

Summary
The emergence of COVID-19 and severe acute respiratory syndrome (SARS) has prioritized understanding bats’ viral tolerance. Myotis bats are exceptionally species rich and have evolved viral tolerance. They also exhibit swarming, a cryptic behavior where large, multi-species assemblages gather for mating, which has been hypothesized to promote interspecific hybridization. To resolve the coevolution of genome architecture and their unusual antiviral tolerance, we undertook a phylogenomic analysis of 60 Old World Myotis genomes. We demonstrate an extensive history of introgressive hybridization that has replaced the species phylogeny across 17%−93% of the genome except for pericentromeric regions of macrochromosomes. Introgression tracts were enriched on microchromosome regions containing key antiviral pathway genes overexpressed during viral challenge experiments. Together, these results suggest that the unusual Myotis karyotype may have evolved to selectively position immune-related genes in high recombining genomic regions prone to introgression of divergent alleles, including a diversity of interleukin loci responsible for the release of pro-inflammatory cytokines.
Graphical abstract
Highlights
-
•
Phylogenomics reveals pervasive introgression among Old World Myotis bats
-
•
A cryptic bat mating behavior called swarming promotes interspecific introgression
-
•
Genomic regions of low recombination repel introgression and retain the species tree
-
•
Immune loci are among those most frequently introgressed
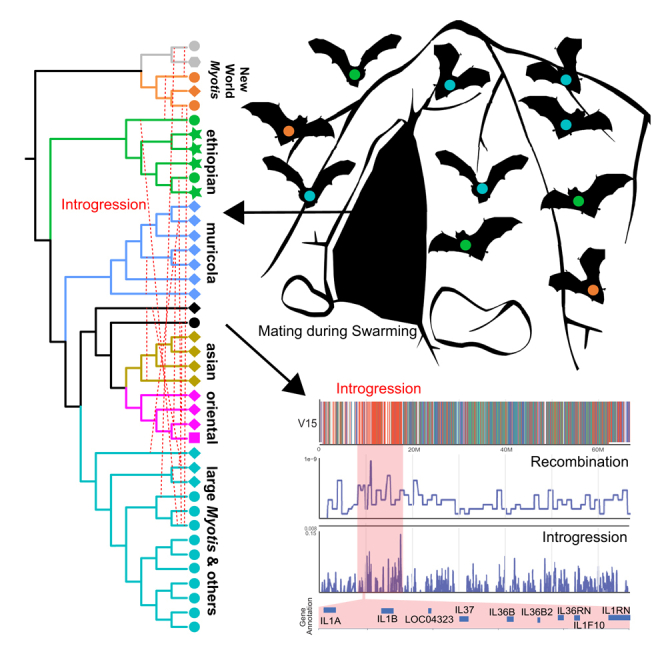
In their recombination-aware phylogenomic analyses, Foley et al. demonstrate pervasive introgression among Myotis bats, likely promoted by their use of cryptic swarming behavior during mating. Introgression has acted to restrict the species tree to low recombining regions of the genome. In contrast, immune loci are enriched in introgressed regions.
Introduction
The observation that several bat species are tolerant to some viruses of significant concern to human and animal health1,2 has led global public health initiatives to prioritize understanding the basis of this phenotype.3 Hypotheses for this enhanced tolerant, antiviral immunity include the coordinated evolution of bat flight,4 unique immune system adaptations, and bat/virus coevolution.2,5,6,7,8,9 The hypothesized coevolution of bats and viruses1,10 suggests that interrogating phylogenomic discordance may be instrumental in understanding the genetic basis of viral tolerance. Resolved species-level phylogenies may also aid in monitoring future disease emergence through the identification of related species that host similar viruses.11 Myotis bats comprise the most species-rich bat genus and have an almost global distribution.12 However, their phylogenomic relationships lack consensus, most likely due to some combination of gene flow and incomplete lineage sorting during their evolution, the resolution of which has been hampered by limited surveys of genomic variation.12,13,14,15
Adaptive introgression of immune loci is frequently observed between species pairs across the mammalian phylogeny.16,17,18,19,20 Additionally, mating strategy has been shown to result in systematic differences in immune responses, conferring enhanced protection again sexually transmitted diseases in promiscuous primates and adenovirus infection in promiscuous bats.21,22 Temperately distributed bats, including numerous Myotis species, exhibit autumnal swarming, a cryptic behavior where large, multi-species assemblages gather for mating.23 During the swarming period between August and October, thousands of bats from multiple bat genera form nightly gatherings at cave entrances.24 At these transient meetings, bats exhibit intense flight activity, chasing, and increased communication, including echolocation and human-audible social calls, which facilitate interspecific communication.24,25,26 Additionally, these assemblages are typically 80% male biased,27,28 suggesting intense competition for mates.29 Although the phenology of swarming varies by species, elevation, and seasonal timing,26,30,31 many bat species and genera still overlap at swarming sites.
Initially, the purpose of swarming was thought to be locating suitable hibernation sites and orienting juveniles to their whereabouts.25 Later, swarming was shown to facilitate gene flow between isolated populations of the same species.32 Most recently, genetic analyses of microsatellites have suggested that swarming may promote interspecific hybridization among Myotis species, but the strength and consistency of this process during the evolution of the genus is unknown.23 We hypothesized that an excess of historical gene flow, facilitated by swarming, has contributed to the acquisition and maintenance of novel and divergent genomic diversity. Some of this novel variation may include immune gene loci that enhance viral tolerance.
Results
Whole-genome phylogenies
To test this hypothesis, we sequenced and aligned 60 new Myotis genomes representing 42 species to a chromosome-level long-read reference genome assembly for Myotis myotis (Tables S1 and S2).33 We improved this assembly by initially using the associated gene annotation and previously generated ZooFISH maps34 to predict the location of unassigned scaffolds.
The original Hi-C data33 were remapped to the assembly, and assignments were made if contacts in the Juicebox Analysis Toolkit35,36 matched predictions made by the ZooFISH and gene annotation analysis. In this way, we placed 37 scaffolds that were previously chromosomally unassigned (Table S3). Notably, these contained large clusters of immune-related genes (e.g., major histocompatibility complex [MHC], immunoglobulin heavy-chain genes, and interleukin receptors) (Figure S1), which have previously been implicated in adaptive introgression.37,38 Our analysis localized these scaffolds predominantly to chromosome ends, which typically possess elevated recombination rates (Figure S2).
To establish a baseline phylogeny for the Old World Myotis, we sampled phylogenomic signal across the chromosome alignments, analyzing 1,463,340 single-nucleotide variants (SNVs) with site pattern frequency-based coalescent methods in SVDquartets.39 The same dataset was also concatenated to generate a maximum likelihood tree using IQ-TREE 2.40 The concatenation- and coalescent-based SNV phylogenies were highly congruent. Both trees identified six primary Old World Myotis clades (Figure 1A) and differed only in the positions of the whiskered and muricola clades and a few rogue taxa, including M. annatessae and M. welwitschii (Figures 1A and S3). By contrast, there was a considerable degree of phylogenomic conflict between the nuclear and mitogenome topologies. Widespread clade switching occurred for several focal species (Figures 1A and 1B), similar to results from small-scale analyses of nuclear and mitochondrial gene fragments (Figures 1B, 1C, and S4).12 Resolving the basis for this discordance was critical to distinguishing species relationships and associations that may arise due to gene flow and other types of phylogenetic noise.41,42,43,44,45
Figure 1.

Clade switching between the whole-genome nuclear and mitogenome topologies
(A) Maximum likelihood cladogram from ∼1.5 million genome-wide SNVs, showing only one sample per species where multiple exist. Bootstrap support is 100% for all nodes unless indicated otherwise. Dashed branches show which relationships differ between concatenation and coalescent analyses of the same dataset. Concatenation bootstrap values are shown in red.
(B) The mitogenome phylogeny is illustrated to highlight, via gray dashed arrows, clade switching relative to the nuclear phylogeny for the species indicated with colored dots.
(C) Robinson-Foulds (RF) distances between the nuclear and mitogenome topologies.
Mitonuclear discordance is generally predictive of gene flow in other mammals, although there are some exceptions.46,47 Among species with a history of post-speciation gene flow, it has been demonstrated that the species tree is not the dominant phylogenomic signal across the genome.42,43 However, majority-rule phylogenomic approaches assume that the most frequent topology represents the species tree and therefore often mask underlying signatures of discordance.41,43,44 In several hybridizing radiations, episodes of post-speciation gene flow have erased the historical branching events of the species tree from large swaths of the genome.42,43,45 Therefore, we implemented a recombination-aware locus tree approach, which has proved crucial to distinguishing the phylogenetic signal of speciation from introgression.
All surveyed Myotis species shared an identical G-banded karyotype (2n = 44) except M. annectans, with (2n = 46),34,48 an interesting trait given that animal radiations with histories of post-speciation gene flow typically possess conserved karyotypes.41,42,43,49 The Myotis karyotype is also unusual among mammals, comprising three large autosomes (∼228–217 Mb) and 18 much smaller chromosomes (∼108–14 Mb) (Figure 2A),34 which superficially resemble macro- and microchromosomes commonly observed in avian and reptilian karyotypes.50,51 We used ReLERNN,52 a deep learning approach, to infer a sex-averaged recombination map for M. myotis (Figure S5). A comparative analysis indicated the recombination landscape is conserved between M. brandtii and M. myotis (Figure S6). Therefore, we proceeded to use the M. myotis map as a representative for all Myotis. Low recombining regions were narrowly restricted to the pericentromeric regions of the largest macrochromosomes (V1, V3, and V5) and the X chromosome (chrX) (Figure 2B). The highest recombination rates were predictably found on the smallest autosomes, which also exhibit elevated gene densities and GC content (Figure 2C), similar to the microchromosomes of birds and some reptiles.50,51
Figure 2.

An unusual mammalian karyotype
(A) The dashed line indicates 30 Mb, which previous studies have used as a cutoff to define microchromosomes.50
(B) Chromosome length is inversely related to recombination rate.
(C–F) Comparison of recombination rate (C), genes per base (D), coding bases (E), and GC content (F) between macro-, mid-sized, and microchromosomes.
Recombination-aware phylogenomics
To investigate phylogenomic variation across the recombination landscape, we computed 36,264 maximum likelihood (ML) locus trees for 60 species from the whole-genome dataset using the GTR + GHOST model of rate heterogeneity53 in IQ-TREE 2 from 50 kilobase (kb) alignment windows covering 91% of the M. myotis genome. The GHOST model was chosen given that recent studies have demonstrated that some tests of introgression are highly sensitive to rate variation across clades.54 Ten subtrees (Table S4) were pruned from these datasets to determine the genomic stability of relationships among the principal clades and between individual species whose positions were difficult to resolve in the whole-genome and mitogenome analyses (Figures 1A and 1B). These analyses revealed widespread locus tree discordance otherwise masked in the whole-genome concatenation and coalescence phylogenies (Figures 3 and S7–S15). Locus tree frequencies varied markedly between chrX and the autosomes and between high and low recombining regions (Figure S16).
Figure 3.

Genome-wide distribution of phylogenomic discordance across the Old World Myotis radiation
(A) Distribution of phylogenomic signal for the whiskered clade visualized using Tree House Explorer61 in 50 kb locus alignment windows across the genome. Vertical bars along each chromosome are color coded to depict the distribution of the most frequent topologies.
(B) Comparison of the frequency of t1, t2, and t3 across the whole genome (left) and between the autosomes and the X (middle) and between high and low recombining regions of the genome (right).
(C) Whole-chromosome view for ChrV3 illustrates local relationship between recombination rate, f(D), the alpha parameter of the gamma model, and the internal branch length (ibl) for relationships between M. mystacinus and M. dasycneme. Note the depletion of topologies associated with introgression (t2 [red], t3 [green]) in the low recombining region at the center of the chromosome.
(D) Distributions of phylogenomic signal for additional Myotis clades in relation to chromosome type and recombination rate (reference the key in B).
Phylogenomic relationships within the whiskered bats and most Myotis clades exhibited asymmetric tree topology frequencies, considered one of the clearest indicators of the presence of gene flow (Figures 3 and S16).55 Previous studies have shown that in the presence of gene flow, the species tree is enriched in low recombining regions of the genome (particularly pericentromeric regions and the chrX) (Figure S17), while trees reflective of gene flow will be enriched in high recombining regions.41,43,44,45 Using a combined approach to infer the species tree, our distributions of phylogenomic signal were validated by three statistical measures that distinguish introgression from incomplete lineage sorting (ILS): (1) whole-genome D-statistics56 (Table S5), (2) windowed f(D) statistics (Figures 3C, S16, and S17), and (3) QuIBL (Table S6). While D-statistics can yield insights into the genome-wide presence of introgression, f(D) can provide information on more precise genomic locations of introgression. QuIBL takes a distribution of internal branch lengths from a set of locus trees and uses a coalescence-based model to infer the proportion of locus trees derived from introgression or ILS. For the whiskered clade, topology 1 (t1) is most frequent in low recombining regions and in regions with reduced introgression, while t2 becomes more frequent in introgressed, high recombining regions (Figure 3B). Similar observations across numerous eukaryotic clades have increasingly demonstrated the predictive power of using regions of low recombination to infer the species tree, as they are depleted of ILS and gene flow.41,43,44,45,57,58,59,60
Remarkably, the inferred species tree uniting the whiskered clade with New World species was restricted to less than ∼60% of the genome, clustering in the pericentromeric regions of the three macrochromosomes, and chrX, which possessed the lowest rates of recombination (Figures 3 and S16). This pattern, as well as patterns of alpha and internal branch length (ibl) parameter distributions along chromosomes (Figure 3C) , was mirrored in a highly predictable manner across other Myotis clades (Figure S18) where the species tree was commonly observed in less than 50% of the genome. Crucially, there are numerous differences between the recombination-aware inferred species tree (Figure 4A) and the whole-genome concatenation and coalescence analyses inferred from SNVs. D-statistics show that analyses of the genome-wide SNV dataset return the most frequent introgressed topology for the relationships between the muricola, large myotis, Asian, and Oriental clades, the respective positions of M. annatessae, M. capaccinii, and M. pilosus, and the relationship among M. pilosus, M. capaccinii, and M. laniger (Figure 4B; Table S5). D-statistics and QuIBL (Table S6) also show that clade switching between nuclear and mitogenome topologies (Figure 1B) is due to historical interclade introgression likely due to range overlap between Asian-distributed M. annectans and M. indochinensis (Figure 4).
Figure 4.

Discordance due to gene flow
(A) Species tree inferred from recombination-aware phylogenomic approaches. Crucially, our analyses show that standard whole-genome analyses in Figure 1 do not recover the species tree and instead depict relationships consistent with introgression for some species (e.g., M. capaccinii). Dashed lines indicate where within and between introgression is detected. Note that we illustrate introgression only for the analyses presented in (B) for the purpose of clarity. Additional instances of genome-wide introgression are depicted in Figure 5.
(B) Whole-genome-based D-statistics generated using ABBA-BABA tests in ANGSD illustrate significant evidence for introgression (Z score > 3) in (A). Trees are represented top to bottom in the format (H1, (H2, H3), O), where the outgroup O (S. latirostris) is not shown. See Table S5 for detailed results.
Previous studies have demonstrated that whole-genome D-statistics maintain sensitivity when there is up to 20% genetic distance among the ingroup species in the test62 and are also relatively robust to the genetic distance between the outgroup and ingroup species. In our analysis, the genetic distance between component species of Old and New World clades is ∼16%, which spans the deepest divergence in our dataset. In conclusion, our findings support a scenario for Old World Myotis evolution where a long history of interspecific hybridization involving species from multiple clades has resulted in massive depletion of the species tree across the arms of the macrochromosomes increasing toward telomere ends, with a near-complete replacement by introgression signatures on the smallest microchromosomes.
A link between introgression and swarming behavior?
Introgression is exceptionally pervasive among Myotis bats, even when compared to other mammalian clades with histories of ancient introgressive hybridization.43,49,63 This may be due to the use of swarming behavior in many Myotis species. Swarming behavior is characterized by multi-species, male-biased mating assemblages often numbering from hundreds to thousands of individuals.23 Many records of species present at swarming sites come from predominantly European studies and overlap our sampling (Figure 5A; Table S7). Therefore, we pruned subtrees derived from the 50 kb windows from the whole-genome 60 species sliding-windows dataset (Table S4) to test the hypothesis that hybridization is common among species within and between clades recorded at swarming sites (Figure 4B). Asymmetry in genome-wide frequency and D-statistics estimated among swarming species indicated that introgression was the dominant driver of phylogenetic discordance in these analyses (Figures 5B, S19, and S20). Additionally, we show that introgressed topologies with the highest f(D) values are the most enriched on microchromosomes (Figure S21). Introgression was observed not only among species within clades but also between species from divergent clades (Figure 5B), like the large Myotis, whiskered, and New World clades. Note, due to our sampling scheme, we could only infer the presence of historical introgression among Myotis species. Population-level data or data sampled from species found contemporaneously at swarming sites would be required to accurately determine the prevalence of ongoing hybridization at swarming sites, which has been documented for some of the species we sampled.15,63,64
Figure 5.

Introgression occurs among species that co-occur at swarming sites
(A) Species assemblages reported at swarming sites throughout Europe (Table S6). The tree inset shows the species tree and clade membership of each species.
(B) Examples of within and between clade introgression. Variation in topological frequency is observed between autosomal, X, and high and low recombining genomic partitions. Whole--genome D-statistics demonstrate phylogenomic conflict is due to introgression.
(C) Comparisons of the genome-wide D-statistic for species representing the large myotis clade, with a promiscuous mating system, and M. bocagei from the Ethiopian clade, which employs a conservative (closed harem) mating system. The same analysis was repeated using M. yumanensis as the reference genome to demonstrate that our results are not confounded by reference bias (Figure S25; Table S5). Trees for introgression tests are represented top to bottom in the format (H1, (H2, H3)), O), where the outgroup O (S. latirostris) is not shown. See Table S5 for detailed results.
The Ethiopian clade (Figure 1A) contains important contrasts to other predominantly swarming Myotis clades. At least one species lacks swarming behavior: M. bocagei employs a harem system where a single male and 6–8 females gather in a banana leaf for mating.65 This behavior should minimize the probability of introgression. Although not all mating strategies employed by the Ethiopian clade species are well characterized, the roughly equal frequency of locus trees for the clade are consistent with ILS, and not introgression, being the dominant source of phylogenomic discordance (Figure 3C). This is notable given that M. bocagei, M. tricolor, and M. welwitschii are sympatric over much of their current distributions. Conversely, two members of the Ethiopian clade, M. emarginatus and M. dasycneme, have a European distribution and are frequently reported at swarming sites (Figures 4A and 5A). Whole-genome D-statistics consistently show significant ancient introgression signatures between M. emarginatus and numerous European distributed species, yet there is minimal evidence of ancient introgression with M. bocagei (Figure 5C). In summary, these contrasting phylogenomic patterns between swarming and non-swarming species support the hypothesis that swarming sites have acted as crucibles of genetic exchange.
Introgression at immune loci
We hypothesized that viral tolerance in Myotis bats may be enhanced by a long history of immune gene introgression involving species from multiple phylogenetic lineages interacting at swarming sites.66 Introgression from Neanderthals into modern humans has had measurable impacts on the expression of genes associated with immunity.16,67,68 Most notable among these is differential expression of an OAS1 isoform, which has previously been shown to contribute to differential susceptibility and severity of COVID-19 outcomes in humans.69 To further examine if the apparent genome-wide intersection between introgression and immune-related processes has had any potential effect on gene expression in Myotis bats, we used a database of differentially expressed genes from a study that performed viral challenge experiments on M. daubentonii cell lines.70 These cells were treated with a modified Rift Valley fever virus or supplemented with interferon, and gene expression was compared 6 h before and after treatment.70 Our results demonstrate that genes that were differentially expressed in response to viral challenges in M. daubentonii were among the most frequently introgressed across microchromosomes (Figure S22) and were also significantly introgressed genome-wide across numerous Myotis clades (Figure S23). Notably, these include microchromosome-linked genes OAS1, OAS3, and OASL, which exhibit up to a 6-fold change in expression in response to viral challenge.70 Because one of the common outcomes of interspecific hybridization is altered gene expression,68,71,72,73 these results suggest that frequent hybridization-induced modulation of immune gene expression in bats might be adaptive and is worthy of further investigation in natural and experimental populations.
Recombination acts to break down introgressed haplotype blocks over time. While long blocks representing more recent introgression events are expected to be rare in our dataset due to low population genomic sampling, one of the largest haplotypes spans ∼10 Mb on the subtelomeric region of V15 and possesses an introgressed topology uniting M. myotis and M. daubentonii (Figures 6 and S24). Measures of recombination rate, ibl, and f(D) were all elevated inside the block relative to the remainder of the chromosome. A STRING analysis of this haplotype block identified a significant enrichment (false discovery rate [FDR] < 0.05) of genes associated with cytokine receptor activity, the interleukin-36 pathway, and the interleukin-1 family signaling pathway (Table S8). IL36 is notable as a member of the IL1 superfamily of pro-inflammatory cytokines, which help regulate the adaptive and innate immune systems.74 IL36 promotes antiviral immunity by increasing cellular sensitivity to interferon75 in human and mouse models to herpes simplex virus75 and a host of other viruses.74
Figure 6.

Evidence for a potential link between immunity and the recombination landscape in Myotis bats
Distribution of phylogenomic signal, recombination rate, ibl, and f(D) values for chromosome V15 for the bechsteinii clade. A large introgressed block for myotis and daubentonii (Figure S24) is highlighted in red and is significantly enriched for IL-36 and IL-1 signaling pathways (FDR < 0.05) (Table S8).
Discussion
Collectively, our results support a scenario where swarming-mediated interspecific hybridization can impact antiviral immune responses in bats. High rates of introgression have also been reported in animal clades where multiple, closely related species are present during mating, including species of Stenella and Tursiops dolphins76,77,78 and Anopheles mosquitoes.42,79 Swarming has specifically contributed to the introgression-mediated evolution of insecticide resistance at several loci in anopheline mosquitoes.80,81 Promiscuous mating behaviors may frequently evolve to promote adaptive interspecific hybridization, providing access to divergent genomic variants and/or quantitative variation in gene expression as a rapid means to counter frequent and novel pathogen exposure.71 The extraordinarily large number of mammalian congeners present in large colonies of bats (sometimes thousands of individuals) and mating during swarming would impose a unique burden on mucosal viral immunity. Under this model, frequent introgression at swarming sites would conceivably afford myriad opportunities for adjusting viral symbiont management and/or defense mechanisms using pooled immunogenomic resources from species across the genus. As such, swarming behavior may have provided a singular opportunity for the evolution of a novel mechanism for viral management/symbiosis through introgression biased toward chromosome termini enriched for immune loci. Such innovations are not unprecedented in nature. Radical and profound innovations in immunogenetic diversification have previously evolved from horizontal genetic transfer in other taxa (Table S9)—for example, the CRISPR-Cas9 prokaryotic adaptive immune system from IscB and IS200/IS605 transposons82 and the somatic cell recombination capability of immunoglobulin and T cell receptor genes via domestication of ProtoRAG transposase83 in an early chordate. Further investigations into understanding how karyotype evolution, the recombination rate landscape, and the introduction of divergent genomic variants via introgression and/or ILS intersect to hone bats’ genomes for viral and cancer tolerance may yield novel innovations for viral immunotherapeutics and even oncology.84 Finally, our results suggest that swarming sites may merit recognition as special sites of conservation given the unique evolutionary processes to which they play host.23,28,31,85
Limitations of the study
All data were mapped to M. myotis, which is part of the ingroup in our analysis, and so may lead to spurious inference of introgression due to reference bias. To account for this potentially confounding factor, we also mapped the data to the long-read M. yumanensis genome assembly86 from the New World clade. Although this analysis confirmed our initial results for the Old World ingroup, ideally a closely related outgroup, which is roughly equidistant to all ingroup species, should be used to mitigate any potential reference bias and data loss due to mapping to a divergent reference genome. Our analysis was also constrained to the detection of historical introgression. Although some studies indicate that contemporary introgression occurs at swarming sites,23 our dataset was not selected to formally test this hypothesis. Further comparative phylogenomic analyses of species with conservative and promiscuous mating behaviors would be useful to fully characterize the contribution of swarming behavior to introgression in bats.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| HLmyoMyo6 | Jebb et al.33 | GCA_014108235.1 |
| Myotis myotis Hi-C data | Jebb et al.33 | SRR11732436 |
| Myotis myotis ZooFISH map | Volleth et al.34 | N/A |
| Myotis yumanensis reference genome | Curti et al.87 | mMyoYum1.0.hap1 |
| Myotis myotis SRA data | Zoonomia Consortium88 | SRS3678524 |
| List of differentially expressed genes from a study that treated Myotis daubentonii cell lines with a modified Rift Valley Fever virus or supplemented with IFN to measure response to a viral challenge | Holzer et al.69 | Supplementary Material |
| WGS phylogenomic data generated in this study | This study | PRJNA1035163 |
| WGS data used to generate Myotis myotis recombination map | This study | PRJNA1035163 |
| WGS data used to generate Myotis brandtii recombination map | Laine et al.89 | SRR18788643, SRR18788644, SRR18788646, SRR18788647 |
| Myotis myotis recombination map | This study | https://doi.org/10.5281/zenodo.7044479 |
| Myotis brandtii recombination map | This study | https://doi.org/10.5281/zenodo.7044479 |
| Alignments | This study | https://doi.org/10.5281/zenodo.7044479 |
| Newick Tree files | This study | https://doi.org/10.5281/zenodo.7044479 |
| Software and algorithms | ||
| Trim Galore! v0.6.5 | Krueger et al.90 | https://github.com/FelixKrueger/TrimGalore |
| bwa-mem v0.7.17 | Li et al.91; Li92 | https://github.com/lh3/bwa |
| Qualimap | Garcia-Alcalde et al.93 | http://qualimap.conesalab.org/ |
| Samtools v1.9 | Li et al.94 | https://github.com/samtools/samtools |
| GATK v4.1.2 | McKenna et al.95; DePriesto et al.96; Poplin et al.97; Van der Auwera et al.98 | https://github.com/broadinstitute/gatk |
| ANGSD v0.916 | Korneliussen et al.56 | http://www.popgen.dk/angsd/index.php/Installation |
| SOAPdenovo2 vr242 | Luo et al.98 | https://github.com/aquaskyline/SOAPdenovo2 |
| Geneious Prime 2019.04 | Dotmatics | https://www.geneious.com/ |
| RYcode | Braun et al.99 | https://github.com/ebraun68/RYcode) |
| IQtree2 v2.2.1 | Minh et al.40 | https://github.com/iqtree/iqtree2 |
| Quast v5.2.0 | Gurevich et al.100 | https://github.com/ablab/quast |
| Bcftools v1.14 | Danecek et al.101 | https://github.com/samtools/bcftools |
| VCFtools v1.16 | Danecek et al.102 | https://vcftools.sourceforge.net/ |
| ReLERNN | Adrion et al.52 | https://github.com/kr-colab/ReLERNN |
| SVDquartets | Chifman and Kubatko39 | https://paup.phylosolutions.com/ |
| SNP-sites v2.5.1 | Page et al.103 | v2.5.1 |
| PAUP∗ v4a168 | Posada et al.104 | https://paup.phylosolutions.com/ |
| trimAl v1.4.1 | Capella-Gutierrez et al.105 | https://vicfero.github.io/trimal/ |
| Tree House Explorer (THEx) | Harris et al.61 | https://github.com/harris-2374/THEx |
| R v4.2.2 | R Core Team106 | https://www.r-project.org/ |
| f(D) | Martin et al.87 | https://github.com/simonhmartin/genomics_general |
| QuIBL | Edelman et al.41 | https://github.com/miriammiyagi/QuIBL |
| STRING v12.0 | Szklarczyk et al.87 | https://string-db.org/ |
| Dfoil | Pease and Hahn88 | https://github.com/jbpease/dfoil |
| Critical commercial assays | ||
| Qiagen Puregene kit | Qiagen | Cat. No./ID: 158063 |
| NEBNext Ultra II DNA Library Prep Kit for Illumina | New England Biolabs | E7805L |
| NEBNext Multiplex Oligos for Illumina | New England Biolabs | E7600S |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, William Murphy (wmurphy@cvm.tamu.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
All code used to generate the analysis is publicly available and are cited in the text. All short-read data generated as part of this study have been submitted to the NCBI Short Read Archive under the following BioProject PRJNA1035163. Recombination maps for M. myotis and M. brandtii, whole-genome SNP datasets used for concatenation and coalescence analyses, a FASTA-formatted version of the updated M. myotis genome assembly, and whole-genome alignments are deposited in the following Zenodo repository: https://doi.org/10.5281/zenodo.7044479. The updated M. myotis genome assembly can be found under BioProject PRJNA628559.
Method details
Taxonomic sampling and sequencing
We isolated genomic DNA from museum specimens, sampling 58 individual bats, putatively representing 58 individuals and 42 putative Old World Myotis species for the ingroup, selected to index the major clades and nodes in the tree.12 An unknown New World species, Myotis spp. (see below), and Submyotodon latirostris were also sequenced as outgroup species (Table S1). BLASTn107 was used to try determine a more specific nomenclature for Myotis spp. using the mitogenome and Cyt-b sequences derived from this sample. The top hit for the mitogenome was Myotis martiniquensis with 94.6% pairwise identity and 91.81% query coverage. The top hit for the Cyt-b sequence was the GenBank: JN020572, a Myotis sp. individual from Suriname which grouped in a clade including Myotis nyctor and Myotis nesopolus in the following analysis.108 Pairwise identity was 94.7% and query coverage was 99.91%.
DNA was extracted using the Qiagen Puregene kit following the manufacturer’s protocol, except 10μL of linear acrylamide was added to the lysis buffer to aid the recovery of DNA from small starting amounts of tissue. Standard Illumina fragment libraries (300-bp) were prepared for each DNA sample using the NEBNext Ultra II DNA Library Prep Kit for Illumina. Samples were sequenced to ∼20× genome-wide depth of coverage with 2 × 150-bp reads on the Illumina HISeq X platform. We combined these data with published genomes from two other bat species: Myotis brandtii109 and Myotis myotis.
Sequence QC and read mapping
Raw reads were trimmed using Trim Galore (http://www.bioinformatics.babraham.ac.uk/projects/trim_galore). All but one species of Myotis possess a karyotype of 2n = 44, the exception being Myotis annectans (2n = 46). G-banding similarities indicate a high degree of collinearity across the genus.48 All trimmed and filtered reads, using default trimgalore parameters,90 were mapped to the Myotis myotis reference genome, HLmyoMyo6. Sequence alignment was carried out using bwa-mem91,92 using the -M and -R parameters. Mapping results were evaluated and summarized using the Qualimap function bamqc.93 Samtools94 was used to remove duplicate reads. Local realignment and variant calling was performed using GATK.95,96,97,98 Consensus pseudohaploid variants were called for each species using ANGSD56 where the minimum required mapping quality was 30 and minimum base quality of 20. Short-read Illumina assemblies struggle to accurately resolve highly repetitive and duplicated regions within a genome. This can introduce erroneous variant calls which can confound branch length inference in downstream phylogenomic analyses. To control for this, we also used ANGSD to identify and exclude all alignment regions with read depth greater than 200% of the average coverage per genome and positions with a minimum depth of 5 reads. Average mapped read depth and the proportion of the reference covered by mapped reads was calculated to assess the degree of missing data per species across the genome (Table S2)
Mitogenome assembly and analysis
To avoid incorporating nuclear copies of mitochondrial genes in our mitogenome assemblies, we aimed to sample small subsets of reads to capture only reads belonging to the mitochondria, as nuclear copies of mitochondrial genes would be rare in such small subsets. Random subsets (2, 4, and 8 million) of raw reads were extracted for each sample and trimmed as described above. SOAPdenovo2110 was used to assemble the mitogenome by evaluating a range of k-mer sizes (71, 81, and 91), with a read length of 150bp and 300bp as the insert size; all other parameters were set to default. Typically, the longest scaffold represented the majority of the assembled mitogenome. The assembly of the highly repetitive D Loop region was incomplete and highly variable across species and so was excluded from further analysis. To orient, annotate and align scaffolds for downstream analyses, each scaffold was mapped to the M. myotis mitogenome,111 with a final alignment of ∼17kb. Coding regions were extracted from the mitogenome using Geneious Prime 2019.04. Coding regions were RY coded using RYcode (https://github.com/ebraun68/RYcode)99 to reduce the potential impact of compositional bias.112 Alignments were then concatenated to form a single matrix. We prepared a second mitochondrial dataset which included the whole mitogenome, D Loop excluded. For both datasets, a Maximum likelihood (ML) tree was generated using IQtree240 under a GTR+I+G4 model of sequence evolution. The tree was evaluated with 1000 bootstrap replicates using the ultrafast bootstrap approximation (Figure S4).113,114
Improving the Myotis myotis reference genome
The reference genomes for bats described in33 reported 96–99% of data assembled to ‘chromosome-level scaffolds’. However, upon examination of the assembly for key regions like the pseudoautosomal region and the major histocompatibility complex (MHC) locus, we determined that many genic regions were not assembled on chromosomes and would limit downstream inferences. We used two complementary approaches to further improve the completeness of the chromosome-level scaffolds. First, we used the gene annotation and a previously published ZOO-FISH map of human autosomal probes painted on Myotis myotis autosomes34 to predict, based on gene content, which scaffolds in the HLmyoMyo6 assembly corresponded to chromosome paints in the ZOO-FISH map. We re-mapped the original Myotis myotis Hi-C (SRR11732436) data to test these predictions and re-scaffolded HLmyoMyo6 (GCA_014108235.1).33 The Hi-C contact map was visualized using Juicebox.36 We searched for contacts uniting scaffold associations identified using the gene annotations and ZOO-FISH maps and adjusted the assembly accordingly. This way, more data were assigned to chromosomes, most often to chromosome ends (Figure S2). Differences between the original assembly and the improved version are summarized via Quast100 in Table S3. Assembling regions with high rates of recombination regions is key to polarizing and accurately interpreting the distribution of phylogenomic signal. Further, we noted that many previously unassigned scaffolds contained immune genes, including the Major Histocompatibility Complex (MHC) and immunoglobin clusters (Figure S1), genomic regions of particular interest given bats’ exceptional immunity115 and role in emerging pathogens.116 Placing these important genomic regions in the correct genomic context furthers our understanding of the evolution of immune genes in bats.
A recombination map for Myotis myotis
We generated a recombination map for M. myotis to help interpret the chromosomal distribution of phylogenomic signal in our data. Studies have shown that hybridization occurs between the closely related sister species M. myotis and Myotis blythii, where they occur in sympatry throughout much of South and Central Europe.117,118 To avoid bias due to potential genomic introgression between the species, we selected bats from Brittany, north-west France, which occur outside the range of M. blythii. Procedures were approved and permits were issued by ‘Arrete’ by the Prefet du Morbihan. M. myotis were sampled in western France, 2013 from three roost locations Ferel (FER), La Roche Bernard (LRB) and Beganne (BEG). Bats were captured in custom harp traps as they left the roost and were placed in individual cloth bags until sampled. A 3mm biopsy punch was obtained from the wing membrane. Samples were flash frozen in liquid nitrogen. Before release all bats were offered food and water. As the recombination landscape of genomes has been shown to vary between different sexes of the same species,119 we selected samples from two males MMY78 (LRB) and MMY243 (BEG) and one female (MMY4) to construct a sex averaged map.
DNA was extracted, and Illumina libraries were prepared and sequenced as described above. Raw reads were filtered, and mapped to the M. myotis reference genome, and the resulting bam files were processed with GATK as before. Variants were called, and all samples were jointly genotyped, then filtered to remove variants in repeatmasked regions using GATK. Variants were further filtered, removing variants within 5bps of an indel and those which did not meet the following quality criteria -e'%QUAL<30 | INFO/DP < 16 | INFO/DP > 62 | QD < 2 | FS > 60 | SOR>10 | ReadPosRankSum <-8 | MQRankSum <-12.5 | MQ < 40′ in bcftools (https://github.com/samtools/bcftools).101 VCFtools (https://vcftools.github.io/man_latest.html) was used to remove indels, leaving 12,273,471 biallelic SNPs (referred to as Mm recombination dataset) for further analysis. ReLERNN, a deep learning approach that uses recurrent neural networks, was used to model the genome-wide recombination rate for M. myotis. The average mammalian mutation rate 2.2 × 10−9120 was used for this analysis. ReLERNN was run using the simulate, train, predict and bscorrect modules with default settings. Finally, inferred recombination rates were averaged in 2Mb blocks in 50kb sliding windows for use in downstream analyses.
To determine if the Myotis myotis recombination landscape was representative of the overall Myotis recombination landscape we used the same methods to generate a recombination map for the New World species, Myotis brandtii.89 Publicly available SRA data were mapped to the Myotis brandtii pseudohaploid assembly generated as part of this study.
Phylogenomics
Coalescent and concatenation whole-genome phylogenies
SVDquartets39 was chosen as the coalescence-based species tree approach to satisfy the assumption of no intra-locus recombination under the Multispecies Coalescent model (MSC). Variant sites were used to impute quartet trees which were then aggregated to infer a species tree. Many published coalescence-based analyses have included or been comprised only of protein coding regions or ultraconserved elements, thereby violating the MSC’s neutrality assumption. Variable sites were extracted from the per chromosome fasta alignments in vcf format using SNP-sites v2.5.1.103 Variable sites were thinned to a minimum of 1000bp between sites using VCFtools v1.16102 and were converted to nexus format using the script vcf2phylip.py (https://github.com/edgardomortiz/vcf2phylip). By using widely spaced variable sites, we aimed to account for the requirement of free recombination between loci, an assumption of coalescent methods like SVDquartets. The dataset was further filtered on missingness, stripping columns from the alignment with more than 10% missingness. While data were initially thinned to a minimum distance of 1000bp apart to avoid intralocus recombination, the final distance between most variable sites is likely much larger. Data from all chromosomes were concatenated using Geneious Prime 2019.04 for a final dataset comprising 1,463,340 SNVs (referred to as the genome-wide SNV dataset). A 50% majority rule tree was estimated using SVDquartets implemented in PAUP∗ v4a168104 using the QFM quartet amalgamation technique, sampling 99% of quartets.
The same genome-wide SNV dataset was concatenated into a supermatrix and analyzed with IQtree2 to generate a corresponding ML tree. Given that our nuclear datasets contained variable sites only, we used the +ASC option to correct for ascertainment bias due to the exclusion of constant sites in our analyses. Trees were analyzed as a single partition using a GTR + I + G model of sequence evolution to facilitate the use of the +ASC option. The +ASC option is currently incompatible with more complex mixture models. Trees were evaluated with 1000 bootstrap replicates using the ultrafast bootstrap approximation.113,114
Sliding windows
Many phylogenomic studies show that phylogenetic signal is non-randomly distributed throughout the genome. To sample phylogenomic signal, we performed a sliding window-based analysis signal across the 60-species whole genome alignment (referred to as the whole-genome dataset). Alignments were divided into 50-kb nonoverlapping, contiguous windows across the entire genome. Alignment columns were filtered in trimAl105 using the setting -gt 0.50 to allow only 50% missing data per alignment column and an alignment filtering script (https://github.com/VCMason/Foley2021) with the following parameters: cutoff = 0.02, Z score = 0.5, windsize = 50, step = 1, Myotis myotis as the reference and PCcutoff = 50, where an alignment is excluded if one species is missing more than 50% than the total alignment length. After filtering, only windows retaining 10kb or more data were retained for further analysis. Maximum Likelihood locus trees were generated using IQtree2 under a GTR model of sequence evolution and the GHOST model of rate variation (GTR∗H4). Trees were evaluated with 1000 bootstrap replicates using the ultrafast bootstrap approximation.113,114 Relationships for the difficult-to-resolve relationships were summarized in subtrees extracted from the whole-genome dataset (Figure 1; Table S4). Data were visualized using Tree House Explorer (THEx).61
Detection of gene flow
Introgression causes phylogenomic distortion.121,122 In a recent review, Hibbins and Hahn stated, “asymmetry in [the frequencies of] discordant tree topologies is one of the clearest signals of introgression”.55 Following this and other studies,41,43 we estimated the relative frequency of each topology for the autosomes and X separately. We also investigated the relationship between recombination rate and phylogenomic signal by calculating the frequencies of topologies in high and low recombining regions, defined as 500 windows at either end of the distribution of recombination rates.
Models of rate heterogeneity are fitted to data as part of some tree-building procedures to account for rate variation among sites. Simulations and empirical studies have shown that genome regions subject to introgression have elevated rate heterogeneity due to the accumulation of discordant donor sites in these regions. Potential introgression regions can be identified via the alpha (or shape) parameter, which is estimated as part of fitting gamma models of rate heterogeneity. Alpha parameters are lower in regions of potential introgression as multiple changes are required to account for apparent homoplasy.122,123 To estimate the alpha parameter per locus, alignments used to generate the 60 species locus trees pruned to represent subtrees (Table S4). As the GHOST model of rate heterogeneity was used to estimate locus trees, we re-ran IQtree2 for alignment loci to fit a gamma model of rate heterogeneity to the data. The alpha parameter, number of parsimony informative sites, and the sum of internal and external branch lengths per locus were extracted from the output using the bash command grep.
The combined effect of introgression and recombination can create local patchworks of phylogenomic histories, resulting in discordant tree topologies and branch lengths. The degree of discordance varies depending on the timing and direction of introgression and the relationship between hybridizing taxa.55 Introgression between sister taxa will not produce discordant topologies but will result in compressed branch lengths or divergence times. Introgression among non-sister species will create discordant topologies with much older or much younger branch lengths or divergence times depending on the direction and age of the introgression events.55 As such, we examined the genome-wide distribution of internal branch lengths extracted from quartet topologies using the ape package124 in R.106 Given the extreme morphological conservation observed among Myotis bats12 we did not formally estimate divergence times given the difficulty in accurately assigning fossils branches in this phylogeny, particularly in the presence of gene flow. In lieu of this, we examined the distribution of internal branch lengths to determine the distorting effect of introgression. In a divergence time analysis, the rate of substitution per site is simply scaled to time, thereby reflecting similar information.
Rather than relying solely upon genome-wide tests of introgression,125 which do not identify specific regions of admixture, we used f (D),87 which provides a point estimate of the admixture proportion at a given locus. More specifically, given a tree (((p1, p2), p3), O), f (D) describes the quantification of excess shared variation between p2 and p3 that is also not shared with p1. This approach also allows for the possibility of bidirectional introgression on a site-by-site basis. To test for evidence of admixture, we calculated f(D) for trios of species and an outgroup corresponding to the two most frequent introgressed topologies from each clade (Table S5). The whole-genome dataset chromosome alignments were pruned to the trio, and the outgroup and biallelic SNPs were called using SNPsites.103 f(D) was calculated using scripts from (https://github.com/simonhmartin/genomics_general) following (https://github.com/simonhmartin/tutorials/blob/master/ABBA_BABA_windows/README.md). Introgression was estimated in 10kb windows requiring a minimum of 250 SNPs per window as a cut-off for analysis.
Given that f(D) can underestimate introgression if all three species in the ingroup taxa hybridize, we also used QuIBL41 to determine to what extent discordance could be explained by ILS and introgression. This approach uses the distribution of internal branch lengths to distinguish between these two processes. Under ILS, internal branch lengths are expected to be exponentially distributed, while internal branches derived from introgressed regions will have a distribution with a non-zero mode.41 The fit of models where all discordance is due to ILS or a mixture of ILS and introgression are compared via likelihood, and the proportion of trees that best fit either model are returned. Trees were pruned to a trio of focal species and an outgroup and run through QuIBL using default parameters.
To determine if there was a difference in the probability of the admixture between related species based on mating behavior, we estimated D the whole genome statistic used to detect introgression among species, in the form (((H1, H2), H3), O), where O is the outgroup S. latirostris in all cases. Within the Ethiopian Clade, M. bocagei uses a harem-based mating strategy where one male and several females gather in a banana leaf for mating. Conversely, M. emarginatus is frequently reported at swarming sites in Europe. Using subsets of the whole-genome dataset, a suite of whole genome D statistics125 were calculated in Dfoil88 using mode = dstat and all other default parameters for all trees shown in Figure 5C which are shown in the format (outgroup, (P1,(P2, P3))). We varied the individual used to represent M. bocagei, the position of species in the tree and varied the third species selected in addition to M. bocagei and M. emarginatus to represent a variety of species which have been found to co-occur with M. emarginatus at swarming sites or are within the Ethiopian clade, but the status of their mating behavior is unknown.
To investigate if our results were impacted by reference bias, we repeated the analysis described above, but instead mapped the data to both the Myotis myotis reference genome and the Myotis yumanensis reference genome (mMyoYum1.0.hap1).86 As a member of the New World clade, M. yumanensis is roughly equally distant from all Old World species and so could be used to detect any possible bias from using a nested ingroup species as the reference. ABBA-BABA or whole genome D-statistics were estimated using ANGSD56 for combinations of BAM files resulting in highly similar estimates of D and Z-scores, indicating the negligible impact of reference bias. As such, the remainder of introgression tests were conducted with data mapped to the M. myotis reference genome. To investigate if sampling a different individual from the same species changed our introgression results, we mapped an additional M. myotis individual (NCBI: SRS3678524)126 and used this individual in our re-analysis. These results also showed negligible differences to our original results (Figures 5C and S25; Table S5).
Hybridization at swarming sites?
Typically swarming is thought to be limited to temperate bat species. However, data on the mating behavior of bats are limited and not widely known in families with large numbers of species. To better investigate this, we performed a literature search and aggregated data from multiple studies reporting species compositions at swarming sites across Europe (Table S7). As before, subtrees corresponding to the species which swarm within and between clades (Figure 5B) were pruned from the 60 species sliding windowed phylogenies derived from analyses of subsets of the whole-genome dataset. The number and frequency of genome-wide topologies were estimated using THEx.61 Given the observed asymmetry in topology frequencies, introgression was determined to be the most likely source of phylogenomic discordance. To test this, we calculated whole genome D-statistics (Figure 5B) and windowed f(D) statistics (Figure S17) for trios of species and an outgroup corresponding to the two most frequent introgressed topologies using data derived from the whole-genome dataset.
Anti-viral immunity and introgression
Given that immune loci are frequently introgressed among mammals,66 we sought to determine if genes associated with anti-viral immunity were introgressed in our dataset. Although the interferon pathway is a conserved anti-viral innate immune pathway, most species possess a repertoire of genes adapted for anti-viral defense. Given that standard gene enrichment analyses rely on data and function information for the best-studied model organisms, a traditional enrichment analysis would likely omit Myotis-specific components or features of anti-viral immunity. Therefore we used a custom database of differentially expressed genes from a study that treated Myotis daubentonii cell lines with a modified Rift Valley Fever virus or supplemented with IFN to look at differential gene expression in response to a viral challenge.70 For our database, we selected all differentially expressed genes with an adjusted p value of 0.005 or less from both treatments after the 6h time point to capture an immediate acute response to viral infection. Of these genes, 731/984 were present in the windows represented by our 50kb sliding windows dataset. The 253 missing genes were likely removed during the filtering of regions of poor alignment quality or were missing from the gene annotation. To determine if this gene set showed significant overlap with introgressed regions of the genome, defined as windows with a non-species tree topology, we performed a chi-squared test with a Yates correction in R using the native stats package.
Large introgressed haplotype blocks
A large introgressed haplotype block was identified through visual examination of genome-wide phylogenomic signal in THEx for the bechsteinii clade on chromosome V15. This corresponded to the topology where M. myotis and M. daubentonii were sister taxa relative to M. bechsteinii (red topology Figure 3C). Genes corresponding to this region were extracted as described previously, and an STRING enrichment analysis127 was performed choosing human for the database, where FDR > 0.05 was considered significant, and all other parameters as default to determine if any function or pathway was enriched in this region.
Acknowledgments
Portions of this research were conducted with the advanced computing resources and consultation provided by Texas A&M High Performance Research Computing. This work was funded by National Science Foundation grants DEB-1753760 and DEB-2150664 awarded to W.J.M. E.C.T. is supported by Irish Research Council Laureate Award IRCLA/2017/58 and Science Foundation Ireland Future Frontiers 19/FFP/6790.
Author contributions
Conceptualization, W.J.M. and N.M.F.; data curation, N.M.F. and K.R.B.; investigation, N.M.F., A.J.H., K.R.B., M.F.C., and W.J.M.; software, A.J.H.; project administration, W.J.M.; samples, M.R., S.J.P., and E.C.T.; writing – original draft, W.J.M. and N.M.F.; writing – review & editing, all authors.
Declaration of interests
The authors declare no competing interests.
Published: January 17, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2023.100482.
Contributor Information
Nicole M. Foley, Email: nfoley@cvm.tamu.edu.
William J. Murphy, Email: wmurphy@cvm.tamu.edu.
Supplemental information
References
- 1.Ruiz-Aravena M., McKee C., Gamble A., Lunn T., Morris A., Snedden C.E., Yinda C.K., Port J.R., Buchholz D.W., Yeo Y.Y., et al. Ecology, evolution and spillover of coronaviruses from bats. Nat. Rev. Microbiol. 2022;20:299–314. doi: 10.1038/s41579-021-00652-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carlson C.J., Albery G.F., Merow C., Trisos C.H., Zipfel C.M., Eskew E.A., Olival K.J., Ross N., Bansal S. Climate change increases cross-species viral transmission risk. Nature. 2022;607:555–562. doi: 10.1038/s41586-022-04788-w. [DOI] [PubMed] [Google Scholar]
- 3.Streicker D.G., Gilbert A.T. Contextualizing bats as viral reservoirs. Science. 2020;370:172–173. doi: 10.1126/science.abd4559. [DOI] [PubMed] [Google Scholar]
- 4.Zhang G., Cowled C., Shi Z., Huang Z., Bishop-Lilly K.A., Fang X., Wynne J.W., Xiong Z., Baker M.L., Zhao W., et al. Comparative analysis of bat genomes provides insight into the evolution of flight and immunity. Science. 2013;339:456–460. doi: 10.1126/science.1230835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O’Shea T.J., Cryan P.M., Cunningham A.A., Fooks A.R., Hayman D.T.S., Luis A.D., Peel A.J., Plowright R.K., Wood J.L.N. Bat flight and zoonotic viruses. Emerg. Infect. Dis. 2014;20:741–745. doi: 10.3201/eid2005.130539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brook C.E., Dobson A.P. Bats as ‘special’reservoirs for emerging zoonotic pathogens. Trends Microbiol. 2015;23:172–180. doi: 10.1016/j.tim.2014.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Calisher C.H., Childs J.E., Field H.E., Holmes K.V., Schountz T. Bats: important reservoir hosts of emerging viruses. Clin. Microbiol. Rev. 2006;19:531–545. doi: 10.1128/CMR.00017-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cui J., Han N., Streicker D., Li G., Tang X., Shi Z., Hu Z., Zhao G., Fontanet A., Guan Y., et al. Evolutionary relationships between bat coronaviruses and their hosts. Emerg. Infect. Dis. 2007;13:1526–1532. doi: 10.3201/eid1310.070448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scheben A., Ramos O.M., Kramer M., Goodwin S., Oppenheim S., Becker D.J., Schatz M.C., Simmons N.B., Siepel A., Richard McCombie W. Long-read sequencing reveals rapid evolution of immunity- and cancer-related genes in bats. bioRxiv. 2021 doi: 10.1093/gbe/evad148. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wynne J.W., Wang L.-F. Bats and viruses: friend or foe? PLoS Pathog. 2013;9 doi: 10.1371/journal.ppat.1003651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gouilh M.A., Puechmaille S.J., Gonzalez J.-P., Teeling E., Kittayapong P., Manuguerra J.-C. SARS-Coronavirus ancestor’s foot-prints in South-East Asian bat colonies and the refuge theory. Infect. Genet. Evol. 2011;11:1690–1702. doi: 10.1016/j.meegid.2011.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ruedi M., Stadelmann B., Gager Y., Douzery E.J.P., Francis C.M., Lin L.-K., Guillén-Servent A., Cibois A. Molecular phylogenetic reconstructions identify East Asia as the cradle for the evolution of the cosmopolitan genus Myotis (Mammalia, Chiroptera) Mol. Phylogenet. Evol. 2013;69:437–449. doi: 10.1016/j.ympev.2013.08.011. [DOI] [PubMed] [Google Scholar]
- 13.Platt R.N., 2nd, Faircloth B.C., Sullivan K.A.M., Kieran T.J., Glenn T.C., Vandewege M.W., Lee T.E., Jr., Baker R.J., Stevens R.D., Ray D.A. Conflicting Evolutionary Histories of the Mitochondrial and Nuclear Genomes in New World Myotis Bats. Syst. Biol. 2018;67:236–249. doi: 10.1093/sysbio/syx070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morales A.E., Ruedi M., Field K., Carstens B.C. Diversification rates have no effect on the convergent evolution of foraging strategies in the most speciose genus of bats, Myotis. Evolution. 2019;73:2263–2280. doi: 10.1111/evo.13849. [DOI] [PubMed] [Google Scholar]
- 15.Morales A.E., Jackson N.D., Dewey T.A., O’Meara B.C., Carstens B.C. Speciation with Gene Flow in North American Myotis Bats. Syst. Biol. 2017;66:440–452. doi: 10.1093/sysbio/syw100. [DOI] [PubMed] [Google Scholar]
- 16.Jagoda E., Xue J.R., Reilly S.K., Dannemann M., Racimo F., Huerta-Sanchez E., Sankararaman S., Kelso J., Pagani L., Sabeti P.C., Capellini T.D. Detection of Neanderthal Adaptively Introgressed Genetic Variants That Modulate Reporter Gene Expression in Human Immune Cells. Mol. Biol. Evol. 2022;39 doi: 10.1093/molbev/msab304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Enard D., Petrov D.A. Evidence that RNA Viruses Drove Adaptive Introgression between Neanderthals and Modern Humans. Cell. 2018;175:360–371.e13. doi: 10.1016/j.cell.2018.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chiou K.L., Bergey C.M., Burrell A.S., Disotell T.R., Rogers J., Jolly C.J., Phillips-Conroy J.E. Genome-wide ancestry and introgression in a Zambian baboon hybrid zone. Mol. Ecol. 2021;30:1907–1920. doi: 10.1111/mec.15858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Upadhyay M., Kunz E., Sandoval-Castellanos E., Hauser A., Krebs S., Graf A., Blum H., Dotsev A., Okhlopkov I., Shakhin A., et al. Whole genome sequencing reveals a complex introgression history and the basis of adaptation to subarctic climate in wild sheep. Mol. Ecol. 2021;30:6701–6717. doi: 10.1111/mec.16184. [DOI] [PubMed] [Google Scholar]
- 20.Song Y., Endepols S., Klemann N., Richter D., Matuschka F.-R., Shih C.-H., Nachman M.W., Kohn M.H. Adaptive introgression of anticoagulant rodent poison resistance by hybridization between old world mice. Curr. Biol. 2011;21:1296–1301. doi: 10.1016/j.cub.2011.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nunn C.L., Gittleman J.L., Antonovics J. Promiscuity and the primate immune system. Science. 2000;290:1168–1170. doi: 10.1126/science.290.5494.1168. [DOI] [PubMed] [Google Scholar]
- 22.Rossetto F., Iglesias-Caballero M., Liedtke H.C., Gomez-Mestre I., Berciano J.M., Pérez-Suárez G., de Paz O., Ibáñez C., Echevarría J.E., Casas I., Juste J. Mating strategy is determinant of adenovirus prevalence in European bats. PLoS One. 2020;15 doi: 10.1371/journal.pone.0226203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bogdanowicz W., Piksa K., Tereba A. Hybridization hotspots at bat swarming sites. PLoS One. 2012;7 doi: 10.1371/journal.pone.0053334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bergmann A., Gloza-Rausch F., Wimmer B., Kugelschafter K., Knörnschild M. Similarities in social calls during autumn swarming may facilitate interspecific communication between Myotis bat species. Front. Ecol. Evol. 2022;10 doi: 10.1002/ece3.9439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fenton M.B. Summer activity of Myotis lucifugus (Chiroptera:Vespertilionidae) at hibernacula in Ontario and Quebec. Can. J. Zool. 1969;47:597–602. [Google Scholar]
- 26.Parsons K.N., Jones G., Greenaway F. Swarming activity of temperate zone microchiropteran bats: effects of season, time of night and weather conditions. J. Zool. 2003;261:257–264. [Google Scholar]
- 27.Thomas R.J., Davison S.P. Seasonal swarming behavior of Myotis bats revealed by integrated monitoring, involving passive acoustic monitoring with automated analysis, trapping, and video monitoring. Ecol. Evol. 2022;12 doi: 10.1002/ece3.9344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Glover A.M., Altringham J.D. Cave selection and use by swarming bat species. Biol. Conserv. 2008;141:1493–1504. [Google Scholar]
- 29.Senior P., Butlin R.K., Altringham J.D. Sex and segregation in temperate bats. Proc. Biol. Sci. 2005;272:2467–2473. doi: 10.1098/rspb.2005.3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Piksa K., Bogdanowicz W., Tereba A. Swarming of Bats at Different Elevations in the Carpathian Mountains. Acta Chiropterol. 2011;13:113–122. [Google Scholar]
- 31.van Schaik J., Janssen R., Bosch T., Haarsma A.-J., Dekker J.J.A., Kranstauber B. Bats Swarm Where They Hibernate: Compositional Similarity between Autumn Swarming and Winter Hibernation Assemblages at Five Underground Sites. PLoS One. 2015;10 doi: 10.1371/journal.pone.0130850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kerth G., Kiefer A., Trappmann C., Weishaar M. High gene diversity at swarming sites suggest hot spots for gene flow in the endangered Bechstein’s bat. Conserv. Genet. 2003;4:491–499. [Google Scholar]
- 33.Jebb D., Huang Z., Pippel M., Hughes G.M., Lavrichenko K., Devanna P., Winkler S., Jermiin L.S., Skirmuntt E.C., Katzourakis A., et al. Six reference-quality genomes reveal evolution of bat adaptations. Nature. 2020;583:578–584. doi: 10.1038/s41586-020-2486-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Volleth M., Heller K.G., Pfeiffer R.A., Hameister H. A comparative ZOO-FISH analysis in bats elucidates the phylogenetic relationships between Megachiroptera and five microchiropteran families. Chromosome Res. 2002;10:477–497. doi: 10.1023/a:1020992330679. [DOI] [PubMed] [Google Scholar]
- 35.Dudchenko O., Shamim M.S., Batra S.S., Durand N.C., Musial N.T., Mostofa R., Pham M., Glenn St Hilaire B., Yao W., Stamenova E., et al. The Juicebox Assembly Tools module facilitates de novo assembly of mammalian genomes with chromosome-length scaffolds for under $1000. bioRxiv. 2018 Preprint at. [Google Scholar]
- 36.Durand N.C., Robinson J.T., Shamim M.S., Machol I., Mesirov J.P., Lander E.S., Aiden E.L. Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell Syst. 2016;3:99–101. doi: 10.1016/j.cels.2015.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abi-Rached L., Jobin M.J., Kulkarni S., McWhinnie A., Dalva K., Gragert L., Babrzadeh F., Gharizadeh B., Luo M., Plummer F.A., et al. The shaping of modern human immune systems by multiregional admixture with archaic humans. Science. 2011;334:89–94. doi: 10.1126/science.1209202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen N., Cai Y., Chen Q., Li R., Wang K., Huang Y., Hu S., Huang S., Zhang H., Zheng Z., et al. Whole-genome resequencing reveals world-wide ancestry and adaptive introgression events of domesticated cattle in East Asia. Nat. Commun. 2018;9:2337. doi: 10.1038/s41467-018-04737-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chifman J., Kubatko L. Quartet inference from SNP data under the coalescent model. Bioinformatics. 2014;30:3317–3324. doi: 10.1093/bioinformatics/btu530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Minh B.Q., Schmidt H.A., Chernomor O., Schrempf D., Woodhams M.D., von Haeseler A., Lanfear R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020;37:1530–1534. doi: 10.1093/molbev/msaa015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Edelman N.B., Frandsen P.B., Miyagi M., Clavijo B., Davey J., Dikow R.B., García-Accinelli G., Van Belleghem S.M., Patterson N., Neafsey D.E., et al. Genomic architecture and introgression shape a butterfly radiation. Science. 2019;366:594–599. doi: 10.1126/science.aaw2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fontaine M.C., Pease J.B., Steele A., Waterhouse R.M., Neafsey D.E., Sharakhov I.V., Jiang X., Hall A.B., Catteruccia F., Kakani E., et al. Extensive introgression in a malaria vector species complex revealed by phylogenomics. Science. 2015;347 doi: 10.1126/science.1258524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li G., Figueiró H.V., Eizirik E., Murphy W.J. Recombination-aware phylogenomics reveals the structured genomic landscape of hybridizing cat species. Mol. Biol. Evol. 2019;36:2111–2126. doi: 10.1093/molbev/msz139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nelson T.C., Stathos A.M., Vanderpool D.D., Finseth F.R., Yuan Y.-W., Fishman L. Ancient and recent introgression shape the evolutionary history of pollinator adaptation and speciation in a model monkeyflower radiation (Mimulus section Erythranthe) PLoS Genet. 2021;17 doi: 10.1371/journal.pgen.1009095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hennelly L.M., Habib B., Modi S., Rueness E.K., Gaubert P., Sacks B.N. Ancient divergence of Indian and Tibetan wolves revealed by recombination-aware phylogenomics. Mol. Ecol. 2021;30:6687–6700. doi: 10.1111/mec.16127. [DOI] [PubMed] [Google Scholar]
- 46.Li G., Davis B.W., Eizirik E., Murphy W.J. Phylogenomic evidence for ancient hybridization in the genomes of living cats (Felidae) Genome Res. 2016;26:1–11. doi: 10.1101/gr.186668.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mao X., Tsagkogeorga G., Thong V.D., Rossiter S.J. Resolving evolutionary relationships among six closely related taxa of the horseshoe bats (Rhinolophus) with targeted resequencing data. Mol. Phylogenet. Evol. 2019;139 doi: 10.1016/j.ympev.2019.106551. [DOI] [PubMed] [Google Scholar]
- 48.O’Brien S.J., Graphodatsky A.S., Perelman P.L. Atlas of Mammalian Chromosomes S. Wiley Blackwell; 2020. [Google Scholar]
- 49.Chafin T.K., Douglas M.R., Douglas M.E. Genome-wide local ancestries discriminate homoploid hybrid speciation from secondary introgression in the red wolf (Canidae: Canis rufus) bioRxiv. 2020 Preprint at. [Google Scholar]
- 50.Perry B.W., Schield D.R., Adams R.H., Castoe T.A. Microchromosomes Exhibit Distinct Features of Vertebrate Chromosome Structure and Function with Underappreciated Ramifications for Genome Evolution. Mol. Biol. Evol. 2021;38:904–910. doi: 10.1093/molbev/msaa253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Waters P.D., Patel H.R., Ruiz-Herrera A., Álvarez-González L., Lister N.C., Simakov O., Ezaz T., Kaur P., Frere C., Grützner F., et al. Microchromosomes are building blocks of bird, reptile, and mammal chromosomes. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2112494118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Adrion J.R., Galloway J.G., Kern A.D. Predicting the Landscape of Recombination Using Deep Learning. Mol. Biol. Evol. 2020;37:1790–1808. doi: 10.1093/molbev/msaa038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Crotty S.M., Minh B.Q., Bean N.G., Holland B.R., Tuke J., Jermiin L.S., Haeseler A.V. GHOST: Recovering historical signal from heterotachously evolved sequence alignments. Syst. Biol. 2020;69:249–264. doi: 10.1093/sysbio/syz051. [DOI] [PubMed] [Google Scholar]
- 54.Frankel L.E., Ané C. Summary tests of introgression are highly sensitive to rate variation across lineages. bioRxiv. 2023 doi: 10.1093/sysbio/syad056. Preprint at. [DOI] [PubMed] [Google Scholar]
- 55.Hibbins M.S., Hahn M.W. Phylogenomic approaches to detecting and characterizing introgression. Genetics. 2022;220:iyab173. doi: 10.1093/genetics/iyab173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Korneliussen T.S., Albrechtsen A., Nielsen R. ANGSD: Analysis of Next Generation Sequencing Data. BMC Bioinf. 2014;15:356. doi: 10.1186/s12859-014-0356-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Feng C., Wang J., Liston A., Kang M. Recombination Variation Shapes Phylogeny and Introgression in Wild Diploid Strawberries. Mol. Biol. Evol. 2023;40 doi: 10.1093/molbev/msad049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ottenburghs J., Honka J., Heikkinen M.E., Madsen J., Müskens G.J.D.M., Ellegren H. Highly differentiated loci resolve phylogenetic relationships in the Bean Goose complex. BMC Ecol. Evol. 2023;23:2. doi: 10.1186/s12862-023-02103-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stubbs R.L., Theodoridis S., Mora-Carrera E., Keller B., Yousefi N., Potente G., Léveillé-Bourret É., Celep F., Kochjarová J., Tedoradze G., et al. Whole-genome analyses disentangle reticulate evolution of primroses in a biodiversity hotspot. New Phytol. 2023;237:656–671. doi: 10.1111/nph.18525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Owens G.L., Huang K., Todesco M., Rieseberg L.H. Re-evaluating Homoploid Reticulate Evolution in Helianthus Sunflowers. Mol. Biol. Evol. 2023;40 doi: 10.1093/molbev/msad013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Harris A.J., Foley N.M., Williams T.L., Murphy W.J. Tree House Explorer: A novel genome browser for phylogenomics. Mol. Biol. Evol. 2022;39 doi: 10.1093/molbev/msac130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zheng Y., Janke A. Gene flow analysis method, the D-statistic, is robust in a wide parameter space. BMC Bioinf. 2018;19:10. doi: 10.1186/s12859-017-2002-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Morales A.E., Carstens B.C. Evidence that Myotis lucifugus “Subspecies” are Five Nonsister Species, Despite Gene Flow. Syst. Biol. 2018;67:756–769. doi: 10.1093/sysbio/syy010. [DOI] [PubMed] [Google Scholar]
- 64.Korstian J.M., Paulat N.S., Platt R.N., 2nd, Stevens R.D., Ray D.A. SINE-Based Phylogenomics Reveal Extensive Introgression and Incomplete Lineage Sorting in Myotis. Genes. 2022;13 doi: 10.3390/genes13030399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zachos F.E. Handbook of the Mammals of the World. Bats. Mamm. Biol. 2020;9:335. 100. [Google Scholar]
- 66.Letko M., Seifert S.N., Olival K.J., Plowright R.K., Munster V.J. Bat-borne virus diversity, spillover and emergence. Nat. Rev. Microbiol. 2020;18:461–471. doi: 10.1038/s41579-020-0394-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Silvert M., Quintana-Murci L., Rotival M. Impact and Evolutionary Determinants of Neanderthal Introgression on Transcriptional and Post-Transcriptional Regulation. Am. J. Hum. Genet. 2019;104:1241–1250. doi: 10.1016/j.ajhg.2019.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dannemann M., Prüfer K., Kelso J. Functional implications of Neandertal introgression in modern humans. Genome Biol. 2017;18:61. doi: 10.1186/s13059-017-1181-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhou S., Butler-Laporte G., Nakanishi T., Morrison D.R., Afilalo J., Afilalo M., Laurent L., Pietzner M., Kerrison N., Zhao K., et al. A Neanderthal OAS1 isoform protects individuals of European ancestry against COVID-19 susceptibility and severity. Nat. Med. 2021;27:659–667. doi: 10.1038/s41591-021-01281-1. [DOI] [PubMed] [Google Scholar]
- 70.Hölzer M., Schoen A., Wulle J., Müller M.A., Drosten C., Marz M., Weber F. Virus- and Interferon Alpha-Induced Transcriptomes of Cells from the Microbat Myotis daubentonii. iScience. 2019;19:647–661. doi: 10.1016/j.isci.2019.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hibbins M.S., Hahn M.W. The effects of introgression across thousands of quantitative traits revealed by gene expression in wild tomatoes. PLoS Genet. 2021;17 doi: 10.1371/journal.pgen.1009892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Michalak P., Noor M.A.F. Genome-wide patterns of expression in Drosophila pure species and hybrid males. Mol. Biol. Evol. 2003;20:1070–1076. doi: 10.1093/molbev/msg119. [DOI] [PubMed] [Google Scholar]
- 73.Mack K.L., Nachman M.W. Gene Regulation and Speciation. Trends Genet. 2017;33:68–80. doi: 10.1016/j.tig.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang X., Yi P., Liang Y. The Role of IL-36 in Infectious Diseases: Potential Target for COVID-19? Front. Immunol. 2021;12 doi: 10.3389/fimmu.2021.662266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang P., Gamero A.M., Jensen L.E. IL-36 promotes anti-viral immunity by boosting sensitivity to IFN-α/β in IRF1 dependent and independent manners. Nat. Commun. 2019;10:1–17. doi: 10.1038/s41467-019-12318-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Moura A.E., Shreves K., Pilot M., Andrews K.R., Moore D.M., Kishida T., Möller L., Natoli A., Gaspari S., McGowen M., et al. Phylogenomics of the genus Tursiops and closely related Delphininae reveals extensive reticulation among lineages and provides inference about eco-evolutionary drivers. Mol. Phylogenet. Evol. 2020;146 doi: 10.1016/j.ympev.2020.106756. [DOI] [PubMed] [Google Scholar]
- 77.Herzing D.L., Elliser C.R. Directionality of Sexual Activities During Mixed-Species Encounters between Atlantic Spotted Dolphins (Stenella frontalis) and Bottlenose Dolphins (Tursiops truncatus) Int. J. Comp. Psychol. 2013;26 [Google Scholar]
- 78.Guo W., Sun D., Cao Y., Xiao L., Huang X., Ren W., Xu S., Yang G. Extensive Interspecific Gene Flow Shaped Complex Evolutionary History and Underestimated Species Diversity in Rapidly Radiated Dolphins. J. Mamm. Evol. 2021;29:353–367. [Google Scholar]
- 79.Mozūraitis R., Hajkazemian M., Zawada J.W., Szymczak J., Pålsson K., Sekar V., Biryukova I., Friedländer M.R., Koekemoer L.L., Baird J.K., et al. Male swarming aggregation pheromones increase female attraction and mating success among multiple African malaria vector mosquito species. Nat. Ecol. Evol. 2020;4:1395–1401. doi: 10.1038/s41559-020-1264-9. [DOI] [PubMed] [Google Scholar]
- 80.Grau-Bové X., Lucas E., Pipini D., Rippon E., van ’t Hof A.E., Constant E., Dadzie S., Egyir-Yawson A., Essandoh J., Chabi J., et al. Resistance to pirimiphos-methyl in West African Anopheles is spreading via duplication and introgression of the Ace1 locus. PLoS Genet. 2021;17 doi: 10.1371/journal.pgen.1009253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Grau-Bové X., Tomlinson S., O’Reilly A.O., Harding N.J., Miles A., Kwiatkowski D., Donnelly M.J., Weetman D., Anopheles gambiae 1000 Genomes Consortium Evolution of the Insecticide Target Rdl in African Anopheles Is Driven by Interspecific and Interkaryotypic Introgression. Mol. Biol. Evol. 2020;37:2900–2917. doi: 10.1093/molbev/msaa128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Altae-Tran H., Kannan S., Demircioglu F.E., Oshiro R., Nety S.P., McKay L.J., Dlakić M., Inskeep W.P., Makarova K.S., Macrae R.K., et al. The widespread IS200/IS605 transposon family encodes diverse programmable RNA-guided endonucleases. Science. 2021;374:57–65. doi: 10.1126/science.abj6856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Escudero J.A., Nivina A., Kemble H.E., Loot C., Tenaillon O., Mazel D. Primary and promiscuous functions coexist during evolutionary innovation through whole protein domain acquisitions. Elife. 2020;9 doi: 10.7554/eLife.58061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.O’Brien S.J. Genomic prospecting. Nat. Med. 1995;1:742–744. doi: 10.1038/nm0895-742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rivers N.M., Butlin R.K., Altringham J.D. Genetic population structure of Natterer’s bats explained by mating at swarming sites and philopatry. Mol. Ecol. 2005;14:4299–4312. doi: 10.1111/j.1365-294X.2005.02748.x. [DOI] [PubMed] [Google Scholar]
- 86.Curti J.N., Fraser D., Escalona M., Fairbairn C.W., Sacco S., Sahasrabudhe R., Nguyen O., Seligmann W., Sudmant P.H., Toffelmier E., et al. A genome assembly of the Yuma myotis bat, Myotis yumanensis. J. Hered. 2023 doi: 10.1093/jhered/esad053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Martin S.H., Davey J.W., Jiggins C.D. Evaluating the Use of ABBA–BABA Statistics to Locate Introgressed Loci. Mol. Biol. Evol. 2015;32:244–257. doi: 10.1093/molbev/msu269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pease J.B., Hahn M.W. Detection and Polarization of Introgression in a Five-Taxon Phylogeny. Syst. Biol. 2015;64:651–662. doi: 10.1093/sysbio/syv023. [DOI] [PubMed] [Google Scholar]
- 89.Laine V.N., Sävilammi T., Wahlberg N., Meramo K., Ossa G., Johnson J.S., Blomberg A.S., Yeszhanov A.B., Yung V., Paterson S., Lilley T.M. Whole-genome Analysis Reveals Contrasting Relationships Among Nuclear and Mitochondrial Genomes Between Three Sympatric Bat Species. Genome Biol. Evol. 2023;15 doi: 10.1093/gbe/evac175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Krueger F. Babraham Institute; 2015. Trim Galore!: A Wrapper Around Cutadapt and FastQC to Consistently Apply Adapter and Quality Trimming to FastQ Files, with Extra Functionality for RRBS Data. [Google Scholar]
- 91.Li H., Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv. 2013 Preprint at. [Google Scholar]
- 93.García-Alcalde F., Okonechnikov K., Carbonell J., Cruz L.M., Götz S., Tarazona S., Dopazo J., Meyer T.F., Conesa A. Qualimap: evaluating next-generation sequencing alignment data. Bioinformatics. 2012;28:2678–2679. doi: 10.1093/bioinformatics/bts503. [DOI] [PubMed] [Google Scholar]
- 94.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M., et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Poplin R., Ruano-Rubio V., DePristo M.A., Fennell T.J., Carneiro M.O., Van der Auwera G.A., Kling D.E., Gauthier L.D., Levy-Moonshine A., Roazen D., et al. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv. 2017 Preprint at. [Google Scholar]
- 98.Van der Auwera G.A., Carneiro M.O., Hartl C., Poplin R., Del Angel G., Levy-Moonshine A., Jordan T., Shakir K., Roazen D., Thibault J., et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Current Protocols in Bioinformatics. 2013;11:11.10.1–11.10.33. doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Braun E.L., Kimball R.T. Data types and the phylogeny of Neoaves. Birds. 2021;2:1–22. [Google Scholar]
- 100.Gurevich A., Saveliev V., Vyahhi N., Tesler G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013;29:1072–1075. doi: 10.1093/bioinformatics/btt086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Danecek P., McCarthy S.A. BCFtools/csq: haplotype-aware variant consequences. Bioinformatics. 2017;33:2037–2039. doi: 10.1093/bioinformatics/btx100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Danecek P., Auton A., Abecasis G., Albers C.A., Banks E., DePristo M.A., Handsaker R.E., Lunter G., Marth G.T., Sherry S.T., et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Page A.J., Taylor B., Delaney A.J., Soares J., Seemann T., Keane J.A., Harris S.R. SNP-sites: rapid efficient extraction of SNPs from multi-FASTA alignments. Microb. Genom. 2016;2 doi: 10.1099/mgen.0.000056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Posada D. Current Protocols in Bioinformatics. Wiley Online Library; 2003. Using MODELTEST and PAUP∗ to select a model of nucleotide substitution. Unit 6.5. [DOI] [PubMed] [Google Scholar]
- 105.Capella-Gutiérrez S., Silla-Martínez J.M., Gabaldón T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25:1972–1973. doi: 10.1093/bioinformatics/btp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.R Core Team . 2022. R: A Language and Environment for Statistical Computing. [Google Scholar]
- 107.Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 108.Larsen R.J., Larsen P.A., Genoways H.H., Catzeflis F.M., Geluso K., Kwiecinski G.G., Pedersen S.C., Simal F., Baker R.J. Evolutionary history of Caribbean species of Myotis, with evidence of a third Lesser Antillean endemic. Mamm. Biol. 2012;77:124–134. [Google Scholar]
- 109.Seim I., Fang X., Xiong Z., Lobanov A.V., Huang Z., Ma S., Feng Y., Turanov A.A., Zhu Y., Lenz T.L., et al. Genome analysis reveals insights into physiology and longevity of the Brandt’s bat Myotis brandtii. Nat. Commun. 2013;4:2212. doi: 10.1038/ncomms3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Luo R., Liu B., Xie Y., Li Z., Huang W., Yuan J., He G., Chen Y., Pan Q., Liu Y., et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience. 2012;1:18. doi: 10.1186/2047-217X-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Jebb D., Foley N.M., Puechmaille S.J., Teeling E.C. The complete mitochondrial genome of the Greater Mouse-Eared bat, Myotis myotis (Chiroptera: Vespertilionidae) Mitochondrial DNA A DNA Mapp. Seq. Anal. 2017;28:347–349. doi: 10.3109/19401736.2015.1122775. [DOI] [PubMed] [Google Scholar]
- 112.Miller W., Drautz D.I., Janecka J.E., Lesk A.M., Ratan A., Tomsho L.P., Packard M., Zhang Y., McClellan L.R., Qi J., et al. The mitochondrial genome sequence of the Tasmanian tiger (Thylacinus cynocephalus) Genome Res. 2009;19:213–220. doi: 10.1101/gr.082628.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Minh B.Q., Nguyen M.A.T., von Haeseler A. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 2013;30:1188–1195. doi: 10.1093/molbev/mst024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hoang D.T., Chernomor O., von Haeseler A., Minh B.Q., Vinh L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018;35:518–522. doi: 10.1093/molbev/msx281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wang L.-F., Gamage A.M., Chan W.O.Y., Hiller M., Teeling E.C. Decoding bat immunity: the need for a coordinated research approach. Nat. Rev. Immunol. 2021;21:269–271. doi: 10.1038/s41577-021-00523-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Irving A.T., Ahn M., Goh G., Anderson D.E., Wang L.-F. Lessons from the host defences of bats, a unique viral reservoir. Nature. 2021;589:363–370. doi: 10.1038/s41586-020-03128-0. [DOI] [PubMed] [Google Scholar]
- 117.Afonso E., Goydadin A.-C., Giraudoux P., Farny G. Investigating Hybridization between the Two Sibling Bat Species Myotis myotis and M. blythii from Guano in a Natural Mixed Maternity Colony. PLoS One. 2017;12 doi: 10.1371/journal.pone.0170534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Berthier P., Excoffier L., Ruedi M. Recurrent replacement of mtDNA and cryptic hybridization between two sibling bat species Myotis myotis and Myotis blythii. Proc. Biol. Sci. 2006;273:3101–3109. doi: 10.1098/rspb.2006.3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Peterson A.L., Payseur B.A. Sex-specific variation in the genome-wide recombination rate. Genetics. 2021;217:1–11. doi: 10.1093/genetics/iyaa019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kumar S., Subramanian S. Mutation rates in mammalian genomes. Proc. Natl. Acad. Sci. USA. 2002;99:803–808. doi: 10.1073/pnas.022629899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Posada D., Crandall K.A. The effect of recombination on the accuracy of phylogeny estimation. J. Mol. Evol. 2002;54:396–402. doi: 10.1007/s00239-001-0034-9. [DOI] [PubMed] [Google Scholar]
- 122.Lemey P., Salemi M., Vandamme A.-M. Cambridge University Press; 2009. The Phylogenetic Handbook: A Practical Approach to Phylogenetic Analysis and Hypothesis Testing. [Google Scholar]
- 123.Schierup M.H., Hein J. Recombination and the molecular clock. Mol. Biol. Evol. 2000;17:1578–1579. doi: 10.1093/oxfordjournals.molbev.a026256. [DOI] [PubMed] [Google Scholar]
- 124.Paradis E., Schliep K. ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics. 2019;35:526–528. doi: 10.1093/bioinformatics/bty633. [DOI] [PubMed] [Google Scholar]
- 125.Green R.E., Krause J., Briggs A.W., Maricic T., Stenzel U., Kircher M., Patterson N., Li H., Zhai W., Fritz M.H.-Y., et al. A draft sequence of the Neandertal genome. Science. 2010;328:710–722. doi: 10.1126/science.1188021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zoonomia Consortium A comparative genomics multitool for scientific discovery and conservation. Nature. 2020;587:240–245. doi: 10.1038/s41586-020-2876-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Szklarczyk D., Kirsch R., Koutrouli M., Nastou K., Mehryary F., Hachilif R., Gable A.L., Fang T., Doncheva N.T., Pyysalo S., et al. The STRING database in 2023: protein–protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023;51:D638–D646. doi: 10.1093/nar/gkac1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All code used to generate the analysis is publicly available and are cited in the text. All short-read data generated as part of this study have been submitted to the NCBI Short Read Archive under the following BioProject PRJNA1035163. Recombination maps for M. myotis and M. brandtii, whole-genome SNP datasets used for concatenation and coalescence analyses, a FASTA-formatted version of the updated M. myotis genome assembly, and whole-genome alignments are deposited in the following Zenodo repository: https://doi.org/10.5281/zenodo.7044479. The updated M. myotis genome assembly can be found under BioProject PRJNA628559.