

Abstract
The AML1-CBFβ transcription factor complex is essential for the definitive hematopoiesis of all lineages and is the most frequent target of chromosomal rearrangements in human leukemia. In the t(8;21) translocation associated with acute myeloid leukemia (AML), the AML1(CBFA2/PEBP2αB) gene is juxtaposed to the MTG8(ETO/CDR) gene. We show here that the resultant AML1-MTG8 gene product specifically and strongly interacts with an 85-kDa phosphoprotein. Molecular cloning of cDNA indicated that the AML1-MTG8-binding protein (MTGR1) is highly related to MTG8 and similar to Drosophila Nervy. Comparison of amino acid sequences among MTGR1, MTG8, and Nervy revealed four evolutionarily conserved regions (NHR1 to NHR4). Ectopic expression of AML1-MTG8 in L-G murine myeloid progenitor cells inhibits differentiation to mature neutrophils and induces cell proliferation in response to granulocyte colony-stimulating factor (G-CSF). Analysis with C-terminal deletion mutants of AML1-MTG8 indicated that the region of 51 residues (488 to 538), which contains NHR2, is essential for the induction of G-CSF-dependent cell proliferation. Immunoprecipitation analysis indicates that this region is required for AML1-MTG8 to form a stable complex with MTGR1. Overexpression of MTGR1 stimulates AML1-MTG8 to induce G-CSF-dependent proliferation of L-G cells and to interfere with AML1-dependent transcription. These results suggest that AML1-MTG8 could function as a complex with MTGR1 and that the complex might be important in promoting leukemogenesis.
Chromosome translocations associated with human leukemia frequently involve genes that code for a variety of transcriptional factors implicated in the regulation of normal hematopoiesis (44). The AML1-CBFβ transcription factor complex is the most frequent target of these translocations. The AML1 gene (on chromosome 21) was identified through its involvement in t(8;21) translocation, which occurs in ∼40% of cases of acute myeloid leukemia with the M2 French-American-British subtype (28). In this translocation, the AML1 gene is juxtaposed to the gene which encodes a zinc finger-containing protein MTG8 (also known as ETO and CDR), resulting in the expression of the AML1-MTG8 chimeric protein (4, 19, 29, 32). In addition, the AML1 gene is fused with the TEL gene, which encodes a member of the Ets family of transcription factors, to form a TEL-AML1 chimeric product by t(12;21) translocation. The resultant chimeric transcripts are detected in pediatric B-cell progenitor acute lymphoblastic leukemia, the most common leukemia seen in children (10, 46). Furthermore, AML1-containing fusion products are formed by t(3;21) translocation, which occurs in myelodysplastic syndrome and the blast crisis phase of chronic myelogenous leukemia (27, 35, 36). Moreover, CBFβ, which forms a heterodimer with AML1, is also the target of leukemia-associated chromosomal rearrangement and makes a fusion protein with smooth muscle myosin heavy chain (MYH11) in inv(16), which is often observed in AML-M4Eo (22).
The AML1 family of transcription factors (AML1, AML2, and AML3 [21]) forms heterodimeric complexes with CBFβ (also known as PEBP2β) and regulates the transcription of target genes by binding to the DNA sequence TGT/cGGT (37, 38, 55). A 128-amino-acid region that is highly homologous to the Drosophila segmentation gene Runt (14) is required for heterodimerization with CBFβ/PEBP2β as well as for DNA binding and has been called the runt homology domain (rhd) (2, 13, 24). At least three forms of AML1 protein are produced by alternative splicing (30). The AML1b isoform (453 amino acids) and the AML1c isoform (480 amino acids) contain the rhd and a C-terminal transcriptional activation domain, whereas the AML1a isoform (250 amino acids) contains the rhd but not the transcriptional activation domain. Possible transcriptional targets include the genes encoding the T-cell antigen receptors (TCRs) (43, 45, 55), the colony-stimulating factor 1 (CSF1/M-CSF) receptor (57), myeloperoxidase, neutrophil elastase (34), interleukin-3 (IL-3) (49), granulocyte-macrophage CSF (8, 50), and granzyme B (56). The targeted disruption demonstrated that both AML1 and CBFβ/PEBP2β are essential for all lineages of definitive hematopoiesis in mouse fetal liver (31, 39, 47, 53, 54).
The MTG8 (ETO) gene encodes a protein with two putative zinc fingers and several proline-rich regions, which is presumed to function as a transcription factor (29). Since the AML1-MTG8 chimeric protein contains the rhd of AML1, it retains the abilities to form a complex with CBFβ/PEBP2β and to bind to the specific DNA sequence. However, AML1-MTG8 lacks the transcriptional activation domain which is present in AML1b/AML1c. AML1-MTG8 has been shown to repress AML1-dependent transcription of the TCRβ enhancer (25) and the granulocyte-macrophage CSF promoter (8). However, the molecular mechanism by which AML1-MTG8 exerts its leukemogenic potential is unclear. In this study we found that AML1-MTG8 forms a stable complex with an MTG8-related protein, MTGR1. In addition, AML1-MTG8 induces granulocyte-CSF (G-CSF)-dependent proliferation of murine hematopoietic cells. We show here that the complex formation between AML1-MTG8 and MTGR1 may be responsible for the repression of AML1-dependent transcription, for the induction of G-CSF-dependent cell growth, and possibly for leukemogenesis.
MATERIALS AND METHODS
Cells and retroviruses.
L-G cells (16) were cultured in RPMI 1640 medium supplemented with 10% fetal calf serum, recombinant mouse IL-3 (0.1 ng/ml) (a generous gift from Kirin Brewery), and 50 μM β-mercaptoethanol. BOSC23 (42) and F19 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum. LNSX and LXSH retrovirus vectors (26) were generously provided by D. Miller. For production of retroviruses, BOSC23 cells were transfected with LNSX- or LXSH-derived retrovirus vectors by calcium phosphate precipitation methods and culture supernatants were saved 48 h after transfection. L-G cells were incubated in the culture supernatant of BOSC23 transfectants for 24 h and then selected with G418 (1 mg/ml) or hygromycin B (2.5 mg/ml). When L-G cells were exposed to G-CSF, the cells maintained in the presence of IL-3 were washed twice with IL-3-free medium and incubated in medium containing human recombinant G-CSF (2 ng/ml) (a generous gift from Chugai Pharmaceutical Co.). Viable cells were counted with a Coulter Counter (Coulter Electronics Ltd.). For cell morphology, cells were stained with May Gruenwald’s solution and Giemsa’s solution (Merck).
cDNA library screening and 5′ rapid amplification of cDNA ends.
A search of the GenBank expressed sequence tag (EST) sequences with the amino acid sequence of human MTG8b showed that several ESTs appeared to encode proteins similar to MTG8. Two ESTs (R85823 and H83692) and three other ESTs (F07076, Z42798 and T04880) could be aligned with the N-terminal and C-terminal regions of MTG8, respectively. A PCR product corresponding to the first two ESTs (about 280 bp) was made with Pr1 and Pr2 as primers (Pr1, GTGAAGATACAGTCCAGATCCTCA; Pr2, CCCAAACTGTTGCCAGAGTGGTAAG), and another PCR product corresponding to the last three ESTs (about 390 bp) was made with Pr3 and Pr4 as primers (Pr3, GGAGGCTATCAAGATGAGTTGGTAGA; Pr4, TCTGTACTTCTGACATGGCCTGAAT). The third PCR product was made with a set of primers (Pr5 and Pr6) from R85823 (Pr5, TTCTTACCACTCTGGCAACAGTTTG) and F07076 (Pr6, TCTACCAACTCATCTTGATAGCCTCC), respectively. Human brain quick-clone cDNA (Clontech) was used as the PCR template. The amino acid sequence deduced from the DNA sequence of this third PCR product could be aligned with that of the middle region of MTG8. A mixture of these three PCR products was used as a probe for the screening of the size-fractionated cDNA library made in pBluescript from poly(A) RNA of human KG-1 cells (33). Since the transcripts of this MTG8-like gene were about 7.5 kb, we screened filters on which pools of plasmids harboring inserts longer than 4.8 kb were blotted. The 5′ end of the MTGR1 cDNA was determined with Marathon Race Ready cDNA from human K562 cells (Clontech) as a template. The first PCR was carried out with Pr2 (described above) and Adaptor Primer 1, included in the kit, as primers. The second PCR was carried out with Pr7 (CAGGATTTATTGGTGGGAGGGGTG) and Adaptor Primer 2, included in the kit, as primers and the diluted (1/100) reaction mixture of the first PCR as a template. PCR products were cloned into pGEM-T vector (Promega), and the sequences of the inserts were determined.
DNA sequencing and protein structural analysis.
Sequencing reactions were carried out with the Prism dye-terminator cycle-sequencing kit (ABI), and the products were applied to an ABI 373S DNA sequencer. Primers made from vector sequences and synthetic oligonucleotides were used as sequencing primers. DNA sequences were analyzed and joined with MacVector (Kodak) and Assembly LIGN (Kodak). The structures of the proteins were analyzed with protein analysis programs in Mac Vector and DNASIS (Hitachi).
Plasmid construction.
The amino-terminal HA tag was fused to the AML1-MTG8 cDNA with the oligonucleotide 5′-CCTAGGCCTCTAGACCATGGCATACCCATACGACGTGCCTGACTACGCCTCCCGTATCCCCGTAGATGCC-3′ as the upstream primer and the oligonucleotide 5′-AGACAGTGATGGTCAGAGTG-3′ as the downstream primer in a PCR. The PCR product was digested with StuI and HindIII and ligated to the HindIII site near the amino terminus of AML1-MTG8. The C-terminal deletion mutant Δ582 was generated by digestion with StuI and ligation of the ClaI linker. Δ538, Δ487, and Δ398 were generated by ligation of the MfeI-ClaI-digested PCR fragments with the following primers to the MfeI site of the C terminus of AML1-MTG8: 5′-ACCCTCGTTCTGGGACTAGTGAACTCCACT-3′ as the common upstream primer, and 5′-GCCGGATCGATTAATTCAATTCTTCCCGGT-3′ for Δ538, 5′-TGATCAATCGATTCTTCTTGACGTGTGCCA-3′ for Δ487, or 5′-GTCAAATCGATTTTCTTTCCTTCTGTCTGG-3′ for Δ398 as the downstream primer. The FLAG-tagged MTGR1 was generated by ligation of the NotI-EcoRI-digested fragment of the PCR products to the EcoRI site of the MTGR1 cDNA sequence with 5′-GGATAAGCATGCGGCCGCACCATGGCAGACTACAAGGACGACGATGACAAGTCCGCTAAAGAATCTGGAATAAGCTTGAAA-3′ as the upstream primer and 5′-AGTGGAATTCCTCAATTGTCACTGTT-3′ as the downstream primer. The sequences of the above constructs were checked by sequencing.
Immunoprecipitations.
Immunoprecipitations were performed as described previously (18). Cells were metabolically labeled for 4 h with [35S]methionine (50 μCi/ml; Amersham) in methionine-free DMEM or 32Pi (300 μCi/ml; Amersham) in phosphorus-free DMEM. The cells were lysed by incubation at 4°C for 30 min in lysis buffer (20 mM sodium phosphate [pH 7.0], 250 mM NaCl, 30 mM sodium pyrophosphate, 0.1% Nonidet P-40, 5 mM EDTA, 10 mM NaF, 0.1 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride) supplemented with 1 μg of leupeptin per ml, 1 μg of pepstatin per ml, and 1 μg of aprotinin per ml. The lysates were cleared by centrifugation at 30,000 × g for 30 min at 4°C, and the supernatants were saved and stored at −80°C. The cell lysates were incubated with anti-HA monoclonal antibody 12CA5 (Boehringer Mannheim), anti-FLAG monoclonal antibody M2 (Eastman Kodak), anti-AML1 polyclonal antibody, anti-MTGR1 polyclonal antibodies, or anti-MTG8 polyclonal antibody on ice for 1 h, followed by addition of protein G-Sepharose beads (Pharmacia) and shaking on a rotary shaker at 4°C for 2 h. The beads were washed with 1 ml of lysis buffer five times. The proteins were separated on sodium dodecyl sulfate (SDS)-polyacrylamide gels and visualized with an imaging analyzer (BAS2000; Fuji). The anti-AML1 antibody, anti-MTG8 antibody, and anti-MTGR1 antibodies were generated by immunizing rabbits with a peptide corresponding to residues 8 to 24 of human AML1a, glutathione S-transferase (GST)-MTG8 (residues 78 to 187 of human MTG8b), and GST-MTGR1 (residues 295 to 335 and 538 to 604 of human MTGR1), respectively, and affinity purified on antigen-conjugated columns.
Western blotting.
Cell lysates or immunoprecipitates were fractionated on SDS-polyacrylamide gels and transferred to membrane filters (Hybond-C ECL; Amersham) by electroblotting. The filters were blocked in 5% low-fat milk and dissolved in phosphate-buffered saline plus 0.1% Tween-20 (PBST) at room temperature for 2 h or at 4°C overnight. After extensive washing in PBST, the filters were incubated for 1 h at room temperature with anti-MTG8 antibody or anti-MTGR1 polyclonal antibodies. After further washes in PBST, the filters were incubated with horseradish peroxidase-conjugated second antibodies and then washed extensively in PBST. The immune complexes were visualized by an enhanced chemiluminescence detection system (Amersham).
CAT assay.
The cells were harvested by centrifugation at 4°C at 3,000 × g for 5 min. The cell pellet was washed in PBS and resuspended in 0.25 M Tris-Cl (pH 7.5). The cells were disintegrated by four freeze-thaw cycles. After centrifugation at 15,000 × g for 10 min at 4°C, aliquots of the clear supernatant were used for the chloramphenicol acetyltransferase (CAT) assay. Cell lysates (30 μl) were incubated in 0.11 M Tris-Cl (pH 7.8)–2.3 mM chloramphenicol–129 μM (1 μCi/ml) [14C]acetyl coenzyme A for 1 h at 37°C and then for 10 min at 65°C. Acetylated chloramphenicol was extracted with ice-cold ethyl acetate, and its radioactivity was measured by liquid scintillation counting.
Northern blotting.
Northern blotting was performed as described previously (17). Poly(A)+ RNA was denatured and fractionated on a 1.0% agarose gel containing 2.2 M formaldehyde, 20 mM 3-(N-morpholino)propanesulfonic acid (MOPS) (pH 7.0), 8 mM sodium acetate, and 1 mM EDTA. The RNAs were transferred to nylon membranes (Amersham) in 20× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate). Hybridization was performed at 42°C for 20 h in 50% formamide–5× SSC–0.1% SDS–5× Denhardt’s solution–50 mM EDTA–100 μg of denatured salmon sperm DNA per ml. The filters were washed several times at 65°C in 0.2× SSC–0.1% SDS, and the hybridized transcripts were visualized with an imaging analyzer (BAS2000; Fuji).
Nucleotide sequence accession number.
The sequence data reported here have been submitted to the DDBJ/EMBL/GenBank databases under accession no. AF013970.
RESULTS
AML1-MTG8 specifically interacts with a cellular phosphoprotein.
The murine L-G cell line is an IL-3-dependent myeloid precursor cell line and can be induced to differentiate into mature neutrophils in response to G-CSF (16). To clarify the effect of expression of the AML1 protein and the leukemia-associated AML1-MTG8 protein on the differentiation of myeloid cells, L-G cells were infected with recombinant retroviruses which encoded either HA-tagged AML1-MTG8, AML1b, or AML1a proteins (Fig. 1A). Neomycin phosphotransferase was used as a selectable marker, and cells were selected with G418. A total of 10 to 30% of cells acquired resistance to G418, and these were further characterized. These infected cells were labeled with [35S]methionine or with [32P]orthophosphate. Then immunoprecipitation was performed with anti-HA monoclonal antibody 12CA5. The AML1-MTG8, AML1b, and AML1a proteins were efficiently expressed in each infected-cell line, and they coprecipitated CBFβ/PEBP2β proteins (Fig. 1B). When the infected cells were labeled with [32P]orthophosphate, an 85-kDa protein (p85) and a 35-kDa protein were precipitated with AML1-MTG8 and AML1b, respectively (Fig. 1C). These proteins were not precipitated with AML1a, suggesting that specific interactions of AML1-MTG8 and AML1b with the cellular proteins took place.
FIG. 1.

Expression of AML1-MTG8 induces G-CSF-dependent proliferation of L-G cells and inhibits their differentiation to mature neutrophils. (A) Structures of AML1-MTG8, AML1b, and AML1a. AML1-MTG8 retains the runt homology domain (Runt) of AML1 and almost all of the region of MTG8. AML1-MTG8 and AML1a lack the C-terminal transactivation domain (PST) which is present in AML1b. (B and C) Immunoprecipitation of AML1-MTG8, AML1b, and AML1a. L-G cells which expressed HA-AML1-MTG8, HA-AML1b, and HA-AML1a were labeled with [35S]methionine (B) or [32P]orthophosphate (C), and immunoprecipitations were performed with the anti-HA monoclonal antibody 12CA5. The immunoprecipitates were subjected to electrophoresis on SDS–10% polyacrylamide gels. The proteins were visualized with a BAS2000 phosphorimager (Fuji). The positions of bands of AML1a, AML1b, AML1-MTG8, CBFβ, p85, and p35 are indicated on the right. (D and E) Growth curve of the infected L-G cells in response to IL-3 (D) or G-CSF (E). The growing cells which express AML1a, AML1b or AML1-MTG8 were washed twice and cultured in the presence of 2 ng of G-CSF per ml (D) or 0.1 ng of IL-3 per ml (E). The relative numbers of viable cells are indicated. (F and G) Morphology of the cells. The vector-control (F) or AML1-MTG8-expressing cells (G) were exposed to G-CSF for 7 days and stained with May-Gruenwald’s and Giemsa’s solutions.
AML1-MTG8 inhibits G-CSF-dependent differentiation of L-G cells.
The cells which expressed AML1a and AML1b proliferated, as did control cells, in the presence of IL-3. However, AML1-MTG8-expressing cells showed slight growth retardation (Fig. 1D). In the presence of G-CSF, the control and AML1b-expressing cells did not proliferate (Fig. 1E) but underwent terminal differentiation to mature neutrophils (Fig. 1F). In contrast, cells which expressed AML1-MTG8 proliferated exponentially in response to G-CSF (Fig. 1E) without differentiating to mature granulocytes, as measured by changes in morphology (Fig. 1G). Expression of AML1a slightly stimulated cell proliferation in response to G-CSF. All of the infectants died within 2 days when deprived of both IL-3 and G-CSF (data not shown). Thus, expression of AML1-MTG8 inhibited differentiation of L-G cells to mature granulocytes and stimulated cell proliferation in response to G-CSF.
AML1-MTG8 sequences that are responsible for stimulation of G-CSF-dependent proliferation of L-G cells and for interaction with p85.
To identify the domain of MTG8 which is required for the induction of G-CSF-dependent proliferation of L-G cells, a series of C-terminal deletion mutants of AML1-MTG8 (Fig. 2A) were constructed and introduced into L-G cells by infection with retroviruses as described above. Immunoblotting analysis with anti-MTG8 polyclonal antibody indicated that each infected-cell line expressed the expected size of mutants of AML1-MTG8, as well as a protein which may be intrinsic MTG8 or its related proteins (Fig. 2B). Each infected-cell line was then tested for its ability to proliferate in the presence of G-CSF. The C-terminal deletion mutants Δ582 and Δ538, as well as wild-type AML1-MTG8, strongly stimulated G-CSF-dependent cell proliferation (Fig. 2C). However, further deletion to residue 487 eliminated this activity. These results indicate that the region of residues 488 to 538 (corresponding to residues 340 to 390 of MTG8b) is essential for the stimulation of G-CSF-dependent proliferation of L-G cells. This region contains the Nervy homology region 2 (NHR2) (see Fig. 6).
FIG. 2.

Identification of the region of AML1-MTG8 required for induction of G-CSF-dependent cell proliferation. (A) Schematic representation of the structure of AML1-MTG8 deletion mutants. The runt homology domain (Runt), the proline-rich regions (P), and the Nervy homology regions (numbered 1 through 4) are indicated. The numbers above the top bar indicate the positions of amino acid residues. (B) Immunoblot analysis of lysate of infected L-G cells which express either wild-type (WT) or mutant AML1-MTG8 with anti-MTG8 polyclonal antibody. The asterisk indicates the position of intrinsic MTG8 or MTG8-like proteins. (C) Growth curve of the L-G cells in response to G-CSF. The growing cells which express wild-type (WT) or mutant AML1-MTG8 were washed twice and cultured in the presence of 2 ng of G-CSF per ml. (D) Repression of AML1-dependent transcriptional activation. P19 cells were cotransfected with 1.0 μg of TCRβ-TK-CAT, 1.0 μg of either pLNSX vector (−) or pLNSX-AML1b (+), 1.0 μg of either wild-type (WT) or mutant pLNSX-AML1-MTG8, and 0.5 μg of TK-luciferase in a 6-cm-diameter plate. The results represent the mean of relative CAT activity from three experiments which were normalized with luciferase expressed from thymidine kinase-luciferase as an internal control.
FIG. 6.


Structure of MTGR1. (A) Comparison of the amino acid sequences of MTGR1, MTG8, and Drosophila Nervy proteins. Residues identical among two or more proteins are shaded. The conserved regions (NHR1 to NHR4) are indicated above the sequence. Cysteine residues of zinc finger motifs are marked underneath the sequence. Epitopes for anti-MTG8 and anti-MTGR1 antibodies are underlined. (B) Schematic representation of the structures of human MTGR1, human MTG8b, and Drosophila Nervy. The conserved regions (NHR1 to NHR4) among these proteins are indicated by shaded boxes. The percentages represent identity to MTGR1. Numbers above bars indicate the positions of amino acid residues. (C) Alignments of TAF homology regions of MTGR1, MTG8, Drosophila Nervy, and human and Drosophila TAFs. Residues identical among four or more proteins are shaded. (D) A helical wheel of the NHR2 of MTG8. The numbers indicate the positions of amino acid residues, where the first residue of the NHR2 is position 1. The wheel of positions 3 to 22 is shown. Hydrophobic and hydrophilic amino acids are indicated by gray and black, respectively. The hydrophobic and hydrophilic sides of the helix are indicated.
AML1-MTG8 can interfere with AML1-mediated activation of a reporter gene containing the consensus AML1-binding site (25). To identify the region that is required for repression of transcription, deletion mutants of AML1-MTG8 were transfected together with AML1b and tested for the activity of AML1-dependent transcription of the TCRβ enhancer. Δ582 and Δ538, as well as wild-type AML1-MTG8, inhibited transactivation by AML1b, but Δ487 and Δ398 did not (Fig. 2D). These results are consistent with those in a previous report (20), which demonstrated that residues 470 to 540 are required for transrepression by AML1-MTG8.
To determine the domain of AML1-MTG8 which is responsible for interaction with p85, the cells which expressed deletion mutants of the HA-tagged AML1-MTG8 were labeled with [32P]orthophosphate and the proteins were immunoprecipitated with anti-HA antibody. As shown in Fig. 3A, serial deletion of the C terminus to residue 538 did not affect the coprecipitation of p85. However, the coprecipitation was abolished by further deletion to residue 487. Thus, the region of residues 488 to 538 is required for interaction with p85. This region is also responsible for AML1-MTG8-mediated stimulation of G-CSF-dependent proliferation of L-G cells and repression of AML1-dependent transcription.
FIG. 3.

p85, which interacts with AML1-MTG8, is recognized by anti-MTG8 antibody. (A) Immunoprecipitation of AML1-MTG8. Infected L-G cells which express either wild-type (WT) or mutant HA-AML1-MTG8 were labeled with [32P]orthophosphate, and immunoprecipitations were performed with the anti-HA monoclonal antibody 12CA5. The immunoprecipitates were subjected to electrophoresis on SDS–10% polyacrylamide gels. The proteins were visualized with a BAS2000 phosphorimager. The position of the p85 band is indicated on the right. (B) Immunoblot of AML1-MTG8 complexes with anti-MTG8 antibody. The immunoprecipitates with the HA antibody, using lysates of cells which express wild-type (WT) or mutant HA-AML1-MTG8, were separated on SDS-polyacrylamide gels and analyzed by immunoblotting with anti-MTG8 polyclonal antibody. The position of the p85 band is indicated on the right.
p85 is recognized by anti-MTG8 antibody.
To analyze the AML1-MTG8 protein complex, immunoprecipitation was performed with anti-HA antibody by using cell lysates from the L-G cells which expressed the HA-tagged AML1-MTG8 protein. The immunoprecipitates were then analyzed by immunoblotting with anti-MTG8 polyclonal antibody. Surprisingly, the band which corresponds to p85, as well as that corresponding to AML1-MTG8, was detected (Fig. 3B). This band was also observed in immunoprecipitates with C-terminal deletions to residue 538 but not in those with further deletions. These results indicate that p85 is recognized by the anti-MTG8 polyclonal antibody.
Cross-reactivity of anti-MTG8 antibody suggests that p85, which coprecipitated with AML1-MTG8, may be an intrinsic MTG8 protein. However, no MTG8 transcript was detected in L-G cells (Fig. 4A), suggesting that p85 is unlikely to be the intrinsic MTG8. Moreover, p85 is not a degradation product of AML1-MTG8 because (i) p85 is coprecipitated with deletion mutants (Δ582 and Δ538) which migrated faster than p85 (Fig. 3) and (ii) the protein which corresponds to p85 is expressed in L-G cells without expression of AML1-MTG8 (Fig. 2B). From these observations, we considered that p85 might be an MTG8-like protein rather than the MTG8 protein itself and that there might be another member of the MTG8 family which could interact with AML1-MTG8 and could be recognized by the anti-MTG8 antibody.
FIG. 4.

Analysis of MTG8 and MTGR1 transcripts in L-G and Kasumi-1 cells. Poly(A)+ RNAs were prepared from L-G cells, L-G cells infected with LNSX-HA-AML1-MTG8, and Kasumi-1 cells which have the t(8;21) translocation and express AML1-MTG8 chimeric transcripts. A 2-μg sample of poly(A)+ RNAs was analyzed by Northern blotting with human MTG8 (A) or human MTGR1 (B) cDNAs as probes. Note that the human MTGR1 transcript (7.5 kb) in Kasumi-1 cells is longer than the mouse MTGR1 transcript (6.7 kb) in L-G cells.
Cloning of a cDNA encoding a new member of the MTG8 family, MTGR1.
To isolate cDNAs encoding MTG8-related proteins as candidates for AML1-MTG8-binding proteins, we initially compared the human MTG8 protein sequence with sequences from various databases. This search revealed significant similarities to randomly sequenced human cDNAs (ESTs). Using these sequences as probes, a size-fractionated cDNA library of human KG-1 cells was screened (see Materials and Methods). From many positive clones, we selected nine clones whose inserts were longer than 6 kb. By combining the sequences of these clones and the sequence obtained by 5′ rapid amplification of cDNA ends, we determined the 6,406-bp cDNA sequence (Fig. 5). Because of the similarity to MTG8, we named it MTGR1 (myeloid translocation gene-related protein 1). The coding sequence started at the first in-frame ATG at nucleotide 11, which was preceded by the termination codon TGA, and terminated at nucleotide 1822. The poly(A) addition signal started from nucleotide 6387 of the cDNA. Alu consensus sequences were found from nucleotides 3207 to 3236 and from nucleotides 3690 to 3960 of cDNA. RNA blotting indicates that transcripts detected by MTGR1 cDNA are ubiquitously expressed in various human tissues and in all the cell lines tested, including L-G cells and Kasumi-1 cells, which have the t(8;21) translocation (Fig. 4B) (30a).
FIG. 5.

Nucleotide sequence of the MTGR1 cDNA and the deduced amino acid sequence of the MTGR1 protein. The 3′-noncoding region (nucleotide 1921 to the 3′ end) is not shown.
The predicted protein product of the cDNA, which consists of 604 amino acids, is highly related to MTG8 (61% identity) and is similar to Nervy (25% identity), a putative Drosophila homolog of MTG8 (7) (Fig. 6A). Comparison of amino acid sequences among MTGR1, MTG8, and Nervy revealed that there are four evolutionarily conserved regions (Fig. 6B). We termed them NHR1 (Nervy homology region) (amino acids 113 to 209 of MTGR1), NHR2 (amino acids 346 to 372), NHR3 (amino acids 436 to 484), and NHR4 (amino acids 507 to 544). While sequence similarity between MTGR1 and MTG8 extended over the entire sequences, similarity between MTGR1/MTG8 and Nervy was restricted to these regions. NHR1 corresponded to the region which shows homology with TAFs (TATA-binding protein-associated factors) (Fig. 6C) (5, 7). NHR2 is found in the domain of MTG8 which is required for interaction with p85. Analysis of the secondary structure suggests that NHR2 is a helical domain with amphipathic characteristics (Fig. 6D). NHR4 corresponded to two zinc finger motifs (-C-x-x-C-7x-C-x-x-C-; -C-x-x-x-C-7x-H-x-x-x-C-). Three Pro-rich regions (amino acids 39 to 116, 257 to 297, and 564 to 603 of MTG8b) found in MTG8 (29) were also present in MTGR1 at the corresponding regions.
AML1-MTG8 specifically interacts with MTGR1.
To analyze the MTGR1 protein, L-G cells were infected with a retrovirus encoding the Flag-tagged human MTGR1 protein (Flag-hMTGR1). Immunoprecipitation analysis with anti-Flag monoclonal antibody M2 indicated that Flag-hMTGR1 is a phosphoprotein and comigrates with p85 (Fig. 7A). MTGR1-specific antibodies were prepared with the specific regions 295 to 335 and 539 to 604 of MTGR1, which were expressed in Escherichia coli as glutathione S-transferase fusion proteins. To determine if MTGR1 forms a complex with AML1-MTG8, infected L-G cells expressing the HA-tagged AML1-MTG8 protein (HA-AML1-MTG8) as described above were further infected with a retrovirus encoding the Flag-hMTGR1 and the hygromycin phosphotransferase selectable marker and then selected with hygromycin B. Cell lysates were prepared, and HA-AML1-MTG8 was immunoprecipitated with anti-HA antibody. Immunoblotting with the anti-MTGR1 antibodies indicated that intrinsic mouse MTGR1 (Fig. 7B, lane 2) and recombinant Flag-hMTGR1 (lane 4) were coprecipitated with the HA-AML1-MTG8. The electrophoretic mobility of Flag-hMTGR1 was very close to but slightly slower than that of the intrinsic mouse MTGR1 protein. This is probably due to additional sequences containing the Flag epitope (10 amino acids) and/or to the difference between mouse and human gene-derived proteins. Immunoprecipitation of the Flag-MTGR1 proteins followed by immunoblotting with anti-MTG8 indicated that MTGR1 coprecipitated with AML1-MTG8 (Fig. 7B, lane 12). This coprecipitation was not detected in lysates from cells which expressed either Flag-hMTGR1 (lane 11) or HA-AML1-MTG8 (lane 10), indicating that there was a specific interaction between MTGR1 and AML1-MTG8. The recombinant Flag-hMTGR1 was recognized by the anti-MTG8 antibody (lanes 11 and 12). Taken together, these results indicate that AML1-MTG8 could form a stable complex with MTGR1.
FIG. 7.

Characterization of MTGR1. (A) MTGR1 is an 85-kDa phosphoprotein. L-G cells were infected with LNSX vector, LNSX-HA-AML1-MTG8, or LNSX-Flag-MTGR1, selected with G418. The infected cells were labeled with [32P]orthophosphate, and immunoprecipitations were performed with the anti-HA monoclonal antibody or the anti-Flag monoclonal antibody. The immunoprecipitates were subjected to electrophoresis on SDS–10% polyacrylamide gels. The proteins were visualized with a BAS2000 phosphorimager. The positions of bands of AML1-MTG8 and MTGR1 are indicated on the right. (B) Interaction between AML1-MTG8 and MTGR1. Cell lysates were prepared from infected L-G cells which express the HA-tagged AML1-MTG8 and/or the Flag-tagged MTGR1. Immunoprecipitations were performed with anti-HA monoclonal antibody (lanes 1 to 8) or anti-Flag monoclonal antibody (lanes 9 to 12). The immunoprecipitates were separated on SDS–10% polyacrylamide gels and analyzed by immunoblotting with anti-MTGR1 polyclonal antibody (lanes 1 to 4) or anti-MTG8 polyclonal antibody (lanes 5 to 12).
To determine the region of AML1-MTG8 responsible for interaction with MTGR1, the deletion mutants above were expressed together with MTGR1. Immunoprecipitation-immunoblotting analyses indicated that when Flag-hMTGR1 was precipitated with anti-Flag antibody, the Δ582 and Δ538 mutants but not the Δ487 or Δ398 mutants were coprecipitated with MTGR1 (Fig. 8B). When each deletion mutant of HA-AML1-MTG8 was precipitated with anti-HA antibody, MTGR1 was coprecipitated with Δ582 and Δ538 but was not precipitated with Δ487 or Δ398 (Fig. 8D). These results indicate that the region of residues 488 to 538 of AML1-MTG8 is required for interaction with MTGR1, as in the case of interaction with p85 (Fig. 2). From these and the above results, we conclude that MTGR1 is the p85 protein.
FIG. 8.

Determination of the region of AML1-MTG8 which is required for interaction with MTGR1. L-G cells were infected with LXSH-Flag-MTGR1 and LNSX-retrovirus, which encodes either wild-type or mutant HA-tagged AML1-MTG8. Control cells (vector) expressed only the Flag-tagged MTGR1. Immunoprecipitations were performed with anti-HA monoclonal antibody (C and D) or anti-Flag monoclonal antibody (B). The immunoprecipitates and total-cell lysates (A) were separated on SDS–10% polyacrylamide gels and analyzed by immunoblotting with anti-MTGR1 polyclonal antibody (D) or anti-MTG8 polyclonal antibody (A to C).
AML1-MTG8 forms complexes with MTGR1 proteins in t(8;21)-containing leukemic cells.
We examined the interaction between AML1-MTG8 and MTGR1 in Kasumi-1, which is an AML cell line with the t(8;21) translocation. Immunoblot analysis with anti-MTG8 antibody indicated that in addition to AML1-MTG8, three protein bands were detected in lysates of Kasumi-1 cells (Fig. 9, lane 2). Since these proteins were also recognized by both of the two MTGR1-specific antibodies (lanes 4, 5, and 8), we consider that they are MTGR1 proteins. We do not know the exact reason for the difference in their electrophoretic mobilities, but they may be isoforms generated by alternative splicing and/or differential modifications such as phosphorylation (Fig. 7A).
FIG. 9.

Interaction of AML1-MTG8 and MTGR1 in Kasumi-1 cells. Cell lysates were prepared from L-G cells which were infected with LNSX-HA-AML1-MTG8 (lane 1) and Kasumi-1 cells (lanes 2 to 9). Immunoprecipitations were performed with anti-HA monoclonal antibody (lane 1), anti-AML1 antibody (lanes 3 and 8), anti-MTGR1 middle region antibody (α-MTGR1M; lane 4), anti-MTGR1 C-terminal region antibody (α-MTGR1C; lane 5), anti-MTG8 antibody (lane 6), and rabbit immunoglobulin G (IgG) (lanes 7 and 9). The immunoprecipitates were separated on SDS–7.5% polyacrylamide gels and analyzed by immunoblotting with anti-MTG8 antibody (lanes 1 to 7) or anti-MTGR1M antibody (lanes 8 and 9).
Immunoprecipitation analysis with anti-AML1 antibody indicated that all of the three MTGR1 proteins in Kasumi-1 cells were coprecipitated with AML1-MTG8 (Fig. 9, lane 3). In addition, AML1-MTG8 was coprecipitated with MTGR1 when either of the two MTGR1-specific antibodies was used (lanes 4 and 5). These results indicated that AML1-MTG8 forms complexes with MTGR1 proteins in Kasumi-1 cells with the t(8;21) translocation.
AML1-MTG8 forms heterocomplexes with MTGR1 in preference to homocomplexes.
Complex formation of AML1-MTG8 and MTGR1 suggests that AML1-MTG8 may form a homocomplex through the regions of MTG8 because MTGR1 is very similar to MTG8. To test this possibility, HA-tagged wild-type AML1-MTG8 and Δ582 mutant without the HA-tag were expressed together in L-G cells. As shown in Fig. 10, immunoblotting analysis with anti-MTG8 antibody indicated that both wild-type and mutant proteins were efficiently expressed as well as endogenous MTGR1 (p85). Immunoprecipitation with anti-HA antibody followed by immunoblotting with anti-MTG8 antibody revealed that coprecipitation of endogenous MTGR1 was detected, but no band or only a very faint band corresponding to Δ582 was observed. These results suggest that AML1-MTG8 forms a heterocomplex with MTGR1 in preference to a homocomplex in the presence of MTGR1.
FIG. 10.

No detectable homocomplex of AML1-MTG8 in L-G cells. Cell lysates were prepared from L-G cells which were infected with LNSX-HA-AML1-MTG8 and/or LXSH-D582 without any tag. Immunoprecipitations were performed with anti-HA monoclonal antibody. The immunoprecipitates were separated on SDS–10% polyacrylamide gels and analyzed by immunoblotting with anti-MTG8 polyclonal antibody.
Overexpression of MTGR1 stimulates G-CSF-dependent cell proliferation and represses AML1-dependent transactivation in concert with AML1-MTG8.
To test the effects of overexpression of MTGR1 on the function of AML1-MTG8, L-G cells, which ectopically expressed AML1-MTG8 and/or MTGR1, were cultured in the presence of G-CSF or IL-3. In the presence of G-CSF, the MTGR1-expressing cells did not proliferate as well as the control L-G cells (Fig. 11A). On the other hand, cells which expressed AML1-MTG8 proliferated exponentially in response to G-CSF, as shown in Fig. 1. Overexpression of MTGR1 together with AML1-MTG8 further stimulated cell proliferation. Overexpression of MTGR1 did not affect the growth response to IL-3 (Fig. 11B).
FIG. 11.

MTGR1 enhances the activities of AML1-MTG8. (A and B) Growth curve of the infected L-G cells in response to G-CSF (A) or IL-3 (B). The growing cells, which were infected with LNSX-AML1-MTG8 and/or LXSH-MTGR1, were washed twice and cultured in the presence of 2 ng of G-CSF per ml (A) or 0.1 ng of IL-3 per ml (B). Relative numbers of viable cells are indicated. (C) Repression of AML1-dependent transcriptional activation. P19 cells were cotransfected with 1.0 μg of TCRβ-TK-CAT, 1.0 μg of either pLNSX vector or pLNSX-AML1b, the indicated amounts (in micrograms) of pLNSX-AML1-MTG8 and pLNSX-MTGR1, and 0.5 μg of thymidine kinase-luciferase in a 6-cm-diameter plate. Results represent the mean relative CAT activity from three experiments which were normalized with luciferase expressed from thymidine kinase-luciferase as an internal control.
To determine if MTGR1 affects the activity of AML1-MTG8, MTGR1 was cotransfected with AML1-MTG8 and the activity of AML1-dependent transcription from the TCRβ enhancer was tested. As shown in Fig. 11C, expression of AML1-MTG8 repressed transcriptional activation by AML1b in a dose-dependent manner. Expression of MTGR1 had no effect on AML1b-dependent transcriptional activation. However, cotransfection of MTGR1 with AML1-MTG8 further repressed AML1b-dependent transcriptional activation. These results indicate that MTGR1 stimulates AML1-MTG8 to induce G-CSF-dependent cell growth and to repress AML1-dependent transcription.
DISCUSSION
The present results indicate that ectopic expression of the AML1-MTG8 gene in L-G mouse myeloid precursor cells induces G-CSF-dependent cell proliferation and inhibits differentiation to mature neutrophils. Analysis with deletion mutants indicates that the MTG8 sequence is required for the fusion protein to stimulate G-CSF-dependent proliferation of myeloid cells. In addition, AML1a, which lacks the C-terminal transactivation domain, does not stimulate G-CSF-dependent cell proliferation under our conditions, although it could stimulate the proliferation of 32Dcl3 cells when expressed at high levels (51). These results support the hypothesis that a simple truncation of the transactivation domain of AML1 is not sufficient to promote leukemia and that the MTG8 sequence is also required.
In an attempt to clarify the mechanism by which AML1-MTG8 promotes leukemogenesis, we investigated proteins which interact with AML1-MTG8 and found that a cellular phosphoprotein, p85, specifically interacts with the region of MTG8 which is essential for the induction of G-CSF-dependent cell proliferation. Since p85 is recognized by anti-MTG8 antibody, we considered p85 to be an the MTG8-related protein and isolated cDNA encoding a new member of the MTG8 family, MTGR1. Three lines of evidence support our conclusion that MTGR1 is at least one of the proteins which are referred to as p85. First, MTGR1 and p85 are phosphoproteins with almost the same mobility on SDS-polyacrylamide gels and are recognized by anti-MTG8 antibody. Second, they specifically interact with the same region of AML1-MTG8. Third, p85 is recognized by two anti-MTGR1 antibodies. These antibodies are prepared with MTGR1-specific sequences as antigens (Fig. 6A) and are highly specific for MTGR1; they did not react with MTG8 (Fig. 7). From these results, we concluded that MTGR1 is one of the components of the AML1-MTG8 complex. However, we do not rule out the possibility of the presence of other factors which interact with AML1-MTG8, as does MTGR1.
Based on the good correlation between activity of AML1-MTG8 and its interaction with MTGR1, AML1-MTG8 is most likely to function as a complex with MTGR1. C-terminal deletion of AML1-MTG8 to residue 538 affects neither its interaction with MTGR1 nor its functions such as stimulation of G-CSF-dependent cell proliferation and repression of AML1b-dependent transcription. Further deletion to residue 487 eliminates its interaction with MTGR1 and its functions. Overexpression of MTGR1 enhances AML1-MTG8-mediated induction of cell proliferation and repression of transcription. Since MTGR1 is highly related to MTG8, we assumed that AML1-MTG8 might also form a homomeric complex through MTG8 sequences. However, coimmunoprecipitation analysis indicated that the homomeric complex of AML1-MTG8 was not obvious, in contrast to the heteromeric complex of AML1-MTG8 and MTGR1. The yeast two-hybrid system indicates that MTGR1 can interact with MTGR1 and MTG8 by using the minimum sequences for protein interaction (30a). These results suggest that AML1-MTG8 forms a heteromeric complex with MTGR1 in preference to a homomeric complex when enough MTGR1 is present and that MTGR1 can form both homomeric and heteromeric complexes with MTG8. Differential interaction profiles have been identified in several transcription factor complexes. In the case of Jun and Fos family proteins, Jun forms both a homocomplex and a heterocomplex with Fos but Fos is unlikely to form a homocomplex. Jun-Fos and Jun-Jun complexes recognize the same DNA sequence, but Jun-Fos is a strong activator of transcription compared with Jun-Jun (reviewed in reference 15). In the case of the retinoic acid receptor (RAR) and the retinoid X receptor (RXR), RAR forms a heterocomplex with RXR in preference to the homocomplex but RXR also functions as a homocomplex. RAR-RXR and RXR-RXR bind the different DNA sequences and probably regulate the transcription of different genes (reviewed in reference 23). Thus, MTG8-MTGR1 might have a different functional activity from the MTGR1 homocomplex.
Comparison of amino acid sequences of MTGR1, MTG8, and Drosophila Nervy reveals that there are four evolutionarily conserved sequence motifs (NHR1 to NHR4) (Fig. 6A), suggesting that they have important functions. NHR1 also shows similarity to sequences of a central 80-amino-acid region of human TAF105, human TAF130, and Drosophila TAF110 (3, 5, 7, 12, 52) (Fig. 6C). The locations of hydrophobic amino acids within this region are highly conserved among MTGR1, MTG8, Nervy, and the three TAFs. The NHR2 is in the region which is essential for interaction between MTG8 family members. By using the Chou-Fasman algorithm (1) and the Robson algorithm (9), the region containing the NHR2 is predicted to present an α-helix. Analysis of helical wheels by the method of Schiffer and Edmundson (48) suggests that the NHR2 is a helical domain with amphipathic characteristics (Fig. 6D). The hydrophobic residues which appear in the hydrophobic side of the helix are conserved among MTG8, MTGR1, and Nervy. This suggests that the hydrophobic sides of the NHR2 α-helices face each other to mediate protein interactions between MTG8 family members. Since hydrophobic residues are located in a 3-4-3-4 spacing (from amino acid 348 of MTGR1 and from amino acid 354 of MTG8: W - - L - - -L - - I - - - V - - T - - - M/L - - L - -), this region of both proteins may form a coiled-coil structure that is frequently observed in oligomerization domains (41). A helical structure is also predicted in the NHR3 by the Chou-Fasman and Robson algorithms. The NHR4 regions correspond to the two zinc finger motifs (Fig. 6A). These regions are the most highly conserved regions among the three proteins. The zinc finger motifs also showed homology to those of programmed cell death-induced RP-8 protein of rat, mouse, and Caenorhabditis elegans (40) and to that of Drosophila DEAF-1 protein, also known as Deformed response element-binding protein (11). Although many of the zinc finger motifs serve as DNA-binding domains, it is not known if zinc finger motifs of MTGR1 and MTG8 are involved in DNA binding. The normal function of MTG8 and MTGR1 is unclear, but these structural characteristics, as well as their localization in the nucleus (6), suggest that they are likely to function as transcriptional regulators.
The molecular mechanism for AML1-MTG8 to promote leukemia has not yet been fully clarified, but two hypotheses are possible. First, AML1-MTG8 may affect the normal function of AML1. Analysis with deletion mutants indicates that repression of AML1-dependent transcription by AML1-MTG8 is closely linked to G-CSF-dependent cell proliferation. Cotransfection of MTGR1 further represses the AML1-dependent transcription. Since AML1-MTG8 retains the rhd, it binds to CBFβ/PEBP2β (Fig. 1B) as well as to the specific DNA sequence for AML1 (25). Thus, AML1-MTG8 is likely to repress transcription as a heterotrimer with MTGR1 and CBFβ/PEBP2β by binding to the AML1-binding site (Fig. 12). It is possible to speculate that the AML1-MTG8/CBFβ/MTGR1 complex may inhibit the formation of a transcriptional initiation complex because of its abnormal structure and/or other functions. Second, AML1-MTG8 may titrate out MTGR1 and/or affect the normal function of MTGR1 by forming the AML1-MTG8/MTGR1 complex. If this hypothesis is true, the AML1-MTG8-mediated induction of cell proliferation should be reversed by overexpression of MTGR1. However, our results indicate that overexpression of MTGR1 stimulates the action of AML1-MTG8. On the other hand, we have found that overexpression of AML1b could reverse the AML1-MTG8-induced cell proliferation (18a). Therefore, we prefer the former hypothesis that AML1-MTG8 might repress AML1-dependent transcription by binding to the DNA sequence for AML1 as the heterotrimer with MTGR1 and CBFβ.
FIG. 12.
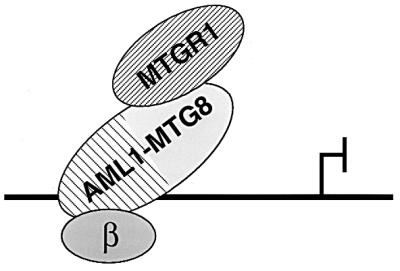
Model for the action of AML1-MTG8 on repression of transcription. AML1-MTG8 forms a heterotrimer complex with MTGR1 and CBFβ/PEBP2β through the regions containing the NHR2 and the rhd, respectively. The complex binds to the DNA sequence for AML1 and represses transcription from the adjacent target genes.
ACKNOWLEDGMENTS
I.K., K.I., and F.M. contributed equally to this work.
We thank T. Honjyo for L-G cells, A. D. Miller for retrovirus vectors, David Baltimore for BOSC23 cells, Y. Ito for TCRβ-CAT plasmid, and T. Kitamura for suggestions about retrovirus infection. We also thank H. Ichikawa for helpful discussions, N. Munakata for suggestions about protein structure, and M. Mori and C. Hatanaka for technical assistance. We thank Kazusa DNA Research Institute for help in screening the KG1 cDNA library.
This work was supported in part by a Grant-in-Aid for Scientific Research on Priority Areas from the Ministry of Education, Science and Culture; by a grant from the Special Coordination Funds for the Promotion of Science and Technology from the Science and Technology Agency; by a Grant-in-Aid for the Comprehensive 10-Year Strategy for Cancer Control and the Grant for Research on Aging and Health from the Ministry of Health and Welfare; and by the Program for Promotion of Fundamental Studies in Health Sciences of the Organization for Drug ADR Relief, R&D Promotion and Product Review of Japan.
REFERENCES
- 1.Chou P Y, Fasman G D. Prediction of the secondary structure of proteins from their amino acid sequence. Adv Enzymol Relat Areas Mol Biol. 1978;47:45–148. doi: 10.1002/9780470122921.ch2. [DOI] [PubMed] [Google Scholar]
- 2.Daga A, Tighe J E, Calabi F. Leukaemia/Drosophila homology. Nature. 1992;356:484. doi: 10.1038/356484b0. [DOI] [PubMed] [Google Scholar]
- 3.Dickstein R, Sharleen Z, Tjian R. Human TAFII105 is a cell type specific TFIID subunit related to hTAF130. Cell. 1996;87:137–146. doi: 10.1016/s0092-8674(00)81330-6. [DOI] [PubMed] [Google Scholar]
- 4.Erickson P, Gao J, Chang K S, Look T, Whisenant E, Raimondi S, Lasher R, Trujilo J, Rowley J, Drabkin H. Identification of breakpoints in t(8;21) acute myelogenous leukemia and isolation of a fusion transcript, AML1/ETO, with similarity to Drosophila segmentation gene, runt. Blood. 1992;80:1825–1831. [PubMed] [Google Scholar]
- 5.Erickson P F, Robinson M, Owens G, Drabkin H A. The ETO portion of acute myeloid leukemia t(8;21) fusion transcript encodes a highly evolutionarily conserved, putative transcription factor. Cancer Res. 1994;54:1782–1786. [PubMed] [Google Scholar]
- 6.Erickson P F, Dessev G, Lasher R S, Philips G, Robinson M, Drabkin H A. ETO and AML1 phosphoproteins are expressed in CD34+ hematopoietic progenitors: implications for t(8;21) leukemogenesis and monitoring residual disease. Blood. 1996;88:1813–1823. [PubMed] [Google Scholar]
- 7.Feinstein P G, Kornfeld K, Hogness D S, Mann R S. Identification of homeotic target genes in Drosophila melanogaster including nervy, a proto-oncogene homologue. Genetics. 1995;140:573–586. doi: 10.1093/genetics/140.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frank R, Zhang J, Meyers S, Hiebert S W, Nimer S D. The AML1/ETO fusion protein blocks transactivation of the GM-CSF promoter by AML1B. Oncogene. 1995;11:2667–2674. [PubMed] [Google Scholar]
- 9.Garnier J, Osguthorpe D J, Robson B. Analysis of the accuracy and implications of simple methods for predicting the secondary structure of globular mproteins. J Mol Biol. 1978;120:97–120. doi: 10.1016/0022-2836(78)90297-8. [DOI] [PubMed] [Google Scholar]
- 10.Golub T R, Barker G F, Bohlander S K, Hiebert S W, Ward D C, Brayward P, Morgan E, Raimondi S C, Rowley J D, Gilliland D G. Fusion of the TEL gene on 12p13 to the AML1 gene on 21q22 in acute lymphoblastic leukemia. Proc Natl Acad Sci USA. 1995;92:4917–4921. doi: 10.1073/pnas.92.11.4917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gross C T, McGinnis W. DEAF-1, a novel protein that binds an essential region in a Deformed response element. EMBO J. 1996;15:1961–1970. [PMC free article] [PubMed] [Google Scholar]
- 12.Hoey T, Weinzieri R O J, Gill G, Chen J-L, Dynlacht B D, Tjian R. Molecular cloning and functional analysis of Drosophila TAF110 reveal properties expected of coactivators. Cell. 1993;72:247–260. doi: 10.1016/0092-8674(93)90664-c. [DOI] [PubMed] [Google Scholar]
- 13.Kagoshima H, Shigesada K, Satake M, Ito Y, Miyoshi H, Ohki M, Pepling M, Gergen J P. The runt domain identifies a new family of heteromeric transcriptional regulators. Trends Genet. 1993;9:338–341. doi: 10.1016/0168-9525(93)90026-e. [DOI] [PubMed] [Google Scholar]
- 14.Kania M A, Bonner A S, Duffy J B, Gergen J P. The Drosophila segmentation gene runt encodes a novel nuclear regulatory protein that is also expressed in the developing nervous system. Genes Dev. 1990;4:1701–1713. doi: 10.1101/gad.4.10.1701. [DOI] [PubMed] [Google Scholar]
- 15.Karin M, Liu Z, Zandi E. AP-1 function and regulation. Curr Opin Cell Biol. 1997;9:240–246. doi: 10.1016/s0955-0674(97)80068-3. [DOI] [PubMed] [Google Scholar]
- 16.Kinashi T, Kwang H L, Ogawa M, Tohyama K, Tashiro K, Fukunaga R, Nagata S, Honjo T. Premature expression of the macrophage-colony stimulating factor receptor on a multipotential stem cell line does not alter differentiation lineages controlled by stromal cells used for coculture. J Exp Med. 1991;173:1267–1279. doi: 10.1084/jem.173.5.1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kitabayashi I, Kawakami Z, Chiu R, Ozawa K, Matsuoka T, Toyoshima S, Umesono T, Evans R M, Gachelin G, Yokoyama K. Transcriptional regulation of the c-jun gene by retinoic acid and E1A during differentiation of F9 cells. EMBO J. 1992;11:167–175. doi: 10.1002/j.1460-2075.1992.tb05039.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kitabayashi I, Eckner R, Arany Z, Chiu R, Gachelin G, Livingstone D M, Yokoyama K. Phosphorylation of the adenovirus E1A-associated 300 kDa protein in response to retinoic acid and E1A during the differentiation of F9 cells. EMBO J. 1995;14:3496–3509. doi: 10.1002/j.1460-2075.1995.tb07356.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18a.Kitabayashi, I., et al. Unpublished data.
- 19.Kozu T, Miyoshi H, Shimizu K, Maseki N, Kaneko Y, Asou H, Kamada N, Ohki M. Junctions of AML1/MTG8(ETO) fusion are constant in t(8;21) acute myeloid leukemia detected by reverse transcription polymerase chain reaction. Blood. 1993;82:1270–1276. [PubMed] [Google Scholar]
- 20.Lenny N, Meyers S, Hiebert S W. Functional domains of the t(8;21) fusion protein, AML1/ETO. Oncogene. 1995;11:1761–1769. [PubMed] [Google Scholar]
- 21.Levanon D, Negreanu V, Bernstein Y, Bar-Am I, Avivi L, Groner Y. AML1, AML2 and AML3, the human members of the runt domain gene-family: cDNA structure, expression, and chromosomal localization. Genomics. 1994;23:425–432. doi: 10.1006/geno.1994.1519. [DOI] [PubMed] [Google Scholar]
- 22.Liu P, Tarle S A, Hajra A, Claxton D F, Marlton P, Freedman M, Siciliano M J, Collins F S. Fusion between transcription factor CBFβ/PEBP2β and a myosin heavy chain in acute myeloid leukemia. Science. 1993;261:1041–1044. doi: 10.1126/science.8351518. [DOI] [PubMed] [Google Scholar]
- 23.Mangelsdorf D J, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, Evans R M. The nuclear receptor superfamily: the second decade. Cell. 1995;83:835–839. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meyers S, Dowing J R, Hiebert S W. Identification of AML-1 and the (8;21) translocation protein (AML-1/ETO) as sequence-specific DNA-binding proteins: the runt homology domain is required for DNA binding and protein-protein interactions. Mol Cell Biol. 1993;13:6336–6345. doi: 10.1128/mcb.13.10.6336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meyers S, Lenny N, Hiebert S W. The t(8;21) fusion protein interferes with AML 1B-dependent transcriptional activation. Mol Cell Biol. 1995;15:1974–1982. doi: 10.1128/mcb.15.4.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miller A D, Miller D G, Garcia J V, Lynch C M. Use of retroviral vectors for gene transfer and expression. Methods Enzymol. 1993;217:581–599. doi: 10.1016/0076-6879(93)17090-r. [DOI] [PubMed] [Google Scholar]
- 27.Mitani K, Ogawa S, Tanaka T, Miyoshi H, Kurokawa M, Mano H, Yazaki Y, Ohki M, Hirai H. Generation of the AML1-Evi-1 fusion gene in the t(3;21)(q26;q22) causes blastic crisis in chronic myelocytic leukemia. EMBO J. 1994;13:504–510. doi: 10.1002/j.1460-2075.1994.tb06288.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miyoshi H, Shimizu K, Kozu T, Maseki N, Kaneko Y, Ohki M. t(8;21) breakpoints on chromosome 21 in acute myeloid leukemia are clustered within a limited region of a single gene, AML1. Proc Natl Acad Sci USA. 1991;88:10431–10434. doi: 10.1073/pnas.88.23.10431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miyoshi H, Kozu T, Shimizu K, Enomoto K, Maseki N, Kaneko Y, Kamada N, Ohki M. The t(8;21) translocation in acute myeloid leukemia results in production of an AML1-MTG8 fusion transcript. EMBO J. 1993;12:2715–2721. doi: 10.1002/j.1460-2075.1993.tb05933.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miyoshi H, Ohira M, Shimizu K, Mitani K, Hirai H, Imai T, Yokoyama K, Soeda E, Ohki M. Alternative splicing and genomic structure of the AML1 gene involved in acute myeloid leukemia. Nucleic Acids Res. 1995;23:2762–2769. doi: 10.1093/nar/23.14.2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30a.Morohoshi, F., et al. Unpublished data.
- 31.Niki M, Okada H, Takano H, Kuno J, Tani K, Hibino H, Asano S, Ito Y, Satake M, Noda T. Hematopoiesis in the fetal liver is impaired by targeted mutagenesis of a gene encoding a non-DNA binding subunit of the transcription factor, polyomavirus enhancer binding protein 2/core binding factor. Proc Natl Acad Sci USA. 1997;94:5697–5702. doi: 10.1073/pnas.94.11.5697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nisson P E, Watkins P C, Sacchi N. Transcriptionally active chimeric gene derived from the fusion of the AML1 gene and a novel gene on chromosome 8 in t(8;21) leukemic cells. Cancer Genet Cytogenet. 1992;63:81–88. doi: 10.1016/0165-4608(92)90384-k. [DOI] [PubMed] [Google Scholar]
- 33.Nomura N, Miyajima N, Sazuka T, Tanaka A, Kawarabayashi Y, Sato S, Nagase T, Seki N, Ishikawa K, Tabata S. Prediction of the coding sequences of unidentified human genes. I. The coding sequences of 40 new genes (KIAA0001–KIAA0040) deduced by analysis of randomly sampled cDNA clones from human immature myeloid cell line KG-1. DNA Res. 1994;1:27–35. doi: 10.1093/dnares/1.1.27. [DOI] [PubMed] [Google Scholar]
- 34.Nuchprayoon I, Meyers S, Scott L M, Suzow J, Hiebert S, Friedman A D. PEBP2/CBF, the murine homolog of the human myeloid AML1 and PEBP2β/CBFβ proto-oncoproteins, regulates the murine myeloperoxidase and neutrophil elastase genes in immature myeloid cells. Mol Cell Biol. 1994;14:5558–5568. doi: 10.1128/mcb.14.8.5558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nucifora G, Begy C R, Erickson P, Drabkin H A, Rowley J D. The 3;21 translocation in myelodysplasia results in a fusion transcript, the AML1 gene and the gene for EAP, a highly conserved protein associated with Epstein-Barr virus small RNA EBER 1. Proc Natl Acad Sci USA. 1993;90:7784–7788. doi: 10.1073/pnas.90.16.7784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nucifora G, Begy C R, Kobayashi H, Roulston D, Claxton D, Pedersen-Bjergaad J, Parganas E, Ihle J N, Rowley J D. Consistent intergenic splicing and production of multiple transcripts between AML1 at 21q22 and unrelated genes at 3q26 in (3;21)(q26;q22) translocations. Proc Natl Acad Sci USA. 1994;91:4004–4008. doi: 10.1073/pnas.91.9.4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ogawa E, Inuzuka M, Maruyama M, Satake M, Naito-Fujimoto M, Ito Y, Shigesada K. Molecular cloning and characterization of PEBP2β, the heterodimeric partner of a novel Drosophila runt-related DNA binding protein PEBP2. Virology. 1993;194:314–331. doi: 10.1006/viro.1993.1262. [DOI] [PubMed] [Google Scholar]
- 38.Ogawa E, Maruyama M, Kagoshima H, Inuzuka M, Lu J, Satake M, Shigesada K, Ito Y. PEBP2/PEA2 represents a family of transcription factors homologous to the Drosophila runt gene and the human AML1 gene. Proc Natl Acad Sci USA. 1993;90:6859–6863. doi: 10.1073/pnas.90.14.6859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Okuda T, van Deusen J, Hiebert S W, Grosveld G, Downing J R. AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell. 1996;84:321–330. doi: 10.1016/s0092-8674(00)80986-1. [DOI] [PubMed] [Google Scholar]
- 40.Owens G P, Hahn W E, Cohen J J. Identification of mRNAs associated with programmed cell death in immature thymocytes. Mol Cell Biol. 1991;11:4177–4188. doi: 10.1128/mcb.11.8.4177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Parry D A D. Coiled-coils in α-helix containing proteins: analysis of the residue types within the heptad repeat and the use of these data in the prediction of coiled-coils in other proteins. Biosci Rep. 1982;2:1017–1024. doi: 10.1007/BF01122170. [DOI] [PubMed] [Google Scholar]
- 42.Pear W S, Nolan G P, Scott M L, Baltimore D. Production of high-titer helper-free retroviruses by transient transfection. Proc Natl Acad Sci USA. 1993;90:8392–8396. doi: 10.1073/pnas.90.18.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Prosser H M, Wotton D, Gegonne A, Ghysdael J, Wang S, Speck N A, Owen M J. A phorbol ester response element within the human T-cell receptor β-chain enhancer. Proc Natl Acad Sci USA. 1992;89:9934–9938. doi: 10.1073/pnas.89.20.9934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rabbitts T H. Chromosomal translocations in human cancer. Nature. 1994;372:143–149. doi: 10.1038/372143a0. [DOI] [PubMed] [Google Scholar]
- 45.Redondo J M, Pfohl J L, Hernandez-Munain C, Wang S, Speck N A, Krangel M S. Indistinguishable nuclear factor binding to functional core sites of the T-cell receptor δ and murine leukemia virus enhancers. Mol Cell Biol. 1992;12:4817–4823. doi: 10.1128/mcb.12.11.4817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Romana S P, Mauchauffe M, Le Coniat M, Chumakov I, Le Paslier D, Berger R, Bernard O A. The t(12;21) of acute lymphoblastic leukemia results in a tel-AML1 gene fusion. Blood. 1995;85:3662–3670. [PubMed] [Google Scholar]
- 47.Sasaki K, Yagi H, Bronson R T, Tominaga K, Matsunashi T, Deguchi K, Tani Y, Kishimoto T, Komori T. Absence of fetal liver hematopoiesis in mice deficient in transcriptional coactivator core binding factor beta. Proc Natl Acad Sci USA. 1996;93:12359–12363. doi: 10.1073/pnas.93.22.12359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schiffer M, Edmundson A B. Use of helical wheels to present the structures of proteins and to identify segments with helical potential. Biophys J. 1967;7:121–135. doi: 10.1016/S0006-3495(67)86579-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shoemaker S G, Hromas R, Kaushansky K. Transcriptional regulation of interleukin 3 gene expression in T lymphocytes. Proc Natl Acad Sci USA. 1990;87:9650–9654. doi: 10.1073/pnas.87.24.9650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Takahashi A, Satake M, Yamaguchi-Iwai Y, Bae S C, Lu J, Maruyama M, Zhang Y M, Oka H, Arai N, Arai K, Ito Y. Positive and negative regulation of granulocyte macrophage colony-stimulating factor promoter activity by AML1-related transcription factor, PEBP2. Blood. 1995;86:607–616. [PubMed] [Google Scholar]
- 51.Tanaka T, Tanaka K, Ogawa S, Kurokawa M, Mitani K, Nishida J, Shibata J, Yazaki Y, Hirai H. An acute myeloid leukemia gene, AML1, regulates hemopoietic myeloid cell differentiation and transcriptional activation antagonistically by two alternative spliced forms. EMBO J. 1995;14:341–350. doi: 10.1002/j.1460-2075.1995.tb07008.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tanese N, Saluja D, Vassallo M F, Chen J-L, Admon A. Molecular cloning and analysis of two subunits of the human TFIID complex: hTAFII130 and hTAFII100. Proc Natl Acad Sci USA. 1996;93:13611–13616. doi: 10.1073/pnas.93.24.13611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang Q, Stacy T, Binder M, Marin-Padilla M, Sharpe A H, Speck N A. Disruption of the Cbfa2 gene causes necrosis and hemorrhaging in the central nervous system and blocks definitive hematopoiesis. Proc Natl Acad Sci USA. 1996;93:3444–3449. doi: 10.1073/pnas.93.8.3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang Q, Stacy T, Miller J D, Lewis A F, Gu T L, Huang X, Bushweller J H, Bories J C, Alt F W, Ryan G, Liu P P, Wynshaw-Boris A, Binder M, Marin-Padilla M, Sharpe A H, Speck N A. The CBFbeta subunit is essential for CBFalpha2 (AML1) function in vivo. Cell. 1996;87:697–708. doi: 10.1016/s0092-8674(00)81389-6. [DOI] [PubMed] [Google Scholar]
- 55.Wang S, Wang Q, Crute B E, Melnikova I N, Keller S R, Speck N A. Cloning and characterization of subunits of the T-cell receptor and murine leukemia virus enhancer core-binding factor. Mol Cell Biol. 1993;13:3324–3339. doi: 10.1128/mcb.13.6.3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wargnier A, Legros-Maida S, Bosselut R, Bourge J F, Lafaurie C, Ghysdael C J, Sasportes M, Paul P. Identification of human granzyme B promoter regulatory elements interacting with activated T-cell-specific proteins: implication of Ikaros and CBF binding sites in promoter activation. Proc Natl Acad Sci USA. 1995;92:6930–6934. doi: 10.1073/pnas.92.15.6930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang D E, Fujioka K, Hetherington C J, Shapiro L H, Chen H M, Look A T, Tenen D G. Identification of a region which directs the monocytic activity of the colony-stimulating factor 1 (macrophage colony-stimulating factor) receptor promoter and binds PEBP2/CBF (AML1) Mol Cell Biol. 1994;14:8085–8095. doi: 10.1128/mcb.14.12.8085. [DOI] [PMC free article] [PubMed] [Google Scholar]