

Abstract
We present a language model Affordable Cancer Interception and Diagnostics (ACID) that can achieve high classification performance in the diagnosis of cancer exclusively from using raw cfDNA sequencing reads. We formulate ACID as an autoregressive language model. ACID is pretrained with language sentences that are obtained from concatenation of raw sequencing reads and diagnostic labels. We benchmark ACID against three methods. On testing set subjected to whole-genome sequencing, ACID significantly outperforms the best benchmarked method in diagnosis of cancer [Area Under the Receiver Operating Curve (AUROC), 0.924 versus 0.853; P < 0.001] and detection of hepatocellular carcinoma (AUROC, 0.981 versus 0.917; P < 0.001). ACID can achieve high accuracy with just 10 000 reads per sample. Meanwhile, ACID achieves the best performance on testing sets that were subjected to bisulfite sequencing compared with benchmarked methods. In summary, we present an affordable, simple yet efficient end-to-end paradigm for cancer detection using raw cfDNA sequencing reads.
Keywords: cell-free DNA, cancer detection, generative language model
INTRODUCTION
Cell-free DNA (cfDNA) is fragmented DNA circulating in the blood [1, 2] through cell necrosis and apoptosis [3, 4]. cfDNA fragments shed into plasma by tumor cells bear cancer-specific genomic and epigenetic alterations that can be leveraged for early cancer detection [5, 6]. Tumor-derived cfDNA fragments are shorter in size than hematopoietic-originated background cfDNA fragments [7]. cfDNA fragments exhibit preferential patterns in patients with and without cancer as well as among different cancer types [8, 9]. The fragmentation patterns of cfDNA have been widely studied in early cancer detection [10–15]. The Circulating Cell-free Genome Atlas (CCGA) is among one of the most ambitious and representative endeavors in investigating the use of cfDNA-based multi-cancer early cancer detection [16–18].
The size profiles of cfDNA are different in patients with and without cancer in which shorter cfDNA fragments are more prevalent in patients with cancer. The median size of background cfDNA is ~166 bp [19–21]. Mouliere and colleagues reported that length selection of short cfDNA fragments increases enrichment of circulating tumor DNA in advanced cancer patients [22]. There are several characteristics manifesting unique fragmentation patterns of cfDNA in patient with cancer, including alterations in nucleosome positions [20, 23] and preference of specific sequence at the end of cfDNA fragments [24]. Whole-genome sequencing (WGS) offers a high-resolution view of the genome. However, it remains challenging to unveil accurate chromosomal aberrations from cfDNA [25–27]. Cristiano and colleagues developed a machine learning approach called DNA evaluation of fragments for early interception (DELFI) [28] by integrating numerous genomic alterations of cfDNA for cancer detection. DELFI incorporates characteristics of cancer-specific profiles with respect to mutation and chromatin organization obtained from low-coverage WGS of cfDNA. Recently, Bae and colleagues proposed an algorithm for cancer detection and tissue-of-origin localization from cfDNA via integrative modeling of tumor genomes and epigenomes to incorporates cancer-specific mutational profiles and chromatin organization as model references [29].
The fragmentation patterns of cfDNA are not random, and certain sequences at the end of cfDNA fragments are more prevalent. This phenomenon is linked to DNA nuclease activities [30]. Serpas and colleagues reported that deoxyribonuclease 1 like 3 is associated with the generation of CCCA end-motif [31]. Jiang and colleagues reported that the diversity of cfDNA end-motifs can be utilized to distinguish patients with and without hepatocellular carcinoma [32]. Recently, Zhou and colleagues used non-negative matrix factorization (NMF) algorithm to deconvolute cfDNA end-motif matrix into distinct fragmentation profiles and observed that the fragmentation signatures can be used to identify hepatocellular carcinoma [30].
Pretrained transformer-based language models have tremendously revolutionized natural language understanding. Large language models (e.g. GPT-3 [33], PaLM [34] and LLaMA [35]) and products developed upon them (e.g. ChatGPT and Alpaca) have attracted enormous attentions from different fields. The core idea underlying these models is generative pretraining, wherein the model uses the preceding input as context to generate new content. Inspired by the success of these transformer-based models [36–41], researchers started to investigate the applications of transformer-based algorithms in addressing biological challenges. Rives and colleagues showed that biological structures and functions of protein were encoded in the language models pretrained on 250 million protein sequences [42]. Madani and colleagues developed a protein language model called ProGen from protein sequences that can generate artificial proteins with different functions [43]. We showed that single-cell transcriptome analysis could be formatted as language understanding task and generative pretraining with single-cell transcriptomes uncovered expression signatures associated with immunotherapy treatment response [44].
Fragmentation of cfDNA is a nonrandom process. The cfDNA molecules exhibit preferred end coordinates under different scenarios. For instance, liver-derived cfDNA ended more frequently at certain genomic positions as compared with non-liver-derived cfDNA molecules [24]. This suggested that cfDNA sequences are different for different disease types; therefore, exploration and exploitation of these differences are helpful for developing computational methods for cancer diagnosis. Inspired by the success of the aforementioned language models, we proposed a generative artificial intelligence (AI) approach, Affordable Cancer Interception and Diagnostics (ACID) that can achieve high classification performance in the diagnosis of cancer exclusively from using a limited number of raw cfDNA sequencing reads. The input to ACID is simply the concatenation of raw cfDNA sequencing reads, enabling ACID to perform end-to-end cancer diagnosis. We developed and evaluated ACID with sequencing data from 2094 samples. We benchmark the classification performance of ACID against three methods. ACID achieved superior performance as compared with the other benchmarked methods on the evaluation dataset that was subjected to WGS. Besides, we observed that ACID also achieved competitive performance on the other data modalities such as bisulfite sequencing.
RESULTS
An overview of the study
The whole procedure consists of three components: data preparation, development of ACID and performance evaluation of ACID in detection of cancer. Figure 1A vividly illustrates the origin of cfDNA and the format of training data for the development of ACID. The ith training data point consists of three attributes: instruction prompt, input content and expected output. The instruction prompt is a short sentence describing the task (denoted as 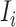 ). The input content is simply the concatenation of sequencing reads that are alphabetically sorted (denoted as
). The input content is simply the concatenation of sequencing reads that are alphabetically sorted (denoted as 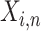 , n is the number of reads). The expected output is a brief description of the clinical phenotype (denoted as
, n is the number of reads). The expected output is a brief description of the clinical phenotype (denoted as  ) of the donor whose cfDNA was subjected to sequencing. The consecutive concatenation of these sentences, i.e.
) of the donor whose cfDNA was subjected to sequencing. The consecutive concatenation of these sentences, i.e.  = {
= {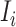 ,
,  ,
,  }, forms a training data point (Figure 1A). The training data points were curated from cfDNA samples subjected to WGS, whole-genome bisulfite sequencing (WGBS), targeted bisulfite sequencing and whole-exome sequencing. ACID was formatted as a generative language model and trained in an autoregressive manner (Figure 1B). Specifically, it makes its output in the context of its preceding inputs (See Materials and Methods). In the training phase, ACID was trained in an autoregressive manner by taking
}, forms a training data point (Figure 1A). The training data points were curated from cfDNA samples subjected to WGS, whole-genome bisulfite sequencing (WGBS), targeted bisulfite sequencing and whole-exome sequencing. ACID was formatted as a generative language model and trained in an autoregressive manner (Figure 1B). Specifically, it makes its output in the context of its preceding inputs (See Materials and Methods). In the training phase, ACID was trained in an autoregressive manner by taking  as input. In the testing phase, ACID predicts the output
as input. In the testing phase, ACID predicts the output 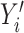 by taking the concatenation of
by taking the concatenation of 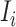 and
and 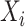 as input, i.e.
as input, i.e.  = {
= {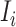 ,
,  }.
}.
Figure 1.

The flowchart depicting the development and evaluation of ACID. The whole procedure consists of three components, including data preparation of cfDNA, development of ACID and performance evaluation of ACID in detection of cancer.
Development of ACID
We collected the sequencing data of 2094 samples from four datasets available at the public databases, covering different diseases and sequencing modalities. For each dataset, we randomly selected 50% of the samples as training set and the remaining 50% as testing set. There is no overlap between donor barcodes in the training set and testing set. For each individual, the number of data points we constructed depend on the sequencing type. For example, we constructed 1 007 866 data points for WGS and 382 206 for targeted bisulfite sequencing. Each data point consists of the instruction prompt, randomly selected number (n) reads and description of clinical phenotype. We developed two ACID models (i.e. ACID–base and ACID-large) that have different number of parameters (See Materials and Methods and Supplementary Table 1). The benchmarked methods include DNA evaluation of fragments for early interception (DELFI), motif diversity score (MDS) and NMF decomposition of fragmentation profiles (See Materials and Methods).
High accuracy of ACID in detection of cancer subjected to whole-genome sequencing
We first evaluated the performance of ACID models on the CUHK dataset that was subjected to WGS. This dataset consists of hepatocellular carcinoma (HCC, n = 20), colorectal cancer (CRC, n = 3), lung cancer (LUCA, n = 6), nasopharyngeal carcinoma (NPC, n = 6), head and neck squamous cell carcinoma (HNSC, n = 4), patients infected with chronic hepatitis B virus (HBV, n = 7) and non-cancerous controls (n = 17). We observed that both ACID–base and ACID-large models achieved higher classification performance as measured by area under the receiver operating characteristic curve (AUROC) as compared with all the benchmarked methods (Figure 2). The ACID-large model outperforms the ACID–base model (Figure 2).
Figure 2.

Receiver operating characteristic curves of ACID versus benchmarked methods in task of cancer diagnosis (A) and HCC detection (B). The benchmarked methods include DELFI, NMF and MDS.
With respect to differentiation of cancer patients from non-cancerous controls, both ACID–base and ACID-large models achieved significantly higher AUROC value as compared with the best benchmarked method (Figure 2A; all P < 0.001). The ACID–base and ACID-large models have an accuracy of 0.857 (0.746–0.933) and 0.889 (0.784–0.954), sensitivity of 0.872 (0.726–0.957) and 0.872 (0.726–0.957), positive predictive value of 0.895 (0.752–0.971) and 0.944 (0.813–0.993), and F1 score of 0.883 and 0.907 (Table 1), respectively. The other classification metrics such as specificity, negative predictive value and performance of the other benchmarked methods were provided in Table 1. Our method showed promising results when stratified by disease types. For example, both ACID–base and ACID-large could detect all four head and neck squamous cell carcinoma and six nasopharyngeal carcinomas. ACID–base could identify six of HBV-positive controls and ACID-large identify all seven HBV-positive controls (Supplementary Figure 1). There are different sequence logos of cfDNA from healthy individuals and patients for each type of cancer (Supplementary Figure 2).
Table 1.
Classification metrics in cancer diagnosis on the WGS dataset
| Methods | Accuracy (95% CI) | Sensitivity (95% CI) | Specificity (95% CI) | Positive predictive value (95% CI) | Negative predictive value (95% CI) | F1 score |
|---|---|---|---|---|---|---|
| DELFI | 0.762 (0.638–0.860) | 0.667 (0.498–0.809) | 0.917 (0.730–0.990) | 0.929 (0.765–0.991) | 0.629 (0.449–0.785) | 0.776 |
| NMF | 0.746 (0.621–0.847) | 0.692 (0.524–0.830) | 0.833 (0.626–0.953) | 0.871 (0.702–0.964) | 0.625 (0.437–0.789) | 0.771 |
| MDS | 0.794 (0.673–0.885) | 0.692 (0.524–0.830) | 0.958 (0.789–0.999) | 0.964 (0.817–0.999) | 0.657 (0.478–0.809) | 0.806 |
| ACID–base | 0.857 (0.746–0.933) | 0.872 (0.726–0.957) | 0.833 (0.626–0.953) | 0.895 (0.752–0.971) | 0.800 (0.593–0.932) | 0.883 |
| ACID-large | 0.889 (0.784–0.954) | 0.872 (0.726–0.957) | 0.917 (0.730–0.990) | 0.944 (0.813–0.993) | 0.815 (0.619–0.937) | 0.907 |
In addition, we tested our method on the dataset reported by Cristiano and colleagues [28] for the identification of cancer. This dataset consists of 231 cancer samples and 246 healthy controls. We used a random subset of 50% of samples as training set and the remaining 50% as testing set. On this testing set, we observed that the AUROC values are 0.939 [95% confidence interval (CI), 0.911–0.968] for ACID–base, 0.945 (0.918–0.972) for ACID-large, 0.890 (0.848–0.932) for DELFI, 0.690 (0.621–0.760) for NMF and 0.659 (0.588–0.730) for MDS (Supplementary Figure 3). When stratified by tumor stages, we observed that both ACID–base and ACID-large models achieved higher AUROC values across different tumor stages as compared with benchmarked methods (i.e. DELFI, NMF and MDS) (Supplementary Figure 4). The AUROC values are higher for tumor at advanced stage than early stage. For ACID–base and ACID-large, AUROC values are, respectively, 0.951 and 0.950 for tumor at stage I, 0.931 and 0.940 at stage II, 0.993 and 0.996 at stage III and 0.973 and 0.991 at stage IV. Among these three benchmarked methods, DELFI achieved higher AUROC values as compared with NMF and MDS. However, it is still lower than ACID (Supplementary Figure 4).
With respect to detection of HCC, both ACID–base and ACID-large models also achieved significantly higher AUROC value as compared with the best benchmarked method (Figure 2B; all P < 0.001). The ACID–base and ACID-large models have an identical accuracy 0.984 (0.915–1.000), sensitivity of 0.950 (0.751–0.999), positive predictive value of 1.000 (0.854–1.000) and F1 score of 0.974 (Table 2). The other classification metrics such as specificity, negative predictive value and performance of the other benchmarked methods were provided in Table 2. At the specificity of 99%, ACID–base has a sensitivity of 95.0% (85.0%–100%), outperforming the best benchmarked method by 35.7% (Supplementary Figure 5). We cannot determine the detection performance for the other specific cancer type due to the limited number of these cancer samples. Therefore, we combined all the non-HCC tumors as a single group. For the identification of non-HCC tumors, ACID–base and ACID-large achieved AUROC values of [0.946 (95% CI, 0.890–1.0)] and [0.965 (95% CI, 0.919–1.0)], respectively (Supplementary Figure 6).
Table 2.
Classification metrics in HCC detection on the WGS dataset
| Methods | Accuracy (95% CI) | Sensitivity (95% CI) | Specificity (95% CI) | Positive predictive value (95% CI) | Negative predictive value (95% CI) | F1 score |
|---|---|---|---|---|---|---|
| DELFI | 0.857 (0.746–0.933) | 0.850 (0.621–0.968) | 0.860 (0.721–0.947) | 0.739 (0.516–0.898) | 0.925 (0.796–0.984) | 0.791 |
| NMF | 0.905 (0.804–0.964) | 0.750 (0.509–0.913) | 0.977 (0.877–0.999) | 0.938 (0.698–0.998) | 0.894 (0.769–0.965) | 0.833 |
| MDS | 0.810 (0.691–0.898) | 0.500 (0.272–0.728) | 0.953 (0.842–0.994) | 0.833 (0.516–0.979) | 0.804 (0.669–0.902) | 0.625 |
| ACID–base | 0.984 (0.915–1.000) | 0.950 (0.751–0.999) | 1.000 (0.933–1.000) | 1.000 (0.854–1.000) | 0.977 (0.880–0.999) | 0.974 |
| ACID-large | 0.984 (0.915–1.000) | 0.950 (0.751–0.999) | 1.000 (0.933–1.000) | 1.000 (0.854–1.000) | 0.977 (0.880–0.999) | 0.974 |
Data-efficiency of ACID
We systematically examined the performance of ACID in relation to data size in evaluation stage. The performance of ACID as measured by AUROC values increased gradually and reached plateau at the number of 20 000 reads (Figure 3 and Supplementary Table 2). At the number of 20 000 reads, ACID–base achieved an AUROC value of 0.893 (0.881–0.904) for diagnosis of cancer (Figure 3A) and 0.973 (0.964–0.981) for detection of HCC (Figure 3B). At the number of 20 000 reads, ACID-large achieved an AUROC value of 0.918 (0.907–0.929) in diagnosis of cancer (Supplementary Figure 7A) and 0.978 (0.971–0.985) in detection of HCC (Supplementary Figure 7B). This result demonstrated that ACID is very data-efficient without sacrificing its performance.
Figure 3.

The performance of ACID in relation to data size. AUROC values of ACID at different number of reads in task of cancer diagnosis (A) and HCC detection (B).
High accuracy of ACID in diagnosis of cancer subjected to bisulfite sequencing
We evaluated the performance of ACID in diagnosis of cancer on three datasets that were subjected to bisulfite sequencing. These three datasets include a subset of samples from the CUHK dataset that subjected to WGBS (CUHK-WGBS, n = 31) and two datasets subjected to targeted bisulfite sequencing (CRC-TBS, n = 622; HCC-TBS, n = 1140). We used NMF and MDS as baseline methods in this section as the DELFI algorithm was not developed for bisulfite sequencing data.
On the CUHK-WGBS dataset, ACID–base and ACID-large models achieved higher AUROC values than the second-best benchmarked method (Figure 4A). On two datasets subjected to targeted bisulfite sequencing for detection of CRC and HCC, ACID–base and ACID-large models achieved significantly higher AUROC values than the second-best benchmarked method, respectively (Figure 4B and C). In addition, ACID also achieved at least comparable or better performance as compared with the alignment-based method reported in the original studies (AUROC, 0.996 versus 0.896 [8]; 0.999 versus 0.944 [42]). The classification metrics such as accuracy, sensitivity, specificity, positive predictive value, negative predictive value and F1 score were provided in Supplementary Table 3.
Figure 4.

Receiver operating characteristic curves of ACID versus benchmarked methods in task of cancer diagnosis subjected to WGBS (A) and CRC (B) and HCC (C) detection subjected to TGBS. The benchmarked methods include NMF and MDS.
On the targeted genome bisulfite sequencing (TGBS)-HCC dataset, we calculated the AUROC values for ACID and the benchmarked methods (i.e. NMF and MDS) stratified by tumor stages. We observed that ACID all achieved higher AUROC values across different tumor stages as compared with NMF and MDS methods (Supplementary Figure 8). The AUROC values for NMF and MDS are lower in tumors at early stage as compared with tumors at advanced stage (Supplementary Figure 8).
On the TGBS-HCC dataset, we deciphered four reference cfDNA signatures (See Materials and Methods and Supplementary Figure 9). We observed that reference Signature 1 had good performance in identification of cancer with an AUROC value of 0.88 (95% CI, 0.865–0.895) (Supplementary Figure 9C). We used attention-based multiple-instance learning (MIL) to identify reads that exhibit strong association with decision made by ACID and subsequently used NMF algorithm to decipher sequence signatures from these high scoring reads (See Materials and Methods). We obtained three signatures from these high scoring reads (denoted as ACID–based signatures; Supplementary Figure 9D). We observed that ACID–based Signature 3 exhibits high correlation with reference Signature 1 (Supplementary Figure 9E). This result suggested that ACID indeed explored and exploited cfDNA fragments that are useful for the identification of cancer.
DISCUSSION
In this study, we presented a new paradigm and introduced a conceptually simple approach entitled ACID toward integrating the raw cfDNA sequencing reads for cancer detection. We demonstrated that ACID is practically simple yet empirically powerful in essence. ACID eliminates the tedious traditional bioinformatic analysis steps for genomic sequencing data including sequence alignment and detection of genomic copy number changes, etc. In contrast, ACID expects the raw sequencing reads as input; therefore, it could perform an end-to-end detection of cancer in a unified single step. Our results showed that ACID outperforms the mainstream state-of-the-art benchmarked methods in the detection of cancer across different sequencing techniques.
It is challenging to identify cancer-specific signals such as somatic mutations, aberrant methylation profiles and copy number changes from cfDNA as the amount of cancer-derived cfDNA is quite low compared with the hematopoietic-originated background cfDNA. Researchers have started to explore new types of signals from cfDNA for early cancer detection including the MDS of end-motifs and deconvolution of end-motif profiles [30, 32]. Our study showed that the transformer-based language model ACID achieved higher classification performance as compared with the two aforementioned methods.
ACID has several advantages. Firstly, ACID has the potential to provide an affordable and cost-effective method for liquid-biopsy-based cancer detection as it only requires super-low WGS of cfDNA. This is exemplified by the result that ACID–base is able to achieve an accuracy of 82.5% (70.9%–90.9%) in the diagnosis of cancer and 98.4% (91.5% − 100%) in the detection of HCC by only using 10 000 raw sequencing reads subjected to WGS. Together with the extraction of DNA and the construction of a sequencing library, the cost of sequencing a plasma cfDNA sample to one-fold coverage is estimated to be 40 US dollars in China. Meanwhile, ACID is designed to receive sequencing reads as input, thus it is agnostic to sequencing techniques. We demonstrated that ACID performs competitively on bisulfite sequencing reads as compared with the results reported by the original studies. Secondly, the performance of ACID is associated with its scalability in that the ACID-large model has higher AUROC value than did the ACID–base model. According to the scaling laws, the loss of transformer-based language model scales as a power-law with model size, dataset size and the amount of compute used for training [45]. Therefore, the performance of ACID is expected to increase as we accumulate more cfDNA sequencing data to train model with higher capacity. Thirdly, ACID eliminates time-consuming steps such as sequence alignment, detection of copy number, characterization of fragmentation profiles and incorporation of chromatin characteristics that are required by DELFI, thus enabling end-to-end detection of cancer from input sequencing reads and speeding up the inference time when deployed. The inference time using central processing unit (CPU) for DELFI is ~83 min for a given sample subjected to 5X WGS, whereas ACID spent 7 min. On graphical processing unit (GPU), the inference time of ACID is 1 min.
ACID was not without limitations. The limitations of ACID lie in its advantages as it expects the raw sequencing reads as input; therefore, it misses well-known cancer-specific information such as size profile [2], preferred end coordinates [24, 46], mutations and copy number changes [6] that would be helpful for detection of cancer. Future works on incorporating these well-known cancer-specific features into ACID are expected to increase the performance of ACID. The quadratic complexity of memory usage in relation to sequence length of the transformer hampers ACID to process long sequence. However, we observed that concatenation of 100 and 200 reads as a sequence produce comparable performance (Supplementary Figure 7). The success of large language models depends on model size, dataset size and amount of compute. In the current study, the amount of data and model size are quite small as compared with GPT-3 [33], LLaMA [35] and PaLM [34]. Therefore, we are planning to perform cfDNA sequencing for a large number of plasma samples to develop a powerful large language model for representing the patterns of cfDNA for early cancer detection.
In summary, we presented a simple yet powerful new paradigm for detection of cancer via directly inspecting the raw sequencing reads of cfDNA. Our work has the potential to facilitate translational research of liquid-biopsy-based early cancer detection in the foreseeable future.
MATERIALS AND METHODS
Data collection
We collected the raw sequencing data of cfDNA from 2094 plasma samples, covering HCC, CRC, LUCA, NPC and HNSC. The sequencing modalities include WGS, WGBS and TGBS. The criteria to collect datasets from the public databases include: (i) plasma samples subjected to cfDNA sequencing with WGS or bisulfite sequencing, (ii) known clinical characteristic (e.g. cancer status and/or cancer subtype) for each sample and (iii) sample size ≥100. We did not perform additional quality control for these collected samples but included all of them in our analysis.
HCC-WGS dataset
The dataset consisted of plasma cfDNA samples subjected to WGS from 74 patients with CRC (n = 10), LUCA (n = 10), NPC (n = 10), HNSC (n = 10), HCC (n = 34) and 55 noncancerous controls. Raw sequencing data were downloaded from European Bioinformatics Institute (Accession code EGAS00001003409) [32].
Cristiano dataset
The dataset consisted of plasma cfDNA samples subjected to WGS from 231 patients with bile duct cancer (n = 25), breast cancer (n = 54), CRC (n = 27), duodenal cancer (n = 1), gastric cancer (n = 27), lung cancer (n = 35), ovarian cancer (n = 28) and pancreatic cancer (n = 34) and healthy individuals (n = 246) [28].
HCC-WGBS dataset
The dataset consisted of plasma cfDNA samples subjected to WGS from 34 patients with HCC and 25 controls without HCC. Raw sequencing data were downloaded it from European Bioinformatics Institute (Accession code EGAS00001003409) [32].
CRC-TGBS dataset
The dataset consisted of plasma cfDNA samples subjected to TGBS from 801 patients with CRC and 1021 healthy controls. Raw sequencing data were downloaded from Sequence Read Archive database (Accession code PRJNA574555) [8].
HCC-TGBS dataset
The dataset consisted of plasma cfDNA samples subjected to TGBS from 1171 patients with HCC and 959 healthy controls. Raw sequencing data were downloaded from Sequence Read Archive (Accession code PRJNA360288) [47].
Data preprocessing
The training dataset consists 1 390 072 data points from 2094 cfDNA samples subjected to WGS, WGBS, targeted bisulfite sequencing. Each training data point includes three attributes: instruction prompt, input content and expected output. The instruction prompt is a brief description providing context for desired task and guiding the model’s response. Instruction prompt for the ith training data point is denoted by  , and specified as ‘Annotate the following sequence’ in this study. The input content is the concatenation of raw sequencing reads. It is denoted as
, and specified as ‘Annotate the following sequence’ in this study. The input content is the concatenation of raw sequencing reads. It is denoted as  , where n is the number of reads. The expected output, denoted as
, where n is the number of reads. The expected output, denoted as  , is a brief description of the clinical phenotype for the donor whose cfDNA was subjected to sequencing. We concatenated these three attributes as a data point, and denoted as
, is a brief description of the clinical phenotype for the donor whose cfDNA was subjected to sequencing. We concatenated these three attributes as a data point, and denoted as  = {
= { ,
,  ,
, 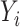 }. The input text is tokenized according to dictionary used by ACID that is consisted of 49 152 tokens.
}. The input text is tokenized according to dictionary used by ACID that is consisted of 49 152 tokens.
The architecture of affordable cancer interception and diagnostics
Embedding layer
The input tokens are transformed into a matrix of real-values using an embedding layer. This matrix contains information about both the token embedding (denoted as Wx) and position encoding (denoted as Wp). The token embedding is obtained by mapping the indices of tokens from the token dictionary to a real-value space using an embedding layer. On the other hand, the position encoding carries information about the sorted tokens. In the case of an input sequence 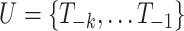 , where k represents the width of the context window, the embedding layer combines the position encoding with token embedding, and denoted as:
, where k represents the width of the context window, the embedding layer combines the position encoding with token embedding, and denoted as:
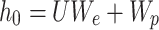 |
Transformer decoder blocks
A multi-layered structure of transformer decoder blocks [48] is employed. These blocks utilize multi-headed masked self-attention to process the input embeddings, followed by position-wise feed-forward layers for further transformation. The outputs from these layers are then passed through a softmax layer.
 |
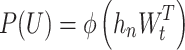 |
where  represent the transformer decoder block and
represent the transformer decoder block and 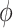 represent the softmax layer, and
represent the softmax layer, and  represent the embedding matrix of the
represent the embedding matrix of the 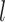 th decoder block.
th decoder block.
Masked self-attention
It is a specialized form of self-attention mechanism [49] that utilizes scale dot-product attention. This attention mechanism involves mapping a query and a set of key-value pairs to generate an output. The input consists of a query and key of dimension  , along with a value of dimension
, along with a value of dimension 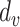 . In the calculation of self-attention, the dot products of the query (
. In the calculation of self-attention, the dot products of the query ( ) with the key (
) with the key (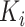 ) are divided by
) are divided by 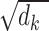 [18]. then multiplied by the value (
[18]. then multiplied by the value ( ) after undergoing softmax transformation [50].
) after undergoing softmax transformation [50].
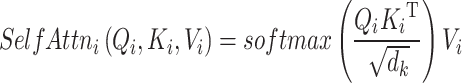 |
The multi-head self-attention is formulated as:
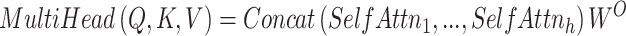 |
where 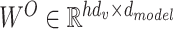 denotes the learned output projection matrix.
denotes the learned output projection matrix.
Position-wise feed forward neural (FFN) network
Position-wise FFN is a layer with fully connected feed-forward layer. The layer consists of two linear transformations with a ReLU activation function in between, which is defined as:
 |
where W1 and W2 are weight matrices and 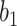 and
and 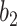 are the bias.
are the bias.
Unsupervised pretraining of ACID
Given a corpus of tokens 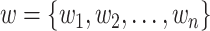 , we use the standard language modeling objective function to maximize this likelihood: [37]
, we use the standard language modeling objective function to maximize this likelihood: [37]
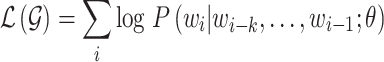 |
Here, 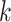 is the width of context,
is the width of context,  are the parameters of ACID that is used to model the conditional probability.
are the parameters of ACID that is used to model the conditional probability.
Development of ACID
We trained the models for 30 epochs with an initial learning rate of 5e – 5, batch size of 16 and weight decay of 0.01. The learning rate was decayed according to cosine scheduling with warmup for 1 epoch. We used AdamW with parameters β1 = 0.9 and β2 = 0.999 as the optimizer. ACID was trained with PyTorch (version 1.13.1) and transformers (version 4.21.1) on NVIDIA DGX A100 with 8 GPUs each with 40 Gb memory.
Benchmarked methods
Benchmarked methods include DELFI [28], MDS [32] and NMF [30].
DNA evaluation of fragments for early interception (DELFI)
We used bwa (version 0.7.17) to map sequencing reads against human reference genome (version hg38). The alignment BAM files were sorted, deduplicated and subsequently merged by samtools (version 0.1.19). DELFI relies on copy number variations to perform cancer detection. We followed pipeline curated at https://github.com/Cancer-Genomics/delfi_scripts.
Motif diversity score of cfDNA end-motifs (MDS)
We obtained 4-kmer end-motif frequencies (e.g. with 256 motifs in total) from sequencing reads and calculated MDS according to the following formula defined by Jiang and collogues [32]:
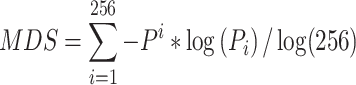 |
where Pi is the frequency of the ith motif.
NMF-based decomposition of cfDNA end-motif profiles (NMF)
We constructed a 4-kmer end-motif matrix from sequencing reads. We denoted this matrix as A whose rows are samples and columns are 256 end-motifs. We factorized A into two non-negative matrices W and H according to:
 |
W is the fragmentation signatures and H is the fragmentation profiles according to Zhou and collegues [30]. We performed NMF with NMF R package (version 0.21.0).
Construction of cfDNA sequence signatures
On the TGBS-HCC dataset, we randomly selected 100 000 sequencing reads for each sample and constructed position frequency matrices (PFM). Subsequently, we converted the PFM into PWM. PWM represents the probabilities of each nucleotide at each position. Let A denoted the PWM whose rows are nucleotides and columns are probabilities for each nucleotide along the sequencing reads. We factorized A into two non-negative matrices W and H (i.e. A ≈ WH). W can be considered the cfDNA sequence signatures whereas H is the cfDNA sequence profiles [30]. We performed NMF with NMF R package (version 0.21.0).
Identification of high scoring reads
We used attention-based MIL with multiple branches to detect high scoring reads. High scoring reads are reads that exhibit strong association with decision made by ACID. This approach is an extension of attention-based deep MIL by Lu and colleagues [51]. For each data point (i.e. concatenation of reads), it is split into multiple tokens. The features of tokens are extracted by ACID–base model. Let  denote feature representation vector of a given data point split into k tokens. A fully connected layer
denote feature representation vector of a given data point split into k tokens. A fully connected layer  projects
projects 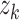 into a 512-dimensional vector
into a 512-dimensional vector 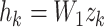 . Suppose the attention network consists of two layers
. Suppose the attention network consists of two layers 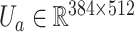 and
and 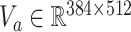 , subsequently the attention network splits into N parallel attention branches
, subsequently the attention network splits into N parallel attention branches 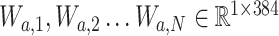 . Then N parallel classifiers (i.e.
. Then N parallel classifiers (i.e.  ) are built to make class-specific prediction for each read. The attention score of the kth token for the ith class
) are built to make class-specific prediction for each read. The attention score of the kth token for the ith class  is calculated as:
is calculated as:
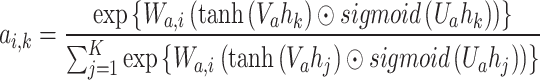 |
The aggregated representation for a read for the ith class is given by:
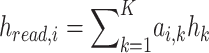 |
The logit value for a read is calculated via:
 |
For each data point, this network is trained to discriminate between cancer and control. The attention vector  is used to identify tokens that exhibits strong association with the prediction labels. A read is considered to be high scoring read if it contains a token ranked first on the top according to
is used to identify tokens that exhibits strong association with the prediction labels. A read is considered to be high scoring read if it contains a token ranked first on the top according to 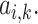 We trained this model for 30 epochs with a learning rate of 2e – 5, batch size of 1. We used AdamW as the optimizer. The model was trained with PyTorch (version 1.13.1) and transformers (version 4.21.1) on NVIDIA DGX A100 with 1 GPUs with 40 Gb memory.
We trained this model for 30 epochs with a learning rate of 2e – 5, batch size of 1. We used AdamW as the optimizer. The model was trained with PyTorch (version 1.13.1) and transformers (version 4.21.1) on NVIDIA DGX A100 with 1 GPUs with 40 Gb memory.
Construction of cfDNA sequence logos
DNA sequence is represented as a PWM, a J × W matrix. In this matrix, the entry at position (j, w) denotes the probability of observing nucleotide j at position w. The information content (IC) profile that quantifies the IC at position w of the sequence is defined as [52]:
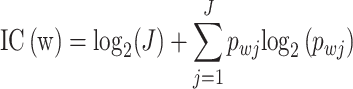 |
IC is measured in bits (ranges from 0 to 2) and reflects substitution tolerance. At a position with equal nucleotide probabilities (0 bits), there is no preference. A position with a single nucleotide (2 bits) indicates maximum specificity. High IC signals low substitution tolerance, marking highly conserved positions, whereas low IC suggests a high tolerance for substitutions.
Statistical analysis
The experiment was conducted using Python (version 3.7.10), R (version 4.2.1), ggplot2 (version 3.3.6) and PROC (version 1.18.0). The area under the receiver operating characteristic curve (AUROC) was calculated using PROC, and the 95% CIs were determined using DeLong method implemented in pROC. Accuracy, sensitivity, and specificity were computed using the caret software package in R (version 6.0.78), with the 95% CIs for these metrics determined using the Clopper-Pearson method [53]. To account for multiple hypothesis testing, P-values were adjusted using the Benjamini-Hochberg procedure when appropriate. Unless otherwise stated, two-sided tests were employed.
Key Points
Cancer detection based on cell-free DNA can be formulated as an end-to-end natural language understanding task.
ACID significantly outperforms the best benchmarked method in diagnosis of cancer and detection of hepatocellular carcinoma.
ACID is insensitive to sequencing depth in that it achieves high prediction accuracy from merely 10 000 number of reads.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful for researchers for their generosity to made their data publicly available.
Author Biographies
Hongru Shen is a PhD student at Tianjin Medical University. She is currently engaged in cancer diagnosis through cell-free DNA analysis using deep learning algorithms.
Jilei Liu is a PhD student at Tianjin Medical University. He is currently working on analyzing cell-free DNA fragmentomics.
Kexin Chen is a Professor at Tianjin Medical University Cancer Institute and Hospital. She is an expert on epidemiology and experienced in data mining.
Xiangchun Li is a Professor at Tianjin Medical University Cancer Institute and Hospital. He has extensive experience in deciphering human cancer genomes via bioinformatics and deep learning.
Contributor Information
Hongru Shen, Tianjin Cancer Institute, Tianjin’s Clinical Research Center for Cancer, National Clinical Research Center for Cancer, Tianjin Medical University Cancer Institute and Hospital, Tianjin Medical University, Tianjin, China.
Jilei Liu, Tianjin Cancer Institute, Tianjin’s Clinical Research Center for Cancer, National Clinical Research Center for Cancer, Tianjin Medical University Cancer Institute and Hospital, Tianjin Medical University, Tianjin, China.
Kexin Chen, Department of Epidemiology and Biostatistics, Key Laboratory of Molecular Cancer Epidemiology of Tianjin, Tianjin’s Clinical Research Center for Cancer, National Clinical Research Center for Cancer, Tianjin Medical University Cancer Institute and Hospital, Tianjin Medical University, Tianjin, China.
Xiangchun Li, Tianjin Cancer Institute, Tianjin’s Clinical Research Center for Cancer, National Clinical Research Center for Cancer, Tianjin Medical University Cancer Institute and Hospital, Tianjin Medical University, Tianjin, China.
AUTHOR CONTRIBUTIONS
Xiangchun Li and Kexin Chen designed and supervised the study; Xiangchun Li and Hongru Shen performed data analysis and wrote the manuscript; Xiangchun Li, Hongru Shen and Jilei Liu developed the model; Xiangchun Li, Hongru Shen and Jilei Liu collected data. Hongru Shen, Xiangchun Li and Kexin Chen revised the manuscript.
FUNDING
This work was supported by the National Key Research and Development Program of China (Grant No. 2021YFC2500400 to K.C.), National Natural Science Foundation of China (Grant Nos. 32270688 and 31801117 to X.L., 31900471 to M.Y.), Program for Changjiang Scholars and Innovative Research Team in University in China (Grant No. IRT_14R40 to K.C.) and Tianjin Key Medical Discipline (Specialty) Construction Project (TJYXZDXK-009A).
DATA AVAILABILITY
Data is publicly available at European Bioinformatics Institute (No.: EGAS00001003409) and Sequence Read Archive (No.: PRJNA574555 and PRJNA360288).
CODE AVAILABILITY
Code will be available at https://github.com/deeplearningplus/ACID.
DECLARATION OF INTEREST
The authors declare that they have no conflict of interest.
References
- 1. Chan KC, Zhang J, Chan AT, et al. Molecular characterization of circulating EBV DNA in the plasma of nasopharyngeal carcinoma and lymphoma patients. Cancer Res 2003;63:2028–32. [PubMed] [Google Scholar]
- 2. Chan KC, Zhang J, Hui AB, et al. Size distributions of maternal and fetal DNA in maternal plasma. Clin Chem 2004;50:88–92. [DOI] [PubMed] [Google Scholar]
- 3. Schwarzenbach H, Hoon DS, Pantel K. Cell-free nucleic acids as biomarkers in cancer patients. Nat Rev Cancer 2011;11:426–37. [DOI] [PubMed] [Google Scholar]
- 4. Zeng C, Stroup EK, Zhang Z, et al. Towards precision medicine: advances in 5-hydroxymethylcytosine cancer biomarker discovery in liquid biopsy. Cancer Commun (Lond) 2019;39:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Diaz LA, Jr, Bardelli A. Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol 2014;32:579–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wan JCM, Massie C, Garcia-Corbacho J, et al. Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat Rev Cancer 2017;17:223–38. [DOI] [PubMed] [Google Scholar]
- 7. Lui YY, Chik K-W, Chiu RW, et al. Predominant hematopoietic origin of cell-free DNA in plasma and serum after sex-mismatched bone marrow transplantation. Clin Chem 2002;48:421–7. [PubMed] [Google Scholar]
- 8. Luo H, Zhao Q, Wei W, et al. Circulating tumor DNA methylation profiles enable early diagnosis, prognosis prediction, and screening for colorectal cancer. Sci Transl Med 2020;12:eaax7533. [DOI] [PubMed] [Google Scholar]
- 9. Chen L, Abou-Alfa GK, Zheng B, et al. Genome-scale profiling of circulating cell-free DNA signatures for early detection of hepatocellular carcinoma in cirrhotic patients. Cell Res 2021;31:589–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Heitzer E, Ulz P, Geigl JB. Circulating tumor DNA as a liquid biopsy for cancer. Clin Chem 2015;61:112–23. [DOI] [PubMed] [Google Scholar]
- 11. Francis G, Stein S. Circulating cell-free tumour DNA in the Management of Cancer. Int J Mol Sci 2015;16:14122–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sozzi G, Conte D, Leon M, et al. Quantification of free circulating DNA as a diagnostic marker in lung cancer. J Clin Oncol 2003;21:3902–8. [DOI] [PubMed] [Google Scholar]
- 13. Sozzi G, Conte D, Mariani L, et al. Analysis of circulating tumor DNA in plasma at diagnosis and during follow-up of lung cancer Patients1. Cancer Res 2001;61:4675–8. [PubMed] [Google Scholar]
- 14. Madhavan D, Wallwiener M, Bents K, et al. Plasma DNA integrity as a biomarker for primary and metastatic breast cancer and potential marker for early diagnosis. Breast Cancer Res Treat 2014;146:163–74. [DOI] [PubMed] [Google Scholar]
- 15. Frattini M, Gallino G, Signoroni S, et al. Quantitative and qualitative characterization of plasma DNA identifies primary and recurrent colorectal cancer. Cancer Lett 2008;263:170–81. [DOI] [PubMed] [Google Scholar]
- 16. Klein EA, Richards D, Cohn A, et al. Clinical validation of a targeted methylation-based multi-cancer early detection test using an independent validation set. Ann Oncol 2021;32:1167–77. [DOI] [PubMed] [Google Scholar]
- 17. Jamshidi A, Liu MC, Klein EA, et al. (2022). Evaluation of cell-free DNA approaches for multi-cancer early detection. Cancer Cell 40, e1512, 1537, 1549.e12. [DOI] [PubMed] [Google Scholar]
- 18. Liu MC, Oxnard GR, Klein EA, et al. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann Oncol 2020;31:745–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ivanov M, Baranova A, Butler T, et al. Non-random fragmentation patterns in circulating cell-free DNA reflect epigenetic regulation. BMC Genomics 2015;16(Suppl 13):S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Snyder MW, Kircher M, Hill AJ, et al. Cell-free DNA comprises an in vivo nucleosome footprint that informs its tissues-of-origin. Cell 2016;164:57–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lo YM, Chan KC, Sun H, et al. Maternal plasma DNA sequencing reveals the genome-wide genetic and mutational profile of the fetus. Sci Transl Med 2010;2:61ra91. [DOI] [PubMed] [Google Scholar]
- 22. Mouliere F, Chandrananda D, Piskorz AM, et al. Enhanced detection of circulating tumor DNA by fragment size analysis. Sci Transl Med 2018;10:eaat4921. 10.1126/scitranslmed.aat4921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ulz P, Thallinger GG, Auer M, et al. Inferring expressed genes by whole-genome sequencing of plasma DNA. Nat Genet 2016;48:1273–8. [DOI] [PubMed] [Google Scholar]
- 24. Jiang P, Sun K, Tong YK, et al. Preferred end coordinates and somatic variants as signatures of circulating tumor DNA associated with hepatocellular carcinoma. Proc Natl Acad Sci USA 2018;115:E10925–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Leary RJ, Kinde I, Diehl F, et al. Development of personalized tumor biomarkers using massively parallel sequencing. Sci Transl Med 2010;2:20ra14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Leary RJ, Sausen M, Kinde I, et al. Detection of chromosomal alterations in the circulation of cancer patients with whole-genome sequencing. Sci Transl Med 2012;4:162ra154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jiang P, Chan CW, Chan KA, et al. Lengthening and shortening of plasma DNA in hepatocellular carcinoma patients. Proc Natl Acad Sci 2015;112:E1317–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cristiano S, Leal A, Phallen J, et al. Genome-wide cell-free DNA fragmentation in patients with cancer. Nature 2019;570:385–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bae M, Kim G, Lee TR, et al. Integrative modeling of tumor genomes and epigenomes for enhanced cancer diagnosis by cell-free DNA. Nat Commun 2023;14:2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhou Z, Ma ML, Chan RWY, et al. Fragmentation landscape of cell-free DNA revealed by deconvolutional analysis of end motifs. Proc Natl Acad Sci USA 2023;120:e2220982120. 10.1073/pnas.2220982120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chan RWY, Serpas L, Ni M, et al. Plasma DNA profile associated with DNASE1L3 gene mutations: clinical observations, relationships to nuclease substrate preference, and in vivo correction. Am J Hum Genet 2020;107:882–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jiang P, Sun K, Peng W, et al. Plasma DNA end-motif profiling as a fragmentomic marker in cancer, pregnancy, and transplantation. Cancer Discov 2020;10:664–73. [DOI] [PubMed] [Google Scholar]
- 33. Brown T, Mann B, Ryder N, et al. Language models are few-shot learners. Adv Neural Inform Processing Syst 2020;33:1877–901. [Google Scholar]
- 34. Chowdhery A, Narang S, Devlin J, et al. Palm: scaling language modeling with pathways. Journal of Machine Learning Research 2023;24:1–113. [Google Scholar]
- 35. Touvron H, Lavril T, Izacard G, et al. Llama: open and efficient foundation language models. arXiv preprint arXiv:230213971 2023.
- 36. Devlin J, Chang M-W, Lee K, Toutanova K. Bert: pre-training of deep bidirectional transformers for language understanding. arXiv preprint arXiv:181004805 2018.
- 37. Radford A, Narasimhan K, Salimans T, Sutskever I. Improving language understanding by generative pre-training, 2018.
- 38. Liu Y, Ott M, Goyal N, et al. Roberta: a robustly optimized bert pretraining approach. arXiv preprint arXiv:190711692 2019.
- 39. Yang Z, Dai Z, Yang Y, et al. Xlnet: generalized autoregressive pretraining for language understanding. Adv Neural Inform Processing Syst 2019;32:5753–5763. [Google Scholar]
- 40. Raffel C, Shazeer N, Roberts A, et al. Exploring the limits of transfer learning with a unified text-to-text transformer. J Mach Learn Res 2020;21:5485–551. [Google Scholar]
- 41. Clark K, Luong M-T, Le QV, Manning CD. Electra: pre-training text encoders as discriminators rather than generators. arXiv preprint arXiv:200310555 2020.
- 42. Rives A, Meier J, Sercu T, et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc Natl Acad Sci 2021;118:e2016239118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Madani A, McCann B, Naik N, et al. Progen: language modeling for protein generation. arXiv preprint arXiv:200403497 2020.
- 44. Shen H, Liu J, Hu J, et al. Generative pretraining from large-scale transcriptomes for single-cell deciphering. Iscience 2023;26:106536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kaplan J, McCandlish S, Henighan T, et al. Scaling laws for neural language models. arXiv preprint arXiv:200108361 2020.
- 46. Chan KC, Jiang P, Sun K, et al. Second generation noninvasive fetal genome analysis reveals de novo mutations, single-base parental inheritance, and preferred DNA ends. Proc Natl Acad Sci USA 2016;113:E8159–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Xu RH, Wei W, Krawczyk M, et al. Circulating tumour DNA methylation markers for diagnosis and prognosis of hepatocellular carcinoma. Nat Mater 2017;16:1155–61. [DOI] [PubMed] [Google Scholar]
- 48. Liu PJ, Saleh M, Pot E, et al. Generating wikipedia by summarizing long sequences. arXiv preprint arXiv:180110198 2018.
- 49. Vaswani A, Shazeer N, Parmar N, et al. Attention is all you need. Adv Neural Inform Processing Syst 2017;30:5998–6008. [Google Scholar]
- 50. Cheng J, Dong L, Lapata M. Long short-term memory-networks for machine reading. arXiv preprint arXiv:160106733 2016.
- 51. Lu MY, Williamson DF, Chen TY, et al. Data-efficient and weakly supervised computational pathology on whole-slide images. Nat Biomed Eng 2021;5:555–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schneider TD. Information content of individual genetic sequences. J Theor Biol 1997;189:427–41. [DOI] [PubMed] [Google Scholar]
- 53. Clopper CJ, Pearson ES. The use of confidence or fiducial limits illustrated in the case of the binomial. Biometrika 1934;26:404–13. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data is publicly available at European Bioinformatics Institute (No.: EGAS00001003409) and Sequence Read Archive (No.: PRJNA574555 and PRJNA360288).