

Abstract
Partial endothelial-to-mesenchymal transition (EndMT) is an intermediate phenotype observed in endothelial cells (ECs) undergoing a transition toward a mesenchymal state to support neovascularization during (patho)physiological angiogenesis. Here, we investigated the occurrence of partial EndMT in ECs under hypoxic/ischemic conditions and identified general transcription factor IIH subunit 4 (GTF2H4) as a positive regulator of this process. In addition, we discovered that GTF2H4 collaborates with its target protein excision repair cross-complementation group 3 (ERCC3) to co-regulate partial EndMT. Furthermore, by using phosphorylation proteomics and site-directed mutagenesis, we demonstrated that GTF2H4 was involved in the phosphorylation of receptor coactivator 3 (NCOA3) at serine 1330, which promoted the interaction between NCOA3 and p65, resulting in the transcriptional activation of NF-κB and the NF-κB/Snail signaling axis during partial EndMT. In vivo experiments confirmed that GTF2H4 significantly promoted partial EndMT and angiogenesis after ischemic injury. Collectively, our findings reveal that targeting GTF2H4 is promising for tissue repair and offers potential opportunities for treating hypoxic/ischemic diseases.
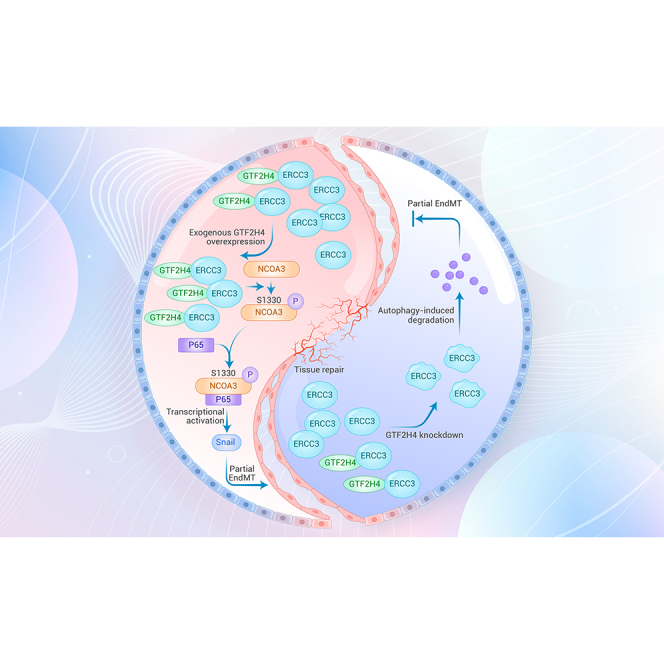
Graphical abstract
Public summary
-
•
Partial endothelial-to-mesenchymal transition (EndMT) helps create new blood vessels and repair tissues after a lack of blood flow.
-
•
GTF2H4 works together with ERCC3 to encourage partial EndMT through the NCOA3/NF-κB/Snail pathway.
-
•
Targeting endothelial GTF2H4 could be a hopeful treatment for diseases caused by insufficient blood supply.
Introduction
Ischemic diseases, which are associated with high morbidity and mortality worldwide, are characterized by obstruction or restriction of blood flow due to hypoxia/ischemia in various tissues and organs. These conditions include ischemic heart disease, cerebral ischemic disease, and peripheral arterial disease.1,2,3 The multiple pathological mechanisms triggered by hypoxia in these ischemic diseases include the inflammatory response, oxidative stress, endothelial dysfunction, necrosis, apoptosis, and fibrosis.4,5 In addition, the endothelial-to-mesenchymal transition (EndMT) is a dynamic biological process in endothelial cells (ECs) that is involved in the occurrence or progression of ischemic diseases, especially in atherosclerosis6,7 and fibrosis after myocardial infarction (MI),8,9 and is characterized by the loss of endothelial properties such as tubule formation and endothelial adherens junctions, the acquisition of mesenchymal features such as migratory capacity, and the ability to detach and migrate away from the endothelium.10 Moreover, EndMT plays a critical role in the formation of endocardial cushions as precursor structures for vasculogenesis and the alignment of cardiac chambers11,12 and is involved in other adult cardiovascular pathologies, such as valvular disease,13 fibroelastosis,14 and pulmonary arterial hypertension.15,16 Under these pathological conditions, hypoxia, inflammation, oxidative stress, high blood sugar, and low shear stress can individually or synergistically trigger EndMT.17
In contrast to complete mesenchymal transition, partial or reversible EndMT results in an intermediate phenotype in ECs undergoing mesenchymal transition and is characterized by the retention or partial loss of endothelial characteristics and the acquisition of mesenchymal characteristics, such as increased motility.18 Notably, neovascularization plays a pivotal role in postischemic functional recovery in ischemic diseases. Coincidentally, a momentarily balancing process occurs in ECs during sprouting angiogenesis known as partial EndMT.18 Previous studies have highlighted the overlapping biological behaviors between angiogenesis and partial or reversible EndMT,18,19,20 suggesting that angiogenesis is a unique example of partial EndMT.21 More recently, single-cell transcriptomic studies have provided insights into the transient and dynamic nature of partial EndMT in ischemic models, including MI and hindlimb ischemia. These studies revealed mesenchymal gene enrichment in regions of clonally expanded vessels, suggesting that hypoxia-induced partial EndMT could contribute to neovascularization under ischemic conditions.22,23
Under ischemic conditions, dynamic time-dependent changes in the gene profile associated with transient mesenchymal activation shows that some genes can participate in pathological microenvironment-induced neovascularization,24 which occurs during conditions such as ischemia, hypoxia, or inflammation. Therefore, discovering the molecules involved in hypoxia-induced partial EndMT may be meaningful in these diseases. In a previous study, time course transcriptome data from peripheral blood samples of patients with ST-segment elevation MI (STEMI) before and after percutaneous coronary intervention (PCI) indicated that general transcription factor IIH subunit 4 (GTF2H4) was an active and responsive transcriptional regulator associated with ischemia/reperfusion.25 TFIIH is a multiprotein complex composed of 10 subunits that is part of the CORE and cyclin-dependent kinase–activating kinase subcomplex. As one of the subunits of the CORE subcomplex, GTF2H4 can collaborate with adjacent subunits to participate in cellular processes, including transcription initiation, DNA repair, chromosome segregation, and cell-cycle regulation.26,27 To further investigate the relationship between GTF2H4 and partial EndMT, this study was performed and first showed that GTF2H4 responded to hypoxia/ischemia to promote partial EndMT and its underlying molecular mechanism.
Results
Ischemia/hypoxia induces partial EndMT
To investigate whether transient or partial EndMT occurs in ischemic/hypoxic tissue, we performed permanent occlusion of the left anterior descending coronary artery in C57BL/6J mice to induce MI. Sham-operated mice that underwent a similar surgical procedure without arterial ligation served as controls. Consistent with previous findings,22,23 immunofluorescence staining revealed a significant increase in double-positive cells expressing CD31 and smooth muscle actin (SMA) 3 and 7 days after MI, confirming the occurrence of partial EndMT following MI. Specifically, the ratio of CD31+/SMA+ double-positive cells to CD31+ cells increased from less than 5% in the sham group to more than 30% 3 days after MI and nearly 45% 7 days after MI (Figures 1A and 1B). Next, we investigated whether partial EndMT occurred in ECs under hypoxic conditions in vitro. HMEC-1 cells were exposed to strictly controlled hypoxic conditions (1% O2, 5% CO2, and balanced N2) and serum deprivation for 1–3 days to simulate the extreme microenvironment of tissue ischemia. As a positive control, HMEC-1 cells were treated with transforming growth factor β (TGF-β) (20 ng/mL) for 3 days to induce complete EndMT. Under hypoxic conditions with serum deprivation, double immunofluorescence staining showed that hypoxic HMEC-1 cells gradually acquired the mesenchymal marker α-SMA and prominent actin stress fibers and lost some endothelial markers (CD31 and vascular endothelial [VE]-cadherin). This resulted in a progressive increase in the number of CD31+/α-SMA+ and VE-cadherin+/phalloidin+ cells (Figures 1C and S1A–S1F), which was consistent with the changes in relative mRNA levels in ECs (Figure 1D), indicating an endothelial-to-mesenchymal phenotypic switch under hypoxic conditions. In addition, exposure to hypoxia significantly upregulated the protein expression of mesenchymal markers, including fibronectin, α-SMA, and fibroblast-specific protein 1 (FSP-1), in HMEC-1 cells in a time-dependent manner, whereas the endothelial markers CD31 and VE-cadherin were downregulated but not completely lost (Figures 1E and S1G). These findings suggested that mesenchymal ECs retained some endothelial phenotypes while undergoing partial EndMT. Interestingly, we observed that hypoxia for up to 3 days had minimal effects on tube formation by HMEC-1 cells (Figures 1F and S1H). In contrast, TGF-β treatment led to the complete loss of endothelial markers and the endothelial phenotype, which is referred to as complete EndMT (Figures 1C, 1F, S1A, S1B, S1D, S1E, and S1H).
Figure 1.

Ischemia/hypoxia induces partial EndMT
(A) Immunofluorescence staining of mouse heart sections isolated from the infarcted border zone 3 and 7 days after MI or sham surgery with antibodies recognizing CD31 (red) and α-SMA (green) was performed. DAPI was used to counterstain the nuclei (blue) (n = 10, scale bars, 100 μm).
(B) The histogram shows the CD31+/α-SMA+/CD31+ cell ratio (%) in the infarcted border zone in each group.
(C) Double immunofluorescence staining for CD31 (green) and α-SMA (red) or VE-cadherin (green) and F-actin (red) in HMEC-1 cells. Nuclei were counterstained with DAPI (blue). HMEC-1 cells were cultured under normoxic or hypoxic conditions for 1–3 days or treated with TGF-β (20 ng/mL) for 3 days (n = 8–10, scale bars, 50 μm).
(D) qRT-PCR analysis of CD31, VE-cadherin, and α-SMA mRNA expression in hypoxic HMEC-1 cells (n = 3).
(E) Western blot analysis of fibronectin, VE-cadherin, CD31, α-SMA, and FSP-1 protein expression relative to that of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) in hypoxic HMEC-1 cells (n = 3).
(F) Tube formation assays of hypoxic (0–3 days) or TGF-β-treated (20 ng/mL, 3 days) HMEC-1 cells (n = 5, scale bar, 500 μm).
The data are presented as means ± SEMs. p values were calculated by 1-way ANOVA (B and D). NS indicates no significant difference.
GTF2H4 mitigates the reduction in the viability of HMEC-1 cells under hypoxic conditions and facilitates hypoxia-induced partial EndMT
The reversible mesenchymal activation of ECs following tissue ischemia/hypoxia has been well established and linked to time-dependent changes in specific gene signatures.22,23 Previous studies have demonstrated that GTF2H4 is an active and responsive transcriptional regulator during the early phase of ischemia-reperfusion injury.25 To explore the involvement of GTF2H4 in ischemic/hypoxic conditions, we performed a comparative analysis of GTF2H4 expression in whole-blood samples from patients with STEMI before and 7 days after PCI and samples from a normal control group.28 The results revealed a significant decrease in GTF2H4 expression in the STEMI group, which subsequently returned to baseline levels 7 days after PCI, indicating that GTF2H4 may be downregulated in response to ischemic/hypoxic conditions (Figure 2A). Further examination using the Genotype-Tissue Expression (GTEx) database: https://storage.googleapis.com/gtex_analysis_v8/rna_seq_data/GTEx_Analysis_2017-06-05_v8_RNASeQCv1.1.9_gene_tpm.gct.gz revealed increased GTF2H4 expression in coronary arteries and ascending aortas compared to the left ventricular myocardium, suggesting a relatively abundant level of GTF2H4 in the endothelium, which is the first layer exposed to fluctuations in oxygen concentrations and hypoxic conditions (Figure 2B). Age was also a factor influencing GTF2H4 expression (Figure S2A). In vitro experiments were subsequently performed to verify the protein expression of GTF2H4 in ECs under serum deprivation and hypoxic conditions, and the results were similar to the findings in the blood samples of MI patients. GTF2H4 was gradually downregulated in both HMEC-1 cells and mouse cardiac microvascular ECs (MCMECs) exposed to hypoxic conditions, and MCMECs showed increased sensitivity to hypoxic stimuli (Figures 2C and S2B). To investigate the impact of hypoxic injury on HMEC-1 cell viability, a CCK-8 assay was used to evaluate the initial increase in cell viability during the early stages of hypoxia, followed by a decrease after 1 day, which was consistent with the hypoxia-induced changes in GTF2H4 expression (Figure S2C), suggesting a biological effect of GTF2H4 on ECs under hypoxic conditions.
Figure 2.

GTF2H4 mitigates the reduction in HMEC-1 cell viability under hypoxic conditions and facilitates hypoxia-induced partial EndMT
(A) The transcription of GTF2H4 in peripheral blood samples from patients with STEMI before and after PCI was evaluated using the published GSE61144 dataset. The x axis shows 24 blood samples divided into the normal, STEMI, and 7-day post-PCI groups. The y axis shows the log2-transformed expression of GTF2H4 normalized by quantile normalization.
(B) The expression of GTF2H4 in different tissues (left ventricle myocardium, ascending coronary aorta, and ascending aorta) from 50- to 59-year-old donors based on data obtained from the GTEx database. The x axis shows the tissue from which the 273 samples were derived, and the y axis shows the log2 (TPM + 1) value. TPM, transcript count per million.
(C) Western blot analysis of GTF2H4 protein expression relative to GAPDH in HEMC-1 cells and MCMECs exposed to hypoxia. HMEC-1 and MCMECs were cultured under normoxic or hypoxic conditions for 1, 2, 4, 6, 8, 12, 24, 48, and 72 h.
(D and E) The viability of HMEC-1 cells exposed to hypoxia for 1, 2, or 3 days in each group was determined by a CCK-8 assay (n = 7).
(F and G) Western blot analysis of Bax, Bcl-xL, and cleaved caspase-3 protein expression relative to GAPDH in each group under normoxic or hypoxic conditions for 3 days (n = 3).
(H and I) Double immunofluorescence staining of CD31 (green) and α-SMA (red) in each group after exposure to hypoxia for 3 days. Nuclei were counterstained with DAPI (blue) (n = 8, scale bars, 50 μm).
(J and K) Western blot analysis of fibronectin, α-SMA, FSP-1, VE-cadherin, CD31, and GTF2H4 protein expression relative to GAPDH in each group under normoxic or hypoxic conditions for 3 days (n = 3).
The data are presented as means ± SEMs. p values were calculated by 1-way ANOVA (A and B) or 2-tailed t tests (D and E).
To determine the role of GTF2H4 in hypoxia-induced cell injury, lentivirus-mediated GTF2H4-overexpressing and -knockdown HMEC-1 cells were generated, and the expression changes in these cells was confirmed by qRT-PCR and western blot analysis (Figures S1I, 2F, and 2G). Subsequent CCK-8 assays showed that GTF2H4 overexpression improved hypoxia-induced injury in HMEC-1 cells, whereas GTF2H4 knockdown exacerbated this effect (Figures 2D and 2E). Flow cytometry further demonstrated that GTF2H4 overexpression inhibited hypoxia-induced EC apoptosis, whereas GTF2H4 knockdown enhanced it (Figures S2D and S2E). This observation was supported by changes in the antiapoptotic protein Bcl-XL and the proapoptotic protein Bax, as well as in the cleaved caspase-3 levels, as detected by western blotting (Figures 2F, 2G, S2F, and S2G).
In a previous study, partial EndMT was observed in ECs that underwent mesenchymal activation while retaining some endothelial characteristics, including intercellular junctions.21 To investigate whether GTF2H4 plays a pivotal role in hypoxia-induced partial EndMT, the expression of endothelial and mesenchymal markers was examined by qRT-PCR and western blotting. GTF2H4 overexpression upregulated the expression of mesenchymal markers (fibronectin, α-SMA, and FSP-1) and downregulated the expression of endothelial markers (CD31 and VE-cadherin), while partially preserving the expression of CD31 and VE-cadherin (Figures 2J, S1J, and S2J). Conversely, GTF2H4 knockdown inhibited the acquisition of mesenchymal markers induced by hypoxia and promoted the expression of endothelial markers (Figures 2K, S1K, and S2K). Under normoxic conditions, neither GTF2H4 overexpression nor knockdown significantly affected the expression of these markers. The effect of GTF2H4 on partial EndMT was further confirmed by double immunofluorescence staining of CD31 (green) and α-SMA (red) in different groups exposed to hypoxia for 3 days (Figures 2H, 2I, S2H, and S2I).
GTF2H4 enhances migration and suppresses tube formation in HMEC-1 cells during partial EndMT
In cases of partial mesenchymal transition, ECs often acquire enhanced mesenchymal traits, leading to increased migratory and invasive abilities that can contribute to the (patho)physiological growth of blood vessels.21 To investigate the role of GTF2H4 in promoting migratory and invasive functions during partial EndMT, we performed migration experiments using scratch and transwell assays. We found that hypoxia significantly increased the migration of HMEC-1 cells compared to that in the normoxia group (Figures 3A and 3B). Moreover, the overexpression of GTF2H4 further increased cell migration under hypoxic conditions, and GTF2H4 knockdown reduced this effect (Figures 3A–3D). Collective migration is a coordinated process in which endothelial tip and stalk cells migrate together, inducing sprouting angiogenesis.29,30,31 During sprouting angiogenesis, angiogenic ECs can undergo partial EndMT and acquire mesenchymal characteristics that are necessary for the formation of new vessel sprouts. To assess the impact of GTF2H4 on this phenomenon, we performed an aortic ring vessel sprouting assay on mouse thoracic aortas infected with GTF2H4-overexpression or GTF2H4-knockdown adeno-associated viruses (AAVs). The results indicated that GTF2H4 reinforced hypoxia-induced sprouting angiogenesis (Figures 3E and 3F). Western blot analysis of the prometastatic enzyme MMP-9 revealed that GTF2H4 overexpression in HEMC-1 cells under hypoxic conditions significantly upregulated MMP-9 (Figure 3G), whereas GTF2H4 knockdown downregulated MMP-9 (Figure 3H). To evaluate endothelial characteristics, we examined tube formation by HMEC-1 cells. Hypoxia had little effect on tube formation within the designated time frame. GTF2H4 overexpression slightly inhibited this process, whereas GTF2H4 knockdown increased this process under hypoxic conditions, suggesting that GTF2H4 could promote the partial loss of endothelial characteristics in HMEC-1 cells during EndMT (Figures 3I–3K).
Figure 3.

GTF2H4 enhances migration and suppresses tube formation during partial EndMT in HMEC-1 cells
(A) Transwell assays of HMEC-1 cells in each group after exposure to normoxic or hypoxic conditions for 3 days (scale bars, 100 μm).
(B) The histogram shows the number of migrated cells per field in each group (n = 13).
(C) Scratch-wound analysis of each group that was exposed to hypoxia for 24 h (scale bars, 500 μm).
(D) The histogram shows the migration rate of each group (n = 6–9).
(E and F) Representative images and quantitative analysis of aortic ring vessel sprouting in mouse aortas after GTF2H4-associated AAV infection and hypoxia for 5 days (n = 5, scale bars, 500 μm).
(G and H) Western blot analysis of MMP-9 and GTF2H4 protein expression relative to GAPDH in each group that was exposed to hypoxia for 3 days or under normoxic conditions (n = 3).
(I) Tube formation analysis of HMEC-1 cells in each group exposed to normoxia or hypoxia for 3 days (scale bar, 500 μm).
(J and K) The histogram shows the number of junctions or total lengths in each group (n = 6).
The data are presented as means ± SEMs. p values were calculated via 2-tailed t tests (B, D, F, J, and K).
GTF2H4 regulates excision repair cross-complementation group 3 (ERCC3) via autophagy-mediated degradation during partial EndMT
To elucidate the molecular mechanism underlying the regulatory effect of GTF2H4 on hypoxia-induced partial EndMT, we collected protein extracts from lentivirus-mediated GTF2H4-overexpressing/knockdown HMEC-1 cells for 4-dimensional (4D) label-free proteomics analysis (Figure S3A). The results revealed 55 differentially expressed genes (DEGs) in the GTF2H4-overexpressing group compared to the control group (fold change ≥2.0 or ≤0.5 and p < 0.05), including 34 upregulated genes and 21 downregulated genes. Similarly, 95 DEGs were identified in GTF2H4 knockdown cells compared to control cells (Figures S3B–S3D), including 43 upregulated genes and 52 downregulated genes. Volcano plots were generated to illustrate the fold changes and p values of the DEGs. Among these DEGs, the top 10 upregulated genes associated with GTF2H4 overexpression were RPS13, DSCC1, ACACA, ALDOC, UBXN6, GFER, ANK1, FXN, ZFYVE19, and ZNF131, and the top 10 upregulated genes associated with GTF2H4 knockdown were UBL5, GTF2H1, RAB17, SATB1, ID1, WASHC2A, CGNL1, JUND, MRPL33, and CCDC42 (Figures 4A and 4B). Two common DEGs were identified by intersecting the DEGs in GTF2H4-overexpressing cells and GTF2H4-knockdown cells, and GTF2H4 could positively regulate the downstream target molecule ERCC3 and negatively regulate MRSP21 (Figure 4C). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis indicated a relationship between ERCC3 and GTF2H4 through the p53 signaling pathway and basal TFs (Figures S4A and S4B). Furthermore, a protein interaction network of a cluster of DEGs in GTF2H4-knockdown cells was constructed using STRING and Cytoscape. The cluster was composed of downregulated DEGs, including GTF2H1, GTF2H3, CHD1L, KCNA5, RPS24, MRPS11, EFL1, ERCC3, and GTF2H4, and upregulated DEGs, including SUMO2 and RPL19 (Figure 4D). These findings align with those of a previous study that showed that GTF2H4 and ERCC3 were part of the CORE subcomplex of TFIIH and that GTF2H4 contained ERCC3 binding regions.26 The interaction between GTF2H4 and ERCC3 in HMEC-1 cells was confirmed through a coimmunoprecipitation (coIP) assay (Figure 4E). Subsequently, qRT-PCR and western blot analyses were performed and revealed that GTF2H4 regulated ERCC3 at the protein level rather than at the transcriptional level. GTF2H4 and ERCC3 were positively related to each other in both normoxic and hypoxic microenvironments (Figures 4F–4H, S3E, and S3F). The discrepancy between the transcription and protein levels suggested that GTF2H4 could influence the degradation of ERCC3. Major intracellular protein degradation pathways include the autophagy‒lysosome pathway and the ubiquitin‒proteasome pathway.32,33 To investigate how GTF2H4 regulates ERCC3 protein expression, we used the macroautophagy/autophagy inhibitor 3-MA and the proteasome inhibitor MG132. In addition, cycloheximide (CHX) was used to block protein synthesis. Our results demonstrated that the autophagy inhibitor 3-MA but not the proteasome inhibitor MG132 abrogated the increase in ERCC3 expression observed in response to GTF2H4 overexpression in HMEC-1 cells (Figure 4I). Similarly, 3-MA but not MG132 inhibited ERCC3 degradation in the context of GTF2H4 knockdown (Figure 4J). These findings suggest that GTF2H4 regulates ERCC3 through autophagy-mediated degradation.
Figure 4.

GTF2H4 regulates ERCC3 via autophagy-mediated degradation during partial EndMT
(A and B) Volcano plots of DEGs identified by 4D label-free proteomics analysis of HEMC-1 cells infected with GTF2H4-overexpressing or GTF2H4-knockdown lentiviruses. The top 10 DEGs and overlapping genes, GTF2H4 and MRPS21, are annotated on the diagram (fold change ≥2.0 or ≤0.5, p < 0.05).
(C) Overlapping DEGs between the GTF2H4 OE/NC and shGTF2H4/shCtrl groups.
(D) The protein interaction network of a cluster of shGTF2H4/shCtrl DEGs was constructed by STRING and Cytoscape. Red indicates upregulated DEGs, and the green represents downregulated DEGs.
(E) CoIP of GTF2H4 and ERCC3 in the lysates of HMEC-1 cells after infection with GTF2H4, ERCC3, or GTF2H4+ERCC3 overexpression lentiviruses.
(F) qRT-PCR analysis of ERCC3 mRNA expression in HMEC-1 cells in each group (n = 9).
(G and H) Western blot analysis of ERCC3 and GTF2H4 protein expression relative to GAPDH in each group (n = 3).
(I) After blocking protein synthesis with CHX (10 μg/mL), GTF2H4 overexpression increased ERCC3 expression by inhibiting autophagy-mediated degradation in the presence or absence of the proteasome inhibitor MG132 (10 μM) or the autophagy inhibitor 3-MA (5 mM), with time following control or GTF2H4 overexpression, as confirmed by western blot analysis.
(J) After blocking protein synthesis with CHX (10 μg/mL), GTF2H4 knockdown decreased ERCC3 expression by promoting autophagy-mediated degradation in the presence or absence of the proteasome inhibitor MG132 (10 μM) or the autophagy inhibitor 3-MA (5 mM), with time following control or GTF2H4 knockdown, as confirmed by western blot analysis.
The data are presented as means ± SEMs.
The interaction between ERCC3 and GTF2H4 is indispensable for GTF2H4-mediated partial EndMT under hypoxic conditions
To explore the biological function of ERCC3 in partial EndMT and the impact of ERCC3 knockdown on GTF2H4-mediated partial EndMT, we performed lentivirus-mediated ERCC3 overexpression or knockdown in HMEC-1 cells. First, ERCC3 knockdown inhibited the expression of GTF2H4, but ERCC3 overexpression did not affect GTF2H4 expression (Figures 5A, 5B, S5A, and S5B). Second, functional assays using transwells and flow cytometry revealed that ERCC3 knockdown inhibited the migration of HMEC-1 cells under hypoxic conditions (Figures 5C and 5D) and exacerbated hypoxia-induced apoptosis (Figures 5E and 5F). Third, western blot analysis confirmed that ERCC3 knockdown inhibited the acquisition of mesenchymal markers induced by hypoxia and promoted the expression of endothelial markers (Figures 5G and S5D), similar to the effect of GTF2H4 knockdown. These results indicated that ERCC3 knockdown inhibited hypoxia-induced partial EndMT, but ERCC3 overexpression did not have a similar effect (Figure S5C). Subsequently, we investigated whether ERCC3 knockdown could reverse partial EndMT mediated by GTF2H4 under hypoxic conditions. HMEC-1 cells were infected with the GTF2H4 overexpression lentivirus and further infected with a lentivirus expressing small hairpin RNA specifically targeting ERCC3. The results demonstrated that after ERCC3 knockdown, migration and the antiapoptotic effect of GTF2H4 overexpression on HMEC-1 cells significantly decreased (Figures 5H–5K). Moreover, changes in endothelial and mesenchymal marker levels were observed (Figures 5L and S5F) and indicated that hypoxia-induced partial EndMT was reversed. To further investigate whether the interaction between GTF2H4 and ERCC3 is involved in GTF2H4-mediated partial EndMT, we explored the biological function of the GTF2H4 mutations E310K/R314E. These mutations have been shown to significantly weaken the interaction between GTF2H4 and ERCC3.34 We constructed HMEC-1 cells overexpressing E310K/R314E-GTF2H4 using a lentivirus (Figure S5E) and performed coIP assays. Compared with those of WT-GTF2H4, the interactions of E310K/R314E-GTF2H4 with ERCC3 were decreased (Figure 5M). Next, we investigated the function of E310K/R314E and found that the expression of ERCC3 and partial EndMT-associated markers did not increase (Figure 5N), highlighting the necessity of the interaction between GTF2H4 and ERCC3 in GTF2H4-mediated partial EndMT under hypoxic conditions. Collectively, our findings suggest that the GTF2H4-ERCC3 complex plays an integral role in hypoxia-induced partial EndMT.
Figure 5.

The interaction between ERCC3 and GTF2H4 is indispensable for GTF2H4-mediated partial EndMT under hypoxic conditions
HMEC-1 cells were infected with an ERCC3-overexpressing lentivirus (ERCC3 OE), an overexpression control lentivirus (NC), an ERCC3-knockdown lentivirus (shERCC3), a knockdown control lentivirus (shCtrl), a GTF2H4-overexpressing lentivirus with shCtrl (GTF2H4 OE + shCtrl), or a GTF2H4-overexpressing lentivirus and a ERCC3-knockdown lentivirus (GTF2H4 OE + shERCC3). For the transwell and apoptosis assays, HMEC-1 cells were cultured under hypoxic conditions for 3 days.
(A and B) Western blot analysis of GTF2H4 protein expression relative to GAPDH in HMEC-1 cells with ERCC3 overexpression or knockdown under normoxic or hypoxic conditions for 3 days (n = 3).
(C and D) Transwell assays of HMEC-1 cells in the shERCC3 and shCtrl groups (n = 13, scale bars, 100 μm).
(E and F) Measurement of apoptosis in the shERCC3 and shCtrl groups by using an annexin V-FITC/PI-PerCP-Cy5.5 double-staining flow cytometric assay (n = 6).
(G) Western blot analysis of fibronectin, VE-cadherin, CD31, α-SMA, FSP-1, GTF2H4, and ERCC3 protein expression relative to GAPDH in the shCtrl and shERCC3 groups that were exposed to hypoxia for 3 days or under normoxic conditions (n = 3).
(H and I) Transwell assays of HMEC-1 cells in the GTF2H4 OE+shCtrl and GTF2H4 OE+shERCC3 groups (n = 13, scale bars, 100 μm).
(J and K) Measurement of apoptosis in the GTF2H4 OE+shCtrl and GTF2H4 over+shERCC3 groups by using an annexin V-FITC/PI-PerCP-Cy5.5 double-staining flow cytometric assay (n = 6).
(L) Western blot analysis of fibronectin, VE-cadherin, CD31, α-SMA, FSP-1, GTF2H4, and ERCC3 protein expression relative to GAPDH in the GTF2H4 OE+shCtrl and GTF2H4 OE + shERCC3 groups exposed to hypoxia for 3 days or under normoxic conditions (n = 3).
(M) CoIP analysis of the GTF2H4 mutation (E310K/R314E) and of ERCC3 compared with GTF2H4 and ERCC3 by using lysates from HMEC-1 cells after infection with GTF2H4, E310K/R314E, or ERCC3 overexpression lentiviruses.
(N) Western blot analysis of fibronectin, VE-cadherin, CD31, α-SMA, FSP-1, GTF2H4, and ERCC3 protein expression relative to GAPDH in the NC group and the E310K/R314E group exposed to hypoxia for 3 days or under normoxic conditions (n = 3).
The data are presented as means ± SEMs. p values were calculated by 2-tailed t tests (D, F, I, and K).
GTF2H4 promotes hypoxia-induced partial EndMT via the nuclear factor κB (NF-κB) signaling axis
To investigate the signal transduction pathway related to GTF2H4 during partial EndMT, we performed 4D label-free phosphorylation proteomics analysis of HMEC-1 cells infected with lentiviruses overexpressing or knocking down GTF2H4 (Figures S6A–S6D). Gene Ontology (GO) analysis demonstrated that GTF2H4 knockdown participated in various biological processes, such as focal adhesions and cadherin binding (Figure 6A). These findings were consistent with our previous results, which showed that GTF2H4 knockdown inhibited the loss of adhesion molecules in HMEC-1 cells during mesenchymal transition. Further analysis revealed that the differentially phosphorylated peptides were primarily enriched in the mechanistic target of mitogen-activated protein kinase,35 Hippo,36 NF-κB,37 and Notch signaling pathway,19 all of which are associated with EndMT (Figure 6B). Given the involvement of GTF2H4/ERCC3 in partial EndMT, we further explored whether GTF2H4 regulated hypoxia-induced partial EndMT via NF-κB signaling. NF-κB II signaling phospho-antibody array analysis revealed that GTF2H4 overexpression in HMEC-1 cells increased the phosphorylation of p65 at Ser536, which is a key activation site (fold change 3.6), and other sites in NF-κB1 (p105/p50), NF-κB2 (p100/p52), and RelA (p65) (Figure 6C). Conversely, GTF2H4 knockdown decreased the phosphorylation of IκB and IKK (Figure 6D), suggesting that GTF2H4 could activate NF-κB signaling in HMEC-1 cells under hypoxic conditions.
Figure 6.

GTF2H4 promotes hypoxia-induced partial EndMT via the NF-κB signaling axis
(A) GO analysis of DEGs between the shGTF2H4 and shCtrl groups (fold change ≥2.0 or ≤0.5, p < 0.05), as determined by 4D label-free phosphorylation proteomics analysis. p values were used to indicate statistical significance. The top 10 GO terms are listed in the diagram.
(B) KEGG pathway enrichment analysis of DEGs between the shGTF2H4 and shCtrl groups (fold change ≥2.0 or ≤0.5, p < 0.05). p values were used to indicate statistical significance. The top 10 cardiovascular diseases or EndMT-related pathways are listed in the diagram.
(C and D) The NF-κB II signaling phospho-antibody array was used to analyze cell lysates in each group that was exposed to hypoxia for 3 days. The results from 6 replicate samples were averaged. The dashed gray line indicates a fold change of 1.5, and proteins with a fold change greater than 1.5 or lower than 0.667 were considered DEPs and are marked in red (blue).
(E and F) Luciferase reporter assays of NF-κB p65 transcriptional activation activity after TNF-α (10 ng/mL; NF-κB activation) stimulation for 4 h in each group (n = 5–8).
(G) EMSA was used to examine the DNA-binding activity of NF-κB (p65/p50) in GTF2H4-overexpressing or -knockdown HMEC-1 cells exposed to normoxia and hypoxia for 1–2 days.
(H and I) Western blot analysis of GTF2H4, Snail, and p-P65/P65 expression relative to GAPDH in GTF2H4-overexpressing or -knockdown HMEC-1 cells exposed to hypoxia for 3 days or under normoxic conditions (n = 3).
(J) Western blot analysis of GTF2H4, Snail, and p-P65/P65 expression relative to GAPDH in E310K/R314E-overexpressing HMEC-1 cells exposed to hypoxia for 3 days or under normoxic conditions (n = 3).
(K) A brief schematic diagram of the role of the NF-κB inhibitor Bay 11-7082 (10 μM).
(L) Western blot analysis showing the effects of Bay 11-7082 on VE-cadherin, CD31, α-SMA, Snail, and GTF2H4 protein expression relative to GAPDH in the NC group and GTF2H4 OE group exposed to hypoxia for 3 days (n = 3).
The data are presented as means ± SEMs. p values were calculated by 2-tailed t tests (E and F).
To further investigate the effect of GTF2H4 on the transcriptional activation of NF-κB, luciferase reporter assays were performed on 293T cells infected with lentiviruses overexpressing or knocking down GTF2H4, and the transfection effects were confirmed by western blotting (Figure S7A). The results indicated that GTF2H4 overexpression markedly promoted tumor necrosis factor α (TNF-α)-induced NF-κB promoter activity (Figure 6E), whereas GTF2H4 knockdown suppressed this activity (Figure 6F). Since the sensitivity of HMEC-1 cells to hypoxia mediated by the NF-κB pathway varies with tissue/cell specificity,38 we performed western blotting and electrophoretic mobility shift assays (EMSAs) to examine hypoxia-induced NF-κB signaling pathway activity. The results demonstrated that hypoxia gradually increased NF-κB p65 binding activity (Figure S6E) and p-P65/P65 expression (Figures S6F and S6G) in a time-dependent manner, indicating that hypoxia induced NF-κB activation in HMEC-1 cells. Next, we explored the impact of GTF2H4 on hypoxia-induced NF-κB activation. Nuclear protein extracts from GTF2H4-overexpressing HMEC-1 cells exhibited increased DNA binding ability (Figure 6G). Conversely, DNA binding of NF-κB p65 was unchanged in GTF2H4 knockdown HMEC-1 cells (Figure S7B), which was consistent with the lack of change in binding activity observed after treatment with spironolactone, which induced ERCC3 degradation in a previous study.39
Previous studies have indicated that the NF-κB/Snail pathway is a canonical pathway that regulates EndMT, and NF-κB binds to the Snail promoter between −194 and −78 bp to promote Snail transcription.40 Therefore, we investigated whether NF-κB/Snail signaling was involved in GTF2H4-mediated partial EndMT. The results indicated that GTF2H4 overexpression significantly increased the expression of p-P65/P65 and Snail under hypoxic conditions, whereas GTF2H4 knockdown inhibited the expression of these genes (Figures 6H, 6I, S7C, and S7D). In addition, the E310K/R314E mutations, which are dysfunctional during hypoxia-induced partial EndMT, did not activate NF-κB/Snail signaling (Figure 6J), suggesting the importance of the interaction between GTF2H4 and ERCC3. Furthermore, we used the NF-κB inhibitor Bay 11-7082 to determine whether GTF2H4 regulated Snail expression via NF-κB (Figure 6K). The results showed that Bay 11-7082 significantly reversed the increase in Snail expression induced by GTF2H4 and reversed the changes in the expression of downstream markers, including CD31, VE-cadherin, and α-SMA (Figures 6L and S7K), suggesting that GTF2H4 partially regulated EndMT through the NF-κB/Snail signaling pathway. To investigate the role of ERCC3 in NF-κB/Snail signaling, luciferase reporter assays and western blotting were performed on cells overexpressing or lacking ERCC3, and the results showed that ERCC3 knockdown significantly inhibited TNF-α-induced NF-κB activation and hypoxia-induced NF-κB/Snail signaling, whereas ERCC3 overexpression had no effect (Figures S7E–S7J).
GTF2H4 promotes NF-κB-induced partial EndMT via receptor coactivator 3 (NCOA3) phosphorylation at S1330
To further determine the mechanisms by which GTF2H4 is involved in NF-κB activation, we analyzed the overlapping differentially expressed proteins (DEPs) and their corresponding phosphorylation sites in the GTF2H4-overexpressing and negative controls, as well as in the presence of shGTF2H4 and shCtrl, by phosphorylation proteomics analysis (Figure 7A). Among these DEPs, NCOA3 Ser1330 and NELFE Ser181 were upregulated in GTF2H4-overexpressing HMEC-1 cells and downregulated in GTF2H4-knockdown HMEC-1 cells. Notably, NCOA3 is a well-known NF-κB coactivator that functions by interacting with NF-κB p65, and specific site phosphorylation of NCOA3 (SRC-3) is required for its function.41,42 The Ser1330 site is located within the domain of unknown function (DUF1518) of NCOA3 (Figure 7B). To examine whether GTF2H4 activates NF-κB via NCOA3 phosphorylation at S1330, we assessed the effect of this modification on the intrinsic activity of NF-κB. We transfected 293T cells with WT-pcDNA3.1-HisA-NCOA3, the phosphorylation-deficient pcDNA3.1-HisA-NCOA3-S1330A mutant, the phosphorylation-mimic pcDNA3.1-HisA-NCOA3-S1330D mutant, and the pcDNA3.1-HisA-NCOA3-S1330E mutant after verifying the site-directed mutagenesis by DNA sequencing (Figure 7C). The results showed that NF-κB promoter activity in the S1330A group was markedly decreased compared to that in the WT-NCOA3, S1330D, and S1330E groups. Although the Asp/Glu substitutions only slightly affected the ability of NCOA3 (SRC-3) to activate NF-κB, possibly due to the limitations of phosphomimetic agents,43 S1330D and S1330E still induce levels of activation similar to that of WT-NCOA3 (Figure 7D). Moreover, we performed coIP assays, which revealed that the interaction between NCOA3 and NF-κB p65 decreased in the presence of NCOA3 1330A, whereas 1330E or 1330D exhibited similar capacities to interact as WT-NCOA3 (Figure 7E). These findings suggested that the phosphorylation of NCOA3 at Ser1330 promoted its interaction with p65 to enhance NF-κB activation. In addition, in the presence of S1330A or S1330E, the changes in NF-κB promoter activity induced by GTF2H4 overexpression/knockdown were abolished, suggesting that GTF2H4 promoted NF-κB activation via NCOA3 phosphorylation at S1330 (Figures 7F–7H). Similarly, the decrease in NF-κB promoter activity induced by ERCC3 knockdown was reversed by S1330A or S1330E (Figure S8A). To determine whether the expression of NCOA3 was affected, we performed western blot analysis and found no difference in NCOA3 expression in the experimental groups (Figures S8B–S8F). These findings indicate that the phosphorylation of NCOA3 but not its expression plays a crucial role in NF-κB activation.
Figure 7.

GTF2H4 promotes NF-κB-induced partial EndMT via NCOA3 phosphorylation at S1330
(A) 4D label-free phosphorylation proteomics results showing the overlapping DEPs and their corresponding phosphorylation sites in the GTF2H4 OE/NC and shGTF2H4/shCtrl groups.
(B) A schematic representation of the NCOA3 protein and its known functional domains. The overlapping phosphopeptide identified by phosphorylation proteomics analysis is shown, and the asterisks indicate the phosphoramide acids (1330S).
(C) DNA-sequencing identification of 3 mutated NCOA3 plasmids: S1330A, S1330D, and S1330E.
(D) Luciferase reporter assays to examine the effects of WT-NCOA3, S1330A, S1330E, or S1330D NCOA3 mutants and TNF-α (10 ng/mL; NF-κB activation) stimulation for 4 h on NF-κB p65 transcriptional activation activity in each group (n = 7–27).
(E) CoIP of p65 after TNF-α (10 ng/mL; NF-κB activation) stimulation for 4 h and after treatment with the WT-NCOA3, S1330A, S1330D, or S1330E NCOA3 mutants. The levels of p65 associated with NCOA3 were detected by immunoblotting.
(F and G) Luciferase reporter assays to examine the effects of GTF2H4 overexpression or knockdown and TNF-α (10 ng/mL; NF-κB activation) stimulation for 4 h on NF-κB p65 transcriptional activation activity in each group after treatment with the WT-NCOA3, S1330A, S1330D, or S1330E NCOA3 mutants (n = 6–18).
(H) Schematic representation of the role of GTF2H4 in hypoxia-induced partial EndMT. The GTF2H4-ERCC3 complex functions as an integral component of this process. Hypoxia-induced p65 transport to the nucleus and subsequent NF-κB activation. Snail is a downstream target gene of NF-κB and regulates EndMT (left side of the dotted line). Upregulation of GTF2H4 resulted in NCOA3 phosphorylation at S1330. Phosphorylated NCOA3 at S1330 significantly promoted its binding to P65 to activate NF-κB/Snail signaling and enhance hypoxia-induced partial EndMT.
The data are presented as means ± SEMs. p values were calculated by 2-tailed t tests (D, F, and G).
GTF2H4 promotes partial EndMT in vivo and improves blood flow recovery after ischemic injury
To assess the in vivo effects of GTF2H4 on partial EndMT in response to ischemia and its role in blood flow recovery after ischemic injury, we induced endothelial-specific GTF2H4 overexpression and knockdown by injecting AAV-Tie-GTF2H4 or AAV-Tie-shGTF2H4, respectively, into the gastrocnemius muscles of mice 4 weeks before hindlimb ischemia surgery (Figures S9A and S9B). Limb perfusion was monitored for 7 days before the gastrocnemius muscle was harvested (Figure 8A). After purifying ECs from mouse gastrocnemius muscle using CD31/PECAM1 antibodies, we confirmed the efficiency of GTF2H4 overexpression or knockdown in the gastrocnemius muscle by western blot analysis (Figure 8B). Laser Doppler ultrasound revealed that the GTF2H4 overexpression group exhibited increased blood flow recovery within 7 days of hindlimb ischemia, whereas the GTF2H4 knockdown group exhibited reduced limb perfusion (Figures 8C–8F). To further investigate the effects of GTF2H4 on blood vessel formation and function, we performed dual staining for CD31 and α-SMA to identify mature blood vessels with perfusion. Double immunofluorescence staining revealed a significant increase in CD31+/α-SMA+ capillary density in GTF2H4-overexpressing mice, and this increase was reduced in GTF2H4-knockdown mice compared to control mice (Figures 8G, S9B, and S9C). In addition, immunohistochemical staining and western blot analysis revealed that GTF2H4 promoted partial EndMT after ischemic injury, which was characterized by increased mesenchymal marker expression and NF-κB signaling activation in GTF2H4-overexpressing mice. However, GTF2H4 knockdown inhibited the expression of mesenchymal markers and NF-κB signaling activation (Figures 8H–8J). Notably, the expression of NCOA3 did not significantly differ among the groups (Figures S8G and S8H). Taken together, our findings reveal that GTF2H4 plays a pivotal role in promoting partial EndMT in vivo, leading to improved blood flow recovery after ischemic injury.
Figure 8.

GTF2H4 promotes partial EndMT in vivo and improves blood flow recovery postischemic injury
(A) Schematic illustration of the in vivo injection of GTF2H4-overexpressing AAV (GTF2H4 OE), overexpression control AAV (NC), GTF2H4-knockdown AAV (shGTF2H4), and knockdown control AAV (shCtrl) into mouse gastrocnemius muscle 4 weeks before hindlimb ischemia surgery. Blood flow recovery was observed at the corresponding time points. Gastrocnemius muscles were harvested on the last day of the observation period.
(B) Western blot analysis of GTF2H4 protein expression normalized to that of GAPDH after the injection of GTF2H4-overexpressing or -knockdown AAVs into the gastrocnemius muscle (n = 6).
(C and E) Representative laser Doppler ultrasound images and quantitative analysis of blood flow recovery after the injection of AAV in the GTF2H4 OE or NC ground (n = 6).
(D and F) Representative laser Doppler ultrasound images and quantitative analysis of blood flow recovery after the injection of shGTF2H4 or shCtrl AAV (n = 6).
(G) Immunofluorescence staining of sections of mouse gastrocnemius muscle 7 days after hindlimb ischemia surgery, with antibodies recognizing CD31 (green) and α-SMA (red). DAPI was used to counterstain the nuclei (blue) (n = 6, scale bars, 100 μm).
(H) Immunohistochemical staining of sections of mouse gastrocnemius muscle with antibodies recognizing α-SMA and vimentin 7 days after hindlimb ischemia surgery (n = 6, scale bars, 50 μm).
(I and J) Western blot analysis of vimentin, α-SMA, P-65, p-P65, Snail, ERCC3, and GTF2H4 protein expression compared to that of GAPDH in each group 7 days after hindlimb ischemia surgery (n = 6).
The data are presented as means ± SEMs. p values were calculated by 2-tailed t tests (E and F).
Discussion
In this study, we reported that GTF2H4 significantly ameliorated the hypoxia-induced viability loss and apoptosis, promoted migration, and inhibited tube formation in ECs under hypoxic conditions; moreover, these cells acquired mesenchymal markers and lost some endothelial markers during GTF2H4-mediated partial EndMT. Mechanistically, GTF2H4 coordinated with ERCC3 to regulate NCOA3 via the phosphorylation of S1330, which subsequently activated the NF-κB/Snail signaling pathway to promote partial EndMT. Subsequently, in vivo findings suggested that GTF2H4 partially enhanced EndMT and angiogenesis postischemia.
To identify molecules associated with transient mesenchymal activation in ECs after tissue ischemia/hypoxia,22,23 an analysis of the GEO database was performed, and the results indicated that the expression of GTF2H4 was significantly decreased in the STEMI group compared to the control group, which was consistent with our previous findings.25 GTF2H4 was gradually downregulated in HMEC-1 cells and primary MCMECs with prolonged hypoxia. Since partial EndMT occurs in a time-dependent manner in hypoxic conditions, we further investigated the correlation between GTF2H4 and hypoxia-induced partial EndMT,18 the loss of cell viability, and apoptosis and found that GTF2H4 plays a positive role in ECs undergoing mesenchymal transition. Functional and phenotypic changes in ECs during mesenchymal transition are fundamental to EndMT.10 Previous studies have shown that collective migration is a biological process that induces sprouting angiogenesis,29 and during this process, angiogenic ECs can undergo partial EndMT to acquire mesenchymal characteristics.18 In vitro and ex vivo experiments indicated that GTF2H4 could enhance the migratory capacity of these cells, which is a mesenchymal characteristic. Although reduced tubule formation has also been associated with EndMT,44,45 GTF2H4 slightly inhibited tube formation but almost sustained this endothelial trait, suggesting that GTF2H4 accelerated the transition of ECs to an intermediate phenotype, preserving the original characteristics of ECs and simultaneously mediating the acquisition of mesenchymal traits. Therefore, GTF2H4-mediated EndMT under hypoxic conditions is a partial or reversible process.
GTF2H4 is 1 of 10 TFIIH subunits that interact with ERCC3 (ERCC excision repair 3, TFIIH core complex helicase subunit) and can stimulate its ATPase activity.26,46 A 4D label-free proteomics analysis revealed that the expression of ERCC3 overlapped with the expression of DEGs between the GTF2H4 overexpression and knockdown groups. Subsequently, MG132 and 3-MA treatments were used, and qRT-PCR and western blot analysis further demonstrated that GTF2H4 modulated the protein expression of ERCC3 via autophagy-mediated degradation rather than transcriptional regulation. Investigating the mechanism by which GTF2H4 regulates ERCC3 is particularly meaningful because blocking this pathway could exclusively target GTF2H4 without affecting ERCC3 levels. This aspect may prove beneficial for future studies. In addition, the knockdown of ERCC3 decreased GTF2H4 expression, inhibited hypoxia-induced partial EndMT, and reversed the promotional effect of GTF2H4 overexpression, whereas ERCC3 overexpression neither influenced the expression of GTF2H4 nor had a similar biological effect on partial EndMT. It is possible that the expression level of endogenous ERCC3 is significantly higher than that of GTF2H4, leading to an oversaturated state of the ERCC3 protein. In this scenario, exogenous overexpression of ERCC3 may have minimal biological effects, since the excess protein may not effectively interact with cellular components. In combination with the finding that the interaction between GTF2H4 and ERCC3 was indispensable for GTF2H4-mediated partial EndMT under hypoxic conditions, we examined the GTF2H4 mutations E310K/R314E and showed that the GTF2H4-ERCC3 complex played an integral role, that GTF2H4 functioned as a rate-limiting enzyme, and that its expression level influenced the overall formation of the GTF2H4-ERCC3 complex, which in turn regulated downstream biological processes.
Next, the downstream signal transduction pathways of GTF2H4-ERCC3 were investigated through 4D label-free phosphorylation proteomics analysis, and NF-kB signaling was the predominant pathway identified by KEGG pathway enrichment analyses; these findings were further validated by using the NF-kB II Signaling Phospho Antibody Array. Moreover, GTF2H4 overexpression markedly increased TNF-α-induced NF-κB promoter activity, and this effect was suppressed by GTF2H4 knockdown. Previous studies have widely demonstrated that the NF-κB pathway is sensitive to hypoxia and cell specific.38,47,48 Moreover, hypoxia significantly activated NF-κB in HMEC-1 cells, and nuclear protein extracts from GTF2H4-overexpressing HMEC-1 cells exhibited increased DNA binding under hypoxic conditions, although the DNA binding of NF-κB p65 was unchanged in GTF2H4-knockdown HMEC-1 cells. Another group showed that spironolactone-induced degradation of ERCC3 suppressed NF-κB promoter activity without affecting binding activity, indicating that a primary mechanism, such as chromatin remodeling or transcriptional initiation, was involved.39 Snail, which is a downstream target gene of the NF-κB signaling pathway, is a critical TF that triggers EndMT. By binding to the Snail promoter between −194 and −78 bp, NF-κB can promote its transcription40 to inhibit the expression of endothelial markers such as VE-cadherin and CD3110 and promote the acquisition of a mesenchymal phenotype, such as by stimulating α-SMA and MMP-9 expression.40,49 Under hypoxic conditions, GTF2H4 significantly upregulated the expression of Snail, and this effect was significantly reversed by treatment with the NF-κB inhibitor Bay 11-7082.50 Similar to the effect of GTF2H4 knockdown, ERCC3 inhibited NF-kB/Snail signaling, and the dysfunction associated with ERCC3 and E310K/R314E overexpression in NF-κB activation may be attributed to the lack of change in the overall level of the GTF2H4-ERCC3 complex. Collectively, these data suggested that the GTF2H4-ERCC3 complex regulated partial EndMT via the NF-κB/Snail signaling pathway.
A 4D label-free phosphorylation proteomics analysis showed that NCOA3 Ser1330 was located within the overlap of differentially expressed phosphopeptides between the GTF2H4 overexpression group and the knockdown group. NCOA3 (SRC-3, steroid receptor coactivator 3) is a member of the SRC/p160 gene family that interacts with multiple nuclear receptors and other TFs to enhance their effects on target gene transcription,47 which was confirmed by the finding that SRC-3 (NCOA3) could interact with p65 to enhance p65-mediated NF-κB reporter activation.42 Moreover, several sites that are phosphorylated by SRC-3, including T24, S505, S543, S587, and S867, have been shown to activate NF-κB in response to TNF-α.41 This study is the first to identify SRC-3 S1330 as a novel phosphorylation site that is required for NF-κB activation, and GTF2H4 indirectly phosphorylates the transcriptional coactivator NCOA3 at S1330, subsequently promoting its interaction with p65 and ultimately causing NF-κB activation; these effects could be reversed by the reintroduction of NCOA3-S1330D or the S1330E mutant, despite the slightly lower activation induced by S1330D and S1330E than WT-NCOA3. This effect may occur because during site-directed mutagenesis for phosphomimetic effects, Asp/Glu substitutions fail to capture the Ser/Thr phosphorylation function completely.43
Angiogenic ECs that undergo partial EndMT during pathological angiogenesis may support neovascularization18,22,23 based on the transient and dynamic nature of EndMT in ischemic diseases involving MI or hindlimb ischemia. Despite the increase in angiogenic factors induced by ischemia/hypoxia in the early stages of MI, the angiogenic response is difficult to trigger in the heart,51 and some clinical trials have shown that proangiogenic pharmacotherapeutics for cardiovascular diseases have little effect.52 Therefore, understanding the mechanism of angiogenesis in detail and exploring the overlapping biological behaviors between angiogenesis and partial EndMT are urgently needed to determine a therapeutic strategy to promote angiogenesis in the clinic. In this study, the occurrence of partial EndMT in post-MI mice was confirmed by immunofluorescence staining, and then, hindlimb ischemia models were used to examine the in vivo function of GTF2H4 in postischemic angiogenesis in ECs by using a laser Doppler ultrasound scanning system. The results showed that AAV-mediated GTF2H4 overexpression in ECs in postischemic hindlimbs improved blood flow perfusion and promoted the formation of new collateral vessels and increased the specific traits of partial EndMT. In contrast, the opposite effects were observed in the AAV-mediated GTF2H4 knockdown group. These in vivo data showed that targeting endothelial GTF2H4 could enhance postischemic partial EndMT, angiogenesis, and tissue repair.
Conclusion
GTF2H4 regulates ischemia/hypoxia-induced partial EndMT in vivo and in vitro. Targeting the GTF2H4/NCOA3/NF-κB signaling pathway may be a promising therapeutic approach for treating ischemic diseases.
Materials and methods
See the supplemental information for details.
Acknowledgments
We thank Shanghai Applied Protein Technology for their technological support of the study. This work was supported by the National Natural Science Foundation of China (82170334 and 81870182).
Author contributions
Hua Li and Junbo Ge conceived the study and its design and gave final approval to the manuscript submitted. Haojie Lu, Y. Zhao, and X.W. made good suggestions that improved the manuscript. Z.F., G.Z., S.Z., X.Y., R.F., You-en Zhang, Haomin Li, and L.H. participated in the data collection and analysis, the drafting of the manuscript, and the submission. Z.G., Z.Z., M.A., H.H., P.L., B.W., J.Z., X. Zhong, D.H., Hao Lu, X. Zhao, Z.C., W.Z., Junjie Guo, H.Z., Y.H., and S.Q. participated in the data collections and additional work.
Declaration of interests
The authors declare no competing interests.
Published Online: January 8, 2024
Footnotes
It can be found online at https://doi.org/10.1016/j.xinn.2024.100565.
Contributor Information
Junbo Ge, Email: ge.junbo@zs-hospital.sh.cn.
Hua Li, Email: lihua199988@hotmail.com.
Lead contact website
Supplemental information
References
- 1.Hu Q., Fang Z., Ge J., et al. Nanotechnology for cardiovascular diseases. Innovation. 2022;3:100214. doi: 10.1016/j.xinn.2022.100214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Benjamin E.J., Virani S.S., Callaway C.W., et al. American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Subcommittee Heart Disease and Stroke Statistics-2018 Update: A Report From the American Heart Association. Circulation. 2018;137:e67–e492. doi: 10.1161/cir.0000000000000558. [DOI] [PubMed] [Google Scholar]
- 3.Zhang T., Liu N., Xu J., et al. Flexible electronics for cardiovascular healthcare monitoring. Innovation. 2023;4:100485. doi: 10.1016/j.xinn.2023.100485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhuang J., Zhang X., Liu Q., et al. Targeted delivery of nanomedicines for promoting vascular regeneration in ischemic diseases. Theranostics. 2022;12:6223–6241. doi: 10.7150/thno.73421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lu H., Wang Y., Yu R. Immune cell membrane-coated nanoparticles for targeted myocardial ischemia/reperfusion injury therapy. Innovat. Med. 2023;1:100015. doi: 10.59717/j.xinn-med.2023.100015. [DOI] [Google Scholar]
- 6.Souilhol C., Harmsen M.C., Evans P.C., et al. Endothelial-mesenchymal transition in atherosclerosis. Cardiovasc. Res. 2018;114:565–577. doi: 10.1093/cvr/cvx253. [DOI] [PubMed] [Google Scholar]
- 7.Evrard S.M., Lecce L., Michelis K.C., et al. Endothelial to mesenchymal transition is common in atherosclerotic lesions and is associated with plaque instability. Nat. Commun. 2016;7:11853. doi: 10.1038/ncomms11853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.von Gise A., Pu W.T. Endocardial and epicardial epithelial to mesenchymal transitions in heart development and disease. Circ. Res. 2012;110:1628–1645. doi: 10.1161/circresaha.111.259960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bischoff J. Endothelial-to-Mesenchymal Transition. Circ. Res. 2019;124:1163–1165. doi: 10.1161/circresaha.119.314813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kovacic J.C., Dimmeler S., Harvey R.P., et al. Endothelial to Mesenchymal Transition in Cardiovascular Disease: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019;73:190–209. doi: 10.1016/j.jacc.2018.09.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang H., Lui K.O., Zhou B. Endocardial Cell Plasticity in Cardiac Development, Diseases and Regeneration. Circ. Res. 2018;122:774–789. doi: 10.1161/circresaha.117.312136. [DOI] [PubMed] [Google Scholar]
- 12.de Vlaming A., Sauls K., Hajdu Z., et al. Atrioventricular valve development: new perspectives on an old theme. Differentiation. 2012;84:103–116. doi: 10.1016/j.diff.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wirrig E.E., Yutzey K.E. Conserved transcriptional regulatory mechanisms in aortic valve development and disease. Arterioscler. Thromb. Vasc. Biol. 2014;34:737–741. doi: 10.1161/atvbaha.113.302071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu X., Friehs I., Zhong Hu T., et al. Endocardial fibroelastosis is caused by aberrant endothelial to mesenchymal transition. Circ. Res. 2015;116:857–866. doi: 10.1161/circresaha.116.305629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gorelova A., Berman M., Al Ghouleh I. Endothelial-to-Mesenchymal Transition in Pulmonary Arterial Hypertension. Antioxidants Redox Signal. 2021;34:891–914. doi: 10.1089/ars.2020.8169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ranchoux B., Antigny F., Rucker-Martin C., et al. Endothelial-to-mesenchymal transition in pulmonary hypertension. Circulation. 2015;131:1006–1018. doi: 10.1161/circulationaha.114.008750. [DOI] [PubMed] [Google Scholar]
- 17.Xu Y., Kovacic J.C. Endothelial to Mesenchymal Transition in Health and Disease. Annu. Rev. Physiol. 2023;85:245–267. doi: 10.1146/annurev-physiol-032222-080806. [DOI] [PubMed] [Google Scholar]
- 18.Fang J.S., Hultgren N.W., Hughes C.C.W. Regulation of Partial and Reversible Endothelial-to-Mesenchymal Transition in Angiogenesis. Front. Cell Dev. Biol. 2021;9:702021. doi: 10.3389/fcell.2021.702021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hultgren N.W., Fang J.S., Ziegler M.E., et al. Slug regulates the Dll4-Notch-VEGFR2 axis to control endothelial cell activation and angiogenesis. Nat. Commun. 2020;11:5400. doi: 10.1038/s41467-020-18633-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alvandi Z., Bischoff J. Endothelial-Mesenchymal Transition in Cardiovascular Disease. Arterioscler. Thromb. Vasc. Biol. 2021;41:2357–2369. doi: 10.1161/atvbaha.121.313788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Welch-Reardon K.M., Wu N., Hughes C.C.W. A role for partial endothelial-mesenchymal transitions in angiogenesis? Arterioscler. Thromb. Vasc. Biol. 2015;35:303–308. doi: 10.1161/atvbaha.114.303220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Manavski Y., Lucas T., Glaser S.F., et al. Clonal Expansion of Endothelial Cells Contributes to Ischemia-Induced Neovascularization. Circ. Res. 2018;122:670–677. doi: 10.1161/circresaha.117.312310. [DOI] [PubMed] [Google Scholar]
- 23.Tombor L.S., John D., Glaser S.F., et al. Single cell sequencing reveals endothelial plasticity with transient mesenchymal activation after myocardial infarction. Nat. Commun. 2021;12:681. doi: 10.1038/s41467-021-20905-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Felmeden D.C., Blann A.D., Lip G.Y.H. Angiogenesis: basic pathophysiology and implications for disease. Eur. Heart J. 2003;24:586–603. doi: 10.1016/s0195-668x(02)00635-8. [DOI] [PubMed] [Google Scholar]
- 25.Liu H., Xu D., Zhong X., et al. LncRNA-mRNA competing endogenous RNA network depicts transcriptional regulation in ischaemia reperfusion injury. J. Cell Mol. Med. 2019;23:2272–2276. doi: 10.1111/jcmm.14163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Compe E., Egly J.M. Nucleotide Excision Repair and Transcriptional Regulation: TFIIH and Beyond. Annu. Rev. Biochem. 2016;85:265–290. doi: 10.1146/annurev-biochem-060815-014857. [DOI] [PubMed] [Google Scholar]
- 27.Bassett J., Rimel J.K., Basu S., et al. Systematic mutagenesis of TFIIH subunit p52/Tfb2 identifies residues required for XPB/Ssl2 subunit function and genetic interactions with TFB6. J. Biol. Chem. 2022;298:102433. doi: 10.1016/j.jbc.2022.102433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park H.J., Noh J.H., Eun J.W., et al. Assessment and diagnostic relevance of novel serum biomarkers for early decision of ST-elevation myocardial infarction. Oncotarget. 2015;6:12970–12983. doi: 10.18632/oncotarget.4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Michaelis U.R. Mechanisms of endothelial cell migration. Cell. Mol. Life Sci. 2014;71:4131–4148. doi: 10.1007/s00018-014-1678-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Okamoto T., Usuda H., Tanaka T., et al. The Functional Implications of Endothelial Gap Junctions and Cellular Mechanics in Vascular Angiogenesis. Cancers. 2019;11:237. doi: 10.3390/cancers11020237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li L., He Y., Zhao M., et al. Collective cell migration: Implications for wound healing and cancer invasion. Burns Trauma. 2013;1:21–26. doi: 10.4103/2321-3868.113331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dikic I. Proteasomal and Autophagic Degradation Systems. Annu. Rev. Biochem. 2017;86:193–224. doi: 10.1146/annurev-biochem-061516-044908. [DOI] [PubMed] [Google Scholar]
- 33.Lu W. Targeted protein degradation bypassing cereblon and von Hippel-Lindau. Innovation. 2023;4:100422. doi: 10.1016/j.xinn.2023.100422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fregoso M., Lainé J.P., Aguilar-Fuentes J., et al. DNA repair and transcriptional deficiencies caused by mutations in the Drosophila p52 subunit of TFIIH generate developmental defects and chromosome fragility. Mol. Cell Biol. 2007;27:3640–3650. doi: 10.1128/mcb.00030-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li J., Xiong J., Yang B., et al. Endothelial Cell Apoptosis Induces TGF-β Signaling-Dependent Host Endothelial-Mesenchymal Transition to Promote Transplant Arteriosclerosis. Am. J. Transplant. 2015;15:3095–3111. doi: 10.1111/ajt.13406. [DOI] [PubMed] [Google Scholar]
- 36.Singh A., Ramesh S., Cibi D.M., et al. Hippo Signaling Mediators Yap and Taz Are Required in the Epicardium for Coronary Vasculature Development. Cell Rep. 2016;15:1384–1393. doi: 10.1016/j.celrep.2016.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ge X., Tang P., Rong Y., et al. Exosomal miR-155 from M1-polarized macrophages promotes EndoMT and impairs mitochondrial function via activating NF-κB signaling pathway in vascular endothelial cells after traumatic spinal cord injury. Redox Biol. 2021;41:101932. doi: 10.1016/j.redox.2021.101932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taylor C.T., Cummins E.P. The role of NF-kappaB in hypoxia-induced gene expression. Ann. N. Y. Acad. Sci. 2009;1177:178–184. doi: 10.1111/j.1749-6632.2009.05024.x. [DOI] [PubMed] [Google Scholar]
- 39.Elinoff J.M., Chen L.Y., Dougherty E.J., et al. Spironolactone-induced degradation of the TFIIH core complex XPB subunit suppresses NF-κB and AP-1 signalling. Cardiovasc. Res. 2018;114:65–76. doi: 10.1093/cvr/cvx198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu Y., Zhou B.P. TNF-α/NF-κB/Snail pathway in cancer cell migration and invasion. Br. J. Cancer. 2010;102:639–644. doi: 10.1038/sj.bjc.6605530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu R.C., Qin J., Yi P., et al. Selective phosphorylations of the SRC-3/AIB1 coactivator integrate genomic reponses to multiple cellular signaling pathways. Mol. Cell. 2004;15:937–949. doi: 10.1016/j.molcel.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 42.Chen W., Zheng W., Liu S., et al. SRC-3 deficiency prevents atherosclerosis development by decreasing endothelial ICAM-1 expression to attenuate macrophage recruitment. Int. J. Biol. Sci. 2022;18:5978–5993. doi: 10.7150/ijbs.74864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen Z., Cole P.A. Synthetic approaches to protein phosphorylation. Curr. Opin. Chem. Biol. 2015;28:115–122. doi: 10.1016/j.cbpa.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Krenning G., Moonen J.R.A.J., van Luyn M.J.A., et al. Vascular smooth muscle cells for use in vascular tissue engineering obtained by endothelial-to-mesenchymal transdifferentiation (EnMT) on collagen matrices. Biomaterials. 2008;29:3703–3711. doi: 10.1016/j.biomaterials.2008.05.034. [DOI] [PubMed] [Google Scholar]
- 45.Moonen J.R.A.J., Krenning G., Brinker M.G.L., et al. Endothelial progenitor cells give rise to pro-angiogenic smooth muscle-like progeny. Cardiovasc. Res. 2010;86:506–515. doi: 10.1093/cvr/cvq012. [DOI] [PubMed] [Google Scholar]
- 46.Coin F., Oksenych V., Egly J.M. Distinct roles for the XPB/p52 and XPD/p44 subcomplexes of TFIIH in damaged DNA opening during nucleotide excision repair. Mol. Cell. 2007;26:245–256. doi: 10.1016/j.molcel.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 47.Koong A.C., Chen E.Y., Giaccia A.J. Hypoxia causes the activation of nuclear factor kappa B through the phosphorylation of I kappa B alpha on tyrosine residues. Cancer Res. 1994;54:1425–1430. [PubMed] [Google Scholar]
- 48.Hu Y., Lu H., Li H., et al. Molecular basis and clinical implications of HIFs in cardiovascular diseases. Trends Mol. Med. 2022;28:916–938. doi: 10.1016/j.molmed.2022.09.004. [DOI] [PubMed] [Google Scholar]
- 49.Manetti M., Romano E., Rosa I., et al. Endothelial-to-mesenchymal transition contributes to endothelial dysfunction and dermal fibrosis in systemic sclerosis. Ann. Rheum. Dis. 2017;76:924–934. doi: 10.1136/annrheumdis-2016-210229. [DOI] [PubMed] [Google Scholar]
- 50.Vonach C., Viola K., Giessrigl B., et al. NF-κB mediates the 12(S)-HETE-induced endothelial to mesenchymal transition of lymphendothelial cells during the intravasation of breast carcinoma cells. Br. J. Cancer. 2011;105:263–271. doi: 10.1038/bjc.2011.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thiagarajan H., Thiyagamoorthy U., Shanmugham I., et al. Angiogenic growth factors in myocardial infarction: a critical appraisal. Heart Fail. Rev. 2017;22:665–683. doi: 10.1007/s10741-017-9630-7. [DOI] [PubMed] [Google Scholar]
- 52.Ylä-Herttuala S., Bridges C., Katz M.G., et al. Angiogenic gene therapy in cardiovascular diseases: dream or vision? Eur. Heart J. 2017;38:1365–1371. doi: 10.1093/eurheartj/ehw547. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.