

Abstract
Relapse remains a major challenge in the clinical management of acute myeloid leukemia (AML) and is driven by rare therapy‐resistant leukemia stem cells (LSCs) that reside in specific bone marrow niches. Hypoxia signaling maintains cells in a quiescent and metabolically relaxed state, desensitizing them to chemotherapy. This suggests the hypothesis that hypoxia contributes to the chemoresistance of AML‐LSCs and may represent a therapeutic target to sensitize AML‐LSCs to chemotherapy. Here, we identify HIFhigh and HIFlow specific AML subgroups (inv(16)/t(8;21) and MLLr, respectively) and provide a comprehensive single‐cell expression atlas of 119,000 AML cells and AML‐LSCs in paired diagnostic‐relapse samples from these molecular subgroups. The HIF/hypoxia pathway signature is attenuated in AML‐LSCs compared with more differentiated AML cells but is more expressed than in healthy hematopoietic cells. Importantly, chemical inhibition of HIF cooperates with standard‐of‐care chemotherapy to impair AML growth and to substantially eliminate AML‐LSCs in vitro and in vivo. These findings support the HIF pathway in the stem cell‐driven drug resistance of AML and unravel avenues for combinatorial targeted and chemotherapy‐based approaches to specifically eliminate AML‐LSCs.
INTRODUCTION
Acute myeloid leukemia (AML) is the most common acute leukemia in adults, constituting a heterogeneous group of disorders characterized by rapid expansion and accumulation of poorly differentiated myeloid cells in bone marrow (BM) and infiltrating tissues. Disease heterogeneity is well documented and patients are stratified based on cytogenetic, molecular, and immunophenotypic features. While our understanding of the characteristics of AML has substantially improved in recent years, many patients fail to respond to standard‐of‐care chemotherapy or show early relapse. 1 , 2
AML is a paradigm of hierarchical cancer stem cell model. 3 Robust experimental data demonstrate that relapse is mediated by a rare subpopulation of cells, termed leukemia stem cells (LSCs), which are chemotherapy‐resistant and drive disease recurrence, leading to a more genetically heterogeneous and clonally evolved AML. 4 , 5 , 6 AML‐LSCs share unique properties with normal hematopoietic stem and progenitor cells (HSPCs), including quiescence, resistance to apoptosis and elevated drug efflux, making them partially refractory to chemotherapy.
Hypoxia represents a self‐security mechanism to maintain cells in a dormant state, preventing their exhaustion and proliferative damage. Recent data suggest that the rate of oxygen consumption and cell metabolism, rather than oxygen perfusion, is responsible for the hypoxic nature of the BM niche where the LSCs/HSPCs reside. 7 , 8 Cells respond to hypoxia by activating specific pathways modulated by the hypoxia‐inducible factors (HIFs), which trigger the expression of hypoxia‐regulated genes with key roles in cell proliferation, survival, apoptosis, angiogenesis, metabolism, and differentiation. 9 At a molecular level, HIFs constitute a family of three related heterodimeric transcription factors (HIF‐1, HIF‐2, and HIF‐3) whose regulation depends on the oxygen‐dependent stabilization of an associated α subunit. Above 5% oxygen, the α subunit is degraded by the proteasome, whereas under hypoxic conditions, it is stabilized posttranslationally, dimerizes with the constitutively expressed β‐subunit, and promotes the transcription of HIF target genes. 9
The HIF/hypoxia pathway is important not only for steady‐state hematopoiesis but also for the initiation, progression, and chemoresistance of solid tumors and leukemias. Indeed, treatment‐resistant AML cells preferentially locate in the hypoxic endosteal BM niche, which offers protection from the pro‐apoptotic effects of the chemotherapeutic agent cytarabine (AraC). 10 Several studies have shown that, similar to HSPCs, 11 the loss of HIF‐1/2 leads to complete abrogation of LSCs in different types of human AML and murine models of chronic myeloid leukemia (CML), 12 , 13 , 14 whereas others reported that it does not impact LSCs in murine models of AML, or could even trigger a more severe leukemic phenotype. 15 , 16 , 17 , 18 , 19 Despite these conflicting observations, HIF‐inhibiting drugs are being actively explored as therapeutic agents for AML. 12 , 13 , 20 However, insufficient information is available regarding HIF in human primary AML‐LSCs, and importantly, the cytogenetic/molecular heterogeneity intrinsic to AML biology makes it plausible that the action of HIF varies among AML patients. Moreover, little is known about whether HIF inhibition sensitizes human AML‐LSCs to chemotherapy.
Here, we used single‐cell RNA sequencing (scRNA‐seq) to survey the transcriptome of human AML‐LSCs in paired diagnostic (Dx) and relapse (REL) samples from risk‐stratified patients with AML. Furthermore, we investigated the role of HIF/hypoxia signaling in LSC quiescence and chemoresistance using cutting‐edge in vitro and in vivo approaches.
MATERIALS/SUBJECTS AND METHODS
Primary human cells
The study was approved by the Institutional Ethical Review Board of Hospital Clinic of Barcelona (HCB/2018/0020). AML mononuclear cells were frozen in liquid nitrogen using fetal bovine serum (FBS) (Sigma) with 10% dimetylsulfoxide (DMSO) (Sigma). The mutational state of AML samples was analyzed on DNA extracted from total cells using Maxwell‐RSC Blood DNA Kit (Promega) and a next‐generation sequencing panel of mutations using the Oncomine Myeloid Research Assay (ThermoFisher).
Healthy HSPCs were obtained from Barcelona Blood and Tissue Bank upon Institutional Review Board approval (HCB/2018/0030). CD34+ cells were isolated using antihuman‐CD34 magnetic beads and the AutoMACS Pro‐Separator (Miltenyi Biotec) after Ficoll‐Hypaque gradient centrifugation (GE Healthcare).
Statistical analysis
Data are represented as mean ± standard error (SEM). Statistical comparisons between groups were assessed using two‐tailed unpaired Student's t‐tests, or paired Student's t‐tests (when analyzing data from the same AML samples subjected to different treatments) unless otherwise stated. Data distribution was assumed to be normal but this was not formally tested. All analyses were performed with Prism software, version 8.0 (GraphPad) and p < 0.05 was considered statistically significant.
Data and code availability
Newly generated scRNA‐seq and bulk RNA‐seq data have been deposited on the European Genome‐Phenome Archive (EGA) and are accessible through accession no. EGAS00001005980. All analyses and code used in this study are available at https://github.com/JLTrincado/scAML.
Additional methods can be found in Supporting Information.
RESULTS
Hypoxia transcriptional signature clusters inv(16) AML subgroup
To capture the contribution of the hypoxia pathway in human AML‐LSCs, we utilized three publicly available RNA‐seq datasets (TARGET, 21 including pediatric and adolescent/young patients, Leucegene 22 and BEAT‐AML, 23 including adult patients) with data from the following AML subgroups: inv(16) (p13q22), t(8;21), and NPM1mut as low‐risk AMLs; and MLL‐rearranged (MLLr), FLT3ITD, and normal karyotype (NK, neither chromosomal rearrangements nor NPM1mut or FLT3ITD) as intermediate‐high‐risk AMLs 24 (Table S1). AML samples mutated for TET2, IDH1, or IDH2 were excluded from analyses as they tend to interfere with HIF signaling. 25 , 26 We used either a multidimensional scaling reduction (MDS) of the genome‐wide information or a specific set of 119 hypoxia target genes (Hypoxia signature) characterized by the presence of functional hypoxia response elements 27 (Table S2, Figure 1A). The hypoxia signature enabled the clustering of inv(16) samples separately from the other cytogenetic groups (Figure 1A). The highest expression of HIF1A and HIF2A (EPAS1) was observed in inv(16) and t(8;21) samples, both core binding factor (CBF)‐rearranged AMLs, whereas MLLr samples showed a trend for the lowest expression (Figure 1B), especially in TARGET and Leucegene. Consistently, and in line with data from BloodSpot, 28 , 29 gene set enrichment analysis (GSEA) revealed a significant enrichment of the hypoxia signature in inv(16) but not in t(8;21) samples, as compared with MLLr samples in all datasets (Figure 1C). Therefore, we specifically focused further analyses on inv(16) and MLLr subgroups as HIFhigh and HIFlow AMLs, and low‐risk and intermediate‐high‐risk AMLs, respectively, although we also included t(8;21) as an additional CBF‐rearranged AML, which reported to cooperate with HIF1A for leukemogenesis. 30
Figure 1.
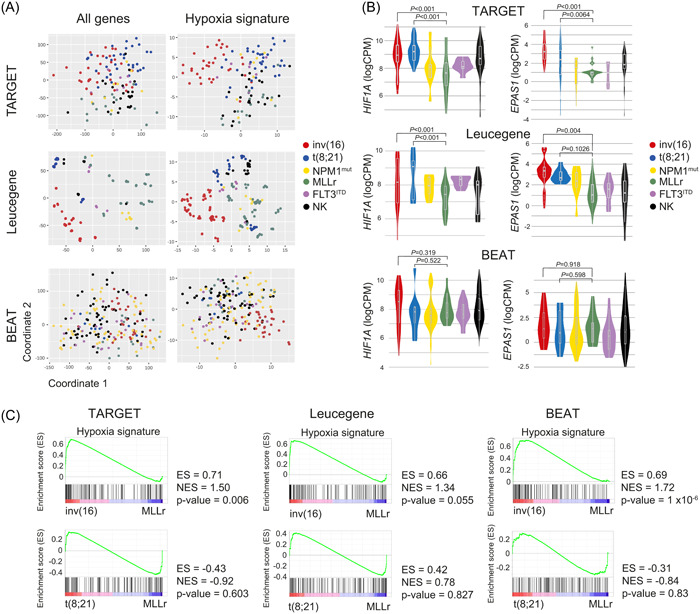
Hypoxia inducible factor (HIF) pathway gene expression signature in different acute myeloid leukemia (AML) cytogenetic subgroups. (A) Multidimensional scaling (MDS) representation of AML samples from TARGET (78 patient samples and 147 runs), Leucegene (72 patient samples and 301 runs), and BEAT‐AML (206 patients and 707 runs) datasets analyzing the expression of all the detected genes (left panels) or, specifically, the 119 HIF target genes (right panels). (B) Expression (LogCPM) of HIF1A and HIF2A (EPAS1) in each cytogenetic AML subgroup from TARGET, Leucegene, and BEAT‐AML. (C) Gene set enrichment analysis (GSEA) of the HIF pathway comparing inv(16) and t(8;21) with MLLr AMLs. CPM, counts per million; ES, enrichment score; NES, normalized enrichment score.
Identification of human AML‐LSCs using scRNA‐seq
To resolve the transcriptional heterogeneity of AML and to survey the expression of HIF pathway genes in AML‐LSCs, we performed scRNA‐seq on 11 Dx samples from pediatric/young adult patients: inv(16) (n = 4), t(8;21) (n = 3), and MLLr (n = 4) (Figure 2A,B, Supporting Information S1: Figure S1). Acknowledging the extremely low frequency of LSCs, 31 , 32 we analyzed two populations: CD34+CD38− cells, enriched in LSCs 3 , 31 , 33 and CD34−CD38+, differentiated cells depleted of LSCs (Figure 2C,D, Supporting Information S1: Figure S1). A total of 26,976, 19,731, and 24,854 cells were sequenced from inv(16), t(8;21), and MLLr subgroups, respectively (Table S3). Samples within each subgroup were computationally integrated and displayed using uniform manifold approximation and projection (UMAP) visualizations (Supporting Information S1: Figure S2A). Consistent with the immunophenotype (Supporting Information S1: Figure S1), inv(16) and t(8;21) samples expressed high CD34 levels (Figure 2E, Supporting Information S1: Figure S2A). By contrast, MLLr samples mainly consisted of CD34− cells, in line with previous studies. 34
Figure 2.
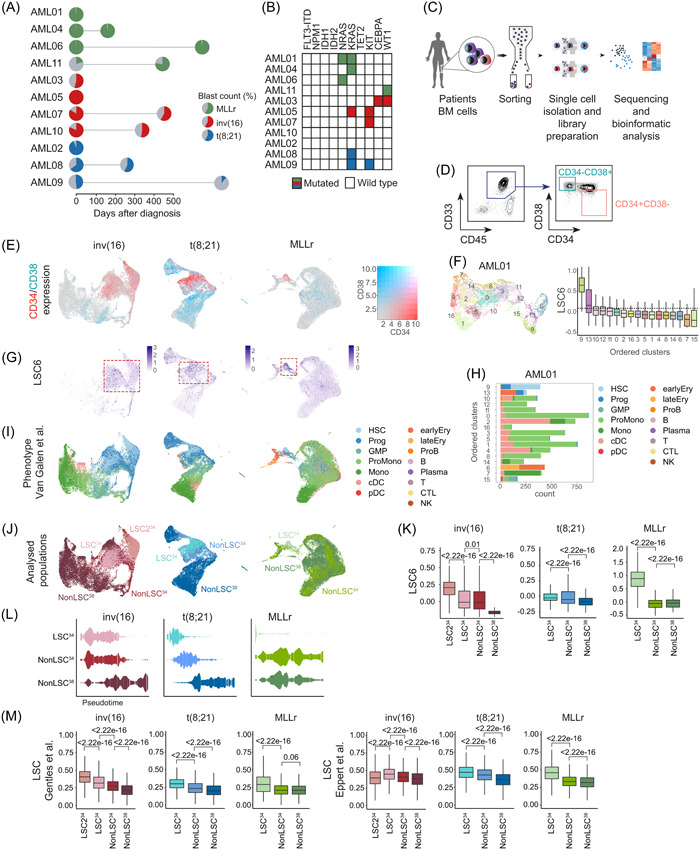
Enrichment and identification of the leukemia stem cell (LSC) compartment in the scRNA‐seq dataset. (A) Overview of the primary acute myeloid leukemia (AML) samples used for the scRNA‐seq analysis. The distinct cytogenetic subgroups are color‐coded. The colored area of the pie charts depicts the percentage of blasts. Paired‐relapsed samples are depicted with a second pie chart at the time of relapse. Further information on each sample can be found in Table S3. (B) Mutational profile of the analyzed samples. (C) Scheme depicting the different steps from sample sourcing to scRNA‐seq analysis. (D) Representative FACS profile depicting how the CD34+CD38− and CD34−CD38+AML cells were FACS‐purified for scRNA‐seq. The specific FACS profiles of each AML sample can be found in Supporting Information S1: Figure S1. (E) Uniform manifold approximation and projection (UMAP) plots showing the expression of CD34 and CD38 among all cells integrated from different samples in each cytogenetic subgroup. (F) UMAP plot showing the random clusterization of the cells from the sample AML01 and boxplot of the LSC6 score (Elsayed et al. 35 ) of each cluster for the identification of the LSC‐enriched cluster. Dotted line marks the 9th decile. (G) UMAP plots depicting the LSC6 score assigned to each cell. All cells from the different samples in each cytogenetic subgroup are integrated. Red square marks the LSC6‐enriched area. (H) Number of cells from each predicted phenotype, according to Van Galen et al., 36 included in each cluster identified in sample AML01. (I, J) UMAP plots showing the predicted phenotype of the cells according to Van Galen et al. 36 (I), and the assigned population (LSC34, nonLSC34, and nonLSC38) (J) for downstream analysis. All cells from the different samples in each cytogenetic subgroup are integrated. (K) LSC6 (Elsayed et al. 35 ) score of each of the defined populations (LSC34, nonLSC34, and nonLSC38). Nonparametric Wilcoxon test p‐values are shown for each comparison. (L) Trajectory/pseudotime analysis of the defined populations from the different cytogenetic subgroups. (M) Expression of the LSC signatures described by Gentles. 37 and Eppert et al. 31 in each of the defined populations for the different cytogenetic subgroups. Nonparametric Wilcoxon test p‐values are shown for each comparison. B, mature B cell; cDC, conventional dendritic cells; CTL, cytotoxic T lymphocyte; Ery, erythroid progenitor; FACS, fluorescence‐activated cell sorting; GMP, granulocyte‐macrophage progenitor; HSC, hematopoietic stem cell; log2FC, log2 fold change; Mono, monocyte; NK, natural killer cell; pDC, plasmacytoid dendritic cells; Plasma, plasma cell; ProB, B cell progenitor; Prog, progenitor; ProMono, promonocyte; T, naïve T cell.
As the LSC definition relies on functional assays, and CD34/CD38 are not absolute markers to identify LSCs, we used scRNA‐seq data to phenotypically categorize bona fide LSCs. We performed unsupervised clustering of all cells (11 patients) and utilized the recently published LSC6‐score, 35 an updated signature of the LSC17‐score adapted for pediatric‐young AML cell annotations. 38 Clusters from each sample were ranked according to LSC6‐score values, and only those with the highest LSC6‐score were considered enriched in LSCs (Figure 2F). When integrating samples from the same cytogenetic subgroup, we observed that cells identified as LSCs clustered together (Figure 2G). Notably, high LSC6‐scoring cells mostly colocalized with CD34+CD38− cells across the three subgroups.
We next queried the normal stem/progenitor phenotypic prediction of the LSC6 signature (Figure 2H,I). For this, we projected our scRNA‐seq data onto an existing reference annotation dataset containing 15 healthy hematologic cell types. 36 Each AML sample was first projected individually (Figure 2H) and then samples within the same subgroup were integrated (Figure 2I). Identically annotated clusters colocalized together, demonstrating a similar identity/phenotype across different patients. Total CD34+ cells and high LSC6‐scoring cells were enriched for hematopoietic stem cells (HSCs) and progenitors (Figure 2I). Data projection on an additional annotated dataset 39 confirmed the stemness phenotype of these cells (Supporting Information S1: Figure S2B,C).
This LSC‐enriched CD34+CD38− cluster (hereinafter, LSC34) was further characterized and compared with the remaining CD34+CD38− cells not identified as LSCs (hereinafter, nonLSC34) and with the CD34−CD38+ cells (hereinafter, nonLSC38) (Figure 2J). Of note, in an individual inv(16) sample (AML10), an additional LSC6high cluster was identified and classified as HSC/progenitors but with high expression of HBB (LSC234) (Supporting Information S1: Figure S2D). When the Dx‐AML samples were integrated, we consistently observed a lower LSC6 score from the LSC34 toward the more differentiated nonLSC38 (Figure 2K), in accord with the observed pseudotime trajectories of these populations along a continuum of differentiation from LSC34 to nonLSC38 (Figure 2L) or along the different predicted phenotypes (Supporting Information S1: Figure S2E,F) irrespective of the AML cytogenetic subgroup. In this line, two additional widely used LSC gene signatures 31 , 37 also correlated with the identified LSC34 clusters (Figure 2M).
Additionally, we analyzed the expression of 18 LSC markers 40 , 41 , 42 , 43 , 44 , 45 being consistently overexpressed in LSC34 in inv(16) samples (CD99, CD82, CD52, CD47, IL3RA), t(8;21) samples (CD99, CD52, and CD96), and in MLLr samples (CD99, CD82, CD52, and CD47), as compared with nonLSC34/nonLSC38 clusters (Supporting Information S1: Figure S2G). Finally, to rule out bias in gene expression analysis due to contaminating healthy HSCs/progenitors, 31 , 46 the expression of specific highly expressed genes in AML was compared against healthy cells from the Human Cell Atlas. 47 Results showed that CLEC12A 48 and JUND were overexpressed in AML cells across cytogenetic groups (Supporting Information S1: Figure S2H), whereas SPARC, or RUNX1T1 and POU4F1, or HOXA9, HOXA10, and PBX3 were specifically highly expressed in inv(16), t(8;21), or MLLr cells, respectively (Supporting Information S1: Figure S2I).
Transcriptional characterization identifies key molecular features of the AML‐LSCs
Recent studies have revealed the existence of dormant and active HSCs in mice 49 , 50 , 51 , 52 and humans, 53 while AML‐LSCs are documented to be quiescent/dormant. To characterize the transcriptional heterogeneity of human AML‐LSCs, we analyzed the cell cycle and quiescence/metabolic dormancy of LSC34 cells (Figure 3A,B). We took advantage of validated signatures defining the G0 phase (Neg‐G0‐to‐G1 [GO:0070317] and G0Mhigh 50 ) (Figure 3B and Table S2). LSC34 were consistently found in the G0/G1 cell cycle phase (Figure 3A), and the Neg‐G0‐to‐G1 and G0Mhigh dormancy signatures were enriched in LSC34 across the distinct subgroups (Figure 3B), revealing homogeneous LSC34 based on the G0 phase and/or quiescence status of the cells. We next analyzed the expression of different metabolic signatures previously related to both HSCs/LSCs and hypoxia signaling (Table S2). Glycolysis 43 signature was less represented in LSC34 in MLLr‐cells, similar to oxidative phosphorylation (OXPHOS) 54 (Figure 3C). However, OXPHOS was increased in LSC34 with respect to nonLSC38 in inv(16) cells. Reactive oxygen species (ROS) 43 and lysosome 55 signatures were less represented in the LSC34 across the distinct AML subgroups, consistent with lower ROS levels reported in HSCs/LSCs (Figure 3C). By contrast, LSC34 cells displayed an enrichment in translation signature consistent with a high protein production rate in these cells. 56 , 57 , 58 However, the ER stress signature 12 differed between LSC34 from distinct subgroups, being enriched in MLLr‐AMLs (Figure 3C).
Figure 3.
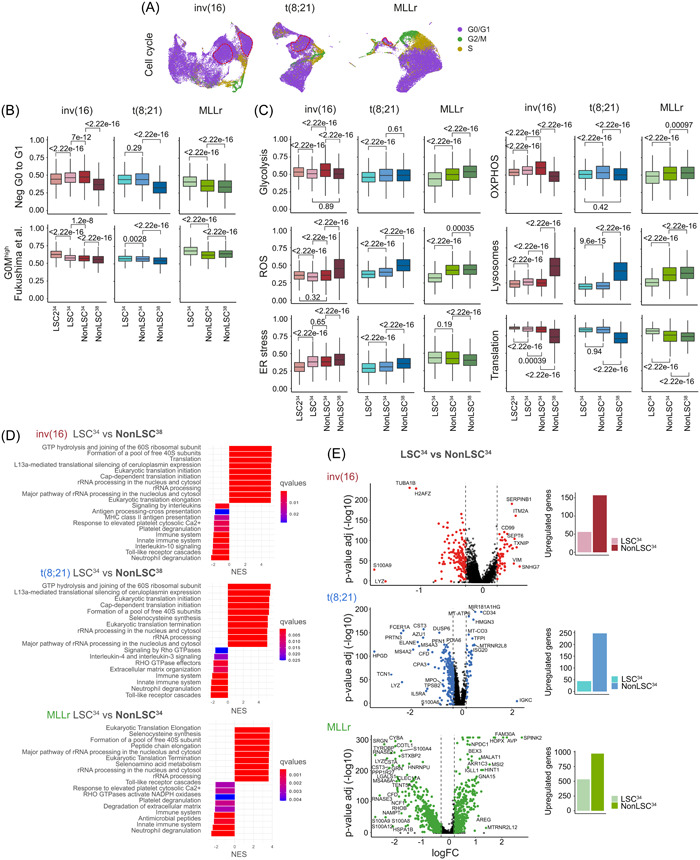
Cell cycle and metabolic characterization of the leukemia stem cells (LSC)34 cluster. (A) Uniform manifold approximation and projection (UMAP) plots showing the cell cycle phase prediction for each cell. Cells from all the different samples in each cytogenetic subgroup are integrated. (B) Quiescence status analysis of the defined populations from the different cytogenetic subgroups using the gene ontology (GO) signature Neg G0 to G1 (GO:0070 317) and the dormancy signature G0Mhigh described in Fukushima et al. 50 Nonparametric Wilcoxon test p‐values are shown for each comparison. (C) Analysis of different metabolic pathways related to stemness and hypoxia (Glycolysis, OXPHOS, reactive oxygen species [ROS], Lysosomes, ER stress, and translation) for the defined populations from the different cytogenetic subgroups. Nonparametric Wilcoxon test p‐values are shown for each comparison. (D) Gene set enrichment analysis (GSEA) showing the enriched biological pathways in the indicated populations of cells. For inv(16) and t(8;21) acute myeloid leukemia (AML), LSC34 cells are compared with nonLSC38 cells. For MLLr AML, LSC34 cells are compared with nonLSC34 cells. Complementary analyses are shown in Supporting Information S1: Figure S3. (E) Volcano plots showing the differentially expressed genes (DEGs) between LSC34 and nonLSC34 cells of each cytogenetic subgroup. Plots on the right show the total number of overexpressed genes in each population.
Unsupervised hierarchical clustering of the differentially expressed genes (DEGs) revealed that in CBF‐rearranged AMLs, LSC34 are transcriptionally closer to nonLSC34 than to nonLSC38 (Supporting Information S1: Figure S3A). Functional enrichment analysis revealed that the main altered functions between LSC34 and nonLSC38 were associated with Translation and other Ribosomal‐related processes (Figure 3D), whereas Mitosis or Cell cycle was the most altered between nonLSC34 and nonLSC38 (Supporting Information S1: Figure S3B). In contrast, the LSC34 in MLLr‐AML differed transcriptionally from both nonLSC34 and nonLSC38, which were transcriptionally closer (Supporting Information S1: Figure S3A), and revealing Translation and Ribosomal‐related processes as the main altered functions between LSC34 and both nonLSC34 and nonLSC38 (Figure 3D, Supporting Information S1: Figure S3B). Overall, a greater number of DEGs were highly expressed in nonLSC34 with respect to LSC34 (Figure 3E), suggesting a greater transcriptional activity. In total, 56, 44, and 573 genes were found to be highly expressed in LSC34 in inv(16), t(8;21), and MLLr‐AMLs, respectively (Supporting Information S1: Figure S4A), being only 10 genes common in the three subgroups (Supporting Information S1: Figure S4B).
Low expression of the hypoxia signature is detected in human AML‐LSCs
Having captured the transcriptional identity of LSC34 across the three subgroups, we sought to analyze the hypoxia signature. LSC34 consistently showed the lowest hypoxia score across all subgroups (Figure 4A,B), in line with the lowest expression of HIF1A (Figure 4C). To rule out potential bias in the selection of the hypoxia signature, we further employed five additional described hypoxia signatures (Figure 4D and Table S2) and confirmed a uniformly lower hypoxia score in LSC34 with a transition toward enrichment in hypoxia signature in nonLSC38. This trend was further confirmed in a different cohort of AML patients 59 (Figure 4E). Notably, while the hypoxia signature showed the lowest enrichment score in LSC34, it was consistently enriched in both total AML cells and LSC34 as compared with both healthy whole BM cells and CD34+ HSPCs, respectively (Figure 4F).
Figure 4.

Low expression of hypoxia signaling signature in human acute myeloid leukemia (AML)‐leukemia stem cells (LSCs). (A) Uniform manifold approximation and projection (UMAP) plots showing expression of the hypoxia signature in all cells integrated from the different samples in each cytogenetic subgroup. (B) Hypoxia signature score in each of the defined populations from the different cytogenetic subgroups. Nonparametric Wilcoxon test p‐values are shown for each comparison. (C) UMAP plots showing expression of the HIF1A gene in all cells integrated from the different samples in each cytogenetic subgroup. (D) Hypoxia signature score of each of the defined clusters comparing the hypoxia signature used in this study with five hypoxia signatures previously reported. (E) Hypoxia signature and HIF1A expression in AML cells from Beneyto‐Calabuig et al. 59 AML cohort. (F) Hypoxia expression signature comparing each AML cytogenetic subgroup with healthy total BM cells (left plot) or healthy hematopoietic stem cells (HSCs)/LSCs (right plot). Nonparametric Wilcoxon test p‐values are shown for each comparison. (G) Expression of the 119 genes from the hypoxia signature in each of the defined clusters. HIF1A targets significantly highly expressed in the LSC34 cluster are highlighted in brown color. (H) Violin plots showing the expression of the significantly overexpressed genes of the hypoxia signaling pathway in the LSC34 cluster in each cytogenetic AML subgroup.
Most of the HIF1A targets were highly expressed in the differentiated nonLSC38 cluster (Figure 4G). However, for several genes, the expression was significantly higher in LSC34: NPM1, CD99, KRT18, and LDHA in inv(16) samples; NPM1, CD99, PMAIP1, and TCF3 in t(8;21) samples; and NPM1, CD99, ANXA1, LDHA, BNIP3L, and NR4A1 in MLLr samples (Figure 4G,H). Together, although AML‐LSCs display a weak hypoxic signature across all the AML subgroups, specific hypoxia‐related genes were highly expressed in LSC34. Notably, the hypoxia signature was overexpressed throughout different tumoral populations compared with healthy HSPCs.
Paired Dx‐REL analysis reveals patient‐specific differential molecular features of the AML‐LSCs
Chemoresistant LSCs display biological features that differ from those of “therapy naïve” LSCs, including more diverse phenotypes, gene expression changes, and increased metabolic flexibility. 4 , 5 , 60 , 61 , 62 To study the evolution of chemoresistant LSCs underlying AML relapse, we performed scRNA‐seq in patient‐matched Dx‐REL samples (Figure 2A). In total, 12,005, 15,909, and 19,506 cells were sequenced from inv(16), t(8;21), and MLLr REL‐AML patients, respectively. LSC34 cluster was identified separately at Dx and REL before data integration (Figure 5A, Supporting Information S1: Figure S5A,B). Of note, the degree of transcriptional overlap between Dx‐REL pairs varied from patient to patient when the total number of cells was integrated (Figure 5A, Supporting Information S1: Figure S5A), suggesting patient‐specific transcriptional changes in Dx‐REL pairs. Similarly, analysis of the LSC6 score and hypoxia score in paired Dx‐REL samples also revealed patient‐specific heterogeneity with a trend toward an increased LSC6 score at REL (4/7) and an inverse Dx‐to‐REL evolution between hypoxia and LSC6 score (6/7) (Figure 5B,C, Supporting Information S1: Figure S5C,D). Finally, dormancy, ER stress, and ROS signatures also revealed a variable, patient‐specific evolution from Dx‐to‐REL in LSC34 irrespective of the AML subgroup (Figure 5D,E, Supporting Information S1: Figure S5E). Notably, the DEGs found in Dx‐LSC34 were highly expressed in REL‐LSC34 and varied between inv(16), t(8;21), and MLLr‐AMLs, highlighting subgroup‐specific differences (Supporting Information S1: Figure S5F).
Figure 5.
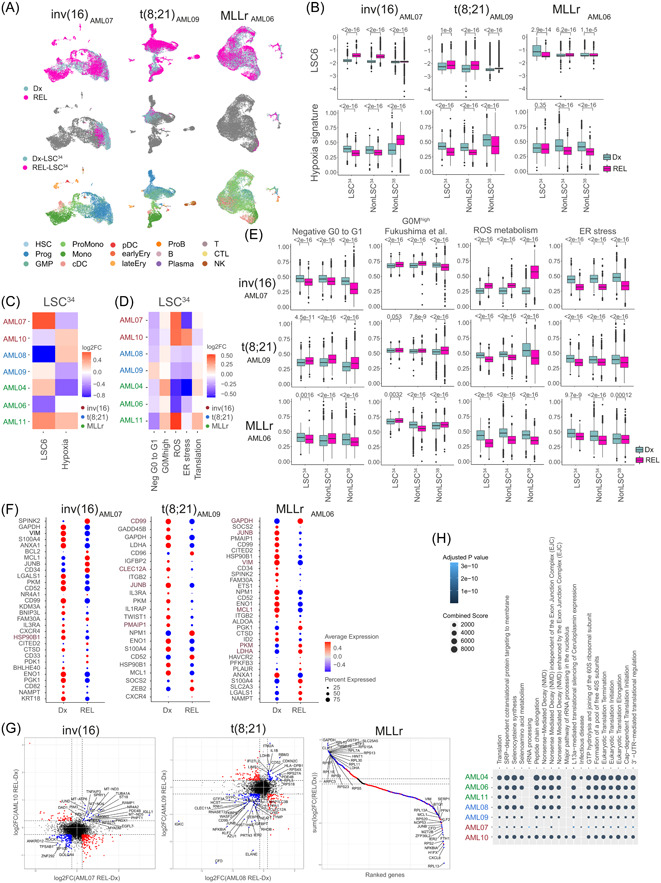
Relapse (REL)‐leukemia stem cells (LSC)34 cluster reveals patient‐specific differential molecular features. (A) Uniform manifold approximation and projection (UMAP) plots integrating patient‐matched acute myeloid leukemia (AML) cells at diagnostic (Dx) and REL (top plots), showing the identified LSC34 cluster at Dx and REL (middle plots) and showing the predicted phenotype according to Van Galen et al. 36 (bottom plots). One pair from each cytogenetic subgroup is shown. Additional paired samples are analyzed in Supporting Information S1: Figure S5A,B. (B) LSC6 score (top plots) and hypoxia signature score (bottom plots) of the defined clusters at Dx and REL for each AML cytogenetic subgroup. Nonparametric Wilcoxon test p‐values are shown for each comparison. (C, D) Heatmap of the variation of the LSC6 and hypoxia (C) and metabolic pathways (D) signature scores in the LSC34 population in the seven Dx‐REL pairs. (E) Score of indicated metabolic pathways related to stemness and hypoxia in the defined clusters at Dx and REL for each AML cytogenetic subgroup. Nonparametric Wilcoxon test p‐values are shown for each comparison. (F) Hypoxia inducible factor (HIF) target genes differentially expressed in the LSC34 population at Dx versus REL in each pair from the indicated patients. Additional paired samples are analyzed in Supporting Information S1: Figure S5G. Genes consistently higher or lower expressed in all patients from each AML subgroup are highlighted in brown color. (G) Comparison of the differentially expressed genes (DEGs) in the LSC34 population of each paired sample in each cytogenetic subgroup. For inv(16) and t(8;21) AMLs, plots compare two AML Dx‐REL pairs (AML07 and AML10 for inv[16]; AML08 and AML09 for t[8;21]). For MLLr AMLs, the plot compares three AML Dx‐REL pairs (AML04, AML06, and AML11). Each dot represents a gene with similar (in blue) or different (in red) expression in paired Dx versus REL samples. (H) Reactome showing biological pathways enriched in REL‐LSC34 cells compared to Dx‐LSC34 cells. B, mature B cell; cDC, conventional dendritic cells; CTL, cytotoxic T lymphocyte; Ery, erythroid progenitor; GMP, granulocyte‐macrophage progenitor; log2FC, log2 fold change; LSC, leukemic stem cell; Mono, monocyte; NK, natural killer cell; pDC, plasmacytoid dendritic cells; Plasma, plasma cell; ProB, B cell progenitor; Prog, progenitor; ProMono, promonocyte; T, naïve T cell.
Specifically, several HIF1A target genes were found differentially expressed in Dx‐ and REL‐LSC34 (Figure 5F, Supporting Information S1: Figure S5G). In inv(16)‐AMLs, HSP90B1 was consistently downregulated at REL in both patients. The same happens in t(8;21)‐AMLs with CD99, JUNB, CLEC12A, and PMAIP1. Finally, in MLLr‐AMLs, six genes showed a consistent Dx‐REL change (downregulation: JUNB, MCL1, and VIM; upregulation: GAPDH, LDHA, and PKM). In addition to the hypoxia targets, we analyzed those DEGs showing a consistent change (up‐ or down‐regulation) in LSC34 for each subgroup (Figure 5G), which identified EGFL7, CD52, as well as many ribosomal proteins consistently highly expressed in REL samples. Functional enrichment analysis using these genes revealed Translation‐related terms as the main altered functions in REL‐LSC34 cells (Figure 5H).
Inhibition of the HIF pathway sensitizes AML‐LSCs to chemotherapy in vitro
HIF signaling and hypoxic niches are reported to protect leukemic cells from chemotherapy by promoting quiescence and low metabolic activity. 63 , 64 , 65 We found that the HIF signature was consistently enriched in AML‐LSCs as compared with healthy HSCs (Figure 4F), prompting us to explore its therapeutic potential. For this, we combined the chemical inhibitor BAY87‐2243 (BAY87), which inhibits both HIF1A and HIF2A by preventing their protein accumulation under hypoxia, 19 , 66 with AraC, a standard‐of‐care chemotherapeutic in AML. 62 , 67 We first tested the effect of BAY87 on the expression of known HIF target genes in AML cell lines and primary cells and observed a significant reduction after 48 h treatment at 5% O2 (Figure 6A). We next performed long‐term culture‐initiating cell (LTC‐IC) assays to assess the impact of HIF inhibition on AML‐LSCs. Primary cells from six AML patients encompassing the three cytogenetic subgroups were treated for 48 h in hypoxic conditions (5% O2) with AraC, BAY87, or the combination, and a significant decrease in AML‐LSC frequency was consistently observed upon treatment with the combo (Figure 6B–D, Supporting Information S1: Figure S6A). We failed to observe a significant difference with BAY87 alone. However, AraC reduced LSC frequency, an effect that was more pronounced upon combination with BAY87 (Figure 6D). We then analyzed the expression of HIF target genes identified as differentially expressed in LSC34 after treatment (Figure 6E, Supporting Information S1: Figure S6B). BAY87‐treated cells (alone or in combination with AraC) showed a decrease in the expression of master genes related to glycolysis (ENO1 and PDK1) and apoptosis (BNIP3L and NR4A1), a decrease in the expression of the tumorigenic‐related gene KRT18, and an increase in ZEB1 expression, in line with its role as a stemness and tumor repressor in AML. 68 The chromosomal abnormalities were detected by FISH and/or qPCR at the end of treatments, confirming that LTC‐ICs originated from the original leukemic clone and not from residual healthy progenitors (Supporting Information S1: Figure S6C,D). Of note, the addition of BAY87 to AraC treatment did not impact apoptosis, cell cycle, or ROS content in therapy‐resistant AML cells (Figure 6F–H). To discard a potential unspecific effect of BAY87, we performed colony unit forming (CFU) assays with four additional primary AMLs, with different levels of HIF1A expression, using two additional HIF inhibitors, echinomycin and PX478 (Figure 6I). The combined treatment of AraC with either echinomycin or PX478 reduced the number of CFUs to a similar or even to a higher extent than BAY87 (Figure 6J). Importantly, however, when healthy cord blood (CB) CD34+ HSPCs were treated similarly, complete abolition of their clonogenic potential was observed upon treatment with AraC but we failed to observe any effect upon treatment with the different HIF inhibitors in comparison to controls (Figure 6K). These data indicate that HIF inhibition may specifically sensitize bulk AML cells and, more importantly, AML‐LSCs, to AraC, independently of the AML cytogenetic subgroup.
Figure 6.
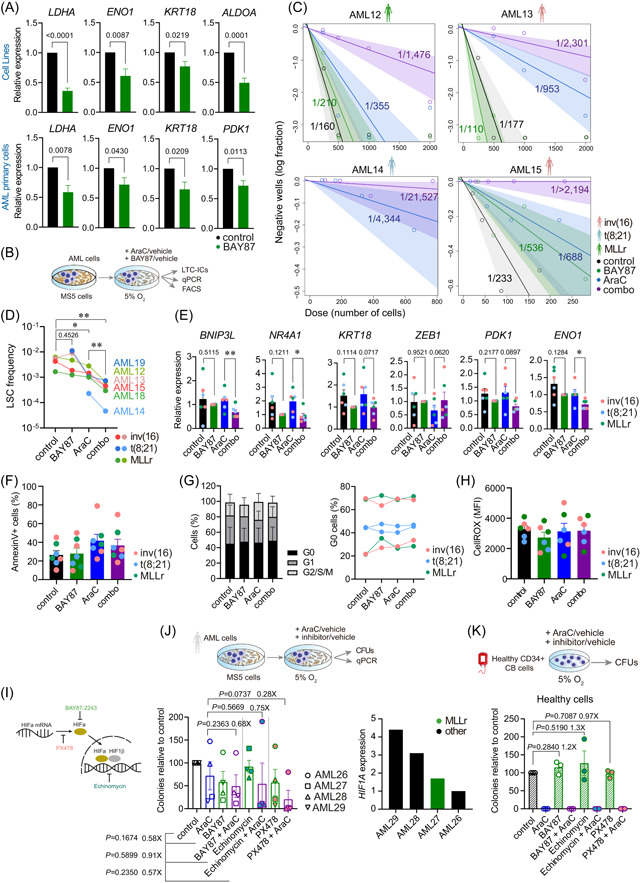
Inhibition of the hypoxia inducible factor (HIF) pathway sensitizes acute myeloid leukemia (AML)‐leukemia stem cells (LSCs) to chemotherapy in vitro. (A) Expression of the indicated HIF target genes after 48 h treatment at 5% O2 in n = 5 cell lines (ME1, Kasumi1, THP1, MV[4;11], and Molm13; upper panels) and in n = 8–9 AML primary samples (lower panels). Statistical significance was calculated using the paired Student's t‐test. Expression is normalized with respect to control samples. (B) Experimental overview for (C–H). Human AML primary cells were cultured over MS5 cells for 4 days and treated afterward with the indicated drugs for 48 h at 5% O2. After the treatment, cells were used for gene expression, flow cytometry, or long‐term culture‐initiating cells (LTC‐IC) assays (n = 15 wells/treatment and AML sample). (C) Estimation of the LSC frequency after the LTC‐IC assay was calculated using the ELDA software. (D) Impact of the indicated treatment on the LSC frequency for all the analyzed samples (n = 6) using data from the LTC‐IC assays. Statistical significance was calculated using the Ratio paired Student's t‐test. p‐Values are indicated for the AraC‐combo groups comparison. (E) Expression of the indicated HIF target genes identified in the scRNA‐seq analysis to be overexpressed in the LSC cluster after 48 h treatment with the indicated drugs at 5% O2 (n = 6 samples, AML03, AML16‐AML20; 2 per cytogenetic group). Statistical significance was calculated using the paired Student's t‐test. Expression is normalized with respect to the BAY87 samples. (F–H) Apoptosis quantification with Annexin V staining (F), cell cycle analysis by fluorescence‐activated cell sorting (FACS) (G), and reactive oxygen species (ROS) content measured using CellROX staining (H), in AML cells treated with the indicated drugs for 48 h at 5% O2 (n = 6 samples, AML03, AML16‐AML20). (I) Scheme depicting the mechanisms of action of the HIF inhibitor drugs used in (J, K). (J) Colony unit forming (CFU)‐assays from AML primary cells (n = 4) treated during 48 h with the indicated drugs at 5% O2. Right plot shows the HIF1A expression levels of each of the four AMLs used for this assay. (K) CFU‐assays from healthy CD34+ cord blood HSPCs (n = 3 donors) treated during 48 h with the indicated drugs at 5% O2. Data are shown as mean ± SEM. *p < 0.1, **p < 0.01, ***p < 0.001. Student's t‐test analysis.
Inhibition of the HIF pathway sensitizes AML‐LSCs to chemotherapy in vivo
We next aimed to address the impact of HIF inhibition alone or combined with AraC on AML‐LSCs in vivo with a special focus on engraftable MLLr‐AMLs (Figure 7A). NSG mice were intra‐BM‐transplanted with primary MLLr‐AML cells and were then randomized into the following treatment groups when AML levels were detectable in BM: control, AraC, BAY87, and combo (Supporting Information S1: Figure S7A). Primary xenografts were treated for five days and mice were sacrificed and analyzed 72 h later (Day 8), ensuring clearance of AraC. 62 , 69 Compared with control mice, peripheral cytopenias (Supporting Information S1: Figure S7B) were observed in AraC‐treated mice, confirming the cytoreductive/cytostatic effect of the treatment. We classified the primary AMLs as AraC‐sensitive if AraC treatment alone produced a reduction in the total tumor burden in BM compared to control mice, and AraC‐resistant if no reduction was observed. Notably, BAY87 with AraC reduces to a higher extent the leukemic burden in BM, both in AraC‐sensitive and AraC‐resistant AMLs (Figure 7A, Supporting Information S1: Figure S7C). A reduction of AML cells was also observed in the peripheral blood (PB), spleen, and liver (Supporting Information S1: Figure S7D). We partially observed this effect in BAY87‐treated samples versus control. However, it seems that BAY87 alone is less effective in reducing the total tumor cell burden, observing a significant reduction in only one sample. The clonogenic and stemness potential of the treated primograft cells were next assessed ex vivo in CFU assays. Primograft cells from combo‐treated mice produced a smaller number of colonies with less cellularity (Figure 7B) than counterparts from AraC‐treated mice, an effect that we did not observe in BAY87 samples compared to control. The presence of the MLLr was detected by FISH in cells collected from the CFUs, confirming that colonies originated from the transplanted MLLr‐leukemic cells (Figure 7B).
Figure 7.
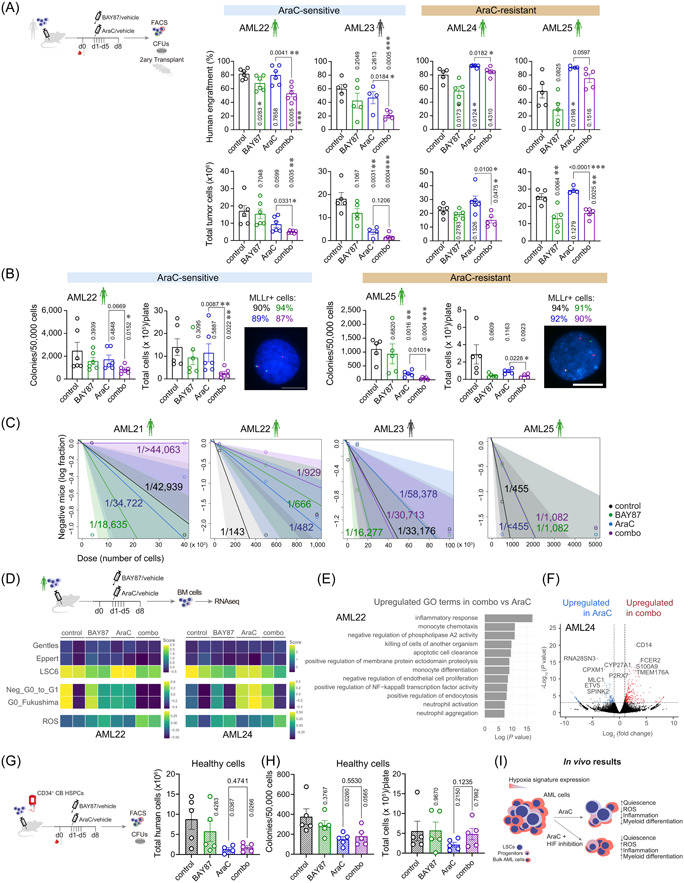
Inhibition of the hypoxia inducible factor (HIF) pathway sensitizes acute myeloid leukemia (AML)‐leukemia stem cells (LSCs) to chemotherapy in vivo. (A) Human AML‐engrafted mice were treated with the indicated drugs for 5 days. After completion of the treatment, organs were collected and analyzed by FACS. Cells from the BM were used for ex vivo CFU assays and secondary transplantations. Percentage (upper plots) of human engraftment in BM after treatment and total tumor cells (bottom plots) in bones (tibias, femurs, and hips) are shown (n = 4–6 mice/group). p‐Values of the comparison to control are shown in vertical. (B) Ex vivo clonogenic capacity of BM cells retrieved from mice treated as indicated (n = 4–6/group). Left plot shows the number of colonies per 50,000 plated cells. Right plot shows the total number of cells collected from each CFU plate. FISH analysis confirmed the leukemic identity of these cells. Percentages of MLLr+ cells are shown on top of the FISH image for each indicated treatment (n = 200 counted cells). Scale bar = 10 µm. p‐Values of the comparison to control are shown in vertical. (C) BM cells from treated primary mice were transplanted into secondary recipients at specific doses. LSC estimation in secondary recipients was calculated using ELDA software. Mice were considered leukemic when presenting >0.1% human cells in BM (n = 1–5 mice/dose and group). (D) RNA sequencing analysis was performed from BM cells from treated animals (n = 2 mice/group). Heatmap showing the expression of different LSCs, quiescence, and reactive oxygen species (ROS) signatures. (E) Gene ontology (GO) terms highly expressed in combo‐treated versus AraC‐treated samples. (F) Volcano plot showing the DEGs in AraC and combo‐treated samples. (G) Human CD34+CB cells‐engrafted mice were treated with the indicated drugs for 5 days. After completion of the treatment, organs were collected and analyzed by FACS. Cells from the BM were used for ex vivo CFU assays. Total human cells in bones (tibias, femurs, and hips) after treatment are shown (n = 4–5 mice/group). p‐Values of the comparison to control are shown in vertical. (H) Ex vivo clonogenic capacity of BM cells retrieved from mice treated as indicated (n = 4–6/group). Left plot shows the number of colonies per 50,000 plated cells. Right plot shows the total number of cells collected from each CFU plate. p‐Values of the comparison to control are shown in vertical. (I) Schematic diagram depicting the impact on AML cells of HIF inhibition over the treatment with AraC. BM, bone marrow; d, day; CFU, colony unit forming; FACS, fluorescence‐activated cell sorting; PB, peripheral blood; RBC, red blood cells; WBC, white blood cells. Data are shown as mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001. Student's t‐test analysis.
Limiting BM cell doses from treated primografts were next serially transplanted into secondary recipients to further assess the impact of the treatments on the long‐term leukemia‐initiating capacity of AML‐LSCs (Figure 7C, Supporting Information S1: Figure S7E). We found a reduction in LSC dose in combo‐treated versus AraC‐treated samples in four out of the five AMLs tested, being the discrepant sample a serially transplantable patient‐derived xenograft rather than a primary sample.
We then characterized the resistant primograft cells by looking at the expression of those HIF targets found differentially expressed in LSC34. We found a higher expression of HIF1A, ENO1, PDK1, ALDOA1, NPM1, BNIP3, and ANXA1 in resistant cells from combo‐treated mice than in AraC‐treated mice, indicating an increased activation of the hypoxia pathway (Supporting Information S1: Figure S7F). In fact, bulk‐RNA sequencing analysis showed a lower expression of LSCs and Quiescence signatures and a higher expression of ROS and Hypoxia signatures in BM cells retrieved from the combo‐treated than from AraC‐treated mice (Figure 7D, Supporting Information S1: Figure S7G). Of note, The LSC signature was very similar between BAY87‐ and combo‐treated cells, despite differences in leukemia burden reduction exerted by each treatment. Gene ontology (GO) analysis using most DEGs revealed many GO pathways related to inflammation and myeloid differentiation in combo‐treated versus AraC‐treated samples (Figure 7E, Supporting Information S1: Figure S7H). In fact, master myeloid genes were found overexpressed (CD14, FCER2, S100A9, and TMEM176A [for AML24] or ELANE, CXCL8, and C1QA [for AML22]) in combo‐treated samples whereas genes related to LSCs and stemness (SPINK2 or CPXM1) were found overexpressed in AraC‐treated samples (Figure 7F, Supporting Information S1: Figure S7I). Finally, we tested the effect of the combo‐treatment on healthy HSPCs in vivo and observed a similar reduction in the number of engrafted cells in AraC and combo‐treated animals (Figure 7G). BM cells from combo‐treated mice revealed a comparable clonogenic capacity to AraC‐treated cells (Figure 7H), indicating a similar sensitivity of the healthy HSPCs to both treatments. Collectively, in vivo assays support the in vitro results and demonstrate sensitization of BAY87 treatment to AraC cytoreductive therapy in debulking AML and eliminating AML‐LSCs in vivo (Figure 7I).
DISCUSSION
Although our understanding of the molecular and phenotypic features of AML is improving, many patients fail to respond to current treatments or exhibit early relapse. From the first reports of LSCs, leukemia ontogeny has been built upon paradigms of healthy hematopoiesis. 3 , 70 However, the classical view that LSCs are both rare and uniform has gradually been revisited based on AML. 70 Furthermore, studies investigating the biology of LSCs in AML use mainly murine models, and, more importantly, do not typically distinguish among the molecular subgroups used to risk‐stratify patients when primary patient samples are used.
Here, we provide an exhaustive analysis at the single‐cell level of the hypoxia/HIF signaling pathway in AML‐LSCs in paired Dx‐REL samples from pediatric/young adult risk‐stratified AML patients. Owing to the great heterogeneity of AML disease and the complex functional interactions of different fusion proteins with HIFs, we focused on three specific cytogenetic subgroups. We resolved the intercellular transcriptional heterogeneity using scRNA‐seq, which enabled us to identify and characterize the LSC population, providing a large comprehensive single‐cell expression atlas (119,000 cells) of AML cells and AML‐LSCs.
We confirmed several features previously described for LSCs, including several LSC signatures, low ROS content, a more quiescence state, and high activation of the translation process. These results are in accord with a published study analyzing 813 LSCs from five AML Dx‐REL matched samples. 71 Strikingly, we consistently found an inverse correlation between the hypoxia signature and cell stemness, manifested as a gradual enrichment in hypoxia signature from LSC34 to differentiating nonLSC34 and nonLSC38 cells. This contrasts with earlier reports showing a higher activation of HIFs in the LSC population. 13 Also, studies in healthy HSCs have shown the preferential expression of Hif‐1α in stem population 11 or in more differentiated fraction 72 of BM cells of different mouse models. This incongruity might be explained by the high heterogeneity of AML patients analyzed in previous studies in the absence of risk‐stratification, the different phenotypic strategies to identify bona fide LSCs, or even by the use of distinct murine‐based LSC readouts/approaches. A lowly expressed pathway may still have a key biological function. Thus, HIF pathway inhibition may sensitize both nonLSCs and LSCs to AraC treatment, thus favoring AML debulking and limiting LSC‐derived relapses.
A hypoxia risk signature with prognostic value has been proposed, 73 linking high HIF expression to shorter overall survival, similar to other studies. 30 , 72 , 74 Comparison of paired Dx‐REL samples enables the analysis of both therapy, naïve and therapy‐resistant LSCs, providing insights into their evolution within the same patient. In this sense, our transcriptomic analysis revealed a patient‐specific heterogeneity of both LSC6 and hypoxia scores in the seven paired Dx‐REL samples. The relatively low number of patients included in the present study, however, does not allow us to draw clinic‐biological conclusions.
Of note, and in line with other studies, 30 , 72 while LSC34 showed the lowest hypoxia enrichment score among the distinct analyzed clusters, it was nevertheless consistently enriched in both total AML cells and LSC34 cells when compared with both healthy whole BM cells and HSPCs, regardless of the cytogenetic subgroup. Moreover, therapy (AraC)‐resistant blasts have been reported to bind pimonidazole, an exogenous marker of hypoxia, 67 encouraging us to explore the chemosensitizer role of HIFs inhibition in human AML. Indeed, targeting HIF1A has been explored as a therapeutic strategy in different malignancies, 13 , 19 and its combination with AraC has also been tested in chronic lymphocytic leukemia, 75 CML, 76 and in JAK2V617F‐positive myeloproliferative neoplasms. 77 We found a reduction in the LSC frequency in vitro when combining BAY87 and AraC. These results are somehow different from the ones obtained with a conditional knock‐out mouse model of Hif‐1α, 17 but this may be explained by the transient inhibition of the pathway with the chemical model versus the permanent deletion of the gene, persistent after the chemotherapy treatment, in the genetic model. The results presented here are also in line with a previous report that tested the LSC dose in AML cells treated with AraC comparing normoxia and hypoxia culture conditions. 78 We observed a similar chemoprotective effect of the low oxygen conditions when chemically manipulating the oxygen‐sensing ability of the cells. We also found a significant effect of the BAY87 and AraC combination in vivo, decreasing not only the presence of total AML cells but also of LSCs, an effect unseen when these compounds were used individually. In line and different from previous studies, 13 , 19 treatment with BAY87 alone only showed limited benefit. Further studies are needed to clarify these discrepancies. The observed effect in vivo was, however, less dramatic than that observed in vitro. We speculate that the BM niche has a protective effect not present in the in vitro assays. Furthermore, optimization of the drug posology will be needed to completely unlock the potential of BAY87 as a chemosensitizer.
In sum, we provide the largest and most comprehensive single‐cell expression atlas (119,000 cells) of AML cells and AML‐LSCs in paired Dx‐REL samples from pediatric/young adult risk‐stratified human AML patients. Our data indicate that the HIF/hypoxia pathway is attenuated in AML‐LSC34 cells as compared with differentiated AML cells but it is enhanced when compared with healthy BM cells and HSPCs. Accordingly, chemical inhibition of the HIF pathway cooperates with standard‐of‐care chemotherapy to impair leukemogenesis in vitro and in vivo, substantially eliminating AML‐LSCs. These findings support the HIF pathway as a stem cell regulator in human AML and open new avenues for combinatorial targeted and chemotherapy‐based treatments to specifically eliminate AML‐LSCs.
AUTHOR CONTRIBUTIONS
Talia Velasco‐Hernandez conceived the study, designed and performed experiments, analyzed and interpreted data, prepared figures, and wrote the manuscript. Juan L. Trincado analyzed and interpreted scRNA‐seq data, prepared figures, and wrote the manuscript. Adria Closa, Narcís Fernández‐Fuentes, and Eduardo Eyras analyzed and interpreted bulk RNA‐seq data. Meritxell Vinyoles, Alba Martínez‐Moreno, Francisco Gutiérrez‐Agüera, Oscar Molina, Virginia C. Rodríguez‐Cortez, Pau Ximeno‐Parpal, Paolo Petazzi, Paola Romecin, Raquel Casquero, Rafael D. de la Guardia, and Patricia Lorden performed experiments. Sergi Beneyto‐Calabuig, Lars Velten, and Fernando Abollo‐Jiménez performed bioinformatic analyses. Alex Bataller, Hélène Lapillonne, Ronald W. Stam, Susana Vives, Montserrat Torrebadell, and Jose L. Fuster provided human primary samples. Clara Bueno, Jean‐Emmanuel Sarry, and Holger Heyn supported the study technically and with scientific discussions. Pablo Menéndez conceived the study, designed experiments, interpreted data, wrote the manuscript, and financially supported the study. All authors have read and agreed to publish the manuscript.
CONFLICT OF INTEREST STATEMENT
Pablo Menéndez is the founder of the spin‐off OneChain Immunotherapeutics, which has no connection with the present research. The other authors declare no conflict of interest.
FUNDING
Competitive financial support for this work was obtained from the Deutsche José Carreras Leukämie‐Stiftung (DJCLS15R/2021) to Pablo Menéndez and Talia Velasco‐Hernandez. This research was also supported by the Spanish Ministry of Economy and Competitiveness (SAF2016‐80481R, PID2019‐108160RB‐I00/AEI/10.13039/50110011033), “la Caixa” Foundation (LCF/PR/HR19/52160011), the Leo Messi Foundation, “Heroes hasta la médula” initiative, Departament de Recerca i Universitats de la Generalitat de Catalunya (2021 SGR 00887) and ISCIII‐RICORS within the Next Generation EU program (plan de recuperación, transformación y resiliencia) to PM and the Health Institute Carlos III (ISCIII/FEDER, PI20/00 822) to Clara Bueno.
Supporting information
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
ACKNOWLEDGMENTS
We thank Dr. Aleix Prat, Dr. Remi Safi, Néstor Tirado, and Mercedes Guerrero for technical help and Dr. Fernando Anjos‐Afonso for technical discussions and advice. We thank the Finnish Hematology Registry and Clinical Biobank (FHRB), Instituto Aragonés de Ciencias de la Salud (IACS), and VIVO Biobank for providing AML samples. We thank CERCA/Generalitat de Catalunya and Fundació Josep Carreras‐Obra Social “la Caixa” for core support. Talia Velasco‐Hernandez was supported by a Marie‐Sklodowska Curie Fellowship (792923) and an investigator fellowship from AECC (INVES223069VELA). Juan L. Trincado was supported by a Juan de la Cierva fellowship (FJC2019‐040868‐I). Oscar Molina was supported by an investigator fellowship from AECC (INVES211226MOLI).
Contributor Information
Talia Velasco‐Hernandez, Email: tvelasco@carreraresearch.org.
Pablo Menéndez, Email: pmenendez@carreraresearch.org.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are openly available in EGA at https://ega.crg.eu/, reference number EGAS00001005980.
REFERENCES
- 1. Döhner H, Wei AH, Löwenberg B. Towards precision medicine for AML. Nat Rev Clin Oncol. 2021;18(9):577‐590. [DOI] [PubMed] [Google Scholar]
- 2. Döhner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424‐447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3(7):730‐737. [DOI] [PubMed] [Google Scholar]
- 4. Ho TC, LaMere M, Stevens BM, et al. Evolution of acute myelogenous leukemia stem cell properties after treatment and progression. Blood. 2016;128(13):1671‐1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shlush LI, Mitchell A, Heisler L, et al. Tracing the origins of relapse in acute myeloid leukaemia to stem cells. Nature. 2017;547(7661):104‐108. [DOI] [PubMed] [Google Scholar]
- 6. Thomas D, Majeti R. Biology and relevance of human acute myeloid leukemia stem cells. Blood. 2017;129(12):1577‐1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature. 2014;505(7483):327‐334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schito L, Rey S, Konopleva M. Integration of hypoxic HIF‐α signaling in blood cancers. Oncogene. 2017;36(38):5331‐5340. [DOI] [PubMed] [Google Scholar]
- 9. Semenza GL. Targeting HIF‐1 for cancer therapy. Nat Rev Cancer. 2003;3(10):721‐732. [DOI] [PubMed] [Google Scholar]
- 10. Ishikawa F, Yoshida S, Saito Y, et al. Chemotherapy‐resistant human AML stem cells home to and engraft within the bone‐marrow endosteal region. Nat Biotechnol. 2007;25(11):1315‐1321. [DOI] [PubMed] [Google Scholar]
- 11. Takubo K, Goda N, Yamada W, et al. Regulation of the HIF‐1α level is essential for hematopoietic stem cells. Cell Stem Cell. 2010;7(3):391‐402. [DOI] [PubMed] [Google Scholar]
- 12. Rouault‐Pierre K, Lopez‐Onieva L, Foster K, et al. HIF‐2α protects human hematopoietic stem/progenitors and acute myeloid leukemic cells from apoptosis induced by endoplasmic reticulum stress. Cell Stem Cell. 2013;13(5):549‐563. [DOI] [PubMed] [Google Scholar]
- 13. Wang Y, Liu Y, Malek SN, Zheng P, Liu Y. Targeting HIF1α eliminates cancer stem cells in hematological malignancies. Cell Stem Cell. 2011;8(4):399‐411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang H, Li H, Xi HS, Li S. HIF1α is required for survival maintenance of chronic myeloid leukemia stem cells. Blood. 2012;119(11):2595‐2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Guitart AV, Subramani C, Armesilla‐Diaz A, et al. Hif‐2α is not essential for cell‐autonomous hematopoietic stem cell maintenance. Blood. 2013;122(10):1741‐1745. [DOI] [PubMed] [Google Scholar]
- 16. Velasco‐Hernandez T, Hyrenius‐Wittsten A, Rehn M, Bryder D, Cammenga J. HIF‐1α can act as a tumor suppressor gene in murine acute myeloid leukemia. Blood. 2014;124(24):3597‐3607. [DOI] [PubMed] [Google Scholar]
- 17. Velasco‐Hernandez T, Soneji S, Hidalgo I, Erlandsson E, Cammenga J, Bryder D. Hif‐1α deletion may lead to adverse treatment effect in a mouse model of MLL‐AF9‐driven AML. Stem Cell Rep. 2019;12(1):112‐121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Velasco‐Hernandez T, Tornero D, Cammenga J. Loss of HIF‐1α accelerates murine FLT‐3ITD‐induced myeloproliferative neoplasia. Leukemia. 2015;29(12):2366‐2374. [DOI] [PubMed] [Google Scholar]
- 19. Vukovic M, Guitart AV, Sepulveda C, et al. Hif‐1α and Hif‐2α synergize to suppress AML development but are dispensable for disease maintenance. J Exp Med. 2015;212(13):2223‐2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Frolova O, Samudio I, Benito JM, et al. Regulation of HIF‐1α signaling and chemoresistance in acute lymphocytic leukemia under hypoxic conditions of the bone marrow microenvironment. Cancer Biol Ther. 2012;13(10):858‐870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bolouri H, Farrar JE, Triche T Jr., et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age‐specific mutational interactions. Nat Med. 2018;24(1):103‐112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lavallée VP, Gendron P, Lemieux S, D'Angelo G, Hébert J, Sauvageau G. EVI1‐rearranged acute myeloid leukemias are characterized by distinct molecular alterations. Blood. 2015;125(1):140‐143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bottomly D, Long N, Schultz AR, et al. Integrative analysis of drug response and clinical outcome in acute myeloid leukemia. Cancer Cell. 2022;40(8):850‐864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Papaemmanuil E, Dohner H, Campbell PJ. Genomic classification in acute myeloid leukemia. N Engl J Med. 2016;375(9):900‐901. [DOI] [PubMed] [Google Scholar]
- 25. Ward PS, Patel J, Wise DR, et al. The common feature of leukemia‐associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting α‐ketoglutarate to 2‐hydroxyglutarate. Cancer Cell. 2010;17(3):225‐234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Young RM, Simon MC. Untuning the tumor metabolic machine: HIF‐α: pro‐ and antitumorigenic? Nat Med. 2012;18(7):1024‐1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Percio S, Coltella N, Grisanti S, Bernardi R, Pattini L. A HIF‐1 network reveals characteristics of epithelial‐mesenchymal transition in acute promyelocytic leukemia. Genome Med. 2014;6(12):84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wierenga ATJ, Cunningham A, Erdem A, et al. HIF1/2‐exerted control over glycolytic gene expression is not functionally relevant for glycolysis in human leukemic stem/progenitor cells. Cancer Metab. 2019;7:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bagger FO, Sasivarevic D, Sohi SH, et al. BloodSpot: a database of gene expression profiles and transcriptional programs for healthy and malignant haematopoiesis. Nucl Acids Res. 2016;44(D1):D917‐D924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gao XN, Yan F, Lin J, et al. AML1/ETO cooperates with HIF1α to promote leukemogenesis through DNMT3a transactivation. Leukemia. 2015;29(8):1730‐1740. [DOI] [PubMed] [Google Scholar]
- 31. Eppert K, Takenaka K, Lechman ER, et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat Med. 2011;17(9):1086‐1093. [DOI] [PubMed] [Google Scholar]
- 32. Sarry JE, Murphy K, Perry R, et al. Human acute myelogenous leukemia stem cells are rare and heterogeneous when assayed in NOD/SCID/IL2Rγc‐deficient mice. J Clin Invest. 2011;121(1):384‐395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lapidot T, Sirard C, Vormoor J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367(6464):645‐648. [DOI] [PubMed] [Google Scholar]
- 34. Zeisig BB, Fung TK, Zarowiecki M, et al. Functional reconstruction of human AML reveals stem cell origin and vulnerability of treatment‐resistant MLL‐rearranged leukemia. Sci Transl Med. 2021;13(582):eabc4822. [DOI] [PubMed] [Google Scholar]
- 35. Elsayed AH, Rafiee R, Cao X, et al. A six‐gene leukemic stem cell score identifies high risk pediatric acute myeloid leukemia. Leukemia. 2020;34(3):735‐745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. van Galen P, Hovestadt V, Wadsworth Ii MH, et al. Single‐cell RNA‐seq reveals AML hierarchies relevant to disease progression and immunity. Cell. 2019;176(6):1265‐1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gentles AJ. Association of a leukemic stem cell gene expression signature with clinical outcomes in acute myeloid leukemia. JAMA. 2010;304(24):2706‐2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ng SWK, Mitchell A, Kennedy JA, et al. A 17‐gene stemness score for rapid determination of risk in acute leukaemia. Nature. 2016;540(7633):433‐437. [DOI] [PubMed] [Google Scholar]
- 39. Triana S, Vonficht D, Jopp‐Saile L, et al. Single‐cell proteo‐genomic reference maps of the hematopoietic system enable the purification and massive profiling of precisely defined cell states. Nat Immunol. 2021;22(12):1577‐1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Garg S, Reyes‐Palomares A, He L, et al. Hepatic leukemia factor is a novel leukemic stem cell regulator in DNMT3A, NPM1, and FLT3‐ITD triple‐mutated AML. Blood. 2019;134(3):263‐276. [DOI] [PubMed] [Google Scholar]
- 41. Herrmann H, Sadovnik I, Eisenwort G, et al. Delineation of target expression profiles in CD34+/CD38‐ and CD34+/CD38+ stem and progenitor cells in AML and CML. Blood Adv. 2020;4(20):5118‐5132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ho JM, Dobson SM, Voisin V, et al. CD200 expression marks leukemia stem cells in human AML. Blood Adv. 2020;4(21):5402‐5413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lagadinou ED, Sach A, Callahan K, et al. BCL‐2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell. 2013;12(3):329‐341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pabst C, Bergeron A, Lavallée VP, et al. GPR56 identifies primary human acute myeloid leukemia cells with high repopulating potential in vivo. Blood. 2016;127(16):2018‐2027. [DOI] [PubMed] [Google Scholar]
- 45. Vetrie D, Helgason GV, Copland M. The leukaemia stem cell: similarities, differences and clinical prospects in CML and AML. Nat Rev Cancer. 2020;20(3):158‐173. [DOI] [PubMed] [Google Scholar]
- 46. Levine JH, Simonds EF, Bendall SC, et al. Data‐driven phenotypic dissection of AML reveals progenitor‐like cells that correlate with prognosis. Cell. 2015;162(1):184‐197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Regev A, Li B, Kowalczyk MS, et al. The Census of Immune Cells. BioStudies; 2020. https://www.ebi.ac.uk/biostudies/studies/S-SUBS12 [Google Scholar]
- 48. Bakker ABH, van den Oudenrijn S, Bakker AQ, et al. C‐type lectin‐like molecule‐1. Cancer Res. 2004;64(22):8443‐8450. [DOI] [PubMed] [Google Scholar]
- 49. Cabezas‐Wallscheid N, Buettner F, Sommerkamp P, et al. Vitamin A‐retinoic acid signaling regulates hematopoietic stem cell dormancy. Cell. 2017;169(5):807‐823. [DOI] [PubMed] [Google Scholar]
- 50. Fukushima T, Tanaka Y, Hamey FK, et al. Discrimination of dormant and active hematopoietic stem cells by G0 marker reveals dormancy regulation by cytoplasmic calcium. Cell Rep. 2019;29(12):4144‐4158. [DOI] [PubMed] [Google Scholar]
- 51. Rodriguez‐Fraticelli AE, Wolock SL, Weinreb CS, et al. Clonal analysis of lineage fate in native haematopoiesis. Nature. 2018;553(7687):212‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wilson A, Laurenti E, Oser G, et al. Hematopoietic stem cells reversibly switch from dormancy to self‐renewal during homeostasis and repair. Cell. 2008;135(6):1118‐1129. [DOI] [PubMed] [Google Scholar]
- 53. Kaufmann KB, Zeng AGX, Coyaud E, et al. A latent subset of human hematopoietic stem cells resists regenerative stress to preserve stemness. Nat Immunol. 2021;22(6):723‐734. [DOI] [PubMed] [Google Scholar]
- 54. Sriskanthadevan S, Jeyaraju DV, Chung TE, et al. AML cells have low spare reserve capacity in their respiratory chain that renders them susceptible to oxidative metabolic stress. Blood. 2015;125(13):2120‐2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Liang R, Arif T, Kalmykova S, et al. Restraining lysosomal activity preserves hematopoietic stem cell quiescence and potency. Cell Stem Cell. 2020;26(3):359‐376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bajaj J, Hamilton M, Shima Y, et al. An in vivo genome‐wide CRISPR screen identifies the RNA‐binding protein Staufen2 as a key regulator of myeloid leukemia. Nat Cancer. 2020;1(4):410‐422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Stevens BM, Khan N, D'Alessandro A, et al. Characterization and targeting of malignant stem cells in patients with advanced myelodysplastic syndromes. Nat Commun. 2018;9(1):3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wu J, Xiao Y, Sun J, et al. A single‐cell survey of cellular hierarchy in acute myeloid leukemia. J Hematol Oncol. 2020;13(1):128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Beneyto‐Calabuig S, Merbach AK, Kniffka JA, et al. Clonally resolved single‐cell multi‐omics identifies routes of cellular differentiation in acute myeloid leukemia. Cell Stem Cell. 2023;30(5):706‐721. [DOI] [PubMed] [Google Scholar]
- 60. Jones CL, Inguva A, Jordan CT. Targeting energy metabolism in cancer stem cells: progress and challenges in leukemia and solid tumors. Cell Stem Cell. 2021;28(3):378‐393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Jones CL, Stevens BM, D'Alessandro A, et al. Inhibition of amino acid metabolism selectively targets human leukemia stem cells. Cancer Cell. 2018;34(5):724‐740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Boyd AL, Aslostovar L, Reid J, et al. Identification of chemotherapy‐induced leukemic‐regenerating cells reveals a transient vulnerability of human AML recurrence. Cancer Cell. 2018;34(3):483‐498. [DOI] [PubMed] [Google Scholar]
- 63. Saito Y, Uchida N, Tanaka S, et al. Induction of cell cycle entry eliminates human leukemia stem cells in a mouse model of AML. Nat Biotechnol. 2010;28(3):275‐280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Rouault‐Pierre K, Hamilton A, Bonnet D. Effect of hypoxia‐inducible factors in normal and leukemic stem cell regulation and their potential therapeutic impact. Expert Opin Biol Ther. 2016;16(4):463‐476. [DOI] [PubMed] [Google Scholar]
- 65. Schito L, Semenza GL. Hypoxia‐inducible factors: master regulators of cancer progression. Trends Cancer. 2016;2(12):758‐770. [DOI] [PubMed] [Google Scholar]
- 66. Ellinghaus P, Heisler I, Unterschemmann K, et al. BAY 87‐2243, a highly potent and selective inhibitor of hypoxia‐induced gene activation has antitumor activities by inhibition of mitochondrial complex I. Cancer Med. 2013;2(5):611‐624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Farge T, Saland E, de Toni F, et al. Chemotherapy‐resistant human acute myeloid leukemia cells are not enriched for leukemic stem cells but require oxidative metabolism. Cancer Discovery. 2017;7(7):716‐735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Almotiri A, Alzahrani H, Menendez‐Gonzalez JB, et al. Zeb1 modulates hematopoietic stem cell fates required for suppressing acute myeloid leukemia. J Clin Invest. 2021;131(1):e129115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Liliemark JO, Gahrton G, Paul CY, Peterson CO. ara‐C in plasma and ara‐CTP in leukemic cells after subcutaneous injection and continuous intravenous infusion of ara‐C in patients with acute nonlymphoblastic leukemia. Semin Oncol. 1987;14(2 suppl 1):167‐171. [PubMed] [Google Scholar]
- 70. Pollyea DA, Jordan CT. Therapeutic targeting of acute myeloid leukemia stem cells. Blood. 2017;129(12):1627‐1635. [DOI] [PubMed] [Google Scholar]
- 71. Stetson LC, Balasubramanian D, Ribeiro SP, et al. Single cell RNA sequencing of AML initiating cells reveals RNA‐based evolution during disease progression. Leukemia. 2021;35(10):2799‐2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Forristal CE, Brown AL, Helwani FM, et al. Hypoxia inducible factor (HIF)‐2α accelerates disease progression in mouse models of leukemia and lymphoma but is not a poor prognosis factor in human AML. Leukemia. 2015;29(10):2075‐2085. [DOI] [PubMed] [Google Scholar]
- 73. Jiang F, Mao Y, Lu B, Zhou G, Wang J. A hypoxia risk signature for the tumor immune microenvironment evaluation and prognosis prediction in acute myeloid leukemia. Sci Rep. 2021;11(1):14657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Deeb G, Vaughan MM, McInnis I, et al. Hypoxia‐inducible factor‐1α protein expression is associated with poor survival in normal karyotype adult acute myeloid leukemia. Leuk Res. 2011;35(5):579‐584. [DOI] [PubMed] [Google Scholar]
- 75. Griggio V, Vitale C, Todaro M, et al. HIF‐1α is over‐expressed in leukemic cells from TP53‐disrupted patients and is a promising therapeutic target in chronic lymphocytic leukemia. Haematologica. 2020;105(4):1042‐1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Qiu S, Sheth V, Yan C, et al. Metabolic adaptation to tyrosine kinase inhibition in leukemia stem cells. Blood. 2023;142(6):574‐588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Baumeister J, Chatain N, Hubrich A, et al. Hypoxia‐inducible factor 1 (HIF‐1) is a new therapeutic target in JAK2V617F‐positive myeloproliferative neoplasms. Leukemia. 2020;34(4):1062‐1074. [DOI] [PubMed] [Google Scholar]
- 78. Griessinger E, Anjos‐Afonso F, Pizzitola I, et al. A niche‐like culture system allowing the maintenance of primary human acute myeloid leukemia‐initiating cells: a new tool to decipher their chemoresistance and self‐renewal mechanisms. Stem Cells Transl Med. 2014;3(4):520‐529. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Data Availability Statement
Newly generated scRNA‐seq and bulk RNA‐seq data have been deposited on the European Genome‐Phenome Archive (EGA) and are accessible through accession no. EGAS00001005980. All analyses and code used in this study are available at https://github.com/JLTrincado/scAML.
Additional methods can be found in Supporting Information.
The data that support the findings of this study are openly available in EGA at https://ega.crg.eu/, reference number EGAS00001005980.