

Abstract
Mice lacking the α-subunit of the heterotrimeric guanine nucleotide binding protein Gq (Gαq) are viable but suffer from ataxia with typical signs of motor discoordination. The anatomy of the cerebellum is not overtly disturbed, and excitatory synaptic transmission from parallel fibers to cerebellar Purkinje cells (PCs) and from climbing fibers (CFs) to PCs is functional. However, about 40% of adult Gαq mutant PCs remain multiply innervated by CFs because of a defect in regression of supernumerary CFs in the third postnatal week. Evidence is provided suggesting that Gαq is part of a signaling pathway that is involved in the elimination of multiple CF innervation during this period.
In a variety of signaling systems, many of the components of the G protein family are highly homologous in structure and activity (1). Thus, for example, there are two similar members of the heterotrimeric Gq family, Gαq and Gα11, that are responsible for coupling receptors to the pertussis toxin-insensitive activation of isoforms of phospholipase C-β (PLC- β) (for review see ref. 2). They share 88% amino acid sequence identity and are expressed together in almost every cell type (3, 4). The receptors activating Gq family members in mammalian systems do not discriminate between Gαq and Gα11 (5–7). Similarly, there appears to be little difference between the abilities of both G protein α-subunits to regulate phospholipase C β isoforms. Thus, Gαq and Gα11 indistinguishably activate the β1, β3, and β4 isoforms of PLC, and both are equally poor regulators of PLC- β2 (6, 8–11). These observations raise fundamental questions about the function of G protein-mediated signaling pathways in the nervous system. Are different isoforms of receptors, G proteins, and effectors used to generate specific signaling pathways in different cells? If they are, they could account for cell-type-specific kinetic and regulatory properties. Do several isoform pathways coexist in the same cell? If so, are their functions redundant and overlapping or do they participate in separable physiological activities? What prevents or maintains cross-talk between these systems?
A clearer picture of the specificity of these pathways requires genetic approaches combined with morphological and physiological analyses, because biochemical reconstitution may eliminate organizational elements, developmental staging, or additional components in the signaling pathway that are necessary for specificity. To explore the biological significance of the diversity among Gαq family members, we inactivated the gene encoding the Gq α-subunit (Gnaq) in mouse embryonic stem cells and generated mice deficient in Gαq. Although both Gαq and Gα11 are found in most cells, the relative levels of expression vary among different tissues. In most regions of the rat central nervous system, Gαq expression levels exceed those of Gα11 by 2- to 5-fold (12). Expression of Gαq/11 in the rat central nervous system is highest in the dendrites of cerebellar Purkinje cells (PCs) and hippocampal CA1 pyramidal cells (13). Both areas are very rich in glutamatergic synapses and show high expression levels of metabotropic glutamate receptors type 1 (mGluR1) and 5 (mGluR5), respectively (14–16). Both mGluRs are known to couple to Gαq and Gα11 and have been implicated in functions required for the modulation of neuronal activity (for review see refs. 17 and 18). The mGluR1 has been shown to be required for postnatal elimination of multiple climbing fiber (CF) innervation of PCs (19). In the cerebellum of newborn rats and mice, each PC is innervated by multiple CFs. Massive elimination of supernumerary CF-PC synapses occurs during the first 3 postnatal weeks, and a state is reached at about postnatal day 20 (P20), in which most PCs are innervated by single CFs. This developmental change appears to depend on the presence of intact granule cells and of functioning synapses between PFs and PCs (20). Failure of CF-PC synapse elimination results in persistent multiple climbing fiber innervation, which correlates with impaired motor coordination (19, 21). Thus, Gαq deficiency might be expected to affect cerebellar development, and our results support this prediction.
MATERIALS AND METHODS
Generation of Gαq Mutant Mice.
A genomic Gαq clone was isolated from a 129/Sv mouse genomic l phage library (Stratagene) with a probe derived from the Gαq cDNA. The genomic clone used for gene targeting contained the two last exons of the Gαq gene. To generate a null mutation, a 2.3-kb BglII fragment containing an exon coding for amino acids 246–297 of Gαq was replaced by the neo gene from plasmid pMC1neo PolyA (Stratagene). The targeting vector contained 5.5 kb of upstream sequence as the 5′ arm and 2.6 kb of intron sequence as the 3′ arm. Cells of the 129/Sv mouse embryonic stem cell line CJ7 were transfected with 20 μg linearized targeting vector by electroporation (Bio-Rad Gene Pulser set at 240 V and 500 μF). G418 selection (150 μg/ml geneticin; GIBCO/BRL) was added 24 h after transfection, and selected cell clones were isolated after 1 week of selection. Correctly targeted embryonic stem (ES) cell clones were identified by PCR using primers hybridizing to the Neo cassette and to the intron sequence just outside the 3′ arm of the targeting construct. Positive clones were confirmed by Southern blot analysis of ES cell DNA. DNA was digested with AflII and probed with a 1.3-kb DNA fragment from the 3′ flanking region (Fig. 1 A and B). Chimeric mice were generated using three independently targeted ES cell clones by aggregation of ES cells with CD1 morulae (22) or by injection of ES cells into C57/BL blastocysts (23). Chimeras were bred with C57/BL and 129/Sv mice to generate hemizygous animals. The outbred mice (C57/BL) were more robust and showed higher fertility than the inbred (129/SV) mice. We used the outbred mice for most of the experiments. However, all of our results were repeated in the inbred animals, and no significant differences were found. Germ-line transmission was confirmed by PCR and Southern blot analysis.
Figure 1.
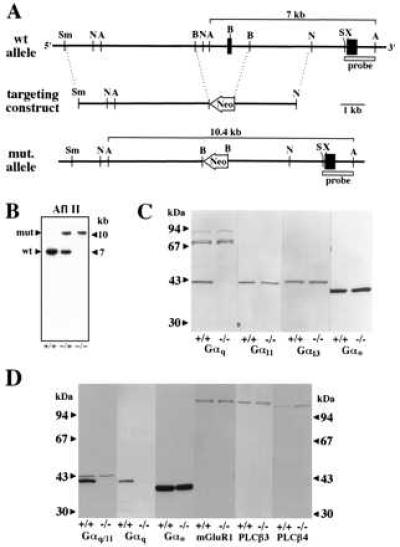
Targeted disruption of the murine Gαq gene. (A) Part of the wild-type Gαq locus containing the two last exons (wt allele), the targeting construct, and the targeted locus (mut. allele) are shown. Neo, neomycin resistance gene. The sizes of the AflII fragments predicted to hybridize to the indicated diagnostic probe are shown. Restriction endonucleases: A, AflII; B, BglII; N, NdeI; S, SacI; Sm, SmaI; and X, XhoI. (B) Southern blot analysis of AflII-digested genomic DNA from wild-type (+/+), hemizygous (−/+), and homozygous mutant mice (−/−) with the diagnostic probe indicated in A. (C) Western blot analysis of whole brain cholate extracts from wild-type (+/+) and homozygous Gαq mutant mice (−/−) with antibodies recognizing the α-subunits of Gq (Gαq), G11 (Gα11), G13 (Gα13), and Go (Gαo). (D) Western blot analysis of cerebellar membrane fractions from wild-type (+/+) and homozygous Gαq mutant animals (−/−) with antibodies recognizing the α-subunits of Gq and G11 (Gαq/Gα11), Go (Gαo), type 1 metabotropic glutamate receptor (mGluR1), phospholipase C-β3 (PLC-β3), and phospholipase C-β4 (PLC-β4).
Rotorod and Runway Test.
The rotorod apparatus is a plastic cylindrical rod 3 cm in diameter and 13 cm long. The rod is supported at the ends by two walls and is 25 cm from the base of the walls. The walls at the ends of the rod are high enough so that the mice cannot climb off the rod. For each trial on the stationary rod test or the rotorod test, the mice are placed on the rod and timed until they fell off the rod. They were timed up to a maximum of 1 min. The intertrial interval was 10 min. For the rotorod test, the rod was turned by an electric motor at 10 rpm.
Immunohistology.
Under deep anesthesia with chloral hydrate (350 mg/kg body weight, intraperitoneally), mice at the second and fifth postnatal months were perfused transcardially with 4% paraformaldehyde in 0.1 M sodium phosphate buffer (pH 7.2). The brains were excised quickly from the skull and immersed overnight in the same fixative. After sectioning at 50 μm in thickness by a microslicer (Dosaka) in the parasagittal plane, the sections were used for Nissl staining with toluidine blue and for immunostaining with rabbit anti-rat spot 35/calbindin antibody (1:1,000) (24), and sections were incubated for 2 h with FITC-labeled goat anti-rabbit IgG (1:100, Jackson ImmunoResearch) and observed by a MRC1024 confocal laser-scanning microscope (Bio-Rad). Paraffin sections (4 μm in thickness) were prepared for Nissl staining with toluidine blue and immunostaining with rabbit anti-Gαq/Gα11 (341–359 aa residues) antibody (0.2 mg/ml, Santa Cruz Biotechnology), prediluted rabbit anti-glial fibrillary acidic protein (GFAP) antibody (DAKO), and guinea pig anti-mGluR1 antibody. Immunoreacted sections were then incubated with biotinylated secondary antibody and ABC complex and observed with a bright-field light microscope (AX 80, Olympus).
Antibodies.
Rabbit polyclonal anti-Gαq/Gα11, anti-PLC-β3, and anti-PLC-β3 antibodies were from Santa Cruz Biotechnology, and anti-mGluR1 antibodies used for Western blot analysis were from Chemicon. Rabbit antisera specific for Gαq, Gα11, Gαo, and Gα13 were raised against specific peptides.
Electrophysiology.
Sagittal cerebellar slices of 200- to 300-μm thickness were prepared from the wild-type and mutant mice as described previously (25–28). Whole cell recordings were made from visually identified PCs using a 40× water immersion objective attached to either a Olympus (BH-2) or Zeiss (Axioskop) upright microscope (25, 26). Resistance of patch pipettes was 3–6 MΩ when filled with an intracellular solution composed of 60 nM CsCl, 30 nM Cs d-gluconate, 20 nM TEA-Cl, 20 nM BAPTA, 4 nM MgCl2, 4 nM ATP, and 30 nM Hepes (pH 7.3, adjusted with CsOH). The composition of standard bathing solution was 125 mM NaCl, 2.5 mM KCl, 2 mM CaCl2, 1 mM MgSO4, 1.25 mM NaH2PO4, 26 mM NaHCO3, and 20 mM glucose, which was bubbled continuously with a mixture of 95% O2 and 5% CO2. Bicuculline (10 μM) was always present in the saline to block spontaneous inhibitory postsynaptic currents (27, 29). Ionic currents were recorded with either Axopatch-1D (Axon Instruments) or EPC-9 patch-clamp amplifier (HEKA Electronics, Lambrecht/Pfalz, Germany) and stored on a DAT data recorder (PC204, Sony, Tokyo) for later analysis. Stimulation and on-line data acquisition were performed using the pulse program on a Macintosh computer (version 7.5, HEKA). The signals were filtered at 3 kHz and digitized at 20 kHz. Fitting of the decay phases of excitatory postsynaptic currents (EPSCs) was done with the pulse-fit program (version 7.5, HEKA). For stimulation of CFs and PFs, a glass pipette with 5- to 10-μm tip diameter filled with standard saline was used. Square pulses (duration, 0.1 ms; amplitude, 0–100 V for CF stimulation, 1–10 V for PF stimulation) were applied for focal stimulation.
RESULTS
Generation and Characterization of Gαq-Deficient Mice.
To disrupt the Gαq gene locus, we constructed a targeting vector containing a 10.4-kb Gαq genomic DNA fragment disrupted by a neomycin phosphotransferase gene (Fig. 1A). Three independently targeted embryonic stem cell clones generated by homologous recombination transmitted the targeted mutation of the Gαq gene through the germ line (Fig. 1B). Western blot analysis of cholate extracts from whole brain membranes (Fig. 1C) and of cerebellar membrane proteins (Fig. 1D) demonstrated that the homozygous mutant mice (−/−) had no detectable Gαq protein, whereas the content of Gα11, Gα13, and Gαo appeared to be unaffected by the Gαq mutation. Similarly, the amounts of mGluR1 metabotropic glutamate receptors and of the β3 isoform of phospholipase C (PLC-β3) in cerebellar membrane fractions were not appreciably affected by the Gαq mutation, whereas the content of the β4 isoform of phospholipase C (PLC-β4) was increased about 2-fold in cerebella from mutant mice when compared with wild-type animals (Fig. 1D).
Mice heterozygous for the targeted mutation of the Gαq gene appeared normal and showed no obvious defects over a 20-month period. In contrast, homozygous mutant mice showed increased mortality during the first postnatal day. The mice have a defect in platelet aggregation that is a result of the absence of Gαq (30), and the mortality appears to result from internal bleeding that occurs during birth trauma. Sixty to 70% of homozygous mutant newborns survived the postnatal period and developed clear signs of motor coordination deficit, which became obvious about 3 weeks after birth. The animals displayed awkward and jerky movements and loss of balance during rearing, and occasionally fell on their side. Homozygous mutant mice had difficulties balancing on the stationary rotorod. When placed on a turning rotorod, the homozygous mutant mice immediately fell off and their performance did not improve with increasing trial numbers. In contrast, heterozygous and wild-type mice managed to stay for some time on the turning rod, and duration of their stay increased with increasing trial numbers (Fig. 2).
Figure 2.
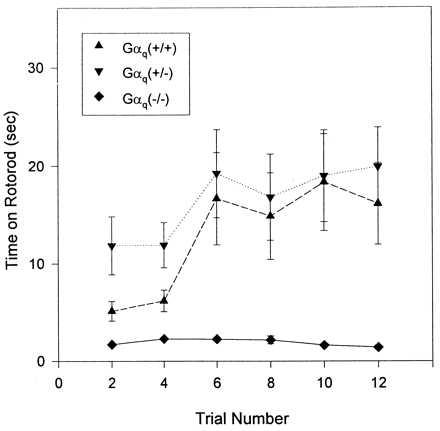
Rotorod test. Each point shows the mean ± SEM of the times each group stayed on the rod from each trail. n = 10 for Gαq (+/+) and Gαq (−/+); n = 12 for Gαq (−/−).
Gαq mutant mice showed an ataxic gait with typical wobbling and tottering steps, which became more severe when the mice increased their walking speed. Mutant mice cannot walk in a straight line and tend to drag their feet along the floor (data not shown). Motor coordination deficits and ataxia have been described in a variety of mice with mutations affecting the morphology or function of the cerebellar cortex. No obvious morphological defects could be observed on examination of the peripheral and central nervous system of Gαq homozygous mutant mice. Extensive morphological examination of the cerebellar cortex by histological, immunohistochemical, and electron microscopic techniques indicated that deletion of the Gαq gene did not affect gross development of the cerebellar anatomy, cell production, cytodifferentiation, and formation of PF-PC synapses (data not shown).
By using affinity-purified antibodies against Gαq/Gα11, the cerebella of wild-type and Gαq mutant mice at P7, P14, and P21 were immunohistochemically examined to reveal the localization of Gαq/Gα11 in the cerebellar cortex. At all of the stages examined, the antibody stained the molecular layer intensely and the granular layer weakly in the wild-type cerebellum (Fig. 3 A–C). At P7, low to moderate levels of the Gαq/Gα11 immunoreactivity were detected in the thin molecular layer between PC somata and the external granular layer. The level gradually increased from P7 to P21, when the immunoreactivity was observed as tiny puncta occupying the molecular layer at high density (Fig. 3C). The immunoreactivity was markedly reduced at each corresponding age (Fig. 3 D–F) in the mutant. However, low levels of residual staining were detected in the molecular layer, representing the Gα11 immunoreactivity. The residual immunoreactivity showed a distribution pattern similar to the wild-type cerebellum. Therefore, it is likely that Gαq and Gα11 are both localized in the molecular layer, with the dominant presence of the Gαq subtype.
Figure 3.
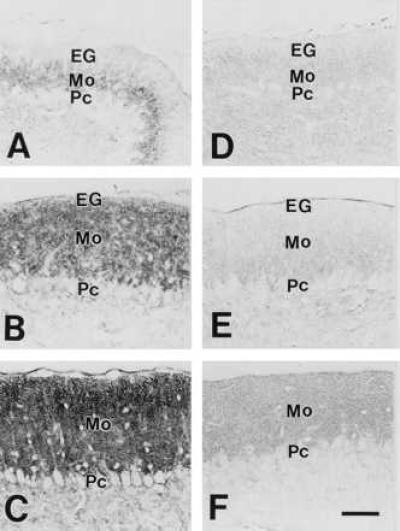
Immunohistochemistry for Gαq/Gα11 in the cerebellum of wild-type (A–C) and Gαq mutant mice (D–F). A and D, P7; B and E, P14; C and F, P21. EG, external granular layer; Pc, Purkinje cell layer. A–F were taken at the same magnification. [Bars = 50 μm (F).]
Gαq Mutant Purkinje Cells Are Multiply Innervated by Climbing Fibers.
In sagittal cerebellar slices prepared from 22- to 57-day-old (P22–P57) wild-type mice or age-matched Gαq mutant animals, CFs were stimulated in the granule cell layer, and evoked responses in single PCs were recorded using patch-clamp techniques in the whole cell configuration (26, 27, 29). As the stimulus intensity was gradually increased (pulse width, 0.1 ms, strength 0–100 V), a large EPSC was elicited in an all-or-none fashion in the majority of wild-type PCs (Fig. 4Aa). In other wild-type PCs, CF-EPSCs showed two or three discrete steps (Fig. 4Ab). The number of discrete steps of CF-EPSCs represents the number of CFs innervating the recorded PC (19–21). Thus, as summarized in the frequency distribution histogram (Fig. 4B Left), 82.6, 14.8, and 2.6% of wild-type PCs recorded in the present study (P22–P57) are innervated by one, two, and three CFs, respectively. On the other hand, in Gαq mutant PCs, 59.6% had single CF-EPSC steps (Fig. 4Ac), whereas 28.2 and 12.1% had two and three discrete steps, respectively (Fig. 4Ad). The frequency distribution histogram (Fig. 5B Left) shows significant differences between wild-type and mutant mice (P < 0.001, χ2 test). Similar experiments have been done with three Gαq heterozygotes (P53–P55), in which 88.7, 9.4, and 1.9% of PCs were innervated by one, two, and three CFs, respectively. These values were indistinguishable from those of wild-type mice (P > 0.05, χ2 test).
Figure 4.
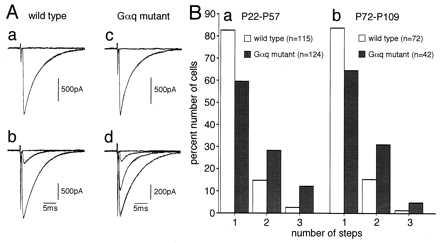
Multiple climbing fiber innervation of Purkinje cells in mature Gαq mutant mice. (A) EPSCs elicited by stimulation of climbing fibers (CFs) in the granule cell layer in wild-type (a, b) and Gαq mutant (c, d) Purkinje cells (PCs). Records in a, b, c, and d were taken from mice at postnatal day 50 (P50), P80, P46, and P46, respectively. One or two traces are superimposed at each threshold intensity. Stimuli were applied at 0.2 Hz. Holding potentials were −10 mV for a, b, and c and 0 mV for d. (B) Summary histograms showing number of discrete steps of CF-EPSCs of the wild-type (open columns) and mutant (hatched columns) PCs. Data obtained from mice at P22–P57 (a) and at P72–P109 (b), respectively. Numbers of tested PCs at P22–P57 (a) are: n = 115 (from seven wild-type mice) and n = 124 (from eight Gαq mutant mice). Those at P60–P109 (b) are: n = 72 (from four wild-type mice) and n = 42 (from three mice). All cells were studied blind to the mouse genotype.
Figure 5.
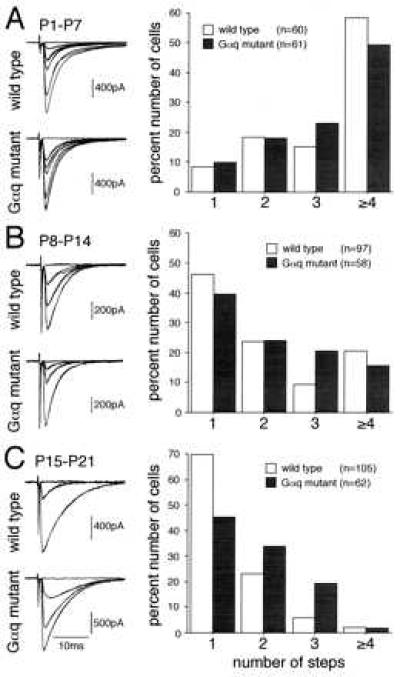
Early postnatal development of CF innervation. (A Left) CF-EPSCs of the wild type (P7, holding potential, −30 mV) and Gαq mutant (P6, holding potential, −60 mV) PCs. One or two traces were superimposed at each threshold intensity. Stimuli were applied at 0.2 Hz. (A Right) Summary graph showing numbers of discrete steps of CF-EPSCs of the wild type (open columns) and mutant (hatched columns) PCs from mice at P1–P7. Numbers of tested PCs are n = 60 (from four wild-type mice, 45 cells studied blind to the mouse genotype) and n = 61 (from five Gαq mutant mice, 46 cells studied blind). (B) Similar to A, but with examples of CF-EPSCs of the wild type (P10, holding potential, −20 mV) and Gαq mutant (P10, holding potential, −20 mV) PCs and summary graph of the data from mice at P8–P14. Numbers of tested PCs are n = 97 (from six wild-type mice) and n = 58 (from four Gαq mutant mice). All cells were studied blind to the mouse genotype. (C) Similar to A and B, but with examples of CF-EPSCs of the wild type (P21, holding potential, −10 mV) and mutant (P18, holding potential, −10 mV) PCs and summary graph of the data from mice at P15–P21. Numbers of tested PCs are n = 105 (from six wild-type mice) and n = 62 (from four Gαq mutant mice). All cells were studied blind to the mouse genotype.
The difference of CF innervation between the wild-type and Gαq mutant mice during P22–P57 could be a result of the retardation of general cerebellar maturation in the Gαq mutant mice. To assess this possibility, we examined CF-EPSCs in older mice (P72–P109) (Fig. 4B Right). The differences between the frequency distributions of PCs during P22–P59 and those during P70–P109 were not significant for either the wild-type or the mutant mice (P > 0.05, χ2 test). Thus, retardation of general maturation is not likely to be the case.
Climbing Fiber Innervation During Early Postnatal Days.
To examine the state in early postnatal development when the abnormality becomes obvious in the Gαq mutant, we followed the developmental course of CF innervation. We observed that during the first postnatal week (P1–P7), the majority of PCs were multiply innervated by CFs in both wild-type and mutant mice (Fig. 5A). However, the frequency distribution of the number of CF-EPSC steps per PC in the mutant mice was not significantly different from that in the wild-type mice (P > 0.05, χ2 test). During the second postnatal week (P8–P14), the percentage of PCs with multiple CF-EPSC steps decreased markedly in both wild-type and mutant mice (Fig. 5B). The distributions were not significantly different between the two strains (P > 0.05, χ2 test). In contrast, the frequency distributions between the two strains during the third postnatal week (P15–P21) were markedly different (P < 0.01, χ2 test). This indicates that a higher percentage of the PCs were multiply innervated by CFs in the Gαq mutant than in the wild-type mice. These results suggest that regression of multiple CF innervation initially occurs normally, but the process is specifically impaired during the third postnatal week in the Gq mutant mice.
Other Electrophysiological Parameters of Gαq Mutant EPSCs Are Normal.
The kinetics of CF-EPSCs were normal in the Gαq mutant mice. There is no significant difference in either the 10–90% rise time or the decay time constant between the Gαq mutant and wild-type mice. Short-term synaptic plasticity examined by applying paired stimuli was normal. CF-EPSCs showed prominent paired-pulse depression in both mono-innervated and poly-innervated PCs derived from the Gαq mutant and wild-type mice. The magnitudes of paired-pulse depression were not significantly different between the wild-type and Gαq mutant mice at varying interpulse intervals. We also verified that the current–voltage relations of EPSCs were linear in the mono-innervated and double-innervated PCs from the wild-type and Gαq mutant. Furthermore, CF-EPSCs in both wild-type and Gαq mutant mice were not affected by an NMDA receptor blocker, DL-2-amino-5-phosphonopentanoate (AP5, 100 μM), but were totally suppressed by an AMPA receptor antagonist, 6-cyano-7-nitroquinoxaline-2,3-dion (CNQX, 10 μM). These results indicate that the CF-EPSCs of mature PCs from wild-type and Gαq mutant mice are exclusively mediated by the non-NMDA subtype of glutamate receptors.
The nature of EPSCs elicited by stimulation of PFs, the other major excitatory input to PCs, was examined in mice at P50–P54. There was no significant difference between the wild-type and Gαq mutant mice in the kinetics of PF-EPSCs. Furthermore, in both wild-type and Gαq mutant PCs, PF-EPSCs were affected little by AP5 (100 μM) but were totally suppressed by CNQX (10 μM). Taken together, the results suggest that basic properties of PF-PC excitatory synaptic transmission are largely normal in the Gαq mutant mice.
DISCUSSION
Impaired Regression of Multiple CFs in Gαq-Deficient Mice.
Our results demonstrate that the absence of Gαq results in motor discoordination, which is a result, at least in part, of cerebellar dysfunction. Deletion of the Gαq gene did not affect gross development of the cerebellar anatomy, cell production, cytodifferentiation, and formation of PF-PC synapses. Basic electrophysiological parameters, pharmacology, and paired-pulse responses of both CF- and PF-mediated excitatory transmission of PCs were largely normal in Gαq mutant mice. The most significant finding is that multiple CF innervation persists to a substantial degree in adult Gαq mutant mice. Regression of multiple CF innervation has been shown to occur in at least two distinct stages both in the rat (31) and in the mouse (21, 32). Our data show that regression of multiple CF innervation during the second stage, which corresponds approximately to P10–P20 (Fig. 5), appears to be specifically impaired in Gαq mutant mice. This indicates that Gαq is involved in the second phase of the CF synapse elimination process. The second phase occurs at the same time as the increased expression of Gαq in the cerebellar cortex (Fig. 3), which also corresponds to the time of the formation of PF-PC synapses. It is interesting that the heterozygous animals do not show defects in CF synapse elimination or motor coordination even though their Gαq levels are reduced. Thus, the effect is not strictly proportional to Gαq concentration. Our data do not bear directly on the mechanism of synapse elimination, but they argue that the assembly of a specific signal processing pathway incorporating Gαq is required for this function.
The Gαq mutant mice resemble the recently described PKCγ and mGluR1 mutant mice (19, 21) in that the regression of multiple CF innervation is impaired with no gross defects in the PF-PC synapses (19, 21). It is particularly significant that regression of multiple CF innervation occurs normally during the first 2 postnatal weeks in these three strains of mutant mice and that the deficiency becomes evident during the third postnatal week. This pattern is also consistent with the expression of PKCγ in the cerebellum (33). Thus, it is likely that both mGluR1 and Gαq mutant mice have defects in signal transduction cascades that lead to activation of PKCγ in PCs during the third postnatal week (see below).
Role of Gαq in Elimination of Multiple Climbing Fiber Innervation.
It has been shown that most cells express Gαq together with its close structural and functional homologue Gα11 (3). Our in situ hybridization results indicate that cerebellar PCs express both Gαq and Gα11 (data not shown). Both Western blot analysis of cerebellar membranes (Fig. 1D) and immunohistochemistry of wild-type and Gαq-deficient cerebellum (Fig. 3) indicate that although the levels of Gαq found in the cerebellar cortex exceeded those of Gα11, both proteins are present in PCs. We conclude that the remaining Gα11 is not able to compensate for the lack of Gαq. This conclusion is further supported by the finding that Gα11-deficient animals do not show any signs of motor discoordination (Tom Wilkie, personal communication) and none of the other G protein-deficient mice thus far generated, including Gαo-deficient (34) and Gαi2-deficient (35) animals, show ataxia. It is possible that Gαq and Gα11 have different subcellular localizations within cerebellar PCs and that localization restricts the interaction of mGluR1 with Gαq.
Gαq is known to couple activated receptors to the regulation of β isoforms of PLC. Activated PLC generates two intracellular messengers: inositol 1,4,5 trisphosphate (IP3) and diacylglycerol (DAG). IP3 releases Ca2+ from intracellular stores, whereas DAG activates PKC, in some cases together with Ca2+. We recently have found that mice lacking the β4 isoform of PLC exhibit motor discoordination and persistent multiple CF innervation of adult cerebellar PCs (M.K., M.W., M.I.S., and D. Wu, unpublished data). These findings suggest that elimination of multiple CF innervation during the third postnatal week involves a defined signaling cascade in cerebellar PCs that is composed of mGluR1, Gαq, PLC-β4, and PKCγ. All of these proteins may be tightly coupled in a multicomponent signaling structure. There is precedence for such complexes involving signaling proteins in other organisms (36, 37) and in the assembly of ion channels.
Both PKCγ- and mGluR1-deficient mice are impaired in motor coordination, and these mutant mice retain multiple CF innervation into adulthood (19, 21, 28, 38). The innervation of PCs by multiple CFs may disrupt the functional one-to-one relationship between the inferior olivery neurons and PCs. This may disturb a proper transfer of the “error” signals that are necessary for motor learning in the cerebellar cortex (38). We have found that Gαq mutant mice have persistent multiple CF innervation and impaired motor coordination similar to PKCγ and mGluR1 mutant mice. This further supports the hypothesis that CF mono-innervation is crucial for motor coordination.
Acknowledgments
We thank S. Pease, V. Mancino, Y.-H. Hu, and J. Silva for expert technical help and Dr. Jeansok J. Kim for help with the behavioral experiments. S.O. was a recipient of a fellowship from the Deutsche Forschungsgemeinschaft and the Guenther Foundation. This work has been partly supported by grants to M.K. from the Japanese Ministry of Education, Science, and Culture, by CREST (Core Research for Evolutional Science and Technology) of Japan Science and Technology Corporation (JST), and by a grant from the National Institutes of Health to M.I.S.
ABBREVIATIONS
- CF
climbing fiber
- PC
Purkinje cell
- PLC-β
phospholipase C-β
- ES
embryonic stem
- EPSC
excitatory postsynaptic current
- Gαq
guanine nucleotide binding protein Gq
References
- 1.Simon M I, Strathmann M P, Gautam N. Science. 1991;252:802–808. doi: 10.1126/science.1902986. [DOI] [PubMed] [Google Scholar]
- 2.Exton J H. Annu Rev Pharmacol Toxicol. 1996;36:481–509. doi: 10.1146/annurev.pa.36.040196.002405. [DOI] [PubMed] [Google Scholar]
- 3.Strathmann M P, Simon M I. Proc Natl Acad Sci USA. 1990;87:9113–9117. doi: 10.1073/pnas.87.23.9113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wilkie T M, Scherle P A, Strathman M P, Slepak V Z, Simon M I. Proc Natl Acad Sci USA. 1991;88:10049–10053. doi: 10.1073/pnas.88.22.10049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wange R L, Smrcka A V, Sternweis P C, Exton J H. J Biol Chem. 1991;266:11409–11412. [PubMed] [Google Scholar]
- 6.Wu D, Katz A, Lee C-H, Simon M I. J Biol Chem. 1992;267:25798–25802. [PubMed] [Google Scholar]
- 7.Offermanns S, Heiler E, Spicher K, Schultz G. FEBS Lett. 1994;349:201–204. doi: 10.1016/0014-5793(94)00667-9. [DOI] [PubMed] [Google Scholar]
- 8.Blank J L, Ross A H, Exton J H. J Biol Chem. 1991;266:18206–18216. [PubMed] [Google Scholar]
- 9.Hepler J R, Kozasa T, Smrcka A V, Simon M I, Rhee S G, Sternweis P C, Gilman A G. J Biol Chem. 1993;268:14367–14375. [PubMed] [Google Scholar]
- 10.Jiang H, Wu D, Simon M I. J Biol Chem. 1994;269:7593–7596. [PubMed] [Google Scholar]
- 11.Lee C-W, Lee K-H, Lee S B, Park D, Rhee S G. J Biol Chem. 1994;269:25335–25338. [PubMed] [Google Scholar]
- 12.Milligan G. J Neurochem. 1993;61:845–851. doi: 10.1111/j.1471-4159.1993.tb03595.x. [DOI] [PubMed] [Google Scholar]
- 13.Mailleux P, Mitchell F, Vanderhaeghen J-J, Milligan G, Erneux C. Neuroscience. 1992;51:311–316. doi: 10.1016/0306-4522(92)90317-u. [DOI] [PubMed] [Google Scholar]
- 14.Abe T, Sugihara H, Nawa H, Shigemoto R, Mizuno N, Nakanishi S. J Biol Chem. 1992;267:13361–13368. [PubMed] [Google Scholar]
- 15.Shigemoto R, Nakanishi S, Mizuno N. J Comp Neurol. 1992;322:121–135. doi: 10.1002/cne.903220110. [DOI] [PubMed] [Google Scholar]
- 16.Shigemoto R, Nomura S, Ohishi H, Sugihara H, Nakanishi S, Mizuno N. Neurosci Lett. 1993;163:53–57. doi: 10.1016/0304-3940(93)90227-c. [DOI] [PubMed] [Google Scholar]
- 17.Nakanishi S. Neuron. 1994;13:1031–1037. doi: 10.1016/0896-6273(94)90043-4. [DOI] [PubMed] [Google Scholar]
- 18.Pin J-P, Duvoisin R. Neuropharmacology. 1995;34:1–26. doi: 10.1016/0028-3908(94)00129-g. [DOI] [PubMed] [Google Scholar]
- 19.Kano M, Hashimoto K, Kurihara H, Watanabe M, Inoue Y, Aiba A, Tonegawa S. Neuron. 1997;18:71–79. doi: 10.1016/s0896-6273(01)80047-7. [DOI] [PubMed] [Google Scholar]
- 20.Crépel F. Trends Neurosci. 1982;5:266–269. [Google Scholar]
- 21.Kano M, Hashimoto K, Chen C, Abeliovich A, Aiba A, Kurihara H, Watanabe M, Inoue Y, Tonegawa S. Cell. 1995;83:1223–1232. doi: 10.1016/0092-8674(95)90147-7. [DOI] [PubMed] [Google Scholar]
- 22.Nagy A, Rossant J, Nagy R, Abramow-Newerly W, Roder J C. Proc Natl Acad Sci USA. 1993;90:8424–8428. doi: 10.1073/pnas.90.18.8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ramírez-Solis R, Davis A N, Bradley A. Methods Enzymol. 1993;225:855–878. doi: 10.1016/0076-6879(93)25054-6. [DOI] [PubMed] [Google Scholar]
- 24.Yamakuni T, Usui H, Iwanaga T, Kondo H, Takahashi Y. Neurosci Lett. 1984;45:235–240. doi: 10.1016/0304-3940(84)90232-5. [DOI] [PubMed] [Google Scholar]
- 25.Edwards F A, Konnerth A, Sakmann B, Takahashi T. Pflügers Arch. 1989;414:600–612. doi: 10.1007/BF00580998. [DOI] [PubMed] [Google Scholar]
- 26.Llano I, Marty A, Armstrong C M, Konnerth A. J Physiol (London) 1991;434:183–213. doi: 10.1113/jphysiol.1991.sp018465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kano M, Konnerth A. In: Practical Electrophysiological Methods. Kettenmann H, Grantyn R, editors. New York: Wiley–Liss; 1992. pp. 54–57. [Google Scholar]
- 28.Aiba A, Kano M, Chen C, Stanton M E, Fox G D, Herrup K, Zwingman T A, Tonegawa S. Cell. 1994;79:377–388. [PubMed] [Google Scholar]
- 29.Konnerth A, Llano I, Armstrong C M. Proc Natl Acad Sci USA. 1990;87:2662–2665. doi: 10.1073/pnas.87.7.2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Offermanns S, Toombs C F, Hu Y-H, Simon M I. Nature (London) 1997;389:183–186. doi: 10.1038/38284. [DOI] [PubMed] [Google Scholar]
- 31.Altmann J. J Comp Neurol. 1972;145:353–398. doi: 10.1002/cne.901450305. [DOI] [PubMed] [Google Scholar]
- 32.Mason C A, Christakos S, Catalano S. J Comp Neurol. 1990;297:77–90. doi: 10.1002/cne.902970106. [DOI] [PubMed] [Google Scholar]
- 33.Hashimoto T, Ase K, Sawamura S, Kikkawa U, Saito N, Tanaka C, Nishizuka Y. J Neurosci. 1988;8:1678–1683. doi: 10.1523/JNEUROSCI.08-05-01678.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Valenzuela D, Han X, Mende U, Fankhauser C, Mashimo H, Huang P, Pfeffer J, Neer E J, Fishman M C. Proc Natl Acad Sci USA. 1997;94:1727–1732. doi: 10.1073/pnas.94.5.1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rudolph U, Finegold M J, Rich S S, Harriman G R, Srinivasan Y, Brabet P, Boulay G, Bradley A, Birnbaumer L. Nat Genet. 1995;10:143–149. doi: 10.1038/ng0695-143. [DOI] [PubMed] [Google Scholar]
- 36.Chevesich J, Kreuz A J, Montell C. Neuron. 1997;18:95–105. doi: 10.1016/s0896-6273(01)80049-0. [DOI] [PubMed] [Google Scholar]
- 37.Tsunoda S, Sierralta J, Sun Y, Bodner R, Suzuki E, Becker A, Socolich M, Zuker C S. Nature (London) 1997;388:243–249. doi: 10.1038/40805. [DOI] [PubMed] [Google Scholar]
- 38.Chen C, Kano M, Abeliovich A, Chen L, Bao S, Kim J J, Hashimoto K, Thompson R F, Tonegawa S. Cell. 1995;83:1233–1242. doi: 10.1016/0092-8674(95)90148-5. [DOI] [PubMed] [Google Scholar]