

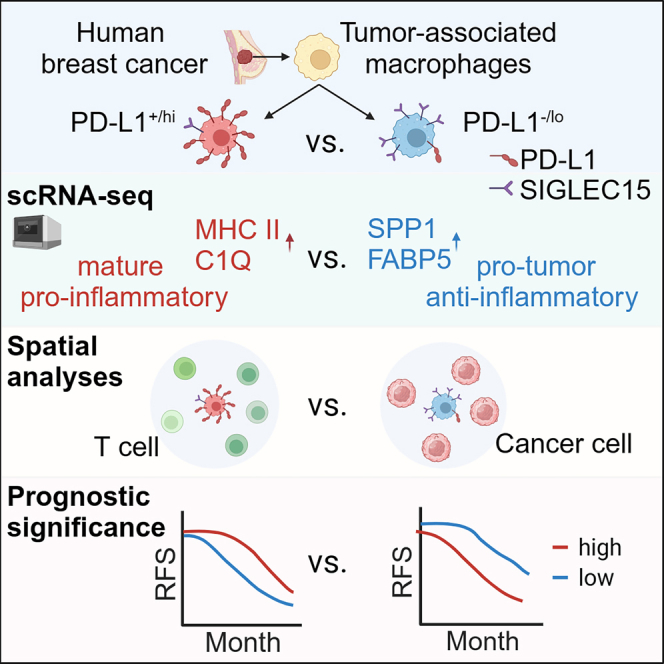
Summary
Tumor-associated macrophages (TAMs) are the predominant cells that express programmed cell death ligand 1 (PD-L1) within human tumors in addition to cancer cells, and PD-L1+ TAMs are generally thought to be immunosuppressive within the tumor immune microenvironment (TIME). Using single-cell transcriptomic and spatial multiplex immunofluorescence analyses, we show that PD-L1+ TAMs are mature and immunostimulatory with spatial preference to T cells. In contrast, PD-L1– TAMs are immunosuppressive and spatially co-localize with cancer cells. Either higher density of PD-L1+ TAMs alone or ratio of PD-L1+/PD-L1– TAMs correlate with favorable clinical outcome in two independent cohorts of patients with breast cancer. Mechanistically, we show that PD-L1 is upregulated during the monocyte-to-macrophage maturation and differentiation process and does not require external IFN-γ stimulus. Functionally, PD-L1+ TAMs are more mature/activated and promote CD8+ T cells proliferation and cytotoxic capacity. Together, our findings reveal insights into the immunological significance of PD-L1 within the TIME.
Graphical abstract
Highlights
-
•
Multi-omics study of PD-L1 expression on human TAMs
-
•
Along with functional analyses reveal PD-L1+ TAMs are mature and immunostimulatory
-
•
In contrast, PD-L1– TAMs are immunosuppressive and pro-tumor
-
•
PD-L1+ TAMs correlate with prolonged RFS in patients with breast cancer
Wang et al. examine the functional significance of PD-L1 expression on macrophages from human breast tumors. They show that PD-L1+ macrophages are immunostimulatory to T cells and associate with good clinical outcome in patients with breast cancer. In contrast, PD-L1– macrophages are immunosuppressive and associate with poor clinical outcome.
Introduction
Programmed cell death ligand 1 (PD-L1), ligand for PD1, may be expressed not only by cancer cells, but also by myeloid cells within the tumor immune microenvironment (TIME).1,2 It is well established that PD-L1-expressing cancer cells could develop adaptive immune resistance via directly inhibiting the cytotoxicity of PD1+ T cells.3 Recent studies demonstrated that tumor-associated macrophages (TAMs) are the predominant cells that express PD-L1 within the human TIME.4 The biological significance and functional role of PD-L1+ TAMs within human tumors remain unclear. Mechanistic studies based on murine tumor models demonstrated that PD-L1-expressing TAMs play important roles in suppression of anti-tumor immunity.5,6,7 However, clinical studies showed that PD-L1-expressing TAMs correlate with better overall survival in non-immunotherapy-treated patients with lung cancer,8 liver cancer,9 or breast cancer (BC).10 Furthermore, PD-L1 expression by myeloid cells is more predictive of response to PD1/PD-L1 blockade than PD-L1 expression by cancer cells in patients with bladder cancer,11 urothelial carcinoma,12 or BC.13 These contrasting findings on PD-L1-expressing TAMs raise important questions about the immunoregulatory functional significance of PD-L1 expression on TAMs.
Transcriptomic profile analyses of human TAMs via single-cell RNA sequencing (scRNA-seq) revealed high heterogeneity, but lack of PD-L1 expression.14,15,16,17,18,19,20 This may be due to PD-L1 gene dropout from scRNA-seq data since it is a low-abundance transcript.21 Studies using bioinformatic simulation with scRNA-seq data reported that human PD-L1+ TAMs upregulate antigen-presenting genes and may be immunosuppressive to T cells.22 On the other hand, studies by ex vivo functional assays reported that PD-L1+ TAMs from patients with lung cancer may not inhibit the cancer cell killing capacity of autologous tumor-specific T cells,23 reflecting inconsistencies about the function of human PD-L1+ TAMs.
In this report, we sought to explore the immunoregulatory functional significance of PD-L1 expression on TAMs from patients with BC via transcriptomic analysis at the single-cell level, coupled with cell-cell spatial interaction analysis via multiplex immunofluorescence (mIF) and ex vivo immunological functional assays with autologous T cells. Our analyses reveal important functional differences between PD-L1+ vs. PD-L1– TAMs, and provide insights into the immunoregulatory roles of PD-L1+ TAMs within the human TIME.
Results
scRNA-seq reveals differential expression profiles of human PD-L1+ vs. PD-L1– TAMs
While recent transcriptomic analysis of human TAMs via scRNA-seq showed clusters of TAMs with potential clinical implications,1,19 these studies did not specifically address PD-L1+/hi vs. PD-L1–/lo TAMs. This may be due to gene dropout in scRNA-seq data as PD-L1 is a low expression transcript.21 To assess the transcriptomic profile of PD-L1 expression in TAMs, we applied scRNA-seq (10X Genomics) to untreated primary human breast tumors (luminal, n = 4, freshly prepared single-cell suspension from digested tumor) and focused on differential expression profiles of PD-L1+/hi vs. PD-L1–/lo TAMs. Utilizing the Seurat scRNA-seq analysis pipeline, including unsupervised clustering and Uniform Manifold Approximation and Projection (UMAP), we first identified 10 cell types including myeloid cells (Figure S1A). Next, by sub-clustering myeloid cells, we identified/annotated TAMs (8 clusters, n = 2,220 cells) (Figure 1A) among all cell types after excluding other myeloid cells such as mast cells and dendritic cells (Figures S1B–S1E). To overcome the PD-L1 dropout hurdle, we utilized SIGLEC15, an immune co-inhibitory ligand whose expression was shown to be mutually exclusive with PD-L1 in tumor-infiltrating myeloid cells,24 to identify clusters of PD-L1+ SIGLEC15– and PD-L1−SIGLEC15+ TAMs (Figure 1B). By examining the blend expression of PD-L1 and SIGLEC15 within each TAM cluster, we confirmed the mutually exclusive expression pattern of PD-L1 and SIGELC15 in TAMs (Figure S1F) and dichotomized TAM clusters into PD-L1+SIGLEC15− (range from 39.4% to 73.5%) vs. PD-L1−SIGLEC15+ (range from 26.5% to 60.6%) populations (Figures 1C and 1D). To further validate this approach of PD-L1+/− TAMs identification via the cluster-based dichotomization of PD-L1/SIGLEC15 gene expression in scRNA-seq, we simultaneously performed flow cytometry to quantify the expression of PD-L1 protein on TAMs (CD14+ and HLA-DR+; gating strategy in Figure S2A) from the same tumor tissues. This confirmed similar percentages of PD-L1+/hi TAMs identified by flow compared with scRNA-seq (Figure 1E).
Figure 1.

Expression profile differences between PD-L1+/hi vs. PD-L1–/lo TAMs from human breast tumors revealed by scRNA-seq
(A) scRNA-seq analysis of TAMs (n = 2,220 cells) from untreated primary breast tumor (n = 5 patients, ER+) shown as a UMAP, highlighting identified clusters.
(B) UMAP showing mutually exclusive expression of PD-L1 and SIGLEC15 in TAMs.
(C) Dichotomization of TAM clusters into PD-L1+/hi and PD-L1–/lo subpopulations.
(D) PD-L1 expression of PD-L1+/hi and PD-L1–/lo subpopulations.
(E) Representative flow cytometry plot showing PD-L1+% TAMs, peripheral monocytes, and intratumoral T cells from the same tumor tissue used for scRNA-seq (left panel), and PD-L1+% quantification was compared between analysis via scRNA-seq and flow cytometry (right panel).
(F) Overlay of the expression of common M1 and M2 signature genes in the PD-L1+/hi vs. PD-L1–/lo TAM dichotomization.
(G) Volcano plot showing differentially expressed genes (DEGs) between PD-L1+/hi vs. PD-L1–/lo TAMs.
(H and I) Expression distribution of selected genes involved in maturation, pro-inflammatory or transcriptional activator (H), and anti-inflammatory, pro-tumor, fatty acid metabolic, or extracellular matrix (I) between PD-L1+/hi and PD-L1–/lo TAMs. ∗∗∗∗p < 0.0001. Wilcoxon rank-sum test.
(J) PD-L1+/hi and PD-L1–/lo TAMs were flow sorted from freshly prepared single-cell suspension of digested breast tumor (n = 4) and supernatants were collected for ELISA after 16 h. Paired t test. ∗p < 0.05, ∗∗p < 0.01.
Next we addressed whether the commonly used M1 vs. M2 classification of macrophages25 overlaps with our dichotomization of PD-L1+/hi vs. PD-L1–/lo TAMs. We found that the expression of M1 and M2 signature genes (Table S1) were common to both PD-L1+/hi and PD-L1–/lo TAMs (Figure 1F) (Figure S2B), consistent with recent reports showing individual TAMs from human tumors generally express both M1 and M2 genes.14,19,26 Therefore, PD-L1+/hi vs. PD-L1–/lo TAMs do not fit within this simple canonical M1 vs. M2 dichotomy.
Differentially expressed gene (DEG) analysis (Figure 1G) revealed that PD-L1+/hi TAMs had higher levels of macrophage maturation marker genes (e.g., CD83, CD74, HLA-DRA/B, and HLA-DQA/B), pro-inflammatory genes, including cytokines/chemokines (e.g., IL1B, CXCL2/3/8, and CCL3/4/18) and complement components (e.g., C1QA/B/C), and transcriptional activators (e.g., FOS, JUNB, and CEBPD) (Figure 1H). In contrast, PD-L1–/lo TAMs had higher expression of anti-inflammatory genes (i.e., CD9, CD52, IL1RN, and CSTB), genes with pro-tumor functions (e.g., osteopontin [SPP1], MMP9, and SPARC), genes involved in fatty acid metabolism (e.g., FABP4/5 and LPL), and genes involved in extracellular matrix organization (e.g., fibronectin 1 [FN1], COL1A1/2, and COL3A1) (Figure 1I). Notably, gene set enrichment analysis identified multiple distinct functional features of PD-L1+/hi vs. PD-L1–/lo TAMs (Figure S2C). As for the most enriched pathways, PD-L1+/hi TAMs were enriched for hallmarks of inflammatory response while PD-L1–/lo TAMs were enriched for hallmarks of epithelial-mesenchymal transition (Figure S2D). To validate the DEGs identified by scRNA-seq, we flow sorted PD-L1+/hi and PD-L1–/lo TAMs from freshly prepared single-cell suspension of human breast tumor and examined the levels of pro-inflammatory cytokines/chemokines in supernatant after 16 h by ELISA. We found that the levels of IL1β and CCL4 were significantly higher for PD-L1+/hi than PD-L1–/lo TAMs (Figure 1J). Given the differential expression profiles revealed by our scRNA-seq, these data suggest that PD-L1+/hi TAMs might be more mature/activated and immunostimulatory, while PD-L1–/lo TAMs might be immunosuppressive and pro-tumor.
To assess the generalizability of the differential expression profiles of human PD-L1+/− TAMs revealed by our scRNA-seq, we also analyzed published scRNA-seq transcriptomic data of human TAMs from luminal breast tumors (n = 6).14 We identified/annotated TAMs (11 clusters, n = 3,130 cells) among all cell types (Figures S3A–S3E). Similar to our findings, the expression pattern of PD-L1 and SIGLEC15 in TAMs was also mutually exclusive in public data (Figures S4A and S4B), allowing us to similarly dichotomize TAMs clusters into PD-L1+SIGLEC15− and PD-L1−SIGLEC15+ TAMs (Figure S4C). DEG analysis also revealed maturation marker genes (i.e., HLA-DQA/B), pro-inflammatory genes (i.e., C1QA/C and IL1B), and transcriptional activator genes (i.e., FOSB and CEBPD) that were upregulated in PD-L1+/hi TAMs. In contrast, anti-inflammatory genes (i.e., CD9 and IL1RN), pro-tumor genes (i.e., SPP1 and TREM2), fatty acid metabolic genes (i.e., FABP4/5 and LPL), and extracellular matrix organization genes (i.e., FN1) were upregulated in PD-L1–/lo TAMs (Figures S4D and S4E).
To further investigate the differential expression profiles of PD-L1+ vs. PD-L1– TAMs from patients with another molecular subtype of BC, we also analyzed published scRNA-seq transcriptomic data of TAMs from human triple-negative BC (TNBC) tumors (n = 8).27 We identified/annotated TAMs (11 clusters, n = 4,484 cells) (Figure 2A) among all cell types (Figures S5A–S5E). After dichotomizing TAM clusters into PD-L1+/hi and PD-L1–/lo TAMs (Figures 2B–2D) based on the expression patterns of PD-L1 and SIGLEC15 among different clusters (Figure S5F), DEG analysis revealed that various maturation and pro-inflammatory genes were upregulated in PD-L1+/hi TAMs while various anti-inflammatory and pro-tumor genes were upregulated in PD-L1–/lo TAMs (Figures 2E and 2F). These findings in TNBC are similar to the differential expression profiles of PD-L1+/− TAMs from luminal breast tumors.
Figure 2.

Expression profile of PD-L1+/− TAMs from patients with TNBC revealed by published scRNA-seq data
(A) Published (Pal et al.27) scRNA-seq transcriptomic clustering of TAMs (n = 4,484 cells) from untreated primary TNBC breast tumors shown as a UMAP.
(B) Mutually exclusive expression of PD-L1 and SIGLEC15 in TAMs.
(C) Dichotomization of TAM clusters into PD-L1+/hi and PD-L1–/lo subpopulations.
(D) PD-L1 expression of PD-L1+/hi and PD-L1–/lo subpopulations.
(E) Volcano plot showing differentially expressed genes (DEGs) between PD-L1+/hi vs. PD-L1−/lo TAMs.
(F) Expression distribution of selected genes involved in maturation, pro-inflammatory, anti-inflammatory, and pro-tumor between PD-L1+/hi and PD-L1–/lo TAMs.
(G) Published (Bassez et al.28) scRNA-seq transcriptomic clustering and dichotomization of TAMs (n = 12,952 cells) from patients with TNBC treated with neoadjuvant anti-PD1 immunotherapy into PD-L1+/hi and PD-L1–/lo subpopulations.
(H) PD-L1 expression of PD-L1+/hi and PD-L1–/lo subpopulations. ∗∗∗∗p < 0.0001. Wilcoxon rank-sum test.
(I) TAMs were annotated based on whether patients exhibited clonal expansion of intratumoral PD1+ T cells after the anti-PD1 treatment as expanded or non-expanded.
(J) The percentages of PD-L1+/hi TAMs were compared between tumors with anti-PD1-induced expanded vs. non-expanded clonal PD1+ T cells. ∗∗p < 0.01. Mann-Whitney test.
Based on another published report of scRNA-seq transcriptomic data of TAMs from patients with BC treated with neoadjuvant anti-PD1 immunotherapy (n = 19),28 we further tested the potential regulatory effects of PD-L1+ TAMs on intratumoral PD1+ T cells. We identified/annotated TAMs (13 clusters, n = 12,952 cells) among all cell types (Figures S6A–S6F). After dichotomizing TAM clusters into PD-L1+/hi and PD-L1–/lo TAMs (Figures 2G and 2H) based on the expression patterns of PD-L1 and SIGLEC15 among different clusters (Figure S6G), we annotated TAMs based on whether patients exhibited clonal expansion of intratumoral PD1+ T cells after the anti-PD1 treatment into TAMs from expanded or non-expanded tumors, respectively (Figure 2I). Interestingly, the abundance of PD-L1+/hi TAMs was significantly higher in tumors with immunotherapy-induced clonal expanded PD1+ T cells (Figure 2J), supporting the notion that PD-L1+/hi TAMs are not immunosuppressive to T cells. Among the observed DEGs in PD-L1+/hi and PD-L1–/lo TAMs identified by the in-house and public scRNA-seq analyses, we found that very few overlapped between DEGs and M1/M2 genes (Figure S7), supporting the notion that PD-L1+/hi vs. PD-L1–/lo TAMs do not fit within this simple canonical M1 vs. M2 dichotomy. Thus, analyses of these public TAM datasets are consistent with our in-house scRNA-seq findings, supporting the notion that PD-L1 is a marker for more mature/activated and immunostimulatory TAMs within human breast tumors.
Clinical outcome significance of PD-L1+/− TAMs in two independent cohorts of patients with BC
Expression of intratumoral PD-L1 was reported to correlate with poor prognosis in patients with BC.29 More recently, PD-L1+ TAMs were reported to associate with improved prognosis in patients with TNBC.10 We sought to further investigate the clinical significance of PD-L1 expression on TAMs by using both public bulk-tumor transcriptomic datasets and in-house patients with BC. We generated gene signatures for PD-L1+/hi or PD-L1–/lo TAMs from top upregulated genes in the DEGs that overlapped between our and public scRNA-seq analyses (Table S2), which may represent distinct immunoregulatory functions of PD-L1+/hi (mature/activated and pro-inflammatory) vs. PD-L1–/lo TAMs (anti-inflammatory and pro-tumor). To explore the prognostic value of PD-L1+/− TAMs, we used these gene signatures to divide patients with luminal BC into high-expressing (top 25%) vs. low-expressing (bottom 25%) groups. In the dataset of the Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) (n = 1098), patients with high gene signature of PD-L1+/hi TAMs (top 25%) had better relapse-free survival (RFS) (log rank test, p = 0.001) vs. the bottom 25%, while patients with high gene signature of PD-L1–/lo TAMs had worse RFS (p = 0.036; Figure 3A). In addition, the PD-L1+/hi TAMs gene signature also showed favorable prognostic significance (p = 0.014) while the PD-L1–/lo TAMs gene signature correlated with worse RFS (p = 0.036) in the dataset of The Cancer Genome Atlas (TCGA) (n = 789) (Figure 3B). Furthermore, the gene signature ratio of PD-L1+/PD-L1– TAMs showed favorable prognostic significance in METABRIC (p < 0.0001) and in TCGA (p = 0.032) (Figures 3A and 3B). PD-L1+/hi TAMs also significantly correlated with better RFS for patients with TNBC in the METABRIC dataset (n = 269) by using the gene signature of PD-L1+/hi TAMs (top 10 upregulated genes) revealed by published scRNA-seq data from patients with TNBC (Figure S8A). On the other hand, expression level of CD68 did not show significant association with clinical outcome in METABRIC or TCGA datasets (Figures S8B and S8C). To further confirm that PD-L1+/hi vs. PD-L1–/lo TAMs do not simply fit within the canonical M1 vs. M2 dichotomy, we used the M1, M2, or the ratio of M1/M2 gene signature to divide patients with BC into high-expressing (top 25%) vs. low-expressing (bottom 25%) groups but found no significance correlation with RFS (Figure 3C).
Figure 3.

PD-L1+ TAMs are associated with favorable clinical outcome in two independent cohorts of patients with BC
(A and B) Kaplan-Meier relapse-free survival (RFS) curves and log rank test generated for the gene signature of PD-L1+ vs. PD-L1– TAMs or the gene signature ratio of PD-L1+/PD-L1– TAMs in the luminal BC cohorts of METABRIC (n = 1098) (A) and TCGA (n = 789) (B) datasets.
(C) Kaplan-Meier analyses for the M1, M2, or the ratio of M1/M2 gene signature in the luminal BC cohorts of METABRIC (n = 1098). Patients were divided into high- and low-expressing groups based on a 25% cutoff of the gene signature.
(D) Schematic summarizing the histological quantification method in cohorts 1 and 2 of patients with luminal BC.
(E) Representative immunofluorescence staining of PD-L1, CD68, and DAPI to identify PD-L1+ and PD-L1– TAMs.
(F–H) Using Kaplan-Meier estimate and log-rank test, relapse-free survival (RFS) was compared between patients with low and high density of PD-L1+ TAMs in cohort 1 (n = 49) (F) and in cohort 2 (n = 93) (G) or low and high density ratio of PD-L1+/PD-L1− TAMs in combined cohorts 1 and 2 (n = 142) (H). Median density was used as the cutoff to divide patients into low vs. high groups.
(I) Univariate and multivariate analysis for the prognostic significance of the density ratio of PD-L1+/PD-L1– TAMs. Hazard ratio calculated with below medium vs. above medium (n = 142).
In relation to T cells, we found that expression of CD8A strongly correlated with the gene signature of PD-L1+/hi TAMs in both METABTRIC (r = 0.52, p < 0.0001) and TCGA (r = 0.6, p < 0.0001) datasets, but not with the gene signature of PD-L1–/lo TAMs (Figures S8D and S8E). Using the immune-related functional gene signatures,30 we found that tumors enriched with PD-L1+ TAMs had significantly higher score for T cell activation and cytotoxicity (Figure S8F), suggesting that PD-L1+ TAMs are not immunosuppressive. Mechanistically, tumors enriched with PD-L1+ TAMs had similar gene signature score for interferon (Figure S8F) and similar expression levels of IFNA/B/G (data not shown), indicating that PD-L1 upregulation on TAMs might be independent of interferons. Moreover, we performed the CIBERSORT deconvolution method to characterize cell composition differences between tumors with high vs. low gene signature of PD-L1+/hi TAMs (top 25% vs. bottom 25%). This showed that M1 macrophages, CD4+ memory resting T cells, and CD8+ T cells were more abundant in tumors with high PD-L1+/hi TAMs gene signature (Figure S8G). Together, these public bulk-tumor transcriptomic analyses are consistent with our scRNA-seq data and support the notion that PD-L1+ TAMs could be immunostimulatory, while PD-L1– TAMs could be immunosuppressive and pro-tumor.
To further validate the clinical significance of PD-L1 expression on TAMs, we examined the association between PD-L1+/− TAMs in archival FFPE tissues and RFS in two independent in-house cohorts of patients with luminal BC who had been clinically followed for at least 36 months via whole-slide quantification or tissue microarray (TMA), respectively (schematic shown in Figure 3D). In cohort 1 (n = 49, clinical and pathological characteristics summarized in Table S3; representative immunofluorescence staining of PD-L1, CD68, and DAPI shown in Figure 3E), we performed Kaplan-Meier survival analysis and log rank test to determine the relationship between PD-L1+/− TAM density and RFS. Patients were divided into two populations using median density of PD-L1+ or PD-L1– TAMs as the cutoff value. We found that patients with PD-L1+ TAM density below median (p = 0.038) and patients with PD-L1– TAMs above median (p = 0.046) had worse RFS (Figure 3F). These results were further validated in a separate cohort in which PD-L1 expression of TAMs was quantified using TMA. In cohort 2 (n = 93, clinical and pathological characteristics summarized in Table S3), we performed the same immunofluorescence staining and also found that patients with below medium density of PD-L1+ TAMs (p = 0.02) and patients with above medium density of PD-L1– TAMs (p = 0.01) had worse RFS (Figure 3G). Furthermore, patients (cohorts 1 and 2 combined) with above medium density ratio of PD-L1+/PD-L1– TAMs had better RFS (p = 0.0003) (Figure 3H). In contrast, density of TAMs alone showed no correlation with clinical outcome in either cohort (data not shown). We also found that patients with above medium density ratio of PD-L1+/PD-L1– TAMs tend to have better overall survival (OS) (p = 0.08) (Figure S8H). In addition, the density of PD-L1+ TAMs was significantly higher in TNBC than in luminal subtype but was not significantly associated with tumor grade, or T status or N status (Figures S8I and S8J). In multivariate analysis adjusted for clinicopathological characteristics (age, tumor stage, grade, and nodal status), the density ratio of PD-L1+/− TAMs retained highly prognostic significance for RFS (p = 0.0099) (Figure 3I). The clinical outcome significance found in these two independent cohorts highlight differences in immunoregulatory potential between PD-L1+ vs. PD-L1– TAMs.
PD-L1+ and PD-L1– TAMs have different cell-to-cell interaction preference
To further explore the immunological function of PD-L1 expression on TAMs, we performed mIF staining to concurrently identify PD-L1−/+ TAMs (PD-L1−/+CD68+), CD8+ T cells (CD3+CD8+), CD4+ T cells (CD3+CD8−), and cancer cells (cytokeratin+) within the TIME of untreated primary breast tumors (n = 36; representative staining image as shown in Figure S9A). Stained slides were scanned, and spatial coordinates of cells were obtained from the corresponding phenotype map (Figure 4A). We quantified the number of PD-L1+ TAMs and cancer cells and found that the ratio of PD-L1+TAM/cancer cells is about 1:1 in the whole-slide total area (Figure 4B), which are consistent with known findings that intratumoral PD-L1 expression is mainly on TAMs and cancer cells. Utilizing the accepted assumption that two cells are more likely to be interacting if the distance between their nuclei is less than 20 μm,31 we quantified the number of CD8+ T cells, CD4+ T cells, and cancer cells within 20 μm of PD-L1– vs. PD-L1+ TAMs (Figure 4C). We found that the number of CD8+ or CD4+ T cells within 20 μm of PD-L1+ TAMs was significantly higher than to PD-L1– TAMs, while the number of cancer cells within 20 μm of PD-L1+TAMs was significantly lower than to PD-L1– TAMs (Figure 4D). These spatial differences indicate that PD-L1+ TAMs have a higher tendency to interact with T cells, while PD-L1– TAMs tend to interact more frequently with cancer cells. Interestingly, we found that PD-L1– TAMs but not PD-L1+ TAMs also tend to self-cluster (Figure 4E), suggesting the possible presence of certain local factors that may suppress maturation and PD-L1 expression on TAMs. Consistent with the differential gene expression profiles revealed via scRNA-seq, these mIF analyses suggest that PD-L1+ TAMs may be immunostimulatory and chemoattract T cells, while PD-L1– TAMs could be pro-tumor by interacting with cancer cells.
Figure 4.

PD-L1+ and PD-L1– TAMs have different cell-to-cell interaction preferences
(A) Multiple immunofluorescence staining and corresponding phenotype map of representative breast tumor tissue section for PD-L1+ TAMs (CD68+PD-L1+), PD-L1– TAMs (CD68+PD-L1–), CD8+ T cells (CD8+), CD4+ T cells (CD3+CD8−), and cancer cells (CK+).
(B) Whole-slide quantification of the ratio of PD-L1+ TAMs/cancer cells in total area.
(C) Schematic representing the calculation of cell-cell interaction based on CD8+ T cells, CD4+ T cells, or cancer cells within a radius of 20 μm from the nuclei of PD-L1+ or PD-L1– TAMs.
(D) CD8+ T cells, CD4+ T cells, or cancer cells within a radius of 20 μm from the nuclei of PD-L1+ or PD-L1– TAMs in untreated primary luminal breast tumors (n = 36). ∗∗∗∗p < 0.0001, ∗∗p < 0.01. Wilcoxon paired test.
(E) The number of TAMs within 20 μm to PD-L1+ TAMs (left) or PD-L1– TAMs (right) (n = 36). Paired t test. ∗∗∗∗p < 0.0001.
(F) Dot plots of ligand-receptor interactions between PD-L1+ or PD-L1– TAMs and CD4/8+ T cells (left panel), and cancer cells (right panel) based on our scRNA-seq transcriptomic analysis. Gray dot represents no significant interaction was found.
To gain further insights into the interactions between PD-L1−/+ TAMs and other cells revealed by our mIF analyses, we performed contact-dependent and secreted factor-mediated cell-cell interaction analysis using CellPhoneDB and CellChat from our scRNA-seq data. Based on known receptor-ligand pairs, we observed 843 and 729 significant ligand-receptor interactions between PD-L1+ or PD-L1– TAMs with other cell types (i.e., T cells, B cells, cancer cells, or stromal cells) within the TIME, respectively. PD-L1+ TAMs displayed more interactions with CD8+ or CD4+ T cells than PD-L1– TAMs, including contact-dependent interactions (such as AREG-ICAM1 and CD162-CD62L) and secreted factors (such as annexin 1 [ANXA1] and MIF) mediated interactions (Figure 4F, left). In contrast, PD-L1– TAMs displayed more interactions with cancer cells than PD-L1+ TAMs, including contact-dependent interactions of fibronectin/collagen with integrin (FN1-integrin aVb1 and COL6A2-integrin a2b1) and secreted factors (such as SPP1 and VEGFA) mediated interactions (Figure 4F, right). These differential cell-cell interaction preferences again support that PD-L1+ TAMs could be immunostimulatory to chemoattract and interact with T cells, while PD-L1– TAMs tend to interact more with cancer cells. Together, these results are in accord with our mIF findings and gene expression patterns revealed by scRNA-seq.
PD-L1 is upregulated during the monocyte-to-macrophage maturation/differentiation process
It is generally accepted that the majority of TAMs are derived from tumor-infiltrating peripheral blood monocytes.32 We hypothesized that PD-L1 could be upregulated during the monocyte-to-macrophage maturation/differentiation process. To test this hypothesis, we examined the expression of PD-L1 on peripheral blood monocytes from untreated patients with BC by flow cytometry. We found that PD-L1 levels were low on fresh monocytes but were upregulated upon ex vivo resting (freshly isolated PBMCs followed by 8 h resting in culture medium) (Figures 5A and 5B, gating strategy in Figure S9B). Notably, PD-L1 levels on adherent monocytes/macrophages were significantly higher than on monocytes in suspension after resting (Figures 5C and 5D). In addition, we found that all monocyte-derived macrophages became PD-L1+ after in vitro differentiation (Figure S9C). These findings suggest that PD-L1 may be upregulated during the monocyte-to-macrophage maturation/differentiation process.
Figure 5.

PD-L1 is upregulated during the monocyte-macrophage maturation/differentiation process
(A and B) Freshly isolated PBMCs from newly diagnosed patients with BC (n = 12) were ex vivo rested in RPMI 1640 with 10% FBS for 8 h. PD-L1+% peripheral blood monocytes are shown in representative flow plots (A) and compared between fresh vs. rested monocytes (B).
(C and D) PD-L1+% between in suspension vs. adherent monocytes after 8 h resting (n = 8) are shown in representative flow plots (C) and compared (D).
(E and F) PD-L1+% monocytes between flow sorted fresh PBMCs vs. 8 h rested PBMCs (n = 5) are shown in representative flow plots (E) and compared (F). Paired t test.
(G and H) MFI ratio of surface protein levels (G) and phosphorylated signal transduction protein levels (H) on PD-L1+ vs. PD-L1– monocytes in 8 h rested PBMCs from patients with BC. Wilcoxon paired test.
(I) Peripheral blood monocytes from patients with BC (n = 6) were treated with small-molecule inhibitors (Selleck) against ERK1/2 (SCH772984 at 0. 5 μM), STAT1 (fludarabine at 50 μM), Akt1/2/3 (MK-2206 2HCl at 0.5 μM), PI3Kα/δ/β (LY294002 at 5 μM), NF-κB (QNZ at 5 μM), and mTOR (rapamycin at 0.1 μM) during the 8 h resting. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗p < 0.0001. Shown are mean ± SEM.
PD-L1 expression can be induced by a number of soluble factors,33 of which IFN-γ is the most prominent inducer of PD-L1.34 To examine whether PD-L1 upregulation is due to the potential presence of IFN-γ in culture medium, we flow sorted PD-L1– monocytes from freshly isolated BC patients’ PBMCs and rested in serum-free medium, along with blocking antibodies against both IFN-γ and IFN-γ receptors. Importantly, we found that PD-L1 was still significantly upregulated on monocytes after resting (Figures 5E and 5F), indicating that PD-L1 on monocytes/macrophages could be upregulated during maturation/differentiation independent of IFN-γ.
Next we examined whether various functional-related proteins in monocytes/macrophages co-upregulate with PD-L1 during the maturation/differentiation process. Levels of maturation markers (such as CD54, CD69, and CD83), M1/M2 markers (such as HLA-DR, MRC1, and CD163), immune co-stimulatory ligands (such as CD40, CD80, and CD86), immune co-inhibitory ligands (such as PD-L2, B7-H3, and B7-H4), Fcγ receptors (CD16, CD32, and CD64), and chemokine receptors (CSF1R and CCR5) were all significantly higher on PD-L1+ than on PD-L1– monocytes from patients with BC after ex vivo resting (Figure 5G; representative flow plots in Figures S10A–S10F). We also examined the activation of intracellular signal transduction pathways via levels of phosphorylated signaling proteins (such as STATs, mTOR, and Akt) by phosflow cytometry (gating strategy in Figure S9D). Notably, levels of phosphorylated STAT1, STAT3, mTOR, and Akt were significantly higher in PD-L1+ than in PD-L1– monocytes (Figure 5H; representative flow plots in Figure S10G). To further explore the molecular mechanisms of PD-L1 upregulation, we used various small-molecule inhibitors against ERK1/2, STAT1, Akt1/2/3, PI3Kα/δ/β, NF-κB, and mTOR signaling pathways during the ex vivo resting of monocytes from patients with BC (Figure 5I). We found that ERK inhibitor significantly suppressed the PD-L1 upregulation, which indicates that the observed PD-L1 upregulation during monocyte-to-macrophage differentiation is partially dependent on ERK signaling pathway. These findings suggest that PD-L1 on monocytes could be upregulated after peripheral blood monocytes infiltrate tumor and that PD-L1 may be a marker of more mature and activated TAMs.
To further examine the functional relationship between PD-L1 upregulation and IFN-γ, we stimulated peripheral monocytes from ex vivo rested BC patients’ PBMCs with IFN-γ for 15 min and examined IFN-γ-induced STAT1 phosphorylation (pSTAT1) via phosflow cytometry (Figure 6A). We found that the percentage of PD-L1+ monocytes responded to IFN-γ stimulation (determined by ΔpSTAT1+% after IFN-γ stimulation) was significantly higher than PD-L1– monocytes (Figure 6B), indicating that PD-L1+ monocytes are primed to respond to IFN-γ stimulation. We observed a significant positive correlation between PD-L1+% and ΔpSTAT1+% in monocytes (Figure S11A) and confirmed that IFN-γ-induced pSTAT1 was higher in flow-sorted PD-L1+ monocytes from PBMCs (Figures S11C and S11D). Importantly, levels of IFN-γ receptor (IFN-γR1) were significantly higher on PD-L1+ than on PD-L1– monocytes (Figure 6C; representative flow plot in Figure S11B). In addition, we further confirmed that IFN-γ stimulation not only dose dependently upregulated PD-L1 but also downregulated IFN-γR1 on monocytes simultaneously (Figures 6D and 6E). Thus, PD-L1+ monocytes/macrophages have higher expression levels of IFN-γ receptors, suggesting that PD-L1 on monocytes/macrophages could be upregulated independent of IFN-γ.
Figure 6.

PD-L1 upregulation in TAMs could be IFN-γ independent
(A–C) PBMCs from patients with BC were rested and stimulated with IFN-γ. Representative flow plots showing IFN-γ (50 ng/mL for 15 min) induced phosphorylation of STAT1 (pY701) in peripheral monocytes (A) from patients with BC. IFN-γ signaling response (n = 28) (B) and levels of IFN-γR1 (n = 20) (C) were compared between PD-L1–/lo vs. PD-L1+/hi monocytes.
(D and E) Rested PBMCs from patients with BC were stimulated with IFN-γ at 0.2, 1, 5, or 25 ng/mL for 16 h. Levels of PD-L1 and IFN-γR1 on monocytes or T cells are shown in the representative flow plots (D) and compared (n = 6) (E). One-way ANOVA.
(F–H) Single-cell suspensions from freshly prepared primary breast tumors were stimulated with IFN-γ. Representative flow plots (F) showing IFN-γ-induced pSTAT1 (G) and levels of IFN-γR1 (H) between PD-L1–/lo vs. PD-L1+/hi TAMs from untreated primary breast tumors (n = 8).
(I and J) Multiplex immunofluorescence staining (I) and quantification of IFN-γR1+ PD-L1+/− cells (J) from untreated primary breast tumor tissues (n = 8). Scale bars, 100 μm.
(K–M) Representative flow plots showing PD-L1 expression (K) and IFN-γ-induced pSTAT1 (L) and levels of IFN-γR1 (M) between PD-L1–/lo vs. PD-L1+/hi breast cancer cells (n = 8). Paired t test. ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. Shown are mean ± SEM.
Next we examined the IFN-γ stimulation responsiveness in TAMs from freshly prepared single-cell suspensions with untreated human breast tumors (Figure 6F; gating strategy in Figure S11E). Similar to the findings in monocytes/macrophages, the percentage of PD-L1+% TAMs responded to IFN-γ stimulation (Figure 6G) and levels of IFN-γR1 (Figure 6H; representative flow plots in Figure S11F) were significantly higher in PD-L1+/hi TAMs than in PD-L1–/lo TAMs. Furthermore, we examined the expression of IFN-γ receptor on PD-L1+ TAMs by mIF (Figure 6I) and confirmed that the IFN-γR1+% was significantly higher for PD-L1+ than for PD-L1– TAMs (Figure 6J). In addition, we examined the responsiveness to IFN-γ stimulation in PD-L1+ BC cells (Figure 6K). Interestingly, the percentage of PD-L1–/lo cancer cells that responded to IFN-γ stimulation was significantly higher than PD-L1+/hi cancer cells (Figures 6L and 6M), which is the opposite to PD-L1+/hi vs. PD-L1–/lo TAMs. Therefore, these findings indicate that PD-L1 could be upregulated independent of IFN-γ, and that PD-L1+/hi monocytes/macrophages are primed for the responsive to IFN-γ stimulation.
Functionally PD-L1+ monocytes/macrophages are mature/activated and immunostimulatory
To investigate immune-related functional differences between PD-L1+ vs. PD-L1– monocytes/macrophages, we performed phagocytosis assay (pH-sensitive pHrodo dye conjugated E. coli) with peripheral blood monocytes after ex vivo resting and PD-L1 upregulation (Figure 7A). We found that PD-L1+/hi monocytes showed significantly higher capacity of phagocytosis than PD-L1–/lo monocytes (Figure 7B), consistent with the notion that PD-L1 may be a marker for more matured/activated monocytes/macrophages. Next we examined the immunoregulatory functions of PD-L1+/− monocytes/macrophages upon co-culture with autologous T cells labeled with CellTrace proliferation dye after flow sorting from ex vivo rested BC patients’ PBMCs (Figure 7C). Remarkably, PD-L1+/hi macrophages had significantly higher stimulatory effects on CD8+ T cell proliferation than PD-L1–/lo macrophages (Figure 7D). In contrast, we observed similar stimulatory effects of PD-L1+ and PD-L1– macrophages on CD4+ T cell proliferation (Figures S11G and S11H). PD-L1 blocking antibody did not suppress the stimulatory of PD-L1+ monocyte/macrophages on T cell proliferation (Figure 7D) despite PD1 expression on T cells (Figure S11I), indicating that the stimulatory effects of PD-L1+/hi macrophages on T cell proliferation may not be mediated via PD-L1:PD1 interaction. We next addressed whether PD-L1+/− macrophages suppress antigen-specific T cell killing by using CD3/CD19 bispecific antibody (BiTE, Blinatumomab). We co-cultured CD8+ T cells with CD19-expressing cancer cells together with autologous PD-L1−/+ monocytes from ex vivo rested BC patients’ PBMCs and examined the killing capacity of CD8+ T cells by flow cytometry (Figure 7E). We found that the presence of BiTE greatly induced the expression of PD1 and CD137 on CD8+ T cells (Figure 7F), confirming that the observed cancer cell killing was antigen specific. Importantly, we found that PD-L1–/lo macrophages but not PD-L1+/hi macrophages significantly suppressed CD8+ T cell killing (Figures 7G and 7H). PD-L1 blocking antibody did not change the effect of PD-L1+/hi macrophages on T cell killing (Figures 7G and 7H). These functional results are consistent with the notion that PD-L1+ monocytes/macrophages are functionally more mature/activated and immunostimulatory, while PD-L1– monocytes/macrophages may be immunosuppressive.
Figure 7.

PD-L1+ TAMs are more activated and pro-inflammatory than PD-L1– TAMs
(A and B) PBMCs from patients with BC (n = 16) were rested and the phagocytosis capacity of monocytes/macrophages were determined by using pHrodo Green E. Coli Bioparticles conjugate as shown in the representative flow plots (A) and compared between PD-L1–/lo vs. PD-L1+/hi monocytes/macrophages (B). Paired t test.
(C and D) Freshly isolated PBMCs from patients with BC (n = 6) were rested and the PD-L1+/− monocytes were flow sorted. CellTrace Violet dilution by CD8+ T cells determined after 4 days of TCR-stimulated coculture with autologous PD-L1+ vs. PD-L1– monocytes/macrophages. (C) Representative flow plots showing percentage of proliferated CD8+ T cells. (D) Proliferation stimulation activity measured by cell number ratio of (CD8/CD4+CD14)/(CD8/CD4) as the stimulatory index.
(E–H) Freshly isolated PBMCs from patients with BC (n = 6) were rested and the PD-L1+/− monocytes were flow sorted. Cytotoxic activity of CD8+ T cells determined using CD19/CD3 bispecific antibody (BiTE) after 2 days of coculture with CD19+ K562 cancer cells in the presence of autologous PD-L1+ or PD-L1– monocytes/macrophages. (E) Schematic representing the experiment setup. (F) Representative flow plots showing PD1 and CD137 expression on CD8+ T cells. (H) Cytotoxic activity calculated by the percentage of K562 cells killed by CD8+ T cells as shown in representative flow plots (G). One-way ANOVA. ∗p < 0.05, ∗∗∗∗p < 0.0001. Shown are mean ± SEM.
Discussion
TAMs represent the dominant infiltrating immune cells in most human tumors and play important immunoregulatory roles within the TME.35,36,37 While TAMs are generally divided along a simplistic M1/M2 dichotomy,32 recent scRNA-seq transcriptomic analysis of tumor-infiltrating myeloid cells from human tumors revealed heterogeneous subsets of TAMs with distinct transcriptomic patterns and diverse functional markers across different cancer types.19 Furthermore, TAMs are now known to the predominant cells that express PD-L1 within the human TIME.4 Whether human PD-L1+ TAMs are immunosuppressive and the functional significance of PD-L1 expression on TAMs remained unclear. Building on these recent reports, we show that PD-L1+ TAMs from human breast tumors are more mature/activated and immunostimulatory similar to M1-like macrophages, while PD-L1– TAMs are less mature and immunosuppressive similar to M2-like macrophages. These findings are collectively supported by our scRNA-seq transcriptomic analysis, mIF and spatial cell-cell interaction analysis, and ex vivo functional assays.
As monocyte-derived macrophages can cross-present antigens to CD8+ T cells, PD-L1 may play a protective role on PD-L1-expressing myeloid cells against killing by PD1+ cytotoxic T cells.23 Similarly, PD-L1 may protect PD-L1-expressing dendritic cells (DCs) from T cell killing, as reported in a mouse tumor study with PD-L1-specific knockout in DCs.38 As such, it is possible that the major functional purpose of PD-L1 expression on TAMs is not immune suppression, but to protect PD-L1-expressing TAMs against T cell killing. Consistent with this notion, our finding that PD-L1 is a maturation marker during the monocyte-macrophage differentiation process is supported by animal studies, which showed that PD-L1 could be upregulated during the maturation of macrophages39 or hematopoietic stem cells.40
In addition to binding to PD1 across cells via trans interaction, PD-L1 can also bind CD80 on the same cell via cis interaction.41 PD-L1:CD80 cis-heterodimerization inhibited both PD-L1:PD-1 and CD80:CTLA-4 interactions through distinct mechanisms, but preserved the ability of CD80 to activate the T cell co-stimulatory receptor CD28.42 Thus, PD-L1 on TAMs can also exert an immunostimulatory function by repressing the CTLA-4 axis.
Recent studies demonstrated that the immunoregulatory roles of PD-L1 expression on macrophages and DCs might be largely different. By using an animal tumor model with PD-L1-specific knockout in DCs or macrophages, it has been shown that PD-L1 knockout in DCs but not in macrophages induced effective tumor immunity.43 Within tumor-draining lymph nodes from melanoma patients, PD-L1:PD1 interactions are mainly established between PD1+ CD8+ T cells and PD-L1+ DCs, whereas PD-L1+ macrophages rarely interacted with T cells.44 Together with our findings described here, these data suggest that the functional significance of PD-L1 expression on TAMs might not be immunosuppressive against PD1+ T cells.
Histological quantification of PD-L1 within tumors remains challenging and provides limited prognostic and predictive value in clinical settings thus far.45 This may be due to heterogeneous expression of PD-L1 on different cell types within tumors, post-translational modifications of PD-L1, or different antibody platforms.46,47,48 PD-L1 expression on TAMs has been reported to associate with better response to immune checkpoint therapies,13,49 whereas PD-L1 expression on peripheral monocytes was associated with shorter survival.50 Our finding that PD-L1 could be rapidly upregulated during the monocyte-macrophage differentiation/maturation process independent of IFN-γ provides additional clues into the reported inconsistencies of intratumoral PD-L1 quantification as a biomarker.
Beyond cancer cells, TAMs are another major source of PD-L1 expression within tumors. It remains unclear what effects anti-PD-L1 immunotherapy has on PD-L1-expressing TAMs. TAMs exert their immunoregulatory roles mainly by secreting various soluble proteins including cytokines/chemokines, pro-angiogenic factors, and immunosuppressive factors.51,52 One recent study53 divided TAMs from human tumor into pro-tumor and anti-tumor subsets based on the expression of soluble factors SPP1 and CXCL9. Consistent with this notion, our ex vivo functional assays showed that PD-L1 blocking antibody did not suppress the T cell stimulatory effects of PD-L1+ monocytes/macrophages. Immunostimulatory functions of PD-L1+ TAMs do not depend on PD-L1-PD1 interactions, and thus anti-PD-L1 immunotherapy may not block the immunoregulatory functions of PD-L1+ TAMs.
In summary, our study provides insights into the functional and clinical significance of PD-L1 expression on TAMs in human breast tumors. Collective findings from our in-house and public scRNA-seq data, cell-cell spatial interaction analysis, and ex vivo functional assays reveal that PD-L1+ TAMs are immunostimulatory rather than immunosuppressive as commonly thought. As such, their presence reflects an immune active TIME and may explain why they predict response to anti-PD1/PD-L1 (ICI) therapy. Further studies are warranted to investigate the impact of anti-PD1/PD-L1 immunotherapies on the function of PD-L1+ TAMs in vivo.
Limitations of the study
A potential limitation of this study is the immunoregulatory roles of PD-L1+ TAMs in ICI immunotherapy remain unexplored. While our functional assays reveal that PD-L1+ TAMs are immunostimulatory and that PD-L1– TAMs are immunosuppressive, these ex vivo experiments do not fully recapitulate the complexities of the TIME. Future studies with longitudinal tumor samples before and after immunotherapy will provide further understanding of how ICIs may modulate PD-L1+ TAMs in mediating clinical efficacy. Another important but unanswered question is whether PD-L1+ and PD-L1– TAMs have similar roles within other human tumor types in addition to BC. Finally, the functional significances of PD-L1 expression on TAMs within primary vs. metastatic tumors need to be further explored.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-human CD3 (clone UCHT1) | BD Biosciences | Cat# 563546; RRID:AB_2744387 |
| Anti-human CD8 (clone SK1) | BD Biosciences | Cat# 561617, RRID:AB_10896281 |
| Anti-human CD16 (clone 3G8) | BD Biosciences | Cat# 563689; RRID:AB_2744299 |
| Anti-human CD33 (clone P67.6) | BD Biosciences | Cat# 333946; RRID:AB_399961 |
| Anti-human CD40 (clone 5C3) | BD Biosciences | Cat# 561211; RRID:AB_10584325 |
| Anti-human CD80 (clone L307.4) | BD Biosciences | Cat# 560926; RRID:AB_10565975 |
| Anti-human CD83 (clone HB15e) | BD Biosciences | Cat# 565336; RRID:AB_2739191 |
| Anti-human CD86 (clone 2331) | BD Biosciences | Cat# 563460; RRID:AB_2744455 |
| Anti-human CSF1R (clone 9-4D2-1E4) | BD Biosciences | Cat# 564945; RRID:AB_2739022 |
| Anti-human CXCR4 (clone 12G5) | BD Biosciences | Cat# 555976; RRID:AB_398616 |
| Anti-human B7-H4 (clone MIH43) | BD Biosciences | Cat# 562787; RRID:AB_2737794 |
| Anti-human VISTA (clone MIH65.rMab) | BD Biosciences | Cat# 566669; RRID:AB_2739762 |
| Anti-human IFNγR1(clone GIR208) | BD Biosciences | Cat# 558934; RRID:AB_397163 |
| Anti-human pSTAT1 (clone pY701; 4a) | BD Biosciences | Cat# 612596; RRID:AB_399879 |
| Anti-human pSTAT3 (clone pS727; 49/p-Stat3) | BD Biosciences | Cat# 558099; RRID:AB_397024 |
| Anti-human pSTAT5 (clone pY694; 47/Stat5) | BD Biosciences | Cat# 560117; RRID:AB_1645546 |
| Anti-human pmTOR (clone pS2448; O21-404) | BD Biosciences | Cat#563489; RRID:AB_2736872 |
| Anti-human pAkt (clone pS473; M89-61) | BD Biosciences | Cat# 560378; RRID:AB_1645328 |
| Anti-human PD-L1 (clone 29E.2A3) | BioLegend | Cat# 329714; RRID:AB_2563852 |
| Anti-human CD14 (clone HCD14) | BioLegend | Cat# 325617; RRID:AB_830690 |
| Anti-human PD-L2 (clone MIH18) | BioLegend | Cat# 345515; RRID:AB_2783233 |
| Anti-human B7-H3 (clone MIH42) | BioLegend | Cat# 351010; RRID:AB_2728323 |
| Anti-human CD45 (clone HI30) | BioLegend | Cat# 304043; RRID:AB_2562498 |
| Anti-human HLA-DR (clone LN3) | BioLegend | Cat# 327007; RRID:AB_893579 |
| Anti-human CD54 (clone HA58) | BioLegend | Cat# 353108; RRID:AB_10900254 |
| Anti-human CD69 (clone FN50) | BioLegend | Cat# 310932; RRID:AB_2563696 |
| Anti-human MRC1 (clone 15-2) | BioLegend | Cat# 321122; RRID:AB_10899411 |
| Anti-human CD163 (clone GHI/61) | BioLegend | Cat# 321122; RRID:AB_10899411 |
| Anti-human CD32 (clone FUN-2) | BioLegend | Cat# 303229; RRID:AB_2894576 |
| Anti-human CD64 (clone 10.1) | BioLegend | Cat# 305028; RRID:AB_2563822 |
| Anti-human CCR2 (clone K036C2) | BioLegend | Cat# 305028; RRID:AB_2563822 |
| Anti-human CCR5 (clone J418F1) | BioLegend | Cat# 359145; RRID:AB_3068173 |
| Anti-human PD-L1 (SP142) | Abcam | Cat# ab228462; RRID:AB_2827816 |
| Anti-human CD3 (clone LN10) | Biocare Medical | Cat#3152 |
| Anti-human CD8 (clone SP16) | Biocare Medical | Cat#CRM 311; RRID:AB_2750579 |
| Anti-human CD68 (clone KP1) | Biocare Medical | Cat#033; RRID:AB_2885063 |
| Anti-human Cytokeratin (clone AE1/AE3) | Biocare Medical | Cat#CM 011; RRID:AB_2811020 |
| LIVE/DEAD Fixable Blue | ThermoFisher | Cat#L23105 |
| Chemicals, peptides, and recombinant proteins | ||
| ERK1/2 inhibitor SCH772984 | Selleck | Cat# S7101 |
| STAT1 inhibitor Fludarabine | Selleck | Cat# S1491 |
| Akt1/2/3 inhibitor MK-2206 2HCl | Selleck | Cat# S1078 |
| PI3Kα/δ/β inhibitor LY294002 | Selleck | Cat# S1105 |
| NF-κB inhibitor QNZ | Selleck | Cat# S4902 |
| mTOR inhibitor Rapamycin | Selleck | Cat# S1039 |
| Critical commercial assays | ||
| pHrodo phagocytosis assay | ThermoFisher | Cat# P35366 |
| Deposited data | ||
| Single-Cell RNA-sequencing data – In-house dataset | This paper | GSE248288 |
| Software and algorithms | ||
| Flowjo v10 | Flowjo software | https://www.flowjo.com/ |
| Graphpad Prism v9 | Graphpad software | https://www.graphpad.com/ |
| EndNote X8 | Endnote software | https://endnote.com/ |
| BioRender | BioRender website | https://www.biorender.com/ |
| QuPath | QuPath software | https://doi.org/10.1038/s41598-017-17204-5 |
| R | R Core Team | https://www.R-project.org/ |
| Python | Python Software Foundation | http://www.python.org |
| Seurat | Stuart et al.54 | https://doi.org/10.1016/j.cell.2019.05.031 |
| icellnet | Noël et al.55 | https://doi.org/10.1038/s41467-021-21244-x |
| CellPhoneDB V2.0 | Efremova et al.56 | https://github.com/ventolab/CellphoneDB |
| Customized scRNA-seq analysis pipeline | This paper | https://doi.org/10.5281/zenodo.10472431 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Peter P. Lee (plee@coh.org).
Materials availability
This study did not generate new unique materials or reagents.
Data and code availability
-
•
The in-house scRNA-seq expression data can be obtained from Gene Expression Omnibus (GEO: GSE248288). All data reported in this paper is available from the lead contact upon request.
-
•
All analysis code has been deposited at Zenodo: https://doi.org/10.5281/zenodo.10472431 and is publicly available. DOIs are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
Experimental model and study participant details
Patient tumor and blood samples
Treatment naive female patients with breast cancer (age range 27–93) were selected for this study. Fresh breast tumors tissues were separated from fat tissues and minced into pieces up to 2 mm in diameter with scalpel blades. Single-cell suspensions were prepared by using the gentleMACS Dissociator (Miltenyl Biotec, Auburn, CA, USA) according to the standard protocol. Tissue homogenates were treated with 0.26 Wunsch U/ml Liberase and 10 U/ml DNase (Sigma-Aldrich, St. Louis, MO, USA) for up to 1 h as needed. The digested tissue homogenates were then filtered through a 100 μm filter. CD45+ cells were enriched with human CD45 positive selection kit (STEMCELL Technologies, Vancouver, BC, Canada). Peripheral blood mononuclear cells (PBMCs) were isolated from heparinized blood by Ficoll-Paque density centrifugation and cryopreserved in 10% DMSO FBS. PBMCs were obtained from patients prior to surgery or any therapy and only PBMC samples with cell viability ≥85% after thawing were selected.
Tissue microarray (TMA) of human breast tumor tissues (cohort #2) were purchased from Shanghai Outdo BioTech Company (Shanghai, China). The study was approved by the Ethics Committee of Shanghai Outdo BioTech Company.
This human study was approved by the Institutional Review Board of City of Hope Comprehensive Cancer Center. All patients had signed written informed consents.
Method details
scRNA-seq analysis of in-house data
Freshly prepared single-cell suspensions of digested tumor tissues were used and single-cell RNA sequencing was performed using the Chromium Single-Cell v2 3′ Chemistry Library, Gel Bead, Multiplex and Chip Kits (10x Genomics) according to the manufacturer’s protocol.
TAMs identification
CellRanger (v3.0) was used to align sequence reads to human genome (GRCh38) and count the aligned transcripts for each cell. The raw counts for each tumor sample were directly filtered, normalized, and scaled by Seurat R package (v3.2.3). Specifically, we first removed the genes detected by less than 3 cells and the cells with less than 5 non-zero expressing genes. Next, we calculated the percentage of all the counts belonging to mitochondrial genes for each cell. Lastly, we set 3 criteria based on Seurat official recommendation to finally remove the “dirty” cells/droplets: 1) potential empty droplets: cells who had less than 200 genes with non-zero expression, 2) potential doublets: cells who had more than 2500 genes with non-zero expression, 3) dying/dead cells: cells whose mitochondrial gene expression percentage larger than 20%.
The normalization was implemented by Seurat with default settings. The top 2,000 most variable protein-encoding genes were selected for principal component analysis. All the tumor samples were integrated with standard integration workflow in Seurat (i.e., “FindIntegrationAnchors” and “IntegrateData” functions in Seurat with default settings). PCA was implemented for the integrated data object. By using the two heuristic methods in Seurat (i.e., modified Jack Straw procedure and ranking variance method), the top 20 principal components were used for the further non-linearly dimensional reduction (i.e., UMAP) and unsupervised clustering analysis. The dimensional reduction and unsupervised clustering were implemented by Seurat with its default settings. Gene markers for each cluster were identified by “FindAllMarkers” function with default settings.
PD-L1/SIGLEC15 dichotomization
All the TAM cells were split by their original sample resources and the cell raw counts of TAMs were re-normalized and re-integrated with the same procedures described above. The top 18 principal components were used for the further non-linearly dimensional reduction (UMAP) and unsupervised clustering analysis. The clustering resolution was set as 0.5, which produced 12 clusters. The blending expression was generated by FeaturePlot function in Seurat with its default settings. Based on the blending expression pattern and clustering results on UMAP, clusters were annotated as PD-L1+/hi TAM or PD-L1-/lo TAM.
Transcriptomic profiling
The differential expression analysis was used to profile the transcriptomic features for PD-L1+/− TAMs, which was implemented by “FindMarkers” function with “MAST” method. The significantly differentially expressed genes (sDEGs) were defined as log2FC > 0.5 or < −0.5 and adjusted p value ≤ 0.10. These sDEGs were further used in gene set enrichment analysis (GSEA) with the pre-ranked mode, in which the log2FC was used as the ranking score. GSEA was implemented with GSEA software and hallmark gene sets in Molecular Signatures Database (v7.4).
Cell-cell interaction (CCI) analysis
CCI analysis was implemented by the cluster annotations for all the cell population in TME, which described above. The CellPhoneDB v2.0 was implemented through its Python package (https://github.com/Teichlab/cellphonedb) and the visualization of the results was implemented by customized R scripts. The ICELLNET was implemented through its R package (https://github.com/sourmelis-lab/ICELLNET) and similarly the result visualization was finished by customized R scripts.
scRNA-seq analysis of public data
Public single-cell RNA-seq data (i.e., raw counts) was downloaded from Gene Expression Omnibus (GSE114725, GSE161529) and from http://biokey.lambrechtslab.org. The raw counts were filtered, normalized, integrated, and scaled by Seurat R package (v4.1.0) with the exact same settings described above. By using the two heuristic methods in Seurat (i.e., modified Jack Straw procedure and ranking variance method), the top 30 principal components were used for the further non-linearly dimensional reduction (UMAP) and unsupervised clustering analysis. Similarly, the clusters with CD14+CD68+HLA-DRA+ cells were annotated as TAM cluster, which was further analyzed to identify the PD-L1+/− TAMs.
Again, the same analysis pipeline was used for the TAM clustering and PD-L1/SIGLEC15 dichotomization for public data. Generally, all the TAM cells were split by their original sample resources and the cell raw counts of TAMs were re-normalized and re-integrated with the same procedures described above. Based on the blending expression pattern and clustering results on UMAP, clusters were annotated as PD-L1+/hi TAM or PD-L1-/lo TAM. The differential expression analysis was used to profile the transcriptomic features for PD-L1+/− TAMs, which was implemented by “FindMarkers” function with “MAST” method. The significantly differentially expressed genes (sDEGs) were defined as log2FC > 0.5 or < −0.5 and adjusted p value ≤ 0.10.
Flow cytometry
The following antibodies were utilized: CD3 (UCHT1), CD8 (SK1), CD16 (3G8), CD33 (P67.6), CD40 (5C3), CD80 (L307.4), CD83 (HB5e), CD86 (2331), CSF1R (9-4D2-1E4), CXCR4 (12G5), B7-H4 (MIH43), VISTA (MIH65.rMab), IFNγR1(GIR208), pSTAT1 (pY701; 4a), pSTAT3 (pS727; 49/p-Stat3), pSTAT5 (pY694; 47/Stat5), pmTOR (pS2448; 021–404), pAkt (pS473; M89-61), PD-L1 (29E.2A3), PD-L2 (MIH18), B7-H3 (MIH42), CD14 (HCD14), CD45 (HI30), HLA-DR (LN3), CD54 (HA58), CD69 (FN50), MRC1 (15-2), CD163 (GHI/61), CD32 (FUN-2), CD64 (10.1), CCR2 (K036C2), CCR5 (J418F1), LIVE/DEAD Fixable Blue Dead Cell Stain.
For the phosphoflow cytometry, PBMCs were stimulated with IFNγ (Peprotech, Rocky Hills, NJ, USA) at 50 ng/mL at 37°C for 15 min followed by fixation with 1.5% paraformaldehyde (PFA) for 10 min at room temperature. Cells were washed with PBS to remove PFA, and permeabilized by the addition of 100% methanol. Flow cytometry was performed using Fortessa Flow Cytometers (BD Biosciences). Flow cytometry data was analyzed using FlowJo software (Tree Star Inc., Ashland, OR, USA.).
Multiplex immunofluorescence staining and image analysis
Formalin-fixed paraffin-embedded (FFPE) breast tumors tissues were cut into 3-μm sections and affixed to microscope slides. They were deparaffinized with xylene and rehydrated with decreasing concentrations of ethanol in water. Heat-induced epitope/antigen retrieval was performed in EnVision FLEX Target Retrieval Solution, High pH (pH 9) (K8004/5, Agilent, Santa Clara, CA, USA) or AR6 buffer (pH 6) (PerkinElmer, Hopkinton, MA, USA) using a microwave oven. Blocking was performed for 10 min using Antibody Diluent, Background Reducing (S3022, Agilent) to minimize non-specific background staining. Tissue slides were stained with the following primary antibodies for 1 h on a shaker at room temperature: CD3 (LN10), CD8 (SP16), CD68 (KP1), PD-L1 (SP142), Cytokeratin (AE1/AE3), and then detected by a horseradish peroxidase (HRP)-conjugated secondary antibody followed Opal fluorescence IHC Kit (PerkinElmer) at a 1:100 dilution following a 10 min incubation. To perform multicolor immunofluorescent staining, the slide would be serially stained with the microwave incubation acting to remove previous antibodies while simultaneously exposing the next epitope of interest. After staining the final marker, cell nuclei were stained with DAPI (PerkinElmer) and the slides were mounted with ProLong Gold Antifade Reagent (P36930, ThermoFisher, Waltham, MA, USA).
Stained sides were whole-slide scanned using the Vectra 3.0 System (PerkinElmer) which initially captured fluorescent spectra of 10x Olympus lens objective (10x) images in five channels (DAPI, FITC, Cy3, Texas Red, Cy5). Using Phenochart whole slide reviewer (PerkinElmer) regions of interest (ROIs) were selected and 25% of the images within the ROIs were systemically gridded and selected to unbiasedly capture tissue heterogeneity. The selected images were then captured at 20x Olympus lens objective (20x) using the Vectra in the same channels. Images of single-stained tissues and unstained tissues were used to extract the spectrum of each fluorophore and tissue autofluorescence in the 20x images and to create a spectral library to perform multispectral unmixing in inForm Cell Analysis (PerkinElmer).
The X and Y coordinates of the center of each cell’s nucleus were acquired by QuPath v0.3.2. The percentages of cells within 40 pixels (20 μm) to neighbor cells of a particular phenotype were determined by the K-nearest neighbor algorithm using R version 3.2.3.
Ex vivo functional assay
Phagocytosis assay
Cryopreserved PBMCs of patients with BC were thawed and rested for 16hrs in RMPI 1640 with 10% FBS. Then PBCSs were incubated with pHrodo Green E.Coli Bioparticles conjugate (ThermoFisher) for 3hrs according to the manufacturer’s protocol. The phagocytotic capacity of PD-L1+/− monocytes/macrophages (pHrodo green+%) were measured by flow cytometry after the incubation.
Autologous T cell proliferation
Freshly isolated PBMCs from patients with BC were rested for 16hrs in RMPI 1640 with 10% FBS and the PD-L1+/− monocytes were flow sorted from rested PBMCs. The autologous T cells were isolated using CD3 positive selection kit (STEMCELL Technologies) and labeled with CellTrace Violet (ThermoFisher). Then the sorted PD-L1+/− monocytes were co-cultured with T cells at 1:1 ratio for 4 days with or without PD-L1 blocking antibody (5 μg/ml) and the proliferation of CellTrace violet-labeled cells were measured by flow cytometry. The proliferation stimulation activity was calculated by the cell number ratio of (CD8/CD4+CD14)/(CD8/CD4) as the stimulatory index.
BiTE killing assay
Freshly isolated PBMCs from patients with BC were rested for 16hrs in RMPI 1640 with 10% FBS and the PD-L1+/− monocytes were flow sorted from rested PBMCs. The PD-L1+/− monocytes and autologous memory CD8+ T cells (CD8+CD45RA-) were flow sorted from rested PBMCs. The CD19+ K562 cancer cells, PD-L1+/− monocytes and CD8+ T cells were co-cultured at 1:1:1 ratio for 2 days in the presence of the CD19/CD3 bispecific antibody (BiTE/Blinatumomab, 5 μg/ml) and the killing ability of CD8+ T cells were measured by flow cytometry.
Quantification and statistical analysis
The statistical analyses were performed using software in R and Graphpad Prism (GraphPad Software, La Jolla, CA, USA). The details of the test significance computation were specified in the figure legends. All tests with p value < 0.05 were considered statistically significant.
Acknowledgments
This work was supported by Stand Up To Cancer (SU2C), the Breast Cancer Research Foundation (BCRF), the V Foundation, China National Natural Science Foundation (92374111 and 82173095), Shenzhen Natural Science Fund (JCYJ20210324094203010 and Stable Support Plan Program 20220810094017001), SZU Top Ranking Project funding (86000000210), Medical-Engineering Interdisciplinary Research Foundation, and Research Team Cultivation Program of Shenzhen University (2023QNT020). We thank Genergy Bio-technology Co. Ltd (Shanghai, China) for assisting with the scRNA-seq analyses. Research reported in this publication included work performed in the Analytical Cytometry Core and Pathology Research Services Core supported by National Cancer Institutes of Health under award number P30CA33572. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author contributions
Conceptualization, L.W., W.G., and P.P.L.; methodology, L.W., W.G., Z.G., J.Y., J.T., D.L.S., Q.Z., X.L., Y.Z., K.H., E.A.C., and J.W.; writing, L.W., W.G., and P.P.L.; funding acquisition, P.P.L. and L.W.
Declaration of interests
The authors declare no competing interests.
Published: February 20, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2024.101420.
Contributor Information
Lei Wang, Email: lei.wang@szu.edu.cn.
Peter P. Lee, Email: plee@coh.org.
Supplemental information
References
- 1.Ma R.Y., Black A., Qian B.Z. Macrophage diversity in cancer revisited in the era of single-cell omics. Trends Immunol. 2022;43:546–563. doi: 10.1016/j.it.2022.04.008. [DOI] [PubMed] [Google Scholar]
- 2.Mantovani A., Allavena P., Marchesi F., Garlanda C. Macrophages as tools and targets in cancer therapy. Nat. Rev. Drug Discov. 2022;21:799–820. doi: 10.1038/s41573-022-00520-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vesely M.D., Zhang T., Chen L. Resistance Mechanisms to Anti-PD Cancer Immunotherapy. Annu. Rev. Immunol. 2022;40:45–74. doi: 10.1146/annurev-immunol-070621-030155. [DOI] [PubMed] [Google Scholar]
- 4.Liu Y., Zugazagoitia J., Ahmed F.S., Henick B.S., Gettinger S.N., Herbst R.S., Schalper K.A., Rimm D.L. Immune Cell PD-L1 Colocalizes with Macrophages and Is Associated with Outcome in PD-1 Pathway Blockade Therapy. Clin. Cancer Res. 2020;26:970–977. doi: 10.1158/1078-0432.Ccr-19-1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lau J., Cheung J., Navarro A., Lianoglou S., Haley B., Totpal K., Sanders L., Koeppen H., Caplazi P., McBride J., et al. Tumour and host cell PD-L1 is required to mediate suppression of anti-tumour immunity in mice. Nat. Commun. 2017;8 doi: 10.1038/ncomms14572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tang H., Liang Y., Anders R.A., Taube J.M., Qiu X., Mulgaonkar A., Liu X., Harrington S.M., Guo J., Xin Y., et al. PD-L1 on host cells is essential for PD-L1 blockade-mediated tumor regression. J. Clin. Invest. 2018;128:580–588. doi: 10.1172/jci96061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin H., Wei S., Hurt E.M., Green M.D., Zhao L., Vatan L., Szeliga W., Herbst R., Harms P.W., Fecher L.A., et al. Host expression of PD-L1 determines efficacy of PD-L1 pathway blockade-mediated tumor regression. J. Clin. Invest. 2018;128:1708. doi: 10.1172/jci120803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gross D.J., Chintala N.K., Vaghjiani R.G., Grosser R., Tan K.S., Li X., Choe J., Li Y., Aly R.G., Emoto K., et al. Tumor and Tumor-Associated Macrophage Programmed Death-Ligand 1 Expression Is Associated With Adjuvant Chemotherapy Benefit in Lung Adenocarcinoma. J. Thorac. Oncol. 2022;17:89–102. doi: 10.1016/j.jtho.2021.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu C.Q., Xu J., Zhou Z.G., Jin L.L., Yu X.J., Xiao G., Lin J., Zhuang S.M., Zhang Y.J., Zheng L. Expression patterns of programmed death ligand 1 correlate with different microenvironments and patient prognosis in hepatocellular carcinoma. Br. J. Cancer. 2018;119:80–88. doi: 10.1038/s41416-018-0144-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang J., Browne L., Slapetova I., Shang F., Lee K., Lynch J., Beretov J., Whan R., Graham P.H., Millar E.K.A. Multiplexed immunofluorescence identifies high stromal CD68(+)PD-L1(+) macrophages as a predictor of improved survival in triple negative breast cancer. Sci. Rep. 2021;11 doi: 10.1038/s41598-021-01116-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Powles T., Eder J.P., Fine G.D., Braiteh F.S., Loriot Y., Cruz C., Bellmunt J., Burris H.A., Petrylak D.P., Teng S.L., et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature. 2014;515:558–562. doi: 10.1038/nature13904. [DOI] [PubMed] [Google Scholar]
- 12.Rosenberg J.E., Hoffman-Censits J., Powles T., van der Heijden M.S., Balar A.V., Necchi A., Dawson N., O'Donnell P.H., Balmanoukian A., Loriot Y., et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet. 2016;387:1909–1920. doi: 10.1016/s0140-6736(16)00561-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ahmed F.S., Gaule P., McGuire J., Patel K., Blenman K., Pusztai L., Rimm D.L. PD-L1 Protein Expression on Both Tumor Cells and Macrophages are Associated with Response to Neoadjuvant Durvalumab with Chemotherapy in Triple-negative Breast Cancer. Clin. Cancer Res. 2020;26:5456–5461. doi: 10.1158/1078-0432.Ccr-20-1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Azizi E., Carr A.J., Plitas G., Cornish A.E., Konopacki C., Prabhakaran S., Nainys J., Wu K., Kiseliovas V., Setty M., et al. Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell. 2018;174:1293–1308.e36. doi: 10.1016/j.cell.2018.05.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Q., He Y., Luo N., Patel S.J., Han Y., Gao R., Modak M., Carotta S., Haslinger C., Kind D., et al. Landscape and Dynamics of Single Immune Cells in Hepatocellular Carcinoma. Cell. 2019;179:829–845.e20. doi: 10.1016/j.cell.2019.10.003. [DOI] [PubMed] [Google Scholar]
- 16.Zhang L., Li Z., Skrzypczynska K.M., Fang Q., Zhang W., O'Brien S.A., He Y., Wang L., Zhang Q., Kim A., et al. Single-Cell Analyses Inform Mechanisms of Myeloid-Targeted Therapies in Colon Cancer. Cell. 2020;181:442–459.e29. doi: 10.1016/j.cell.2020.03.048. [DOI] [PubMed] [Google Scholar]
- 17.Sharma A., Seow J.J.W., Dutertre C.A., Pai R., Blériot C., Mishra A., Wong R.M.M., Singh G.S.N., Sudhagar S., Khalilnezhad S., et al. Onco-fetal Reprogramming of Endothelial Cells Drives Immunosuppressive Macrophages in Hepatocellular Carcinoma. Cell. 2020;183:377–394.e21. doi: 10.1016/j.cell.2020.08.040. [DOI] [PubMed] [Google Scholar]
- 18.Sun Y., Wu L., Zhong Y., Zhou K., Hou Y., Wang Z., Zhang Z., Xie J., Wang C., Chen D., et al. Single-cell landscape of the ecosystem in early-relapse hepatocellular carcinoma. Cell. 2021;184:404–421.e16. doi: 10.1016/j.cell.2020.11.041. [DOI] [PubMed] [Google Scholar]
- 19.Cheng S., Li Z., Gao R., Xing B., Gao Y., Yang Y., Qin S., Zhang L., Ouyang H., Du P., et al. A pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells. Cell. 2021;184:792–809.e23. doi: 10.1016/j.cell.2021.01.010. [DOI] [PubMed] [Google Scholar]
- 20.Wu S.Z., Al-Eryani G., Roden D.L., Junankar S., Harvey K., Andersson A., Thennavan A., Wang C., Torpy J.R., Bartonicek N., et al. A single-cell and spatially resolved atlas of human breast cancers. Nat. Genet. 2021;53:1334–1347. doi: 10.1038/s41588-021-00911-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kharchenko P.V., Silberstein L., Scadden D.T. Bayesian approach to single-cell differential expression analysis. Nat. Methods. 2014;11:740–742. doi: 10.1038/nmeth.2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mulder K., Patel A.A., Kong W.T., Piot C., Halitzki E., Dunsmore G., Khalilnezhad S., Irac S.E., Dubuisson A., Chevrier M., et al. Cross-tissue single-cell landscape of human monocytes and macrophages in health and disease. Immunity. 2021;54:1883–1900.e5. doi: 10.1016/j.immuni.2021.07.007. [DOI] [PubMed] [Google Scholar]
- 23.Singhal S., Stadanlick J., Annunziata M.J., Rao A.S., Bhojnagarwala P.S., O'Brien S., Moon E.K., Cantu E., Danet-Desnoyers G., Ra H.J., et al. Human tumor-associated monocytes/macrophages and their regulation of T cell responses in early-stage lung cancer. Sci. Transl. Med. 2019;11 doi: 10.1126/scitranslmed.aat1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang J., Sun J., Liu L.N., Flies D.B., Nie X., Toki M., Zhang J., Song C., Zarr M., Zhou X., et al. Siglec-15 as an immune suppressor and potential target for normalization cancer immunotherapy. Nat. Med. 2019;25:656–666. doi: 10.1038/s41591-019-0374-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martinez F.O., Gordon S., Locati M., Mantovani A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J. Immunol. 2006;177:7303–7311. doi: 10.4049/jimmunol.177.10.7303. [DOI] [PubMed] [Google Scholar]
- 26.Cassetta L., Fragkogianni S., Sims A.H., Swierczak A., Forrester L.M., Zhang H., Soong D.Y.H., Cotechini T., Anur P., Lin E.Y., et al. Human Tumor-Associated Macrophage and Monocyte Transcriptional Landscapes Reveal Cancer-Specific Reprogramming, Biomarkers, and Therapeutic Targets. Cancer Cell. 2019;35:588–602.e10. doi: 10.1016/j.ccell.2019.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pal B., Chen Y., Vaillant F., Capaldo B.D., Joyce R., Song X., Bryant V.L., Penington J.S., Di Stefano L., Tubau Ribera N., et al. A single-cell RNA expression atlas of normal, preneoplastic and tumorigenic states in the human breast. EMBO J. 2021;40 doi: 10.15252/embj.2020107333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bassez A., Vos H., Van Dyck L., Floris G., Arijs I., Desmedt C., Boeckx B., Vanden Bempt M., Nevelsteen I., Lambein K., et al. A single-cell map of intratumoral changes during anti-PD1 treatment of patients with breast cancer. Nat. Med. 2021;27:820–832. doi: 10.1038/s41591-021-01323-8. [DOI] [PubMed] [Google Scholar]
- 29.Muenst S., Schaerli A.R., Gao F., Däster S., Trella E., Droeser R.A., Muraro M.G., Zajac P., Zanetti R., Gillanders W.E., et al. Expression of programmed death ligand 1 (PD-L1) is associated with poor prognosis in human breast cancer. Breast Cancer Res. Treat. 2014;146:15–24. doi: 10.1007/s10549-014-2988-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chu Y., Dai E., Li Y., Han G., Pei G., Ingram D.R., Thakkar K., Qin J.J., Dang M., Le X., et al. Pan-cancer T cell atlas links a cellular stress response state to immunotherapy resistance. Nat. Med. 2023;29:1550–1562. doi: 10.1038/s41591-023-02371-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang L., Simons D.L., Lu X., Tu T.Y., Solomon S., Wang R., Rosario A., Avalos C., Schmolze D., Yim J., et al. Connecting blood and intratumoral Treg cell activity in predicting future relapse in breast cancer. Nat. Immunol. 2019;20:1220–1230. doi: 10.1038/s41590-019-0429-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cassetta L., Pollard J.W. A timeline of tumour-associated macrophage biology. Nat. Rev. Cancer. 2023;23:238–257. doi: 10.1038/s41568-022-00547-1. [DOI] [PubMed] [Google Scholar]
- 33.Kuang D.M., Zhao Q., Peng C., Xu J., Zhang J.P., Wu C., Zheng L. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J. Exp. Med. 2009;206:1327–1337. doi: 10.1084/jem.20082173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun C., Mezzadra R., Schumacher T.N. Regulation and Function of the PD-L1 Checkpoint. Immunity. 2018;48:434–452. doi: 10.1016/j.immuni.2018.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Engblom C., Pfirschke C., Pittet M.J. The role of myeloid cells in cancer therapies. Nat. Rev. Cancer. 2016;16:447–462. doi: 10.1038/nrc.2016.54. [DOI] [PubMed] [Google Scholar]
- 36.Mantovani A., Marchesi F., Malesci A., Laghi L., Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017;14:399–416. doi: 10.1038/nrclinonc.2016.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kloosterman D.J., Akkari L. Macrophages at the interface of the co-evolving cancer ecosystem. Cell. 2023;186:1627–1651. doi: 10.1016/j.cell.2023.02.020. [DOI] [PubMed] [Google Scholar]
- 38.Peng Q., Qiu X., Zhang Z., Zhang S., Zhang Y., Liang Y., Guo J., Peng H., Chen M., Fu Y.X., Tang H. PD-L1 on dendritic cells attenuates T cell activation and regulates response to immune checkpoint blockade. Nat. Commun. 2020;11:4835. doi: 10.1038/s41467-020-18570-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hartley G., Regan D., Guth A., Dow S. Regulation of PD-L1 expression on murine tumor-associated monocytes and macrophages by locally produced TNF-alpha. Cancer Immunol. Immunother. 2017;66:523–535. doi: 10.1007/s00262-017-1955-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tober J., Maijenburg M.M.W., Li Y., Gao L., Hadland B.K., Gao P., Minoura K., Bernstein I.D., Tan K., Speck N.A. Maturation of hematopoietic stem cells from prehematopoietic stem cells is accompanied by up-regulation of PD-L1. J. Exp. Med. 2018;215:645–659. doi: 10.1084/jem.20161594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chaudhri A., Xiao Y., Klee A.N., Wang X., Zhu B., Freeman G.J. PD-L1 Binds to B7-1 Only In Cis on the Same Cell Surface. Cancer Immunol. Res. 2018;6:921–929. doi: 10.1158/2326-6066.CIR-17-0316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhao Y., Lee C.K., Lin C.H., Gassen R.B., Xu X., Huang Z., Xiao C., Bonorino C., Lu L.F., Bui J.D., Hui E. PD-L1:CD80 Cis-Heterodimer Triggers the Co-stimulatory Receptor CD28 While Repressing the Inhibitory PD-1 and CTLA-4 Pathways. Immunity. 2019;51:1059–1073.e9. doi: 10.1016/j.immuni.2019.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oh S.A., Wu D.C., Cheung J., Navarro A., Xiong H., Cubas R., Totpal K., Chiu H., Wu Y., Comps-Agrar L., et al. PD-L1 expression by dendritic cells is a key regulator of T-cell immunity in cancer. Nat. Can. (Ott.) 2020;1:681–691. doi: 10.1038/s43018-020-0075-x. [DOI] [PubMed] [Google Scholar]
- 44.Dammeijer F., van Gulijk M., Mulder E.E., Lukkes M., Klaase L., van den Bosch T., van Nimwegen M., Lau S.P., Latupeirissa K., Schetters S., et al. The PD-1/PD-L1-Checkpoint Restrains T cell Immunity in Tumor-Draining Lymph Nodes. Cancer Cell. 2020;38:685–700.e8. doi: 10.1016/j.ccell.2020.09.001. [DOI] [PubMed] [Google Scholar]
- 45.Doroshow D.B., Bhalla S., Beasley M.B., Sholl L.M., Kerr K.M., Gnjatic S., Wistuba I.I., Rimm D.L., Tsao M.S., Hirsch F.R. PD-L1 as a biomarker of response to immune-checkpoint inhibitors. Nat. Rev. Clin. Oncol. 2021;18:345–362. doi: 10.1038/s41571-021-00473-5. [DOI] [PubMed] [Google Scholar]
- 46.McLaughlin J., Han G., Schalper K.A., Carvajal-Hausdorf D., Pelekanou V., Rehman J., Velcheti V., Herbst R., LoRusso P., Rimm D.L. Quantitative Assessment of the Heterogeneity of PD-L1 Expression in Non-Small-Cell Lung Cancer. JAMA Oncol. 2016;2:46–54. doi: 10.1001/jamaoncol.2015.3638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee H.H., Wang Y.N., Xia W., Chen C.H., Rau K.M., Ye L., Wei Y., Chou C.K., Wang S.C., Yan M., et al. Removal of N-Linked Glycosylation Enhances PD-L1 Detection and Predicts Anti-PD-1/PD-L1 Therapeutic Efficacy. Cancer Cell. 2019;36:168–178.e4. doi: 10.1016/j.ccell.2019.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rimm D.L., Han G., Taube J.M., Yi E.S., Bridge J.A., Flieder D.B., Homer R., West W.W., Wu H., Roden A.C., et al. A Prospective, Multi-institutional, Pathologist-Based Assessment of 4 Immunohistochemistry Assays for PD-L1 Expression in Non-Small Cell Lung Cancer. JAMA Oncol. 2017;3:1051–1058. doi: 10.1001/jamaoncol.2017.0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Madonna G., Ballesteros-Merino C., Feng Z., Bifulco C., Capone M., Giannarelli D., Mallardo D., Simeone E., Grimaldi A.M., Caracò C., et al. PD-L1 expression with immune-infiltrate evaluation and outcome prediction in melanoma patients treated with ipilimumab. OncoImmunology. 2018;7 doi: 10.1080/2162402X.2017.1405206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ando K., Hamada K., Shida M., Ohkuma R., Kubota Y., Horiike A., Matsui H., Ishiguro T., Hirasawa Y., Ariizumi H., et al. A high number of PD-L1(+) CD14(+) monocytes in peripheral blood is correlated with shorter survival in patients receiving immune checkpoint inhibitors. Cancer Immunol. Immunother. 2021;70:337–348. doi: 10.1007/s00262-020-02686-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pittet M.J., Michielin O., Migliorini D. Clinical relevance of tumour-associated macrophages. Nat. Rev. Clin. Oncol. 2022;19:402–421. doi: 10.1038/s41571-022-00620-6. [DOI] [PubMed] [Google Scholar]
- 52.Ren X., Zhang L., Zhang Y., Li Z., Siemers N., Zhang Z. Insights Gained from Single-Cell Analysis of Immune Cells in the Tumor Microenvironment. Annu. Rev. Immunol. 2021;39:583–609. doi: 10.1146/annurev-immunol-110519-071134. [DOI] [PubMed] [Google Scholar]
- 53.Bill R., Wirapati P., Messemaker M., Roh W., Zitti B., Duval F., Kiss M., Park J.C., Saal T.M., Hoelzl J., et al. CXCL9:SPP1 macrophage polarity identifies a network of cellular programs that control human cancers. Science (New York, N.Y.) 2023;381:515–524. doi: 10.1126/science.ade2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., 3rd, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive Integration of Single-Cell Data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Noël F., Massenet-Regad L., Carmi-Levy I., Cappuccio A., Grandclaudon M., Trichot C., Kieffer Y., Mechta-Grigoriou F., Soumelis V. Dissection of intercellular communication using the transcriptome-based framework ICELLNET. Nat. Commun. 2021;12:1089. doi: 10.1038/s41467-021-21244-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Efremova M., Vento-Tormo M., Teichmann S.A., Vento-Tormo R. CellPhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat. Protoc. 2020;15:1484–1506. doi: 10.1038/s41596-020-0292-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The in-house scRNA-seq expression data can be obtained from Gene Expression Omnibus (GEO: GSE248288). All data reported in this paper is available from the lead contact upon request.
-
•
All analysis code has been deposited at Zenodo: https://doi.org/10.5281/zenodo.10472431 and is publicly available. DOIs are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.