

Summary
Colorectal cancer (CRC) is a common malignancy involving multiple cellular components. The CRC tumor microenvironment (TME) has been characterized well at single-cell resolution. However, a spatial interaction map of the CRC TME is still elusive. Here, we integrate multiomics analyses and establish a spatial interaction map to improve the prognosis, prediction, and therapeutic development for CRC. We construct a CRC immune module (CCIM) that comprises FOLR2+ macrophages, exhausted CD8+ T cells, tolerant CD8+ T cells, exhausted CD4+ T cells, and regulatory T cells. Multiplex immunohistochemistry is performed to depict the CCIM. Based on this, we utilize advanced deep learning technology to establish a spatial interaction map and predict chemotherapy response. CCIM-Net is constructed, which demonstrates good predictive performance for chemotherapy response in both the training and testing cohorts. Lastly, targeting FOLR2+ macrophage therapeutics is used to disrupt the immunosuppressive CCIM and enhance the chemotherapy response in vivo.
Keywords: colorectal cancer, artificial intelligence, tumor microenvironment, FOLR2+ macrophages, immuno module
Graphical abstract
Highlights
-
•
A FOLR2+ macrophage-oriented cell module is uncovered in colorectal cancer
-
•
Deep learning models utilize the macrophage-oriented spatial interaction map
-
•
Targeting FOLR2+ resident macrophages enhances the response to chemotherapy
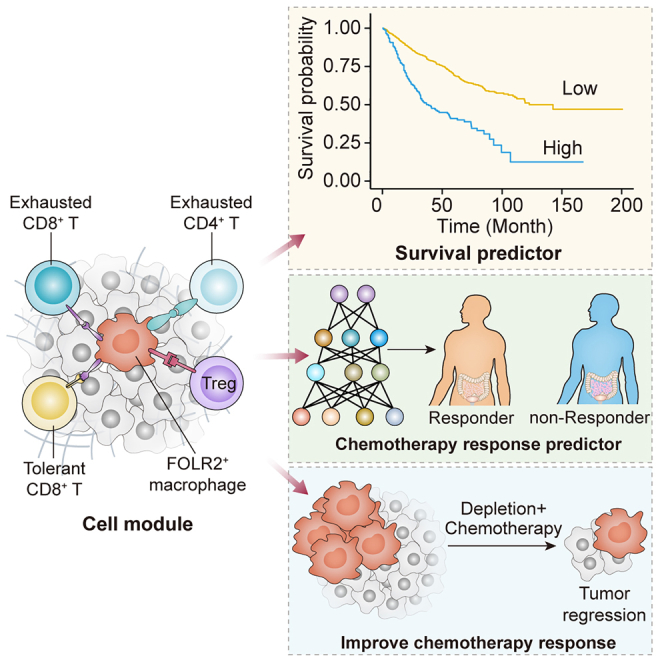
Bao et al. utilize a deep learning model supported by multiomics analysis to establish a FOLR2+ macrophage-oriented cell module. This module serves as both a therapeutic target and a prognostic predictor in colorectal cancer. Additionally, the spatial interaction map not only predicts chemotherapy response but also identifies potential therapeutic targets.
Introduction
Colorectal cancer (CRC) is one of the most common malignancies worldwide. Mortality from CRC has decreased slightly over the past 30 years due to earlier diagnosis and developments of chemotherapy, targeted therapy and immunotherapy. XELOX (combination of oxaliplatin and capecitabine), FOLFOX (combination of 5-fluorouracil, leucovorin, and oxaliplatin), and FOLFIRI (combination of 5-fluorouracil, leucovorin, and irinotecan) are the first-line chemotherapy regimens in late-stage CRC. Cetuximab and bevacizumab exhibit good efficacy, mainly in combination with systematic chemotherapy.1,2 In addition, patients with microsatellite instability-high (MSI-H) or mismatch repair-deficient (dMMR) CRC are susceptible to immune checkpoint inhibitor (ICI)-based immunotherapy with objective clinical responses,3 providing an alternative treatment for a subset of CRC patients with advanced disease.
Nevertheless, a heterogeneous treatment response has been found in CRC patients, possibly due to heterogeneity of the tumor microenvironment (TME). The TME consists of distinctive and interacting cell populations, including myeloid cells, T cells, B cells, natural killer (NK) cells, and other cell types. Studies of the diversity and reprogramming of the TME have shown its potential to influence immunotherapy response and prognosis.4,5,6,7,8,9,10,11 The development of a suppressive TME limits the response to chemotherapy and immunotherapy treatment and is characterized by increased levels of pro-tumor macrophages, exhausted T cells, and regulatory T (Treg) cells. A high level of macrophages is associated with a poor prognosis in various tumor types. Targeting macrophages is a promising strategy for cancer therapy.12,13 The colony-stimulating factor 1 (CSF1)/CSF1 receptor (CSF1R) axis has gained the most attention in the context of macrophage-targeting therapies. A variety of monoclonal antibodies and small molecules targeting CSF1R or its ligand CSF1 are in phase I studies as monotherapies or in combination with chemotherapy as well as other cancer immunotherapy approaches.8 Nevertheless, anti-CSF1R therapy has shown limited therapeutic effects and significant toxicity in several tumor types, including CRC,8 which warrants the exploration of the mechanism underlying the resistance to macrophage-targeting therapy.
The CRC TME has been characterized well at single-cell resolution.14,15,16,17,18 However, most of the previous studies focused on the identification of different cell populations. A spatial interaction map of the CRC TME is still elusive. Moreover, the generation of high-throughput bulk and single-cell transcriptome data relies on fresh tumor tissues rather than paraffin sections. Transcriptomics data-driven patient stratification is not pathology friendly. The prognosis and the therapeutic sensitivity prediction of CRC patients are also not established well by pathological methods. To solve these problems, in this study, we integrated multiomics data (bulk transcriptome, single-cell transcriptome, and multiplex immunohistochemistry [mIHC] staining) to uncover an immunospatial interaction map of CRC. Our work highlights that the FOLR2+ macrophage-mediated CRC immune module (CCIM) orchestrates a suppressive CRC TME and leads to therapy resistance in CRC. A seven-color CCIM mIHC panel was designed. Deep learning models were applied to the CCIM mIHC panel to develop a CCIM-Net for artificial intelligence (AI)-assisted diagnosis, which showed the properties of being efficient, pathology friendly, and cost effective. Last, we evaluated the possibility of the multicellular module being a promising therapeutic target.
Results
Deeping learning-assisted multiomics integration workflow for CRC
The detailed workflow of our research is shown in Figure 1A. We performed the integration analysis on single-cell RNA sequencing (scRNA-seq) (public, n = 18; in house, n = 8), bulk transcriptome (total, n = 2,081; n = 1,673 for the integrative cohort, and n = 408 for four independent cohorts), and mIHC staining (n = 226) to construct a cell module for prognosis stratification and chemotherapy prediction. A multicellular module was identified as the CCIM based on the integrative analysis of scRNA-seq and bulk RNA-seq. With the established CCIM, a CCIM score was calculated to stratify patients with a poor prognosis in the training (n = 1,405) and 5 testing cohorts (total, n = 682) (total n = 2,081). Furthermore, we developed an mIHC imaging panel based on the CCIM to spatially detect the CCIM pattern. A CCIM deep learning model (CCIM-Net) based on the previous established CCIM pattern was developed with two cohorts from different centers (training cohort, n = 181; testing cohort, n = 45) to facilitate precision chemotherapy. In vivo experiments were performed for validation.
Figure 1.

Depiction of the workflow for multiomics-assisted deep learning analysis
(A) Schematic of this research.
(B) The uniform manifold approximation and projection (UMAP) plot identifies the main cell types in colorectal cancer (CRC).
(C) Diverse cell types of CRC and the expression of markers.
We first explored the TME heterogeneity and molecular signature for the cellular components in CRC using scRNA-seq (Figures 1B and 1C). T-distributed stochastic neighbor embedding (t-SNE) was performed on variably expressed genes across all cells for dimensionality reduction and cell clustering. The cells were obtained from 18 patients by integrating the public dataset and our in-house cohort (Figure S1). To define the identity of each cell cluster, the gene signature was determined by performing differential gene expression analysis (Figure 1C). Cell type signatures were used to identify cell clusters using the "SingleR" package with manual assistance. For instance, the expression of NKG7, CCL5, CD3D, and CD3E was significantly higher in T cells than in other cell clusters. Myeloid cells highly expressed S100A8, S100A9, and PLAUR. CD79A and CD79B were used to annotate B cells. In total, we found six cell types, which are shown in Figure 1C. Altogether, the integrated scRNA-seq landscape covers the major cellular components of the CRC TME, including various immune cells from all stages of CRC tumor tissues, based on which we applied a deep learning-assisted multiomics integration workflow for CRC.
Myeloid subpopulations and their immune activities in CRC
Next, we determined the myeloid populations in CRC. The tumor myeloid populations were further subclustered into dendritic cells (DCs), mast cells, FCN1+ monocytes,19 OAS+ macrophages,20 OLR1+ macrophages,21 HSPA6+ monocytes,22 and FOLR2+ macrophages (see Figure 2A and Table S1 for the featured genes in each population). The key lineage markers are shown in Figure 2B. For instance, CD1C is highly expressed in DCs, while KIT is expressed on mast cells. CD14, CD16, and CD68 were also investigated for monocyte and macrophage populations. With the help of the lineage markers, the DC and mast cell populations were identified. In the next step, we investigated the subpopulations of monocytes and macrophages. Monocytes could be divided into FCN1+ and HSPA6+ monocytes, respectively, characterized by expression of classic monocyte markers (S100A8/9/12), pro-inflammatory cytokines (IL1B, IL6, CCL3, and CCL4), and heat shock proteins (HSPA6 and HSPA1A/B).22 FCN1+ monocytes have been identified in bronchoalveolar immune cells,23 which fuel inflammation during severe disease. We also identified FCN1+ and HSPA6+ monocyte subpopulations (Figure S2A). OLR1 was more highly expressed in pro-inflammatory M1-like macrophages and DCs than M2-like macrophage subpopulations.21 OAS1 is upregulated by type I interferons (IFNs) and is also involved in the innate immune response. Therefore, we named the two macrophage subpopulations that highly express OLR1 or OAS1 OLR1+ macrophages and OAS+ macrophages, respectively (Figure S2A). Resident macrophages featured high expression of FOLR2, C1QA, C1QB, and APOE.24 Previous studies have shown that FOLR2 is highly expressed in resident macrophages and related to the activation of resident macrophages.25,26,27 We therefore considered that FOLR2+ macrophage exhibited a resident phenotype.
Figure 2.

The properties of myeloid subpopulations
(A) The t-distributed stochastic neighbor embedding (t-SNE) shows seven subclusters of myeloid cells (patients, n = 26).
(B) Stacked violin plot showing the scaled expression level of markers for each population.
(C) The enriched Gene Ontology (GO) terms for each monocyte/macrophage population. The GO analysis was performed with the gene signature for each monocyte and macrophage subpopulation. The height of each column represents the –log10(p) value of enriched biological processes for each subpopulation.
(D) Heatmap showing the immune pathway activities in myeloid populations. Purple indicates low values, while yellow indicates high values.
In the next step, Gene Ontology (GO) analysis was performed to characterize the underlying biological processes of each monocyte/macrophage population. FOLR2+ macrophages were enriched in lipid metabolism-related processes, while OLR1+ macrophages were enriched in G protein-coupled receptor and growth factor receptor binding (Figure 2C). Gene set variation analysis (GSVA) was performed with an immune gene set28 on the identified myeloid populations (Figure 2D), which indicated significantly lower anti-tumor immunity activities in FOLR2+ macrophage populations than in the other monocyte and macrophage populations (Figure 2D). To determine the effects of myeloid cell populations on the prognosis of CRC patients, we combined bulk transcriptome and scRNA-seq data for further analysis. An integrated cohort that included 1,673 CRC patients was selected. With the gene signature of each myeloid subpopulation identified from scRNA-seq, the association of the myeloid population and recurrence-free survival (RFS) of CRC patients was analyzed. We observed that patients with enrichment of FCN+ monocytes, DCs, mast cells, OLR1+ macrophages, HSPA6+ monocytes, and FOLR2+ macrophages exhibited worse RFS survival (Figure S2B). Taken together, the myeloid populations and their immune activities were identified in CRC patients, which helped to identify the CCIM in our study.
Functionality difference of FOLR2+ macrophages compared with other macrophage populations
To further analyze the functionality difference of the identified macrophage populations (Figure 3A), we performed the analyses below. Single-cell trajectory analysis revealed the pseudotime differentiation trajectory of monocytes/macrophages. In particular, the FOLR2+ macrophage subpopulation is located at the terminal differentiation site (Figure 3B). Distinct macrophage-related signatures were associated with aspects of macrophage activity, including antigen presentation, the M1 pathway, complement, the proteasome, FC receptor signaling, and interferon (IFN)-gamma response function, and were observed in the different monocyte/macrophage populations (Figure 3C). M1 pathway, complement, FC receptor signaling and IFN-gamma response functions were downregulated in FOLR2+ macrophages compared with other macrophage populations (Figure 3C). Furthermore, the transcription factor (TF) analysis suggested significantly distinct TF activities among FOLR2+ macrophages and other monocyte/macrophage subpopulations (Figure 3D). For instance, CEBPB is a TF that plays crucial roles in macrophages differentiating into the pro-inflammatory spectrum.29 FOLR2+ macrophages showed significantly less expression of CEBPB compared with the other populations. In contrast, macrophage TCF4, which acts as a crucial Wnt pathway regulator and contributes hypoinflammation M2 polarization,30 was significantly more expressed in FOLR2+ macrophages (Figure 3D). In parallel, distinct pro-inflammatory and anti-inflammatory expression patterns were observed in the monocyte/macrophage populations with a previously defined gene set (Figure 3E). We therefore confirmed that FOLR2+ macrophages showed a more M2-like phenotype than other macrophage subpopulations (Figure 3E). M2-like macrophages exhibit anti-inflammatory characteristics, while M1-like macrophages feature pro-inflammatory characteristics. We investigated the inflammatory properties in each monocyte/macrophage subpopulation. The density plot showed that a large proportion of FOLR2+ macrophages exhibited lower inflammatory response GSVA scores than the other monocyte and macrophage populations, indicating a hypoinflammatory status of FOLR2+ macrophages compared with other macrophage populations (Figure 3F). To validate the results from the single-sample gene set enrichment analysis (ssGSEA), we selected one typical inflammation marker, PTGS2 (commonly known as COX2), as well as one anti-inflammation marker, CTSD, for validation using mIHC technology. FOLR2+ macrophages exhibited lower levels of the inflammatory markers PTGS2 while showing higher levels of the anti-inflammatory markers CTSD compared with other monocytes/macrophages (Figures 3G–3I). Taken together, the terminal differentiation trajectory location, distinct TF activities, gene expression pattern, and macrophage-related pathway alterations in the FOLR2+ macrophage population suggested their special properties, which makes it possible to develop new therapeutics that employ tumor-associated macrophage (TAM) subset targeting. We then focused on FOLR2+ macrophages and their relationship with T cell populations for CCIM construction.
Figure 3.

The anti-inflammatory properties of FOLR2+ macrophages
(A) The UMAP plot shows five monocyte/macrophage subpopulations (patients, n = 26).
(B) Pseudotime analysis of five subclusters of monocytes/macrophages (patients, n = 26). The color key from deep blue to light blue indicates the pseudotime score from low to high.
(C) Heatmap of the single-sample gene set enrichment analysis (ssGSEA) scores for gene sets in monocyte/macrophage subtypes, calculated according to single-cell RNA sequencing (scRNA-seq) data.
(D) Transcription factor (TF) activity in monocyte/macrophage subpopulations.
(E) Heatmap of the expression of M1-related and M2-related genes from the scRNA-seq data.
(F) Density plot indicating the density distribution of the inflammatory response activity score in monocyte and macrophage populations and boxplot indicating the inflammatory response activity score in monocyte and macrophage populations. The inflammatory response activity score was calculated with the ssGSEA method using the inflammatory response signature on the single-cell transcriptome. The p value was calculated with a Kruskal-Wallis test.
(G) Multiplex immunohistochemistry (mIHC) staining for CD68 (green), FOLR2 (red), PTGS2 (purple), CTSD (purple), and DAPI (blue). Scale bar, 10 μm. Biological replicates, n = 5.
(H) The percentage of FOLR2+ CD68+ cells and FOLR2− CD68+ cells expressing PTGS2. A total of 100 cells were included for analysis. Significance was evaluated by a chi-square test.
(I) The percentage of FOLR2+ CD68+ cells and FOLR2− CD68+ cells expressing CTSD. Significance was evaluated by a chi-squared test.
The components and pathway heterogeneity of T cells in CRC
We next determined the T cell subpopulations and key genes expressed in T cells. CD4+ T cells were further subclustered into 5 cell populations: exhausted CD4+ T cells, memory CD4+ T cells, naive CD4+ T cells, helper T cell 17 (Th17) cells, and Treg cells (Figure 4A; see Table S2 for the featured genes in each population). Treg cells showed high expression of FOXP3. Naive CD4+ T cells exhibited high CCR7 and SELL expression. Th17 cells expressed GIMAP-family genes, and exhausted T cells were identified to have high PDCD1 and TIGIT expression. CD8+ cells were subclustered into tolerant CD8+ T cells, effector memory CD8+ T cells, exhausted CD8+ T cells, exhausted CD8+ NK T cells, and tissue-resident CD8+ T cells (Figure 4B; see Table S3 for the featured genes in each population). In terms of the feature genes, effector memory CD8+ T cells showed high expression of GZMK and GIMAP4. Exhausted NK T cells highly expressed LAG3 and NCR3. Tissue-resident CD8+ T cells exhibited high S100A4 expression. Great expression of DKK3 was found on tolerant CD8+ T cells and TMIGD2, and PDCD1 was identified as the feature gene for exhausted CD8+ T cells.
Figure 4.

The subpopulations of T cells showed distinct gene expression patterns and pathway activation levels
(A) The t-SNE plot identifies five subclusters for CD4+ T cells (patients, n = 26).
(B) The t-SNE plot identifies five subclusters for CD8+ T cells (patients, n = 26).
(C) Heatmap showing gene expression in key pathways regulating T cell activities. The color key from blue to red indicates relative expression levels from low to high.
(D) Heatmap representation of the gene set variation analysis (GSVA) results based on immunologic signature C7. The color key from purple to yellow indicates the relative GSVA score from low to high.
(E) Trajectory inference by monocle2 method on five subclusters of CD8+ T cells.
(F) Trajectory inference with the pseudotime score for five subclusters of CD8+ T cells. The color key from black to gold indicates the pseudotime score from low to high.
(G) Trajectory inference with PDCD1 expression for five subclusters of CD8+ T cells. The color key from blue to white indicates PDCD1 expression from low to high.
Next, the key genes involved in T cell regulation pathways, such as cytolytic, cytokine, and inhibitory pathways, were identified (Figure 4C), revealing the heterogeneity of T cell activation. For instance, ZNF683 acted as a TF and was mainly expressed in tissue-resident CD8+ T cells. GZMA, GZMB, NKG7, and other cytolytic genes were mainly expressed in CD8+ T cell subpopulations. Exhausted NK T cells and tissue-resident CD8+ T cells still had the capacity to release IFNG. The C7 immunological signature GSVA score alterations further revealed a suppressive TME in CRC and the modulation of T cell exhaustion (Figure 4D). Last, the pseudotime analysis suggested that PDCD1 was highly expressed in CD8+ T cells that underwent the exhaustion process (Figures 4E–4G). In parallel, high expression of PDCD1 was also observed during the exhaustion process of CD4+ T cells (Figures S3A–S3C). Taken together, the main T cell populations were identified in our RNA-seq analysis, which showed distinct gene expression pattern and pathway activation. We therefore investigated the relationship between the T cell subpopulations and FOLR2+ macrophages.
The FOLR2+ macrophage-oriented CCIM is composed of immunosuppressive T cell populations
The complex cell crosstalk was investigated to reveal the regulation of the suppressive CRC TME. The cell-cell interactions (CCIs) between each cell type were identified.31 FOLR2+ macrophages showed strong CCIs activities with multiple cell populations (Figure 5A). We next focused on the interaction between macrophage populations and T cell populations. FOLR2+ macrophages showed the greatest number of CCIs with both CD4+ T and CD8+ T cells (Figures 5B and 5C; Table S4). In terms of the T cell subpopulations, exhausted CD4+ T cells, Treg cells, tolerant CD8+ T cells, and exhausted CD8+ T cells showed the most CCIs with FOLR2+ macrophages than other T cells. Taken together, these findings indicate that FOLR2+ macrophages have abundant interactions with exhausted CD4+ cells, exhausted CD8+ T cells, tolerant CD8+ T cells, and Treg cells, which may affect the formation of a suppressive CRC TME. The topological pattern of the FOLR2+ macrophages, exhausted CD4+ cells, exhausted CD8+ T cells, tolerant CD8+ T cells, and Treg cells in the CRC TME was validated by mIHC staining (Figure 5D). Large proportions of exhausted CD4+ cells (20.3%), exhausted CD8+ T cells (25.2%), tolerant CD8+ T cells (16.6%), and Treg cells (24.1%) cells were found in the area around FOLR2+ macrophages (Figure 5E), which further confirmed the spatial interaction of these cells. Last, the Voronoi plot suggested aggregation of FOLR2+ macrophages with Tregs cells, exhausted CD4+ T cells, tolerant CD8+ T cells, and exhausted CD8+ T cells in CRC tissues (Figure 5F). Therefore, a CCIM was defined as a module that contained at least one FOLR2+ macrophage, one tolerant CD8+ T cell, one Treg cell, and one exhausted CD4+ T cell (Figure 5G). The high density of the CCIM numbers implied the important influence of CCIM in the suppressive CRC TME (Figure 5H). The interaction between FOLR2+ macrophages and the other CCIM cell populations was identified, showing the suppressive interaction effects from FOLR2+ macrophages on others (Figure 5I). Significant associations of FOLR2+ macrophages with other CCIM cell components were identified, showing the aggregation effects of CCIM (Figures 5J–5L).
Figure 5.

Construction of the CRC immune module (CCIM)
(A) The cell-cell interaction (CCI) network for all cell subpopulations in CRC. The color of edges indicates the type of ligand cell, and the size of edges represents the CCI numbers between cells.
(B) The number of interactions between monocytes/macrophages and CD4+ T cells inferred from single-cell RNA sequencing (scRNA-seq).
(C) The number of interactions between monocytes/macrophages and CD8+ T cells.
(D) mIHC staining for FOLR2 (red), DKK3 (white), FOXP3 (yellow), PDCD1 (orange), CD4 (green), CD8 (light blue), and DAPI (blue) reveals the CCIM image pattern. Scale bar, 50 μm. The dotted line indicates the margin between tumor tissue and para-tumor tissue.
(E) The proportion of cells from CCIM and other cell types around FOLR2+ macrophages. The statistics were determined with the area of 800 pixels around FOLR2+ macrophages. Biological replicates, n = 5.
(F) Voronoi plot for the CCIM topological pattern. The green dot represents the center of each part.
(G) Depiction of the CCIM module. A CCIM module contains at least one FOLR2+ macrophage, one tolerant CD8+ T cell, one Treg cell, and one exhausted CD4+ T cell.
(H) The density plot for the CCIM in CRC.
(I) Heatmap of cell-type-specific receptor-ligand interactions inferred by SingleCellSignalR. Shown are inferred interactions between FOLR2+ macrophages and Treg cells, exhausted CD4+ T cells, exhausted CD4+ T cells, and tolerant CD8+ T cells. Circle color indicates the inferred interaction score, and circle size indicates the mean expression of receptor and ligand genes for each pair.
(J–L) The association of exhausted T cells (p < 0.01) (J), tolerant T cells (p <0.01) (K), and Treg cells (p<0.01) (L) with FOLR2+ macrophages. The p value was calculated with Pearson’s coefficient, and p < 0.05 was considered to indicate significance.
(M) The CCIM score stratifies CRC patients with poor prognosis (patients, n = 1,405).
The weighted gene co-expression network analysis (WGCNA) method infers the co-expression network and related biological mechanisms in high-throughput data. We therefore applied WGCNA to CRC bulk transcriptome data to explore the potential regulation roles underlying CCIM. A power of β = 3 was chosen as the optimal soft threshold to ensure scale-free co-expression (Figures S4A–S4C). The FOLR2+ macrophage score was calculated with the GSVA method for each patient. The unsupervised clustering method was used for obtaining the clustered gene modules. Eleven non-gray (meaningful) gene modules were identified, and the yellow module showed the most significant correlation with the FOLR2+ macrophage GSVA score (Figure S4D). Therefore, the genes involved in the yellow module were identified as the key genes associated with the FOLR2+ macrophage score. A total of 1,052 genes were identified, which suggested a significant association between the module membership score (the central point score for each module calculated by the WCGNA method) and gene significance (the Pearson’s coefficient between the target gene expression and the FOLR2+ macrophage score in the cohort), indicating the importance of those genes in the FOLR2+ macrophage regulating network (Figures S4E and S4F). Using those genes as input for Kyoto Encyclopedia of Genes and Genomes (KEGG) and GO analysis, we further found that T cell-related pathways were altered, which confirmed the immunosuppressive regulation potential of FOLR2+ macrophages (Figures S4G and S4H). Briefly, we utilized the scRNA-seq technology to identify a CCIM cell module whose function was further confirmed by bulk transcriptome analysis.
Lastly, we developed a CCIM scoring system to stratify CRC patients with a distinct prognosis. The integrated cohort (GSE39582, GSE17536, GSE17537, GSE14333, GSE56699, GSE37892, and GSE33113) was used as the training cohort. The top 20 featured genes in each CCIM cell component and the ligand-receptor pairs between FOLR2+ macrophage and other CCIM cell components (Figure 5I) were used as inputs for the least absolute shrinkage and selection operator (LASSO) Cox regression model. A total of 17 features (genes) were selected by the regression model to build the CCIM scoring system (Figure S5A). With the coefficients (Figure S5B) and the distinct expression pattern of the 17 features, the CCIM scores were calculated for each patient. The results indicated that patients with high CCIM scores exhibited poorer survival probability than patients with low CCIM scores (Figure 5M; best-cutoff method for threshold of high and low score). Multivariate Cox analysis suggested that the CCIM score is an independent risk factor for patients with CRC in The Cancer Genome Atlas (TCGA) colon adenocarcinoma (COAD) cohort (Figures S5C and S5D). Furthermore, five independent cohorts (GSE28722, GSE12945, GSE72970, GSE5851, and TCGA-CRC) validated the robustness of the CCIM score in stratifying CRC patients (Figures S5E–S5I). Pooled estimates revealed that a lower CCIM score was significantly associated with improved survival probability (hazard ratio [HR] 2.04, 95% confidence interval [CI] 1.56–2.67, p < 0.001; Figure S5J). In addition, the GSVA algorithm showed significantly increased anti-PD1 resistance in patients with greater CCIM scores by calculating the anti-PD1 resistance-related gene set activities32 (Figure S5K). Through the integration analysis of scRNA-seq and bulk RNA-seq, we identified a cell pattern consisting of FOLR2+ macrophages, Treg cells, exhausted CD4+ T cells, tolerant CD8+ T cells, and exhausted CD8+ T cells. We named this cell pattern the CCIM module, with its features depicted through mIHC and related image analysis.
The FOLR2+ macrophage-oriented CCIM module contributes to AI-guided precision chemotherapy
Several studies have shown the capacity of AI-assisted analysis of transcriptome data to predict therapy response, molecular subtyping, and pathway activation. Nevertheless, the efficiency and accuracy were unstable among different cohorts.33,34,35 We next assessed the potential relationship between FOLR2+ macrophages and the chemotherapy response. With the application of a neural network, we built an FOLR2-RM model based on the FOLR2+ macrophage signature to predict the chemotherapy response in the training cohort and then assessed the prediction efficiency of the FOLR2-RM model in an external cohort (Figure S6A). The confusion matrix and receiver operating characteristic (ROC) plot revealed an acceptable predictive efficiency of the FOLR2-RM model with a relative high area under the curve (AUC) value (AUC = 0.797), which also indicated potential important roles of FOLR2+ macrophages in the response to chemotherapy in CRC patients (Figures S6B and S6C).
However, the AUC value of FOLR2-RM is not as great enough to help with clinical decisions. We then assessed the potential application of the CCIM module to precision chemotherapy. Deep learning technology enabled the extraction of hidden information directly from image patterns and provided potentially clinically useful information. We therefore tested several advanced deep learning models for predicting the response to systemic first-line chemotherapy by CCIM image pattern. To further utilize the CCIM module and improve the prediction efficiency, we built a diagram using the AI-guided CCIM imaging pattern (Figure 6A). We established two medical center cohorts as training and validation cohorts (n = 181, 80% for the training cohort and 20% for the validation cohort) and an independent testing cohort (n = 45). The tumor tissues were obtained by colonoscopy and subjected to mIHC staining depicting the CCIM module before systemic chemotherapy. Then, the patients were treated with systemic chemotherapy, and the chemotherapy response was assessed according to the Response Evaluation Criteria in Solid Tumors (RECIST) standard. Typical computed tomography (CT) scans before and after chemotherapy and CCIM mIHC staining images are shown in Figure 6B. The tumor areas in the mIHC slides were aligned with the tumor areas in the corresponding HE staining slides. Only the tumor areas were used for the subsequent analysis in the CCIM pipeline. The non-responders exhibited greater enrichment of the CCIM per area than the responders among the total patients (Figure 6C). We trained 8 models (ResNet18, ResNet34, ResNet50, ResNet101, Xception, DenseNet121, and ResNext50) in the training and validation cohorts. The predictive accuracy of each model was tested in the independent testing cohort. The AUC value varied from 0.77 to 0.99 in the independent testing cohort (Figure 6D). Finally, the ResNext50 model was selected as the CCIM-Net. CCIM-Net showed the greatest AUC value (AUC = 0.99) and highest accuracy rate (98%) by the confusion matrix (Figures 6E and 6F). The detailed work flow of CCIM-Net construction is depicted in Figure S6D, which shows the four stages from input of the CCIM module to output of decision. Thus, we confirmed that the CCIM-Net is a robust AI-guided tool for clinical chemotherapy decision-making by integrating two independent cohorts.
Figure 6.

Artificial intelligence (AI)-guided precision chemotherapy based on CCIM imaging pattern
(A) Schematic of the AI-guided precision chemotherapy process.
(B) Representative computed tomography (CT) scan and CCIM mIHC pattern images for patients who responded or did not respond to chemotherapy. mIHC staining of FOLR2 (red), DKK3 (white), FOXP3 (yellow), PDCD1 (orange), CD4 (green), CD8 (light-blue) and DAPI (blue) was applied to portray the CCIM. Scale bar, 50 μm. The dotted line indicates the margin between tumor tissue and para-tumor tissue.
(C) Boxplot showing the CCIM numbers in the non-responder group and responder group in the training and validation cohorts. The significance was calculated by a Wilcoxon test, with p < 0.05 considered significant.
(D) Comparison of eight deep learning models in the independent testing cohort. The y axis indicates the area under the curve (AUC) value of each model in the independent testing cohort.
(E) The receiver operating characteristic (ROC) analysis shows the sensitivity and specificity of the CCIM-Net in the independent testing cohort.
(F) The confusion matrix for predicting the chemotherapy response by the CCIM-Net in the independent testing cohort. Columns indicate the predicted response, while rows represent the actual response.
Disruption of the CCIM by targeting FOLR2+ macrophages improved the chemotherapy response
We next assessed the effect of the abundance of FOLR2+ macrophages on chemotherapy outcomes by an in vivo model (Figure 7A). We developed a FOLR2-EGFP (enhanced GFP)-diphtheria toxin receptor (DTR) mouse strain to deplete the FOLR2+ macrophages (Figure S7). The DTR-EGFP fusion sequence was integrated into the region between the 6th exon and the noncoding region of the FOLR2 gene in a homologous recombination way after the genomic cutting from recombinant Cas9 nucleases and designed guide RNAs. The FOLR2-DTR-ApcMin mice were bred by crossing FOLR2-DTR-EGFP mice with B6/JGpt-Apcem1Cin(Min)/Gpt mice. Four groups were designated as control (Ctrl), 5-Fu treatment, diphtheria toxin (DT) treatment, and 5-Fu + DT treatment, and the 5-Fu + DT treatment group exhibited the lowest tumor burden (Figures 7B and 7C). H&E staining and Ki-67 IHC staining were used to assess the properties of the CRC tumor model (Figure 7D). Ki-67 IHC staining suggested reduced proliferation in the 5-Fu + DT treatment group compared with the other groups (Figure 7E). Precise ablation of FOLR2+ (EGFP+) macrophages could be observed after DT injection (Figure 7F). Taken together, these results indicated that anti-FOLR2+ macrophage therapeutics led to disruption of the immunosuppressive CCIM module and enhanced chemotherapy response.
Figure 7.

FOLR2+ macrophage depletion disrupts the immunosuppressive tumor microenvironment (TME)
(A) The workflow of the in vivo experiment.
(B) The tumor burden from mice of four groups was assessed (n = 5).
(C) The statistics for tumor burden in each group. Significance was evaluated by a Kruskal-Wallis test (n = 5).
(D) H&E and Ki-67 IHC staining for the tumor tissues from four groups of mice (biological replicates, n = 5). Scale bars, 100 μm (H&E) and 40 μm (IHC)
(E) The statistics for Ki-67-positive cells in each group. Significance was evaluated by a Kruskal-Wallis test (n = 5).
(F) Multiplex immunohistochemistry staining of F4/80 (green) and FOLR2 (red) on tumor tissues in the in vivo model.
Discussion
In this study, we comprehensively analyzed the molecular and cellular signature of CRC components through depiction of the single-cell landscape and integration of the bulk transcriptome and mIHC. Importantly, we determined the importance of FOLR2+ macrophages, which showed distinct properties compared with other macrophage populations, in the formation of the immunosuppressive module affecting prognosis. A CCIM consisting of FOLR2+ macrophages, exhausted CD4+ T cells, exhausted CD8+ T cells, tolerant CD8+ T cells, and Treg cells was identified. We therefore developed a CCIM scoring system to serve as a clinically tractable tool for survival prediction and treatment guidance in patients with CRC. With the CCIM spatial interaction map-based mIHC pattern, we developed an AI-guided precision chemotherapy model, which showed robust prediction efficiency for chemotherapy sensitivity. The in vivo model further indicated that targeting FOLR2+ macrophages disrupted the CCIM and improved the chemotherapy response; thus, we considered that disruption of the FOLR2+ macrophage-mediated CCIM could be a promising therapy for CRC.
Zhang et al.36 highlighted that myeloid populations and conventional dendritic cell subsets are key mediators of cellular crosstalk in the CRC TME.36 In our study, we observed that the majority of DCs were type 2 conventional DCs (cDC2s), as evidenced by high expression levels of CD1c and CLEC10A. cDC2s were associated with Treg cell abundance and promoted Treg cell-mediated immunosuppression in hepatocellular carcinoma through a hypoxia-dependent mechanism.37 Regarding CRC, cDC2s were found to be heterogeneous and could be further subdivided into several subpopulations.38 Specifically, C1QC+ cDC2s were associated with worse overall survival in TCGA COAD and rectum adenocarcinoma (READ) datasets.38 The functions and heterogeneity of cDC2s in CRC warrant further exploration. TAMs are one of the main populations in myeloid cells and have been a promising target of immunotherapy. Macrophages in tumor tissues are traditionally classified into two subsets, the M1 and M2 populations, based on their distinguished molecular features and pathway differences.39,40 M1 macrophages show proinflammatory properties and are considered an antitumor subset. In contrast, M2 macrophages, which feature anti-inflammatory and proangiogenic properties, promote tumor progression in various solid tumor types.41,42,43 Nevertheless, we consider that the M1 and M2 classification may not be suitable in CRC considering the heterogeneity of CRC immune populations. A variety of TAMs in the TME confer both protection and vulnerability.44 Targeting protumorigenic macrophages is a priority in cancer treatment, but in most tumor types, it is still unclear which TAM subsets have a protumorigenic role and when they should be targeted. Identifying the protumorigenic TAM subsets and their biological functions in CRC is thus crucial to develop new therapeutics that employ TAM subset targeting. We therefore explored the monocyte/macrophage populations in CRC by integrated scRNA-seq. FCN1+ monocyte, HSPA6+ monocyte, OAS+ macrophage, OLR1+ macrophage, and FOLR2+ macrophage populations with distinct gene expression signatures were identified.
Macrophages are typically distributed throughout the villi of the lamina propria.45 In our study, we obtained tumor tissues that contained both the stromal area and intraepithelial layer. The majority of macrophages we obtained were from the stromal area, which explains the high abundance of macrophages in our dataset. Resident macrophages within the stromal area exhibit a persistent anti-inflammatory phenotype, even in inflammatory settings, which may be essential for mucosal healing.46,47 In agreement with this, depletion of resident macrophages has been shown to exacerbate experimental colitis.48 In our dataset, we also observed the low inflammatory properties of FOLR2+ macrophages. In tumor tissues, it is still not clear what the exact functions of resident macrophages in the TME are. One study showed that recruitment of macrophages in CCR2−/− mice to pancreatic ductal adenocarcinoma tumors was reduced.49 Nevertheless, pancreatic ductal tumor growth is not affected by a reduction in macrophages, implying that pancreatic ductal tumor progression may be regulated by a CCR2+ macrophage-independent mechanism. They then found that resident macrophages are able to self-renew in tumors with the aid of tumor-derived CSF-1 and that this process can promote tumor progression.49 Nalio Ramos et al.50 revealed that FOLR2+ macrophages reside in a perivascular niche in the tumor stroma of breast cancer. The density of FOLR2+ macrophages positively correlate with T cell infiltration and better prognosis. In our study, we found distinct TF, pathway activation, and expression patterns of FOLR2+ macrophages compared with other macrophage populations. Active lipid metabolism-related pathways were enriched in FOLR2+ macrophages. Given that M2-phenotype macrophages are macrophages that have anti-inflammatory properties and significant lipid metabolism activation, we considered that FOLR2+ macrophages exhibited an M2-like resident phenotype in CRC and were correlated with a poor prognosis of CRC. Regarding the contradictory results between breast cancer and CRC, we investigated our signature of FOLR2+ macrophages in breast cancer and found a positive association between FOLR2+ macrophages and prognosis. Several factors may contribute to the observed inconsistencies between the two cancers. First, in CRC, FOLR2+ macrophages exhibit numerous cell communication pairs with Treg, exhausted T, and tolerant T cells, which may act as immunosuppressive factors. In contrast, FOLR2+ macrophages in breast cancer demonstrate the functional ability to activate CD8+ T cells. Additionally, the distinct pathological features of breast cancer and CRC may influence the spatial distribution of FOLR2+ macrophages and other immune cells. Specifically, FOLR2+ macrophages reside in a perivascular niche in the tumor stroma of breast cancer, whereas the observed FOLR2+ macrophages in this study predominantly originate from the stromal area. Therefore, FOLR2+ macrophages from different organs may have distinct effects on the TME and prognosis of tumors of different origins.
In the next step, we detected abundant FOLR2+ macrophages, Treg cells, exhausted CD4+ T cells, exhausted CD8+ T cells, and tolerant CD8+ T cells in CRC tissues, which constitute a cell module named CCIM. Colocalization of FOLR2+ macrophages, Treg cells, exhausted CD4+ T cells, exhausted CD8+ T cells, and tolerant CD8+ T cells was observed in tumor tissues, indicating the potential roles of CCIM topology in reprogramming the suppressive TME in CRC. Most previous studies infer interaction networks between cell populations based on the expression of receptor-ligand pairs. However, the full spectrum of network relationships cannot be captured by discrete cell clusters considering the spatial distribution. Recently, imaging-based studies have highlighted CCI networks based on the colocalization of neighboring cell populations.51 The so-called TME hub (or module) showed the capacity to regulate the suppressive immune TME and predict the immunotherapy response. For instance, Leader et al.52 revealed a cellular module consisting of PDCD1+CXCL13+ activated T cells, immunoglobulin G (IgG)+ plasma cells, and SPP1+ macrophages, referred to as the lung cancer activation module (LCAM).52 High baseline LCAM scores were correlated with enhanced immunotherapy response even in patients with above-median tumor mutation burden (TMB), suggesting that immune cell composition, while correlated with TMB, may be a nonredundant biomarker of the response to immunotherapy.52 Pelka et al.18 discovered a myeloid cell-attracting hub at the tumor-luminal interface associated with tissue damage. In contrast, a dMMR-enriched immune hub within the tumor was identified, featuring activated T cells together with malignant and myeloid cells expressing T cell-attracting chemokines.18 Nevertheless, most of the previous TME module research focused on depiction of the image pattern.
In recent years, AI-guided risk stratification and precision treatment for cancer have been promising methods for personized medicine.4,6,53,54,55,56 Considering the special topological pattern of CCIM shown by the Voronoi plot, we developed a workflow for building an scRNA-seq-assisted and CCIM-based precision chemotherapy model. We applied cutting-edge deep learning technology to train 8 AI models and finally built a CCIM-Net, which showed high prediction accuracy in the testing cohort. Therefore, we demonstrate that the CCIM-Net based on the seven-colour mIHC panel is a robust tool for helping oncologists predict chemotherapy response. The CCIM-Net did not rely on high-throughput data generated from fresh tumor tissues and showed the properties of being efficient, cost effective, and pathology friendly. The AI-assisted tool may aid in the development of precise treatment strategies for CRC.
The anti-CSF1R antibody is a decent way to target TAMs and lead to their depletion.57 Nevertheless, various solid tumors showed limited response to the CSF1R antibody. Zhang et al.36 identified the persistence of the anti-inflammatory TAM population, and the loss of proinflammatory TAM populations is involved in resistance to anti-CSF1R therapy. FOLR2+ macrophages showed a hypoinflammatory property in CRC that was not observed for other macrophage populations. Thus, targeting the distinct FOLR2+ macrophage population may be more advantageous to overcome the drawback of general TAM blockade therapy. As expected, the depletion of the FOLR2+ macrophages in vivo also showed a considerable antitumor effect. Therefore, we considered that targeting FOLR2+ macrophages may be a promising method in combination with chemotherapy for CRC.
Taken together, our study reveals the CCIM topological pattern and uncovers the importance of CCIM components in reprogramming the suppressive CRC TME. The CCIM score serves as a clinically tractable tool for survival prediction. The CCIM-Net acts as a promising and effective opportunity for therapeutic intervention and precision chemotherapy. Targeting FOLR2+ macrophages can alter the enrichment of the CCIM and holds promise as a therapeutic strategy. Therefore, we believe that the immune spatial interaction map of CCIM can serve as a more direct measurement of the immunosuppressive TME and provide information for clinical decision-making.
Limitations of the study
There are still some limitations to our study. First, background differences caused by the biopsy methods and length of data sequencing from the in-house cohort and the public cohort may exist. Second, two in-house samples make a predominant contribution to the myeloid and epithelial cell populations, which may affect the generalizability of these findings, although we applied large series of mIHC sections for validation. Last, we proposed that our AI-guided precision chemotherapy model contained information for clinical decision-making and obtained samples from our clinic to further validate this system. Multicenter cohort validation and further clinical trials should be considered.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal Anti-F4/80 | Abcam | Cat#ab300421; RRID:AB_2936298 |
| Rabbit monoclonal Anti-Cathepsin D | Abcam | Cat#ab75852; RRID:AB_1523267 |
| CD68 (D4B9C) XP® Rabbit mAb | Cell Signaling Technology | Cat#76437; RRID:AB_2799882 |
| Rabbit monoclonal Anti-CD4 | Abcam | Cat#ab183685; RRID:AB_2686917 |
| InVivoMAb anti-mouse CSF1R (CD115) | Bioxcell | Cat#BE0213; RRID:AB_2687699 |
| CD4 (EP204) Rabbit mAb | Cell Signaling Technology | Cat#48274; RRID:AB_3076699 |
| CD8α (D8A8Y) Rabbit mAb | Cell Signaling Technology | Cat#85336s; RRID:AB_2800052 |
| Rabbit monoclonal Anti-CD8a | Abcam | Cat#ab217344; RRID:AB_2890649) |
| Mouse monoclonal Anti-FOLR2 | Abcam | Cat#ab103988; RRID:AB_10711133 |
| Rabbit polyclonal Anti-FOLR2 | Abcam | Cat#ab228643 |
| Rabbit monoclonal Anti-PTGS2 | Abcam | Cat#ab179800; RRID:AB_2894871 |
| Rabbit monoclonal Anti-PD-1 | Abcam | Cat# ab214421; RRID:AB_294180 |
| Mouse monoclonal Anti-PD-1 | Abcam | Cat#ab52587; RRID:AB_881954 |
| Rabbit polyclonal Anti-DKK3 | Invitrogen | Cat#PA5-102626; RRID:AB_2852023 |
| Rabbit monoclonal Anti-FOXP3 | Invitrogen | Cat#700914; RRID:AB_2532349 |
| BD Pharmingen™ Purified Mouse Anti-Ki-67 | BD Biosciences | Cat#550609; RRID:AB_393778 |
| Chemicals, peptides, and recombinant proteins | ||
| Dextran sulfate (sodium salt) | Cayman Chemical | Cat#23250 |
| RPMI 1640 | Gibco | Cat#11875–093 |
| Fluorouracil | MCE | Cat#HY-90006 |
| Protease inhibitor | Solarbio | Cat#P6730 |
| Dispase II | Sigma‒Aldrich | Cat#42613-33-2 |
| Type VIII Collagenase | Sigma‒Aldrich | Cat#C2139 |
| DNase I | NEB | Cat#M0303S |
| Fetal bovine serum | Gibco | Cat#16000–044 |
| PBS | Solarbio | Cat#P1020 |
| Nylon cell strainer | Falcon | Cat#352340 |
| Red blood cell lysis buffer | Invitrogen | Cat#00-4333-57 |
| Bovine serum albumin | Sigma‒Aldrich | Cat#B2064 |
| Dead Cell Removal Kit | Miltenyi Biotec | Cat#130-090-101 |
| TBST | Solarbio | Cat#T1082 |
| Opal Polymer HRP Ms+Rb | AKOYA Biosciences | Cat#NEL820001KT |
| Blocking solution | Proteintech | Cat#B900780 |
| Diphtheria toxin | MCE | Cat#HY-108851 |
| 4% Paraformaldehyde | Solarbio | Cat#P1110 |
| Modified Hematoxylin-Eosin (HE) Stain Kit | Solarbio | Cat#G1121 |
| 0.1% ammonia water | Solarbio | Cat#G1823 |
| Parafin | Solarbio | Cat#YA0011 |
| 10% neutral buffered formalin (NBF) | Solarbio | Cat#G2161 |
| Xylene | Sinopharm Chemical ReagentCo., Ltd | Cat#10023418 |
| Alcohol | Sinopharm Chemical ReagentCo., Ltd | Cat#10009218 |
| Epitope retrieval agent | Solarbio | Cat#C1032 |
| Anti-mouse/rabbit universal immunohistochemical detection kit | Proteintech | Cat#pk10006 |
| Biological samples | ||
| B6/JGpt-Apcem1Cin(Min)/Gpt mice | Gempharmatech Co., Ltd | N/A |
| Critical commercial assays | ||
| Single-cell sequencing | OE Biotech Co., Ltd | N/A |
| Deposited data | ||
| Single-cell sequencing data | This paper | HRA003569 |
| TCGA-COAD cohort RNA sequencing data | TCGA database | N/A |
| TCGA-READ cohort RNA sequencing data | TCGA database | N/A |
| GEO cohorts | GEO | GSE56699, GSE14333, GSE39582, GSE17536, GSE17537, GSE33113, GSE37892, GSE146771, GSE72970, GSE19860 |
| Software and algorithms | ||
| R software (version 4.0.4) | R Core | https://www.r-project.org/ |
| Seurat (version 3.1.1)58 | Stuart T. et al., 201858 | https://satijalab.org/seurat/ |
| Cell Ranger software (version 3.1.0) | 10× genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/overview/welcome |
| sva R package (version 3.44.0)59 | Leek, J.T. et al.59 | https://bioconductor.org/packages/release/bioc/html/sva.html |
| ConsensusClusterPlus (version 3.16)60 | Matt, W., Peter, W.60 | https://git.bioconductor.org/packages/ConsensusClusterPlus |
| CMScaller R package61 | Peter, W.E. et al.61 | https://github.com/peterawe/CMScaller |
| SingleCellSignalR R package31 | Simon, C.A. et al.31 | https://git.bioconductor.org/packages/SingleCellSignalR |
| Monocle2 package (v2.8.0)62 | Trapnell, C. et al.62 | |
| WGCNA R package63 | Peter, L. et al.63 | https://github.com/cran/WGCNA |
| ClusterProfiler R package (version 4.5.2)64 | Yu, G. et al.64 | https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html |
| GSVA R package (version 1.45.5)65 | Hänzelmann, S. et al.65 | https://bioconductor.org/packages/release/bioc/html/GSVA.html |
| Image-Pro Plus software (version 6.0) | Media Cybernetics | https://www.mediacy.com/78-products/image-pro-plus |
| ImageJ software (version 4.0) | NIH | https://imagej.nih.gov/ij/ |
| Countess | Thermo | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Peng Zhao (zhaop@zju.edu.cn).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The RNA sequencing data were deposited at Gene Expression Omnibus (GEO) (GSE56699, GSE14333, GSE39582, GSE17536, GSE17537, GSE33113, GSE37892, GSE146771, GSE72970, and GSE19860) and The Cancer Genome Atlas (TCGA), which are publicly available. All raw data generated by this study have been deposited in the Chinese national genomics data center (https://ngdc.cncb.ac.cn), under accession number HRA003569. The software and algorithms for data analyses used in this study are published and referenced throughout the STAR Methods section. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and study participant details
Animal objects
B6/JGpt-Apcem1Cin(Min)/Gpt mice were purchased from the Gempharmatech Co., Ltd (China), and FOLR2-DTR mice were a kind gift from Prof. Jianpeng Sheng. All mice were housed in the SPF facility of the First Affiliated Hospital, Zhejiang University School of Medicine, with approval from the Institutional Animal Care and Use Committee (IACUC2022-71) and Zhejiang Center of Laboratory Animals, with approval from the Institutional Animal Care and Use Committee (ZJCLA-IACUC-20020173), respectively. The FOLR2-DTR-ApcMin mice was bred by crossing the FOLR2-DTR-eGFP mice with the B6/JGpt-Apcem1Cin(Min)/Gpt mice. To establish spontaneous CRC, 3% Dextran sulfate (sodium salt) (DSS) (Cayman Chemical, #23250, USA) was given in the drinking water for 1 week followed by ordinary drinking water. Mice were treated with diphtheria toxin (MCE, #HY-108851, USA) or 50 mg/kg 5-Fluorouracil (5FU) (MCE, #HY-90006, USA) through intraperitoneal injection for 2 weeks before sacrificed, and the injections were performed every 3 days. On day 42, all mice were sacrificed, and tumors were collected for H&E, IHC, and mIHC staining.
Human subjects
The bulk transcriptome data and clinical information were obtained from the Gene Expression Omnibus (GEO) repository (https://www.ncbi.nlm.nih.gov/geo/) and The Cancer Genome Atlas (TCGA) (colon cancer and rectal cancer, https://xenabrowser.net/datapages/). GSE39582, GSE17536, GSE17537, GSE14333, GSE56699, GSE37892, and GSE33113 expression profile data were integrated as an integrated cohort after removing batch effects using the sva R package.59 A total of 2081 patients were included in the survival analysis. 1405 patients were used as the integrative training cohorts and 682 patients were included in the four independent validation cohorts. The detailed clinical information is provided in Table S5. The transcriptome and the chemotherapy response data from GSE72970 were used to build the FOLR2+ resident-phenotype macrophage (RM) model, and data from GSE19860 were used to test the efficiency.
The scRNA-seq data for our in-house cohort and the GSE146771 cohort from the GEO repository were integrated. This study was approved by the Ethics Committee of the First Affiliated Hospital, Zhejiang University School of Medicine and the Ethics Committee of the Affiliated Hospital of Southwest Medical University (IIT20220758A) and the Ethics Committee of the Affiliated Hospital of Southwest Medical University (KY2023312). Tumor tissues were obtained from biopsies of 8 patients diagnosed with CRC. All samples for scRNA-seq were verified by pathological examinations as adenocarcinoma. Among the eight samples, three samples came from right-side (ascending colon and transverse colon), three samples came from left-side (sigmoid colon and descending colon) and two samples came from rectum. Each sample was cut into 5 mm3 size with a sterile scalpel to keep the consistency between each sample and followed by dissociation. The detailed clinical information for scRNA-seq patient is provided in Table S6. The inclusion criteria for both the training cohort and validation cohort were as follows: patients who received first-line therapy, with the first-line chemotherapy regimens being CAPOX (Capecitabine + Oxaliplatin) or FOLFOX (Leucovorin Calcium + Fluorouracil + Oxaliplatin). The age range of the patients was from 18 to 75 years, and they had an ECOG performance status ranging from 0 to 2, along with adequate renal, hepatic, and bone marrow function. The major exclusion criteria included active inflammation, prior or concurrent malignant disease within the last 5 years, previous use of study drugs, and the presence of poorly controlled hypertension, diabetes, serious cardiovascular disease, or other chronic diseases, among others. Tumor response was regularly assessed every 2 months, following the Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1. A total of 226 patients were included, with 181 in the training cohort and 45 in the testing cohort. More detailed clinical information can be found in Tables S7 and S8. The tumor tissues contained both the stromal area and intraepithelial layer for the next step analysis.
Method details
scRNA-seq processing
RPMI 1640 (Gibco, #11875–093, USA) with 1 mM protease inhibitor (Solarbio, #P6730, China) was used to transport CRC tissues. Tissues were digested with a dissociation enzyme cocktail prepared by dissolving 2 mg/mL Dispase II (Sigma‒Aldrich, #42613-33-2, USA), 1 mg/mL Type VIII Collagenase (Sigma‒Aldrich, #C2139, USA), and 1 unit/mL DNase I (NEB, #M0303S, USA) in PBS with 5% fetal bovine serum (FBS; Gibco, #16000–044, USA) for 40 min at 37°C. The cells were dissociated and collected every 20 min and then filtered using a 40 μm nylon cell strainer (Falcon, #352340, USA). Red blood cell lysis buffer (Invitrogen, #00-4333-57, USA) with 1 unit/mL DNase I was used to remove red blood cells. Finally, the cells were washed in PBS (Solarbio, #P1020, China) with 0.04% bovine serum albumin (BSA; Sigma‒Aldrich, #B2064, USA). The concentration of the single-cell suspension was computed with Countess (Thermo) and adjusted to 1000 cells/μL. After removing the dead cells by Dead Cell Removal Kit (Miltenyi Biotec, #130-090-101, Germany), the cells were loaded according to the Chromium single-cell 3′ kit standard protocol to capture 5,000–10,000 cells/chip position (V2 chemistry). Library construction and all the other processes were performed according to the standard manufacturer’s protocol.
Illumina HiSeq X Ten was used to obtain single-cell libraries using 150 nt paired-end sequencing. The Cell Ranger software pipeline (version 3.1.0) provided by 10×Genomics was used to demultiplex cellular barcodes, map reads to the genome and transcriptome using the STAR aligner, and down-sample reads as required to generate normalized aggregate data across samples, producing a matrix of gene counts versus cells. We processed the unique molecular identifier (UMI) count matrix using the R package Seurat (version 3.1.1). To remove low-quality cells and likely multiplet captures, which is a major concern in microdroplet-based experiments, a set of criteria were conducted: (1) removing cells with less than 500 UMIs; (2) removing high mitochondrial RNA UMIs (more than four times of the median number of mitochondrial UMIs across cells); (3) filtering genes expressed in less than five cells. Also, the doublets were excluded by the parameter: nFeature_RNA <6000 of subset function in Seurat package. Library size normalization was performed with NormalizeData function in Seurat to obtain the normalized count. Specifically, the global-scaling normalization method “LogNormalize” normalized the gene expression measurements for each cell by the total expression, multiplied by a scaling factor (10,000 by default), and the results were log transformed. Subclusters were identified by principal component analysis (PCA) and t-distributed stochastic neighbor embedding (t-SNE) using the Seurat R package and the Seurat FindVariableGenes function.66 The featured gene signature was constructed with the Seurat R package. Single-cell trajectories in CRC were constructed using the Monocle2 package (v2.8.0) with the features of each populations as the input.67 The transcription factor (TF) analysis was performed with dorothea package using the default parameters.68
Functional enrichment analysis
The GSVA R package was applied for single-sample GSEA (ssGSEA) for each sample.65 The gene sets obtained from scRNA-seq and from Broad Institute (http://www.gsea-msigdb.org/gsea/msigdb/index.jsp, C7 represents cell states and perturbations within the immune system) were used for the enrichment analysis. The results were plotted with the ggplot2 R package and pheatmap R package.69
Cell‒cell interaction (CCI) analysis using SingleCellSignalR
To systematically analyze CCIs, we adopted the SingleCellSignalR package to explore the ligand and target gene pairs.31 Gene expression data of interacting cells were input into SingleCellSignalR and combined with a prior model that integrated existing knowledge on ligand-to-target signaling paths. Then, the ligand‒receptor interactions that drive gene expression changes in the cells of interest were predicted.
Weighted gene co-expression network analysis (WGCNA)
A gene co-expression network was built by the WGCNA model.63 Raising the co-expression similarity to a power β defined a weighted network adjacency. By evaluating the correlations between the FOLR2+ macrophage abundance of patients from the integrated cohort and the module memberships, it was possible to identify highly correlated modules. The hub gene in the yellow module was selected and subjected to further analysis. Gene ontology (GO) analysis was performed by the clusterProfiler R package,64 and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis was performed by Proteomaps, a bionic visualization method of all pathways (https://bionic-vis.biologie.uni-greifswald.de/).
Multiplex immunohistochemistry (mIHC) staining of tissue sections
Tumor tissues (4 μm) from CRC patients were fixed with 4% paraformaldehyde (Solarbio, #P1110, China), and then embedded in paraffin (Solarbio, #YA0011, China). The specificity and optimal dilution of each antibody were individually determined using slides from CRC tissues before being used in combination, following the SITC multiplex immunohistochemistry guidance.70 To overcome the issue of signal-to-noise, a tyramide signal amplification (TSA) approach was applied. In summary, several key steps should be followed before performing mIHC. The first step is to select the appropriate monoclonal antibody that matches the target protein intended for labeling. Next, mIHC staining was performed. In brief, the prepared tissue sections (4 μm) were baked in an oven at 65°C for 1 h to improve sample adhesion to the slide. Then, they were dewaxed with fresh xylene twice for 15 min each and rehydrated with graded alcohol (100%, 95%, 85%, 75%) for 5 min each, respectively. After rehydration, the sections were fixed in 10% neutral buffered formalin (NBF) (Solarbio, #G2161, China) for 20 min at room temperature. Subsequently, the sections were transferred to an appropriate antigen retrieval (AR) buffer and placed in a microwave for 1 min at 100% power, followed by an additional 15 min at 20% power. Then, the slides were blocked after cooling to room temperature and subsequently incubated with a primary antibody at room temperature for 10 min. To remove any excess antibody, the slides were washed three times with TBST (Solarbio, #T1082, China). Following this, the slides were incubated with Opal Polymer HRP Ms+Rb (AKOYA Biosciences, #NEL820001KT, USA) at room temperature for 10 min. To remove any remaining wash buffer, the slides were rinsed three times with TBST before incubation with Opal Signal Generation. The process of microwave treatment, blocking, primary antibody incubation, introduction of Opal Polymer HRP, and signal amplification were repeated before each subsequent antibody incubation. Primary antibodies, including CD68 (1:100, #76437, Cell Signaling Technology), F4/80 (1:200, #ab300421, Abcam), FOLR2 (1:300, #ab103988, Abcam), FOLR2 (1:300, #ab228643, Abcam), PTGS2 (1:100, #ab179800, Abcam), CD4 (1:200, #48274, Cell Signaling Technology), CD4 (1:100, #ab183685, Abcam), CD8α (1:200, #85336s, Cell Signaling Technology), CD8a (1:100, #ab217344, Abcam), PD-1 (1:200, #ab214421, Abcam), PD-1 (1:100, #ab52587, Abcam), DKK3 (1:100, #PA5-102626, Invitrogen), FOXP3 (1:100, #700914, Invitrogen), CTSD (1:100, #ab75852, Abcam) were labeled. Next, the slides were incubated with DAPI working solution in the dark for 5 min at room temperature. Afterward, the slides were washed with distilled water and TBST before mounting. Finally, a confocal microscope (Nikon, Japan) or a Vectra Polaris Quantitative Pathology Imaging Systems was used to capture images of those tissue samples, and the acquired images were analyzed using ImageJ (version 4.0).
AI-guided chemotherapy model construction
The FOLR2+ macrophage gene signature was applied to build the neural network model for predicting the chemotherapy response for CRC. Briefly, the transcriptome data and the chemotherapy response data from GSE72970 were used as the input and output for training the FOLR2-RM model. GSE19860 was applied as the testing cohort. And the traditional neural network was applied with 25 layers for construction.
The mIHC staining slide and paired HE staining slide were prepared for model construction. HE staining was scanned by 3D Histech and used for classifying tumor areas. The classifiers tab function of HALO digital pathology system was utilized to identify tumor areas. Subsequently, the tumor areas in the mIHC slides were aligned with the tumor areas in the corresponding HE staining slides. Only tumor areas were considered for the subsequent analysis in the CCIM pipeline (detailed pipeline shown in Figure S8). In the image preprocessing stage, in order to preserve the original information of the image as much as possible, only local pixel resampling and color normalization are adopted. Since the sensitivity of color features in the task of determining chemotherapeutic response from multi-color fluorescence images, techniques for color enhancement other than color normalization are not used during preprocessing or even during training. For CCIM-net, 8 models (ResNet18, ResNet34, ResNet50, ResNet101, InceptionV3, Xception, DenseNet121, ResNext50) are pre-trained on ImageNet, which makes the model have better initialization parameters for deep feature extraction. To help the model more effectively adapt to the initial learning rate, we use one round of warm-up prior to the training phase. At 30, 50, and 80 epochs, the learning rate then declines using a multi-step method to 0.8 times its initial value. The learning rate and batch size hyperparameters for these models were set at 0.001 and 4, respectively. After training each model for 100 epochs, save the model parameters for the best-performing epoch. The ResNet several models introduced the conception of residual network. For a block, let the function that can be fitted be and its expected latent mapping be . ResNet network model learns the residual namely , so that the rise path becomes , and is used to fit H(x). In ResNet, the block formed by is called Residual block. Multiple similar Residual blocks in series formed the ResNet. The ResNet18, ResNet34, ResNet50 and ResNet101 are the different versions of ResNet. The main difference of each version came from the composition of residual block and the number of convolution layers. The ResNet18 and ResNet34 were constructed with BasicBlock while ResNet50 and ResNet101 were constructed with Bottleneck. The ResNext model can improve the accuracy in the case of not improving the parameter complexity and reducing the number of super parameters. For instance, the algorithm of traditional fully connected layer is:
where .
is a D-channel input vector to the neuron and is a filter’s weight for the i-th channel. The ResNext replaced the to a more common function as:
where can be an arbitrary function. C is cardinality (the size of the set of transformations). have the same topological pattern. The ResNext50 was selected for building the CCIM-net for the highest accuracy in the testing cohort.
These models all adopt the cross-entropy loss function, which is frequently used to measure the difference between two probability distributions and aims to describe how difficult it is to express a probability distribution p by a probability distribution q. Assuming that p and q are two probability distributions of the data x, the cross-entropy loss of N samples of p represented by q can be calculated as follows:
According to the formula, the closer the probability distributions p and q are, the smaller the cross-entropy loss. In the training process, by constructing the loss constraint between the prediction result and the ground truth and multiple rounds of optimization iterations, the cross-entropy loss will converge, and the model has the ability to characterize the chemotherapy response results corresponding to the multi-color fluorescence images. Finally, the probability distribution output by the model on the test samples is close to the probability distribution of the training samples, which makes the prediction results more accurate.
Haematoxylin and eosin (HE) staining
The tumor tissue sections (4μm) were deparaffinized using xylene (Sinopharm Chemical Reagent Co., Ltd, #10023418, China), followed by rehydration with graded alcohol (Sinopharm Chemical Reagent Co., Ltd, #10009218, China). To stain the nuclei, the sections were subjected to hematoxylin staining at room temperature for 30 min, using hematoxylin (Solarbio, #G1121, China). After staining, the sections were washed with PBS to remove any excess hematoxylin and prevent over-staining. Next, 0.1% ammonia water (Solarbio, #G1823, China) was applied to the sections to change the color of the hematoxylin-stained nuclei from reddish to a distinct blue-purple hue. To prepare the sections for cytoplasm staining, they were rinsed with 75% alcohol at room temperature for 2 min. Cytoplasm staining was performed using eosin (Solarbio, #G1121, China) at room temperature for 1 h. After eosin staining, the sections were directly rinsed with graded alcohol to remove excess dye and prepare them for further analysis. Finally, the sections were treated with xylene, which acted as an anhydrous alcohol, before being mounted on slides. The sections were examined using a light microscope (Leica), and the images were analyzed with Image-Pro Plus software (version 6.0).
Immunohistochemistry (IHC) staining
The sections were deparaffinized using xylene, a widely-used solvent known for its ability to effectively remove wax from tissue sections. Subsequently, the sections were rehydrated using graded alcohol to ensure optimal preparation for subsequent procedures. Microwave heating was employed to enhance the detection of specific proteins and retrieve antigen epitopes. A specific epitope retrieval agent (Solarbio, #C1032, China) was utilized to achieve this goal. By subjecting the sections to microwave heating alongside the epitope retrieval agent, the antigens were effectively unmasked, resulting in improved efficiency of antibody binding. To prevent non-specific binding, the sections were then treated with a blocking solution and allowed to incubate for 1 h at room temperature. Following this, the primary antibody specific to Ki67 (1:200, #550609, BD Biosciences) was introduced and allowed to incubate overnight at 4°C. The next day, the sections were thoroughly washed three times with PBS to eliminate any excess primary antibody and unbound molecules. For immunostaining, a versatile anti-mouse/rabbit universal immunohistochemical detection kit (Proteintech, #pk10006, China) was employed. The staining procedure was meticulously carried out according to the manufacturer’s instructions, ensuring superior detection and visualization of the Ki67 protein. Subsequently, the mounted sections were meticulously examined using a Leica microscope specifically calibrated for light microscopy. Finally, the obtained images were subjected to detailed analysis using Image-Pro Plus software (version 6.0).
Tandem mass tag (TMT)-based proteomic analysis
The tissues were dewaxed, rehydrated and then acidic hydrolysis with formic acid (FA) was performed. Proteins were denatured with 6 M urea (Sigma–Aldrich, Germany) and 2 M thiourea (Sigma–Aldrich, Germany) before being digested into peptides with trypsin (1:20; Hualishi, Beijing, China) and Lys-C (1:80; Hualishi, Beijing, China) using pressure-cycling technology (PCT).71,72 TMTpro 16 plex (Thermo Fisher Scientific, San Jose, USA) was used to label peptides.73 In the TMT126 channel, each batch contained 15 experimental samples and one pooled sample for normalization. The fractions (60 per batch) were separated using offline high-pH reversed-phase fractionation with a Thermo Dionex Ultimate 3000 RSLC nano System and then merged to produce a total of 30 fractions per batch. The fractionated samples were subsequently separated using a Thermo Dionex Ultimate 3000 RSLC nano System before being examined with an HF mass spectrometer in data-dependent acquisition (DDA) mode (Thermo Fisher Scientific, San Jose, USA). Using Proteome Discoverer (version 2.4, Thermo Fisher Scientific, Waltham, MA), all reviewed human entries from UniProt (downloaded on 14 April 2020, containing 20,365 proteins) were searched. The detailed parameters were previously described without modification.74,75
Construction of the CCIM scoring system
A CCIM scoring system was constructed with modifications from previous studies.76,77 A CCIM signature was built with the featured genes in FOLR2+ macrophages, exhausted CD4+ T cells, Treg cells, exhausted CD8+ T cells, tolerance NK cells and the genes from CCI ligand‒receptor pairs. The least absolute shrinkage and selection operator (LASSO) was applied to construct the CCIM model.78 The CCIM scoring system was built by including individual normalized gene expression values weighted by their LASSO Cox coefficients as follows: .
Quantification and statistical analysis
All experimental data were analyzed using R software (R version 4.0.4). Measurement data are expressed as the mean ± standard error. A p value <0.05 was considered to indicate significance. Univariate and multivariate Cox analysis was performed to determine whether a variable significantly affected disease-free survival (DFS).
Acknowledgments
This work was supported in part by National Natural Science Foundation of China grants 82101830 (to X.B.), 81874173 (to J.R.), 81472346 (to P.Z.), 82074208 (to P.Z.), and 82102817 (to X.D.) and by Natural Science Foundation of Zhejiang Province grants Y23H160106 (to X.B.), LY20H160033 (to P.Z.), LQ22H160041 (to X.D.), and LY22H160019 (to J.R.). The results here, whole or in part, are based on data generated by the TCGA Research Network (https://www.cancer.gov/tcga).
Author contributions
Conceptualization, X.B.; methodology, X.B., Q.L., D.C., X.D., H.Z., Y.J., C.L., Y.W., C.Y., S.X., X.L., W.T., Y.D., Y.X., V.T., Y.W., W. Fu, S.D., and W. Fang; investigation, X.B., Q.L., D.C., X.D., and H.Z.; visualization, X.B., Q.L., X.D., and H.Z.; supervision, X.B., J.S., J.R., and P.Z.; writing – original draft, X.B.; writing – review & editing, X.B., J.S., J.R., and P.Z.
Declaration of interests
The authors declare no competing interests.
Published: February 1, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2024.101399.
Contributor Information
Xuanwen Bao, Email: xuanwen.bao@zju.edu.cn.
Jianpeng Sheng, Email: shengjp@zju.edu.cn.
Jian Ruan, Email: software233@zju.edu.cn.
Peng Zhao, Email: zhaop@zju.edu.cn.
Supplemental information
References
- 1.Alberts S.R., Sargent D.J., Nair S., Mahoney M.R., Mooney M., Thibodeau S.N., Smyrk T.C., Sinicrope F.A., Chan E., Gill S., et al. Effect of oxaliplatin, fluorouracil, and leucovorin with or without cetuximab on survival among patients with resected stage III colon cancer: a randomized trial. JAMA. 2012;307:1383–1393. doi: 10.1001/jama.2012.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.de Gramont A., Van Cutsem E., Schmoll H.-J., Tabernero J., Clarke S., Moore M.J., Cunningham D., Cartwright T.H., Hecht J.R., Rivera F., et al. Bevacizumab plus oxaliplatin-based chemotherapy as adjuvant treatment for colon cancer (AVANT): a phase 3 randomised controlled trial. Lancet Oncol. 2012;13:1225–1233. doi: 10.1016/S1470-2045(12)70509-0. [DOI] [PubMed] [Google Scholar]
- 3.Dudley J.C., Lin M.-T., Le D.T., Eshleman J.R. Microsatellite instability as a biomarker for PD-1 blockade. Clin. Cancer Res. 2016;22:813–820. doi: 10.1158/1078-0432.CCR-15-1678. [DOI] [PubMed] [Google Scholar]
- 4.Bao X., Shi R., Zhao T., Wang Y., Anastasov N., Rosemann M., Fang W. Integrated analysis of single-cell RNA-seq and bulk RNA-seq unravels tumour heterogeneity plus M2-like tumour-associated macrophage infiltration and aggressiveness in TNBC. Cancer Immunol. Immunother. 2021;70:189–202. doi: 10.1007/s00262-020-02669-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu L., Zhang R., Deng J., Dai X., Zhu X., Fu Q., Zhang H., Tong Z., Zhao P., Fang W. Construction of TME and Identification of crosstalk between malignant cells and macrophages by SPP1 in hepatocellular carcinoma. Cancer Immunol. Immunother. 2021;71:121–136. doi: 10.1007/s00262-021-02967-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bao X., Zhang H., Wu W., Cheng S., Dai X., Zhu X., Fu Q., Tong Z., Liu L., Zheng Y., et al. Analysis of the molecular nature associated with microsatellite status in colon cancer identifies clinical implications for immunotherapy. J. Immunother. Cancer. 2020;8 doi: 10.1136/jitc-2020-001437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DeBerardinis R.J. Tumor microenvironment, metabolism, and immunotherapy. N. Engl. J. Med. 2020;382:869–871. doi: 10.1056/NEJMcibr1914890. [DOI] [PubMed] [Google Scholar]
- 8.Frankel T., Lanfranca M.P., Zou W. The role of tumor microenvironment in cancer immunotherapy. Adv. Exp. Med. Biol. 2017;1036:51–64. doi: 10.1007/978-3-319-67577-0_4. [DOI] [PubMed] [Google Scholar]
- 9.Chen D., Bao X., Zhang R., Ding Y., Zhang M., Li B., Zhang H., Li X., Tong Z., Liu L., et al. Depiction of the genomic and genetic landscape identifies CCL5 as a protective factor in colorectal neuroendocrine carcinoma. Br. J. Cancer. 2021;125:994–1002. doi: 10.1038/s41416-021-01501-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bao X., Li Q., Chen J., Chen D., Ye C., Dai X., Wang Y., Li X., Rong X., Cheng F. Molecular Subgroups of Intrahepatic Cholangiocarcinoma Discovered by Single-Cell RNA Sequencing-Assisted Multi-Omics Analysis. Cancer Immunol. Res. 2022;10:811–828. doi: 10.1158/2326-6066.CIR-21-1101. [DOI] [PubMed] [Google Scholar]
- 11.Bao X., Wang D., Dai X., Liu C., Zhang H., Jin Y., Tong Z., Li B., Tong C., Xin S., et al. An immunometabolism subtyping system identifies S100A9+ macrophage as an immune therapeutic target in colorectal cancer based on multiomics analysis. Cell Rep. Med. 2023;4 doi: 10.1016/j.xcrm.2023.100987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Geeraerts X., Bolli E., Fendt S.-M., Van Ginderachter J.A. Macrophage metabolism as therapeutic target for cancer, atherosclerosis, and obesity. Front. Immunol. 2017;8:289. doi: 10.3389/fimmu.2017.00289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barkal A.A., Brewer R.E., Markovic M., Kowarsky M., Barkal S.A., Zaro B.W., Krishnan V., Hatakeyama J., Dorigo O., Barkal L.J., Weissman I.L. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature. 2019;572:392–396. doi: 10.1038/s41586-019-1456-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bian S., Hou Y., Zhou X., Li X., Yong J., Wang Y., Wang W., Yan J., Hu B., Guo H., et al. Single-cell multiomics sequencing and analyses of human colorectal cancer. Science. 2018;362:1060–1063. doi: 10.1126/science.aao3791. [DOI] [PubMed] [Google Scholar]
- 15.Roerink S.F., Sasaki N., Lee-Six H., Young M.D., Alexandrov L.B., Behjati S., Mitchell T.J., Grossmann S., Lightfoot H., Egan D.A., et al. Intra-tumour diversification in colorectal cancer at the single-cell level. Nature. 2018;556:457–462. doi: 10.1038/s41586-018-0024-3. [DOI] [PubMed] [Google Scholar]
- 16.Zhang L., Yu X., Zheng L., Zhang Y., Li Y., Fang Q., Gao R., Kang B., Zhang Q., Huang J.Y., et al. Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature. 2018;564:268–272. doi: 10.1038/s41586-018-0694-x. [DOI] [PubMed] [Google Scholar]
- 17.Lee H.-O., Hong Y., Etlioglu H.E., Cho Y.B., Pomella V., Van den Bosch B., Vanhecke J., Verbandt S., Hong H., Min J.-W., et al. Lineage-dependent gene expression programs influence the immune landscape of colorectal cancer. Nat. Genet. 2020;52:594–603. doi: 10.1038/s41588-020-0636-z. [DOI] [PubMed] [Google Scholar]
- 18.Pelka K., Hofree M., Chen J.H., Sarkizova S., Pirl J.D., Jorgji V., Bejnood A., Dionne D., Ge W.H., Xu K.H., et al. Spatially organized multicellular immune hubs in human colorectal cancer. Cell. 2021;184:4734–4752.e20. doi: 10.1016/j.cell.2021.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xue D., Tabib T., Morse C., Yang Y., Domsic R., Khanna D., Lafyatis R. Expansion of FCGR3A+ macrophages, FCN1+ mo-DC, and plasmacytoid dendritic cells associated with severe skin disease in systemic sclerosis. Arthritis Rheumatol. 2021;74:329. doi: 10.1002/art.41813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li J., Wang Y., Wang X., Ye L., Zhou Y., Persidsky Y., Ho W. Immune activation of human brain microvascular endothelial cells inhibits HIV replication in macrophages. Blood, The Journal of the American Society of Hematology. 2013;121:2934–2942. doi: 10.1182/blood-2012-08-450353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chou C.-H., Jain V., Gibson J., Attarian D.E., Haraden C.A., Yohn C.B., Laberge R.-M., Gregory S., Kraus V.B. Synovial cell cross-talk with cartilage plays a major role in the pathogenesis of osteoarthritis. Sci. Rep. 2020;10:10868. doi: 10.1038/s41598-020-67730-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wauters E., Van Mol P., Garg A.D., Jansen S., Van Herck Y., Vanderbeke L., Bassez A., Boeckx B., Malengier-Devlies B., Timmerman A., et al. Discriminating mild from critical COVID-19 by innate and adaptive immune single-cell profiling of bronchoalveolar lavages. Cell Res. 2021;31:272–290. doi: 10.1038/s41422-020-00455-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liao M., Liu Y., Yuan J., Wen Y., Xu G., Zhao J., Cheng L., Li J., Wang X., Wang F., et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 2020;26:842–844. doi: 10.1038/s41591-020-0901-9. [DOI] [PubMed] [Google Scholar]
- 24.Dick S.A., Wong A., Hamidzada H., Nejat S., Nechanitzky R., Vohra S., Mueller B., Zaman R., Kantores C., Aronoff L., et al. Three tissue resident macrophage subsets coexist across organs with conserved origins and life cycles. Sci. Immunol. 2022;7 doi: 10.1126/sciimmunol.abf7777. [DOI] [PubMed] [Google Scholar]
- 25.Sharma A., Seow J.J.W., Dutertre C.-A., Pai R., Blériot C., Mishra A., Wong R.M.M., Singh G.S.N., Sudhagar S., Khalilnezhad S., et al. Onco-fetal reprogramming of endothelial cells drives immunosuppressive macrophages in hepatocellular carcinoma. Cell. 2020;183:377–394.e21. doi: 10.1016/j.cell.2020.08.040. [DOI] [PubMed] [Google Scholar]
- 26.Cresswell G.M., Wang B., Kischuk E.M., Broman M.M., Alfar R.A., Vickman R.E., Dimitrov D.S., Kularatne S.A., Sundaram C.P., Singhal S., et al. Folate Receptor Beta Designates Immunosuppressive Tumor-Associated Myeloid Cells That Can Be Reprogrammed with Folate-Targeted Drugs. Cancer Res. 2021;81:671–684. doi: 10.1158/0008-5472.CAN-20-1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Puig-Kröger A., Sierra-Filardi E., Domínguez-Soto A., Samaniego R., Corcuera M.T., Gómez-Aguado F., Ratnam M., Sánchez-Mateos P., Corbí A.L. Folate receptor β is expressed by tumor-associated macrophages and constitutes a marker for M2 anti-inflammatory/regulatory macrophages. Cancer Res. 2009;69:9395–9403. doi: 10.1158/0008-5472.CAN-09-2050. [DOI] [PubMed] [Google Scholar]
- 28.Zou Y., Ye F., Kong Y., Hu X., Deng X., Xie J., Song C., Ou X., Wu S., Wu L., et al. The Single-Cell Landscape of Intratumoral Heterogeneity and The Immunosuppressive Microenvironment in Liver and Brain Metastases of Breast Cancer. Adv. Sci. 2023;10 doi: 10.1002/advs.202203699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ruffell D., Mourkioti F., Gambardella A., Kirstetter P., Lopez R.G., Rosenthal N., Nerlov C. A CREB-C/EBPβ cascade induces M2 macrophage-specific gene expression and promotes muscle injury repair. Proc. Natl. Acad. Sci. USA. 2009;106:17475–17480. doi: 10.1073/pnas.0908641106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cosín-Roger J., Ortiz-Masiá D., Calatayud S., Hernández C., Alvarez A., Hinojosa J., Esplugues J.V., Barrachina M.D. M2 macrophages activate WNT signaling pathway in epithelial cells: relevance in ulcerative colitis. PLoS One. 2013;8 doi: 10.1371/journal.pone.0078128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cabello-Aguilar S., Alame M., Kon-Sun-Tack F., Fau C., Lacroix M., Colinge J. SingleCellSignalR: inference of intercellular networks from single-cell transcriptomics. Nucleic Acids Res. 2020;48:e55. doi: 10.1093/nar/gkaa183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hugo W., Zaretsky J.M., Sun L., Song C., Moreno B.H., Hu-Lieskovan S., Berent-Maoz B., Pang J., Chmielowski B., Cherry G., et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell. 2016;165:35–44. doi: 10.1016/j.cell.2016.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Azarkhalili B., Saberi A., Chitsaz H., Sharifi-Zarchi A. DeePathology: deep multi-task learning for inferring molecular pathology from cancer transcriptome. Sci. Rep. 2019;9:16526–16614. doi: 10.1038/s41598-019-52937-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cieślik M., Chinnaiyan A.M. Cancer transcriptome profiling at the juncture of clinical translation. Nat. Rev. Genet. 2018;19:93–109. doi: 10.1038/nrg.2017.96. [DOI] [PubMed] [Google Scholar]
- 35.Way G.P., Sanchez-Vega F., La K., Armenia J., Chatila W.K., Luna A., Sander C., Cherniack A.D., Mina M., Ciriello G., et al. Machine learning detects pan-cancer ras pathway activation in the cancer genome atlas. Cell Rep. 2018;23:172–180.e3. doi: 10.1016/j.celrep.2018.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang L., Li Z., Skrzypczynska K.M., Fang Q., Zhang W., O’Brien S.A., He Y., Wang L., Zhang Q., Kim A., et al. Single-cell analyses inform mechanisms of myeloid-targeted therapies in colon cancer. Cell. 2020;181:442–459.e29. doi: 10.1016/j.cell.2020.03.048. [DOI] [PubMed] [Google Scholar]
- 37.Suthen S., Lim C.J., Nguyen P.H.D., Dutertre C.A., Lai H.L.H., Wasser M., Chua C., Lim T.K.H., Leow W.Q., Loh T.J., et al. Hypoxia-driven immunosuppression by Treg and type-2 conventional dendritic cells in HCC. Hepatology. 2022;76:1329–1344. doi: 10.1002/hep.32419. [DOI] [PubMed] [Google Scholar]
- 38.Liu Y., Zhang Q., Xing B., Luo N., Gao R., Yu K., Hu X., Bu Z., Peng J., Ren X., Zhang Z. Immune phenotypic linkage between colorectal cancer and liver metastasis. Cancer Cell. 2022;40:424–437.e5. doi: 10.1016/j.ccell.2022.02.013. [DOI] [PubMed] [Google Scholar]
- 39.Wang Y., Smith W., Hao D., He B., Kong L. M1 and M2 macrophage polarization and potentially therapeutic naturally occurring compounds. Int. Immunopharmacol. 2019;70:459–466. doi: 10.1016/j.intimp.2019.02.050. [DOI] [PubMed] [Google Scholar]
- 40.Yunna C., Mengru H., Lei W., Weidong C. Macrophage M1/M2 polarization. Eur. J. Pharmacol. 2020;877 doi: 10.1016/j.ejphar.2020.173090. [DOI] [PubMed] [Google Scholar]
- 41.Martínez V.G., Rubio C., Martínez-Fernández M., Segovia C., López-Calderón F., Garín M.I., Teijeira A., Munera-Maravilla E., Varas A., Sacedón R., et al. BMP4 induces M2 macrophage polarization and favors tumor progression in bladder cancer. Clin. Cancer Res. 2017;23:7388–7399. doi: 10.1158/1078-0432.CCR-17-1004. [DOI] [PubMed] [Google Scholar]
- 42.Aljabery F., Olsson H., Gimm O., Jahnson S., Shabo I. Vol. 4. Elsevier; 2018. M2-macrophage infiltration and macrophage traits of tumor cells in urinary bladder cancer; pp. 159.e119–159.e126. (Urologic Oncology: Seminars and Original Investigations). [DOI] [PubMed] [Google Scholar]
- 43.Suárez-Sánchez F.J., Lequerica-Fernández P., Suárez-Canto J., Rodrigo J.P., Rodriguez-Santamarta T., Domínguez-Iglesias F., García-Pedrero J.M., de Vicente J.C. Macrophages in oral carcinomas: Relationship with cancer stem cell markers and PD-L1 expression. Cancers. 2020;12:1764. doi: 10.3390/cancers12071764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li X., Rawat K., Jakubzick C.V. Targeting resident macrophages in cancer. Nat. Immunol. 2021;22:1078–1079. doi: 10.1038/s41590-021-01002-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Viola M.F., Boeckxstaens G. Intestinal resident macrophages: Multitaskers of the gut. Neuro Gastroenterol. Motil. 2020;32 doi: 10.1111/nmo.13843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weber B., Saurer L., Schenk M., Dickgreber N., Mueller C. CX3CR1 defines functionally distinct intestinal mononuclear phagocyte subsets which maintain their respective functions during homeostatic and inflammatory conditions. Eur. J. Immunol. 2011;41:773–779. doi: 10.1002/eji.201040965. [DOI] [PubMed] [Google Scholar]
- 47.Bain C.C., Oliphant C.J., Thomson C.A., Kullberg M.C., Mowat A.M. Proinflammatory Role of Monocyte-Derived CX3CR1(int) Macrophages in Helicobacter hepaticus-Induced Colitis. Infect. Immun. 2018;86 doi: 10.1128/IAI.00579-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qualls J.E., Kaplan A.M., van Rooijen N., Cohen D.A. Suppression of experimental colitis by intestinal mononuclear phagocytes. J. Leukoc. Biol. 2006;80:802–815. doi: 10.1189/jlb.1205734. [DOI] [PubMed] [Google Scholar]
- 49.Zhu Y., Herndon J.M., Sojka D.K., Kim K.-W., Knolhoff B.L., Zuo C., Cullinan D.R., Luo J., Bearden A.R., Lavine K.J., et al. Tissue-resident macrophages in pancreatic ductal adenocarcinoma originate from embryonic hematopoiesis and promote tumor progression. Immunity. 2017;47 doi: 10.1016/j.immuni.2017.08.018. 597–338.e326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nalio Ramos R., Missolo-Koussou Y., Gerber-Ferder Y., Bromley C.P., Bugatti M., Núñez N.G., Tosello Boari J., Richer W., Menger L., Denizeau J., et al. Tissue-resident FOLR2(+) macrophages associate with CD8(+) T cell infiltration in human breast cancer. Cell. 2022;185:1189–1207.e25. doi: 10.1016/j.cell.2022.02.021. [DOI] [PubMed] [Google Scholar]
- 51.Schürch C.M., Bhate S.S., Barlow G.L., Phillips D.J., Noti L., Zlobec I., Chu P., Black S., Demeter J., McIlwain D.R., et al. Coordinated cellular neighborhoods orchestrate antitumoral immunity at the colorectal cancer invasive front. Cell. 2020;182:1341–1359.e19. doi: 10.1016/j.cell.2020.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Leader A.M., Grout J.A., Maier B.B., Nabet B.Y., Park M.D., Tabachnikova A., Chang C., Walker L., Lansky A., Le Berichel J., et al. Single-cell analysis of human non-small cell lung cancer lesions refines tumor classification and patient stratification. Cancer Cell. 2021;39:1594–1609.e12. doi: 10.1016/j.ccell.2021.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.D’Amore B., Smolinski-Zhao S., Daye D., Uppot R.N. Role of Machine Learning and Artificial Intelligence in Interventional Oncology. Curr. Oncol. Rep. 2021;23:70–78. doi: 10.1007/s11912-021-01054-6. [DOI] [PubMed] [Google Scholar]
- 54.Kann B.H., Hosny A., Aerts H.J. Artificial intelligence for clinical oncology. Cancer Cell. 2021;39:916–927. doi: 10.1016/j.ccell.2021.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Elkhader J., Elemento O. Elsevier; 2021. Artificial Intelligence in Oncology: From Bench to Clinic. [DOI] [PubMed] [Google Scholar]
- 56.Tong Z., Liu Y., Ma H., Zhang J., Lin B., Bao X., Xu X., Gu C., Zheng Y., Liu L., et al. Development, validation and comparison of artificial neural network models and logistic regression models predicting survival of unresectable pancreatic cancer. Front. Bioeng. Biotechnol. 2020;8:196. doi: 10.3389/fbioe.2020.00196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cannarile M.A., Weisser M., Jacob W., Jegg A.-M., Ries C.H., Rüttinger D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J. Immunother. Cancer. 2017;5:53. doi: 10.1186/s40425-017-0257-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., 3rd, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive Integration of Single-Cell Data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Leek J.T., Johnson W.E., Parker H.S., Jaffe A.E., Storey J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012;28:882–883. doi: 10.1093/bioinformatics/bts034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wilkerson M.D., Hayes D.N. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics. 2010;26:1572–1573. doi: 10.1093/bioinformatics/btq170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Eide P.W., Bruun J., Lothe R.A., Sveen A. CMScaller: an R package for consensus molecular subtyping of colorectal cancer pre-clinical models. Sci. Rep. 2017;7 doi: 10.1038/s41598-017-16747-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Trapnell C., Cacchiarelli D., Grimsby J., Pokharel P., Li S., Morse M., Lennon N.J., Livak K.J., Mikkelsen T.S., Rinn J.L. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 2014;32:381–386. doi: 10.1038/nbt.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Langfelder P., Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinf. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wu T., Hu E., Xu S., Chen M., Guo P., Dai Z., Feng T., Zhou L., Tang W., Zhan L. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation. 2021;2 doi: 10.1016/j.xinn.2021.100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hänzelmann S., Castelo R., Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinf. 2013;14:7. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., III, Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Qiu X., Hill A., Packer J., Lin D., Ma Y.-A., Trapnell C. Single-cell mRNA quantification and differential analysis with Census. Nat. Methods. 2017;14:309–315. doi: 10.1038/nmeth.4150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Garcia-Alonso L., Holland C.H., Ibrahim M.M., Turei D., Saez-Rodriguez J. Benchmark and integration of resources for the estimation of human transcription factor activities. Genome Res. 2019;29:1363–1375. doi: 10.1101/gr.240663.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wickham H. ggplot2. Wiley Interdisciplinary Reviews: Comput. Stat. 2011;3:180–185. [Google Scholar]
- 70.Taube J.M., Akturk G., Angelo M., Engle E.L., Gnjatic S., Greenbaum S., Greenwald N.F., Hedvat C.V., Hollmann T.J., Juco J., et al. The Society for Immunotherapy of Cancer statement on best practices for multiplex immunohistochemistry (IHC) and immunofluorescence (IF) staining and validation. J. Immunother. Cancer. 2020;8 doi: 10.1136/jitc-2019-000155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhu Y., Weiss T., Zhang Q., Sun R., Wang B., Yi X., Wu Z., Gao H., Cai X., Ruan G., et al. High-throughput proteomic analysis of FFPE tissue samples facilitates tumor stratification. Mol. Oncol. 2019;13:2305–2328. doi: 10.1002/1878-0261.12570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gao H., Zhang F., Liang S., Zhang Q., Lyu M., Qian L., Liu W., Ge W., Chen C., Yi X., et al. Accelerated Lysis and Proteolytic Digestion of Biopsy-Level Fresh-Frozen and FFPE Tissue Samples Using Pressure Cycling Technology. J. Proteome Res. 2020;19:1982–1990. doi: 10.1021/acs.jproteome.9b00790. [DOI] [PubMed] [Google Scholar]
- 73.Li J., Van Vranken J.G., Pontano Vaites L., Schweppe D.K., Huttlin E.L., Etienne C., Nandhikonda P., Viner R., Robitaille A.M., Thompson A.H., et al. TMTpro reagents: a set of isobaric labeling mass tags enables simultaneous proteome-wide measurements across 16 samples. Nat. Methods. 2020;17:399–404. doi: 10.1038/s41592-020-0781-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shen B., Yi X., Sun Y., Bi X., Du J., Zhang C., Quan S., Zhang F., Sun R., Qian L., et al. Proteomic and Metabolomic Characterization of COVID-19 Patient Sera. Cell. 2020;182:59–72.e15. doi: 10.1016/j.cell.2020.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nie X., Qian L., Sun R., Huang B., Dong X., Xiao Q., Zhang Q., Lu T., Yue L., Chen S., et al. Multi-organ proteomic landscape of COVID-19 autopsies. Cell. 2021;184:775–791.e14. doi: 10.1016/j.cell.2021.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Braun D.A., Street K., Burke K.P., Cookmeyer D.L., Denize T., Pedersen C.B., Gohil S.H., Schindler N., Pomerance L., Hirsch L., et al. Progressive immune dysfunction with advancing disease stage in renal cell carcinoma. Cancer Cell. 2021;39:632–648.e8. doi: 10.1016/j.ccell.2021.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bao X., Shi R., Zhao T., Wang Y. Mast cell-based molecular subtypes and signature associated with clinical outcome in early-stage lung adenocarcinoma. Mol. Oncol. 2020;14:917–932. doi: 10.1002/1878-0261.12670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Engebretsen S., Bohlin J. Statistical predictions with glmnet. Clin. Epigenetics. 2019;11:123. doi: 10.1186/s13148-019-0730-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA sequencing data were deposited at Gene Expression Omnibus (GEO) (GSE56699, GSE14333, GSE39582, GSE17536, GSE17537, GSE33113, GSE37892, GSE146771, GSE72970, and GSE19860) and The Cancer Genome Atlas (TCGA), which are publicly available. All raw data generated by this study have been deposited in the Chinese national genomics data center (https://ngdc.cncb.ac.cn), under accession number HRA003569. The software and algorithms for data analyses used in this study are published and referenced throughout the STAR Methods section. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.