

Abstract
A liquid chromatography tandem mass spectrometry method was developed for quantifying ten cannabinoids in oral fluid (OF). This method utilizes OF collected by the Quantisal™ device and concurrently quantifies cannabinol (CBN), cannabidiol (CBD), Δ9-tetrahydrocannabinol (THC), 11-hydroxy-Δ9-THC (11-OH-THC), 11-nor-9-carboxy-Δ9-THC (THC-COOH), 11-nor-9-carboxy-Δ9-THC glucuronide (THC-COOH-gluc), Δ9-THC glucuronide (THC-gluc), cannabigerol (CBG), tetrahydrocannabiverin (THCV), and Δ9-tetrahydrocannabinolic acid A (THCA-A). Solid phase extraction was optimized using Oasis Prime HLB 30 mg 96-well plates. Cannabinoids were separated by liquid chromatography over a BEH C18 column and detected by a Waters TQ-S micro tandem mass spectrometer. The lower limits of quantification (LLOQ) were 0.4 ng/mL for CBN, CBD, THC, 11-OH-THC, THC-gluc, and THCV; and 1.0 ng/mL for THC-COOH, THC-COOH-gluc, CBG and THCA-A. Linear ranges extended to 2000 ng/mL for THC and 200 ng/mL for all other analytes. Inter-day analytical bias and imprecision at three levels of quality control (QC) was within ±15%. Mean extraction efficiencies ranged from 26.0–98.8%. Applicability of this method was tested using samples collected from individuals randomly assigned to smoke either a joint containing <0.1%, 5.9%, or 13.4% THC content. This method was able to identify and calculate the concentration of 6 of 10 cannabinoids validated in this method.
Keywords: Cannabinoid, THC, Oral fluid, Tandem mass spectrometry
1. Introduction
The majority of the US population lives in a State where it is legal to consume marijuana for medical or recreational purposes [1]. With increasing prevelance of marijuana use, there are concerns about the potential for marijuana to impair driving performance [2]. Epidemiological findings based on motor vehicle crash reports have been inconclusive with regards to the extent that marijuana consumption increases an individual’s risk (odds ratio (OR)) of crashing [3]. Many of the drivers included in the studies were often impaired through a combination of marijuana and other drugs, such as alcohol, making it harder to tease out the effects of THC alone on crash risk [4]. Depending on the inclusion/exclusion criteria, the OR of crashing ranged from 2 to 14. Several studies have concluded, after adjusting for numerous factors (age, other drugs, etc.), that the OR for increased crash risk following use of marijuana was only moderately increased with an OR of 1.2–1.4 [5,6].
The main psychoactive component of marijuana is Δ9-tetrahydrocannabinol (THC). Driving under the influence (DUI) of marijuana is known to impair tracking ability, attention, reaction time, hand-eye coordination, and perception of time and distance in a dose dependent manner [7,8]. Time to peak impairment after smoking marijuana is variable, but is thought to be around 1 h post-smoking and appears to be dependent on multiple factors including frequency of use and smoking technique [4].
Unlike blood alcohol, currently there are no blood concentrations of THC (or metabolites) that society recognizes as causing impairment. The lack of an established marker of marijuana impairment makes it difficult to craft objective legislation for road safety. Most states with per se driving laws can be separated into zero tolerance, very low tolerance (< 2 ng/mL THC), or low tolerance (< 5 ng/mL THC) limits in whole blood. The difficulty in using a zero tolerance approach is that some cannabinoids are present in chronic users blood for > 30 days after abstaining [9]. Another difficulty with using blood specimens to prosecute DUI suspects is the delay between the time of a traffic stop and blood draw. Generally it takes about 1.5 h after a suspect is pulled over to obtain a blood sample. In this timeframe THC concentrations may decrease by as much as 90% [10]. These limitations with whole blood specimens make the use of alternative matrices like oral fluid attractive.
Oral fluid has clear advantages over whole blood because collection is less-invasive and can be performed at the roadside immediately after an individual is determined impaired. Currently, no significant association between oral fluid and whole blood cannabinoid concentrations exist after smoking [11,12]. Oral fluid has demonstrated a temporal association with cannabis intake suggesting it would make a better matrix for assessing recent intake compared with whole blood or urine [13].
Methods exist for extraction of cannabinoids from oral fluid using the Quantisal device [14,15]. However, the goal of this manuscript is to combine previous methods into a single extraction procedure without a hydrolysis step that would allow for the quantification of the following compounds by one method: THC, cannabidiol (CBD), cannabinol (CBN), cannabigerol (CBG), Δ9-tetrahydrocannabinolic acid (THCA-A), tetrahydrocannabiverin (THCV), 11-hydroxy-Δ9-THC (11-OH-THC), 11-nor-9-carboxy-Δ9-THC (THC-COOH), 11-nor-9-carboxy-Δ9-THC-glucuronide (THC-COOH-gluc), and Δ9-THC-glucuronide (THC-gluc). This assay will be useful for OF cannabinoid analysis in the establishment of a cannabinoid concentration associated with driving impairment.
2. Material and methods
2.1. Chemicals, materials, and sample collection
Stock solutions containing 1 mg/mL or 100 μg/mL of CBN, CBD, THC, 11-OH-THC, THC-COOH, THC-COOH-gluc, CBG, THCV, THCA-A, and the internal standards cannabidiol-D3, (−)-Δ9-THC-D3, cannabinol-D3, ( ± )-11-hydroxy-Δ9-THC-D3, and ( ± )-11-nor-9-carboxy-Δ9-THC-D3 were purchased from Cerilliant (Round Rock, TX, USA). THC-gluc (10 μg/mL) was purchased from ElSohly Laboratories (Oxford, MS, USA).
Oasis prime HLB (30 mg) 96-well extraction plates were purchased from Waters (Milford, MA, USA). Mass spectrometry grade methanol (MeOH), acetonitrile (ACN), and formic acid were purchased from Fisher Scientific (Hampton, NH, USA). Blank synthetic OF matrix used to prepare calibrators and quality control specimens was purchased from Immunalysis (Pomona, CA, USA). OF was collected with the Quantisal™ device also from Immunalysis. Participants refrained from food or drink for 10 min, then the absorptive cellulose pad was placed under their tongue until the indicator turned blue or 5 min had passed. The collection pad was then placed into the plastic collection device containing 3 mL of extraction/stabilization buffer. The extraction buffer is supplied with the Quantisal™ device. The capped tube was placed at room temperature for at least 4 h but not > 24 h. The pad was then removed from the stem using fisherbrand standard serum filters (Fisher Scientific) and decanted into nunc 3 mL cryovials from Wheaton (Millville, NJ, USA). The samples were then stored at 4 °C. Each sample was weighed in attempt to derive a short sample correction factor before being analyzed within 2 months of collection [16].
2.2. Preparation of standard solutions
Methanol calibrator solutions were prepared using class A glass volumetric pipettes and glass volumetric flasks. A 100 μg/mL stock was prepared from 1 mg/mL stock solutions of 11-OH-THC, THC, CBD, CBN, CBG, THC-V and THCA-A. A 10 μg/mL stock was prepared by adding 1 mL of the 100 μg/mL stock plus 1 mL of each 100 μg/mL solutions of THC-COOH, THC-COOH-glucuronide and quantum satis (q.s.) to 10 mL with MeOH. A 1000 ng/mL stock was prepared from the 10 μg/mL stock plus 1 mL of the 10 μg/mL THC-glucuronide. The 100 ng/mL stock was made from the 1000 ng/mL stock. Parallel dilutions were made from the 100 ng/mL stock to make up the remaining 50, 20, 5 and 2 ng/mL stock solution. All stocks were aliquoted and stored at −20 °C in amber glass bottles with a teflon lined screw caps. When spiked into OF the calibrators correspond to seven levels of standard (0.4, 1, 4, 10, 20, 200, and 2000 ng/mL) in each batch of oral fluid samples for THC and six levels of standard (0.4, 1, 4, 10, 20, and 200 ng/mL) for every other analyte.
A 1000 ng/mL stock solution of deuterium labeled internal standards (IS) was made from CBD-D3, (−)-Δ9-THC-D3, CBN-D3, ( ± )-11-hydroxy-Δ9-THC-D3, and ( ± )-11-nor-9-carboxy-Δ9-THC-D3 stock solutions in methanol. A 100 ng/mL working IS solution was prepared from the 1000 ng/mL stock solution in methanol. The working IS solution was aliquotted into 20 mL amber glass screw top vials with teflon lined screw caps. The internal standard solutions were sealed with parafilm between each use and stored at −20 °C.
2.3. Quality control materials
The blank matrix for calibrators and QCs (“synthetic OF”) is a mixture of 1 part BSA (0.1% in PBS) with 3 parts extraction buffer. Positive QC standard solutions of 300, 60, and 12 ng/mL were prepared by parallel dilutions from a 1000 ng/mL stock in methanol made the same as described for the calibrator solution except using stock solutions from a different lot than the calibrators. Each solution was aliquoted into amber glass auto sampler vials, sealed with parafilm and stored at −20 °C. The solutions correspond to three levels of QC at 60, 12, and 2.4 ng/mL when processed in synthetic OF. The lower level of QC was chosen to reflect a low concentration that was closer to per se cut off values adapted by several states [17]. QC results were reviewed according to an absolute criteria of ± 20% of target values.
2.4. Solid-phase extraction (SPE)
All calibrators and QCs were prepared by adding 50 μL of calibrator, 50 μL of working IS, followed by one mL of blank matrix to corresponding borosilicate tubes. Subject specimens were treated in a similar manner except methanol was substituted for the calibrator. Samples were then acidified with 400 μL of 4% phosphoric acid. Samples were vortexed briefly then contents were transferred to a well of a 96 well Oasis Prime HLB C18 SPE plate. Samples were forced through the wells using a positive pressure manifold on low pressure until all liquid was pushed through. This took approximately five minutes to drip through. Each well was washed with 500 μL of SPE wash buffer (25% MeOH with 5% ammonium hydroxide) twice under low pressure. The pressure was switched to max flow for one minute following the second wash to push any excess liquid through the well. Compounds of interest were eluted into 750 μL glass inserts (Waters Corporation) with three successive 100 μL aliquots of 98% ACN with 2% formic acid for a total of 300 μL eluant. Extracts were evaporated under nitrogen at 40 °C with gas flow set to 70 psi for 30 min. Dried extracts were reconstituted with 200 μL of 50% ACN containing 0.1% formic acid. Plates were covered with a silicone/PTFE treated, pre-slit cap mat and vortexed using the Fisher Scientific Ana Multi-tube vortexer (Cat #: 02215450) on speed setting of 4 for 5 min. Plates were centrifuged at 1962 x g for 10 min in Sorvall legend XFR centrifuge (Thermo) and then transferred to the sample organizer for LC-MS/MS analysis.
2.5. Liquid chromatography
Chromatography was performed using a Waters Aquity i-class UPLC system equipped with sample organizer, binary solvent manager, autosampler, and a column oven (Waters Corporation). Separation of analytes was achieved using a Waters 2.1 × 50 mm Acquity UPLC BEH C18 column packed with 1.7 μm sized particles. The analytical column was attached to a 2.1 mm × 5 mm ACQUITY UPLC BEH C18 VanGuard Pre-column packed with 1.7 μm particle size to prevent column degradation due to sample buildup. Guard columns were replaced after every 1000 injections. The autosampler was set to 10 °C. The column heater was set to 40 °C. A full-loop 10 μL injection was made for each sample. Gradient elution was performed using a mobile phase A (MPA) of 5 mM ammonium formate buffer with 0.1% formic acid and a mobile phase B (MPB) of acetonitrile with 0.1% formic acid at a constant flow rate of 400 μL/min. The initial gradient conditions were 50% MPB, held for 30 s, and then linearly increased to 90% MPB over 3.5 min. The final MPB concentration was maintained for 15 s, before returning to initial conditions and holding for 45 s. The maximum pressure was set to 15,000 psi.
2.6. Mass spectrometry
The LC system was coupled to a Waters TQ-S-micro triple quadrupole mass spectrometer interfaced with an electrospray ionization (ESI) probe. Negative ionization was used for THC-COOH-gluc. All other compounds used positive ionization. The mass spectrometry transition ions were collected using a scheduled multiple reaction monitoring mode (MRM) with four separate time windows. The first time window (TW) was collected in negative ion mode from 1.00 to 1.50 min. The subsequent windows were collected in positive ion mode from 1.51 to 2.59 min for TW-2, 2.60 to 3.22 min for TW-3, and 3.23 to 4.20 min for TW-4. The selected precursor and product ions, collision energy, retention times and associated windows are displayed in Table 1. The source temperature was set to 550 °C for both modes. The instrument was controlled with Masslynx V4.1 SCN945 SCN960 software (Waters Inc.) and peaks were processed using TargetLynxs XS. A representative reconstructed chromatogram of all quantifier ions from a 20 ng/mL calibrator is displayed in Fig. 1.
Table 1.
MRM parameters for cannabinoids in oral fluid.
| Compound name | Precursor ion (m/z) | Product ion (m/z) | DT (s) | CE (V) | RT (min) | TW |
|---|---|---|---|---|---|---|
|
| ||||||
| THC-COOH-gluc | 519.1 | 192.9 | 0.110 | −18 | 1.20 | 1 |
| 519.1 | 299.2 | 0.110 | −38 | 1.20 | 1 | |
| 519.1 | 343.2 | 0.110 | −22 | 1.20 | 1 | |
| THC-gluc | 491.2 | 193.1 | 0.040 | 34 | 1.71 | 2 |
| 491.2 | 315.3 | 0.040 | 20 | 1.71 | 2 | |
| 11-OH-THC | 331.1 | 193.1 | 0.040 | 22 | 2.22 | 2 |
| 331.1 | 201.1 | 0.040 | 24 | 2.22 | 2 | |
| THC-COOH | 345.1 | 193.1 | 0.040 | 26 | 2.29 | 2 |
| 345.1 | 327.2 | 0.040 | 14 | 2.29 | 2 | |
| CBG | 317.1 | 94.9 | 0.040 | 36 | 2.85 | 3 |
| 317.1 | 122.9 | 0.040 | 34 | 2.85 | 3 | |
| 317.1 | 193.1 | 0.040 | 16 | 2.85 | 3 | |
| CBD | 315.1 | 122.9 | 0.040 | 32 | 2.88 | 3 |
| 315.1 | 193.1 | 0.040 | 20 | 2.88 | 3 | |
| THC-V | 287.1 | 122.9 | 0.040 | 32 | 2.88 | 3 |
| 287.1 | 165.1 | 0.040 | 24 | 2.88 | 3 | |
| CBN | 311.1 | 195.1 | 0.035 | 24 | 3.35 | 4 |
| 311.1 | 223.1 | 0.035 | 16 | 3.35 | 4 | |
| THC | 315.1 | 135.1 | 0.035 | 20 | 3.60 | 4 |
| 315.1 | 193.1 | 0.035 | 18 | 3.60 | 4 | |
| THCA-A | 359.2 | 77.0 | 0.035 | 70 | 3.91 | 4 |
| 359.2 | 219.0 | 0.035 | 32 | 3.91 | 4 | |
| 359.2 | 341.2 | 0.035 | 14 | 3.91 | 4 | |
| THC-COOH-D3 | 348.1 | 302.1 | 0.040 | 20 | 2.28 | 2 |
| 11-OH-THC-D3 | 334.3 | 196.3 | 0.040 | 22 | 2.19 | 2 |
| CBD-D3 | 318.2 | 196.1 | 0.040 | 18 | 2.87 | 3 |
| CBN-D3 | 314.2 | 223.1 | 0.035 | 20 | 3.33 | 4 |
| THC-D3 | 318.2 | 196.1 | 0.035 | 18 | 3.60 | 4 |
Boldface denotes quantifier ion.
DT dwell time, CE collision energy, RT retention time, TW time window.
Fig. 1.

Representative chromatogram for 20 ng/mL OF calibrator. Numbers correspond to the following compounds (1) THCCOOH-gluc, (2) THC-gluc, (3) 11-OH-THC-d3, (4) 11-OH-THC, (5) THCCOOH-d3, (6) THCCOOH, (7) CBG, (8) CBD-d3, (9) CBD, (10) THC-V, (11) CBN-D3, (12) CBN, (13) THC-D3, (14) THC, and (15) THCA-A. The y-axis was set to 1 ×106 counts per second for all compounds.
2.7. Method validation
Method validation was performed according to Clinical & Laboratory Standards Institute (CLSI) 62-A guidance on liquid chromatography-mass spectrometry methods and included establishing linearity, within-run and between-run precision, trueness and bias, limits of detection, extraction recovery, interferences, and matrix effects.
2.7.1. Sensitivity, limits of detection, and quantification
The LLOQ was established as the lowest concentration that exhibited acceptable trueness (< 20% bias) and precision (CV < 20%), n = 6. It also must exhibit a signal-to-noise (S/N) ratio of at least 10 with a quantifier to qualifier ion ratio within 20% of mean calibrator ratios and have a visibly acceptable peak shape. Analyte peak identification criteria included relative retention time 1.02 ± 0.02 and ion ratio (± 20% of calibrators). Compounds without deuterated IS had to have matching retention times to that of calibrators within ± 0.1 min.
2.7.2. Linearity, trueness and precision
The calibration curve to establish linear fit included six calibrators (0.4, 1, 4, 10, 20, and 200 ng/mL) for all analytes and a seventh calibrator for THC that extends the analytical measuring interval to 2000 ng/mL. Each batch also included a double blank (standard containing matrix only) and a zero (blank matrix with IS). Linearity was investigated by calculating the regression using the least-squares with a weighting factor of 1/x applied for each analyte. Linearity was established with five sets of calibrators, which were required to quantify within ± 15%, except at the LLOQ, which was required to quantify within ± 20% of target concentration with precision (% CV) of < 20%. Correlation coefficients (R2) were required to exceed 0.995.
2.7.3. Intra-day and inter-day imprecision and bias
The intra-day bias and imprecision were evaluated with 8 replicates at three levels of QC concentrations on the same day. The bias was determined by comparing the mean measured concentration of each analyte with that of the target value and was expressed as a percentage of the target concentration. Acceptable values were within 20% of the target concentration for the bias and within 20% for the CV. QC values for each analyte were reassigned based on the mean of the intra-day concentrations.
The inter-day imprecision and bias were evaluated with 2 replicates at three levels of QC over 10 days (n = 20 for each level of QC). The mean values for each level of QC were acceptable if the bias was within 20% of the intra-day concentration and imprecision was < 20%.
2.7.4. Selectivity, interferences, extraction recovery, and matrix effects
Extraction efficiencies and matrix effects were determined for each analyte at the lowest level of QC according to the three sample set method proposed by Matuszewski et al. (n = 5 for each analyte set) [18]. In set A, synthetic blank matrix was fortified with the low QC and IS prior to SPE. In set B, synthetic blank matrix was fortified with low QC and IS after SPE. In set C, elution solvent was fortified with low QC and IS. Percent Extraction efficiency was calculated by dividing analyte mean peak areas of set A by set B and multiplied by 100. The matrix effect was calculated by dividing the analyte mean peak areas of set B by set C and multiplied by 100 to express as a percentage.
To demonstrate that the synthetic OF was a valid substitute for OF specimens from humans, OF from 10 drug free (by self report) volunteers was collected and processed such that a one mL aliquot was fortified with low QC and IS, while another one mL aliquot was processed unaltered (to demonstrated absence of cannabinoids). The percent bias was calculated by dividing the difference between the averaged concentration of the low QC in human OF samples from the average concentration of low QC in blank matrix by the averaged concentration of low QC in blank matrix. A qualitative assessment of matrix interference was also performed by injecting each of the unspiked OF samples while simultaneously infusing a calibrator solution containing 10 ng/mL of each analyte [16,19]. See Fig. 2 for total ion chromatograms of the blank OF samples.
Fig. 2.

Representative chromatograms for the quantifier transitions for THC-COOH-gluc, THC-gluc, THC-COOH, CBG, THC, and THCA-A in authentic drug free oral sample (left column) and the same sample spiked with the low QC (right column). Middle column demonstrates the chromatography at the LLOQ for each compound displayed in synthetic oral fluid. Values in graph correspond to maximal peak height of each compound.
Potential drug interferences were assessed by generating 5 pools of 10 different drugs belonging to opiates, benzodiazepines, and other common drugs of abuse that could be present in a suspected DUI subject (Supplemental Table 1). Superphysiological concentrations of the pools of drugs were added to blank OF samples fortified with low QC. Recovery of the QC within ± 20% of expected concentration in the presence of superphysiological concentrations of drug pools was required to demonstrate lack of interference.
2.7.5. Stability studies and carryover
Autosampler stability was assessed by comparing the average area counts from the low QC to the area counts of an injection at 24 and 48 h post-extraction. Acceptable stability was set at ± 20% CV in area of a 1.25 ng/mL stock compared to the initial injection. Lack of carryover was established by injecting a blank matrix fortified with IS immediately after the highest calibrator and then comparing the area counts to the same blank matrix with IS injected prior to the calibration curve. The acceptable level of carryover was a set to < 20% increase in area counts of the blank matrix following reinjection after the highest calibrator.
2.8. Determination of cannabinoids in authentic oral fluid samples collected from participants in the California Assembly Bill (AB) 266 study
Proof of applicability is demonstrated by evaluating the concentrations of cannabinoids in three participants enrolled in an Institutional Review Board-approved study evaluating the effects of inhaled cannabis containing either placebo (0.02%), 5.9% or 13.4% THC by weight. Inclusion criteria for participation were individuals had to be at a minimum an occasional user (≥3× per week), abstain from marijuana use 48 h prior to testing, and have a valid drivers license. Oral fluid samples were collected upon arrival to the laboratory which was tested to demonstrate THC < 5 ng/mL using the Alere OF point of care instrument. Individuals whose OF screened negative on the Alere device then smoked a joint containing either placebo, 5.9%, or 13.4% THC. Oral fluid was then collected 15, 90, 210, and 280 min after smoking and processed as described above. The complete study design and detailed methods will be published after the target enrollment of 180 subjects is complete.
2.8.1. Dilution verification
Dilution integrity was evaluated by fortifying blank matrix (n = 3) to a final concentration of 4000 ng/mL and then diluting 1:10 (v/v) with blank matrix. Samples were then processed as described. Dilution integrity was maintained if specimens quantified within ± 20% of the expected diluted concentration.
3. Results
3.1.1. Sensitivity and linearity
The LLOQ, defined as the concentration with a CV < 20% for the bias and precision values, was 0.4 ng/mL for CBN, CBD, THC, 11-OH-THC, THC-gluc, and THC-V (Table 2). The LLOQ was 1 ng/mL for THC-COOH, THC-COOH-gluc, CBG, and THCA-A. The linear range was demonstrated for each analyte from the LLOQ to 200 ng/mL (CV < 20%), except for THC where the upper limit was extended to 2000 ng/mL (Table 2). The R2 values with the 1/x weighting were acceptable for all cannabinoids (R2 > 0.995). Fig. 2 shows the chromatography at the LLOQ isolated from synthetic oral fluid and the low QC in native oral fluid.
Table 2.
Linearity for each cannabinoid in the method. Calibrators percent accuracy range and average correlation coefficient (R2) were established during the Inter-day validation (n = 10).
| LLOQ (ng/mL) | ULOQ (ng/mL) | Calibrators % accuracy range | Average R2 | |
|---|---|---|---|---|
|
| ||||
| THC-COOH-gluc | 1.0 | 200 | 80–102 | 0.9990 |
| THC-gluc | 0.4 | 200 | 96–103 | 0.9977 |
| 11-OH-THC | 0.4 | 200 | 93–112 | 0.9995 |
| THC-COOH | 1.0 | 200 | 96–103 | 0.9996 |
| CBG | 1.0 | 200 | 92–114 | 0.9986 |
| CBD | 0.4 | 200 | 93–110 | 0.9996 |
| THC-V | 0.4 | 200 | 89–122 | 0.9993 |
| CBN | 0.4 | 200 | 90–107 | 0.9994 |
| THC | 0.4 | 2000 | 85–116 | 0.9988 |
| THCA-A | 1.0 | 200 | 95–113 | 0.9982 |
3.1.2. Intra-day and inter-day precision and bias
The intra-day biases ranged from 85 to 103% with an imprecision (CV) ≤7% for the low, middle, and high levels of QC (n = 8). The inter-day biases ranged from 95 to 114% with an imprecision ≤15% for all analytes in the low, middle, and high levels of QC (n = 20 over 10 days) (Table 3).
Table 3.
Intra- and Inter-day bias and precision for each level of QC.
| Analyte | Intra-day bias (%) |
Intra-day precision (%CV) |
Inter-day bias (%) |
Inter-day precision (% CV) |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Low | Mid | High | Low | Mid | High | Low | Mid | High | Low | Mid | High | |
|
| ||||||||||||
| THC-COOH-gluc | 7 | 2 | 8 | 7 | 7 | 5 | −14 | 2 | −3 | 15 | 8 | 11 |
| THC-gluc | 2 | 0 | 1 | 2 | 2 | 2 | −9 | −2 | 0 | 5 | 5 | 7 |
| 11-OH-THC | 4 | 1 | 3 | 4 | 4 | 2 | 0 | 0 | −5 | 5 | 5 | 6 |
| THC-COOH | 5 | 1 | 2 | 3 | 3 | 4 | −3 | 0 | −1 | 3 | 5 | 4 |
| CBG | 15 | 11 | 10 | 4 | 4 | 4 | 0 | 0 | −2 | 8 | 7 | 9 |
| CBD | 6 | 1 | 5 | 2 | 3 | 5 | 1 | 1 | −4 | 7 | 4 | 6 |
| THC-V | 5 | −3 | 2 | 3 | 2 | 5 | 5 | 4 | −1 | 6 | 5 | 8 |
| CBN | 7 | 2 | 5 | 4 | 4 | 4 | −3 | −1 | −5 | 6 | 7 | 6 |
| THC | 6 | 1 | 3 | 3 | 2 | 3 | 2 | 3 | −1 | 6 | 4 | 6 |
| THCA-A | 14 | 14 | 7 | 6 | 6 | 5 | −10 | 0 | 1 | 11 | 11 | 10 |
3.1.3. Matrix effects, extraction recovery, and interferences
A qualitative matrix effect study was performed by infusing a 10 ng/mL calibrator solution during an injection of an extracted oral fluid specimen from drug-free volunteers (n = 10). There was no observable ion suppression or enhancement across any of the analytes peaks in the human oral fluid specimens (Fig. 3). Quantitative assessment of matrix effects was performed by fortifying the human drug-free oral fluid specimens with low QC and calculating the percent recovery to expected values established in the inter-day validation (Table 4). Acceptable critera were set as a percent difference < 20% from expected. No matrix effect exceeding this criteria was observed in the human oral fluid specimens when compared with the synthetic oral fluid used for calibrators and controls.
Fig. 3.
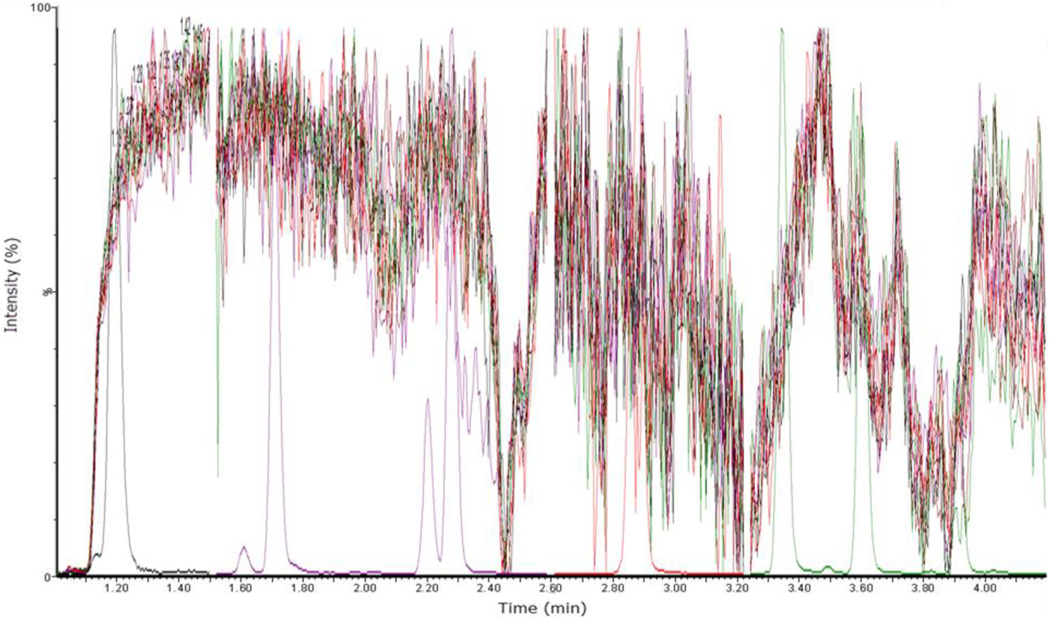
Qualitative matrix effect assessment of ten individually injected blank OF samples infused with a 10 ng/mL calibrator. All ten samples are overlaid on this total ion chromatogram along with a calibrator to more easily identify expected peak retention times for each compound. Identical to Fig. 1 the peaks correspond to the following compounds retention time (minutes) THCCOOH-gluc (1.20), THC-gluc (1.71), 11-OH-THC (2.22), THCCOOH (2.29), CBG (2.85), CBD (2.88), THC-V (2.88), CBN (3.35), THC (3.60), and THCA-A (3.91).
Table 4.
Quantitative matrix interferences between real OF and synthetic OF.
| % Bias | |
|---|---|
|
| |
| THC-COOH-gluc | 17 |
| THC-gluc | 4 |
| 11-OH-THC | 4 |
| THC-COOH | 9 |
| CBG | 10 |
| CBD | 0 |
| THC-V | 4 |
| CBN | 0 |
| THC | 9 |
| THCA-A | 20 |
Extraction efficiency was determined by comparing average peak areas of extracted blank matrix samples fortified with low, mid, or high QC divided by peak areas of blank matrix samples fortified post-extraction with QC. All analytes had less than a 9% difference in extraction efficiency between any level of QC with a range of efficiencies from 26.0–98.8% (Table 5). Percent matrix bias were determined by comparing average peak areas of blank matrix samples fortified post-extraction with low, mid, or high QC divided by neat solutions of QC. The range of percent matrix bias was −37.6–23.7%. THCA-A observed the worst ion suppression followed by THC-V (Table 5). THC-COOH observed the greatest ion enhancement due to matrix effects observed to be > 20% in the mid QC, whereas all other analytes had percent differences < 20%. The THC-COOH internal standard compensated for the matrix enhancement providing results within ± 20% of target values.
Table 5.
Mean extraction efficiency and percent matrix bias for cannabinoids extracted from blank matrix by SPE.
| % Extraction efficiency |
% Matrix bias |
|||||
|---|---|---|---|---|---|---|
| Low | Mid | High | Low | Mid | High | |
|
| ||||||
| THC-COOH-gluc | 37.5 | 36.6 | 37.6 | −4.1 | 5.9 | 3.6 |
| THC-gluc | 83.2 | 84.6 | 82.0 | 1.8 | 9.2 | 14.1 |
| 11-OH-THC-D3 | 87.2 | 87.8 | 84.1 | 14.5 | 19.9 | 16.9 |
| 11-OH-THC | 82.0 | 84.6 | 81.2 | 14.3 | 19.9 | 16.4 |
| THC-COOH-D3 | 64.7 | 63.5 | 62.9 | 17.4 | 23.7 | 19.8 |
| THC-COOH | 61.7 | 60.4 | 58.3 | 14.8 | 22.7 | 20.3 |
| CBG | 50.2 | 57.9 | 53.3 | 13.2 | 17.5 | 15.3 |
| CBD-D3 | 87.3 | 94.3 | 90.5 | −10.6 | −8.8 | −4.7 |
| CBD | 79.3 | 87.4 | 86.6 | −11.4 | −9.7 | −4.4 |
| THC-V | 90.2 | 97.0 | 98.8 | −32.6 | −27.9 | −18.2 |
| CBN-D3 | 55.2 | 61.4 | 56.3 | 7.3 | 12.2 | 12.4 |
| CBN | 51.1 | 58.4 | 55.5 | 5.4 | 12.3 | 12.2 |
| THC-D3 | 63.8 | 71.5 | 68.2 | −11.7 | −6.6 | 1.0 |
| THC | 60.2 | 67.2 | 63.1 | −12.2 | −5.2 | −2.6 |
| THCA-A | 27.4 | 26.0 | 27.4 | −37.6 | −21.2 | −21.7 |
Interferences were assessed from five pools of ten drugs (Supplemental Table 1) in blank OF fortified with low QC. The percent bias for all cannabinoids ranged from −17.4–12.7% (Table 6). Thus, no drugs that were tested caused any interference in calculating the low QC concentration.
Table 6.
Drug interferences assessed by % Bias from interday assigned low QC values.
| Blanka % bias | Drug pool 1% bias | Drug pool 2% bias | Drug pool 3% bias | Drug pool 4% bias | Drug pool 5% bias | |
|---|---|---|---|---|---|---|
|
| ||||||
| THC-COOH-gluc | −5.4 | −8.3 | −4.2 | 0.0 | 4.2 | −12.5 |
| THC-gluc | −6.3 | 0.0 | 8.3 | 4.2 | 4.2 | 4.2 |
| 11-OH-THC | −4.8 | 0.0 | −4.5 | 0.0 | 9.1 | 0.0 |
| THC-COOH | 2.1 | −16.7 | −8.3 | −4.2 | −4.2 | −8.3 |
| CBG | 12.7 | −4.3 | −17.4 | −13.0 | 0.0 | −8.7 |
| CBD | 3.6 | −13.0 | 0.0 | 0.0 | 4.3 | −8.7 |
| THC-V | 6.0 | −8.7 | −4.3 | −4.3 | 0.0 | −4.3 |
| CBN | 0.0 | −8.7 | −8.7 | −4.3 | 0.0 | 4.3 |
| THC | −0.5 | −4.5 | 0.0 | −4.5 | 4.5 | −4.5 |
| THCA-A | 1.7 | −13.0 | −8.7 | −17.4 | −4.3 | 0.0 |
Denotes low QC fortified with methanol (vehicle).
3.1.4. Autosampler stability, dilution integrity, and carry-over
The areas for all the compounds were within ± 20% upon reinjection at 24 h in the autosampler. The 48 h injection of samples had a percent difference within 20% for CBN, CBD, THC, 11-OH-THC, THCCOOH, and CBG, but a percent difference < 28% for THC-gluc, THCCOOH-gluc, THC-V, and THCA-A. In the 48 h reinjections, the internal standards compensated for changes in area counts so quantitative results were within 20% of the initial values.
Dilution integrity was acceptable within ± 20% of target concentrations for THC after diluting 1:10 with blank matrix. THC quantified within 3.1% of expected concentrations.
There was no evidence of carry-over for any of the cannabinoids following injection of a sample containing 2000 ng/mL.
3.1.5. Analysis of authentic oral fluid samples
Quantification of THC and related metabolites is part of a research project that aims to establish the concentration of cannabinoids associated with driving impairment following consumption of a low does (5.9% THC), high dose (13.4% THC), or placebo (0.02% THC). Participants have their OF samples collected prior to and immediately after smoking one of the randomly assigned joints. The study is a double-blinded approach, thus the laboratory is blinded to which participants have smoked which kind of joint until the conclusion of the study. To demonstrate proof of applicability, the laboratory was unblinded to identify the first three participants in this study that smoked either the placebo, low or high dose joint.
The purpose of including data from three subjects who smoked marijuana is to demonstrate the analytical method is capable of measuring these compounds in specimens obtained from human volunteers, and not to draw any conclusions regarding pharmacokinetics or determining severity of impairment. One participant from each group had their oral fluid samples assessed by this method with concentrations of each cannabinoid listed in Table 7. The placebo joint participant had detectable levels of CBN and THC following consumption. The low dose and high dose particpants had detectable levels of CBN, THC, CBG, THC-V, and THCA-A following consumption of the joint. CBD was detected in the low dose participant immediately after smoking. THC was also detected in the placebo participant prior to smoking the joint. THCCOOH-gluc, THC-gluc, 11-OH-THC, and THCCOOH were not detected in any of the oral fluid samples tested.
Table 7.
Concentrations of cannabinoids in the oral fluid of the first participant enrolled in each of the smoking groups (placebo (0.02%), low (5.9%), and high (13.4%) THC joint content).
| Compound | THC Content (%) | Baseline | 15 min | 90 min | 210 min | 280 min |
|---|---|---|---|---|---|---|
|
| ||||||
| CBN | < 0.1% | < LLOQ | 22.9 | 1.4 | < LLOQ | < LLOQ |
| 5.90% | < LLOQ | 87.7 | 4.2 | 0.7 | 0.6 | |
| 13.40% | < LLOQ | 15.4 | 0.5 | < LLOQ | < LLOQ | |
| CBD | < 0.1% | < LLOQ | < LLOQ | < LLOQ | < LLOQ | < LLOQ |
| 5.90% | < LLOQ | 0.5 | < LLOQ | < LLOQ | < LLOQ | |
| 13.40% | < LLOQ | < LLOQ | < LLOQ | < LLOQ | < LLOQ | |
| THC | < 0.1% | 0.7 | 2.6 | < LLOQ | 0.4 | < LLOQ |
| 5.90% | < LLOQ | 142.4 | 9.7 | 0.5 | 0.6 | |
| 13.40% | < LLOQ | 96.6 | 4.5 | 0.4 | 0.5 | |
| 11-OH-THC | < 0.1% | < LLOQ | < LLOQ | < LLOQ | < LLOQ | < LLOQ |
| 5.90% | < LLOQ | < LLOQ | < LLOQ | < LLOQ | < LLOQ | |
| 13.40% | < LLOQ | < LLOQ | < LLOQ | < LLOQ | < LLOQ | |
| THC-COOH | < 0.1% | < LLOQ | < LLOQ | < LLOQ | < LLOQ | < LLOQ |
| 5.90% | < LLOQ | < LLOQ | < LLOQ | < LLOQ | < LLOQ | |
| 13.40% | < LLOQ | < LLOQ | < LLOQ | < LLOQ | < LLOQ | |
| THC-Gluc | < 0.1% | < LLOQ | < LLOQ | < LLOQ | < LLOQ | < LLOQ |
| 5.90% | < LLOQ | < LLOQ | < LLOQ | < LLOQ | < LLOQ | |
| 13.40% | < LLOQ | < LLOQ | < LLOQ | < LLOQ | < LLOQ | |
| THC-COOH-Gluc | < 0.1% | < LLOQ | < LLOQ | < LLOQ | < LLOQ | < LLOQ |
| 5.90% | < LLOQ | < LLOQ | < LLOQ | < LLOQ | < LLOQ | |
| 13.40% | < LLOQ | < LLOQ | < LLOQ | < LLOQ | < LLOQ | |
| CBG | < 0.1% | < LLOQ | < LLOQ | < LLOQ | < LLOQ | < LLOQ |
| 5.90% | < LLOQ | 7.6 | < LLOQ | < LLOQ | < LLOQ | |
| 13.40% | < LLOQ | 2.4 | < LLOQ | < LLOQ | < LLOQ | |
| THC-V | < 0.1% | < LLOQ | < LLOQ | < LLOQ | < LLOQ | < LLOQ |
| 5.90% | < LLOQ | 1.2 | < LLOQ | < LLOQ | < LLOQ | |
| 13.40% | < LLOQ | 0.8 | < LLOQ | < LLOQ | < LLOQ | |
| THCA-A | < 0.1% | < LLOQ | < LLOQ | < LLOQ | < LLOQ | < LLOQ |
| 5.90% | < LLOQ | 56.5 | 1.3 | < LLOQ | < LLOQ | |
| 13.40% | < LLOQ | 30.3 | 5 | < LLOQ | < LLOQ | |
4. Discussion
This work expands upon the prior knowledge of working with OF collected in the Quantisal devices and LC-MS/MS methods [14,15] to simultaneously quantify ten cannabinoids. The simplistic design of this method was intentional to demonstrate feasibility in future use in driving under the influence of cannabis testing. Various parameters were optimized during method development. We evaluated multiple SPE bed volumes (Waters Oasis Prime HLB μElution, 10 mg, or 30 mg 96-well plates), with multiple combinations of washing and elution conditions (alkalinized, neutral, or acidified solvents, data not shown). Additional LC columns evaluated included XSelect HSS C18 2.5 μm beads 2.1 mm × 150 mm, XSelect HSS T3 2.5 μm beads 2.1 mm × 75 mm, and HSS PFP 1.8 μm beads 2.1 mm × 50 mm using either acidified methanol or acetonitrile based mobile phase buffers. Electrospray ionization in positive and negative ion mode was completed on each analyte. Ultimately, each parameter described in the method was selected based on largest peak area with highest signal to noise, while providing sufficient chromatographic separation.
All analytes in this method had an inter-day analytical bias ± 15% with an imprecision ≤15% CV. Extraction efficiencies and matrix effects were similar to previous studies [14,15]. The deuterated internal standards accounted for any extraction or matrix effects allowing for the quantification of analytes within ± 20%. Similar to Desrosiers et al. [14], CBD-d3 was selected as the internal standard for THC-V and CBG, since at the time of validation no deuterated internal standards were available for these two compounds. A quantifier to qualifier ion ratio flag was observed at 0.4 ng/mL for CBG resulting in an elevated LLOQ for CBG to 1 ng/mL.
THC-d3 was employed as the deuterated internal standard for THCA-A due to the closeness in retention time. THCA-A had the lowest extraction efficiency and largest matrix effect of all the cannabinoids tested. This is likely due to the adhesiveness of this molecule to glass and plastics used throughout the procedure.
We did not detect 11-OH-THC, THCCOOH, THC-gluc or THCCOOH-gluc in the first three participants of each group using this method. However it does seem unlikely that glucuronidated molecules will be present above those lower limits in oral fluid, since concentrations of THC-gluc in blood following controlled cannabis smoking were <1.1 ng/mL [20,21]. Negligible amounts of THC-gluc in OF has been suggested by the lack of increased THC concentrations following glucuronidase treatment [22]. The lack of 11-OH-THC and THCCOOH in OF collected using quantisal devices is not unexpected as other published works have measured these analytes in the 0.01–0.35 ng/mL range, which is below our LLOQ in this method [13,14]. Furthermore, THCCOOH has been notoriously difficult to quantify using OF collected from the quantisal device with most methods utilizing 2-dimensional GC–MS or atmospheric pressure chemical ionization LC-MS/MS with a enzymatic hydrolysis to enrich the THCCOOH pool [14,22–25]. The inclusion of THCCOOH in OF was suggested to confirm direct inhalation and help establish a limit to rule out passive exposure [25,26]. However, due to the analytical difficulties of measuring to such a small concentration THCCOOH is likely to be only useful to rule in consumption with a negative result unable to accurately rule out.
We included THC-V, CBG, and THCA-A in this method to incorporate as many available cannabinoid markers as possible since this method will be used to support pharmacokinetic and pharmacodynamics studies of marijuana use. This method differs from previous methods measuring cannabinoids in OF after solid phase extraction such that this method quantifies 10 cannabinoids, whereas others have measured 6–8 in one method [14,15].
5. Conclusions
This LC-MS/MS method expands upon previous methods by quantifying 10 cannabinoids in OF with a LLOQ of 0.4 ng/mL for THC, CBN, 11-OH-THC, CBD, THC-V and THC-glucuronide, and 1.0 ng/mL for THC-COOH, THC-COOH-glucuronide, CBG, and THCA-A. The main advantages of this method include the utilization of a simplified sample preparation procedure and its validation over a clinicaly relevant analytical measurement range of concentrations for each cannabinoid.
Supplementary Material
Acknowledgements
The authors would like to acknowledge the support of Waters corporation for technical expertise (Michael Wakefield and Erik Todd) and for the use of the Waters TQS Micro tandem mass spectrometer used in this project.
Primary support for this study was provided by the State of California via Assembly Bill 266, the Medical Marijuana Regulation and Safety Act, which included a study on impaired driving due to cannabis.
This project was also supported by the Center for Medicinal Cannabis Research (CMCR) at the University of California, San Diego. Additional support for the CMCR is provided by State of California Proposition 64, The Control, Regulate and Tax Adult Use of Marijuana Act, and a philanthropic gift from the Ray and Tye Noorda, and Wholistic Research and Education, Foundations.
The CMCR is directed by Igor Grant, M.D. and Co-Directors Thomas D. Marcotte, Ph.D. and J. Hampton Atkinson, M.D., Department of Psychiatry, UC San Diego.
Abbreviations:
- OF
oral fluid
- OR
odds ratio
- CBN
cannabinol
- CBD
cannabidiol
- THC
Δ9-tetrahydrocannabinol (THC)
- 11-OH-THC
11-hydroxy-Δ9-THC
- THCCOOH
11-nor-9-carboxy-Δ9-THC
- THC-COOH-gluc
11-nor-9-carboxy-Δ9-THC glucuronide
- THC-gluc
Δ9-THC glucuronide
- CBG
cannabigerol
- THCV
tetrahydrocannabiverin
- THCA-A
Δ9-tetrahydrocannabinolic acid A
- ESI
electrospray ionization
- LLOQ
lower limit of quantification
- QC
quality control
- DUI
driving under the influence
- MeOH
methanol
- ACN
acetonitrile
- SPE
solid-phase extraction
- MPA
mobile phase A
- MPB
mobile phase B
- MRM
multiple reaction monitoring
- CV
coefficient of variation
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.cca.2019.01.002.
References
- [1].Klieger SB, Gutman A, Allen L, Pacula RL, Ibrahim JK, Burris S, Mapping medical marijuana: state laws regulating patients, product safety, supply chains and dispensaries, 2017, Addiction 112 (2017) 2206–2216, 10.1111/add.13910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Ramaekers JG, Driving under the influence of cannabis: an increasing public health concern, JAMA 319 (2018) 1433–1434, 10.1001/jama.2018.1334. [DOI] [PubMed] [Google Scholar]
- [3].Compton R, Marijuana-Impaired Driving - A Report to Congress. (DOT HS 812 440), National Highway Traffic Safety Administration, Washington, DC, 2017. [Google Scholar]
- [4].Sewell RA, Poling J, Sofuoglu M, The effect of cannabis compared with alcohol on driving, Am. J. Addict. 18 (2009) 185–193, 10.1080/10550490902786934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Rogeberg O, Elvik R, Response: cannabis intoxication, recent use and road traffic crash risks, Addiction 111 (2016) 1495–1498, 10.1111/add.13443. [DOI] [PubMed] [Google Scholar]
- [6].Gjerde H, Mørland J, Risk for involvement in road traffic crash during acute cannabis intoxication, Addiction 111 (2016) 1488–1499, 10.1111/add.13435. [DOI] [PubMed] [Google Scholar]
- [7].Kelly E, Darke S, Ross J, A review of drug use and driving: epidemiology, impairment, risk factors and risk perceptions, Drug Alcohol Rev. 23 (2004) 319–344, 10.1080/09595230412331289482. [DOI] [PubMed] [Google Scholar]
- [8].Ramaekers JG, Berghaus G, Van Laar M, Drummer OH, Dose related risk of motor vehicle crashes after cannabis use, Drug Alcohol Depend. 73 (2004) 109–119, 10.1016/j.drugalcdep.2003.10.008. [DOI] [PubMed] [Google Scholar]
- [9].Bergamaschi MM, Karschner EL, Goodwin RS, Scheidweiler KB, Hirvonen J, Queiroz RHC, Huestis MA, Impact of prolonged cannabinoid excretion in chronic daily cannabis smokers’ blood on per se drugged driving laws, Clin. Chem. 59 (2013) 519–526, 10.1373/clinchem.2012.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Hartman RL, Brown TL, Milavetz G, Spurgin A, Gorelick DA, Gaffney GR, Huestis MA, Effect of blood collection time on measured δ9-Tetrahydrocannabinol concentrations: implications for driving interpretation and drug policy, Clin. Chem. 62 (2016) 367–377, 10.1373/clinchem.2015.248492. [DOI] [PubMed] [Google Scholar]
- [11].Langel K, Gjerde H, Favretto D, Lillsunde P, Øiestad EL, Ferrara SD, Verstraete AG, Comparison of drug concentrations between whole blood and oral fluid, Drug Test. Anal. 6 (2014) 461–471, 10.1002/dta.1532. [DOI] [PubMed] [Google Scholar]
- [12].Vandrey R, Herrmann ES, Mitchell JM, Bigelow GE, Flegel R, LoDico C, Cone EJ, Pharmacokinetic profile of oral cannabis in humans: blood and oral fluid disposition and relation to pharmacodynamic outcomes, J. Anal. Toxicol. 41 (2017) 83–99, 10.1093/jat/bkx012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Lee D, Huestis MA, Current knowledge on cannabinoids in oral fluid, Drug Test. Anal. 6 (2014) 88–111, 10.1002/dta.1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Desrosiers NA, Scheidweiler KB, Huestis MA, Quantification of six cannabinoids and metabolites in oral fluid by liquid chromatography-tandem mass spectrometry, Drug Test. Anal. 7 (2015) 684–694, 10.1002/dta.1753. [DOI] [PubMed] [Google Scholar]
- [15].Fabritius M, Staub C, Mangin P, Giroud C, Analysis of cannabinoids in oral fluid by liquid chromatography-tandem mass spectrometry, Forensic Toxicol. (2013) 151–163, 10.1007/s11419-012-0168-z. [DOI] [Google Scholar]
- [16].Scheidweiler KB, Andersson M, Swortwood MJ, Sempio C, Huestis MA, Long-term stability of cannabinoids in oral fluid after controlled cannabis administration, Drug Test. Anal. 9 (2017) 143–147, 10.1002/dta.2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Doucette ML, Frattaroli S, Vernick JS, Oral fluid testing for marijuana intoxication: enhancing objectivity for roadside DUI testing, Inj. Prev. (2017), 10.1136/injuryprev-2016-042264. [DOI] [PubMed]
- [18].Matuszewski BK, Constanzer ML, Chavez-Eng CM, Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS, Anal. Chem. 75 (2003) 3019–3030, 10.1021/ac020361s. [DOI] [PubMed] [Google Scholar]
- [19].Côté C, Bergeron A, Mess J-N, Furtado M, Garofolo F, Matrix effect elimination during LC–MS/MS bioanalytical method development, Bioanalysis 1 (2009) 1243–1257, 10.4155/bio.09.117. [DOI] [PubMed] [Google Scholar]
- [20].Desrosiers NA, Himes SK, Scheidweiler KB, Concheiro-Guisan M, Gorelick DA, Huestis MA, Phase i and ii cannabinoid disposition in blood and plasma of occasional and frequent smokers following controlled smoked cannabis, Clin. Chem. 60 (2014) 631–643, 10.1373/clinchem.2013.216507. [DOI] [PubMed] [Google Scholar]
- [21].Schwope DM, Karschner EL, Gorelick DA, Huestis MA, Identification of recent cannabis use: Whole-blood and plasma free and glucuronidated cannabinoid pharmacokinetics following controlled smoked cannabis administration, Clin. Chem. 57 (2011) 1406–1414, 10.1373/clinchem.2011.171777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Moore C, Rana S, Coulter C, Day D, Vincent M, Soares J, Detection of conjugated 11-nor-Delta9-tetrahydrocannabinol-9-carboxylic acid in oral fluid, J. Anal. Toxicol. 31 (2007) 187–194, 10.1093/jat/31.4.187. [DOI] [PubMed] [Google Scholar]
- [23].Day D, Kuntz DJ, Feldman M, Presley L, Detection of THCA in oral fluid by GCMS-MS, J. Anal. Toxicol. 30 (2006) 645–650, 10.1093/jat/30.9.645. [DOI] [PubMed] [Google Scholar]
- [24].Moore C, Coulter C, Rana S, Vincent M, Soares J, Analytical procedure for the determination of the marijuana metabolite 11-nor-Delta9-tetrahydrocannabinol-9-carboxylic acid in oral fluid specimens, J. Anal. Toxicol. 30 (2006) 409–412, 10.1093/jat/30.7.409. [DOI] [PubMed] [Google Scholar]
- [25].Moore C, Coulter C, Uges D, Tuyay J, van der Linde S, van Leeuwen A, Garnier M, Orbita J, Cannabinoids in oral fluid following passive exposure to marijuana smoke, Forensic Sci. Int. 212 (2011) 227–230, 10.1016/j.forsciint.2011.06.019. [DOI] [PubMed] [Google Scholar]
- [26].Milman G, Barnes AJ, Schwope DM, Schwilke EW, Darwin WD, Goodwin RS, Kelly DL, Gorelick DA, Huestis MA, Disposition of cannabinoids in oral fluid after controlled around-the-clock oral THC administration, Clin. Chem. 56 (2010) 1261–1269, 10.1373/clinchem.2009.141853. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.