposomes results in increased membrane “roughness” on the surface of liposomes37. These observations indicate that the yeast ortholog Mgm1 may utilize a similar mechanism to perturb membrane properties. Together, our structural and functional analyses suggest that OPA1-mediated leaflet perturbations support the membrane remodelling activity of the protein. We note that these conclusions are based on our current structural knowledge and are limited to lipids in synthetic membranes and reconstitution assays in vitro. To what extent these findings apply to mitochondrial membrane remodelling in human cells remains elusive. Our work provides the foundation to further study the complex mechanisms that regulate mitochondrial morphology and function.
Extended Data Table 1.
Cryo-EM data collection, refinement, and validation statistics
| Human S-0PA1 membrane-proximal conformation (EMDB-26977) (PDB 8CT1) | Human S-OPA1 membrane-distal conformation (EMDB-26984) (PDB 8CT9) | |
|---|---|---|
| Data collection and processing | ||
| Microscope | FEI Titan Krios | |
| Camera | Gatan K3 Summit | |
| Magnification | 105,000x | |
| Voltage (kV) | 300 | |
| Electron exposure (e−/Å2) | 65 | |
| Defocus (μm) | −0.5 to −1.2 | |
| Pixel size (Å) | 0.833 | |
| Symmetry imposed | helical | |
| Micrographs (no.) | 12,237 | |
| Initial particle images (no.) | 623,000 | |
| Symmetry expanded particle images (no.) | 4,248,873 | |
| Final particle images (no.) | 139,018 | 96,152 |
| Map resolution (Å) | 4.8 | 6.8 |
| FSC threshold | 0.143 | 0.143 |
| Map resolution range (Å) | 3.7 to >10.0 | 5.6 to >10.0 |
| Refinement | ||
| Initial model used (PDB code) | de novo - G domain (6JTG) | de novo - G domain (6JTG) |
| Model resolution (Å) | 4.7 | 7.8 |
| FSC threshold | 0.143 | 0.143 |
| Map sharpening B factor (Å2) | −198.51 | −200.00 |
| Model composition | ||
| Non hydrogen atoms | 193,290 | 198,307 |
| Protein residues | 23,732 | 23,732 |
| Lipids | 91 | |
| B factors (Å2) - min | ||
| Protein | 97.85 | 202.86 |
| Ligands | 390.35 | |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.001 | 0.006 |
| Bond angles (Å) | 0.309 | 0.570 |
| Validation | ||
| MolProbity score | 1.19 | 1.17 |
| Clashscore | 4.06 | 3.82 |
| Poor rotamers (%) | 0.0 | 0.0 |
| Ramachandran plot | ||
| Favored (%) | 99.57 | 98.60 |
| Allowed (%) | 0.43 | 1.40 |
| Disallowed (%) | 0.0 | 0.0 |
Supplementary Material
Acknowledgements
We thank L. Doan for reagent preparation; the staff of the W.M. Keck Foundation Advanced Microscopy laboratory of the University of California, San Francisco, A. Myasnikov, D. Bulkley and Z. Yu for help with data collection; E. Paraskevi Tsiolaki for technical assistance with mass photometry; P. Thomas for computational support; E. Krause for assistance with cell sorting; S. Jungbluth for acquiring the TEM images of cells; G. Morgan and C. Ozzello for electron microscopy training and support; the staff at the Shared Instrument Pool (SIP) core facility (SCR_018986) of the Department of Biochemistry at the University of Colorado Boulder for the use of the shared research instrumentation infrastructure; A. Erbse for assistance with biophysical instruments and support; the members of the Biofrontiers Advanced Light Microscopy Core for the use of laser confocal microscopes; J. Dragavon for training and support; K. Luger and J. Rudolph for their support and for sharing the microplate reader for the fluorescence-based assays; M. Ford, K. Faelber, V. Gama and C. Hayes for reading the manuscript; and O. Daumke and the members of the Aydin and Kasinath laboratories for technical advice and discussions. This work was supported in part by American Heart Association Postdoctoral Fellowship 23POST1020756 (to K.E.Z.), Boettcher Foundation Webb-Waring Biomedical Research Award (H.A.), a National Institute of Health grant R35 GM150942 (to H.A.), Deutsche Forschungsgemeinschaft (DFG), Collaborative Research Center 894, project A20 (to M.v.d.L.), a National Institute of Health grant R01 GM127673 (to A.F.), a Faculty Scholar Grant from the HHMI (to A.F.) and a QBI-FUN Collaborative Integrative Structural Biology Grant (to A.F.). A.F. is an alumni investigator of the Chan Zuckerburg Biohub.
Footnotes
Competing interests A.F. is a shareholder and employee of Altos Labs and a shareholder and consultant for Relay Therapeutics. The other authors declare no competing interests.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41586-023-06441-6.
Peer review information Nature thanks Thomas Langer and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Reprints and permissions information is available at http://www.nature.com/reprints.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All of the 3D cryo-EM data supporting the findings of this study have been deposited in Electron Microscopy Data Bank under accession codes EMD-26977 and EMDB-26984. The model coordinates have been deposited at the PDB under accession codes 8CT1 and 8CT9. Protein sequence data for sequence alignments are available from UniProt (see the figure legends for accession codes). OPA1 sequences used in this study are as follows: human (UniProt: O60313), Chlorocebus sabaeus (green monkey; UniProt: A0A0D9R952), Macaca mulatta (rhesus macaque; UniProt: F6Y1N8), Pan troglodytes (Chimpanzee; UniProt: A0A2I3SKT2), Gorilla gorilla (gorilla; UniProt: G3S1U3), Pan paniscus (bonobo; UniProt: A0A2R9BDG8), Papio anubis (baboon; UniProt: A0A096N399), Callithrix jacchus (marmoset; UniProt: A0A2R8PC53), Oryctolagus cuniculus (rabbit; UniProt: G1TAB7), Ictidomys tridecemlineatus (squirrel; UniProt: I3MI89), Cavia porcellus (guinea pig; UniProt: H0V6M3), Mus musculus (mouse; UniProt: P58281), Rattus norvegicus (rat; UniProt: Q2TA68), Canis familiaris (dog, UniProt: F1PK93), Vulpes vulpes (red fox, UniProt: A0A3Q7T0T6), Felis catus (cat; UniProt: A0A337SN50), Ailuropoda melanoleuco (cat; UniProt: G1MBN4), Sus scrofa (pig; UniProt: A0A5G2QQR2), Loxodonta africana (African elephant; UniProt: G3SNG0), Equus caballus (horse; UniProt: F6Z2C8), Vicugna pacos (alpaca; UniProt: A0A6I9I1B0), Bos taurus (cow; UniProt: E1BBC4), Capra hircus (goat; UniProt: A0A452EKR4), Ovis aries (sheep; UniProt: A0A6P7D299), Desmodus rotundus (vampire bat; UniProt: K9J3D6), Tursiops truncatus (dolphin; UniProt: A0A2U4ACH9), Delphinapterus leucas (beluga whale; UniProt: A0A2Y9MT19), Danio rerio (zebrafish; UniProt: Q5U3A7), Oncorhynchus masou (salmon; UniProt: O93248), Gallus gallus (chicken; UniProt: Q5F499) and Meleagris gallopavo (wild turkey; UniProt: G3UT81). Full versions of all of the gels and blots are provided in Supplementary Fig. 1. Source data are provided with this paper.
References
- 1.Wai T & Langer T Mitochondrial dynamics and metabolic regulation. Trends Endocrinol. Metab. 27, 105–117 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Quintana-Cabrera R & Scorrano L Determinants and outcomes of mitochondrial dynamics. Mol. Cell 83, 857–876 (2023). [DOI] [PubMed] [Google Scholar]
- 3.Eisner V, Picard M & Hajnóczky G Mitochondrial dynamics in adaptive and maladaptive cellular stress responses. Nat. Cell Biol. 20, 755–765 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Olichon A et al. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J. Biol. Chem. 278, 7743–7746 (2003). [DOI] [PubMed] [Google Scholar]
- 5.Griparic L, van der Wel NN, Orozco IJ, Peters PJ & van der Bliek AM Loss of the intermembrane space protein Mgm1/OPA1 induces swelling and localized constrictions along the lengths of mitochondria. J. Biol. Chem. 279, 18792–18798 (2004). [DOI] [PubMed] [Google Scholar]
- 6.Carlton JG, Jones H & Eggert US Membrane and organelle dynamics during cell division. Nat. Rev. Mol. Cell Biol. 21, 151–166 (2020). [DOI] [PubMed] [Google Scholar]
- 7.Stefan CJ et al. Membrane dynamics and organelle biogenesis—lipid pipelines and vesicular carriers. BMC Biol. 15, 102 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aydin H, Sultana A, Li S, Thavalingam A & Lee JE Molecular architecture of the human sperm IZUMO1 and egg JUNO fertilization complex. Nature 534, 562–565 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Giacomello M, Pyakurel A, Glytsou C & Scorrano L The cell biology of mitochondrial membrane dynamics. Nat. Rev. Mol. Cell Biol. 21, 204–224 (2020). [DOI] [PubMed] [Google Scholar]
- 10.Kalia R et al. Structural basis of mitochondrial receptor binding and constriction by DRP1. Nature 558, 401–405 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cao Y-L et al. MFN1 structures reveal nucleotide-triggered dimerization critical for mitochondrial fusion. Nature 542, 372–376 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chan DC Mitochondrial dynamics and its involvement in disease. Annu. Rev. Pathol. Mech. Dis. 15, 235–259 (2020). [DOI] [PubMed] [Google Scholar]
- 13.Alexander C et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat. Genet. 26, 211–215 (2000). [DOI] [PubMed] [Google Scholar]
- 14.Delettre C et al. Nuclear gene OPA1, encoding a mitochondrial dynamin- related protein, is mutated in dominant optic atrophy. Nat. Genet. 26, 4 (2000). [DOI] [PubMed] [Google Scholar]
- 15.Cipolat S, de Brito OM, Dal Zilio B & Scorrano L OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl Acad. Sci. USA 101, 15927–15932 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cogliati S et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 155, 160–171 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wai T et al. Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science 350, aad0116 (2015). [DOI] [PubMed] [Google Scholar]
- 18.Herkenne S et al. Developmental and tumor angiogenesis requires the mitochondria-shaping protein Opa1. Cell Metab. 31, 987–1003 (2020). [DOI] [PubMed] [Google Scholar]
- 19.Gómez-Valadés AG et al. Mitochondrial cristae-remodeling protein OPA1 in POMC neurons couples Ca2+ homeostasis with adipose tissue lipolysis. Cell Metab. 33, 1820–1835 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen L et al. OPA1 mutation and late-onset cardiomyopathy: mitochondrial dysfunction and mtDNA instability. J. Am. Heart Assoc. 1, e003012 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zerem A et al. Metabolic stroke in a patient with bi-allelic OPA1 mutations. Metab. Brain Dis. 34, 1043–1048 (2019). [DOI] [PubMed] [Google Scholar]
- 22.Bonneau D et al. Early-onset Behr syndrome due to compound heterozygous mutations in OPA1. Brain 137, e301 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Carelli V et al. Syndromic Parkinsonism and dementia associated with OPA1 missense mutations. Ann. Neurol. 78, 21–38 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Del Dotto V et al. OPA1 isoforms in the hierarchical organization of mitochondrial functions. Cell Rep. 19, 2557–2571 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Mishra P, Carelli V, Manfredi G & Chan DC Proteolytic cleavage of Opa1 stimulates mitochondrial inner membrane fusion and couples fusion to oxidative phosphorylation. Cell Metab. 19, 630–641 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anand R et al. The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J. Cell Biol. 204, 919–929 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ban T, Heymann JAW, Song Z, Hinshaw JE & Chan DC OPA1 disease alleles causing dominant optic atrophy have defects in cardiolipin-stimulated GTP hydrolysis and membrane tubulation. Hum. Mol. Genet. 19, 2113–2122 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Song Z, Chen H, Fiket M, Alexander C & Chan DC OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J. Cell Biol. 178, 749–755 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Glytsou C et al. Optic atrophy 1 is epistatic to the core MICOS component MIC60 in mitochondrial cristae shape control. Cell Rep. 17, 3024–3034 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cipolat S et al. Mitochondrial rhomboid PARL regulates cytochrome c release during apoptosis via OPA1-dependent cristae remodeling. Cell 126, 163–175 (2006). [DOI] [PubMed] [Google Scholar]
- 31.Ban T et al. Molecular basis of selective mitochondrial fusion by heterotypic action between OPA1 and cardiolipin. Nat. Cell Biol. 19, 856–863 (2017). [DOI] [PubMed] [Google Scholar]
- 32.Zhang D et al. Cryo-EM structures of S-OPA1 reveal its interactions with membrane and changes upon nucleotide binding. eLife 9, e50294 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ge Y et al. Two forms of Opa1 cooperate to complete fusion of the mitochondrial inner-membrane. eLife 9, e50973 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu C et al. Structural insights into G domain dimerization and pathogenic mutation of OPA1. J. Cell Biol. 219, e201907098 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Faelber K et al. Structure and assembly of the mitochondrial membrane remodelling GTPase Mgm1. Nature 571, 429–433 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yan L et al. Structural analysis of a trimeric assembly of the mitochondrial dynamin-like GTPase Mgm1. Proc. Natl Acad. Sci. USA 117, 4061–4070 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rujiviphat J et al. Mitochondrial genome maintenance 1 (Mgm1) protein alters membrane topology and promotes local membrane bending. J. Mol. Biol. 427, 2599–2609 (2015). [DOI] [PubMed] [Google Scholar]
- 38.Faelber K et al. Crystal structure of nucleotide-free dynamin. Nature 477, 556–560 (2011). [DOI] [PubMed] [Google Scholar]
- 39.Low HH, Sachse C, Amos LA & Löwe J Structure of a bacterial dynamin-like protein lipid tube provides a mechanism for assembly and membrane curving. Cell 139, 1342–1352 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ramachandran R & Schmid SL Real-time detection reveals that effectors couple dynamin’s GTP-dependent conformational changes to the membrane. EMBO J. 27, 27–37 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Daumke O et al. Architectural and mechanistic insights into an EHD ATPase involved in membrane remodelling. Nature 449, 923–927 (2007). [DOI] [PubMed] [Google Scholar]
- 42.Low HH & Löwe J Dynamin architecture—from monomer to polymer. Curr. Opin. Struct. Biol. 20, 791–798 (2010). [DOI] [PubMed] [Google Scholar]
- 43.Gao S et al. Structural basis of oligomerization in the stalk region of dynamin-like MxA. Nature 465, 502–506 (2010). [DOI] [PubMed] [Google Scholar]
- 44.Alvarez FJD et al. CryoEM structure of MxB reveals a novel oligomerization interface critical for HIV restriction. Sci. Adv. 3, e1701264 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu-Wai-Man P et al. Multi-system neurological disease is common in patients with OPA1 mutations. Brain 133, 771–786 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu T et al. CHCHD10-regulated OPA1-mitofilin complex mediates TDP-43-induced mitochondrial phenotypes associated with frontotemporal dementia. FASEB J. 34, 8493–8509 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chappie JS et al. A pseudoatomic model of the dynamin polymer identifies a hydrolysis-dependent powerstroke. Cell 147, 209–222 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Antonny B et al. Membrane fission by dynamin: what we know and what we need to know. EMBO J. 35, 2270–2284 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Studier FW Protein production by auto-induction in high-density shaking cultures. Protein Expr. Purif. 41, 207–234 (2005). [DOI] [PubMed] [Google Scholar]
- 50.Ardail D et al. Mitochondrial contact sites. Lipid composition and dynamics. J. Biol. Chem. 265, 18797–18802 (1990). [PubMed] [Google Scholar]
- 51.Bennett JA, Steward LR, Rudolph J, Voss AP & Aydin H The structure of the human LACTB filament reveals the mechanisms of assembly and membrane binding. PLoS Biol. 20, e3001899 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.von der Malsburg A, Abutbul-Ionita I, Haller O, Kochs G & Danino D Stalk domain of the dynamin-like MxA GTPase protein mediates membrane binding and liposome tubulation via the unstructured L4 Loop. J. Biol. Chem. 286, 37858–37865 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schindelin J et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Manicki M et al. Structure and functionality of a multimeric human COQ7:COQ9 complex. Mol. Cell 82, 4307–4323 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rohou A & Grigorieff N CTFFIND4: fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zivanov J et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mastronarde DN Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005). [DOI] [PubMed] [Google Scholar]
- 58.Zheng SQ et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.He S & Scheres SHW Helical reconstruction in RELION. J. Struct. Biol. 198, 163–176 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shaikh TR et al. SPIDER image processing for single-particle reconstruction of biological macromolecules from electron micrographs. Nat. Protoc. 3, 1941–1974 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Emsley P, Lohkamp B, Scott WG & Cowtan K Features and development of Coot. Acta Crystallogr. D 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pettersen EF et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 63.Liebschner D et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D 75, 861–877 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Alford RF et al. The Rosetta all-atom energy function for macromolecular modeling and design. J. Chem. Theory Comput. 13, 3031–3048 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Williams CJ et al. MolProbity: more and better reference data for improved all-atom structure validation. Protein Sci. 27, 293–315 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jurrus E et al. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 27, 112–128 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Krissinel E & Henrick K Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 (2007). [DOI] [PubMed] [Google Scholar]
- 68.Holm L & Rosenström P Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pettersen EF et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bui KH et al. Integrated structural analysis of the human nuclear pore complex scaffold. Cell 155, 1233–1243 (2013). [DOI] [PubMed] [Google Scholar]
- 71.Leitner A et al. Expanding the chemical cross-linking toolbox by the use of multiple proteases and enrichment by size exclusion chromatography. Mol. Cell. Proteom. 11, M111.014126 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bern M, Kil YJ & Becker C Byonic: advanced peptide and protein identification software. Curr. Protoc. Bioinform. 40, 13.20.1–13.20.14 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Combe CW, Fischer L & Rappsilber J xiNET: cross-link network maps with residue resolution. Mol. Cell. Proteom. 14, 1137–1147 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kosinski J et al. Xlink Analyzer: software for analysis and visualization of cross-linking data in the context of three-dimensional structures. J. Struct. Biol. 189, 177–183 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jo S, Kim T, Iyer VG & Im W CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 29, 1859–1865 (2008). [DOI] [PubMed] [Google Scholar]
- 76.Abraham MJ et al. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25 (2015). [Google Scholar]
- 77.Humphrey W, Dalke A & Schulten K VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38 (1996). [DOI] [PubMed] [Google Scholar]
- 78.Fischer CA et al. MitoSegNet: easy-to-use deep learning segmentation for analyzing mitochondrial morphology. iScience 23, 101601 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sievers F et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Robert X & Gouet P Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ashkenazy H et al. ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 44, W344–W350 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lee J & Lentz BR Outer leaflet-packing defects promote poly(ethylene glycol)-mediated fusion of large unilamellar vesicles. Biochemistry 36, 421–431 (1997). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All of the 3D cryo-EM data supporting the findings of this study have been deposited in Electron Microscopy Data Bank under accession codes EMD-26977 and EMDB-26984. The model coordinates have been deposited at the PDB under accession codes 8CT1 and 8CT9. Protein sequence data for sequence alignments are available from UniProt (see the figure legends for accession codes). OPA1 sequences used in this study are as follows: human (UniProt: O60313), Chlorocebus sabaeus (green monkey; UniProt: A0A0D9R952), Macaca mulatta (rhesus macaque; UniProt: F6Y1N8), Pan troglodytes (Chimpanzee; UniProt: A0A2I3SKT2), Gorilla gorilla (gorilla; UniProt: G3S1U3), Pan paniscus (bonobo; UniProt: A0A2R9BDG8), Papio anubis (baboon; UniProt: A0A096N399), Callithrix jacchus (marmoset; UniProt: A0A2R8PC53), Oryctolagus cuniculus (rabbit; UniProt: G1TAB7), Ictidomys tridecemlineatus (squirrel; UniProt: I3MI89), Cavia porcellus (guinea pig; UniProt: H0V6M3), Mus musculus (mouse; UniProt: P58281), Rattus norvegicus (rat; UniProt: Q2TA68), Canis familiaris (dog, UniProt: F1PK93), Vulpes vulpes (red fox, UniProt: A0A3Q7T0T6), Felis catus (cat; UniProt: A0A337SN50), Ailuropoda melanoleuco (cat; UniProt: G1MBN4), Sus scrofa (pig; UniProt: A0A5G2QQR2), Loxodonta africana (African elephant; UniProt: G3SNG0), Equus caballus (horse; UniProt: F6Z2C8), Vicugna pacos (alpaca; UniProt: A0A6I9I1B0), Bos taurus (cow; UniProt: E1BBC4), Capra hircus (goat; UniProt: A0A452EKR4), Ovis aries (sheep; UniProt: A0A6P7D299), Desmodus rotundus (vampire bat; UniProt: K9J3D6), Tursiops truncatus (dolphin; UniProt: A0A2U4ACH9), Delphinapterus leucas (beluga whale; UniProt: A0A2Y9MT19), Danio rerio (zebrafish; UniProt: Q5U3A7), Oncorhynchus masou (salmon; UniProt: O93248), Gallus gallus (chicken; UniProt: Q5F499) and Meleagris gallopavo (wild turkey; UniProt: G3UT81). Full versions of all of the gels and blots are provided in Supplementary Fig. 1. Source data are provided with this paper.