

Abstract
INTRODUCTION
Although large‐scale genome‐wide association studies (GWAS) have been conducted on AD, few have been conducted on continuous measures of memory performance and memory decline.
METHODS
We conducted a cross‐ancestry GWAS on memory performance (in 27,633 participants) and memory decline (in 22,365 participants; 129,201 observations) by leveraging harmonized cognitive data from four aging cohorts.
RESULTS
We found high heritability for two ancestry backgrounds. Further, we found a novel ancestry locus for memory decline on chromosome 4 (rs6848524) and three loci in the non‐Hispanic Black ancestry group for memory performance on chromosomes 2 (rs111471504), 7 (rs4142249), and 15 (rs74381744). In our gene‐level analysis, we found novel genes for memory decline on chromosomes 1 (SLC25A44), 11 (BSX), and 15 (DPP8). Memory performance and memory decline shared genetic architecture with AD‐related traits, neuropsychiatric traits, and autoimmune traits.
DISCUSSION
We discovered several novel loci, genes, and genetic correlations associated with late‐life memory performance and decline.
Highlights
Late‐life memory has high heritability that is similar across ancestries.
We discovered four novel variants associated with late‐life memory.
We identified four novel genes associated with late‐life memory.
Late‐life memory shares genetic architecture with psychiatric/autoimmune traits.
Keywords: Alzheimer's disease, genetics, GWAS, memory
1. BACKGROUND
Over the last several years, multiple genome‐wide association studies (GWAS) have explored the genetic characteristics of late onset Alzheimer's disease (AD) dementia, 1 , 2 , 3 , 4 and converging evidence demonstrates that it is a highly heritable (≈60% to 80%) polygenic disease. 5 , 6 , 7 While clinical AD diagnosis has been the focus of most AD‐related GWAS, memory performance has received less attention even though it is a strong endophenotype for AD. Memory performance is a particularly interesting cognitive trait to investigate because it is a robust clinical feature of AD and is often one of the first signs of cognitive impairment to clinically manifest. Memory is also a highly heritable trait 8 , 9 that appears to have a genetic architecture linked to AD, with a recent study on verbal short‐term memory and learning in healthy adults identifying several AD‐relevant loci (eg, apolipoprotein E [APOE]/APOC1/TOMM40, CDH18) 8 and another study suggesting a role of cytoskeleton dynamics in episodic memory maintenance. 10 Therefore, disentangling the genetic architecture of memory performance over the course of normal aging and AD may provide insight into the molecular pathways that contribute to differential risk and resilience to AD.
A major challenge in performing large‐scale genomic analysis of memory performance is that many studies use disparate measures to quantify memory abilities, making integration and meta‐analysis challenging. Recently, the Phenotype Harmonization Consortium (PHC) was established within the Alzheimer's Disease Sequencing Project (ADSP) to provide robust harmonization of phenotypes including cognition across the studies of ADSP, including a recent flagship publication demonstrating a robust latent variable modeling approach to cross‐cohort harmonization that provided the foundation for the present analysis. 11 In the present study, we included harmonized memory performance measures from multiple cohorts (Adult Changes in Thought [ACT], Alzheimer's Disease Neuroimaging Initiative [ADNI], National Alzheimer's Coordinating Center [NACC], Religious Orders Study/Rush Memory and Aging Project/Minority Aging Research Study [ROS/MAP/MARS]) to perform the largest longitudinal GWAS to date on memory performance and memory decline in aging adults with and without cognitive impairment. This cross‐ancestry GWAS on memory performance (n = 27,633) and memory decline (n = 22,365) included self‐identified non‐Hispanic White (NHW, n = 24,216) and non‐Hispanic Black (NHB, n = 3417) individuals to provide a comprehensive picture of the genetic architecture of memory performance in late life. Our analyses include narrow‐sense heritability estimates, common variant associations, gene‐ and pathway‐level analyses, and genetic correlation analyses. We hypothesized that the genetic architecture of memory performance would partially reflect the genetic architecture of AD, while also highlighting novel loci that contribute to normal aging and AD.
RESEARCH IN CONTEXT
Systematic review: We used PubMed and Google Scholar to review literature that had reported genome‐wide associations studies (GWAS) on memory performance and decline. Although prior research has suggested that memory is a highly heritable trait, a large‐scale, cross‐ancestry GWAS has yet to be conducted in older adults.
Interpretation: We demonstrated that memory performance and decline are both highly heritable traits across ancestries and these traits are highly associated with AD. We identified several novel variants and genes that associated with memory performance and decline.
Future directions: Our study emphasizes the importance of incorporating different ancestries into large‐scale GWAS of continuous measures of memory performance and decline. Future studies that continue to increase sample size will facilitate the discovery of potential treatment targets.
2. METHODS
2.1. Participants
The present study leveraged multiple cognitive aging cohorts from the ADSP, including the ACT, ADNI, NACC, and ROS/MAP/MARS cohorts. ACT began in Seattle in 1994 and has since then amassed a cohort of 4960 cognitively unimpaired individuals. 12 ADNI (https://adni.loni.usc.edu) began in 2003 as a public‐private partnership, led by Principal Investigator, Michael W. Weiner, MD. The primary goal of the ADNI cohort is to test whether serial magnetic resonance imaging (MRI), positron emission tomography (PET), other biological markers, and clinical and neuropsychological assessment can be combined to measure the progression of mild cognitive impairment and early AD. 13 Since 2003, the ADNI cohort has progressed through four different phases (ADNI 1, ADNI‐GO, ADNI 2, and ADNI 3), all of which are included in the present study. ADNI recruits cognitively unimpaired, mild cognitive impairment, and AD dementia participants. A full list of ADNI investigators can be found in Appendix 1. The NACC cohort began in 1999 and is comprised of dozens of Alzheimer's Disease Research Centers that collect multimodal AD data. 14 The overall intention of the NACC cohort is to collate a large database of standardized clinical/neuropathological data. 15 , 16 , 17 , 18 The ROS is an ongoing longitudinal study which started in 1994 with the goal of building a large clinical‐pathologic cohort of aging and AD. 19 Recruitment for ROS includes 65+ year‐old Catholic nuns, priests, and brothers from more than 40 groups throughout the United States. 19 The MAP began in 1997 and is an ongoing longitudinal study that enrolls and follows cognitively unimpaired participants. 20 The MARS began in 2004 and enrolls and follows 65+ year‐old African American participants who are cognitively unimpaired at study entry. The ROS/MAP/MARS cohorts are all actively collecting longitudinal data. Across all cohorts, written informed consent was provided by participants and research was conducted in accordance with approved Institutional Review Board protocols. Secondary analysis of these data was approved by the Vanderbilt University Medical School Institutional Review Board. Table 1 provides an overview of the ACT, ADNI, NACC, and ROS/MAP/MARS cohorts.
TABLE 1.
Participant characteristics by cohort and ancestry.
| non‐Hispanic White (NHW) | non‐Hispanic Black (NHB) | |||||||
|---|---|---|---|---|---|---|---|---|
| Measure | ACT | ADNI | NACC | ROS/MAP/MARS | ACT | ADNI | NACC | ROS/MAP/MARS |
| Number of participants | 3585 | 1363 | 17,159 | 2109 | 102 | 50 | 2742 | 523 |
| Number of sessions | 18,337 | 7217 | 68,628 | 19,888 | 481 | 254 | 10,374 | 4022 |
| Number of visits | 6.79 (3.00) | 7.29 (3.30) | 6.37 (3.58) | 12.91 (5.77) | 6.29 (2.86) | 7.12 (3.13) | 6.22 (3.58) | 10.07 (4.23) |
| Follow‐up time (years) | 7.33 (4.94) | 3.06 (2.72) | 4.06 (2.92) | 6.87 (5.02) | 7.05 (4.76) | 3.21 (2.89) | 4.15 (2.98) | 5.80 (4.02) |
| Baseline age (years) | 74.34 (6.50) | 74.33 (6.61) | 73.97 (8.22) | 78.78 (7.43) | 73.39 (5.58) | 72.07 (5.80) | 73.27 (7.80) | 72.99 (6.40) |
| Education (years) | 15.02 (3.21) | 16.06 (2.78) | 15.93 (2.84) | 16.36 (3.53) | 13.38 (3.66) | 15.08 (3.26) | 14.33 (3.14) | 14.90 (3.53) |
| APOE ε4 (% positive) | 26.16 | 44.16 | 40.81 | 24.99 | 33 | 46 | 46.41 | 35.95 |
| Baseline diagnosis (% CU/MCI/AD) | 100/0/0 | 38.5/47.3/14.2 | 50.6/26.3/23.1 | 70.8/23.9/5.3 | 100/0/0 | 46.0/40.0/14.0 | 54.3/26.1/19.6 | 71.6/26.7/1.7 |
Note: Values denoted as mean (standard deviation) or frequency.
Abbreviations: ACT, Adult Changes in Thought; AD, Alzheimer's disease; ADNI, Alzheimer's Disease Neuroimaging Initiative; APOE, apolipoprotein E; CU, cognitively unimpaired; MAP, Memory and Aging Project; MARS, Minority Aging Research Study; MCI, mild cognitive impairment; NACC, national Alzheimer's coordinating center; ROS, religious orders study.
2.2. Cognitive harmonization
Neuropsychological data were collected independently for each cohort and subsequently harmonized. We have published methods for our cognitive data harmonization. 11 This harmonization process involved experts assigning test item‐level data into memory, executive function, language, visuospatial, or “none of” domains. Investigators ensured identical scoring of anchor items across studies and a confirmatory factor analysis was conducted to choose the best single factor or bi‐factor model. Anchor items were items identified as having been administered and scored precisely the same way in two or more cohorts. All items had freely estimated parameters, with anchor items forced to have the same parameters across studies. We used these co‐calibrated parameters for anchor and study‐specific items to generate cognitive scores that were on the same scale across cohorts and across waves within each cohort. 11 Although harmonized cognitive scores were created for the memory, executive function, language, and visuospatial domains, the present study focused on memory. Full details on the items used in the memory co‐calibration analysis can be found in the Supplemental Materials (Tables S1‐S5).
Two memory outcomes were included in this study: baseline memory performance and memory decline. For the baseline memory performance analysis, we considered the memory score from the first cognitive visit for each participant available in the dataset. For the memory decline analysis, we conducted a linear mixed‐effects regression to calculate a longitudinal trajectory for each participant. Importantly, participants were only included in the linear mixed‐effects regression analysis if they had at least two cognitive visits. Memory slopes (ie, memory decline) were calculated with a null linear mixed‐effects regression model, letting slope and intercept vary for each participant. These baseline memory performance and memory decline scores were then used as endophenotypes for all GWAS and post‐GWAS analyses.
2.3. Genetic data quality control and imputation
Raw genetic data were collected with a variety of genotyping arrays across—and within—cohorts. For ACT, genetic data were collected with two arrays (Illumina Human660W‐Quad Array and Infinium Global Screening Array‐24 BeadChip). For ADNI, genetic data were collected with four different arrays (Illumina Human610‐Quad BeadChip, Illumina HumanOmniExpress BeadChip, Illumina Omni 2.5 M, and Illumnia Global Screening Array v2). NACC is a consortium of 37+ Alzheimer's Disease Research Centers (ADRCs), and several different arrays were used to collect genetic data—acquisition of all genetic data is outlined on the NACC website (https://naccdata.org/nacc‐collaborations/partnerships). The ROS/MAP/MARS cohort data were collected with three different arrays (Global Screening Array‐24 v3.0 BeadChip, Affymetrix GeneChip 6.0, Illumina HumanOmniExpress). Identical and robust quality control and imputation pipelines were performed for each chip/cohort. 21 First, variants which had a low genotype rate (<95%), low minor allele frequency (MAF; <1%), or were outside of Hardy‐Weinberg equilibrium (p < 1 × 10−6) were removed. Participants were excluded if the reported and genotypic sex differed, or there was poor genotyping efficiency (missing >1% of variants), or cryptic relatedness was present (PIHAT > 0.25). Imputation was performed on the University of Michigan Imputation Server using the TOPMed reference panel (hg38) 22 with SHAPEIT phasing.
Following imputation, datasets were filtered to exclude variants with low imputation quality (R2 < 0.8), duplicated/multi‐allelic variants, and MAF < 1%. Within each self‐identified racial group (NHW and NHB), principal component analysis was conducted and genetic ancestry outliers were excluded.
2.4. Statistical analyses
2.4.1. Single nucleotide polymorphism‐heritability tests
We conducted ancestry‐aware single nucleotide polymorphism (SNP)‐heritability tests using the Genome‐Wide Complex Trait Analysis (GCTA) pipeline. 23 For the NHW and NHB meta‐analyses of memory performance and decline, we used the restricted maximum likelihood with the genetic relatedness matrices tool to calculate heritability estimates. We then used an equation, , to determine if heritability estimates differed by ancestry, 24 and the p‐value was extracted from the normal distribution.
2.4.2. Genome‐wide association testing and meta‐analysis
Memory performance and decline GWAS were conducted in each cohort and ancestry group (ie, NHW or NHB) separately using PLINK (Version 1.9, https://www.cog‐genomics.org/plink/1.9). 25 Covariates included age, sex, and the first five genetic ancestry principal components. Significance was set a priori to p = 5 × 10−8 and we also evaluated suggestive loci which approach significance at p = 1 × 10−5. NHW and NHB memory performance and memory decline GWAS were followed with an ancestry‐specific fixed effects meta‐analysis using GWAMA, 26 and variants were filtered to only include those present in at least three of the four cohorts. Following ancestry‐specific meta‐analyses, a cross‐ancestry fixed effects meta‐analysis was performed across NHW and NHB GWAS for baseline memory performance and decline, and variants were filtered to only include those present in both ancestry groups. Importantly, GWAS were only included in ancestry‐specific meta‐analyses if at least 50 participants were present in cohort specific GWAS. For this reason, ADNI was not included in the NHB memory decline meta‐analysis or any subsequent analyses.
2.4.3. Expression quantitative trait locus analyses
Variants reaching genome‐wide significance—and suggestive variants approaching significance—were mapped to genes and functionally annotated using several databases, including GTEx (https://gtexportal.org), 27 , 28 eQTLGen Consortium (whole blood; https://www.eqtlgen.org), 29 Brain xQTLServe (http://mostafavilab.stat.ubc.ca/xqtl/), 30 BrainSeq (dorsolateral prefrontal cortex and hippocampus [DLPFC]; http://eqtl.brainseq.org), 31 and MetaBrain (https://www.metabrain.nl). 32 The expression quantitative trait locus (eQTL) significance threshold was set a priori at p < 0.05, and eQTL significance was determined by listed p‐values in each respective database.
2.4.4. Gene‐ and pathway‐level analysis
Gene‐ and pathway‐level analyses were conducted using the Multi‐marker Analysis of GenoMic Annotation (MAGMA v1.09) 33 software on each ancestry‐specific meta‐analysis and the cross‐ancestry meta‐analysis for both memory performance and memory decline. All results were corrected for multiple comparisons using the false discovery rate (FDR) procedure (p < 0.05).
2.4.5. Genetic correlation analysis
The NHW within‐ancestry meta‐analysis results for memory performance and decline were used to perform genetic correlation analysis with the GWAS of 65 other complex traits using the Genetic Covariance Analyzer (GNOVA) program. 34 For example, one complex trait included cognitive performance from a prior meta‐analysis of the COGENT and UK Biobank cohorts. 35 Genetic correlations analysis results were corrected for multiple comparisons using the FDR approach. We focused solely on the NHW within‐ancestry meta‐analysis given that all prior complex traits focused on NHW ancestry.
2.4.6. Sensitivity analyses
Sensitivity analyses included stratifications based on clinical diagnosis at baseline, in which analyses were subset to cognitively unimpaired participants only and cognitively impaired participants only (ie, mild cognitive impairment or AD dementia diagnosis). Additionally, all analyses were repeated after removing participants with any of a number of 17 comorbidities (eg, frontotemporal dementia, depression—see Table S6 for a full description). Detailed results from our main analysis and all sensitivity analyses can be found in the Supplemental Tables.
2.4.7. Replication analyses
All variant‐ and gene‐level associations that reached genome‐wide significance were replicated using publicly available data from FinnGen (https://r8.finngen.fi/). The FinnGen study is focused on establishing genotype‐phenotype correlations in the Finnish population, and the current database includes data from 342,499 individuals. Several outcomes in the FinnGen database were evaluated, including “Alzheimer's disease, wide definition,” “Alzheimer's disease (Late onset),” “Alzheimer disease,” “Dementia in Alzheimer disease,” “Alzheimer's disease (Early onset),” “Alzheimer's disease (undefined),” and “Alzheimer's disease (Atypical or mixed).” Full details on the derivation and sample size of these phenotypes can be found at https://r8.risteys.finngen.fi/phenocode. Moreover, we extracted reported SNPs from prior memory‐specific GWAS from the Cohorts for Heart and Aging Research in GEnomic Genomic Epidemiology (CHARGE), 8 , 36 UK Biobank (UKBB), 37 and Cognitive Genomics Consoirtum (COGENT) 38 cohorts.
2.5. Data availability
All phenotype and genetic data used in this analysis are available on NIAGADS (https://dss.niagads.org/). Other phenotype data available through the ADSP‐PHC may be browsed on a data curation tool housed at Vanderbilt (https://vmacdata.org/adsp‐phc). All summary statistics are also available on NIAGADS. The results published here are in whole or in part based on data obtained from the Accelerating Medicines Partnerships ‐ Alzheimer's Disease Target Discovery and Preclinical Validation Project (AMP‐AD) ADKnowledgePortal.
3. RESULTS
3.1. Heritability estimates
Significant heritability was found in both the NHW and NHB meta‐analyses for memory performance and decline. For the NHW, we observed statistically significant heritability for baseline memory performance (h2 = 0.151–0.181) and memory decline (h2 = 0.123–0.159). Similar estimates were observed in NHB participants for both baseline memory performance (h2 = 0.194–0.398) and memory decline (h2 = 0.051–0.295). No differences between NHW and NHB in heritability were observed. Heritability for all main analyses and stratified analyses can be found in Table S7.
3.2. Single‐variant associations
Results of the cross‐ancestry GWAS of baseline memory performance are presented in Figure 1. As expected, there was a strong genome‐wide signal at the APOE locus on chromosome 19 (Figure 1A)—(index SNP rs10119, MAF = 0.351; β = −0.137 ± 0.007, p = 5.22 × 10−99). While there were no genome‐wide significant signals outside of chromosome 19 for baseline memory in our main analysis, there were several regions approaching significance in previous AD‐associated regions, including those on chromosomes 1 (rs7537669, CR1) and 2 (rs6733839, BIN1 Figure 1B,C).
FIGURE 1.
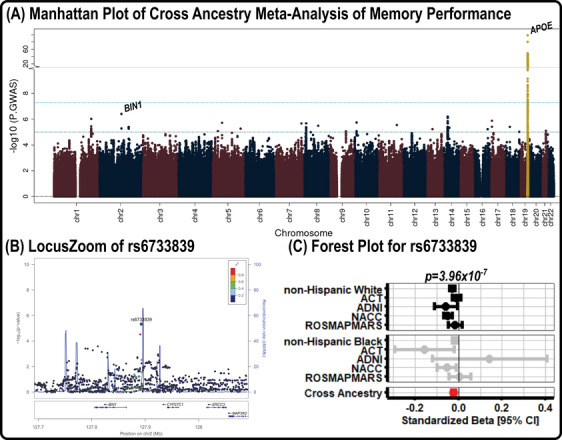
Baseline memory performance GWAS results. (A) Manhattan plot of the results from the GWAS on memory performance, in which genome‐wide significance (5.0 × 10−8) and suggestive significance (1.0 × 10−5) are marked by cyan and teal lines, respectively. (B) LocusZoom plot for the top locus (rs6733839) outside of the APOE region, in which colors highlight the locus disequilibrium. (C) A forest plot for rs6733839, which shows the direction and magnitude of effect for all NHW and NHB datasets. The summary estimate for the NHW, NHB, and cross‐ancestry meta‐analyses are also presented. ACT, Adult Changes in Thought; ADNI, Alzheimer's Disease Neuroimaging Initiative; APOE, apolipoprotein E; CI, confidence interval; GWAS, genome‐wide association studies; NACC, National Alzheimer's Coordinating Center; NHB, non‐Hispanic Black; NHW, non‐Hispanic White; ROSMAPMARS, Religious Orders Study / Memory and Aging Project /Minority Aging Research Study.
Similarly, results from the cross‐ancestry GWAS of memory decline are presented in Figure 2. We again observed a strong signal at the APOE locus (index SNP rs10119, MAF = 0.348; β = −0.016 ± 0.001, p = 6.87 × 10−104; Figure 2A) and also observed two additional genome‐wide signals, including in chromosome 1 near CR1 (index SNP rs4562624, MAF = 0.178; β = −0.005 ± 0.001, p = 2.94 × 10−8) and chromosome 2 near BIN1 (index SNP rs6733839, MAF = 0.402; β = −0.005 ± 0.001, p = 2.21 × 10−11). For both variants, results were consistent across NHW and NHB participants (Figure 2B,C).
FIGURE 2.
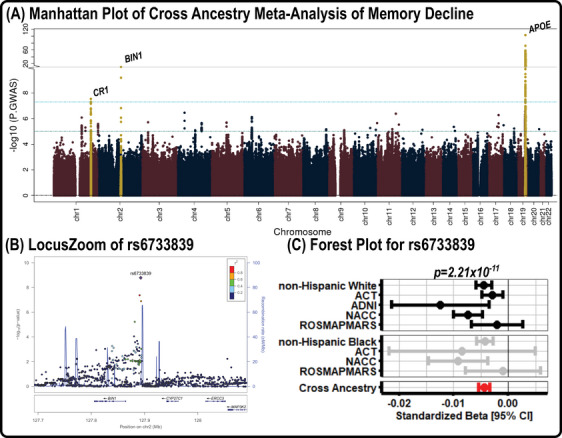
Memory decline GWAS results. (A) Manhattan plot of the results from the GWAS on memory decline, in which genome‐wide significance (5.0 × 10−8) and suggestive significance (1.0 × 10−5) are marked by cyan and teal lines, respectively. (B) LocusZoom plot for the top locus (rs6733839) outside of the APOE region, in which colors highlight the locus disequilibrium. (C) A forest plot for rs6733839, which shows the direction and magnitude of effect for all NHW and NHB datasets. The summary estimate for the NHW, NHB, and cross‐ancestry meta‐analyses are also presented. ACT, Adult Changes in Thought; ADNI, Alzheimer's Disease Neuroimaging Initiative; APOE, apolipoprotein E; CI, confidence interval; GWAS, genome‐wide association studies; NACC, National Alzheimer's Coordinating Center; NHB, non‐Hispanic Black; NHW, non‐Hispanic White; ROSMAPMARS, Religious Orders Study /Memory and Aging Project /Minority Aging Research Study.
Sensitivity analyses are presented in Table S8. Association near the known AD‐loci APOE, BIN1, and CR1 were largely similar in sensitivity analyses. We also observed a novel genome‐wide signal on chromosome 4 when removing participants with comorbid conditions (index SNP rs6848524, MAF = 0.034; β = −0.011 ± 0.002, p = 1.95 × 10−8), and two novel associations in the NHB impaired analysis on chromosome 7 (index SNP rs4142249, MAF = 0.106; β = −0.289 ± 0.051, p = 1.97 × 10−8) and chromosome 15 (index SNP rs74381744, MAF = 0.014; β = −0.963 ± 0.170, p = 1.38 × 10−8) at baseline, and chromosome 1 in longitudinal analysis (index SNP rs116675675, MAF = 0.012; β = −0.096 ± 0.017, p = 3.37 × 10−8). However, none of these novel signals replicated in the FinnGen database when looking at AD phenotypes (p > 0.08). Further, these novel signals did not replicate in prior GWAS of memory.
3.3. Single‐variant gene mapping and replication
We evaluated eQTL evidence for the known AD loci in our primary analysis including rs7537669 (chromosome 1), rs6733839 (chromosome 2), and rs10119 (chromosome 19) for baseline memory performance, and rs4562624 (chromosome 1) for longitudinal decline. We found that rs7537669 was an eQTL for CD46 in 18 different tissues and was also an eQTL for CR1 and CD46 in the cortex. We found that rs6733839 was an eQTL for BIN1 in artery‐aorta tissue and was replicated for AD in the FinnGen database (p = 3.5 × 10−10). The rs4562624 variant is an eQTL for CR1 and CR2 in the cortex (eg, DLPFC) and replicated in the FinnGen database (p = 7.4 × 10−6). Neither of these variants replicated in prior GWAS of memory. The rs10119 variant was an eQTL for NECTIN2 in whole blood in addition to TOMM40 in several tissues—it also replicated for AD in the FinnGen database (p = 1.10 × 10−204) and in a prior CHARGE cohort analysis of cognitive function (p = 5.67 × 10−9).
We then characterized the functional evidence for novel variants that reached genome‐wide significance in sensitivity analyses. We found that the intronic variant rs116675675 within CEP350 was an eQTL for CEP350 in whole blood. No eQTLs were found for rs111471504 (located in an intron in SLC8A1) or for rs6848524 (located upstream of BEND4). The rs4142249 variant was an eQTL for HERPUD2 in whole blood, tibial artery, and skin, and was additionally an eQTL for SEPTIN7‐AS1 in tibial nerve. Additionally, we found that rs4142249 was an eQTL for HERPUD2 in cortex and SEPTIN7 in DLPFC and hippocampus. The rs74381744 variant is an eQTL for ATP10A in whole blood.
3.4. AD risk loci associations
We curated a list of 94 SNPs previously associated with AD from multiple GWAS, 1 , 2 , 3 , 4 , 39 and evaluated their association with memory performance and decline. Table 2 summarizes the ten most significant associations in the cross‐ancestry GWAS and provides the summary statistics for the respective NHW and NHB meta‐analyses; full results are reported in Table S9. Interestingly, only three AD risk variants exhibited genome‐wide (p < 5 × 10−8) significance or a level approaching significance (p < 1 × 10−5) with baseline memory performance, including rs429358 (APOE, p = 2.03 × 10−33), rs6733839 (BIN1, p = 3.96 × 10−7), and rs4844610 (CR1, p = 8.43 × 10−6). Further, the rs7920721 (ECHDC3) locus demonstrated significance in the NHW (p = 4.55 × 10−6) meta‐analysis but was not significant in the NHB meta‐analysis (p = 0.614). Similar results were observed for the longitudinal memory decline GWAS, in which four variants exhibited or approached genome‐wide significance, including rs429358 (APOE, p = 3.20 × 10−59), rs6733839 (BIN1, p = 2.21 × 10−11), rs4844610 (CR1, 7.14 × 10−8), and rs9473117 (CD2AP, p = 1.03 × 10−6). In the NHW analyses, we found that the rs7920721 locus (ECHDC3, p = 5.83 × 10−6) approached significance.
TABLE 2.
Top 10 Associations between AD loci and memory performance and memory decline GWAS.
| Non‐Hispanic white (NHW) | Non‐Hispanic black (NHB) | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Variant | Gene a | Chr | BP b | RA | OA | Cross Ancestry p‐value | EAF c | β (βSE) | p‐value | N | EAFc | β (βSE) | p‐value | N |
| Memory performance | ||||||||||||||
| rs429358 | APOE | 19 | 44908684 | C | T | 2.03 ×€€10−33 | 0.16 | −1.34 ×€€10−1 (1.13 ×€€10−2) | 3.32 ×€€10−32 | 7046 | 0.204 | −8.20 ×€€10−2 (2.83 ×€€10−2) | 3.74 ×€€10−3 | 625 |
| rs6733839 | BIN1 | 2 | 127135234 | T | C | 3.96 ×€€10−7 | 0.402 | −2.97 ×€€10−2 (6.49 ×€€10−3) | 4.70 ×€€10−6 | 24209 | 0.405 | −3.61 ×€€10−2 (1.63 ×€€10−2) | 2.69 ×€€10−2 | 3367 |
| rs4844610 | CR1 | 1 | 207629207 | A | C | 8.43 ×€€10−6 | 0.193 | −3.38 ×€€10−2 (8.11 ×€€10−3) | 3.04 ×€€10−5 | 24214 | 0.039 | −1.25 ×€€10−1 (5.52 ×€€10−2) | 2.40 ×€€10−2 | 2844 |
| rs7920721 | ECHDC3 | 10 | 11678309 | G | A | 1.32 ×€€10−5 | 0.389 | −2.99 ×€€10−2 (6.51 ×€€10−3) | 4.55 ×€€10−6 | 24215 | 0.16 | 1.43 ×€€10−2 (2.84 ×€€10−2) | 6.14 ×€€10−1 | 2844 |
| rs6931277 | HLA‐DRB1 | 6 | 32615580 | T | A | 1.81 ×€€10−4 | 0.166 | 3.02 ×€€10−2 (8.57 ×€€10−3) | 4.32 ×€€10−4 | 24216 | 0.097 | 3.43 ×€€10−2 (2.66 ×€€10−2) | 1.98 ×€€10−1 | 3367 |
| rs9473117 | CD2AP | 6 | 47463548 | C | A | 6.28 ×€€10−4 | 0.28 | −2.01 ×€€10−2 (7.03 ×€€10−3) | 4.20 ×€€10−3 | 24216 | 0.214 | −4.14 ×€€10−2 (1.94 ×€€10−2) | 3.26 ×€€10−2 | 3367 |
| rs2632516 | MIR142/TSPOAP1‐AS1 | 17 | 58331728 | G | C | 2.03 ×€€10−3 | 0.443 | 1.60 ×€€10−2 (6.31 ×€€10−3) | 1.10 ×€€10−2 | 24216 | 0.402 | −4.71 ×€€10−2 (2.08 ×€€10−2) | 2.39 ×€€10−2 | 2844 |
| rs3848143 | SNX1 | 15 | 64131307 | G | A | 3.82 ×€€10−3 | 0.222 | −2.33 ×€€10−2 (7.66 ×€€10−3) | 2.36 ×€€10−3 | 24214 | 0.389 | −2.22 ×€€10−3 (2.10 ×€€10−2) | 9.15 ×€€10−1 | 2844 |
| rs113260531 | SCIMP | 17 | 5235685 | A | G | 3.86 ×€€10−3 | 0.124 | −2.67 ×€€10−2 (9.62 ×€€10−3) | 5.45 ×€€10−3 | 24216 | 0.206 | −1.74 ×€€10−2 (1.92 ×€€10−2) | 3.66 ×€€10−1 | 3367 |
| rs11218343 | SORL1 | 11 | 121564878 | C | T | 6.20 ×€€10−3 | 0.038 | 4.14 ×€€10−2 (1.65 ×€€10−2) | 1.20 ×€€10−2 | 24185 | 0.084 | 3.22 ×€€10−2 (2.87 ×€€10−2) | 2.62 ×€€10−1 | 3365 |
| Memory decline | ||||||||||||||
| rs429358 | APOE | 19 | 44908684 | C | T | 3.20 ×€€10−59 | 0.159 | −1.98 ×€€10−2 (1.23 ×€€10−3) | 3.87 ×€€10−58 | 6425 | 0.204 | −1.15 ×€€10−2 (3.67 ×€€10−3) | 1.78 ×€€10−3 | 588 |
| rs6733839 | BIN1 | 2 | 127135234 | T | C | 2.21 ×€€10−11 | 0.401 | −4.52 ×€€10−3 (7.49 ×€€10−4) | 1.66 ×€€10−9 | 19700 | 0.405 | −6.21 ×€€10−3 (2.06 ×€€10−3) | 2.61 ×€€10−3 | 2617 |
| rs4844610 | CR1 | 1 | 207629207 | A | C | 7.14 ×€€10−8 | 0.192 | −4.77 ×€€10−3 (9.36 ×€€10−4) | 3.65 ×€€10−7 | 19705 | 0.039 | −1.65 ×€€10−2 (6.60 ×€€10−3) | 1.24 ×€€10−2 | 2122 |
| rs9473117 | CD2AP | 6 | 47463548 | C | A | 1.03 ×€€10−6 | 0.28 | −3.62 ×€€10−3 (8.13 ×€€10−4) | 8.66 ×€€10−6 | 19707 | 0.214 | −5.23 ×€€10−3 (2.47 ×€€10−3) | 3.41 ×€€10−2 | 2617 |
| rs7920721 | ECHDC3 | 10 | 11678309 | G | A | 1.51 ×€€10−5 | 0.389 | −3.40 ×€€10−3 (7.49 ×€€10−4) | 5.83 ×€€10−6 | 19706 | 0.16 | 1.63 ×€€10−3 (3.45 ×€€10−3) | 6.37 ×€€10−1 | 2122 |
| rs113260531 | SCIMP | 17 | 5235685 | A | G | 1.24 ×€€10−4 | 0.124 | −4.19 ×€€10−3 (1.11 ×€€10−3) | 1.52 ×€€10−4 | 19707 | 0.207 | −2.28 ×€€10−3 (2.42 ×€€10−3) | 3.45 ×€€10−1 | 2617 |
| rs73223431 | PTK2B | 8 | 27362470 | T | C | 6.04 ×€€10−4 | 0.361 | −2.32 ×€€10−3 (7.61 ×€€10−4) | 2.23 ×€€10−3 | 19707 | 0.259 | −3.96 ×€€10−3 (2.30 ×€€10−3) | 8.50 ×€€10−2 | 2617 |
| rs112403360 | ANKH | 5 | 14724304 | A | T | 8.56 ×€€10−4 | 0.075 | −4.28 ×€€10−3 (1.36 ×€€10−3) | 1.66 ×€€10−3 | 19707 | 0.096 | −3.83 ×€€10−3 (3.45 ×€€10−3) | 2.67 ×€€10−1 | 2617 |
| rs11218343 | SORL1 | 11 | 121564878 | C | T | 1.14 ×€€10−3 | 0.038 | 6.23 ×€€10−3 (1.88 ×€€10−3) | 9.31 ×€€10−4 | 19683 | 0.084 | 2.48 ×€€10−3 (3.65 ×€€10−3) | 4.96 ×€€10−1 | 2616 |
| rs62374257 | COX7C | 5 | 86927378 | C | T | 1.24 ×€€10−3 | 0.231 | −3.05 ×€€10−3 (8.62 ×€€10−4) | 4.21 ×€€10−4 | 19707 | 0.046 | 1.11 ×€€10−2 (5.95 ×€€10−3) | 6.13 ×€€10−2 | 2122 |
Note: Boldface indicates p < 0.05.
Abbreviations: AD, Alzheimer's disease; BP, base pair; Chr, chromosome; EAF, effect allele frequency; GWAS, genome‐wide associations studies; OA, other allele; RA, reference allele.
aGene previously reported in prior AD GWAS.
b GRCh38 assembly.
cFrequency across all cohorts included in analysis.
3.5. Gene‐level and pathway results
Genetic architecture of baseline memory performance and memory decline was also investigated at the gene and pathway level. For baseline memory performance, several genes exhibited significance after correction for multiple comparisons, including nine genes in the APOE region of chromosome 19 (eg, APOE, TOMM40), which was consistent across all sensitivity analyses. For the pathway level analysis, one biological process was significantly enriched for memory performance (calcium ion dependent exocytosis; β = 1.26 ± 0.27, p‐corrected = 0.03), but was not significant in any sensitivity analyses. After removing participants with comorbidities, we found that 1‐alkyl‐2 acetylglycerophosphocholine esterase activity was enriched for memory performance (β = 1.44 ± 0.32, p‐corrected = 0.04), but was not significant in any other analysis.
Gene‐ and pathway‐level analysis was also conducted for memory decline, and we found that—like the memory performance analysis—there was high involvement in the APOE region of chromosome 19 which was consistent across all sensitivity analyses. Additionally, we found significant genes in chromosomes 1 (SLC25A44, p‐corrected = 0.012), 6 (CD2AP, p‐corrected = 0.010), 11 (BSX, p‐corrected = 0.022), 15 (DPP8, p‐corrected = 0.038), and 16 (ITGAX, p‐corrected = 0.024). After removing participants with comorbidities, the p‐values were again significant for SLC25A44 (p‐corrected = 0.020), CD2AP (p‐corrected = 0.021), and BSX (p‐corrected = 0.022). Moreover, we found significance in the CR1L gene (p‐corrected = 0.022). We found several variants within these genes that replicated for the 7 AD phenotypes evaluated in the FinnGen database—6 replicated for SLC25A44 (all p < 0.007), 6 for CD2AP (all p < 0.001), 6 for BSX (all p < 0.004), 7 for DPP8 (all p < 0.001), 7 for ITGAX (all p < 0.001), and 5 for CR1L (all p < 0.003). For the pathway level analysis, no pathways were enriched for memory decline in the main analysis; however, after removing participants with comorbidities there were two significant pathways, including one related to low‐density lipoprotein assembly (p‐corrected = 0.03) and one related to presynaptic membrane binding (p‐corrected = 0.03). In the NHW meta‐analysis, the low‐density lipoprotein assembly pathway was significant in the analysis with and without participants with comorbidities, while the presynaptic membrane binding pathway was only significant in the analysis including participants with comorbidities. All gene‐level results are shown in Table S10 and pathway‐level results are shown in Table S11.
3.6. Genetic correlation
Genetic correlation analysis was performed to determine the extent of shared genetic architecture between memory and other complex traits (n = 65). Results from this analysis for memory performance are shown in Figure 3 and presented in Table S12. We found that baseline memory was associated with cognitive performance (genetic correlation = 0.47, p‐corrected = 5.55 × 10−24), educational attainment (genetic correlation = 0.44, p‐corrected = 5.61 × 10−22), and AD (genetic correlation = −0.66, p‐corrected = 4.60 × 10−16), all of which remained significant when removing the APOE region (see Table S13).
FIGURE 3.
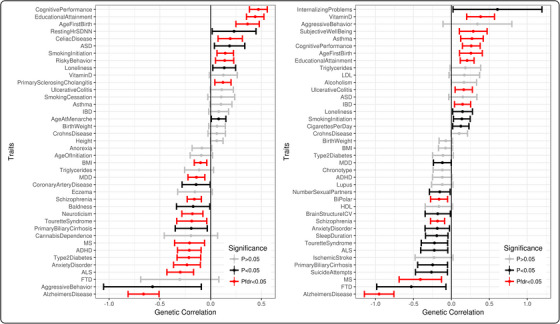
Genome‐wide genetic correlation results. Genetic correlation between memory performance (A) and memory decline (B) with 65 complex traits. Error bars represent 95% confident intervals. ADHD, attention deficit hyperactivity disorder; ALS, amyotrophic lateral sclerosis; ASD, autism spectrum disorders; BMI, body mass index; FTD, frontotemporal dementia; HLD, high‐density lipoprotein; IBD, inflammatory bowel disease; ICV, intracranial volume; LDL, low‐density lipoprotein; MDD, major depressive disorder; MS, multiple sclerosis; SDNN, standard deviation of the NN interval (ie, interval between two heart beats).
Genetic correlation analysis was also conducted on memory decline—results are presented in Figure 3 and Table S12. We found comparable correlations with cognitive performance (genetic correlation = 0.26, p‐corrected = 2.46 × 10−4), educational attainment (genetic correlation = 0.21, p‐corrected = 2.46 × 10−4), and AD (genetic correlation = −0.95, p‐corrected = 6.27 × 10−20), all of which remained significant when removing the APOE region (see Table S13). Other notable genetic correlations included multiple neuropsychiatric traits such as schizophrenia and bipolar disorder and autoimmune traits such as multiple sclerosis, and in all cases genetic risk for worse outcomes was associated with faster memory decline. In contrast, genetic risk for inflammatory conditions (eg, asthma) demonstrated counter‐intuitive correlations in which higher genetic risk was associated with a slower rate of cognitive decline (ie, better memory).
4. DISCUSSION
This study leveraged a cross‐ancestry GWAS on memory performance (n = 27,633) and decline (n = 22,365; nobs = 129,201) in older adults. We found that both traits are heritable across ancestral groups and that the genetic architecture of memory is strongly influenced by AD. Our top associations came from well‐established AD loci. We observed a novel cross‐ancestry locus on chromosome 4 (rs6848524), and three novel NHB‐specific loci on chromosomes 2 (rs111471504), 7 (rs4142249), and 15 (rs74381744). The gene‐level analysis identified novel signals on chromosomes 1 (SLC25A44), 11 (BSX), and 15 (DPP8)— these displayed some regional evidence of AD relevance in our replication cohort. Finally, genetic correlation analysis demonstrated strong associations with cognitive performance, educational attainment, and AD, in addition to several neuropsychiatric and autoimmune traits. These results deepen our understanding of the genetic architecture of late‐life memory performance and decline and highlight the value of detailed cognitive harmonization to expand genomic analyses to larger and more representative samples.
4.1. Heritability of memory in late life
We observed stable heritability estimates for memory ranging from 17% to 35%, which are similar to previous estimates from the Health and Retirement Study (HRS) and the CHARGE consortiums, 8 although twin studies suggest higher estimates ranging from 30% to 80%. 40 When we deconvolved the heritability estimates by disease stage and race, we did not see evidence of differences in heritability. Given the differences in the environmental contributors to cognitive decline across socially constructed racial/ethnic groups, and different environmental contributors across the AD continuum, it will be important to deconvolve genetic contribution to cognitive decline with larger sizes.
4.2. Novel genetic drivers of memory
Our gene‐level analysis identified several novel loci including SLC25A44, BSX, and DPP8. Solute Carrier Family 25 Member 44 (SLC25A44) has not been previously identified in AD GWAS; however, it has demonstrated involvement in cerebral small vessel disease and hypertension. 41 , 42 RNA‐seq analysis of post mortem AD brains found that this gene was significantly expressed in several brain regions and is associated with Braak staging (https://agora.adknowledgeportal.org). The brain specific homeobox (BSX) gene is involved in double stranded DNA binding activity, is expressed in the pineal gland, and has been shown to have a role in circadian rhythm. 43 The DPP8 (ie, serine dipeptidase 8) gene is involved in T‐cell activation and induces a form of cell death called pyroptosis in monocytes/macrophages. 44 It is also expressed in several regions in post mortem AD brains.
We identified several variants that had not been reported previously, though none showed supporting evidence of an association with AD in FinnGen. Among the novel loci, we had eQTL evidence implicating CEP350 on chromosome 1, three genes in the chr7 locus (HERPUD2, SEPTIN7, SEPTIN7‐AS1), and ATP10A on chromosome 15. The CEP350 gene is involved in microtubule organization, is expressed in the brain, and is upregulated in AD in the AMP‐AD cohorts in several brain regions. Evidence suggests that the minor allele is associated with lower expression of CEP350 and a faster rate of cognitive decline. Interestingly, CEP350 protein expression in blood is implicated as a potential biomarker of memory performance in several neuropsychiatric traits. 45 However, the relationship between blood expression and brain expression, as well as the connection between transcript abundance and protein function, remains unclear. Even so, our findings add to the evidence that CEP350 is an exciting potential biomarker for memory decline. Among the genes implicated on chromosome 7, the SEPTIN7 gene stands out as particularly intriguing as the minor allele is linked to elevated levels of SEPTIN7 in the prefrontal cortex, and it experiences downregulation at the transcript and protein level in AD brain prefrontal cortex. This gene also codes a protein that is localized to the centromere and is critical for microtubule function. In AD, SEPTIN7 has been implicated in p25 regulation and dendritic spine formation and morphology, particularly during memory formation. 46 (p7) Finally, we had eQTL evidence implicating ATP10A—an aminophospholipid transporting ATPase involved in Angelman syndrome. ATP10A acts as a flippase and was also reported to be downregulated in endothelial cells in the AD brain along with a floppase ABCB1. 47 Our work, 48 along with that of others, 49 has implicated other P4‐ATPases in cognitive susceptibility and AD, and numerous ABC cassette genes are floppases that have been implicated in AD, highlighting the potential importance of these phospholipid translocase proteins.
4.3. Genetic drivers of AD strongly contribute to memory decline in late life
Several strong associations with prior AD loci (ie, APOE, BIN1, CR1, ECHDC3, CD2AP) were found. As expected, the APOE region demonstrated a particularly strong association, and this was present across all sensitivity analyses. For BIN1, we found a strongly suggestive signal in our main memory performance GWAS and a genome‐wide signal in our memory decline GWAS. BIN1 also exhibited strong signals in several of our NHW sensitivity analyses, particularly in analyses among participants with mild cognitive impairment and/or AD. For CR1, we found a signal approaching significance for memory performance and a genome‐wide signal for memory decline. This signal remained when excluding participants with comorbidities. For ECHDC3, there was a signal approaching significance in the memory decline GWAS. Finally, we found a signal approaching significance for CD2AP in the memory decline GWAS. The CD2AP gene was also significant in the gene‐level analysis but was only significant in the memory decline analysis with and without the inclusion of participants with comorbidities. Previous evidence has also suggested that the FASTKD2 gene has a protective effect on memory and hippocampal volume in carriers. 50 Although significant associations were not detected in our primary analyses, we did find evidence for nominal protection of memory performance and memory decline in our cross‐ancestry impaired sensitivity analyses (all p < 0.04). Together, our results highlight strong associations between several known AD loci and late‐life memory performance.
4.4. Novel genetic correlations with memory
We observed an association between memory and AD genetic architecture in addition to educational attainment and cognitive performance. Additionally, we found correlations with several neuropsychiatric traits, including schizophrenia and bipolar disorder, whereby worse memory performance and more rapid memory decline were associated with higher risk of these traits. These findings support the hypothesis that biological pathways are shared across neuropsychiatric traits. 51 Prior GWAS studies have indicated a genetic correlation between short‐term working memory and schizophrenia, but not with bipolar disorder or AD. 1 , 8
Our analysis identified several genetic correlations between memory and autoimmune traits. For memory performance, we found that genetic architecture was positively associated with an increased risk for celiac disease and primary sclerosing cholangitis, but negatively associated an increased risk for multiple sclerosis. For memory decline, we found that genetic architecture was positively associated with an increased risk for asthma, ulcerative colitis, and vitamin D levels, but negatively associated with an increased risk for irritable bowel syndrome. While these results support the notion that inflammatory pathways play a role in cognitive decline, 52 , 53 the directionality of these correlations are counter‐intuitive. For example, our results suggest that individuals who are predisposed to memory decline have less risk for asthma, which conflicts with prior evidence demonstrating that AD genetic architecture is positively associated with asthma diagnosis. 1
4.5. Strengths and limitations
The most significant novelty of the present study is that it is the largest GWAS on memory performance and decline to date including cognitively unimpaired participants. To accomplish this feat, memory scores were harmonized across four well‐established cohorts of aging. Importantly, our sample encompassed all phases of the AD clinical spectrum (cognitively unimpaired, mild cognitive impairment, AD). An additional strength of this study is that we incorporated NHW and NHB meta‐analyses into a cross‐ancestry analysis. This study, however, has some limitations. Specifically, although this is the largest longitudinal memory GWAS to date, our sample included many highly educated participants; thus, we did not include an education covariate in our analysis. Future studies using data from heterogeneous educational backgrounds should consider the inclusion of an education covariate given its strong association with longitudinal cognitive decline. We also used a slope calculation for cognitive decline as opposed to alternative longitudinal methods; thus, the ability to generalize our results may be limited. Additionally, our GWAS considered a single cognitive domain, and the assessment of other cognitive domains is critical to our understanding of cognitive decline in AD. Another limitation of this study is that we considered self‐reported race/ethnicity to be synonymous with ancestry. Newer tools which consider population structure at the SNP level will allow for robust admixed GWAS. While we were well‐powered to detect small variant effects in the NHW analyses (f2 ≈0.003 across all MAFs), we were only powered to detect borderline small effects in the NHB analyses (f2 ≈0.03 across all MAFs). Finally, we utilized the FinnGen study database in addition to four prior GWAS of memory performance as replication cohorts. Given that the FinnGen study is comprised solely of individuals from Finland, this homogeneity restricts the generalizability of our findings. Hence, it is imperative for future research to replicate our results using more diverse cohorts, ensuring broader applicability and robustness of our results. Ongoing efforts to harmonize cognitive and genetic data across multiple cohorts will assist in addressing this statistical limitation and will allow for the assessment of rare variants.
5. CONCLUSIONS
The present study conducted the largest memory performance and memory decline GWAS to date leveraging several well‐established cohorts of aging. We found that these GWAS are similar to AD GWAS, demonstrating that memory performance and decline are suitable endophenotypes for AD. Incorporating larger sample sizes into GWAS of memory may allow for the discovery of candidate genes for the treatment of AD.
CONFLICT OF INTEREST STATEMENT
T.J.H. is a member of the scientific advisory board for Vivid Genomics. P.M.T. received partial research grant support from Biogen, Inc., for research unrelated to this manuscript. No other authors report a conflict of interest relevant to this research. Author disclosures are available in the supporting information.
CONSENT STATEMENT
All participants provided informed consent in their respective cohort studies.
Supporting information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
The authors have nothing to report. Data collection was supported through funding by NIA grants P30AG010161 (ROS), P30AG072975, R01AG015819 (ROSMAP; genomics and RNAseq), R01AG017917 (MAP), R01AG022018, R01AG030146, R01AG036042 (5hC methylation, ATACseq), RC2AG036547 (H3K9Ac), R01AG036836 (RNAseq), R01AG048015 (monocyte RNAseq) RF1AG057473 (single nucleus RNAseq), U01AG032984 (genomic and whole exome sequencing), U01AG046152 (ROSMAP AMP‐AD, targeted proteomics), U01AG046161 (TMT proteomics), U01AG061356 (whole genome sequencing, targeted proteomics, ROSMAP AMP‐AD), the Illinois Department of Public Health (ROSMAP), and the Translational Genomics Research Institute (genomic). The results published here are in whole or in part based on data obtained from the AD Knowledge Portal (https://adknowledgeportal.synapse.org). Study data were provided by the Rush Alzheimer's Disease Center, Rush University Medical Center, Chicago. Additional phenotypic data can be requested at (https://www.rushu.rush.edu/research/departmental‐research/rush‐alzheimers‐disease‐center). Data collection and sharing for ADNI were supported by National Institutes of Health Grant U01AG024904 and Department of Defense (award number W81XWH‐12‐2‐0012). ADNI is also funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer's Association; Alzheimer's Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol‐Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann‐La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California. The NACC database is funded by NIA/NIH Grant U24AG072122. NACC data are contributed by the NIA‐funded ADRCs: P30AG062429 (PI James Brewer, MD, PhD), P30AG066468 (PI Oscar Lopez, MD), P30AG062421 (PI Bradley Hyman, MD, PhD), P30AG066509 (PI Thomas Grabowski, MD), P30AG066514 (PI Mary Sano, PhD), P30AG066530 (PI Helena Chui, MD), P30AG066507 (PI Marilyn Albert, PhD), P30AG066444 (PI John Morris, MD), P30AG066518 (PI Jeffrey Kaye, MD), P30AG066512 (PI Thomas Wisniewski, MD), P30AG066462 (PI Scott Small, MD), P30AG072979 (PI David Wolk, MD), P30AG072972 (PI Charles DeCarli, MD), P30AG072976 (PI Andrew Saykin, PsyD), P30AG072975 (PI David Bennett, MD), P30AG072978 (PI Ann McKee, MD), P30AG072977 (PI Robert Vassar, PhD), P30AG066519 (PI Frank LaFerla, PhD), P30AG062677 (PI Ronald Petersen, MD, PhD), P30AG079280 (PI Eric Reiman, MD), P30AG062422 (PI Gil Rabinovici, MD), P30AG066511 (PI Allan Levey, MD, PhD), P30AG072946 (PI Linda Van Eldik, PhD), P30AG062715 (PI Sanjay Asthana, MD, FRCP), P30AG072973 (PI Russell Swerdlow, MD), P30AG066506 (PI Todd Golde, MD, PhD), P30AG066508 (PI Stephen Strittmatter, MD, PhD), P30AG066515 (PI Victor Henderson, MD, MS), P30AG072947 (PI Suzanne Craft, PhD), P30AG072931 (PI Henry Paulson, MD, PhD), P30AG066546 (PI Sudha Seshadri, MD), P20AG068024 (PI Erik Roberson, MD, PhD), P20AG068053 (PI Justin Miller, PhD), P20AG068077 (PI Gary Rosenberg, MD), P20AG068082 (PI Angela Jefferson, PhD), P30AG072958 (PI Heather Whitson, MD), P30AG072959 (PI James Leverenz, MD). The Alzheimer's Disease Genetics Consortium supported genotyping, and data processing of samples through National Institute on Aging (NIA) grants U01AG032984. Data for this study were prepared, archived, and distributed by the National Institute on Aging Alzheimer's Disease Data Storage Site (NIAGADS) at the University of Pennsylvania (U24AG041689). The Adult Changes in Thought Study, based at Kaiser Permanente Health Research Institute of Washington and the University of Washington, is supported through U19AG066567. Additional support includes K01AG073584, R01AG073439, U24AG074855, K01AG049164, R01AG059716, R01AG061518, R21AG005994, K12HD043483, K24AG046373, HHSN311201600276P, S10OD023680, R01AG034962, R01NS100980, R01AG056534, P30AG010161, R01AG015819, R01AG017917, U01AG046152, R01AG048927, U19AG068753, U01AG058654, R01AG062634, R01AG048927, U19AG068753, K23AG045966, the Nancy and Buster Alvord Endowment (C. D. K.), the Vanderbilt Clinical Translational Science Award (UL1TR000445), the Vanderbilt Memory and Alzheimer's Center, and the Vanderbilt Alzheimer's Disease Research Center (P20AG068082). Additional funding also includes F31AG077791. The ADSP Phenotype Harmonization Consortium (ADSP‐PHC) is funded by NIA (U24AG074855, U01AG068057 and R01AG059716). The ADSP‐PHC cohorts include: Adult Changes in Thought (ACT, U01AG006781, U19AG066567), the Alzheimer's Disease Centers (ADC, P30AG062429 (PI James Brewer, MD, PhD), P30AG066468 (PI Oscar Lopez, MD), P30AG062421 (PI Bradley Hyman, MD, PhD), P30AG066509 (PI Thomas Grabowski, MD), P30AG066514 (PI Mary Sano, PhD), P30AG066530 (PI Helena Chui, MD), P30AG066507 (PI Marilyn Albert, PhD), P30AG066444 (PI John Morris, MD), P30AG066518 (PI Jeffrey Kaye, MD), P30AG066512 (PI Thomas Wisniewski, MD), P30AG066462 (PI Scott Small, MD), P30AG072979 (PI David Wolk, MD), P30AG072972 (PI Charles DeCarli, MD), P30AG072976 (PI Andrew Saykin, PsyD), P30AG072975 (PI David Bennett, MD), P30AG072978 (PI Neil Kowall, MD), P30AG072977 (PI Robert Vassar, PhD), P30AG066519 (PI Frank LaFerla, PhD), P30AG062677 (PI Ronald Petersen, MD, PhD), P30AG079280 (PI Eric Reiman, MD), P30AG062422 (PI Gil Rabinovici, MD), P30AG066511 (PI Allan Levey, MD, PhD), P30AG072946 (PI Linda Van Eldik, PhD), P30AG062715 (PI Sanjay Asthana, MD, FRCP), P30AG072973 (PI Russell Swerdlow, MD), P30AG066506 (PI Todd Golde, MD, PhD), P30AG066508 (PI Stephen Strittmatter, MD, PhD), P30AG066515 (PI Victor Henderson, MD, MS), P30AG072947 (PI Suzanne Craft, PhD), P30AG072931 (PI Henry Paulson, MD, PhD), P30AG066546 (PI Sudha Seshadri, MD), P20AG068024 (PI Erik Roberson, MD, PhD), P20AG068053 (PI Justin Miller, PhD), P20AG068077 (PI Gary Rosenberg, MD), P20AG068082 (PI Angela Jefferson, PhD), P30AG072958 (PI Heather Whitson, MD), P30AG072959 (PI James Leverenz, MD), the Alzheimer's Disease Neuroimaging Initiative (ADNI), funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01AG024904) and DOD ADNI (Department of Defense award number W81XWH‐12‐2‐0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer's Association; Alzheimer's Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol‐Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann‐La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California, the Memory ang Aging Project at the Knight‐ADRC (Knight‐ADRC), supported by NIH grants R01AG064614, R01AG044546, RF1AG053303, RF1AG058501, U01AG058922 and R01AG064877 to Carlos Cruchaga. The recruitment and clinical characterization of research participants at Washington University was supported by NIH grants P30AG066444, P01AG003991, and P01AG026276. Data collection and sharing for this project was supported by NIH grants RF1AG054080, P30AG066462, R01AG064614 and U01AG052410. This work was supported by access to equipment made possible by the Hope Center for Neurological Disorders, the Neurogenomics and Informatics Center (NGI: https://neurogenomics.wustl.edu/) and the Departments of Neurology and Psychiatry at Washington University School of Medicine; the Minority Aging Research Study (MARS, R01AG022081, R01AG042210), the National Alzheimer's Coordinating Center (NACC, U01AG016976, U24AG072122),the National Institute on Aging Late Onset Alzheimer's Disease Family Study (NIA‐ LOAD, U24AG056270), the Religious Orders Study (ROS, P30AG010161, P30AG072975, R01AG015819, R01AG042210), the RUSH Memory and Aging Project (MAP, R01AG017917, R01AG42210), the National Institute on Aging Genetics of Alzheimer's Disease Data Storage Site (NIAGADS, U24AG041689) at the University of Pennsylvania, and the Genome Center for Alzheimer's Disease (U54AG052427), funded by NIA. The Alzheimer's Disease Genetics Consortium supported collection and genotyping of samples used in this study through National Institute on Aging (NIA) grants U01AG032984 and RC2AG036528. Data for this study were prepared, archived, and distributed by the National Institute on Aging Alzheimer's Disease Data Storage Site (NIAGADS) at the University of Pennsylvania (U24AG041689), The Center for Applied Genomics at the Children's Hospital of Philadelphia Research Institute performed genotyping of samples. The NACC database is funded by NIA/NIH Grant U24AG072122. NACC data are contributed by the NIA‐funded ADRCs: P30AG062429 (PI James Brewer, MD, PhD), P30AG066468 (PI Oscar Lopez, MD), P30AG062421 (PI Bradley Hyman, MD, PhD), P30AG066509 (PI Thomas Grabowski, MD), P30AG066514 (PI Mary Sano, PhD), P30AG066530 (PI Helena Chui, MD), P30AG066507 (PI Marilyn Albert, PhD), P30AG066444 (PI John Morris, MD), P30AG066518 (PI Jeffrey Kaye, MD), P30AG066512 (PI Thomas Wisniewski, MD), P30AG066462 (PI Scott Small, MD), P30AG072979 (PI David Wolk, MD), P30AG072972 (PI Charles DeCarli, MD), P30AG072976 (PI Andrew Saykin, PsyD), P30AG072975 (PI David Bennett, MD), P30AG072978 (PI Neil Kowall, MD), P30AG072977 (PI Robert Vassar, PhD), P30AG066519 (PI Frank LaFerla, PhD), P30AG062677 (PI Ronald Petersen, MD, PhD), P30AG079280 (PI Eric Reiman, MD), P30AG062422 (PI Gil Rabinovici, MD), P30AG066511 (PI Allan Levey, MD, PhD), P30AG072946 (PI Linda Van Eldik, PhD), P30AG062715 (PI Sanjay Asthana, MD, FRCP), P30AG072973 (PI Russell Swerdlow, MD), P30AG066506 (PI Todd Golde, MD, PhD), P30AG066508 (PI Stephen Strittmatter, MD, PhD), P30AG066515 (PI Victor Henderson, MD, MS), P30AG072947 (PI Suzanne Craft, PhD), P30AG072931 (PI Henry Paulson, MD, PhD), P30AG066546 (PI Sudha Seshadri, MD), P20AG068024 (PI Erik Roberson, MD, PhD), P20AG068053 (PI Justin Miller, PhD), P20AG068077 (PI Gary Rosenberg, MD), P20AG068082 (PI Angela Jefferson, PhD), P30AG072958 (PI Heather Whitson, MD), P30AG072959 (PI James Leverenz, MD) Samples from the National Centralized Repository for Alzheimer's Disease and Related Dementias (NCRAD), which receives government support under a cooperative agreement grant (U24AG021886) awarded by the National Institute on Aging (NIA), were used in this study. The authors thank contributors who collected samples used in this study, as well as patients and their families, whose help and participation made this work possible. Data collection and sharing for this project was funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01AG024904) and DOD ADNI (Department of Defense award number W81XWH‐12‐2‐0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer's Association; Alzheimer's Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol‐Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann‐La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California. Rush University grants P30AG010161, R01AG019085, R01AG015819, R01AG017917, R01AG030146, P30AG010161 RC2AG036650, R01AG022081, U01AG006781, U01HG004610.
APPENDIX 1. COLLABORATORS
1.1.
Data used in preparation of this article were obtained from the Alzheimer's Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: https://adni.loni.usc.edu/wp‐content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf
Archer DB, Eissman JM, Mukherjee S, et al. Longitudinal change in memory performance as a strong endophenotype for Alzheimer's disease. Alzheimer's Dement. 2024;20:1268–1283. 10.1002/alz.13508
REFERENCES
- 1. Kunkle BW, Grenier‐Boley B, Sims R, et al. Genetic meta‐analysis of diagnosed Alzheimer's disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat Genet. 2019;51(3):414‐430. 10.1038/s41588-019-0358-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lambert JC, Ibrahim‐Verbaas CA, Harold D, et al. Meta‐analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet. 2013;45(12):1452‐1458. 10.1038/ng.2802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bellenguez C, Küçükali F, Jansen IE, et al. New insights into the genetic etiology of Alzheimer's disease and related dementias. Nat Genet. 2022;54(4):412‐436. 10.1038/s41588-022-01024-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wightman DP, Jansen IE, Savage JE, et al. A genome‐wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer's disease. Nat Genet. 2021;53(9):1276‐1282. 10.1038/s41588-021-00921-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gatz M, Reynolds CA, Fratiglioni L. Role of genes and environments for explaining alzheimer disease. Arch Gen Psychiatry. 2006;63(2):168‐174. [DOI] [PubMed] [Google Scholar]
- 6. Ridge PG, Mukherjee S, Crane PK, Kauwe JSK. Alzheimer's disease: analyzing the missing heritability. PLoS One. 2013;8(11):e79771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Karlsson IK, Escott‐Price V, Gatz M, et al. Measuring heritable contributions to Alzheimer's disease: polygenic risk score analysis with twins. Brain Commun. 2022;4(1):fcab308. 10.1093/braincomms/fcab308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lahti J, Tuominen S, Yang Q, et al. Genome‐wide meta‐analyses reveal novel loci for verbal short‐term memory and learning. Mol Psychiatry. 2022;27(11):4419‐4431. 10.1038/s41380-022-01710-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Davies G, Lam M, Harris SE, et al. Study of 300,486 individuals identifies 148 independent genetic loci influencing general cognitive function. Nat Commun. 2018;9(1):2098. 10.1038/s41467-018-04362-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gao Y, Felsky D, Reyes‐Dumeyer D, et al. Integration of GWAS and brain transcriptomic analyses in a multiethnic sample of 35,245 older adults identifies DCDC2 gene as predictor of episodic memory maintenance. Alzheimers Dement. 2022;18(10):1797‐1811. 10.1002/alz.12524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mukherjee S, Choi SE, Lee ML, et al. Cognitive domain harmonization and cocalibration in studies of older adults. Neuropsychology. 2023;37(4):409‐423. 10.1037/neu0000835. Published online 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kukull WA, Higdon R, Bowen JD, et al. Dementia and Alzheimer disease incidence: a prospective cohort study. Arch Neurol. 2002;59(11):1737‐1746. [DOI] [PubMed] [Google Scholar]
- 13. Jack CR, Bernstein MA, Fox NC, et al. The Alzheimer's disease neuroimaging initiative (ADNI): mRI methods. J Magn Reson Imaging. 2008;27(4):685‐691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Beekly DL, Ramos EM, van Belle G, et al. The National Alzheimer's Coordinating Center (NACC) Database: an Alzheimer disease database. Alzheimer Dis Assoc Disord. 2004;18(4):270‐277. [PubMed] [Google Scholar]
- 15. Beekly DL, Ramos EM, Lee WW, et al. The National Alzheimer's Coordinating Center (NACC) Database: the Uniform Data Set. Alzheimer Dis Assoc Disord. 2007;21(3):249. 10.1097/WAD.0b013e318142774e [DOI] [PubMed] [Google Scholar]
- 16. Besser LM, Kukull WA, Teylan MA, et al. The Revised National Alzheimer's Coordinating Center's neuropathology form‐available data and new analyses. J Neuropathol Exp Neurol. 2018;77(8):717‐726. 10.1093/jnen/nly049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Weintraub S, Salmon D, Mercaldo N, et al. The Alzheimer's Disease Centers’ Uniform Data Set (UDS): the neuropsychological test battery. Alzheimer Dis Assoc Disord. 2009;23(2):91‐101. 10.1097/WAD.0b013e318191c7dd [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Weintraub S, Besser L, Dodge HH, et al. Version 3 of the Alzheimer disease centers’ neuropsychological test battery in the uniform data set (UDS). Alzheimer Dis Assoc Disord. 2018;32(1):10‐17. 10.1097/WAD.0000000000000223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bennett DA, Buchman AS, Boyle PA, Barnes LL, Wilson RS, Schneider JA. Religious orders study and rush memory and aging project. JAD. 2018;64(s1):S161‐S189. 10.3233/JAD-179939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Barnes LL, Shah RC, Aggarwal NT, Bennett DA, Schneider JA. The Minority Aging Research Study: ongoing efforts to obtain brain donation in African Americans without dementia. Curr Alzheimer Res. 2012;9(6):734‐745. 10.2174/156720512801322627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Eissman JM, Dumitrescu L, Mahoney ER, et al. Sex differences in the genetic architecture of cognitive resilience to Alzheimer's disease. Brain. 2022;145(7):2541‐2554. 10.1093/brain/awac177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Taliun D, Harris DN, Kessler MD, et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature. 2021;590(7845):290‐299. 10.1038/s41586-021-03205-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome‐wide complex trait analysis. Am J Hum Genet. 2011;88(1):76‐82. 10.1016/j.ajhg.2010.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Martin J, Khramtsova EA, Goleva SB, et al. Examining sex‐differentiated genetic effects across neuropsychiatric and behavioral traits. Biol Psychiatry. 2021;89(12):1127‐1137. 10.1016/j.biopsych.2020.12.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second‐generation PLINK: rising to the challenge of larger and richer datasets. GigaScience. 2015;4(1). 10.1186/s13742-015-0047-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mägi R, Morris AP. GWAMA: software for genome‐wide association meta‐analysis. BMC Bioinf. 2010;11(1):288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. GTEx Consortium . Genetic effects on gene expression across human tissues. Nature. 2017;550(7675):204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. GTEx Consortium . The genotype‐tissue expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348(6235):648‐660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Võsa U, Claringbould A, Westra HJ, et al. Large‐scale cis‐ and trans‐eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat Genet. 2021;53(9):1300‐1310. 10.1038/s41588-021-00913-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ng B, White CC, Klein HU, et al. An xQTL map integrates the genetic architecture of the human brain's transcriptome and epigenome. Nat Neurosci. 2017;20(10):1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. BrainSeq: A Human Brain Genomics Consortium . Electronic address: drweinberger@libd.org, BrainSeq: a Human Brain Genomics Consortium. BrainSeq: neurogenomics to Drive Novel Target Discovery for Neuropsychiatric Disorders. Neuron. 2015;88(6):1078‐1083. doi: 10.1016/j.neuron.2015.10.047 [DOI] [PubMed] [Google Scholar]
- 32. de Klein N, Tsai EA, Vochteloo M, et al. Brain expression quantitative trait locus and network analyses reveal downstream effects and putative drivers for brain‐related diseases. Nat Genet. 2023:1‐12. 10.1038/s41588-023-01300-6. Published online February 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. de Leeuw CA, Mooij JM, Heskes T, Posthuma D. MAGMA: generalized gene‐set analysis of GWAS data. PLoS Comput Biol. 2015;11(4):e1004219‐e1004219. 10.1371/journal.pcbi.1004219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lu Q, Li B, Ou D, et al. A powerful approach to estimating annotation‐stratified genetic covariance via GWAS summary statistics. Am Hum Genet. 2017;101(6):939‐964. 10.1016/j.ajhg.2017.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lee JJ, Wedow R, Okbay A, et al. Gene discovery and polygenic prediction from a 1.1‐million‐person GWAS of educational attainment. Nat Genet. 2018;50(8):1112‐1121. 10.1038/s41588-018-0147-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Davies G, Armstrong N, Bis JC, et al. Genetic contributions to variation in general cognitive function: a meta‐analysis of genome‐wide association studies in the CHARGE consortium (N = 53 949). Mol Psychiatry. 2015;20(2):183‐192. 10.1038/mp.2014.188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Davies G, Marioni RE, Liewald DC, et al. Genome‐wide association study of cognitive functions and educational attainment in UK Biobank (N = 112 151). Mol Psychiatry. 2016;21(6):758‐767. 10.1038/mp.2016.45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Trampush JW, Yang MLZ, Yu J, et al. GWAS meta‐analysis reveals novel loci and genetic correlates for general cognitive function: a report from the COGENT consortium. Mol Psychiatry. 2017;22(3):336‐345. 10.1038/mp.2016.244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jansen IE, Savage JE, Watanabe K, et al. Genome‐wide meta‐analysis identifies new loci and functional pathways influencing Alzheimer's disease risk. Nat Genet. 2019;51(3):404‐413. 10.1038/s41588-018-0311-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Swan GE, Reed T, Jack LM, et al. Differential genetic influence for components of memory in aging adult twins. Arch Neurol. 1999;56(9):1127‐1132. 10.1001/archneur.56.9.1127 [DOI] [PubMed] [Google Scholar]
- 41. Lee B, Yao X, Shen L. For the Alzheimer's Disease Neuroimaging Initiative. Genome‐Wide association study of quantitative biomarkers identifies a novel locus for alzheimer's disease at 12p12.1. Bmc Genomics [Electronic Resource]. 2022;23(1):85. 10.1186/s12864-021-08269-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chung J, Marini S, Pera J, et al. Genome‐wide association study of cerebral small vessel disease reveals established and novel loci. Brain. 2019;142(10):3176‐3189. 10.1093/brain/awz233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Carstensen MB, Hertz H, Bering T, et al. Circadian regulation and molecular role of the Bsx homeobox gene in the adult pineal gland. J Pineal Res. 2020;68(2):e12629. 10.1111/jpi.12629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Johnson DC, Taabazuing CY, Okondo MC, et al. DPP8/DPP9 inhibitor‐induced pyroptosis for treatment of acute myeloid leukemia. Nat Med. 2018;24(8):1151‐1156. 10.1038/s41591-018-0082-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Niculescu AB, Le‐Niculescu H, Roseberry K, et al. Blood biomarkers for memory: toward early detection of risk for Alzheimer disease, pharmacogenomics, and repurposed drugs. Mol Psychiatry. 2020;25(8):1651‐1672. 10.1038/s41380-019-0602-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang X, Fei F, Qu J, Li C, Li Y, Zhang S. The role of septin 7 in physiology and pathological disease: a systematic review of current status. J Cell Mol Med. 2018;22(7):3298. 10.1111/jcmm.13623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sun N, Akay LA, Murdock MH, et al. Single‐cell multi‐region dissection of brain vasculature in Alzheimer's Disease. Published online February 10 2022:2022.02.09.479797. doi: 10.1101/2022.02.09.479797 bioRxiv [DOI]
- 48. Dumitrescu L, Mahoney ER, Mukherjee S, et al. Genetic variants and functional pathways associated with resilience to Alzheimer's disease. Brain. 2020;143(8):2561‐2575. 10.1093/brain/awaa209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Holstege H, Hulsman M, Charbonnier C, et al. Exome sequencing identifies rare damaging variants in ATP8B4 and ABCA1 as risk factors for Alzheimer's disease. Nat Genet. 2022;54(12):1786‐1794. 10.1038/s41588-022-01208-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ramanan VK, Nho K, Shen L, et al. FASTKD2 is associated with memory and hippocampal structure in older adults. Mol Psychiatry. 2015;20(10):1197‐1204. 10.1038/mp.2014.142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ahmad K, Baig MH, Mushtaq G, Kamal MA, Greig NH, Choi I. Commonalities in biological pathways, genetics, and cellular mechanism between Alzheimer disease and other neurodegenerative diseases: an in silico‐updated overview. Curr Alzheimer Res;14(11):1190‐1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bettcher BM, Kramer JH. Longitudinal inflammation, cognitive decline, and Alzheimer's disease: a mini‐review. Clin Pharmacol Ther. 2014;96(4):464‐469. 10.1038/clpt.2014.147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Annweiler C, Llewellyn DJ, Beauchet O. Low serum vitamin D concentrations in Alzheimer's disease: a systematic review and meta‐analysis. J Alzheimers Dis. 2013;33(3):659‐674. 10.3233/JAD-2012-121432 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Data Availability Statement
All phenotype and genetic data used in this analysis are available on NIAGADS (https://dss.niagads.org/). Other phenotype data available through the ADSP‐PHC may be browsed on a data curation tool housed at Vanderbilt (https://vmacdata.org/adsp‐phc). All summary statistics are also available on NIAGADS. The results published here are in whole or in part based on data obtained from the Accelerating Medicines Partnerships ‐ Alzheimer's Disease Target Discovery and Preclinical Validation Project (AMP‐AD) ADKnowledgePortal.