

Abstract
Sturge–Weber Syndrome (SWS) is a sporadic (non-inherited) syndrome characterized by capillary vascular malformations in the facial skin, leptomeninges, or the choroid. A hallmark feature is the mosaic nature of the phenotype. SWS is caused by a somatic mosaic mutation in the GNAQ gene (p.R183Q), leading to activation of the G protein, Gαq. Decades ago, Rudolf Happle hypothesized SWS as an example of “paradominant inheritance”, that is, a “lethal gene (mutation) surviving by mosaicism”. He predicted that the “presence of the mutation in the zygote will lead to death of the embryo at an early stage of development”. We have created a mouse model for SWS using gene targeting to conditionally express the GNAQ p.R183Q mutation. We have employed two different Cre-drivers to examine the phenotypic effects of expression of this mutation at different levels and stages of development. As predicted by Happle, global, ubiquitous expression of this mutation in the blastocyst stage results in 100% embryonic death. The majority of these developing embryos show vascular defects consistent with the human vascular phenotype. By contrast, global but mosaic expression of the mutation enables a fraction of the embryos to survive, but those that survive to birth and beyond do not exhibit obvious vascular defects. These data validate Happle's paradominant inheritance hypothesis for SWS and suggest the requirement of a tight temporal and developmental window of mutation expression for the generation of the vascular phenotype. Furthermore, these engineered murine alleles provide the template for the development of a mouse model of SWS that acquires the somatic mutation during embryonic development, but permits the embryo to progress to live birth and beyond, so that postnatal phenotypes can also be investigated. These mice could then also be employed in pre-clinical studies of novel therapies.
Keywords: G proteins, mouse models, rare disease
Sturge-Weber Syndrome (SWS) is a sporadic vascular malformation syndrome caused by a mutation in the GNAQ gene (p.R183Q). SWS is hypothesized as “paradominant inheritance,” meaning a lethal gene that survives through mosaicism. Here, Wetzel-Stron, Galeffi et al. conditionally express the mutation in mouse. Global expression results in embryonic death with vascular defects; however, global mosaic expression enables limited survival to birth. These results validate paradominant inheritance, suggesting a tight temporal and developmental window for mutation expression for the vascular phenotype.
Introduction
Sturge–Weber Syndrome (SWS) is a rare vascular malformation syndrome characterized by capillary vascular malformations that may be present in the facial skin, leptomeninges, or the choroid. A characteristic feature of SWS is the mosaic nature of the clinical phenotype. The facial capillary malformation is usually unilateral (Van Trigt et al. 2022), as are the leptomeningeal and choroidal vascular lesions. Together, these vascular malformations cause a severe decrease in the quality of life due to the stigma associated with the facial vascular lesions, early vision loss due to glaucoma when choroidal lesions are present, as well as debilitating headaches, stroke-like ischemic events, and seizures due to the leptomeningeal lesions. To date, the phenotype appears sporadically, and importantly, there are no confirmed cases of inheritance from parent to child.
In 1987, Dr. Rudolf Happle proposed that SWS and other select mosaic phenotypes were due to somatic mutations in essential developmental genes and that germline inheritance of these mutations would be lethal (Happle 1987); he coined the term “paradominant inheritance” to describe the concept of a “lethal mutation surviving because of mosaicism”. In 2013, the causative mutation of SWS was identified as the somatic, mosaic mutation c.548G > A, exchanging arginine 183 for glutamine (p.R183Q) in the gene GNAQ, encoding the G protein subunit alpha q protein (Shirley et al. 2013). Functional analysis has confirmed that this mutation activates the G protein, Gαq (GNAQ) (Shirley et al. 2013; Galeffi et al. 2022). However, the full consequences of the expression of this genetic mutation during embryonic development remain a point of conjecture because no study has addressed this critical question. Thus, the “paradominant inheritance” hypothesis for SWS has yet to be experimentally validated.
Animal models are essential tools for biomedical research and are especially important for understanding disease processes in developmental disorders. Yet generating an animal model that faithfully recapitulates the SWS phenotype has been challenging. The somatic mutation is presumably acquired in a particular progenitor cell, likely of mesodermal origin (Uchiyama et al. 2016), but the precise identity of this progenitor cell remains unknown, thereby hindering animal model development. Two mouse models of SWS have been published to date (Huang et al. 2022; Sasaki et al. 2022). Both rely on the implantation of mutant endothelial cells into the flank of athymic nude (nu/nu) mice, resulting in dilated vessels in the implanted xenograft. Although these models have provided important information about the consequences of these mutations within endothelial cells in vivo, they are unable to provide insight into the role of the p.R183Q GNAQ mutation during in utero development—a hallmark feature of the human syndrome. Thus, these models cannot test Happle's “paradominant inheritance” hypothesis.
In this study, we developed a mouse model that allows for conditional expression of p.R183Q GNAQ from the endogenous mouse Gnaq locus. Through this approach, we tested whether early embryonic expression of this mutant is compatible with life and performed an initial examination of the phenotypes associated with expression of this mutation during embryonic development.
Methods and materials
Mice
Mice with conditional expression of p.R183Q GNAQ were generated through gene targeting techniques by the Duke Transgenic and Knockout Mouse core. The targeting vector (shown in Supplementary Figure 1) contained a cDNA segment encoding exons 4 through 7 of mouse Gnaq flanked by loxP sites, a neomycin resistance cassette flanked by FRT sites, and a cDNA segment encoding exons 4 through 7 of mouse Gnaq with the p.R183Q mutation. The 3′UTR and poly(A) site from Bovine growth hormone 1 (BGH1) was ligated to the 3′-end of exon 7 substituting for the endogenous 3′UTR and poly(A) site to provide a way to distinguish transcripts arising from the conditional allele from transcripts arising from the untouched endogenous Gnaq. Because R183 is encoded in exon 4 of Gnaq, the homology arms were designed in the introns flanking exon 4 to direct replacement of the endogenous Gnaq exon 4 with the targeting construct. The resulting mice were named B6-GnaqRQtmR183Q, referred to as GnaqRQ. The conditional allele will be denoted as GnaqRQfl and the endogenous wild-type (WT) allele as GnaqRQwt. To remove the neo cassette, the GnaqRQ mice were crossed with ACTBFLPe/FLPe mice [JAX stock #003800, (Rodriguez et al. 2000)]. PCR genotyping confirmed removal of the neo cassette. Mice were backcrossed to C57Bl6/J mice for five generations to ensure a homogeneous genetic background for the experiments. GnaqRQfl/wt mice were first crossed to R26-CreERT2 [JAX stock #008463, (Ventura et al. 2007)] to determine the level of recombination on the conditional allele. One hundred µg of Tamoxifen (Sigma, T5648) was injected intra-gastrically to pups on P3, and the animals were euthanized and analyzed at various ages for the presence of mutant and WT transcript. In further experiments, GnaqRQfl/wt mice were crossed to either the β-actin-Cre [JAX stock # 019099, (Lewandoski et al. 1997)] or the E2a-Cre [JAX stock #003724, (Lakso et al. 1996)] lines to drive constitutive, global expression of p.R183Q GNAQ. Timed matings were performed and embryos were collected at different times during embryonic development or postnatal animals were assessed. Mice were genotyped for the presence of the conditional Gnaq allele, for R26-CreERT2, β-actin-Cre, and E2a-Cre by Transnetyx (Cordova, TN). All animal procedures were performed according to protocols approved by the Duke University Institutional Animal Care and Use Committee.
RNA isolation and cDNA synthesis
Mouse tissue was flash-frozen in liquid Nitrogen immediately after dissection, and RNA was extracted using TRIzol after manual tissue disruption following the manufacturer's protocol (Invitrogen, 15596026). RNA concentrations were measured using a Nanodrop (Nanodrop Onec, Thermo Scientific, Waltham, MA, USA). Prior to cDNA synthesis, 1 µg of total RNA was DNase-treated (Invitrogen, 18068015). RNA was then used for cDNA synthesis with M-MLV primed by oligo(dT) following the manufacturer's protocol (Invitrogen, 28025013).
SNaPshot assay
Full-length Gnaq transcript was amplified from cDNA using Platinum Taq DNA Polymerase High Fidelity (Invitrogen, 113040110) with the following primers (Forward: 5′-CAGCTGCGCAGGGACAAG-3′; Reverse: 5′−AGGAAAGGACAGTGGGAGTG-3′) with the forward primer in exon 1 of Gnaq and the reverse primer in the 3′ BGH1 UTR, according to the manufacturer's protocol. The PCR product was separated on a 1% agarose gel and gel-purified using GeneClean Turbo (MP Biomedical, 111102200CF). The SNaPshot single-base extension reaction was performed following the manufacturer's protocol using the following primers: (top strand: 5′AAGACGTGCTTAGAGTTC-3′; bottom strand: 5′CCCTGTAGTGGGGACT-3′). The sequencing data were analyzed on an ABI Prism 3130 Genetic Analyzer sequencer, and peaks were determined using GeneMapper software. Percent recombination was calculated with the following formula: percent mutant allele frequency = ([mutant peak height]/[mutant + WT peak heights]) × 100%.
Quantitative reverse transcription PCR
The mRNA levels of Gnaq (of both the endogenous and conditional alleles) and Angpt2 in GnaqRQfl/wt;β-actin-Cre+ embryos and GnaqRQfl/wt littermate controls were measured by quantitative PCR (qPCR) as previously described (Keum et al. 2013; Lee et al. 2022). Briefly, to determine transcript levels of Gnaq and Angpt2 for each mouse embryo, mRNA was isolated from e14.5 mouse embryo tissue using TRIzol (Invitrogen, 15596026), and cDNA was synthesized as described above. Transcript levels of Gnaq and Angpt2 from individual mouse embryos were quantitated using a dye-based qPCR method (iTaq Univeral SYBR Green Supermix, Bio-Rad, CA). All samples were run in triplicate, and an additional assay for endogenous Gapdh was performed to control for input cDNA template quantity. The primers used for qPCR reactions were as follows: Gnaq, exon 3 (Forward): 5′-TACGACAGACGACGGGAATA-3′, (endogenous and conditional alleles); 3′ UTR (Reverse): 5′-CCCTCTTGTTTATCTTCAAC-3′ (endogenous allele); BGH1 UTR (Reverse): 5′-GGCAAACAACAGATGGCTGG-3′(conditional allele); Angpt2: (Forward): 5′-ACAGCTGTGATGATAGAGATTGG-3′, (Reverse): 5′-GTTGGAGAAGCTGCAGCTCG-3′; Gapdh (Forward): 5′-TCCCACTCTTCCACCTTCGA-3′, (Reverse): 5′-AGTTGGGATAGGGCCTCTCTTG-3′.
Histology
For histological analysis, mouse embryos were removed at e14.5, fixed in 10% buffered formalin overnight, and then processed for paraffin embedding according to standard protocols. Sagittal sections of the entire embryo (6-μm thick) were stained with Hematoxylin and Eosin. Alternating sections were incubated with anti-CD31 (PECAM-1) antibody (Abcam, Cambridge, CB2 0AX UK. Cat #ab28364) as a specific marker for vascular endothelial cells and then incubated with HRP-conjugated multimer antibody reagent. HRP-conjugated multimers (Dics Omnimap anti-Rb HRP RUO, Roche, 1211 Vienna, Austria, Cat #760-4311) were used followed by DAB staining with the avidin-biotin complex peroxidase kit (Vectastain Elite, Newark, CA, 94560) and counterstained with Hematoxylin. Tissue processing for paraffin embedding and staining was performed by the Pathology and Histology Laboratory Research Core Facility at Duke. Images were obtained using a Zeiss microscope and Zen 3.5 Pro software. ImageJ software was used for measuring the subcutaneous vessel diameter.
Results
Generating a mouse model of conditional p.R183Q GNAQ expression
To determine the effect of the p.R183Q GNAQ mutation during development, we generated a conditional mouse model where we can modulate the expression of this mutation. As shown in Fig. 1a, the engineered construct was integrated into the endogenous mouse Gnaq locus to place it under the gene's normal regulation and level of expression. The WT segment of the construct is flanked by loxP sites, which upon Cre recombination is deleted from the locus. This Cre-mediated deletion of the WT genomic sequence from the conditional allele leaves the mutant construct intact, and the resulting transcript will be spliced onto the first three exons of Gnaq from the endogenous gene (floxed allele). Thus, expression of WT or mutant transcript is regulated by Cre recombination. To determine the amount of genomic recombination, we amplified the region surrounding p.183. These primers amplify both the WT and mutant DNA from the conditional allele but not from the endogenous Gnaq allele as the primers are located within exons 4 and 5, which are separated by a large intron in the endogenous locus (Fig. 1a and b). A similar assay was designed so that when cDNA is used as the template, we could determine the ratio of WT to mutant transcript expression. Forward primers from the endogenous Gnaq exons 1 or 3 were paired with reverse primers specific to the novel 3′UTR found only on the conditional allele ensuring that these assays only monitor molecular events within the engineered construct—neither genomic DNA nor cDNA can amplify from the endogenous non-engineered allele (Fig. 1a and c). We then designed a single-base extension assay at the position of the mutation (SNaPshot) to distinguish the extent of recombination in the construct along with the level of WT vs mutant transcript. Using the genomic DNA assay, Fig. 1b shows that the conditional expression system performs as expected; without Cre recombination, both WT and mutant are seen in roughly equal amounts, and with Cre recombination, the mutant transcript level is increased (Fig. 1b). In cDNA from animals with Cre recombination, transcription is only seen from the mutant allele; there is no mutant transcript detectable in the Cre-negative animals (Fig. 1c).
Fig. 1.

Demonstrating conditional p.R183Q GNAQ expression. a) The engineered construct was integrated into the endogenous Gnaq exon 4 as described in Supplementary Figure 1. Cre-mediated recombination deletes the WT genomic sequence from the construct resulting in expression of the mutant Gnaq. b) To measure genomic recombination of the construct, primers 1 and 2 (black triangles) were designed to amplify the DNA surrounding codon 183 from both the WT and the mutant R183Q sections of the engineered construct. The endogenous Gnaq locus cannot amplify under the conditions used since Primers 1 and 2 are located in separate exons; Primer 1 in exon 4 is separated from Primer 2 in exon 5 by almost 3 kb. SNaPshot analysis (a single base extension assay) at the site of the mutation shows equal amounts of both WT (G, left peak) and mutant (A, right peak) in DNA from Cre− animals. In contrast, SNaPshot analysis from Cre+ animals shows that after Cre recombination, the WT section of the construct has been removed from the majority of cells leaving the mutant portion of the construct intact. c) Primers 3 and 4 were designed to measure the levels of WT and mutant transcription from the engineered construct. Primer 4 is located in the 3-UTR poly(A) site unique to the construct ensuring amplification from transcripts arising only from the engineered allele, not from the endogenous Gnaq allele. SNaPshot analysis of cDNA from Cre− animals shows that nearly all of the transcription comes from the WT portion of the construct while Cre+ animals show almost exclusive expression from the mutant portion of the construct.
Although we inserted the floxed engineered construct into the endogenous Gnaq locus, it is formally possible that this additional DNA might influence the gene's expression level. To determine whether the introduction of the construct into the endogenous Gnaq locus alters Gnaq transcript expression, we performed quantitative (q)RT-PCR for Gnaq endogenous and conditional alleles on mouse embryonic tissue from GnaqRQfl/wt;β-actin-Cre+ and GnaqRQfl/wt littermate controls. We found that in both groups, the levels of Gnaq conditional allele transcripts were significantly higher than the endogenous allele (P*** = 0.0002 and P** = 0.007 conditional vs endogenous in GnaqRQfl/wt and GnaqRQfl/wt;β-actin-Cre+, respectively, two-tailed paired t-test) (Supplementary Figure 2) with a greater increase in GnaqRQfl/wt;β-actin-Cre+ (P*** < 0.001 GnaqRQfl/wt;β-actin-Cre+ vs GnaqRQfl/wt, two-tailed unpaired t-test) (Supplementary Figure 2).
Another technical possibility is that due to the nature of the engineered conditional allele, the Gnaq mutant allele might undergo exon skipping. Using the GeneMapper software, we are able to detect allele frequencies of 1% and higher [(Shirley et al. 2013) and unpublished data] and to date, SNaPshot results from Cre-negative animals using any of the Cre drivers do not show the mutant Gnaq allele at measurable levels. Additionally, we have been breeding the GnaqRQ line for more than 10 generations and have not seen any overt phenotypic consequences: the mice are phenotypically normal, fertile, and there is no sex bias at birth or in longevity. Thus, if there is some nominal amount of mutant Gnaq expression, it is not having any demonstrable effect on the mice.
Effect of global but mosaic recombination of Gnaq mutant expression
We first crossed our GnaqRQ strain to a global Cre transgene, E2a-Cre, under the control of the adenovirus E2a promoter. E2a-Cre-mediated recombination occurs early in the mouse embryo in a broad range of tissues, but recombination efficiency varies widely across tissues and animals (Lakso et al. 1996). We reasoned that if Happle was correct, and the mutation would be lethal if acquired in all tissues, then mosaic recombination might permit survival of some of the developing embryos. In this cross, GnaqRQfl/wt×E2a-Cre+/+, we did observe survival to birth of some GnaqRQfl/wt;E2a-Cre+ mice, but survival of this genotype was statistically below expected values (Fig. 2). Genotyping of the offspring revealed that of 93 live-born mice, 15 were GnaqRQfl/wt;E2a-Cre+ vs 78 GnaqRQwt/wt;E2a-Cre+, representing a significant reduction from the expected 50% ratio for the cross (P*** < 0.001; chi-square test).
Fig. 2.
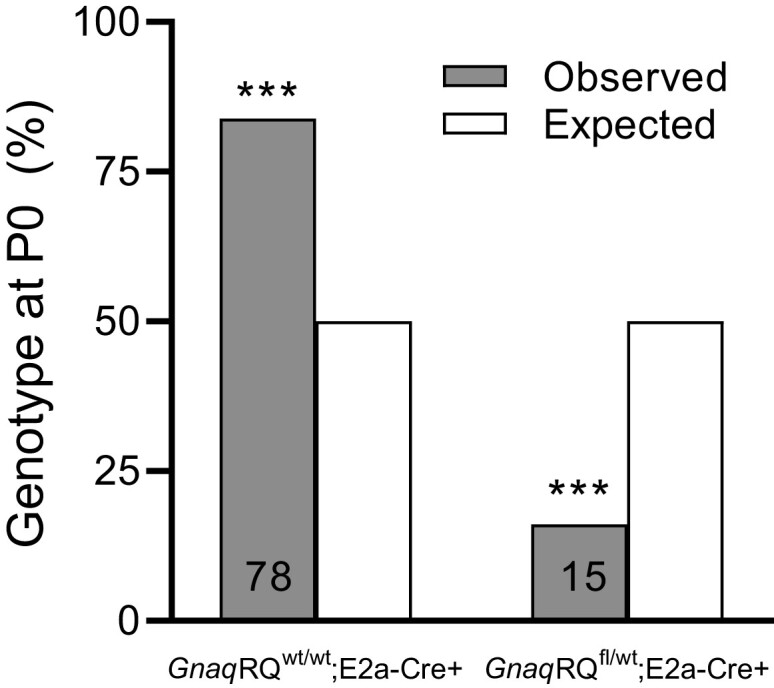
Postnatal characterization of global but mosaic Gnaq expression. GnaqRQfl/wt animals were crossed to homozygous E2a-Cre+/+ animals, and the resulting offspring were genotyped postnatally. The number of animals genotyped is shown on the bars for each category. *** P < 0.001, by chi-square test.
Some of the live-born pups were sacrificed at weaning (P21) for molecular analysis and phenotype assessment, while others were allowed to age up to 1 year for observation of any overt phenotypes. In analysis of RNA from the liver, (the brains were used for other phenotypic analyses), the percentage of the mutant transcript ranged from 5.3 to 51.5% in GnaqRQfl/wt;E2a-Cre+ animals of a range of ages, confirming mosaic Cre-mediated recombination and transcription of the mutant allele (Fig. 3a). As expected with this Cre driver, recombination was incomplete and varied widely, leading to varied ratios of mutant to WT transcript in the survivors.
Fig. 3.

Postnatal phenotypes of GnaqRQ;E2a-Cre mice. a) The mutant allele frequency from liver tissue cDNA was assessed by SNaPshot. The age range of the animals was P21 to P391. b) Representative images of brains from postnatal animals with the leptomeninges intact. The brains from P21 animals are labeled, and the other brains shown are from animals ranging in age from 4 to 6 months. Arrows indicate regions of increased pigmentation in GnaqRQfl/wt;E2a-Cre+ animals.
Because SWS patients can develop capillary malformations involving both the skin and/or brain leptomeninges, we performed a gross examination of the skin and the surface of the brains of GnaqRQfl/wt;E2a-Cre+ and GnaqRQfl/wt littermates. During gross examination of newborn pups and later of 21-day-old animals, we did not observe any obvious skin vascular phenotype or vascular abnormalities on the surface of the brains. However, in 11 out of 15 GnaqRQfl/wt;E2a-Cre+ animals, we observed significantly increased pigmentation on the leptomeninges between the cerebellum and cortex, and between the cortex and the olfactory bulb (Fig. 3b). Although low in numbers, melanocytes are present in the leptomeninges of the brain (Gudjohnsen et al. 2015). These cells are vulnerable to GNAQ mutations, which can potentially induce abnormal proliferation (Urtatiz et al. 2020). Therefore, this phenotype of enhanced melanocytic pigmentation in these particular areas of the brain, observed in the brains of older GnaqRQfl/wt;E2a-Cre+ mice, is consistent with altered GNAQ signaling.
Due to the decreased number of live-born animals of the GnaqRQfl/wt;E2a-Cre+ genotype, we sought to determine if there were any embryonic phenotypes associated with the observed loss of these mice during development. Therefore, we dissected embryos at e13.5–14.5 for gross morphological evaluation and genotyping. At this stage of development, we found normal, Mendelian ratios for all the expected genotypes (differences not significant by chi-square test) (Fig. 4a). However, in GnaqRQfl/wt;E2a-Cre+ embryos, we observed prominent edema and some dilated small vessels in 50% (5 out of 10) and 20% (2 of 10) of the animals, respectively (Fig. 4b). These findings indicate that mosaic expression of the mutant allele results in a severe embryonic phenotype in a significant number of GnaqRQfl/wt;E2a-Cre+ mice that can eventually result in premature death.
Fig. 4.

Characterization of embryonic GnaqRQ × E2a-Cre mice. a) GnaqRQfl/wt animals were crossed to heterozygous E2a-Cre+ animals, and the resulting offspring were dissected and genotyped at e13.5. The bars show the percentage of embryos present at e13.5 for each genotype. The number of animals genotyped is shown on the bars for each category. b) Representative images of embryos that were dissected at e13.5. The upper panel images of each timepoint show the embryo within the yolk sac with the placenta attached. Lower panel images show the same embryo removed from the yolk sac. Arrows point to areas of possible vascular malformations. Asterisks indicate edema. c) The mutant allele frequency from cDNA from whole e14.5 embryonic tissue was assessed by SNaPshot.
Additionally, we examined Gnaq transcript levels in the GnaqRQfl/wt;E2a-Cre+ embryos and found that levels of mutant transcript ranged from 43 to 100% (n = 3) while the GnaqRQfl/wt embryos had 0% mutant transcript detectable (n = 3) (Fig. 4c). This range of mutant allele expression is expected due to the mosaic expression of the E2a Cre. We have previously noted that the highest mutant transcript levels in live-born animals that we found are approximately 50%; it is probable, that although e14.5 embryos are present with mutant transcript levels greater than 50%, these embryos are not surviving to birth.
Global expression of p.R183Q GNAQ is incompatible with survival
Our findings in the GnaqRQfl/wt;E2a-Cre+ mice suggest that mosaic expression of the Gnaq mutant allele (p.R183Q) during development causes different degrees of pathology, the extent of which are likely due to the tissue location and the expression level of the mutant allele. Therefore, as a formal test of Happle's hypothesis, we next investigated the effect of global p.R183Q expression to determine if early global embryonic expression of the p.R183Q GNAQ mutation is compatible with survival. For this experiment, we crossed our GnaqRQ conditional mutant strain with a transgenic line expressing a global (ubiquitous) β-actin Cre driver where the recombination occurs in the entire soma as early as the blastocyst stage (Lewandoski, Meyers, and Martin 1997).
Genotyping of mice at P0 in the cross between the β-actin Cre driver and our Gnaq inducible construct showed that no live-born mice contained both the Cre driver and the Gnaq conditional allele (Table 1), indicating that the p.R183Q GNAQ mutation causes embryonic lethality when globally expressed during the earliest stages of development.
Table 1.
Viable mice at e14.5 and P0 from GnaqRQwt/wt;β-actin-Cre+ × GnaqRQfl/wt cross.
| Number of mice/genotype | |||||
|---|---|---|---|---|---|
| Age | Total number | GnaqRQwt/wt |
GnaqRQwt/wt; β-actin-Cre+ |
GnaqRQfl/wt |
GnaqRQfl/wt; β-actin-Cre+ |
| e14.5 | 70 | 15 | 22 | 19 | 14 |
| P0 | 77 | 24 | 28 | 25 | 0 |
To determine if and when the embryos expressing the mutant allele were present and if they displayed any phenotype, we examined mouse embryos at different stages of development. In initial observations of e11.5 and e14.5 embryos from crosses that would only yield β-actin-Cre-positive embryos, we saw equal numbers of GnaqRQwt/wt;Cre+ and GnaqRQfl/wt;Cre+ embryos (data not shown). At e11.5, none of the GnaqRQfl/wt;Cre+ embryos appeared different than littermate controls, but at e14.5, GnaqRQfl/wt;Cre+ embryos did show an overt vascular phenotype. Therefore, we decided to undertake a more systematic study with crosses designed to yield all four possible genotypes and to examine the embryos at e14.5. Genotypic analysis of larger numbers of embryos at e14.5 showed that the distribution of the four possible genotypes from the GnaqRQwt/wt;β-actin-Cre+ × GnaqRQfl/wt cross was normal (Table 1). We did note that of the resorbing embryos seen at e14.5 that we were able to genotype, a large percentage (72.7%, 8/11) was GnaqRQfl/wt;β-actin-Cre+.
Using the SNaPshot transcript assay, we showed that the mutant Gnaq allele represented 100% of the total Gnaq transcript from GnaqRQfl/wt;β-actin-Cre+ e14.5 embryos (Fig. 5). These data demonstrate complete recombination of the targeted allele in GnaqRQfl/wt;β-actin-Cre+ e14.5 embryos. These results from the e14.5 embryos, combined with the absence of any GnaqRQfl/wt;β-actin-Cre+ pups born live, indicate that the GnaqRQfl/wt;β-actin-Cre+ embryos are resorbed at some point during the third week of embryonic development. These results strongly suggest that global expression of p.R183Q GNAQ early during development is not compatible with survival, further supporting Happle's paradominant inheritance hypothesis.
Fig. 5.

Mutant transcript analysis of e14.5 GnaqRQfl/wt and GnaqRQfl/wt;β-actin-Cre+ embryos. SNaPshot analysis was employed to assay the mutant allele transcript levels from whole embryo tissue. All of the GnaqRQfl/wt embryos had less than 5% mutant allele expression. Conversely, the GnaqRQfl/wt;β-actin-Cre + embryos all had mutant allele expression levels of 100%.
Embryonic death in GnaqRQfl/wt;β-actin-Cre+ animals is associated with hemorrhage and vasodilation
Gross examination of GnaqRQfl/wt;β-actin-Cre+ embryos demonstrated a variable phenotype. Although a small fraction of the GnaqRQfl/wt;β-actin-Cre+ embryos appeared similar to their control littermates, a much higher percentage of GnaqRQfl/wt;β-actin-Cre+ embryos demonstrated subcutaneous hemorrhage, edema, or were pale in color compared to controls (Fig. 6b). Genotype-blinded examination of e14.5 embryos of all four possible genotypes showed hemorrhage in 86% of the GnaqRQfl/wt;β-actin-Cre+ animals, which was often associated with subcutaneous edema (71%) (Table 2). By contrast, control littermates of all other genotypes did not exhibit any strong pathologies (Fig. 6a). Mild hemorrhage and edema was reported only in one GnaqRQwt/wt embryo and in one GnaqRQwt/wt;β-actin-Cre+ embryo, confirming that the severe phenotypes observed in GnaqRQfl/wt;β-actin-Cre+ mice were associated with the expression of the mutant R183Q allele during development.
Fig. 6.

GnaqRQfl/wt;β-actin-Cre+ embryos have hemorrhagic lesions and dilated vessels. a) GnaqRQfl/wt embryo (control) at e14.5 exhibits normal morphology. b) GnaqRQfl/wt;β-actin-Cre + embryo exhibits extensive hemorrhage, dilated capillaries (as indicated by arrows), and subcutaneous edema (asterisk). c) and d) Micrographs of parasagittal sections of e14.5 embryos stained with Hematoxylin and Eosin (H&E). The embryo of the GnaqRQfl/wt;β-actin-Cre+ mouse shows extravasation and dilated vessels in different areas of the body including subcutaneous regions and in the leptomeninges. e) and f) Higher power images (20×) of the lower back region of the embryo (as indicated by the lower box in panels c and d) of control and GnaqRQfl/wt;β-actin-Cre+ embryos. The arrow in f indicates dilated vessels, the asterisk indicates extravasation. g) and h) PECAM-1 immunostaining, depicted in brown, confirmed the presence of dilated and abnormal vessels (arrow) in e14.5 embryos expressing the mutant GnaqRQ allele compared to control. i) to l) Higher power images (20×) of the leptomeningeal region (as indicated by the upper box in panels c and d) demonstrate severe vascular alterations in the GnaqRQfl/wt;β-actin-Cre+ embryo compared to control (arrows). Bar in panel c represents 1 mm (scale for c and d). Bar in panel e represents 100 μm (scale for panels e–l).
Table 2.
Phenotypes observed at e14.5 from GnaqRQwt/wt;β-actin-Cre+ × GnaqRQfl/wt cross.
| Genotype (e14.5) |
Total number of embryos | Embryos with vascular anomaly |
Embryos with edema |
|---|---|---|---|
| GnaqRQwt/wt | 15 | 1 (7%) | 0 |
| GnaqRQwt/wt;β-actin-Cre+ | 22 | 0 | 1 (4%) |
| GnaqRQfl/wt | 19 | 0 | 0 |
| GnaqRQfl/wt;β-actin-Cre+ | 14 | 12 (86%) | 10 (71%) |
As most of these e14.5 embryos exhibited vascular abnormalities during gross examination, we performed histological assessment. Sagittal sections of the whole embryo revealed areas of extravasation in the subcutaneous region in GnaqRQfl/wt;β-actin-Cre+ embryos, indicating severe alteration of blood vessel formation and circulation (Fig. 6d, f and j). Moreover, PECAM-1 immunostaining confirmed the presence of dilated blood vessels filled with red blood cells at sites of apparent hemorrhage in GnaqRQfl/wt;β-actin-Cre+ embryos (Fig. 6h and l), and was not seen in controls (Fig. 6g and k). Instead, the blood vessels in GnaqRQfl/wt littermate controls were small, with only a few red blood cells visible in each vessel (Fig. 6c, e and i).
Downstream signaling effects
Constitutive activation of GNAQ caused by the p.R183Q somatic GNAQ mutation alters signaling pathways and distinct transcriptional programs implicated in vasculature development, angiogenesis, and blood vessel morphogenesis including angiopoietin-2 expression (Galeffi et al. 2022). A recent study reported that ANGPT2 expression was significantly increased both in human endothelial cultured cells expressing the R183Q mutation and in capillary malformations of SWS patients (Huang et al. 2022). Therefore, we performed qRT-PCR to measure Angpt2 expression in e14.5 GnaqRQfl/wt;β-actin-Cre+ embryos. We found that the level of Angpt2 transcript was significantly higher in GnaqRQfl/wt;β-actin-Cre+ embryos as compared to their GnaqRQfl/wt littermate controls (Fig. 7).
Fig. 7.
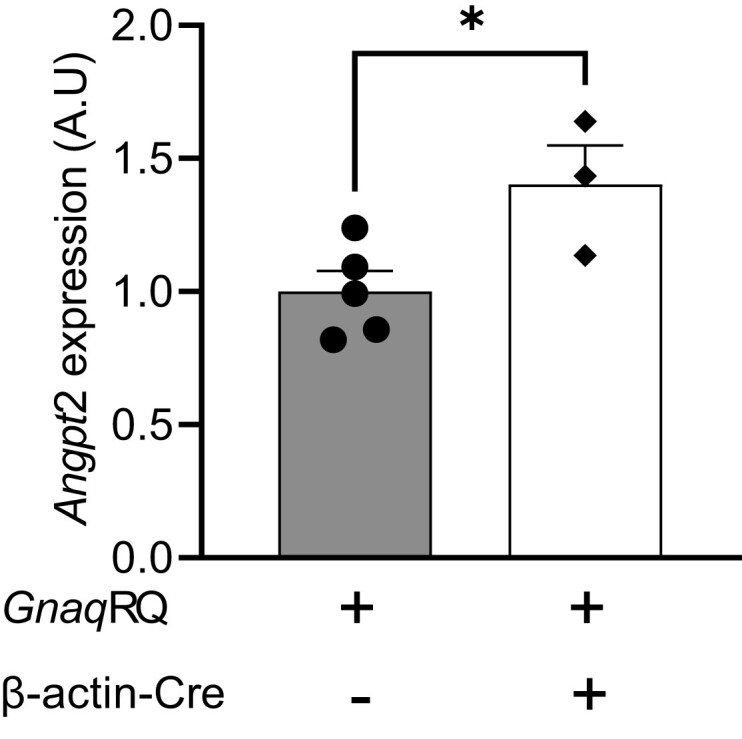
Downstream signaling effect of mutant Gnaq expression. qRT-PCR results showing Angpt2 expression in tissue from e14.5 embryos. Expression of Angpt2 is significantly increased in embryos expressing mutant Gnaq. All embryos examined contained one copy of the conditional allele. Data represent the mean ± SEM, n = 5 and 3 for GnaqRQfl/wt and GnaqRQfl/wt;β-actin-Cre+, respectively. *P = 0.035 unpaired two-tailed Student's t-test.
Discussion
In our efforts to create a mouse model of SWS, we found that global mosaic expression of p.R183Q GNAQ with the “mosaic” E2a-Cre driver mouse line resulted in high levels of embryonic lethality. Among the postnatally viable GnaqRQfl/wt;E2a-Cre+ animals, the levels of mutant Gnaq transcript expression were variable, reproducing the variable, mosaic recombination reported for this Cre line. The highest level of mutant transcript recorded among the postnatally viable GnaqRQfl/wt;E2a-Cre+ animals was approximately 50%, suggesting that levels greater than 50% of mutant Gnaq transcript expression lead to prenatal lethality. This is further supported by the high levels of mutant transcript detected in GnaqRQfl/wt;E2a-Cre+ embryos, strengthening the idea that high levels of p.R183Q GNAQ expression contribute to the embryonic lethality observed. We then found that ubiquitous global expression of p.R183Q GNAQ with the β-actin-Cre driver mouse line resulted in complete embryonic lethality in GnaqRQfl/wt;β-actin-Cre+ animals and variable, but disease-relevant vascular phenotypes, in developing embryos. Vascular abnormalities in the embryos included grossly dilated vessels and hemorrhage. The combined data using the two Cre drivers support Happle's hypothesis that this mutation would be lethal if passed through the germline.
The Genome Aggregation Database (gnomAD) of human DNA sequence variation in various populations provides further support of germline p.R183Q GNAQ lethality, albeit in the form of indirect evidence. As of January 5, 2023, the p.R183Q GNAQ mutation has not been reported in gnomAD, which includes sequence information from over 100,000 unrelated individuals. The embryonic lethality of the mutation is consistent with the known biochemical consequences of the mutation. The p.R183Q mutation in GNAQ has been shown to activate the G protein Gαq leading to increased downstream signaling (Shirley et al. 2013; Galeffi et al. 2022). Thus, this mutation might be postulated to be highly detrimental if globally expressed during development. The combined data from the human population, the biochemical consequences of the mutation, and the mouse model data presented here all support Happle's “paradominant inheritance” hypothesis—that this is a “lethal mutation surviving because of mosaicism”.
Similar to the p.R183Q GNAQ mutation, mutations of glutamine 209 (Q209) result in activation of the GNAQ protein. While the p.R183Q GNAQ mutation results in a slowing of the GTP hydrolysis rate, p.Q209 GNAQ mutations result in irreversible, constitutive activation of the subunit (Kimple et al. 2011). Interestingly, the p.Q209 GNAQ mutation occurs frequently in uveal melanoma, a cancer of melanocytes in the uvea, the middle layer of the eye (Van Raamsdonk et al. 2009; Van Raamsdonk et al. 2010). In our model, we observed increased pigmentation of the leptomeninges in GnaqRQfl/wt;E2a-Cre+ adults, presumably in the melanocytes that are normally present in that location, further suggesting that our mouse model produces functional mutant protein. We note that the orthologous human p.R183Q GNAQ somatic mutation is also often found in uveal melanoma, (Tate et al. 2019; Van Trigt, Kelly, and Hughes 2022). We surmise then that melanocytes are particularly sensitive to this mutation. Although the murine leptomeningeal melanocyte phenotype is not a SWS-associated phenotype that has been reported in patients, it is not clear whether this phenotype has ever been explored.
To date, two other animal models have been reported that examine mutant GNAQ in endothelial cells. In one approach, Matrigel plugs containing human endothelial cells expressing either WT or p.R183Q GNAQ were introduced into nude mice (Huang et al. 2022). This approach resulted in enlarged vascular lumens that resembled capillary malformations. In another approach, Matrigel plugs containing mouse Ms1 endothelial cells expressing p.Q209L GNAQ, a more strongly activating mutation, were introduced into nude mice (Sasaki et al. 2022). Through this approach, vascular tumors were identified and characterized. Although both models are useful in characterizing the role of GNAQ mutations in endothelial cells in the formation of vascular lesions, neither of these models are able to characterize the role and consequences of the p.R183Q GNAQ mutation during embryonic development. Other published models of mutant GNAQ endothelial cells rely on CMV promoters to drive expression of GNAQ, resulting in over-expression of this transcript relative to endogenous levels. Therefore, we targeted our conditional construct to the endogenous Gnaq locus. Moreover, to distinguish the conditional allele transcripts from those arising from the endogenous allele, we inserted the poly(A) element from the BGH1 gene into the conditional allele. Despite our effort at maintaining physiological regulation of gene expression, allele-specific qRT-PCR analysis revealed a significant increase in the levels of the conditional allele transcripts in both GnaqRQfl/wt and GnaqRQfl/wt;Cre+ embryos, compared to the endogenous allele (Supplementary Figure 2). Previous studies indicated that introduction of different poly(A) regions can have a significant effect on gene expression (Pfarr et al. 1986; Wang et al. 2022). Moreover, the introduction of the BGH poly(A) region results in a higher increase of recombinant mRNA expression (Pfarr et al. 1986; Wang et al. 2022) compared to other poly(A) elements tested due to efficient RNA stabilization (Pfarr et al. 1986). Despite the over-expression of both the wild-type and mutant transcripts from the conditional allele, we do not believe that overexpression per se (i.e. overexpression of the wild-type allele) causes the vascular malformation phenotypes that we have observed. Even with a significant increase in transcript expression from the conditional wild-type allele, GnaqRQfl/wt mice were born at the expected ratio and were phenotypically normal. We only observed embryonic lethality and severe vascular phenotypes in GnaqRQfl/wt;Cre+ mice, strongly suggesting that only the expression of mutant transcript is correlated with the formation of the severe phenotype.
Here we have described the first mouse model with inducible expression of p.R183Q GNAQ, the causative mutation for SWS. We have also provided the first in vivo experimental evidence supporting the lethality of germline p.R183Q GNAQ. Intriguingly, deletion of both copies of murine GNAQ does not cause embryonic lethality, resulting in viable mice with relatively minor defects (impaired platelet activation and motor coordination) (Offermanns, Toombs, et al. 1997b) (Offermanns, Hashimoto, et al. 1997a). Viability is likely due to compensation by the highly homologous and widely expressed GNA11 gene/protein (Offermanns et al. 1998). Thus, the normal function of GNAQ is not essential for development. By contrast, per Happle's paradominant inheritance hypothesis, inheritance of even a single copy of an activated (gain-of-function) GNAQ mutation leads to embryonic lethality, and thus, the p.R183Q mutation is only ever seen in a mosaic state. This inducible mouse model could be crucial in understanding the pathophysiology of SWS. Further studies using this conditional allele will be required to identify the developmental timeframe and the precise precursor cell(s) that must express the mutant transcript to fully recapitulate the SWS human phenotype.
Supplementary Material
Acknowledgements
The authors would like to acknowledge the Duke Transgenic Mouse Facility for generating the GnaqRQ mouse model. We thank Drs. Christopher Counter and Özgün Le Roux for the kind gift of the ACTBFLPe/FLPe mice. This work was supported by the following grants: US Dept of Defense Medical Research Program Discovery Award W81XWH2110061 to DAM; 2020 Sturge–Weber Foundation Catalyst Award, and postdoctoral trainee position on T32 HL007101-45 to SEWS.
Contributor Information
Sarah E Wetzel-Strong, Department of Molecular Genetics and Microbiology, Duke University School of Medicine, Durham, NC 27710, USA.
Francesca Galeffi, Department of Molecular Genetics and Microbiology, Duke University School of Medicine, Durham, NC 27710, USA.
Christian Benavides, Department of Molecular Genetics and Microbiology, Duke University School of Medicine, Durham, NC 27710, USA.
Mary Patrucco, Department of Molecular Genetics and Microbiology, Duke University School of Medicine, Durham, NC 27710, USA.
Jessica L Bullock, Department of Molecular Genetics and Microbiology, Duke University School of Medicine, Durham, NC 27710, USA.
Carol J Gallione, Department of Molecular Genetics and Microbiology, Duke University School of Medicine, Durham, NC 27710, USA.
Han Kyu Lee, Department of Molecular Genetics and Microbiology, Duke University School of Medicine, Durham, NC 27710, USA.
Douglas A Marchuk, Department of Molecular Genetics and Microbiology, Duke University School of Medicine, Durham, NC 27710, USA.
Data availability
Mouse strains are available upon request. The authors affirm that all data necessary for confirming the conclusions of the article are present within the article, figures, and tables.
Supplemental material available at GENETICS online.
Literature cited
- Galeffi F, Snellings DA, Wetzel-Strong SE, Kastelic N, Bullock J, Gallione CJ, North PE, Marchuk DA. A novel somatic mutation in GNAQ in a capillary malformation provides insight into molecular pathogenesis. Angiogenesis. 2022;25(4)2;493–502. doi: 10.1007/s10456-022-09841-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudjohnsen SA, et al. Meningeal melanocytes in the mouse: distribution and dependence on MITF. Front Neuroanat. 2015;9:149. doi: 10.3389/fnana.2015.00149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Happle R. Lethal genes surviving by mosaicism: a possible explanation for sporadic birth defects involving the skin. J Am Acad Dermatol. 1987;16(4):899–906. doi: 10.1016/S0190-9622(87)80249-9. [DOI] [PubMed] [Google Scholar]
- Huang L, Bichsel C, Norris AL, Thorpe J, Pevsner J, Alexandrescu S, Pinto A, Zurakowski D, Kleiman RJ, Sahin M, et al. Endothelial GNAQ p.R183Q increases ANGPT2 (Angiopoietin-2) and drives formation of enlarged blood vessels. Arterioscler Thromb Vasc Biol. 2022;42(1):e27–e43. doi: 10.1161/ATVBAHA.121.316651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keum S, Lee HK, Chu PL, Kan MJ, Huang MN, Gallione CJ, Gunn MD, Lo DC, Marchuk DA. Natural genetic variation of integrin alpha L (Itgal) modulates ischemic brain injury in stroke. PLoS Genet. 2013;9(10):e1003807. doi: 10.1371/journal.pgen.1003807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimple AJ, Bosch DE, Giguere PM, Siderovski DP. Regulators of G-protein signaling and their Galpha substrates: promises and challenges in their use as drug discovery targets. Pharmacol Rev. 2011;63(3):728–749. doi: 10.1124/pr.110.003038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakso M, Pichel JG, Gorman JR, Sauer B, Okamoto Y, Lee E, Alt FW, Westphal H. Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc Natl Acad Sci U S A. 1996;93(12):5860–5865. doi: 10.1073/pnas.93.12.5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Kwon DH, Aylor DL, Marchuk DA. A cross-species approach using an in vivo evaluation platform in mice demonstrates that sequence variation in human RABEP2 modulates ischemic stroke outcomes. Am J Hum Genet. 2022;109(10):1814–1827. doi: 10.1016/j.ajhg.2022.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewandoski M, Meyers EN, Martin GR. Analysis of Fgf8 gene function in vertebrate development. Cold Spring Harb Symp Quant Biol. 1997;62:159–168. doi: 10.1101/SQB.1997.062.01.021. [DOI] [PubMed] [Google Scholar]
- Offermanns S, Hashimoto K, Watanabe M, Sun W, Kurihara H, Thompson RF, Inoue Y, Kano M, Simon MI. Impaired motor coordination and persistent multiple climbing fiber innervation of cerebellar Purkinje cells in mice lacking Galphaq. Proc Natl Acad Sci U S A. 1997a;94(25):14089–14094. doi: 10.1073/pnas.94.25.14089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Offermanns S, Toombs CF, Hu YH, Simon MI. Defective platelet activation in G alpha(q)-deficient mice. Nature. 1997b;389(6647):183–186. doi: 10.1038/38284. [DOI] [PubMed] [Google Scholar]
- Offermanns S, Zhao LP, Gohla A, Sarosi I, Simon MI, Wilkie TM. Embryonic cardiomyocyte hypoplasia and craniofacial defects in G alpha q/G alpha 11-mutant mice. EMBO J. 1998;17(15):4304–4312. doi: 10.1093/emboj/17.15.4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfarr DS, Rieser LA, Woychik RP, Rottman FM, Rosenberg M, Reff ME. Differential effects of polyadenylation regions on gene expression in mammalian cells. DNA. 1986;5(2):115–122. doi: 10.1089/dna.1986.5.115. [DOI] [PubMed] [Google Scholar]
- Rodriguez CI, Buchholz F, Galloway J, Sequerra R, Kasper J, Ayala R, Stewart AF, Dymecki SM. High-efficiency deleter mice show that FLPe is an alternative to Cre-loxP. Nat Genet. 2000;25(2):139–140. doi: 10.1038/75973. [DOI] [PubMed] [Google Scholar]
- Sasaki M, Jung Y, North P, Elsey J, Choate K, Toussaint MA, Huang C, Radi R, Perricone AJ, Corces VG, et al. Introduction of mutant GNAQ into endothelial cells induces a vascular malformation phenotype with therapeutic response to imatinib. Cancers (Basel). 2022;14(2):413. doi: 10.3390/cancers14020413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirley MD, Tang H, Gallione CJ, Baugher JD, Frelin LP, Cohen B, North PE, Marchuk DA, Comi AM, Pevsner J. Sturge–Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N Engl J Med. 2013;368(21):1971–1979. doi: 10.1056/NEJMoa1213507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, Boutselakis H, Cole CG, Creatore C, Dawson E, et al. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res. 2019;47(D1):D941–D947. doi: 10.1093/nar/gky1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchiyama Y, Nakashima M, Watanabe S, Miyajima M, Taguri M, Miyatake S, Miyake N, Saitsu H, Mishima H, Kinoshita A, et al. Ultra-sensitive droplet digital PCR for detecting a low-prevalence somatic GNAQ mutation in Sturge–Weber syndrome. Sci Rep. 2016;6:22985. doi: 10.1038/srep22985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urtatiz O, Cook C, Huang JL, Yeh I, Van Raamsdonk CD. GNAQ(Q209l) expression initiated in multipotent neural crest cells drives aggressive melanoma of the central nervous system. Pigment Cell Melanoma Res. 2020;33(1):96–111. doi: 10.1111/pcmr.12843. [DOI] [PubMed] [Google Scholar]
- Van Raamsdonk CD, Bezrookove V, Green G, Bauer J, Gaugler L, O’Brien JM, Simpson EM, Barsh GS, Bastian BC. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature. 2009;457(7229):599–602. doi: 10.1038/nature07586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Raamsdonk CD, Griewank KG, Crosby MB, Garrido MC, Vemula S, Wiesner T, Obenauf AC, Wackernagel W, Green G, Bouvier N, et al. Mutations in GNA11 in uveal melanoma. N Engl J Med. 2010;363(23):2191–2199. doi: 10.1056/NEJMoa1000584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Trigt WK, Kelly KM, Hughes CCW. GNAQ mutations drive port wine birthmark-associated Sturge–Weber syndrome: a review of pathobiology, therapies, and current models. Front Hum Neurosci. 2022;16:1006027. doi: 10.3389/fnhum.2022.1006027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, Newman J, Reczek EE, Weissleder R, Jacks T. Restoration of p53 function leads to tumour regression in vivo. Nature. 2007;445(7128):661–665. doi: 10.1038/nature05541. [DOI] [PubMed] [Google Scholar]
- Wang XY, Du QJ, Zhang WL, Xu DH, Zhang X, Jia YL, Wang TY. Enhanced transgene expression by optimization of poly a in transfected CHO cells. Front Bioeng Biotechnol. 2022;10:722722. doi: 10.3389/fbioe.2022.722722. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Mouse strains are available upon request. The authors affirm that all data necessary for confirming the conclusions of the article are present within the article, figures, and tables.
Supplemental material available at GENETICS online.